Настоящая заявка испрашивает приоритет по китайской патентной заявке 201911241526.8, поданной 6 декабря 2019 года, китайской патентной заявке 201911249226.4, поданной 9 декабря 2019 года, китайской патентной заявке 202010165349.6, поданной 11 марта 2020 года, китайской патентной заявке 202010799506.9, поданной 11 августа 2020 года, китайской патентной заявке 202011164541.X, поданной 27 октября 2020 года, и китайской патентной заявке 202011371550.6, поданной 30 ноября 2020 года, которые включены в настоящий документ посредством ссылки в полном объеме.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской химии и, в частности, к классу аналогов эхинокандина, которые могут быть использованы для борьбы с грибковыми инфекциями.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Разработка схем противогрибковой терапии является задачей, постоянно актуальной для современного общества. Доступные в настоящее время лекарственные средства для лечения грибковых инфекций включают амфотерицин B, макролидный полиен, который взаимодействует со стеролами мембран грибков; флуцитозин, фторпиримидин, который взаимодействует с белком грибка и биосинтезом ДНК; и различные азольные противогрибковые препараты, которые ингибируют биосинтез мембраных стеролов грибка (например, кетоконазол, итраконазол и флуконазол) (Alexander и др., Drugs, 54:657, 1997). Применение амфотерицина В ограничено инфузионными реакциями и почечной токсичностью, несмотря на то, что он обладает широким спектром активности и считается «золотым стандартом» в противогрибковой терапии (Warnock, J. Antimicrob. Chemother., 41:95, 1998). Применение флуцитозина также ограничено из-за развития устойчивых к лекарственным препаратам микроорганизмов и узкого спектра его активности. Широкое применение азольных противогрибковых препаратов приводит к возникновению устойчивых к лекарственным препаратам клинических штаммов видов Candida. Эхинокандины представляет собой новый класс противогрибковых препаратов. Как правило, они содержат циклический гексапептид и липофильный хвост, причем последний связан с ядром гексапептида амидной связью. Такие лекарственные средства препятствуют синтезу β-1,3-глюкозы в клеточных стенках грибов за счет неконкурентного ингибирования β-1,3-глюкозосинтазы, что приводит к изменению проницаемости клеточных стенок грибов и лизису и, следовательно, гибели клеток. Из-за отсутствия клеточных стенок в клетках человека и наличия клеточных стенок в клетках грибов противогрибковые препараты эхинокандина могут непосредственно воздействовать на компоненты клеточных стенок грибов, тем самым обладая низкой токсичностью для человека. Поэтому они до настоящего момента являются одними из самых безопасных противогрибковых препаратов.
В настоящее время такие лекарственные средства на рынке включают каспофунгин, микафунгин и анидулафунгин. Каспофунгин, первый противогрибковый препарат на основе эхинокандина, был разработан компанией Merck Sharp & Dohme, США, и одобрен USFDA (управление по контролю качества пищевых продуктов и лекарственных средств США) для лечения грибковых инфекций в 2004 году и одобрен для лечения инфекций Candida у детей в 2008 году. Микафунгин (Mycamine) представляет собой новый полусинтетический противогрибковый препарат, выпуск которого бьл начат в Японии в 2002 году. Анидулафунгин является полусинтетическим противогрибковым препаратом третьего поколения эхинокандинов, выпуск которого был начат в 2006 году.
WO2017049102A и WO2018102407A раскрывают противогрибковые средства на основе эхинокандина, показанные в виде формулы 1.

КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли или изомеру,
 ,
,
где R1 выбран из группы, состоящей из гидрокси, O(C(RA1)(RA2))a(C(RA3)(RA4))jX1, NH(C(RA1)(RA2))a(C(RA3)(RA4))jX1, O(CH2CH2O)bCH2CH2X1, O(CH2CH2CH2O)bCH2CH2X1, O(CH2CH2NH)bCH2CH2X1, NH(CH2CH2O)bCH2CH2X1, NH(CH2CH2NH)bCH2CH2X1, NH(CH2CH2CH2O)bCH2CH2X1, NH[(CH2(CH2)cO)]bCH{CH2[OCH2(CH2)c]dX1}2, O[(CH2(CH2)cO)]bCH{CH2[OCH2(CH2)c]dX1}2 и (OCH2CH2)b(NHCH2CH2)eX2;
R2 выбран из группы, состоящей из водорода, RB1RB2N-, CH2CH2NRB1RB2, CH2C(O)NRB1RB2, C1-10 низшего алкила, C2-10 алкенила, C2-10 алкинила, арила, гетероарила, циклогидрокарбила, гетероциклила и PEG (полиэтиленгликоль);
R3 выбран из группы, состоящей из H, OSO3H и CH2NRB1RB2;
G представляет собой C10-42 жирный фрагмент;
RA1, RA2, RA3 и RA4 независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, низшего алкила, циклогидрокарбила и циклогидрокарбилена ( );
);
RB1 и RB2 независимо выбраны из группы, состоящей из H, -C(O)RJ и низшего алкила;
X1 независимо представляет собой N(RC1RC2RC3) или структуру  ,
,
где кольцо A представляет собой необязательно замещенное, насыщенное или ненасыщенное, моноциклическое или конденсированное кольцо, содержащее один или более атомов N;
RC1, RC2 и RC3 независимо выбраны из группы, состоящей из H, C1-6 алкила, C1-6 низшего галогеналкила и дейтерированного C1-6 низшего алкила, и по меньшей мере один из RC1, RC2 и RC3 не является водородом;
каждый RF независимо выбран из группы, состоящей из H, дейтерия, гидрокси, гидроксиалкила, амино, алкокси, низшего алкила, алкенила, алкинила, галогена, SR', SOR', SO2R', NR'(R''), COOR' и CONR'(R''), где низший алкил необязательно замещен заместителем, замещенным одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, циклоалкила, алкокси, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, нитрильных групп, гидрокси, галогена, SR', NR'(R''), COOR' и CONR'(R');
X2 представляет собой N(RD1RD2RD3) или структуру X1;
RD1, RD2 и RD3 независимо выбраны из группы, состоящей из H, C1-6 низшего алкила, C1-6 низшего галогеналкила и дейтерированного C1-6 низшего алкила;
R' и R'' независимо выбраны из группы, состоящей из водорода, гидрокси, алкила, алкокси, алкенила и -C(O)RJ;
RJ выбран из группы, состоящей из водорода, дейтерия, C1-10 низшего алкила, циклогидрокарбила и циклогидрокарбилена;
a представляет собой целое число от 0 до 5;
b представляет собой целое число от 1 до 5;
c представляет собой целое число от 1 до 2;
d представляет собой целое число от 0 до 3;
e представляет собой целое число от 1 до 5;
k представляет собой целое число от 0 до 20;
j представляет собой целое число от 0 до 5 и
n представляет собой целое число от 1 до 7.
"Независимо выбранный из группы, состоящей из" или "независимо представляет собой/представляют собой" означает, что вариабельные группы в каждом случае независимо выбраны из группы, состоящей из определенных заместителей.
В некоторых вариантах осуществления R1 может быть выбран из группы, состоящей из O(C(RA1)(RA2))a(C(RA3)(RA4))jX1, NH(C(RA1)(RA2))a(C(RA3)(RA4))jX1, O(CH2CH2O)bCH2CH2X1, O(CH2CH2CH2O)bCH2CH2X1, O(CH2CH2NH)bCH2CH2X1, NH(CH2CH2O)bCH2CH2X1, NH(CH2CH2NH)bCH2CH2X1, NH(CH2CH2CH2O)bCH2CH2X1, NH[(CH2(CH2)cO)]bCH{CH2[OCH2(CH2)c]dX1}2, O[(CH2(CH2)cO)]bCH{CH2[OCH2(CH2)c]dX1}2 и (OCH2CH2)b(NHCH2CH2)eX2;
R2 может быть выбран из группы, состоящей из водорода, RB1RB2N-, CH2CH2NRB1RB2, CH2C(O)NRB1RB2, C1-10 низшего алкила, C2-10 алкенила, C2-10 алкинила, арила, гетероарила, циклогидрокарбила, гетероциклила и PEG;
R3 может быть выбран из группы, состоящей из H, OSO3H и CH2NRB1RB2;
G может представлять собой C10-36 липофильный фрагмент;
RA1, RA2, RA3 и RA4 независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, низшего алкила, циклогидрокарбила и циклогидрокарбилена ();
RB1 и RB2 каждый независимо выбран из группы, состоящей из H, -C(O)RJ и низшего алкила;
X1 независимо представляет собой N (RC1RC2RC3) или структуру ,
где кольцо A представляет собой необязательно замещенное, насыщенное или ненасыщенное, моноциклическое или конденсированное кольцо, содержащее один или более атомов N;
RC1, RC2 и RC3 каждый независимо выбран из группы, состоящей из H, C1-6 низшего галогеналкила и дейтерированного C1-6 низшего алкила, и по меньшей мере один из RC1, RC2 и RC3 не является водородом;
каждый RF независимо выбран из группы, состоящей из H, дейтерия, гидрокси, гидроксиалкила, амино, алкокси, низшего алкила, алкенила, алкинила, галогена, SR', SOR', SO2R ', NR'(R''), COOR' и CONR'(R''), где низший алкил может быть необязательно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, циклоалкила, алкокси, гидроксиалкила, алкенила и алкинила;
X2 представляет собой N (RD1RD2RD3) или структуру X1;
RD1, RD2 и RD3 независимо выбраны из группы, состоящей из H, C1-6 низшего алкила, C1-6 низшего галогеналкила и дейтерированного C1-6 низшего алкила;
R' и R'' независимо выбраны из группы, состоящей из водорода, гидрокси, алкила, алкокси, алкенила и -C(O)RJ;
RJ независимо выбран из группы, состоящей из водорода, C1-10 низшего алкила, циклогидрокарбила и циклогидрокарбилена;
a представляет собой целое число от 0 до 5;
b представляет собой целое число от 1 до 5;
c представляет собой целое число от 1 до 2;
d представляет собой целое число от 0 до 3;
e представляет собой целое число от 1 до 5;
k представляет собой целое число от 0 до 20;
j независимо представляет собой целое число от 0 до 5 и
n представляет собой целое число от 1 до 7.
В некоторых вариантах осуществления X1 может быть выбран из группы, состоящей из следующих структур:
 ,
,  ,
,  и
и  ,
,
где каждый RF независимо выбран из группы, состоящей из H, дейтерия, гидрокси, гидроксиалкила, амино, алкокси, низшего алкила, алкенила, алкинила, галогена, SR', SOR', SO2R ', NR'(R''), COOR' и CONR'(R''), где низший алкил может быть необязательно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, циклоалкила, алкокси, гидроксиалкила, алкенила и алкинила;
Rq1 и Rq2 могут быть независимо H или C1-6 низшим алкилом, необязательно замещенным одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, циклоалкила, алкокси, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, нитрильных групп, гидрокси, галогена, SR', NR'(R''), COOR' и CONR'(R'');
R' и R'' независимо выбраны из группы, состоящей из водорода, гидрокси, алкила, алкокси, алкенила и -C(O)RJ;
RJ выбран из группы, состоящей из водорода, дейтерия, C1-10 низшего алкила, циклогидрокарбила и циклогидрокарбилена;
f представляет собой целое число от 0 до 16;
g представляет собой целое число от 0 до 16;
h представляет собой целое число от 0 до 9;
i представляет собой целое число от 0 до 4;
n представляет собой целое число от 1 до 7 и
p представляет собой целое число от 1 до 3.
В некоторых вариантах осуществления G может быть выбран из группы, состоящей из
 ,
,  и
и  ,
,
где X независимо выбран из группы, состоящей из O, C(RB1) (RB2), NRp4- и S; RT может быть C1-5 линейным или разветвленным алкилом, где алкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, галогена, алкила, циклогидрокарбила и циклогидрокарбилена ();
Rp1, Rp2 и Rp3, в каждом случае, независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, C1-10 низшего алкила, C1-10 низшего галогеналкила, C2-10 алкенила, C2-10 алкинила, арила, гетероарила, циклогидрокарбила, гетероциклила и PEG; m представляет собой целое число от 0 до 4; n представляет собой целое число от 1 до 7; Rp4 представляет собой водород или C1-6 низший алкил; RB1 и RB2 каждый независимо выбран из группы, состоящей из H, -C(O) и C1-10 алкила, где RJ представляет собой группу, состоящую из водорода, дейтерия, C1-10 низшего алкила, циклогидрокарбила и циклогидрокарбилена.
В некоторых вариантах осуществления G может быть выбран из группы, состоящей из:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
В некоторых вариантах осуществления R1 может быть выбран из группы, состоящей из:
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
В других вариантах осуществления R1 может быть выбран из группы, состоящей из:
 и
и  .
.
Настоящее изобретение относится к соединению формулы II или его фармацевтически приемлемой соли или изомеру,
 ,
,
G1 может быть выбран из группы, состоящей из
, и ,
где Rp1, Rp2 и Rp3 независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, C1-6 низшего алкила, C1-6 низшего галогеналкила, C2-10 алкенила, C2-10 алкинила, арила, гетероарила, циклогидрокарбила, гетероциклила и PEG;
X независимо выбран из группы, состоящей из O, C(RB1)(RB2), NRp4- и S;
Rp4 представляет собой водород или C1-3 низший алкил;
RT может быть C1-5 линейным или разветвленным алкилом, где алкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, гидрокси, амино, алкокси, амино, NR'(R''), галогена, циклогидрокарбила и циклогидрокарбилена
(),
и когда Rp1, Rp2 и Rp3 все представляют собой H и X представляет собой O, RT не является -C5H11; когда X представляет собой O и RT представляет собой -C5H11, по меньшей мере один из Rp1, Rp2 и Rp3 не является H; когда RT представляет собой -C5H11 и Rp1, Rp2 и Rp3 все представляют собой H, X не является O;
RB1 и RB2 независимо выбраны из группы, состоящей из H, -C(O)RJ и C1-10 низшего алкила;
RJ выбран из группы, состоящей из водорода, C1-10 низшего алкила, циклогидрокарбила и циклогидрокарбилена;
m представляет собой целое число от 0 до 4 и
n представляет собой целое число от 1 до 7.
Настоящее изобретение также относится к соединению формулы III или его фармацевтически приемлемой соли или изомеру

где RG1, RG2, RG3 и RG4 независимо выбраны из группы, состоящей из водорода, дейтерия, галогена и низшего алкила, и по меньшей мере один из RG1, RG2, RG3 и RG4 не является водородом.
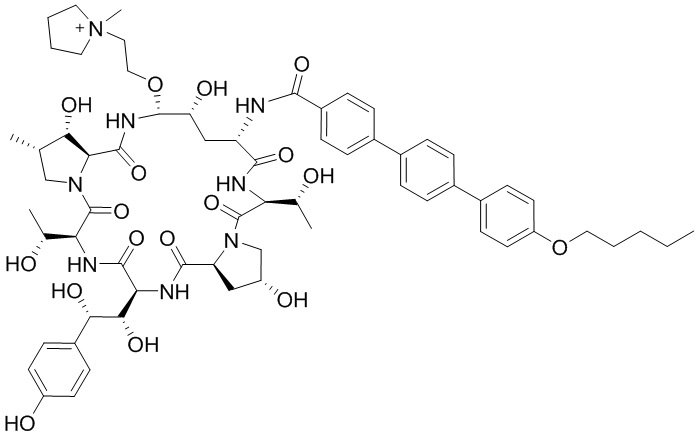
Настоящее изобретение относится к следующим соединениям или их фармацевтически приемлемым солям или изомерам,
 ,
,  ,
,  ,
, 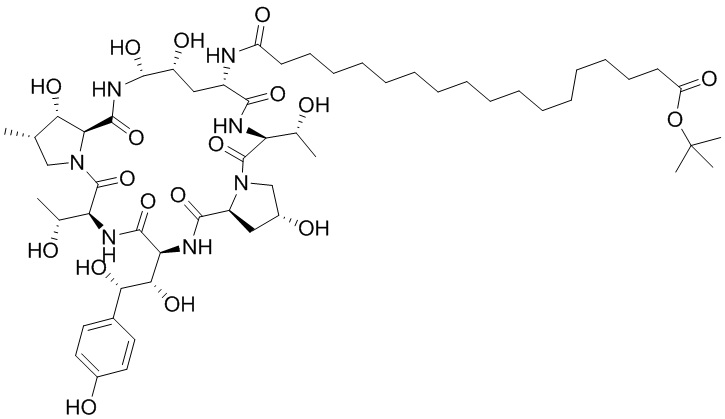 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и 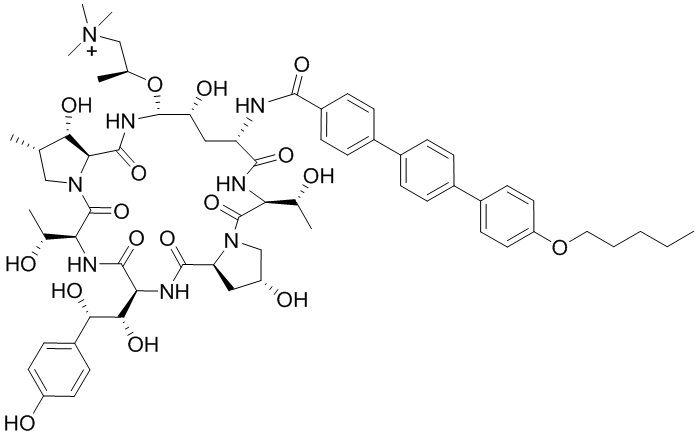 .
.
В некоторых вариантах осуществления фармацевтически приемлемая соль соединения выбрана из группы, состоящей из ацетатной соли, трифторацетатной соли и формиатной соли.
Настоящее изобретение также относится к способу получения соединения или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
В настоящем изобретении также предложен способ лечения грибковой инфекции у пациента путем введения пациенту количества фармацевтической композиции по настоящему изобретению, достаточного для лечения инфекции. В конкретных вариантах осуществления фармацевтическую композицию вводят внутривенно, местно или перорально. Фармацевтическая композиция может быть введена для лечения инфекции кровотока, инфекции ткани (например, инфекции легких, почек или печени) или других типов инфекции у пациента. Грибковая инфекция, подлежащая лечению, может представлять собой инфекцию, выбранную из группы, состоящей из: дерматомикоза головы, дерматомикоза туловища, дерматомикоза стоп, дерматомикоза ногтей, перионихомикоза, отрубевидного лишая, кандидоза, вагинального кандидоза, респираторного кандидоза, кандидоза желчевыводящих путей, кандидоза пищевода, кандидоза уретры, системный кандидоз, кандидоз слизистых оболочек и кожи, аспергиллез, мукормикоз, паракокцидиоидомикоз, североамериканский бластомикоз, гистоплазмоз, кокцидиоидомикоз, споротрихоз, грибковый синусит и хронический параназальный синусит. В некоторых вариантах осуществления инфекция, подлежащая лечению, представляет собой инфекцию, вызванную Candida albicans, C. parapsilosis, C. glabrata, C. guilliermondii, C. krusei, C. lusitaniae, C. tropicalis, Aspergillus fumigatus, A. flavus, A. terreus, A. niger, A. candidus, A. clavatus или A. ochraceus.
В настоящем изобретении также предложен способ предотвращения грибковой инфекции у пациента путем введения пациенту количества фармацевтической композиции по настоящему изобретению, достаточного для предотвращения инфекции. Например, способ по настоящему изобретению может быть использован для профилактического лечения пациентов, которые готовятся к инвазивным медицинским процедурам (например, пациентов, которые готовятся к операции, такой как трансплантация, терапия стволовыми клетками, трансплантация, реконструктивная хирургия или долгосрочные или частые внутривенные катетеризации, или для лечения в отделениях интенсивной терапии), пациентов со сниженным иммунитетом (например, у пациентов, которые имеют рак или ВИЧ/СПИД, или принимают иммунодепрессанты), или пациентов, которые получают долгосрочную антибиотикотерапию.
В одном конкретном варианте осуществления способа по настоящему изобретению фармацевтическая композиция содержит соединение 1 или любое другое соединение, описанное в настоящем документе, или его фармацевтически приемлемую соль.
Настоящее изобретение также относится к способу предотвращения, стабилизации или ингибирования роста грибков или уничтожения грибков путем приведения в контакт грибков или участка, благоприятного для роста грибков, с соединением по настоящему изобретению или его фармацевтически приемлемой солью.
Термин "достаточное количество" относится к количеству лекарственного средства, необходимому для лечения или предотвращения инфекции. Достаточное количество для осуществления настоящего изобретения для терапевтического или профилактического лечения состояния, вызванного или опосредованного инфекцией, варьируется в зависимости от пути введения, типа инфекции, а также возраста, веса и общего состояния здоровья пациента.
Под "грибковой инфекцией" подразумевается, что патогенные грибки поражают хозяина. Например, инфекция может включать чрезмерный рост грибков, которые обычно обнаруживаются у пациента или на коже пациента, или рост грибков, которые обычно не обнаруживаются у пациента или на коже пациента. В более общем плане грибковая инфекция может быть любым случаем, в котором присутствие грибковой популяции наносит вред организму-хозяину. Таким образом, пациент «поражен» грибковой инфекцией, когда избыток популяции грибков присутствует у пациента или на коже пациента, или когда популяция грибков вызывает повреждение клеток или других тканей пациента.
Термин "лечение" относится к введению фармацевтической композиции в профилактических и/или терапевтических целях. Термин "предотвращение заболевания" означает профилактическое лечение субъекта, у которого еще не развилось заболевание, но который является восприимчивым к конкретному заболеванию или подвержен риску его развития. Под "лечением заболевания" подразумевается лечение пациента, страдающего от заболевания, для улучшения или стабилизации состояния пациента.
Настоящее изобретение также относится к применению соединения или его фармацевтически приемлемой соли для получения лекарственного средства для лечения грибковых инфекций.
Настоящее изобретение также относится к применению соединения или его фармацевтически приемлемой соли для получения лекарственного средства для предотвращения грибковых инфекций.
Настоящее изобретение также относится к применению соединения или его фармацевтически приемлемой соли для получения лекарственного средства для предотвращения, стабилизации или ингибирования роста грибков или уничтожения грибков. В некоторых вариантах осуществления изобретения применение включает приведение в контакт грибков или участка, благоприятного для роста грибков, с соединением или его фармацевтически приемлемой солью или изомером.
Соединения по настоящему изобретению могут быть синтезированы, например, путем приведения в реакцию соединений эхинокандина с соответствующими ацильными, алкильными, карбоксильными, гидрокси- и/или аминогруппами в стандартных условиях реакции, как описано в примерах.
Для полусинтетического способа получения раскрытых соединений, стереохимия соединений будет определяться исходными материалами. Таким образом, производные эхинокандина, не встречающиеся в природе, как правило, по стереохимии идентичны встречающимся в природе структурам эхинокандина, из которых они получены (репрезентативная стереохимия описана в примерах).
Соединения по настоящему изобретению могут быть синтезированы, например, с использованием способов, описанных в примерах.
Если не указано иное, следующие термины, используемые в описании и формуле изобретения, имеют приведенные ниже значения.
"Алкил" относится к линейной или разветвленной алкановой группе, содержащей предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода; неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, гексил, гептил, октил, нонил, децил и т.д. Если в описании специально не указано иное, алкил может быть необязательно замещен одним или более из следующих заместителей: галогеном, циано, нитро, оксо, тио, триметилсилилом и т.д. Если не указано иное, "низший алкил" относится к линейной или разветвленной алкановой группе, содержащей от 1 до 10 атомов углерода; неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, гексил, гептил, октил, нонил, децил и т.д. Если в описании специально не указано иное, низший алкил может быть необязательно замещен одним или более из следующих заместителей: галогеном, циано, нитро, оксо, тио, триметилсилилом и т.д.
"Алкенил" относится к алкильному соединению, содержащему углерод-углеродные двойные связи в молекуле, где алкил является таким, как определено выше. Неограничивающие примеры включают винил, 1-пропен-2-ил, 1-бутен-4-ил, 1-пентен-5-ил, 1-бутен-1-ил и т.д. Если в описании не указано иное, алкенил может быть необязательно замещен одним или более из следующих заместителей: галогеном, циано, нитро, оксо, тио, триметилсилилом и т.д.
"Алкинил" относится к алкильному соединению, содержащему углерод-углеродные тройные связи в молекуле, где алкил является таким, как определено выше. Неограничивающие примеры включают этинил, пропинил, пентинил, бутинил и т.д. Если в описании не указано иное, алкинил может быть необязательно замещен одним или более из следующих заместителей: галогеном, циано, нитро, оксо, тио, триметилсилилом и т.д.
"C10-36 липофильный фрагмент" относится к замещенному или незамещенному алкилу, замещенному или незамещенному алкенилу, замещенному или незамещенному алкинилу, замещенному или незамещенному циклогидрокарбилу, замещенному или незамещенному арилу, замещенному или незамещенному гетероарилу, замещенному или незамещенному гетероциклилу и т.д., содержащему 10-36 атомов углерода.
"Арил" относится к группе углеводородной кольцевой системы, содержащей атомы водорода, от 6 до 14 атомов углерода и по меньшей мере одно ароматическое кольцо. Он может представлять собой моноциклическую, бициклическую или трициклическую кольцевую систему и может включать спирокольцевую систему. Арильные группы включают, но не ограничиваются ими, группы, полученные из аценафтена, антрацена, азулена, бензола, 6,7,8,9-тетрагидро-5H-бензо [7]аннулена, флуорена, индена, нафталина, феналена и фенантрена. Если в описании специально не указано иное, арил может быть необязательно замещен одним или более заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, циано, нитро и т.д.
"Циклогидрокарбил" относится к стабильной неароматической моноциклической или полициклической гидрокарбильной группе, состоящей только из атомов углерода и водорода, которая может содержать спиро- или мостиковую кольцевую систему и содержит от 3 до 15 атомов углерода, от 3 до 10 атомов углерода или от 5 до 7 атомов углерода; она является насыщенной или ненасыщенной и связана с остальной частью молекулы одной связью. Моноциклические циклогидрокарбильные группы включают немостиковые циклогидрокарбильные группы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Полициклические группы включают конденсированные, спиро- или мостиковые циклогидрокарбильные группы, например, группы C10, такие как адамантил (мостиковый) и декалинил (конденсированный), и группы C7, такие как бицикло[3.2.0]гептил (конденсированный), норборнил и норборненил (мостиковый); и замещенные полициклические группы, например, замещенные группы C7, такие как 7,7-диметилбицикло[2.2.1]гептил (мостиковый). Если в описании специально не указано иное, циклогидрокарбил может быть необязательно замещен одним или более заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, оксо, тио, циано, нитро и т.д.
"Циклоалкил" относится к насыщенному моноциклическому или полициклическому углеводородному заместителю, содержащему от 3 до 15 атомов углерода, от 3 до 10 атомов углерода или от 5 до 7 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.д.; полициклические циклоалкильные группы включают спиро, конденсированные и мостиковые циклоалкильные группы.
"Галоген" означает бром, хлор, фтор или йод.
"Гетероциклил" относится к стабильной 3-18-членной неароматической кольцевой группе, содержащей от 1 до 12 атомов углерода и от 1 до 6 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. Если специально не указано иное в описании, гетероциклил может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую систему и может включать спиро- или мостиковые кольцевые системы; атомы азота, углерода или серы в гетероциклиле необязательно могут быть окислены; атомы азота необязательно могут быть кватернизированы; и гетероциклил может быть частично или полностью насыщен. Если в описании специально не указано иное, гетероциклильные группы включают те, которые необязательно замещены одним или более заместителями, выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, оксо, тио, циано, нитро и т.д.
"Гетероарил" относится к 5-14-членной группе кольцевой системы, содержащей атомы водорода, от 1 до 13 атомов углерода, от 1 до 6 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и по меньшей мере одно ароматическое кольцо. Гетероарил может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую систему и может включать спирокольцевые системы; атомы азота, углерода или серы в гетероариле необязательно могут быть окислены; и атомы азота необязательно могут быть кватернизированы. Ароматическое кольцо гетероарила необязательно содержит гетероатомы, при условии, что одно кольцо гетероарила содержит гетероатомы. Например, 1,2,3,4-тетрагидроизохинолин-7-ил считается «гетероарильной» группой. Если в описании специально не указано иное, гетероарильные группы включают те, которые необязательно замещены одним или более заместителями, выбранными из группы, состоящей из алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, оксо, тио, циано, нитро и т.д.
Термин "PEG" относится к полиэтиленгликолю, и, если не указано иное, "PEG" включает этиленоксидные полимеры любой длины. PEG также может быть необязательно замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, алкила, алкенила, алкинила, галогена, галогеналкила, галогеналкенила, галогеналкинила, оксо, тио, циано, нитро и т.д.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 показаны изменения концентрации гистамина после внутривенного введения соединений.
На фиг. 2 показано сравнение концентраций гистамина через 30 мин после внутривенного введения соединений.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение дополнительно описано ниже со ссылкой на примеры, но эти примеры не предназначены для ограничения объема настоящего изобретения.
Экспериментальные процедуры без конкретных условий, указанных в следующих примерах, как правило, проводят в соответствии с общепринятыми условиями или в соответствии с условиями, рекомендованными производителями исходных материалов или коммерческих продуктов. Реагенты без указания конкретного происхождения являются коммерчески доступными обычными реагентами.
Анидулафунгин и каспофунгин были приобретены у компании Taizhou KEDE Chemical. Резафунгин синтезировали в соответствии с CN103889221A.
Способ анализа чистоты методом HPLC (высокоэффективная жидкостная хроматография):
Программа градиента:
Способ анализа LC-MS (жидкостная хроматография с масс-спектрометрией):
Программа градиента
Пример 1:
Этап 1

Соединение SM1 (510 мг, 5,04 ммоль) растворяли в ацетоне (5,1 мл) и по каплям добавляли метил-п-толуолсульфонат (938 мг, 5,04 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 ч, и белое твердое вещество выпадало в осадок. Реакционную смесь фильтровали, и осадок на фильтре сушили in vacuo с получением соединения SM2 (385 мг, 98% чистоты, 26,7% выхода). MS (масспектрометрия): 116,1[M+].
Этап 2

Анидулафунгин (114 мг, 0,1 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота и добавляли фенилбороновую кислоту (24 мг, 0,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 10 мл ацетонитрила с последующим добавлением соединения SM2 (170 мг, 0,6 ммоль) и п-толуолсульфоновой кислоты (86 мг, 0,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (79 мг, чистота 97,6%, выход 60,8%). HRMS (масс-спектрометрия высокого разрешения): 1237,6021[M+].
1H ЯМР (ядерный магнитный резонанс) (400 МГц, METHANOL (метанол)-d4) δ 7,98-8,01 (m, 2H), 7,82 (d, J = 8,3 Гц, 2H), 7,71-7,78 (m, 4H), 7,63 (d, J = 8,8 Гц, 2H), 7,17 (d, J = 8,6 Гц, 2H), 7,03 (d, J = 8,8 Гц, 2H), 6,79 (d, J = 8,6 Гц, 2H), 5,40-5,42 (m, 1H), 5,00-5,05 (m, 1H), 4,58-4,80 (m, 7H), 4,41 (d, J = 4,4 Гц, 1H), 4,34-4,36 (m, 2H), 4,25-4,32 (m, 2H), 3,97-4,24 (m, 8H), 3,89-3,96 (m, 1H), 3,82-3,87 (m, 1H), 3,48-3,53 (m, 1H), 3,12-3,17 (m, 6H), 2,45-2,57 (m, 4H), 2,27-2,35 (m, 1H), 2,05-2,15 (m, 2H), 1,79-1,87 (m, 5H), 1,40-1,53 (m, 4H), 1,26-1,32 (m, 6H), 1,10 (d, J = 6,8 Гц, 3H), 0,99 (t, J = 7,2 Гц, 3H).
Пример 2:
Этап 1

Соединение SM3 (1,29 г, 10 ммоль) растворяли в ацетоне (13 мл) и по каплям добавляли йодметан (1,42 г, 10 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 ч, и белое твердое вещество выпадало в осадок. Реакционную смесь фильтровали, и осадок на фильтре сушили in vacuo с получением соединения SM4 (2,54 г, чистота 98% , выход 89,2%). MS: 144,1[M+].
Этап 2

Анидулафунгин (114 мг, 0,1 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота и добавляли фенилбороновую кислоту (24 мг, 0,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 10 мл ацетонитрила с последующим добавлением соединения SM4 (171 мг, 0,6 ммоль) и D- (+) -камфорсульфоновой кислоты (120 мг, 0,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (61 мг, чистота 97,0% , выход 46,2%). HRMS: 1265,6327[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 8,01 (d, J = 8,3 Гц, 2H), 7,83 (dd, J = 0,49, 8,3 Гц, 2H), 7,71-7,79 (m, 4H), 7,63 (d, J = 8,80 Гц, 2H), 7,17 (d, J = 8,6 Гц, 2H), 7,03 (d, J = 8,80 Гц, 2H), 6,79 (d, J = 8,6 Гц, 2H), 5,34-5,41 (m, 1H), 5,00-5,05 (m, 1H), 4,58-4,80 (m, 7H), 4,40 (d, J = 4,4 Гц, 1H), 4,32-4,38 (m, 2H), 4,25-4,32 (m, 2H), 4,18-4,22 (m, 1H), 4,15-4,24 (m, 1H), 3,81-4,15 (m, 8H), 3,43-3,52 (m, 2H), 3,21 (d, J = 6,6 Гц, 3H), 3,04 (d, J = 6,6 Гц, 3H), 2,42-2,59 (m, 2H), 2,24-2,36 (m, 1H), 2,05-2,12 (m, 2H), 1,80-1,88 (m, 9H), 1,42-1,53 (m, 5H), 1,28 (d, J = 6,4 Гц, 6H), 1,10 (d, J = 6,9 Гц, 3H), 0,99 (t, J = 7,1 Гц, 3H).
Пример 3:

Анидулафунгин (114 мг, 0,1 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота и добавляли фенилбороновую кислоту (24 мг, 0,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 10 мл диоксана с последующим добавлением соединения SM5 (73 мг, 0,6 ммоль) и п-толуолсульфоновой кислоты (86 мг, 0,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (68 мг, чистота 97,8% , выход 52,3%). HRMS: 1243,5924[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,97 (d, J = 8,0 Гц, 2H), 7,79 (d, J = 8,0 Гц, 2H), 7,69-7,75 (m, 4H), 7,61 (d, J = 8,4 Гц, 2H), 7,15 (d, J = 8,0 Гц, 2H), 7,01 (d, J = 8,4 Гц, 2H), 6,76 (d, J = 8,0 Гц, 2H), 5,45 (d, J = 17,6 Гц, 2H), 5,36 (s, 1H), 5,05 (s, 1H), 4,73-4,77 (m, 1H), 4,58-4,61 (m, 3H), 4,16-4,40 (m, 6H), 3,81-4,10 (m, 8H), 3,63-3,73 (m, 2H), 3,46-3,50 (m, 1H), 3,20 (s, 6H), 2,42-2,53 (m, 2H), 2,26-2,29 (m, 1H), 2,05-2,11 (m, 2H), 1,78-1,84 (m, 2H), 1,40-1,52 (m, 4H), 1,25-1,30 (m, 7H), 1,07 (d, J = 6,8 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H).
Пример 4:

Эхинокандин B
Эхинокандин B (50 мг, 0,06 ммоль) и моно-трет-бутил октадекандиоат (24,43 мг, 1,1 экв.) растворяли в DMF (N,N-диметилформамид) (2 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния тетрафторборат) (28,9 мг, 1,5 экв.) и DIPEA (диизопропилэтиламин) (15,5 мг, 2 экв.). Реакционную смесь перемешивали на ледяной водяной бане в течение еще 1,5 ч, гасили водой (5,0 мл) и экстрагировали этилацетатом (5 мл×5). Органические фракции объединяли, промывали насыщенным солевым раствором (5 мл×2), сушили над безводным натрия сульфатом и концентрировали. Остаток очищали с помощью препаративной HPLC с получением продукта (55,2 мг, чистота 97,1%, выход 80%). HRMS: 1150,6482[M+1].
1H ЯМР (400 МГц, DMSO (диметилсульфоксид)d6): δ 9,31 (s, 1H), 8,06-7,94 (m, 3H), 7,40 (s, br, 1H), 7,29 (d, 1H, J = 9,2 Гц), 7,02 (d, 2H, J = 8,4 Гц), 6,68 (d, 2H, J = 8,0 Гц), 5,45 (d, 1H, J = 6,4 Гц), 5,19 (d, 1H, J = 3,2 Гц), 5,14 (d, 1H, J = 4,4 Гц), 5,10 (d, 1H, J = 5,2 Гц), 5,04-4,91 (m, 3H), 4,75-4,64 (m, 4H), 4,42 (s, 1H), 4,36-4,28 (m, 2H), 4,21-4,16 (m, 3H), 4,04-4,01 (m, 1H), 4,00-3,93 (m, 3H), 3,88-3,84 (m, 1H), 3,79-3,77 (m, 1H), 3,71-3,68 (m, 1H), 3,19 (t, 1H, J = 8,0 Гц), 2,35-2,34 (m, 1H), 2,24-2,15 (m, 3H), 2,08-2,08 (m, 2H), 1,90-1,82 (m, 2H), 1,69-1,59 (m, 1H), 1,47-1,43 (m, 5H), 1,39 (s, 9H), 1,24 (s, 24H), 1,08-1,06 (m, 6H), 0,96 (d, 3H, J = 6,8 Гц).
Пример 5:
Этап 1

N-(2-гидроксиэтил)-пирролидин (2,30 г, 20 ммоль) растворяли в 40 мл ацетона и медленно добавляли йодметан (2,84 г, 1,0 экв.). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 4 ч и концентрировали до тех пор, пока не осталась половина растворителя, и твердое вещество не выпадало в осадок. Твердое вещество собирали фильтрованием и сушили с получением соединения SM6 в виде белого твердого вещества (4,88 г, выход 95% ). MS: 130,0[M+].
1H ЯМР (400 МГц, D2O): δ 4,09 (d, 2H, J = 2,0 Гц), 3,63-3,56 (m, 6H), 3,15 (s, 3H), 2,26 (s, 4H).
Этап 2

Анидулафунгин (300 мг, 0,26 ммоль) и фенилбороновую кислоту (64,2 мг, 2 экв.) растворяли в THF (тетрагидрофуран) (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и концентрировали досуха. Добавляли безводный ацетонитрил (10 мл) с последующим добавлением соединения SM6 (422,7 мг, 6 экв.) и п-толуолсульфоновой кислоты (340 мг, 7,5 экв.). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 часов, гасили водным раствором ацетата натрия и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением ацетатной соли (259 мг, чистота 96,5%, выход 76%). HRMS: 1251,6174 [М+].
1H ЯМР (400 МГц, CD3OD): δ 8,00 (d, 2H, J = 8,4 Гц), 7,83 (d, 2H, J = 8,0 Гц), 7,77 (m, 4H), 7,63 (d, 2H, J = 8,8 Гц), 7,17 (d, 2H, J = 8,4 Гц), 7,03 (d, 2H, J = 8,8 Гц), 6,78 (d, 2H, J = 8,8 Гц), 5,45 (d, 1H, J = 2,0 Гц), 5,06 (d, 1H, J = 3,2 Гц), 4,81-4,76 (m, 1H), 4,62-4,60 (m, 3H), 4,41 (d, 1H, J = 4,4 Гц), 4,36-4,33 (m, 2H), 4,28-4,25 (m, 2H), 4,22-4,18 (m, 1H), 4,11-3,83 (m, 8H), 3,68-3,42 (m, 8H), 3,09 (s, 3H), 2,55-2,44 (m, 2H), 2,33-2,27 (m, 1H), 2,23-2,03 (m, 6H), 1,92 (s, 3H), 1,87-1,80 (m, 2H), 1,55-1,40 (m, 4H), 1,28 (d, 6H, J = 5,6 Гц), 1,09 (d, 3H, J = 7,2 Гц), 0,92 (t, 3H, J = 6,4 Гц).
Пример 6:

Эхинокандин B (100 мг, 0,12 ммоль) и боковую цепь семаглутида (100,9 мг, 1,0 экв.) растворяли в DMF (4 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (58 мг, 1,5 экв.) и DIPEA (31 мг, 2 экв.). Реакционную смесь перемешивали на ледяной водяной бане в течение еще 1,5 ч и по каплям добавляли к 100 мл ледяной воды, и твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием, сушили и суспендировали с ацетонитрилом с получением продукта (120 мг, чистота 95,8%, выход 61,5%). HRMS: 1625,8989[M+1].
1H ЯМР (400 МГц, DMSOd6): δ 9,31 (s, 1H), 8,05 (d, 2H, J = 7,6 Гц), 7,95 (d, 1H, J = 8,4 Гц), 7,90-7,88 (m, 1H), 7,73-7,69 (m, 2H), 7,43-7,31 (m, 2H), 7,02 (d, 2H, J = 8,4 Гц), 6,69 (d, 2H, J = 8,0 Гц), 5,52 (d, 1H, J = 5,6 Гц), 5,20 (d, 1H, J = 2,8 Гц), 5,15 (d, 1H, J = 3,6 Гц), 5,10 (d, 1H, J = 5,6 Гц), 5,01-4,91 (m, 3H), 4,80-4,60 (m, 4H), 4,42 (s, 1H), 4,37-4,33 (m, 3H), 4,22-4,17 (m, 2H), 4,05-3,57 (m, 11H), 3,57 (s, br, 9H), 3,48-3,45 (m, 2H), 3,43-3,40 (m, 2H), 3,30-3,28 (m, 2H), 3,21-3,18 (m, 3H), 2,36-2,33 (m, 1H), 2,24-2,05 (m, 7H), 1,94-1,84 (m, 3H), 1,76-1,62 (m, 2H), 1,47 (s, br, 5H), 1,39 (s, 18H), 1,24 (s, 24H), 1,08-1,06 (m, 6H), 0,96 (d, 3H, J = 6,8 Гц).
Пример 7:

Соединение примера 6 (100 мг, 0,0615 ммоль) и фенилбороновую кислоту (15 мг, 2 экв.) растворяли в THF (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и концентрировали досуха. Добавляли соединение SM7 (101,6 мг, 6 экв.) и п-толуолсульфоновую кислоту (53 мг, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота еще в течение 5 часов, гасили водным раствором ацетата натрия и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением трифторацетатной соли (44,9 мг, чистота 96,5%, выход 40%). HRMS: 1710,9867[M+].
1H ЯМР (400 МГц, CD3OD): δ 8,48 (d, 1H, J = 8,4 Гц), 8,41 (d, 1H, J = 8,8 Гц), 8,23 (d, 1H, J = 7,6 Гц), 8,04-7,99 (m, 2H), 7,60-7,54 (m, 2H), 7,15 (d, 2H, J = 8,0 Гц), 6,77 (d, 2H, J = 8,4 Гц), 5,51 (d, 1H, J = 9,6 Гц), 5,05-4,89 (m, 1H), 4,60-4,45 (m, 4H), 4,37-4,32 (m, 3H), 4,28-4,25 (m, 3H), 4,13-3,82 (m, 10H), 3,76-3,63 (m, 10H), 3,59-3,56 (m, 3H), 3,54-3,56 (m, 3H), 3,53-3,46 (m, 3H), 3,41-3,38 (m, 3H), 3,21 (m, 9H), 2,55-2,44 (m, 2H), 2,36-2,21 (m, 6H), 2,17-2,06 (m, 2H), 1,92-1,83 (m, 2H), 1,64-1,56 (m, 4H), 1,49 (s, 9H), 1,46 (s, 9H), 1,31 (s, 24H), 1,22 (d, 6H, J = 6,4 Гц), 1,08 (d, 3H, J = 6,4 Гц).
Пример 8:

Трифторацетатную соль соединения примера 7 (24 мг, 0,014 ммоль) растворяли в TFA (1 мл) и полученный раствор перемешивали на ледяной водяной бане в течение 5 часов и концентрировали досуха. Остаток очищали с помощью препаративной HPLC с получением трифторацетатной соли (7,2 мг, чистота 97,8%, выход 30%). HRMS: 1598,8629 [M+].
Пример 9:
Этап 1

Эхинокандин B (200 мг, 0,2397 ммоль) и SM8 (89 мг, 1,0 экв.) растворяли в DMF (5 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (115 мг, 1,5 экв.) и DIPEA (62 мг, 2 экв.). Реакционную смесь перемешивали на ледяной водяной бане в течение еще 1,5 ч и очищали с помощью колоночной обращенно-фазовой хроматографии с получением продукта (234 мг, выход 85%). MS: 1152,5[M+1].
1H ЯМР (400 МГц, CD3OD): δ 7,96 (d, 2H, J = 6,0 Гц), 7,77-7,69 (m, 6H), 7,62 (d, 2H, J = 8,4 Гц), 7,17 (d, 2H, J = 8,4 Гц), 7,02 (d, 2H, J = 8,8 Гц), 6,78 (d, 2H, J = 8,4 Гц), 5,37 (d, 1H, J = 2,8 Гц), 5,05-5,02 (m, 1H), 4,89-4,88 (m, 1H), 4,70-4,58 (m, 5H), 4,39-4,33 (m, 3H), 4,28-4,22 (m, 3H), 4,10-4,07 (m, 2H), 4,03-4,00 (m, 1H), 3,93-3,83 (m, 2H), 3,45-3,40 (m, 1H), 2,55-2,44 (m, 2H), 2,25-2,06 (m, 3H), 1,98-1,91 (m, 2H), 1,46-1,37 (m, 2H), 1,31-1,27 (m, 6H), 1,08 (d, 3H, J = 6,8 Гц), 0,77-0,74 (m, 1H), 0,50-0,46 (m, 2H), 0,10-0,06 (m, 2H).
Этап 2

SM9 (140 мг, 0,1214 ммоль) и фенилбороновую кислоту (0,728 мг, 2 экв.) растворяли в THF (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и концентрировали досуха. Добавляли соединение SM7 (200 мг, 6 экв.), п-толуолсульфоновую кислоту (105 мг, 5 экв.) и безводный ацетонитрил (5 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 часов, гасили водным раствором ацетата натрия и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением ацетатной соли (102 мг, чистота 95,8%, выход 65%). HRMS: 1237,6022[M+].
1H ЯМР (400 МГц, CD3OD): δ 7,99 (d, 2H, J = 8,4 Гц), 7,82 (d, 2H, J = 8,0 Гц), 7,78-7,71 (m, 4H), 7,62 (d, 2H, J = 8,4 Гц), 7,17 (d, 2H, J = 8,4 Гц), 7,03 (d, 2H, J = 8,4 Гц), 6,78 (d, 2H, J = 8,4 Гц), 5,46 (d, 1H, J = 8,4 Гц), 5,08-5,05 (m, 1H), 4,90-4,77 (m, 2H), 4,63-4,59 (m, 3H), 4,41-4,33 (m, 3H), 4,29-4,26 (m, 2H), 4,21-4,18 (m, 1H), 4,11-3,90 (m, 7H), 3,84 (d, 1H, J = 11,2 Гц), 3,64-3,62 (m, 1H), 3,67-3,48 (m, 2H), 3,17 (s, 9H), ), 2,55-2,44 (m, 2H), 2,34-2,27 (m, 1H), 2,13-2,02 (m, 2H), 1,97-1,90 (m, 2H), 1,46-1,41 (m, 2H), 1,29 (s, 3H), 1,27 (s, 3H), 1,10 (d, 3H, J = 6,8 Гц) 0,80-0,74 (m, 1H), 0,50-0,46 (m, 2H), 0,10-0,06 (m, 2H).
Пример 10:
Этап 1

N-(2-гидроксиэтил)-пирролидин (6,5 г, 50 ммоль) растворяли в 25 мл ацетонитрила и медленно добавляли йодметан (7,09 г, 1,0 экв.). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 4 ч и концентрировали до тех пор, пока не осталась половина растворителя, и твердое вещество не выпадало в осадок. Твердое вещество собирали фильтрованием и сушили с получением соединения SM10 в виде белого твердого вещества (12,1 г, выход 90% ). MS: 144,0 [M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота и добавляли фенилбороновую кислоту (21,4 мг, 0,1754 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 10 мл диоксана с последующим добавлением соединения SM10 (162,6 мг, 0,6 ммоль) и п-толуолсульфоновой кислоты (75,5 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (47,6 мг, чистота 97,1%, выход 41%). HRMS: 1265,6324 [M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,98 (d, J = 8,4 Гц, 2H), 7,81 (d, J = 8,4 Гц, 2H), 7,70-7,76 (m, 4H), 7,61 (d, J = 8,8 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,01 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,42 (d, J = 1,6 Гц, 1H), 5,04 (d, J = 3,2 Гц, 1H), 4,74-4,78 (m, 1H), 4,58-4,61 (m, 3H), 4,16-4,40 (m, 6H), 3,81-4,10 (m, 8H), 3,38-3,65 (m, 7H), 3,09 (s, 3H), 2,42-2,53 (m, 2H), 2,26-2,28 (m, 1H), 2,03-2,12 (m, 2H), 1,90 (s, 3H), 1,78-1,83 (m, 6H), 1,38-1,63 (m, 6H), 1,26-1,30 (m, 7H), 1,08 (d, J = 7,2 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H).
Пример 11:
Этап 1

Эхинокандин B (300 мг, 0,36 ммоль) и SM11 (147 мг, 1,1 экв.) растворяли в DMF (15 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (174 мг, 1,5 экв.) и DIPEA (141 мг, 3 экв.). Реакционную смесь перемешивали на лядной водяной бане еще час и очищали колоночной хроматографией с обращенной фазой (MECN (ацетонитрил)/H2O) с получением соединения SM12 (392 мг, чистота 96%, выход 70,6%). MS: 1158,5[M+1].
Этап 2

SM12 (100 мг, 0,086 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота и добавляли фенилбороновую кислоту (21,3 мг, 0,173 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 10 мл диоксана с последующим добавлением соединения SM7 (108 мг, 0,777 ммоль) и камфорсульфоновой кислоты (100 мг, 0,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением формиатной соли (39,5 мг, чистота 97,0%, выход 35,5%). HRMS: 1243,5936[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 8,54 (s, 1H), 7,98 (d, J = 8,0 Гц, 2H), 7,81 (d, J = 8,0 Гц, 2H), 7,70-7,76 (m, 4H), 7,61 (d, J = 8,4 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,02 (d, J = 8,0 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,42 (s, 1H), 5,04 (s, 1H), 4,71-4,78 (m, 1H), 4,48-4,58 (m, 4H), 4,16-4,42 (m, 7H), 4,05 (t, J = 6,4 Гц, 3H), 3,81-4,01 (m, 3H), 3,47-3,65 (m, 4H), 3,22 (s, 9H), 2,42-2,53 (m, 2H), 2,25-2,32 (m, 1H), 2,00-2,11 (m, 2H), 1,73-1,88 (m, 4H), 1,59-1,66 (m, 2H), 1,26-1,37 (m, 8H), 1,08 (d, J = 6,8 Гц, 3H).
Пример 12:

Анидулафунгин (200 мг, 0,175 ммоль) растворяли в тетрагидрофуране (20 мл) в атмосфере азота и добавляли фенилбороновую кислоту (42,8 мг, 0,351 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 20 мл диоксана с последующим добавлением соединения SM13 (235 мг, 1,58 ммоль) и п-толуолсульфоновой кислоты (151 мг, 0,877 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (2 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (48,3 мг, чистота 97,7% , выход 21,3%). HRMS: 1234,6581[М+].
Пример 13:
Этап 1

N-метил-D-пролинол (1,15 г, 10 ммоль) растворяли в 20 мл ацетона и медленно добавляли йодметан (1,56 г, 1,1 экв.). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 4 ч и концентрировали до тех пор, пока не осталась половина растворителя, и твердое вещество не выпадало в осадок. Твердое вещество собирали фильтрованием и сушили с получением соединения SM14 в виде белого твердого вещества (2,44 г, выход 95% ). MS: 130,0[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в THF (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и концентрировали досуха. Добавляли соединение SM14 (135,2 мг, 6 экв.), п-толуолсульфоновую кислоту (75,6 мг, 5 экв.) и безводный диоксан (5 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 часов, гасили водным раствором ацетата натрия и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением ацетатной соли (57,5 мг, чистота 95,8%, выход 50%). HRMS: 1251,6173[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,98 (d, J = 8,8 Гц, 2H), 7,81 (d, J = 8,0 Гц, 2H), 7,69-7,76 (m, 4H), 7,61 (d, J = 9,2 Гц, 2H), 7,15 (d, J = 8,8 Гц, 2H), 7,01 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,42 (d, J = 2,4 Гц, 1H), 5,03 (d, J = 3,2 Гц, 1H), 4,92-4,93 (m, 1H), 4,74-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,38 (d, J = 4,0 Гц, 1H), 4,32-4,34 (m, 2H), 4,24-4,28 (m, 2H), 4,16-4,20 (m, 1H), 4,06-4,10 (m, 1H), 3,97-4,04 (m, 4H), 3,81-3,92 (m, 4H), 3,46-3,63 (m, 3H), 3,21 (s, 3H), 3,00 (s, 3H), 2,42-2,52 (m, 2H), 2,26-2,31 (m, 2H), 1,92-2,15 (m, 5H), 1,90 (s, 3H), 1,78-1,85 (m, 2H), 1,40-1,52 (m, 4H), 1,25-1,28 (m, 6H), 1,08 (d, J = 6,8 Гц, 3H), 0,97 (t, J = 6,8 Гц, 3H).
Пример 14:
Этап 1

Соединение SM15 (1,02 г, 10,08 ммоль) растворяли в ацетонитриле (10 мл) и по каплям добавляли метил-п-толуолсульфонат (1,88 г, 10,08 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 часов. Растворитель удаляли путем ротационного выпаривания с получением неочищенного соединения SM16, которое непосредственно использовали на следующем этапе. MS: 116,1[M+].
Этап 2

Анидулафунгин (1,14 мг, 1 ммоль) растворяли в тетрагидрофуране (40 мл) в атмосфере азота и добавляли фенилбороновую кислоту (244 мг, 2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 50 мл диоксана с последующим добавлением соединения SM16 (2,86 г, 10 ммоль) и камфорсульфоновой кислоты (1,16 г, 5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (10 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (563 мг, чистота 95,4% , выход 43,4%). HRMS: 1237,6023[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,97 (d, J = 8,3 Гц, 2H), 7,80-7,82 (m, 2H), 7,69-7,76 (m, 4H), 7,61 (d, J = 8,8 Гц, 2H), 7,14 (d, J = 8,6 Гц, 2H), 7,01 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,6 Гц, 2H), 5,35-5,36 (m, 1H), 5,02-5,03 (m, 1H), 4,86 (d, J = 5,1 Гц, 1H), 4,74 (dd, J = 5,3, 12,1 Гц, 1H), 4,44-4,65 (m, 5H), 4,39 (d, J = 4,4 Гц, 1H), 4,31-4,33 (m, 2H), 4,22-4,26 (m, 2H), 4,06-4,20 (m, 4H), 4,02 (t, J = 6,5 Гц, 3H), 3,81-3,98 (m, 4H), 3,51-3,69 (m, 2H), 3,43-3,50 (m, 1H), 3,19 (s, 3H), 2,60-2,72 (m, 1H), 2,34-2,56 (m, 3H), 2,21-2,33 (m, 1H), 1,97-2,14 (m, 2H), 1,75-1,87 (m, 5H), 1,39-1,52 (m, 4H), 1,24-1,28 (m, 6H), 1,07 (d, J = 6,8 Гц, 3H), 0,92-1,01 (m, 3H).
Пример 15:
Этап 1

Эхинокандин B (250 мг, 0,3 ммоль) и SM17 (114 мг, 1,0 экв.) растворяли в DMF (2,5 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (145 мг, 1,5 экв.) и DIPEA (78 мг, 2 экв.). Реакционную смесь перемешивали на лядной водяной бане еще час и очищали колоночной хроматографией с обращенной фазой (MECN/H2O) с получением соединения SM18 (298 мг, чистота 97%, выход 72,6%). MS: 1158,5[M+1].
Этап 2

SM18 (150 мг, 0,13 ммоль) растворяли в тетрагидрофуране (7,5 мл) в атмосфере азота и добавляли фенилбороновую кислоту (31,6 мг, 0,259 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 7,5 мл диоксана с последующим добавлением соединения SM7 (108 мг, 0,777 ммоль) и камфорсульфоновой кислоты (150 мг, 0,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением формиатной соли (57 мг, чистота 97,8%, выход 34,1%). HRMS: 1243,5928[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 8,55 (s, 1H), 7,61-7,81 (m, 9H), 7,15 (d, J = 8,8 Гц, 2H), 7,02 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,37 (d, J = 2,4 Гц, 1H), 5,03 (d, J = 3,2 Гц, 1H), 4,71-4,76 (m, 1H), 4,58-4,61 (m, 4H), 4,14-4,40 (m, 6H), 3,81-4,08 (m, 8H), 3,46-3,65 (m, 3H), 3,16 (s, 9H), 2,42-2,54 (m, 2H), 2,25-2,31 (m, 1H), 2,01-2,12 (m, 2H), 1,78-1,85 (m, 2H), 1,38-1,53 (m, 4H), 1,25-127 (m, 6H), 1,08 (d, J = 6,8 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H).
Пример 16:
Этап 1

Эхинокандин B (400 мг, 1,12 ммоль) и SM19 (930 мг, 1,0 экв.) растворяли в DMF (8 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (359 мг, 1,0 экв.) и DIPEA (288 мг, 2,0 экв.). Реакционную смесь перемешивали на лядной водяной бане еще час и очищали колоночной хроматографией с обращенной фазой (MECN/H2O) с получением соединения SM20 (890 мг, чистота 89,6%, выход 70,1%). MS: 1138,5[M+1].
Этап 2

SM20 (200 мг, 0,18 ммоль) растворяли в тетрагидрофуране (4 мл) в атмосфере азота и добавляли фенилбороновую кислоту (42,8 мг, 0,351 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 8 мл диоксана с последующим добавлением соединения SM7 (146 мг, 1,05 ммоль) и камфорсульфоновой кислоты (204 мг, 0,88 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (123 мг, чистота 97,6% , выход 54,6%). HRMS: 1223,6224[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,98 (d, J = 8,4 Гц, 2H), 7,80 (d, J = 8,4 Гц, 2H), 7,72-7,78 (m, 4H), 7,59 (d, J = 8,0 Гц, 2H), 7,28 (d, J = 8,0 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 6,76 (d, J = 8,8 Гц, 2H), 5,42 (d, J = 2,0 Гц, 1H), 5,04 (d, J = 3,2 Гц, 1H), 4,87 (s, 1H), 4,74-4,78 (m, 1H), 4,56-4,61 (m, 3H), 4,16-4,40 (m, 6H), 3,81-4,11 (m, 6H), 3,46-3,62 (m, 3H), 3,14 (s, 9H), 2,64-2,68 (m, 2H), 2,42-2,53 (m, 2H), 2,26-2,29 (m, 1H), 2,04-2,12 (m, 2H), 1,91 (s, 3H), 1,62-1,68 (m, 2H), 1,35-1,40 (m, 6H), 1,26-1,27 (m, 6H), 1,07 (d, J = 7,2 Гц, 3H), 0,91 (t, J = 6,8 Гц, 3H).
Пример 17:
Этап 1

Эхинокандин B (162 мг, 0,195 ммоль) и SM21 (70 мг, 1,0 экв.) растворяли в DMF (1,4 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (84,6 мг, 1,5 экв.) и DIPEA (50 мг, 2 экв.). Реакционную смесь перемешивали на лядной водяной бане еще час и очищали колоночной хроматографией с обращенной фазой (MECN/H2O) с получением соединения SM22 (94 мг, чистота 73%, выход 32,9%). MS: 1139,5[M+1].
Этап 2

SM22 (100 мг, 0,18 ммоль) растворяли в тетрагидрофуране (2 мл) в атмосфере азота и добавляли фенилбороновую кислоту (21,4 мг, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM7 (73,5 мг, 6,0 экв.) и камфорсульфоновой кислоты (102 мг, 5,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (31 мг, чистота 96,1% , выход 27,5%). HRMS: 1224,6163[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,96 (d, J = 8,4 Гц, 2H), 7,79 (d, J = 8,0 Гц, 2H), 7,65-7,72 (m, 4H), 7,48 (d, J = 8,8 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 6,71-6,78 (m, 4H), 5,43 (d, J = 2,0 Гц, 1H), 5,04 (d, J = 3,6 Гц, 1H), 4,91-4,93 (m, 1H), 4,73-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,16-4,40 (m, 6H), 3,81-4,10 (m, 6H), 3,46-3,62 (m, 3H), 3,11-3,14 (m, 11H), 2,42-2,53 (m, 2H), 2,26-2,31 (m, 1H), 2,02-2,12 (m, 2H), 1,91 (s, 3H), 1,62-1,67 (m, 2H), 1,38-1,44 (m, 4H), 1,26 (d, J = 6,0 Гц, 6H), 1,08 (d, J = 6,8 Гц, 3H), 0,96 (t, J = 6,8 Гц, 3H).
Пример 18:

Анидулафунгин (100 мг, 0,088 ммоль) растворяли в тетрагидрофуране (4 мл) в атмосфере азота и добавляли фенилбороновую кислоту (21,4 мг, 0,175 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM23 (75,6 мг, 0,526 ммоль) и п-толуолсульфоновой кислоты (75,5 мг, 0,438 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (2 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (35 мг, чистота 97,3%, выход 30,9%). HRMS: 1229,6268[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,97 (d, J = 8,4 Гц, 2H), 7,79 (d, J = 8,4 Гц, 2H), 7,69-7,76 (m, 4H), 7,61 (d, J = 8,8 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,00 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,42 (d, J = 2,4 Гц, 1H), 5,04 (d, J = 2,8 Гц, 1H), 4,88 (s, 1H), 4,74-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,15-4,40 (m, 6H), 3,81-4,11 (m, 6H), 3,46-3,50 (m, 1H), 3,14 (s, 9H), 2,42-2,54 (m, 2H), 2,25-2,32 (m, 1H), 2,01-2,11 (m, 2H), 1,91 (s, 3H), 1,77-1,84 (m, 2H), 1,38-1,53 (m, 4H), 1,27 (d, J = 5,6 Гц, 6H), 1,07 (d, J = 7,6 Гц, 3H), 0,97 (t, J = 6,8 Гц, 3H).
Пример 19:
Этап 1

Эхинокандин B (161 мг, 0,193 ммоль) и SM24 (80 мг, 1,0 экв.) растворяли в DMF (3,2 мл) и полученный раствор перемешивали на ледяной водяной бане. Добавляли TBTU (93 мг, 1,5 экв.) и DIPEA (50 мг, 2 экв.). Реакционную смесь перемешивали на лядной водяной бане еще час и очищали колоночной хроматографией с обращенной фазой (MECN/H2O) с получением соединения SM25 (165 мг, чистота 95,9%, выход 71,6%). MS: 1194,5[M+1].
Этап 2

SM25 (100 мг, 0,084 ммоль) растворяли в тетрагидрофуране (4 мл) в атмосфере азота и добавляли фенилбороновую кислоту (20,4 мг, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM7 (70 мг, 6,0 экв.) и камфорсульфоновой кислоты (97,6 мг, 5,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (33 мг, чистота 97,2% , выход 29,4%). HRMS: 1279,5720[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,97 (d, J = 8,0 Гц, 2H), 7,80 (d, J = 8,4 Гц, 2H), 7,69-7,76 (m, 4H), 7,62 (d, J = 8,4 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,03 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,42 (d, J = 2,0 Гц, 1H), 5,04 (d, J = 3,2 Гц, 1H), 4,88 (s, 1H), 4,73-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,16-4,40 (m, 6H), 3,81-4,11 (m, 8H), 3,46-3,62 (m, 3H), 3,15 (s, 9H), 2,42-2,53 (m, 2H), 2,23-2,32 (m, 3H), 2,04-2,11 (m, 2H), 1,93 (s, 3H), 1,87-1,90 (m, 2H), 1,76-1,82 (m, 2H), 1,26 (d, J = 6,4 Гц, 6H), 1,08 (d, J = 6,8 Гц, 3H).
Пример 20:
Этап 1

SM-26 (590 мг, 1 экв.) растворяли в 10 мл ацетонитрила и добавляли метил п-толуолсульфонат (1,2 г, 1,1 экв.). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 4 ч и концентрировали, а остаток суспендировали с петролейным эфиром ацетона с получением соединения SM-27 в виде белого твердого вещества (1,6 г, выход 95%). MS: 117,1[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в THF (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и концентрировали досуха. Добавляли соединение SM27 (151 мг, 6 экв.), п-толуолсульфоновую кислоту (75,4 мг, 5 экв.) и безводный диоксан (5 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 часов, гасили водным раствором ацетата натрия и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением формиатной соли (60 мг, чистота 97,8%, выход 55,6%). HRMS: 1237,6024[M+].
1H ЯМР (400 МГц, CD3OD): δ 8,56 (s, 1H), 8,02 (d, 2H, J = 10,8 Гц), 7,82 (d, 2H, J = 8,4 Гц), 7,77 (m, 4H), 7,63 (d, 2H, J = 8,4 Гц), 7,17 (d, 2H, J = 8,8 Гц), 7,03 (d, 2H, J = 8,8 Гц), 6,78 (d, 2H, J = 8,8 Гц), 5,42 (d, 1H, J = 2,4 Гц), 5,06 (d, 1H, J = 2,8 Гц), 4,92-4,88 (m, 2H), 4,81-4,72 (m, 2H), 4,63-4,58 (m, 3H), 4,42 (d, 1H, J = 4,0 Гц), 4,29-3,81 (m, 14H), 3,52-3,38 (m, 1H), 3,19 (s, 6H), 2,57-2,43 (m, 4H), 2,34-2,27 (m, 1H), 2,13-2,04 (m, 2H), 1,87-1,80 (m, 2H), 1,55-1,40 (m, 4H), 1,29 (d, 6H, J = 6,0 Гц), 1,08 (d, 3H, J = 6,8 Гц), 0,99 (t, 3H, J = 6,8 Гц).
Пример 21:

Трифторацетатную соль соединения примера 6 (24 мг, 0,014 ммоль) растворяли в TFA (1 мл) и полученный раствор перемешивали на ледяной водяной бане в течение 5 часов и концентрировали досуха. Остаток очищали с помощью препаративной HPLC с получением трифторацетатной соли (7,2 мг, чистота 97,8%, выход 30%). HRMS: 1513,7743[M+1].
Пример 22:
Этап 1

Соединение SM-29 (5,4 г, 52,3 ммоль) растворяли в ацетоне (54 мл) и по каплям добавляли метил п-толуолсульфонат (10,23 г, 54 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 ч, и белое твердое вещество выпадало в осадок. Реакционную смесь охлаждали до комнатной температуры и фильтровали, а осадок на фильтре сушили in vacuo с получением соединения SM30 (6,5 г, чистота 98%, выход 42,9%). MS: 118,12[M+].
Этап 2

Анидулафунгин
Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в тетрагидрофуране (4 мл) в атмосфере азота, и полученный раствор перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM30 (152,3 мг, 0,526 ммоль) и камфорсульфоновой кислоты (102 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (53 мг, чистота 96,1% , выход 46,5%). HRMS: 1239,6225[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,98 (d, J = 8,4 Гц, 2H), 7,81 (d, J = 7,2 Гц, 2H), 7,69-7,76 (m, 4H), 7,61 (d, J = 8,8 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,01 (d, J = 8,8 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,44 (s, 1H), 5,04 (d, J = 3,2 Гц, 1H), 4,85 (m, 1H), 4,74-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,39 (d, J = 4,0 Гц, 1H), 4,32-4,34 (m, 2H), 4,24-4,27 (m, 2H), 4,16-4,20 (m, 1H), 3,81-4,07 (m, 8H), 3,74 (m, 1H), 3,46-3,50 (m, 1H), 3,11 (s, 9H), 2,42-2,53 (m, 2H), 2,28-2,36 (m, 1H), 2,02-2,11 (m, 2H), 1,90 (s, 3H), 1,78-1,85 (m, 2H), 1,43-1,52 (m, 7H), 1,25-1,27 (m, 6H), 1,08 (d, J = 6,4 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H).
Пример 23
Этап 1

Соединение SM-31 (2,15 г, 20,84 ммоль) растворяли в ацетоне (21,5 мл) и по каплям добавляли метил-п-толуолсульфонат (4,08 г, 22 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 ч, и белое твердое вещество выпадало в осадок. Реакционную смесь охлаждали до комнатной температуры и фильтровали, а осадок на фильтре сушили in vacuo с получением соединения SM32 (3,2 г, чистота 98%, выход 53%). MS: 118,12[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в тетрагидрофуране (4 мл) в атмосфере азота, и полученный раствор перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM32 (152,3 мг, 0,526 ммоль) и камфорсульфоновой кислоты (102 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (39 мг, чистота 99,6% , выход 34,2%). HRMS:1239,6226[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,97 (d, J = 8,4 Гц, 2H), 7,69-7,81 (m, 6H), 7,61 (d, J = 8,4 Гц, 2H), 7,15 (d, J = 8,4 Гц, 2H), 7,01 (d, J = 8,4 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,34 (d, J = 2,8 Гц, 1H), 5,02 (d, J = 2,4 Гц, 1H), 4,85 (m, 1H), 4,73-4,78 (m, 1H), 4,57-4,61 (m, 3H), 4,39 (d, J = 4,4 Гц, 1H), 4,32-4,34 (m, 2H), 4,24-4,28 (m, 2H), 4,17-4,21 (m, 1H), 3,79-4,10 (m, 8H), 3,66 (m, 1H), 3,46-3,50 (m, 1H), 3,12 (s, 9H), 2,42-2,53 (m, 2H), 2,25-2,31 (m, 1H), 2,03-2,11 (m, 2H), 1,89 (s, 3H), 1,78-1,84 (m, 2H), 1,37-1,53 (m, 7H), 1,25-1,28 (m, 6H), 1,08 (d, J = 6,8 Гц, 3H), 0,97 (t, J = 7,2 Гц, 3H).
Пример 24
Этап 1
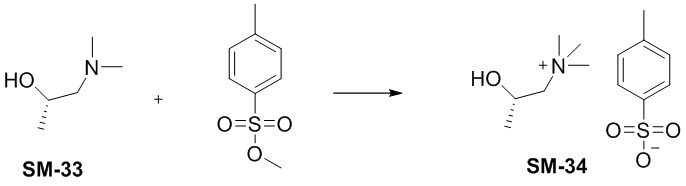
Соединение SM-33 (2,2 г, 21,3 ммоль) растворяли в ацетоне (22 мл) и по каплям добавляли метил-п-толуолсульфонат (4,17 г, 22,4 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 ч, и белое твердое вещество выпадало в осадок. Реакционную смесь охлаждали до комнатной температуры и фильтровали, а осадок на фильтре сушили in vacuo с получением соединения SM34 (2,05 г, чистота 98%, выход 33%). MS: 118,12[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в тетрагидрофуране (4 мл) в атмосфере азота, и полученный раствор перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM34 (152,3 мг, 0,526 ммоль) и камфорсульфоновой кислоты (102 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали путем ротационного выпаривания для удаления растворителя. Полученный неочищенный продукт очищали с помощью препаративной хроматографии с получением ацетатной соли (29 мг, чистота 97,5% , выход 25,4%). HRMS: 1239,6223[M+].
1H ЯМР (400 МГц, METHANOL-d4) δ 7,96 (d, J = 7,2 Гц, 2H), 7,69-7,80 (m, 6H), 7,61 (d, J = 8,4 Гц, 2H), 7,15 (d, J = 8,8 Гц, 2H), 7,01 (d, J = 7,6 Гц, 2H), 6,76 (d, J = 8,4 Гц, 2H), 5,50 (s, 1H), 5,04 (d, J = 3,2 Гц, 1H), 4,85 (m, 1H), 4,78-4,79 (m, 1H), 4,55-4,60 (m, 3H), 4,30-4,36 (m, 4H), 4,22-4,27 (m, 2H), 4,16-4,20 (m, 1H), 3,80-4,10 (m, 6H), 3,52-3,55 (m, 1H), 3,34-3,43 (m, 2H), 3,18 (s, 9H), 2,42-2,51 (m, 2H), 2,24-2,30 (m, 1H), 2,05-2,13 (m, 2H), 1,90 (s, 3H), 1,78-1,84 (m, 2H), 1,40-1,51 (m, 4H), 1,27 (t, J = 7,2 Гц, 6H), 1,20 (d, J = 5,2 Гц, 3H), 1,07 (d, J = 7,2 Гц, 3H), 0,97 (t, J = 6,8 Гц, 3H).
Пример 25
Этап 1

SM35 (505 мг, 1 экв.) растворяли в 5 мл ацетона и добавляли метил п-толуолсульфонат (1,02 г, 1,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и фильтровали, а осадок на фильтре промывали ацетоном с получением соединения SM36 в виде белого твердого вещества (1,22 г, выход 85%). MS: 116,2[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в тетрагидрофуране (4 мл) в атмосфере азота, и полученный раствор перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM36 (151,2 мг, 0,526 ммоль) и камфорсульфоновой кислоты (102 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением ацетатной соли (50 мг, чистота 96,2%, выход 44%). HRMS: 1238,4225[M+].
1H ЯМР (400 МГц, METHANOL-d4): δ 8,02 (d, 2H, J = 8,4 Гц), 7,84 (d, 2H, J = 8,0 Гц), 7,78-7,71 (m, 4H), 7,63 (d, 2H, J = 6,8 Гц), 7,17 (d, 2H, J = 8,8 Гц), 7,03 (d, 2H, J = 8,8 Гц), 6,81 (d, 2H, J = 7,2 Гц), 5,52 (d, 1H, J = 1,6 Гц), 5,08 (d, 1H, J = 3,2 Гц), 4,84-4,76 (m, 1H), 4,63-4,60 (m, 6H), 4,42 (d, 1H, J = 4,4 Гц), 4,36-4,33 (m, 2H), 4,27-4,23 (m, 2H), 4,18-4,15 (m, 1H), 4,06-4,00 (m, 4H), 3,96-3,90 (m, 2H), 3,86-3,83 (m, 1H), 3,74-3,46 (m, 4H), 3,18 (s, 3H), 3,11 (s, 3H), 2,60-2,30 (m, 4H), 2,10-2,04 (m, 2H), 1,91 (s, 3H), 1,85-1,80 (m, 2H), 1,53-1,40 (m, 4H), 1,29-1,26 (m, 6H), 1,10 (d, 3H, J = 7,2 Гц), 0,99 (t, 3H, J = 7,2 Гц).
Пример 26:
Этап 1

SM37 (505 мг, 1 экв.) растворяли в 5 мл ацетона и добавляли метил п-толуолсульфонат 1,02 г, 1,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и фильтровали, а осадок на фильтре промывали ацетоном с получением соединения SM38 в виде белого твердого вещества (1,29 г, выход 90%). MS: 116,2[M+].
Этап 2

Анидулафунгин (100 мг, 0,0877 ммоль) и фенилбороновую кислоту (21,39 мг, 2 экв.) растворяли в тетрагидрофуране (4 мл) в атмосфере азота, и полученный раствор перемешивали при комнатной температуре в течение 2 часов и концентрировали in vacuo досуха для удаления растворителя. Добавляли 4 мл диоксана с последующим добавлением соединения SM38 (151,2 мг, 0,526 ммоль) и камфорсульфоновой кислоты (102 мг, 0,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, гасили 1 н. водным раствором ацетата натрия (1 мл) и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной HPLC с получением ацетатной соли (40 мг, чистота 96,2%, выход 34%). HRMS: 1238,4225[M+].
1H ЯМР (400 МГц, METHANOL-d4): δ 8,00 (d, 2H, J = 8,4 Гц), 7,82 (d, 2H, J = 8,4 Гц), 7,78-7,71 (m, 4H), 7,63 (d, 2H, J = 8,8 Гц), 7,17 (d, 2H, J = 8,8 Гц), 7,03 (d, 2H, J = 8,8 Гц), 6,78 (d, 2H, J = 8,4 Гц), 5,44 (d, 1H, J = 2,4 Гц), 5,06 (d, 1H, J = 2,8 Гц), 4,83-4,77 (m, 1H), 4,63-4,60 (m, 6H), 4,42 (d, 1H, J = 4,0 Гц), 4,36-4,33 (m, 2H), 4,27-4,25 (m, 2H), 4,20-4,18 (m, 1H), 4,06-4,00 (m, 4H), 3,94-3,90 (m, 2H), 3,86-3,78 (m, 1H), 3,71-3,46 (m, 4H), 3,25 (s, 3H), 3,19 (s, 3H), 2,58-2,41 (m, 4H), 2,32-2,28 (m, 2H), 1,92 (s, 3H), 1,86-1,80 (m, 2H), 1,53-1,40 (m, 4H), 1,32-1,27 (m, 6H), 1,10 (d, 3H, J = 7,2 Гц), 0,99 (t, 3H, J = 7,2 Гц).
Способ масс-спектрометрии высокого разрешения:
133-2000 м/з
Пример испытания 1: Способ испытания на противогрибковую активность
После серийного разведения тестируемого соединения выполняли анализ MIC (минимальная ингибирующая концентрация) на стандартном штамме Candida и анализ MEC (минимальная эффективная концентрация) на стандартном штамме Aspergillus. Анализ MIC проводили в соответствии с рекомендациями Института клинических и лабораторных стандартов (CLSI M27-A3) и анализ MEC в соответствии с рекомендациями Института клинических и лабораторных стандартов (CLSI M38-A2).
Получение грибковой инокуляционной жидкости
Candida:
Замороженный штамм пассировали по меньшей мере два раза, а одну колонию отбирали и ресуспендировали в физиологическом растворе или стерильной воде в пробирке. Суспензию перемешивали на вортексе и доводили до 0,5 МкФ (единицы мутности по МакФарланду) (от 1×106 до 5×106 КОЕ (колониеобразующая единица/мл) с помощью спектрофотометра при длине волны 530 нм. Суспензию в 50 раз разбавляли физиологическим раствором, а затем в 20 раз разбавляли бульоном 1×RPMI (среда мемориального института Розуэлл-Парка) 1640 (от 1×103 до 5×103 КОЕ/мл). 10 мкл суспензии наносили на чашку с SDA (декстрозный агар Сабуро) для подсчета колоний в диапазоне от около 10 до 50 одиночных колоний.
После достижения полного растворения при комнатной температуре в подготовленной чашке для испытания на чувствительность, бактериальную суспензию добавляли в 96-луночный планшет по 100 мкл на лунку с помощью многоканальной пипетки. При этом концентрация бактерий в каждой лунке должна составлять от 0,5×103 до 2,5×103 КОЕ/мл.
Aspergillus (работа в шкафу биологической безопасности класса II):
Aspergillus пассировали на чашке с SDA и культивировали при 35 °C в течение от 48 ч до 7 дней для индуцирования спорообразования. Колонии на планшете покрывали приблизительно 1 мл 0,85% физиологического раствора или стерильной воды (добавляли полисорбат 20 в конечной концентрации 0,1%-0,01%). Среду осторожно протирали на ее поверхности наконечником или стерильным ватным тампоном (будьте осторожны, чтобы не сломать среду), а суспензию спорных гиф переносили в стерильную пробирку и оставляли стоять в течение 3-5 минут, чтобы тяжелые частицы осели. Гомогенный верхний слой суспензии переносили в новую стерильную пробирку, которую затем закрывают и перемешивали на вортексе в течение 15 с (необходимо соблюдать осторожность, поскольку суспензия может образовывать аэрозоль при снятии крышки). Концентрацию суспензии доводили до значения ОП (оптическая плотность) 0,09-0,13 с помощью спектрофотометра при 530 нм. Суспензию разбавляли в 50 раз 1×RPMI 1640. 100 мкл образца добавляли в каждую лунку 96-луночного планшета в течение 2 ч после разведения (конечная концентрация спор в чашке для определения чувствительности составляла от 0,4×104 до 5×104 КОЕ/мл).
Подсчет колоний: Суспензию, разведенную RPMI 1640, дополнительно разбавляли в 10 раз, и 10 мкл разведения наносили на плашку с SDA, культивировали при 28 °C и наблюдали каждый день; колонии немедленно подсчитывали после того, как они были видны невооруженным глазом.
Культура
Аналитический планшет для дрожжевых грибков инкубировали в инкубаторе при 35 °С с влажностью 85% в течение 24 ч, а затем считывали значение MIC. Для лекарственных средств на основе эхинокандина Aspergillus инкубировали при 28 °C в течение 21-26 ч, а затем считывали результаты MEC.
Интерпретация MIC или MEC
Грибки дрожжевого типа: одноразовую герметизирующую пленку наносили на 96-луночный планшет, и смесь хорошо перемешивали путем встряхивания. Наблюдение проводили с помощью планшетного ридера и невооруженным глазом. Было проведено сравнение с контролем роста, и минимальная концентрация соединения, соответствующая ингибированию роста, составляющего 50% или более, была определена как MIC. Фотографии были сделаны и сохранены с помощью автоматического планшетного ридера.
Aspergillus: Для лекарственных средств эхинокандина сравнивали с контролем роста с помощью планшетного ридера, а минимальная концентрация лекарственного средства, которая может вызвать образование гифами небольших, круглых, компактных гифалических частиц, определялась как MEC. Для точного определения значений MEC планшет не следует перемешивать вортексом до считывания.
(мкг/мл)
90029
15126
22019
6258
6260
750
28539
Примечание: 1. Candida parapsilosis ATCC 22019 и Candida krusei ATCC6258 были штаммами контроля качества. Согласно CLSI-M60 (CLSI - институт клинических и лабораторных стандартов), 24-часовой MIC ANI (анидулафунгин) для ATCC 22019 составляет (0,25-2) мкг/мл, а CAS (каспофунгин) составляет (0,25-1) мкг/мл; 24-часовой MIC ANI для ATCC6258 составляет (0,03-0,12) мкг/мл, а CAS составляет (0,12-1) мкг/мл.
(мкг/мл)
90028
90028
44858
36583
200956
Данные анализа показали, что значительное количество примеров соединений по настоящему изобретению обладают превосходной противогрибковой активностью, а некоторые из соединений обладают более превосходной противогрибковой активностью по сравнению с положительными лекарственными средствами.
Пример испытания 2: Концентрации соединений гистамина в плазме и фармакокинетическое исследование
Способ проведения испытания:
12 крыс SD (Спрег-Доули) были разделены на 2 группы по 6 особей (половина самцов и половина самок). Крысы наблюдались не реже одного раза в сутки. Массу тела измеряли один раз перед введением. Введение осуществляли путем однократной внутривенной инъекции в течение 20 минут на животное. Измерение PK (фармакокинетика) проводили один раз перед введением и через 5 мин, 30 мин, 1 ч, 4 ч, 8 ч, 24 ч, 48 ч, 72 ч и 96 ч после введения. Измерение гистамина проводили один раз перед введением и через 30 мин, 4 ч, 8 ч и 24 ч после введения.
Схема дозирования показана в таблице ниже:
(мг/кг)
(мг/мл)
(мл/кг)
Результаты в основном следующие:
Наблюдение за общим состоянием
Незначительное временное снижение активности наблюдалось у 2 самок в группе 2 (2/3 крыс) в день введения.
Кроме того, крысы SD в каждой группе были в хорошем общем состоянии, демонстрировали нормальную спонтанную двигательную активность, имели чистую кожу и шерсть, а также демонстрировали нормальную дефекацию и мочеиспускание, и никаких других отклонений не наблюдалось.
Измерение гистамина
Временное повышение уровня гистамина было вызвано внутривенным введением как в группе 1, так и в группе 2. Концентрация гистамина в плазме достигала максимума через 30 мин, продемонстрировала тенденцию к восстановлению через 4 ч и по существу вернулась к нормальному уровню через 8-24 ч, как показано на фиг. 1. 30 мин после введения, средняя концентрация гистамина в плазме крыс в группе 2 составляла 1333,0 нг/мл, что в 4,5 раза (p = 0,046) выше, чем в группе 1 (296,6 нг/мл), как показано на фиг. 2. Способность соединения группы 1 индуцировать повышение уровня гистамина у крыс значительно ниже, чем у соединения группы 2, когда их вводят в той же дозе.
Фармакокинетика
Фармакокинетические параметры у животных после введения в группу 1 или группу 2 приведены в таблице ниже:
Данные анализа показали, что после введения одной и той же дозы путем однократной внутривенной инъекции группа 1 и группа 2 близки друг к другу по уровням воздействия препарата в плазме (Cmax и AUC) и не демонстрировали существенной разницы, связанной с полом, а остальные фармакокинетические параметры имеют по существу эквивалентные значения для обеих групп.
В заключение, после введения в той же дозе (10 мг/кг) путем однократной внутривенной инъекции, уровни воздействия лекарственного средства в плазме конечного продукта примера 13 и ацетата резафунгина близки, в то время как способность конечного продукта примера 13 индуцировать повышение уровня гистамина у крыс значительно ниже, чем у ацетата резафунгина, что позволяет предположить, что конечный продукт примера 13 при клиническом применении не будет легко вызывать аллергию по сравнению с резафунгином.

Изобретение относится к аналогам эхинокандина формулы I, где R1, R2, R3 и G определены в формуле изобретения. Соединение может быть использовано для предотвращения или лечения грибковой инфекции или для предотвращения, стабилизации или ингибирования роста грибков или уничтожения грибков. 4 н. и 6 з.п. ф-лы, 2 ил., 4 табл., 26 пр.

1. Соединение формулы I или его фармацевтически приемлемая соль или изомер

где R1 представляет собой O(C(RA1)(RA2))a(C(RA3)(RA4))jX1;
R2 представляет собой -СН3 или CH2CH2NH2;
R3 представляет собой Н;
G выбран из группы, состоящей из:

RA1, RA2, RA3 и RA4 независимо выбраны из группы, состоящей из водорода;
X1 выбран из группы, состоящей из следующих структур:
 ,
,  ,
,
каждый RF представляет собой Н;
Rq1 и Rq2 независимо представляют собой Н или C1-6 алкил;
f представляет собой 0;
g представляет собой 0;
h представляет собой 0;
р представляет собой целое число от 1 до 3;
а представляет собой целое число от 0 до 5;
j представляет собой целое число от 0 до 5 и
n представляет собой целое число от 1 до 7.
2. Соединение формулы I или его фармацевтически приемлемая соль или изомер по п. 1, где R1 выбран из группы, состоящей из:

3. Соединение формулы I или его фармацевтически приемлемая соль или изомер по п. 1, где R1 выбран из группы, состоящей из:

4. Соединение формулы I или его фармацевтически приемлемая соль или изомер по п. 1, где R1 выбран из группы, состоящей из:

5. Соединение формулы I или его фармацевтически приемлемая соль или изомер по п. 1, где R1 выбран из группы, состоящей из:

6. Соединение формулы I или его фармацевтически приемлемая соль или изомер по п. 1, где соединение формулы I представляет собой:



7. Соединение по любому из пп. 1-6, где его фармацевтически приемлемая соль выбрана из группы, состоящей из ацетатной соли, трифторацетатной соли и формиатной соли.
8. Фармацевтическая композиция, обладающая противогрибковой активностью, содержащая эффективное количество соединения или его фармацевтически приемлемой соли или изомера по любому из пп. 1-7 и фармацевтически приемлемый эксципиент.
9. Применение соединения или его фармацевтически приемлемой соли или изомера по любому из пп. 1-7 для получения лекарственного средства для лечения и/или предотвращения грибковых инфекций.
10. Применение соединения или его фармацевтически приемлемой соли или изомера по любому из пп. 1-7 для получения лекарственного средства для предотвращения, стабилизации или ингибирования роста грибков или уничтожения грибков.
| US 5652213 A1, 29.07.1997 | |||
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2129562C1 |
| WO 2016201283 A1, 15.12.2016 | |||
| WO 2017120471 A1, 13.07.2017 | |||
| WO 1996008267 A1, 21.03.1996 | |||
| ТРЁХМЕРНАЯ ПАРКОВОЧНАЯ СИСТЕМА | 2021 |
|
RU2803584C1 |

