ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым кристаллическим формам нового соединения {5-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-6-аминопиридазин-3-ил}-N-{4-[((3S,5R)-3,5-диметилпиперазинил)карбонил]фенил}карбоксамида гидрохлорида и его гидрата или сольвата. Настоящее изобретение также относится к способу получения данного соединения, и кристаллических форм, и родственных промежуточных соединений, к фармацевтическим композициям, содержащим описанное соединение, и их применению для ингибирования активности протеинкиназы (PK). Настоящее изобретение также относится к способу применения по меньшей мере одного из описанных выше соединений или одной из описанных выше кристаллических форм и фармацевтических композиций для лечения заболевания, расстройства или состояния, ассоциированного с модулированием протеинкиназы.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Протеинкиназы представляют собой ферменты, которые катализируют фосфорилирование белков, в большинстве случаев фосфорилирование происходит по остаткам серина (ser), треонина (thr) и тирозина (tyr) в белках. Многие аспекты жизни клеток (такие как рост, дифференцировка, пролиферация клеток, клеточный цикл и выживаемость клеток) зависят от активности протеинкиназ. Кроме того, аберрантную активность протеинкиназы ассоциируют со многими расстройствами, такими как рак и воспаление.
К настоящему времени открыто более 500 видов протеинкиназ. Их можно разделить на пять категорий в соответствии с типами аминокислотных остатков фосфорилируемых субстратов-белков: (1) сериновая/треониновая (Ser/Thr) протеинкиназа: фосфорилирует гидроксильную группу белка; (2) тирозиновая (Tyr) протеинкиназа: в качестве рецептора фосфата служит гидроксил фенольной группы белка; (3) гистидиновая протеинкиназа: фосфорилирует основную группу остатка гистидина, аргинина или лизина белка; (4) триптофановая протеинкиназа: в качестве рецептора фосфата служат остатки триптофана белка; (5) аспартильная/глутамильная протеинкиназа: в качестве рецептора фосфата служит ацильная группа белка.
Тирозиновая протеинкиназа (PTK) (в настоящее время открыто более 100 членов семейства) играет важную роль в регуляции дифференцировки, роста и активации клеток. РТК можно разделить на две категории, киназы рецепторного типа и нерецепторного типа, в соответствии с их структурой, при этом первые известны как трансмембранные РТК, последние известны как внутриклеточные PTK.
Возникновение и развитие опухолей у человека зависит от активации ряда онкогенов и инактивации генов онкосупрессоров. При исследовании эпителиальных опухолей обнаружено, что трансмембранный белок рецепторных тирозинкиназ (RTK) играет фундаментальную роль в регуляции роста, дифференцировки и выживаемости клеток и играет жизненно важную роль в возникновении и развитии опухолей.
Среди RTK подсемейство протоонкогена МЕТ содержит 2 члена, МЕТ и RON (рецептор "нантского происхождения").
Протоонкоген C-Met кодирует рецепторную тирозинкиназу Met. Рецептор Met, гликозилированный димерный комплекс с молекулярной массой (ММ) 190 кДа, состоит из α-цепи с ММ 50 кДа, которая соединена дисульфидной связью с β-цепью с ММ 145 кДа. Установлено, что α-цепь является внеклеточной, в то время как β-цепь включает в себя трансмембранный и цитоплазматический домены. Met принимает участие в онкогенезе и метастазировании, и Met изменяется при экспрессии своего лиганда - фактора роста гепатоцитов (HGF), вызывая онкогенез и метастазирование (Jefferson М. et al., Oncogene, 1996, 13, 853-856; Michieli P. et al., Oncogene, 1999, 18, 5221-5231). C-Met сверхэкспрессируется в значительном числе случаев рака человека и амплифицируется при переходе от первичных опухолей к метастазам. В многочисленных исследованиях показана связь экспрессии C-Met и/или HGF/SF с прогрессированием рака разных типов. Помимо этого показано, что сверхэкспрессия C-Met или HGF коррелирует с неблагоприятным прогнозом и прогнозом заболевания рядом основных видов рака человека (включая рак легкого, рак печени, рак желудка и рак молочной железы). С-Met также непосредственно вовлечен в те виды рака, которые не поддаются успешному лечению, такие как рак поджелудочной железы, нейроглиома и гепатоклеточная карцинома.
Гомологи RON включают Stk (тирозинкиназа стволовых клеток) (мышь) и Sea (цыпленок). Его лигандом является белок, стимулирующий макрофаги (MSP), который представляет собой белок сыворотки крови, гомологичный HGF. Ген RON расположен на хромосоме 3р21 человека, содержит 20 экзонов и 19 интронов. Зрелый белок RON представляет собой гетеродимер, составленный из α- и β-субъединиц с молекулярной массой примерно 185 кДа. Продукт гена RON может быть обнаружен в различных нормальных тканях человека. RON экспрессируется в эпителиальных клетках человека, гранулоцитах, мононуклеарных макрофагах, мегакариоцитах, остеокластах, клетках тонзиллярного зародышевого слоя, тонкого кишечника, толстой кишки, почки, легкого и костного мозга. В последние годы исследованиями показано, что во многих первичных опухолях человека и линиях опухолевых клеток, включая пищеварительную систему, мочевыделительную систему, легкое и молочную железу, экспрессия RON значительно изменяется в качественном и количественном отношениях. Онкогенная активность RON коррелирует с киназной активностью, и киназную активность RON можно существенно активировать посредством механизмов сверхэкспрессии, мутирования и расщепления, приводящих к малигнизации, росту и движению клеток. Кроме того, RON может функционировать самостоятельно или совместно с другими факторами, вызывая инвазию и метастазирование опухолей (International Journal of Pathology and Clinical Medic, 2005, 25(5): 441-443).
CSF1R (рецептор колониестимулирующего фактора 1), также известный как C-fms (рецептор макрофагального колониестимулирующего фактора), представляет собой состоящую из одной цепи трансмембранную рецепторную тирозинкиназу и является членом семейства RTK, которое содержит иммуноглобулиновые (Ig) мотивы. CSF1R в большинстве случаев экспрессируется в клетках моноцитарных линий, а также клетках женской репродуктивной системы и плаценты. Также обнаружено, что CSF1R экспрессируется в клетках Лангерганса, подгруппах клеток гладких мышц, В-клетках и клетках микроглии в коже. В основе главных биологических эффектов передачи сигнала с участием CSF1R лежит дифференцировка, пролиферация, миграция и выживаемость предшественников макрофагов и остеокластов в моноцитарных линиях.
Axl принадлежит к подсемейству рецепторных тирозинкиназ, которое также включает в себя Tyro 3 и Mer. Сверхэкспрессия Axl отмечена во многих случаях рака у человека и ассоциирована с инфицированием и метастазированием в случае рака легкого, рака предстательной железы, рака молочной железы, рака желудка, почечноклеточной карциномы и глиобластомы. Недавними исследованиями показано, что сверхэкспрессия Axl посредством "тирозинкиназного переключателя" вызывает устойчивость к иматинибу в гастроинтестинальной стромальной опухоли. Экспрессия Axl индуцируется химиотерапевтическими лекарственными средствами, и сверхэкспрессия Axl приводит к устойчивости при острых миелоидных лейкозах, что позволяет предположить, что Axl может быть вовлечена в регуляцию различных аспектов онкогенеза (Oncogene, 2009, 28: 3442).
EphA2 (эфриновый рецептор А2) принадлежит к крупнейшей подгруппе эфриновых рецепторных тирозинкиназ (ЕРН RTK), и исследованиями показано, что EphA2 ассоциирован с каскадом регуляции патологических состояний, включая опухоли (Pasquale ЕВ. Eph receptors and ephrins in cancer: bidirectional signaling and beyond. Nat. Rev. Cancer, 2010, 10: 165-80). Недавними исследованиями показано, что путем блокирования EphA2 в случае рака легкого можно преодолеть приобретенную устойчивость к ингибиторам киназы, являющейся рецептором фактора эпидермального роста (EGFR) (Amato et al., Cancer Res., 2016, 76(2): 305-18).
ROS1 является членом семейства рецепторов инсулина. Недавно у небольшого числа пациентов с раком легкого была обнаружена перестройка ROS1, и казолиниб, как ингибитор ROS1, очень эффективен в лечении таких пациентов (Bergethon et al., J. Clin. Oncol., 2012, 30(8), 863).
В случае рака легкого также обнаружены перестройки NTRK1 (нейротрофная рецепторная тирозинкиназа 1), приводящие к онкогенности и восприимчивости к лекарственным средствам (Vaishnavi et al., Nature Medicine, 2013, 19(11), 1469). Ген NTRK1 кодирует белок - высокоаффинный рецептор фактора роста нервов (TRKA (тропомиозин-рецепторная киназа А)).
Киназа анапластической лимфомы (ALK) принадлежит к суперсемейству RTK. Вследствие гетеротопии t2 хромосомы, при анапластической крупноклеточной лимфоме (ALCL) и воспалительной миофибробластной опухоли (IMT) экспрессируются онкогенные конститутивно активные слитые с ALK белки. В последнее время ALK рассматривается в качестве протоонкогена в небольшом числе случаев немелкоклеточного рака легкого и нейроцитомы (Choi et al., Cancer Res., 2008, 68:(13); Webb et al., Expert Rev. Anticancer Then, 2009, 9(3), 331-356).
Недавно было обнаружено, что в 11% случаев меланомы и в отдельных других типах рака человека, но не в нормальных тканях, экспрессируется новый изотип ALK (Wiesner et al., Nature, 2015, 526, 453). Новый транскрипт ALK вызывает инициацию альтернативной транскрипции (ATI) интрона в ALK 19 и известен как ALKATI.
ALK также вовлечена в заболевания нервной системы. Показано, что ALK регулирует функции фронтального отдела коры и гиппокампа головного мозга у взрослого человека, и ALK идентифицируют как новую мишень для выявления психических симптомов (таких как шизофрения, депрессия и наркотическая (кокаиновая) зависимость).
Сообщалось о применении кризотиниба в качестве эффективного ингибитора принадлежащей семейству HGF рецепторной тирозинкиназы (C-Met) и ALK (WO 2004076412, WO 2006021881, WO 2006021886).
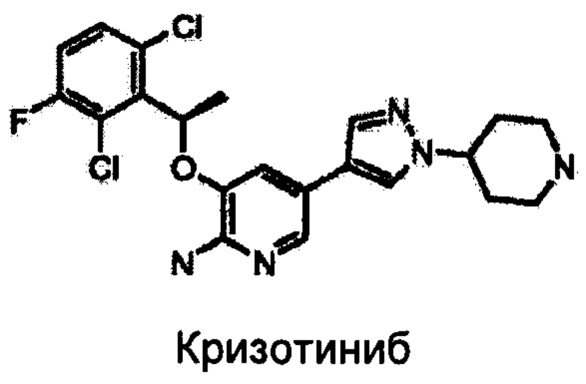
В фазе I клинического испытания кризотиниба достигали частоты объективных ответов (ORR), составляющей 64%, а регулирования заболевания достигали в 90% случаев (J. Clin. Oncology, 2010, 28: 7S, Suppl; abstr. 3). К сожалению, сильный ответ на кризотиниб носит только временный характер. У большинства пациентов развивается устойчивость и наблюдается прогрессирование заболевания через 6-18 месяцев лечения. В частности, значительная часть пациентов имеет метастазы в головной мозг, не излечиваемые кризотинибом.
В предыдущих патентных публикациях (WO 2009/154769 A1, WO 2012/048259 A2, CN 103298806 В) описываются замещенные соединения пиридазинкарбоксамидов в качестве ингибиторов протеинкиназ, большинство из которых эффективно ингибируют c-Met и ALK с IC50 (концентрация, вызывающая половину от максимального ингибирования) ниже 100 нМ. Поскольку все еще существуют неудовлетворенные потребности в выборе лечения для опосредованных киназами заболеваний, авторы изобретения провели дополнительный скрининг полиморфных форм замещенных соединений пиридазинкарбоксамидов с целью удовлетворения медицинских потребностей пациентов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящим изобретением решается задача разработки {5-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-6-аминопиридазин-3-ил)-N-{4-[((3S,5R)-3,5-диметилпиперазинил)карбонил]фенил}карбоксамида гидрохлорида формулы I:
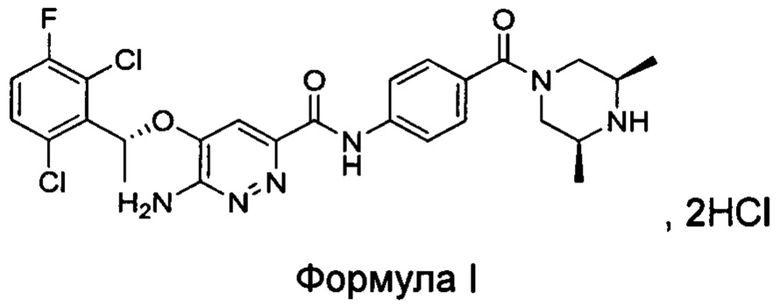
Изобретение также относится к ряду относительно чистых кристаллических форм соединения формулы I, его гидратов и/или сольватов.
В настоящем изобретении кристаллические формы соединения формулы I, его гидратов и/или сольватов существуют в одной или нескольких кристаллических формах.
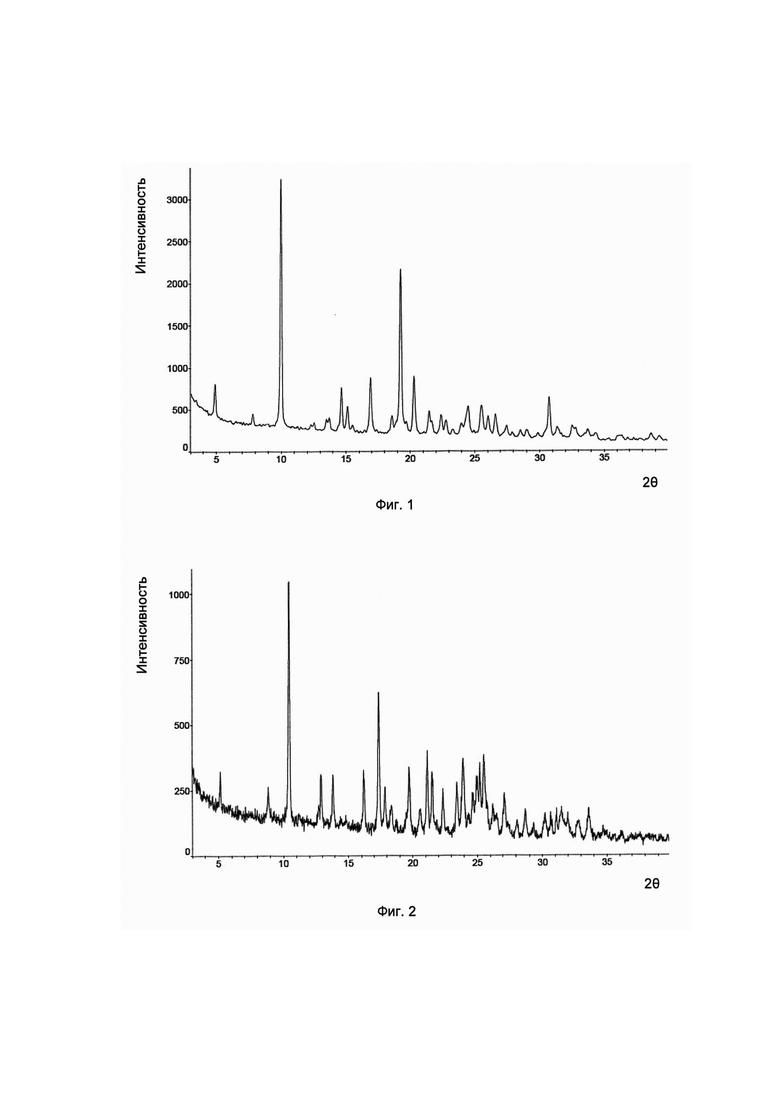
Во-первых, согласно изобретению предложена кристаллическая форма соединения формулы I, его гидратов и/или сольватов, и соответствующая ей картина дифракции рентгеновских лучей на порошке имеет характерные пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2° и 19,3±0,2°. Для удобства данный предмет изобретения рассматривается как кристаллическая форма А.
Во-вторых, согласно настоящему изобретению дополнительно предложены предпочтительные воплощения упомянутой выше кристаллической формы А.
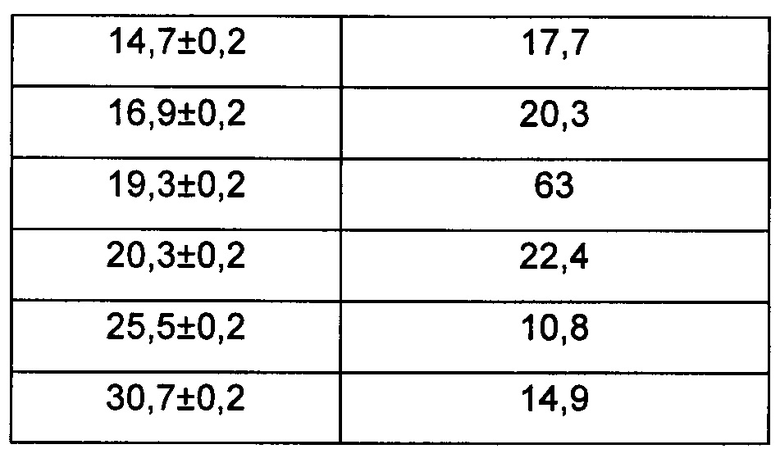
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы А имеет характерные пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2°, 14,7±0,2°, 16,9±0,2°, 19,3±0,2° и 20,3±0,2°.
Предпочтительно, картина дифракции рентгеновских лучей на порошке кристаллической формы А имеет характерные пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2°, 14,7±0,2°, 16,9±0,2°, 19,3±0,2°, 20,3±0,2°, 25,5±0,2° и 30,7±0,2°.
Предпочтительно, упомянутая выше кристаллическая форма А имеет картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 1.
В настоящем изобретении суммированы данные картины дифракции рентгеновских лучей на порошке для кристаллической формы А так, как показано в Таблице 1.

Предпочтительно, кристаллическая форма А имеет чистоту ≥85%.
Предпочтительно, кристаллическая форма А имеет чистоту ≥95%.
Предпочтительно, кристаллическая форма А имеет чистоту ≥99%.
Предпочтительно, кристаллическая форма А имеет чистоту ≥99,5%.
Предпочтительно, кристаллическая форма А представляет собой дигидрат.
Согласно изобретению дополнительно предложена другая кристаллическая форма соединения формулы I, его гидратов и/или сольватов, и соответствующая ей картина дифракции рентгеновских лучей на порошке имеет характерные пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2° и 21,1±0,2°. Для удобства данный предмет изобретения рассматривается как кристаллическая форма В.
Согласно настоящему изобретению дополнительно предложены предпочтительные воплощения упомянутой выше кристаллической формы В.
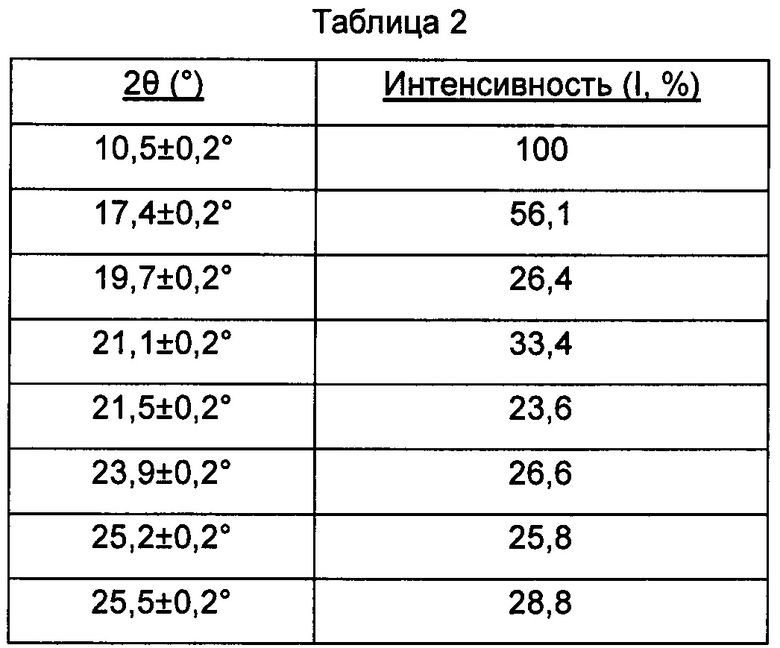
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы В имеет характерные пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2°, 19,7±0,2°, 21,1±0,2°, 23,9±0,2° и 25,5±0,2°.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы В имеет характерные пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2°, 19,7±0,2°, 21,1±0,2°, 21,5±0,2°, 23,9±0,2°, 25,2±0,2° и 25,5±0,2°.
Предпочтительно, упомянутая выше кристаллическая форма В имеет картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 2.
В настоящем изобретении суммированы данные картины дифракции рентгеновских лучей на порошке для кристаллической формы В так, как показано в Таблице 2.
Предпочтительно, кристаллическая форма В имеет чистоту ≥85%.
Предпочтительно, кристаллическая форма В имеет чистоту ≥95%.
Предпочтительно, кристаллическая форма В имеет чистоту ≥99%.
Предпочтительно, кристаллическая форма В имеет чистоту ≥99,5%.
Предпочтительно, кристаллическая форма В представляет собой тригидрат.
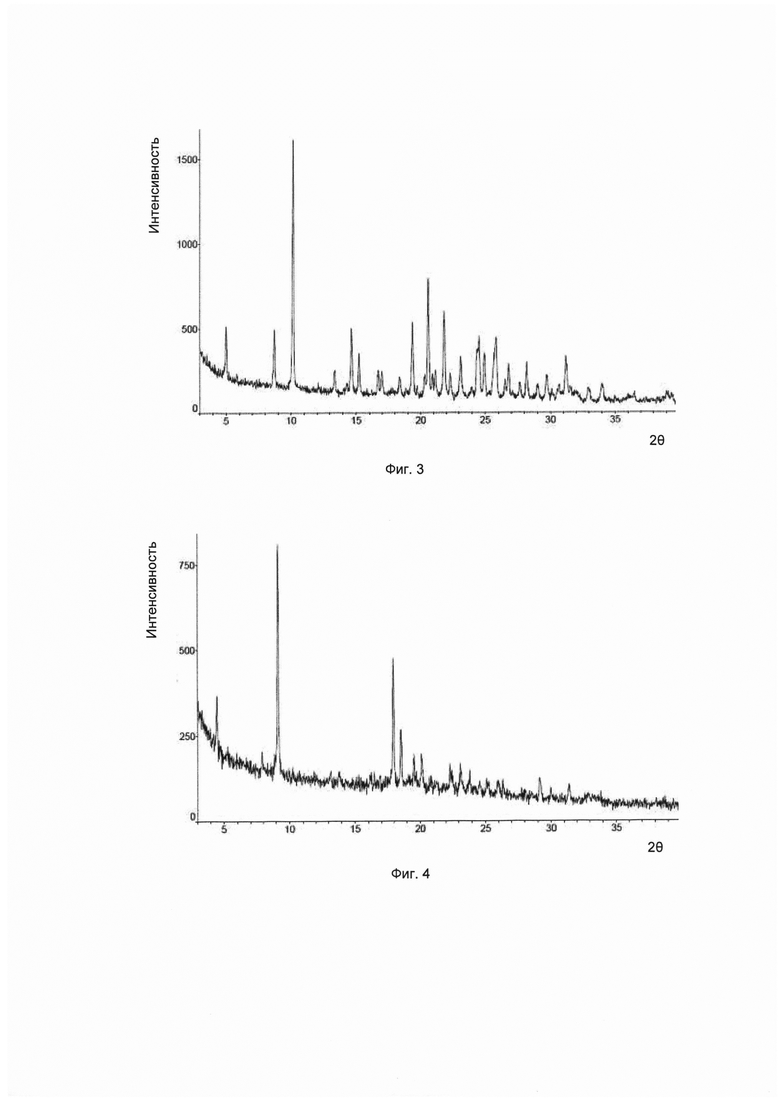
Согласно изобретению дополнительно предложена другая кристаллическая форма соединения формулы I, его гидратов и/или сольватов, и соответствующая ей картина дифракции рентгеновских лучей на порошке имеет характерные пики при углах дифракции 2θ, составляющих 10,2±0,2°, 20,6±0,2° и 21,8±0,2°. Для удобства данный предмет изобретения рассматривается как кристаллическая форма С.
Во-вторых, согласно настоящему изобретению дополнительно предложены предпочтительные воплощения упомянутой выше кристаллической формы С.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы С имеет характерные пики при углах дифракции 2θ, составляющих 10,2±0,2°, 14,7±0,2°, 19,4±0,2°, 20,6±0,2°, 21,8±0,2° и 24,5±0,2°.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы С имеет характерные пики при углах дифракции 2θ, составляющих 8,7±0,2°, 10,2±0,2°, 14,7±0,2°, 19,4±0,2°, 20,6±0,2°, 21,8±0,2°, 24,5±0,2° и 25,9±0,2°.
Предпочтительно, упомянутая выше кристаллическая форма С имеет картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 3.
В настоящем изобретении суммированы данные картины дифракции рентгеновских лучей на порошке для кристаллической формы С так, как показано в Таблице 3.
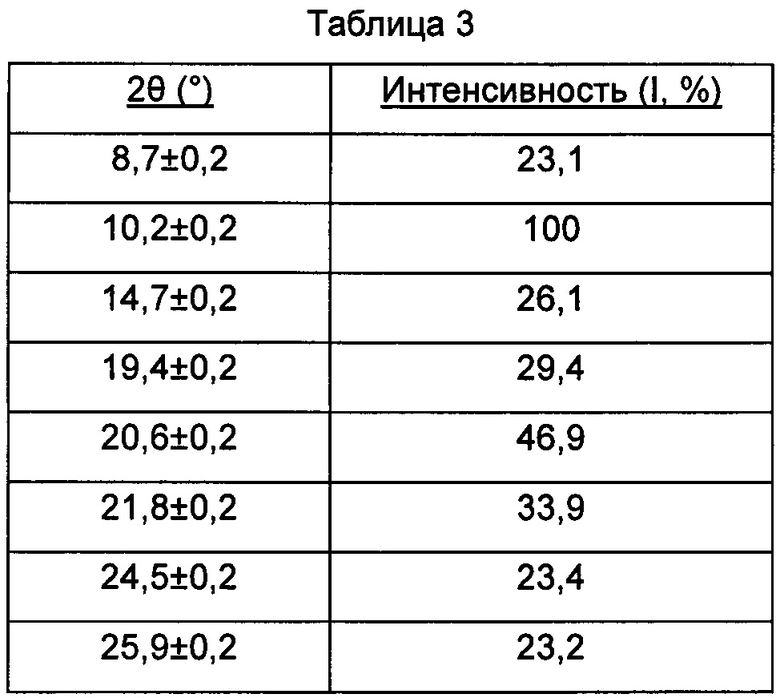
Предпочтительно, кристаллическая форма С имеет чистоту ≥85%.
Предпочтительно, кристаллическая форма С имеет чистоту ≥95%.
Предпочтительно, кристаллическая форма С имеет чистоту ≥99%.
Предпочтительно, кристаллическая форма С имеет чистоту ≥99,5%.
Предпочтительно, кристаллическая форма С представляет собой соединение, полученное с использованием метанольного растворителя.
Согласно изобретению дополнительно предложена другая кристаллическая форма соединения формулы I, его гидратов и/или сольватов, и соответствующая ей картина дифракции рентгеновских лучей на порошке имеет характерные пики при углах дифракции 2θ, составляющих 9,2±0,2°, 18,0±0,2° и 18,5±0,2°. Для удобства данный предмет изобретения рассматривается как кристаллическая форма D.
Во-вторых, согласно настоящему изобретению дополнительно предложены предпочтительные воплощения упомянутой выше кристаллической формы D.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы D имеет характерные пики при углах дифракции 2θ, составляющих 4,5±0,2°, 9,2±0,2°, 18,0±0,2°, 18,510,2", 19,5±0,2° и 20,1±0,2°.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы D имеет характерные пики при углах дифракции 2θ, составляющих 4,5±0,2°, 9,2±0,2°, 18,0±0,2°, 18,5±0,2°, 19,5±0,2°, 20,1±0,2°, 22,3±0,2° и 23,1±0,2°.
Предпочтительно, упомянутая выше кристаллическая форма D имеет картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 4.
В настоящем изобретении суммированы данные картины дифракции рентгеновских лучей на порошке для кристаллической формы D так, как показано в Таблице 4.
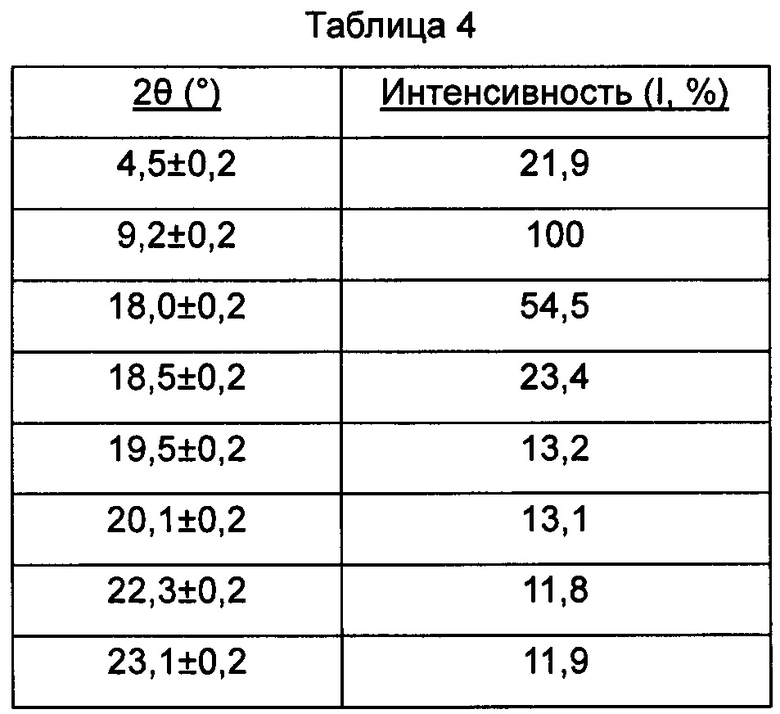
Предпочтительно, кристаллическая форма D имеет чистоту ≥285%. Предпочтительно, кристаллическая форма D имеет чистоту ≥95%. Предпочтительно, кристаллическая форма D имеет чистоту ≥99%. Предпочтительно, кристаллическая форма D имеет чистоту ≥99,5%. Предпочтительно, кристаллическая форма D представляет собой соединение, полученное с использованием диметилсульфоксидного растворителя.
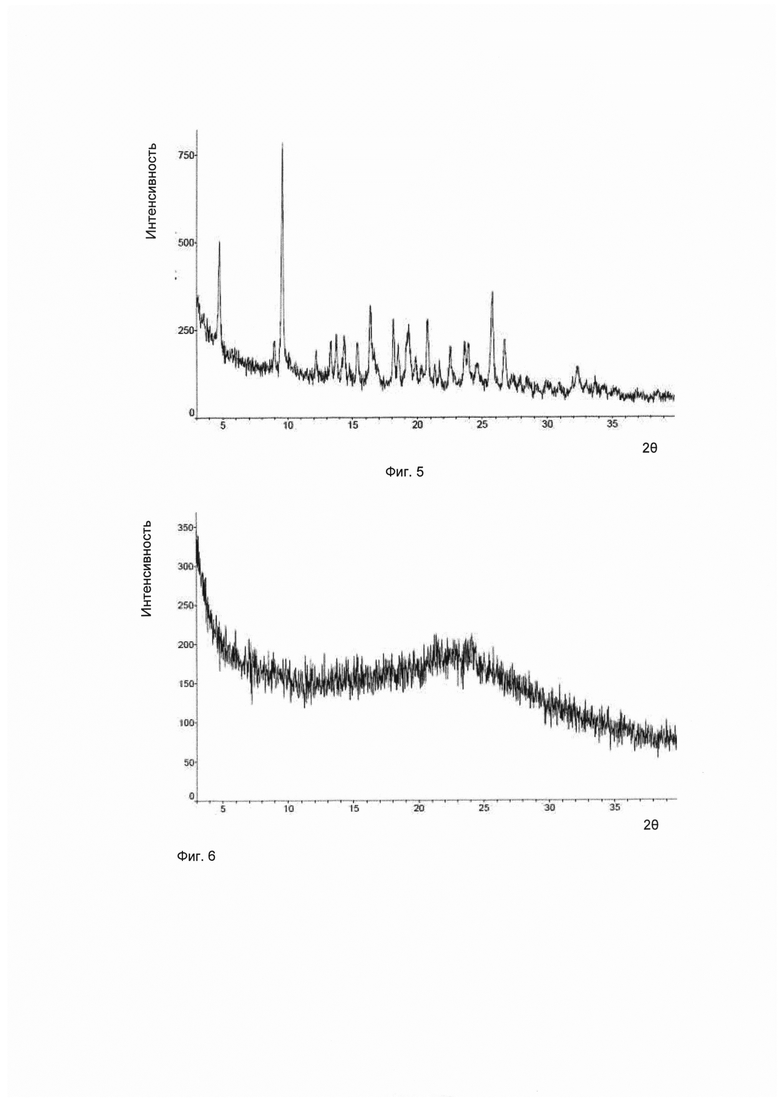
Согласно изобретению дополнительно предложена другая кристаллическая форма соединения формулы I, его гидратов и/или сольватов, и соответствующая ей картина дифракции рентгеновских лучей на порошке имеет характерные пики при углах дифракции 2θ, составляющих 4,8±0,2°, 9,6±0,2° и 25,8±0,2°. Для удобства данный предмет изобретения рассматривается как кристаллическая форма Е.
Во-вторых, согласно настоящему изобретению дополнительно предложены предпочтительные воплощения упомянутой выше кристаллической формы Е.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы Е имеет характерные пики при углах дифракции 2θ, составляющих 4,8±0,2°, 9,6±0,2°, 16,3±0,2°, 18,1±0,2°, 20,8±0,2° и 25,8±0,2°.
Предпочтительно, картина дифракции рентгеновских лучей на порошке упомянутой выше кристаллической формы Е имеет характерные пики при углах дифракции 2θ, составляющих 4,8±0,2°, 9,6±0,2°, 16,3±0,2°, 18,1±0,2°, 19,3±0,2°, 20,8±0,2°, 25,8±0,2° и 26,7±0,2°.
Предпочтительно, упомянутая выше кристаллическая форма Е имеет картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 5.
В настоящем изобретении суммированы данные картины дифракции рентгеновских лучей на порошке для кристаллической формы Е так, как показано в Таблице 5.
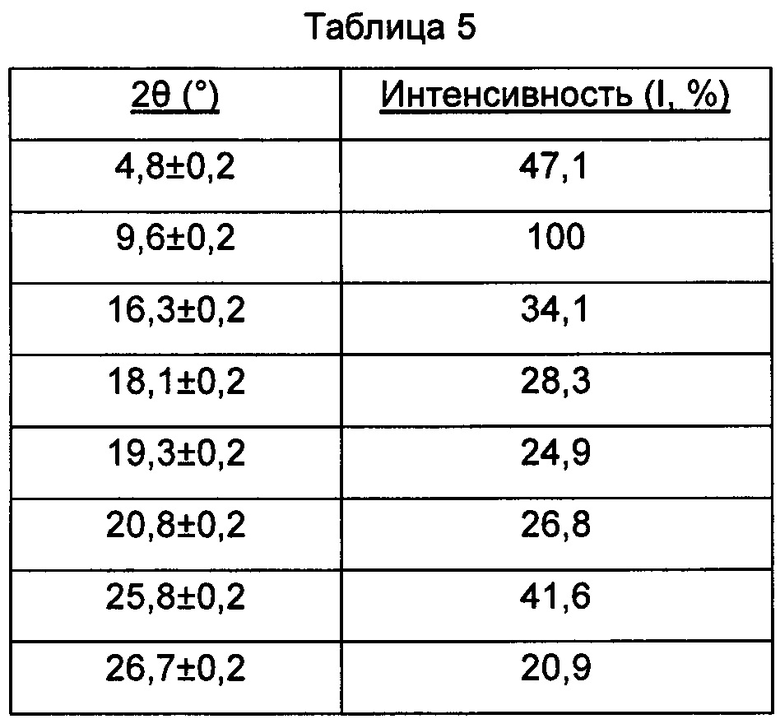
Предпочтительно, кристаллическая форма Е имеет чистоту ≥85%.
Предпочтительно, кристаллическая форма Е имеет чистоту ≥95%.
Предпочтительно, кристаллическая форма Е имеет чистоту ≥99%.
Предпочтительно, кристаллическая форма Е имеет чистоту ≥99,5%.
Предпочтительно, кристаллическая форма Е представляет собой дигидрат.
Согласно настоящему изобретению дополнительно предложены аморфные формы соединений формулы I, их гидратов и/или сольватов, имеющие картину дифракции рентгеновских лучей на порошке приблизительно такую, которая показана на Фиг. 6.
Согласно изобретению дополнительно предложена фармацевтическая композиция, содержащая терапевтически эффективное количество упомянутой выше кристаллической формы А и/или кристаллической формы В.
Согласно настоящему изобретению также предложено предпочтительное воплощение упомянутых выше фармацевтических композиций.
Предпочтительно, фармацевтическая композиция содержит терапевтически эффективное количество кристаллической формы А или кристаллической формы В, предложенной в данной заявке, и фармацевтически приемлемый эксципиент, адъювант или носитель.
Предпочтительно, фармацевтическая композиция содержит терапевтически эффективное количество кристаллической формы А и кристаллической формы В, предложенных в данной заявке, и фармацевтически приемлемый эксципиент, адъювант или носитель.
Предпочтительно, фармацевтическая композиция содержит терапевтически эффективное количество кристаллической формы А или кристаллической формы В по изобретению в комбинации по меньшей мере с одним другим эффективным компонентом.
Предпочтительно, фармацевтическая композиция содержит терапевтически эффективное количество кристаллической формы А и кристаллической формы В по изобретению в комбинации по меньшей мере с одним другим эффективным компонентом.
Предпочтительно, фармацевтическая композиция находится в форме перорального препарата.
Предпочтительно, фармацевтическая композиция находится в форме таблетки или капсулы.
Предпочтительно, фармацевтическая композиция содержит от 20 мг до 150 мг кристаллической формы А и/или кристаллической формы В, и ее готовят в общей сложности в количестве примерно от 50 мг до 500 мг по меньшей мере с одним эксципиентом, адъювантом и/или носителем.
Предпочтительно, эксципиентом, адъювантом и/или носителем в фармацевтической композиции являются микрокристаллическая целлюлоза, маннит, кросповидон, натриевая соль кроскармелозы, карбоксиметилкрахмал натрия, повидон, гидроксипропилцеллюлоза и/или стеариновая кислота.
Предпочтительно, фармацевтическая композиция содержит от 0,01 масс. % до 99 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 0,01 масс. % до 99 масс. % кристаллической формы А и кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 0,1 масс. % до 70 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 0,1 масс. % до 70 масс. % кристаллической формы А и кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 70 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 70 масс. % кристаллической формы А и кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 50 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 50 масс. % кристаллической формы А и кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 30 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 1 масс. % до 30 масс. % кристаллической формы А и кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 10 масс. % до 30 масс. % кристаллической формы А или кристаллической формы В.
Предпочтительно, фармацевтическая композиция содержит от 10 масс. % до 30 масс. % кристаллической формы А и кристаллической формы В.
Согласно настоящему изобретению также предложено применение кристаллической формы А и/или кристаллической формы В в приготовлении лекарственного средства для лечения заболевания, расстройства или состояния у пациента, при этом данное заболевание, расстройство или состояние опосредуется c-Met, RON, Axl, CSF1R, EphA2, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK.
Согласно изобретению также предложено предпочтительное воплощение упомянутого выше применения кристаллической формы А и/или кристаллической формы В.
Предпочтительно, чтобы слитый белок на основе ALK представлял собой EML4(белок иглокожих, ассоциированный с микротрубочками, тип 4)-ALK или NPM(нуклеофосмин)-ALK киназу.
Предпочтительно, заболевание, расстройство или состояние представляет собой рак и/или пролиферативное расстройство.
Предпочтительно, заболевание, расстройство или состояние представляет собой рак легкого, меланому, рак толстой кишки, рак молочной железы, рак печени, рак поджелудочной железы, рак головного мозга, рак почки, рак яичников, рак желудка, рак кожи, рак кости, глиому, лимфому, нейробластому, гепатоклеточную карциному, папиллярную почечноклеточную карциному и/или плоскоклеточную карциному головы и шеи.
Предпочтительно, заболевание, расстройство или состояние представляет собой немелкоклеточный рак легкого (NSCLC), резистентный к терапии кризотинибом.
Предпочтительно, заболевание, расстройство или состояние представляет собой меланому.
Предпочтительно, заболевание, расстройство или состояние представляет собой неврологическое заболевание, психическое заболевание, ожирение, диабет и/или сердечно-сосудистое заболевание.
Предпочтительно, психическое заболевание представляет собой шизофрению, депрессию и/или зависимость от психоактивных веществ или злоупотребление ими.
Предпочтительно, зависимость от психоактивных веществ или злоупотребление ими представляет собой зависимость от кокаина, табака или алкоголя или злоупотребление ими.
Согласно настоящему изобретению также предложен способ лечения заболевания, расстройства или состояния путем введения кристаллической формы А и/или кристаллической формы В, предложенной в данной заявке, пациенту.
Согласно настоящему изобретению дополнительно предложены предпочтительные воплощения описанного выше способа лечения заболевания, расстройства или состояния у пациента с использованием кристаллической формы А и/или кристаллической формы В.
Предпочтительно, заболевание, расстройство или состояние опосредуется c-Met, RON, Axl, CSF1R, EphA2, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK.
Предпочтительно, чтобы слитый белок на основе ALK представлял собой EML4-ALK или NPM-ALK киназу.
Предпочтительно, заболевание, расстройство или состояние представляет собой рак и/или пролиферативное расстройство.
Предпочтительно, заболевание, расстройство или состояние представляет собой рак легкого, меланому, рак толстой кишки, рак молочной железы, рак печени, рак поджелудочной железы, рак головного мозга, рак почки, рак яичников, рак желудка, рак кожи, рак кости, глиому, лимфому, нейробластому, гепатоклеточную карциному, папиллярную почечноклеточную карциному и/или плоскоклеточную карциному головы и шеи.
Предпочтительно, заболевание, расстройство или состояние представляет собой немелкоклеточный рак легкого (NSCLC), резистентный к терапии кризотинибом.
Предпочтительно, заболевание, расстройство или состояние представляет собой меланому.
Предпочтительно, заболевание, расстройство или состояние представляет собой неврологическое заболевание, психическое заболевание, ожирение, диабет и/или сердечно-сосудистое заболевание.
Предпочтительно, психическое заболевание представляет собой шизофрению, депрессию и/или зависимость от психоактивных веществ или злоупотребление ими.
Предпочтительно, зависимость от психоактивных веществ или злоупотребление ими представляет собой зависимость от кокаина, табака или алкоголя либо злоупотребление ими.
Согласно настоящему изобретению дополнительно предложены способы получения соединения формулы I, приведенные ниже.
Способ 1:
Способ 2:
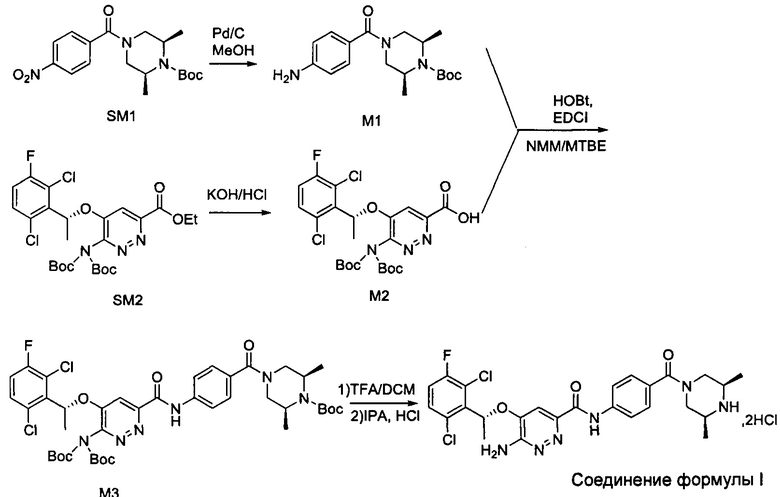
Способ 3:
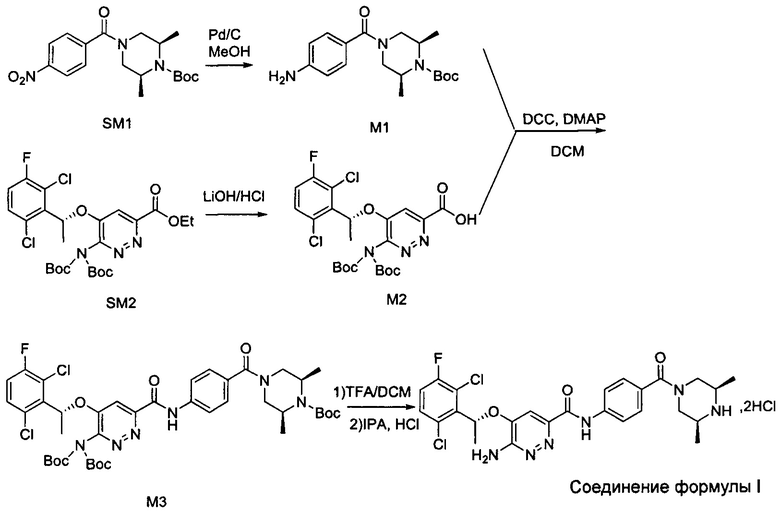
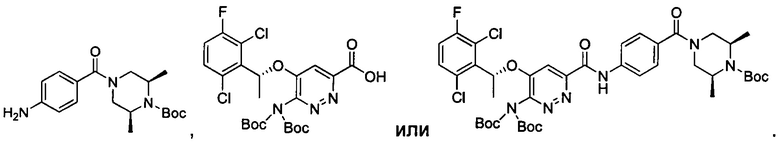
Согласно изобретению дополнительно предложены промежуточные соединения при получении соединения формулы I, приведенные ниже:
Согласно изобретению дополнительно предложены способы получения кристаллической формы А и кристаллической формы В соединений формулы I, их гидратов и/или сольватов,
при этом кристаллическую форму А получают так, как приведено ниже:
аморфный образец соединения формулы I помещали в центрифужные пробирки и хранили без доступа воздуха в этаноле или ацетонитриле в течение 6-10 суток при комнатной температуре, получая кристаллическую форму А; или
аморфный образец соединения формулы I добавляли в этанол, перемешивали при 4°С-25°С и фильтровали, получая кристаллическую форму А; или
аморфный образец соединения формулы I добавляли в этанол при 4°С-25°С и растворяли, получая прозрачный раствор, раствор фильтровали, получая фильтрат; затем к фильтрату при перемешивании добавляли н-гептан до тех пор, пока не наблюдали образование большого количества кристаллов, затем фильтровали, получая кристаллическую форму А; или
аморфный образец соединения формулы I добавляли в метил-трет-бутиловый эфир/этанол или н-гептан/этанол при 55°С-70°С и растворяли, получая прозрачный раствор; раствор фильтровали, получая фильтрат; затем фильтрат перемешивали при -20°С до тех пор, пока не наблюдали образование твердого вещества, и фильтровали, получая кристаллическую форму А; или
аморфный образец соединения формулы I добавляли во втор-бутиловый спирт и растворяли, получая прозрачный раствор, который фильтровали, затем выдерживали при 35°С-50°С для выпаривания растворителя, получая кристаллическую форму А; или
аморфный образец соединения формулы I добавляли в метанол и растворяли, получая прозрачный раствор, раствор фильтровали, получая фильтрат, затем к фильтрату добавляли карбоксиметилцеллюлозу и выдерживали при комнатной температуре для выпаривания растворителя, получая кристаллическую форму А;
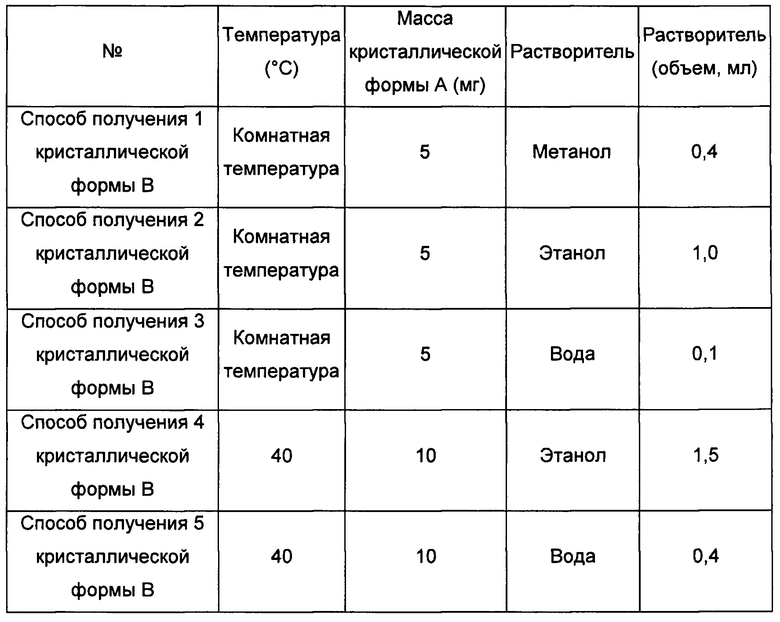
при этом способ получения кристаллической формы В приведен ниже:
кристаллическую форму А добавляли в метанол, этанол или воду, растворяли, получая прозрачный раствор, фильтровали, затем выдерживали при температуре от комнатной температуры (20°С) до 40°С для выпаривания растворителя, получая кристаллическую форму В; или
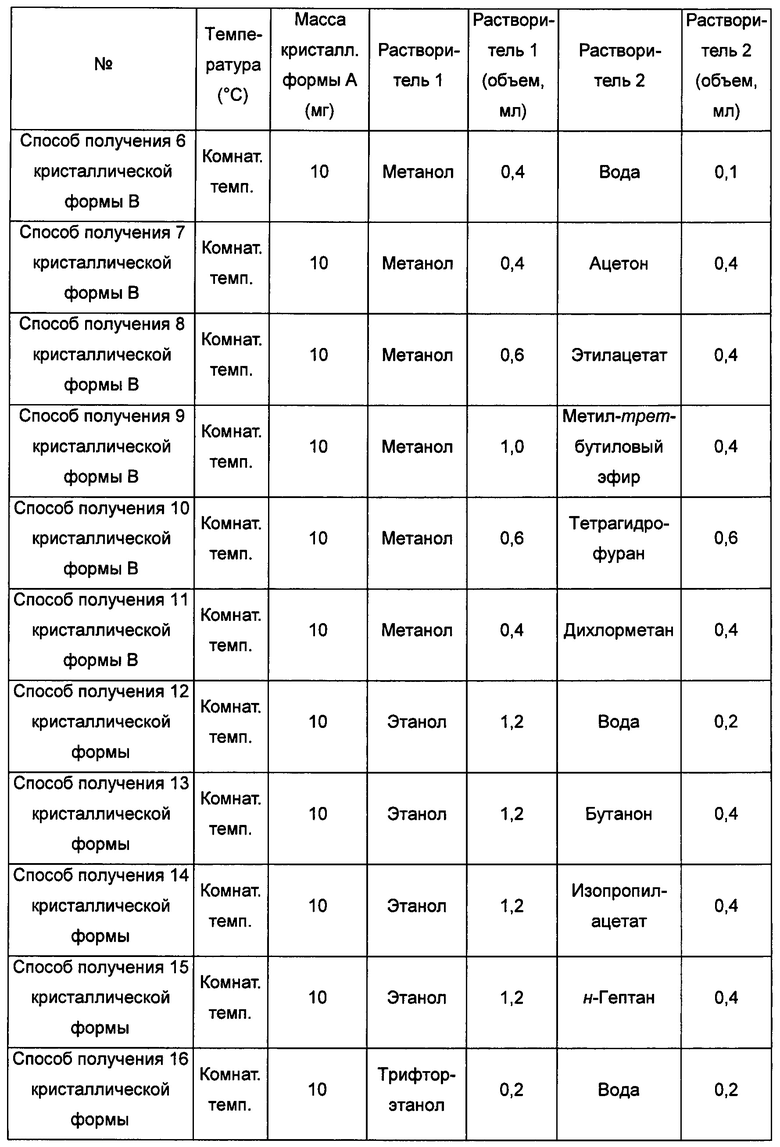
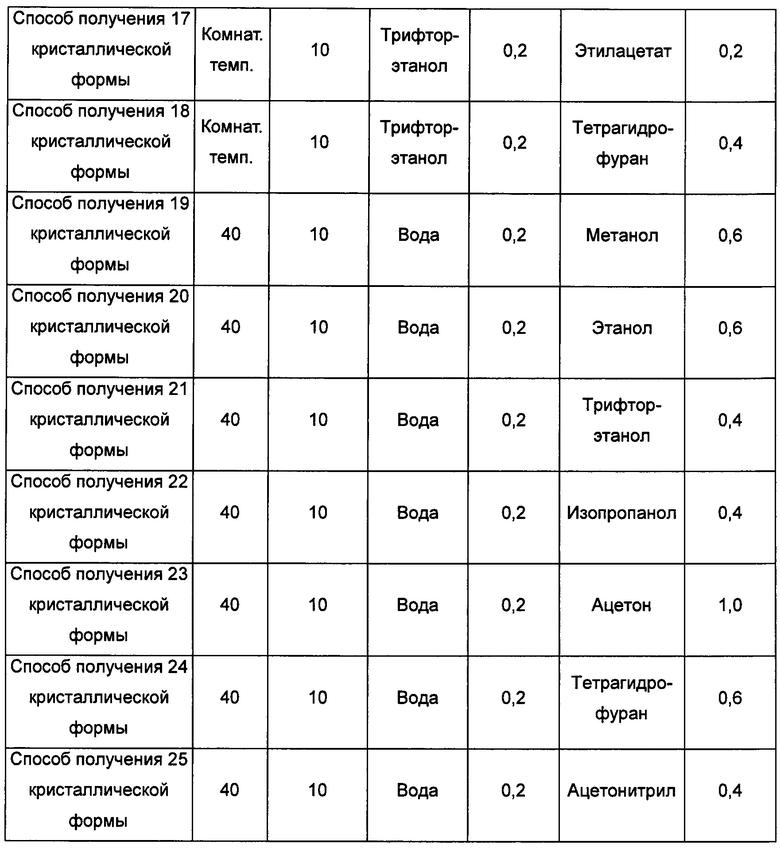
кристаллическую форму А добавляли в метанол/воду, метанол/ацетон, метанол/этилацетат, метанол/метил-трет-бутиловый эфир,
метанол/тетрагидрофуран, метанол/дихлорметан, этанол/воду, этанол/бутанон, этанол/изопропилацетат, этанол/н-гептан, трифторэтанол/воду,
трифторэтанол/этилацетат, трифторэтанол/тетрагидрофуран, воду/метанол, воду/этанол, воду/трифторэтанол, воду/изопропанол, воду/ацетон, воду/тетрагидрофуран или воду/ацетонитрил, растворяли, получая прозрачный раствор, затем раствор фильтровали и выдерживали при температуре в диапазоне от комнатной температуры (20°С) до 40°С для выпаривания растворителя, получая кристаллическую форму В; или
кристаллическую форму А добавляли в низший спирт, воду, нитрометан, бутанон, этиловый эфир, этилацетат, тетрагидрофуран, толуол или н-гептан с образованием суспензии; затем суспензию перемешивали в течение 4-5 суток при температуре от комнатной температуры до 40°С и центрифугировали, получая кристаллическую форму В; или
кристаллическую форму А добавляли в насыщенный водой слой этилацетата, насыщенный этилацетатом водный слой, в этанол/диэтиловый эфир, толуол/ацетонитрил, бутанон/этанол или толуол/изопропиловый эфир с образованием суспензии; затем суспензию перемешивали при 4°С-40°С в течение 4-5 суток и центрифугировали, получая кристаллическую форму В; или
кристаллическую форму А добавляли в метанол, ацетон/воду (3:1, об./об.) или в ацетонитрил/воду (3:2, об./об.) при комнатной температуре и растворяли, получая прозрачный раствор; затем к этому раствору добавляли гидроксипропилцеллюлозу, этилцеллюлозу, повидон K30, полиаллиламина гидрохлорид, карбоксиметилцеллюлозу или поливиниловый спирт, выдерживали при комнатной температуре для выпаривания растворителя, получая кристаллическую форму В; или
кристаллическую форму А добавляли в низший спирт или воду при 60°С-70°С, растворяли, получая прозрачный раствор, и перемешивали при 4°С до тех пор, пока не наблюдали образование кристаллов, получая кристаллическую форму В; или
кристаллическую форму А добавляли в ацетон/трифторэтанол, ацетон/воду, диоксан/воду, ацетонитрил/воду или метил-трет-бутиловый эфир/н-пропанол при 55°С-70°С, растворяли, получая прозрачный раствор; раствор фильтровали, получая фильтрат, затем фильтрат перемешивали при -20°С до тех пор, пока не наблюдали образование кристаллов, затем фильтровали, получая кристаллическую форму В; или
кристаллическую форму А добавляли в нитрометан/метанол, ацетонитрил/метанол, бутанон/этанол, этилацетат/этанол, 1,4-диоксан/этанол или тетрагидрофуран/воду при 60°С-70°С и растворяли, получая прозрачный раствор, фильтровали и выдерживали при комнатной температуре для выпаривания растворителя, получая кристаллическую форму В; или
кристаллическую форму А добавляли в метанол, этанол, воду, трифторэтанол, н-пропанол или диметилсульфоксид при комнатной температуре и растворяли, получая прозрачный раствор; раствор фильтровали и к фильтрату по каплям добавляли ацетон, этилацетат, метил-трет-бутиловый эфир, изопропиловый эфир, изопропилацетат, тетрагидрофуран, 1,4-диоксан, ацетонитрил, н-гептан, дихлорметан или хлороформ до тех пор, пока не наблюдали образование большого количества кристаллов, получая кристаллическую форму В; или
кристаллическую форму А добавляли в метанол или этанол при комнатной температуре и растворяли, получая прозрачный раствор; после этого раствор фильтровали, получая фильтрат; затем к фильтрату при перемешивании добавляли дихлорметан или тетрагидрофуран и выдерживали при комнатной температуре для выпаривания растворителя, получая кристаллическую форму В; или
аморфный образец соединения формулы I помещали в центрифужные пробирки и затем центрифужные пробирки помещали в атмосферу н-бутанола, воды, нитрометана, этилацетата, метил-трет-бутилового эфира, тетрагидрофурана, метиленхлорида, хлороформа или толуола для осуществления диффузии, получая кристаллическую форму В; или
аморфный образец соединения формулы I добавляли в н-пропанол, воду, бутанон, этилацетат, тетрагидрофуран, дихлорметан, этанол, изопропанол, н-бутанол, ацетон, этиловый эфир, изопропилацетат, 1,4-диоксан, ацетонитрил, хлороформ, втор-бутанол, нитрометан или толуол, перемешивали при 4°С-40°С в течение 30 минут, затем фильтровали, получая кристаллическую форму В; или
аморфный образец соединения формулы I добавляли в изопропиловый эфир/метанол, этилацетат/метанол, 1,4-диоксан/метанол, бутанон/этанол, ацетонитрил/этанол, н-гептан/трифторэтанол, нитрометан/трифторэтанол, эфир/трифторэтанол, тетрагидрофуран/трифторэтанол, ацетон/воду, тетрагидрофуран/воду, ацетонитрил/воду, метил-трет-бутиловый эфир/изопропанол, изопропилацетат/н-пропанол, метилциклогексан/н-бутанол, ацетон/диметилсульфоксид, этилацетат/диметилсульфоксид, ацетонитрил/диметилсульфоксид, метил-трет-бутиловый эфир/хлороформ или толуол/этилацетат с образованием суспензии, перемешивали при 4°С-40°С, затем фильтровали, получая кристаллическую форму В; или
аморфный образец соединения формулы I помещали при комнатной температуре в атмосферу с относительной влажностью (RH) 85% на 10 суток, получая кристаллическую форму В; или
кристаллическую форму А добавляли в воду или метанол и растворяли, получая прозрачный раствор; раствор фильтровали, получая фильтрат; затем фильтрат упаривали на роторном испарителе с целью сушки при 40°С-60°С, получая кристаллическую форму В.
Кристаллические формы по настоящему изобретению являются относительно чистыми.
Термин "относительно чистый", использованный в данном описании, означает, что по меньшей мере 85 масс. %, предпочтительно по меньшей мере 95 масс. %, более предпочтительно по меньшей мере 99 масс. %, наиболее предпочтительно по меньшей мере 99,5 масс. % соединения, показанного формулой I, существует в кристаллической форме по настоящему изобретению. В частности, в кристаллической форме А и/или кристаллической форме В.
Вышеупомянутые кристаллические формы представлены в виде суммы только основных пиков. Основные пики воспроизводятся и находятся в пределах ошибки измерения (±0,2).
Использованный в настоящем изобретении термин "картина дифракции рентгеновских лучей на порошке, показанная на Фиг. 1" или "картина дифракции рентгеновских лучей на порошке, показанная на Фиг. 2", относится к картине дифракции рентгеновских лучей на порошке, на которой показаны основные пики, присутствующие на Фиг. 1 или Фиг. 2, где основными пиками обозначают пики с относительной интенсивностью более 10%, предпочтительно более 30%, от интенсивности самого большого пика (при этом его относительную интенсивность принимают за 100%) на Фиг. 1 или Фиг. 2.
Применяемый в настоящем изобретении термин "добавляли в метанол/ацетон", который используется при описании способа получения кристаллической формы А или кристаллической формы В, означает, что в этом способе сначала добавляли метанол, а затем добавляли ацетон. Аналогично, "этанол/вода" означает, что сначала добавляли этанол, а затем добавляли воду; а "трифторэтанол/этилацетат" означает, что сначала добавляли трифторэтанол, а затем добавляли этилацетат. Аналогичным образом, например, "растворитель 1/растворитель 2" означает, что сначала добавляли растворитель 1, а затем добавляли растворитель 2; а "растворитель 2/растворитель 1" означает, что сначала добавляли растворитель 2, а затем добавляли растворитель 1.
Использованный в настоящем изобретении термин "терапевтически эффективное количество" относится к количеству соединения, которого при введении субъекту для лечения заболевания или по меньшей мере одного из клинических симптомов заболевания или расстройства достаточно для осуществления такого лечения в отношении данного заболевания, расстройства или симптома. "Терапевтически эффективное количество" может меняться в зависимости от соединения, заболевания, расстройства и/или симптомов заболевания или расстройства, тяжести заболевания, расстройства и/или симптомов заболевания или расстройства, возраста подвергаемого лечению субъекта и/или массы подвергаемого лечению субъекта. В любом заданном случае соответствующее количество может быть очевидно специалистам в данной области техники или может быть определено посредством рутинного экспериментирования. В случае комбинированной терапии термин "терапевтически эффективное количество" относится к общему количеству составляющих комбинацию объектов, применяемых для эффективного лечения заболевания, расстройства или состояния.
Все готовые формы фармацевтической композиции по настоящему изобретению могут быть приготовлены традиционными в фармацевтической области методами. Например, активный ингредиент можно смешивать с одним или более эксципиентами, затем готовить желаемую композицию.
Термин "фармацевтически приемлемые носители" относится к традиционным фармацевтическим носителям, подходящим для желаемой фармацевтической композиции, например: к разбавителю, эксципиенту, такому как вода, различные органические растворители и т.д.; наполнителю, такому как крахмал, сахароза и т.д.; связующему веществу, такому как производные целлюлозы, альгинаты, желатин и поливинилпирролидон (PVP); увлажняющему агенту, такому как глицерин; разрыхлителю, такому как агар, карбонат кальция и бикарбонат натрия; усилителю всасывания, такому как соединение четвертичного аммония; поверхностно-активному веществу, такому как гексадеканол; абсорбирующему наполнителю, такому как каолин и мыльная глина; смазывающему веществу, такому как тальк, стеарат кальция, стеарат магния, полиэтиленгликоль и т.д. Помимо этого, фармацевтическая композиция дополнительно содержит другие фармацевтически приемлемые эксципиенты, такие как дезинтегрирующий агент, стабилизатор, загуститель, комплексообразующий агент, буферный агент, усилитель проникновения, полимер, ароматизаторы, подсластитель и краситель. Предпочтительно, чтобы эксципиент подходил для желаемой композиции и типа введения.
Термин "заболевание", или "расстройство", или "состояние" относится к любому заболеванию, дискомфорту, болезни, симптомам или показаниям.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1. Картина дифракции рентгеновских лучей на порошке для кристаллической формы А соединения, показанного формулой I.
Фиг. 2. Картина дифракции рентгеновских лучей на порошке для кристаллической формы В соединения, показанного формулой I.
Фиг. 3. Картина дифракции рентгеновских лучей на порошке для кристаллической формы С соединения, показанного формулой I.
Фиг. 4. Картина дифракции рентгеновских лучей на порошке для кристаллической формы D соединения, показанного формулой I.
Фиг. 5. Картина дифракции рентгеновских лучей на порошке для кристаллической формы Е соединения, показанного формулой I.
Фиг. 6. Картина дифракции рентгеновских лучей на порошке для аморфного образца соединения, показанного формулой I.
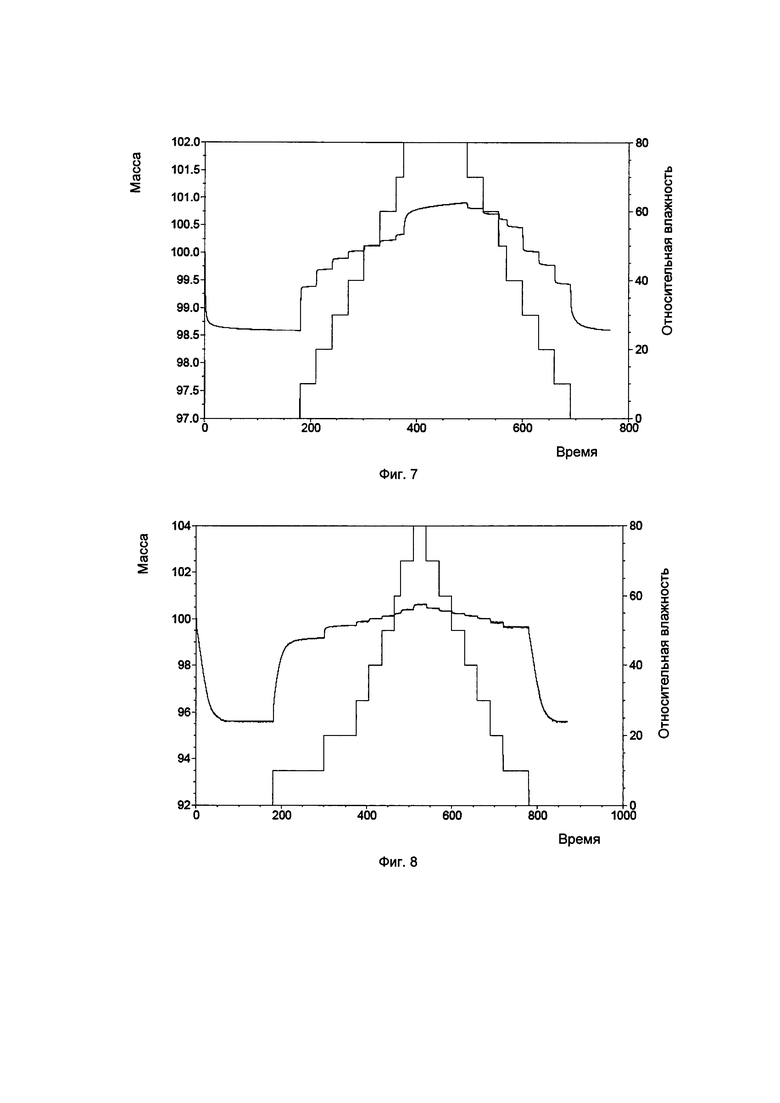
Фиг. 7. Картина динамической адсорбции паров воды для кристаллической формы А соединения, показанного формулой I.
Фиг. 8. Картина динамической адсорбции паров воды для кристаллической формы В соединения, показанного формулой I.
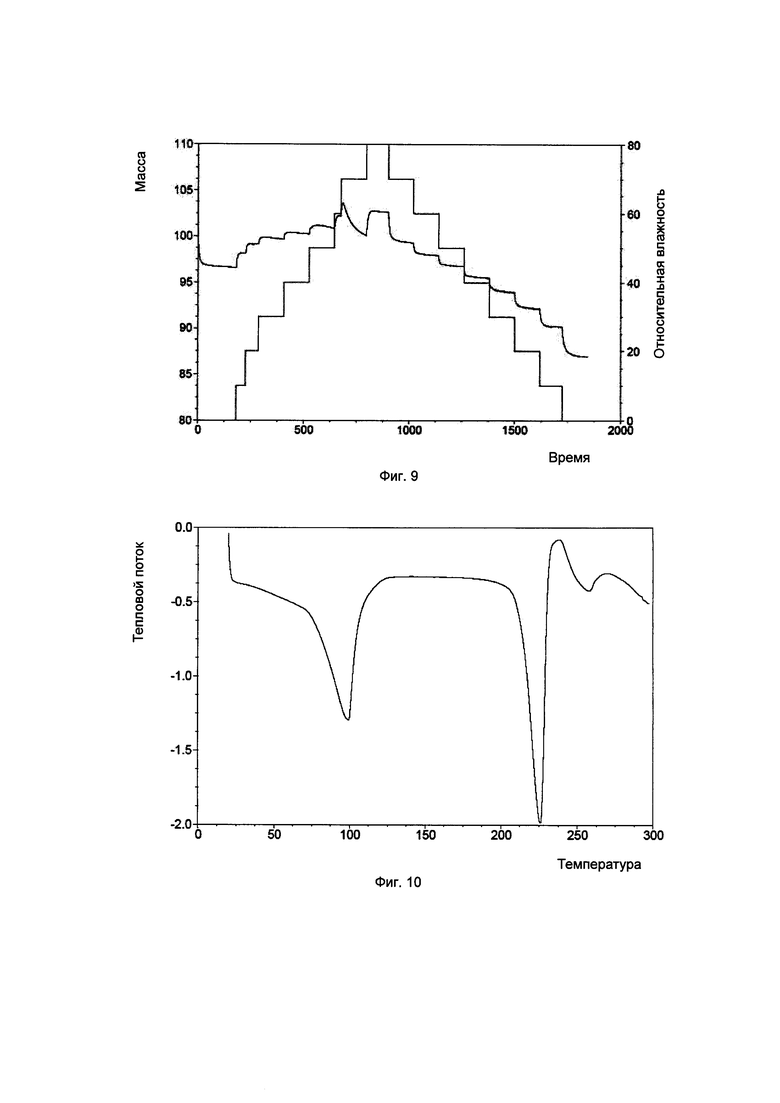
Фиг. 9. Картина динамической адсорбции паров воды для аморфного образца соединения, показанного формулой I.
Фиг. 10. Результаты термогравиметрического анализа для кристаллической формы А соединения, показанного формулой I.
Фиг. 11. Результаты термогравиметрического анализа для кристаллической формы В соединения, показанного формулой I.
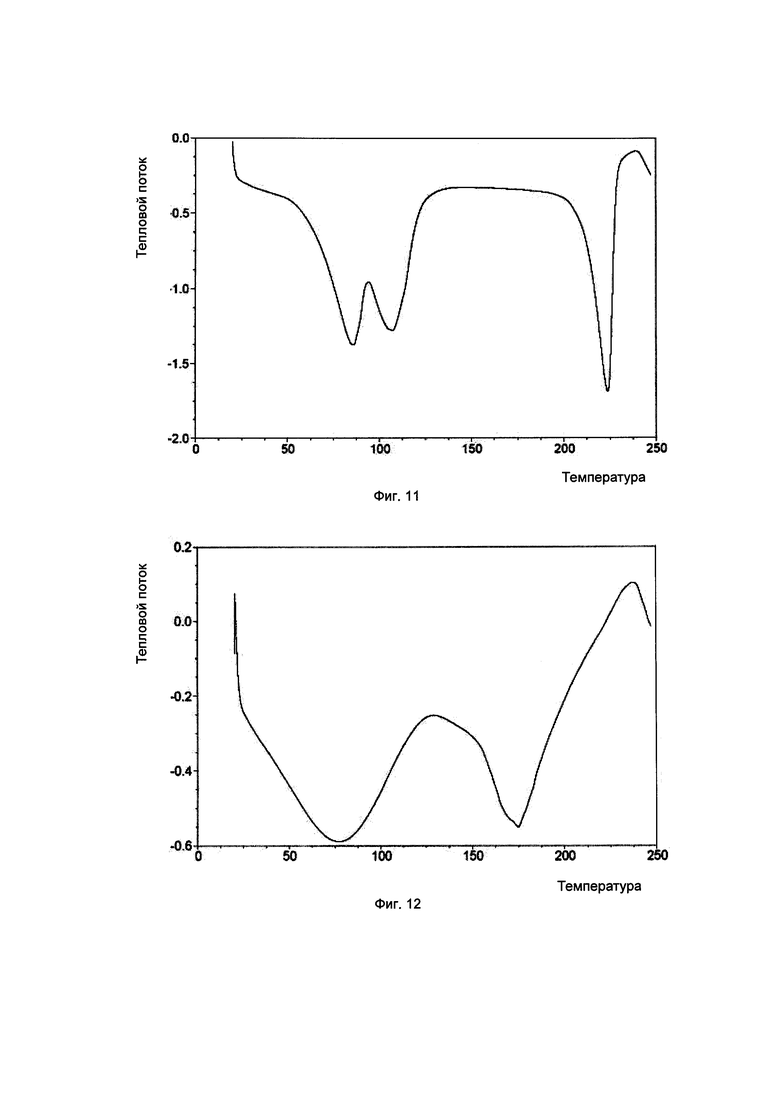
Фиг. 12. Результаты термогравиметрического анализа для кристаллической формы для аморфного образца соединения, показанного формулой I.
В Таблице 6 суммированы данные касательно оборудования и методов для детектирования картин дифракции рентгеновских лучей на порошке, показанных на Фиг. 1-6.
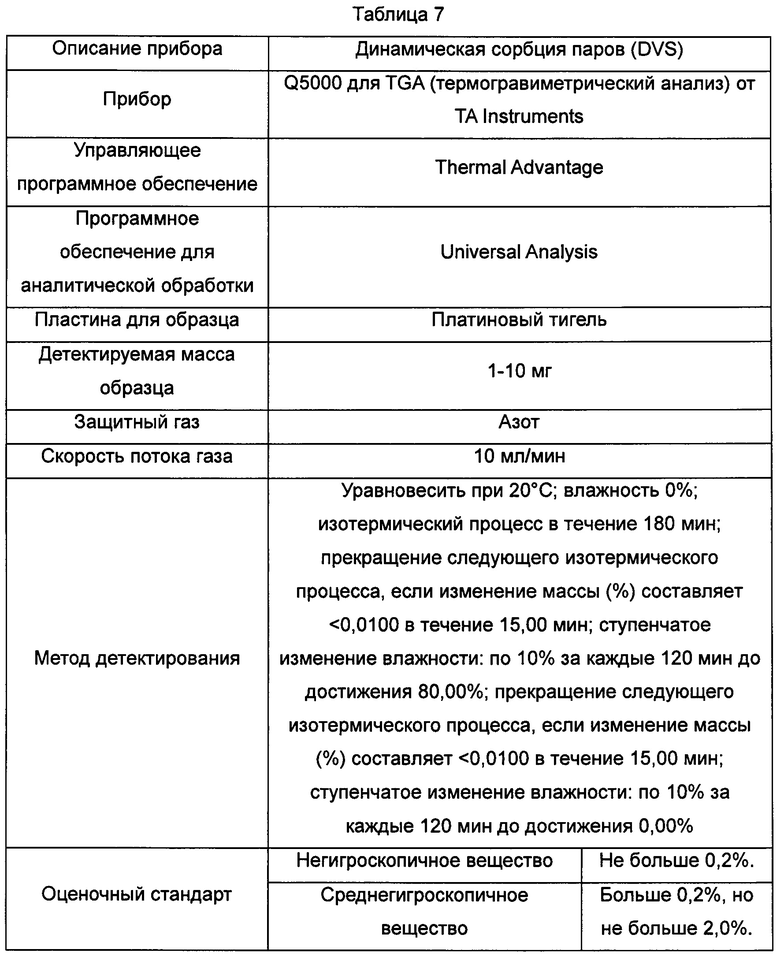
В Таблице 7 суммированы данные касательно оборудования и методов для детектирования картин динамической адсорбции паров воды, показанных на Фиг. 7-9.
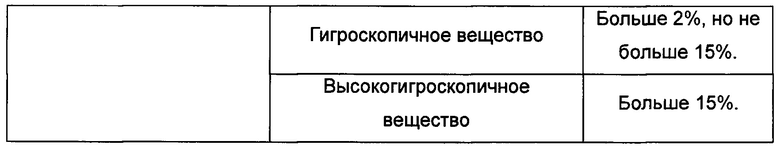
В Таблице 8 суммированы данные касательно оборудования и методов для детектирования результатов термогравиметрического анализа сканированных картин, показанных на Фиг. 10-12.

ПРИМЕРЫ
Настоящее изобретение дополнительно поясняется, но не ограничивается этим, приведенными далее примерами, которые иллюстрируют изобретение. В примерах, приведенных в настоящем изобретении, методы или способы, если специально не оговорено иным образом, являются традиционными в данной области техники методами или способами.
Сокращения:
Вос: бутоксикарбонил;
DCC: дициклогексилкарбодиимид;
DCM: дихлорметан;
DIPEA: диизопропилэтиламин;
DMAP: 4-диметиламинопиридин;
DMF: N,N-диметилформамид;
EDCI: 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид;
HATU: 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат;
НОВТ: 1-гидроксибензотриазол;
IPA: изопропиловый спирт;
МеОН: метанол;
МТВЕ: метил-трет-бутиловый эфир;
NMM: N-метилморфолин;
н.: моль/л;
TFA: трифторацетат;
THF: тетрагидрофуран.
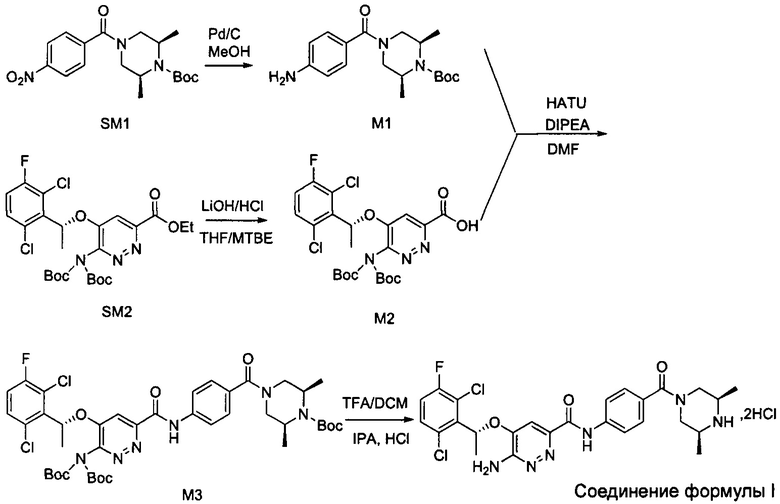
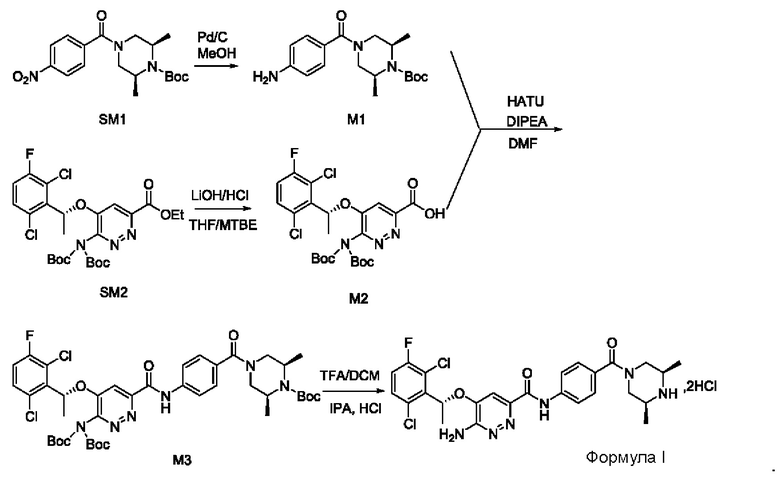
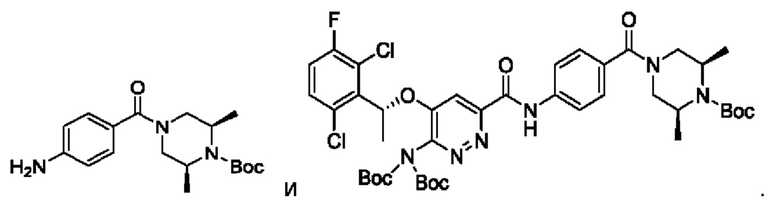
Пример 1. Синтез соединений формулы I
Синтез соединения М1
8,57 г (0,024 моль; 1,00 экв.) соединения SM1 растворяли в 85,7 мл безводного метанола и в атмосфере азота добавляли 5%-ный палладий на угле (0,86 г). Смесь три раза загружали водородом и проводили взаимодействие в атмосфере водорода в течение 3 часов. Когда реакция между исходными веществами полностью завершалась, фильтрат собирали фильтрованием, затем концентрировали до твердого вещества и сушили под вакуумом, получая белое твердое вещество, которое представляет собой соединение М1, с выходом 100% и чистотой 97,32%.
LC-MS (жидкостная хроматография в сочетании с масс-спектрометрией) [М+Н+]: 334.
Синтез соединения М2
9,60 г (0,017 моль; 1,00 экв.) соединения SM2 растворяли в 53 мл THF, реакционную смесь охлаждали до температуры от -5 до 5°С. По каплям добавляли 1 н. водный раствор KOH (1,40 г KOH и 25 мл воды), смесь инкубировали при 1-10°С и перемешивали в течение 4 часов. Добавляли 1 н. раствор разбавленной соляной кислоты для подведения рН до значения примерно 5. Смесь дважды экстрагировали этилацетатом (50 мл × 2). Органические слои объединяли и насыщенным водным солевым раствором, сушили над безводным сульфатом натрия в течение 1 часа, затем фильтровали и концентрировали под вакуумом, получая вязкое масло, добавляли 23 мл дихлорметана для растворения вышеупомянутого вязкого масла и затем концентрировали под вакуумом, вновь получая густое масло. Добавляли 69 мл дихлорметана для повторного растворения этого густого масла и концентрировали под вакуумом, получая 9,10 г промежуточного соединения М2 в виде желтого твердого вещества с выходом 99,7%.
LC-MS [М+Н+]: 546.
Синтез соединения М2
9,60 г (0,017 моль; 1,00 экв.) соединения SM2 растворяли в 53 мл THF и реакционную смесь охлаждали до 0-10°С. По каплям добавляли 1 н. водный раствор LiOH (1,05 г LiOH+25 мл воды). После добавления в течение 30 минут, температура постепенно повышалась до комнатной температуры, и реакционную смесь перемешивали в течение ночи. Реакция завершалась полностью, и реакционную смесь концентрировали под вакуумом. Полученный остаток растворяли в 100 мл воды, добавляли 50 мл метил-трет-бутилового эфира, перемешивали и проводили разделение фаз. К водной фазе по каплям добавляли 1 н. раствор разбавленной соляной кислоты для подведения рН до значения примерно 5. Смесь дважды экстрагировали этилацетатом (50 мл × 2). Органические слои объединяли и промывали насыщенным водным солевым раствором, сушили над безводным сульфатом натрия в течение 1 часа. Смесь фильтровали и упаривали под вакуумом, получая 8,58 г промежуточного соединения М2 в виде желтого твердого вещества. Выход составлял 94%.
LC-MS [М+Н+]: 546.
Синтез соединения М3
8,23 г (0,015 моль; 1,15 экв.) соединения М2 растворяли в 66 мл дихлорметана и охлаждали до 10-20°С. По очереди добавляли 4,30 г (0,013 моль; 1,00 экв.) соединения М1, 3,00 г (0,022 моль; 1,69 экв.) гидроксибензотриазола (НОВТ), 4,58 г (0,024 моль; 1,85 экв.) 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорида (EDCI) и 6,90 г (0,068 моль; 5,23 экв.) N-метилморфолина (NMM). Смесь инкубировали и перемешивали при 20-30°С в течение 16 часов, к реакционной смеси добавляли 16 мл воды, перемешивали в течение 10-15 минут, после отстаивания проводили разделение жидкости: к органическому слою добавляли 16 мл 1 н. раствора разбавленной соляной кислоты, перемешивали в течение 5 минут, после отстаивания проводили разделение жидкости: органический слой промывали 16 мл 1 н. раствора разбавленной соляной кислоты, 5 мл 1 н. водного раствора KOH и 30 мл насыщенного водного солевого раствора, соответственно, и проводили разделение отстоявшейся жидкости. Органическую фазу сушили с использованием безводного сульфата натрия, затем фильтровали и концентрировали в вакууме. Полученный остаток три раза промывали метил-трет-бутиловым эфиром (20 мл + 20 мл + 6 мл) и проводили фильтрование для сбора твердого вещества, твердое вещество сушили, получая 7,95 г соединения М3 в виде желтоватого твердого вещества с выходом 71,6%.
LC-MS [М+Н+]: 861.
Синтез соединения М3
7,96 г (0,015 моль; 1,15 экв.) соединения М2 растворяли в 40 мл DMF, к нему добавляли 5,23 г (0,014 моль; 1,08 экв.) HATU, 4,17 г (0,013 моль; 1,00 экв.) соединения М1 и 2,17 мл диизопропилэтиламина. Реакционную смесь перемешивали в течение ночи при комнатной температуре. К реакционной системе добавляли 50 мл насыщенного водного раствора карбоната натрия, затем перемешивали и фильтровали. Полученное твердое вещество взбивают с 60 мл воды и затем фильтруют. Твердое вещество растворяли в 30 мл дихлорметана, дважды промывали водой, органическую фазу сушили, фильтровали, концентрировали в вакууме и сушили, получая 10,03 г соединения М3 в виде коричневого твердого вещества с выходом 93,2%.
LC-MS [М+Н+]: 861.
Синтез соединения М3
9,77 г (0,018 моль; 1,20 экв.) соединения М2 растворяли в 75 мл дихлорметана, к нему добавляли 6,21 г (0,030 моль; 2,00 экв.) дициклогексилкарбодиимида, 0,96 г (0,0079 моль; 0,53 экв.) DMAP и 5,00 г (0,015 моль; 1,00 экв.) соединения М1. Реакционную смесь перемешивали в течение ночи при комнатной температуре. К реакционной системе добавляли 20 мл воды, перемешивали и после отстаивания проводили разделение жидкости: к органической фазе добавляли 20 мл 1 н. разбавленного раствора соляной кислоты, перемешивали в течение 5 минут, после отстаивания проводили разделение жидкости. Органический слой промывали 20 мл разбавленного раствора соляной кислоты, 6 мл 1 н. водного раствора KOH и 35 мл насыщенного водного солевого раствора, соответственно, и проводили разделение отстоявшейся жидкости. Органическую фазу сушили с использованием безводного сульфата натрия, затем фильтровали и концентрировали в вакууме. Полученный остаток три раза промывали, добавляя метил-трет-бутиловый эфир (25 мл + 25 мл + 8 мл). Проводили фильтрование для сбора твердого вещества и твердое вещество сушили, получая 10,62 г соединения М3 в виде коричневого твердого вещества с выходом 82,3%.
LC-MS [М+Н+]: 861.
Синтез соединения формулы I
7,95 г (0,092 моль) соединения М3 растворяли в 30 мл дихлорметана, реакционный раствор охлаждали до температуры от -5°С до +5°С, к нему по каплям с перемешиванием при комнатной температуре в течение 2 часов добавляли 15 мл трифторацетата. После охлаждения смеси до 10-20°С к этой реакционной смеси медленно добавляли 60 мл насыщенного водного раствора карбоната калия, затем перемешивали и проводили расслоение в стационарных условиях. Органический слой промывали насыщенным водным солевым раствором и сушили над безводным сульфатом натрия, затем фильтровали и концентрировали. Полученный остаток растворяли в 41,2 г изопропанола, после охлаждения смеси до 10-20°С к этой реакционной смеси добавляли 5 мл концентрированной соляной кислоты с перемешиванием при комнатной температуре. Проводили фильтрование для сбора твердого вещества и это твердое вещество промывали изопропанолом, сушили в вакууме, получая 5,75 г соединения формулы I.
LC-MS [М+Н+]: 561.
1Н-ЯМР (300 МГц, CDCl3): δ=1.25-1.43 (m, 6Н), 1.91 (d, 3Н), 3.15-3.48 (m, 4Н), 3.66-3.89 (m, 0.5Н), 4.55-4.78 (m, 0.5Н), 5.49 (s, 2Н), 6.26 (q, 1Н), 7.10 (t, 1H), 7.33-7.44 (m, 4Н), 7.78 (d, 2Н), 9.93 (s, 1H).
Пример 2. Способ получения кристаллической формы А
Способ получения 1 кристаллической формы А
Примерно 5-10 мг аморфного соединения формулы I растворяли во втор-бутаноле, получая прозрачный раствор, после чего фильтровали; этот раствор выдерживали при 40°С, получая кристаллическую форму А.
Способ получения 2 кристаллической формы А
Примерно 10 мг аморфного соединения формулы I растворяли в метаноле, получая прозрачный раствор, после чего фильтровали; к раствору добавляли 1 мг карбоксиметилцеллюлозы. Смесь выдерживали при комнатной температуре, получая кристаллическую форму А.
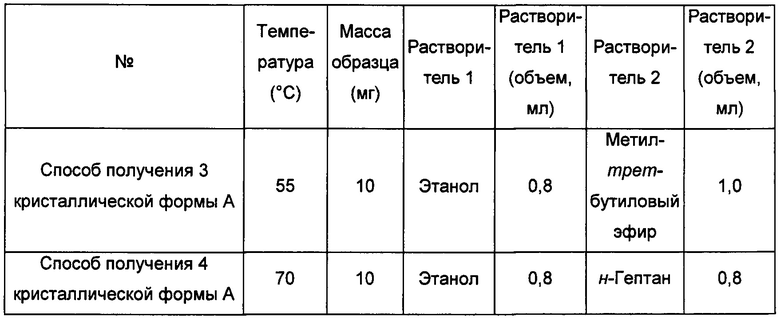
Способы получения 3 и 4 кристаллической формы А
Примерно 10 мг аморфного соединения формулы I при соответствующей температуре добавляли в растворитель 2, затем добавляли в растворитель 1, получая прозрачный раствор, и далее фильтровали, перемешивали при -20°С для осаждения твердого вещества. Осадок собирали фильтрованием, получая кристаллическую форму А.
Способ получения 5 кристаллической формы А
При комнатной температуре примерно 10 мг аморфного соединения формулы I растворяли в этаноле, получая прозрачный раствор, затем фильтровали. К этому раствору по каплям при перемешивании добавляли н-гептан до тех пор, пока не наблюдали образование большого количества твердого вещества, фильтровали и извлекали кристаллическую форму А.
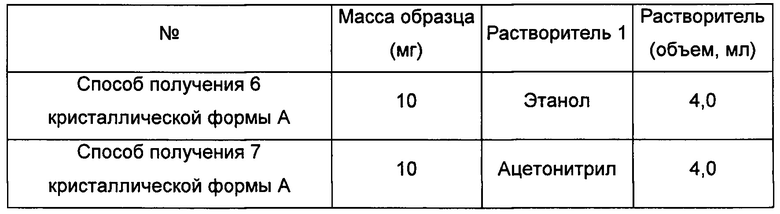
Способы получения 6 и 7 кристаллической формы А
Приблизительно 10 мг аморфного соединения формулы I помещали в центрифужную пробирку емкостью 2,0 мл и выдерживали при комнатной температуре в течение 6 суток в закрытой атмосфере соответствующего растворителя. Получали кристаллическую форму А.
Способ получения 8 кристаллической формы А
Приблизительно 15 мг аморфного соединения формулы I добавляли к 0,2 мл этанола при 4°С, смесь перемешивали при соответствующей температуре в течение 30 минут и фильтровали, получая кристаллическую форму А.
Пример 3. Способ получения кристаллической формы В
Первый экспериментальный способ
Примерно 5-10 мг кристаллической формы А растворяли в соответствующем растворителе, получая прозрачный раствор, и затем фильтровали. Смесь выдерживали при соответствующей температуре для выпаривания растворителя, получая кристаллическую форму В.
Второй экспериментальный способ
Примерно 10 мг кристаллической формы А добавляли в растворитель 1, после чего добавляли в растворитель 2, получая прозрачный раствор, и фильтровали, затем выдерживали при соответствующей температуре для выпаривания растворителей, получая кристаллическую форму В.
Третий экспериментальный способ
Примерно 15-30 мг кристаллической формы А добавляли в соответствующий растворитель с образованием суспензии, перемешивали при комнатной температуре в течение 5 суток, фильтровали, получая кристаллическую форму В.
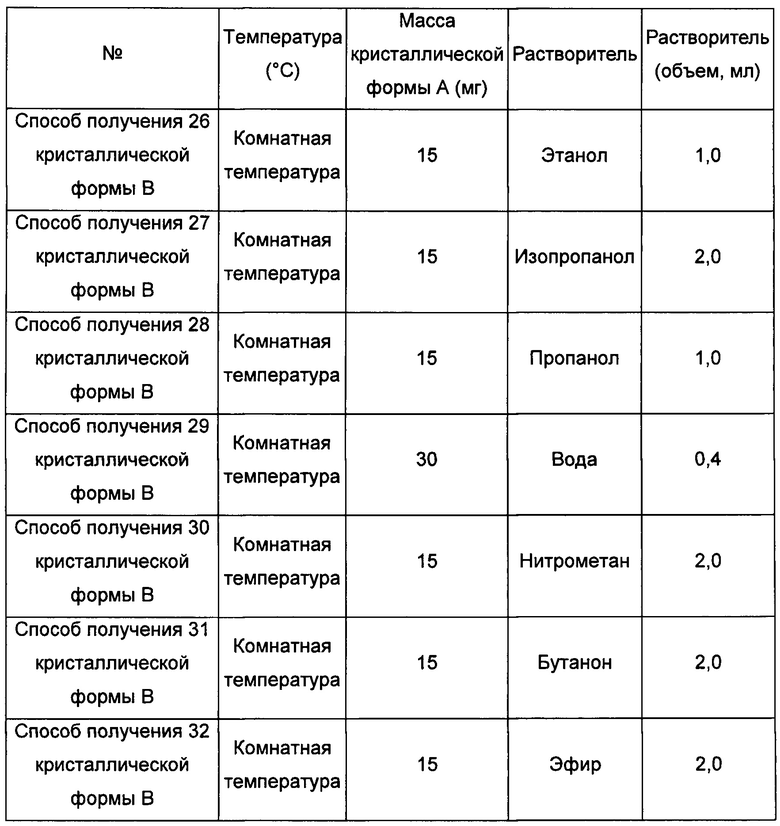
Четвертый экспериментальный способ
Примерно 15-30 мг кристаллической формы А добавляли в соответствующий растворитель с образованием суспензии, перемешивали в течение 5 суток при 40°С, фильтровали, получая кристаллическую форму В.


Пятый экспериментальный способ
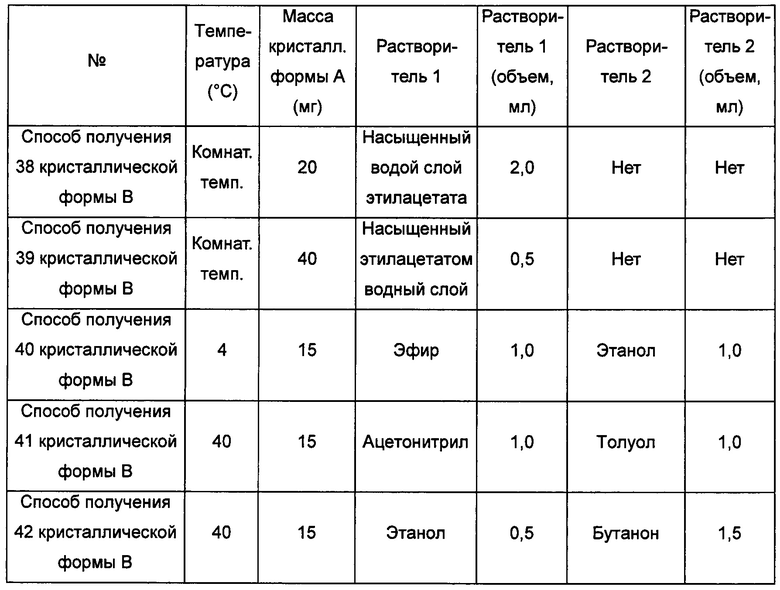
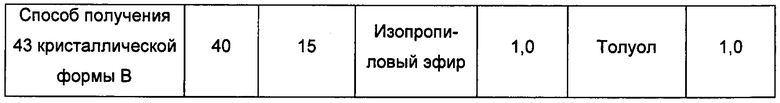
Примерно 15-40 мг кристаллической формы А сначала добавляли в растворитель 2, затем добавляли в растворитель 1 с образованием суспензии и перемешивали при соответствующей температуре в течение 4 суток, фильтровали, получая кристаллическую форму В.
Шестой экспериментальный способ
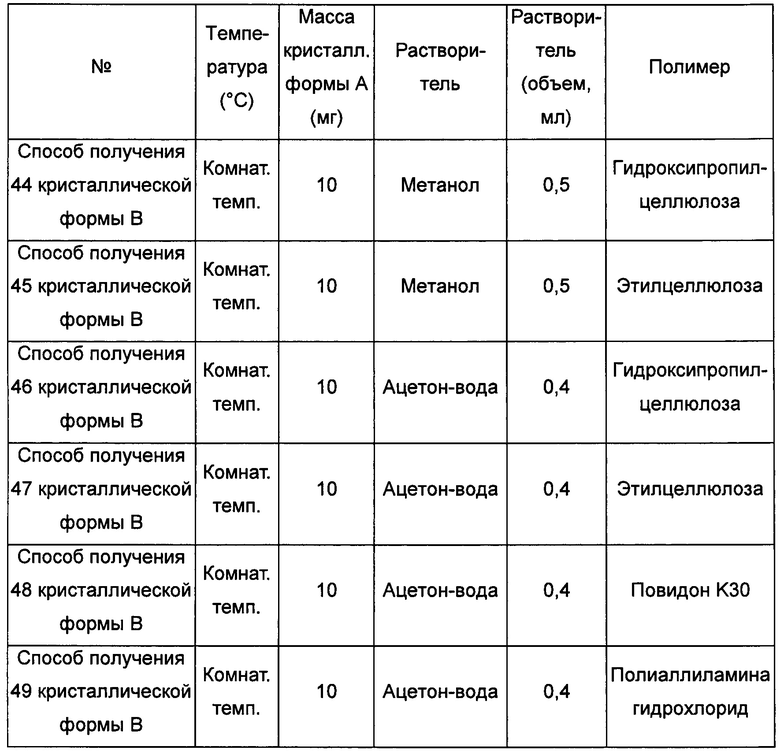
Примерно 10 мг кристаллической формы А добавляли в соответствующий растворитель, при этом соотношение ацетон : вода составляло 3:1 (объемное соотношение), ацетонитрил : вода составляло 3:2 (объемное соотношение), получая при обработке ультразвуком прозрачный раствор, который затем фильтровали и к которому добавляли 1 мг полимера. Смесь выдерживали при комнатной температуре для выпаривания растворителей, получая кристаллическую форму В.
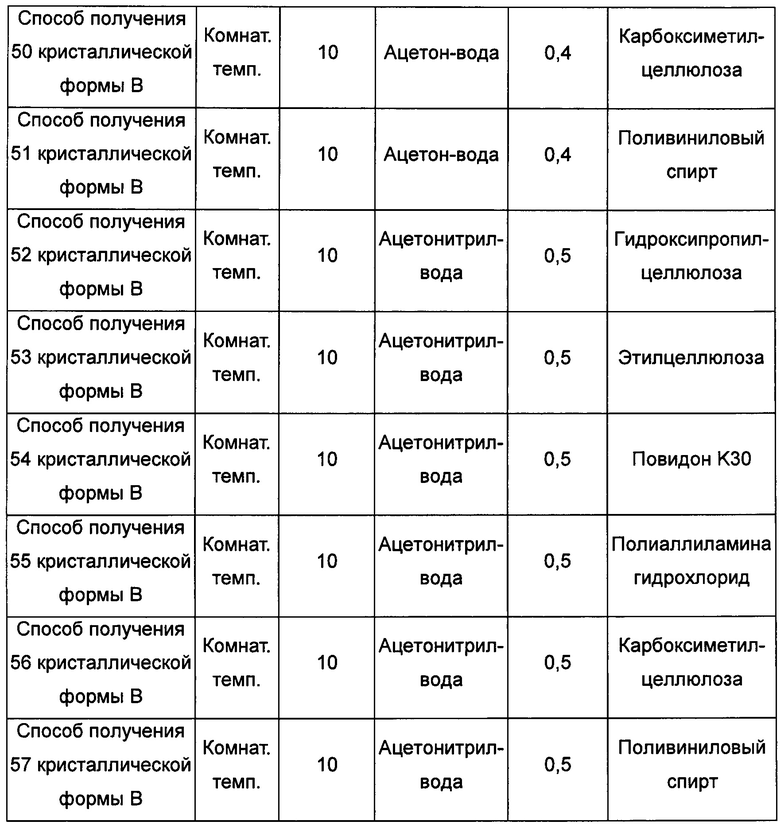
Седьмой экспериментальный способ
Примерно 15-50 мг кристаллической формы А растворяли в соответствующем растворителе при соответствующей температуре, получая прозрачный раствор, затем фильтровали, фильтрат помещали на 4°С при постоянном перемешивании, выдерживая до тех пор, пока не наблюдали образование твердого вещества, и фильтровали, получая кристаллическую форму В. В условиях, когда образования кристаллической формы в изопропаноле при 4°С с перемешиванием не наблюдается, тогда выдерживали при комнатной температуре для выпаривания растворителя, получая кристаллическую форму В.
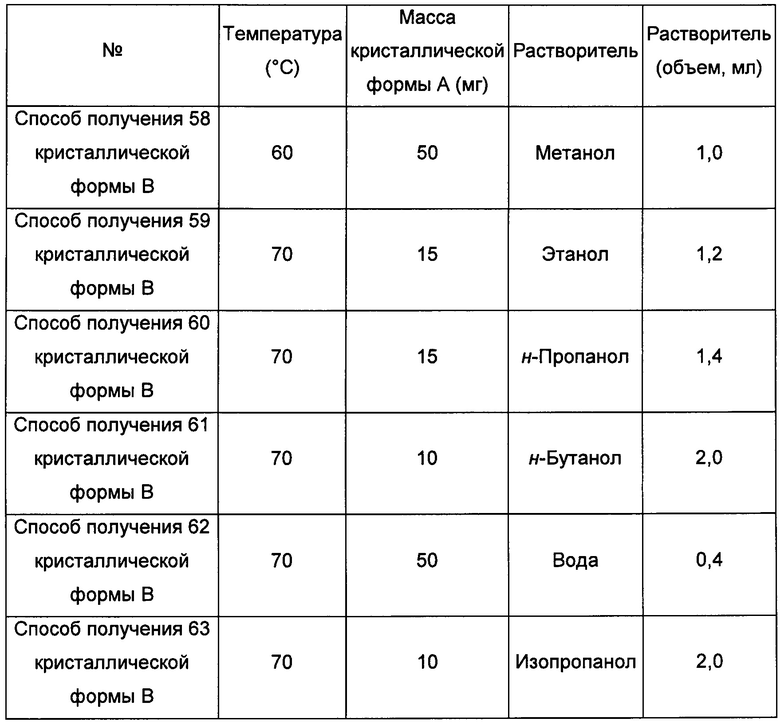
Восьмой экспериментальный способ
Примерно 10-15 мг кристаллической формы А добавляли сначала в растворитель 2, затем добавляли в растворитель 1, получая прозрачный раствор при соответствующей температуре, после этого фильтровали, затем фильтрат перемешивали при -20°С до тех пор, пока не наблюдали образование твердого вещества, извлекая кристаллическую форму В. В условиях, когда не наблюдается образования кристаллической формы при перемешивании с использованием способов получения 69-74 кристаллической формы В, фильтрат выдерживали при комнатной температуре для выпаривания растворителей, получая кристаллическую форму В.
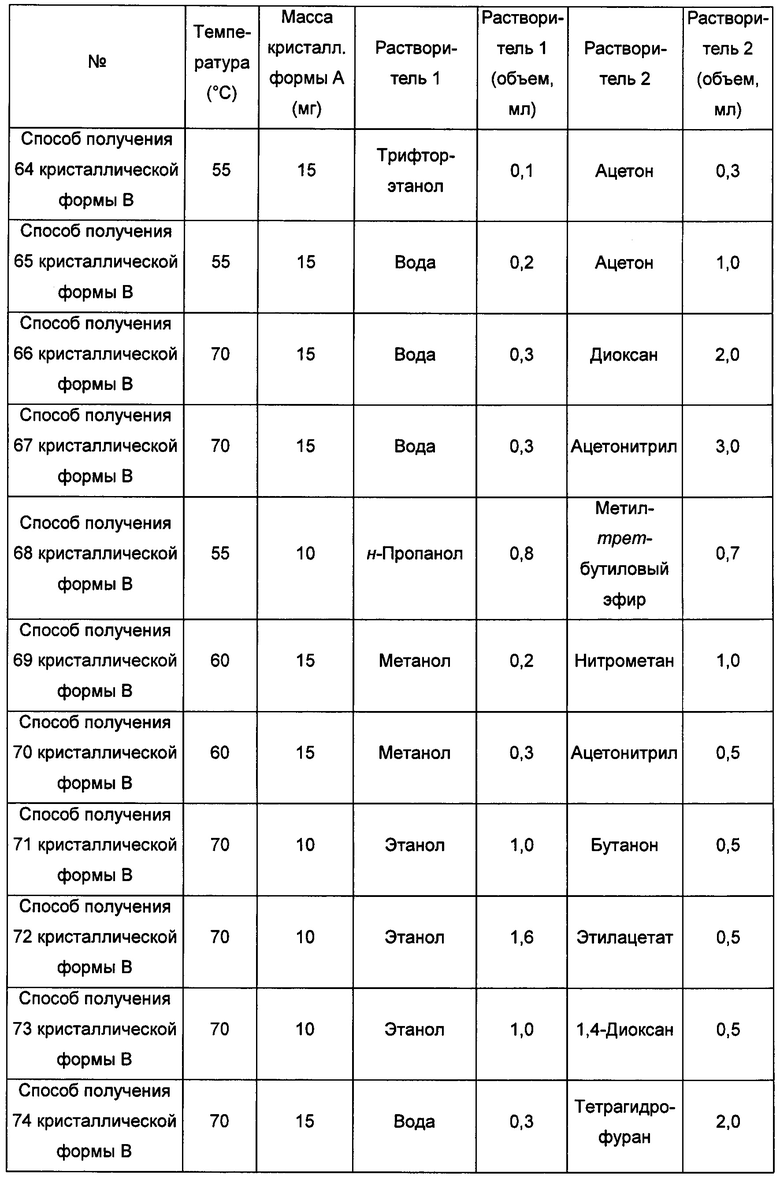
Девятый экспериментальный способ
Примерно 10-15 мг кристаллической формы А добавляли в растворитель 1, получая при обработке ультразвуком при комнатной температуре прозрачный раствор, который затем фильтровали и по каплям с перемешиванием добавляли в растворитель 2 до тех пор, пока не наблюдали образование большого количества твердого вещества, и фильтровали, получая кристаллическую форму В. В условиях, когда при использовании способов получения 89 и 90 кристаллической формы В образования твердого вещества не наблюдается, смесь выдерживали при комнатной температуре для выпаривания растворителей, получая кристаллическую форму В.
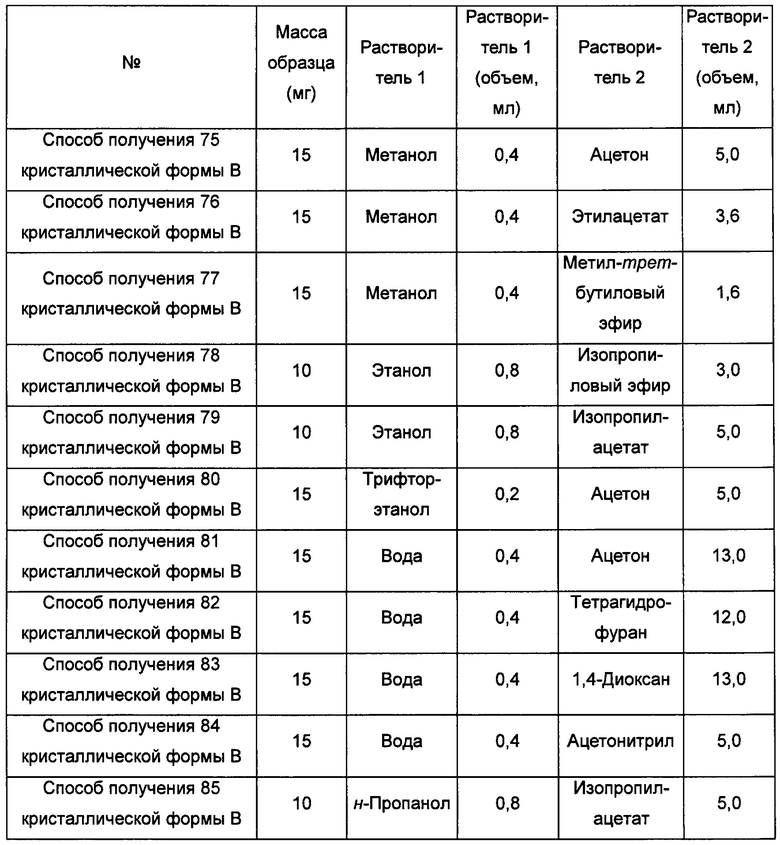
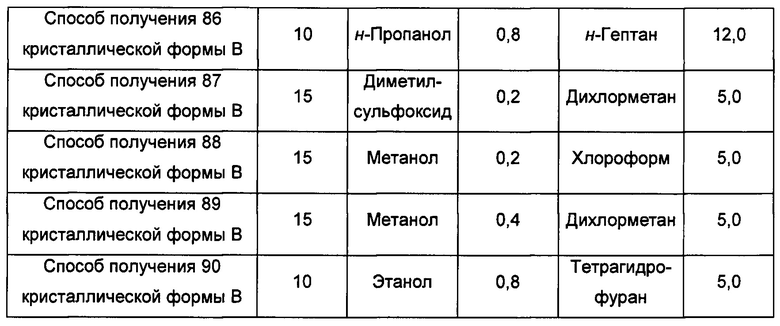
Десятый экспериментальный способ
Примерно 10 мг аморфного образца соединения формулы I помещали в центрифужную пробирку емкостью 2,0 мл. Затем центрифужную пробирку помещали в атмосферу соответствующего растворителя при комнатной температуре на 6 суток, получая кристаллическую форму В.
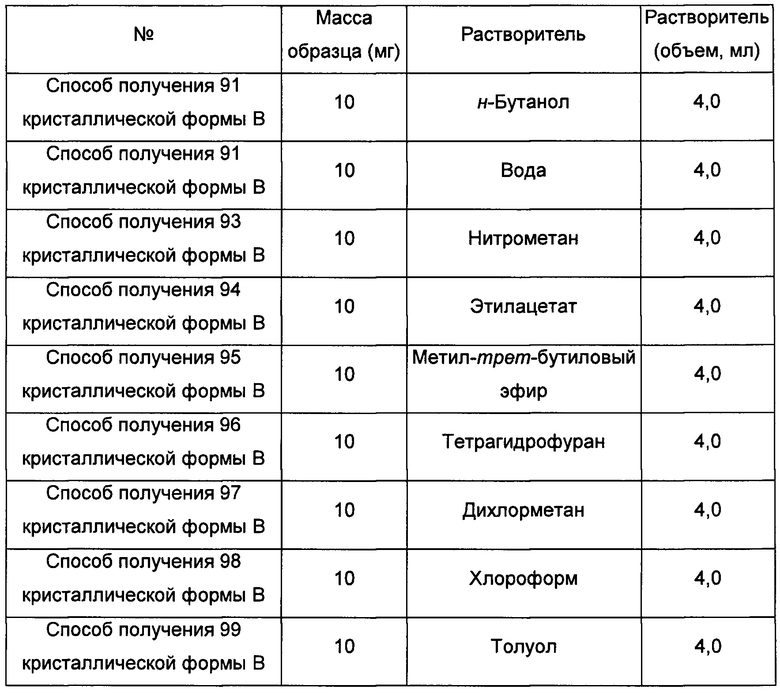
Одиннадцатый экспериментальный способ
Примерно 15-30 мг аморфного образца соединения формулы I добавляли в соответствующий растворитель при соответствующей температуре и перемешивали в течение 30 минут суток, затем фильтровали, получая кристаллическую форму В.
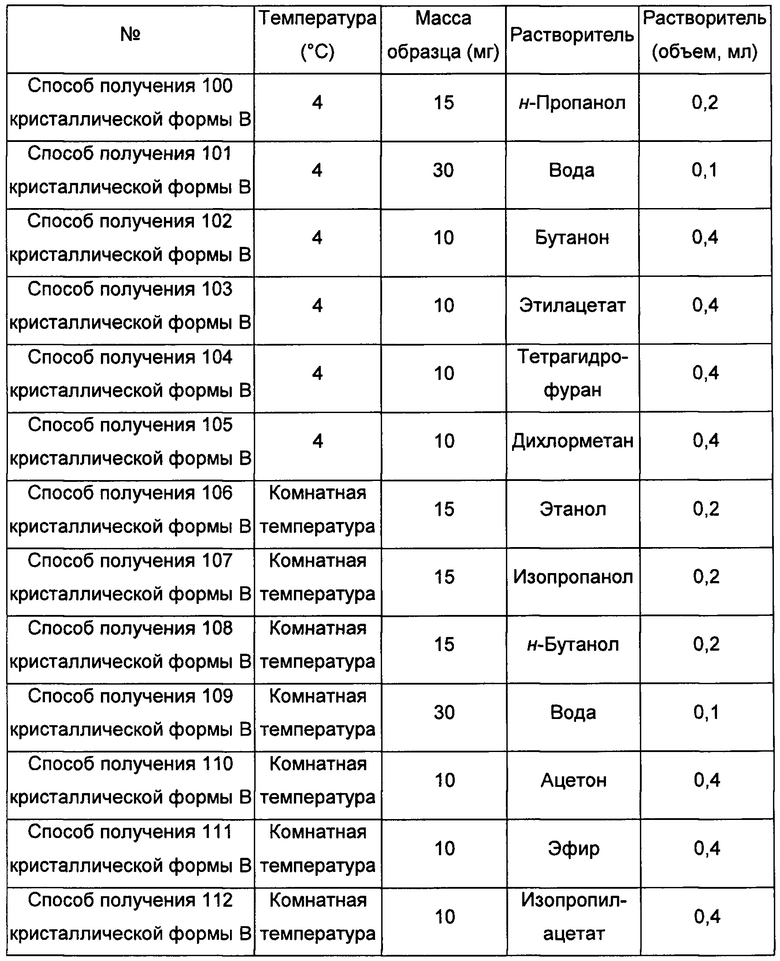
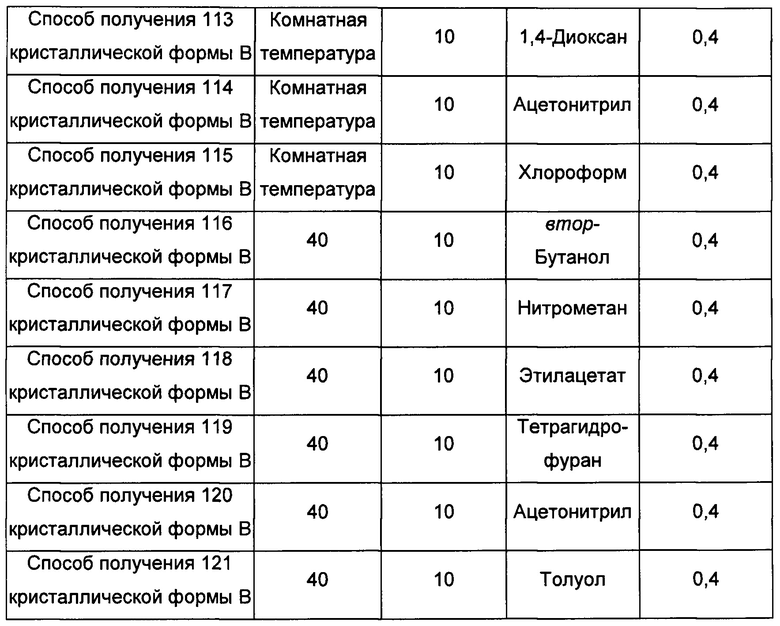
Двенадцатый экспериментальный способ
Примерно 15-30 мг аморфного образца соединения формулы I добавляли сначала в растворитель 2, а затем добавляли в растворитель 1, получая суспензию, перемешивали в течение 30 минут при соответствующий температуре, фильтровали, получая кристаллическую форму В.
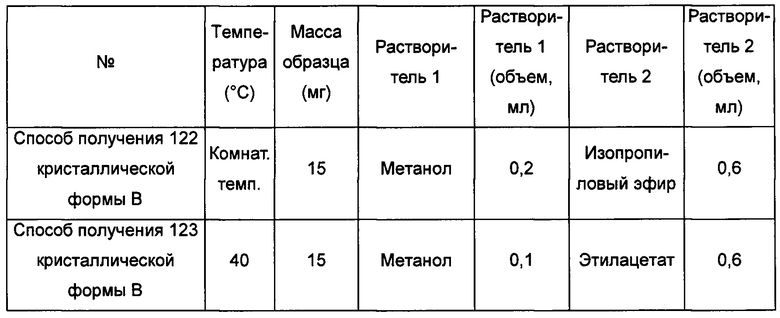
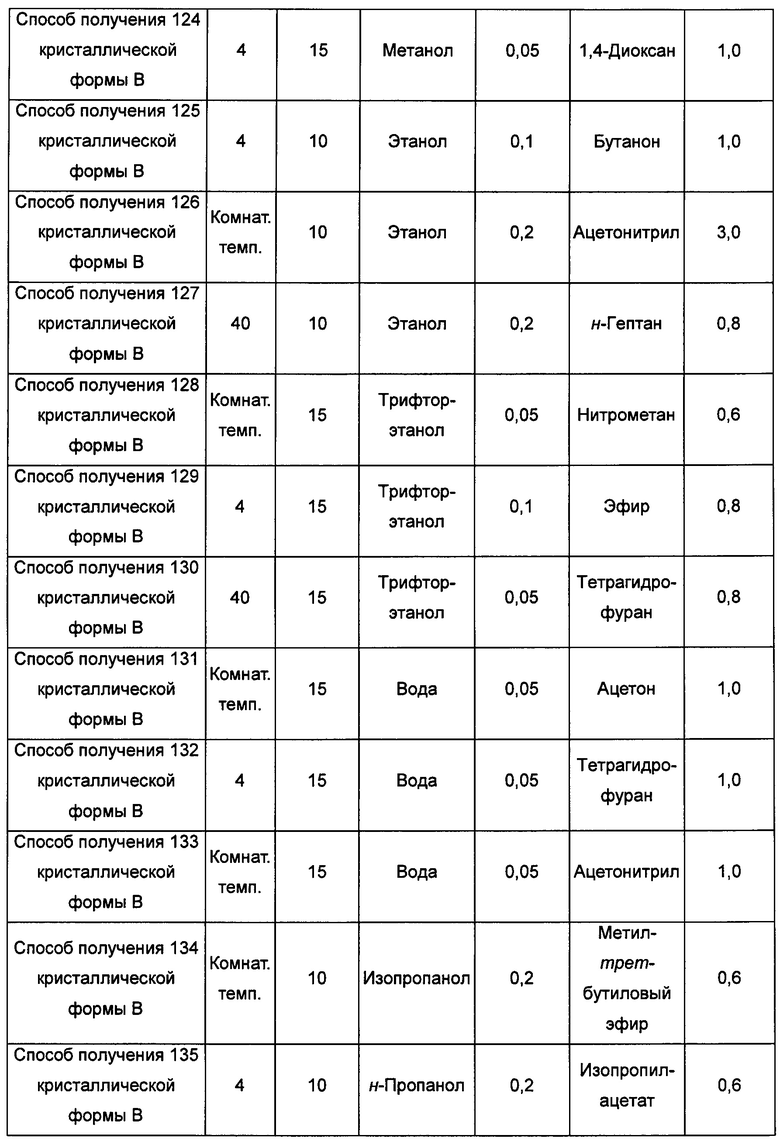
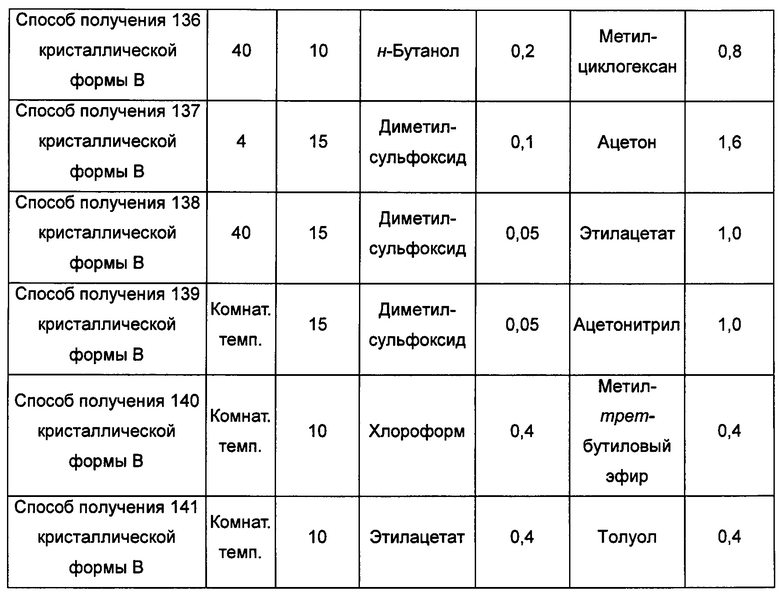
Тринадцатый экспериментальный способ. Способ получения 142 кристаллической формы В
Примерно 10 мг аморфного образца соединения формулы I помещали в атмосферу с относительной влажностью RH 85% на 10 суток при комнатной температуре, получая кристаллическую форму В.
Четырнадцатый экспериментальный способ
Соответствующее количество образца добавляли в соответствующий растворитель, получая при обработке ультразвуком прозрачный раствор, который после этого фильтровали, затем фильтрат быстро упаривали на роторном испарителе при соответствующей температуре водяной бани, получая кристаллическую форму В.
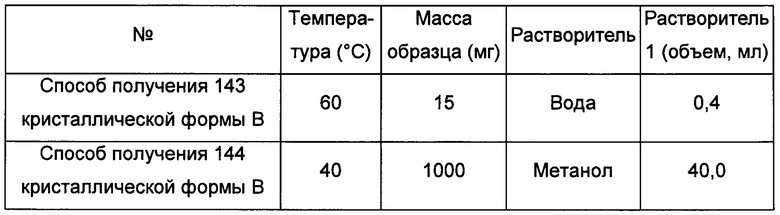
Пример 4. Способ получения кристаллической формы С
Способ получения 1 кристаллической формы С
100 мг кристаллической формы А соединения формулы I добавляли к 2,8 мл изопропилового эфира, затем добавляли к 2,8 мл метанола с образованием суспензии. Смесь перемешивали в течение 4 суток при комнатной температуре и фильтровали под вакуумом, получая 73 мг кристаллической формы С.
Способ получения 2 кристаллической формы С
100 мг кристаллической формы А соединения формулы I добавляли к 1,0 мл изопропилового эфира, затем добавляли к 3,0 мл метанола с образованием суспензии. Смесь перемешивали при комнатной температуре в течение 4 суток и фильтровали под вакуумом, получая 63 мг кристаллической формы С.
Способы получения 3 и 4 кристаллической формы С
Примерно 15 мг кристаллической формы А соединения формулы I растворяли в растворителе 2, затем растворяли в растворителе 1, получая прозрачный раствор, который фильтровали и перемешивали при -20°С до тех пор, пока не наблюдали образование твердого вещества, извлекая кристаллическую форму С.
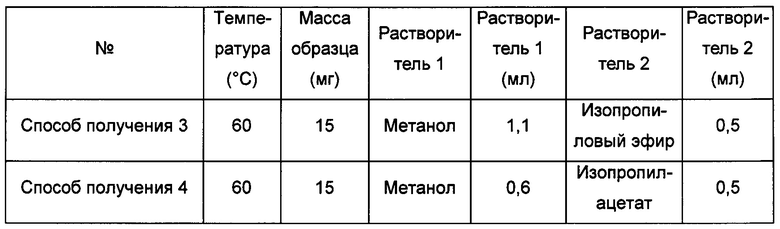
Способ получения 5 кристаллической формы С
Примерно 10 мг кристаллической формы А соединения формулы I помещали в центрифужную пробирку емкостью 2,0 мл и выдерживали в закрытой атмосфере растворителя метанола (4 мл) в течение 6 суток при комнатной температуре, получая кристаллическую форму С.
Пример 5. Способ получения кристаллической формы D
Способ получения 1 кристаллической формы D
100 мг кристаллической формы А соединения формулы I добавляли к 2,0 мл этилацетата, затем добавляли к 1,0 мл диметилсульфоксида с образованием суспензии. Суспензию перемешивали при комнатной температуре в течение 1 суток, получая кристаллическую форму D.
Способ получения 2 кристаллической формы D
Примерно 5 мг кристаллической формы А соединения формулы I растворяли в 0,1 мл диметилсульфоксида, получая прозрачный раствор, который затем фильтровали и выдерживали при 40°С для выпаривания растворителя, получая кристаллическую форму D.
Способ получения 3 кристаллической формы D
Примерно 20 мг кристаллической формы А соединения формулы I добавляли в растворитель 2, затем добавляли в растворитель 1 с образованием суспензии. Смесь перемешивали при соответствующей температуре в течение 4 суток, получая кристаллическую форму D.

Способы получения 4 и 5 кристаллической формы D
Примерно 15 мг кристаллической формы А соединения формулы I добавляли в растворитель 1, получая при обработке ультразвуком при комнатной температуре прозрачный раствор, который после этого фильтровали, и затем к этому раствору при перемешивании по каплям добавляли растворитель 2 до тех пор, пока не наблюдали образование большого количества твердого вещества, извлекая кристаллическую форму D.
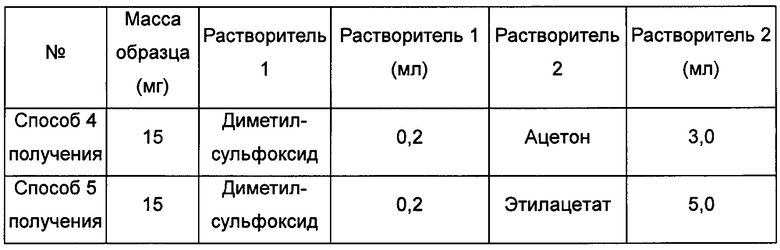
Пример 6. Способ получения кристаллической формы Е
Примерно 20 мг кристаллической формы А соединения формулы I помещали в аппарат с относительной влажностью RH 85% на 26 суток при комнатной температуре, получая кристаллическую форму Е.
Пример 7. Способ получения аморфной формы
Способ получения 1 аморфной формы
200 мг кристаллической формы А соединения формулы I добавляли к 0,6 мл трифторэтанола, получая при обработке ультразвуком прозрачный раствор, который затем фильтровали, и фильтрат быстро упаривали на роторном испарителе в вакууме при 40°С, получая аморфную форму.
Способ получения 2 аморфной формы
200 мг кристаллической формы А соединения формулы I добавляли к 7,0 мл метанола, получая при обработке ультразвуком прозрачный раствор, который после этого фильтровали и быстро упаривали на роторном испарителе в вакууме при 40°С, получая аморфную форму.
Способы получения 3-5 аморфной формы
5-10 мг кристаллической формы А соединения формулы I растворяли в соответствующем растворителе, получая прозрачный раствор, который после этого фильтровали и выдерживали при соответствующей температуре для выпаривания растворителя, получая аморфную форму.
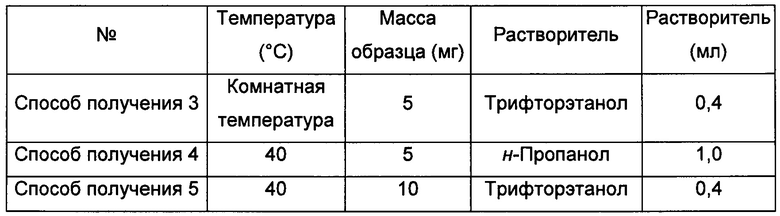
Способы получения 6-9 аморфной формы
Примерно 10 мг кристаллической формы А соединения формулы I растворяли в соответствующем растворителе, получая прозрачный раствор, который после этого фильтровали и выдерживали при соответствующей температуре для выпаривания растворителей, получая аморфную форму.
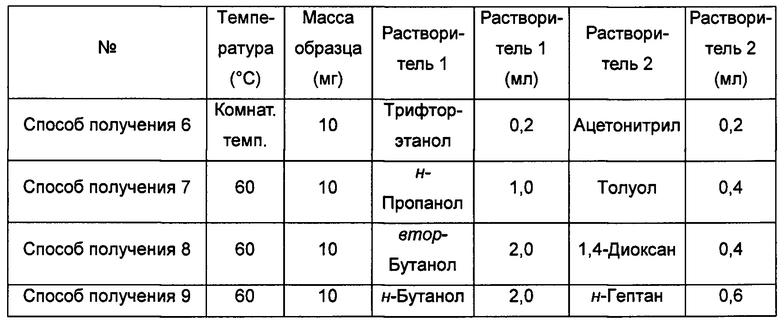
Способ получения 10 аморфной формы
Примерно 15 мг кристаллической формы А соединения формулы I добавляли к 2,0 мл н-гептана, смесь перемешивали при комнатной температуре в течение 5 суток, получая аморфную форму.
Способы получения 11 и 12 аморфной формы
Примерно 10-15 мг кристаллической формы А соединения формулы I растворяли в соответствующем растворителе, получая при соответствующей температуре прозрачный раствор, который после этого фильтровали и перемешивали при -20°С до тех пор, пока не наблюдали образование твердого вещества, извлекая аморфную форму.
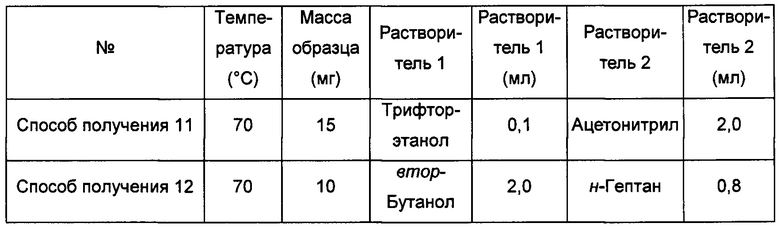
Способ получения 13 аморфной формы
Примерно 15 мг кристаллической формы А соединения формулы I добавляли к 0,2 мл трифторэтанола, получая при обработке ультразвуком при комнатной температуре прозрачный раствор, который затем фильтровали, к этому раствору при перемешивании по каплям добавляли 1,0 мл изопропилового эфира до тех пор, пока не наблюдали образование большого количества твердого вещества, извлекая аморфную форму.
Способы получения 14-17 аморфной формы
Примерно 10-15 мг кристаллической формы А соединения формулы I растворяли в соответствующем растворителе, получая прозрачный раствор, который затем фильтровали, и фильтрат быстро упаривали на роторном испарителе при соответствующей температуре, получая аморфную форму.
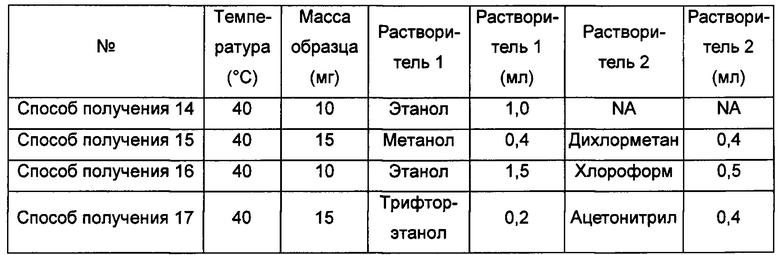
"NA" означает "данные отсутствуют".
Пример 8. Стабильность кристаллической формы
Образцы кристаллической формы А и В помещали в условия: 80°С на 24 часа, 25°C -RH 60% на 10 суток и 40°C -RH 75% на 10 суток, соответственно, при этом никакого изменения в кристаллической форме не наблюдали.
Кристаллическая форма С почти полностью превращалась в кристаллическую форму А в условиях сушки в вакууме при комнатной температуре в течение ночи.
Большая часть кристаллической формы D в условиях сушки при комнатной температуре или в условиях сушки в вакууме при комнатной температуре превращалась в кристаллическую форму В, которая не является стабильной.
Большая часть кристаллической формы Е превращалась в условиях выдерживания в эксикаторе в течение 2 суток в кристаллическую форму А, которая является нестабильной формой.
Пример 9. Анализ динамической сорбции паров (DVS)
Кристаллическая форма А: изменение массы составляло 2,3% в диапазоне RH от 0% до 80%. Из гидрата кристаллической формы А удалялось примерно 1,5% влаги на стадии сушки в условиях RH 0% и поглощалось примерно 2,3% влаги в диапазоне RH0%-RH80%. На стадии десорбции в условиях RH 30% может быть удалено 1,5% влаги, и изменение массы составляло меньше 2% в диапазоне RH 30-80%.
Кристаллическая форма В: изменение массы составляло 2,3% в диапазоне RH от 0% до 80%. Из гидрата кристаллической формы В может быть удалено примерно 4% влаги в условиях RH 10% RH и поглощалось примерно 4% влаги в условиях RH 10% RH на стадии адсорбции, и изменение массы составляло меньше 2% в диапазоне RH 10-80%.
Аморфная форма: изменение массы составляло примерно 15,7% в диапазоне RH 0-80%, что соответствует высокогигроскопичному веществу.
Пример 10. Определение растворимости
Растворимость кристаллической формы А соединения, показанного формулой I, в воде составляет 20-100 мг/мл при комнатной температуре, а растворимость используемой в виде свободного основания аморфной формы соединения, показанного формулой I, в воде составляет меньше 1 мг/мл.
Пример 11. Композиция в форме капсул
В качестве конкретного воплощения перорального лекарственного средства готовят композицию, содержащую примерно 20-150 мг полиморфов, описанных в примере 1 и/или примере 2, вместе с тонкоизмельченной микрокристаллической целлюлозой и/или стеариновой кислотой, получая в общей сложности количество, составляющее примерно от 50 мг до 500 мг, для внесения в капсулу 0-го типа.
Пример 12. Композиция в форме таблеток или капсул
В качестве конкретного воплощения перорального лекарственного средства готовят композицию, содержащую примерно 20-150 мг полиморфов, описанных в примере 1 и/или примере 2, вместе с двумя или более из следующих эксципиентов: тонкоизмельченной микрокристаллической целлюлозы, маннита, кросповидона, натриевой соли кроскармелозы, натрия крахмала гликолята, повидона, гидроксипропилцеллюлозы и/или стеариновой кислоты, распределяя общее количество по таблеткам или капсулам с содержанием примерно от 50 мг до 500 мг.
Пример 13. Фармакокинетические данные
6 самок крыс SD (Sprague Dawley) делили на две группы, по три крысы в каждой группе. По 50 мг/кг кристаллической формы А соединения формулы I и аморфной формы соединения формулы I в виде свободного основания вводили посредством внутрижелудочного зонда в разовой дозе каждой самке крысы SD, соответственно; в конкретный момент времени отбирали образцы крови через яремную вену, из этих образцов готовили плазму и хранили в холодильнике при -80°С.
В упомянутой выше плазме, приготовленной из этих образцов, проводили осаждение белков с использованием ацетонитрила, супернатант разбавляли в 3 раза водой, затем отбирали 5 мкл раствора для тестирования посредством LC-MS/MS, и данные показаны в Таблице 9.
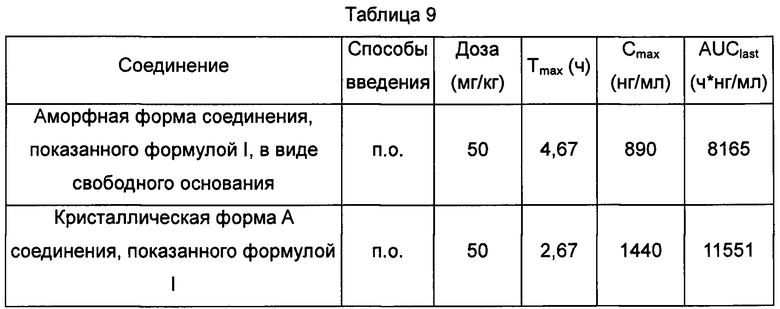
"п.о." означает "перорально".
По 50 мг/кг кристаллической формы А соединения, показанного формулой I, и аморфной формы соединения, показанного формулой I, в виде свободного основания вводили перорально крысам, при этом значения Tmax составляли 2,67 и 4,67 ч, соответственно, Cmax составляли 1440 и 890 нг/мл, соответственно, и AUClast (площадь под кривой зависимости концентрации в плазме крови от времени в диапазоне от нулевой отметки до последней временной точки) составляли 11551 и 8165 ч*нг/мл, соответственно.
На основании приведенных выше результатов, высказано предположение, что кристаллическая форма А соединения, показанного формулой I, демонстрировала более высокую степень всасывания in vivo, чем аморфная форма соединения, показанного формулой I, в виде свободного основания.
Пример 14. Биохимическая киназная активность соединения формулы I
Биохимическую киназную активность соединения, показанного формулой I, тестировали в Reaction Biology Corp., расположенной в г. Малверн, штат Пенсильвания (Ра.), США. Правила тестирования описаны Anastassiadis и др. в Nat.
Biotechnol., 2011, 29(11): 1039-45. Было обнаружено, что соединение формулы I потенциально может ингибировать приведенные далее киназы.
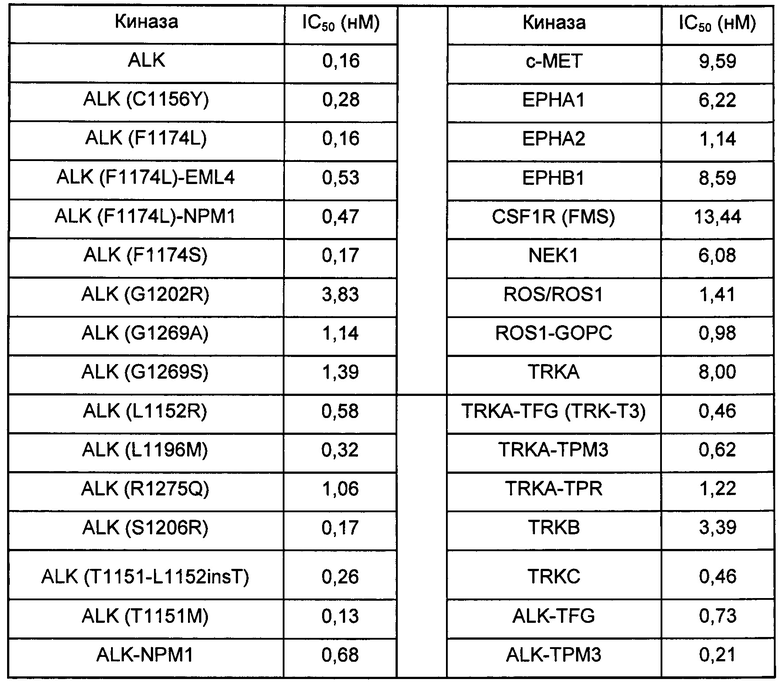
Несмотря на то, что настоящее изобретение описано полно вместе со своими предпочтительными воплощениями и со ссылкой на прилагаемые графические материалы, необходимо отметить, что специалистам в данной области техники очевидны различные изменения и модификации. Следует понимать, что такие изменения и модификации включены в объем настоящего изобретения.
Изобретение относится к соединению, представленному структурной формулой (I) ({5-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-6-аминопиридазин-3-ил}-N-{4-[((3S,5R)-3,5-диметилпиперазинил)карбонил]фенил}карбоксамида гидрохлорид), и его новым кристаллическим формам дигидрату и тригидрату, а также способу получения соединения и кристаллической формы, соответствующим промежуточным соединениям, фармацевтической композиции, содержащей соединение по изобретению, применению для приготовления фармацевтического продукта для лечения заболевания, расстройства или состояния у пациента, где заболевание, расстройство или состояние опосредовано c-Met, CSF1R, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK. Технический результат: получены новые кристаллические формы соединения формулы (I), показывающие лучшую растворимость и более высокую степень всасывания. 9 н. и 26 з.п. ф-лы, 12 ил., 32 табл., 14 пр.
1. Способ получения соединения формулы I, включающий следующие стадии:
2. Способ получения соединения формулы I, включающий следующие стадии:
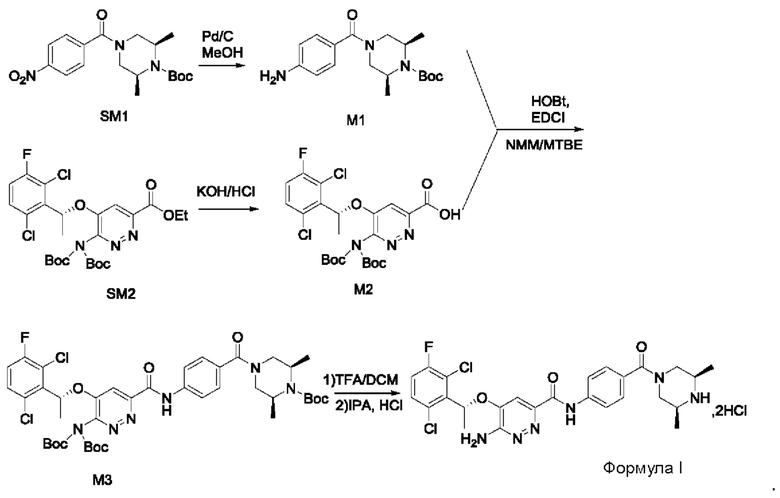
3. Способ получения соединения формулы I, включающий следующие стадии:
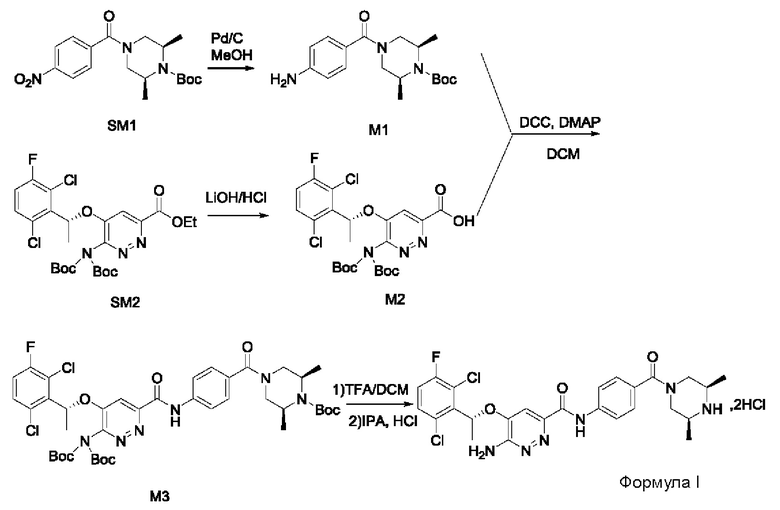
4. Промежуточные соединения:
5. Кристаллическая форма соединения формулы I, его гидратов и/или сольватов
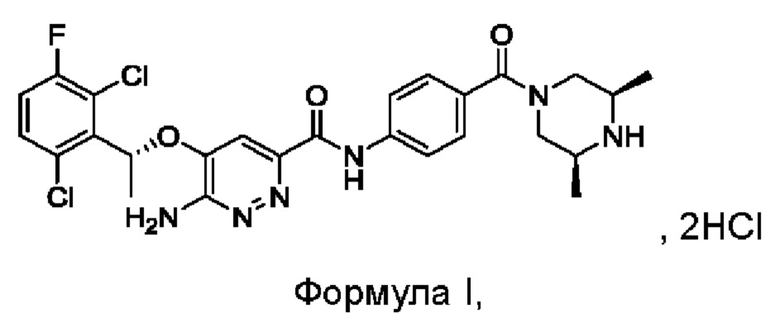
причем порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2° и 19,3±0,2°, и кристаллическая форма представляет собой дигидрат.
6. Кристаллическая форма соединения формулы I по п. 5, где порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2°, 14,7±0,2°, 16,9±0,2°, 19,3±0,2° и 20,3±0,2°.
7. Кристаллическая форма соединения формулы I по п. 5, где порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 4,9±0,2°, 10,0±0,2°, 14,7±0,2°, 16,9±0,2°, 19,3±0,2°, 20,3±0,2°, 25,5±0,2° и 30,7±0,2°.
8. Кристаллическая форма соединения формулы I по п. 5, где кристаллическая форма имеет порошковую дифракционную рентгенограмму приблизительно такую, как показана на Фиг. 1.
9. Кристаллическая форма соединения формулы I, его гидратов и/или сольватов
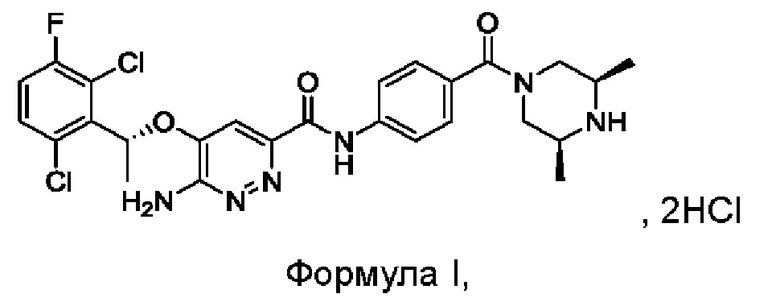
где порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2° и 21,1±0,2°, и кристаллическая форма представляет собой тригидрат.
10. Кристаллическая форма соединения формулы I по п. 9, где порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2°, 19,7±0,2°, 21,1±0,2°, 23,9±0,2° и 25,5±0,2°.
11. Кристаллическая форма соединения формулы I по п. 9, где порошковая дифракционная рентгенограмма кристаллической формы имеет характеристические пики при углах дифракции 2θ, составляющих 10,5±0,2°, 17,4±0,2°, 19,7±0,2°, 21,1±0,2°, 21,5±0,2°, 23,9±0,2°, 25,2±0,2° и 25,5±0,2°.
12. Кристаллическая форма соединения формулы I по п. 9, где порошковая дифракционная рентгенограмма является приблизительно такой, как показано на Фиг. 2.
13. Фармацевтическая композиция для лечения заболевания, расстройства или состояния, опосредованного c-Met, CSF1R, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK, содержащая терапевтически эффективное количество кристаллической формы по п. 7 и фармацевтически приемлемые эксципиенты, адъюванты и/или носители.
14. Фармацевтическая композиция по п. 13, где фармацевтическая композиция находится в форме перорального препарата.
15. Фармацевтическая композиция по п. 13, где фармацевтическая композиция находится в форме таблетки или капсулы.
16. Фармацевтическая композиция по п. 13, содержащая от 20 мг до 150 мг указанной кристаллической формы и по меньшей мере один эксципиент, адъювант и/или носитель, что в общей сложности составляет примерно от 50 мг до 500 мг.
17. Фармацевтическая композиция по п. 13, где эксципиент, адъювант и/или носитель представляет собой микрокристаллическую целлюлозу, маннит, кросповидон, натриевую соль кроскармеллозы, целлюлозу, натрия гликолят крахмала, повидон, гидроксипропилцеллюлозу и/или стеариновую кислоту.
18. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 0,01%-99% (по массе) кристаллической формы по п. 7.
19. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 0,1%-70% (по массе) кристаллической формы по п. 7.
20. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 1%-70% (по массе) кристаллической формы по п. 7.
21. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 1%-50% (по массе) кристаллической формы по п. 7.
22. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 1%-30% (по массе) кристаллической формы по п. 7.
23. Фармацевтическая композиция по п. 13, где фармацевтическая композиция содержит 10%-30% (по массе) кристаллической формы по п. 7.
24. Применение кристаллической формы по п. 7 или фармацевтической композиции по п. 13 для изготовления лекарственного средства для лечения заболевания, расстройства или состояния у пациента, где заболевание, расстройство или состояние опосредовано c-Met, CSF1R, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK.
25. Применение по п. 24, где слитый белок на основе ALK представляет собой EML4-ALK или NPM-ALK киназу.
26. Применение по п. 24, где заболевание, расстройство или состояние представляет собой рак и/или пролиферативное расстройство.
27. Применение по п. 24, где заболевание, расстройство или состояние представляет собой рак легкого, меланому, рак толстой кишки, рак молочной железы, рак печени, рак поджелудочной железы, рак головного мозга, рак почки, рак яичников, рак желудка, рак кожи, рак кости, глиому, лимфому, нейробластому, гепатоклеточную карциному, папиллярную почечноклеточную карциному и/или плоскоклеточную карциному головы и шеи.
28. Применение по п. 24, где заболевание, расстройство или состояние представляет собой немелкоклеточный рак легкого, резистентный к терапии кризотинибом.
29. Применение по п. 24, где заболевание, расстройство или состояние представляет собой меланому.
30. Способ лечения заболевания, расстройства или состояния у пациента путем введения пациенту кристаллической формы по п. 7 или фармацевтической композиции по п. 13, где заболевание, расстройство или состояние опосредовано c-Met, CSF1R, ROS1 или слитым белком на основе ROS1, TRKA или слитым белком на основе TRKA, TRKB, TRKC, ALK, ALKATI или слитым белком на основе ALK.
31. Способ лечения заболевания, расстройства или состояния у пациента по п. 30, где слитый белок на основе ALK представляет собой EML4-ALK или NPM-ALK киназу.
32. Способ лечения заболевания, расстройства или состояния у пациента по п. 30, где заболевание, расстройство или состояние представляет собой рак и/или пролиферативное расстройство.
33. Способ лечения заболевания, расстройства или состояния у пациента по п. 30, где заболевание, расстройство или состояние представляет собой рак легкого, меланому, рак толстой кишки, рак молочной железы, рак печени, рак поджелудочной железы, рак головного мозга, рак почки, рак яичников, рак желудка, рак кожи, рак кости, глиому, лимфому, нейробластому, гепатоклеточную карциному, папиллярную почечноклеточную карциному и/или плоскоклеточную карциному головы и шеи.
34. Способ лечения заболевания, расстройства или состояния у пациента по п. 30, где заболевание, расстройство или состояние представляет собой немелкоклеточный рак легкого, резистентный к терапии кризотинибом.
35. Способ лечения заболевания, расстройства или состояния у пациента по п. 30, где заболевание, расстройство или состояние представляет собой меланому.
| WO 2012048259 A2, 12.04.2012 | |||
| Mino R | |||
| Caira, Crystalline Polymorphism of Organic Compounds, Topics in Current Chemistry, т | |||
| Складная решетчатая мачта | 1919 |
|
SU198A1 |
| Клиническая фармакокинетика: теоретические, прикладные и аналитические аспекты: руководство / Под ред | |||
| В.Г | |||
| Кукеса | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| ВЗАИМОСВЯЗЬ КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ СУБСТАНЦИИ | |||

