Ссылка на родственные заявки
[1] Данная заявка заявляет приоритет следующих заявок:
[2] CN201710900542.8, дата подачи 28.09.2017;
[3] CN201810239824.2, дата подачи 21.03.2018.
Область техники, к которой относится изобретение
[4] Настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли и его применению в изготовлении лекарственного препарата для лечения заболевания, связанного с рецептором A2A.
Предшествующий уровень техники
[5] Аденозиновый рецептор A2A широко распространен в тканях человека и высоко экспрессируется в тканях и органах, например, в селезенке, тимусе, лейкоцитах, тромбоцитах, GABA-эргических нейронах и обонятельной луковице. Он также экспрессируется в других частях, таких как сердце, легкие, кровеносные сосуды и головной мозг. Аденозиновый рецептор A2A, как правило, существует совместно с другими GPCR, и они связываются вместе с образованием гетеродимеров, например, рецептор A2A может образовывать гетеродимеры с дофаминовым рецептором D2, каннабиноидным рецептором CB1, глутаматным рецептором mGluR5 и т. д. Аденозиновый рецептор A2A играет важную роль в регулировании расширения кровеносных сосудов, поддержке образования новых кровеносных сосудов и защите тканей организма от повреждения, обусловленного воспалением; аденозиновый рецептор A2A также влияет на активность непрямого пути базальных ганглий.
[6] В солидных опухолях разложение клеточных тканей и бескислородная среда обуславливают разложение большого количества ATP, что, таким образом, приводит к внеклеточному обогащению аденозином до аномально высокой концентрации, которая в 10-20 раз выше обычного значения. Высокая концентрация аденозина обеспечивает связывание с рецептором A2A, за счет чего обеспечивается активация аденозинового сигнального пути. Данный сигнальный путь представляет собой механизм, который защищает ткань организма посредством иммуносупрессии при повреждении ткани организма. Активация аденозинового сигнального пути приводит к долговременному подавлению врожденного иммунного ответа, что обуславливает иммунологическую толерантность и приводит к неконтролируемому росту злокачественных опухолей. Связывание аденозина с рецептором A2A в лейкоцитах (например, лимфоцитах, T-лимфоцитах, естественных клетках-киллерах, дендритных клетках и т. д.) подавляет эффекторную функцию этих лейкоцитов в иммунной системе. Связывание аденозина с рецептором A2A увеличивает экспрессию CD39, CD73 и CTLA4 (T-клеточные контрольные точки), за счет чего обеспечивается большее количество Treg-клеток с более сильной иммуносупрессией. Блокирование аденозинового сигнального пути через рецептор A2A может снизить подавляющий эффект на иммунную систему и усилить иммунную функцию T-клеток, таким образом, оно считается перспективным механизмом отрицательной обратной связи, который может подавлять рост опухоли.
[7] Моноклональное антитело CS1003 представляет собой полноразмерное полностью гуманизированное моноклональное антитело к PD-1, представляющее собой иммуноглобулин G4 (IgG4).
Содержание изобретения
[8] В настоящем изобретении предусмотрены соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
 ,
,
[9] где
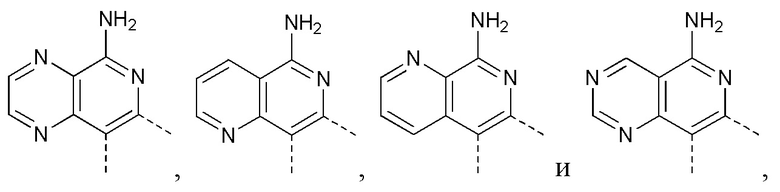
[10] 1 или 2 из T1, T2, T3 и T4 представляют собой N, при этом остальные независимо представляют собой CH;
[11] каждый R1 независимо выбран из H, галогена, OH, NH2 или C1-3алкила, необязательно замещенного 1, 2 или 3 R;
[12] каждый R2 независимо выбран из H, галогена, OH, NH2 или C1-3алкила, необязательно замещенного 1, 2 или 3 R;
[13] n выбран из 0, 1, 2 и 3;
[14] m выбран из 0, 1, 2 и 3;
[15] кольцо A выбрано из 6-10-членного арила и 5-10-членного гетероарила;
[16] кольцо B выбрано из фенила и 5-6-членного гетероарила;
[17] R выбран из F, Cl, Br, I, OH, NH2 и CN;
[18] каждый «гетеро» в 5-6-членном гетероариле и 5-10-членном гетероариле независимо выбран из N, O, S, NH, -C(=O)-, -C(=O)O- и -C(=O)NH-;
[19] число гетероатомов или гетероатомных групп независимо выбрано из 1, 2, 3 и 4.
[20] В некоторых вариантах осуществления настоящего изобретения каждый R1 независимо выбран из H, F, Cl, Br, I, OH, NH2, Me и Et, где Me и Et необязательно замещены 1, 2 или 3 R, другие переменные определены в настоящем изобретении.
[21] В некоторых вариантах осуществления настоящего изобретения каждый R1 независимо выбран из H, F, Cl, Br, I, OH, NH2, Me, CF3 и Et, другие переменные определены в настоящем изобретении.
[22] В некоторых вариантах осуществления настоящего изобретения каждый R2 независимо выбран из H, F, Cl, Br, I, OH, NH2 и Me, необязательно замещенного 1, 2 или 3 R, другие переменные определены в настоящем изобретении.
[23] В некоторых вариантах осуществления настоящего изобретения каждый R2 независимо выбран из F, Cl, Br, I, OH, NH2 и Me, другие переменные определены в настоящем изобретении.
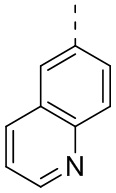
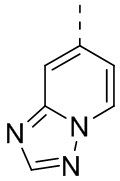
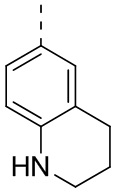
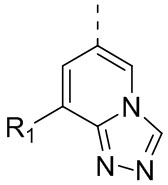
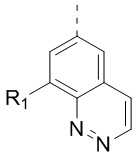
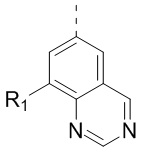
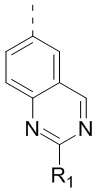
[24] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из фенила, пиридила, хинолила, хиноксалила, 1,2,3,4-тетрагидрохинолила, 3,4-дигидро-2H-бензо[b][1,4]оксазинила, [1,2,4]триазоло[1,5-a]пиридила, 1H-индазолила, бензо[d]изоксазолила, [1,2,4]триазоло[4,3-a]пиридила и 1H-бензо[d][1,2,3]триазолила, другие переменные определены в настоящем изобретении.
[25] В некоторых вариантах осуществления настоящего изобретения кольцо A выбрано из фенила, пиридила, хинолила, хиноксалила, 1,2,3,4-тетрагидрохинолила, 3,4-дигидро-2H-бензо[b][1,4]оксазинила, [1,2,4]триазоло[1,5-a]пиридила, 1H-индазолила, бензо[d]изоксазолила, [1,2,4]триазоло[4,3-a]пиридила, 1H-бензо[d][1,2,3]триазолила, циннолинила, хиназолинила, хинолила, изохинолила, имидазо[1,2-a]пиридила, [1,2,4]триазоло[1,5-a]пиридила, 1H-индазолила и бензо[d]тиазолила, другие переменные определены в настоящем изобретении.
[26] В некоторых вариантах осуществления настоящего изобретения структурное звено  выбрано из
выбрано из 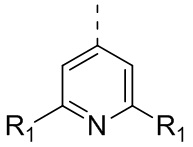 ,
,  ,
,  ,
,  ,
,  ,
,  и
и  , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[27] В некоторых вариантах осуществления настоящего изобретения структурное звено выбрано из ,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 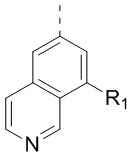 ,
, 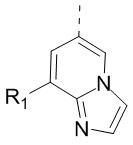 ,
,  ,
,  ,
,  ,
,  и
и  , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[28] В некоторых вариантах осуществления настоящего изобретения структурное звено выбрано из 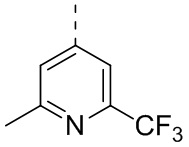 ,
, 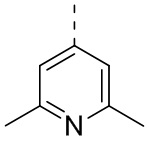 ,
,  ,
, 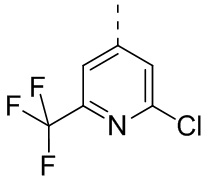 , , , , , , ,
, , , , , , ,  ,
,  ,
, 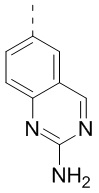 ,
, 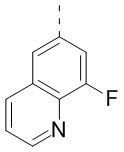 ,
, 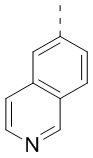 ,
,  ,
, 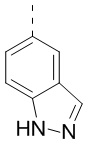 ,
, 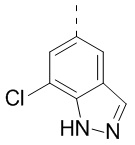 ,
, 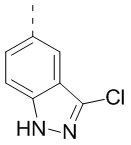 ,
,  ,
, 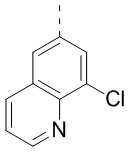 и
и 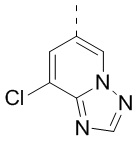 , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[29] В некоторых вариантах осуществления настоящего изобретения структурное звено 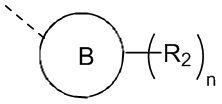 представляет собой
представляет собой  , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[30] В некоторых вариантах осуществления настоящего изобретения кольцо B выбрано из фенила, фуранила, тиенила и пиразолила, другие переменные определены в настоящем изобретении.
[31] В некоторых вариантах осуществления настоящего изобретения структурное звено выбрано из  ,
,  ,
,  и
и 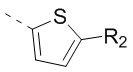 , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[32] В некоторых вариантах осуществления настоящего изобретения структурное звено выбрано из 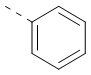 , ,
, ,  ,
,  ,
, 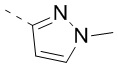 ,
,  и
и 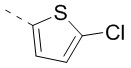 , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[33] В некоторых вариантах осуществления настоящего изобретения структурное звено 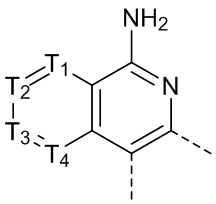 выбрано из
выбрано из  и
и  , другие переменные определены в настоящем изобретении.
, другие переменные определены в настоящем изобретении.
[34] В некоторых вариантах осуществления настоящего изобретения структурное звено выбрано из
другие переменные определены в настоящем изобретении.
[35] В некоторых вариантах осуществления настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из

[36] где
[37] R1 и R2 определены в настоящем изобретении;
[38] кольцо C выбрано из 5-6-членного гетероарила и 5-6-членного гетероциклоалкила;
[39] кольцо D выбрано из 5-6-членного гетероарила;
[40] «гетеро» в 5-6-членном гетероариле выбран из N, S и NH;
[41] «гетеро» в 5-6-членном гетероциклоалкиле представляет собой NH;
[42] число гетероатомов или гетероатомных групп независимо выбрано из 1, 2, 3 и 4.
[43] В некоторых вариантах осуществления настоящего изобретения структурное звено  выбрано из , , , , , , , , ,
выбрано из , , , , , , , , ,  и .
и .
[44] В некоторых вариантах осуществления настоящего изобретения структурное звено  выбрано из , и .
выбрано из , и .
[45] Другие варианты осуществления настоящего изобретения получают путем произвольной комбинации указанных выше переменных.
[46] В настоящем изобретении также предусмотрены соединение, представленное следующей формулой, или его фармацевтически приемлемая соль:





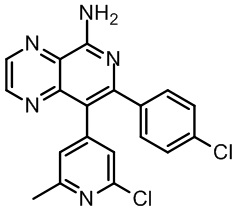
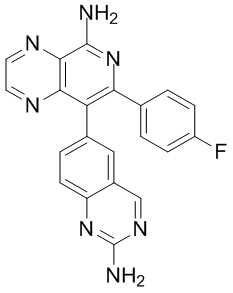
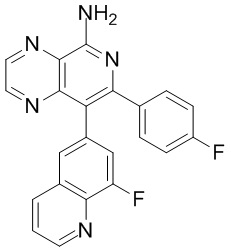
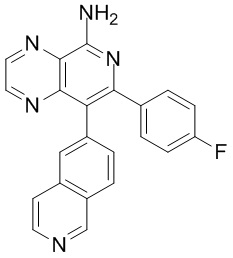


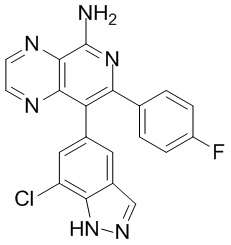

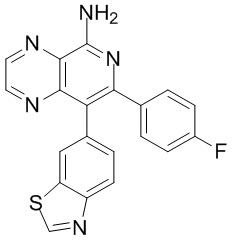
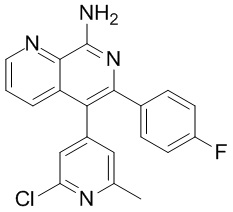



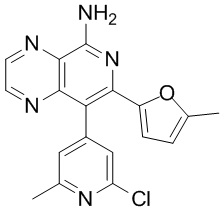




 и
и  .
.
[47] В настоящем изобретении также предусмотрено применение соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата для лечения заболевания, связанного с рецептором A2A.
[48] Технический эффект
[49] В настоящей заявке представлен синтез соединения формулы (I), которое является новым антагонистом аденозинового рецептора A2A. Соединение можно применять для иммунотерапии опухолей в виде одного лекарственного средства или в комбинации с антителом. Соединение по настоящему изобретению обладает лучшей растворимостью и явно улучшенными фармакокинетическими характеристиками.
[50] Комбинация соединения по настоящему изобретению и CS1003 обеспечивает достижение лучшего эффекта в отношении подавления роста опухоли и имеет синергический эффект.
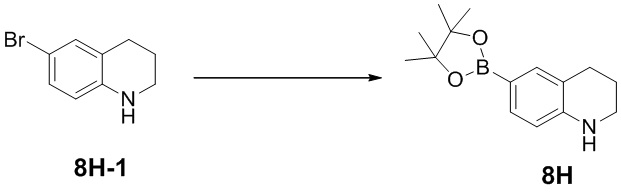
[51] Соединение по настоящему изобретению характеризуется достаточным содержанием в плазме крови и тканях опухолей.
[52] Определения и описание
[53] Если не указано иное, следующие термины при использовании в описании и формуле настоящего изобретения имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не следует считать неопределенным или неясным, а следует понимать в соответствии с общепринятым значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту. Термин «фармацевтически приемлемый» используется в данном документе применительно к тем соединениям, материалам, композициям и/или лекарственным формам, которые в пределах тщательной медицинской оценки являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений в соответствии с обоснованным соотношением польза/риск.
[54] Термин «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которую получают путем осуществления реакции соединения, содержащего конкретный заместитель по настоящему изобретению, с относительно нетоксичными кислотой или основанием. Если соединение по настоящему изобретению содержит относительно кислотную функциональную группу, то соль присоединения основания может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния, или подобные соли. Если соединение по настоящему изобретению содержит относительно основную функциональную группу, то соль присоединения кислоты может быть получена посредством приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т. п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту и метансульфоновую кислоту и т. п.; и соль аминокислоты (такой как аргинин и т. п.), и соль органической кислоты, такой как глюкуроновая кислота и т. п. Определенные конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, можно превратить в любую соль присоединения основания или соль присоединения кислоты.
[55] Фармацевтически приемлемую соль по настоящему изобретению можно получать из исходного соединения, которое содержит кислотный или основный фрагмент, с помощью традиционного химического способа. Как правило, такая соль может быть получена путем осуществления реакции свободных кислотной или основной форм соединения со стехиометрическим количеством соответствующих основания или кислоты в воде, или в органическом растворителе, или в их смеси.
[56] В дополнение к солевой форме соединение, предусмотренное в настоящем изобретении, также находится в форме пролекарства. Пролекарство на основе соединения, описанного в данном документе, представляет собой соединение, которое легко подвергается химическим изменениям в физиологических условиях с превращением в соединение по настоящему изобретению. Кроме того, пролекарство можно превращать в соединение по настоящему изобретению посредством химического или биохимического способа в условиях in vivo.
[57] Определенные соединения по настоящему изобретению могут находиться в несольватированной форме или сольватированной форме, в том числе в гидратированной форме. Как правило, сольватированная форма является эквивалентной несольватированной форме, и обе формы включены в объем настоящего изобретения.
[58] Соединение по настоящему изобретению может находиться в форме конкретного геометрического или стереоизомера. В настоящем изобретении подразумеваются все такие соединения, в том числе цис- и транс-изомер, (-)- и (+)-энантиомер, (R)- и (S)-энантиомер, диастереоизомер, (D)-изомер, (L)-изомер, и рацемическая смесь, и другие смеси, например, энантиомерно или диастереоизомерно обогащенная смесь, все из которых включены в объем настоящего изобретения. Заместитель, такой как алкил, может содержать дополнительный асимметричный атом углерода. Все такие изомеры и их смеси включены в объем настоящего изобретения.
[59] Если не указано иное, термин «энантиомер» или «оптический изомер» относится к стереоизомерам, которые представляют собой зеркальные отражения друг друга.
[60] Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» определяется неспособностью к свободному вращению вокруг двойной связи или одинарной связи между атомами углерода в кольце.
[61] Если не указано иное, термин «диастереомер» относится к стереоизомерам, молекулы которых имеют два или более хиральных центра и не являются зеркальными отражениями друг друга.
[62] Если не указано иное, «(D)» или «(+)» относится к правостороннему вращению, «(L)» или «(-)» относится к левостороннему вращению, «(DL)» или «(±)» относится к рацемической смеси.
[63] Если не указано иное, абсолютная конфигурация стереогенного центра представлена обозначенной клиновидной сплошной линией связью ( ) и обозначенной клиновидной пунктирной линией связью (
) и обозначенной клиновидной пунктирной линией связью (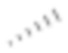 ), а относительная конфигурация стереогенного центра представлена обозначенной прямой сплошной линией связью (
), а относительная конфигурация стереогенного центра представлена обозначенной прямой сплошной линией связью ( ) и обозначенной прямой пунктирной линией связью (
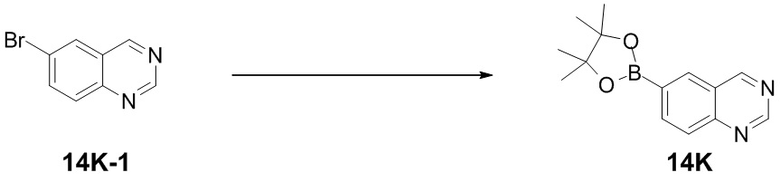
) и обозначенной прямой пунктирной линией связью (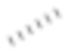 ). Волнистая линия (
). Волнистая линия ( ) означает связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или означает связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
) означает связь, обозначенную клиновидной сплошной линией (), или связь, обозначенную клиновидной пунктирной линией (), или означает связь, обозначенную прямой сплошной линией (), или связь, обозначенную прямой пунктирной линией ().
[64] Соединения по настоящему изобретению могут находиться в конкретной форме. Если не указано иное, термины «таутомер» или «таутомерная форма» относятся к тому факту, что различные функциональные изомеры находятся в состоянии динамического равновесия при комнатной температуре и могут быстро превращаться друг в друга. Если существует возможность образования таутомеров (например, в растворе), таутомеры могут достигать состояния химического равновесия. Например, протонные таутомеры (также известные как прототропные таутомеры) предусматривают взаимопревращения посредством протонного переноса, такие как кето-енольная изомеризация и имино-енаминовая изомеризация. Валентный таутомер предусматривает взаимное преобразование с участием некоторых связывающих электронов. Конкретным примером кето-енольной таутомеризации является взаимопревращение двух таутомеров пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
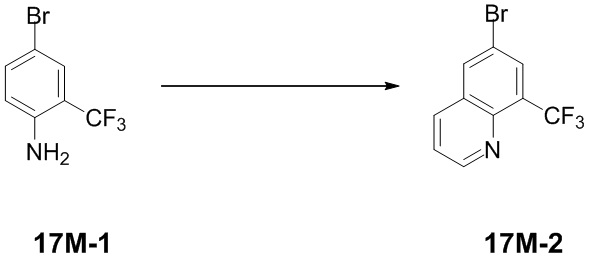
[65] Если не указано иное, термины «обогащенный одним изомером», «обогащенный изомером», «обогащенный одним энантиомером» или «обогащенный энантиомером» относятся к содержанию одного из изомеров или энантиомеров, которое составляет менее 100%, и при этом содержание изомера или энантиомера составляет 60% или больше, или 70% или больше, или 80% или больше, или 90% или больше, или 95% или больше, или 96% или больше, или 97% или больше, или 98% или больше, или 99% или больше, или 99,5% или больше, или 99,6% или больше, или 99,7% или больше, или 99,8% или больше, или 99,9% или больше.
[66] Если не указано иное, термины «избыток изомера» или «избыток энантиомера» относятся к разнице между значениями относительного процентного содержания двух изомеров или энантиомеров. Например, если содержание одного из изомеров или энантиомеров составляет 90%, а другого составляет 10%, то избыток изомера или энантиомера (значение ee) составляет 80%.
[67] Оптически активный (R)- и (S)-изомер или D- и L-изомер можно получить с применением хирального синтеза или хиральных реагентов, или других общепринятых методик. Если требуется получение одного типа энантиомера конкретного соединения по настоящему изобретению, чистый необходимый энантиомер может быть получен путем асимметрического синтеза или дериватизации с помощью хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. В качестве альтернативы, если молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксильная), соединение реагирует с соответствующими оптически активными кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают диастереомерному разделению посредством общепринятого способа, известного из уровня техники, с получением чистого энантиомера. Кроме того, энантиомеры и диастереоизомеры обычно выделяют посредством хроматографии, в которой используется хиральная неподвижная фаза, необязательно в комбинации со способом химической дериватизации (например, карбамат, полученный из амина). Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов при одном или более атомах, которые составляют соединение. Например, соединение может быть мечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). В качестве другого примера, водород может быть заменен тяжелым водородом с образованием дейтерированного лекарственного средства, и при этом связь, образуемая между дейтерием и углеродом, является более прочной, чем связь, образуемая между обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами дейтерированные лекарственные средства характеризуются менее выраженными побочными эффектами и более высокой стабильностью лекарственного средства с усилением эффективности и продлением биологического периода полувыведения лекарственного средства. Все изотопные варианты соединения по настоящему изобретению, вне зависимости от радиоактивности, включены в объем настоящего изобретения. Термин «фармацевтически приемлемый носитель» относится к любому средству или несущей среде, которые способны доставлять эффективное количество активного вещества по настоящему изобретению, не оказывают отрицательного воздействия на биологическую активность активного вещества и не вызывают какого-либо токсичного побочного эффекта у хозяина или пациента. Иллюстративный носитель включает воду, растительное и минеральное масло, основу для крема, основу для лосьона, основу для мази и т. п. Основа содержит суспендирующее средство, загуститель, вещество, способствующее проникновению, и т. п. Их составы хорошо известны специалисту в области косметических средств или в области фармацевтических препаратов для местного применения.
[68] Термин «вспомогательное вещество» обычно относится к носителю, разбавителю и/или среде, необходимым для составления эффективной фармацевтической композиции.
[69] Применительно к лекарственному препарату или фармакологически активному средству термин «эффективное количество» или «терапевтически эффективное количество» относится к нетоксичному, но достаточному количеству для достижения необходимого эффекта лекарственного препарата или средства. Применительно к лекарственной форме по настоящему изобретению для перорального применения «эффективное количество» активного вещества в композиции относится к количеству, необходимому для достижения необходимого эффекта при объединении с другим активным веществом в композиции. Эффективное количество отличается для каждого человека и определяется в зависимости от возраста и общего состояния реципиента, а также от конкретного активного вещества. Соответствующее эффективное количество в каждом отдельном случае может быть определено специалистом в данной области техники на основе обычного эксперимента.
[70] Термины «активный ингредиент», «терапевтическое средство», «активное вещество» или «активное средство» относятся к химическому соединению, с помощью которого можно эффективно лечить целевое нарушение, заболевание или состояние.
[71] «Необязательный» или «необязательно» означает, что последующее событие или условие может реализовываться, но не является необходимым, и что термин включает случаи, в которых событие или условие реализуется, и случаи, в которых событие или условие не реализуется.
[72] Термин «замещенный» означает, что один или более атомов водорода при конкретном атоме замещены заместителем, в том числе дейтерием и вариантами водорода, при условии, что валентность конкретного атома является нормальной, и замещенное соединение является стабильным. Если заместитель представляет собой атом кислорода (т. е. =O), то это означает, что два атома водорода являются замещенными. Положения в ароматическом кольце не могут быть замещены кетогруппой. Термин «необязательно замещенный» означает, что атом может быть замещенным или не замещенным заместителем, если не указано иное, причем тип и число заместителей могут быть произвольными при условии, что это химически достижимо.
[73] Если любая переменная (такая как R) встречается более одного раза в составе или структуре соединения, то определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, то данная группа может быть необязательно замещена не более чем двумя R, где определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта является допустимой, только если такая комбинация приводит к образованию стабильного соединения.
[74] Если количество линкерных групп равно 0, например, -(CRR)0-, это означает, что линкерная группа представляет собой одинарную связь.
[75] Если одна из переменных выбрана из одинарной связи, это означает, что две группы, соединенные одинарной связью, соединены непосредственно. Например, если L в A-L-Z представляет собой одинарную связь, то структура A-L-Z фактически представляет собой A-Z.
[76] Если заместитель не указан, это означает, что заместитель отсутствует. Например, если X не указан в A-X, то структура A-X фактически представляет собой A. Если связь заместителя может быть перекрестно соединена с более чем одним атомом в кольце, то такой заместитель может быть связан с любым атомом кольца. Например, структурное звено 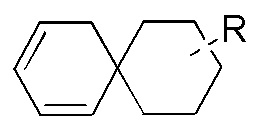 или
или 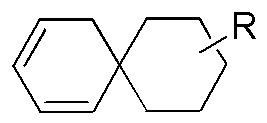 означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть связан с замещаемой группой посредством любого атома углерода в пиридиновом кольце. Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в
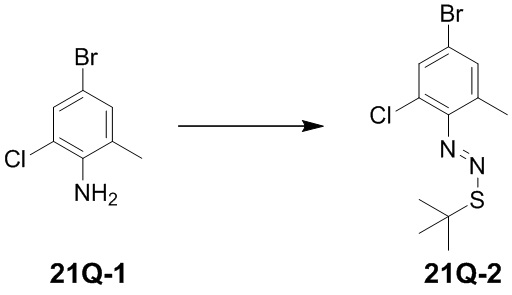
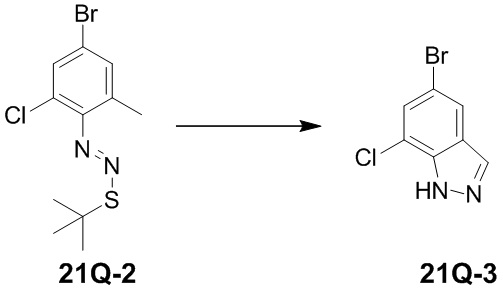
означает, что заместитель R может находиться в любом положении в циклогексиле или циклогексадиене. Если в перечисленном заместителе не указано, посредством какого атома он связан с замещаемой группой, такой заместитель может быть связан посредством любого его атома. Например, если пиридил выполняет функцию заместителя, он может быть связан с замещаемой группой посредством любого атома углерода в пиридиновом кольце. Если в перечисленной линкерной группе не указано направление связывания, то направление связывания является произвольным; например, если линкерная группа L, содержащаяся в  , представляет собой -MW-, то -MW- может связывать кольцо A и кольцо B с образованием
, представляет собой -MW-, то -MW- может связывать кольцо A и кольцо B с образованием 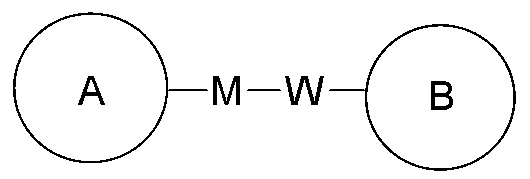 в направлении, соответствующем порядку чтения слева направо, и с образованием
в направлении, соответствующем порядку чтения слева направо, и с образованием 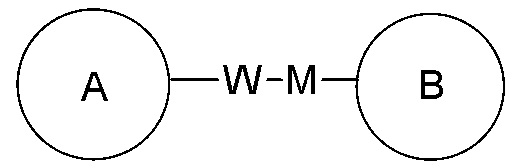 в направлении, противоположном порядку чтения слева направо. Комбинация заместителей и/или их вариантов является допустимой, только если такая комбинация может приводить к образованию стабильного соединения.
в направлении, противоположном порядку чтения слева направо. Комбинация заместителей и/или их вариантов является допустимой, только если такая комбинация может приводить к образованию стабильного соединения.
[77] Если не указано иное, термин «гетеро» означает гетероатом или гетероатомную группу (например, группу атомов, содержащую гетероатом), в том числе атом, отличный от атома углерода (C) и водорода (H), и группу атомов, содержащую вышеуказанный гетероатом, например, в том числе атом кислорода (O), азота (N), серы (S), кремния (Si), германия (Ge), алюминия (Al), бора (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- и группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждый из которых необязательно замещен.
[78] Если не указано иное, термин «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одно кольцо, бициклическое кольцо, спирокольцо, конденсированное кольцо или кольцо с мостиковой связью. Число атомов в кольце обычно определено как число членов в кольце, например, «5-7-членное кольцо» означает, что от 5 до 7 атомов расположены в виде кольца. Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Следовательно, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует вышеуказанному определению.
[79] Если не указано иное, термин «гетероцикл» или «гетероциклил» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим), и может содержать атомы углерода и 1, 2, 3 или 4 гетероатома в кольце, независимо выбранных из N, O и S, где любой из вышеуказанного гетероцикла может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, p равняется 1 или 2). Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероцикл может быть присоединен к боковой группе посредством любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, гетероцикл, описанный в данном документе, может быть замещен в положении, соответствующему атому углерода или азота. Атом азота в гетероцикле необязательно кватернизирован. В предпочтительном варианте осуществления, если общее число атомов S и O в гетероцикле превышает 1, то гетероатомы не являются смежными друг с другом. В другом предпочтительном варианте осуществления общее число атомов S и O в гетероцикле не превышает 1. Как используется в данном документе, термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 гетероатома кольца, независимо выбранных из N, O и S. Атом азота может быть замещенным или незамещенным (т. е. N или NR, где R представляет собой H или другие заместители, уже определенные в данном документе). Гетероатомы, представляющие собой азот и серу, необязательно могут быть окислены (т. е. NO и S(O)p, p равняется 1 или 2). Следует отметить, что общее количество атомов S и O в ароматическом гетероцикле не превышает один. Кольцо с мостиковой связью также включено в определение гетероцикла. Кольцо с мостиковой связью образуется, если один или более атомов (т. е. C, O, N или S) соединяют два несмежных атома углерода или азота. Предпочтительное кольцо с мостиковой связью включает без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостиковая связь всегда превращает моноциклическое кольцо в трициклическое кольцо. В кольце с мостиковой связью заместитель в кольце также может присутствовать при мостиковой связи.
[80] Примеры гетероциклического соединения включают без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксиндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазолил, пиридоимидазолил, пиридотиазолил, пиридинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил, 4H-1,2,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиросоединения.
[81] Если не указано иное, термин «гидрокарбил» или его гипонимы (например, алкил, алкенил, алкинил и арил и т. д.), сами по себе или как часть другого заместителя, относятся к линейной, разветвленной цепи, или циклическому углеводородному радикалу, или любой их комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или поливалентными (например, метенил), могут также включать двухвалентную или поливалентную группу, имеют конкретное число атомов углерода (например, C1-C12 означает от 1 до 12 атомов углерода, причем C1-12 выбран из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12; C3-12 выбран из C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12). Термин «гидрокарбил» включает без ограничения алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности, включает без ограничения алкил, алкенил и алкинил. Ароматический гидрокарбил включает без ограничения 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и т. п. В некоторых вариантах осуществления термин «гидрокарбил» относится к линейной или разветвленной группе, или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной, и может включать двухвалентную или поливалентную группу. Примеры насыщенной гидрокарбильной группы включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амила, н-гексила, н-гептила, н-октила и других групп атомов. Ненасыщенный гидрокарбил содержит одну или более двойных или тройных связей. Примеры ненасыщенного алкила включают без ограничения винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и гомологи и изомеры более высокого порядка.
[82] Если не указано иное, термин «гетерогидрокарбил» или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил и гетероарил и т. д.), сами по себе или как часть другого заместителя, относятся к стабильной линейной, разветвленной или циклической углеводородной группе или любой их комбинации, которая содержит конкретное число атомов углерода и по меньшей мере один гетероатом. В некоторых вариантах осуществления термин «гетероалкил» сам по себе или в комбинации с другим термином относится к стабильной линейной цепи, разветвленному углеводородному радикалу или их комбинации, которая содержит конкретное число атомов углерода и по меньшей мере один гетероатом. В конкретном варианте осуществления гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероатом или гетероатомная группа могут находиться в любом внутреннем положении гетерогидрокарбила, в том числе в положении, где гидрокарбил присоединяется к остальной части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкил) используются в их общепринятом значении и означают алкильную группу, соединенную с остальной частью молекулы посредством атома кислорода, аминогруппы или атома серы соответственно. Примеры включают без ограничения -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Могут присутствовать не более двух последовательно расположенных гетероатомов, например -CH2-NH-OCH3.
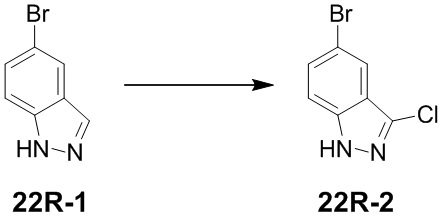
[83] Если не указано иное, термины «циклогидрокарбил», «гетероциклогидрокарбил» или их гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т. д.), применяемые сами по себе или в комбинации с другим термином, относятся к циклизированному «гидрокарбилу» или «гетерогидрокарбилу». Кроме того, в случае гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкил и гетероциклоалкил) один гетероатом может занимать положение, в котором гетероцикл присоединяется к остальной части молекулы. Примеры циклогидрокарбила включают без ограничения циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т. п. Неограничивающие примеры гетероциклила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[84] Если не указано иное, термин «алкил» означает линейную цепь или разветвленную насыщенную углеводородную группу, которая может быть монозамещенной (например, -CH2F) или полизамещенной (например, -CF3), может быть одновалентной (например, метил), двухвалентной (например, метилен) или поливалентной (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т. п.
[85] Если не указано иное, термин «алкенил» относится к алкильной группе, содержащей одну или более углерод-углеродных двойных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или поливалентной. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т. п.
[86] Если не указано иное, термин «алкинил» относится к алкильной группе, содержащей одну или более углерод-углеродных тройных связей в любом положении в цепи, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или поливалентной. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и т. п.
[87] Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил и любой атом углерода, который является насыщенным, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают без ограничения циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и т. п.
[88] Если не указано иное, циклоалкенил включает любой стабильный циклический или полициклический гидрокарбил, содержащий одну или более ненасыщенных углерод-углеродных одинарных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкенила включают без ограничения циклопентенил, циклогексенил и т. п.
[89] Если не указано иное, циклоалкинил включает любой стабильный циклический или полициклический гидрокарбил, содержащий одну или более углерод-углеродных тройных связей в любом положении в кольце, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным.
[90] Если не указано иное, термин «галоген», сам по себе или как часть другого заместителя, относится к атому фтора, хлора, брома или йода. Кроме того, подразумевается, что термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген(C1-C4)алкил» включает без ограничения трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т. п. Примеры галогеналкила включают без ограничения трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[91] Термин «алкокси» означает алкил, определенный выше, содержащий конкретное число атомов углерода, присоединенных посредством кислородного мостика. Если не указано иное, C1-6алкокси включает C1-, C2-, C3-, C4-, C5- и C6алкокси. Примеры алкокси включают без ограничения метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
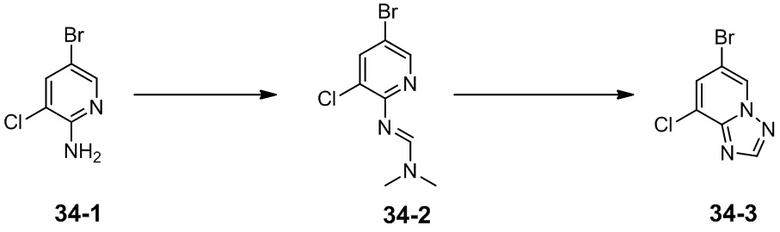
[92] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, который может быть моно-, ди- или полизамещенным, может быть одновалентным, двухвалентным или многовалентным, может представлять собой одно кольцо или несколько колец (например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые являются вместе конденсированными или соединенными ковалентно. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизирован. Гетероарил может быть присоединен к остальной части молекулы посредством гетероатома. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель в любой из указанных выше арильных и гетероарильных кольцевых систем выбран из приемлемых заместителей, описанных ниже.
[93] Если не указано иное, при использовании арила в сочетании с другими терминами (например, арилокси, арилтио, арилалкил) арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, подразумевается, что термин «аралкил» включает группу (например, бензил, фенэтил, пиридилметил и т. д.), где арил присоединен к алкилу, в том числе к алкилу, где атом углерода (например, метилен) был заменен таким атомом, как атом кислорода, например, феноксиметил, 2-пиридилокси, 3-(1-нафтилокси)пропил и т. п.
[94] Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть заменены другой функциональной группой или атомом посредством реакции замещения (такой как реакция замещения по аффинности). Например, иллюстративные уходящие группы включают трифлат; хлор, бром и йод; сульфонатную группу, такую как мезилат, тозилат, п-бромбензолсульфонат, п-толуолсульфонаты и т. п.; ацилоксигруппу, такую как ацетокси, трифторацетокси и т. п.
[95] Термин «защитная группа» включает без ограничения «защитную группу для аминогруппы», «защитную группу для гидроксигруппы» или «защитную группу для тиогруппы». Термин «защитная группа для аминогруппы» относится к защитной группе, подходящей для блокирования побочной реакции с участием атома азота аминогруппы. Иллюстративные защитные группы для аминогруппы включают без ограничения формил, ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т. п. Термин «защитная группа для гидроксигруппы» относится к защитной группе, подходящей для блокирования побочной реакции с участием гидроксигруппы. Иллюстративные защитные группы для гидроксигруппы включают без ограничения алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и т. п.
[96] Соединение по настоящему изобретению можно получать посредством различных способов синтеза, хорошо известных специалистам в данной области техники, в том числе посредством следующего перечисленного варианта осуществления, варианта осуществления, образованного следующим перечисленным вариантом осуществления в комбинации с другими способами химического синтеза и эквивалентных замен, хорошо известных специалистам в данной области техники. Предпочтительный вариант осуществления включает без ограничения вариант осуществления настоящего изобретения.
[97] Все растворители, используемые в настоящем изобретении, являются коммерчески доступными. В настоящем изобретении применяются следующие сокращения: «водн.» означает водный; «HATU» означает гексафторфосфат O-(7-азабензoтриазол-1-ил)-N,N,N',N'-тетраметилурония; «EDC» означает гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида; «m-CPBA» означает 3-хлорпероксибензойную кислоту; «экв.» означает эквивалент; «CDI» означает карбонилдиимидазол; «DCM» означает дихлорметан; «PE» означает петролейный эфир; «DIAD» означает диизопропилазодикарбоксилат; «DMF» означает N,N-диметилформамид; «DMSO» означает диметилсульфоксид; «EtOAc» означает этилацетат; «EtOH» означает этанол; «MeOH» означает метанол; «CBz» означает бензилоксикарбонил, который представляет собой защитную группу для амина; «BOC» означает трет-бутоксикарбонил, который представляет собой защитную группу для амина; «HOAc» означает уксусную кислоту; «NaCNBH3» означает цианоборогидрид натрия; «к. т.» означает комнатную температуру; «O/N» означает в течение ночи; «THF» означает тетрагидрофуран; «Boc2O» означает ди-трет-бутилдикарбонат; «TFA» означает трифторуксусную кислоту; «DIPEA» означает диизопропилэтиламин; «SOCl2» означает тионилхлорид; «CS2» означает сероуглерод; «TsOH» означает п-толуолсульфоновую кислоту; «NFSI» означает N-фтор-N-(фенилсульфонил)бензолсульфонамид; «NCS» означает 1-хлорпирролидин-2,5-дион; «n-Bu4NF» означает фторид тетрабутиламмония; «iPrOH» означает 2-пропанол; «т. пл.» означает точку плавления; «LDA» означает диизопропиламид лития; NBS означает N-бромсукцинимид.
[98] Названия соединениям даны вручную или с помощью программного обеспечения ChemDraw®, а для коммерчески доступных соединений используются их названия в соответствии с каталогом поставщика.
[99] Синтез промежуточных соединений является таким, как описано далее.
Подробное описание предпочтительного варианта осуществления
[100] Следующие варианты осуществления дополнительно иллюстрируют настоящее изобретение, но настоящее изобретение ими не ограничивается. Настоящее изобретение было описано подробно в данном документе, а также были раскрыты конкретные варианты осуществления. Специалистам в данной области техники будет очевидно, что в отношении конкретных вариантов осуществления настоящего изобретения можно осуществлять различные изменения и модификации без отступления от сущности и объема настоящего изобретения.
[101] Синтез промежуточного соединения 3C

[102] Соединение бис(пинаколато)дибора (25,61 г, 100,85 ммоль, 0,65 экв.), димер (1,5)-циклооктадиенметоксииридия (308,56 мг, 465,48 мкмоль, 0,003 экв.) и 4,4-ди-трет-бутил-2,2-бипиридин (249,88 мг, 930,96 мкмоль, 0,006 экв.) растворяли в н-гексане (250 мл) и реакционную смесь перемешивали при 50°C в защитной атмосфере азота до тех пор, пока цвет реакционной смеси не становился темно-красным. К указанному выше раствору добавляли соединение 3C-1 и затем реакционную смесь перемешивали в течение 3 часов при 50°C в защитной атмосфере азота. LCMS показала полное превращение в продукт гидролиза, представляющий собой соединение эфира бороновой кислоты 3C. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой соединение 3C, который непосредственно применяли на следующей стадии без очистки.
[103] Соответствующие данные характеристик: LCMS масса/заряд: 206,1 [M+H] (что демонстрирует, что эфир бороновой кислоты гидролизовался до борной кислоты).
[104] 1H ЯМР (400 МГц, CDCl3): δ 7,83 (s, 1H), 7,70 (s, 1H), 1,37 (s, 12H).
[105] Синтез промежуточного соединения 5E

[106] Соединение 5E-1 (0,5 г, 2,39 ммоль, 1 экв.), бис(пинаколато)дибор (668,12 мг, 2,63 ммоль, 1,1 экв.), дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия(II) (87,51 мг, 119,59 мкмоль, 0,05 экв.), ацетат калия (352,11 мг, 3,59 ммоль, 1,5 экв.) растворяли в 10 мл диоксана и проводили реакцию при 80°C в течение 12 часов в защитной атмосфере азота. LCMS показала полное превращение в продукт. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой соединение 5E, который непосредственно применяли на следующей стадии без очистки.
[107] Соответствующие данные характеристик: LCMS масса/заряд: 257,2 [M+H].
[108] 1H ЯМР (400 МГц, CDCl3): δ 8,87 (d, J = 4,8 Гц, 2H), 8,61 (s, 1H), 8,15 (dd, J = 20,4, 8,4 Гц, 2H), 1,40 (s, 12H).
[109] Синтез промежуточного соединения 6F
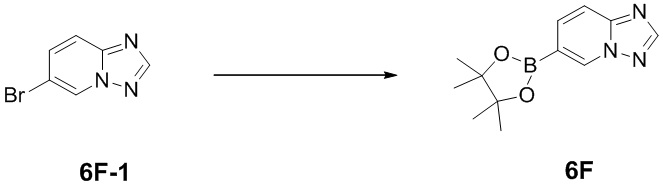
[110] Соединение 6F-1 (0,5 г, 2,39 ммоль, 1 экв.), бис(пинаколато)дибор (769,43 мг, 3,03 ммоль, 1,2 экв.), дихлорид [1,1-бис(дифенилфосфино)ферроцен]палладия(II) (92,38 мг, 126,25 мкмоль, 0,05 экв.), ацетат калия (743,43 мг, 7,57 ммоль, 3 экв.) растворяли в 5 мл диоксана и проводили реакцию при 80°C в течение 12 часов в защитной атмосфере азота. LCMS показала полное превращение в продукт. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой соединение 6F, который непосредственно применяли на следующей стадии без очистки.
[111] Соответствующие данные характеристик: LCMS масса/заряд: 246,1 [M+H].
[112] 1H ЯМР (400 МГц, CDCl3): δ 8,97 (s, 1H), 8,36 (s, 1H), 8,15 (dd, J = 8,0, 23,6 Гц, 2H), 1,35 (s, 12H).
[113] Синтез промежуточного соединения 7G

[114] Соединение 7G-1 (0,135 г, 681,75 мкмоль, 1 экв.), бис(пинаколато)дибор (190,43 мг, 749,92 мкмоль, 1,1 экв.), ацетат калия (100,36 мг, 1,02 ммоль, 1,5 экв.), трис(дибензилиденацетон)дипалладий (31,21 мг, 34,09 мкмоль, 0,05 экв.), PCy3 (19,12 мг, 68,17 мкмоль, 22,10 мкл, 0,1 экв.) растворяли в 2 мл диоксана и перемешивали при 80°C в течение 12 часов в защитной атмосфере азота. LCMS показала полное превращение в продукт. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, представляющего собой соединение 7G, который непосредственно применяли на следующей стадии без очистки.
[115] Соответствующие данные характеристик: LCMS масса/заряд: 164,1 [M+H] (MS продукта гидролиза, представляющего собой соединение 8H)
[116] 1H ЯМР (400 МГц, CDCl3): δ 8,59 (d, J = 6,8 Гц, 1H), 8,39 (d, J = 6,8 Гц, 1H), 7,62 (s, 1H), 8,36 (s, 1H), 1,40 (s, 12H).
[117] Синтез промежуточного соединения 8H
[118] Дихлорид (1,1'-бис(дифенилфосфино)ферроцен)палладия(II) (345,00 мг, 471,51 мкмоль, 0,2 экв.), ацетат калия (694,12 мг, 7,07 ммоль, 3,0 экв.) и соединение 8H-1 (500 мг, 2,36 ммоль, 1,0 экв.) добавляли в раствор бис(пинаколато)дибора (718,40 мг, 2,83 ммоль, 1,2 экв.) в 1,4-диоксане (3 мл), смесь нагревали до 90°C в защитной атмосфере азота и перемешивали в течение 10 часов. Смесь фильтровали, и концентрировали, и очищали с помощью колоночной хроматографии (SiO2, градиентное элюирование с помощью PE/этилацетат с объемным соотношением от 20/1 до 10/1) с получением соединения 8H.
[119] Соответствующие данные характеристик: LCMS масса/заряд: 260,1 [M+H].
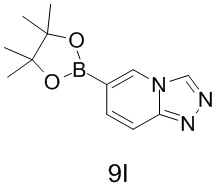
[120] Синтез промежуточного соединения 9I

[121] Стадия 1 (синтез соединения 9I-2)

[122] п-Толуолсульфоновую кислоту (228,96 мг, 1,33 ммоль, 0,05 экв.) добавляли в раствор соединения 9I-1 (5 г, 26,59 ммоль, 1 экв.) в триэтилортоформиате (50 мл) и реакционную смесь перемешивали при 110°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали и выпаривали до сухого состояния. Остаток разбавляли водой (50 мл), регулировали до pH=9 насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом. Органические фазы объединяли и промывали насыщенным солевым раствором (30 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния. Неочищенный продукт отделяли и очищали с помощью колоночной хроматографии (градиентное элюирование с помощью PE/этилацетат с объемным соотношением от 3/1 до чистого этилацетата) с получением соединения 9I-2.
[123] Соответствующие данные характеристик: LCMS масса/заряд: 197,9 [M+H].
[124] 1H ЯМР (400 МГц, CDCl3): δ 8,80 (s, 1H), 8,32 (d, J = 1,2 Гц, 1H), 7,73 (t, J = 13,8 Гц, 1H), 7,35 (dd, J = 1,6, 9,6 Гц, 1H).
[125] Стадия 2 (синтез соединения 9I)

[126] В защитной атмосфере азота раствор соединения 9I-2 (0,5 г, 2,52 ммоль, 1 экв.), пинаколового эфира бороновой кислоты (705,31 мг, 2,78 ммоль, 1,1 экв.), Pd(dppf)2Cl2.CH2Cl2 (103,10 мг, 126,25 мкмоль, 0,05 экв.) и ацетата калия (371,71 мг, 3,79 ммоль, 1,5 экв.) в диоксане (5 мл) перемешивали при 80°C в течение 12 часов. LCMS показала, что реакция была завершена, затем реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до сухого состояния с получением неочищенного продукта, представляющего собой соединение 9I.
[127] Соответствующие данные характеристик: LCMS масса/заряд: 197,9 [M+H].
[128] 1H ЯМР (400 МГц, CDCl3): δ 8,81 (s, 1H), 8,53 (s, 1H), 7,75 (t, J = 9,6 Гц, 1H), 7,58 (t, J = 9,6 Гц, 1H), 1,37 (s, 12H).
[129] Синтез промежуточного соединения 13J

[130] Соединение 13J-1 (50 мг, 239,19 мкмоль, 1 экв.), бис(пинаколато)дибор (66,81 мг, 263,10 мкмоль, 1,1 экв.), ацетат калия (46,95 мг, 478,37 мкмоль, 2 экв.) и Pd(dppf)2Cl2 (15,59 мг, 23,92 мкмоль, 0,1 экв.) добавляли в 1,4-диоксан (2 мл). Смесь нагревали до 80°C в защитной атмосфере азота и перемешивали при 80°C в течение 10 часов. LC-MS показала, что исходные вещества исчезли и выявленное значение MS для борной кислоты соответствует 13J. На основании TLC определили, что продукт представляет собой эфир бороновой кислоты. Реакционную смесь фильтровали и концентрировали с получением неочищенного продукта, представляющего собой соединение 13J, который непосредственно применяли на следующей стадии без дополнительной очистки.
[131] Соответствующие данные характеристик: LCMS масса/заряд: 175,1 [M+H].
[132] Синтез промежуточного соединения 14K
[133] Исходное вещество 14K-1 (200 мг, 956,74 мкмоль, 1 экв.), бис(пинаколато)дибор (267,25 мг, 1,05 ммоль, 1,1 экв.), ацетат калия (187,79 мг, 1,91 ммоль, 2 экв.), Pd(dppf)2Cl2 (62,36 мг, 95,67 мкмоль, 0,1 экв.) добавляли в 1,4-диоксан, смесь нагревали до 80°C в защитной атмосфере азота и перемешивали при 80°C в течение 4 часов. LCMS показала, что исходные вещества исчезли и выявленное значение MS для борной кислоты соответствует 14K. Реакционную смесь фильтровали, концентрировали и очищали с помощью пластины с силикагелем для препаративной хроматографии (SiO2, EA/PE = 3/1) с получением 14K.
[134] Соответствующие данные характеристик: LCMS масса/заряд: 175,2 [M+H].
[135] Синтез промежуточного соединения 17M

[136] Стадия 1 (синтез соединения 17M-2)
[137] Глицерин (2,88 г, 31,25 ммоль, 2,34 мл, 1,5 экв.) добавляли к раствору соединения 17M-1 (5 г, 20,83 ммоль, 2,92 мл, 1 экв.) и йодида натрия (62,45 мг, 416,63 мкмоль, 0,02 экв.) в концентрированной серной кислоте (20 мл) и реакционную смесь перемешивали при 110°C в течение 5 часов, затем нагревали до 120°C и перемешивали в течение 12 часов. Реакционную смесь медленно выливали в ледяную воду (30 мл), экстрагировали этилацетатом (25 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором (25 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением неочищенного продукта, представляющего собой соединение 17M-2.
[138] Соответствующие данные характеристик: LCMS масса/заряд: 275,9 [M+H].
[139] Стадия 2 (синтез соединения 17M)

[140] В защитной атмосфере азота раствор соединения 17M-2 (1 г, 3,62 ммоль, 1 экв.), пинаколового эфира бороновой кислоты (4,60 г, 18,11 ммоль, 5 экв.), ацетата калия (1,07 г, 10,87 ммоль, 3 экв.) и Pd(dbcp)2Cl2 (118,05 мг, 181,13 мкмоль, 0,05 экв.) в диоксане (50 мл) перемешивали при 90°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток отделяли и очищали с помощью колоночной хроматографии (PE/этилацетат = 40/1, силикагель 100-200 меш) с получением неочищенного продукта, представляющего собой соединение 17M.
[141] Соответствующие данные характеристик: LCMS масса/заряд: 324,1 [M+H].
[142] Синтез промежуточного соединения 18N

[143] Исходное вещество 18N-1 (500 мг, 2,40 ммоль, 1 экв.), бис(пинаколато)дибор (610,27 мг, 2,40 ммоль, 1 экв.), Pd(dppf)2Cl2 (175,85 мг, 240,32 мкмоль, 0,1 экв.), ацетат калия (471,70 мг, 4,81 ммоль, 2 экв.) растворяли в 1,4-диоксане (22 мл), смесь нагревали до 90°C в защитной атмосфере азота и перемешивали при 90°C в течение 10 часов. LCMS показала, что исходные вещества исчезли и выявленное значение MS для борной кислоты соответствует 18N. На основании TLC определили, что продукт представляет собой эфир бороновой кислоты. Реакционную смесь фильтровали и концентрировали с получением неочищенного продукта, представляющего собой соединение 18N.
[144] Соответствующие данные характеристик: LCMS масса/заряд: 174,2 [M+H].
[145] Синтез промежуточного соединения 19O

[146] Пинаколовый эфир бороновой кислоты (3,09 г, 12,18 ммоль, 1,2 экв.) и ацетат калия (2,99 г, 30,45 ммоль, 3 экв.) добавляли к раствору соединения 19O-1 (2,0 г, 10,15 ммоль, 1 экв.) в 1,4-диоксане (30 мл) и смесь три раза продували азотом, затем добавляли Pd(pph3)2Cl2 (712,47 мг, 1,02 ммоль, 0,1 экв.). Систему продували несколько раз азотом, затем нагревали до 90°C и перемешивали в течение 1,5 часа. LCMS показала, что исходные вещества исчезли и выявленное значение MS для борной кислоты соответствует 19O. На основании TLC определили, что продукт представляет собой эфир бороновой кислоты. Реакционную смесь охлаждали и фильтровали и фильтрат концентрировали с получением неочищенного продукта, представляющего собой соединение 19O, который непосредственно применяли на следующей стадии без дополнительной очистки.
[147] Соответствующие данные характеристик: LCMS масса/заряд: 163,3 [M+H].
[148] Синтез промежуточного соединения 21Q

[149] Стадия 1 (синтез соединения 21Q-2)
[150] Водный раствор (2 мл) нитрита натрия (1,31 г, 19,05 ммоль, 1,05 экв.) медленно по каплям добавляли к водному раствору соединения 21Q-1 (4 г, 18,14 ммоль, 1 экв.) в хлористоводородной кислоте (5 мл, весовое содержание 24%) при -5°C. Смесь перемешивали в течение 30 минут и регулировали до pH 5 путем добавления твердого ацетата натрия. Затем в реакционную смесь добавляли этанольный раствор (20 мл) трет-бутилмеркаптана (1,64 г, 18,14 ммоль, 2,04 мл, 1 экв.) при 0°C. Реакционную смесь перемешивали в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь гасили путем добавления ледяной воды и образовывался осадок. Смесь фильтровали и осадок на фильтре промывали водой и высушивали с получением неочищенного продукта, представляющего собой соединение 21Q-2, который непосредственно применяли на следующей стадии без дополнительной очистки.
[151] Соответствующие данные характеристик: LCMS масса/заряд: 322,9 [M+H].
[152] Стадия 2 (синтез соединения 21Q-3)
[153] Добавляли трет-бутоксид калия (10,26 г, 91,40 ммоль, 10 экв.) к раствору соединения 21Q-2 (3 г, 9,14 ммоль, 1 экв.) в диметилсульфоксиде (35 мл) и смесь перемешивали при 25°C в течение 30 минут. TLC показала, что исходные вещества исчезли и с помощью LCMS выявили образование продукта. Реакционную смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (35 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором (35 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением неочищенного продукта, представляющего собой соединение 21Q-3, который непосредственно применяли на следующей стадии без дополнительной очистки.
[154] Соответствующие данные характеристик: LCMS масса/заряд: 232,9 [M+H].
[155] Стадия 3 (синтез соединения 21Q)

[156] Раствор соединения 21Q-3 (0,5 г, 2,16 ммоль, 1 экв.), пинаколового эфира бороновой кислоты (877,63 мг, 3,46 ммоль, 1,6 экв.), ацетата калия (635,97 мг, 6,48 ммоль, 3 экв.) и Pd(dppf)2Cl2 (79,03 мг, 108,00 мкмоль, 0,05 экв.) в диметилсульфоксиде (2 мл) перемешивали при воздействии микроволнового излучения при 120°C в течение 1 часа. С помощью LCMS выявили образование продукта. Реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до сухого состояния. Неочищенный продукт отделяли и очищали с помощью колоночной хроматографии (PE/этилацетат = от 10/1 до 5/1) с получением неочищенного продукта, представляющего собой соединение 21Q, который непосредственно применяли на следующей стадии.
[157] Соответствующие данные характеристик: LCMS масса/заряд: 279,1 [M+H].
[158] Синтез промежуточного соединения 22R

[159] Стадия 1 (синтез соединения 22R-2)
[160] Добавляли NCS (745,49 мг, 5,58 ммоль, 1,1 экв.) к раствору соединения 22R-1 (1 г, 5,08 ммоль, 1 экв.) в ацетонитриле (10 мл) и реакционную смесь перемешивали при 60°C в течение 5 часов. LC-MS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором (20 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением неочищенного продукта, представляющего собой соединение 22R-2.
[161] Соответствующие данные характеристик: LCMS масса/заряд: 232,9 [M+H].
[162] Стадия 2 (синтез соединения 22R)

[163] В защитной атмосфере азота раствор соединения 22R-2 (0,2 г, 864,02 мкмоль, 1 экв.), пинаколового эфира бороновой кислоты (438,81 мг, 1,73 ммоль, 2 экв.), ацетата калия (169,59 мг, 1,73 ммоль, 2 экв.) и Pd(dppf)2Cl2 (56,31 мг, 86,40 мкмоль, 0,1 экв.) в диоксане (10 мл) перемешивали при 90°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до сухого состояния. Остаток отделяли и очищали с помощью колоночной хроматографии (PE/этилацетат = от 10:1 до 5:1) с получением неочищенного продукта, представляющего собой соединение 22R.
[164] Соответствующие данные характеристик: LCMS масса/заряд: 279,1 [M+H].
[165] Синтез промежуточного соединения 23S

[166] Исходное вещество 23S-1 (500 мг, 2,34 ммоль, 1 экв.), бис(пинаколато)дибор (652,39 мг, 2,57 ммоль, 1,1 экв.), Pd(dppf)2Cl2 (170,89 мг, 233,56 мкмоль, 0,1 экв.), ацетат калия (458,43 мг, 4,67 ммоль, 2 экв.) растворяли в 1,4-диоксане (22 мл), смесь нагревали до 80°C в защитной атмосфере азота и перемешивали при 80°C в течение 10 часов. LC-MS показала, что исходные вещества исчезли и образовался продукт. Смесь фильтровали и концентрировали с получением неочищенного продукта 23S, который непосредственно применяли на следующей стадии без дополнительной очистки.
[167] Соответствующие данные характеристик: LCMS масса/заряд: 262,0 [M+H].
[168] Синтез промежуточного соединения 34-3
[169] Стадия 1 (синтез соединения 34-2)
[170] Соединение 34-1 (2,0 г, 9,64 ммоль, 1 экв.) и DMF-DMA (1,72 г, 14,46 ммоль, 1,5 экв.) растворяли в метаноле (50 мл) и реакционную смесь нагревали до 70°C и перемешивали в течение 6,5 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь охлаждали и выпаривали при пониженном давлении с удалением метанола с получением соединения 34-2. Неочищенный продукт непосредственно применяли на следующей стадии без дополнительной очистки.
[171] Соответствующие данные характеристик: LCMS масса/заряд: 264,0 [M+H].
[172] Стадия 2 (синтез соединения 34-3)
[173] Соединение 34-2 (2,49 г, 9,48 ммоль, 1 экв.) растворяли в метаноле (40 мл) и охлаждали до 0°C, затем добавляли пиридин (1,50 г, 18,97 ммоль, 1,53 мл, 2 экв.) и гидроксиламинсульфоновую кислоту (1,39 г, 12,33 ммоль, 1,3 экв.) в защитной атмосфере азота. Смесь медленно нагревали до 25°C и перемешивали в течение 4 часов, затем нагревали до 70°C и перемешивали в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь концентрировали и разбавляли этилацетатом (20 мл) и добавляли насыщенный раствор бикарбоната натрия (15 мл). Смешанный раствор экстрагировали этилацетатом (20 мл * 3). Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали, концентрировали и очищали на колонке с силикагелем (PE: EA = от 8/1 до 6/1) с получением соединения 34-3.
[174] Соответствующие данные характеристик: LCMS масса/заряд: 234,1 [M+H].
[175] Вариант осуществления 1

[176] В синтезе варианта осуществления 1 применяли (1-1) в качестве исходного вещества, подробный путь синтеза 1 являлся следующим.

[177] Стадия 1 (синтез соединения 1-2)
[178] Исходное вещество 1-1 (15,0 г, 72,11 ммоль, 1 экв.) растворяли в метаноле (350 мл), затем охлаждали до 0-3°C, в смесь порциями добавляли метантиолат натрия (6,30 г, 89,89 ммоль, 5,73 мл, 1,25 экв.). Добавление завершали через 15 минут (наблюдали явный экзотермический эффект). После добавления внутреннюю температуру реакционной смеси повышали до 15°C, затем осаждалось все большее и большее количество желтого твердого вещества. Смесь перемешивали при 25°C в течение 15 минут. Смесь выливали в ледяную воду (400 мл), перемешивали и фильтровали посредством отсасывания. Твердое вещество растворяли в этилацетате (400 мл) и промывали водой (100 мл). Водные фазы объединяли и дважды экстрагировали этилацетатом (100 мл). Фазы на основе этилацетата объединяли и промывали насыщенным солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением соединения 1-2.
[179] Соответствующие данные характеристик: LCMS масса/заряд: 219,9 [M+H].
[180] Стадия 2 (синтез соединения 1-3)
[181] Соединение 1-2 (15,7 г, 71,48 ммоль, 1 экв.), 4-фторфенилбороновую кислоту (12,00 г, 85,77 ммоль, 1,2 экв.), карбонат натрия (22,7 г, 214,17 ммоль, 3 экв.) добавляли в смесь 1,4-диоксана (200 мл) и воды (20 мл) и смесь три раза продували азотом. Затем добавляли Pd(PPh3)4 (4 г, 3,46 ммоль, 4,84 e-2 экв.) и систему несколько раз продували азотом и затем нагревали до 90-100°C и перемешивали в течение 12 часов. Реакционную смесь охлаждали и выпаривали при пониженном давлении с удалением 1,4-диоксана, остаток разбавляли водой (200 мл) и этилацетатом (300 мл). Водную фазу отделяли и дважды экстрагировали этилацетатом (200 мл). Фазы на основе этилацетата объединяли и промывали солевым раствором (200 мл), высушивали над сульфатом натрия, фильтровали, затем объединяли с предыдущей партией, концентрировали, растирали со смесью PE/этилацетат (10:1, 400 мл), затем фильтровали и высушивали с получением соединения 1-3, которое непосредственно применяли на следующей стадии.
[182] Соответствующие данные характеристик: LCMS масса/заряд: 280,2 [M+H].
[183] Стадия 3 (синтез соединений 1-4)
[184] Соединение 1-3 (17,4 г, 50,04 ммоль, 1 экв.) добавляли к ацетонитрилу (350 мл), затем охлаждали до 0°C. Порциями добавляли NBS (11,5 г, 64,61 ммоль, 1,29 экв.) в защитной атмосфере азота и смесь сразу становилась темно-коричневой. Смесь медленно нагревали до 25°C и проводили реакцию в течение 15 часов. Реакционную смесь гасили с помощью 10% раствора тиосульфата натрия (200 мл) и осажденное желтое твердое вещество фильтровали и ополаскивали водой (30 мл) с получением части продукта, представляющего собой соединение 1-4. Фильтрат один раз экстрагировали этилацетатом (100 мл). Органическую фазу промывали один раз водой (100 мл) и один раз солевым раствором (100 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением желтого твердого вещества, которое растирали с ацетонитрилом (200 мл), фильтровали и высушивали с получением продукта. Две части твердого вещества объединяли и высушивали в вакууме с получением соединения 1-4.
[185] Соответствующие данные характеристик: LCMS масса/заряд: 358,1 [M+H].
[186] Стадия 4 (синтез соединения 1-5)
[187] Соединение 1-4 (14,2 г, 39,64 ммоль, 1 экв.) суспендировали в метаноле (150 мл), добавляли порошок железа (11,0 г, 196,97 ммоль, 4,97 экв.). Затем по каплям добавляли концентрированную хлористоводородную кислоту (60 мл, 15,24 экв.) при 0-15°C и завершали добавление через 15 минут. После добавления реакционную смесь нагревали до 25-30°C и перемешивали в течение 14 часов, затем добавляли дополнительное количество порошка железа (2,2 г, 39,39 ммоль) и концентрированной хлористоводородной кислоты (10 мл, 2,54 экв.) и реакционную смесь непрерывно перемешивали при 25-30°C в течение 2 часов. Смесь фильтровали через диатомовую землю, затем ополаскивали метанолом (20 мл). Фильтрат регулировали до pH 8 с помощью 20% раствора гидроксида натрия (300 мл) (в это время осаждалось синее твердое вещество и твердое вещество получали путем фильтрации), затем к фильтрату добавляли этилацетат (500 мл) и смесь перемешивали в течение 30 минут. Смесь фильтровали через диатомовую землю. Органическую фазу отделяли и промывали солевым раствором (200 мл), высушивали над сульфатом натрия, затем фильтровали и концентрировали с получением твердого вещества. Две части твердого вещества объединяли и высушивали в вакууме с получением соединения 1-5.
[187] Соответствующие данные характеристик: LCMS масса/заряд: 328,0 [M+H].
[188] Стадия 5 (синтез соединений 1-6)
[189] Соединение 1-5 (21,0 г, 63,98 ммоль, 1 экв.) суспендировали в этаноле (250 мл), к суспензии добавляли 40% раствор ацетальдегида (13,88 г, 95,63 ммоль, 12,5 мл, 1,49 экв.) в защитной атмосфере азота. Реакционную смесь перемешивали при 90-100°C в течение 2 часов, затем охлаждали до 25°C и твердое вещество осаждали. Смесь фильтровали и осадок на фильтре промывали холодным этанолом (20 мл) и высушивали в вакууме с получением соединения 1-6.
[190] Соответствующие данные характеристик: LCMS масса/заряд: 351,0 [M+H].
[191] 1H ЯМР (DMSO-d6, 400 МГц) δ 9,33 (s, 1H), 9,07 (d, J = 1,6 Гц, 1H), 7,95-7,92 (m, 2H), 7,42-7,37 (m, 2H), 2,61 (s, 3H).
[192] Стадия 6 (синтез соединения 1-7)
[193] При комнатной температуре м-хлорпероксибензойную кислоту (17,39 г, 85,66 ммоль, чистота 85%, 3,0 экв.) добавляли к смеси дихлорметана (200 мл) и соединения 1-6 (10 г, 28,55 ммоль, 1 экв.). Смесь перемешивали при комнатной температуре в течение 10 минут, затем добавляли дихлорметан (150 мл). Смесь фильтровали и фильтрат промывали водой (100 мл * 3), насыщенным раствором сульфита натрия (50 мл * 3) и солевым раствором (50 мл), высушивали над твердым сульфатом натрия, затем фильтровали и концентрировали с получением соединения 1-7.
[194] Соответствующие данные характеристик: LCMS масса/заряд: 383,9 [M+1].
[195] Стадия 7 (синтез соединения 1-8)
[196] Добавляли N,N-диизопропилэтиламин (6,59 г, 51,02 ммоль, 8,89 мл, 2,5 экв.) и 2,4-диметоксибензиламин (4,09 г, 24,49 ммоль, 3,69 мл, 1,2 экв.) к смеси изопропанола (300 мл) и соединения 1-7 (7,8 г, 20,41 ммоль, 1 экв.), смесь нагревали до 80°C и перемешивали при 80°C в течение 8 часов. TLC (SiO2, PE/этилацетат = 3/1) показала, что исходные вещества полностью исчезли, и выявила образование нового соединения, что указывало на завершение реакции. Смесь концентрировали, затем добавляли этанол (20 мл) и получали большое количество желтого твердого вещества. Смесь фильтровали и осадок на фильтре промывали этанолом (10 мл * 3) с получением твердого вещества. Фильтрат концентрировали с получением желтого твердого вещества, которое также промывали этанолом (10 мл * 3). Две части твердого вещества объединяли и высушивали в вакууме с получением соединения 1-8, которое непосредственно применяли на следующей стадии.
[197] Стадия 8 (синтез соединения 1-9)
[198] 1,4-Диоксан (10 мл) и воду (2 мл) добавляли к соединению 1-8 (1,0 г, 2,13 ммоль, 1,0 экв.) с последующим добавлением 2,6-диметил-4-пиридинбороновой кислоты (386,03 мг, 2,56 ммоль, 1,2 экв., соединение 1A), комплекса дихлорида [1,1-бис(дифенилфосфино)ферроцен]палладия(II) и дихлорметана (348,02 мг, 426,16 мкмоль, 0,2 экв.) и карбоната калия (883,50 мг, 6,39 ммоль, 3,0 экв.). Смесь нагревали до 90°C в защитной атмосфере азота и перемешивали при 90°C в течение 30 минут. Смесь концентрировали и очищали на колонке (SiO2, PE/этилацетат с объемным соотношением 1/1) с получением соединения 1-9 в виде коричневого масла.
[199] Соответствующие данные характеристик: LCMS масса/заряд: 496,1 [M+H].
[200] Стадия 9 (синтез варианта осуществления 1)
[201] Соединение 1-9 (200 мг, 403,59 мкмоль, 1 экв.) добавляли к трифторуксусной кислоте (3 мл), смесь нагревали до 70°C и перемешивали при 70°C в течение 2 часов. Смесь концентрировали и очищали на колонке с обращенной фазой (колонка: Phenomenex Luna Phenyl-Hexyl 150 * 30 мм, 5 мкм; подвижная фаза: [вода (10 мМ бикарбонат аммония)-ацетонитрил]; B%: 35%-65%, 3 мин.)) с получением варианта осуществления 1.
[202] Соответствующие данные характеристик: LCMS масса/заряд: 346,0 [M+H].
[203] 1H ЯМР (400 MГц, CD3OD) δ 8,87 (d, J = 2,0 Гц, 1H), 8,77 (d, J = 2,0 Гц, 1H), 7,45-7,30 (m, 2H), 6,98 (m, 2H), 6,92 (s, 2H), 2,38 (s, 6H).
[204] Соединения вариантов осуществления, перечисленные в таблице 1, получали с помощью стадий, аналогичных стадиям пути 1 для получения варианта осуществления 1, за исключением того, что применяли производное борной кислоты в следующей таблице в качестве исходного вещества на стадии 8 вместо исходного вещества 1A с получением соответствующего соединения.
Таблица 1
тор исходного вещества
заряд продукта: [M+H]
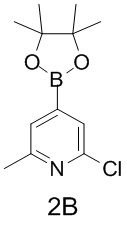
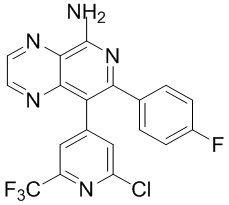

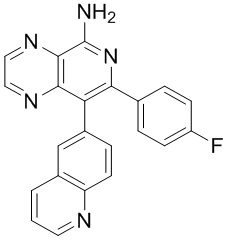
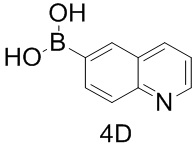
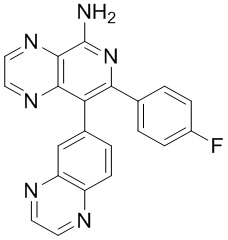

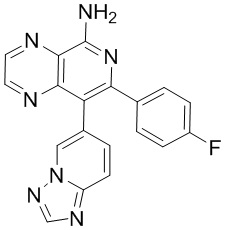




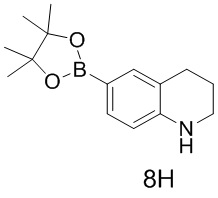
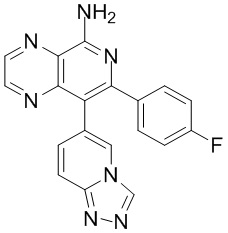
[205] Получение гидрохлорида соединения в качестве примера для варианта осуществления 6
[206] 200 мл ацетонитрила и 200 мл воды добавляли в реакционную колбу при 25°C, затем добавляли вариант осуществления 6 в виде свободного основания (6 г) с последующим добавлением 1 M разбавленной хлористоводородной кислоты с регулированием значения pH до 3-5. Реакционную смесь перемешивали при 25°C в течение 0,5 часа. Получали гидрохлорид варианта осуществления 6.
[207] LCMS масса/заряд продукта: [M+H] 358,2.
[208] 1H ЯМР (400 МГц, DMSO-d6) δ = 9,24 (t, J = 3,6 Гц, 1H), 9,12 (d, J = 2,0 Гц, 1H), 8,87 (s, 1H), 8,59 (t, J = 5,2 Гц, 1H), 7,84 (t, J = 8,4 Гц, 1H), 7,58-7,55 (m, 2H), 7,50 (d, J = 15,6 Гц, 1H), 7,27 (t, J = 9,2 Гц, 2H).
[209] Вариант осуществления 10

[210] В синтезе варианта осуществления 2 применяли (1-6) в качестве исходного вещества, подробный путь синтеза являлся следующим.

[211] Стадия 1 (синтез соединения 10-2)
[212] Дихлорид ферроценпалладия (208,94 мг, 285,54 мкмоль, 0,1 экв.), карбонат калия (789,30 мг, 5,71 ммоль, 2 экв.), 2-метил-6-трифторметил-4-пинаколовый эфир бороновой кислоты (983,71 мг, 3,43 ммоль, 1,2 экв.) добавляли к раствору соединения 1-6 (1,0 г, 2,86 ммоль, 1 экв.) в 1,4-диоксане (20 мл) и воде (4 мл), смесь три раза продували азотом и перемешивали при 90°C в течение 2 часов. Когда LCMS показала, что реакция была завершена, реакционную смесь концентрировали с получением неочищенного продукта, который очищали на колонке с силикагелем (силикагель 100-200 меш, градиентное элюирование с помощью PE/этилацетат с объемным соотношением от 10/1 до 5/1) с получением соединения 10-2.
[213] Соответствующие данные характеристик: LCMS масса/заряд: 431,0 [M+H].
[214] Стадия 2 (синтез соединения 10-3)
[215] м-Хлорпероксибензойную кислоту (1,71 г, 8,42 ммоль, чистота 85%, 2,5 экв.) добавляли в раствор соединения 10-2 (1,45 г, 3,37 ммоль, 1 экв.) в дихлорметане (2 мл) при 10°C и реакционную смесь перемешивали при 25°C в течение 20 минут. После того как LCMS показала, что реакция была завершена, к реакционной смеси добавляли насыщенный раствор бикарбоната натрия (20 мл) и насыщенный раствор сульфита натрия (20 мл) и полученную смесь экстрагировали дихлорметаном (30 мл * 2). Органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 10-3.
[216] Соответствующие данные характеристик: LCMS масса/заряд: 463,0 [M+H].
[217] Стадия 3 (синтез соединения 10-4)
[218] Диизопропилэтиламин (877,61 мг, 6,79 ммоль, 1,18 мл, 2 экв.) и 2,4-диметоксибензиламин (851,54 мг, 5,09 ммоль, 767,15 мкл, 1,5 экв.) добавляли к раствору соединения 10-3 (1,57 г, 3,40 ммоль, 1 экв.) в изопропаноле (20 мл) и реакционную смесь перемешивали при 85°C в течение 17 часов. После того как LCMS показала, что реакция была завершена, реакционную смесь концентрировали до сухого состояния, затем добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл * 2). Объединенную органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, представляющего собой соединение 10-4 (2,4 г).
[219] Соответствующие данные характеристик: LCMS масса/заряд: 550,0 [M+H].
[220] Стадия 4 (синтез варианта осуществления 10)
[221] Раствор соединения 10-4 (2,4 г, 4,37 ммоль, 1 экв.) в трифторуксусной кислоте (20 мл) перемешивали при 90°C в течение 2 часов. Смесь концентрировали при пониженном давлении до сухого состояния, затем регулировали до pH 8 насыщенным раствором карбоната калия, экстрагировали этилацетатом (30 мл * 2). Органическую фазу промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия, фильтровали, концентрировали и очищали на колонке для препаративной хроматографии с получением варианта осуществления 10.
[222] Соответствующие данные характеристик: LCMS масса/заряд: 400,0 [M+H].
[223] 1H ЯМР (DMSO-d6) δ 9,10 (d, J = 1,6 Гц, 1H), 9,01 (d, J = 1,6 Гц, 1H), 7,69 (s, 2H), 7,42 (s, 2H), 7,36-7,34 (m, 2H), 7,14-7,09 (m, 2H), 2,48 (s, 3H).
[224] Вариант осуществления 11
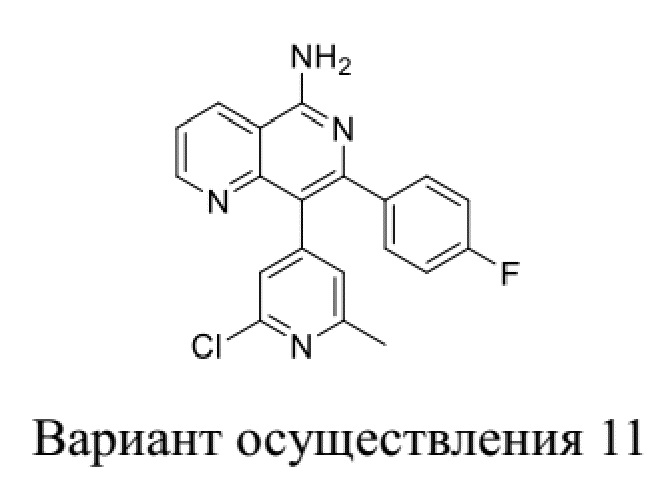
[225] В синтезе варианта осуществления 11 применяли (11-1) в качестве исходного вещества, подробный путь синтеза являлся следующим.
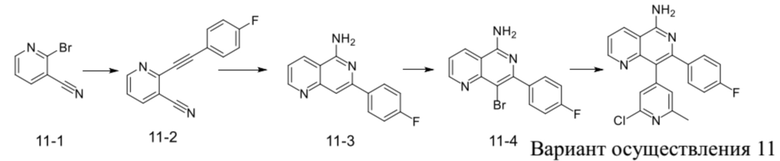
[226] Стадия 1 (синтез соединения 11-2)
[227] Раствор соединения 11-1 (0,4 г, 2,19 ммоль, 1 экв.), 4-фторфенилацетилена (525,11 мг, 4,37 ммоль, 500,11 мкл, 2 экв.), Pd(PPh3)2Cl2 (306,83 мг, 437,15 мкмоль, 0,2 экв.), йодида меди (83,25 мг, 437,15 мкмоль, 0,2 экв.) и триэтиламина (884,70 мг, 8,74 ммоль, 1,22 мл, 4 экв.) в N,N-диметилформамиде (10 мл) три раза продували азотом в вакууме и затем перемешивали при 25°C в течение 1,5 часа в атмосфере азота. После того как LC-MS показала, что реакция была завершена, к реакционной смеси добавляли 20 мл воды и 30 мл этилацетата и смесь разделяли. Органическую фазу высушивали над безводным сульфатом натрия, фильтровали, концентрировали и остаток очищали на колонке с силикагелем (подвижная фаза: PE/этилацетат с объемным соотношением от 9/1 до 6/1) с получением соединения 11-2.
[228] Соответствующие данные характеристик: LCMS масса/заряд: 223,0 [M+H].
[229] 1H ЯМР (DMSO-d6) δ 8,8 (d, J = 4,8 Гц, 1H), 8,0 (d, J = 7,6 Гц, 1H), 7,7 (dd, J = 5,6, 8 Гц, 2H), 7,4 (dd, J = 4,8, 7,6 Гц, 1H), 7,1 (t, J = 8,4 Гц, 2H).
[230] Стадия 2 (синтез соединения 11-3)
[231] Карбонат калия (355,75 мг, 2,57 ммоль, 1,3 экв.) добавляли к раствору соединения 11-2 (0,44 г, 1,98 ммоль, 1 экв.) в N,N-диметилформамиде (10 мл) и водному раствору аммиака (10 мл) и полученную смесь перемешивали при 80°C в течение 16 часов. Когда LCMS показала, что реакция была завершена, реакционную смесь охлаждали до 25°C, затем разбавляли с помощью 30 мл воды и экстрагировали с помощью 40 мл этилацетата. Органическую фазу дважды промывали с помощью 20 мл воды каждый раз, затем высушивали над безводным сульфатом натрия, фильтровали, концентрировали в вакууме с получением соединения 12-3, соединение 11-3 непосредственно применяли на следующей стадии без дополнительной очистки.
[232] Соответствующие данные характеристик: LCMS масса/заряд: 240,2 [M+H].
[233] Стадия 3 (синтез соединения 11-4)
[234] При 0°C N-бромсукцинимид (357,08 мг, 2,01 ммоль, 1,2 экв.) добавляли к раствору соединения 12-3 (0,4 г, 1,67 ммоль, 1 экв.) в ацетонитриле (30 мл) и полученную смесь перемешивали при 25°C в течение 2,5 часа. Когда LCMS показала, что реакция была завершена, реакционную смесь концентрировали и полученный остаток растирали со смешанным раствором (20 мл, дихлорметан/PE = 1/5) с получением соединения 11-4, которое непосредственно применяли на следующей стадии без дополнительной очистки.
[235] Соответствующие данные характеристик: LCMS масса/заряд: 319,8 [M+H].
[236] Стадия 4 (синтез варианта осуществления 11)
[237] После продувания три раза азотом в вакууме смешанного раствора соединения 11-4 (0,05 г, 157,16 мкмоль, 1 экв.) и 2-метил-6-хлор-4-пиридинбороновой кислоты (47,81 мг, 188,59 мкмоль, 1,2 экв.) в диоксане (10 мл) и воде (2 мл) добавляли фосфат калия (66,72 мг, 314,32 мкмоль, 2 экв.) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (20,49 мг, 31,43 мкмоль, 0,2 экв.), полученную смесь перемешивали при 90°C в течение 2 часов в защитной атмосфере азота. После того как LCMS показала, что реакция была завершена, реакционную смесь концентрировали в вакууме, полученный остаток очищали на колонке с силикагелем (PE/этилацетат = 1/1) и затем остаток дополнительно очищали на колонке для препаративной хроматографии (Phenomenex Synergi C18, 150 * 30 мм*4 мкм; подвижная фаза: [вода (0,05% хлористоводородной кислоты)-ацетонитрил]; B%: 20%-41%, 7 мин.) с получением варианта осуществления 11.
[238] Соответствующие данные характеристик: LCMS масса/заряд: 364,9 [M+H].
[239] 1H ЯМР (400 МГц, DMSO-d6) δ = 9,23 (dd, J = 1,4, 8,4 Гц, 1H), 9,14 (d, J = 4,8 Гц, 1H), 7,84 (dd, J = 4,4, 8,4 Гц, 1H), 7,50-7,43 (m, 2H), 7,29 (t, J = 8,8 Гц, 2H), 7,15 (s, 1H), 7,10 (s, 1H), 2,37 (s, 3H).
[240] Вариант осуществления 12

[241] Вариант осуществления 12 получали в соответствии со стадиями, аналогичными стадиям пути для получения варианта осуществления 11, за исключением того, что применяли исходное вещество 6F на стадии 4 вместо исходного вещества 2B (2-метил-6-хлор-4-пиридинбороновой кислоты) с получением соответствующего варианта осуществления 12.
[242] Соответствующие данные характеристик: LCMS масса/заряд: 357,3 [M+H].
[243] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,90 (dd, J = 1,6, 4,4 Гц, 1H), 8,77-8,68 (m, 2H), 8,45 (s, 1H), 7,74 (d, J = 9,2 Гц, 1H), 7,56 (dd, J = 4,4, 8,4 Гц, 1H), 7,50 - 7,37 (m, 5H), 7,14 - 7,02 (m, 2H).
[244] Соединения вариантов осуществления, перечисленные в таблице 2, получали в соответствии со стадиями, аналогичными стадиям пути 1 для получения варианта осуществления 1, за исключением того, что применяли производные борной кислоты в следующей таблице в качестве исходного вещества на стадии 8 вместо исходного вещества 1A с получением соответствующих соединений.
Таблица 2
заряд продукта: [M+H]+
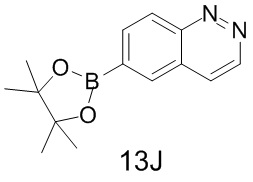
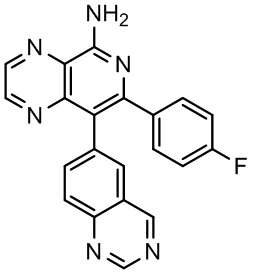

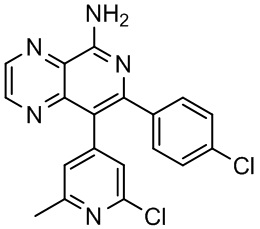


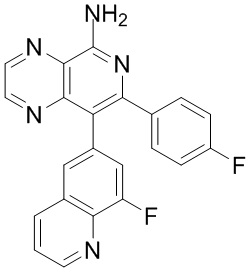
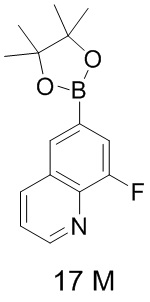




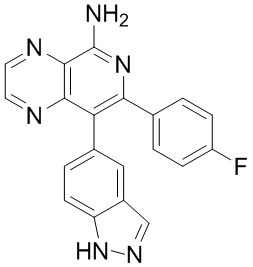


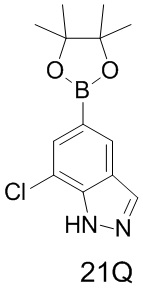



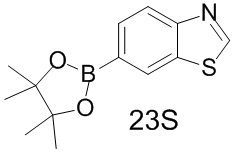
[245] Вариант осуществления 24

[246] В синтезе варианта осуществления 24 применяли (24-1) в качестве исходного вещества, подробный путь синтеза являлся следующим.
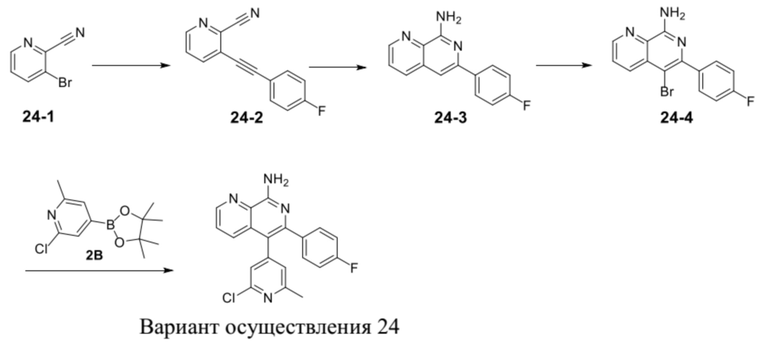
[247] Стадия 1 (синтез соединения 24-2)
[248] Соединение 24-1 (1,0 г, 5,46 ммоль, 1 экв.), триэтиламин (1,66 г, 16,39 ммоль, 2,28 мл, 3,0 экв.), йодид меди (208,14 мг, 1,09 ммоль, 0,2 экв.) и дихлорид бис(трифенилфосфино)палладия (767,08 мг, 1,09 ммоль, 0,2 экв.) добавляли в раствор п-фторфенилацетилена (722,04 мг, 6,01 ммоль, 687,65 мкл, 1,1 экв.) в N,N-диметилформамиде (20 мл), реакционную смесь несколько раз продували азотом, а затем нагревали до 90°C и перемешивали в течение 3 часов. LCMS показала, что исходные вещества исчезли и образовался продукт. В смесь добавляли воду (50 мл), затем смесь экстрагировали этилацетатом (50 мл * 2). Органические фазы объединяли и промывали водой (30 мл * 2) и насыщенным солевым раствором (30 мл), высушивали над сульфатом натрия, фильтровали, концентрировали и очищали с помощью колоночной хроматографии (SiO2, PE/этилацетат = 5/1) с получением соединения 24-2.
[249] Соответствующие данные характеристик: LCMS масса/заряд: 223,1 [M+H]+.
[250] Стадия 2 (синтез соединения 24-3)
[251] Водный раствор аммиака (18,59 г, 148,50 ммоль, 20,43 мл, 28% по весу, 30 экв.) и карбонат калия (1,37 г, 9,90 ммоль, 2,0 экв.) добавляли в раствор соединения 24-2 (1,1 г, 4,95 ммоль, 1 экв.) в N,N-диметилформамиде (10 мл) и смесь нагревали до 80°C и перемешивали в течение 15 часов. LCMS показала образование продукта. В смесь добавляли воду (50 мл), затем смесь экстрагировали этилацетатом (15 мл * 3). Объединенную органическую фазу промывали водой (20 мл * 3) и насыщенным солевым раствором (20 мл), высушивали над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, представляющего собой соединение 24-3.
[252] Соответствующие данные характеристик: LCMS масса/заряд: 240,1 [M+H]+.
[253] Стадия 3 (синтез соединения 24-4)
[254] N-Бромсукцинимид (818,33 мг, 4,60 ммоль, 1,1 экв.) добавляли к раствору соединения 24-3 (1,0 г, 4,18 ммоль, 1 экв.) в ацетонитриле (5 мл) при 0°C в атмосфере азота и смесь нагревали до 25°C и перемешивали в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь концентрировали и разбавляли водой (20 мл), затем экстрагировали этилацетатом (15 мл * 3). Объединенную органическую фазу промывали насыщенным раствором сульфита натрия (15 мл * 2) и насыщенным солевым раствором (15 мл), высушивали над сульфатом натрия, фильтровали и фильтрат концентрировали с получением неочищенного продукта, представляющего собой соединение 24-4. Неочищенный продукт непосредственно применяли на следующей стадии без дополнительной очистки.
[255] Соответствующие данные характеристик: LCMS масса/заряд: 318,0 [M+H]+.
[256] Стадия 4 (синтез варианта осуществления 24)
[257] Соединение 24-4 (200 мг, 628,65 мкмоль, 1 экв.), фосфат калия (400,32 мг, 1,89 ммоль, 3,0 экв.) и дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия (81,94 мг, 125,73 мкмоль, 0,2 экв.) добавляли к раствору соединения 2B (159,38 мг, 628,65 мкмоль, 1,0 экв.) в диоксане (10 мл) и воде (2 мл), смесь нагревали до 90°C в атмосфере азота и перемешивали в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь фильтровали и фильтрат концентрировали. Остаток отделяли и очищали с помощью пластины для препаративной TLC (SiO2, PE/этилацетат = 2/1) с получением варианта осуществления 24.
[258] Соответствующие данные характеристик: LCMS масса/заряд: 365,1 [M+H]+.
[259] 1H ЯМР (400 МГц, CDCl3) δ = 8,73 (d, J = 3,0 Гц, 1H), 7,74 (d, J = 8,0 Гц, 1H), 7,46 (dd, J = 4,2, 8,6 Гц, 1H), 7,29-7,14 (m, 2H), 6,98-6,83 (m, 3H), 6,79 (s, 1H), 6,14 (br s, 2H), 2,34 (s, 3H).
[260] Вариант осуществления 25

[261] Вариант осуществления 25 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 24, за исключением того, что применяли исходное вещество 4-хлор-5-цианопиримидин на стадии 1 вместо исходного вещества 24-1 с получением соответствующего варианта осуществления 25.
[262] Соответствующие данные характеристик: LCMS масса/заряд: 366,2 [M+H] +.
[263] 1H ЯМР (400 МГц, DMSO-d6) δ = 9,80 (s, 1H), 9,26 (s, 1H), 7,99 (s, 2H), 7,37 (dd, J = 5,6, 8,8 Гц, 2H), 7,15 (t, J = 9,2 Гц, 2H), 7,11 (s, 1H), 7,06 (s, 1H), 2,36 (s, 3H).
[264] Вариант осуществления 26
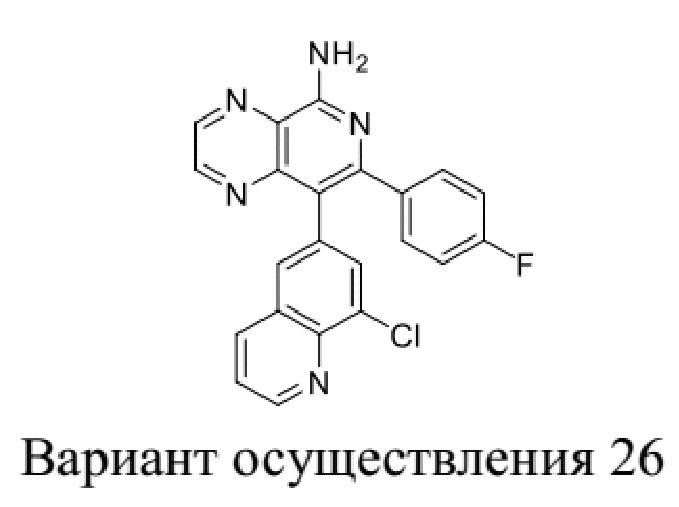
[265] В синтезе варианта осуществления 26 применяли (1-8) в качестве исходного вещества, подробный путь синтеза являлся следующим.

[266] Стадия 1 (синтез соединения 26-1)
[267] В защитной атмосфере азота раствор соединения 1-8 (0,9 г, 1,92 ммоль, 1 экв.), пинаколового эфира бороновой кислоты (2,43 г, 9,59 ммоль, 5 экв.), Pd(dbcp)2Cl2 (124,99 мг, 191,77 мкмоль, 0,1 экв.) и ацетата калия (564,63 мг, 5,75 ммоль, 3 экв.) в диоксане (20 мл) перемешивали при 90°C в течение 30 минут. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до сухого состояния. Неочищенный продукт отделяли и очищали с помощью колоночной хроматографии (PE/этилацетат = от 10:1 до 5:1, силикагель 100-200 меш) с получением соединения 26-1.
[268] Соответствующие данные характеристик: LCMS масса/заряд: 517,3 [M+H]+.
[269] Стадия 2 (синтез соединения 26-3)
[270] В защитной атмосфере азота раствор соединения 26-1 (0,2 г, 132,58 мкмоль, 1 экв.), соединения 26-2 (64,30 мг, 265,16 мкмоль, 2 экв.), Pd(dbcp)2Cl2 (8,64 мг, 13,26 мкмоль, 0,1 экв.) и карбоната калия (54,97 мг, 397,74 мкмоль, 3 экв.) в диоксане (2 мл) и воде (0,4 мл) перемешивали при 90°C в течение 30 минут. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до сухого состояния. Неочищенный продукт отделяли и очищали с помощью колоночной хроматографии (PE/этилацетат = от 5/1 до 1/1, силикагель 100-200 меш) с получением соединения 26-3.
[271] Соответствующие данные характеристик: LCMS масса/заряд: 552,2 [M+H]+.
[272] Стадия 3 (синтез варианта осуществления 26)
[273] Раствор соединения 26-3 (0,06 г, 103,63 мкмоль, 1 экв.) в трифторуксусной кислоте (3 мл) перемешивали при 70°C в течение 30 минут. LCMS показала, что исходные вещества исчезли и образовался продукт. Реакционную смесь концентрировали и выпаривали до сухого состояния, разбавляли водой (10 мл), регулировали до pH 9 насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (15 мл * 3). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния. Неочищенный продукт очищали с помощью препаративной тонкослойной хроматографии (PE/этилацетат = 1/1) с получением варианта осуществления 26.
[274] Соответствующие данные характеристик: LCMS масса/заряд: 402,1 [M+H]+.
[275] 1H ЯМР (400 МГц, DMSO-d6) δ = 9,02-8,97 (m, 2H), 8,85 (d, J = 2,0 Гц, 1H), 8,31-8,26 (m, 1H), 7,84 (d, J = 1,8 Гц, 1H), 7,72 (d, J = 1,8 Гц, 1H), 7,62-7,57 (m, 3H), 7,39 (dd, J = 5,6, 8,8 Гц, 2H), 7,05 (t, J = 9,0 Гц, 2H).
[276] Вариант осуществления 27

[277] Вариант осуществления 27 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 24, за исключением того, что исходное вещество 24-1 было заменено на 2-бром-3-цианопиридин на стадии 1, и п-фторфенилацетилен был заменен на фенилацетилен; исходное вещество 2B было заменено на 6F на стадии 4 с получением соответствующего варианта осуществления 27.
[278] Соответствующие данные характеристик: LCMS масса/заряд: 339,0 [M+H]+.
[279] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,90 (dd, J = 1,2, 4,0 Гц, 1H), 8,74 (dd, J = 1,2, 8,0 Гц, 1H), 8,64 (s, 1H), 8,42 (s, 1H), 7,71 (d, J = 9,2 Гц, 1H), 7,55 (dd, J = 4,4, 8,4 Гц, 1H), 7,47 (dd, J = 1,6, 9,2 Гц, 1H), 7,42 (s, 2H), 7,37 (dd, J = 2,0, 8,0 Гц, 2H), 7,25-7,17 (m, 3H).
[280] Вариант осуществления 28
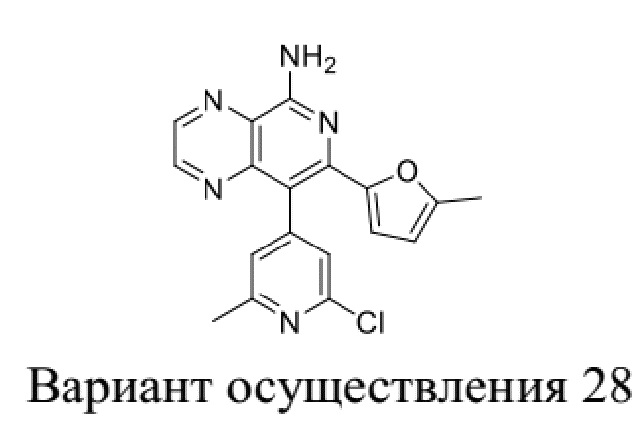
[281] В синтезе варианта осуществления 28 применяли (28-1) в качестве исходного вещества, подробный путь синтеза являлся следующим.
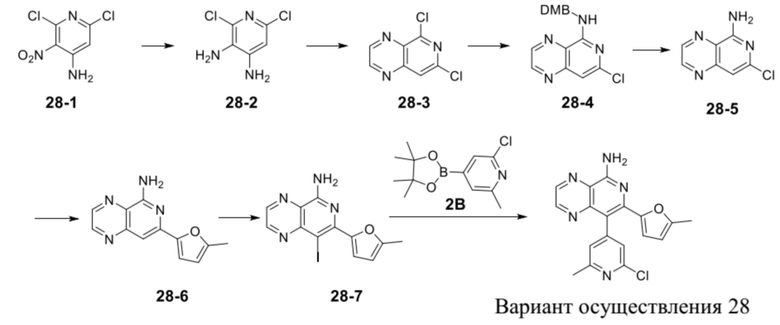
[282] Стадия 1 (синтез соединения 28-2)
[283] Порошок железа (6,71 г, 120,19 ммоль, 5,0 экв.) добавляли к раствору соединения 28-1 (5,00 г, 24,04 ммоль, 1 экв.) в этаноле (100 мл), затем добавляли хлористоводородную кислоту (36,52 г, 360,57 ммоль, 35,80 мл, весовое содержание 36%, 15 экв.) при 0°C. Смесь перемешивали при 0-15°C в течение 10 минут, затем нагревали до 25°C и перемешивали при 25°C в течение 10 часов. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь фильтровали, осадок на фильтре промывали водой (15 мл * 3) и фильтраты объединяли. Фильтрат регулировали до pH 10 (водным раствором NaOH, 6 M) и образовывалось большое количество осадка, затем к смеси добавляли этилацетат (200 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали с получением фильтрата. Фильтрат экстрагировали этилацетатом (50 мл * 4), объединенную органическую фазу промывали насыщенным солевым раствором (50 мл) и концентрировали в вакууме с получением соединения 28-2.
[284] Соответствующие данные характеристик: LCMS масса/заряд: 178,1 [M+H]+.
[285] Стадия 2 (синтез соединения 28-3)
[286] Глиоксаль (5,13 г, 35,39 ммоль, 4,63 мл, 1,5 экв.) добавляли к раствору соединения 28-2 (4,2 г, 23,59 ммоль, 1 экв.) в этаноле (40 мл) и смесь нагревали до 80°C и перемешивали при 80°C в течение 2 часов. LCMS показала, что исходные вещества исчезли и образовался продукт. В системе образовывалось большое количество желтого осадка и смесь фильтровали и осадок на фильтре высушивали с получением соединения 28-3.
[287] Соответствующие данные характеристик: LCMS масса/заряд: 200,0 [M+H].
[288] Стадия 3 (синтез соединения 28-4)
[289] При комнатной температуре 2,4-диметоксианилин (835,92 мг, 5,00 ммоль, 753,08 мкл, 1,0 экв.) и N,N-диизопропилэтиламин (775,36 мг, 6,00 ммоль, 1,04 мл, 1,2 экв.) добавляли к раствору соединения 28-3 (1,0 г, 5,00 ммоль, 1 экв.) в тетрагидрофуране (20 мл), смесь перемешивали при 25°C в течение 4 часов, затем нагревали до 66°C и перемешивали в течение 1 часа. TLC показала исчезновение исходных веществ, и с помощью LCMS выявили образование продукта. Смесь концентрировали с получением соединения 28-4.
[290] Соответствующие данные характеристик: LCMS масса/заряд: 331,0 [M+H].
[291] Стадия 4 (синтез соединения 28-5)
[292] Соединение 28-4 (500 мг, 1,51 ммоль, 1 экв.) добавляли к трифторуксусной кислоте (3 мл), систему нагревали до 70°C и перемешивали в течение 3 часов. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь гасили насыщенным водным раствором карбоната (30 мл), экстрагировали этилацетатом (15 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором (10 мл), высушивали над сульфатом натрия, фильтровали, концентрировали с получением неочищенного продукта, который очищали на колонке (SiO2, PE/этилацетат = 3/1) с получением соединения 28-5.
[293] Соответствующие данные характеристик: LCMS масса/заряд: 181,1 [M+H].
[294] Стадия 5 (синтез соединения 28-6)
[295] Соединение 28-5 (200 мг, 1,11 ммоль, 1 экв.) растворяли в смешанном растворителе из 1,4-диоксана (5 мл) и воды (1 мл), затем добавляли пинаколовый сложный эфир 5-метил-2-фуранбороновой кислоты (276,50 мг, 1,33 ммоль, 1,2 экв.), хлорид 1,1-бис(дифенилфосфино)ферроценпалладия (162,07 мг, 221,49 мкмоль, 0,2 экв.) и фосфат калия (705,23 мг, 3,32 ммоль, 3,0 экв.), смесь нагревали до 90°C в атмосфере азота и перемешивали при 90°C в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь фильтровали с получением фильтрата, осадок на фильтре промывали этилацетатом (15 мл * 3). Фильтраты объединяли и концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (SiO2, PE/этилацетат = 3/1) с получением соединения 28-6.
[296] Соответствующие данные характеристик: LCMS масса/заряд: 227,1 [M+H].
[297] Стадия 6 (синтез соединения 28-7)
[298] N-Йодсукцинимид (59,67 мг, 265,21 мкмоль, 1,2 экв.) добавляли к раствору соединения 28-6 (50 мг, 221,01 мкмоль, 1 экв.) в ацетонитриле (3 мл) и смесь нагревали до 70°C и перемешивали в течение 1 часа. LCMS показала, что исходные вещества исчезли и образовался продукт. Смесь концентрировали с получением неочищенного продукта, представляющего собой соединение 28-7. Неочищенный продукт непосредственно применяли на следующей стадии без дополнительной очистки.
[299] Соответствующие данные характеристик: LCMS масса/заряд: 353,0 [M+H].
[300] Стадия 7 (синтез варианта осуществления 28)
[301] Соединение 2B (60,48 мг, 238,55 мкмоль, 1,2 экв.) добавляли к смешанному растворителю из 1,4-диоксана (3 мл) и воды (0,05 мл) с последующим добавлением соединения 28-7 (70 мг, 198,79 мкмоль, 1 экв.), фосфата калия (126,59 мг, 596,37 мкмоль, 3,0 экв.) и хлорида 1,1-бис(дифенилфосфино)ферроценпалладия (29,09 мг, 39,76 мкмоль, 0,2 экв.), смесь нагревали до 90°C в атмосфере азота и перемешивали в течение 2 часов. LCMS показала, что исходные вещества исчезли и образовался продукт. После завершения реакции смесь фильтровали, осадок на фильтре промывали этилацетатом (10 мл * 3). Объединенный фильтрат концентрировали и очищали с помощью колоночной хроматографии и дополнительно очищали с помощью пластины для препаративной TLC (PE/этилацетат = 3/1) с получением неочищенного продукта. Неочищенный продукт отделяли с помощью препаративной хроматографии с обращенной фазой (колонка: Phenomenex Gemini 150 * 25 мм * 10 мкм; подвижная фаза: [вода (10 мМ бикарбонат аммония)-ацетонитрил]; B%: 30%-60%, 10 мин.) и концентрировали с получением варианта осуществления 28.
[302] Соответствующие данные характеристик: LCMS масса/заряд: 352,0 [M+H].
[303] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,92 (d, J = 1,6 Гц, 1H), 8,76 (s, 1H), 7,54 (s, 2H), 7,21 (d, J = 13,6 Гц, 2H), 6,43 (d, J = 3,2 Гц, 1H), 6,15 (s, 1H), 2,48 (s, 3H), 2,10 (s, 3H).
[304] Вариант осуществления 29
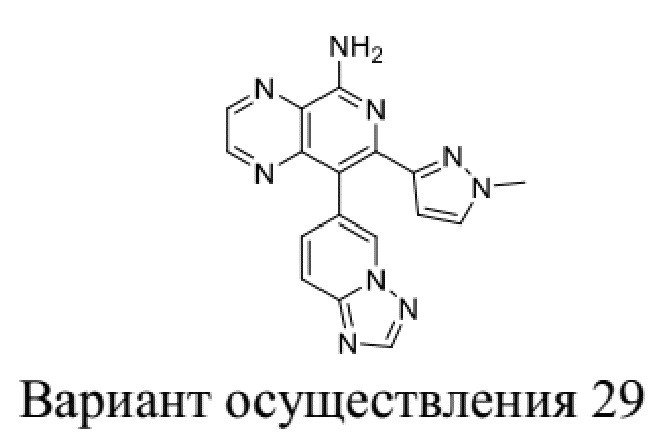
[305] Вариант осуществления 29 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 28, с применением 28-5 в качестве исходного вещества, за исключением того, что применяли исходное вещество, представляющее собой пинаколовый сложный эфир 1-метилпиразол-3-бороновой кислоты, вместо пинаколового сложного эфира 5-метил-2-фуранбороновой кислоты на стадии 5; применяли исходное вещество 6F вместо исходного вещества 2B на стадии 7 с получением соответствующего варианта осуществления 29.
[306] Соответствующие данные характеристик: LCMS (M-1): 342,2; LCMS масса/заряд: 344,1 [M+H]+.
[307] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,94 (d, J = 1,8 Гц, 1H), 8,80 (d, J = 1,8 Гц, 1H), 8,74 (s, 1H), 8,48 (s, 1H), 7,78 (d, J = 9,0 Гц, 1H), 7,55 (d, J = 2,0 Гц, 1H), 7,51-7,42 (m, 3H), 6,33 (d, J = 2,0 Гц, 1H), 3,61 (s, 3H).
[308] Вариант осуществления 30


[309] Вариант осуществления 30 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 28, за исключением того, что применяли исходное вещество, представляющее собой 2-фуранбороновую кислоту, вместо пинаколового сложного эфира 5-метил-2-фуранбороновой кислоты на стадии 5; и исходное вещество 6F применяли вместо исходного вещества 2B на стадии 7 с получением соответствующего соединения 30-3.
[310] Стадия 8 (синтез варианта осуществления 30)
[311] Добавляли NCS (26,13 мг, 195,72 мкмоль, 1,05 экв.) к раствору соединения 30-3 (0,08 г, 186,40 мкмоль, 1 экв.) в DMF (2 мл) и смесь перемешивали при 60°C в течение 3 часов. LCMS показала, что осталось небольшое количество исходного вещества, и образовался продукт. Реакционную смесь разбавляли водой (10 мл), экстрагировали этилацетатом (15 мл * 3). Объединенную органическую фазу промывали насыщенным солевым раствором (15 мл * 2), высушивали над безводным сульфатом натрия, фильтровали и выпаривали до сухого состояния. Неочищенный продукт отделяли и очищали с помощью тонкослойной хроматографии TLC (этилацетат/метанол = 25/1) и препаративной высокоэффективной жидкостной хроматографии (колонка: Gemini 150 * 25 мм, 5 мкм; подвижная фаза: [вода (0,05% водный раствор аммиака об./об.)-ацетонитрил]; B%: 20%-50%, 12 мин.) с получением варианта осуществления 30.
[312] Соответствующие данные характеристик: LCMS масса/заряд: 364,1 [M+H]+.
[313] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,94 (d, J = 2,0 Гц, 2H), 8,81 (d, J = 1,8 Гц, 1H), 8,53 (s, 1H), 7,90 (d, J = 9,2 Гц, 1H), 7,63 (s, 2H), 7,57 (d, J = 9,0 Гц, 1H), 6,52-6,46 (m, 2H).
[314] Вариант осуществления 31

[315] Вариант осуществления 31 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 28 с применением 28-7 в качестве исходного вещества, за исключением того, что применяли исходное вещество 6F на стадии 7 вместо исходного вещества 2B с получением соответствующего варианта осуществления 31.
[316] Соответствующие данные характеристик: LCMS масса/заряд: 344,2 [M+H].
[317] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,89 (br d, J = 12,4 Гц, 2H), 8,76 (s, 1H), 8,52 (s, 1H), 7,87 (br d, J = 8,8 Гц, 1H), 7,59-7,47 (m, 3H), 6,39 (s, 1H), 6,09 (s, 1H), 2,00 (s, 3H).
[318] Вариант осуществления 32

[319] Вариант осуществления 32 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 28 с применением 28-5 в качестве исходного вещества, за исключением того, что применяли исходное вещество, представляющее собой 5-хлортиофен-2-бороновую кислоту, на стадии 5 вместо пинаколового сложного эфира 5-метил-2-фуранбороновой кислоты; применяли исходное вещество 6F на стадии 7 вместо исходного вещества 2B с получением соответствующего варианта осуществления 32.
[320] Соответствующие данные характеристик: LCMS масса/заряд: 380,1 [M+H].
[321] 1H ЯМР (400 МГц, DMSO-d6) δ = 9,02 (s, 1H), 8,93 (d, J = 1,8 Гц, 1H), 8,80 (d, J = 1,8 Гц, 1H), 8,59 (s, 1H), 7,98 (d, J = 9,0 Гц, 1H), 7,69-7,56 (m, 3H), 6,91 (d, J = 4,0 Гц, 1H), 6,45 (d, J = 4,0 Гц, 1H).
[322] Вариант осуществления 33
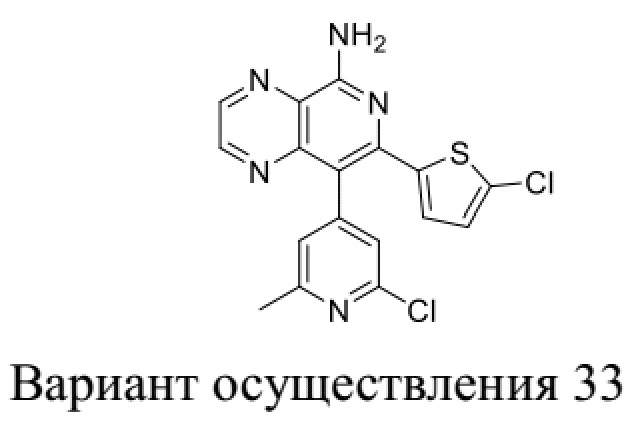
[323] Вариант осуществления 33 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 28, за исключением того, что применяли исходное вещество, представляющее собой 5-хлортиофен-2-бороновую кислоту, на стадии 5 вместо пинаколового сложного эфира 5-метил-2-фуранбороновой кислоты с получением соответствующего варианта осуществления 33.
[324] Соответствующие данные характеристик: LCMS масса/заряд: 388,0 [M+H].
[325] 1H ЯМР (400 МГц, DMSO-d6) δ = 8,94 (d, J = 1,8 Гц, 1 H), 8,79 (d, J = 2,0 Гц, 1 H), 7,62 (s, 2 H), 7,32 (d, J = 18,8 Гц, 2 H), 6,96 (d, J = 4,0 Гц, 1 H), 6,30 (d, J = 4,2 Гц, 1 H), 3,30 (s, 3 H).
[326] Вариант осуществления 34

[327] Вариант осуществления 34 получали в соответствии со стадиями, аналогичными стадиям получения варианта осуществления 26, за исключением того, что применяли исходное вещество 34-3 на стадии 2 вместо исходного вещества 26-2 с получением соответствующего варианта осуществления 34.
[328] Соответствующие данные характеристик: LCMS масса/заряд: 392,1 [M+H].
[329] 1H ЯМР (400 МГц, CDCl3) δ = 8,94 (d, J = 1,8 Гц, 1H), 8,82-8,71 (m, 1H), 8,44-8,30 (m, 2H), 7,51 (d, J = 1,2 Гц, 1H), 7,41 (dd, J = 5,6, 8,8 Гц, 2H), 6,99 (t, J = 8,8 Гц, 2H), 6,20 (br s, 2H).
[330] Данные биологического теста
[331] Экспериментальный пример 1. Тестовый эксперимент в отношении активности соединений по настоящему изобретению in vitro
[332] Эксперимент по обнаружению потока кальция через аденозиновый рецептор A2a человека
[333] Источник клеток:
[334] клеточную линию, устойчиво экспрессирующую A2a, сконструировали Shanghai Wuxi Apptec, при этом клетка-хозяин представляла собой CHO.
[335] Тестовый набор:
[336] набор Fluo-4 Direct (Invitrogen, номер товара: F10471). После того как реагент для флуоресцентного обнаружения (специфически связывающийся с ионами кальция и обуславливающий увеличение флуоресцентного сигнала) в наборе инкубировали с клетками в течение соответствующего периода времени, добавляли соединение для стимуляции клеток и индуцирования изменений во внутриклеточном потоке кальция, что таким образом вызывало изменения во флуоресцентном сигнале, которые могут отображать интенсивность агонистической или ингибирующей активности соединений.
[337] Среда для культивирования клеток:
[338] F12 + 10% эмбриональная бычья сыворотка + 300 мкг/мл генетицина + 2 мкг/мл бластицидина.
[339] Буфер для разведения соединений:
[340] сбалансированный солевой буфер Хенкса(Invitrogen) + 20 мМ HEPES, который получали перед каждым применением.
[341] Агонист:
[342] NECA (Sigma-E2387).
[343] Эталонное соединение (антагонист):
[344] CGS-15943 (Sigma-C199).
[345] Разведение соединений
[346] Соединения, подлежащие тестированию, растворяли в DMSO с получением 10 мМ исходного раствора. Тестируемое соединение разбавляли до 0,2 мМ с помощью DMSO и эталонное соединение CGS-15943 разбавляли до 0,015 мМ с помощью DMSO. Затем осуществляли 10-точечное 3-кратное серийное разведение с помощью ECHO, затем 900 нл разведений переносили в планшет для соединений (Greiner-781280) и добавляли 30 мкл буфера для разведения соединений. Конечная исходная концентрация соединений, подлежащих тестированию, составляла 1 мкМ, и концентрация CGS-15943 составляла 0,075 мкМ.
[347] Способ определения
[348] Получение клеток
[349] Замороженные клетки, экспрессирующие A2A, после восстановления суспендировали до 1 × 106 клеток/мл с применением среды для культивирования, высевали в количестве 20 мкл/лунка в 384-луночный покрытый полилизином планшет для клеток (Greiner-781946), затем инкубировали при 5% CO2 при 37°C в течение ночи.
[350] Планшет для клеток, полученный за один день перед этим, вынимали из инкубатора, в каждую лунку добавляли 20 мкл буфера 2X Fluo-4 DirectTM, затем его инкубировали при 5% CO2 при 37°C в инкубаторе в течение 50 минут и оставляли отстояться при комнатной температуре в течение 10 минут.
[351] Определение EC80 агониста NECA
[352] Разведение агониста NECA. Обеспечивали 3-кратное серийное разведение NECA с исходной концентрацией 0,15 мМ с помощью Echo с получением 10 разведений, а затем 900 нл разведений переносили в соответствующий планшет для соединений; затем в соответствующий планшет для соединений добавляли 30 мкл буфера для разведения соединений. Конечная исходная концентрация составляла 750 нМ.
[353] Обеспечивали работу прибора и программного обеспечения FLIPR в соответствии с установленной программой, добавляли в планшет для клеток 10 мкл буфера для разведения соединения и считывали флуоресцентный сигнал. В планшет для клеток добавляли 10 мкл эталонного соединения, представляющего собой агонист, с предварительно определенной концентрацией и считывали флуоресцентный сигнал. После считывания данные экспортировали с помощью методов «Max-Min» и «Read 90 to Maximum allowed» в программном обеспечении, рассчитывали EC80 для клеточной линии, экспрессирующей A2A, и получали концентрацию агониста, составляющую 6 × EC80. Получали эталонное соединение, представляющее собой агонист, с концентрацией 6 × EC80 с применением буферного солевого раствора и добавляли 30 мкл/лунка в виде аликвот в соответствующий планшет для соединений для последующего применения.
[354] Определение IC50 соединений, подлежащих тестированию
[355] Обеспечивали работу прибора и программного обеспечения FLIPR в соответствии с установленной программой, в планшет для соединений добавляли 10 мкл тестируемых соединений и эталонного соединения с предварительно определенной концентрацией и считывали флуоресцентный сигнал. В планшет для клеток добавляли 10 мкл эталонного соединения, представляющего собой агонист, с концентрацией 6 × EC80 и считывали флуоресцентный сигнал. Для обнаружения агонистической активности соединений данные экспортировали с помощью методов «Max-Min» и «Read 1 to 90» в программном обеспечении. Для обнаружения антагонистической активности соединений данные экспортировали с помощью методов «Max-Min» и «Read 90 to Maximum allowed» в программном обеспечении. Данные анализировали с помощью GraphPad Prism 5.0 и рассчитывали значения IC50 тестируемых соединений.
[356] Таблица 3. Результаты скринингового теста in vitro соединений по настоящему изобретению
[357] Заключение: как показано в таблице 3, соединения по настоящему изобретению проявляют отличную антагонистическую активность в отношении аденозинового рецептора A2A.
[358] Экспериментальный пример 2. Фармакокинетическая оценка
[359] Экспериментальные материалы: использовали мышей линии Balb/C (самки, 15-30 г, возрастом 7-9 недель, Shanghai Linchang).
[360] Экспериментальный способ: фармакокинетические характеристики соединения после внутривенной инъекции и перорального введения тестировали на грызунах с помощью стандартной схемы, при этом в эксперименте получали соединения-кандидаты в виде прозрачных растворов и вводили их мышам посредством однократной внутривенной инъекции и однократного перорального введения. Растворитель для внутривенной инъекции (IV) представлял собой смешанный растворитель из 5% DMSO/5% гидроксистеарата полиэтиленгликоля/90% воды, а растворитель для перорального введения (PO) представлял собой смешанный растворитель из 1% Tween 80, 9% PEG400 и 90% воды (pH = 3). Через 48 часов образцы цельной крови собирали, центрифугировали при 3000 g при 4°C в течение 15 минут и надосадочную жидкость отделяли с получением образца плазмы крови. Для осаждения белка добавляли раствор ацетонитрила, содержащий внутренний стандарт, в объеме в 20 раз больше, чем объем образца плазмы крови, центрифугировали надосадочную жидкость и добавляли равный объем воды, затем отбирали надосадочную жидкость для инъекции. Концентрацию лекарственного средства в крови количественно анализировали с помощью способа анализа LC-MS/MS и рассчитывали фармакокинетические параметры, такие как пиковая концентрация, время достижения пиковой концентрации, скорость выведения, период полувыведения, площадь под кривой зависимости концентрации лекарственного средства от времени, биодоступность и т. д.
[361] Таблица 4. PK-параметры в плазме крови соединений вариантов осуществления
[362] Заключение: соединения по настоящему изобретению очевидно могут обладать улучшенными фармакокинетическими параметрами у мышей.
[363] Экспериментальный пример 3. Фармакодинамический тест соединений по настоящему изобретению in vivo
[364] Экспериментальные материалы. Использовали мышей линии BALB/C (самок). Клетки рака толстой кишки мыши CT26 (клеточный банк Комитета по хранению типовых культур, Китайская академия наук) культивировали in vitro в монослое со средой RPMI-1640, содержащей 10% эмбриональной бычьей сыворотки, в инкубаторе при 37°C, 5% CO2. Использовали трипсин-EDTA для обычной процедуры открепления клеток и пассирования. Когда клетки находились в экспоненциальной фазе роста и насыщение составляло 80%-90%, клетки собирали и подсчитывали.
[365] Получение соединения: вариант осуществления 19 взвешивали и добавляли в растворитель (10% PEG400 + 90% (10% водный раствор Cremophor)) с получением образцов с концентрацией 2,5 мг/мл, 5 мг/мл и 10 мг/мл соответственно. Отбирали 72 мкл раствора CS1003 (антитело к PD-1) (25 мг/мл) и добавляли 1,728 мл фосфатного буфера Дульбекко (DPBS) с получением 1 мг/мл раствора, затем добавляли 16,2 мл DPBS с получением 0,1 мг/мл прозрачного раствора.
[366] Экспериментальная процедура. Клетки повторно суспендировали в фосфатном буфере Дульбекко при плотности 3 × 106клеток/мл. 0,1 мл DPBS (содержащий 3 × 105 клеток CT26) подкожно инокулировали в правую часть спины каждой мыши. В день инокуляции мышей произвольным образом разделяли на группы по 9 мышей в каждой группе в соответствии с весом мышей, затем начинали введение и его продолжали в течение 20 дней. Во время всего эксперимента животных взвешивали и здоровье животных контролировали каждый день, при этом в случае какой-либо особой ситуации, лицо, ответственное за соответствующий проект, было бы своевременно проинформировано и были бы сделаны соответствующие записи. Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля с нониусом. Формула для расчета объема опухоли представляла собой: V = 0,5×a × b2, где a и b представляют собой соответственно длинный и короткий диаметры опухоли.
[367] Таблица 5. Режим дозирования для фармакодинамического теста in vivo соединений по настоящему изобретению
(мг/кг)
(мл/кг)
[368] Противоопухолевый эффект соединений оценивали с помощью GI(%) или относительной скорости роста опухоли T/C(%). Относительная скорость роста опухоли T/C(%) = Vt/VC × 100% (Vt: средний объем опухоли в группе обработки; Vc: средний объем опухоли в группе отрицательного контроля). Vt и VC представляли собой данные в один и тот же день.
[369] GI(%), степень подавления опухоли. GI(%) = 1 - Vt/VC × 100%.
[370] Проводили статистический анализ с применением программного обеспечения SPSS на основе относительного объема опухоли и веса опухоли в конце эксперимента. Сравнение между двумя группами анализировали с помощью критерия Стьюдента, а сравнение между тремя или более группами анализировали с помощью однофакторного ANOVA, при этом если дисперсия была однородной (значения F существенно не отличались), то для анализа применяли способ Тьюки, и если дисперсия была неоднородной (значения F имели значительное отличие), для исследования применяли способ Геймса-Ховелла. P < 0,05 считали значительным отличием.
[371] На 20-й день после начала введения объем опухоли в группе с растворителем достиг 847,09±79,65 мм3, тогда как объем в группе с CS1003 (1 мг/кг) составлял 487,34±109,07 мм3, со степенью подавления роста опухоли 42,47% (отсутствие значительного отличия по сравнению с контрольной группой). По сравнению с группой с растворителем, группы с введением комбинации лекарственных средств могут в значительной степени подавлять рост трансплантированной опухоли in vivo, и эффективность гидрохлорида варианта осуществления 6 в комбинации с CS1003 имеет положительную корреляцию с дозой и частотой введения. Объемы опухоли в конце эксперимента составляли 312,06±80,17 мм3, 246,48±62,57 мм3 и 233,10±59,55 мм3 для 25 мг/кг, 50 мг/кг и 100 мг/кг гидрохлорида варианта осуществления 6 в комбинации с 1 мг/кг CS1003 соответственно; значения степени подавления роста опухоли составляли 63,16%, 70,90% и 72,48% соответственно (p < 0,001). Однако гидрохлорид варианта осуществления 6 (50 мг/кг), вводимый дважды в сутки в комбинации с CS1003, продемонстрировал более сильный эффект в отношении подавления роста опухоли, при этом средний объем опухоли в данной группе в конце эксперимента составлял 142,17±40,30 мм3, и степень подавления опухоли составляла 83,22% (P<0,001). Таким образом, гидрохлорид варианта осуществления 6 может в значительной степени подавлять рост аллотрансплантата опухоли из клеток рака толстой кишки мыши CT26 in vivo в комбинации с CS1003 в случае профилактического введения.
[372] Применение гидрохлорида варианта осуществления 6 (50 мг/кг) + CS1003 (1 мг/кг), гидрохлорида варианта осуществления 6 (100 мг/кг) + CS1003 и гидрохлорида варианта осуществления 6 (50 мг/кг дважды в сутки) + CS1003 демонстрирует значительное отличие по сравнению с монотерапией с применением гидрохлорида варианта осуществления 6, при этом P-значения трех групп составляли 0,032, 0,023 и 0,002 соответственно по сравнению с применением 50 мг/кг гидрохлорида варианта осуществления 6; по сравнению с применением 100 мг/кг гидрохлорида варианта осуществления 6 P-значения трех групп составляли 0,038, 0,027 и 0,002 соответственно. Кроме того, рассчитывали значение Q путем применения формулы Джина. Было установлено, что гидрохлорид варианта осуществления 6 (50 мг/кг) с CS1003 обладали определенным аддитивным эффектом, тогда как гидрохлорид варианта осуществления 6 (100 мг/кг) с CS1003 обладали синергическим эффектом.
[373] Заключение: с помощью комбинации соединений по настоящему изобретению и CS1003 можно достигать лучшего эффекта в отношении подавления роста опухоли, а также комбинация соединений по настоящему изобретению и CS1003 обладает синергическим эффектом.
[374] Экспериментальный пример 4. PK-тест эффективности соединений по настоящему изобретению in vivo
[375] Эксперимент проводили на основе экспериментального примера 3, при этом на 20-й день после введения образцы крови и тканей собирали из каждой группы в различные моменты времени (через 0 ч., 0,25 ч., 0,5 ч., 1 ч., 2 ч., 4 ч., 8 ч. и 24 ч.) после введения.
[376] Таблица 6. Фармакокинетические параметры каждой экспериментальной группы, измеренные с помощью тестов
[377] Таблица 7. Концентрация лекарственного средства в ткани опухоли и биологическая доля концентрации лекарственного средства в ткани опухоли и концентрации лекарственного средства в плазме крови в соответствующие точки сбора крови в каждой экспериментальной группе, измеренные с помощью тестов
(нМ)
[378] Заключение: соединения по настоящему изобретению характеризуются достаточным содержанием в плазме крови и ткани опухоли.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ HDAC6, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2772274C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ПИРИМИДОИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДНК-PK | 2020 |
|
RU2796163C1 |
| ИНГИБИТОРЫ FXIA, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2020 |
|
RU2802878C1 |
Настоящее изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли,

В соединении формулы (I): 1 или 2 из T1, T2, T3 и T4 представляют собой N, при этом остальные независимо представляют собой CH; каждый R1 независимо выбран из H, галогена, NH2 или C1-3алкила, необязательно замещенного 1, 2 или 3 R; каждый R2 независимо выбран из H, галогена или C1-3алкила; n выбран из 0 и 1; m выбран из 0, 1 и 2; кольцо A выбрано из 5-10-членного гетероарила; каждый «гетеро» в 5-10-членном гетероариле независимо выбран из N, S и NH; число гетероатомов или гетероатомных групп в 5-10-членном гетероариле независимо выбрано из 1, 2 и 3; кольцо B выбрано из фенила и 5-6-членного гетероарила; каждый «гетеро» в 5-6-членном гетероариле независимо выбран из N, O и S; число гетероатомов или гетероатомных групп в 5-6-членном гетероариле независимо выбрано из 1 и 2; R выбран из F, Cl, Br и I. Также предложены конкретные соединения и применение в изготовлении лекарственного препарата, предназначенного для лечения заболевания, связанного с рецептором A2A. Технический результат: предложены новые органические соединения формулы (I), которые являются антагонистами аденозинового рецептора A2A. 3 н. и 14 з.п. ф-лы, 4 пр., 7 табл.
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
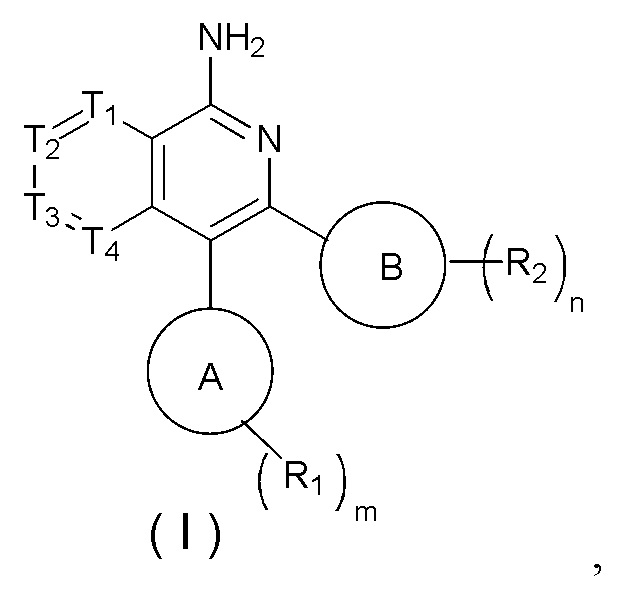
где 1 или 2 из T1, T2, T3 и T4 представляют собой N, при этом остальные независимо представляют собой CH;
каждый R1 независимо выбран из H, галогена, NH2 или C1-3алкила, необязательно замещенного 1, 2 или 3 R;
каждый R2 независимо выбран из H, галогена или C1-3алкила;
n выбран из 0 и 1;
m выбран из 0, 1 и 2;
кольцо A выбрано из 5-10-членного гетероарила; каждый «гетеро» в 5-10-членном гетероариле независимо выбран из N, S и NH; число гетероатомов или гетероатомных групп в 5-10-членном гетероариле независимо выбрано из 1, 2 и 3;
кольцо B выбрано из фенила и 5-6-членного гетероарила; каждый «гетеро» в 5-6-членном гетероариле независимо выбран из N, O и S; число гетероатомов или гетероатомных групп в 5-6-членном гетероариле независимо выбрано из 1 и 2;
R выбран из F, Cl, Br и I.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый R1 независимо выбран из H, F, Cl, Br, I, NH2, Me и Et, где Me и Et необязательно замещены 1, 2 или 3 R.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где каждый R1 независимо выбран из H, F, Cl, Br, I, NH2, Me, CF3 и Et.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый R2 независимо выбран из H, F, Cl, Br, I и Me.
5. Соединение или его фармацевтически приемлемая соль по п. 4, где каждый R2 независимо выбран из F, Cl, Br, I и Me.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где кольцо A выбрано из пиридила, хинолила, хиноксалила, 1,2,3,4-тетрагидрохинолила, 3,4-дигидро-2H-бензо[b][1,4]оксазинила, [1,2,4]триазоло[1,5-a]пиридила, 1H-индазолила, бензо[d]изоксазолила, [1,2,4]триазоло[4,3-a]пиридила, 1H-бензо[d][1,2,3]триазолила, циннолинила, хиназолинила, изохинолила, имидазо[1,2-a]пиридила и бензо[d]тиазолила.
7. Соединение или его фармацевтически приемлемая соль по п. 5, где структурное звено  выбрано из
выбрано из
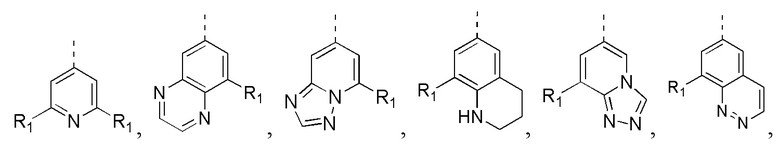


8. Соединение или его фармацевтически приемлемая соль по п. 3 или 7, где структурное звено выбрано из


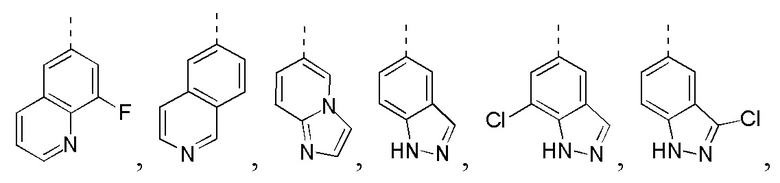
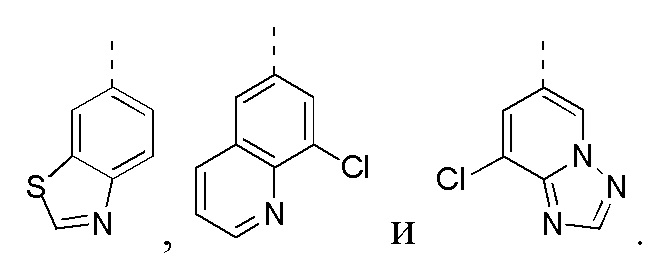
9. Соединение или его фармацевтически приемлемая соль по п. 1, где кольцо B выбрано из фенила, фуранила, тиенила и пиразолила.
10. Соединение или его фармацевтически приемлемая соль по п. 9, где структурное звено  выбрано из
выбрано из 
11. Соединение или его фармацевтически приемлемая соль по п. 5 или 10, где структурное звено выбрано из
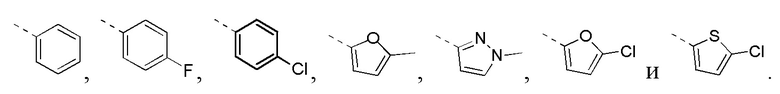
12. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-5, где структурное звено  выбрано из
выбрано из
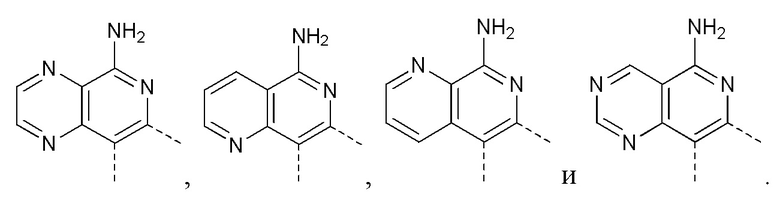
13. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-5, которые выбраны из группы, состоящей из

где R1 и R2 определены в любом из пп. 1-5;
кольцо C выбрано из 5-6-членного гетероарила и 5-6-членного гетероциклоалкила;
кольцо D представляет собой 5-6-членный гетероарил;
«гетеро» в 5-6-членном гетероариле выбран из N, S и NH;
«гетеро» в 5-6-членном гетероциклоалкиле представляет собой NH;
число гетероатомов или гетероатомных групп независимо выбрано из 1, 2 и 3.
14. Соединение или его фармацевтически приемлемая соль по п. 13, где структурное звено  выбрано из
выбрано из
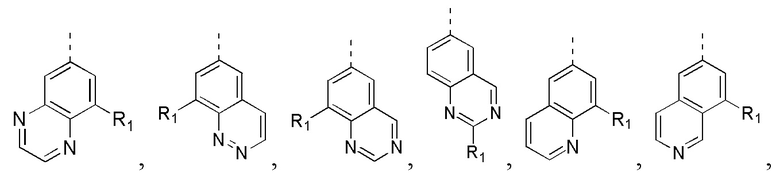

15. Соединение или его фармацевтически приемлемая соль по п. 13, где структурное звено  выбрано из
выбрано из 
16. Соединение, представленное следующей формулой, или его фармацевтически приемлемая соль,
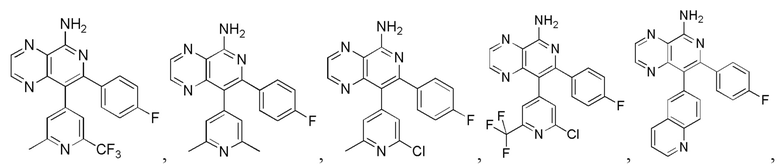
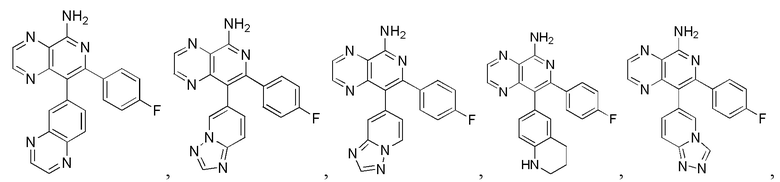

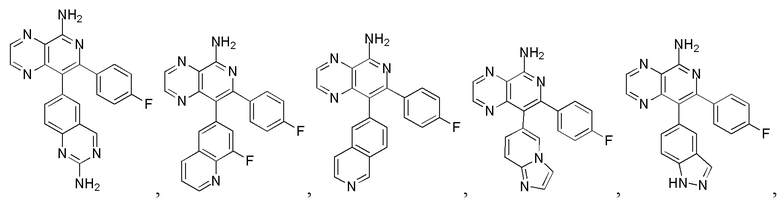
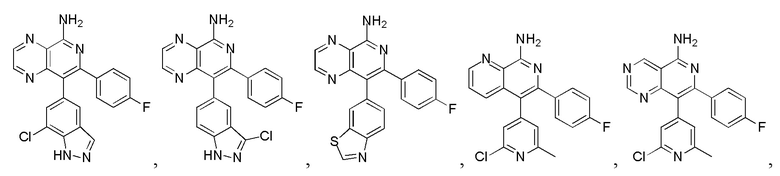


17. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-16 в изготовлении лекарственного препарата, предназначенного для лечения заболевания, связанного с рецептором A2A.
| CN 105025899 A, 04.11.2005 | |||
| Причальное устройство морского сооружения | 1986 |
|
SU1437471A1 |
| WO 2015020565 A1, 12.02.2015 | |||
| WO 2005095360 A1, 12.10.2005 | |||
| ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ МОДУЛЯЦИИ ГАМК - РЕЦЕПТОРНОГО КОМПЛЕКСА | 1996 |
|
RU2136676C1 |

