Ссылка на связанные заявки
Согласно настоящей заявке испрашивается приоритет в соответствии с предварительной заявкой США №62/460562, поданной 17 февраля 2017 г.; предварительной заявкой США №62/479169, поданной 30 марта 2017 г.; предварительной заявкой США №62/551645, поданной 29 августа 2017 г.; предварительной заявкой США №62/551647, поданной 29 августа 2017 г.; и предварительной заявкой США №62/551668, поданной 29 августа 2017 г.. Все из этих заявок включены посредством ссылки, как если бы они были полностью переписаны в данном документе.
Предшествующий уровень техники настоящего изобретения
STING (стимулятор генов интерферона) представляет собой сигнальную молекулу во врожденном ответе на dsDNA в цитозоле. Делеция STING была описана при многочисленных видах рака человека. Помимо этого, дерегуляция передачи сигнала с участием STING при видах рака человека также была описана при меланоме (Xia Т, et al., "Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis" Cancer Res. 2016) и раке толстой кишки. (Xia Т, et al., "Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis" Cell Rep. 2016; 14:282-97). Интересно отметить, что в этих исследованиях результаты геномного анализа показали, что меньшая экспрессия STING обусловлена не делецией или мутацией гена, а эпигенетическими изменениями. (Xia, Cancer Res. 2016; Xia, Cell Rep.2016). Защитная активность STING против рака также подтверждена фактами, полученными в результате исследований на мышиных моделях. Было показано, что мыши с нокаутом STING характеризуются нарушением опухолевого контроля. (Woo SR, et al. "STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors" Immunity 2014; 41:830-42).
Помимо этого, роль STING в защите онтогенеза была продемонстрирована в нескольких спонтанных мышиных моделях, включая глиому (Ohkuri Т, et al., "Protective role of STING against gliomagenesis: Rational use of STING agonist in anti-glioma immunotherapy" Oncoimmunology. 2015;4:e999523) и рак толстой кишки (Zhu Q, et al., "Cutting edge: STING опосредует защиту против колоректального онкогенеза с помощью регуляции величины воспаления кишечника " J. Immunol. 2014; 193:4779-82). Указанный противоопухолевый эффект может быть связан с его способностью преодолевать сверхактивацию NF-kB и STAT3. (Okihuri 2015). Активация пути с участием STING также характеризуется высокой активностью в доклинических мышиных моделях опухолей. (Woo 2014; Chandra D, et al. "STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer" Cancer Immunol Res. 2014;2:901-10; Corrales L, et al., "Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity" Cell Rep. 2015; 11:1018-30; Curran E, et al. "STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia" Cell Rep. 2016; 15:23 57-66; Tang CH, et al. "Agonist-Mediated Activation of STING Induces Apoptosis in Malignant В Cells" Cancer Res. 2016; 76:2137-52). Указанная противоопухолевая активность вероятно связана с нарушением сосудистой сети опухоли и возникает после индукции адаптивного иммунного ответа. (Corrales L, et al., "The host STING pathway at the interface of cancer and immunity" J. Clin. Invest. 2016; 126:2404-11). Соответственно, прямая стимуляция STING в опухолевом микроокружении с помощью агониста может представлять собой новый подход в лечении многочисленных типов рака.
Краткое описание настоящего изобретения
Варианты осуществления могут обеспечивать соединение формулы (I):
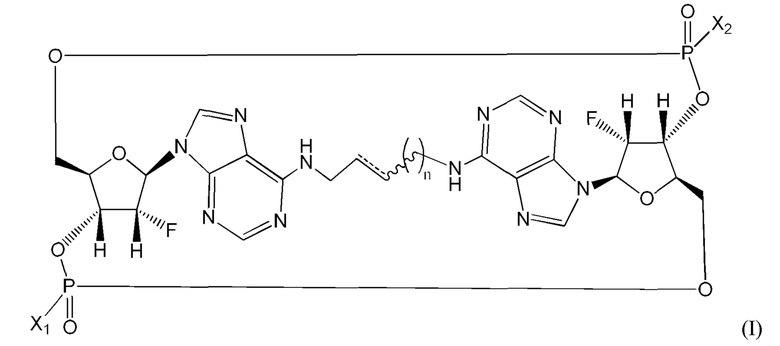
(где P1 представляет собой левый нижний фосфор, а P2 представляет собой верхний правый фосфор, как показано выше), содержащее заместители и стереохимию, как указано в таблице 1 ниже, или его фармацевтически приемлемую соль.  означает простую связь или двойную связь.
означает простую связь или двойную связь.
Если фосфорный атом содержит четыре заместителя, которые отличаются, такой фосфорный атом будет стереоцентром. SpSp/RpRp/SpRp/RpSp относится к фосфорной стереохимии, как отмечено.
Варианты осуществления могут дополнительно обеспечивать соединение формулы (II):
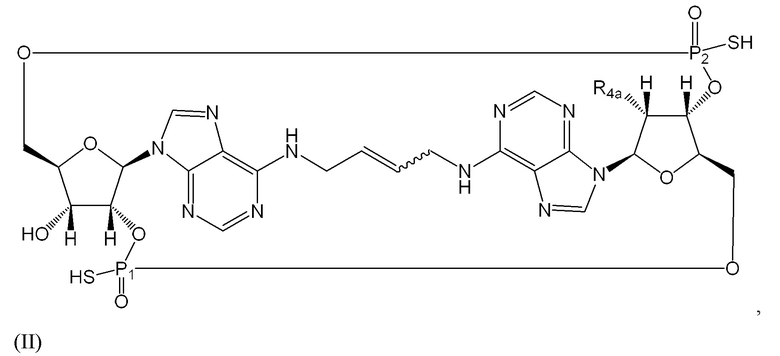
содержащее заместители и стереохимию, как отмечено в таблице 2 ниже, или его фармацевтически приемлемую соль. Если фосфорный атом содержит четыре заместителя, которые отличаются, такой фосфорный атом будет стереоцентром.

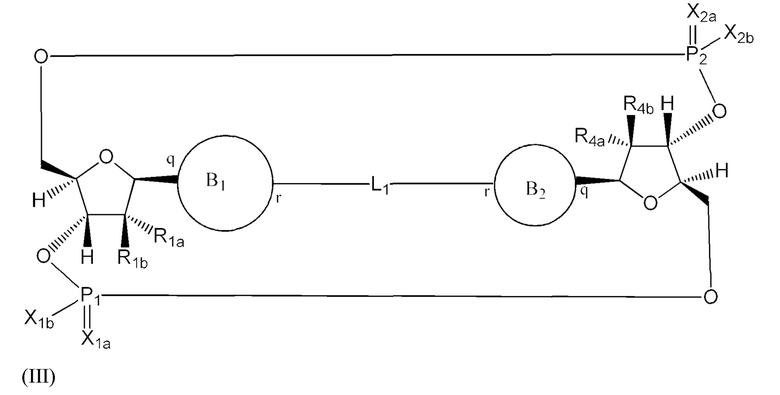
Варианты осуществления могут обеспечивать соединение формулы (III):
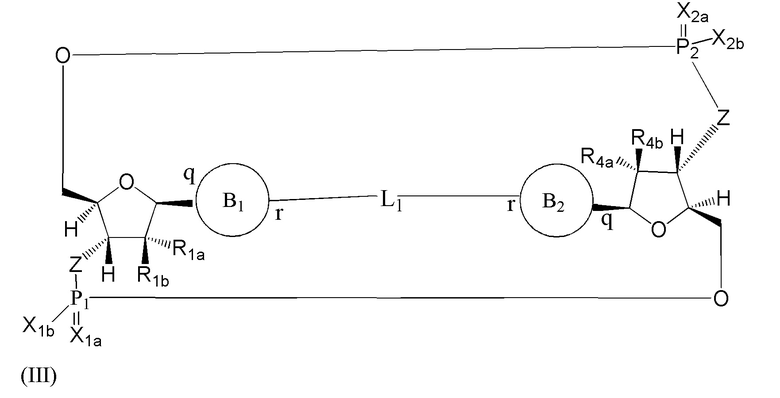
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н, -ОН и -F;
R1b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из -Н, -ОН и -F;
R4b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и P2 каждый независимо обладает S или R стереохимической конфигурацией;
Z представляет собой -О- или -NH-;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила, -С(O)C1-6алкила и -CH2OC(O)OC1-6алкила;
L1 в формуле (III) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой 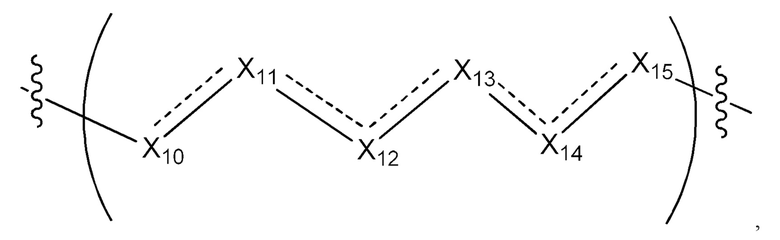
где  означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости в L1 означает двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость в L1 означает тройную связь, 0 встречаемостей в L1 означает двойную связь; и (iv) где, если 2 встречаемости в L1 означает двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости в L1 означает двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость в L1 означает тройную связь, 0 встречаемостей в L1 означает двойную связь; и (iv) где, если 2 встречаемости в L1 означает двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, X12, X13, Х14 и X15 независимо выбраны из связи, -СН2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН, (ii) -F, (iii) -Cl, (iv) -NH2 или (v) -D и если Х10 или X15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
и где любые два смежных члена группы, включая Х10, Х11, X12, X13, Х14 и X15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N или О атом;
где B1 и В2 независимо выбраны из:
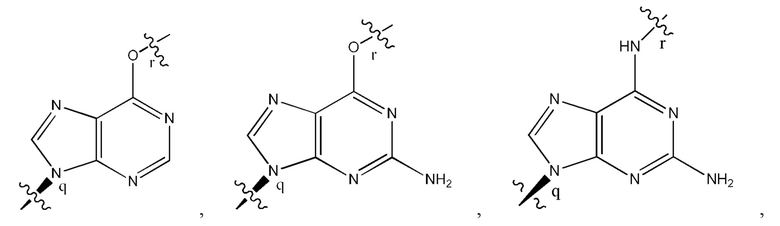
 , где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (III).
, где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (III).
Согласно некоторым вариантам осуществления, где L1 включает в себя тройную связь или более одной двойной связи, L1 может быть, например, 
Варианты осуществления также могут обеспечивать соединение формулы (III):
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н и -F;
R1b выбран из группы, состоящей из -Н и -F, где R1a и R1b оба не могут быть -F;
R4a выбран из группы, состоящей из -H и -F;
R4b выбран из группы, состоящей из -Н и -F, где R4a и R4b оба не могут быть -F;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила и -С(O)C1-6алкила;
L1 в формуле (III) представляет собой четыре или пять атомов углерода длиной и представляет собой 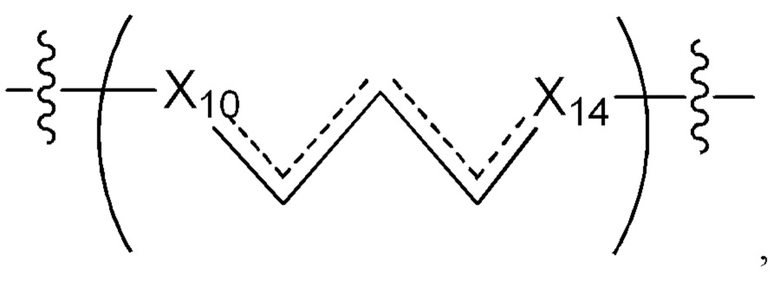
где означает простую связь или двойную связь и где или 0, или 1 встречаемость в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и X14 независимо выбраны из связи, -CH- или -CH2- и где, если Х10 или Х14 представляет собой связь, такая связь не представляет собой двойную связь;
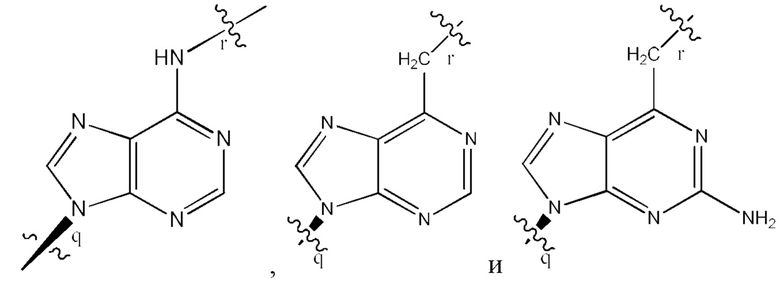
где B1 и В2 независимо выбраны из:
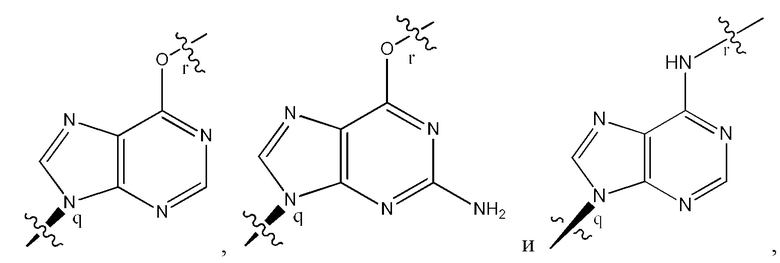 где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (III).
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (III).
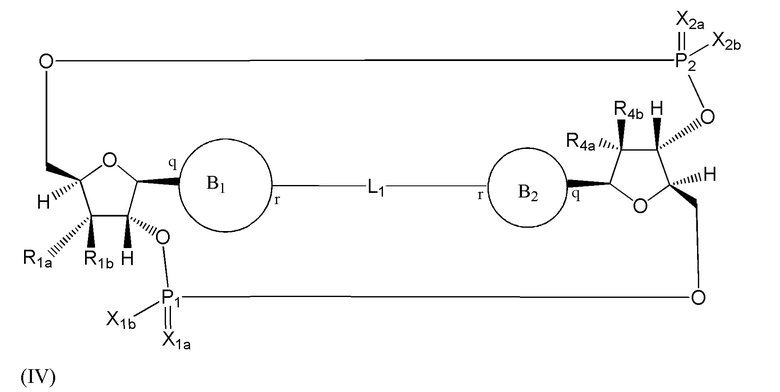
Варианты осуществления могут обеспечивать соединение формулы (IV):
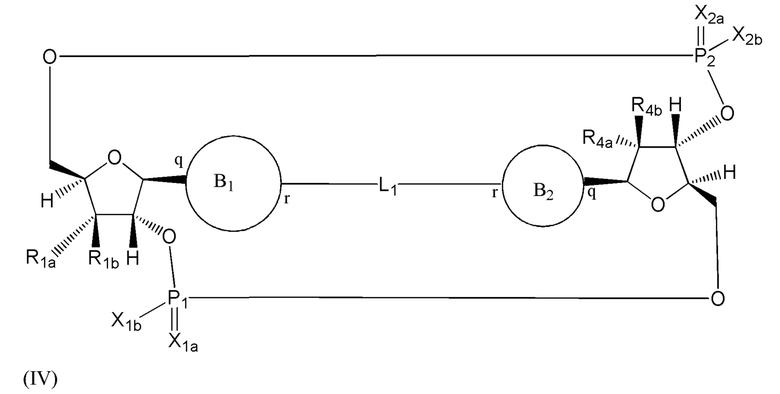
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н, -ОН и -F;
R1b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из -Н, -ОН и -F;
R4b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и P2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила, -С(O)C1-6алкила и -CH2OC(O)OC1-6алкила;
L1 в формуле (IV) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой 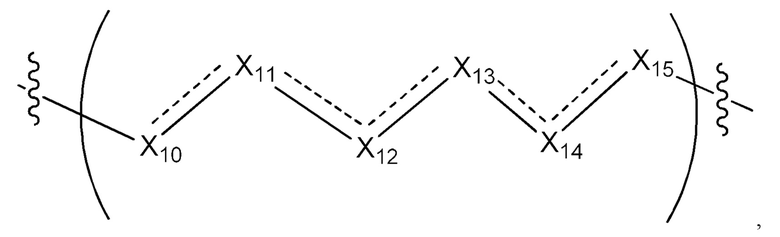
где означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости в L1 означает двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость в L1 означает тройную связь, 0 встречаемостей в L1 означает двойную связь; и (iv) где, если 2 встречаемости в L1 означает двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, X12, X13, Х14 и X15 независимо выбраны из связи, -СН2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН, (ii) -F, (iii) -Cl, (iv) -NH2 или (v) -D и если Х10 или X15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
и где любые два смежных члена группы, включая Х10, Х11, X12, X13, Х14 и X15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N или О атом;
где B1 и В2 независимо выбраны из:
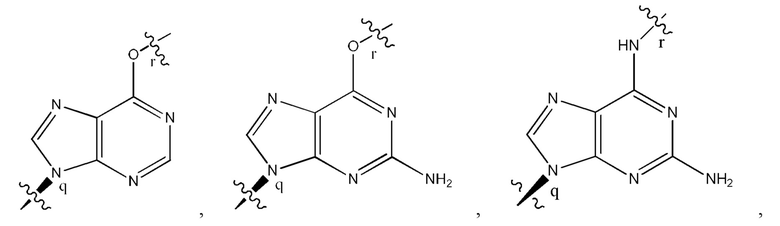 , где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (IV).
, где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (IV).
Варианты осуществления также могут обеспечивать соединение формулы (IV):
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н и -F;
R1b выбран из группы, состоящей из -Н и -F, где R1a и R1b оба не могут быть -F;
R4a выбран из группы, состоящей из -H и -F;
R4b выбран из группы, состоящей из -Н и -F, где R4a и R4b оба не могут быть -F;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила и -С(O)C1-6алкила;
L1 в формуле (IV) представляет собой четыре или пять атомов углерода длиной и представляет собой
где означает простую связь или двойную связь и где или 0, или 1 встречаемость в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и X14 независимо выбраны из связи, -CH- или -CH2- и где, если Х10 или Х11 представляет собой связь, такая связь не представляет собой двойную связь;
где B1 и В2 независимо выбраны из:
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (IV).
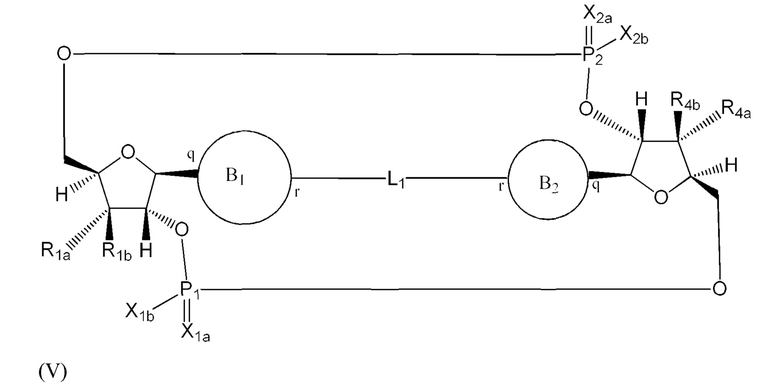
Варианты осуществления могут обеспечивать соединение формулы (V):
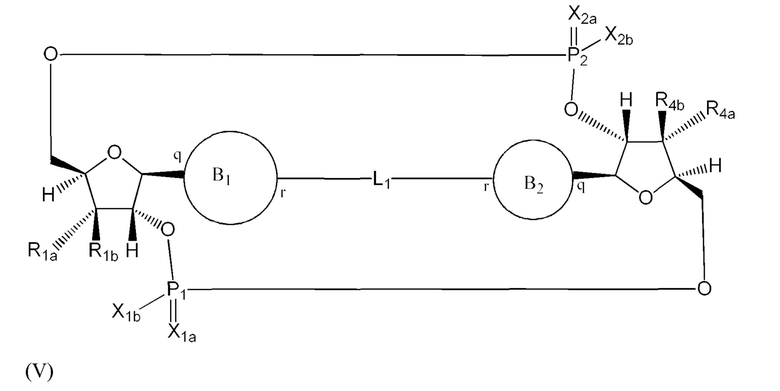
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н, -ОН и -F;
R1b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из -Н, -ОН и -F;
R4b выбран из группы, состоящей из -Н, -ОН и -F и где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и P2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила, -С(O)C1-6алкила и -CH2OC(O)OC1-6алкила;
L1 в формуле (V) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой 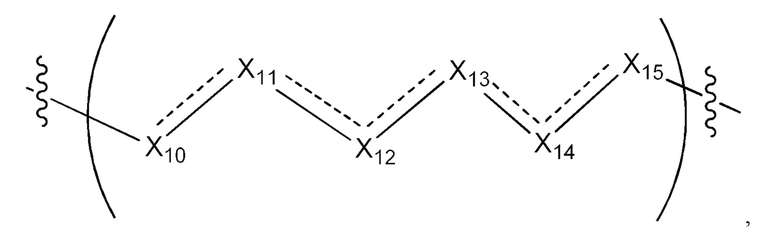
где означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости в L1 означает двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость в L1 означает тройную связь, 0 встречаемостей в L1 означает двойную связь; и (iv) где, если 2 встречаемости в L1 означает двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, X12, X13, Х14 и X15 независимо выбраны из связи, -CH2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН, (ii) -F, (iii) -Cl, (iv) -NH2 или (v) -D и если Х10 или X15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
и где любые два смежных члена группы, включая Х10, Х11, X12, X13, Х14 и X15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N или О атом;
где B1 и В2 независимо выбраны из:
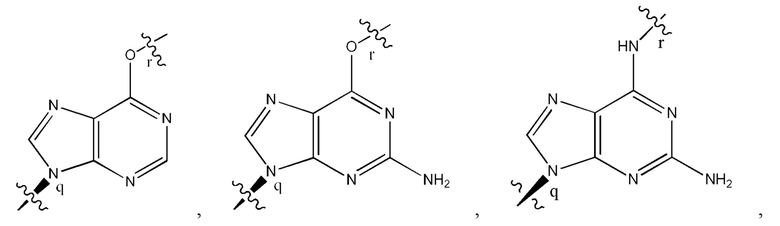 , где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (V).
, где связи в точках q и r на В1 и В2 присоединены в точках q и r на формуле (V).
Варианты осуществления также могут обеспечивать соединение формулы (V):
или его фармацевтически приемлемую соль, где
R1a выбран из группы, состоящей из -Н и -F;
R1b выбран из группы, состоящей из -Н и -F, где R1a и R1b оба не могут быть -F;
R4a выбран из группы, состоящей из -H и -F;
R4b выбран из группы, состоящей из -Н и -F, где R4a и R4b оба не могут быть -F;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила и -С(O)C1-6алкила;
L1 в формуле (V) представляет собой четыре или пять атомов углерода длиной и представляет собой
где означает простую связь или двойную связь и где или 0, или 1 встречаемость в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и X14 независимо выбраны из связи, -CH- или -CH2- и где, если Х10 или Х14 представляет собой связь, такая связь не представляет собой двойную связь;
где B1 и В2 независимо выбраны из:
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (V).
Согласно некоторым вариантам осуществления соединений и/или солей, как было сообщено в формулах выше, (i) стереохимическая конфигурация P1 и Р2 обоих представляет собой R, и стереохимическая конфигурация P1 представляет собой R и Р2 представляет собой S, или стереохимическая конфигурация P1 представляет собой S и Р2 представляет собой R; (ii) одна встречаемость в L1 означает двойную связь, где геометрия вокруг двойной связи является транс; и (iii) Z представляет собой -O-.
Дополнительные варианты осуществления соединений и/или солей, как было сообщено в формулах выше, могут быть обнаружены в других аспектах настоящего изобретения. Например, некоторые варианты осуществления обеспечивают соединение или фармацевтически приемлемую соль, где R1a и R4a каждый представляет собой -F. Согласно некоторым вариантам осуществления R1b и R4b каждый представляет собой -F. Согласно некоторым вариантам осуществления B1 и В2 каждый представляет собой 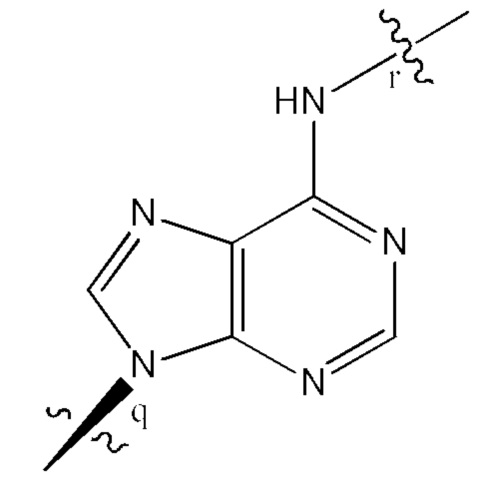 . Согласно некоторым вариантам осуществления X1a и Х2а оба представляют собой =O и X1b и X2b оба представляют собой -SH. Согласно некоторым вариантам осуществления L1 представляет собой
. Согласно некоторым вариантам осуществления X1a и Х2а оба представляют собой =O и X1b и X2b оба представляют собой -SH. Согласно некоторым вариантам осуществления L1 представляет собой 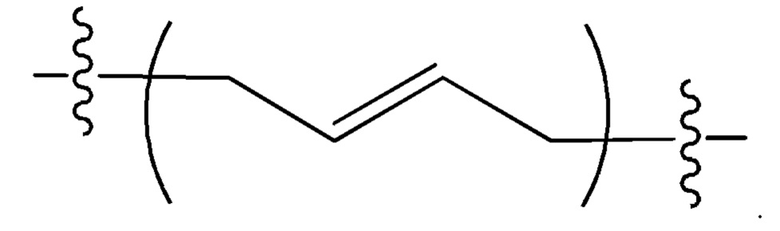
Согласно некоторым вариантам осуществления линкер представляет собой четыре атома углерода длиной. Согласно некоторым вариантам осуществления линкер представляет собой пять атомов углерода длиной.
Некоторые варианты осуществления обеспечивают соединение, выбранное из группы, состоящей из:
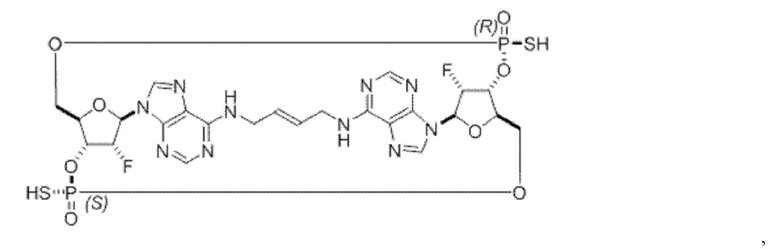
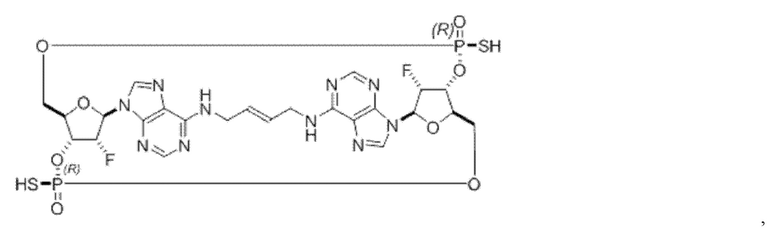
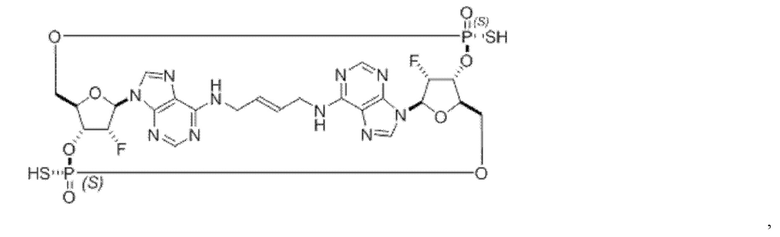
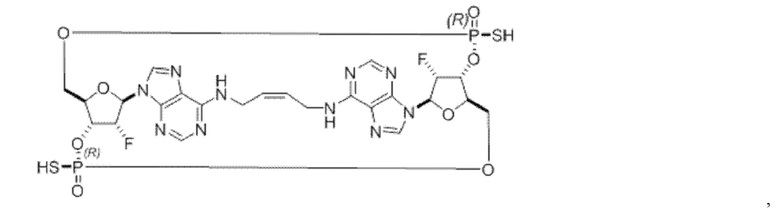
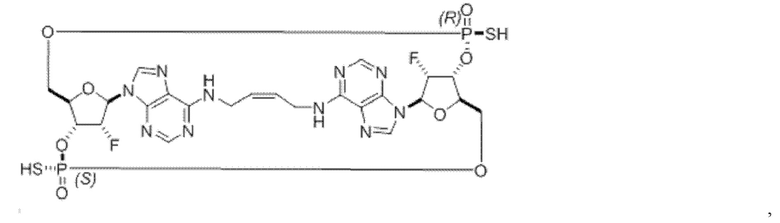
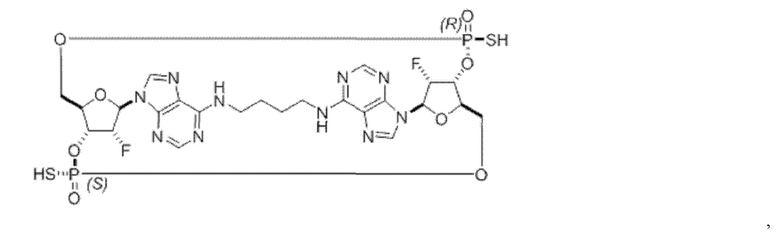
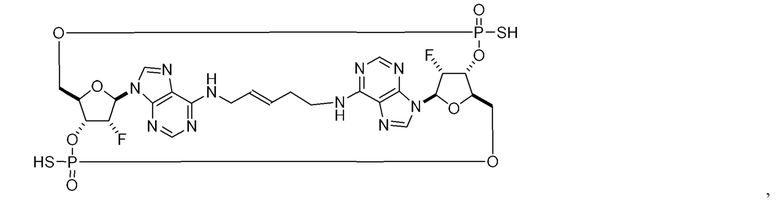
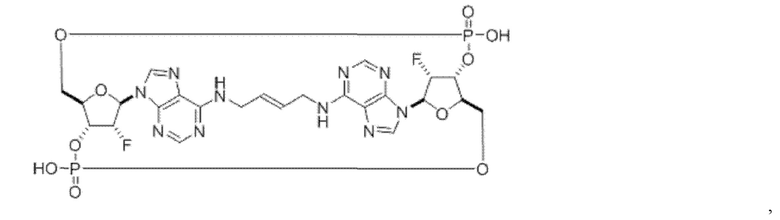
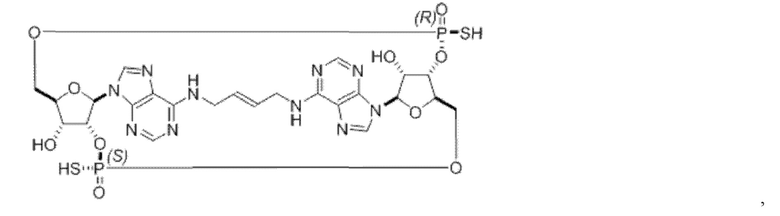
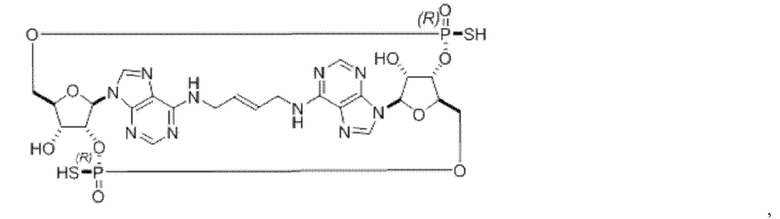
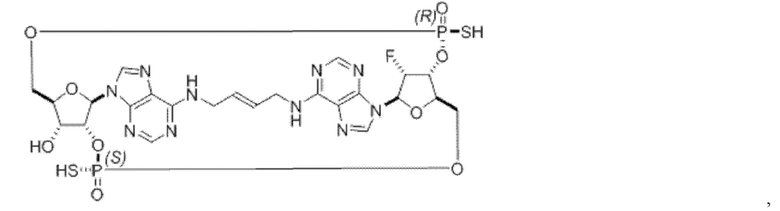
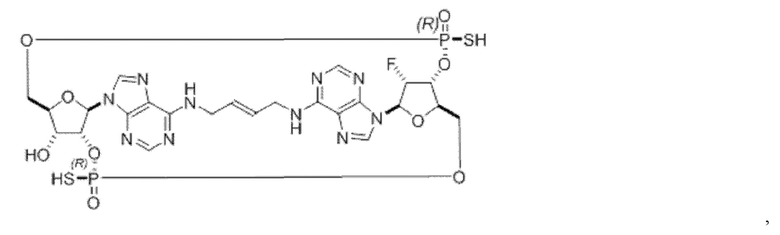
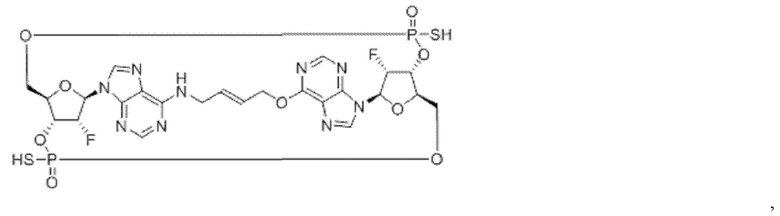
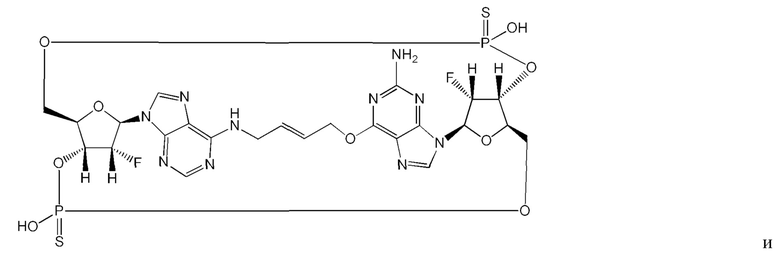
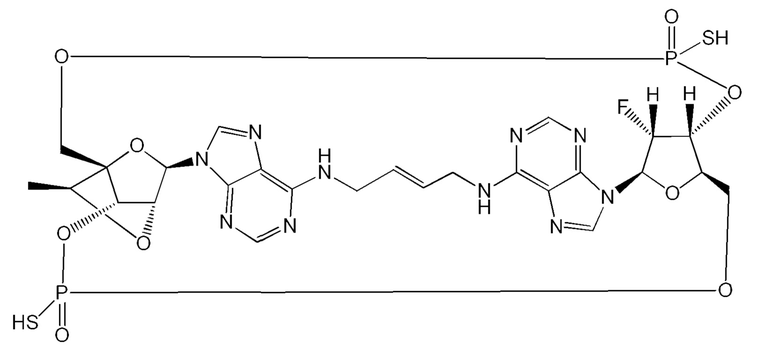
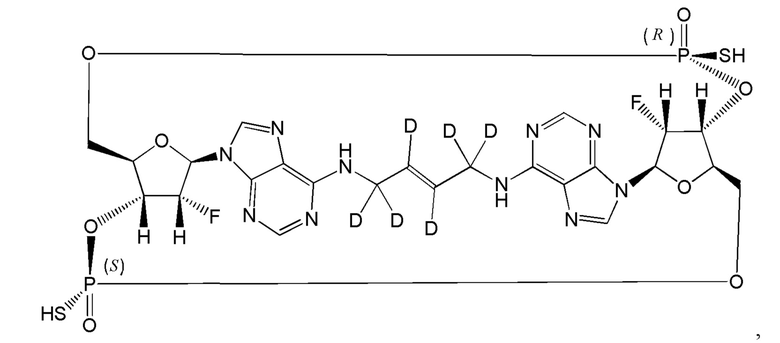
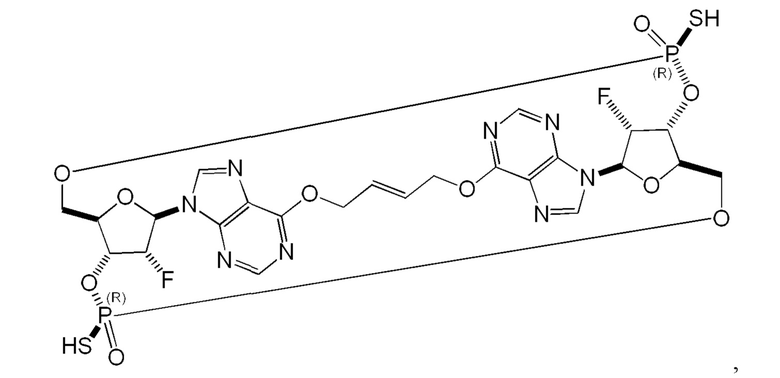
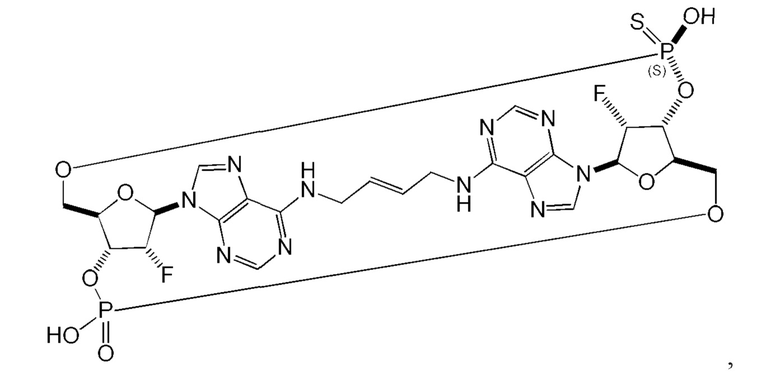
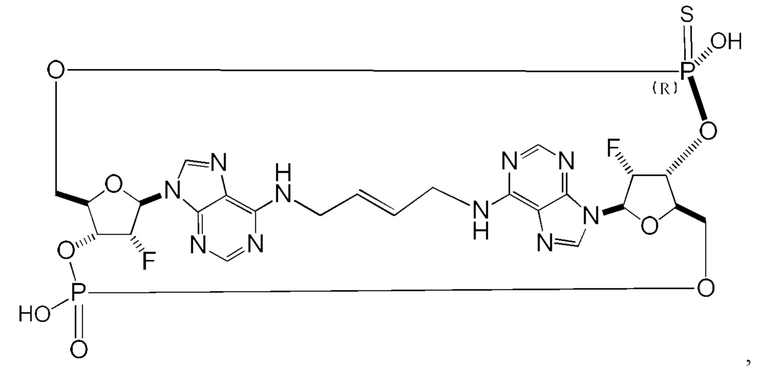
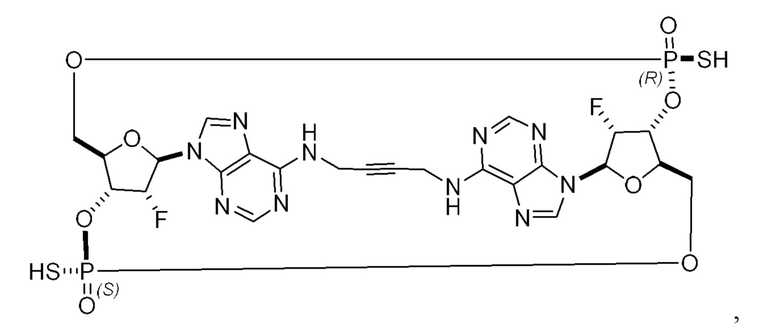
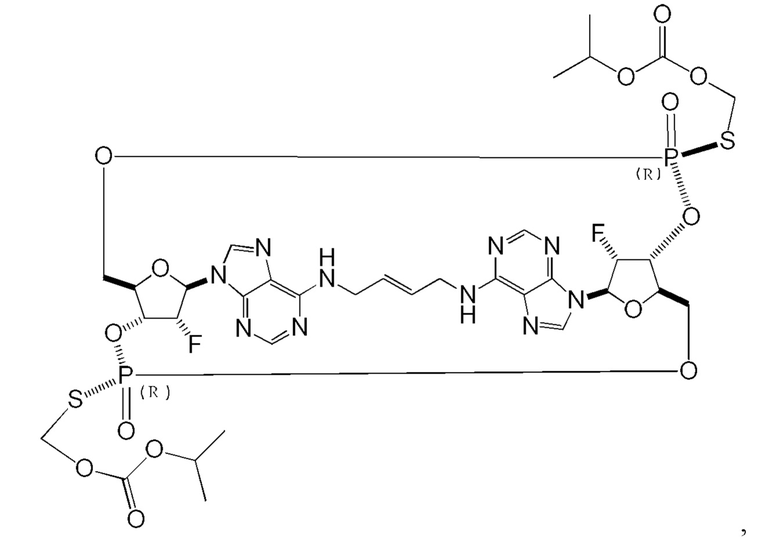
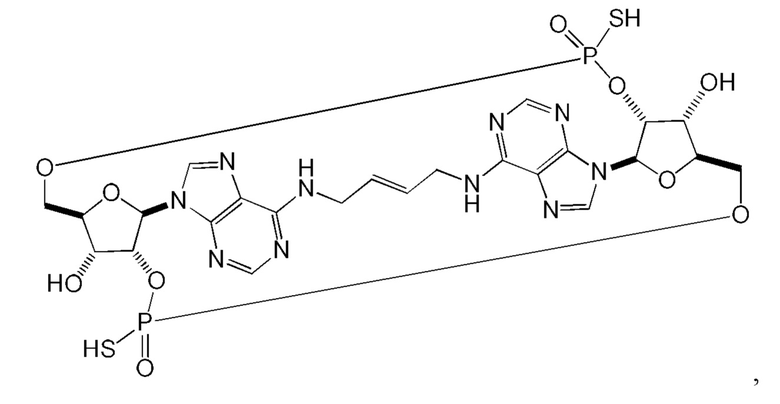
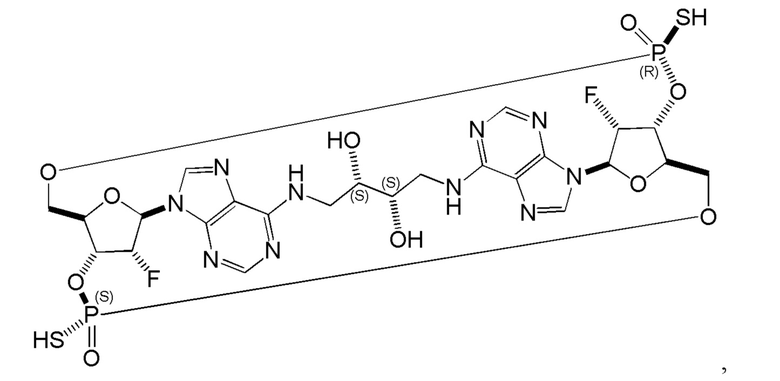
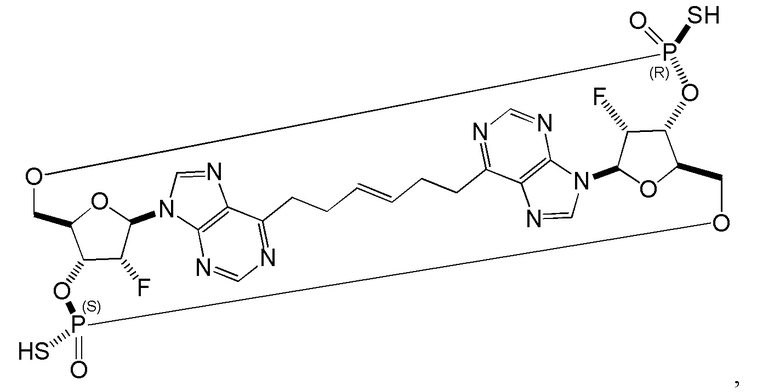
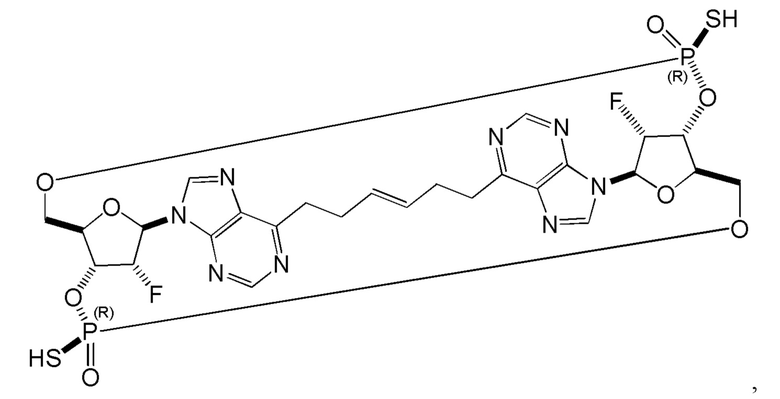
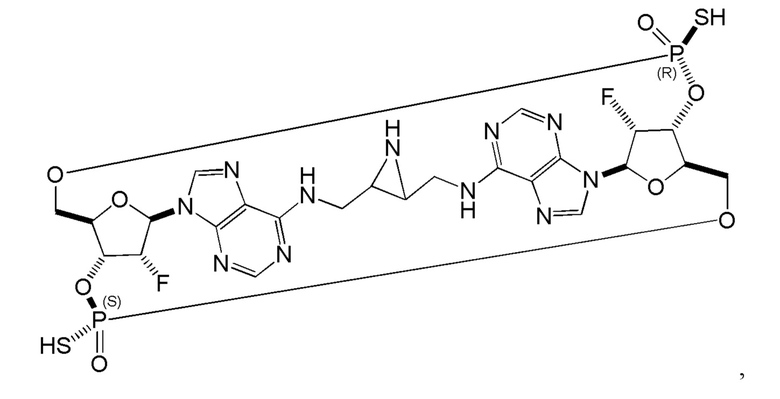
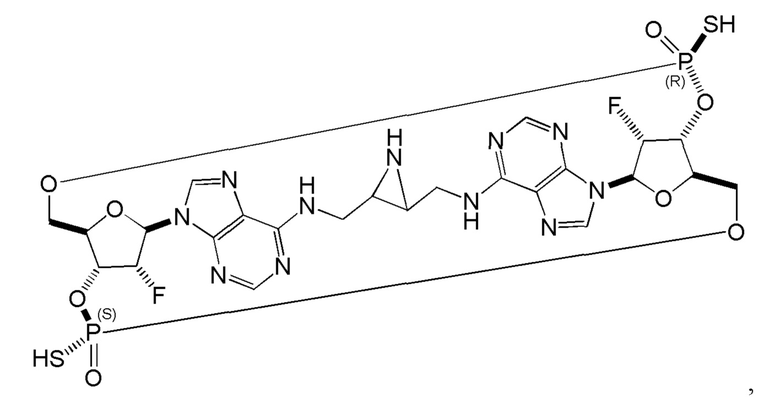
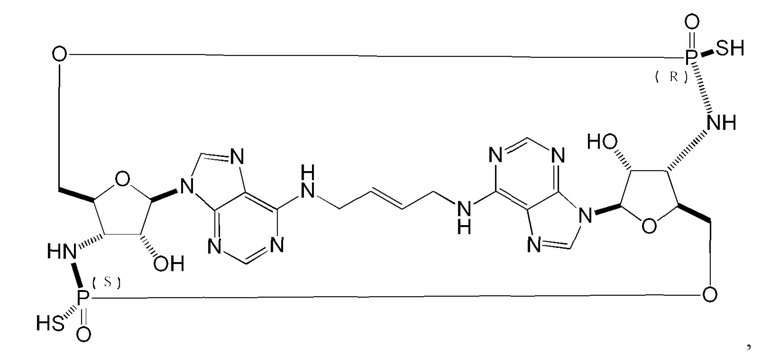
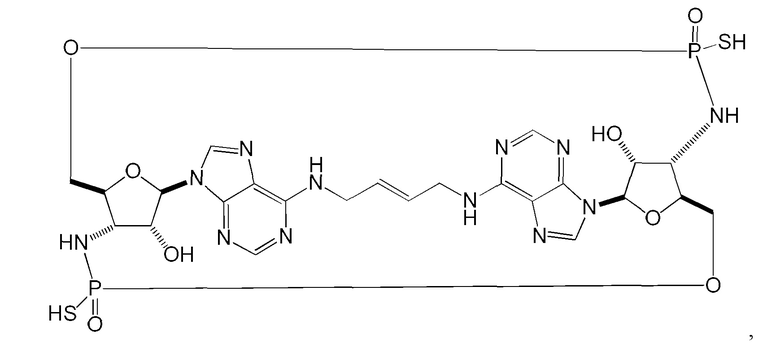
или его фармацевтически приемлемую соль.
В соответствии с вариантами осуществления может быть предусмотрено соединение или фармацевтически приемлемая соль с одним или несколькими из (i) значения EC50 ниже 100 микромоль в клеточном репортерном анализе, в котором экспрессируется генетический вариант STING HAQ; (ii) значения ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING AQ человека; (iii) значения ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING WT человека; и (iv) значения ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING REF. В соответствии с одним вариантом осуществления предусмотрено соединение, имеющее следующую структуру:
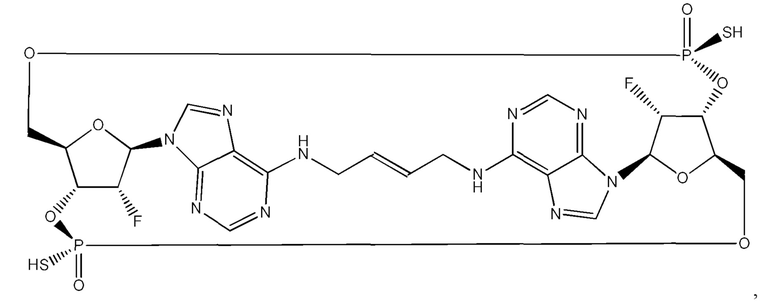
или его фармацевтически приемлемая соль.
В соответствии с дополнительными вариантами осуществления может быть предусмотрена фармацевтически приемлемая соль соединения, описанного в данном документе, при этом соль представляет собой диаммониевую соль. В соответствии с дополнительными вариантами осуществления могут быть предусмотрены соединения, описанные в данном документе, в виде триэтиламиновой (TEA) соли. В соответствии с дополнительными вариантами осуществления может быть предусмотрена фармацевтическая композиция, содержащая соединение или соль, описанные в данном документе, и фармацевтически приемлемый наполнитель.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака, предусматривающий введение пациенту соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
Предусмотрено применение соединения или его фармацевтически приемлемой соли, описанных в данном документе, для получения фармацевтической композиции для лечения рака.
В соответствии с вариантами осуществления может быть предусмотрено соединение, фармацевтически приемлемая соль или фармацевтическая композиция, описанные в данном документе, для лечении рака.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака, предусматривающий идентификацию индивидуума, имеющего рак, подлежащий лечению с помощью соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе; и введение указанному индивидууму фармацевтически эффективного количества соединения, фармацевтически приемлемой соли или фармацевтической композиции, с помощью которых рак был идентифицирован как подлежащий лечению.
В соответствии с некоторыми вариантами осуществления индивидуума идентифицируют как имеющего рак, подлежащий лечению с помощью соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе, в результате присутствия аллеля варианта STING REF у пациента.
В соответствии с некоторыми вариантами осуществления предусмотрен способ лечения рака у пациента, имеющего аллель STING REF, предусматривающий введение указанному пациенту соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с некоторыми вариантами осуществления предусмотрен способ лечения рака у пациента, имеющего аллель STING WT, предусматривающий введение указанному пациенту соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с некоторыми вариантами осуществления предусмотрен способ лечения рака у пациента, имеющего аллель STING AQ, предусматривающий введение указанному пациенту соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с некоторыми вариантами осуществления предусмотрен способ лечения рака у пациента, имеющего аллель STING HAQ, предусматривающий введение указанному пациенту соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с некоторыми вариантами осуществления рак выбирают из группы, состоящей из лимфомы, меланомы, колоректального рака, рака молочной железы, острого миелоидного лейкоза, рака толстой кишки, рака печени, рака предстательной железы, рака поджелудочной железы, рака почки и глиомы. В соответствии с некоторыми вариантами осуществления рак является метастатическим.
Согласно некоторым вариантам осуществления соединений формул, представленных в настоящем изобретении, одна из связей в L1 представляет собой двойную связь. Согласно дополнительным вариантам осуществления такая двойная связь обладает транс-геометрией. Согласно дополнительным вариантам осуществления L1 является насыщенным. Согласно определенным вариантам осуществления L1 включает в себя пять атомов углерода. Согласно другим вариантам осуществления L1 включает в себя 4 атома углерода.
Варианты осуществления могут дополнительно обеспечивать смеси соединений, как описано в настоящем изобретении, включая смеси стереоизомеров таких соединений. Например, может быть представлена смесь из соединения 11 и соединения 12 или может быть представлена смесь из соединения 2 и соединения 4. Разумеется, это не ограничивающие примеры и другие смеси являются возможными.
Конкретные варианты осуществления изложены в таблице 3 ниже.
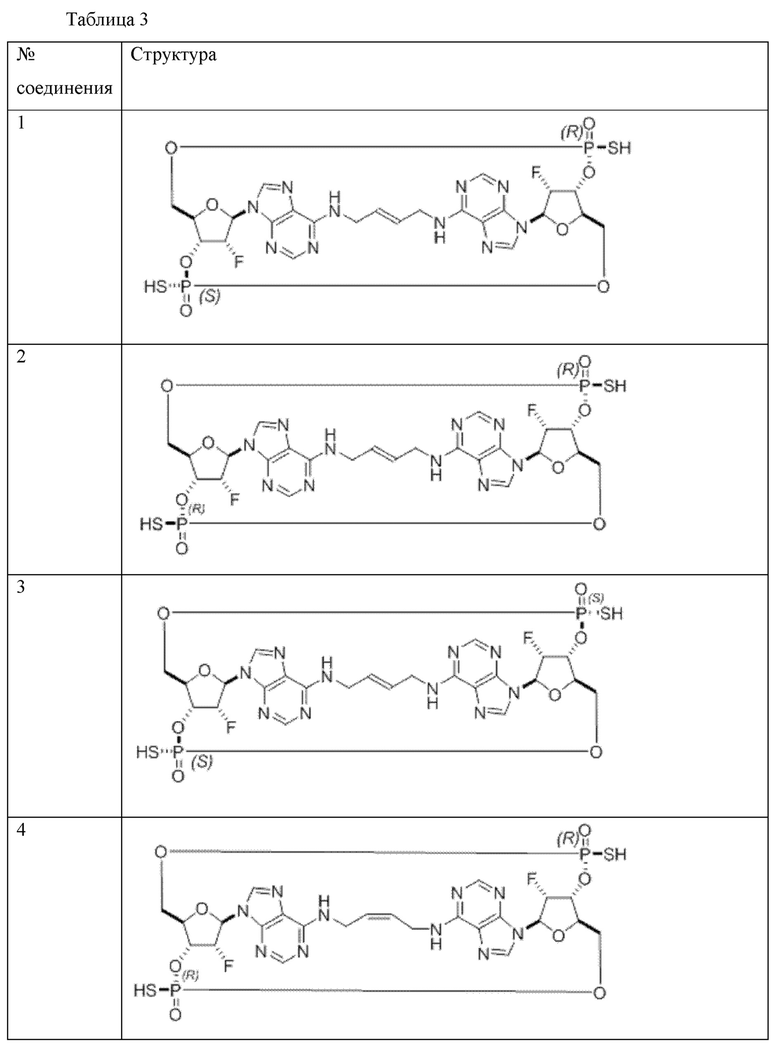
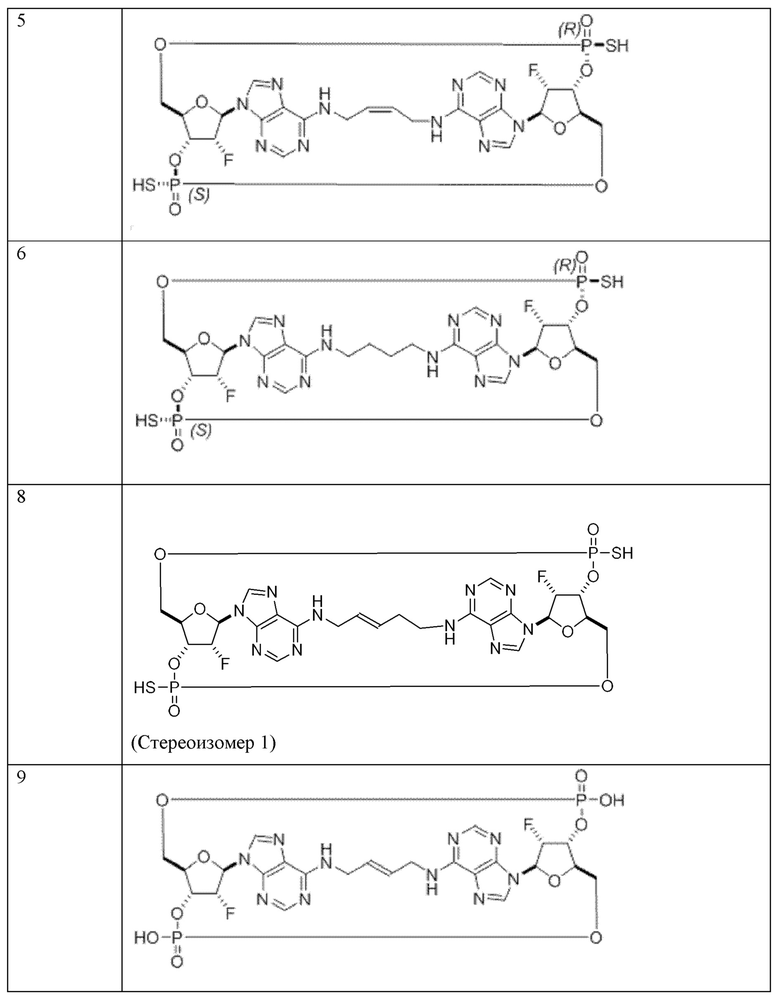
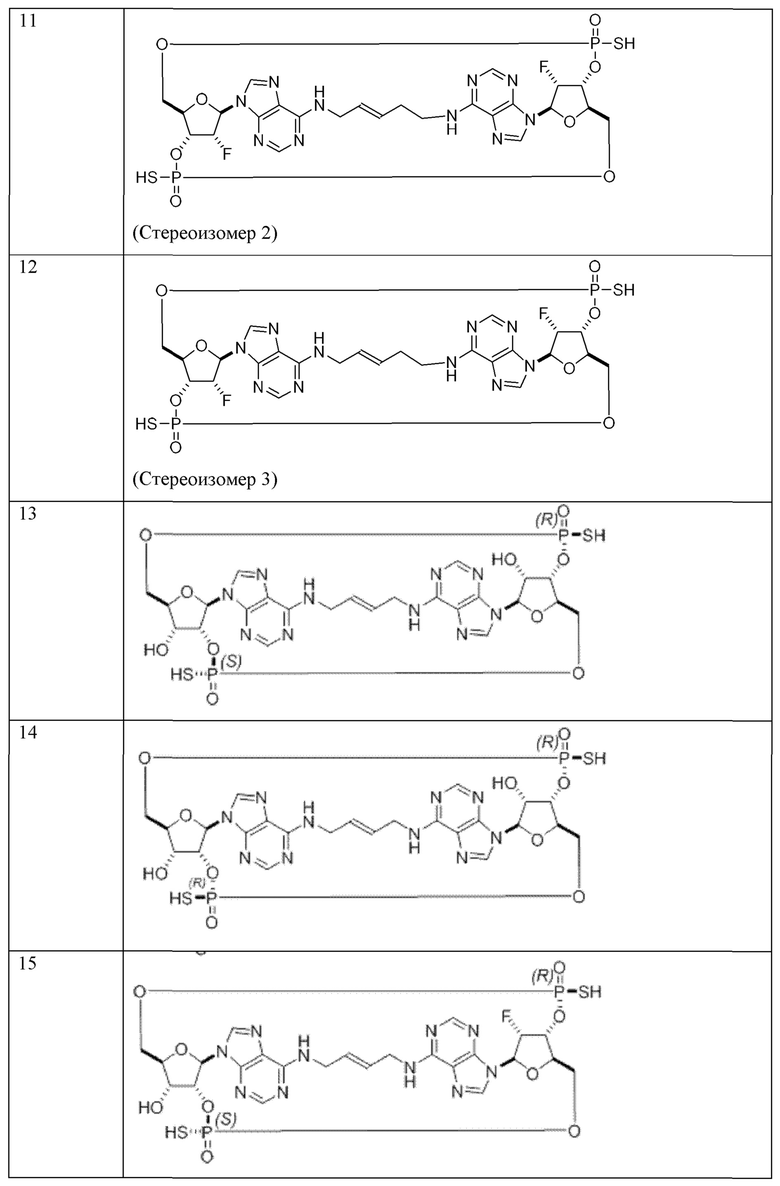
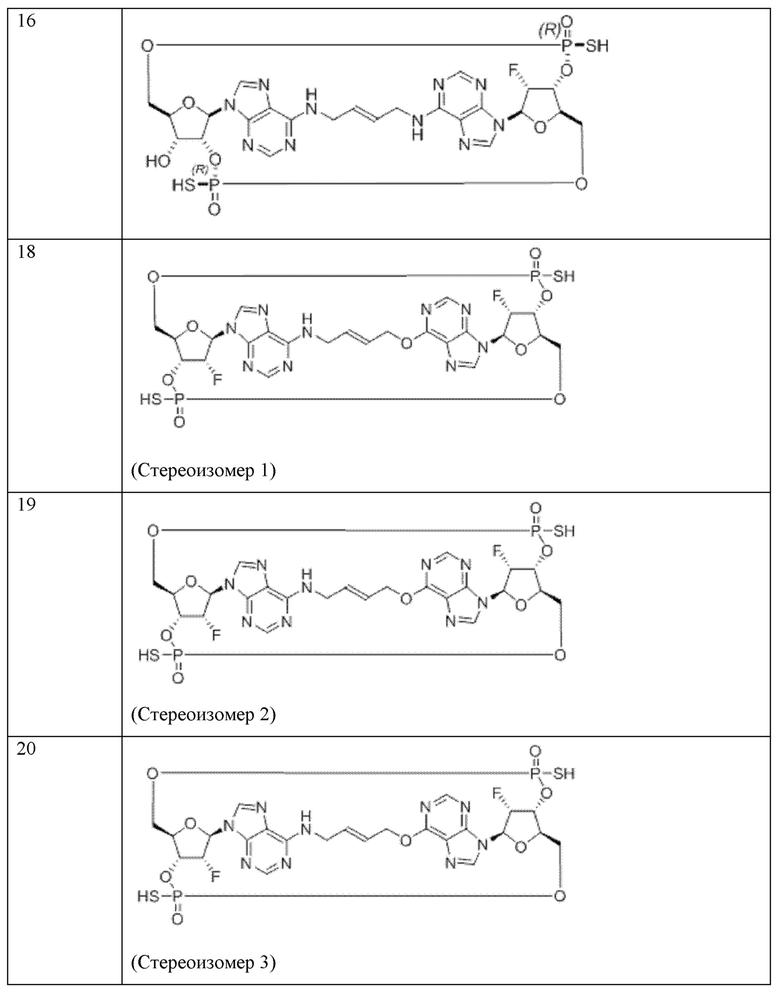
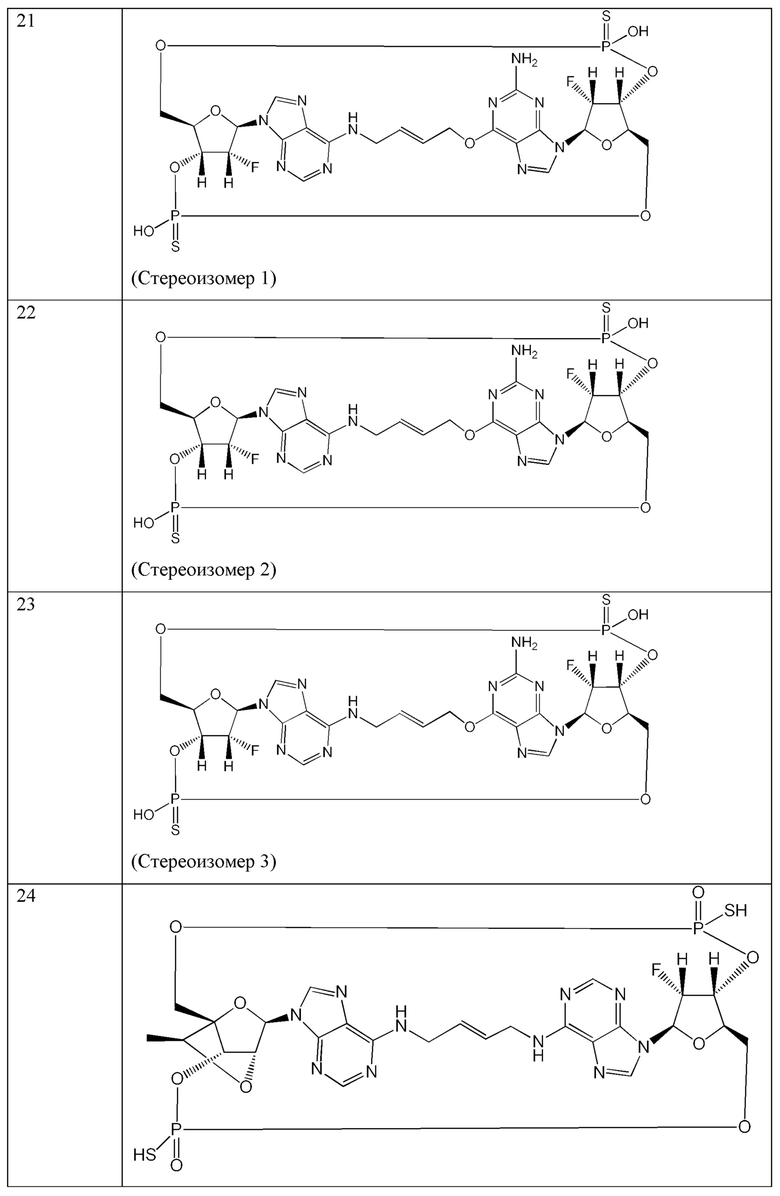
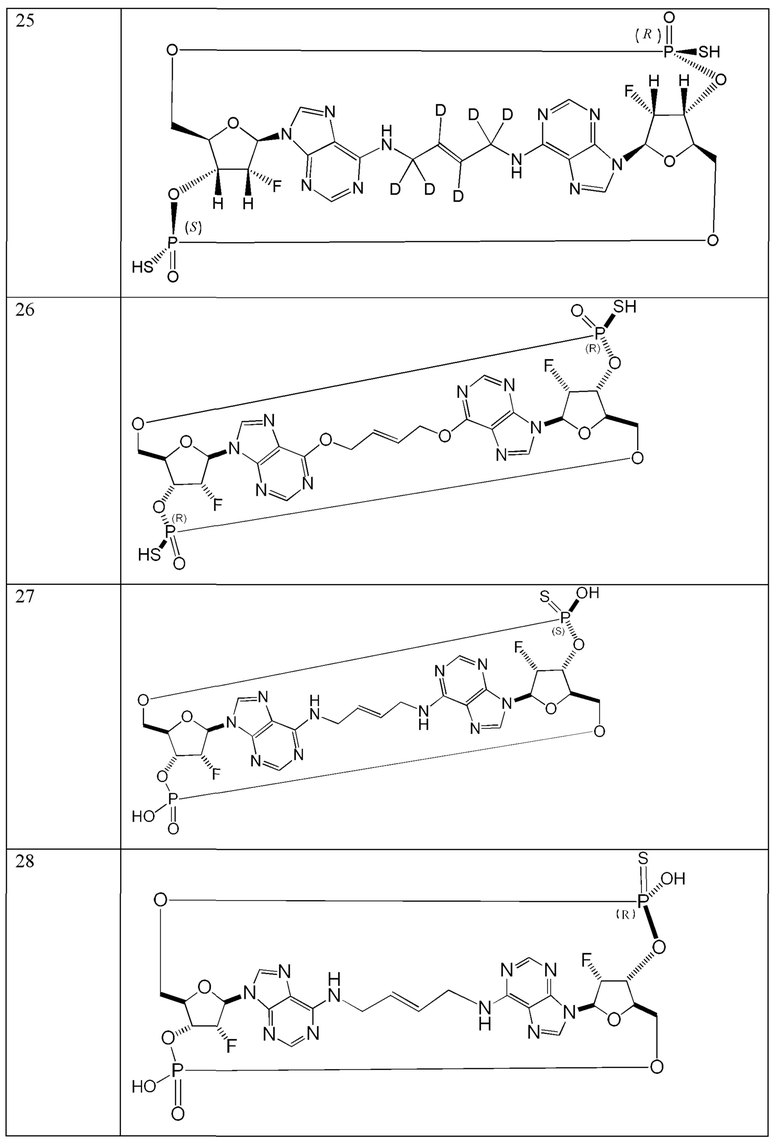
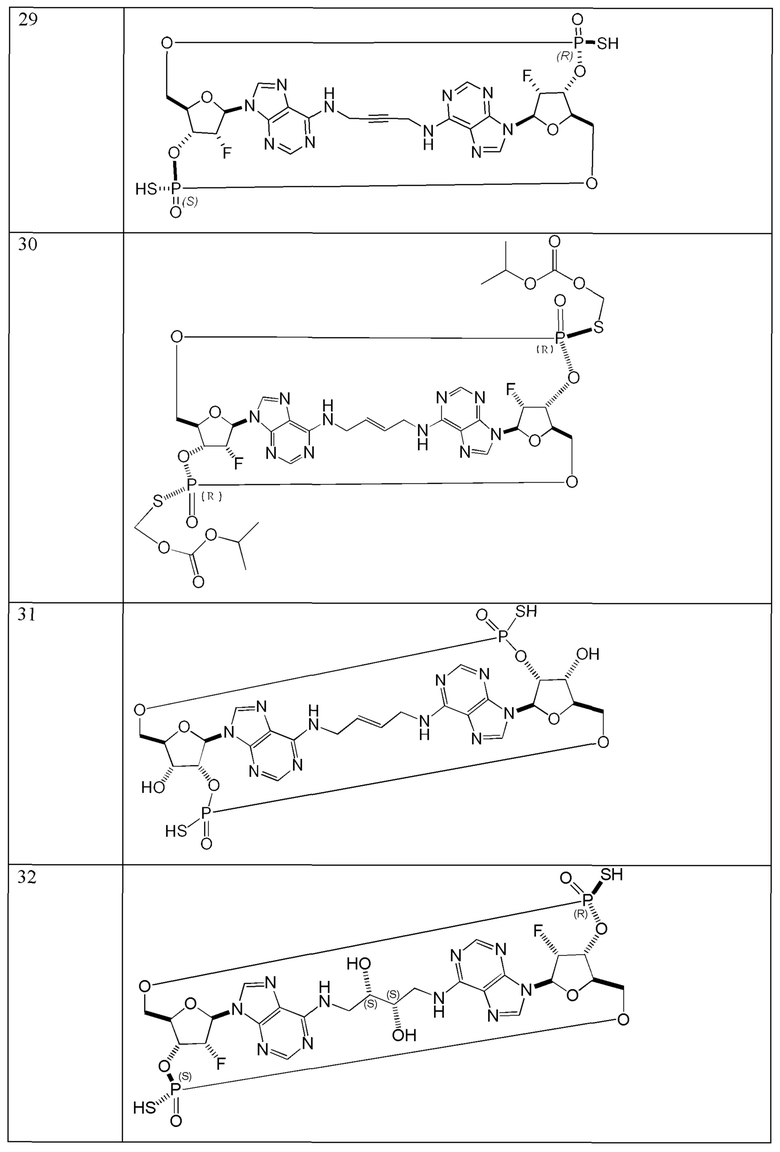
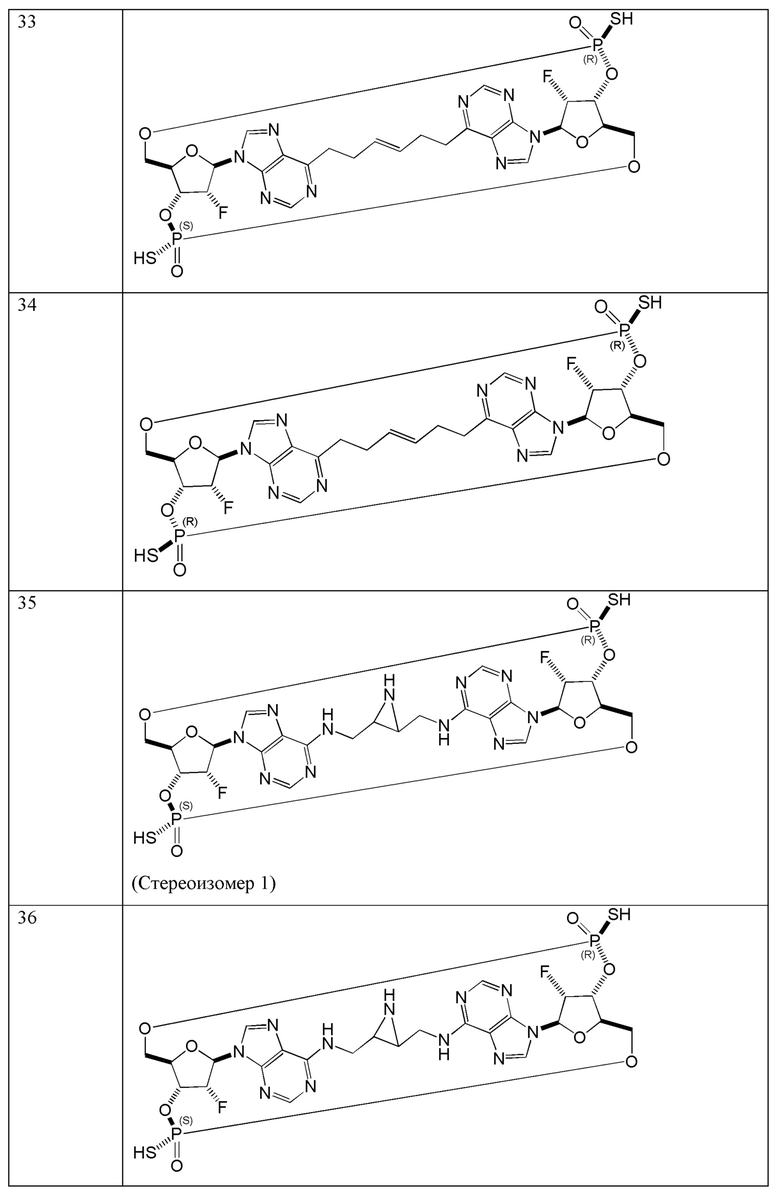
В таблице выше соединения 8, 11 и 12 изображены при помощи той же структурной формулы и относятся к трем отдельным стереоизомерам. Тем не менее, заявителем будет отмечено, что фосфорная хиральность соединения 8 не является обязательно такой же, что и фосфорная хиральность для других соединений, отмеченных как «Стереоизомер I», так, например, Соединение 21. Это же справедливо по отношению к другим стереоизомерам.
Варианты осуществления могут относится к C4-C6 линкерам, которые могут быть ковалентно связаны на любом конце с пуриновыми или пиримидиновыми основаниями, которые образуют часть циклического динуклеотида. Согласно варианту осуществления линкерами являются бутеновые, пентеновые или гексеновые линкеры, связанные на любом конце с пуриновыми основаниями. Согласно другому варианту осуществления линкерами являются бутеновые линкеры, связанные на любом конце с пуриновыми основаниями; согласно другому варианту осуществления линкерами являются трансбутеновые линкеры с двойной связью, расположенной между центральными двумя атомами углерода.
Следующие пронумерованные варианты осуществления являются иллюстративными для применения таких С4-С6 линкеров:
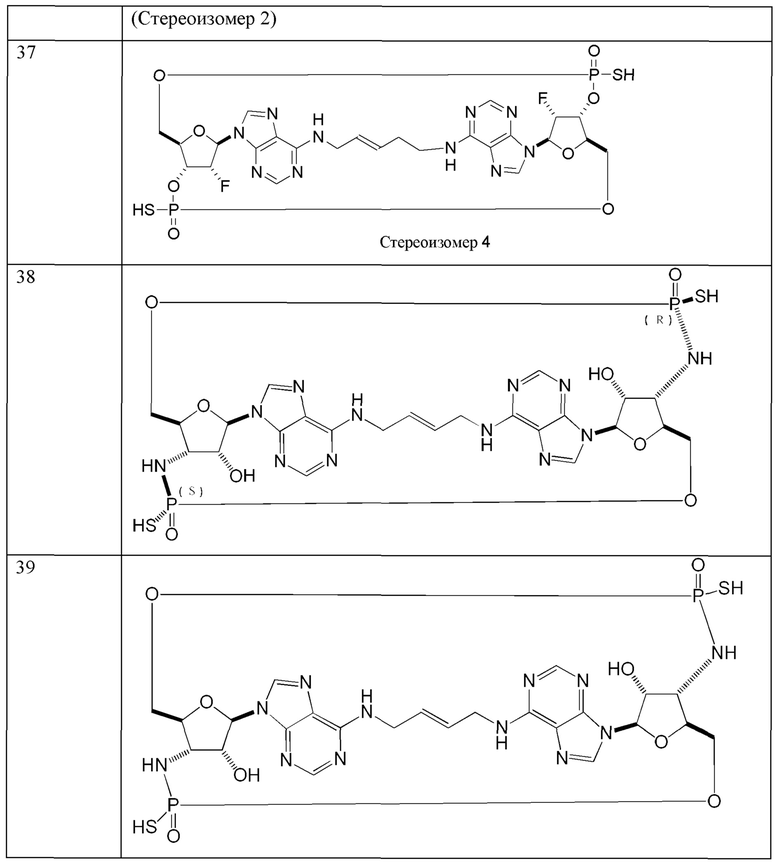
1. Соединение формулы (X):
или его фармацевтически приемлемая соль, где:
A1 и А2 являются фрагментами сахаров и могут быть одинаковыми или разными;
В3 и В4 представляют собой пуриновые или пиримидиновые основания, которые могут быть одинаковыми или разными и которые образуют нуклеотиды с, соответственно, A1 и А2;
L представляет собой алкильный линкер;
X19 и Х20 одинаковые или разные и выбраны из группы, состоящей из -O-, -СН-, -NH- и -S-, где -СН- и -NH- могут быть замещенными или незамещенными.
2. Соединение или фармацевтически приемлемая соль пронумерованного варианта осуществления 1, где -СН- и -NH- X19 и Х20 могут быть замещены C1-6 алкилом.
3. Соединение или фармацевтически приемлемая соль пронумерованного варианта осуществления 1, где L представляет собой бутен, пентен или гексан.
4. Соединение или фармацевтически приемлемая соль пронумерованного варианта осуществления 3, где L представляет собой трансбутеновый линкер с двойной связью в своем центре.
Соединения или фармацевтически приемлемые соли пронумерованных вариантов осуществления могут быть применимы, например, для лечения рака. A1, А2, В3 и В4 могут быть дополнительно замещены, например, гидроксилом, галогеном или метокси. Каждый фосфор в формуле (X) может быть замещен, например, -SH, -ОН, =O, или =S, до насыщения его валентности.
Примеры циклических динуклеотидных аналогов, которые могут извлекать пользу от линкеров по настоящему описанию, включают в себя без ограничения аналоги, определенные в заявке на патент США №2014/0205653 А1; заявке на патент США №2014/0329889 А1; заявке на патент США №2014/0341976 А1; заявке на патент США №2015/0056224 А1; заявке на патент США №2016/0362441 А1; заявке на патент США №2017/0158724 А1; заявке на патент США №2017/044206 А1; патенте США №5547941; патенте США №7569555 В2; патенте США №7592326 В2; патенте США №7709458 В2; патенте США №9549944 В2; WO 2009/133560 А1; WO 2015/074145 А1; WO 2015/077354 А1; WO 2015/185565 А1; WO 2016/100261 А1; WO 2016/120305 A1;WO 2016/145102 А1; WO 2017/027645 А1; WO 2017/027646 А1; WO 2017/075477 А1; WO 2017/093933 A1; WO 2017/123657 А1; WO 2017/175156 А1; EP 1740,192 В1; CN 102199183 A; Corrales, L. et al., «Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity», Cell Reports, 11: 1018-1030 (2015); и Lioux, Т. et al., «Design, Synthesis и Biological Evaluation of Novel Cyclic Adenosine-Inosine Monophosphate (cAIMP) Analogs That Activate Stimulator of Interferon Genes (STING)», J. Med. Chem., 59: 10253-10267 (2016). Все такие документы, включая соединения по настоящему изобретению, включены при помощи ссылки в настоящее описание; если любой компонент любого из таких документов противоречит или иным образом несовместим с любым из настоящего описания, тогда настоящее описание является контрольным.
Согласно некоторым вариантам осуществления соединение, описанное в настоящем изобретении, представлено в виде свободной кислоты. Согласно некоторым вариантам осуществления соединение представлено в виде NH4 соли или в виде триэтиламиновой (TEA) соли.
Варианты осуществления могут обеспечивать способ лечения рака у пациента при необходимости такого лечения, который предусматривает введение пациенту терапевтически эффективного количества соединения, описанного в настоящем изобретении, или его фармацевтически приемлемой соли, как описано выше.
Согласно некоторым вариантам осуществления соединение вводили в виде свободной кислоты. Согласно некоторым вариантам осуществления соединение вводили в виде диаммонийной соли (NH4). Предусмотрены фармацевтические композиции, содержащие соединение формулы I, формулы II, формулы III, формулы IV, формулы V, таблицы 3 или его фармацевтически приемлемую соль, а также фармацевтически приемлемое вспомогательное вещество. Варианты осуществления могут обеспечивать соединение формулы I, в которой n представляет собой 1; геометрия вокруг двойной связи является транс; X1 и Х2 каждый представляет собой SH; стереохимия при P1 представляет собой S; и стереохимия при P2 представляет собой R; или его фармацевтически приемлемую соль.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, предусматривающий введение указанному пациенту соединения или его фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе. Виды рака, подлежащие лечению, описанные в данном документе, могут представлять собой метастатические виды рака. Они могут быть выбраны, например, из лимфомы, меланомы, колоректального рака, рака молочной железы, острого миелоидного лейкоза, рака толстой кишки, рака печени, рака предстательной железы, рака поджелудочной железы, рака почки и глиомы. Также предусмотрены варианты применения соединений, солей и фармацевтических композиций для лечения рака и/или получения лекарственного препарата для лечения рака.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака, предусматривающий идентификацию индивидуума, имеющего рак, подлежащий лечению с помощью соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе, и введение индивидууму соединения, фармацевтически приемлемой соли или фармацевтической композиции, с помощью которых пациент был идентифицирован как подлежащий лечению. В соответствии с некоторыми вариантами осуществления индивидуума идентифицируют как имеющего рак, подлежащий лечению с помощью соединения, фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе, в результате присутствия аллеля варианта STING REF человека у пациента.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, имеющего аллель STING REF, предусматривающий введение указанному пациенту соединения или его фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, имеющего аллель STING WT, предусматривающий введение указанному пациенту соединения или его фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, имеющего аллель STING AQ, предусматривающий введение указанному пациенту соединения или его фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, имеющего аллель STING HAQ, предусматривающий введение указанному пациенту соединения или его фармацевтически приемлемой соли или фармацевтической композиции, описанных в данном документе.
Краткое описание чертежей
На Фиг. 1 изображен синтез Соединения 1а и Соединения 2а.
На Фиг. 2А и Фиг. 2В изображен альтернативный синтез соединения 1 и соединения 1а. Указанный альтернативный синтез также изображен на Фиг. 2С - Фиг. 2Е.
На Фиг. 3 изображен спектрограф 1H NMR для Соединения 1.
На Фиг. 4А, Фиг. 4В и Фиг. 4С изображены результаты рентгенокристаллографии (чертежи ORTEP) соответственно для асимметрического кристалла Соединения 1, первой молекулы из асимметрического кристалла и второй молекулы из асимметрического кристалла.
На Фиг. 4D изображены результаты рентгенокристаллографии (чертеж ORTEP) для кристалла Соединения 2.
На Фиг. 5А и Фиг. 5В изображен путь синтеза для Соединений 18, 19 и 20.
На Фиг. 6 изображена карта вектора экспрессии для STING WT (pLenti-STING WT человека-Puro).
Фиг. 7 и Фиг. 8 сопровождают Пример 108 и на них изображена терапевтическая активность Соединения 1а в двойной модели опухоли СТ26.
Фиг. 9 сопровождает Пример 109 и на ней изображен график объема опухоли для опухолей, претерпевших лечение, а также кривая выживаемости.
Фиг. 10 сопровождает Пример 110 и на ней изображен график объема опухоли для опухолей, претерпевших лечение, а также кривая выживаемости.
На Фиг. 11 представлено изображение рентгенокристаллографической структуры STING WT человека в комплексе с Соединением 1.
На Фиг. 12 изображен С-концевой домен STING REF в комплексе с Соединением 1.
На Фиг. 13 изображен пример синтеза Соединения 38 и Соединения 39.
Подробное описание вариантов осуществления
В данном документе предусмотрены соединения, которые могут быть пригодны в лечении рака. Указанные соединения могут активировать стимулятор генов интерферона (STING).
В соответствии с некоторыми вариантами осуществления указанное соединение предусмотрено в виде свободной кислоты. В соответствии с некоторыми вариантами осуществления указанное соединение предусмотрено, например, в виде соли NH4 или соли TEA. Отсылка на номер соединения, который следует за «а» будет обозначать диаммониевую соль определенного соединения. Например, «Соединение 1а» означает диаммониевую соль Соединения 1.
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, нуждающегося в этом, предусматривающий введение пациенту терапевтически эффективного количества соединения формулы I, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли.
В соответствии с некоторыми вариантами осуществления указанное соединение вводят в виде свободной кислоты. В соответствии с некоторыми вариантами осуществления соединение вводят, например, в виде диаммониевой соли (NH4). Также могут быть предусмотрены фармацевтические композиции для лечения рака, содержащие соединение, описанное в данном документе или его фармацевтически приемлемую соль, а также фармацевтически приемлемый наполнитель.
Варианты осуществления, описанные в данном документе, могут быть использованы для лечения рака или для получения лекарственных препаратов, используемых для лечения рака. Термин «рак» может включать в себя без ограничения рак толстой кишки, рак печени, меланому, колоректальный рак, рак молочной железы, острый миелоидный лейкоз и глиому.
Специалистами настоящей области техники будет установлено, что если заместители, связанные с атомами фосфора (P1, P2), содержат как простые, так и двойные связи, они могут быть подвержены таутомеризации. Например, соединения могут таутомеризировать в состоянии равновесия. Один пример показан ниже:
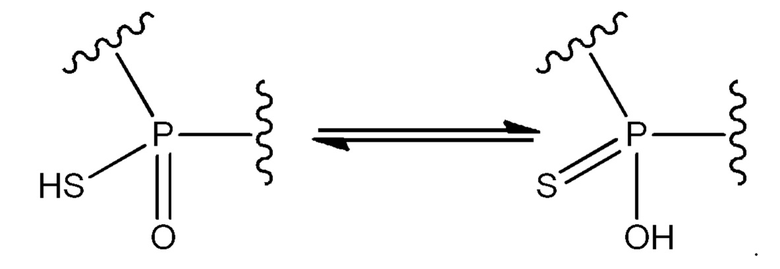
Такие таутомеры следует рассматривать как находящиеся в пределах объема формулы изобретения. Структурное представление любого таутомера для представленного соединения будет представлять то же соединение.
Согласно некоторым вариантам осуществления соединение, выбранное из группы, состоящей из соединений, описанных в настоящем изобретении, представлено в виде свободной кислоты или его фармацевтически приемлемой соли. Согласно некоторым вариантам осуществления соединение, выбранное из группы, состоящей из соединений, описанных в настоящем изобретении, представлено в виде NH4 соли, которая может быть диаммонийной солью.
Предусмотрено, что используемый в настоящем описании «C1-6алкил» или «C1-С6» алкил включает в себя C1, C2, C3, С4, C5 или C6 с неразветвленной цепью (линейной) насыщенные алифатические углеводородные группы и С3, С4, С5 или С6 с разветвленной цепью насыщенные алифатические углеводородные группы. Например, предусмотрено, что C1-6 алкил включает в себя C1, C2, C3, С4, C5 или C6 алкильные группы. Примеры алкила включают в себя фрагменты, содержащие от одного до шести атомов углерода, такие как без ограничения метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил или н-гексил. Подобным образом, предусмотрено, что «С1-3алкил» или «C1-C3алкил» включает в себя C1, C2 или С3 с неразветвленной цепью (линейной) насыщенные алифатические углеводородные группы и С3 с разветвленной цепью насыщенные алифатические углеводородные группы.
Используемый в настоящем описании термин «C3-6циклоалкил» или «C3-C6циклоалкил» относится к насыщенному или ненасыщенному неароматическому углеводородному кольцу, содержащему от 3 до 6 атомов углерода (например, C3-C6). Примеры циклоалкила включают в себя без ограничения циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил и циклогексенил. Термин «C5-6циклоалкил» или «C5-C6циклоалкил» относится к насыщенному или ненасыщенному неароматическому углеводородному кольцу, содержащему 5 или 6 атомов углерода (например, C5-C6).
Термин «C5-6гетероциклоалкил» или «C5-C6гетероциклоалкил» относится к насыщенному или ненасыщенному неароматическому 5-6-членному моноциклическому, содержащему один или несколько гетероатомов (таких как О, N или S), если конкретно не отмечено иное. Термин «С4-6гетероциклоалкил» или «С4-C6гетероциклоалкил» относится к насыщенному или ненасыщенному неароматическому 4-6-членному моноциклическому, содержащему один или несколько гетероатомов (таких как О, N или S), если конкретно не отмечено иное. Примеры гетероциклоалкильных групп включают в себя без ограничения пиперидинил, пиперазинил, пирролидинил, диоксанил, тетрагидрофуранил, изоиндолинил, индолинил, имидазолидинил, пиразолидинил, оксазолидинил, изоксазолидинил, триазолидинил, оксиранил, азетидинил, оксетанил, тиетанил, 1,2,3,6-тетрагидропиридинил, тетрагидропиранил, тетрагидротиофен, дигидропиранил, пиранил, морфолинил, 1,4-диазепанил, 1,4-оксазепанил и т.п.
Дополнительные примеры гетероциклоалкильных групп включают в себя без ограничения акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензоксазолинил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изатионил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил; 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,2,4-оксазол5(4Н)-он, оксазолидинил, оксазолил, оксиндолил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил.
Используемый в настоящем описании термин «C5-6арил» или «C5-С6арил» относится к ароматическому углеводородному кольцу, содержащему от 5 до 6 атомов углерода (например, C5-С6), которое не содержит никакого гетероатома в кольцевой структуре.
Термин «С3-6гетероарил» или «С3-С6гетероарил» относится к ароматическому 3-6-членному моноциклическому, содержащему один или несколько гетероатомов (таких как О, N или S), если конкретно не отмечено иное, за исключением того, что гетероарильное кольцо будет включать в себя не более одного атома кислорода или одного атома серы.
Термин «С2-6алкенил» включает в себя ненасыщенные алифатические группы, содержащие 2, 3, 4, 5, или 6 атомов углерода и которые содержат по меньшей мере одну двойную связь. Например, термин «С2-6алкенил» включает в себя алкенильные группы с неразветвленной цепью (например, этенил, пропенил, бутенил, пентенил, гексенил) и неразветвленные алкенильные группы. Согласно определенным вариантам осуществления алкенильная группа с неразветвленной цепью или разветвленной содержит шесть или меньше атомов углерода в своей основной цепи (например, С2-С6 для неразветвленной цепи, С3-С6 для разветвленной цепи). Термин «С2-6алкенил» включает в себя алкенильные группы, содержащие от двух до шести атомов углерода.
Термин «С2-6алкинил» включает в себя ненасыщенные алифатические группы, содержащие 2, 3, 4, 5 или 6 атомов углерода, но которые содержат по меньшей мере одну тройную связь. Например, «алкинил» включает в себя алкинильные группы с неразветвленной цепью (например, этинил, пропинил, бутинил, пентинил, гексинил) и разветвленные алкинильные группы. Согласно определенным вариантам осуществления алкинильная группа с неразветвленной цепью или разветвленной содержит шесть или меньше атомов углерода в своей основной цепи (например, С2-С6 для неразветвленной цепи, С3-С6 для разветвленной цепи). Термин «С2-6алкинил» включает в себя алкинильные группы, содержащие от двух до шести атомов углерода.
Способы лечения
В соответствии с вариантами осуществления может быть предусмотрен способ лечения рака у пациента, нуждающегося в этом, предусматривающий введение пациенту терапевтически эффективного количества соединения, описанного в данном документе, или его фармацевтически приемлемой соли.
В соответствии с некоторыми вариантами осуществления вводимое соединение предусмотрено в виде свободной кислоты или ее фармацевтически приемлемой соли. В соответствии с некоторыми вариантами осуществления вводимое соединение предусмотрено в виде соли NH4, свободной кислоты или ее фармацевтически приемлемой соли. В соответствии с некоторыми вариантами осуществления указанное соединение предусмотрено в виде соли NH4.
Термин «необязательно замещенный» относится к фрагменту, имеющему обозначенные заместители, замещающие один или несколько атомов водорода в одном или нескольких атомах, несущих водород в фрагменте.
Обозначенные или описанные химические соединения включают в себя все встречающиеся в природе изотопы атомов, встречающихся в соединениях настоящего изобретения. Изотопы включают в себя атомы, имеющие одинаковое атомное число, но разные массовые числа. В качестве общего примера и без ограничения изотопы водорода 1H включают в себя тритий и дейтерий, а изотопы углерода 12С включают в себя 13С и 14С.
Дозы
Оптимальная доза для лечения рака может быть определена эмпирически для каждого индивидуума с помощью известных способов и будет зависеть от ряда факторов, в том числе активности средств; возраста, массы тела, общего состояния здоровья, пола и питания индивидуума; времени и способа введения; и других лекарственных препаратов, который индивидуум принимает. Оптимальные дозы могут быть установлены с помощью стандартного тестирования и процедур, которые хорошо известны в данной области техники. Введение вышеуказанных соединений может происходить с помощью любого подходящего способа.
Термин «фармацевтически приемлемая соль», используемый в данном документе, относится к солям присоединения кислоты или солям присоединения основания соединений в настоящем раскрытии. Фармацевтически приемлемая соль представляет собой любую соль, которая сохраняет активность родительского соединения и не оказывает какого-либо чрезмерно вредного или нежелательного влияния на субъекта, которому ее вводят и в контексте которого ее вводят. Фармацевтически приемлемые соли включают в себя без ограничения комплексы металлов и соли как неорганических, так и карбоновых кислоты. Фармацевтически приемлемые соли также включают в себя соли металлов, такие как соли алюминия, кальция, железа, магния и комплексные соли. Помимо этого, фармацевтически приемлемые соли включают в себя без ограничения кислые соли, такие как, уксусные, аспарагиновые, алкилсульфоновые, арилсульфоновые, аксетиловые, бензолсульфоновые, бензойные, бикарбоновые, бисерные, бивинные, масляные, кальций-эдетатные, камзиловые, карбоновые, хлорбензойные, лимонные, этилендиаминтетрауксусные, эдизиловые, эстоловые, эзиловые, эзилиновые, муравьиные, фумаровые, глюцептовые, глюконовые, глутаминовые, гликолевые, гликолиларсаниловые, гексаминовые, гексилрезорциновые, гидрабамовые, бромистоводородные, хлористоводородные, йодистоводородные, гидроксинафтойные, изэтиновые, молочные, лактобионовые, малеиновые, яблочные, малоновые, миндальные, метансульфоновые, метилазотные, метилсерные, слизевые, муконовые, напсиловые, азотные, щавелевые, п-нитрометансульфоновые, памовые, пантотеновые, фосфорные, моногидрофосфорные, дигидрофосфорные, фталевые, полигалактоуроновые, пропионовые, салициловые, стеариновые, янтарные, сульфаминовые, сульфаниловые, сульфоновые, серные, таниновые, винные, теоклиновые, толуолсульфоные и т.п. Также могут быть получены натриевые соли и калиевые соли.
В вариантах осуществления могут быть предусмотрены диаммониевые соли. Фармацевтически приемлемые соли могут быть получены из аминокислот, в том числе без ограничения цистеина. Способы получения соединений в виде солей известны специалистам в данной области техники (см., например, Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66: 1, 1977).
Термин «эффективное количество» терапевтического средства представляет собой количество, достаточное для получения наблюдаемой терапевтической пользы по сравнению с раком, который не претерпел лечения у субъекта или пациента.
Активные средства, описанные в данном документе, можно сочетать с фармацевтически приемлемым носителем для получения их фармацевтических составов. Конкретный выбор носителя и состава будет зависеть от определенного способа введения, для которого композиция предусмотрена.
Термин «фармацевтически приемлемый носитель», используемый в данном документе, относится к нетоксичному носителю, адъюванту или основе, которые не нарушают фармакологической активности соединения, с которым его составляют. Фармацевтически приемлемые носители, адъюванты или основы, которые могут быть использованы в композициях по настоящему изобретению, включают в себя без ограничения сорбиновую кислоту, сорбат калия, смеси неполного глицерида насыщенных растительных жирных кислот, воду, соли или электролиты, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воска, полиэтиленгликоль и шерстяной жир.
Композиции по настоящему изобретению могут быть пригодны для парентерального, перорального введения, ингаляции с помощью спрея, местного, ректального, назального, буккального, вагинального введения или введения с помощью имплантируемого резервуара. В соответствии с некоторыми вариантами осуществления состав содержит ингредиенты, которые происходят из природных или неприродных источников. В соответствии с некоторыми вариантами осуществления состав или носитель могут быть предусмотрены в стерильной форме. Неограничивающие примеры стерильного носителя включают в себя не содержащую эндотоксинов воду или апирогенную воду.
Термин «парентеральный», используемый в данном документе, включает в себя подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, интрастернальные, интратекальные, внутрипеченочные, внутриочаговые и внутричерепные способы введения инъекций или инфузий. В соответствии с определенным вариантом осуществления соединения вводят внутривенно, перорально, подкожно или посредством внутримышечного введения. Стерильные инъекционные формы композиций по настоящему изобретению могут представлять собой водную или маслянистую суспензию. Эти суспензии могут быть составлены в соответствии с методиками, известными в данной области техники с использованием подходящих диспергирующих или смачивающих средств и суспендирующих средств. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе. Среди приемлемых основ и растворителей, которые могут быть использованы, присутствуют вода, раствор Рингера и изотонический раствор хлорида натрия. Помимо этого, стерильные нелетучие масла традиционно используются в качестве растворителя или суспендирующей среды.
С этой целью может быть использовано любое мягкое нелетучее масло, в том числе синтетические моно- или диглицериды. Жирные кислоты и их глицеридные производные являются пригодными для получения инъекционных препаратов, поскольку представляют собой природные фармацевтически приемлемые масла, такие как оливковое или касторовое масло, особенно в своих полиоксиэтилированных версиях. Эти масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или диспергент, такие как карбоксиметилцеллюлоза, или аналогичные диспергирующие средства, которые широко используются при составлении фармацевтически приемлемых лекарственных форм, в том числе эмульсий и суспензий. Другие широко используемые поверхностно-активные вещества, такие как Tween, Span и другие эмульгирующие средства, которые широко используются при получении фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть использованы с целью составления.
Для перорального введения соединение или соль могут быть предусмотрены в приемлемой лекарственной форме для перорального введения, в том числе без ограничения капсулах, таблетках, водных суспензиях или растворах. В случае таблеток для перорального применения широко используемые носители включают в себя лактозу и кукурузный крахмал. Также могут быть добавлены скользящие вещества, такие как стеарат магния. В случае перорального применения в капсульной форме пригодные разбавители включают в себя лактозу и высушенный кукурузный крахмал. Если для перорального применения требуются водные суспензии, то активный ингредиент можно комбинировать с эмульгирующими и суспендирующими средствами. При необходимости также могут быть добавлены определенные подсластители, ароматизаторы или красители. Помимо этого, могут быть добавлены консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают в себя без ограничения различные антибактериальные и противогрибковые средства, такие как разбавители, например, этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, соли четвертичного аммония и парабены (такие как метилпарабен, этилпарабен, пропилпарабен и т.д.).
«Немедленное высвобождение» подразумевает стандартное высвобождение, при котором высвобождение лекарственного средства начинается непосредственно после введения. Используемый в данном документе термин «немедленное высвобождение» предусматривает лекарственные формы, которые способствуют растворению лекарственного средства в содержимом желудочно-кишечного тракта, при этом не предполагается задержка или продление растворения или всасывания лекарственного средства. Целью является быстрое высвобождение лекарственного средства после введения, например, чтобы было возможным высвобождение по меньшей мере 80% лекарственного средства в течение примерно 30 минут после начала растворения в тесте растворения.
«Замедленное высвобождение» или «длительное высвобождение» предусматривает лекарственные формы, временные и/или пространственные характеристики высвобождения лекарственного средства которых выбирают для осуществления терапевтических целей или с целью удобства, не обеспечиваемых стандартными лекарственными формами, такими как раствор или лекарственная форма немедленного высвобождения.
Термин «устойчивое состояние» означает, что уровень в плазме крови определенного активного средства был достигнут и поддерживается с помощью последующих доз активного средства на уровне, который находится на минимальном эффективном терапевтическом уровне или выше него и ниже минимального токсического уровня в плазме крови для определенного активного средства.
Термин «единичный состав», используемый в данном документе, относится к одному носителю или основе, составляемым для доставки эффективных количеств обеих терапевтических средств пациенту. Единичный носитель конструируют для доставки эффективного количества каждого из средств наряду с любыми фармацевтически приемлемыми носителями или наполнителями. В соответствии с некоторыми вариантами осуществления основа представляет собой таблетку, капсулу, пилюлю или пластырь.
Термин «унифицированная доза» используется в данном документе для обозначения одновременного введения обоих средств совместно, в одной лекарственной форме, пациенту, подлежащему лечению. В соответствии с некоторыми вариантами осуществления унифицированная доза представляет собой единичный состав. В соответствии с определенными вариантами осуществления унифицированная доза содержит одну или несколько основ, в результате чего каждая основа содержит эффективное количество средства наряду с фармацевтически приемлемыми носителями и наполнителями. В соответствии с некоторыми вариантами осуществления унифицированная доза представляет собой одну или несколько таблеток, капсул, пилюль или пластырей, вводимых пациенту в одном и то же время.
Термин «диапазон дозы», используемый в данном документе, относится к верхней и нижней границе допустимой вариации количества определяемого средства. В типичном случае доза средства в любом количестве в пределах определяемого диапазона может быть введена пациентам, подвергающимся лечению.
Термин «лечить» используется в данном документе для облегчения, ослабления или нормализации по меньшей мере одного симптома заболевания у субъекта. Например, по отношению к раку, термин «лечить» может означать остановку, задержку наступления (т.е., периода до клинического проявления заболевания или симптома заболевания) и/или снижение риска развития или обострения симптома заболевания. Термин «оказывать защитное действие», используемый в данном документе, означает предупреждение, задержку или лечение или все, при необходимости, развития или продолжения или обострения симптомов заболевания у субъекта.
Термин «субъект» или «пациент» включает в себя животных, которые способны страдать от рака или быть пораженными им. Примеры субъектов или пациентов включают в себя млекопитающих, например, людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных не относящихся к человеку животных. В соответствии с определенными вариантами осуществления субъект представляет собой человека, например, человека, страдающего, имеющего риск страдать или потенциально способного страдать от рака.
Термин «приблизительно или «примерно» обычно означает в пределах 20%, более предпочтительно в пределах 10% и наиболее предпочтительно даже в пределах 5% определенного значения или диапазона. В качестве альтернативы особенно в биологических системах термин «приблизительно» означает примерно в пределах логарифма (т.е., порядка величины), предпочтительно в пределах множителя двух от определенного значения.
Использование терминов в единственном числе в контексте описания настоящего изобретения (особенно в контексте следующей далее формулы изобретения) подразумевает включение единственного и множественного числа, если в данном документе не указано иное или явно не противоречит контексту. Термины «содержащий», «имеющий», «включающий в себя» и «включающий» рассматриваются в виде открытых терминов (т.е., означающих «включающий в себя без ограничения»), если не указано иное. Предполагается, что перечисление диапазонов значений в данном документе будет использоваться только в виде сокращенного способа отдельного упоминания каждого отдельного значения, входящего в диапазон, если в данном документе не указано иное, и каждое отдельное значение включено в описание так, как если бы оно было отдельно упомянуто в данном документе.
Иллюстративные нарушения пролиферации клеток, которые можно лечить с помощью одного или нескольких соединений, раскрываемых в данном документе, включают в себя без ограничения рак, предрак или предраковое состояние, а также метастатические поражения в ткани и органах в организме. Нарушения пролиферации клеток могут включать в себя гиперплазию, метаплазию и дисплазию.
Соединение, раскрываемое в данном документе, или его фармацевтически приемлемая соль могут быть использованы для лечения или предупреждения нарушения пролиферации клеток или для лечения или предупреждения рака у субъекта, имеющего повышенный риск развития рака по отношению к популяции, или использованы для идентификации подходящих кандидатов для таких целей.
Фармацевтические составы и способы введения
В данном документе предусмотрены фармацевтические составы, содержащие соединение или его фармацевтически приемлемую соль для лечения рака. Фармацевтические составы могут дополнительно содержать носитель или наполнитель, стабилизатор, ароматизатор и/или краситель.
Соединение или его фармацевтически приемлемая соль могут быть введены с помощью ряда способов введения, известных специалистам в данной области техники. Способы введения включают в себя пероральное введение. В соответствии с определенными вариантами осуществления фармацевтический состав, содержащий соединение или его фармацевтически приемлемую соль, могут быть приняты перорально в форме жидкости, сиропа, таблетки, капсулы, порошка, аэрозоли, жевательной таблетки или растворимого диска. В качестве альтернативы фармацевтические составы по настоящему изобретению могут быть введены внутривенно или трансдермально. Дополнительные способы введения известны специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, Gennaro A. R., Ed., 20th Edition, Mack Publishing Co., Easton, Pa.).
В соответствии с некоторыми вариантами осуществления соединение или фармацевтически приемлемую соль составляют в виде пасты, геля или суспензии. Например, лекарственное средство растворяют, улавливают или суспендируют в форме частиц лекарственного средства, микроинкапсулированных частиц или частиц лекарственное средство-полимер в гелеобразном растворе или мягкой форме. Преимуществом гелеобразного состава для перорального применения является более легкое введение лекарственного средства пациентам, которые испытывают сложность с проглатыванием таблеток, капсул или пилюль. В соответствии с определенными вариантами осуществления соединение тщательно смешивают и суспендируют в соответствующей среде с образованием пасты или геля. Дополнительные средства могут необязательно быть смешаны с целью получения запаха во время перорального введения. Арахисовое масло или альгинат, ароматизированные малиной или подсластителем, представляют собой примеры из множества подходящих средств, маскирующих вкус лекарственного средства. В соответствии с различными вариантами осуществления паста или гель также могут быть составлены с подходящими связующими веществами или наполнителями, известными в данной области техники для местного применения.
Способы получения составов с замедленным высвобождением в форме таблеток, капсул или пилюль известны в данной области техники. В соответствии с некоторыми вариантами осуществления состав с замедленным высвобождением получают в результате покрытия активного ингредиента лекарственного средства полимером, предпочтительно нерастворимым в воде полимером. Например, нерастворимый в воде полимер, используется в фармацевтической области в виде покрывающего средства для замедленного высвобождения, кишечнорастворимого покрывающего средства или желудочнорастворимого покрывающего средства. Нерастворимый в воде полимер может включать в себя, например, этилцеллюлозу, очищенный шеллак, белый шеллак, сополимер аминоалкилметакрилата RS, гидроксипропилметилцеллюлозы фталат, гидроксипропилметилцеллюлозы ацетата сукцинат, сополимер метакриловой кислоты L, сополимер метакриловой кислоты LD, сополимер метакриловой кислоты S, сополимер аминоалкилметакрилата Е или поливинилацетальдиэтиламиноацетат.
Тип, степень замещения и молекулярная масса нерастворимых в воде полимеров может зависеть от растворимости активного ингредиента в воде или спирте, необходимого уровня замедленного высвобождения и т.п. Нерастворимые в воде полимеры могут быть использованы либо в отдельности, либо в комбинации. Они могут быть дополнительно включены в гидрогенизированное масло, стеариновую кислоту или цетанол в качестве покрывающего вспомогательного средства и среднецепочечный триглицерид, триацетин, триэтилцитрат или цетанол в качестве пластификатора.
В соответствии с некоторыми вариантами осуществления состав с замедленным высвобождением представляет собой таблетку или гранулу матричного типа. Активные ингредиент может быть покрыт до 3 различных типов полимеров. Эти три различные типы полимеров могут включать в себя: 1) нерастворимый в воде полимер, такой как этилцеллюлоза; 2) не зависимый от рН гелеобразующий полимер, такой как гидроксипропилметилцеллюлоза; и 3) рН-зависимый гелеобразующий полимер, такой как альгинат натрия. Эти три различных типа полимеров могут быть использованы вместе для замедления скорости высвобождения лекарственных средств.
Лекарственные формы: свойства высвобождения
Составы с замедленным высвобождением могут достигать степени замедленного эффекта. В то же время содержание и/или биодоступность активного ингредиента могут варьировать на основании ряда факторов, таких как, например, окно всасывания, носители или наполнители, используемые в составе, способ доставки состава и/или время прохождения активного ингредиента через желудочно-кишечный тракт пациента.
Лекарственный препарат может содержать по меньшей мере одну часть с замедленным высвобождением для осуществления функции замедленного высвобождения и одну часть немедленного высвобождения для осуществления функции немедленного высвобождения. В соответствии с определенными вариантами осуществления в случае, если лекарственный препарат находится в единичной лекарственной форме, он может находиться в форме таблеток, образованных из смеси гранул с замедленным высвобождением, содержащих часть с замедленным высвобождением, и гранул с немедленным высвобождением, содержащих часть с немедленным высвобождением, капсульного препарата, полученного с помощью заполнения капсулы гранулами с замедленным высвобождением и гранулами с немедленным высвобождении, или таблеток с прессованным покрытием, в которых наружный слой, состоящий из части с немедленным высвобождением, образуется на внутреннем слое, содержащим часть с замедленным высвобождением. В то же время ограничение в отношении вышеуказанных вариантов осуществления отсутствует.
Более того, отсутствуют определенные ограничения в отношении состояния удерживания лекарственного средства в композиции или в части с немедленным высвобождением или части с замедленным высвобождением; соединение может быть диспергировано однородно в композиции, часть с немедленным высвобождением или часть с замедленным высвобождением, или может содержать только в одной части композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может содержаться таким образом, что имеет место градиент концентрации.
Часть с замедленным высвобождением в композиции в соответствии с настоящим изобретением содержит по меньшей мере одно не зависимое от рН полимерное вещество или одно рН-зависимое полимерное вещество для контроля высвобождения лекарственного средства.
Не зависимое от рН полимерное вещество, используемое в данном документе, может представлять собой полимерное вещество, состояние заряда которого крайне незначительно изменяется в условиях рН, как правило, встречающихся в желудочно-кишечном тракте, в частности, от рН 1 до рН 8. Это означает, например, полимерное вещество, которое не имеет функциональных групп, изменения состояния заряда которых зависят от рН, например, основных функциональных групп, таких как аминогруппы, или кислотных функциональных групп, таких как группы карбоновых кислот. Следует обратить внимание, что не зависимое от рН полимерное вещество может быть включено с целью придания композиции по настоящему изобретению функции замедленного высвобождения, но также может быть включено для другой цели. Более того, не зависимое от рН полимерное вещество, используемое в настоящем изобретении, может быть водорастворимым или может набухать в воде или растворяться в воде с образованием геля.
Примеры нерастворимых в воде не зависимых от рН полимерных веществ включают в себя без ограничения эфиры целлюлозы, сложные эфиры целлюлозы и сополимеры метакриловой кислоты и акриловой кислоты (торговое наименование Eudragit, производится Rohm GmbH & Co. KG, Дармштадт, Германия). Примеры включают в себя без ограничения эфиры алкилэфиры целлюлозы, такие как этилцеллюлоза (торговое наименование Ethocel, производится Dow Chemical Company, США), этилметилцеллюлоза, этилпропилцеллюлоза и изопропилцеллюлоза и бутилцеллюлоза, аралкилэфиры целлюлозы, такие как бензилцеллюлоза, эфиры цианоалкилцеллюлозы, такие как цианоэтилцеллюлоза, сложные эфиры целлюлозы и органических кислот, такие как ацетобутират целлюлозы, ацетат целлюлозы, пропионат целлюлозы или бутират целлюлозы и ацетопропионат целлюлозы, сополимеры этилакрилата и метилметакрилата (торговое наименование Eudragit NE, производится Rohm GmbH & Co. KG, Дармштадт, Германия) и сополимер аминоалкилметакрилата RS (торговые наименования Eudragit RL, Eudragit RS). Отсутствуют определенные ограничения в отношении среднего диаметра частиц нерастворимого в воде полимера, используемого в настоящем изобретении, однако обычно чем ниже этот средний диаметр частиц, тем эффективнее функциональные характеристики, при этом средний диаметр частиц предпочтительно составляет от 0,1 до 100 мкм, более предпочтительно от 1 до 50 мкм, особенно предпочтительно от 3 до 15 мкм, наиболее предпочтительно от 5 до 15 мкм. Более того, примеры водорастворимых или набухающих в воде не зависимых от рН полимерных веществ включают в себя без ограничения полиэтиленоксид (торговое наименование Polyox, производится Dow Chemical Company, молекулярная масса от 100000 до 7000000), гидроксипропилцеллюлоза с низкой степенью замещения (торговое наименование L-HPC, производится Shin-Etsu Chemical, Япония), гидроксипропилцеллюлоза (торговое наименование НРС, производится Nippon Soda, Co., Ltd, Япония), гидроксипропилметилцеллюлоза (торговые наименования Metolose 60SH, 65SH, 90SH, производятся Shin-Etsu Chemical, Япония) и метилцеллюлоза (торговое наименование Metolose SM, производится Shin-Etsu Chemical, Япония).
В соответствии с некоторыми вариантами осуществления в композиции может содержаться одно не зависимое от рН полимерное вещество или может содержаться несколько не зависимых от рН полимерных веществ. Не зависимое от рН полимерное вещество, при использовании в соответствии с вариантами осуществления, описанными в данном документе, может представлять собой нерастворимое в воде полимерное вещество, наиболее предпочтительно этилцеллюлозу, сополимер этилакрилата и метилметакрилата (торговое наименование Eudragit NE) или сополимер аминоалкилметакрилата RS (торговое наименование Eudragit RL, Eudragit RS). Особенно предпочтительным является одно из этилцеллюлозы и сополимера аминоалкилметакрилата RS. Наиболее предпочтительной является этилцеллюлоза. Отсутствуют определенные ограничения в отношении количества не зависимого от рН полимерного вещества, содержащегося в композиции; это количество можно регулировать подходящим образом в соответствии с целью, такой как контролируемое замедленное высвобождение лекарственного средства.
рН-Зависимое полимерное вещество, которое может быть использовано в вариантах осуществления, описанных в данном документе, может представлять собой полимерное вещество, состояние заряда которого изменяется в условиях рН, как правило, встречающихся в желудочно-кишечном тракте, в частности, от рН 1 до рН 8. Это означает, например, полимерное вещество, имеющее функциональные группы, изменения состояния заряда которых зависят от рН, например, основных функциональных групп, таких как аминогруппы, или кислотных функциональных групп, таких как группы карбоновых кислот. рН-Зависимые функциональные группы рН-зависимого полимерного вещества предпочтительно представляют собой кислотные функциональные группы, при этом рН-зависимое полимерное вещество наиболее предпочтительно имеет группы карбоновых кислот.
рН-Зависимое полимерное вещество, используемое в настоящем изобретении, может быть водорастворимым или может набухать в воде или растворяться в воде с образованием геля. Примеры рН-зависимых полимерных веществ, используемых в настоящем изобретении, включат в себя без ограничения кишечнорастворимые полимерные вещества. Примеры кишечнорастворимых полимерных веществ включают в себя без ограничения сополимеры метакриловой кислоты и метилметакрилата (Eudragit L100, Eudragit S100, производятся Rohm GmbH & Co. KG, Дармштандт, Германия), сополимеры метакриловой кислоты и этилакрилата (Eudragit L100-55, Eudragit L30D-55, производятся Rohm GmbH & Co. KG, Дармштадт, Германия), гидроксипропилметилцеллюлозы фталат (НР-55, НР-50, производится Shin-Etsu Chemical, Япония), гидроксипропилметилцеллюлозы ацетата сукцинат (AQOAT, производится Shin-Etsu Chemical, Япония), карбоксиметилцеллюлозу (СМЕС, производится Freund Corporation, Япония) и ацетатфталат целлюлозы.
Примеры рН-зависимых полимерных веществ, которые набухают в воде или растворяются в воде с образованием геля, включают в себя без ограничения альгиновую кислоту, пектин, карбоксивиниловый полимер и карбоксиметилцеллюлозу. В соответствии с настоящим изобретением в композиции может содержаться одно рН-зависимое полимерное вещество или может содержаться несколько рН-зависимых полимерных веществ. рН-Зависимое полимерное вещество, используемое в настоящем изобретении, предпочтительно представляют собой кишечнорастворимое полимерное вещество, более предпочтительно сополимер метакриловой кислоты и этилакрилата, сополимер метакриловой кислоты и метилметакрилата, гидроксипропилметилцеллюлозы фталат или гидроксипропилметилцеллюлозы ацетата сукцинат, особенно предпочтительно сополимер метакриловой кислоты и этилакрилата.
При использовании рН-зависимых полимерных веществ в способе получения композиции в соответствии с настоящим изобретением, коммерчески доступный продукт порошкообразного типа или гранулярного типа или суспензионного типа, в котором рН-зависимое полимерное вещество было диспергировано в растворителе заранее, может быть использован, как есть, или такой коммерчески доступный продукт может быть использован диспергированным в воде или органическом растворителе. Чем ниже диаметр частицы рН-зависимого полимерного вещества, тем более эффективными являются функциональные характеристики, при этом рН-зависимое полимерное вещество предпочтительно относится к порошкообразному типу. В случае сополимера метакриловой кислоты и этилакрилата примером является Eudragit L100-55. Отсутствуют определенные ограничения в отношении среднего диаметра частиц рН-зависимого полимерного вещества, используемого в настоящем изобретении, однако средний диаметр частиц предпочтительно составляет от 0,05 до 100 мкм, более предпочтительно от 0,05 до 70 мкм, наиболее предпочтительно от 0,05 до 50 мкм. Более того, отсутствуют определенные ограничения в отношении количества рН-зависимого полимерного вещества, например, в случае кишечнорастворимого полимерного вещества количество, как правило, составляет от 1 до 90 массовых частей, предпочтительно от 1 до 70 массовых частей, более предпочтительно от 5 до 60 массовых частей, особенно предпочтительно от 10 до 50 массовых частей, исходя из 100 массовых частей композиции.
Лекарственный препарат в соответствии с вариантами осуществления, описанными в данном документе, может дополнительно содержать любое из различных вспомогательных средств, таких как любое из фармакологически приемлемых разбавителей, скользящих веществ, связующих веществ и дезинтегрантов, а также консервантов, красителей, подсластителей, пластификаторов, пленкообразующих средств и т.д., при необходимости. Примеры разбавителей включат в себя без ограничения лактозу, маннит, двухзамещенный фосфат кальция, крахмал, прежелатинизированный крахмал, кристаллическую целлюлозу, неуплотненный коллоидный диоксид кремния, синтетический силикат алюминия, метасиликат алюмината магния и т.д. Примеры скользящих веществ включают в себя без ограничения стеарат магния, стеарат кальция, тальк, стеарилфумарат натрия и т.д. Примеры связующих веществ включают в себя без ограничения гидроксипропилцеллюлозу, метилцеллюлозу, карбоксиметилцеллюлозу натрия, гидроксипропилметилцеллюлозу, поливинилпирролидон и т.д. Примеры дезинтегрантов включают в себя без ограничения карбоксиметилцеллюлозу, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, карбоксиметилкрахмал натрия, гидроксипропилцеллюлозу с низкой степенью замещения и т.д. Примеры консервантов включают в себя без ограничения сложные эфиры параоксибензойной кислоты, хлорбутанол, бензиловый спирт, фенетиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту и т.д. Предпочтительные примеры красителей включают в себя без ограничения нерастворимые в воде красочные пигменты, природные пигменты (например, бета-каротин, хлорофилл, оксид железа красный), оксид железа желтый, оксид железа красный, оксид железа желтый и т.д. Предпочтительные примеры подсластителей включают в себя без ограничения сахарин натрия, дикалия глицирризат, аспартам, стевию или т.п. Примеры пластификаторов включают в себя без ограничения сложные эфиры глицерина и жирных кислот, триэтилцитрат, пропиленгликоль, полиэтиленгликоль или т.п. Примеры пленкообразующих средств включают в себя без ограничения гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу и т.д.
Способы получения
С целью получения вариантов осуществления, описанных в данном документе, могут быть использованы один стандартных способ или комбинация стандартных способов. Например, при получении содержащих лекарственное средство гранул в виде части с замедленным высвобождением или части с немедленным высвобождением основной операцией является гранулирование, однако его можно комбинировать с другими операциями, такими как смешивание, сушка, просеивание и классификация. В качестве способа гранулирования, например, осуществляют способ влажного гранулирования, в котором связующее вещество и растворитель добавляют к порошку и выполняют гранулирование, способ сухого гранулирования, в котором порошок прессуют и выполняют гранулирование, способ гранулирования плавлением, в котором связующее вещество, которое плавится при нагревании, добавляют и нагревают и выполняют гранулирование, или могут быть использованы подобные способы.
Кроме того, в соответствии со способом гранулирования может быть использован действующий способ, такой как способ гранулирования смешиванием с помощью с помощью планетарной мешалки, винтовой мешалки или т.п., способ высокоскоростного гранулирования смешиванием с помощью мешалки Henschel, мешалки Super или т.п., способ гранулирования экструзией с помощью цилиндрического гранулятора, ротационного гранулятора, винтового экструзионного гранулятора, брикетировочного гранулятора или т.п., способ влажного гранулирования с большим усилием сдвига, способ гранулирования в псевдоожиженном слое, способ гранулирования с последующим прессованием, способ гранулирования раздавливанием или способ гранулирования распылением. После гранулирования можно осуществлять сушку с помощью сушилки, псевдоожиженного слоя и т.п., дробление и просеивание с получением гранул или мелкодисперсных гранул для применения. Более того, растворитель для гранулирования может быть использован при получении композиции в соответствии с настоящим изобретением. Отсутствуют определенные ограничения в отношении такого растворителя для гранулирования, который может представлять собой воду или любой из органических растворителей, например, воду, низший спирт, такой как метанол или этанол, кетон, такой как ацетон или метилэтилкетон, метиленхлорид или их смеси.
В случае гранул с замедленным высвобождением, содержащихся в вариантах осуществления, по меньшей мере одно лекарственное средство и по меньшей мере одно, выбранное из не зависимых от рН полимерных веществ и рН-зависимых полимерных веществ, смешивают вместе, разбавитель и связующее вещество добавляют при необходимости, и выполняют гранулирование с получением гранулированного вещества. Полученное гранулированное вещество сушат с помощью полочной сушилки, сушилки псевдоожиженного слоя и т.п., и осуществляют просеивание с помощью мельницы или осциллятора, в результате чего могут быть получены гранулы с замедленным высвобождением. В качестве альтернативы в качестве способа получения гранул с замедленным высвобождением в настоящем изобретении можно добавлять по меньшей мере одно лекарственное средство, по меньшей мере одно выбранное из не зависимых от рН полимерных веществ и рН-зависимых полимерных веществ, и, при необходимости, разбавитель и связующее вещество с помощью пресса для сухого прессования или роликового пресса или таблеточной машины для заполнения, и осуществлять прессование в форме при смешивании, а затем осуществлять гранулирование с помощью дробления до подходящего размера. Гранулированное вещество, полученное с помощью такого гранулятора, может быть использовано, как есть, в виде гранул или мелкодисперсных гранул в соответствии с настоящим изобретением или может быть дополнительно измельчено с помощью электрической мельницы, валикового гранулятора, скоростной мельницы и просеяно с получением гранул с замедленным высвобождением. Следует обратить внимание, что гранулы с замедленным высвобождением также могут быть получены в виде гранул с замедленным высвобождением.
Прессованный в форме препарат может быть получен в виде содержащей лекарственное средство части с замедленным высвобождением или части с немедленным высвобождением или в виде композиции, описанной в данном документе, с помощью одного стандартного способа или комбинации стандартных способов. Например, используют по меньшей мере одно лекарственное средство, по меньшей мере одно выбранное из не зависимых от рН полимерных веществ и рН-зависимых полимерных веществ, разбавитель, такой как маннит или лактоза, связующее вещество, такое как поливинилпирролидон или кристаллическая целлюлоза, дезинтегрант, такой как кармеллоза натрия или кросповидон, и скользящее вещество, такое как стеарат магния или тальк, и выполняют таблетирование с помощью стандартного способа, при этом может быть получен прессованный в форме препарат. В этом случае таблетирование представляет собой основную операцию в способе получения прессованного в форме препарата, однако его можно комбинировать с другими операциями, такими как смешивание, сушка, покрытие сахарной оболочкой и покрытие оболочкой.
Примеры способа таблетирования включают в себя без ограничения прямое прессование в форме, при котором по меньшей мере одно лекарственное средство и фармакологически приемлемые вспомогательные средства смешивают вместе и затем смесь непосредственно прессуют в форме в таблетки в таблеточной машине, а также сухое прессование гранул или влажное прессование гранул, при которых гранулы с замедленным высвобождением или гранулы с немедленным высвобождением в соответствии с настоящим изобретением подвергаются прессованию в форме после добавления скользящего вещества или дезинтегранта, при необходимости. Отсутствуют определенные ограничения в отношении таблеточной машины, используемые в прессовании в форме; например, могут быть использованы одноштамповая таблеточная машина, роторная таблеточная машина или таблеточная машина с прессованием покрытия.
Содержащие лекарственное средство гранулы с замедленным высвобождением или гранулы с немедленным высвобождением или прессованный в форме препарат в соответствии с вариантами осуществления в данном документе, могут быть использованы, как есть, в виде гранул или таблетки в виде композиции, однако также могут быть подвергнуты дополнительной обработке с получением композиции. Например, прессованный в форме препарат или гранулы могут быть покрыты пленочным покрытием с помощью вещества на пленочной основе, такого как этилцеллюлоза, казеин, метилцеллюлоза, гидроксипропилметилцеллюлоза, сополимер метакриловой кислоты L, ацетатфталат целлюлозы, шеллак или т.п., или покрыты сахарным покрытием с помощью жидкости для покрытия сахарной оболочкой, содержащей сахарозу, сахарный спирт, порошок аравийской камеди, тальк или т.п., при этом образуются таблетки с пленочным покрытием или таблетки с сахарным покрытием. Одним растворителем в этой методике покрытия может быть очищенная вода, однако также может быть использован органический растворитель, такой как спирт, кетон, эфир или хлорированный углеводород, или их смесь. Например, этанол, ацетон, метиленхлорид или т.п. могут быть использованы в качестве органического растворителя. Кроме того, в качестве установки для нанесения покрытий может быть использована установка, обычно используемая в методиках покрытия для получения лекарственных препаратов, при этом примеры включают установку для покрытия распылением, в которой покрытие может выполняться с помощью распыления жидкости для покрытия или т.п., и роторный гранулятор с псевдоожиженным слоем для нанесения слоев.
В случае получения капсульных препаратов капсульные препараты могут быть получены с помощью заполнения гранул с замедленным высвобождением или гранул с немедленным высвобождением, описанных выше, или минитаблеток в твердые желатиновые капсулы или капсулы НРМС с помощью автоматической машины для заполнения капсул. В качестве альтернативы в случае препаратов для введения через зонд или в виде сухого сиропа, который используют смешанным с водой или подобным при приеме, гранулы с замедленным высвобождением или гранулы с немедленным высвобождением, описанные выше, можно смешивать с загустителем или диспергирующим средством с целью диспергирования этих гранул, затем смесь превращают в гранулы или таблетки. Помимо этого, жидкость или гель могут быть получены с помощью воды и веществ, выбранных из диспергирующих средств, эмульгаторов, загустителей, консервантов, регуляторов рН, подсластителей, ароматизаторов, отдушек и т.д. В то же время в отношении других способов получения отсутствуют ограничения в отношении вышеуказанного.
Для того, чтобы варианты осуществления, описанные в данном документе, можно было более полно понять, далее изложены примеры. Следует понимать, что эти примеры представлены только для иллюстративных целей и не предполагают ограничительного характера.
Примеры
Следующие аббревиатуры могут быть использованы в любых примерах.
All: аллил
ВОР: (бензотриазол-1-илокси)трис(диметиламино)фосфоний гексафторфосфат
DMT: 4,4'-диметокситритил
(DMTO-:
Bz: бензоил
ib: изобутирил
Основание Хунига: i-Pr2NEt (диизопропилэтиламин)
АллилОН: аллиловый спирт
OAll: -ОСН2СНСН2
ACN: ацетонитрил
All: -СН2СНСН2
2-НитроВnВr: 2-нитробензилбромид
Bz: бензоил
ib: изобутирил
i-Pr: изопропил
СЕ: цианоэтил
DEAD: диэтилазодикарбоксилат
DIAD: диизопропилазодикарбоксилат
DCM: дихлорметан
DDTT: N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамид
DMOCP: 2-хлор-5,5-диметил-1,3,2-диоксафосфинан 2-оксид
TBS: t-бутилдиметилсилил
3Н-бензо[с][1,2]дитиол-3-он:
Пример 1 - Синтез соединения 1а
Полная схема такого синтеза доступна на фиг. 1.
Стадия А
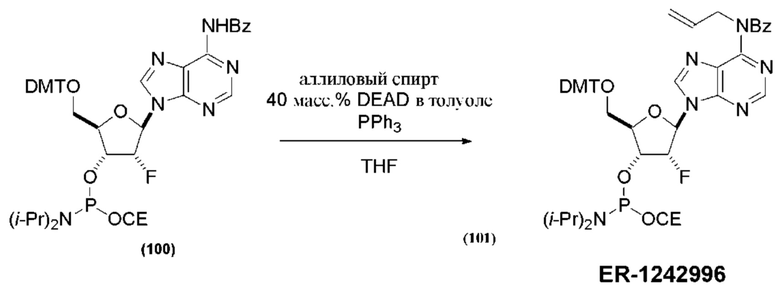
К смеси (2R,3R,4R,5R)-5-(6-бензамидо-9Н-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ил (2-цианоэтил) диизопропилфосфорамидита (соединение 100) (смесь фосфорных диастереомеров; 80,0 г, 91,332 ммоль, 1 экв., ChemGenes Corporation № в каталоге ANP-9151), аллилового спирта (9,63 мл, 142 ммоль, 1,55 экв.) и трифенилфосфина (38,3 г, 146 ммоль, 1,60 экв.) в THF (1,1 л) добавляли DEAD (40 масс. % раствор в толуоле; 54,2 мл, 137 ммоль, 1,5 экв.) при температуре окружающей среды. Перемешивание продолжали при температуре окружающей среды и за реакцией наблюдали при помощи LC/MS. После завершения (19 ч) смесь концентрировали в вакууме (35°С) и полученную смесь очищали методом колоночной хроматографии на силикагеле (800 г × 2 колонки, 40 - 60% EtOAc в н-гептане, буферизированном 0,5% триэтиламином) с получением соединения 101 в виде белой пены (84,2 г, количественный выход, смесь фосфорных диастереомеров).
1Н ЯМР (3:2 смесь фосфорных диастереомеров, 400 МГц, CDCl3) δ 1.14-1.21 (m, 12 Н) 2.40 (t, J=6.2 Гц, 1.2Н) 2.59 (t, J=6.2 Гц, 0.8Н) 3.27 (d, J=8.6 Гц, 1Н) 3.52-3.66 (m, 5Н) 3.78 (s 2.4Н) 3.79 (s 3.6Н) 4.28-4.34 (m, 1Н) 4.84-4.96 (m, 0.4Н) 4.99 (d, J=5.5 Гц, 2Н) 4.95-5.10 (m, 0.6Н) 5.05 (d, J=10.9 Гц, 1Н) 5.22 (br d, J=17.6 Гц, 1H) 5.64 (br d, J=53.2 Гц, 0.6H) 5.70 (br d, J=51.6 Гц, 0.4H) 5.96-6.75 (m, 1H) 6.20 (d, J=16.0 Гц, 0.6H) 6.24 (d, 1=17.2 Гц, 0.4H) 6.74-6.79 (m, 4H) 7.02-7.06 (m, 2H) 7.17-7.24 (m, 8H) 7.32-7.34 (m, 2H) 7.41-7.44 (m, 2H) 8.11 (s, 1H) 8.52 (s, 0.4H) 8.54 (s, 0.6H).
Стадия В
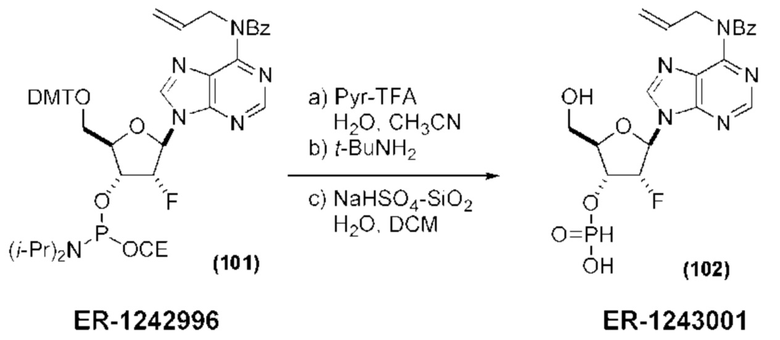
К раствору соединения 101 (3,00 г, 3,28 ммоль, 1 экв.) в ацетонитриле (30 мл) добавляли воду (0,118 мл, 6,55 ммоль, 2,0 экв.) и пиридиновую трифторацетатную соль (0,759 г, 3,93 ммоль, 1,2 экв.). После перемешивания при температуре окружающей среды в течение 1 минуты добавляли трет-бутиламин (14,5 г, 21,0 мл, 0,20 моль, 60 экв.). После завершения отщепления цианоэтильной группы (наблюдали при помощи LC/MS) реакционную смесь концентрировали в вакууме и азеотропировали дважды ацетонитрилом. Неочищенную смесь растворяли в DCM (45,0 мл) и обрабатывали водой (0,118 мл, 6,55 ммоль, 2,0 экв.) и NaHSO4-SiO2 (1,18 г, 6,55 ммоль, 2 экв.) при температуре окружающей среды. После завершения отщепления DMT группы (наблюдали при помощи LC/MS, приблизительно 1 час), реакционную смесь фильтровали и дважды промывали DCM/MeOH (9/1, 20 мл). Объединенные фильтраты концентрировали в вакууме и обрабатывали 1:1 смесью н-гептана/толуола (~30 мл). Верхний слой удаляли декантацией. Ту же операцию еще раз повторяли при помощи н-гептана/толуола (1/1, 30 мл) и нижний слой азеотропировали дважды ацетонитрилом с получением соединения 102 (100% предположительного теоретического выхода). Продукт использовали на следующей стадии без дополнительной очистки.
Стадия С
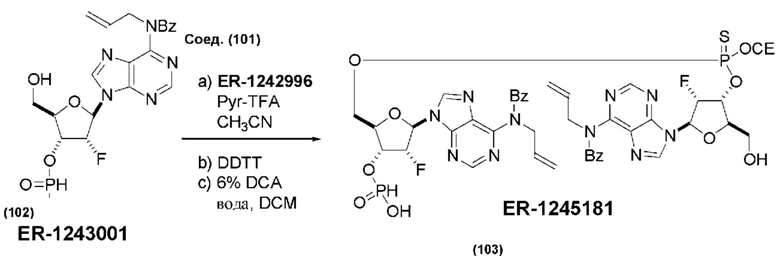
К смеси соединения 102 (1,56 г, 3,27 ммоль, 1 экв.; и соединения 101 (3,00 г, 3,28 ммоль, 1 экв.) в ацетонитриле (30 мл) добавляли пиридиновую трифторацетатную соль (азеотропично сушили с пиридином; 0,760 г, 3,94 ммоль, 1,25 экв.). Через 5 минут добавляли DDTT (0,840 г, 4,09 ммоль, 1,30 экв., ChemGenes Corporation № в каталоге RN-1588) и после завершения сульфурирования (наблюдали при помощи LC/MS) реакционную смесь концентрировали в вакууме. Остаток растворяли в DCM (30 мл) и обрабатывали водой (0,57 мл, 32 ммоль, 10 экв.) и 6% дихлоруксусной кислотой (1,56 мл, 18,9 ммоль, 6,0 экв.) в DCM (30 мл). Через 20 минут реакцию гасили пиридином (20 мл) и конценрировали в вакууме. Остаток азеотропировали с пиридином с получением соединения 103 (3,22 г, 100% предположительного теоретического выхода). Продукт использовали на следующей стадии без дополнительной очистки.
Стадия D
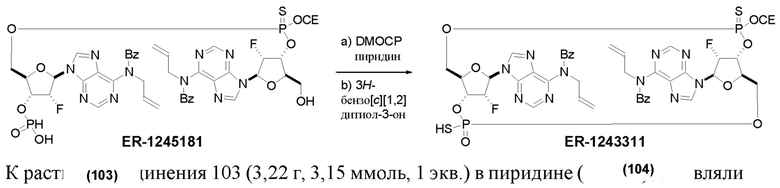
DMOCP (1,45 г, 7,88 ммоль, 2,50 экв.) при температуре окружающей среды. После завершения макроциклизации (наблюдали при помощи LC/MS) добавляли воду (1,7 мл, 94,5 ммоль, ×10 кратно по отношению к DMOCP), а затем 3Н-бензо[с][1,2]дитиол-3-он (0,795 г, 4,73 ммоль, 1,5 экв.). После завершения сульфурирования (приблизительно 40 минут) реакционную смесь частично концентрировали в вакууме до приблизительно 15 мл и выливали в смесь насыщенного водного NaHCO3 (50 мл) и воды (30 мл). Через 10 мин перемешивания при температуре окружающей среды смесь экстрагировали 1:1 смесью EtOAc/МТВЕ (60 мл × 3 раза). Органические слои объединяли, промывали солевым раствором (25 мл), сушили над Mg2SO4 и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (0-20% МеОН в DCM) с получением соединения 104 (3,31 г, 3,20 ммоль, 100% предположительного теоретического выхода) в виде коричневого масла. Продукт использовали на следующей стадии без дополнительной очистки.
Стадия Е
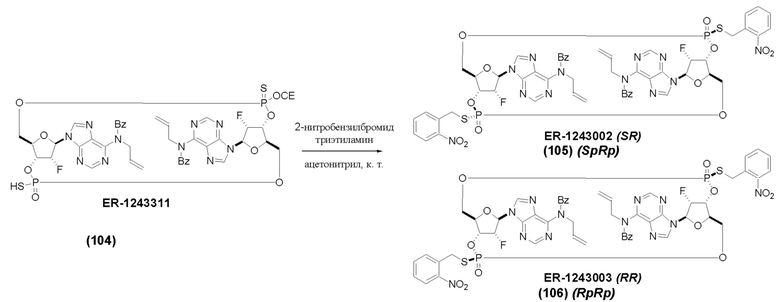
К раствору соединения 104 (3,31 г, 3,20 ммоль, 1 экв.) в ацетонитриле (66,2 мл) добавляли 2-нитробензилбромид (2,42 г, 11,2 ммоль, 3,50 экв.) и триэтиламин (1,78 мл, 12,8 ммоль, 4,00 экв.). После завершения реакции (наблюдали при помощи LC/MS, приблизительно 20 часов при температуре окружающей среды) реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (60% этилацетата/н-гептана до 100% этилацетата) с получением 0,568 г продукта в виде смеси фосфорных диастереомеров. Разделением диастереомеров методом препаративной HPLC получали соединение 105 (SR изомер; 0,225 г, 0,180 ммоль, 5,6% общий выход из соединения 101) и соединение 106 (RR изомер; 0,187 г, 0,149 ммоль, 4,7% общий выход из соединения 1).
Соединение 105 (SpRp) 1Н ЯМР (400 МГц, CDCl3) δ=8.63 (s, 1H), δ=8.61 (s, 1H), 8.04-8.00 (m, 2Н), 7.99 (s, 1H), 7.90 (s, 1H), 7.65-7.44 (m, 8Н), 7.40-7.31 (m, 4Н), 7.25-7.21 (m, 4Н), 6.15-5.89 (m, 5Н), 5.61 (dd, J=52.0, 5.1 Гц, 1H), 5.55 (ddd, J=51.2, 4.7, 2.7 Гц, 1Н) 5.51-5.42 (m, 1H), 5.31-5.22 (m, 2H), 5.11 (dd, J=3.9, 9.8 Гц, 2H), 5.04-4.95 (m, 4H), 4.55-4.37 (m, 7H), 4.29-4.12 (m, 3Н)
Соединение 106 (RpRp) 1Н ЯМР (400 МГц, CDCl3) δ=8.65 (s, 2Н), 8.06 (dd, J=1.4, 8.0 Гц, 2Н), 7.98 (s, 2Н), 7.57-7.52 (m, 6Н), 7.47-7.32 (m, 6Н), 7.25-7.21 (m, 4Н), 6.15 (d, J=18.7 Гц, 2Н), 6.09-5.99 (m, 2Н), 5.82-5.76 (m, 2Н), 5.60 (dd, J=51.8, 4.9 Гц, 2Н), 5.27 (dd, J=1.2, 17.2 Гц, 2Н), 5.12 (dd, J=1.0, 10.4 Гц, 2Н), 5.06-4.96 (m, 4Н), 4.55-4.40 (m, 4Н), 4.36-4.24 (m, 4Н), 4.21-4.02 (m, 2Н)
Условия препаративной HPLC:
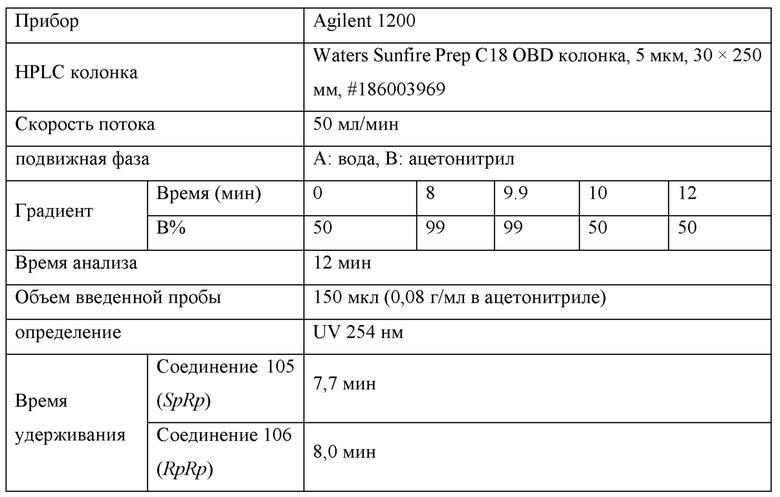
Стадия F
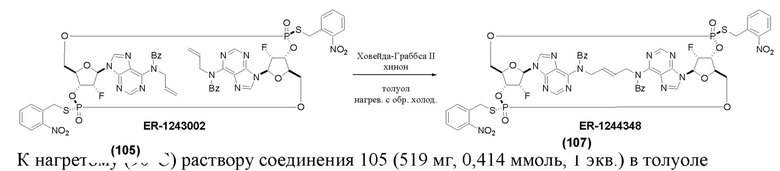
(519 мл) добавляли катализатор Ховейда-Граббса™ 2-го поколения ((1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(орто-изопропоксифенилметилен)рутений; доступный у SIGMA-ALDRITCH® № каталога 569755; CAS 301224-40-8; 91 мг, 0,15 ммоль, 0,35 экв.) и хинон (0,102 мл, 1,243 ммоль, 3,0 экв.). Смесь нагревали до температуры образования флегмы и за ходом реакции наблюдали при помощи LC/MS. Через 3 часа добавляли дополнительный катализатор (91 мг, 0,15 ммоль, 0,35 экв.) и реакцию продолжали еще 3 часа. После охлаждения смесь обрабатывали DMSO (0,59 мл, 8,3 ммоль, 20 экв.) при температуре окружающей среды в течение 15 часов, концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 66% этилацетата в н-гептане до 100% этилацетата) с получением соединения 107 (200 мг, 0.163 ммоль, 39% выход) в виде коричневой сухой пены.
1Н ЯМР (400 МГц, CDCl3) δ=8.19 (s, 1H), 8.12 (dd, J=7.8 Гц, 1.9 Гц, 1H), 8.10 (s, 1H), 8.02 (d, J=8.2 Гц, 1H), 7.89 (s, 1H), 7.63 (br d, J=7.0 Гц, 1H), 7.53-7.41 (m, 10Н), 7.35-7.30 (m, 2Н), 7.25-7.20 (m, 4Н), 6.23 (d, J=17.6 Гц, 1H), 6.14 (d, J=18.8 Гц, 1H), 5.86-5.75 (m, 1H), 5.75 (dt, J=15.3, 5.0 Гц, 1H), 5.67 (dt, J=15.3, 4.7 Гц, 1H), 5.60 (dd, J=52.0, 3.9 Гц. 1H), 5.48 (dd, J=50.4, 3.9 Гц. 1Н) 5.50-5.39 (m, 1H), 4.91-4.64 (m, 4Н), 4.57-4.25 (m, 9Н), 4.15 (d, J=7.03 Гц, 1H), 4.11 (d, J=7.03 Гц, 1H).
Стадия G
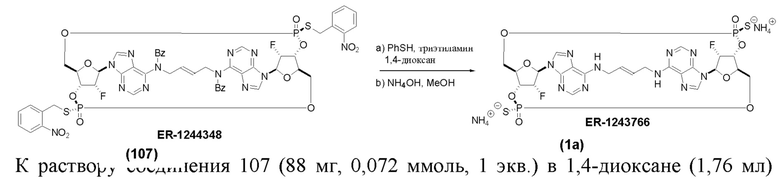
добавляли тиофенол (0,88 мл, 8,55 ммоль, 119 экв.) и триэтиламин (0,88 мл, 6,31 ммоль, 88 экв.). Полученную смесь перемешивали при температуре окружающей среды. После завершения реакции (наблюдали при помощи LC/MS, 13 часов) добавляли метанол (5,28 мл) и 28% гидроксид аммония (3,52 мл) и полученную смесь нагревали до 50°С. После завершения реакции (наблюдали при помощи LC/MS, 5 часов) смесь охлаждали до температуры окружающей среды и полученную коричневатую взвесь фильтровали и промывали водой (15 мл). Фильтрат снова фильтровали с удалением дополнительных твердых веществ. Конечный фильтрат экстрагировали дважды смесью 1:1 толуола и гептана (30 мл). Водный слой концентрировали в вакууме, а затем повторно суспендировали в воде (6 мл). Полученное твердое вещество отфильтровывали и фильтрат подвергали методу препаративной HPLC с получением соединения 1 диаммонийной соли (также называемого соединение 1а) (39 мг, 0,050 ммоль, 70% выход) в виде белого твердого вещества.
Соединение 1a (SpRp, транс) 1Н ЯМР (400 МГц, CD3OD) δ=9.05 (s, 1H), 8.33 (s, 1H), 8.25 (s, 1H), 8.12 (s, 1H), 6.34 (br s, 2Н), 5.88 (br s, 2H), 5.66 (br d, J=51.6 Гц, 1H), 5.59 (br d, J=52.2 Гц, 1H) 5.01 (br s, 2H), 4.68-4.34 (m, 6H), 4.07-3.82 (m, 2H), 3.79-3.55 (m, 2H); 31P ЯМР (162 МГц, CD3OD) δ=55.48 (s, 1P), 55.16 (s, 1P).
Соединение 1a. Условия препаративной HPLC:
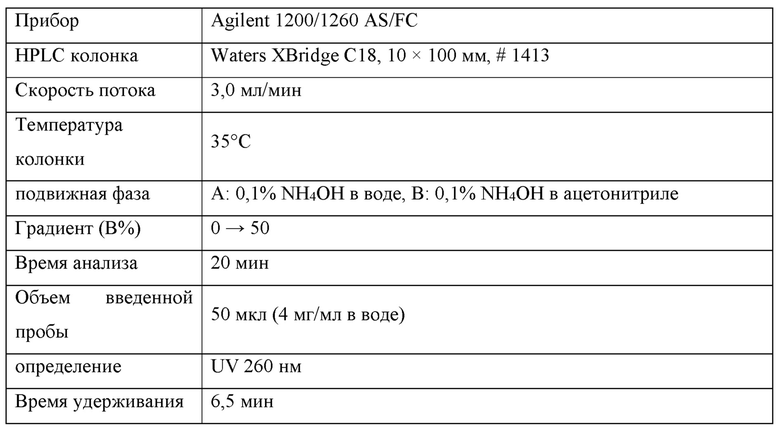
Пример 1.1 - Альтернативный синтез для соединения 1а
Путь альтернативного синтеза соединения 1а изложен на фиг. 2А и фиг. 2В, а также на фиг. 2С и представлен ниже.
Стадия 1
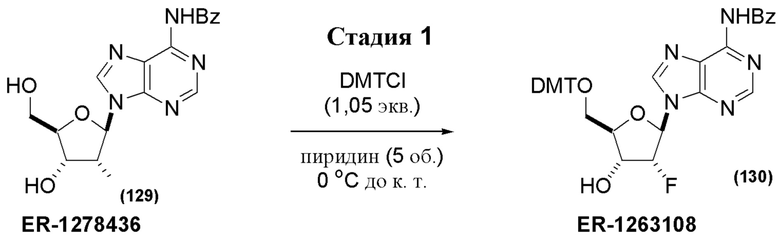
Соединение 129 (570 г, 1,53 мол., 1 масс., 1 об., 1 экв.) растворяли в пиридине (2,85 л, 35,2 мол., 4,89 масс., 5,0 об., 23 экв.). Смесь охлаждали до 2,6°С и обрабатывали 4,4'-диметокситритилхлоридом (DMTCl; 543 г, 1,60 мол., 0,953 масс., 1,05 экв.). Смесь перемешивали при 0-5°С в течение 2 ч, а затем оставляли нагреваться до температуры окружающей среды. За реакцией наблюдали при помощи LC/MS и полное превращение было подтверждено после перемешивания всю ночь. Реакционную смесь охлаждали ниже 5°С и гасили обработкой МеОН (124 мл, 3,05 мол., 0,172 масс., 0,217 об., 2,0 экв.) в течение 15 минут. Смесь совместно выпаривали с тоуолом (2,00 л, 3,04 масс., 3,51 об.) в вакууме, а затем разбавляли смесью EtOAc (2,850 л, 4,5 масс., 5,0 об.) и н-гептана (2,85 л, 3,42 масс., 5,0 об.). Органический слой промывали насыщенным NaHCO3 (9 масс. % раствор в воде; 2,0 л, 3,5 об.). Дополнительное количество EtOAc (2,85 л, 4,5 масс., 5,0 об.) добавляли для полного растворения неочищенного продукта. После перемешивания в течение 5 минут два слоя разделяли. Органический слой промывали водой (2,0 л, 3,5 масс., 3,5 об.). Твердое вещество начинало медленно осаждаться из органического слоя. Водный слой отделяли. Органический слой затем концентрировали до приблизительно 1 об. Неочищенный продукт переводили во взвесь при помощи смеси н-гептана (2,00 л, 2,40 масс., 3,51 об.) и толуола (0,50 л, 0,76 масс., 0,88 об.). После перемешивания в течение 15 минут бледно-желтое твердое вещество собирали вакуумной фильтрацией. Фильтрационный кек последовательно промывали: (1) смесью н-гептана (0,60 л, 0,72 масс., 1,05 об.) и толуола (0,30 л, 0,46 масс., 0,53 об.), а затем (2) н-гептаном (3,00 л, 3,6 масс., 5,26 об.). Твердое вещество сушили без нагревания в течение 30 минут, а затем переносили в ванночки для сушки при 50°С в вакуумной печи всю ночь с получением соединения 130 в виде бледно-желтого твердого вещества (996,7 г, 1,47 мол., 1,75 масс., 97% выход).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.99 (s, 1H), 8.76 (s, 1H), 8.21 (s, 1H), 8.04-8.00 (m, 2Н), 7.64-7.59 (m, 1H), 7.57-7.50 (m, 2Н), 7.41-7.36 (m, 2Н), 7.32-7.15 (m, 7Н), 6.83-6.76 (m, 4Н), 6.31 (dd, J=2.5, 17.0 Гц, 1H), 5.68 (ddd, J=2.3, 4.7, 52.7 Гц, 1H), 4.88-4.77 (m, 1H), 4.26-4.21 (m, 1H), 3.77 (s, 6Н), 3.57 (dd, J=3.1, 10.9 Гц, 1H), 3.43 (dd, J=4.1, 10.7 Гц, 1H), 2.60 (br s, 1Н)
Стадия 1'

Соединение 129 (430 г, 1,15 мол., 1 масс., 1 об., 1 экв.) и имидазол (118 г, 1,73 мол., 0,274 масс., 1,50 экв.) растворяли в DMF (1,72 л, 3,78 масс., 4,0 об.) и полученную смесь охлаждали до 5°С. Добавляли TBS-Cl (191 г, 1,27 мол., 0,444 масс., 1,10 экв.). Смесь перемешивали при от 0 до 11°С в течение 2 ч, оставляли медленно нагреваться до температуры окружающей среды (за прогрессом наблюдали при помощи LCMS). Реакцию завершали через 6 ч после добавления TBS-Cl, еще перемешивали при температуре окружающей среды еще 20 ч. Смесь охлаждали до 2°С и обрабатывали метанолом (93 мл, 74 г, 2,3 мол., 0,17 масс., 0,22 масс., 2,0 экв.) в течение 10 минут. Реакционную смесь разбавляли смесью МТВЕ (1,72 л, 1,23 кг, 2,96 масс., 4,0 об.) и EtOAc (1,72 л, 1,55 кг, 3,60 масс., 4,0 об.), а затем насыщенным NH4Cl (28 масс. % раствор в воде; 2,15 л, 5,0 об.). Твердые вещества медленно начинали выпадать из раствора. Смесь оставляли нагреваться до 24°С и туда добавляли воду (1,08 л, 1,08 кг, 2,5 масс., 2,5 об.) (Т-внутренняя = 22°С). Больше твердых веществ начинали осаждаться из смеси. К смеси добавляли дополнительное количество воды (1,08 л, 1,08 кг, 2,5 масс., 2,5 об.) и МТВЕ (1,40 л, 1,04 кг, 2,4 масс., 3,3 об.). Грязно-белое твердое вещество собирали вакуумной фильтрацией. Реактор промывали водой (320 мл, 0,74 об.), а затем МТВЕ (1,80 л, 1,33 кг, 3,10 масс., 4,19 об.) для переноса любого оставшегося твердого вещества на фильтр. Фильтрационный кек последовательно промывали: (1) водой (1,80 л, 1,80 кг, 4,2 масс., 4,2 об.), (2) водой (1,80 л, 1,80 кг, 4,2 масс., 4,2 об.), (3) смесью МТВЕ (0,90 л, 0,67 кг, 1,5 масс., 2,1 об.) и н-гептана (0,90 л, 0,62 кг, 1,4 масс., 2,1 об.), (4) смесью МТВЕ (0,90 л, 0,67 кг, 1,5 масс., 2,1 об.) и н-гептана (0,90 л, 0,62 кг, 1,4 масс., 2,1 об.). Восстановленное твердое вещество сушили в вакууме при 40°С в течение 2 суток с получением соединения 133 в виде белого твердого вещества (483 г, 0,991 мол., 1,12 масс., 86% выход).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.97 (s, 1H), 8.82 (s, 1H), 8.36 (s, 1H), 8.04-8.00 (m, 2Н), 7.64-7.58 (m, 1H), 7.56-7.51 (m, 2Н), 6.40 (dd, J=2.3, 16.0 Гц, 1H), 5.45 (ddd, J=2.7, 4.3, 53.1 Гц, 1H), 4.75-4.66 (m, 1H), 4.22-4.17 (m, 1H), 4.07 (dd, J=2.3, 11.7 Гц, 1H), 3.91 (dd, J=2.7, 11.7 Гц, 1H), 2.38 (dd, J=2.7, 7.0 Гц, 1H), 0.92 (s, 9Н), 0.11 (s, 3Н), 0.11 (s, 3Н).
Стадия 2

Соединение 130 (993 г, 1,47 мол., 1 масс., 1 об., 1 экв.) и имидазол (150 г, 2,20 мол., 0,151 масс., 1,5 экв.) растворяли в DMF (3,48 л, 3,28 кг, 3,3 масс., 3,5 об.) и смесь охлаждали до 5°С. Добавляли TBS-Cl (244 г, 1,62 мол., 0,245 масс., 1,10 экв.). Реакционную смесь перемешивали при от 0 до 5°С в течение 2 ч, оставляли медленно нагреваться до температуры окружающей среды и наблюдали при помощи LCMS. Через 17 ч добавляли дополнительное количество имидазола (100 г, 1,47 мол., 0,10 масс., 1,0 экв.) и TBS-Cl (111 г, 735 ммоль, 0,112 масс., 0,50 экв.) и перемешивание продолжали при температуре окружающей среды в течение 2 ч и при 35°С в течение 2 ч. Полученную смесь охлаждали до 13,6°С и обрабатывали МеОН (119 мл, 2,94 мол., 2 экв.) в течение 10 минут. В отдельный реактор добавляли лед (5 кг, 5 масс.) и насыщенный NH4Cl (28 масс. % раствор в воде; 5,0 л, 5 об.). Реакционную смесь добавляли в смесь льда/NH4Cl. Грязно-белое твердое вещество сразу начало осаждаться из раствора. К смеси добавляли дополнительно 2 кг льда (2 кг, 2 масс.) и воду (3,0 л, 3 об.). Реакционный сосуд промывали водой (0,50 л, 0,5 об.) и пробу для промывки добавляли к смеси. Н-гептан (2,00 л, 2 об.) добавляли к смеси и перемешивание продолжали в течение 10 минут. Грязно-белое твердое вещество собирали вакуумной фильтрацией. Фильтрационный кек промывали: (1) водой (4,0 л, 4,0 об.), (2) водой (4,0 л, 4,0 об.), (3) н-гептаном (4,0 л, 4,0 об.), (4) н-гептаном (4,0 л, 4,0 об.). Восстановленное твердое вещество сушили в вакууме при 45°С в течение 4 суток с получением соединения 131 в виде грязно-белого твердого вещества (1,095 кг, 1,39 мол., 1,10 масс., 94% выход).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=9.09 (s, 1H), 8.78 (s, 1H), 8.28 (s, 1H), 8.02 (d, J=7.4 Гц, 2Н), 7.63-7.59 (m, 1H), 7.55-7.50 (m, 2Н), 7.37 (d, J=7.1 Гц, 2Н), 7.29-7.17 (m, 7Н), 6.79 (d, J=7.9 Гц, 4Н), 6.29 (dd, J=2.9, 16.2 Гц, 1H), 5.60 (ddd, J=2.7, 3.9, 53.1 Гц, 1H), 4.78 (ddd, J=4.7, 6.4, 15.8 Гц, 1H), 4.26-4.22 (m, 1H), 3.77 (s, 6Н), 3.58 (dd, J=3.1, 10.9 Гц, 1H), 3.26 (dd, J=3.7, 10.7 Гц, 1H), 0.85 (s, 9Н), 0.10 (s, 3Н), 0.02 (s, 3Н)
Стадия 3
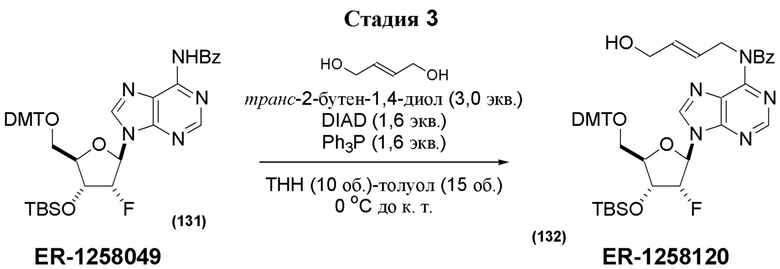
Соединение 131 (1000 г, 1,27 мол., 1 масс., 1 об., 1 экв.) и транс-2-бутен-1,4-диол (олефиновая геометрия подтверждена методом 1H-ЯМР; 335 г, 3,80 мол. 0,335 масс., 3,0 экв.) азеотропировали дважды THF (3,0 л, 3,0 об.). Остаток растворяли в смеси THF (10 л, 10 об.) и толуола (15 л, 15 об.). Трифенилфосфин (432 г, 1.65 мол., 0.432 масс., 1.3 экв.) добавляли, а затем реакционную смесь охлаждали до -5°С. Медленно добавляли DIAD (0,320 л, 1,65 мол., 333 г, 0,333 масс., 0,320 об., 1,3 экв.) в течение 20 минут при поддержании Т-внутренней ниже 5°С. Реакционную смесь перемешивали при 0-5°С в течение 1 ч и наблюдали при помощи LCMS. Ледяную баню удаляли и смесь оставляли нагреваться до к.т. После перемешивания всю ночь (17 ч) добавляли трифенилфосфин (83 г, 0,32 мол., 0,083 масс., 0,25 экв.) и DIAD (62 мл, 0,32 мол., 64 г, 0,064 масс., 0,062 об., 0,25 экв.). После дополнительного 1 ч при к.т. реакционную смесь разбавляли МТВЕ (10 л, 10 об.), дважды промывали полунасыщенным NaCl (18 масс. % раствор в воде; 2×4 л) и концентрировали в вакууме до густого масла. Смесь повторно растворяли в смеси МТВЕ (4,00 л, 4 об.) и н-гептана (0,50 л, 0,5 об.), а затем охлаждали до 0°С. Затравочный кристалл трифенилфосфиноксида добавляли к раствору. Твердые вещества медленно начинали осаждаться из раствора и перемешивали всю ночь. Белое твердое вещество собирали вакуумной фильтрацией и промывали МТВЕ (2 л, 2 об.) для выделения 540 г трифенилфосфиноксида. Фильтрат концентрировали и очищали на Biotage 150L KP-Sil (SiO2 5 кг; предварительно обрабатывали 1% TEA в Hep/etOAc; элюенты: гептан/EtOAc (48 л 33% EtOAc с 1% TEA, 24 л 50% EtOAc с 1% TEA, 24 л 66% EtOAc с 1% TEA) → 100% EtOAc с 1% TEA). За колонкой наблюдали при помощи TLC (2:1 EtOAc/н-гептан). Чистые фракции продукта объединяли и концентрировали в вакууме с получением соединения 132 в виде бледно-белого пенящегося твердого вещества (634 г, содержащего 14 масс. % DIAD полученного побочного продукта, чистого 545 г, 0,63 мол., 50% вычисленный выход). Фракции смеси объединяли и концентрировали в вакууме с получением бледно-желтого пенящегося твердого вещества (750 г), которое подвергали повторной очистке при помощи Biotage 150М HP-Sphere (2,5 кг SiO2; предварительно обрабатывали 1% TEA в Нер/EtOAc; загруженная проба с толуолом, элюенты: Нер/EtOAc/1% ТЕА (12 л 50% EtOAc с 1% TEA, 16 л 66% EtOAc с 1% TEA) → EtOAc с 1% TEA). За колонкой наблюдали при помощи TLC (2/1/0,03 EtOAc/n-hep/TEA). Чистые фракции продукта объединяли и концентрировали в вакууме с получением дополнительного соединения 132 в виде бледно-белого пенящегося твердого вещества (206 г, 0,24 мол., 18% выход).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.58 (s, 1H), 8.10 (s, 1H), 7.43-7.37 (m, 2Н), 7.32-7.28 (m, 2Н), 7.24-7.15 (m, 8Н), 7.03-6.98 (m, 2Н), 6.78-6.73 (m, 4Н), 6.18 (dd, J=2.7, 17.2 Гц, 1H), 5.88 (td, J=5.5, 15.6 Гц, 1H), 5.77 (td, J=5.1, 15.6 Гц, 1H), 5.60 (ddd, J=2.7, 4.3, 53.1 Гц, 1H), 5.03-4.96 (m, 2Н), 4.91 (ddd, J=4.5, 6.6, 16.6 Гц, 1H), 4.18-4.14 (m, 1H), 3.88-3.82 (m, 2Н), 3.78 (s, 6Н), 3.52 (dd, J=2.7, 10.9 Гц, 1H), 3.14 (dd, J=3.5, 10.9 Гц, 1H), 0.85 (s, 9Н), 0.10 (s, 3Н), 0.01 (s, 3Н).
Стадия 4
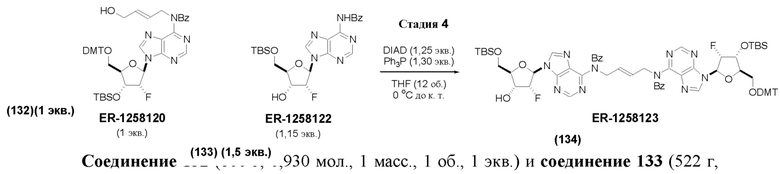 1,07 мол., 0,652 масс., 1,15 экв.) азеотропически сушили при помощи THF (2×3 л, 2×3,8 об.) и повторно растворяли в THF (9,60 л, 8,45 кг, 12,0 об.) при к.т. Трифенилфосфин (317 г, 1,21 мол., 0,396 масс., 1,30 экв.) добавляли и смесь охлаждали ниже -5°С. Добавляли DIAD (226 мл, 1,16 мол., 235 г, 0,294 масс., 0,283 об., 1,25 экв.), Т-внутренняя ниже 7°С. Реакционную смесь оставляли нагреваться до к.т. медленно. За реакцией наблюдали при помощи LCMS. Через 21 ч реакционную смесь концентрировали в вакууме до густого масла, азеотропировали н-гептаном (2,00, 1,37 кг, 1,71 масс., 2,50 об.), а затем повторно растворяли в смеси МТВЕ (2,40 л, 1,78 кг, 2,2 масс., 3,0 об.) и н-гептана (800 мл, 547 г, 0,68 масс., 1,0 об.). Раствор затравляли трифенилфосфиноксидом и охлаждали до 5°С, разбавляли н-гептаном (400 мл, 274 г, 0,34 масс., 0,50 об.) и перемешивали при 5°С в течение 30 минут. Осадок белого твердого вещества собирали вакуумной фильтрацией и промывали 2:1 (объем/объем) смесью МТВЕ и н-гептана (1,8 л) с получением трифенилфосфиноксида (455 г). Фильтрат концентрировали в вакууме и очищали при помощи Biotage 150 L KP-Sil (SiO2 5 кг; предварительно обрабатывали 1% TEA; загружали пробы растворением в толуоле, элюенты: 9:1 гептан/EtOAc (16 л) и 15 TEA, 3.6:1 (46 л), 2:1 (20 л) и 1% TEA, 1:1 (30 л) и 1% TEA и 100% EtOAc (16 л) и 1% TEA). Объединенные чистые фракции продукта концентрировали в вакууме с получением соединения 134 в виде грязно-белой твердой пены (662,2 г). Фракции смеси объединяли и концентрировали в вакууме (480 г). Белое нерастворимое твердое вещество, образованное разбавлением толуолом (300 мл) перед загрузкой в Biotage 150L, удаляли вакуумной фильтрацией. Вещество, растворимое в толуоле, очищали при помощи Biotage 150М HP-Sphere (SiO2 2,5 кг (предварительно обработанное 1% TEA); загрузка пробы при помощи толуола; элюенты: 2:1 гептан/EtOAc (26 л) масс/ 1% TEA, 1:1 (25 л) масс/ 1% TEA, 1:4 (34 л) масс/ 1% TEA). За колонкой наблюдали при помощи TLC (1:1 гептан/EtOAc). Объединенные чистые фракции продукта концентрировали в вакууме с получением дополнительного соединения 134 в виде грязно-белой твердой пены (165,5 г. Всего 662,2+165,5 г=827,7 г, 930 ммоль, 1,03 масс., 67% выход).
1,07 мол., 0,652 масс., 1,15 экв.) азеотропически сушили при помощи THF (2×3 л, 2×3,8 об.) и повторно растворяли в THF (9,60 л, 8,45 кг, 12,0 об.) при к.т. Трифенилфосфин (317 г, 1,21 мол., 0,396 масс., 1,30 экв.) добавляли и смесь охлаждали ниже -5°С. Добавляли DIAD (226 мл, 1,16 мол., 235 г, 0,294 масс., 0,283 об., 1,25 экв.), Т-внутренняя ниже 7°С. Реакционную смесь оставляли нагреваться до к.т. медленно. За реакцией наблюдали при помощи LCMS. Через 21 ч реакционную смесь концентрировали в вакууме до густого масла, азеотропировали н-гептаном (2,00, 1,37 кг, 1,71 масс., 2,50 об.), а затем повторно растворяли в смеси МТВЕ (2,40 л, 1,78 кг, 2,2 масс., 3,0 об.) и н-гептана (800 мл, 547 г, 0,68 масс., 1,0 об.). Раствор затравляли трифенилфосфиноксидом и охлаждали до 5°С, разбавляли н-гептаном (400 мл, 274 г, 0,34 масс., 0,50 об.) и перемешивали при 5°С в течение 30 минут. Осадок белого твердого вещества собирали вакуумной фильтрацией и промывали 2:1 (объем/объем) смесью МТВЕ и н-гептана (1,8 л) с получением трифенилфосфиноксида (455 г). Фильтрат концентрировали в вакууме и очищали при помощи Biotage 150 L KP-Sil (SiO2 5 кг; предварительно обрабатывали 1% TEA; загружали пробы растворением в толуоле, элюенты: 9:1 гептан/EtOAc (16 л) и 15 TEA, 3.6:1 (46 л), 2:1 (20 л) и 1% TEA, 1:1 (30 л) и 1% TEA и 100% EtOAc (16 л) и 1% TEA). Объединенные чистые фракции продукта концентрировали в вакууме с получением соединения 134 в виде грязно-белой твердой пены (662,2 г). Фракции смеси объединяли и концентрировали в вакууме (480 г). Белое нерастворимое твердое вещество, образованное разбавлением толуолом (300 мл) перед загрузкой в Biotage 150L, удаляли вакуумной фильтрацией. Вещество, растворимое в толуоле, очищали при помощи Biotage 150М HP-Sphere (SiO2 2,5 кг (предварительно обработанное 1% TEA); загрузка пробы при помощи толуола; элюенты: 2:1 гептан/EtOAc (26 л) масс/ 1% TEA, 1:1 (25 л) масс/ 1% TEA, 1:4 (34 л) масс/ 1% TEA). За колонкой наблюдали при помощи TLC (1:1 гептан/EtOAc). Объединенные чистые фракции продукта концентрировали в вакууме с получением дополнительного соединения 134 в виде грязно-белой твердой пены (165,5 г. Всего 662,2+165,5 г=827,7 г, 930 ммоль, 1,03 масс., 67% выход).
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.47 (s, 1H), 8.39 (s, 1H), 8.20 (s, 1H), 8.01 (s, 1H), 7.38-7.31 (m, 5H), 7.27-7.19 (m, 6H), 7.14-7.06 (m, 3H), 6.93-6.87 (m, 2H), 6.76 (d, J=8.6 Гц, 4H), 6.26 (dd, J=2.0, 16.0 Гц, 1H), 6.15 (dd, J=2.7, 17.2 Гц, 1H), 5.86 (dd, J=4.7, 15.2 Гц, 1H), 5.80 (dd, J=4.7, 15.2 Гц, 1H), 5.51 (ddd, J=2.7, 4.3, 52.8 Гц, 1H), 5.31 (ddd, J=2.0, 4.3, 52.8 Гц, 1H), 4.87 (d, J=4.7 Гц, 2H), 4.85-4.81 (m, 1H), 4.79 (d, J=4.3 Гц, 2H), 4.71-4.59 (m, 1H), 4.20-4.13 (m, 2H), 4.06 (dd, J=2.7, 11.3 Гц, 1H), 3.90 (dd, J=2.7, 11.7 Гц, 1H), 3.77 (s, 6H), 3.52 (dd, J=3.1, 10.9 Гц, 1H), 3.18 (dd, J=3.9, 10.9 Гц, 1H), 0.92 (s, 9H), 0.84 (s, 9H), 0.10 (s, 3H), 0.09 (s, 6H), 0.07 (s, 3H)
Стадия 5-6
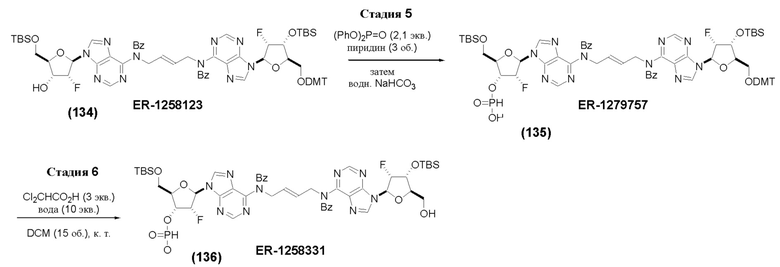
К раствору соединения 134 (410.7 г, 309 ммоль, 1 масс., 1 об., 1 экв.) в пиридине (1,23 л, 1,21 кг, 15,2 мол., 2,9 масс., 3,0 об., 49 экв.) добавляли дифенилфосфит (90 мл, 109 г, 0,46 мол., 0,26 масс., 0,22 об., 1,5 экв.). Реакционную смесь перемешивали при к.т. и наблюдали при помощи LCMS. Через 2 ч (80% превращения) добавляли дополнительное количество дифенилфосфита (29,9 мл, 36,2 г, 155 ммоль, 0,088 масс., 0,073 об., 0,50 экв.). После дополнительного 1 ч добавляли дополнительно дифенилфосфит (6,0 мл, 7,2 г, 31 ммоль, 0,018 масс., 0,015 об., 0,10 экв.) и реакцию продолжали еще 0,5 ч (98% превращения). Реакционную смесь добавляли к смеси насыщенного NaHCO3 (9 масс. % раствор в воде; 2,1 л, 5 об.) и воды (1,0 л мл, 2,5 об.) при поддерживании Т-внутренней 4,7-12°С. Реактор промывали небольшим объемом EtOAc. Перемешивание продолжали при к.т. в течение 30 минут и за реакцией наблюдали при помощи LCMS (100% превращения). Реакционную смесь экстрагировали дважды 1:1 смесью EtOAc и МТВЕ (2×8,2 л, 2×20 об.). Объединенные органические слои промывали водой (4,1 л, 10 об.), концентрировали в вакууме и азеотропировали толуолом (3×4,1 л, 3×10 об.; непрерывная подача) для удаления пиридина с получением соединения 135 (0,55 экв. оставшегося пиридина).
Стадия 6 - Неочищенное соединение 135 растворяли в дихлорметане (3,08 л, 4,07 кг, 9,9 масс., 7,5 об.) при температуре окружающей среды. Добавляли воду (55,7 мл, 0,136 об., 10 экв.), а затем раствор дихлоруксусной кислоты (77 мл, 120 г, 0,93 мол., 0,29 масс., 0,19 об., 3,0 экв.) в DCM (3,08 л, 7,5 об.) при поддержании внутренней Т ниже 25°С. (Превращалось в оранжевый раствор). Через 30 мин добавляли триэтилсилан (Et3SiH; 494 мл, 359 г, 3,09 мол., 0,875 масс., 1,20 об., 10,0 экв.) (Т-внутренняя менялась от 18,2°С до 17°С) и перемешивание продолжали в течение 20 мин. Триэтиламин (431 мл, 313 г, 3,09 мол., 0,762 масс., 1,05 об., 10,0 экв.) добавляли (Т-внутренняя менялась от 17,8°С до 22°С). Смесь концентрировали до 1,55 кг (3,8 масс), повторно растворяли в EtOAc (6,2 л, 5,5 кг, 14 масс., 15 об.), последовательно промывали: (1) водой (1,0 л, 2,5 об.) и насыщенным NaHCO3 (9 масс. % раствор в воде, 0,82 л, 2,0 об.). Неочищенный продукт EtOAc раствор хранили при -20°С всю ночь (0,82 л, 2,0 об.), а на следующий день раствор концентрировали в вакууме при 25°С. Полученную таким образом неочищенную смесь (654 г) растирали в порошок с: (1) н-гептаном (3,01 л, 7,5 об.), (2) смесью н-гептана (2,46 л, 6,0 об.) и толуола (0,82 л, 2,0 об.). Часть раствора (супернатант) декантировали и оставшееся на дне твердое вещество растворяли в ацетонитриле (4,1 л, 10 об.). Смесь концентрировали в вакууме при 25°С и азеотропировали ацетонитрилом дважды с получением соединения 136. Продукт использовали на следующей стадии без очистки (допустимый теоретический 100% выход).
Стадия 7
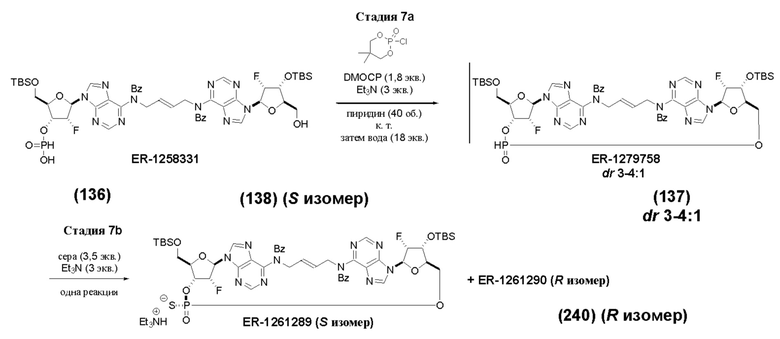
Стадия 7а Соединение 136 (337 г, 309 ммоль, 1 масс., 1 об., 1 экв.) растворяли в безводном пиридине (13,5 л, 13,2 кг, 39 масс., 40 об.) при к.т. Триэтиламин (129 мл, 927 ммоль, 94 г, 0,28 масс., 0,38 об., 3,0 экв.) добавляли, а затем 2-хлор-5,5-диметил-1,3,2-диоксафосфинана 2-оксид (DMOCP; 103 г, 556 ммоль, 0,31 масс., 1,80 экв.). Полученную смесь перемешивали при температуре окружающей среды в течение 30 минут и наблюдали при помощи LCMS (100% превращения) с образованием соединения 137.
Стадия 7b TEA (129 мл, 927 ммоль, 94 г, 0,28 масс., 0,38 об., 3,0 экв.), воду (100 мл, 5,56 мол., 0,30 масс., 0,30 масс., 18 экв.) и серу (34,7 г, 1,08 мол., 0,10 масс., 3,5 экв.) добавляли к вышеуказанной смеси соединения 137. Через 90 минут (100% превращения) добавляли NaHCO3 (9 масс. % раствор в воде; 3,37 л, 10 об.) при поддержании Т-внутренней ниже 30°С (16,6°С-27°С). Полученную смесь фильтровали с удалением солей. Фильтрат концентрировали в смеси в вакууме, разбавляли МТВЕ (5,1 л, 15 об.) и промывали дважды NaCl (30 масс. % раствор в воде; 2×1,35 л, 2×4 об.). Нерастворимые твердые вещества отфильтровывали и фильтрат концентрировали в вакууме и азеотропировали толуолом (4,0 л, 12 об.). Полученное твердое вещество удаляли фильтрацией и неочищенную смесь растворяли в толуоле и очищали при помощи Biotage 150L KP-Sil (SiCh 5 кг; предварительно обрабатывали Нер/EtOAc/ТЕА (1.5/1.5/0.03 CV); элюировали: EtOAc/ТЕА (3/0,03 CV), EtOAc/MeOH/TEA (4/0,2/0,04 CV), EtOAC/MeOH/TEA (2/0,2/0,02CV) 3a колонкой наблюдали при помощи TLC (EtOAC/MeOH/TEA=9/1/0,1). Фракции, содержащие Sp изомер, объединяли и концентрировали в вакууме с получением соединения 138 в виде светло-розового пенящегося твердого вещества (Sp изомер; 154 г, 128 ммоль, 0,46 масс., 41,3% выход). = 9/1/0,1). Фракции, содержащие Rp изомер, объединяли и концентрировали в вакууме с получением соединения 240 в виде светло-розового пенящегося твердого вещества (Rp изомер; 64 г, 53 ммоль, 0,19 масс., 17% выход).
Соединение 138 (Sp изомер):
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.51 (s, 1Н), 8.50 (s, 1Н), 8.22 (s, 1Н), 8.14 (s, 1Н), 7.49-7.44 (m, 2Н), 7.38-7.27 (m, 4Н), 7.25-7.21 (m, 2Н), 7.14 (t, J=7.1 Гц, 2Н), 6.44 (dd, J=2.5, 13.9 Гц, 1H), 6.18 (d, J=15.2 Гц, 1H), 5.78 (td, J=6.3, 15.6 Гц, 1H), 5.69 (td, J=4.7, 15.6 Гц, 1H), 5.56 (dd, J=3.9, 50.8 Гц, 1H), 5.20-5.06 (m, 1H), 4.95-4.79 (m, 4Н), 4.69 (dd, J=4.3, 16.0 Гц, 1H), 4.54-4.38 (m, 3Н), 4.35 (d, J=5.5 Гц, 1H), 4.32-4.29 (m, 1H), 4.05 (dd, J=1.6, 11.7 Гц, 1H), 3.91 (dd, J=3.1, 11.7 Гц, 1H), 3.14-3.06 (m, 6Н), 1.30 (t, J=7.4 Гц, 9Н), 0.91 (s, 9Н), 0.90 (s, 9Н), 0.12 (s, 3Н), 0.08 (s, 3Н), 0.06 (s, 3Н), 0.05 (s, 3Н)
Соединение 240 (Rp изомер):
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.54 (s, 1Н), 8.38 (s, 1H), 8.33 (s, 1H), 8.01 (s, 1H), 7.39-7.09 (m, 10H), 6.39 (dd, J=2.3, 14.1 Гц, 1H), 6.13 (d, J=17.2 Гц, 1H), 5.72 (d, J=3.1 Гц, 2H), 5.68 (dd, J=4.3, 51.2 Гц, 1H), 5.43-5.29 (m, 1H), 5.10-4.96 (m, 3H), 4.90-4.83 (m, 2H), 4.78-4.72 (m, 1H), 4.52 (ddd, J=3.9, 6.6, 17.2 Гц, 1H), 4.44-4.35 (m, 2H), 4.31-4.26 (m, 1H), 4.20-4.12 (m, 2H), 3.87 (dd, J=3.5, 11.7 Гц, 1H), 3.79-3.77 (m, 1H), 3.15-3.09 (m, 6H), 1.33 (t, J=7.4 Гц, 9H), 0.94 (s, 9H), 0.89 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H)
Стадия 8
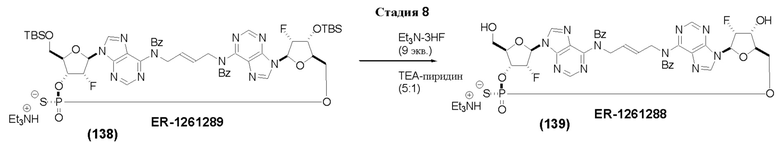
Соединение 138 (221 г, 183 ммоль, 1 масс., 1 об., 1 экв.) растворяли в смеси пиридина (530 мл, 6,56 мол., 519 г, 2,3 масс., 2,4 об.) и TEA (2,65 л, 19,0 мол., 1,93 кг, 8,7 масс., 12 об., 104 экв.). Триэтиламина тригидрофторид (264 мл, 1,62 мол., 262 г, 1,2 масс., 1,2 об., 8,9 экв. в виде комплекса, 27 экв. HF) добавляли и смесь перемешивали при к.т. , при этом за превращением наблюдали при помощи LCMS. Через 3 ч (97% превращения) метокситриметилсилан (TMSOMe; 1,40 л, 10,2 мол., 1,06 кг, 4,8 масс., 6,3 об., 55 экв.) добавляли и перемешивание продолжали в течение 30 минут. Реактор с липким твердым покрытием. Часть раствора (супернатант) декантировали. Твердое вещество растирали в порошок дважды с толуолом (2×2,2 л, 2×10 об.; супернатант декантировали). Неочищенное твердое вещество, оставшееся в реакторе, растворяли в дихлорметане (2,2 л, 10 об.) и промывали NH4Cl (28 масс. % раствор в воде; 2,2 л, 10 об.). Водный слой снова экстрагировали при помощи дихлорметана (2,2 л, 10 об.). Объединенные органические слои промывали смесью NaCl (36 масс. % раствор в воде; 1,1 л, 5 об.) и воды (1,1 л, 5 об.), а затем концентрировали в вакууме с получением соединения 139 в виде коричневой сухой пены (152 г, 155 ммоль, 0,70 масс., 85% выход). Неочищенный продукт переносили на следующую стадию без очистки.
Стадия 9
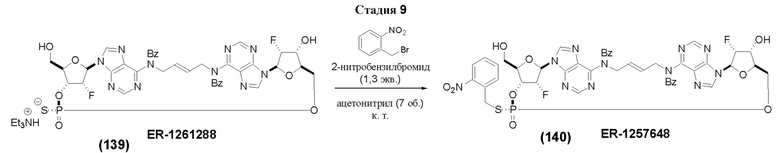
Соединение 139 (150 г, 153 ммоль, 1 масс., 1 об., 1 экв.) азеотропировали с ацетонитрилом (4 л, 27 об.), а затем повторно растворяли в ацетонитриле (1,05 л, 0,83 кг, 5,5 масс., 7,0 об.) при к.т. 2-Нитробензилбромид (44,4 г, 205 ммоль, 0,30 масс., 1,34 экв.) добавляли при к.т. и за реакцией наблюдали при помощи LCMS. Через 23 ч (100% превращения) EtOAc (1,50 л, 10 об.), NH4Cl (28 масс. % раствора в воде; 300 мл, 2 об.) и воду (300 мл, 2 об.) добавляли (рН=6) и полученную смесь частично концентрировали в вакууме при 25°С до массы 1,11 кг. EtOAc (2,25 л, 15 об.) добавляли и смесь перемешивали в течение 5 минут. Два слоя разделяли. Водный слой экстрагировали с этилацетатом (750 мл, 5 об.). Объединенные органические слои последовательно промывали: (1) смесью NaCl (36 масс. % раствор в воде; 300 мл, 2 об.) и воды (300 мл, 2 об.) и (2) водой (600 мл, 4 об.). Органический слой затем концентрировали в вакууме и азеотропировали с н-гептаном (1,50 л, 10 об.). МТВЕ (0,95 л, 6,3 об.) добавляли к неочищенному твердому веществу и смесь нагревали при 40°С. Смесь разбавляли EtOAc (300 мл, 2 об.) и медленно охлаждали до 0°С. Плотное твердое вещество оставляли оседать и супернатант откачивали через фриттовую трубку фильтра. Твердое вещество дважды промывали МТВЕ (2×300 мл, 2×2 об.; супернатант каждый раз откачивали через фриттовую трубку фильтра) и сушили в вакууме при 40°С всю ночь с получением соединения 140 в виде бледно-желтого твердого вещества (156 г). Фильтрат концентрировали в вакууме с получением коричневого масла (17,8 г), которое подвергали очистке при помощи Biotage Snap-Ultra 340 g (элюенты: 0-5% МеОН в EtOAc) с получением дополнительного соединения 140 в виде бледно-желтого твердого вещества (5,8 г). Всего 156 г + 5,8 г = 161,8 г (чистого 152 ммоль, 95% чистоты, 99% выход)
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.46 (s, 1H), 8.15 (s, 1H), 8.10 (s, 1H), 8.09-8.06 (m, 1H), 7.89 (s, 1H), 7.54-7.51 (m, 1H), 7.49-7.45 (m, 4Н), 7.37-7.28 (m, 3Н), 7.24-7.19 (m, 3Н), 7.16-7.11 (m, 2Н), 6.22 (d, J=16.8 Гц, 1H), 6.14 (dd, J=2.7, 17.2 Гц, 1H), 5.83-5.61 (m, 3Н), 5.60-5.48 (m, 1H), 5.07 (dd, J=3.5, 51.6 Гц, 1H), 5.06-4.96 (m, 1H), 4.79 (dd, J=4.9, 15.8 Гц, 1H), 4.69 (d, J=5.9 Гц, 2Н), 4.67-4.56 (m, 1H), 4.48-4.40 (m, 3Н), 4.37-4.30 (m, 1H), 4.27 (d, J=5.9 Гц, 2Н), 4.19-4.13 (m, 1H), 3.93-3.85 (m, 1H), 3.85-3.78 (m, 1Н)
Стадия 10-11
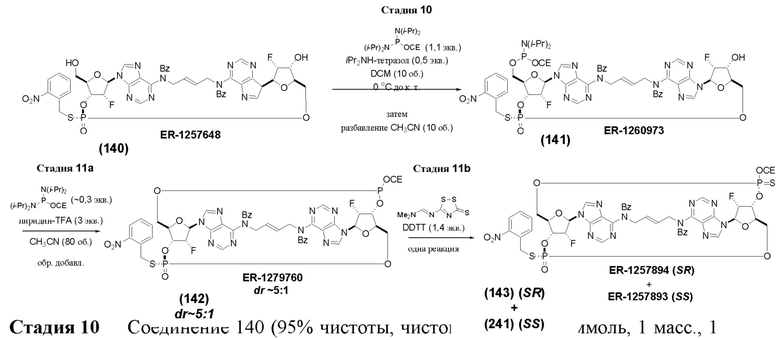 об., 1 экв.) и 2-цианоэтил-N,N,N',N'-тетраизопропилфосфородиамидит (25,3 мл, 79,5 ммоль, 0,33 масс., 0,35 об., 1,10 экв.) азеотропировали с безводным ацетонитрилом трижды (3×2 л), повторно растворяли в дихлорметане (0,73 л, 10 об.) и охлаждали до 0-5°С. Добавляли диизопропиламмония тетразолид (6,19 г, 36,1 ммоль, 0,085 масс., 0,50 экв.). Полученную реакционную смесь перемешивали при 0°С в течение 10 ч, нагревали до 10°С в течение 2 ч, сохраняли при 10°С в течение 10 ч и нагревали до к.т. в течение 2 ч. За реакцией наблюдали при помощи LCMS и TLC (EtOAc с 0,5% TEA). Через 18 ч добавляли безводный ацетонитрил (0,73 л, 10 об.) и смесь хранили при -20°С в течение 3 суток.
об., 1 экв.) и 2-цианоэтил-N,N,N',N'-тетраизопропилфосфородиамидит (25,3 мл, 79,5 ммоль, 0,33 масс., 0,35 об., 1,10 экв.) азеотропировали с безводным ацетонитрилом трижды (3×2 л), повторно растворяли в дихлорметане (0,73 л, 10 об.) и охлаждали до 0-5°С. Добавляли диизопропиламмония тетразолид (6,19 г, 36,1 ммоль, 0,085 масс., 0,50 экв.). Полученную реакционную смесь перемешивали при 0°С в течение 10 ч, нагревали до 10°С в течение 2 ч, сохраняли при 10°С в течение 10 ч и нагревали до к.т. в течение 2 ч. За реакцией наблюдали при помощи LCMS и TLC (EtOAc с 0,5% TEA). Через 18 ч добавляли безводный ацетонитрил (0,73 л, 10 об.) и смесь хранили при -20°С в течение 3 суток.
Стадия 11а Смесь из стадии 10 нагревали до температуры окружающей среды и добавляли при помощи капельной воронки частями (100 мл каждые 30 минут в течение 9 ч) в смесь пиридиновой трифторацетатной соли (азеотропировали заранее дважды с пиридином; 41,9 г, 217 ммоль, 0,57 масс., 3,0 экв.) и ацетонитрила (5,85 л, 80 об.). За реакцией наблюдали при помощи LCMS. Через 13 ч раствор 2-цианоэтил-N,N,N',N'-тетраизопропилфосфородиамидита (5,8 мл, 18 ммоль, 0,25 экв.) в ацетонитриле (24 мл) добавляли в течение 4 ч. Количество дополнительного реагента определяли на основе оставшегося соединения 140 (~30% на основе LCMS). Больше превращения диола наблюдали через 6 ч.
Стадия 11b ((Диметиламинометилиден)амино)-3Н-1,2,4-дитиазолин-3-тион (DDTT; 20,8 г, 101 ммоль, 0,28 масс., 1,4 экв.) добавляли и перемешивание продолжали в течение 1 ч. Реакционную смесь частично концентрировали до ~800 мл и разбавляли МТВЕ (1,46 л, 20 об.), NaHCO3 (9 масс. % раствор в воде; 1,1 л, 15 об.) и водой (0,37 л, 5 об.). рН=8. Слои разделяли и водный слой экстрагировали смесью МТВЕ (1,46 л, 20 об.) и EtOAc (1,10 л, 15 об.). Объединенные органические слои дважды промывали 30% водн. NaCl (2×0,73 л, 2×10 об.), концентрировали в вакууме при 35°С и азеотропировали с толуолом (1,46 л, 20 об.). LCMS и TLC (EtOAc) показывали соотношение соединения 143 (SpRp, требуемый): соединения 241 (SpSp) = 5:1.
Неочищенный продукт очищали на Biotage 150М KP-Sil, (SiO2 2,5 кг; элюенты: EtOAc/Hep: 2:1 (4 CV), 3:1 (2.5 CV), 4:1 (2.5 CV), 100% ЕА (3 CV), 5-10% МеОН в ЕА 4 CV) с получением соединения 143 (36 г, 31,5 ммоль, 44% выход).
Соединение 143 (SpRp): 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.59 (s, 1H), 8.10 (s, 1H), 8.03-7.99 (m, 1H), 7.91 (s, 1H), 7.56-7.53 (m, 2Н), 7.49-7.40 (m, 5Н), 7.35-7.28 (m, 2Н), 7.24-7.16 (m, 4Н), 6.92 (s, 1H), 6.29 (d, J=14.9 Гц, 1H), 6.08 (d, J=20.7 Гц, 1H), 5.97-5.83 (m, 1H), 5.76 (td, J=4.7, 15.6 Гц, 1H), 5.61-5.51 (m, 2Н), 5.40 (d, J=4.3 Гц, 1H), 5.29-5.17 (m, 1H), 4.91 (dd, J=7.4, 14.9 Гц, 1H), 4.86-4.75 (m, 3Н), 4.63 (dd, J=3.7, 9.2 Гц, 1H), 4.58-4.43 (m, 5Н), 4.34-4.19 (m, 4Н), 2.79 (td, J=5.9, 16.8 Гц, 1H), 2.66 (td, J=6.3, 16.8 Гц, 1H).
Соединение 241 (SpSp) 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.11 (s, 1H), 8.03 (d, J=8.2 Гц, 1H), 7.94 (s, 1H), 7.90 (s, 1H), 7.61 (s, 1H), 7.56-7.40 (m, 7Н), 7.33-7.28 (m, 2Н), 7.23-7.17 (m, 4Н), 6.22 (d, J=17.6 Гц, 1H), 6.15 (d, J=18.8 Гц, 1H), 5.85 (dd, J=3.5, 51.2 Гц, 1H), 5.75-5.45 (m, 5Н), 4.95-4.23 (m, 14Н), 2.82 (t, J=6.1 Гц, 2H).
Стадия 12
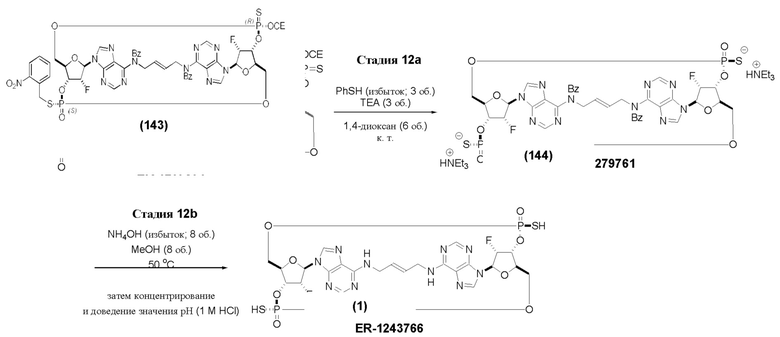
Соединение 143 (71,6 г, 62,6 ммоль, 1 масс., 1 об., 1 экв.) растворяли в 1,4-диоксане (0,43 л, 6 об.). Добавляли тиофенол (215 мл, 2,09 мол., 230 г, 3,2 масс., 3 об., >30 экв.), а затем триэтиламин (215 мл, 1,54 мол., 156 г, 2,2 масс., 3 об.). Наблюдали некоторый экзотермический эффект (Т-внутренняя повышена на ~7°С), таким образом, баню вода/лед использовали для охлаждения и контроля Т-внутренней ниже 27°С. За реакцией наблюдали при помощи LCMS. Через 2 ч МеОН (0,57 л, 8 об.) и NH4OH (28 масс. %; 15 мол., 0,57 л, 8 об., >200 экв.) добавляли. Полученную смесь нагревали при 50°С в течение 5 ч, охлаждали до к.т. и перемешивали всю ночь. Через 14 ч добавляли воду (0,72 л, 10 об.) (твердое вещество не наблюдалось) и смесь экстрагировали трижды 1:1 (объем/объем) смесью н-гептана и толуола (3×0,86 л, 3×12 об.), а затем толуолом (0,57 л, 8 об.). Водный слой концентрировали в вакууме при 40-50°С и разбавляли водой (1,07 л, 15 об.). Полученную взвесь сохраняли всю ночь при к.т. Полученное твердое вещество отфильтровывали, промывали водой (0,36 л, 5 об.). Фильтрат оставался еще мутным и фильтровали через целит и фильтр Kuno. Мутность все еще присутствовала. HCl (1,0 М раствор в воде; 132 мл, 132 ммоль, 2,1 экв.) добавляли в течение 1 ч и значение рН проверяли (рН <2). Перемешивание продолжали при к.т. в течение 1 ч и смесь фильтровали. Фильтрационный кек промывали водой (8×0,20 л), сушили в вакуумной печи при 35°С в течение 2 суток и без нагревания в течение 1 суток с получением соединения 1 в виде бледно-оранжевого твердого вещества (44,88 г, 60,1 ммоль, 0,63 масс., 96% выход).
Стадия 13
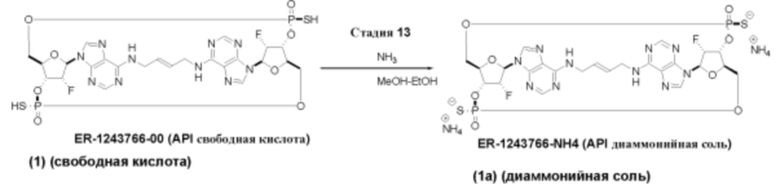
К соединению свободной кислоты 1 (22,42 г, 1 экв.) добавляли аммиак (2,0 М раствор в МеОН; 220 мл, 440 ммоль, 10 об., 15 экв.). EtOH (55 мл, 2,5 об.) добавляли и полученный раствор фильтровали через фильтр Kuno (0,45 микрон; PTFE), промывали 1:1 (объем/объем) смесью МеОН и EtOH (90 мл, 4 об.). Фильтрат концентрировали в вакууме при 30°С с получением грязно-белого твердого вещества, которое сушили при к.т. всю ночь, измельчали лопаткой (легко разбить) и сушили дополнительно в вакууме при к.т. Выделенное твердое вещество затем суспендировали в толуоле (250 мл) и перемешивали при к.т. в течение 30 минут. Твердое вещество затем собирали вакуумной фильтрацией и промывали дважды толуолом (2×50 мл). Твердое вещество затем сушили в вакууме в вакуумной печи с получением 22,4 г соединения 1а (диаммонийная соль соединения 1).
Перекристаллизация: Соединение 1а (22,14 г, 28,36 ммоль, 1 масс, 1 об., 1 экв.) растворяли в смеси воды (664 мл, 30 об.) и гидроксида аммония (28 масс. %; 2,5 мл, 18 ммоль, 0,63 экв.) (рН=9-10) и экстрагировали трижды толуолом (3×300 мл, 3×14 об.), трижды EtOAc (3×200 мл, 3×9 об.) и трижды толуолом (3×300 мл, 3×14 об.). Полученный водный слой обрабатывали HCl (1,0 М раствор в воде; 90 мл, 90 ммоль, 3,2 экв.) в течение 3,5 часов (рН≤2). Смесь перемешивали в течение 30 минут, а затем осадок твердого вещества собирали вакуумной фильтрацией. Фильтрационный кек промывали трижды водой (3×200 мл, 3×9 об.) и сушили в вакууме всю ночь. Аммиак (2,0 М раствор в МеОН; 250 мл, 500 ммоль, 17,6 экв.) и этанол (100 мл) добавляли к твердому веществу и полученную смесь концентрировали в вакууме до появления кристаллов (~100 мл), в это время концентрирование останавливали и смесь перемешивали в течение 20 минут. Этанол (45 мл) добавляли и смесь частично концентрировали (45 мл удаляли). Ту же операцию повторяли еще два раза, а затем смесь охлаждали до 0°С и перемешивали в течение 3,5 ч. Белое твердое вещество собирали вакуумной фильтрацией и промывали холодным этанолом (20 мл), а затем этилацетатом (2×50 мл). Белое твердое вещество сушили в вакууме при к. т.в течение 3 суток с получением соединения 1а в виде белого твердого вещества (16,6 г, 21,3 ммоль, 0,75 масс., 75% выход). Фильтрат концентрировали в вакууме и сушили в вакууме при к. т.в течение 3 суток с получением соединения 1а в виде грязно-белого твердого вещества (4,16 г, 5,3 ммоль, 18% выход).
Пример 1.2 - 1Н ЯМР анализ соединения 1
1Н ЯМР спектрограмма соединения 1а показана на фиг.3. Полученный спектр:
1Н-ЯМР спектр (400 МГц, DMSO-d6, δН 2.49 ppm, 80°С)
δ (ppm): 3.05-3.13 (4Н, m), 3.70 (1Н, dd, J=13, 5 Гц), 3.78 (1Н, dd, J=12, 4 Гц), 4.21-4.24 (2Н, m), 4.28 (1Н, m), 4.38 (1Н, m), 4.53-4.68 (2Н, m), 5.22 (1H, m), 5.76 (2Н, s), 5.78 (1Н, m), 6.26 (1Н, m), 6.29 (1Н, m), 8.13 (1Н, s), 8.14 (1Н, s), 8.36 (1Н, brs), 8.59 (1Н, brs).
Пример 1.3 - Рентгеноструктурный анализ соединения 1
Приблизительно 2 мг соединения 1 растворяли в 600 мкл воды. 120 мкл этого раствора помещали в другой стеклянный сосуд, а затем этот сосуд хранили в закрепленном контейнере с 3 мл MeCN при комнатной температуре в течение 1 недели. Это представляет собой Н2О/MeCN способ диффузии пара получения образца.
Бесцветный блоковый монокристалл (0,1×0,1×0,1 мм), полученный в кристаллизационном растворе, диспергировали в жидком Parabar 10312 и устанавливали на Dual-Thickness MicroMounts™ (MiTeGen). Данные дифракции собирали при -160°С на XtaLAB PRO Р200 MM007HF (Rigaku) при помощи метода колебаний ω осей с применением многослойного зеркального монохромированого Cu-Kα излучения.
На фиг. 4А показана ORTEP фигура молекул соединения 1 в асимметрической единице вместе с числом беспорядочных молекул воды. На фиг. 4В показана кристаллическая структура одной из молекул соединения 1 из фиг. 4А. На фиг. 4С показана кристаллическая структура другой молекулы соединения 1, показанной на фиг. 4А.
Кристаллическая структура соединения 1 была объяснена при помощи конечного R-фактора 0,1354. Параметр Flack был равен приблизительно нулю (0,083(17)), обозначая, что абсолютная конфигурация соединения 1 представляет собой (R, S). Анализ кристаллической структуры также показывает, что многие молекулы воды присутствовали в большом канале соединения 1, что означает, что молекулы воды способны легко ускользать из канала. Анализ также подтверждает, что конформации обеих кристаллографически независимых молекул асимметричной единицы были почти одинаковыми.
Дополнительные параметры рентгеноструктурного анализа показаны ниже:
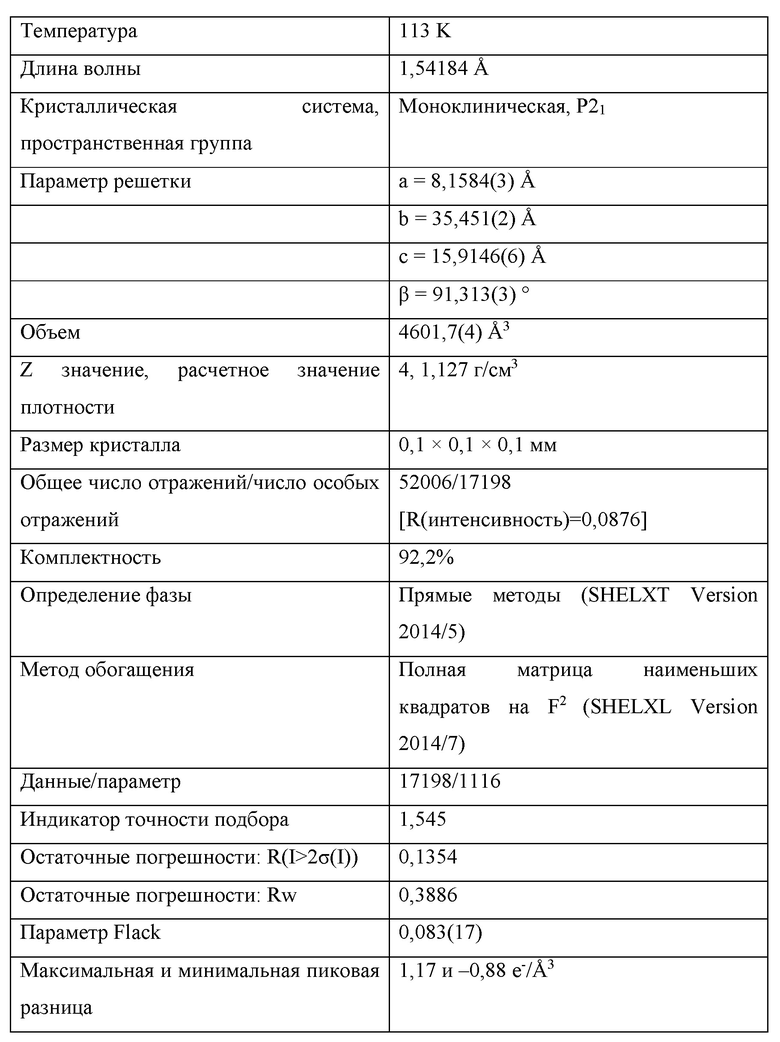
Пример 2 - Синтез соединения 2
Соединение 106 (RpRp изомер соединения 105), полученное из примера 1, стадия Е, обрабатывали отдельно как в примере 1, стадия F и примере 1, стадия G, с получением соединения 2а (RR изомер соединения 1а):
 7.99 (s, 2H), 7.96 (s, 2H), 7.65-7.48 (m, 10Н), 7.38-7.33 (m, 2H), 7.26-7.24 (m, 4H), 6.22 (d, J=17.6 Гц, 2H), 5.95-5.84 (m, 2H), 5.71 (dd, J=50.8, 3.9 Гц, 2H) 5.73-5.71 (m, 2H), 4.88-4.77 (m, 4H), 4.59-4.38 (m, 8H), 4.19 (m, 2H).
7.99 (s, 2H), 7.96 (s, 2H), 7.65-7.48 (m, 10Н), 7.38-7.33 (m, 2H), 7.26-7.24 (m, 4H), 6.22 (d, J=17.6 Гц, 2H), 5.95-5.84 (m, 2H), 5.71 (dd, J=50.8, 3.9 Гц, 2H) 5.73-5.71 (m, 2H), 4.88-4.77 (m, 4H), 4.59-4.38 (m, 8H), 4.19 (m, 2H).
Соединение 2а (RpRp, транс) 1Н ЯМР (400 МГц, CD3OD) δ=8.70 (s, 1H), 8.49 (s, 1H), 8.23 (s, 1H), 8.09 (s, 1H), 6.34 (br s, 2Н), 5.83 (br s, 2H), 5.73-5.53 (m, 2H), 5.38-5.01 (m, 2H), 4.76-4.32 (m, 6H), 3.95 (br s, 2H), 3.69-3.64 (m, 2H); 31P ЯМР (162 МГц, CD3OD) δ=55.57 (s, 1P), 55.32 (s, 1P).
Пример 2.1 - Рентгеноструктурный анализ соединения 2
Соединение 2 (0,5 мг) взвешивали и растворяли в ацетонитриле/28% аммиачном растворе. Затем такой раствор хранили при комнатной температуре со свободно закрепленным колпачком. Через 2 недели появляется кристалл столбчатой формы.
Бесцветный блоковый монокристалл (0,1×0,1×0,5 мм), полученный в кристаллизационном растворе, диспергировали в жидком Parabar 10312 и устанавливали на Dual-Thickness MicroMounts™ (MiTeGen). Данные дифракции собирали при -160°С на XtaLAB PRO Р200 MM007HF (Rigaku) при помощи метода колебаний со осей с применением многослойного зеркального монохромированого Cu-Kα излучения.
На фиг. 4D показана ORTEP фигура молекулы соединения 2.
Дополнительные параметры этого рентгеноструктурного анализа показаны ниже:
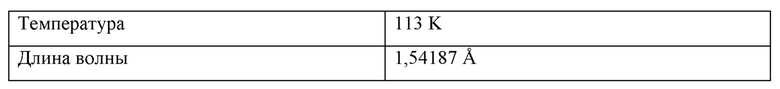
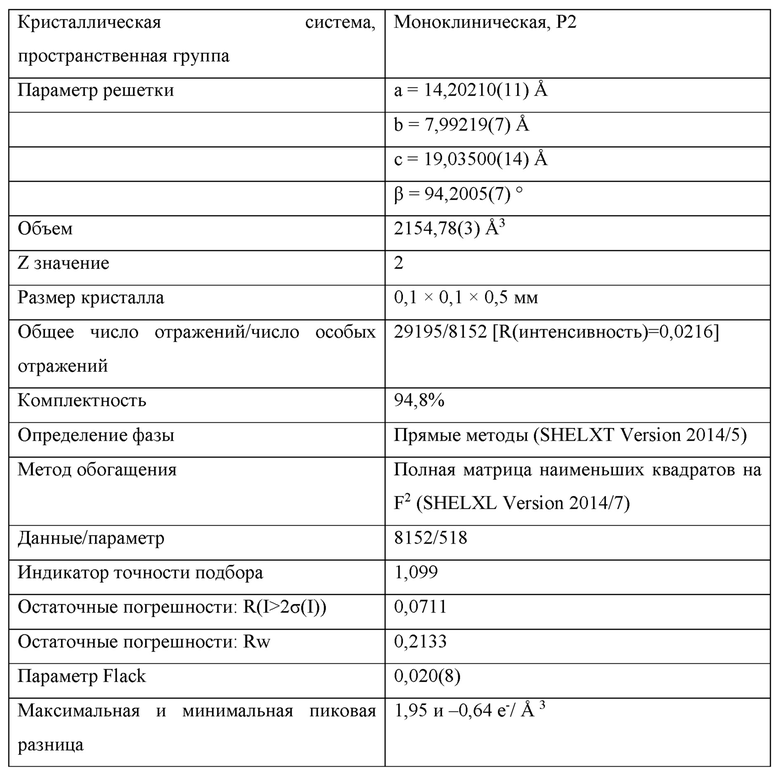
Пример 3 - Синтез соединения 3
Соединение 109 (SpSp изомер соединения 105), полученное из примера 1, стадия Е, обрабатывали отдельно как в примере 1, стадия F и примере 1, стадия G, с получением соединения 3а (SS изомер соединения 1а):
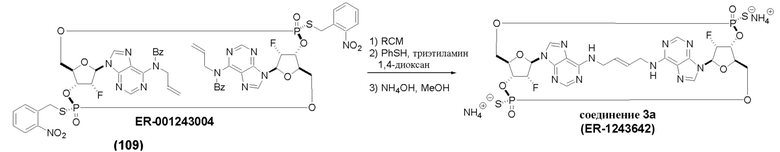
Соединение 109 (SpSp изомер соединения 105): 1НЯМР (400 МГц, CHLOROFORM-d) δ=8.52 (s, 2Н), 7.99 (d, J=8.2 Гц, 2Н), 7.91 (s, 2Н), 7.63-7.40 (m, 10Н), 7.35-7.28 (m, 2Н), 7.23-7.16 (m, 4Н), 6.10-5.93 (m, 4Н), 5.92-5.75 (m, 2Н), 5.62-5.50 (m, 2Н), 5.26-5.16 (m, 2H), 5.09-5.03 (m, 2H), 4.98-4.91 (m, 4H), 4.61-4.25 (m, 10Н)
Соединение 3а (SpSp, транс): 1Н ЯМР (400 МГц, CD3OD) δ=8.97 (br s, 1H), 8.84 (br s, 1H), 8.21 (br s, 2H), 6.31 (br s, 2H), 6.08 (br d, J=53.5 Гц, 1H), 5.89 (br s, 2H), 5.63 (br d, J=52.4 Гц, 1H), 5.13-4.96 (m, 2H), 4.72-4.32 (m, 6H), 4.01 (br d,J=9.8 Гц, 2H), 3.67 (br s, 2H).
Пример 4 - Синтез соединения 4a
Соединение 107 полученное из примера 2, стадия F, выделяли в виде единственного цис-изомера при помощи хроматографии на силикагеле, обрабатывали отдельно как в примере 1, стадия G, с получением соединения 4а.
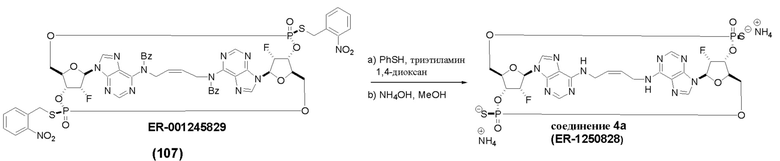
Соединение 107: 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.13 (s, 2H), 8.12-8.08 (m, 2H), 7.91 (s, 2H), 7.65-7.55 (m, 4H), 7.54-7.45 (m, 6H), 7.33-7.27 (m, 2H), 7.23-7.16 (m, 4H), 6.14 (d, J=17.6 Гц, 2H), 5.88 (dd, J=3.9, 50.8 Гц, 2H), 5.76-5.61 (m, 4H), 5.21-4.99 (m, 4H), 4.60-4.46 (m, 4H), 4.45-4.37 (m, 2H), 4.30-4.13 (m, 4H), 3.49 (d, J=5.1 Гц, 2H)
Соединение 4а (RpRp, цис 1a, Соединение 4а): 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.48 (br s, 2Н), 8.02 (br s, 2H), 6.29 (d, J=14.8 Гц, 2H), 5.99 (br s, 2H), 5.43 (d, J=51.2 Гц, 2H), 5.03-4.88 (m, 2H), 4.43 (br d, J=11.7 Гц, 2H), 4.32 (br d, J=9.4 Гц, 2H), 4.27-4.17 (m, 2H), 4.21-4.02 (m, 2H), 3.97 (br dd, J=6.1, 12.3 Гц, 2H).
Пример 5 - Синтез соединения 5
Цис-изомер, полученный из примера 1, стадия F, обрабатывали отдельно как в примере 1, стадия G, с получением соединения 5а.
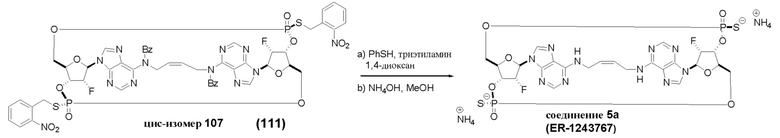
Соединение 111 (цис-изомер соединения 107): 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.48 (s, 1H), 8.15-8.09 (m, 1H), 8.03 (d,J=8.2 Гц, 1H), 7.94 (s, 1H), 7.75 (s, 1H), 7.65-7.06 (m, 11H), 6.07 (d, J=17.2 Гц, 1H), 5.98 (d, J=20.3 Гц, 1H), 5.97-5.79 (m, 1H), 5.84 (dd, J=3.5, 51.2 Гц, 1H), 5.54-5.47 (m, 1H), 5.50 (dd, J=3.9, 52.0 Гц, 1H), 5.38-5.21 (m, 2H), 5.18-5.02 (m, 2H), 5.02-4.95 (m, 1H), 4.78-4.69 (m, 1H), 4.60-4.16 (m, 10H)
Соединение 5a (SpRp, цис 1a): 1H ЯМР (400 МГц, METHANOL-d4) δ=8.88 (br s, 1H), 8.51 (br s, 1H), 8.16 (s, 1H), 8.05 (s, 1H), 6.38 (d, J=15.6 Гц, 1H), 6.33 (d, J=14.1 Гц, 1H), 6.14-6.09 (m, 2H), 6.01 (d, J=49.2 Гц, 1H), 5.42 (d, J=49.6 Гц, 1H), 5.02-4.87 (m, 2H), 4.76-3.92 (m, 8H), 3.75-3.56 (m, 2H)
Пример 6 - Синтез соединения 6a (SpRp)
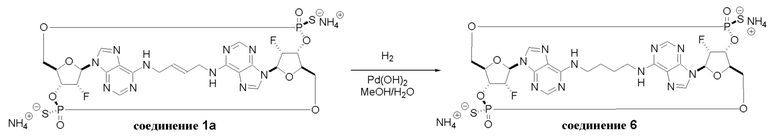
К раствору соединения 1a (1,5 мг, 2,0 мкмоль) в МеОН/Н2О (0,6/0,5 мл) при температуре окружающей среды добавляли гидроксид палладия на углеродном носителе (20 масс. % сухая масса, 2 мг). Полученную смесь обрабатывали водородом (баллон), при этом за реакцией наблюдали при помощи LCMS. После завершения израсходования исходного вещества смесь фильтровали и промывали метанолом, пока весь продукт не очистили. Фильтрат концентрировали в вакууме и остаток растворяли в воде (1 мл). Методом очистки RHPLC получали соединение 6а (0,9 мг).
LCMS (MS m/z 749,2 [М+Н]+)
Пример 7 - Синтез соединения 9
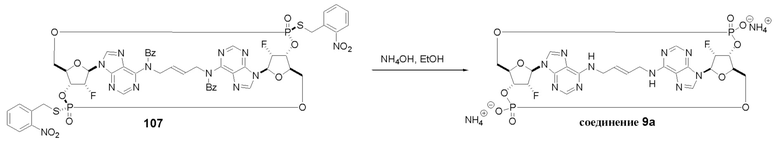
К раствору соединения 107 (3,7 мг, 3,02 мкмоль) в EtOH (1,5 мл) добавляли гидроксид аммония (1 мл). Полученную смесь нагревали при 50°С в течение 8 ч и охлаждали до температуры окружающей среды. Растворитель удаляли при пониженном давлении. Остаток обрабатывали 2 мл воды и полученное твердое вещество отфильтровывали. Фильтрат подвергали методу препаративной HPLC с получением соединения 9а (2,5 мг).
Соединение 9а: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.67 (br s, 1H), 8.50 (br s, 1H), 8.25 (br s, 1H), 8.12 (br s, 1H), 6.47-6.28 (m, 2H), 5.92-5.79 (m, 2H), 5.74 (d, J=50.8 Гц, 1H), 5.32 (d, J=52.4 Гц, 1H), 5.16-4.95 (m, 2H), 4.71-4.25 (m, 6H), 4.17-3.97 (m, 2H), 3.83-3.61 (m, 2H).
LCMS: MS m/z 715.2 [M+H]+.
Пример 8 - Пути синтеза для соединений 8а, 11а и 12а
С соединением 112 и соединением 102 в качестве исходных веществ, соединения 8, 11 и 12 получали такими же реакционными последовательностями, как описано в примере 1.
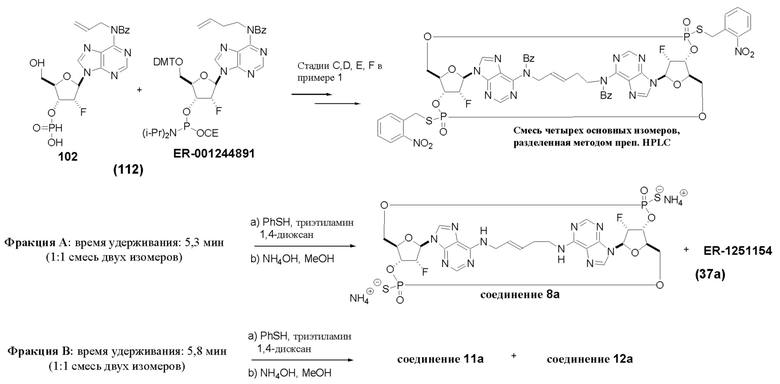
Условия препаративной HPLC для фракций А и В
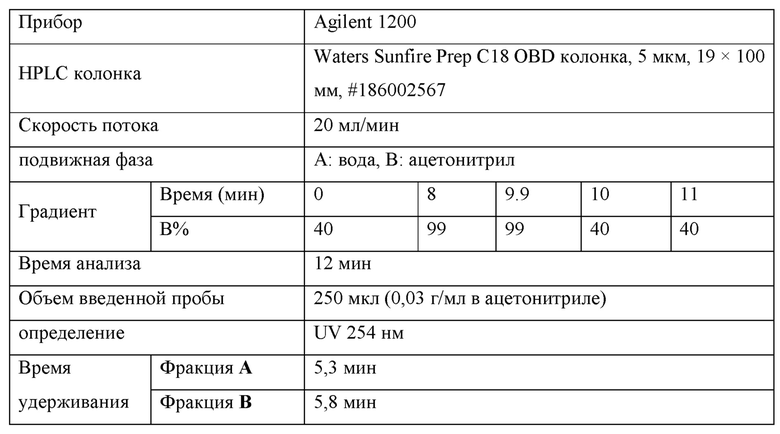
Соединение 8а: Фракцию А обрабатывали как на стадии g, пример 1 с получением двух изомеров, которые разделяли методом HPLC. Соединение 8а было изомером наиболее быстрого вращения (время удерживания: 4.1 мин) и соединение 37а (время удерживания: 4,5 мин) было изомером более медленного вращения.
1Н ЯМР (400 МГц, METHANOL-d4) δ=9.01 (s, 1H), 8.61 (s, 1H), 8.22 (s, 1H), 8.20 (s, 1H), 6.34 (d, J=10.2 Гц, 1H), 6.31 (d, J=10.9 Гц, 1H), 5.92 (dd, J=2.7, 51.2 Гц, 1H), 5.85-5.71 (m, 2Н), 5.37 (d, J=51.6 Гц, 1H), 4.81-4.56 (m, 6Н), 4.52 (br d, J=12.1 Гц, 1H), 4.42 (br d, J=9.4 Гц, 3Н), 4.05 (dd, J=4.1, 11.9 Гц, 1H), 3.95 (br dd, J=4.3, 12.1 Гц, 1H), 3.94-3.84 (m, 1H), 3.62 (br dd, J=4.9, 15.4 Гц, 1H), 2.71-2.58 (m, 1H), 2.46-2.33 (m, 1H)
31P ЯМР (162 МГц, METHANOL-d4) δ=55.36 (s, 1P), 55.18 (s, 1P)
LCMS: MS m/z 761.2 [M+H]+
Соединение 37a: 1H ЯМР (400 МГц, METHANOL-d4) δ=8.95 (s, 1H), 8.40 (s, 1H), 8.18 (s, 1H),8.15(brs, 1H), 6.34 (d, J=5.5 Гц, 1H), 6.31 (d, J=6.6 Гц, 1H), 5.96-5.68 (m, 3H), 5.40 (dd, J=2.7, 51.6 Гц, 1H), 4.85 (s, 3H), 4.58 (br d, J=12.1 Гц, 1H), 4.49 (br d, J=12.1 Гц, 1H), 4.43-4.33 (m, 3H), 4.29 (dd, J=8.6, 14.1 Гц, 1H), 4.05 (dd, J=4.5, 11.9 Гц, 1H), 3.95 (dd, J=4.9, 12.3 Гц, 1H), 3.94-3.86 (m, 1H), 2.67-2.55 (m, 1H), 2.52-2.40 (m, 1H).
Соединение 8a/37a Условия препаративной HPLC:
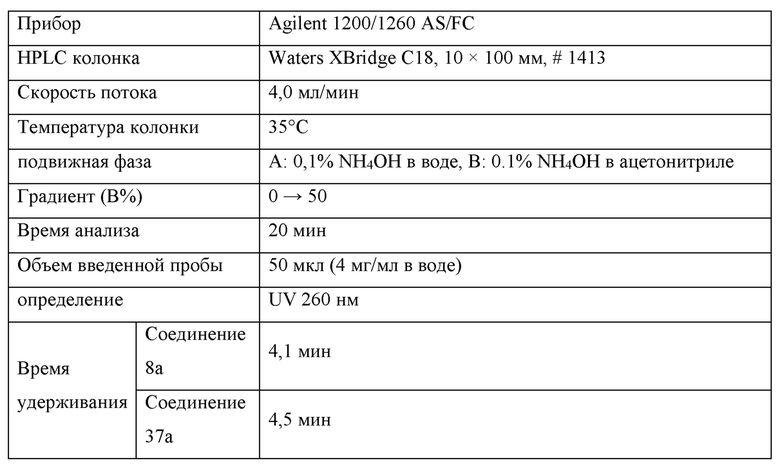
Фракцию В обрабатывали как на стадии g, пример 1 с получением двух изомеров, которые разделяли методом преп. -HPLC, описанным ниже. Соединение 11а было изомером наиболее быстрого вращения (время удерживания: 11,2 мин) и соединение 12а было изомером более медленного вращения (время удерживания: 12,1 мин)
Соединение 11а: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.63 (br s, 2Н), 8.18 (br s, 1H), 8.17 (br s, 1H), 6.34 (d, J=13.3 Гц, 1H), 6.32 (d, J=12.9 Гц, 1H), 5.86-5.65 (m, 2H), 5.48 (d, J=48.5 Гц, 1H), 5.35 (d, J=43.8 Гц, 1H), 4.84-4.53 (m, 6H), 4.47-4.36 (m, 2H), 4.05-3.91 (m, 2H), 3.96-3.85 (m, 1H), 3.70-3.54 (m, 1H), 2.66-2.54 (m, 1H), 2.43-2.30 (m, 1H)
Соединение 12а: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.56 (br s, 1H), 8.41 (s, 1H), 8.17 (s, 1H), 7.95 (br s, 1H), 6.35 (d, J=14.8 Гц, 1H), 6.31 (d, J=15.2 Гц, 1H), 5.82-5.65 (m, 2H), 5.50 (d, J=51.2 Гц, 1H), 5.36 (d, J=53.9 Гц, 1H), 4.74-4.33 (m, 8H), 4.26-4.16 (m, 1H), 4.07-3.93 (m, 3H), 2.64-2.44 (m, 2H).
Соединение 11a/12a Условия препаративной HPLC:

Пример 9 - Пути синтеза для соединения 13 и 14
С соединением 114 и соединением 115 в качестве исходных веществ соединения 13 и 14 получали такими же реакционными последовательностями, как описано в примере 1 плюс стадия снятия защитных групп с TBS.
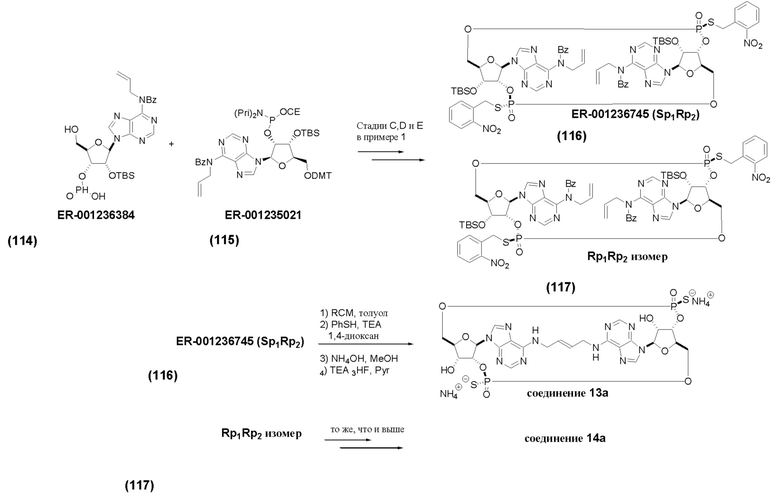
Соединение 16: 1H ЯМР (400 МГц, CHLOROFORM-d) δ=9.36 (s, 1H), 9.18 (s, 1H), 8.77 (s, 1H), 8.76 (s, 2Н), 8.07 (br dd, J=1.8, 7.6 Гц, 3Н), 8.05-8.00 (m, 2Н), 7.99 (s, 1H), 7.87-7.82 (m, 1H), 7.70-7.65 (m, 1H), 7.63-7.55 (m, 3Н), 7.55-7.41 (m, 6Н), 7.39-7.35 (m, 1H), 7.30-7.26 (m, 1H), 6.37 (d, J=8.6 Гц, 1H), 5.89 (d, J=4.7 Гц, 1H), 5.36-5.27 (m, 2Н), 5.15 (ddd, J=3.7, 8.4, 11.7 Гц, 1H), 4.64-4.43 (m, 4Н), 4.43-4.38 (m, 1H), 4.37-4.30 (m, 1H), 4.23-4.13 (m, 2Н), 4.11-3.98 (m, 2Н), 3.97-3.86 (m, 1H), 0.97 (s, 9Н), 0.83 (s, 9Н), 0.25 (s, 3Н), 0.18 (s, 3Н), 0.16 (s, 3Н), -0.13 (s, 3Н).
Соединение 13а: 1H ЯМР (400 МГц, METHANOL-d4) δ=8.78 (br s, 1H), 8.47 (br s, 1H), 8.21 (br s, 1H), 8.08 (br s, 1H), 6.44-6.21 (m, 1H), 6.19-6.03 (m, 1H), 6.01-5.78 (m, 2H), 5.38-4.95 (m, 2H), 4.67-4.45 (m, 5H), 4.45-4.35 (m, 1H), 4.34-4.29 (m, 1H), 4.25 (br d, J=11.3 Гц, 1H), 4.18-3.89 (m, 2H), 3.88-3.54 (m, 2H).
31P ЯМР (162 МГц, METHANOL-d4) δ=56.89 (br s, 1P), 56.37 (br s, 1P)
Соединение 117 (Rp1Rp2 изомер соединения 116): 1H ЯМР (400 МГц, CHLOROFORM-d) δ=9.36 (br s, 1H), 9.11 (br s, 1H), 8.98 (s, 1H), 8.87 (s, 1H), 8.78 (s, 1H), 8.14-7.96 (m, 6H), 7.72-7.27 (m, 13H), 6.44 (d, J=8.6 Гц, 1H), 5.84 (d, J=6.6 Гц, 1H), 5.59-5.50 (m, 1H), 5.48-5.44 (m, 1H), 5.33-5.24 (m, 1H), 4.75-3.85 (m, 11H), 0.90 (s, 9H), 0.79 (s, 9H), 0.20 (s, 3H), 0.11 (s, 3H), 0.07 (s, 3H), -0.21 (s, 3H)
Соединение 14а (Rp1Rp2 изомер соединения 13а)
LCMS: MS m/z 743.17 [М+Н]+
Пример 10 - Пути синтеза для соединения 15 и 16
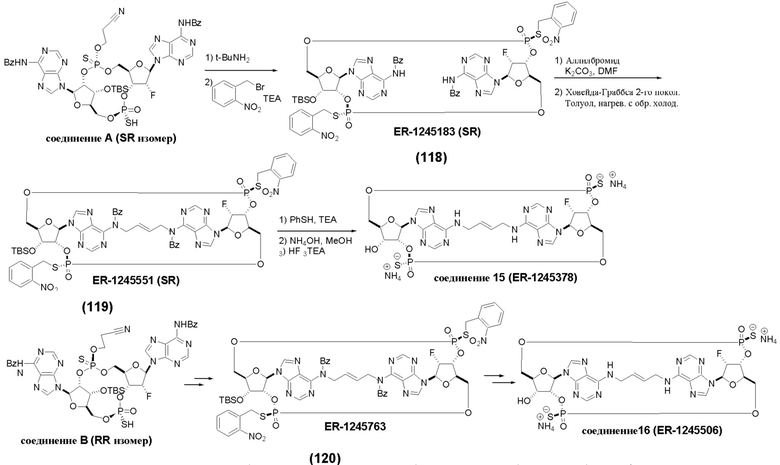
К раствору соединения A (2,05 г, 2,40 ммоль) в MeCN (26,3 мл) добавляли трет-бутиламин (13,15 мл, 124,1 ммоль). После перемешивания в течение 1 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме и азеотропировали с MeCN. Остаток растворяли в MeCN (52,6 мл) и обрабатывали 1-(бромметил)-2-нитробензолом (1,064 г, 4,925 ммоль) и триэтиламином (0,755 мл, 5,42 ммоль). После завершения реакции (наблюдали при помощи LC/MS) реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (66% этилацетата/н-гептана до 100% этилацетата) с получением 0,226 г соединения 118. Выделенный продукт растворяли в DMF (5 мл) и добавляли аллилбромид (0,046 мл, 0,528 ммоль) и карбонат калия (0,073 г, 0,528 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за прогрессом наблюдали при помощи LCMS. Через 19 ч 1/1 добавляли смесь МТВЕ/EtOAc (12/12 мл), насыщенный водный NH4Cl раствор (15 мл) и воду (10 мл). Органический слой отделяли, промывали солевым раствором (5 мл) дважды, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (50% этилацетата/н-гептана до 100% этилацетата) с получением 37 мг бис-аллилированного продукта. Выделенный продукт (37 мг, 0,027 ммоль) растворяли в толуоле (55 мл) и нагревали до умеренной температуры образования флегмы (масляная баня 120-125°С). Добавляли раствор катализатора Ховейда-Граббса 2-го поколения ((1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(орто-изопропоксифенилметилен)рутения; доступного от SIGMA-ALDRITCH® № каталога 569755; CAS 301224-40-8; 8,5 мг, 0,014 ммоль) и хинона (5,9 мг, 0,054 ммоль) в толуоле (10 мл). Смесь нагревали до температуры образования флегмы и за ходом реакции наблюдали при помощи LC/MS. После завершения израсходования исходного вещества смесь охлаждали до температуры окружающей среды и обрабатывали DMSO (0,059 мл, 0,81 ммоль) в течение 15 часов. Полученную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 66% этилацетата в н-гептане до 100% этилацетата) с получением соединения 119 (2,2 мг).
Соединение 119: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.49 (s, 1H), 8.12 (d, J=7.8 Гц, 1H), 8.05-8.02 (m, 1H), 8.02 (s, 1H), 7.82 (s, 1H), 7.70-7.10 (m, 17Н), 6.27 (d, J=16.8 Гц, 1H), 6.15-6.05 (m, 1H), 5.97 (d, J=8.2 Гц, 1H), 5.94-5.72 (m, 3Н), 4.94 (br s, 2H), 4.86-4.69 (m, 2H), 4.67-4.60 (m, 1H), 4.60-4.44 (m, 4H), 4.39-4.33 (m, 1H), 4.31-4.10 (m, 6H), 0.96 (s, 9H), 0.25 (s, 3Н), 0.19 (s, 3Н)
Соединение 119 обрабатывали как на стадии G в примере с последующим снятием защитных групп с TBS при помощи TEA 3HF с получением соединения 15а.
Соединение 15а: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.76 (br s, 1H), 8.44 (br s, 1H), 8.24 (br s, 1H), 8.07 (br s, 1H), 6.45-6.19 (m, 2H), 6.12-5.75 (m, 2H), 5.54 (br d, J=51.6 Гц, 1H), 5.67-5.06 (m, 2H), 4.66-4.37 (m, 3H), 4.47-4.37 (m, 1H), 4.36-4.22 (m, 2H), 4.17-3.94 (m, 2H), 3.94-3.80 (m, 1H) 3.80-3.59 (m, 2H)
Соединение 16 получали из соединения В (RR изомер соединения А) теми же последовательностями, что описаны для соединения 15.
Соединение 120 (Rp1Rp2 изомер соединения 119): 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.89 (s, 1H), 8.10 (d, J=8.2 Гц, 1H), 8.01 (d, J=8.2 Гц, 1H), 7.85-7.74 (m, 4Н), 7.69-7.61 (m, 2Н), 7.58 (s, 1H), 7.55-7.48 (m, 1H), 7.45-7.30 (m, 6Н), 7.22-7.15 (m, 1H), 7.13-7.04 (m, 3Н), 6.76-6.68 (m, 1H), 6.31-6.22 (m, 1H), 6.22 (d, J=16.0 Гц, 1H), 6.12-5.99 (m, 2Н), 5.94 (d, J=8.2 Гц, 1H), 5.92-5.81 (m, 1H), 5.67 (dd, J=3.5, 51.2 Гц, 1H), 5.08-4.93 (m, 2Н), 4.87-4.77 (m, 1H), 4.60-4.22 (m, 8Н), 4.21-4.16 (m, 1H), 4.11-4.02 (m, 1H), 3.94 (br d, J=11.7 Гц, 1H), 3.66 (dd, J=4.7, 11.3 Гц, 1H), 0.90 (s, 9Н), 0.09 (s, 3Н), 0.08 (s, 3Н)
Соединение 16а: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.54 (br s, 1H), 8.24 (br s, 1H), 8.17 (br s, 1H), 8.08 (br s, 1H), 6.44-6.23 (m, 2H), 6.07-5.68 (m, 2H), 5.43 (d, J=50.4 Гц, 1H), 5.31-5.05 (m, 2H), 4.69-4.26 (m, 6H), 4.19-3.90 (m, 2H), 3.90-3.57 (m, 3H)
Пример 11 - Пути синтеза соединений адениновых/гуаниновых (A/G) аналогов - соединение 21, соединение 22 и соединение 23
С соединением 121 и соединением 101 в качестве исходных веществ адениновые/гуаниновые аналоги получали такими же реакционными последовательностями, как описано в примере 1.
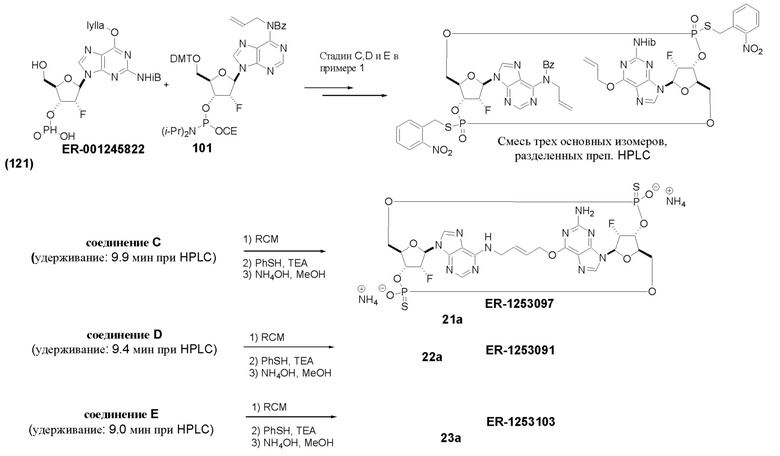
Условия препаративной HPLC для разделения соединений С, D и Е


Соединение D: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.60 (s, 1H), 8.27 (s, 1H), 8.04-8.01 (m, 1H), 8.01 (s, 1H), 7.99-7.95 (m, 1H), 7.80 (s, 1H), 7.63-7.51 (m, 5Н), 7.46-7.41 (m, 1H), 7.39-7.30 (m, 3Н), 7.25-7.20 (m, 2Н), 6.96-6.84 (m, 1H), 6.19-5.99 (m, 4Н), 5.67 (dd, J=4.3, 52.3 Гц, 1H), 5.47 (dd, J=1.4, 17.4 Гц, 1H), 5.36-5.24 (m, 4Н), 5.14-5.09 (m, 1H), 5.08 (d, J=5.5 Гц, 2Н), 5.04-4.98 (m, 2Н), 4.51-4.30 (m, 10Н), 4.21-4.13 (m, 1H), 2.83-2.67(m, 1H), 1.16 (d, J=7.0 Гц, 3Н), 1.14 (d, J=6.6 Гц, 3Н)
Соединение D обрабатывали как на стадиях F и G, пример 1, с получением соединения 22.
Соединение 22: LCMS: MS m/z 763.07 [М+Н]+
Соединение С: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=9.08 (s, 1H), 8.60 (s, 1H), 8.07-8.02 (m, 2Н), 8.02 (s, 1H), 7.80 (s, 1H), 7.55-7.30 (m, 9Н), 7.24-7.19 (m, 2Н), 6.58-6.45 (m, 1H), 6.23-5.97 (m, 4Н), 5.97-5.86 (m, 1H), 5.63-5.57 (m, 1H), 5.54-5.47 (m, 1H), 5.48-5.44 (m, 1H), 5.35-5.30 (m, 1H), 5.29-5.22 (m, 1H), 5.18-5.13 (m, 2Н), 5.11-5.07 (m, 1H), 5.02-4.97 (m, 2Н), 4.51-4.22 (m, 9Н), 4.14-4.07 (m, 1H), 2.99-2.85 (m, 1H), 1.20 (d, J=6.6 Гц, 3Н), 1.17 (d, J=7.0 Гц, 3Н)
Соединение С обрабатывали как на стадиях F и G в примере 1 с получением соединения 21
Соединение 21: LCMS: MS m/z 763.13 [М+Н]+
Соединение Е: 1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.99 (s, 1H), 8.57 (s, 1H), 8.02-7.94 (m, 2Н), 7.93 (s, 1H), 7.82 (s, 1H), 7.68-7.63 (m, 1H), 7.60-7.55 (m, 1H), 7.54-7.49 (m, 2Н), 7.45-7.31 (m, 5Н), 7.25-7.21 (m, 2Н), 6.22-5.94 (m, 6Н), 5.56 (br dd, J=5.1, 51.5 Гц, 1H), 5.54-5.46 (m, 1H), 5.34-5.29 (m, 1H), 5.29-5.22 (m, 2Н), 5.16-5.08 (m, 3Н), 5.02-4.96 (m, 2Н), 4.62-4.54 (m, 1H), 4.52-4.26 (m, 9Н), 2.93-2.81 (m, 1H), 1.21 (d, J=2.3 Гц, 3Н), 1.20 (d, J=2.0 Гц, 3Н)
Соединение Е обрабатывали как на стадиях F и G в примере 1 с получением соединения 23
Соединение 23: LCMS: MS m/z 763.18 [М+Н]+
Пример 12 - Синтез соединения 24
С соединением F соединение 24а получали такими же реакционными последовательностями, как описано в примере 1.
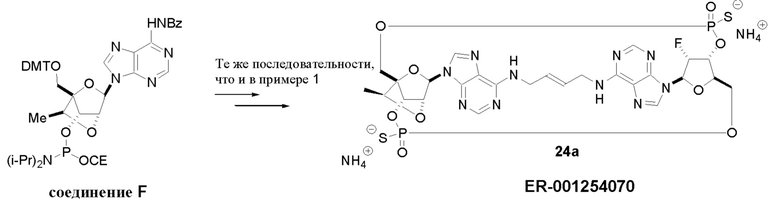
Соединение 24: 1Н ЯМР (400 МГц, METHANOL-d4) δ=8.95 (br s, 1H), 8.29 (br s, 1H), 8.23 (br s, 1H), 8.08 (br s, 1H), 6.31 (br d, J=12.9 Гц, 2H), 6.23-6.07 (m, 1H), 6.04-5.84 (m, 2H), 5.71 (br d, J=50.8 Гц, 1H), 5.27-4.32 (m, 7H), 4.27-4.16 (m, 1H), 4.12 (dd, J=5.7, 11.9 Гц, 1H), 4.07-3.98 (m, 1H), 3.78-3.62 (m, 2H), 1.40 (d, J=7.0 Гц, 3Н)
Пример 13 - Синтез соединения 18, соединения 19 и соединения 20
Путь такого синтеза показан на фиг. 3.
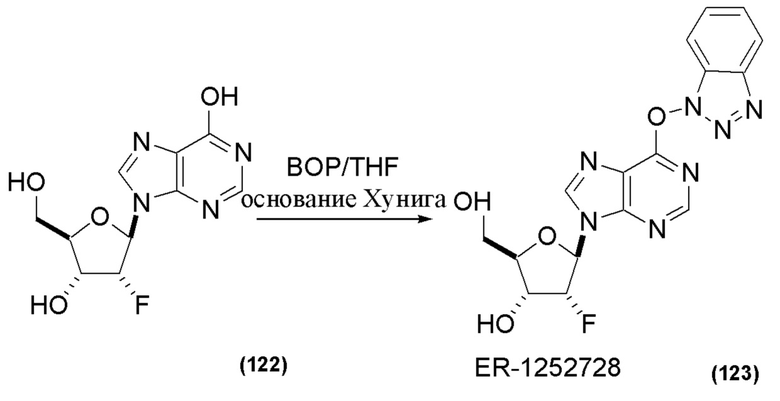
К раствору 9-((2R,3R,4R,5R)-3-фтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-9Н-пурин-6-ола (100 мг, 37 ммоль) и основания Хунига (0,129 мл, 0,74 ммоль) в DMF (1 мл) добавляли ВОР (327 мг, 74 ммоль) при 20°С. Смесь перемешивали при 20°С всю ночь. При помощи UPLC-MS наблюдали завершение реакции. Растворитель удаляли на ротационном испарителе highvac. Остаток растворяли в DCM и очищали с применением Biotage (24 g Si-gel колонка, EtOAc в гептане = 0-100%, 10 об., 100%, 10 об.) с получением 105 мг ВОР аддукта с 73% выходом.
1H ЯМР (400 МГц, METHANOL-d4) δ ppm 3.74-3.84 (m, 1Н) 3.92-4.00 (m, 1Н) 4.12-4.19 (m, 1Н) 4.62-4.74 (m, 1Н) 5.37-5.57 (m, 1Н) 6.41-6.52 (m, 1Н) 7.47-7.55 (m, 1Н) 7.57-7.64 (m, 2Н) 8.03-8.14 (m, 1Н) 8.36-8.46 (m, 1Н) 8.86 (s, 1Н)
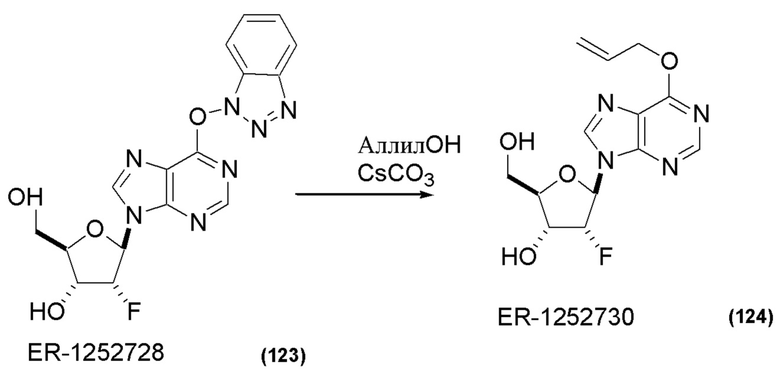
К раствору (2R,3R,4R,5R)-5-(6-((1Н-бензо[d][1,2,3]триазол-1-ил)окси)-9Н-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ола (135 мг, 0,349 ммоль) в аллиловом спирте (3 мл) добавляли карбонат цезия (500 мг, 1,535 ммоль) при 20°С. Смесь перемешивали при 20°С в течение 1 ч. При помощи UPLC-MS наблюдали завершение реакции. Реакционную смесь обрабатывали нас. NHCO3/солевым раствором и экстрагировали EtOAc/Hept. Органический слой сушили над Na2SO4 и фильтровали. Растворитель и летучие органические вещества в фильтрате удаляли на ротационном испарителе. Остаток очищали с применением Biotage (12 g Si-gel колонка, EtOAc в гептане = 0-100%, 10 об., 100%, 10 об.) с получением 100 мг требуемого аллильного аддукта с 92% выходом.
1Н ЯМР (400 МГц, METHANOL-d4) δ ppm 8.56 (s, 1Н) 8.47 (s, 1Н) 6.29-6.41 (m, 1Н)6.06-6.21 (m, 1Н) 5.33-5.53 (m, 2Н) 5.22-5.31 (m, 1Н) 5.02-5.14 (m, 2Н) 4.56-4.71 (m, 1Н) 4.10-4.18 (m, 1Н) 3.88-3.96 (m, 1Н) 3.70-3.79 (m, 1Н)
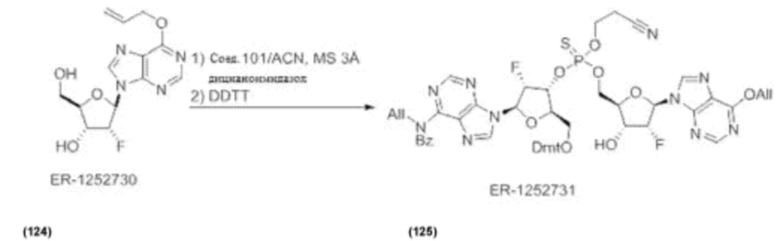
К раствору диизопропилфосфорамидита (150 мг, 0,164 ммоль) и (2R,3R,4R,5R)-5-(6-(аллилокси)-9Н-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ола (76 мг, 0,246 ммоль) в ацетонитриле (1069 мкл, 20,47 ммоль) добавляли молекулярные сита (3 А, 150 мг) при 20°С. Смесь перемешивали при 20°С в течение 1 ч перед добавлением 1H-имидазол-4,5-дикарбонитрила (38,7 мг, 0,328 ммоль) при к.т. Смесь перемешивали при к.т. в течение 1 ч. При помощи UPLC-MS наблюдали завершение реакции. Добавляли (Е)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамид (37,0 мг, 0,18 ммоль). При помощи UPLC-MS наблюдали завершение сульфурирования за 30 мин. Реакционную смесь обрабатывали нас. NHCO3/солевым раствором и экстрагировали EtOAc/Hept. Органический слой сушили над Na2SO4 и фильтровали. Растворитель и летучие органические вещества в фильтрате удаляли на ротационном испарителе. Остаток очищали с применением Biotage (25 g Si-gel колонка, EtOAc в гептане = 0-100%, 15 об., 100%, 10 об.).
1Н ЯМР (500 МГц, METHANOL-d4, два диастереомера ~1:1) δ ppm 8.62-8.63 (m, 1Н) 8.57-8.58 (m, 1Н) 8.48-8.50 (m, 1Н) 8.44-8.46 (m, 1Н) 8.40-8.41 (m, 1Н) 8.38-8.39 (m, 1Н) 8.33-8.35 (m, 1Н) 8.25-8.27 (m, 1Н) 7.22-7.33 (m, 11Н) 7.11-7.20 (m, 17Н) 7.05-7.11 (m, 4Н) 6.83-6.92 (m, 6Н) 6.65-6.77 (m, 11Н) 6.28-6.41 (m, 5Н) 6.23-6.28 (m, 2Н) 6.10-6.22 (m, 5Н) 6.00-6.10 (m, 4Н) 5.89-6.00 (m, 5Н) 5.52-5.57 (m, 2Н) 5.45-5.52 (m, 3Н) 5.41-5.45 (m, 1Н) 5.24-5.34 (m, 4Н) 5.13-5.17 (m, 3Н) 5.07-5.12 (m, 5Н) 4.93-4.98 (m, 4Н) 4.88-4.93 (m, 5Н) 4.73-4.82 (m, 3Н) 4.50-4.59 (m, 2Н) 4.36-4.48 (m, 4Н) 4.27-4.36 (m, 5Н) 4.16-4.27 (m, 5Н) 3.83-4.04 (m, 5Н) 3.73-3.78 (m, 13Н) 3.64-3.73 (m, 4 Н) 3.52-3.58 (m, 1Н) 3.45-3.51 (m, 2Н) 3.35-3.41 (m, 2Н) 3.16-3.23 (m, 2Н) 3.08-3.15 (m, 2Н) 2.84-2.93 (m, 2Н) 2.79-2.83 (m, 2Н) 2.70-2.77 (m, 2Н) 2.61-2.69 (m, 3Н)
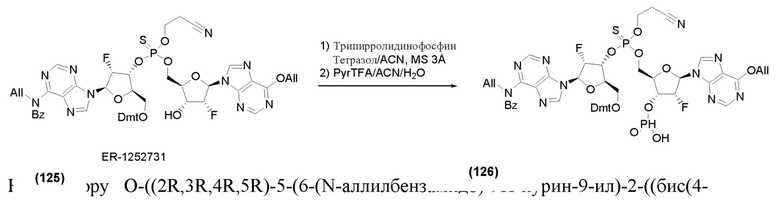 метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ил) O-(((2R,3R,4R,5R)-5-(6-(аллилокси)-9Н-пурин-9-ил)-4-фтор-3-гидрокситетрагидрофуран-2-ил)метил)-O-(2-цианоэтил)фосфоротиоат (120 мг, 0,104 ммоль) в ацетонитриле (4171 мкл, 79,852 ммоль) добавляли молекулярные сита (3А, 1 масс.) при 20°С. Смесь перемешивали при 20°С в течение 1 ч перед добавлением трипирролидинофосфина (71,6 мкл, 0,311 ммоль). 0,45 М тетразол в ACN (253 мкл, 0,114 ммоль) затем добавляли в 7 частях с 2 мин интервалом. TLC (EtOAc/Hept=2:1, RfSM=0,7, RfProd=0,0-0,6) было неполным. Добавляли 2× фосфорилирующего реагента и тетразола. Реакционную смесь перемешивали при к.т. В течение 10 мин. Ни при помощи UPLC-MS, ни TLC не наблюдали остатка SM. Реакционную смесь переносили в колбу, содержащую ацетонитрил (20,8 мл), воду (104 мкл, 5,776 ммоль) и пиридиновую трифторацетатную соль (421 мг, 2,178 ммоль). Смесь перемешивали в течение 10 мин. При помощи UPLC-MS наблюдали образование требуемого продукта. Реакционную смесь смешивали с EtOAc и промывали HCl [0,1 н]/солевым раствором, а затем нас. NHCO3/солевым раствором для предотвращения снятия защитных групп с DMT. Водный слой снова экстрагировали EtOAc (1×). Объединенный органический слой сушили над Na2SO4, а затем фильтровали. Растворители и летучие вещества в фильтрате удаляли на ротационном испарителе с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки.
метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ил) O-(((2R,3R,4R,5R)-5-(6-(аллилокси)-9Н-пурин-9-ил)-4-фтор-3-гидрокситетрагидрофуран-2-ил)метил)-O-(2-цианоэтил)фосфоротиоат (120 мг, 0,104 ммоль) в ацетонитриле (4171 мкл, 79,852 ммоль) добавляли молекулярные сита (3А, 1 масс.) при 20°С. Смесь перемешивали при 20°С в течение 1 ч перед добавлением трипирролидинофосфина (71,6 мкл, 0,311 ммоль). 0,45 М тетразол в ACN (253 мкл, 0,114 ммоль) затем добавляли в 7 частях с 2 мин интервалом. TLC (EtOAc/Hept=2:1, RfSM=0,7, RfProd=0,0-0,6) было неполным. Добавляли 2× фосфорилирующего реагента и тетразола. Реакционную смесь перемешивали при к.т. В течение 10 мин. Ни при помощи UPLC-MS, ни TLC не наблюдали остатка SM. Реакционную смесь переносили в колбу, содержащую ацетонитрил (20,8 мл), воду (104 мкл, 5,776 ммоль) и пиридиновую трифторацетатную соль (421 мг, 2,178 ммоль). Смесь перемешивали в течение 10 мин. При помощи UPLC-MS наблюдали образование требуемого продукта. Реакционную смесь смешивали с EtOAc и промывали HCl [0,1 н]/солевым раствором, а затем нас. NHCO3/солевым раствором для предотвращения снятия защитных групп с DMT. Водный слой снова экстрагировали EtOAc (1×). Объединенный органический слой сушили над Na2SO4, а затем фильтровали. Растворители и летучие вещества в фильтрате удаляли на ротационном испарителе с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки.
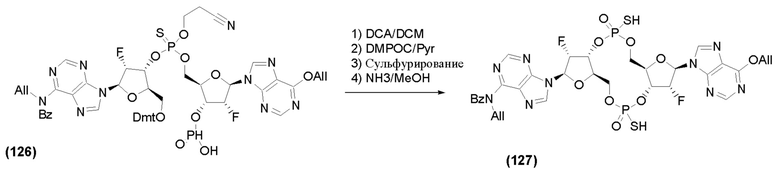
Неочищенное промежуточное соединение гидрофосфоната растворяли в DCM (2635 мкл, 40,946 ммоль) и воде (29,5 мкл, 1,638 ммоль) перед добавлением дихлоруксусной кислоты (135 мкл, 1,638 ммоль) в DCM (2635 мкл, 40,946 ммоль) при 20°С. Смесь перемешивали при 20°С в течение 5 мин. При помощи UPLC-MS наблюдали завершение реакции. Реакцию нейтрализовали пиридином (510 мкл, 6,308 ммоль). Летучие вещества удаляли на ротационном испарителе, а затем на ротационном испарителе highvac. Остаток азеотропировали с пиридином еще раз перед использованием для циклизации.
Неочищенный без защитных групп DMT гидрофосфонат (80 мг, 0,087 ммоль) растворяли в пиридине (1831 мкл, 22,639 ммоль) перед добавлением 2-хлор-5,5-диметил-1,3,2-диоксафосфинан 2-оксида (48,2 мг, 0,261 ммоль) при к.т. Смесь перемешивали при к.т. в течение 10 мин. При помощи UPLC не наблюдали SM пика при 2,1 мин. Реакцию гасили добавлением воды (47,1 мкл, 2,612 ммоль), а затем сразу добавляли 3Н-бензо[с][1,2]дитиол-3-он (21,97 мг, 0,131 ммоль). Через 10 мин при помощи UPLC наблюдали, что не осталось исходного вещества. Реакционную смесь перемешивали при к.т. в течение 1 ч. При помощи UPLC-MS не наблюдали изменений. Реакционную смесь разбавляли EtOAc и промывали нас. NaHCO3 и солевым раствором. Органический слой сушили над Na2SO перед фильтрацией. Остаток после выпаривания растворителя растворяли в МеОН перед добавлением NH3-H2O. Смесь перемешивали в течение 5 мин перед тем, как весь растворитель и летучие вещества удаляли на ротационном испарителе и ротационном испарителе highvac.
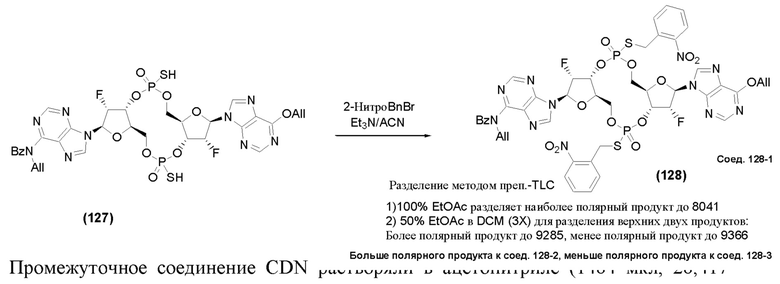 ммоль) и 1,4-диоксане (496 мкл, 5,797 ммоль) перед добавлением TEA (39,6 мкл, 0,284 ммоль) и 1-(бромметил)-2-нитробензола (49,1 мг, 0,227 ммоль) при 20°С. Смесь перемешивали при 20°С в течение 16 ч. При помощи UPLC-MS наблюдали, что реакция не завершилась. Ни при помощи TLC (EtOAc), ни HPLC не наблюдали, что остался орто-нитробензилбромид. Добавляли 1-(бромметил)-2-нитробензол (49,1 мг, 0,227 ммоль) и TEA (39,6 мкл, 0,284 ммоль). Реакционную смесь перемешивали в течение 5 ч. Реакционную смесь обрабатывали нас. NHCO3/солевым раствором и экстрагировали EtOAc/гепт. Органический слой сушили над Na2SO4 и фильтровали. Растворитель и летучие органические вещества в фильтрате удаляли на ротационном испарителе. Остаток очищали с применением Biotage (12 г Si-gel колонка, EtOAc в гептане = 0-100%, 15 об., 100%, 10 об., 10% МеОН в EtOAc, 6 об.). Фракции, содержащие требуемый продукт, объединяли. Выпариванием растворителя получали требуемый продукт. Преп.-TLC (EtOAc) использовали для выделения наиболее полярного изомера, что приводило к конечному продукту соединения 18. Другие два изомера разделяли с применением преп.-TLC (EtOAc/DCM=1:1, проводили 3 раза). Более полярный изомер превращали в соединение 19, а менее полярный изомер превращали в соединение 20.
ммоль) и 1,4-диоксане (496 мкл, 5,797 ммоль) перед добавлением TEA (39,6 мкл, 0,284 ммоль) и 1-(бромметил)-2-нитробензола (49,1 мг, 0,227 ммоль) при 20°С. Смесь перемешивали при 20°С в течение 16 ч. При помощи UPLC-MS наблюдали, что реакция не завершилась. Ни при помощи TLC (EtOAc), ни HPLC не наблюдали, что остался орто-нитробензилбромид. Добавляли 1-(бромметил)-2-нитробензол (49,1 мг, 0,227 ммоль) и TEA (39,6 мкл, 0,284 ммоль). Реакционную смесь перемешивали в течение 5 ч. Реакционную смесь обрабатывали нас. NHCO3/солевым раствором и экстрагировали EtOAc/гепт. Органический слой сушили над Na2SO4 и фильтровали. Растворитель и летучие органические вещества в фильтрате удаляли на ротационном испарителе. Остаток очищали с применением Biotage (12 г Si-gel колонка, EtOAc в гептане = 0-100%, 15 об., 100%, 10 об., 10% МеОН в EtOAc, 6 об.). Фракции, содержащие требуемый продукт, объединяли. Выпариванием растворителя получали требуемый продукт. Преп.-TLC (EtOAc) использовали для выделения наиболее полярного изомера, что приводило к конечному продукту соединения 18. Другие два изомера разделяли с применением преп.-TLC (EtOAc/DCM=1:1, проводили 3 раза). Более полярный изомер превращали в соединение 19, а менее полярный изомер превращали в соединение 20.
Наиболее полярный продукт из первой p-TLC (EtOAc), обозначенный как соединение 128-1: 1Н ЯМР (500 МГц, chloroform-d) δ ppm 8.62 (s, 1Н) 8.55 (s, 1Н) 8.00-8.07 (m, 2Н) 7.99 (s, 1Н) 7.96 (s, 1Н) 7.56-7.64 (m, 2Н) 7.48-7.56 (m, 3Н) 7.42-7.48 (m, 1Н) 7.34-7.42 (m, 2Н) 7.29-7.34 (m, 1Н) 7.18-7.25 (m, 2Н) 6.09-6.21 (m, 3Н) 5.98-6.09 (m, 2Н) 5.63-5.75 (m, 1H) 5.52-5.63 (m, 1H) 5.42-5.51 (m, 2H) 5.32 (dd, J=10.27, 0.98 Гц, 1H) 5.27 (dd, J=17.12, 1.47 Гц, 1Н) 5.13 (d, J=5.87 Гц, 2 Н) 5.09-5.12 (m, 1Н) 4.95-5.05 (m, 2Н) 4.36-4.54 (m, 7Н) 4.23-4.31 (m, 2Н) 4.15-4.21 (m, 1Н)
Более полярный продукт из второго p-TLC, обозначенный как соединение 128-2 (EtOAc/DCM=1:1, 3Х) 1Н ЯМР (400 МГц, methanol-d4) δ ppm 8.75 (s, 1Н) 8.59 (s, 1Н) 8.43 (s, 1Н) 8.39 (s, 1Н) 8.01-8.07 (m, 2Н) 7.38-7.56 (m, 7Н) 7.27-7.35 (m, 1Н) 7.15-7.26 (m, 3Н) 6.45 (d, J=19.91 Гц, 1Н) 6.38 (d, J=19.52 Гц, 1Н) 5.95-6.26 (m, 4Н) 5.83 (dd, J=51.54, 4.69 Гц, 1Н) 5.73 (dd, J=51.54, 4.69 Гц, 1Н) 5.46-5.57 (m, 1Н) 5.28-5.36 (m, 1Н) 5.19-5.28 (m, 1Н) 5.16 (dt, J=5.56, 1.51 Гц, 2Н) 5.03-5.10(m, 1Н)4.95 (br d, J=5.50 Гц, 2Н) 4.41-4.62 (m, 4Н) 4.28-4.40 (m, 2Н) 4.12-4.27 (m, 4Н)
Менее полярный продукт из второго p-TLC, обозначенный как соединение 128-3 (EtOAc/DCM=1:1, 3Х) 1НЯМР (400 МГц, chloroform-d) δ ppm 8.53 (s, 1Н) 8.36 (s, 1Н) 7.95-8.02 (m, 2Н) 7.95 (s, 1Н) 7.89 (s, 1Н) 7.53-7.61 (m, 2Н) 7.44-7.53 (m, 4Н) 7.36-7.44 (m, 2Н) 7.25-7.31 (m, 1Н) 7.12-7.20 (m, 2Н) 5.91-6.18 (m, 4Н) 5.85-5.91 (m, 1Н) 5.72-5.79 (m, 1Н) 5.61-5.72 (m, 1Н) 5.47-5.57 (m, 1Н) 5.40-5.47 (m, 1Н) 5.25-5.32 (m, 1Н) 5.15-5.23 (m, 1Н) 4.99-5.14 (m, 3Н) 4.92 (d, J=5.47 Гц, 2Н) 4.52-4.60 (m, 2Н) 4.37-4.52 (m, 4Н) 4.35 (d, J=3.13 Гц, 1Н) 4.30 (d, J=5.47 Гц, 1Н) 4.25 (d, J=2.74 Гц, 1Н) 4.20 (d, J=4.69 Гц, 1Н)
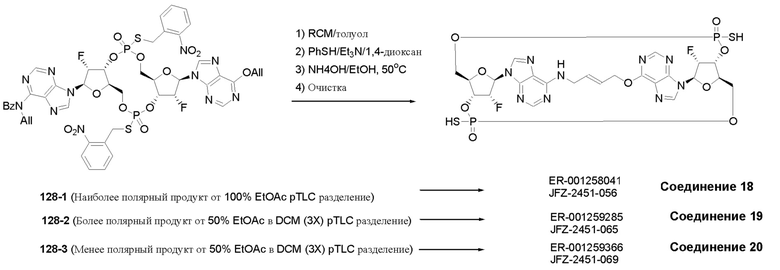
К нагреваемому с обратным холодильником раствору 2-нитробензил-защищенного CDN (10 мг, 8,696 мкмоль) в толуоле (20 мл, 187,761 ммоль) добавляли раствор катализатора Ховейда-Граббса 2-го поколения ((1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(орто-изопропоксифенилметилен)рутений; доступный от SIGMA-ALDRITCH® № каталога 569755; CAS 301224-40-8; 2,73 мг, 4,348 мкмоль) и бензохинона (2,350 мг, 0,022 ммоль) в толуоле (2 мл). Полученный раствор нагревали с обратным холодильником в течение 4 ч (темп, масляной бани 120-125°С). При помощи TLC (EtOAc, Rfsm=0,8, RfProd=0,3) и UPLC-MS наблюдали, что реакция не завершалась. Добавляли 1 мл раствора катализатора Ru. Реакционную смесь нагревали с обратным холодильником еще 2 ч. Все еще оставалось много непрореагировавшего SM. Добавляли еще 1 мл раствора катализатора Ru. Реакционную смесь нагревали с обратным холодильником еще 2 ч. Не наблюдали SM на HPLC с обращенной фазой. Реакционную смесь охлаждали до к.т. и гасили DMSO (0,019 мл, 0,261 ммоль). Выпариванием растворителя получали неочищенный продукт, который очищали на колонке с силикагелем (10 г колонка Si-gel, элюент от 50 до 100%, 15 об., 100%, 10 об.). Фракции, содержащие продукты с требуем. MS, объединяли и выпаривали на ротационном испарителе. Остаток очищали еще раз методом преп.-TLC (EtOAc).
К pTLC очищенному с закрытой мостиковой связью CDN (2,5 мг, 2,228 мкмоль) (транс-изомер) в 1,4-диоксане (0,5 мл, 5,845 ммоль) добавляли тиофенол (0,25 мл, 2,428 ммоль) и TEA (0,25 мл, 1,794 ммоль) при 20°С. Смесь перемешивали при к.т. в течение 3 ч. При помощи UPLC наблюдали завершение превращения.
Добавляли метанол (1,5 мл), за которым следовало добавление 29% NH3 в H2O (1,0 мл). Полученную смесь нагревали при 50°С в течение 6 ч и охлаждали до к.т.
Суспензию реакционной смеси фильтровали и промывали водой (25 мл). Осадки образовывались в фильтрате после промывки водой. Фильтрат снова фильтровали с удалением некоторой части твердого вещества.
Полученный фильтрат экстрагировали смесью тол./геп. (3×, 1/1, 25 мл каждого). Водный слой концентрировали в вакууме, а затем растворяли в воде (6 мл). Осадки фильтровали еще раз. При помощи UPLC наблюдали образование требуемого продукта. Продукт очищали методом HPLC с обращенной фазой с применением того же способа для очистки других основных закрытых CDN аналогов.
Для соединения 18, LCMS: MS m/z 748,11 [М+Н]+ 746,15 [М-Н]-
Для соединения 19, LCMS: MS m/z 748,06 [М+Н]+ 746,17 [М-Н]-
Для соединения 20, LCMS: MS m/z 748,09 [М+Н]+ 746,21 [М-Н]-
Пример 13 - Синтез соединения 26
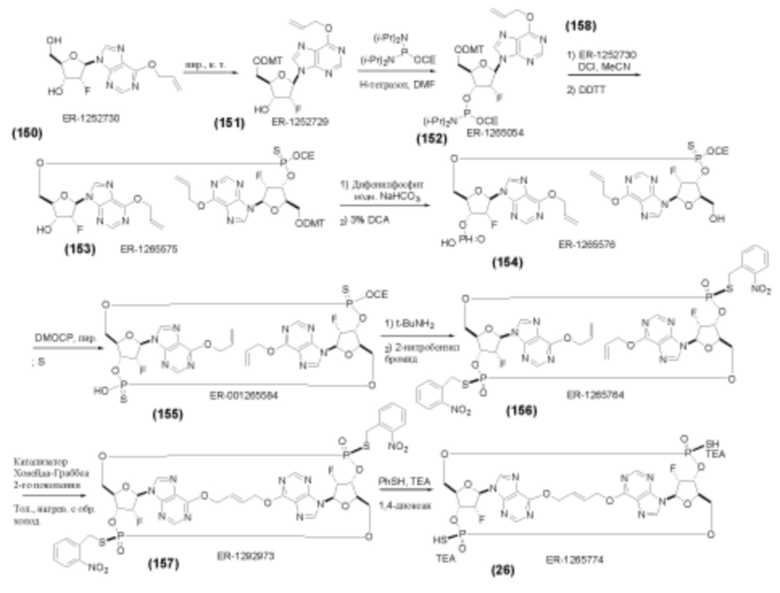
Соединение 151
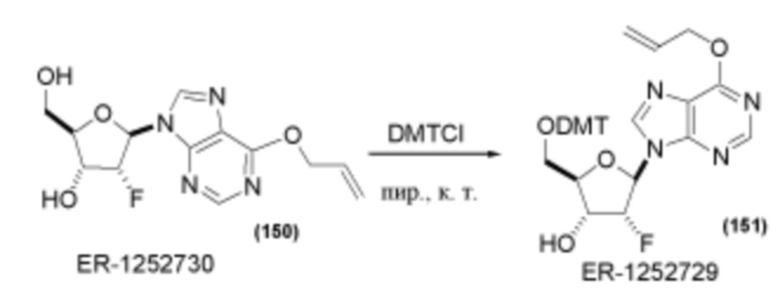
К раствору соединения 150 (0,70 г, 2,256 ммоль) в пиридине (10,5 мл) при 0°С добавляли 4,4'-диметокситритилхлорид (0,803 г, 2,369 ммоль). Полученный раствор перемешивали всю ночь, при том нагревали до температуры окружающей среды. После завершения (наблюдали при помощи LCMS) добавляли насыщенный раствор NH4Cl (10 мл) и МТВЕ (20 мл). Слои разделяли и водный слой экстрагировали МТВЕ/EtOAc (3/1, 12 мл). Объединенные органические слои промывали 30% водным раствором NaCl (5 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой неочищенного продукта методом колоночной хроматографии на силикагеле (SiO2 25 г, 50% - 70% EtOAc в н-гептане) получали 0,942 г соединения 151.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.47 (s, 1H), 8.11 (s, 1H), 7.43-7.35 (m, 2Н), 7.30-7.26 (m, 4Н), 7.25-7.17 (m, 3Н), 6.82-6.75 (m, 4Н), 6.28 (dd, J=2.3, 17.6 Гц, 1H), 6.22-6.10 (m, 1H), 5.63 (ddd, J=2.7, 4.7, 53.1 Гц, 1H), 5.47 (qd, J=1.4, 17.3 Гц, 1H), 5.31 (dd, J=1.2, 10.6 Гц, 1H), 5.13 (t, J=1.4 Гц, 1H), 5.12 (t, J=1.2 Гц, 1H), 4.89-4.76 (m, 1H), 4.21 (td, J=3.2, 6.8 Гц, 1H), 3.78 (s, 6Н), 3.55 (dd, J=2.7, 10.9 Гц, 1H), 3.44 (dd, J=4.3, 10.9 Гц, 1H), 2.29-2.21 (m, 1Н).
Соединение 152
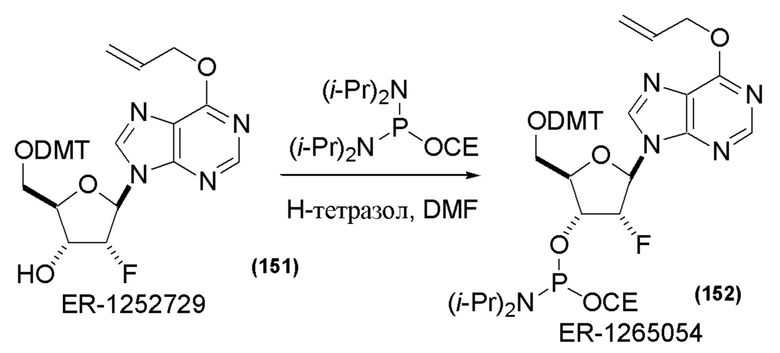
К раствору соединения 151 (0,942 г, 1,538 ммоль) в DMF (7,5 мл) при температуре окружающей среды добавляли 3-((бис(диизопропиламино)фосфино)окси)пропаннитрил (0,927 г, 3,075 ммоль), 0,45 М 1,2,3,4-тетразол (2,8 мл, 1,2 ммоль) и 1-метилимидазол (30 мкл, 0,38 ммоль). Полученный раствор перемешивали при температуре окружающей среды в течение 4 ч. После завершения (наблюдали при помощи LCMS) добавляли TEA (0,50 мл, 3,6 ммоль), DMF (11,3 мл) и воду (1,9 мл). Полученную смесь экстрагировали трижды н-гептаном (3 мл каждый раз). Слой DMF разбавляли водой (4 мл) и экстрагировали смесью толуола/н-гептана (1/1, 10 мл). Объединенные органические слои дважды промывали 30% водным раствором NaCl (3 мл каждый раз), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 50 г, 33% - 40% EtOAc в н-гептане с 1% TEA) получали 1,115 г соединения 152 (3:2 диастереомерная смесь) в виде белого пенящегося твердого вещества.
Соединение 153
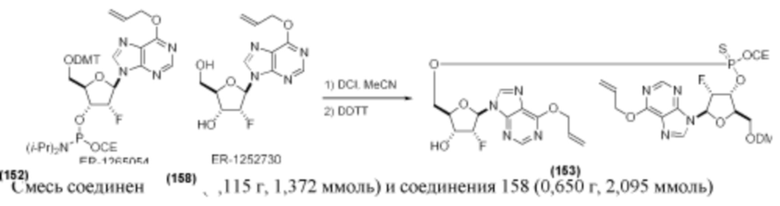 дважды азеотропировали MeCN (20 мл каждый раз). К полученному остатку добавляли MeCN (20,0 мл) и 1Н-имидазол-4,5-дикарбонитрил (0,243 г, 2,058 ммоль). После завершения реакции (наблюдали при помощи LCMS) реакционную смесь обрабатывали (Е)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамидом (0,422 г, 2,058 ммоль). После завершения сульфурирования добавляли насыщенный раствор NaHCO3 (20 мл) и МТВЕ (30 мл). Слои разделяли и водный слой экстрагировали смесью МТВЕ/EtOAc (15/15 мл). Объединенные органические слои промывали 30% водным раствором NaCl (10 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 50 г, 33% - 100% EtOAc в н-гептане с 1% TEA) получали 0,88 г соединения 153.
дважды азеотропировали MeCN (20 мл каждый раз). К полученному остатку добавляли MeCN (20,0 мл) и 1Н-имидазол-4,5-дикарбонитрил (0,243 г, 2,058 ммоль). После завершения реакции (наблюдали при помощи LCMS) реакционную смесь обрабатывали (Е)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамидом (0,422 г, 2,058 ммоль). После завершения сульфурирования добавляли насыщенный раствор NaHCO3 (20 мл) и МТВЕ (30 мл). Слои разделяли и водный слой экстрагировали смесью МТВЕ/EtOAc (15/15 мл). Объединенные органические слои промывали 30% водным раствором NaCl (10 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 50 г, 33% - 100% EtOAc в н-гептане с 1% TEA) получали 0,88 г соединения 153.
LC/MS (ESI) m/z 1076,48 [M+Na]+.
Соединение 154
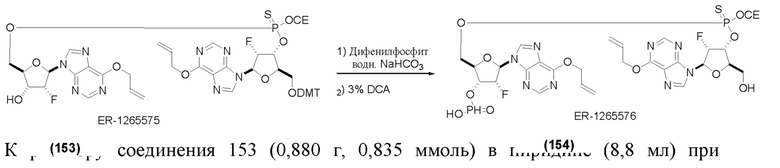 температуре окружающей среды добавляли дифенилфосфит (0,323 мл, 1,67 ммоль). Реакционную смесь перемешивали в течение 1 ч и наблюдали при помощи LCMS. После завершения смесь добавляли в смесь насыщенного водного раствора NaHCO3 (13,2 мл) и воды (4,4 мл), при этом поддерживали внутреннюю Т ниже 30°С, промывали EtOAc (8,8 мл). Полученную смесь перемешивали при температуре окружающей среды и гидролиз наблюдали при помощи LCMS. После завершения смесь дважды экстрагировали смесью EtOAc/МТВЕ (1/1, 26 мл). Объединенные органические слои промывали 30% водным раствором NaCl (3 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток дважды азеотропировали толуолом (7 мл каждый раз). Неочищенный продукт растворяли в дихлорметане (6,6 мл) и обрабатывали водой (0,15 мл, 8,35 ммоль) и 6% дихлоруксусной кислотой (0,41 мл, 5,0 ммоль) в дихлорметане (6,6 мл) при температуре окружающей среды. После завершения снятия защитных групп с DMT (наблюдали при помощи LCMS) добавляли триэтилсилан (2,7 мл, 17 ммоль). Через 10 мин перемешивания полученную смесь обрабатывали пиридином (4,4 мл) и TEA (1 мл) и концентрировали в вакууме. Остаток трижды растирали в порошок с н-гептаном (8,8 мл каждый раз) и дважды азеотропировали MeCN. Неочищенный продукт (соединение 154) использовали на следующей стадии без дополнительной очистки.
температуре окружающей среды добавляли дифенилфосфит (0,323 мл, 1,67 ммоль). Реакционную смесь перемешивали в течение 1 ч и наблюдали при помощи LCMS. После завершения смесь добавляли в смесь насыщенного водного раствора NaHCO3 (13,2 мл) и воды (4,4 мл), при этом поддерживали внутреннюю Т ниже 30°С, промывали EtOAc (8,8 мл). Полученную смесь перемешивали при температуре окружающей среды и гидролиз наблюдали при помощи LCMS. После завершения смесь дважды экстрагировали смесью EtOAc/МТВЕ (1/1, 26 мл). Объединенные органические слои промывали 30% водным раствором NaCl (3 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток дважды азеотропировали толуолом (7 мл каждый раз). Неочищенный продукт растворяли в дихлорметане (6,6 мл) и обрабатывали водой (0,15 мл, 8,35 ммоль) и 6% дихлоруксусной кислотой (0,41 мл, 5,0 ммоль) в дихлорметане (6,6 мл) при температуре окружающей среды. После завершения снятия защитных групп с DMT (наблюдали при помощи LCMS) добавляли триэтилсилан (2,7 мл, 17 ммоль). Через 10 мин перемешивания полученную смесь обрабатывали пиридином (4,4 мл) и TEA (1 мл) и концентрировали в вакууме. Остаток трижды растирали в порошок с н-гептаном (8,8 мл каждый раз) и дважды азеотропировали MeCN. Неочищенный продукт (соединение 154) использовали на следующей стадии без дополнительной очистки.
LC/MS (ESI) m/z 814,29 [М-Н]-.
Соединение 155
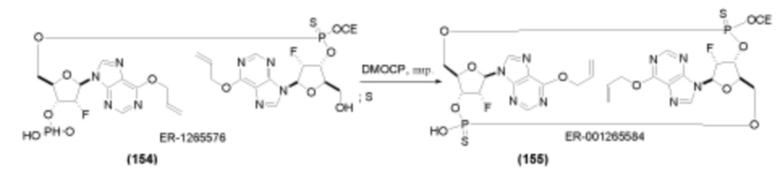
К раствору соединения 154 (0,681 г, 0,835 ммоль) в пиридине (68 мл) при температуре окружающей среды добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфоринан-2-оксид (0,462 г, 2,505 ммоль). Полученный раствор перемешивали в течение 1 ч при температуре окружающей среды и наблюдали при помощи LCMS. После завершения добавляли воду (0,45 мл, 25 ммоль) (10 экв. DMOCP) и серу (0,134 г, 4,175 ммоль). После завершения сульфурирования (наблюдали при помощи LCMS) реакционную смесь обрабатывали насыщенным водным раствором NaHCO3 (13,6 мл) и концентрировали в вакууме. Остаток обрабатывали EtOAc (27 мл) и водой (13.6 мл). Слои разделяли и водный слой экстрагировали смесью EtOAc/МТВЕ (1/1, 27 мл). Объединенные органические слои промывали 30% водным раствором NaCl (13,6 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 25 г, 0% - 10% МеОН в EtOAc) получали 0,693 г (теоретический выход) соединения 155.
LC/MS (ESI) m/z 830,23 [М+Н]+.
Соединение 156
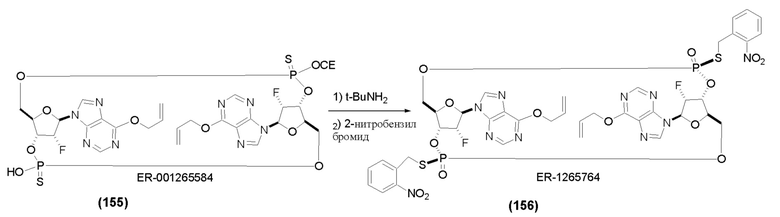
К раствору соединения 155 (0,693 г, 0,835 ммоль) в ацетонитриле (14 мл) при температуре окружающей среды добавляли трет-бутиламин (14 мл, 131 ммоль). Полученный раствор перемешивали в течение 10 мин и наблюдали при помощи LCMS. После завершения, реакционную смесь концентрировали в вакууме и дважды азеотропироовали MeCN. К остатку добавляли ацетонитрил (14 мл) и 2-нитробензилбромид (0,541 г, 2,51 ммоль). После перемешивания всю ночь реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 100 г, 75% - 100% EtOAc в н-гептане и 0% - 10% МеОН в EtOAc) с получением 0,103 г соединения 156.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.59 (s, 2Н), 8.05 (br d, J=8.2 Гц, 2H), 8.03 (s, 2H), 7.58 (d, J=7.4 Гц, 2H), 7.48 (t, J=7.4 Гц, 2H), 7.45-7.37 (m, 2H), 6.20 (br d, J=18.8 Гц, 2H), 6.21-6.08 (m, 2H), 5.98-5.83 (m, 2H), 5.69 (dd, J=4.7, 52.0 Гц, 1H), 5.47 (br d, J=17.2 Гц, 2H), 5.31 (br d, J=10.6 Гц, 2H), 5.14 (br d, J=5.5 Гц, 4H), 4.54 - 4.46 (m, 2H), 4.45-4.40 (m, 2H), 4.37-4.26 (m, 4H), 4.21-4.11 (m, 2H).
На основе методики синтеза, данных ЯМР (симметричные) и времени удерживания HPLC (самый медленный элюирующий изомер), заявители полагали, что соединение 156 обладает RR фосфорной стереохимией. Такое стереохимическое определение будет подвержено подтверждению методом рентгеновской кристаллографии.
Соединение 157
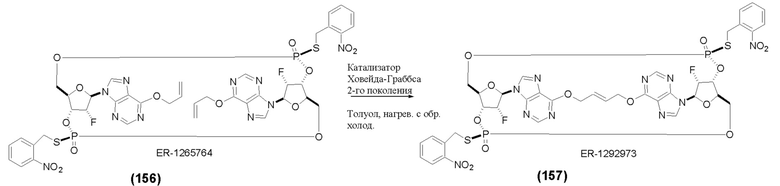
К раствору соединения 156 (0,103 г, 0,098 ммоль) в толуоле (103 мл) с обратным холодильником добавляли раствор катализатора Ховейда-Граббса 2-го поколения ((1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(орто-изопропоксифенилметилен)рутений; доступный от SIGMA-ALDRITCH® № каталога 569755; CAS 301224-40-8; 31 мг, 0,049 ммоль) и хинона (32 мг, 0,295 ммоль) в толуоле (10 мл). Смесь перемешивали с обратным холодильником в течение 2 ч и добавляли дополнительный катализатор (16 мг, 0,025 ммоль). После завершения реакционную смесь охлаждали до температуры окружающей среды и обрабатывали DMSO (0,14 мл, 2,0 ммоль) всю ночь. Полученную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 0% - 10% метанола в EtOAc) с получением требуемого продукта, который дополнительно очищали методом преп. TLC (EtOAc) с получением 3,6 мг соединения 157.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.11 (dd, J=1.2, 8.2 Гц, 2Н), 8.09 (s, 2Н), 7.68 (s, 2Н), 7.67-7.58 (m, 4Н), 7.52-7.48 (m, 2Н), 6.26 (d, J=17.6 Гц, 2Н), 6.02-5.97 (m, 2Н), 5.97-5.88 (m, 2Н), 5.68 (dd, J=3.9, 50.8 Гц, 2Н), 5.15 (br d, J=10.9 Гц, 2Н), 4.99 (br d, J=10.9 Гц, 2H), 4.56-4.44 (m, 8Н), 4.25 (br t, J=6.1 Гц, 2H).
Соединение 26
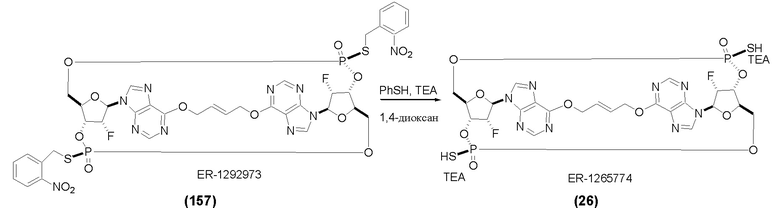
К соединению 157 (3,6 мг, 3,5 мкмоль) добавляли 1,4-диоксан (0,11 мл), тиофенол (0,045 мл, 0,44 ммоль) и TEA (0,054 мл, 0,39 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения превращения добавляли воду (0,5 мл). Полученную смесь экстрагировали трижды смесью н-гептана/толуола (1/1, 0,4 мл каждый раз), а затем толуолом (0,3 мл). Водный слой концентрировали в вакууме и обрабатывали водой (0,5 мл). Полученное твердое вещество отфильтровывали, промывали водой (0,5 мл). Сублимационной сушкой объединенных фильтратов получали 2,0 мг бис-ТЕА соли соединения 26 в виде белого пенящегося твердого вещества.
1Н ЯМР (400 МГц, METHANOL-d4) δ=8.67 (s, 2Н), 8.18 (s, 2Н), 6.41 (d, J=14.8 Гц, 2Н), 6.07-6.01 (m, 2Н), 5.56 (dd, J=3.1, 51.2 Гц, 2Н), 5.48-5.41 (m, 2Н), 5.08 (br d, J=12.1 Гц, 2H), 4.99-4.88 (m, 2Н), 4.52 (br d, J=12.5 Гц, 2H), 4.40 (br d, J=9.8 Гц, 2H), 3.98 (dd, J=5.7, 12.3 Гц, 2H), 3.14 (q, J=7.4 Гц, 12H), 1.27 (t, J=7.4 Гц, 18H).
Пример 15 - Синтез соединения 27
Соединение 27 получали из соединения 159, побочного продукта стадия 11b пути для соединения 1, показанного на фиг. 2В.
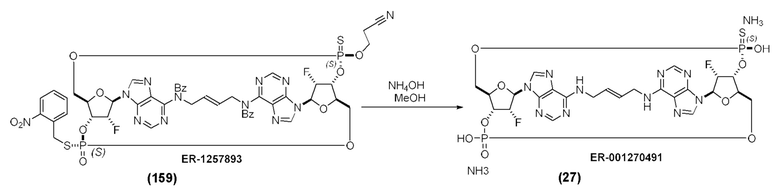
К раствору соединения 159 (2,0 г, 1,75 ммоль) в метаноле (24 мл) добавляли 28% гидроксид аммония (12 мл). Полученную смесь перемешивали при 50°С в течение 5 ч и охлаждали до температуры окружающей среды. После завершения (наблюдали при помощи LCMS) реакционную смесь охлаждали до температуры окружающей среды, обрабатывали водой (20 мл) и экстрагировали трижды смесью толуола/EtOAc (1/1, 24 мл каждый раз). Водный слой подкисляли 1,0 М хлористоводородной кислотой (3,5 мл, 3,5 ммоль), перемешивали при температуре окружающей среды в течение 30 мин и 0°С в течение 30 мин. Полученную взвесь фильтровали, промывали водой (20 мл). Кек сушили в вакуумной печи при 35°С всю ночь и растворяли в смеси 28% NH4OH (2 мл) и МеОН (20 мл). Полученный раствор концентрировали в вакууме и обрабатывали EtOH (4 мл) с получением взвеси. Полученное твердое вещество собирали фильтрацией и сушили в вакууме. Получали 70 мг соединения 27 в виде белого твердого вещества.
1Н ЯМР (400 МГц, METHANOL-d4) δ=9.03 (br s, 1H), 8.31 (br s, 1H), 8.26 (br s, 1H), 8.13 (br s, 1H), 6.43-6.24 (m, 2H), 5.97-5.83 (m, 2H), 5.71 (br d, J=51.6 Гц, 1H), 5.64 (br d, J=50.0 Гц, 1H), 5.12-4.99 (m, 1H), 4.96-4.90 (m, 1H), 4.68-4.32 (m, 6H), 4.14-3.97 (m, 2H), 3.77-3.62 (m, 2H).
Пример 16 - Синтез соединения 28
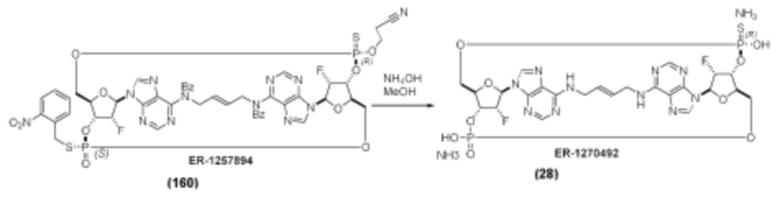
Соединение 28 получали из соединения 160, продукта стадии 11 пути для соединения 1, показанного на фиг.2 В, с применением того же способа для соединения 27.
1Н ЯМР (400 МГц, METHANOL-d4) δ=8.70 (br s, 1H), 8.49 (br s, 1H), 8.24 (br s, 1H), 8.10 (br s, 1H), 6.52-6.26 (m, 2H), 5.94-5.78 (m, 2H), 5.69 (br d, J=54.7 Гц, 1H), 5.48-5.26 (m, 1H), 5.14 (br d, J=53.1 Гц, 2H), 5.06-4.94 (m, 1H), 4.74-4.19 (m, 4H), 4.10-3.93 (m, 2H), 3.76-3.58 (m, 2H)
Пример 17 - Синтез соединения 29
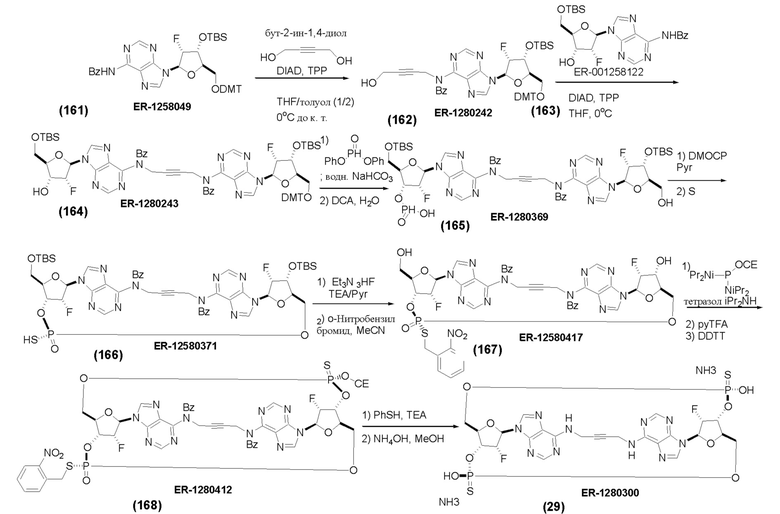
Соединение 162
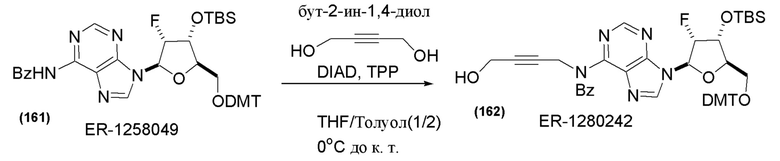
Соединение 161 (5,0 г, 6,329 ммоль) и бут-2-ин-1,4-диол (1,907 г, 22,153 ммоль) дважды азеотропировали THF. Остаток растворяли в THF (50,0 мл) и толуоле (75 мл). Добавляли трифенилфосфин (2,158 г, 8,228 ммоль) и полученный раствор охлаждали ниже 5°С. По каплям добавляли DIAD (1,60 мл, 8,23 ммоль), при этом поддерживали внутреннюю Т ниже 10°С. Полученный раствор перемешивали при 0-5°С, при этом за прогрессом наблюдали при помощи LCMS. После завершения реакционную смесь концентрировали в вакууме с удалением большей части THF. Остаток разбавляли МТВЕ (50,0 мл) и дважды промывали 30% водным раствором NaCl (40,0 мл каждый раз) и дважды водой (25 мл каждый раз). Органический раствор концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 100 г, 50% - 70% EtOAc в н-гептане, буферизированном 1% TEA) с получением 4,671 г соединения 162.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.61 (s, 1H), 8.14 (s, 1H), 7.43 (d, J=7.2 Гц, 2Н), 7.33-7.27 (m, 3Н), 7.24-7.18 (m, 8Н), 7.05-7.01 (m, 2Н), 6.76 (d, J=8.6 Гц, 4Н), 6.20 (dd, J=2.7, 17.2 Гц, 1H), 5.59 (td, J=3.1, 53.1 Гц, 1H), 5.19 (br d, J=2.3 Гц, 2H), 4.93-4.83 (m, 1H), 4.18-4.15 (m, 1H), 4.01 (br d, J=6.3 Гц, 2H), 3.78 (s, 6H), 3.53 (dd, J=2.7, 10.9 Гц, 1H), 3.17 (dd, J=3.7, 11.1 Гц, 1H), 0.85 (s, 9H), 0.10 (s, 3H), 0.02 (s, 3H).
Соединение 164
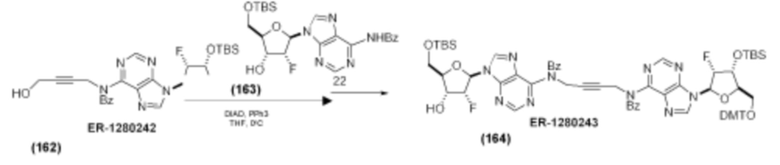
Соединение 162 (4,671 г, 5,444 ммоль), соединение 163 (3,32 г, 6,805 ммоль) и трифенилфосфин (1,856 г, 7,077 ммоль) растворяли в THF (56,1 мл) и охлаждали ниже 5°С. К полученному раствору добавляли DIAD (1,3 мл, 6,8 ммоль), при этом поддерживали внутреннюю Т ниже 10°С. После завершения израсходования соединения 162 (наблюдали при помощи LCMS) реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 340 г, 55% - 70% EtOAc в н-гептане, буферизированном 1% TEA) с получением 3,879 г соединения 164 в виде белого пенящегося твердого вещества.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.51 (s, 1H), 8.45 (s, 1H), 8.20 (s, 1H), 8.05 (s, 1H), 7.41 (d, J=7.1 Гц, 2Н), 7.36-7.32 (m, 4Н), 7.28-7.13 (m, 8Н), 6.98 (t, J=7.8 Гц, 2Н), 6.79-6.74 (m, 4Н), 6.29 (dd, J=2.0, 16.4 Гц, 1H), 6.19 (dd, J=2.7, 17.2 Гц, 1H), 5.53 (ddd, J=2.7,4.7, 53.1 Гц, 1H), 5.33 (ddd, J=2.0, 3.9, 53.1 Гц, 1H), 5.23-5.21 (m, 1H), 5.05-5.01 (m, 2Н), 4.99-4.94 (m, 2Н),4.81 (ddd, J=4.3, 6.7, 16.7 Гц, 1H), 4.71-4.60 (m, 1H), 4.21-4.14 (m, 2Н), 4.08-4.03 (m, 1H), 3.89 (dd, J=3.1, 11.7 Гц, 1H), 3.77 (s, 6Н), 0.91 (s, 9Н), 0.84 (s, 9Н), 0.09 (s, 3Н), 0.08 (s, 6Н), 0.00 (s, 3Н).
Соединение 165
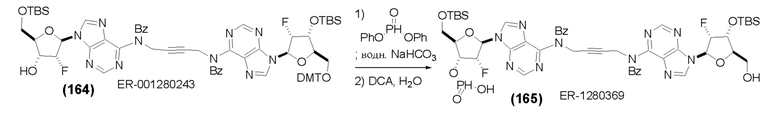
К раствору соединения 164 (1,5 г, 1,13 ммоль) в пиридине (12,0 мл) добавляли дифенилфосфит (0,35 мл, 1,8 ммоль) при температуре окружающей среды. Полученный раствор перемешивали при температуре окружающей среды, за этим наблюдали при помощи LCMS. После завершения реакционную смесь добавляли в смесь насыщенного водного NaHCO3 (22,5 мл) и воды (7,5 мл), при этом поддерживали внутреннюю Т ниже 30°С. После завершения гидролиза (наблюдали при помощи LCMS) полученную смесь дважды экстрагировали смесью EtOAc/МТВЕ (30,0/30,0 мл). Объединенные органические слои промывали 30% водн. NaCl (15,0 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток трижды азеотропировали толуолом и растворяли в дихлорметане (11 мл), воде (0,20 мл, 11 ммоль) и 6% дихлоруксусной кислоте (0,57 мл, 6,9 ммоль) в дихлорметане (11 мл) при температуре окружающей среды. После завершения удаления группы DMT (наблюдали при помощи LCMS) добавляли триэтилсилан (1,8 мл, 11 ммоль). После 10 мин перемешивания добавляли TEA (1,6 мл, 11 ммоль) и полученную смесь концентрировали в вакууме. Остаток растворяли в EtOAc (22,5 мл), промывали водой (3,8 мл) и насыщенным водным NaHCO3 (8%) (3,0 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт дважды растирали в порошок с н-гептаном (33,8 мл) и смесью н-гептана (9,0 мл) и толуола (3,0 мл). Супернатант декантировали и твердое вещество, оставшееся на дне, растворяли в ацетонитриле. Полученный раствор концентрировали в вакууме и азеотропировали дважды в ацетонитриле. Неочищенный продукт, соединение 165, использовали на следующей стадии без очистки (допустимый теоретический выход 100%).
LC/MS (ESI) m/z 1089.74 [М+Н]+.
Соединение 166

К раствору соединения 165 (1,231 г, 1,13 ммоль) в пиридине (100 мл) при температуре окружающей среды добавляли TEA (0,47 мл, 3,4 ммоль) и DMOCP (0,375 мг, 2,03 мл). Полученный раствор перемешивали при температуре окружающей среды, за этим наблюдали при помощи LCMS. После завершения добавляли воду (0,41 мл, 22,6 ммоль), а затем серу (0,181 г, 5,651 ммоль). После завершения сульфурирования (наблюдали при помощи LCMS) добавляли насыщенный водный NaHCO3 (8%) (24,6 мл) и воду (10 мл). Полученную смесь концентрировали в вакууме до ~50 мл и дважды экстрагировали смесью МТВЕ/EtOAc (31/31 мл) каждый раз. Объединенные органические слои промывали 30% водн. NaCl (25 мл), сушили над MgSO4, фильтровали, концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 100 г, 0-10% метанола в этилацетате, буферизированном 1% TEA) с получением соединения 166 (0,484 г).
LC/MS (EST) m/z 1103.65 [М+Н]+.
Соединение 167
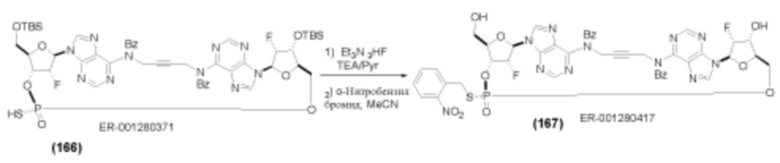
К раствору соединения 166 (0,484 г, 402 ммоль) в пиридине (1,2 мл) при температуре окружающей среды добавляли Et3N 3HF (0,581 мл, 3,563 ммоль). Полученную смесь перемешивали при температуре окружающей среды, за этим наблюдали при помощи LCMS. После завершения превращения добавляли метокситриметилсилан (3,9 мл, 28 ммоль). Через 20 мин перемешивания при температуре окружающей среды супернатант декантировали. К остатку добавляли толуол (5 мл) и через 10 мин перемешивания слой толуола декантировали. Ту же операцию повторяли еще раз. Полученный неочищенный продукт растворяли в дихлорметане (10 мл), промывали насыщенным водным NH4Cl (27 масс. %) (5 мл) и 30% водн. NaCl (2,4 мл) и сушили над MgSO4. Фильтрацией после концентрирования полученного органического слоя получали 0,386 г бледно-коричневого твердого вещества, которое дважды азеотропировали MeCN. Полученный неочищенный продукт растворяли в MeCN (4,6 мл) и обрабатывали 2-нитробензилбромидом (0,103 г, 0,475 ммоль) при температуре окружающей среды. После завершения алкилирования (наблюдали при помощи LCMS) реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 0-5% МеОН в EtOAc) с получением 0,251 г соединения 167 в виде белого твердого вещества.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.42 (br s, 1H), 8.31 (br s, 1H), 8.22 (br s, 1H), 8.10-8.04 (m, 1H), 7.93 (s, 1H), 7.53-7.37 (m, 8H), 7.32-7.27 (m, 3H), 7.24-7.16 (m, 2H), 6.18 (br d, J=16.8 Гц, 1H), 6.26-6.14 (m, 1H), 5.56 (d, J=53.1 Гц, 1H), 5.48-5.37 (m, 1H), 5.31 (brs, 1H), 5.15 (brd, J=17.6 Гц, 1H), 4.92 (d, J=17.6 Гц, 1H), 4.82 (d, J=17.2 Гц, 1H), 4.88-4.77 (m, 1H), 4.76-4.66 (m, 1H), 4.58-4.33 (m, 3H), 4.29 (br d, J=5.9 Гц, 1H), 4.21 (br s, 1H), 4.13-4.06 (m, 1H), 3.93-3.79 (m, 2H).
Соединение 168
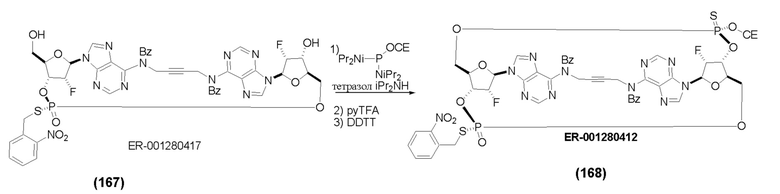
Соединение 167 (0,224 г, 0,222 ммоль) и 3-
(бис(диизопропиламино)фосфиноокси)пропаннитрил (0,081 мл, 0,255 ммоль) растворяли в MeCN (5 мл) и концентрировали. Ту же операцию повторяли еще два раза. Полученную смесь растворяли в дихлорметане (2,2 мл), охлаждали до 0°С и обрабатывали диизопропиламмония тетразолидом (0,019 г, 0,111 ммоль). Реакционную смесь доводили до температуры окружающей среды в течение ночи и разбавляли MeCN (2,2 мл). Полученный раствор добавляли в течение 10 ч через шприцевой насос к раствору пиридиновой трифторацетатной соли (0,129 г, 0,665 ммоль) в MeCN (18 мл). После дополнительного перемешивания в течение 1 ч 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (0,04 мл, 0,12 ммоль) в MeCN (1 мл) добавляли в течение 4 ч. Добавляли (Е)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамид (0,064 г, 0,311 ммоль). После завершения сульфурирования (наблюдали при помощи LCMS) реакционную смесь концентрировали в вакууме и полученный остаток поглощали МТВЕ (4,5 мл). Полученный органический раствор промывали насыщенным водным NaHCO3 (8%) (4 мл) и водой (2 мл). Объединенные водные слои снова экстрагировали смесью МТВЕ/EtOAc (4/4 мл). Объединенные органические слои дважды промывали 30% водным NaCl (2 мл каждый раз), сушили над MgSO4, фильтровали, концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 50% - 100% EtOAc в н-гептане) с получением 64 мг соединения 168.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.57 (s, 1H), 8.18 (s, 1H), 8.06-8.01 (m, 1H), 7.91 (s, 1H), 7.62-7.56 (m, 2Н), 7.55-7.50 (m, 2Н), 7.49-7.43 (m, 3Н), 7.42-7.34 (m, 2Н), 7.30-7.18 (m, 5Н), 6.33 (d, J=15.2 Гц, 1H), 6.10 (d, J=20.7 Гц, 1H), 5.95-5.77 (m, 1H), 5.54 (dd, J=4.3, 52.8 Гц, 1H), 5.46 (dd, J=3.9, 50.8 Гц, 1H), 5.32-5.18 (m, 1H), 5.10 (d, J=17.6 Гц, 1H), 4.89 (d, J=17.6 Гц, 1H), 4.85-4.81 (m, 2Н), 4.78 (br d, J=12.1 Гц, 1H), 4.64 (br dd, J=4.1, 9.2 Гц, 1H), 4.55 (dd, J=2.7, 12.1 Гц, 1H), 4.52-4.44 (m, 3Н), 4.38-4.20 (m, 4Н), 2.75 (td, J=6.3, 17.6 Гц, 1H), 2.58 (td, J=5.9, 17.2 Гц, 1Н).
Соединение 29
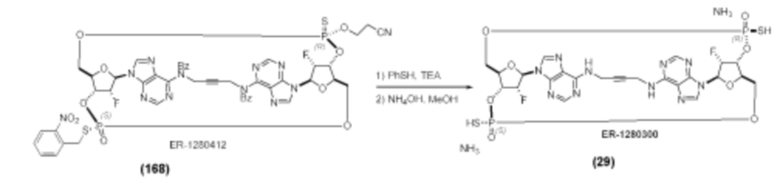
К раствору соединения 168 (40 мг, 0,035 ммоль) в 1,4-диоксане (0,5 мл) добавляли тиофенол (0,24 мл, 2,3 ммоль) и TEA (0,24 мл, 1,7 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения добавляли метанол (0,64 мл) и 28% гидроксид аммония (0,64 мл). Полученную смесь нагревали до 50°С, перемешивали в течение 5 ч и охлаждали до температуры окружающей среды. После завершения снятия защитных групп (наблюдали при помощи LCMS) полученную смесь обрабатывали водой (0,80 мл) и трижды экстрагировали смесью н-гептана/толуола (1/1, 0,5 мл каждый раз), а затем толуолом (0,3 мл). Водный слой концентрировали в вакууме при 40-50°С и обрабатывали водой (1 мл). Полученное твердое вещество отфильтровывали, промывали водой (0,3 мл). Объединенные фильтраты обрабатывали 1,0 М HCl (0,07 мл, 0,07 ммоль). Полученную взвесь фильтровали и промывали водой (1 мл). Фильтрационный кек растворяли в 2,0 М растворе аммиака (1,0 мл, 2,0 ммоль) в МеОН. Полученный раствор концентрировали в вакууме до 0,5 мл и обрабатывали EtOH (0,5 мл). Ту же операцию повторяли еще два раза. К полученному раствору по каплям добавляли EtOAc (0,9 мл). Возникало осаждение. Полученное твердое вещество собирали фильтрацией, промывали 4/1 смесью EtOAc/EtOH (0,4 мл) и сушили в вакууме всю ночь при температуре окружающей среды. Получали 12 мг соединения 29.
1H ЯМР (400 МГц, METHANOL-d4) δ=8.88 (br s, 1H), 8.50 (br s, 1H), 8.20 (br s, 1H), 8.12 (br s, 1H), 6.42-6.17 (m, 2H), 5.84-5.54 (m, 1H), 5.52 (d, J=51.6 Гц, 1H), 5.06-4.78 (m, 2H), 4.77-4.55 (m, 3H), 4.49 (br d, J=10.2 Гц, 1H), 4.42 (br s, 2H),4.09-3.77 (m, 4H).
Та же реакционная последовательность
Пример 18 - Синтез соединения 25
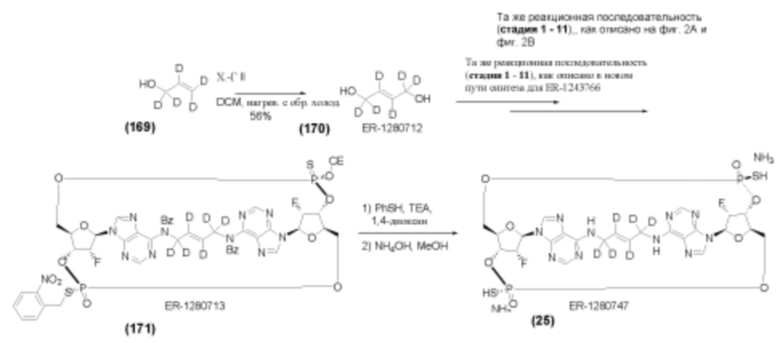
Соединение 170

К раствору катализатора Ховейда-Граббса 2-го поколения (0,996 г, 1,585 ммоль) в дихлорметане (40,0 мл) с обратным холодильником добавляли проп-2-ен-d5-1-ол (10,0 г, 158 ммоль). Через 1 ч добавляли дополнительно 1 мол. % катализатор Ховейда-Граббса 2-го поколения (0,996 г, 1,585 ммоль). Полученный раствор перемешивали с обратным холодильником всю ночь, охлаждали до температуры окружающей среды и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (Si02 340 г 0% - 10% МеОН в EtOAc) с получением 4,2 г соединения 170 в виде коричневого масла.
13С ЯМР (101 МГц, METHANOL-d4) δ=130.87 (t, J=23.8 Гц, 2С), 62.22 (квин., J=21.9 Гц, 2С).
Соединение 171
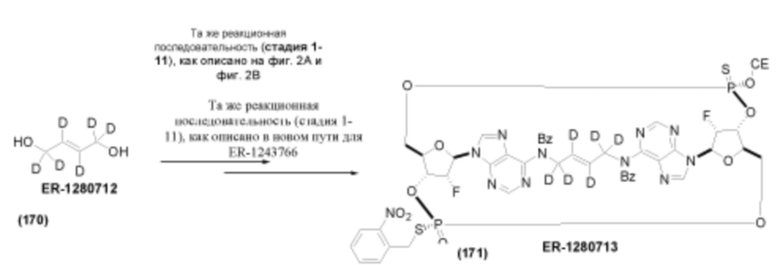
С соединением 170 в качестве исходного вещества соединение 171 получали такой же реакционной последовательностью (стадия 1-11), как описано в пути синтеза для соединения 1 на фиг. 2А и фиг. 2В.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.59 (s, 1H), 8.10 (s, 1H), 8.02-7.96 (m, 1H), 7.93 (s, 1H), 7.58-7.50 (m, 2Н), 7.48-7.37 (m, 5Н), 7.34-7.27 (m, 2Н), 7.24-7.14 (m, 4Н), 6.90 (s, 1H), 6.28 (d, J=14.8 Гц, 1H), 6.14-6.03 (m, 1H), 5.97-5.81 (m, 1H), 5.49 (dd, J=4.7, 52.3 Гц, 2Н), 5.43 (dd, J=3.9, 50.8 Гц, 1H), 5.30-5.15 (m, 1H), 4.75 (br d, J=12.1 Гц, 1H), 4.61 (brdd, J=4.3, 9.4 Гц, 1H), 4.53 (dd, J=2.9, 11.9 Гц, 1H), 4.47-4.41 (m, 3Н), 4.32-4.18 (m, 4Н), 2.79 (td, J=7.0, 17.2 Гц, 1H), 2.66 (td, J=6.3, 16.8 Гц, 1H)
Соединение 25
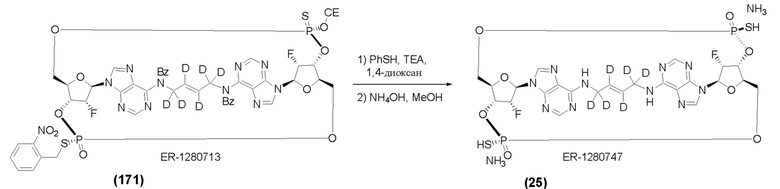
С соединением 171 в качестве исходного вещества соединение 25 получали такой же реакционной последовательностью (стадия 12-13), как описано на фиг. 2А и фиг. 2В.
1Н ЯМР (400 МГц, CD3OD) δ=9.03 (br s, 1H), 8.30 (br s, 1H), 8.24 (br s, 1H), 8.10 (br s, 1H), 6.44-6.13 (m, 2H), 0.00 (d, J=52.4 Гц, 1H), 0.00 (d, J=51.2 Гц, 1H), 4.71-4.32 (m, 6Н), 4.09-3.98 (m, 1H), 3.97-3.87 (m, 1H).
Пример 19 - Синтез соединения 31 (или RpRp, или SpSp)
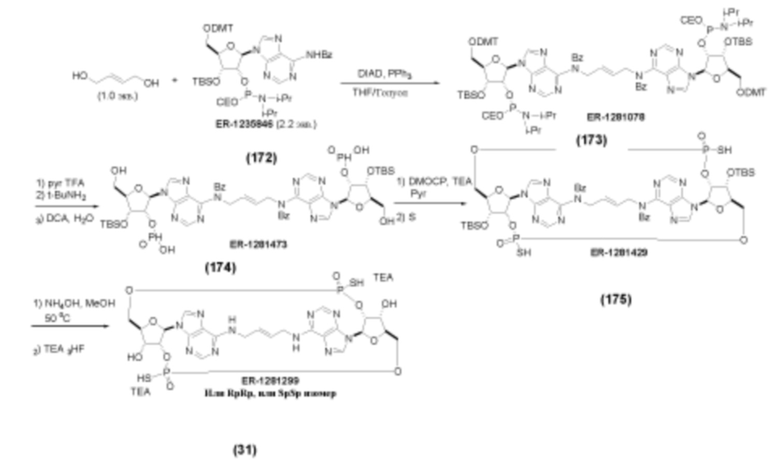
Соединение 17
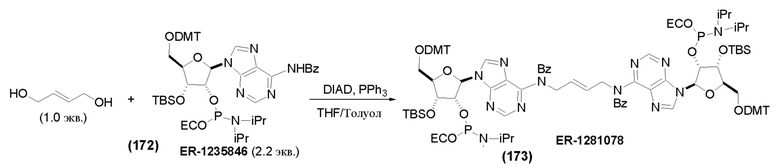
Смесь (Е)-бут-2-ен-1,4-диола (0,10 г, 1,135 ммоль) и соединения 172 (2,52 г, 2,55 ммоль) дважды азеотропировали THF и растворяли в THF (5,0 мл) и толуоле (7,5 мл). К полученному раствору добавляли трифенилфосфин (0,714 г, 2,724 ммоль). Полученный раствор охлаждали ниже 5°С и обрабатывали DIAD (0,53 мл, 2,72 ммоль) при поддерживании внутренней Т ниже 10°С. Полученную реакционную смесь перемешивали всю ночь при нагревании до температуры окружающей среды. После завершения реакции (наблюдали при помощи LCMS) реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 50 г, предварительно обрабатывали 1% TEA в н-гептане/EtOAc (1/1), 50% - 66% EtOAc в н-гептане) с получением 2,46 г соединения 173 в виде белого пенящегося твердого вещества. Это вещество использовали на следующей стадии без дополнительной очистки.
Соединение 174
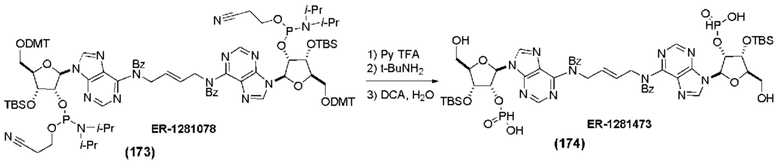
К раствору соединения 173 (2,387 г, 1,177 ммоль) в ацетонитриле (28,6 мл) добавляли воду (0,085 мл, 4,7 ммоль), пиридиновую трифторацетатную соль (0,500 г, 2,589 ммоль) при температуре окружающей среды. Через 1 мин добавляли 2-амино-2-метилпропан (19,1 мл, 182 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения реакционную смесь концентрировали в вакууме и азеотропировали MeCN. Остаток растворяли в дихлорметане (24 мл) и обрабатывали водой (0,42 мл) и 6% дихлоруксусной кислотой (1,2 мл, 14 ммоль) в дихлорметане (24 мл). После завершения снятия защитных групп с DMT (наблюдали при помощи LCMS) реакцию гасили пиридином (12 мл) и реакционную смесь концентрировали в вакууме. Полученный остаток обрабатывали смесью н-гептана/толуола (15/15 мл) и верхний слой декантировали. Ту же операцию повторяли еще раз. Сушкой оставшегося остатка в вакууме получали соединение 174, которое использовали на следующей стадии без дополнительной очистки.
LC/MS (ESI) m/z 1151.64 [М+Н]+.
Соединение 175
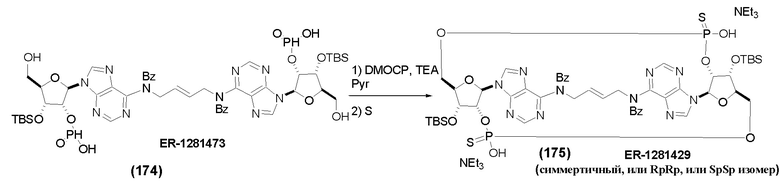
К раствору соединения 174 (1,355 г, 1,177 ммоль) в пиридине (271 мл) при температуре окружающей среды добавляли TEA (0,98 мл, 7,1 ммоль) и 2-хлор-5,5-диметил-1,3,2-диоксафосфинан 2-оксид (0,869 г, 4,708 ммоль). Полученный раствор перемешивали при температуре окружающей среды до израсходования всего исходного вещества (наблюдали при помощи LCMS). После завершения реакции добавляли воду (0,85 мл, 47 ммоль) (10 экв. DMOCP), а затем серу (0,377 г, 11,77 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS После завершения сульфурирования реакционную смесь обрабатывали насыщенным водным NaHCO3 (27 мл) и водой (10 мл) и концентрировали в вакууме. К полученному остатку добавляли смесь МТВЕ/EtOAc (34/34 мл), насыщенный водный раствор NaHCO3 (27 мл) и воду (20 мл). Слои разделяли и водный слой экстрагировали смесью EtOAc/МТВЕ (34/34 мл). Объединенные органические слои промывали 30% водным раствором NaCl (7 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 50 г, 0%-20% МеОН в EtOAc с 1% TEA) с получением 56 мг соединения 175 (симметричные на основе ЯМР; или RpRp, или SpSp изомер).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=9.09 (s, 2Н), 8.46 (s, 2Н), 7.56-7.05 (m, 10Н), 6.37 (d, J=8.2 Гц, 2Н), 6.28-6.22 (m, 2Н), 5.29 (ddd, J=4.7, 7.8, 11.7 Гц, 2Н), 5.00 (br d, J=13.7 Гц, 2H), 4.84 (d, J=4.3 Гц, 2H), 4.76 (br d, J=13.3 Гц, 2H), 4.31 (br s, 2H), 4.28-4.20 (m, 2H), 4.16-4.08 (m, 2H), 3.00-2.77 (m, 12H), 1.12-0.99 (m, 18H), 0.96 (s, 18H), 0.34 (s, 6H), 0.25 (s, 6H).
Соединение 31
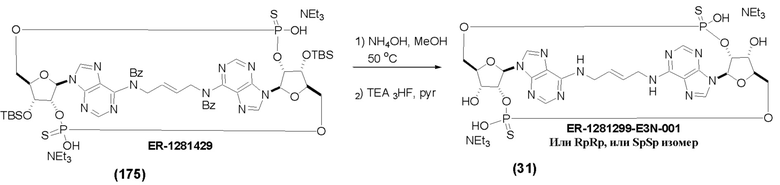
К раствору соединения 175 (56 мг, 0,0405 ммоль) в метаноле (1,1 мл) добавляли 28% гидроксид аммония (0,22 мл). Полученную смесь перемешивали при 50°С в течение 5 ч, охлаждали до температуры окружающей среды, концентрировали в вакууме и дважды азеотропировали MeCN. К полученному остатку добавляли пиридин (0,22 мл), TEA (0,11 мл) и TEA 3HF (0,11 мл, 0,69 ммоль). Полученную смесь перемешивали при 50° в течение 5 ч, охлаждали до температуры окружающей среды, обрабатывали метокситриметилсиланом (0,90 мл, 6,50 ммоль) в течение 30 мин. Полученную смесь концентрировали в вакууме и растворяли в воде (5 мл). Полученный водный раствор экстрагировали трижды толуолом (5 мл каждый раз) и концентрировали в вакууме. Неочищенный продукт растворяли в 1 мл воды, фильтровали через шприцевой фильтр и подкисляли 1 н HCl до значения рН<3. Полученную смесь хранили в морозилке (5°С) в течение 2 суток и полученное твердое вещество собирали фильтрацией. Твердое вещество растворяли в 2 М NH3 в МеОН (1,5 мл) и концентрировали в вакууме с получением 2,8 мг соединения 31 в виде смеси NH3 и TEA соли. Продукт растворяли в EtOH (1 мл), обрабатывали 0,1 мл TEA, концентрировали до 0,5 мл и хранили в морозилке (-20°С) всю ночь. Супернатант удаляли и оставшееся твердое вещество дополнительно сушили в вакууме с получением 0,8 мг бис TEA соли соединения 31 (симметричные на основе ЯМР; или RpRp, или SpSp изомер).
1Н ЯМР (400 МГц, METHANOL-d4) δ=8.26-7.83 (m, 4Н), 6.37 (br s, 2Н), 5.89 (br s, 2H), 5.36-5.07 (m, 2H), 4.95 (br d, J=3.5 Гц, 2H), 4.58 (br dd, J=6.3, 10.6 Гц, 2H), 4.43 (br s, 2H), 4.12-4.00 (m, 4H), 3.89-3.70 (m, 2H), 3.19 (q, J=7.2 Гц, 12H), 1.30 (t, J=7.4 Гц, 18H)
Пример 20 - Синтез соединения 32
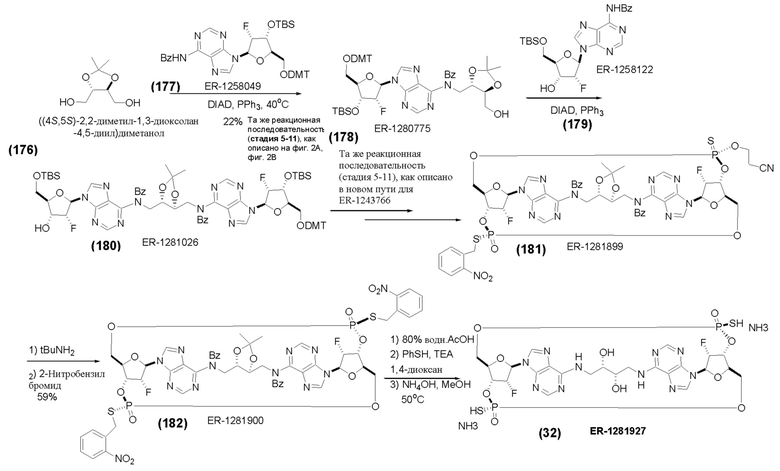
Соединение 178
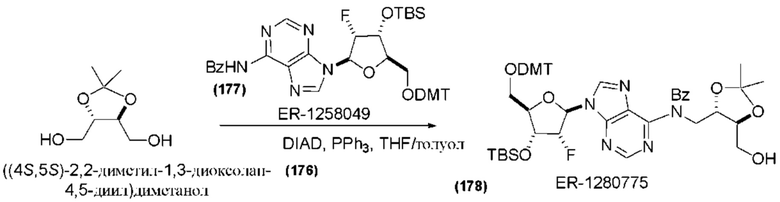
К раствору соединения 177 (2,0 г, 2,532 ммоль) и (+)-2,3-орто-изопропилиден-L-треитола (соединение 176) (1,232 г, 7,595 ммоль) в THF (20,0 мл) и толуоле (30,0 мл) добавляли трифенилфосфин (0,930 г, 3,544 ммоль). Полученный раствор охлаждали ниже 5°С и обрабатывали DIAD (0,64 мл, 3,3 ммоль). Реакционную смесь нагревали до температуры окружающей среды. Через 8 ч перемешивания при температуре окружающей среды добавляли дополнительно DIAD (0,64 мл) и PPh3 (0,93 г). После перемешивания всю ночь при температуре окружающей среды реакционную смесь перемешивали при 40°С в течение 2 суток и концентрировали в вакууме. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 100 г, предварительно обрабатывали 1% TEA в н-гептане/EtOAc (1/1), 50%-70% EtOAc в н-гептане) получали 0,524 г соединения 178 в виде белого пенящегося твердого вещества.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.62 (s, 1H), 8.09 (s, 1H), 7.40-7.09 (m, 12Н), 7.01-6.92 (m, 2Н), 6.80-6.73 (m, 4Н), 6.16 (dd, J=2.7, 16.8 Гц, 1H), 5.49 (ddd, J=3.1, 4.3, 53.1 Гц, 1H), 4.85-4.75 (m, 2Н), 4.63 (dd, J=3.9, 14.8 Гц, 1H), 4.30-4.19 (m, 2Н), 4.18-4.12 (m, 1H), 3.94 (tt, J=3.5, 8.2 Гц, 1H), 3.84 (td, J=5.5, 12.1 Гц, 1H), 3.78 (s, 6Н), 3.53 (dd, J=2.7, 10.9 Гц, 1H), 3.17 (dd, J=3.5, 10.9 Гц, 1H), 2.94 (br t, J=5.5 Гц, 1H), 0.89 (s, 6Н), 0.83 (s, 9Н), 0.08 (s, 3Н), -0.01 (s, 3Н)
Соединение 180
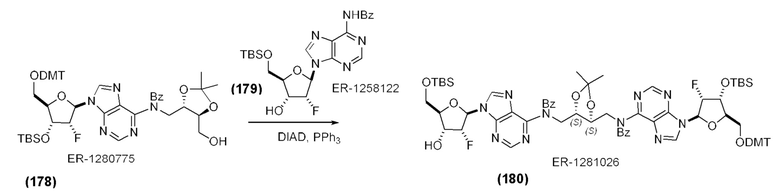
К раствору соединения 178 (0,409 г, 0,84 ммоль) в THF (6 мл) добавляли трифенилфосфин (0,191 г, 0,728 ммоль) и DIAD (0,142 мл, 0,728 ммоль). Через 40 ч перемешивания при температуре окружающей среды реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии (SiO2 50 г, предварительно обрабатывали 1% TEA в н-гептане/EtOAc (1/1), 50%-66% EtOAc в н-гептане) с получением 0,533 г соединения 180.
LC/MS: LRMS (ESI) m/z 1403.86 [М+Н]+.
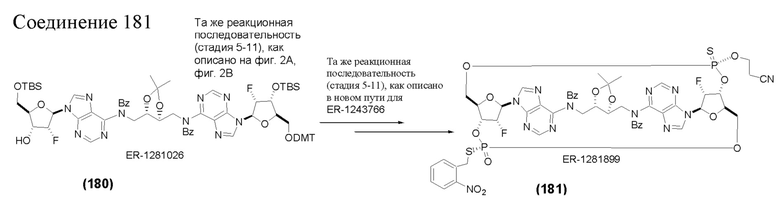
С соединением 180 в качестве исходного вещества соединение 181 получали такой же реакционной последовательностью (стадия 5-11), как описано на фиг. 2А и фиг. 2В.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.72 (s, 1H), 8.14-8.06 (m, 1H), 7.93 (s, 1H), 7.69 (s, 1H), 7.62-7.53 (m, 6Н), 7.53-7.45 (m, 1H), 7.43-7.33 (m, 2Н), 7.30-7.24 (m, 3Н), 7.19-7.12 (m, 2Н), 6.11 (d, J=16.8 Гц, 1H), 6.05 (d, J=19.5 Гц, 1H), 5.89 (dd, J=3.5, 52.3 Гц, 1H), 5.57-5.46 (m, 1H), 5.41 (dd, J=3.5, 53.1 Гц, 1H), 5.18-5.04 (m, 1Н), 4.83 (dd, J=4.7, 14.4 Гц, 1H), 4.78-4.73 (m, 1H), 4.70 (br d, J=12.1 Гц, 1H), 4.64-4.48 (m, 4Н), 4.46-4.35 (m, 4Н), 4.33-4.27 (m, 2Н), 4.16-4.06 (m, 2Н), 4.05-3.96 (m, 1H), 2.55 (t, J=6.1 Гц, 2H), 1.26 (s, 6Н).
Соединение 182
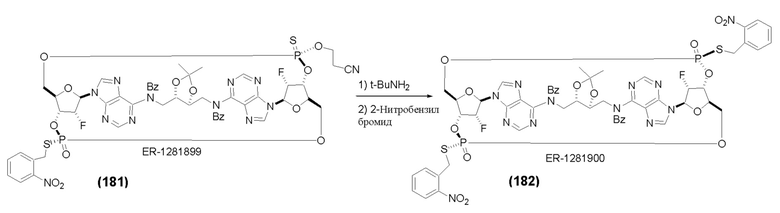
К раствору соединения 181 (3,2 мг, 2,629 мкмоль) в дихлорметане (0,5 мл) добавляли трет-бутиламин (0,5 мл, 4,7 ммоль). Через 30 мин перемешивания реакционный раствор концентрировали в вакууме и дважды азеотропировали MeCN. Остаток растворяли в ацетонитриле (1 мл) и обрабатывали 1-(бромметил)-2-нитробензолом (1,7 мг, 7,9 мкмоль). После завершения алкилирования (наблюдали при помощи LCMS) реакционную смесь концентрировали продуванием азотом. Очисткой остатка методом колоночной хроматографии на силикагеле (SiO2 4 г, 80%-100% EtOAc в н-гептане) получали 2 мг соединения 182.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.12 (dd, J=1.4, 8.0 Гц, 1H), 8.03-8.00 (m, 1H), 8.00 (s, 1H), 7.93 (s, 1H), 7.70 (s, 1H), 7.70-7.29 (m, 13Н), 7.21 (dt, J=2.7, 7.6 Гц, 4H), 6.07 (d, J=19.9 Гц, 1H), 5.94 (d, J=21.1 Гц, 1H), 5.91-5.77 (m, 1H), 5.81 (dd, J=4.3, 51.5 Гц, 1H), 5.62-5.49 (m, 1H), 5.54 (dd, J=3.1, 52.7 Гц, 2Н), 4.83 (q, J=5.9 Гц, 1H), 4.72-4.63 (m, 2Н), 4.61-4.28 (m, 13Н), 1.42 (s, 3Н), 1.28 (s, 3Н).
Соединение 32
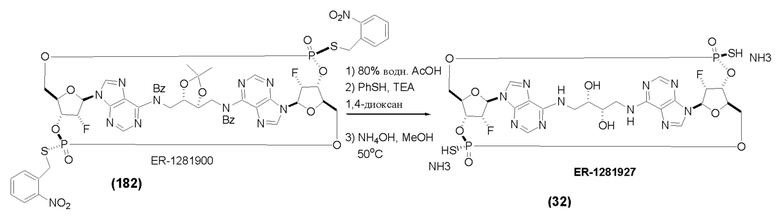
К соединению 182 (2,0 мг, 1,5 мкмоль) добавляли уксусную кислоту (0,8 мл) и воду (0,2 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 14 ч и при 45-50°С в течение 24 ч, концентрировали в вакууме и дважды азеотропировали толуолом. К остатку добавляли 1,4-диоксан (0,12 мл), тиофенол (60 мкл), а затем TEA (60 мкл). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения добавляли метанол (160 мкл) и 28% гидроксид аммония (160 мкл). Полученную смесь перемешивали при 50°С до завершения дебензоилирования (наблюдали при помощи LCMS). После завершения добавляли воду (0,3 мл). Полученное твердое вещество отфильтровывали, промывали водой (0,2 мл). Фильтрат дважды экстрагировали толуолом (0,5 мл каждый раз) и концентрировали в вакууме при 40-50°С. Остаток обрабатывали водой (0,5 мл) и полученное твердое вещество отфильтровывали, промывали водой (0,1 мл), очисткой методом HPLC объединенных водных слоев при условиях, описанных ниже, получали 1,5 мг соединения 32.
LC/MS: LRMS (ESI) m/z 781.23 [М+Н]+.
Условия препаративной HPLC для соединения 32:
Пример 21 - Синтез соединения 33 и соединения 34
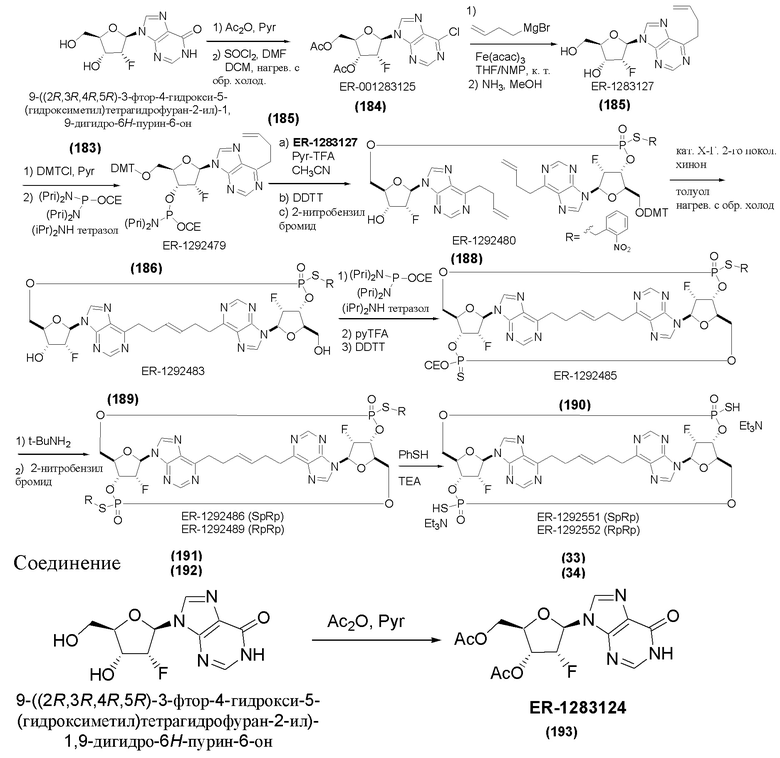
К раствору 9-((2R,3R,4R,5R)-3-фтор-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-1,9-дигидро-6Н-пурин-6-она, соединение 183 (5,00 г, 18,5 ммоль) в пиридине (75 мл) при 0°С добавляли Ас2О (7,0 мл, 74 ммоль) и DMAP (0,565 г, 4,626 ммоль). Полученную смесь нагревали до температуры окружающей среды и перемешивали, при этом за реакцией наблюдали при помощи LCMS. После завершения реакционную смесь концентрировали в вакууме и обрабатывали EtOAc (200 мл) и водой (50 мл). Происходило осаждение. Полученное твердое вещество собирали фильтрацией и промывали МТВЕ. Сушкой в вакууме при 40°С всю ночь получали 5,81 г соединения 193 в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ=12.53 (br s, 1H), 8.33 (s, 1H), 8.15 (d, J=4.3 Гц, 1H), 6.37 (dd, J=2.7, 19.1 Гц, 1H), 5.86 (ddd, J=2.7, 5.1, 51.6 Гц, 1H), 5.62 (ddd, J=5.5, 7.0, 16.0 Гц, 1H), 4.49-4.38 (m, 2Н), 4.26 (dd, J=4.7, 12.1 Гц, 1H), 2.19 (s, 3Н), 2.04 (s, 3Н).
Соединение 184
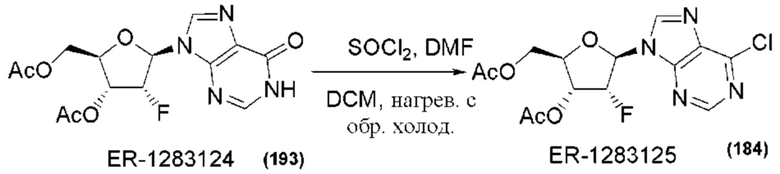
К раствору соединения 193 в дихлорметане (40,0 мл) при 0°С медленно добавляли DMF (1,3 мл, 16,9 ммоль) и тионилхлорид (1,28 мл, 17,5 ммоль). Полученную смесь нагревали до температуры образования флегмы и перемешивали до израсходования всего исходного вещества (наблюдали при помощи LCMS). После завершения реакционную смесь охлаждали до температуры окружающей среды и обрабатывали насыщенным водным раствором NaHCO3 (40 мл). Слои разделяли и водный слой дважды экстрагировали дихлорметаном (30 мл каждого). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением 2,81 г соединения 184 в виде бледно-коричневого масла.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.79 (s, 1H), 8.30 (s, 1H), 6.29 (dd, J=2.0, 18.0 Гц, 1H), 5.79 (ddd, J=1.6, 4.7, 51.6 Гц, 1H), 5.53 (ddd, J=5.1, 7.6, 17.8 Гц, 1H), 4.58-4.48 (m, 2Н), 4.33 (dd, J=3.9, 12.5 Гц, 1H), 2.20 (s, 3Н), 2.07 (s, 3Н).
Соединение 194
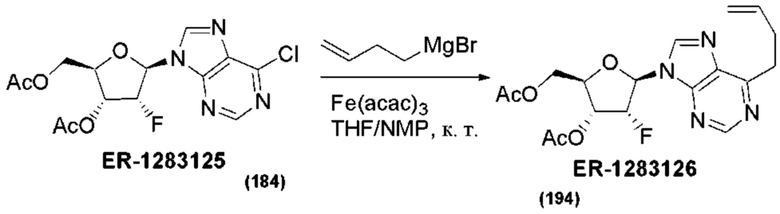
К раствору соединения 184 (0,22 г, 0,59 ммоль) в смеси THF (7,7 мл) и NMP (0,77 мл) при температуре окружающей среды добавляли ацетилацетонат железа (III) (0,021 г, 0,059 ммоль) и 0,5 М 3-бутенилмагния бромид (1,77 мл, 0,885 ммоль) в THF. После завершения реакции (наблюдали при помощи LCMS) добавляли МТВЕ (10 мл) и 0,1 н HCl (10 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 10 мин. Слои разделяли и водный слой экстрагировали и смесью EtOAc/МТВЕ (1/1, 10 мл). Объединенные органические слои промывали 30% водным раствором NaCl (5 мл), сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.90 (s, 1H), 8.16 (s, 1H), 6.26 (dd, J=2.0, 18.4 Гц, 1H), 5.98-5.85 (m, 1H), 5.90-5.73 (m, 1H), 5.63 (ddd, J=4.9, 7.5, 17.7 Гц, 1H), 5.12-5.05 (m, 1H), 4.98 (dd, J=1.6, 10.2 Гц, 1H), 4.54-4.50 (m, 2H), 4.31 (dd, J=5.1, 12.9 Гц, 1H), 3.30 (t, J=7.4 Гц, 2H), 2.70-2.64 (m, 2H), 2.19 (s, 3H), 2.05 (s, 3H).
Соединение 185
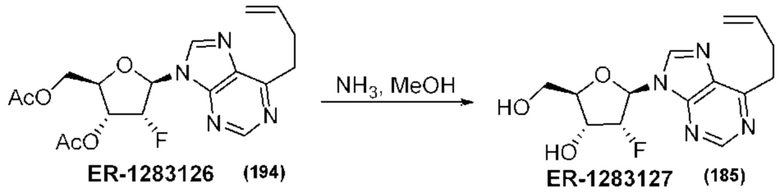
Неочищенный продукт соединения 194 (0,232 г, 0,590 ммоль в теории) растворяли в метаноле (1,5 мл) и обрабатывали 2,0 М аммиаком (1,5 мл, 3,0 ммоль) в МеОН при температуре окружающей среды. После завершения диацетилирования реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 0%-5% МеОН в EtOAc) с получением 0,17 г соединения 185.
LCMS: MS (ESI) m/z 309.20 [М+Н]+
Соединение 195
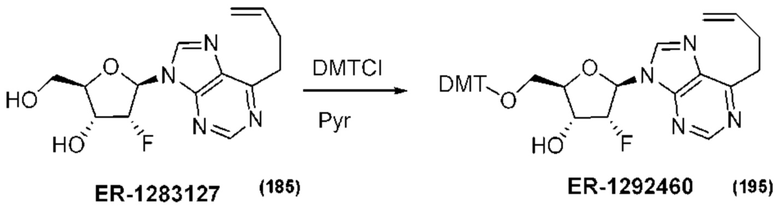
К раствору соединения 185 (1,22 г, 2,928 ммоль) в пиридине (9,0 мл) при 0° добавляли 4,4'-(хлор(фенил)метилен)бис(метоксибензол) (1,042 г, 3,075 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения добавляли МТВЕ (18 мл) и насыщенный водный NaHCO3 (9,0 мл). Через 10 мин перемешивания слои разделяли и водный слой экстрагировали МТВЕ (18,0 мл). Объединенные органические слои промывали 30% водным раствором NaCl (10 мл), фильтровали и концентрировали в вакууме. Очисткой методом колоночной хроматографии на силикагеле (SiO2 50 г, 50%-100% этилацетата в гептане с 1% TEA) с получением 1,53 г соединения 195 в виде оранжевого твердого вещества.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.86-8.78 (m, 1H), 8.21 (s, 1H), 7.40-7.36 (m, 2Н), 7.31-7.18 (m, 7Н), 6.79 (d, J=8.6 Гц, 4Н), 6.29 (dd, J=2.3, 17.2 Гц, 1H), 5.92 (tdd, J=6.4, 10.4, 17.0 Гц, 1H), 5.70 (ddd, J=2.7, 4.7, 52.8 Гц, 1H), 5.09 (dd, J=1.6, 17.2 Гц, 1H), 4.97 (dd, J=1.6, 10.2 Гц, 1H), 4.90-4.77 (m, 1H), 4.26-4.19 (m, 1Н), 3.78 (s, 6H), 3.56 (dd, J=3.1, 10.9 Гц, 1H), 3.43 (dd, J=4.3, 10.9 Гц, 1H), 3.33-3.28 (m, 2H), 2.70-2.64 (m, 2H), 2.20 (dd, J=2.5, 7.2 Гц, 1H).
Соединение 186
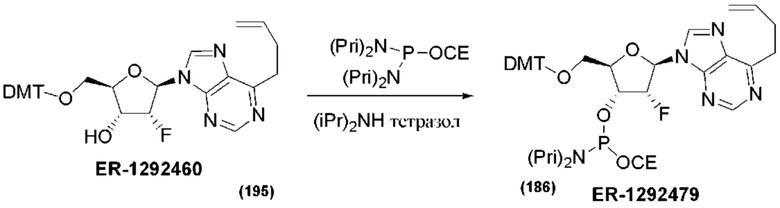
К раствору соединения 195 (1,530 г, 2,505 ммоль) в дихлорметане (23 мл) при температуре окружающей среды добавляли 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (1,20 мл, 3,76 ммоль) и диизопропиламмония тетразолид (0,493 г, 2,881 ммоль). Реакционную смесь перемешивали при температуре окружающей среды, при этом за прогрессом наблюдали при помощи LCMS. После завершения реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 50 г, 40%-66% EtOAc в н-гептане с 1% TEA) с получением 1,80 г соединения 186 в виде бледно-оранжевого пенящегося твердого вещества.
Соединения 196 и 197
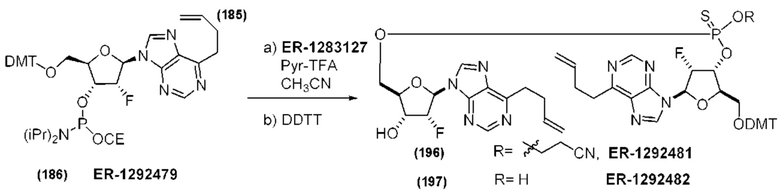
Соединение 186 (1,8 г, 2,22 ммоль) и соединение 185 (1,110 г, 2,664 ммоль) растворяли в ацетонитриле (21,6 мл) и концентрировали в вакууме. Ту же операцию повторяли еще раз. Полученную смесь растворяли в ацетонитриле (21,6 мл) и обрабатывали трифторацетатом пиридиния (0,514 г, 2,664 ммоль) при температуре окружающей среды. После завершения реакции (наблюдали при помощи LCMS) добавляли ((диметиламинометилиден)амино)-3Н-1,2,4-дитиазолин-3-тион (0,911 г, 4,439 ммоль) и полученную смесь перемешивали при температуре окружающей среды. После завершения сульфурирования (наблюдали при помощи LCMS) добавляли насыщенный водный раствор NaHCO3 (30 мл) и полученную смесь экстрагировали трижды МТВЕ (30 мл каждый раз). Объединенные органические слои промывали 30% водным раствором NaCl (20 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой методом колоночной хроматографии на силикагеле (SiO2 100 г, 0%-10% МеОН в EtOAc с 1% TEA) получали 0,481 г соединения 196 и 1,315 г соединения 197.
Соединение 196: LCMS: MS (ESI) m/z 1073.25 [M+Na]+
Соединение 197: LCMS: MS (ESI) m/z 995.23 [M-H]-
Соединение188
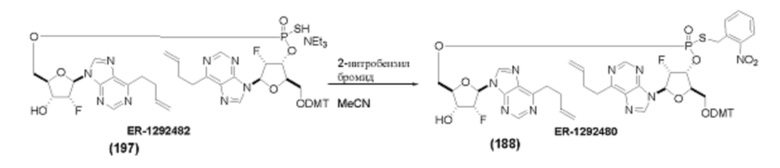
К раствору TEA соли соединения 197 (1,315 г, 1,197 ммоль) в MeCN (20 мл) при температуре окружающей среды добавляли 2-нитробензилбромид (0,388 г, 1,796 ммоль). Полученный раствор перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 100 г, 0%-5% МеОН в EtOAC) с получением 0,90 г соединения 188.
LCMS: MS (ESI) m/z 1154.29 [M+Na]+
Соединение 198
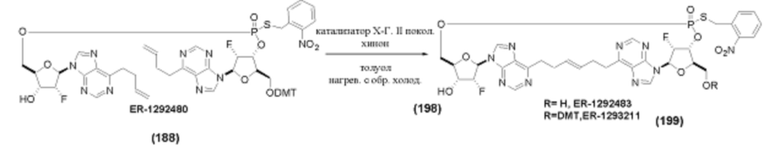
К раствору соединения 188 (1,35 г, 1,192 ммоль) в толуоле (540 мл) при мягком нагревании с обратным холодильником (110-112°С внутренняя Т) добавляли раствора катализатора Ховейда-Граббса 2-го поколения (0,187 г, 0,298 ммоль) и хинона (0,322 г, 2,981 ммоль)) в толуоле (20 мл). Полученный раствор перемешивали при 110-112°С, при этом за реакцией наблюдали при помощи LCMS. Через 4 ч добавляли дополнительный катализатор (0,10 г, 0,16 ммоль) и перемешивание продолжали с обратным холодильником. После завершения реакционную смесь охлаждали до температуры окружающей среды и обрабатывали DMSO (2,54 мл, 35,8 ммоль). Через 3 ч перемешивания реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 50 г, 0%-15% MeOH в EtOAC) с получением смеси соединения 198 и соединения 199. Смесь растворяли в дихлорметане (2 мл) и обрабатывали водой (0,021 мл, 1,2 ммоль) и 6% дихлоруксусной кислотой (0,098 мл, 1,2 ммоль) в дихлорметане (2 мл). Через 10 мин реакцию гасили пиридином (1 мл) и концентрировали в вакууме. Остаток растворяли в дихлорметане (50 мл) и дважды промывали 30% водным раствором NaCl (15 мл каждый раз) и сушили над MgSO4. Очисткой неочищенного продукта методом колоночной хроматографии на силикагеле (SiO2 50 г, 5%-10% МеОН в DCM) получали 0,25 г соединения 198.
Соединение 199: LCMS: MS (ESI) m/z 1104.31 [M+H]+
Соединение 198: LCMS: MS (ESI) m/z 802.16 [M+H]+
Соединение 190
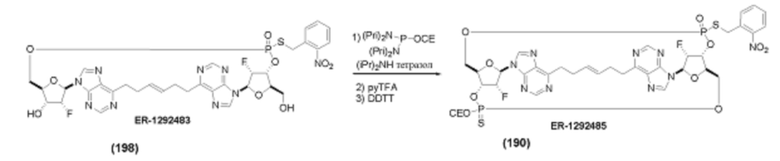
Соединение 198 (0,233 г,  ммоль) и 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (0,111 мл, 0,349 ммоль) растворяли в MeCN (10 мл) и концентрировали в вакууме. Ту же операцию повторяли еще два раза. Полученную смесь растворяли в дихлорметане (2,3 мл), охлаждали до 0-5°С и обрабатывали диизопропиламмонием тетразолидом (0,025 г, 0,145 ммоль). Реакционную смесь нагревали до температуры окружающей среды всю ночь, а затем разбавляли ацетонитрилом (2,3 мл). Полученный раствор добавляли в течение 7 ч при помощи шприцевого насоса к раствору пиридиновой трифторацетатной соли (0,168 г, 0,872 ммоль) в MeCN (18,6 мл). Затем добавляли 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (40 мг, 0,133 ммоль) в MeCN (2 мл) в течение 2 часов. После 1 ч перемешивания добавляли (E)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамид (0,119 г, 0,581 ммоль) и реакционный раствор перемешивали до завершения сульфурирования (наблюдали при помощи LCMS). После завершения смесь концентрировали в вакууме, растворяли в МТВЕ (5 мл) и обрабатывали насыщенным водным NaHCO3 (3,50 мл) и водой (1,2 мл). Слои разделяли и водный слой экстрагировали смесью МТВЕ/EtOAc (5/3 мл). Объединенные органические слои промывали дважды 30% водным раствором NaCl (2,3 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой неочищенного продукта методом колоночной хроматографии на силикагеле (SiO2 25 г, 0%-20% МеОН в EtOAc) получали 119 мг соединения 190 (58 мг более быстро элюирующих изомеров и 61 мг более медленно элюирующих изомеров).
ммоль) и 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (0,111 мл, 0,349 ммоль) растворяли в MeCN (10 мл) и концентрировали в вакууме. Ту же операцию повторяли еще два раза. Полученную смесь растворяли в дихлорметане (2,3 мл), охлаждали до 0-5°С и обрабатывали диизопропиламмонием тетразолидом (0,025 г, 0,145 ммоль). Реакционную смесь нагревали до температуры окружающей среды всю ночь, а затем разбавляли ацетонитрилом (2,3 мл). Полученный раствор добавляли в течение 7 ч при помощи шприцевого насоса к раствору пиридиновой трифторацетатной соли (0,168 г, 0,872 ммоль) в MeCN (18,6 мл). Затем добавляли 3-(бис(диизопропиламино)фосфиноокси)пропаннитрил (40 мг, 0,133 ммоль) в MeCN (2 мл) в течение 2 часов. После 1 ч перемешивания добавляли (E)-N,N-диметил-N'-(3-тиоксо-3Н-1,2,4-дитиазол-5-ил)формимидамид (0,119 г, 0,581 ммоль) и реакционный раствор перемешивали до завершения сульфурирования (наблюдали при помощи LCMS). После завершения смесь концентрировали в вакууме, растворяли в МТВЕ (5 мл) и обрабатывали насыщенным водным NaHCO3 (3,50 мл) и водой (1,2 мл). Слои разделяли и водный слой экстрагировали смесью МТВЕ/EtOAc (5/3 мл). Объединенные органические слои промывали дважды 30% водным раствором NaCl (2,3 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Очисткой неочищенного продукта методом колоночной хроматографии на силикагеле (SiO2 25 г, 0%-20% МеОН в EtOAc) получали 119 мг соединения 190 (58 мг более быстро элюирующих изомеров и 61 мг более медленно элюирующих изомеров).
Соединение 191 (SpRp изомер)
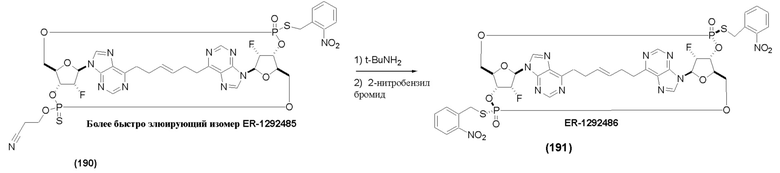
К раствору более быстро элюирующих изомеров соединения 190 (58 мг, 0,062 ммоль) в дихлорметане (1,8 мл) добавляли трет-бутиламин (1,2 мл, 11 ммоль). Полученный раствор перемешивали при температуре окружающей среды до израсходования всего исходного вещества (наблюдали при помощи LCMS). После завершения реакционную смесь концентрировали в вакууме, дважды азеотропировали ацетонитрилом и растворяли в ацетонитриле (1,7 мл). К полученному раствору добавляли 1-(бромметил)-2-нитробензол (40,3 мг, 0,187 ммоль). Реакционную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения алкилирования реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 0%-5% МеОН в EtOAC) с получением 19 мг соединения 191 (SpRp изомер).
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.42 (s, 1H), 8.27 (s, 1H), 8.13 (dd, J=1.2, 8.2 Гц, 1H), 8.05 (s, 1H), 7.98 (d, J=7.8 Гц, 1H), 7.69-7.67 (m, 1H), 7.61-7.34 (m, 5Н), 6.33 (d, J=17.2 Гц, 1H), 6.19 (d, J=19.9 Гц, 1H), 6.09-5.93 (m, 1H), 5.82-5.66 (m, 2Н), 5.67 (dd, J=3.5, 50.8 Гц, 1H), 5.35-5.32 (m, 2Н), 4.62-4.29 (m, 11Н), 3.35-3.28 (m, 1H), 3.18-3.14 (m, 2Н), 3.01-2.91 (m, 1H), 2.84-2.78 (m, 1H), 2.60-2.49 (m, 3Н).
Соединение 192 (RpRp изомер)
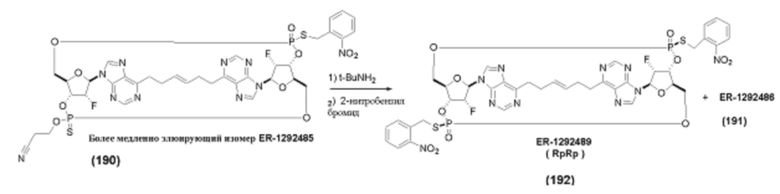
Более медленно элюирующий изомер соединения 190  обрабатывали такой же реакционной последовательностью, как описано для соединения 191, с получением 14 мг соединения 192 и 7 мг соединения 191.
обрабатывали такой же реакционной последовательностью, как описано для соединения 191, с получением 14 мг соединения 192 и 7 мг соединения 191.
1H ЯМР (400 МГц, CHLOROFORM-d) δ=8.18 (s, 2Н), 8.10 (s, 2Н), 8.10 (dd, J=1.0, 8.0 Гц, 2Н), 7.66-7.57 (m, 4Н), 7.53-7.46 (m, 2Н), 6.31 (d, J=17.6 Гц, 2Н), 6.19-6.05 (m, 2Н), 5.84 (dd, J=4.3, 50.8 Гц, 2Н), 5.39-5.30 (m, 2Н), 4.58-4.46 (m, 6Н), 4.45-4.39 (m, 2Н), 4.19 (ddd, J=3.1, 6.3, 10.2 Гц, 2Н), 3.32 (td, J=3.5, 15.6 Гц, 2Н), 3.06 (td, J=6.3, 15.2 Гц, 2Н), 2.67-2.57 (m, 4Н).
На основе методики синтеза, данных ЯМР (симметричные), времени удерживания HPLC (самый медленный элюирующий изомер) и биологических активностей соединения 34, которое получали из соединения 192, заявители полагали, что соединение 192 обладает RR фосфорной стереохимией. Такое стереохимическое определение будет подвержено подтверждению методом рентгеновской кристаллографии
Соединение 33
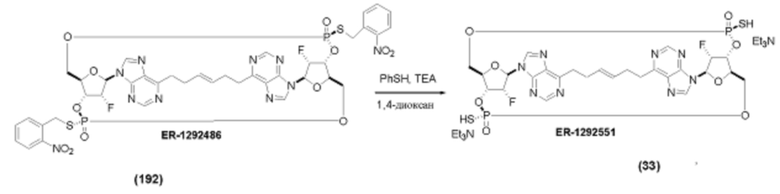
К раствору соединения 192 (26 мг, 0,026 ммоль) в 1,4-диоксане  добавляли тиофенол (0,26 мл, 2,5 ммоль), а затем TEA (0,26 мл, 1,9 ммоль). Полученную смесь перемешивали при температуре окружающей среды для завершения снятия защитных групп (наблюдали при помощи LCMS). После завершения добавляли воду (2 мл). Полученную смесь трижды экстрагировали толуолом (2 мл каждый раз). Водный слой концентрировали в вакууме при 40-50°С и растворяли в воде (2 мл). Полученную смесь экстрагировали четыре раза смесью EtOAc/МТВЕ (1/1 мл каждый раз). Водный слой концентрировали в вакууме с получением бис-ТЕА соли соединения 33.
добавляли тиофенол (0,26 мл, 2,5 ммоль), а затем TEA (0,26 мл, 1,9 ммоль). Полученную смесь перемешивали при температуре окружающей среды для завершения снятия защитных групп (наблюдали при помощи LCMS). После завершения добавляли воду (2 мл). Полученную смесь трижды экстрагировали толуолом (2 мл каждый раз). Водный слой концентрировали в вакууме при 40-50°С и растворяли в воде (2 мл). Полученную смесь экстрагировали четыре раза смесью EtOAc/МТВЕ (1/1 мл каждый раз). Водный слой концентрировали в вакууме с получением бис-ТЕА соли соединения 33.
1Н ЯМР (400 МГц, METHANOLS) δ=9.06 (s, 1H), 8.73 (s, 1H), 8.56 (s, 1H), 8.47 (s, 1H), 6.43 (d, J=14.4 Гц, 1H), 6.38 (d, J=15.2 Гц, 1H), 6.27 (dd, J=3.5, 51.1 Гц, 1H), 5.63 (dd, J=2.7, 51.5 Гц, 1H), 5.30-5.14 (m, 2Н), 5.06-4.91 (m, 2Н), 4.58 (br d, J=12.5 Гц, 1H), 4.52-4.39 (m, 3Н), 4.07 (dd, J=4.7, 11.7 Гц, 1H), 3.98 (dd, J=5.1, 12.5 Гц, 1H), 3.43-3.34 (m, 2Н), 3.22 (q, J=7.2 Гц, 12H), 2.98-2.86 (m, 2H), 2.79-2.45 (m, 4H), 1.32 (t, J=7.4 Гц, 18H)
Соединение 34
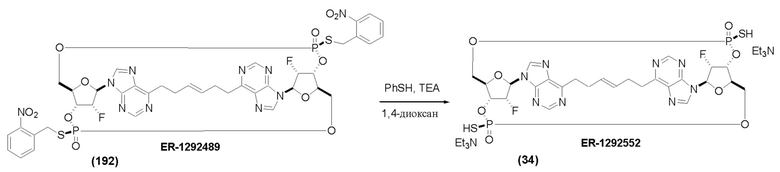
С соединением 192 в качестве исходного вещества соединение 34 получали такой же реакционной последовательностью, как описано для соединения 33.
1Н ЯМР (400 МГц, METHANOL-d4) δ=8.77 (s, 2Н), 8.49 (s, 2Н), 6.39 (d, J=15.2 Гц, 2Н), 5.71 (dd, J=3.1, 51.2 Гц, 2Н), 5.24-5.13 (m, 2Н), 5.02 (dtd, J=3.1, 9.0, 25.4 Гц, 2Н), 4.54 (br d, J=12.1 Гц, 2Н), 4.42 (br d, J=9.8 Гц, 2Н), 3.99 (dd, J=5.9, 12.1 Гц, 2Н), 3.20 (q, J=7.3 Гц, 12Н), 2.90 (ddd, J=3.5, 10.2, 14.1 Гц, 2Н), 2.81-2.70 (m, 2Н), 2.55-2.43 (m, 2Н), 1.30 (t, J=7.4 Гц, 18Н).
Пример 22 - Синтез соединения 35 и соединения 36
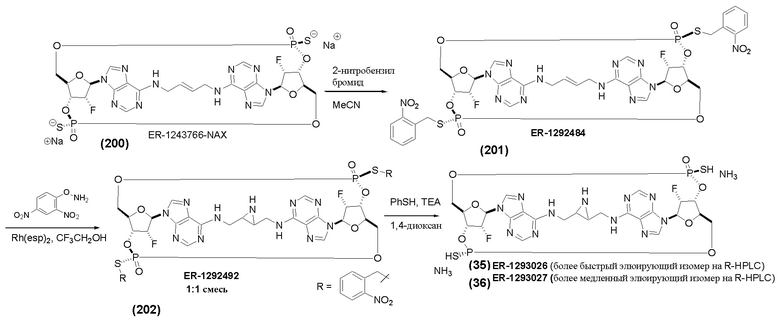
Соединение 201
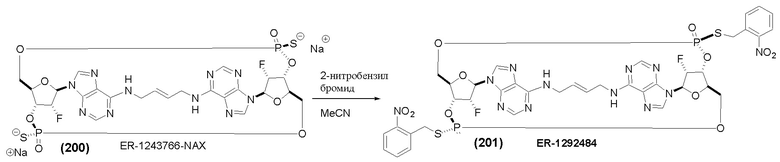
К динатриевой соли соединения 200 (0,10 г, 0,126 ммоль) добавляли 2-нитробензилбромид (0,068 г, 0,316 ммоль) и MeCN (2,0 мл). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения, реакционную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 0%-5% МеОН в DCM) с получением 115 мг соединения 201.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.13 (dd, J=0.8, 8.2 Гц, 1H), 8.06 (s, 1H), 8.03 (dd, J=1.0, 8.0 Гц, 1H), 7.80 (br s, 1H), 7.75 (s, 1H), 7.70-7.65 (m, 1H), 7.62-7.54 (m, 2H), 7.53-7.47 (m, 2H), 7.45-7.39 (m, 1H), 7.34-7.27 (m, 1H), 6.23 (br d, J=16.8 Гц, 1H), 6.14 (brd, J=18.8 Гц, 1H), 5.95 (br s, 2H), 5.77 (td, J=5.1, 15.2 Гц, 2H), 5.71 (td, J=5.1, 15.6 Гц, 1H), 5.63-5.55 (m, 1H), 5.47 (br d, J=11.7 Гц, 1H), 4.58-4.27 (m, 11H), 4.18-4.10 (m, 1H), 4.05-3.71 (m, 2H)
Соединение 202
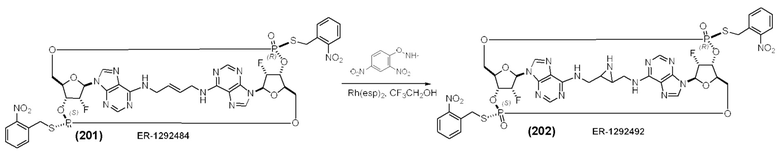
К раствору соединения 201 (77 мг, 0,076 ммоль) в 2,2,2-трифторэтаноле (2 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (36,5 мг, 0,183 ммоль) и бис[родия(α,α,α',α'-тетраметил-1,3-бензолдипропионовая кислота)] (5,74 мг, 0,015 ммоль) одну часть. Смесь перемешивали при температуре окружающей среды всю ночь, разбавляли DCM (10 мл) и обрабатывали насыщенным водным раствором NaHCO3 (10 мл). Слои разделяли и водный слой дважды экстрагировали DCM (10 мл каждого). Объединенные органические слои сушили над MgSO4 и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 0%-15% МеОН в EtOAc) с получением 3,3 мг соединения 202.
LCMS: MS m/z 1032,07 [М+Н]+
Соединение 35 и соединение 36
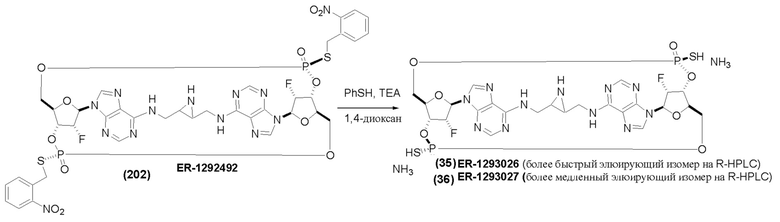
К раствору соединения 202 (3,3 мг, 3,2 мкмоль) в 1,4-диоксане (0,40 мл) добавляли тиофенол (0,2 мл, 1,9 ммоль) и TEA (0,2 мл, 1,4 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения реакционную смесь обрабатывали водой (2 мл) и трижды экстрагировали толуолом (2 мл каждый раз). Водный слой концентрировали в вакууме при 40-50°С и растворяли в воде (1,5 мл). Полученное твердое вещество отфильтровывали, промывали водой (0,5 мл). Разделением методом HPLC объединенных фильтратов при условиях, описанных ниже, получали соединение 35 (время удерживания: 8,4 мин) и соединение 36 (время удерживания: 8,9 мин).
Соединение 35
LCMS: MS m/z 762.08 [М+Н]+
Соединение 36
LCMS: MS m/z 762.22 [M+H]+
Соединение 35/соединение 36. Условия препаративной HPLC:
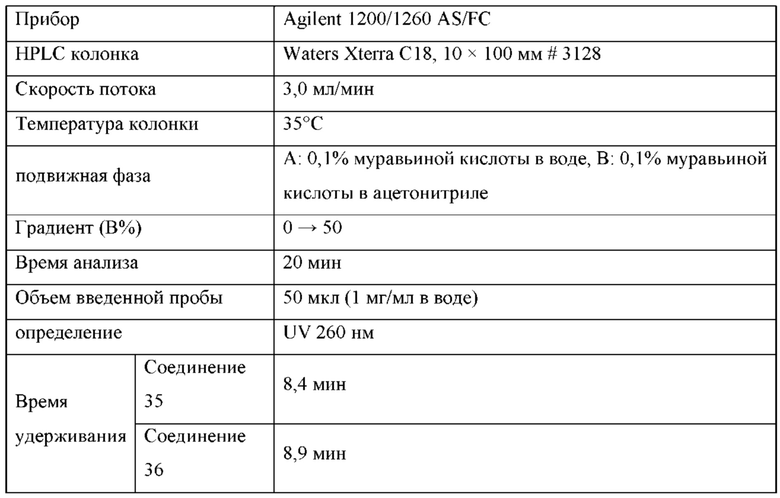
Пример - 23 - Синтез соединения 38 и соединения 39
На фиг. 13 показан пример синтеза соединения 38 и соединения 39.
Стадия 1
К раствору соединения 203 (5,82 г, 9,314 ммоль) и аллилового спирта (0,95 мл, 14 ммоль) в THF (50 мл) при 0°С добавляли раствор DIAD (2,4 мл, 11,6 ммоль) и трифенилфосфин (2,93 г, 11,2 ммоль) в толуоле (25 мл), при этом поддерживали внутреннюю Т ниже 10°С. Полученный раствор нагревали до температуры окружающей среды и перемешивали, при этом за реакцией наблюдали при помощи LCMS. После завершения (5 ч) смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (100 г, 20%-60% EtOAc в н-гептане) с получением 4,56 г соединения 204 в виде розоватого масла.
Соединение 204: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.55 (s, 1H), 8.24 (s, 1H), 7.46 (d, J=8.2 Гц, 2Н), 7.31-7.23 (m, 1H), 7.20-7.10 (m, 2Н), 6.02 (d, J=5.1 Гц, 1H), 6.07-5.95 (m, 1H), 5.21 (dd, J=1.6, 17.2 Гц, 1H), 5.05 (dd, J=1.4, 10.4 Гц, 1H), 5.00 (d, J=5.5 Гц, 2Н), 4.82 (t, J=4.9 Гц, 1H), 4.22-4.16 (m, 1H), 4.05-4.00 (m, 2Н), 3.82 (dd, J=2.5, 11.5 Гц, 1H), 0.95 (s, 9Н), 0.81 (s, 9Н), 0.14 (s, 3Н), 0.13 (s, 3Н), 0.00 (s, 3Н), -0.23 (s, 3Н).
Стадия 2
К соединению 204 (4,56 г, 6,858 ммоль) добавляли уксусную кислоту (80 мл) и воду (20 мл). Полученную смесь перемешивали при температуре окружающей среды всю ночь и при 35°С в течение 1 суток. Полученную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2, 50 г, 33%-60% EtOAc в н-гептане) с получением 3,22 г соединения 205 в виде белого пенящегося твердого вещества.
Соединение 205: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.56 (s, 1H), 7.88 (s, 1H), 7.51-7.47 (m, 2Н), 7.32-7.27 (m, 1H), 7.20-7.16 (m, 2Н), 6.06-5.95 (m, 1H), 5.72 (d, J=7.8 Гц, 1H), 5.26 (dd, J=5.5, 7.8 Гц, 1H), 5.20 (dd, J=1.6, 17.2 Гц, 1H), 5.06 (dd, J=1.6, 10.2 Гц, 1H), 5.03-4.98 (m, 2Н), 4.23 (d, J=5.5 Гц, 1H), 4.14 (s, 1H), 3.92 (dd, J=1.6, 12.9 Гц, 1H), 3.68 (br d, J=12.9 Гц, 1H), 0.75 (s, 9Н), -0.14 (s, 3Н), -0.62 (s, 3Н).
Стадия 3
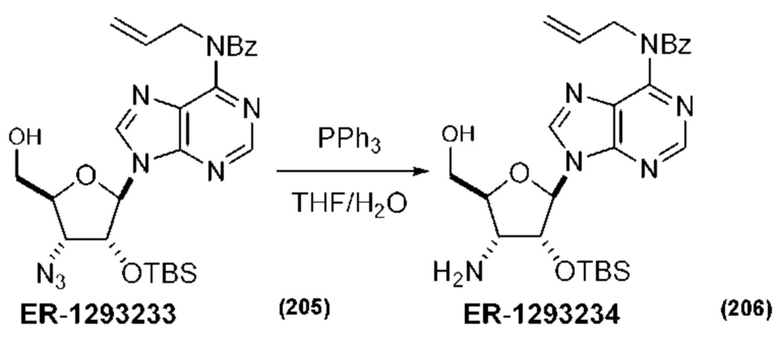
К раствору соединения 205 (3,22 г, 5,847 ммоль) в THF (120 мл) при температуре окружающей среды добавляли воду (30 мл) и трифенилфосфин (2,454 г, 9,355 ммоль). Полученную смесь перемешивали при температуре окружающей среды, при этом за реакцией наблюдали при помощи LCMS. После завершения (18 ч) реакционную смесь концентрировали в вакууме и азеотропировали MeCN трижды. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2(NH) 55 г, 20%-100% EtOAc в н-гептане) с получением 4,82 г соединения 206 (63% допустимая чистота). Продукт использовали на следующей стадии без дополнительной очистки.
Соединение 206: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.56 (s, 1H), 7.92 (s, 1H), 7.54 (qd, J=1.6, 7.6 Гц, 2Н), 7.31-7.27 (m, 1H), 7.19-7.15 (m, 2Н), 6.07-5.94 (m, 1H), 5.84 (d, J=6.6 Гц, 1H), 5.50 (brd, J=10.6 Гц, 1H), 5.20 (dd, J=1.6, 17.2 Гц, 1H), 5.05 (dd, J=1.4, 10.4 Гц, 1H), 5.02-4.98 (m, 2Н), 4.95-4.91 (m, 1H), 4.16-4.08 (m, 2Н), 3.96 (dd, J=1.6, 12.9 Гц, 1H), 3.73 (dd, J=2.0, 5.5 Гц, 1H), 3.73-3.67 (m, 1H), 0.78 (s, 9Н), -0.20 (s, 3Н), -0.46 (s, 3Н).
Стадия 4
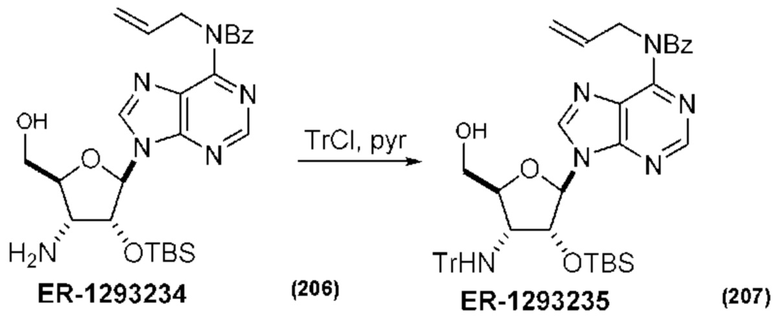
К раствору соединения 206 (4,82 г, 63% чистоты, 5,72 ммоль) в пиридине (39,0 мл) при температуре окружающей среды добавляли TEA (1,3 мл, 8,6 ммоль) и тритил-Cl (1,753 г, 6,289 ммоль). После завершения реакции (наблюдали при помощи LC/MS) добавляли насыщенный раствор NaHCO3 (60 мл). Полученную смесь экстрагировали трижды смесью МТВЕ/EtOAc (1/1, 70 мл каждый раз). Объединенные органические слои промывали 30% водным раствором NaCl (30 мл) и сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 50 г, предварительно обрабатывали 1% TEA в EtOAc/н-гептане, 20%-100% EtOAc в н-гептане с 1% TEA) с получением 1,484 г соединения 207 в виде белого твердого вещества.
Соединение 207: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.49 (s, 1H), 8.10 (s, 1H), 7.50-7.46 (m, 8Н), 7.31-7.22 (m, 7Н), 7.20-7.14 (m, 5Н), 6.11 (d, J=6.3 Гц, 1H), 6.07-5.94 (m, 1H), 5.20 (dd, J=1.2, 17.2 Гц, 1H), 5.06 (dd, J=1.4, 10.4 Гц, 1H), 5.03-4.98 (m, 2Н), 4.84 (dd, J=3.1, 10.6 Гц, 1H), 4.52 (t, J=5.7 Гц, 1H), 3.78 (d, J=1.6 Гц, 1H), 3.64-3.57 (m, 1H), 3.26-3.16 (m, 3Н), 0.77 (s, 9Н), -0.18 (s, 3Н), -0.63 (s, 3Н).
Стадия 5
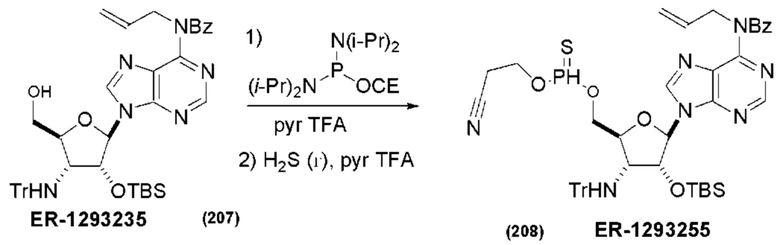
К раствору соединения 207 (0,50 г, 0,652 ммоль) в MeCN (6 мл) при температуре окружающей среды добавляли 3-((бис(диизопропиламино)фосфанеил)окси)пропаннитрил (0,393 г, 1,304 ммоль) и трифторацетат пиридиния (0,113 г, 0,587 ммоль). Полученный раствор перемешивали при температуре окружающей среды, за чем наблюдали при помощи LCMS. После завершения (1 ч) полученный раствор обрабатывали газообразным сульфидом водорода (барботирование) в течение 1 мин, а затем трифторацетатом пиридиния (0,252 г, 1,304 ммоль). Полученный раствор перемешивали при температуре окружающей среды, за чем наблюдали при помощи LCMS. После завершения (1 ч) полученную смесь разбавляли МТВЕ (60 мл). Полученный раствор промывали водой (15 мл каждый раз) дважды и 30% водным раствором NaCl (15 мл) и сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 25%-40% EtOAc в н-гептане) с получением 0,411 г соединения 208 (1:1 Р диастереомерная смесь) в виде белого пенящегося твердого вещества.
Соединение 208 (1:1 Р диастереомерная смесь) 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.49 (s, 1H), 8.49 (s, 1H), 8.11 (s, 1H), 8.06 (s, 1H), 7.54-7.47 (m, 4Н), 7.46-7.38 (m, 14Н), 7.32-7.27 (m, 2Н), 7.23-7.04 (m, 20Н), 6.11-5.99 (m, 2H), 5.98 (d, J=2.0 Гц, 1H), 5.91 (d, J=1.6 Гц, 1H), 5.27 (dd, J=1.6, 6.3 Гц, 1H), 5.23 (dd, J=1.6, 6.3 Гц, 1H), 5.12 (dd, J=1.6, 6.3 Гц, 1H), 5.09 (dd, J=1.2, 6.3 Гц, 1H), 5.02 (br d, J=5.5 Гц, 4H), 4.57-4.45 (m, 2Н), 4.43-4.34 (m, 1H), 4.32-4.04 (m, 8Н), 3.23-3.19 (m, 1H), 3.10 (br s, 2H), 3.06-2.99 (m, 1H), 2.96-2.89 (m, 2H), 2.73 (dt, J=1.4, 6.4 Гц, 2H), 2.65 (t, J=6.3 Гц, 2H), 0.85 (s, 9H), 0.83 (s, 9H), 0.01 (s, 3Н), -0.09 (s, 6H), -0.11 (s, 3Н).
Стадия 6
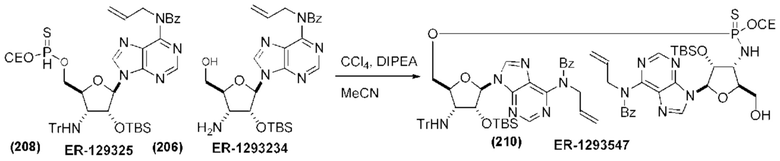
К раствору соединения 208 (0,411 г,,457 ммоль) и соединения 206 (0,293 г, 0,502 ммоль) в ацетонитриле (5 мл) при температуре окружающей среды добавляли DIEA (0,16 мл, 0,91 ммоль) и CCl4 (0,18 мл, 1,8 ммоль). Реакционную смесь перемешивали при температуре окружающей среды, за чем наблюдали при помощи LCMS. После завершения (1 ч) полученную смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 33%-100% EtOAc в н-гептане) с получением 0,461 г соединения 210 (1:1 Р диастереомерная смесь) в виде белого пенящегося твердого вещества.
Соединение 210: 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.52 (s, 1H), 8.51 (s, 1H), 8.49 (s, 1H), 8.49 (s, 1H), 8.24 (s, 1H), 8.19 (s, 1H), 8.13 (s, 1H), 8.01 (s, 1H), 7.55-7.37 (m, 20Н), 7.33-6.99 (m, 30Н), 6.09-5.97 (m, 4Н), 5.96 (d, J=2.7 Гц, 1H), 5.87 (d, J=1.2 Гц, 1H), 5.82 (d, J=4.7 Гц, 1H), 5.80 (d, J=3.9 Гц, 1H), 5.28-5.17 (m, 4Н), 5.11-5.04 (m, 4Н), 5.03-4.96 (m, 8Н), 4.75 (t, J=5.1 Гц, 1H), 4.70 (t, J=4.5 Гц, 1H), 4.46-4.33 (m, 3Н), 4.31-4.18 (m, 5Н), 4.12-3.96 (m, 8Н), 3.89 (br d, J=11.7 Гц, 1H), 3.78-3.67 (m, 3Н), 3.57 (dd, J=2.9, 4.1 Гц, 1H), 3.56-3.44 (m, 2Н), 3.08-3.01 (m, 2Н), 2.99-2.93 (m, 1H), 2.91 (d, J=8.6 Гц, 1H), 2.77-2.73 (m, 1H), 2.72-2.53 (m, 4Н), 0.84 (s, 9Н), 0.84 (s, 9Н), 0.84 (s, 9Н), 0.82 (s, 9Н), 0.00 (s, 3Н), -0.05 (s, 6Н), -0.08 (s, 3Н), -0.12 (s, 3Н), -0.15 (s, 3Н), -0.18 (s, 3Н), -0.18 (s, 3Н).
Стадия 7
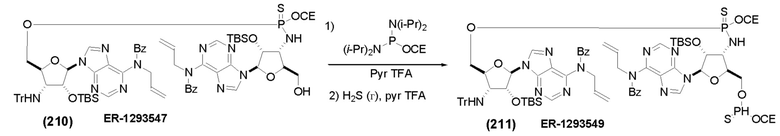
К раствору соединения 210 (0,461 г, 0,324 ммоль) в MeCN (5,5 мл) при температуре окружающей среды добавляли 3-((бис(диизопропиламино)фосфанеил)окси)пропаннитрил (0,195 г, 0,648 ммоль) и трифторацетат пиридиния (0,050 г, 0,259 ммоль). Полученный раствор перемешивали при температуре окружающей среды, за чем наблюдали при помощи LCMS. После завершения (40 мин) полученный раствор барботировали газообразным сульфидом водорода в течение 1 мин, а затем обрабатывали трифторацетатом пиридиния (0,138 г, 0,713 ммоль). После завершения (наблюдали при помощи LCMS) полученный раствор разбавляли МТВЕ (30 мл), промывали водой (10 мл каждого) дважды и 30% водным раствором NaCl (10 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 33%-60% EtOAc в н-гептане) с получением 0,329 г соединения 211 в виде белого пенящегося твердого вещества.
Соединение 211: LC/MS (ESI) m/z 1555.48 [М+Н]+.
Стадия 8
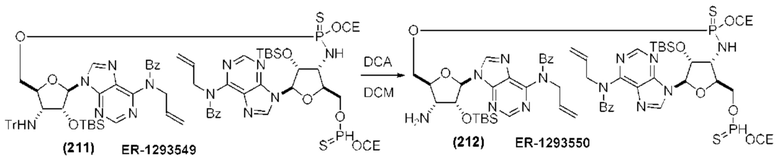
К раствору соединения 211 (0,329 г, 0,211 ммоль) в дихлорметане (7,2 мл) при температуре окружающей среды добавляли воду (0,032 мл, 1,8 ммоль) и дихлоруксусгую кислоту (0,29 мл, 3,6 ммоль). Полученную смесь перемешивали при температуре окружающей среды, за чем наблюдали при помощи LCMS. После завершения (30 мин) реакционную смесь нас. раствором NaHCO3 (25 мл). Полученную смесь экстрагировали дважды DCM (30 мл каждый раз). Объединенные органические слои промывали 30% водным раствором NaCl (20 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт соединения 212 (0,278 г теоретический выход) использовали на следующей стадии без дополнительной очистки.
Соединение 212: LC/MS (ESI) m/z 1313.40 [М+Н]+.
Стадия 9
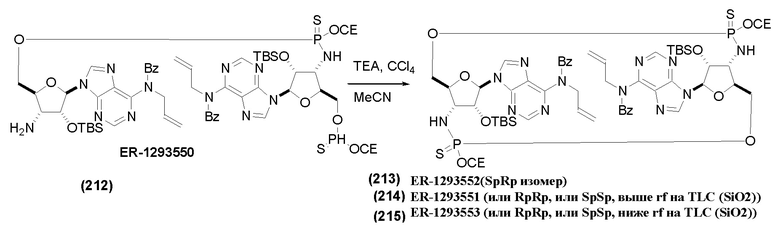
К раствору соединения 212 (0,278 г, 0,212 ммоль) в MeCN (55,6 мл) при температуре окружающей среды добавляли триэтиламин (1,0 мл, 7,2 ммоль) и CCl4 (1,0 мл, 10 ммоль). Полученный раствор перемешивали при температуре окружающей среды, за этим наблюдали при помощи LCMS. После завершения (30 мин) реакционную смесь концентрировали до ~20 мл в вакууме и разбавляли МТВЕ (30 мл). Полученную смесь промывали водой (10 мл) и 30% водным раствором NaCl (10 мл каждого) дважды, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали методом колоночной хроматографии на силикагеле (SiO2 25 г, 20%-75% EtOAc в н-гептане) с получением 71 мг соединения 213 (SpRp изомер), 43 мг соединения 214 (или RpRp, или SpSp изомер, более высокий rf на TLC (SiO2)) и 24 мг соединения 215 (или RpRp, или SpSp изомер, более низкий rf на TLC (SiO2)).
Соединение 213 (SpRp изомер) 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.58 (s, 1H), 8.44 (s, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 7.46 (dd, J=7.2, 15.4 Гц, 4Н), 7.36-7.27 (m, 2Н), 7.18 (q, J=7.6 Гц, 4Н), 6.09 (s, 1H), 5.98 (s, 1H), 6.06-5.95 (m, 2Н), 5.25 (d, J=11.3 Гц, 1H), 5.21 (dd, J=1.4, 10.4 Гц, 1H), 5.12-5.03 (m, 3Н), 5.00-4.92 (m, 3Н), 4.87 (d, J=4.7 Гц, 1H), 4.60 (br d, J=11.7 Гц, 1H), 4.55 (d, J=3.9 Гц, 1H), 4.44 (br d, J=11.3 Гц, 1H), 4.39-4.32 (m, 2Н), 4.26-4.08 (m, 8Н), 3.75 (dd, J=11.3, 16.0 Гц, 1H), 3.49 (dd, J=8.2, 10.6 Гц, 1H), 2.82-2.72 (m, 3Н), 2.61 (td, J=5.9, 17.2 Гц, 1H), 0.99 (s, 9Н), 0.96 (s, 9Н), 0.32 (s, 3Н), 0.23 (s, 6Н), 0.22 (s, 3Н).
Соединение 214 (или RpRp, или SpSp изомер, более высокий rf на TLC (SiO2) изомер): 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.60 (s, 2Н), 8.18 (s, 2Н), 7.50 (d, J=7.0 Гц, 4Н), 7.37-7.31 (m, 2Н), 7.25-7.21 (m, 4Н), 6.05-5.93 (m, 4Н), 5.22 (dd, J=1.6, 17.2 Гц, 2Н), 5.09 (dd, J=1.0, 10.4 Гц, 2Н), 5.00-4.83 (m, 6Н), 4.61 (dd, J=1.2, 11.7 Гц, 2Н), 4.25 (dq, J=4.3, 10.8 Гц, 2Н), 4.09 (br dd, J=3.7, 10.4 Гц, 2H), 4.04 (td, J=5.5, 10.9 Гц, 2Н), 3.99-3.88 (m, 4Н), 3.70 (dd, J=11.9, 15.0 Гц, 2Н), 2.71 (td, J=5.5, 17.1 Гц, 2Н), 2.49-2.37 (m, 2Н), 0.98 (s, 18Н), 0.25 (s, 6Н), 0.24 (s, 6Н).
Соединение 215 (или RpRp, или SpSp изомер, более низкий rf на TLC (SiO2) изомер): 1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.54 (s, 2Н), 8.06 (s, 2Н), 7.47-7.44 (m, 4Н), 7.32-7.27 (m, 2Н), 7.20-7.15 (m, 4Н), 6.06-5.95 (m, 2Н), 5.92 (d, J=3.5 Гц, 2Н), 5.22 (dd, J=1.2, 17.2 Гц, 2Н), 5.07 (dd, J=1.4, 10.4 Гц, 2Н), 5.04-4.94 (m, 6Н), 4.62-4.52 (m, 2Н), 4.46-4.40 (m, 2Н), 4.40-4.22 (m, 8Н), 3.81 (dd, J=6.6, 12.1 Гц, 2Н), 2.89-2.71 (m, 4Н), 0.88 (s, 18Н), 0.08 (s, 6Н), -0.07 (s, 6Н).
Стадия 10
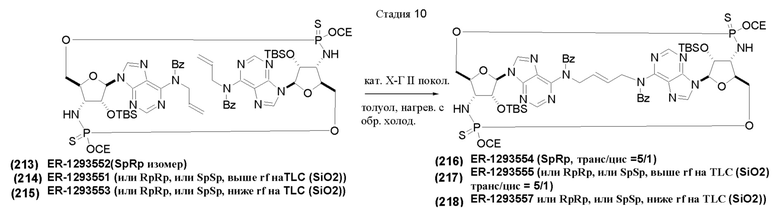
К раствору соединения 213 (71 мг, 0,054 ммоль) в толуоле (28,4 мл) с обратным холодильником добавляли раствор катализатора Ховейда-Граббса 2-го поколения (17,0 мг, 0,027 ммоль) и Р-бензохинон (11,70 мг, 0,108 ммоль) в толуоле (8 мл). Смесь нагревали до температуры образования флегмы и за ходом реакции наблюдали при помощи LC/MS. Через 3 ч добавляли дополнительный катализатор (8,5 мг, 0,0135 ммоль) в толуоле (2,5 мл) и реакцию продолжали еще 2,5 часа. После охлаждения смесь концентрировали в вакууме и очищали методом колоночной хроматографии на силикагеле (SiO2 10 г, 33%-66% этилацетата в н-гептане) с получением 17 мг соединения 216 (транс/цис=5/1) в виде коричневой сухой пены.
Соединение 216: 1Н ЯМР (только транс-изомер, 400 МГц, CHLOROFORM-d) δ=8.37 (s, 1H), 8.34 (s, 1H), 8.24 (s, 1H), 8.20 (s, 1H), 7.41 (d, J=7.1 Гц, 2Н), 7.36-7.27 (m, 3Н), 7.24-7.13 (m, 5Н), 5.97 (d, J=5.9 Гц, 2Н), 5.75 (td, J=5.5, 15.2 Гц, 1H), 5.69 (td, J=5.5, 15.6 Гц, 1H), 5.07 (d, J=3.9 Гц, 1H), 5.03 (dd, J=5.1, 15.2 Гц, 1H), 4.94 (t, J=5.5 Гц, 2Н), 4.85 (dd, J=5.1, 15.2 Гц, 1H), 4.74 (d, J=3.9 Гц, 1H), 4.67 (br d, J=12.1 Гц, 1H), 4.45 (br d, J=10.2 Гц, 1H), 4.42-4.34 (m, 2H), 4.28-4.19 (m, 2H), 4.19-4.06 (m, 4H), 3.96-3.84 (m, 1H), 3.82-3.68 (m, 1H), 3.56 (br dd, J=12.1, 14.5 Гц, 1H), 3.33 (dd, J=9.0, 10.9 Гц, 1H), 2.83-2.78 (m, 2H), 2.72 (td, J=5.6, 16.6 Гц, 1H), 2.39 (td, J=6.3, 17.2 Гц, 1H), 1.00 (s, 18Н), 0.42 (s, 3Н), 0.40 (s, 3Н), 0.34 (s, 3Н), 0.30 (s, 3Н).
Соединение 217
Соединение 214, полученное на стадии 9, обрабатывали отдельно на основе стадии 10 с получением соединения 217 (5/1 смесь транс/цис-изомеров).
1Н ЯМР (только транс-изомер, 400 МГц, CHLOROFORM-d) δ=8.47 (s, 2Н), 8.14 (s, 2Н), 7.43-7.38 (m, 4Н), 7.36-7.29 (m, 2Н), 7.22 (t, J=7.0 Гц, 4Н), 5.83 (s, 2Н), 5.77 (t, J=3.3 Гц, 2Н), 5.17 (d, J=3.5 Гц, 2Н), 5.11-5.04 (m, 2Н), 4.70 (br dd, J=3.5, 16.4 Гц, 2Н), 4.62 (br d, J=12.1 Гц, 2Н), 4.16-4.08 (m, 6Н), 3.93 (br dd, J=3.7, 11.9 Гц, 2Н), 3.74-3.64 (m, 2Н), 3.49 (t, J=12.9 Гц, 2H), 2.42 (td, J=5.9, 18.4 Гц, 2Н), 2.13 (ddd, J=5.5, 7.8, 16.8 Гц, 2Н), 1.00 (s, 18Н), 0.40 (s, 6Н), 0.34 (s, 6Н).
Соединение 218
Соединение 215, полученное на стадии 9, обрабатывали отдельно на основе стадии 10 с получением соединения 218.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=8.14 (s, 2Н), 8.12 (s, 2Н), 7.49-7.43 (m, 2Н), 7.36-7.27 (m, 4Н), 7.19-7.12 (m, 4Н), 5.96 (s, 2Н), 5.67-5.54 (m, 2Н), 4.95-4.82 (m, 4Н), 4.69 (d, J=4.3 Гц, 2Н), 4.60-4.09 (m, 12Н), 3.63 (dd, J=8.8, 16.2 Гц, 2Н), 2.93 (td, J=5.9, 17.2 Гц, 2Н), 2.82-2.72 (m, 2Н), 0.99 (s, 18Н), 0.30 (s, 6Н), 0.27 (s, 6Н).
Стадия 11
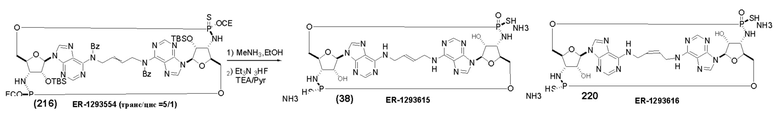
К соединению 216 (транс/цис=5/1, 17 мг, 0,013 ммоль) добавляли раствор метиламинового раствора (2 мл) (33% в EtOH) при температуре окружающей среды. Полученный раствор перемешивали при температуре окружающей среды, что наблюдали при помощи LCMS. После завершения (1 ч) реакционную смесь концентрировали в вакууме. К остатку добавляли пиридин (0,9 мл), TEA (0,45 мл) и триэтиламин тригидрофторид (0,36 мл, 2,2 ммоль). Полученную смесь перемешивали при 50-60°С в течение 4 ч и охлаждали до температуры окружающей среды. После завершения снятия защитных групп с TBS (наблюдали при помощи LCMS) реакционную смесь обрабатывали метокситриметилсиланом (1,5 мл, 12 ммоль) и перемешивали в течение 1 ч. Добавляли воду (3 мл) и полученную смесь дважды экстрагировали толуолом (3 мл каждый раз) и дважды EtOAc (2 мл каждый раз). Водный слой фильтровали при помощи шприцевого фильтра и фильтрат подвергали методу препаративной HPLC с получением 5,3 мг соединения 38 и 1,1 мг соединения 220.
Соединение 38 (SpRp, транс): 1Н ЯМР (400 МГц, METHANOL-d4) δ=9.27 (br s, 1H), 8.47 (br s, 1H), 8.21 (br s, 1H), 8.09 (br s, 1H), 6.19-5.96 (m, 2H), 5.94-5.71 (m, 2H), 5.13-4.68 (m, 2H), 4.55-4.39 (m, 2H), 4.38-4.23 (m, 1H), 4.20-3.91 (m, 5H), 3.75-3.50 (m, 4H).
Соединение 220 (SpRp, цис): 1H ЯМР (400 МГц, METHANOL-d4) δ=9.08 (s, 1H), 8.72 (br s, 1H), 8.55 (s, 1H), 8.24 (s, 1H), 8.18 (s, 1H), 6.22-6.14 (m, 2H), 6.10 (d, J=1.6 Гц, 2H), 5.01 (d, J=3.9 Гц, 1H), 4.41 (br d, J=9.8 Гц, 1H), 4.34-4.25 (m, 3H), 4.24-4.18 (m, 1H), 4.15 (dd, J=6.1, 11.5 Гц, 1H), 4.08-3.99 (m, 4H), 3.71-3.57(m, 3H).
Соединение 39 и соединение 222
Соединение 217 обрабатывали отдельно на основе стадии 11 с получением соединения 39 и соединения 222.
Соединение 39 (транс-изомер): 1H ЯМР (400 МГц, METHANOL-d4) δ=8.83 (br s, 1Н), 8.43 (br s, 1H), 8.17 (br s, 1H), 8.11 (br s, 1H), 6.17-5.95 (m, 2H), 5.93-5.63 (m, 2H), 5.10-4.78 (m, 2H), 4.69-4.53 (m, 1H), 4.52-4.35 (m, 2H), 4.12-3.92 (m, 5H), 3.76-3.44 (m, 4H).
Соединение 222 (цис-изомер): 1H ЯМР (400 МГц, METHANOL-d4) δ=8.71 (br s, 2H), 8.18 (s, 2H), 6.17-6.06 (m, 4H), 4.37 (br d, J=9.8 Гц, 2H), 4.33 (d, J=3.9 Гц, 2H), 4.27 (br dd, J=7.0, 13.7 Гц, 2H), 4.06 (dd, J=7.0, 11.3 Гц, 2H), 4.19-4.03 (m, 2H), 4.01 (br d, J=9.8 Гц, 2H), 3.74 (ddd, J=3.9, 6.4, 10.5 Гц, 2H).
Соединение 40
Соединение 218 обрабатывали отдельно на основе стадии 11 с получением соединения 40: 1Н ЯМР (400 МГц, METHANOL-d4) δ=9.34-8.88 (m, 2Н), 8.73 (br s, 1Н), 8.34-8.02 (m, 2H), 6.20-5.96 (m, 2H), 5.89 (br s, 2H), 5.25-4.95 (m, 2H), 4.76-4.64 (m, 1H), 4.47-4.22 (m, 2H), 4.20-4.07 (m, 2H), 4.07-3.93 (m, 2H), 3.82-3.55 (m, 3H), 3.54-3.38 (m, 2H)
Пример 24 - Синтез соединения 30
Соединение 2 (1,1 мг, 1,4 мкмоль) добавляли в раствор йодметилизопропилкарбоната (13,75 мг, 0,056 ммоль) в ацетоне/воде (0,4/0,10 мл) при температуре окружающей среды. Полученную смесь перемешивали при температуре окружающей среды в темноте, при этом за реакцией наблюдали при помощи LCMS. После завершения (2 ч) реакционную смесь разбавляли водой (0,4 мл) и экстрагировали н-гептаном трижды (0,5 мл каждого). Очисткой неочищенного продукта в водном слое получали 0,5 мг соединения 30.
1Н ЯМР (400 МГц, CHLOROFORM-d) δ=7.92 (s, 2Н), 7.72 (br s, 2Н), 6.22 (d, J=17.2 Гц, 2H), 6.19-6.03 (m, 2Н), 5.98 (br t, J=6.3 Гц, 2H), 5.82-5.77 (m, 2H), 5.68 (dd, J=2.3, 51.6 Гц, 1H), 5.54 (dd, J=10.9, 13.7 Гц, 2H), 5.48 (dd, J=10.9, 12.9 Гц, 2H), 4.94 (квин., J=6.2 Гц, 2Н), 4.71-4.63 (m, 2Н), 4.63-4.56 (m, 2Н), 4.50-4.43 (m, 2Н), 4.22-4.12 (m, 2Н), 4.05-3.90 (m, 2Н), 1.33 (d, J=4.3 Гц, 6Н), 1.32 (d, J=4.3 Гц, 6Н).
Пример 103. Репортерный анализ активности агонистов STING HAQ
Для определения EC50 использовали клетки THP1-Dual™ (InvivoGen, № в кат. thpd-nfis). Поставщиком Invivogen (Insight 201402-1) клетки ТНР1 Dual™ характеризовались как несущие генотип STING HAQ. Клетки выращивали и поддерживали в условиях, рекомендованных производителем. Индукцию пути регуляторного фактора интерферонов (IRF), описанную в руководстве производителя, контролировали для определения EC50. Вкратце, клетки высевали и обрабатывали различными концентрациями соединения в течение 20 часов при инкубации при 37°С, 5% СО2. Клетки ресуспендировали и добавляли раствор QUANTI-Luc™ (№ в кат.: rep-qlc1). Образующееся в результате излучение света измеряли с помощью люминометра (Envision, Perkin Elmer). Полученные сигналы откладывали на графике и рассчитывали EC50 с помощью компьютерной программы GraphPad Prism7.
Значения EC50 описаны в Табл. 4-8 ниже. Значения EC50 могут быть представлены на основании одного анализа или в виде среднего многих анализов. Перед каждой таблицей представлена структура, используемая для описания данной таблицы.
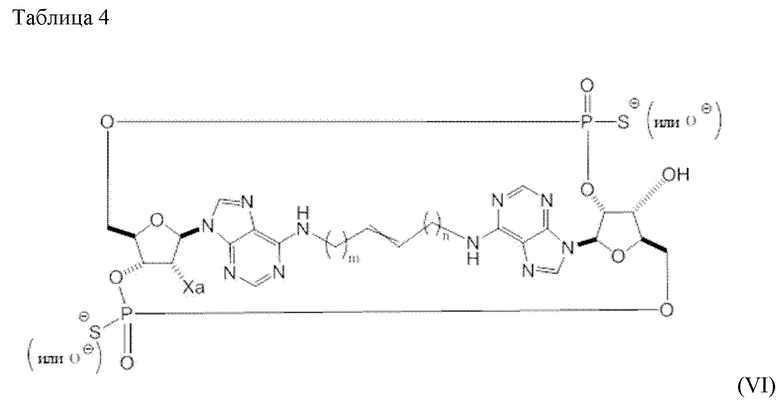
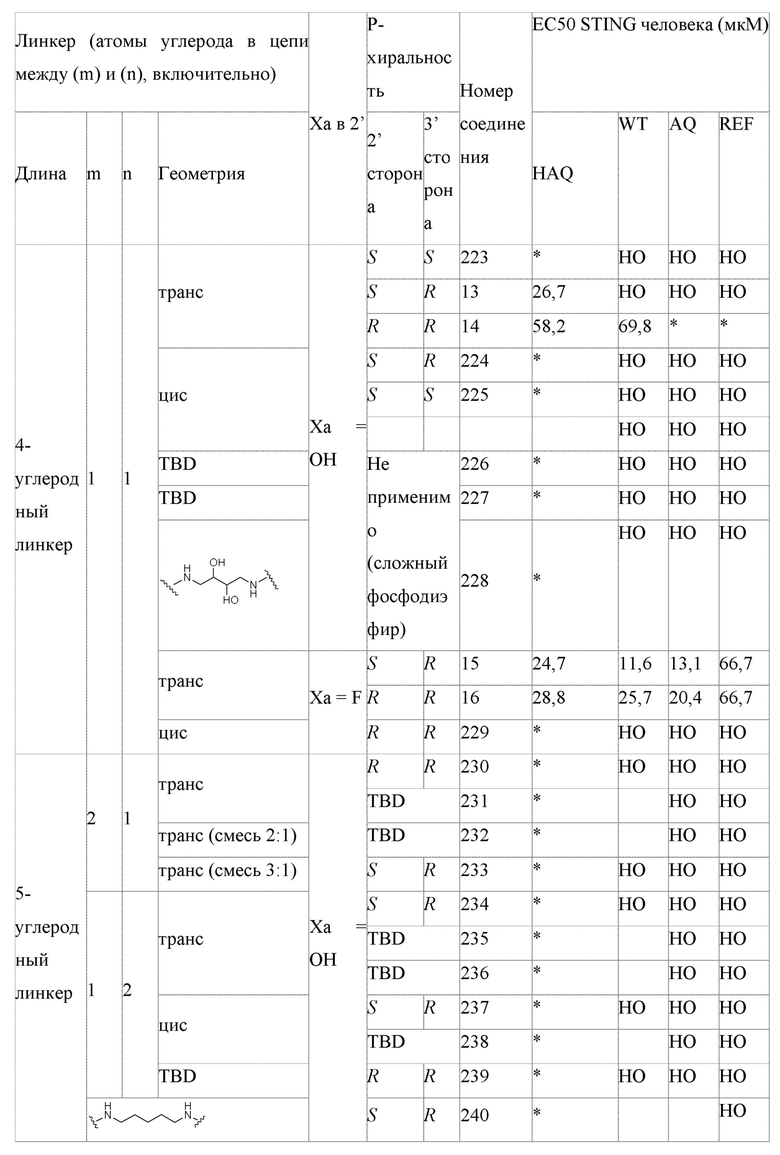

ЕС50 STING человека (мкМ) измеряли с помощью формы аммониевой соли каждого соединения в Табл. 4.
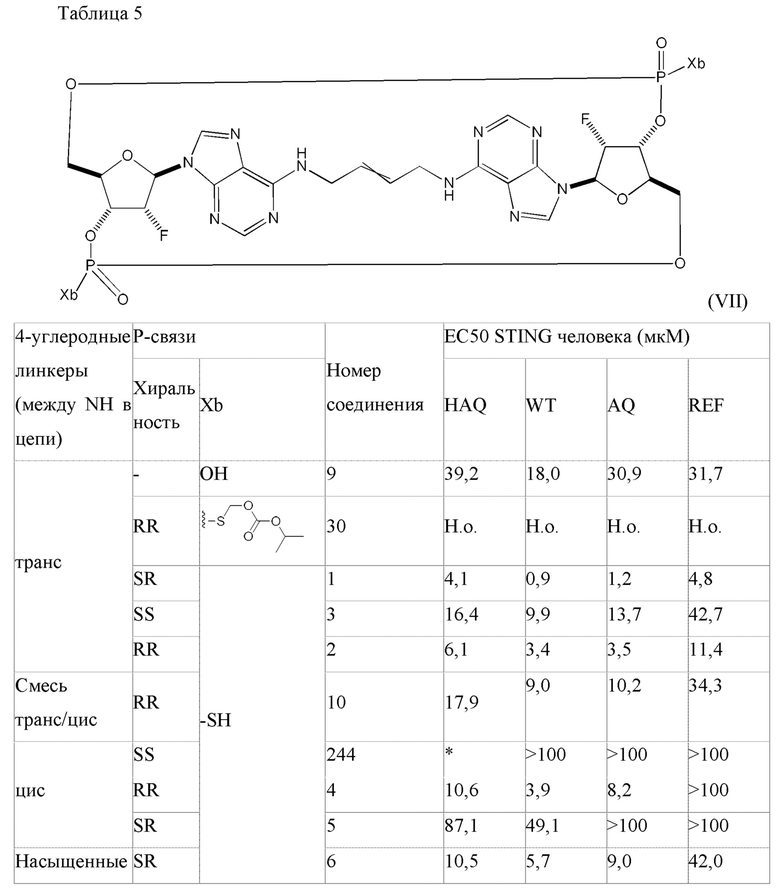

ЕС50 STING человека (мкМ) измеряли с помощью формы аммониевой соли каждого соединения, приведенного в Табл. 5.
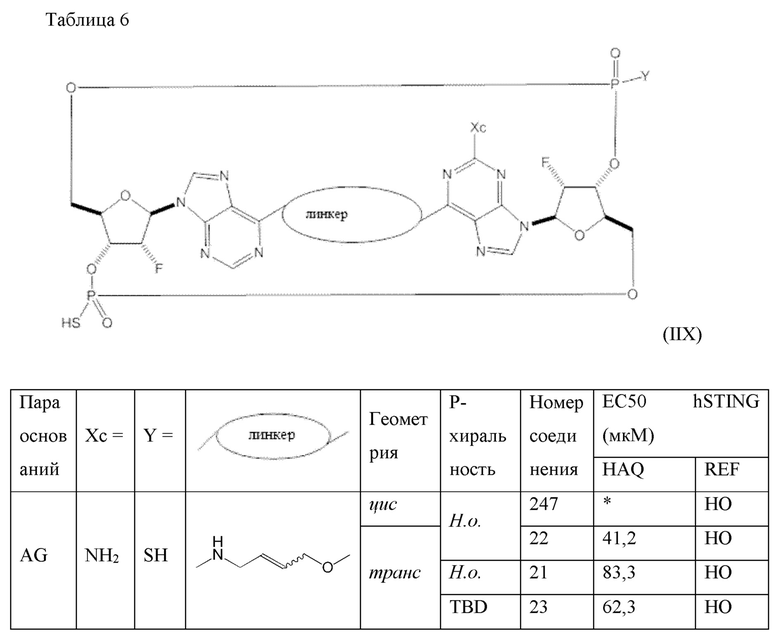
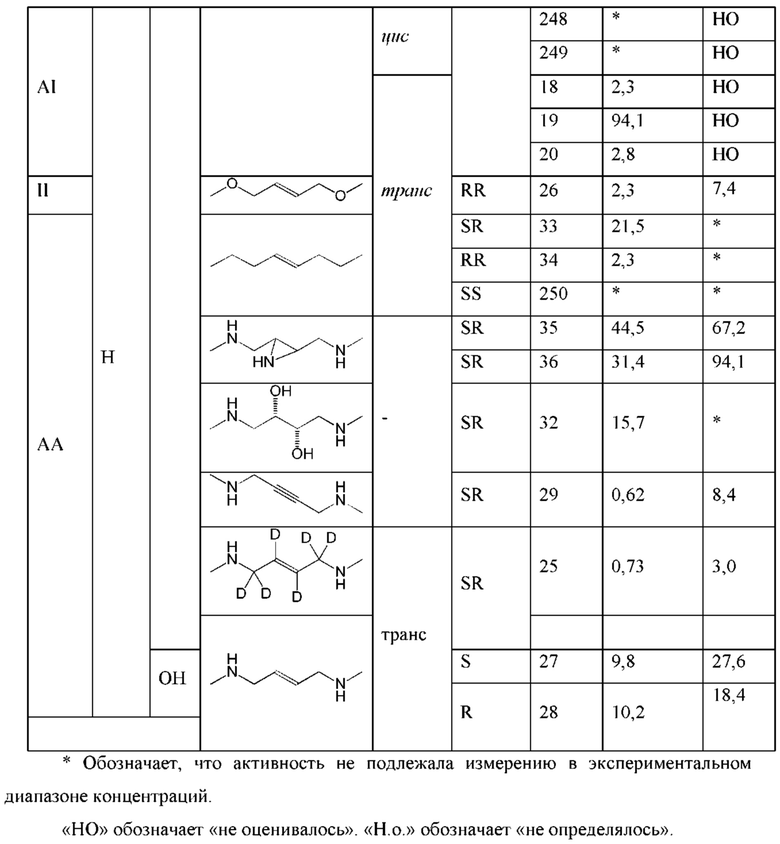
ЕС50 STING человека (мкМ) измеряли с помощью формы аммонийной соли каждого соединения, приведенного в Табл. 6, за исключением Соединения 26, Соединения 31, Соединения 33 и Соединения 34, все из которых исследовали в виде бис-ТЕА солей.
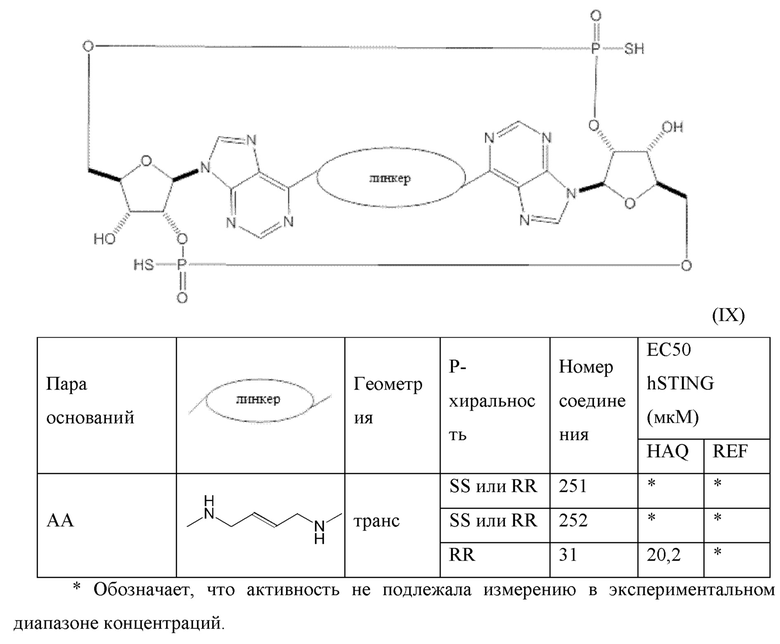
ЕС50 STING человека (мкМ) измеряли с помощью формы аммониевой соли каждого соединения в Табл. 7.
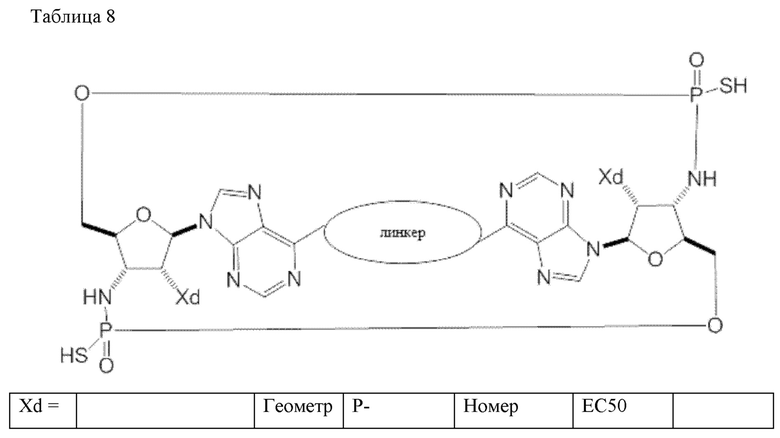
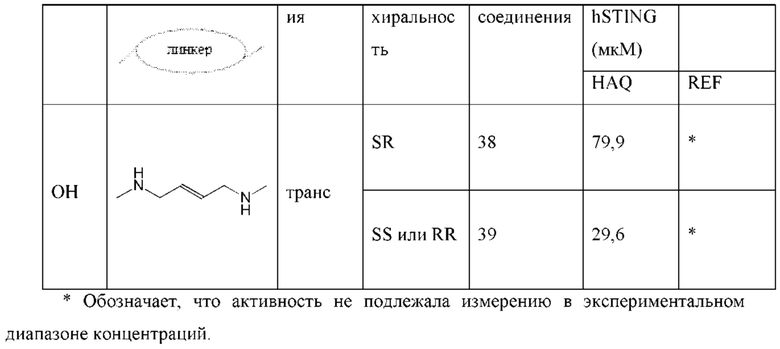
ЕС50 STING человека (мкМ) измеряли с помощью формы аммониевой соли каждого соединения в Табл. 8.
Пример 104. Специфический в отношении вариантов STING репортерный анализ
STING человека имеет 4 основных варианта, в том числе варианты WT, HAQ, REF и AQ. REF-STING, также обозначаемый R232H, например, встречается у приблизительно 14% человеческой популяции. По сравнению с аллелем дикого типа,, R232H характеризовался сниженным ответом в отношении циклических динуклеотидов бактерий и многоклеточных животных. Детали касательно этих 4 основных вариантов, а также других редких вариантов, описаны Yi G, et al., "Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides" PLoS One 2013; 8:e77846. Специфические в отношении вариантов STING репортерные клеточные линии определяли с помощью клеток THP1-Dual™ KO-STING (InvivoGen, № в кат. thpd-kostg) и экспрессионных векторов трех вариантов белка STING. Карта экспрессионного вектора для STING WT изображена на Фиг. 6. Для двух других экспрессионных векторов в этом векторе использовали последовательности различных вариантов STING, при этом STING WT замещали подходящей нуклеотидной последовательностью.
Векторы, экспрессирующие варианты STING, для WT-STING, REF-STING и AQ-STING получали и стабильно трансформировали в клетки THP1-Dual™ KO-STING для подготовки специфических в отношении вариантов STING репортерных анализов для WT-STING, REF-STING и AQ-STING соответственно. Значения EC50 определяли, как описано выше в Примере 103 для репортерного анализа активности агонистов STING HAQ. Результаты показаны ниже в Табл. 9. Последовательности ДНК, используемые для этих вариантов STING, показаны в SEQ ID NO: 1 (нуклеотидная последовательность STING WT человека), SEQ ID NO: 2 (нуклеотидная последовательность STING REF человека) и SEQ ID NO: 3 (нуклеотидная последовательность STING AQ человека).
STING WT человека: 
STING REF человека:

STING AQ человека:

Пример 105. Репортерный анализ активности агонистов STING мыши
Клетки RAW-Lucia™ ISG (InvivoGen, № в кат. rawl-isg) использовали для репортерного анализа агонистов STING мыши. Значения ЕС50 определяли, как описано выше в Примере 103 в репортерном анализе активности агонистов STING HAQ. Результаты показаны ниже в Табл. 9.
Пример 106. Анализ на основе дифференциальной сканирующей флуориметрии (DSF)
Анализ на основе DSF использовали для измерения физического взаимодействия между соединением и рекомбинантным белком STING. Усеченный рекомбинантный белок STING (а.к. 155-341) (SEQ ID NO: 4) экспрессировали в Е. coli и выделяли для анализа, как описано ниже. Матрикс для анализа готовили в 384-луночных планшетах до конечного объема 10 мкл на лунку, состоящего из 1 мкМ рекомбинантного белка STING (а.к. 155-341) (SEQ ID NO: 4), 100 мМ PBS, рН 7,4, с добавлением 100 мМ KCl, 5Х оранжевого красителя SYPRO и 50 мкМ соединения (конечная конц. DMSO 0-1%). Анализы выполняли в системе ПЦР в реальном времени QuantStudio 12K Flex с помощью температурного градиента от 25°С до 95°С при скорости 0,05°С/мин., и фильтрах возбуждения и излучения при 470 и 586 нм соответственно. В соответствии с кривыми производных флуоресценции, определенными с помощью компьютерной программы Applied Biosystems® Protein Thermal Shift (версия алгоритма 1.3.), рассчитывали температуру плавления (Tm) несвязанного и связанного лигандом рекомбинантного белка STING и рассчитывали разницу температуры плавления (dTm D).
Как правило, соединения со значениями ΔTm, большими 0, рассматривали в качестве таких, при которых имело место физическое взаимодействие с исследуемым белком, и значение ΔTm положительно ассоциировано с аффинностью связывания соединений. В данном документе, Соединение 1а характеризовалось ΔTm, составляющим 17,6 (Табл. 9), что указывало на физическое взаимодействие с белком STING.
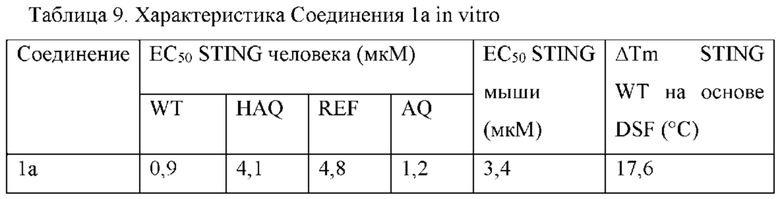
Пример 107. Анализ стимуляции РВМС ex vivo
Кровь человека от 5 здоровых доноров собирали с помощью 10,0 мл пробирок с гепарином BD Vacutainer Sodium (№ в кат. 367874). Выделение мононуклеарных клеток периферической крови (РВМС) выполняли с помощью 50 мл пробирок SIGMA ACCUSPIN (№ в кат. А2055) и Sigma ACCUSPIN System-HISTOPAQUE-1077 (№ в кат. А7054) с помощью протокола, предусмотренного производителем. Слой РВМС извлекали и промывали 1х фосфатно-солевым буферным раствором (PBS), как предложено Sigma. РВМС подсчитывали и в конечном итоге суспендировали при 1×10е6/мл в RPMI (№ в кат. Corning 10-041-CV) с добавлением 10% фетальной бычьей сыворотки (FBS) (№ в кат. Gibco 20140.79). 1 мл клеток (1×10е6) переносили в 5 мл круглодонную полипропиленовую лабораторную пробирку Falcon (№ в кат. 352063) и стимулировали с помощью различных концентраций (0, 0,1, 1, 10 мкМ) в течение 24 часов в 5% CO2 инкубаторе при 37°С.
Через 24 часа инкубации пробирки центрифугировали при 1400 об./мин. в течение 5 минут и супернатанты извлекали. Супернатанты хранили при -80°С для последующего измерения IFNβ. Измерение IFNβ выполняли с помощью базового набора для определения IFN-β человека (№ в кат. Meso Scale Diagnostics K151ADA) и использовали протокол, предусмотренный производителем. Оценку IFN-бета выполняли с помощью считывания аналитического планшета в MESO SECTOR Imager 2400 и использования программы MSD Discovery Workbench 4.0. Через 24 часа белок IFNβ анализировали. Результаты показали, что Соединение 1а может индуцировать продуцирование IFNβ первичными РВМС человека дозозависимым образом.
Результаты, показанные в Табл. 10, отражают среднее значение измерений, проведенных с помощью пяти различных доноров.
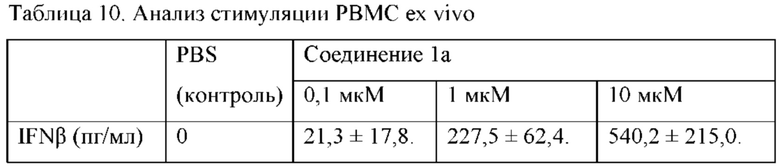
Для количественной оценки мРНК IFNβ суммарную РНК выделяли с помощью набора RNeasy Mini (Qiagen, Германия) в соответствии с протоколом производителя. мРНК IFNβ количественно оценивали с помощью анализа qPCR. Вкратце, суммарную РНК (от 400 нг до 1000 нг) превращали в cDNA в 60 мкл реакционном объеме с помощью SuperScript VILO MasterMix (Life Technologies, США). Полученные cDNA (10 нг) затем амплифицировали с помощью анализов экспрессии Applied Biosystems TaqMan с использованием РНК-специфических праймеров для IFNB1 (Hs01077958_s1) и GAPDH (Hs99999905_m1). Анализ qPCR выполняли с помощью TaqMan Fast Advanced Master Mix (Life Technologies, США) на системе ПЦР в реальном времени Applied Biosystems Quantstudio 12K Flex, при этом выполняли начальный 2-минутный этап при 50°С, затем следовали условия: 95°С в течение 2 сек. и 40 циклов по 95°С в течение 1 сек. и 60°С в течение 20 сек. Относительную экспрессию генов рассчитывали после нормализации по отношению к эталонному гену GAPDH с помощью способа 2-ΔΔСТ. Расчеты выполняли с помощью компьютерной программы Applied Biosystems Quantstudio 12K Flex v1.2.2. Кратность изменений мРНК IFNβ по отношению к образцам, обработанным носителем, представлена в Табл. 11. Результаты показали, что Соединение 1а может индуцировать мРНК IFNβ в первичных РВМС дозозависимым и зависимым от времени образом. В Табл. 11 представлено среднее значение, рассчитанное на основании пяти различных доноров.
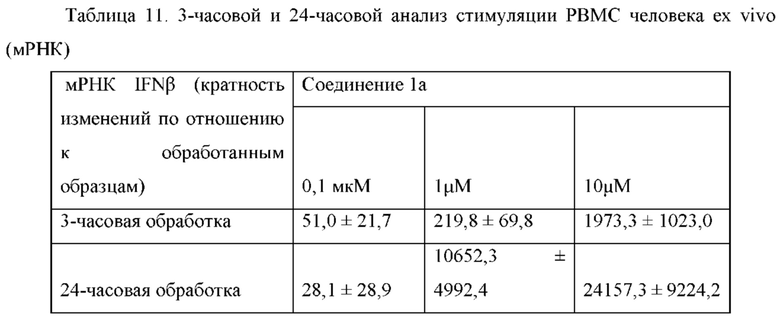
Пример 108. Противоопухолевый эффект Соединения 1а в двойной опухолевой
модели СТ26
Соединение 1a исследовали в отношении противоопухолевой активности в двойной опухолевой модели СТ26, которая представляет собой модель рака толстой кишки мыши. Самкам мышей Balb/cJ в возрасте 5-6 недель (Jackson Labs, Бар-Харбор, Мэн) иплантировали подкожно опухолевые клетки СТ26 на обе стороны каждого животного, по 105 клеток на каждую сторону. Для исследования А обработку начинали через 5 дней (1,25 мг/кг, 2,5 мг/кг и 5 мг/кг) после имплантации опухоли, когда средние объемы опухоли достигали примерно 100 мм3. Для исследования В обработку начинали через 8 дней (0,6 мг/кг и 10 мг/кг) после имплантации опухоли, когда средние объемы опухоли достигали примерно 120 мм3. Схема обработки описана в Табл. 12 и Табл. 13.


Все мыши в исследовании имели две подкожные опухоли СТ26. «Обработанная опухоль» означает опухоль, в отношении которой осуществляли прямое введение соединения, в то время как «необработанная опухоль» означает опухоль, в отношении которой не осуществляли прямое введение соединения. Объем опухоли контролировали в течение всего эксперимента. Объем опухоли измеряли ежедневно после начала эксперимента. Опухолевую нагрузку рассчитывали на основании измерений с помощью штангенциркуля с использованием формулы для объема вытянутого эллипсоида (L×W2)/2, где L и W представляют собой соответствующие ортогональные величины длины и ширины (мм).
Соединение 1а характеризовалось высокой и терапевтической активность в двойной модели опухоли СТ26 (Фиг. 7 и Фиг. 8). В случае обработанных опухолей уровень излечения, составляющий 20%, выявляли даже при наиболее низкой исследуемой дозе в исследовании (Фиг. 8, доза 0,6 мг/кг). В то же самое время наиболее высокая доза (10 мг/кг) приводила к излечению 100% животных от указанной опухоли в конце исследования. В случае необработанных опухолей также был очевидным дозозависимый противоопухолевый эффект. Группа с максимальной дозой (10 мг/кг) характеризовалась 80% терапевтическими эффектами; все более низкие дозы также характеризовались ингибирующей активностью опухолевого роста. Таким образом, наблюдали терапевтическое окно, составляющее от 0,6 мг/кг до 10 мг/кг для Соединения 1а, при этом противоопухолевую активность наблюдали не только локально, но также и системно, на основании эффектов в не подвергшемся инъекции дистальном опухолевом очаге. В заключение следует отметить, что эти результаты свидетельствовали о том, что локальное введение Соединения 1а может индуцировать как локальную, так и системную (ингибирующую) противоопухолевую активность.
Пример 109. Противоопухолевый эффект Соединения 1а в модели метастазов печени СТ26
Соединение 1а исследовали в отношении его противоопухолевой активности в модели метастазов печени СТ26. Находящимся под анестезией самкам мышей BALB/cJ в возрасте 5-6 недель (Jackson Labs, Бар-Харбор, Мэн) внутриселезеночно имплантировали опухолевые клетки СТ26, экспрессирующие люциферазу (5×105 клеток на мышь). В течение последующего десятиминутного периода опухолевым клеткам давали поступить по кровотоку в печени животных. Затем селезенки удаляли и животным накладывали швы и давали восстановиться. Через три дня опухолевые клетки СТ26 (105 клеток на мышь) имплантировали повторно, в этот раз подкожно (sc) в области под правой передней конечностью, способствуя развитию опухолевой массы для введения соединений. Через девять дней после внутриселезеночной инъекции соединение (10 мг/кг) вводили интратуморально, однократно, в sc опухоль.
Локальный противоопухолевый эффект соединения измеряли с помощью его влияния на sc опухоль, при этом ингибирующий эффект соединения оценивали по суммарной выживаемости обработанных мышей по сравнению с обработанными основой контрольными мышами, на основании неблагоприятного влияния растущей опухолевой массы в печени каждой мыши. Соединение 1a характеризовалось как высокой активностью в отношении локальных sc опухолей, равно как и терапевтической системной активностью у 9 из 10 обработанных животных (Фиг. 9). Эти результаты свидетельствовали о том, что локальное введение Соединения 1а может индуцировать как локальную, так и системную (ингибирующую) противоопухолевую активность, в том числе в отношении удаленного поражения, например, в печени.
Пример 110. Противоопухолевый эффект Соединения 1а в ортотопической модели головного мозга GL261
Соединение 1а исследовали в отношении его противоопухолевой активности в ортотопической модели головного мозга GL261. GL261 представляет собой линию клеток глиомы мышей. Клетки глиомы мышей GL261, экспрессирующие люциферазу (2×104 клеток/мышь), интракраниально имплантировали самкам мышей-альбиносов В6 в возрасте 5-6 недель (Jackson Labs, Бар-Харбор, Мэн). Через 3-4 дня клетки GL261 имплантировали подкожно (106клеток/мышь) в области под правой передней конечностью с целью способствования развитию опухолевой массы для введения соединений. Через десять дней после интракраниальной имплантации соединение (10 мг/кг) вводили интратуморально, однократно, в sc опухоль. Локальный противоопухолевый эффект соединения измеряли с помощью его влияния на sc опухоль, при этом ингибирующий эффект соединения оценивали по суммарной выживаемости обработанных мышей по сравнению с обработанными основой контрольными мышами, на основании неблагоприятного влияния растущей опухолевой массы в головной мозг каждой мыши. Соединение 1а характеризовалось как высокой активностью в отношении локальных sc опухолей, как и терапевтической системной активностью у 5 из 8 обработанных животных (Фиг. 10). Эти результаты свидетельствовали о том, что локальное введение Соединения 1а может индуцировать как локальную, так и системную (ингибирующую) противоопухолевую активность, в том числе в отношении удаленного поражения, например, в головном мозге.
Пример 111. Рентгенокристаллографическая структура, подтверждающая комплекс с STING WT
С целью дополнительного понимания механизма связывания с мишенью новых исследуемых соединений определяли рентгенокристаллографическую структуру STING WT в комплексе с соединениями.
A. Экспрессия и очистка С-концевого домена STING WT (остаток 155-341)
Последовательность ДНК, кодирующую белок STING WT человека от аминокислоты 155 до 341 (SEQ ID NO: 4) клонировали в вектор pET21b, после метки His-TEV-Sumo на его N-конце (SEQ ID NO: 5). Последовательность pET21b депонирована в Addgene и доступна по адресу addgene.org/vector-database/2550/; указанная последовательность включена в данный документ посредством ссылки.
Клетки BL21 (DE3) Codon Plus Е. coli трансформировали указанной плазмидой и экспрессию рекомбинантного белка индуцировали 0,1 мМ изопропил-β-D-1-тиогалактопиранозидом (IPTG). Белок очищали из растворимой фракции клеточного лизата с помощью аффинной хроматографии Ni-NTA. Метку His-TEV-Sumo удаляли с помощью протеазы Sumo и отделяли от не содержащей метки последовательности 155-341 STING WT с помощью второй колонки для аффинной хроматографии Ni-NTA. Белок дополнительно очищали с помощью анионообменной хроматографии и хранили в буфере, содержащем 20 мМ Tris⋅HCl, рН 7,5, и 150 мМ NaCl при концентрации 35 мг/мл.
B. Кристаллизация и определение структуры С-концевого домена STING WT в комплексе с Соединением 1
С целью сокристаллизации последовательности 155-341 STING WT с Соединением 1 белок STING WT разводили до 10 мг/мл с помощью буфера для хранения (20 мМ Tris⋅HCl, рН 7,5, и 150 мМ NaCl) и смешивали с Соединением 1 (100 мМ стоковый раствор в DMSO) в молярном соотношении 1:5. Смесь инкубировали в течение 4 часов при 4°С и центрифугировали при 13000 об./мин. в течение 20 минут до кристаллизации. Сетчатые поддоны для кристаллизации устанавливали с помощью метода висячей капли посредством диффузии в парах при 18°С. Кристаллы выращивали с помощью смешивания 1 мкл раствора STING WT/Соединения 1 с равным объемом раствора из лунки, содержащего 100 мМ HEPES, рН 7,5, 200 мМ CaCl2 и 15% (мас./об.) PEG 8000. 20% (мас./об.) PEG 400 использовали в качестве криопротекторного реагента при мгновенной заморозке в жидком азоте. Массивы данных для дифракции собирали с помощью детектора Pilatus с пучком излучения SSRF BL19U1 и обрабатывали с помощью HKL3000 и программы SCALEPACK2MTZ в пакете программного обеспечения ССР4.
Структуру последовательности 155-341 STING WT, связанной с Соединением 1, определяли с помощью программы молекулярного замещения PHASER (молекулярное замещение с максимальным правдоподобием) с использованием PDB ID 4F9E в качестве модели исходного поиска. Присутствие Соединения 1 между поверхностью раздела димера STING WT подтверждали в разностной карте Fo-Fc, рассчитанной с использованием модельных фаз. Модель строили и завершали вручную с помощью программы Coot и совершенствовали с помощью программы Refmac5 в пакете программного обеспечения ССР4. Конечную усовершенствованную структуру описывали при разрешении 2,38  в пространственной группе Р212121 с использованием элементарной ячейки, измеренной при а=33,820, b=78,110, с=132,212, α=90,00, β=90,00, γ=90,00. Две копии последовательности 155-341 STING WT идентифицировали в каждой асимметричной ячейке, связанной с одной молекулой Соединения 1 на поверхности раздела димера.
в пространственной группе Р212121 с использованием элементарной ячейки, измеренной при а=33,820, b=78,110, с=132,212, α=90,00, β=90,00, γ=90,00. Две копии последовательности 155-341 STING WT идентифицировали в каждой асимметричной ячейке, связанной с одной молекулой Соединения 1 на поверхности раздела димера.
С. Взаимодействие Соединения 1 с STING WT, наблюдаемое в рентгенокристаллической структуре
На Фиг. 11 представлено изображение рентгенокристаллографической структуры STING WT человека в комплексе с Соединением 1. Исследовали рентгенокристаллографическую структуру STING WT человека в комплексе с Соединением 1, которое сокристаллизовали из образца Соединения 1а. Соединение связывалось в кармане поверхности раздела, образованном димером белка STING WT. Две грани аденинового основания соединения образовывали π-π стэкинг-взаимодействие с Tyr240 и гуаниновой группой Arg238 соответственно. Транс-олефиновый линкер образовывал ван-дер-ваальсовое взаимодействие с алифатической частью боковой цепи Arg238. Фтористый заместитель в С2' положении рибозной группы соединения встраивался в гидрофобное отверстие, определенное с помощью Thr263, Pro264 и Tyr163. Отрицательно заряженная трифосфатная группа соединения образовывала солевой мостик с помощью Arg238 и водородных связей с Ser162 и Thr267 соответственно. Кроме этого, триофосфатная группа также образовывала электростатическое взаимодействие с гуанидиновой группой Arg232. Область петли LID STING WT, состоящая из остатков 226-243, охватывал две группы оснований и транс-олеиновый линкер.
Пример 112. Определение рентгенокристаллографической структуры STING REF в комплексе с Соединением 1.
А. Экспрессия и очистка С-концевого домена STING REF (остаток 155-341,, SEQ ID NO: 6)
Последовательность ДНК, кодирующую белок STING REF человека от аминокислоты 155 до 341 (SEQ ID NO: 6) клонировали в вектор pET21b, после метки His-TEV-Sumo на его N-конце (SEQ ID NO: 7). Последовательность pET21b депонирована в Addgene и доступна по адресу addgene.org/vector-database/2550/; указанная последовательность включена в данный документ посредством ссылки.
Клетки BL21 (DE3) Codon Plus Е. coli трансформировали указанной плазмидой и экспрессию рекомбинантного белка индуцировали 0,1 мМ изопропил-β-D-1-тиогалактопиранозидом (IPTG). Белок очищали из растворимой фракции клеточного лизата с помощью аффинной хроматографии Ni-NTA. Метку His-TEV-Sumo удаляли с помощью протеазы Sumo и отделяли от не содержащей метки последовательности 155-341 STING REF с помощью второй колонки для аффинной хроматографии Ni-NTA. Белок дополнительно очищали с помощью анионообменной хроматографии и хранили в буфере, содержащем 20 мМ Tris⋅HCl, рН 7,5, и 150 мМ NaCl при концентрации 24 мг/мл.
В. Кристаллизация и определение структуры С-концевого домена STING REF в комплексе с Соединением 1
С целью сокристаллизации последовательности 155-341 STING REF с Соединением 1 белок STING REF разводили до 10 мг/мл с помощью буфера для хранения (20 мМ Tris⋅HCl, рН 7,5, и 150 мМ NaCl) и смешивали с Соединением 1 (100 мМ стоковый раствор в DMSO) в молярном соотношении 1:5. Смесь инкубировали в течение 4 часов при 4°С и центрифугировали при 13000 об./мин. в течение 20 минут до кристаллизации. Сетчатые поддоны для кристаллизации устанавливали с помощью метода висячей капли посредством диффузии в парах при 18°С. Кристаллы выращивали с помощью смешивания 1 мкл раствора STING REF/Соединения 1 с равным объемом раствора из лунки, содержащего 100 мМ HEPES, рН 7,5, 200 мМ CaCl2 и 15% (мас./об.) PEG 8000. 20% (мас./об.) PEG 400 использовали в качестве криопротекторного реагента при мгновенной заморозке в жидком азоте. Массивы данных для дифракции собирали с помощью детектора Pilatus с пучком излучения SSRF BL18U1 и обрабатывали с помощью HKL3000 и программы SCALEPACK2MTZ в пакете программного обеспечения ССР4. Указанная структура изображена на Фиг. 12.
Структуру последовательности 155-341 STING REF, связанной с Соединением 1, определяли с помощью программы молекулярного замещения PHASER (молекулярное замещение с максимальным правдоподобием) с использованием ранее определенной структуры последовательности 155-341 STING WT (как описано выше) в качестве модели исходного поиска. Присутствие Соединения 1 между поверхностью раздела димера STING REF подтверждали в разностной карте Fo-Fc, рассчитанной с использованием модельных фаз. Модель строили и завершали вручную с помощью программы Coot и совершенствовали с помощью программы Refmac5 в пакете программного обеспечения ССР4. Конечную усовершенствованную структуру описывали при разрешении 2,76 в пространственной группе Р212121 с использованием элементарной ячейки, измеренной при а=33,733, b=77,831, с=131,689, α=90,00, β=90,00, γ=90,00. Две копии последовательности 155-341 STING REF идентифицировали в каждой асимметричной ячейке, связанной с одной молекулой Соединения 1 на поверхности раздела димера.
С. Взаимодействие Соединения 1 с STING REF, наблюдаемое в рентгенокристаллической структуре
На Фиг. 12 изображена рентгенокристаллографическая структура STING REF человека в комплексе с Соединением 1, которое сокристаллизовали из образца Соединения 1а. Соединение связывалось в кармане поверхности раздела, образованном димером белка STING. Две грани аденинового основания соединения образовывали π-π стэкинг-взаимодействие с Tyr240 и гуаниновой группой Arg238 соответственно. Транс-олефиновый линкер образовывал ван-дер-ваальсовое взаимодействие с алифатической частью боковой цепи Arg238, в то время как гуанидиновая часть боковой цепи Arg238 образовывала π-π стэкинг-взаимодействие с имидазольной группой боковой цепи His232 снаружи. Олефиновый линкер находился в контакте с взаимодействующей парой боковых цепей Arg238 и His232. Фтористый заместитель в С2' положении рибозной группы соединения встраивался в гидрофобное отверстие, определенное с помощью Thr263, Pro264 и Tyr163. Отрицательно заряженная трифосфатная группа соединения образовывала солевой мостик с помощью Arg238 и водородных связей с Ser162 и Thr267 соответственно. Область петли LID STING REF, состоящая из остатков 226-243, охватывал две группы оснований и трансолеиновый линкер.
Пример 113. Сравнения
Значения ЕС50 рассчитывали для анализов STING человека в случае STING WT, STING HAQ, STING AQ и STING REF при прямых сравнениях с использованием Соединения 1а по настоящему исследованию, природного лиганда STING (2'3' cGAMP) и предполагаемом агонисте STING ML RR-S2 CDA, описанном в Corrales, et al., "Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity," Cell Reports (2015) 11:1018-1030, которая включена в данный документ посредством ссылки. Анализы проводили, как описано в примерах, изложенных выше. Следует обратить внимание, что значения в Табл. 14 были ограничены анализами, проводимыми в прямых сравнениях и могли не отражать усредненных значений, определенных в большем количестве исследований, описанных в Табл. 5 или в других разделах. Дополнительно следует обратить внимание, что «2'3' cGAMP» означает то же самое, что и «ML cGAMP», как описано в публикации Cell Reports.

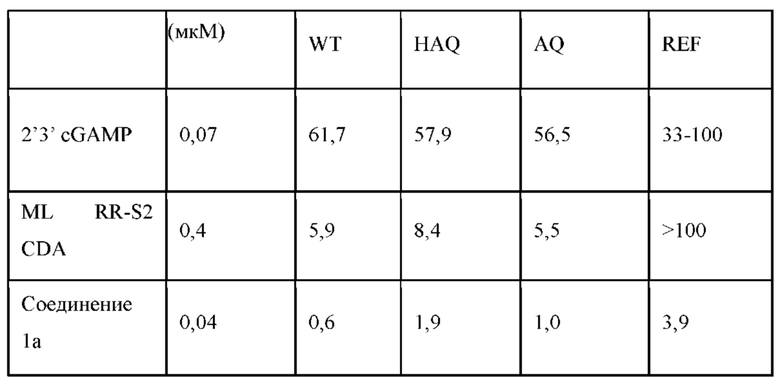
В Табл. 14 также описаны константы диссоциации (Kd) в случае связывания STING WT человека с каждым из трех исследуемых соединений, измеренные с помощью изотермической титрационной калориметрии (ITC). ITC представляет собой микрокалориметрическую титрационную методику, с помощью которой измеряют термодинамические свойства, ассоциированные с межмолекулярными взаимодействиями. На основании этих исследований, Соединение 1а, по-видимому, образовало наиболее сильную связь с STING WT из исследуемых соединений.
Материалы
Рекомбинантный белок дикого типа человека STING (а.к. 139-379, H232R) получали с помощью экспрессии конструкции в Е. coli, кодирующей цитозольный домен STING WT человека, содержащий аминокислоты 139-379.
Реагенты
Источники реагентов, используемых в данном исследовании, показаны ниже.
Приготовление белкового буфера
Белок STING хранили при -60°С в 90 мкл и 100 мкл аликвот, каждую в концентрации 3,0 мг/мл и 20 мг/мл соответственно в PBS, рН 7,5, содержащем 5% глицерина. В день анализа аликвоты белка размораживали, разводили в 400 мкл и обменивали с помощью буфера в PBS с использованием центрифужного фильтровального блока Amicon Ultra (MW пороговое значение 10k, 0,5 мл) с применением по меньшей мере четырех 10 минутных 14000 × g центрифугирований с использованием микроцентрифуги Eppendorf и затем окончательно разводили до 20-30 мкМ (в зависимости от эксперимента) с помощью 1X PBS. Концентрацию белка определяли с помощью спектрофотометра Nanodrop 2000 и коэффициент экстинкции белка 22140 (М-1 см-1).
Приготовление образцов
Двести мл 1 мМ стоковых растворов Соединения 1a, 2'3' cGAMP и ML RR-S2CDA помещали в 1,5 мл микроцентрифужные пробирки. Перед каждым экспериментом образцы разводили до концентраций 200-500 мкМ (в зависимости от эксперимента).
Способы
Анализы выполняли на блоке Affinity ITC (ТА Instruments. №609003.901), оснащенном приспособлением для очистки при выполнении ITC (ТА Instruments, №601800.901). Раствор белка STING в количестве примерно 400 мкл, содержащий 20-30 мкМ белка STING, отмеряли пипеткой в 185 мкл калориметрическую ячейку, обеспечивая некоторый допустимый избыток. Эталонная ячейка содержала эквивалентное количество воды Milli-Q. Инкубирование выполняли при 25°С с помощью 20×2,5 мкл инъекций 100-300 мкМ соединений. Контрольную программу ITC Run Ver. 3.3.0.0 (ТА Instruments) использовали для получения термограмм, состоящих из многочисленных пиков исходного тепла (мккал/сек), представляющих тепловой поток при каждой инъекции. Аналитическую программу Nano Analyze Ver. 3.70 (ТА Instruments) использовали с целью поправки на исходный уровень, поправки на холостую теплоту разведения или теплоту разведения образца (при насыщении) и с целью интеграции пиков теплового потока, приводя к образованию значений «Q» на графиках. Образующиеся изотермы подгоняли с помощью независимой модели для получения термодинамических параметров.
Получали и описывали значения Kd и n (молярные соотношения в точках перегиба кривой). Оптимальные условия для концентраций белка и лиганда получили из предварительных экспериментов.
Результаты
Определяли термограммы теплового потока и полученные на основе них изотермы связывания исследуемых соединений с рекомбинантных белком дикого типа человека STING (а.к. 139-379, H232R). Связывание каждого соединения со STING было эндотермическим, как обозначено отрицательными тепловыми потоками с наличием экзотермической (положительное направление) теплоты разведения (наблюдаемой после того, как соединение достигло насыщения белка). Было показано, что 2', 3' cGAMP приводили к аналогичному эндотермическому ответу в отношении различных вариантов STING. Соединение 1а характеризовалось наименьшим значением Kd, составляющим 0,04 мкМ, за ним следовали 2'3' cGAMP с Kd, составляющей 0,07 мкМ и затем ML RR-S2 CDA с Kd, составляющей 0,40 мкМ. Все соединения характеризовались значениями n, близкими к 0,5, обозначая, что белок STING присутствовали в виде димера и был связан с 1 молем соединения на 2 моля STING.
Пример 114. Идентификация потенциальных метаболитов
Соединение 1а инкубировали в гепатоциах мыши CD-1, крысы Sprague Dawley, собаки Beagle, яванского макака и человека с целью оценки образования основных метаболитов.
Материалы
Криоконсервированные объединенные гепатоциты приобретали в ThermoFisher Scientific (Уолтем, Массачусетс), Xenotech, LLC (Канзас-Сити, Канзас) и In Vitro ADMET Laboratories (Колумбия, Мериленд), а соответствующие среды приобретали в In Vitro ADMET Laboratories (Колумбия, Мериленд) и Life Technologies (Карлсбад, Калифорния). Раствор для окрашивания на основе AOPI и фосфатный буфер приобретали в Corning Life Sciences (Тьюксбери, Массачусетс) и Nexcelom Bioscience (Лоренс, Массачусетс) соответственно. Все химические вещества, реагенты и растворители, используемых в анализе, были аналитической степени чистоты или степени чистоты для ВЭЖХ.
Схемы и процедуры экспериментов
Инкубация гепатоцитов
Соединение 1а взвешивали и растворяли в воде для ВЭЖХ, содержащей 0,12% муравьиную кислоту в PBS с получением 1020 ммоль/л. Затем раствор разводили в 2,5 раза отдельно до 4 ммоль/л и после этого дополнительно разводили в 21000 раз средой Вильямса Е, содержащей 0,1% человеческий сывороточный альбумин и 2 ммоль/л L-глутамина с образованием рабочего стокового раствора с концентрацией 20 мкмоль/л.
Перед инкубацией криоконсервированные гепатоциты размораживали в воде при 37°С. Одну пробирку криоконсервированных гепатоцитов добавляли к каждой 50 мл конической пробирке со средой для извлечения криоконсервированных гепатоцитов (UCRM), полученной в In Vitro ADMET Laboratories (Колубмия, Мериленд). Клетки центрифугировали в центрифуге Beckman (Брея, Калифорния) с ротором GH 3.8 при 740 об./мин. в течение 10 минут при комнатной температуре 4°С. Супернатант удаляли и клетки ресуспендировали в среде для культивирования для подсчета. После того, как клетки ресуспендировали в среде для культивирования, 20 мкл ресуспензии переносили и смешивали с 20 мкл раствора для окрашивания на основе AOPI. Раствор осторожно смешивали и клетки подсчитывали с помощью Cellometer (Nexcelom, Лоренс, Массачусетс). После подсчета клетки ресуспендировали при концентрации 1 или 2 миллиона жизнеспособных клеток/мл в среде Вильямса Е, содержащей 2 ммоль/л L-глутамиона (рН 7,4).
Суспензию гепатоцитов (50 мкл/лунка) добавляли в 48-луночный планшет. Пятьдесят микролитров рабочего стокового раствора, содержащего Соединение 1а (20 мкмоль/л), добавляли к началу реакции. Планшет помещали в инкубатор для культивирования тканей (атмосфера увлажненного воздуха 5% CO2/95% и 37°С) и реакции завершали добавлением 200 мкл останавливающего раствора из смеси 100% метанол/ацетонитрил (1/1, об./об.) с 2010 нг/мл фуросемида и 0,2 мкмоль/л (R)-пропранолола через 5, 30, 60, 120, 180 и 240 минут. Смесь центрифугировали и фильтровали, а супернатант собирали для анализа. Конечные концентрации криоконсервированных гепатоцитов составляли 1×106 клеток/мл. Конечная инкубационная концентрация Соединения 1а составляла 10 мкмоль/л.
Условия LC-MS/MS для идентификации метаболитов
Система LC-MS/MS состояла из системы для ВЭЖХ Shimadzu и AB-SCIEX TripleTOF 5600 Hybrid Quadrupole и масс-спектрометра TOF (Фрамингем, Массачусетс). Система для ВЭЖХ Shimadzu (Киото, Япония) состояла из модуля коммуникационной шины (СВМ-20А), автодозатора (SIL-30AC) с прикрепленным устройством для смены штативов (Rack Changer II), двух насосов (LC-30AD) и термостата колонок (СТО-30А). Масс-спектрометр калибровали с помощью как отрицательных, так и положительных калибровочных растворов AB-SCIEX APCI (Фрамингем, Массачусетс). Образцы, полученные в результате инкубации с гепатоцитами, анализировали как при отрицательных, так и при положительных режимах сканирования. Основной аналитический метод и инструментальные условия представлены ниже. Модификация настроек спектрометра зависела от потребности аналита.
Условия LC-MS/MS:

Анализ данных активности
Данные масс-спектрометрии получали с помощью AB-Sciex Analyst TF (версия 1.5.1; Фрамингем, Массачусетс). Хроматограммы и спектры получали с помощью AB-Sciex PeakView (версия 2.2.0.1; Фрамингем, Массачусетс). Сравнение относительных площадей пика для экстракционных ионных хроматограмм было основано на ± 0,0002 Да ожидаемого точного отношения массы к заряду (m/z) для каждого аналита, представляющего интерес.
Результаты
В результате инкубации с гепатоцитами никакого метаболита не выявляли. В представленных условиях анализа Соединение 1а характеризовалось временем удерживания, составляющим примерно 7,8 минут. В режиме отрицательного сканирования Соединение 1а характеризовалось m/z депротонированного молекулярного иона 745 (C24H25F2N10O8P2S2-) и m/z дважды депротонированного молекулярного иона 372 (C24H24F2N10O8P2S22-). Наблюдали основные ионы-продукты MS/MS с m/z 533 (C19H19FN10O4PS-) и m/z 186 (C9H8N5-). В условиях положительного режима сканирования Соединение 1а характеризовалось m/z депротонированного молекулярного иона 747 (C24H27F2N10O8P2S2+) и основными ионами-продуктами MS/MS с m/z 651 (C24H26F2N10O6PS+), m/z 252 (C10H11FN5O2+) и m/z 188 (C9H10N5+). Данные MS и MS/MS подтверждали структуру Соединения 1а.
Соединение 1а было стабильным при инкубациях с гепатоцитами мыши, крысы, собаки, обезьяны и человека. Никакого видимого метаболита Соединения 1а в данном исследования не идентифицировали. В образцах, полученных в результате инкубации с гепатоцитами, только самое Соединение 1а могли выявлять и подтверждать с использованием фрагментов тандемной масс-спектрометрии (MS/MS).
Все документы, на которые идет отсылка в настоящем раскрытии, включены в данный документ посредством ссылки, хотя в случае, если какой-либо включенный документ противоречит текстовому описанию, то указанное текстовое описание необходимо проверить. Специалистам в данной области техники будет понятно, что различные изменения и модификации могут быть выполнены в отношении материала, предусмотренного в данном документе, и что материал находится в пределах объема и сущности настоящего раскрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| НОВЫЕ СОЕДИНЕНИЯ КОНДЕНСИРОВАННОГО ИМИДАЗОЛА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АГОНИСТОВ РЕЦЕПТОРА СВ2 | 2002 |
|
RU2312864C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| Композиции для лечения заболевания почек и/или печени | 2016 |
|
RU2712624C2 |
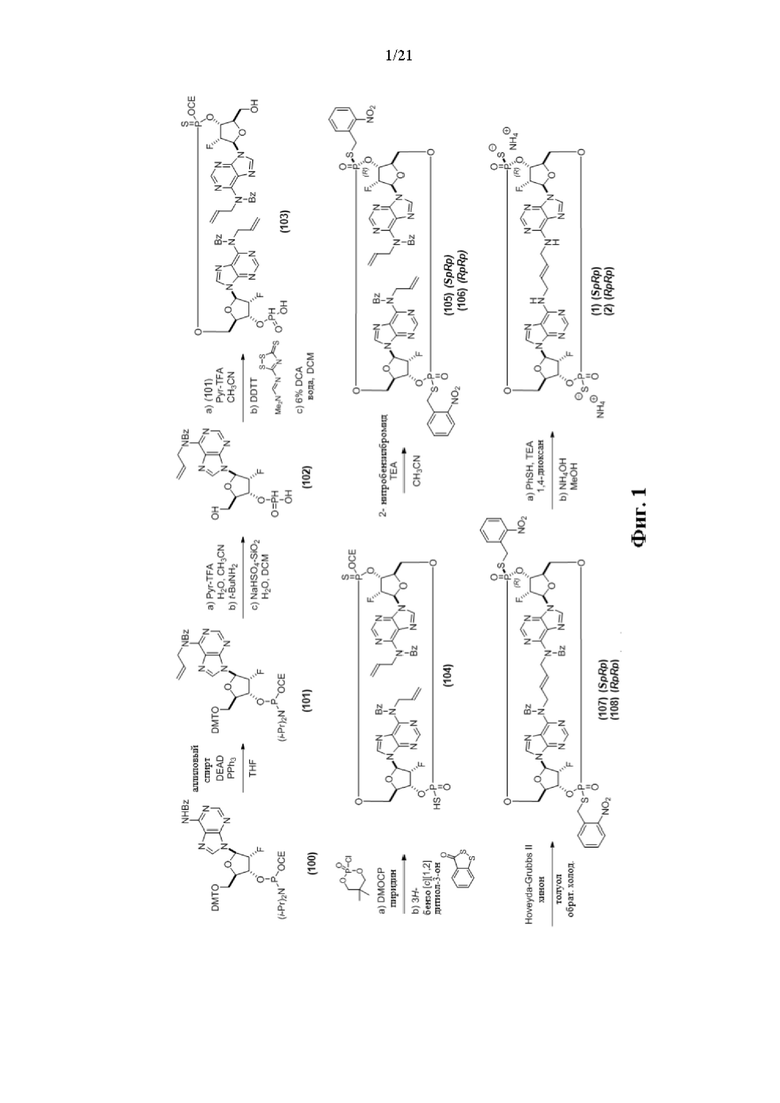
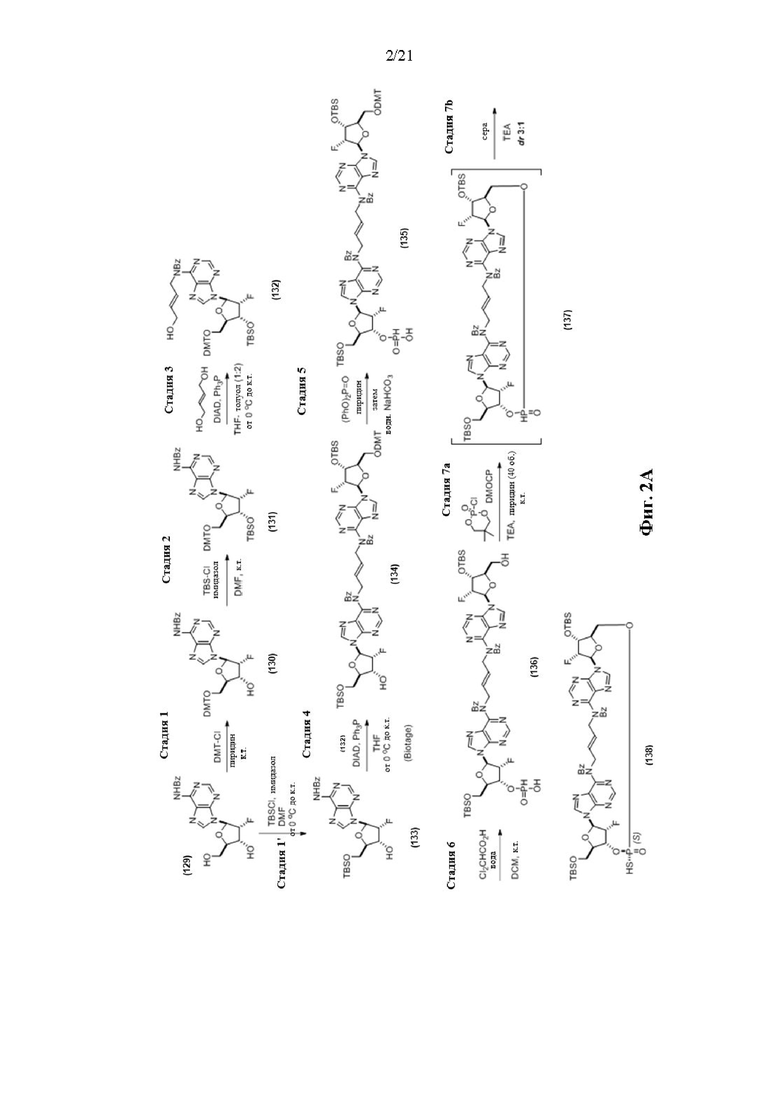
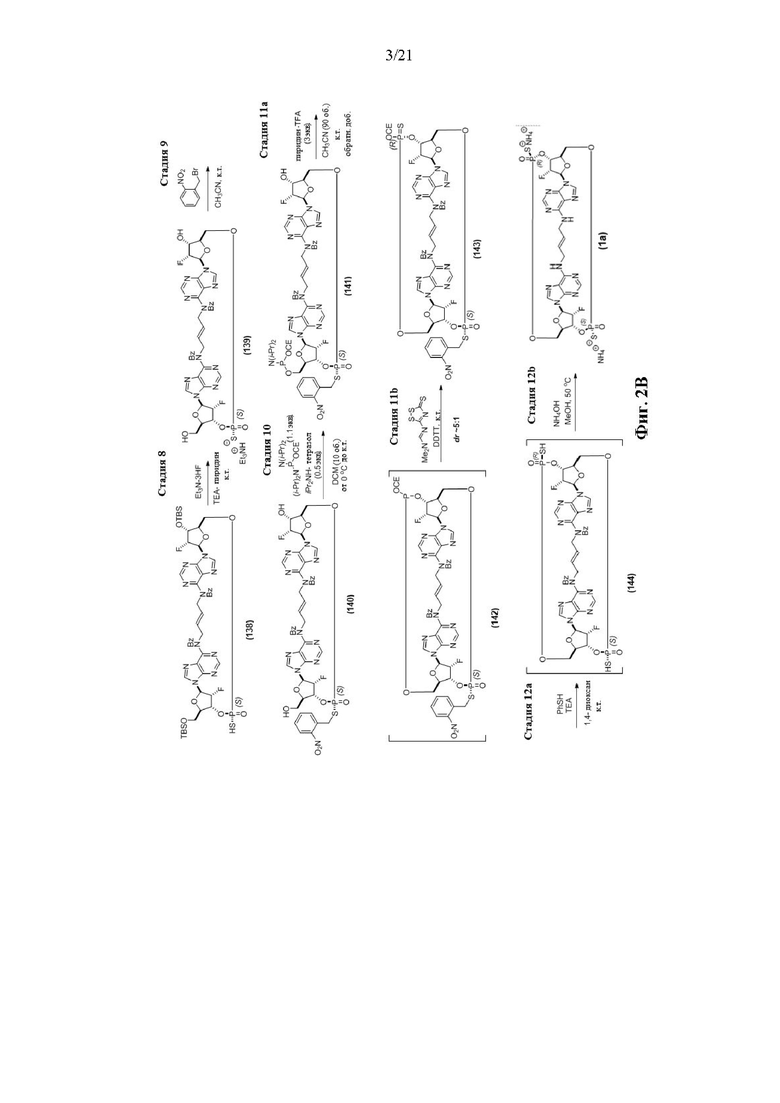
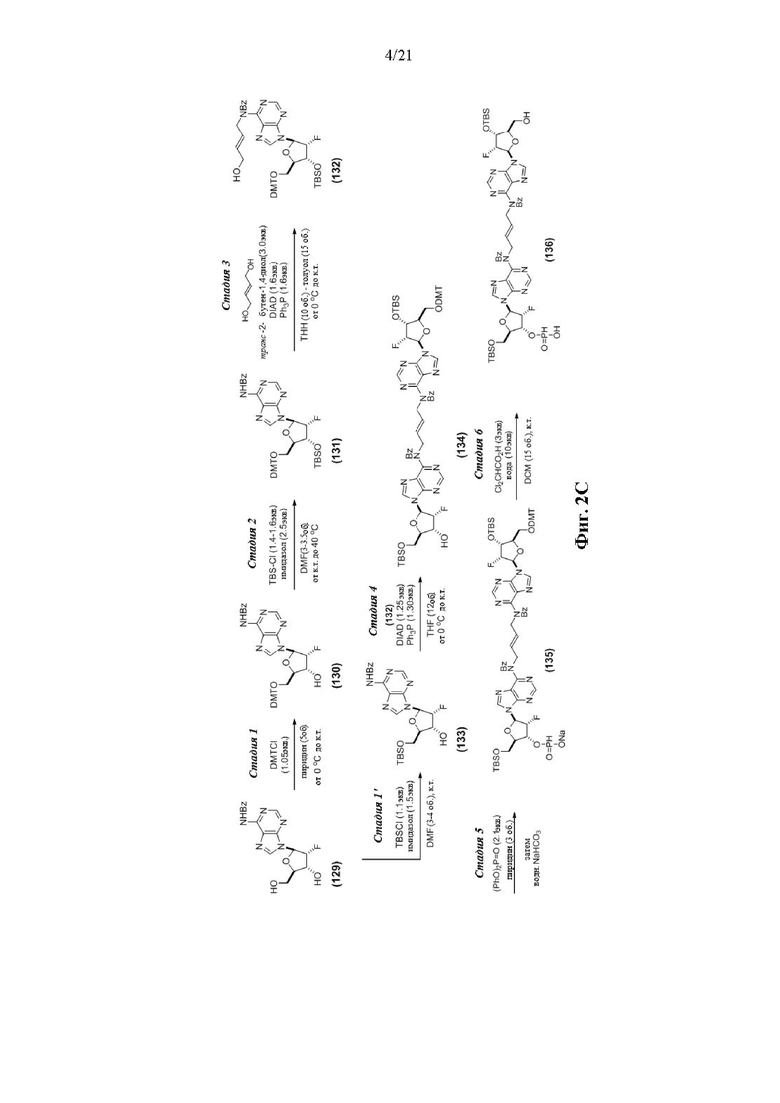
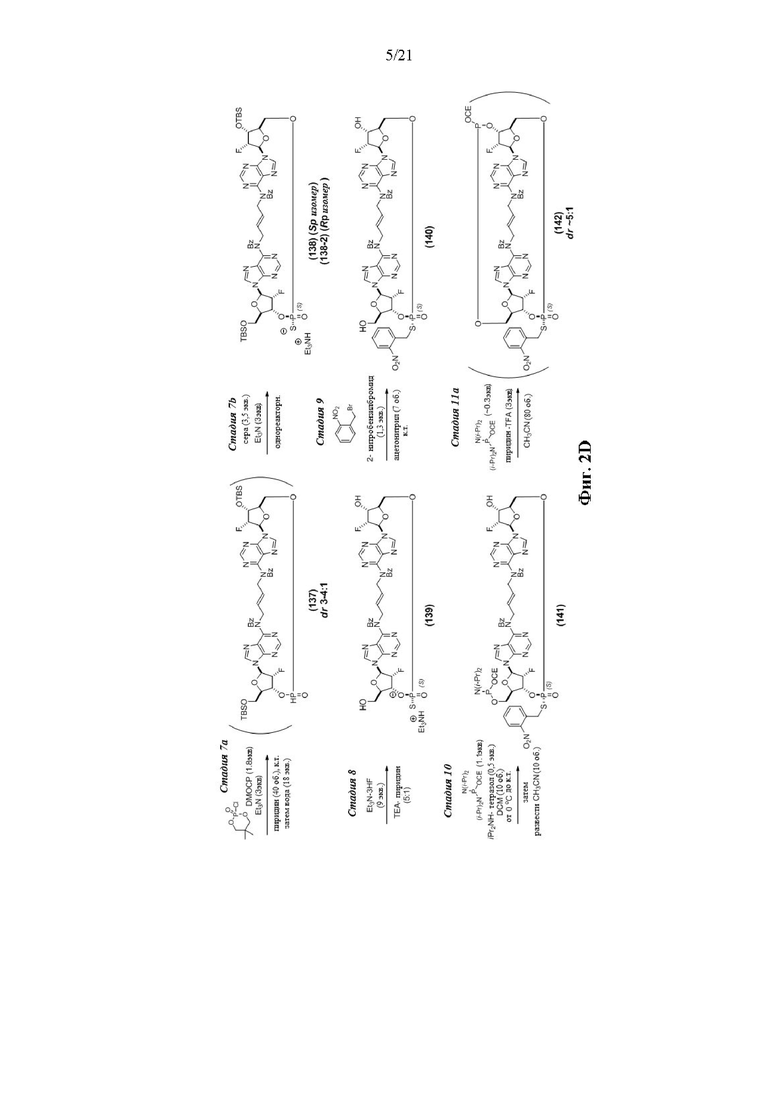
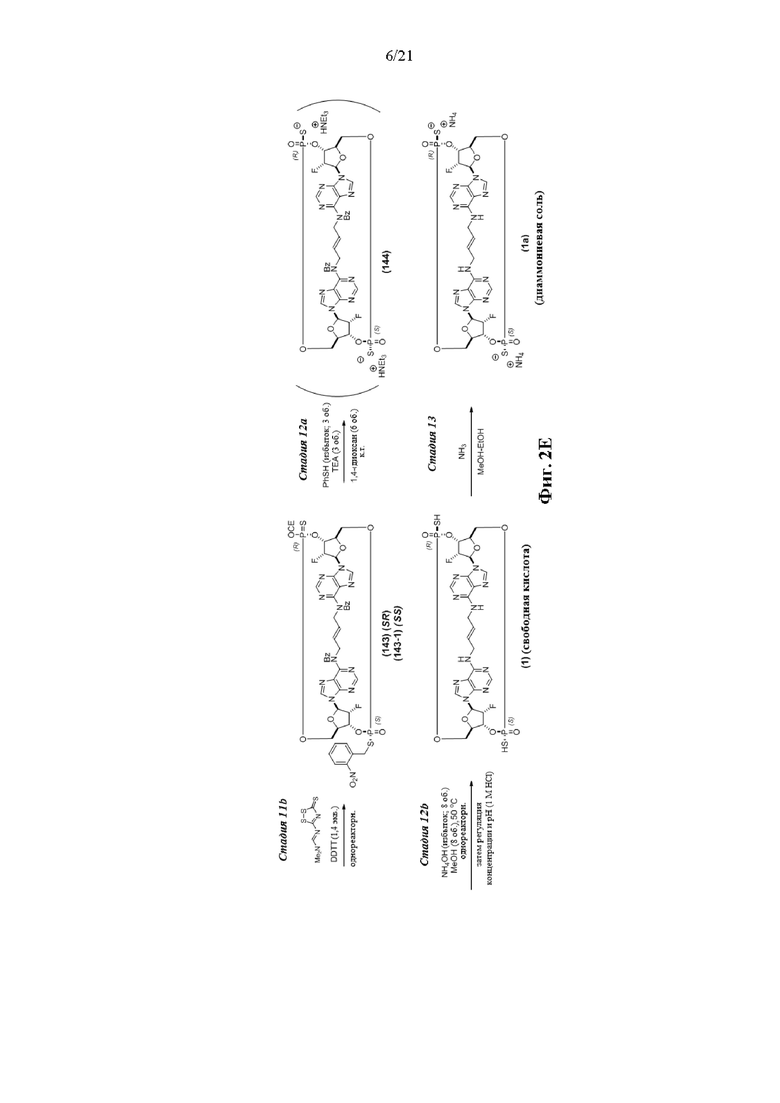

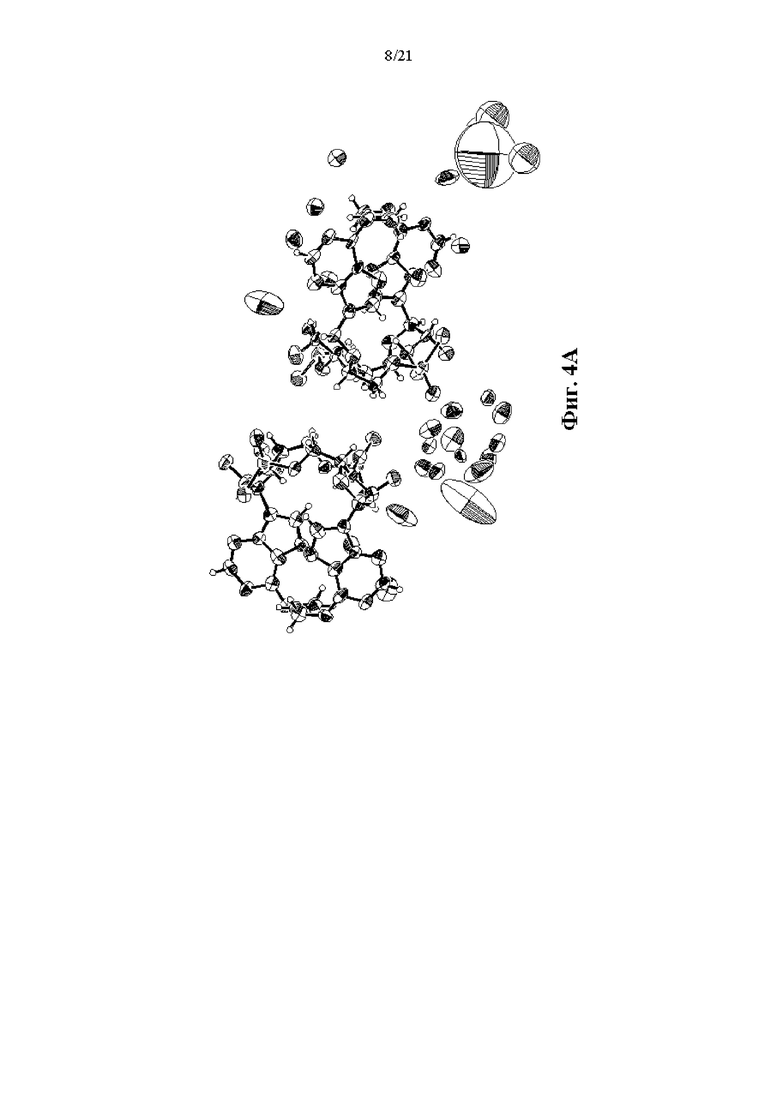
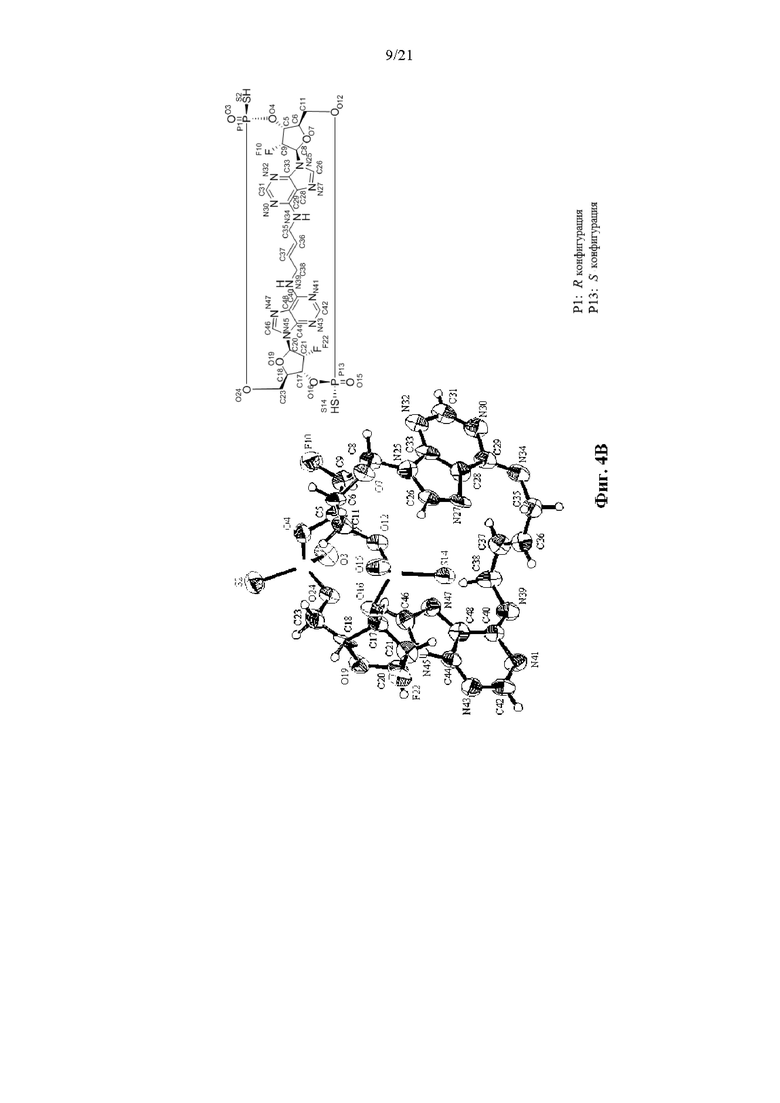
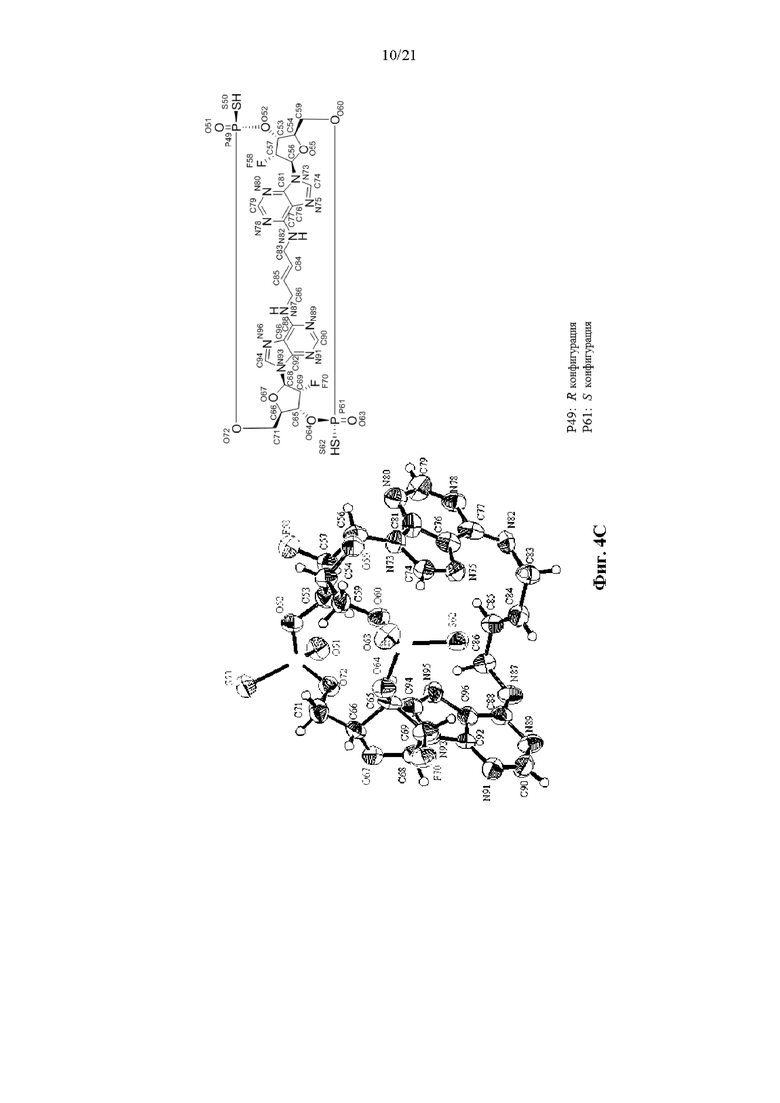
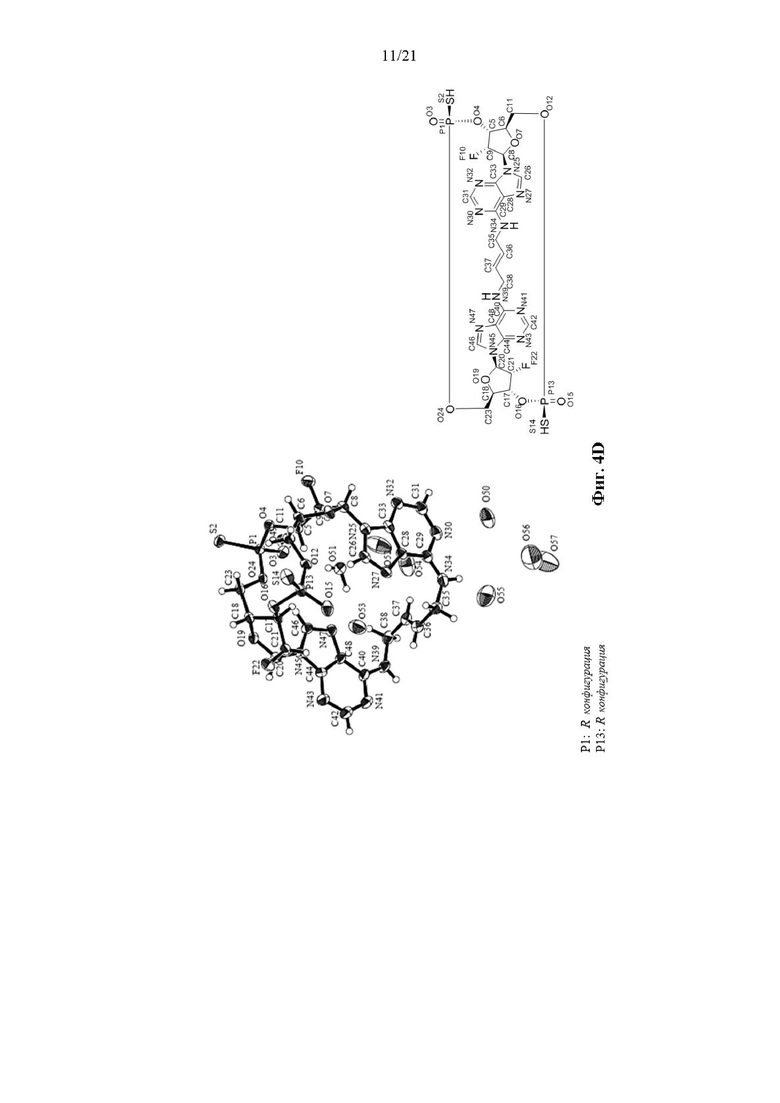

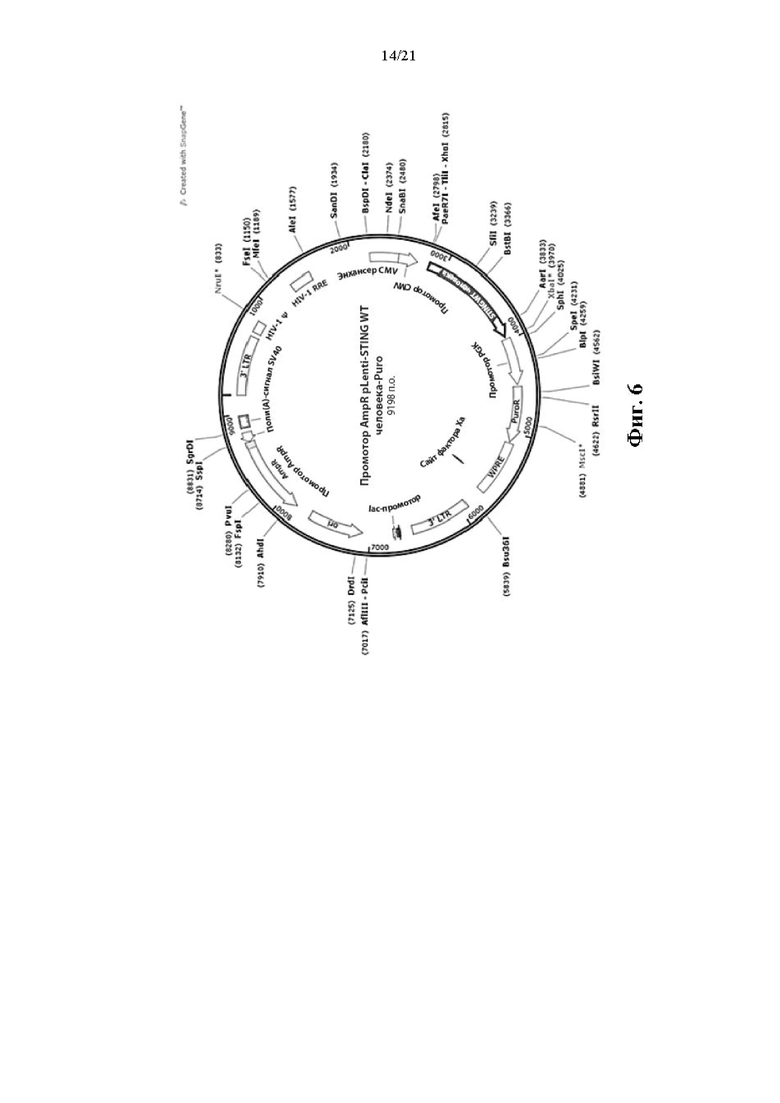
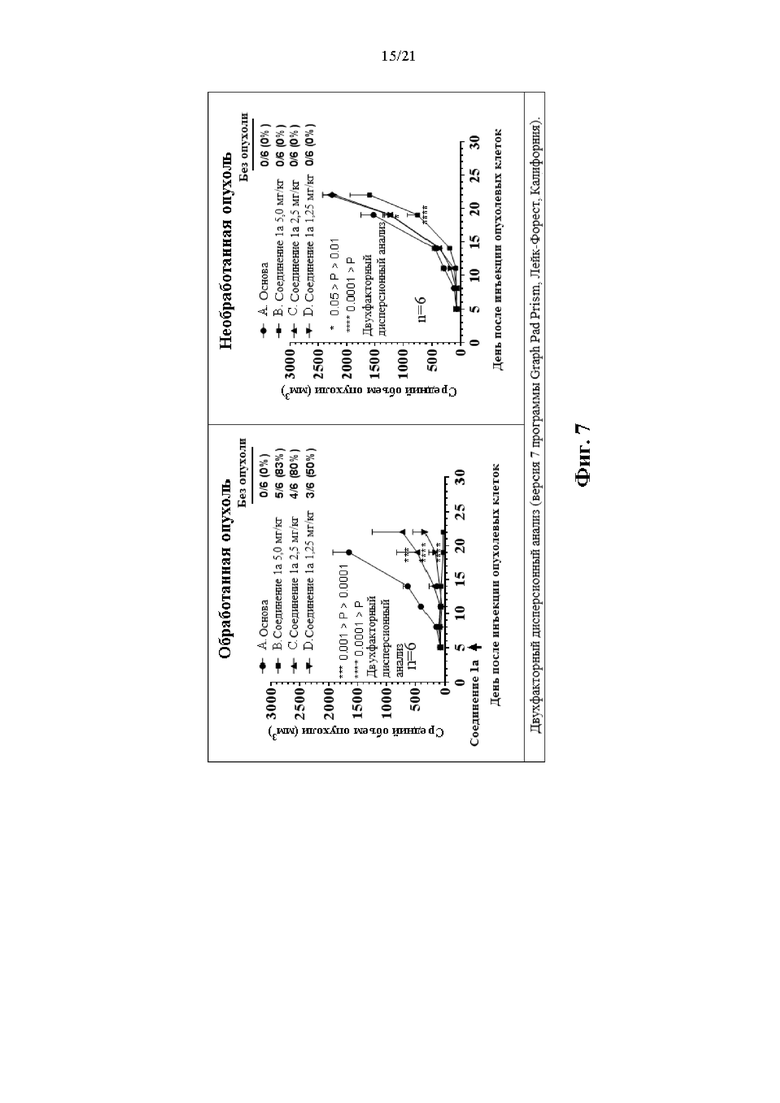
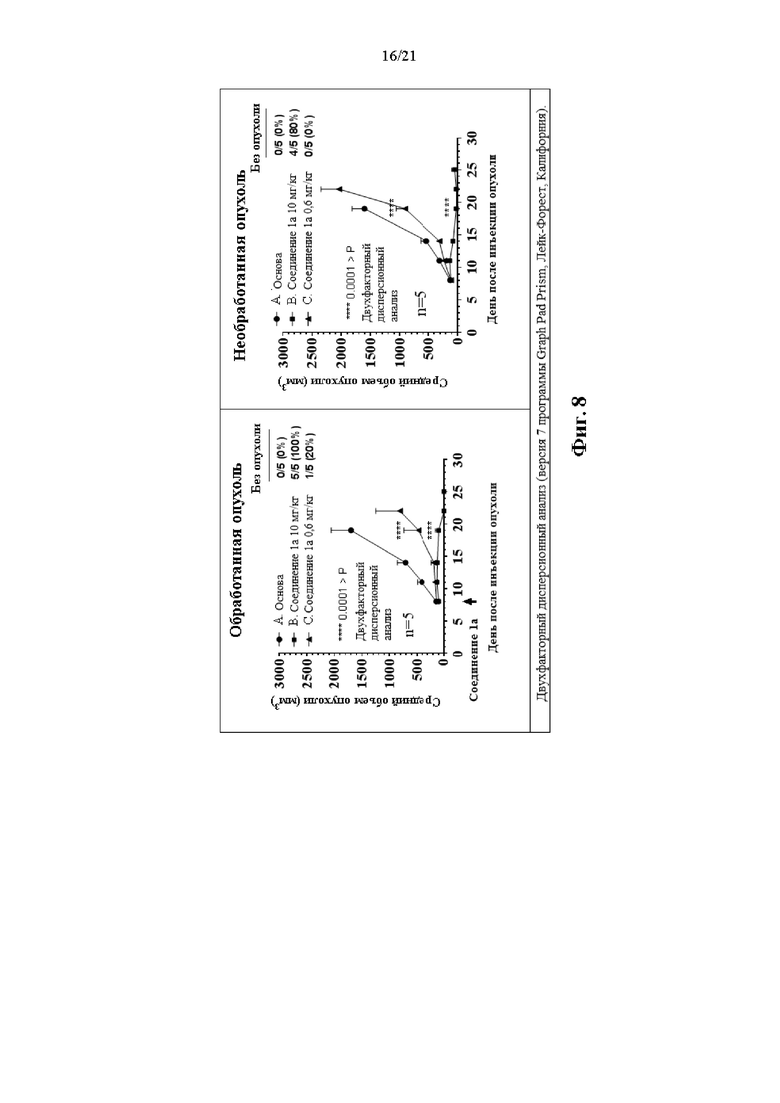
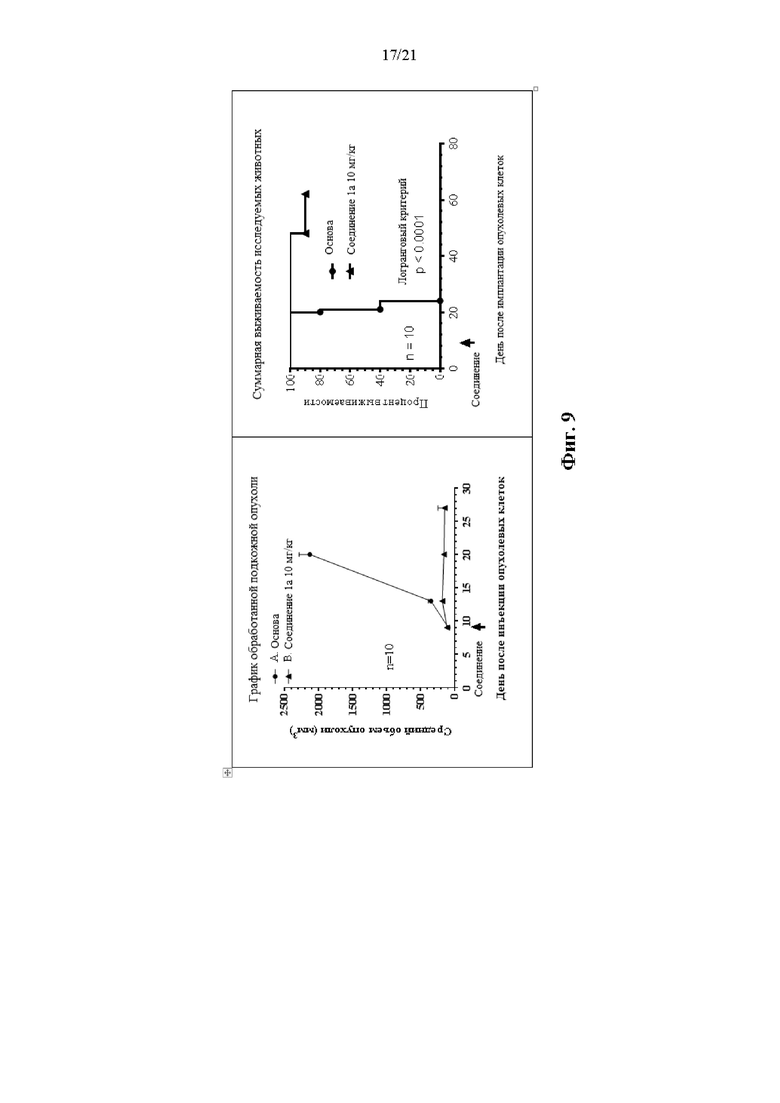
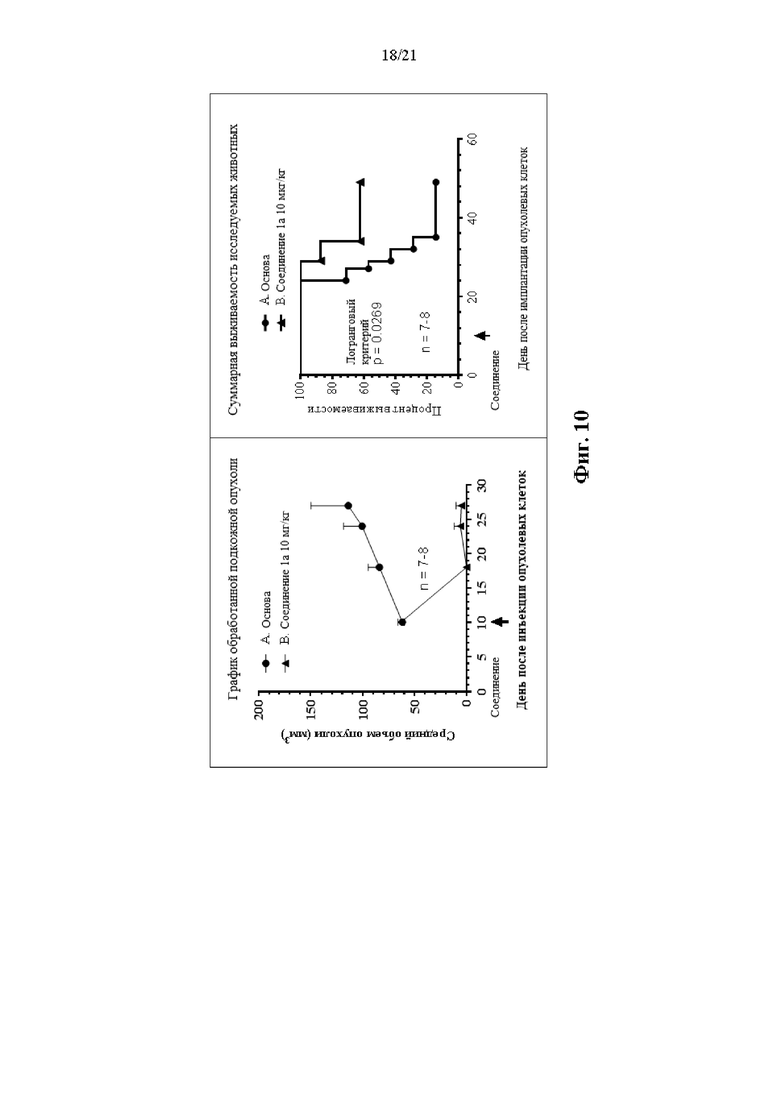
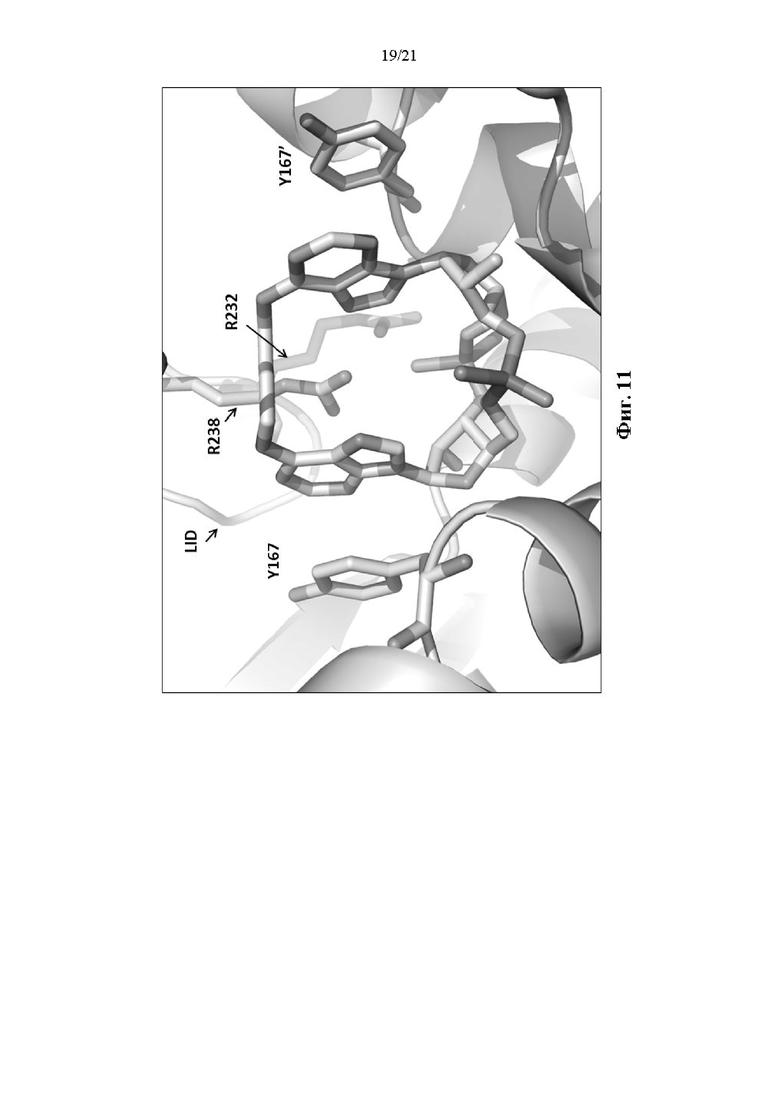
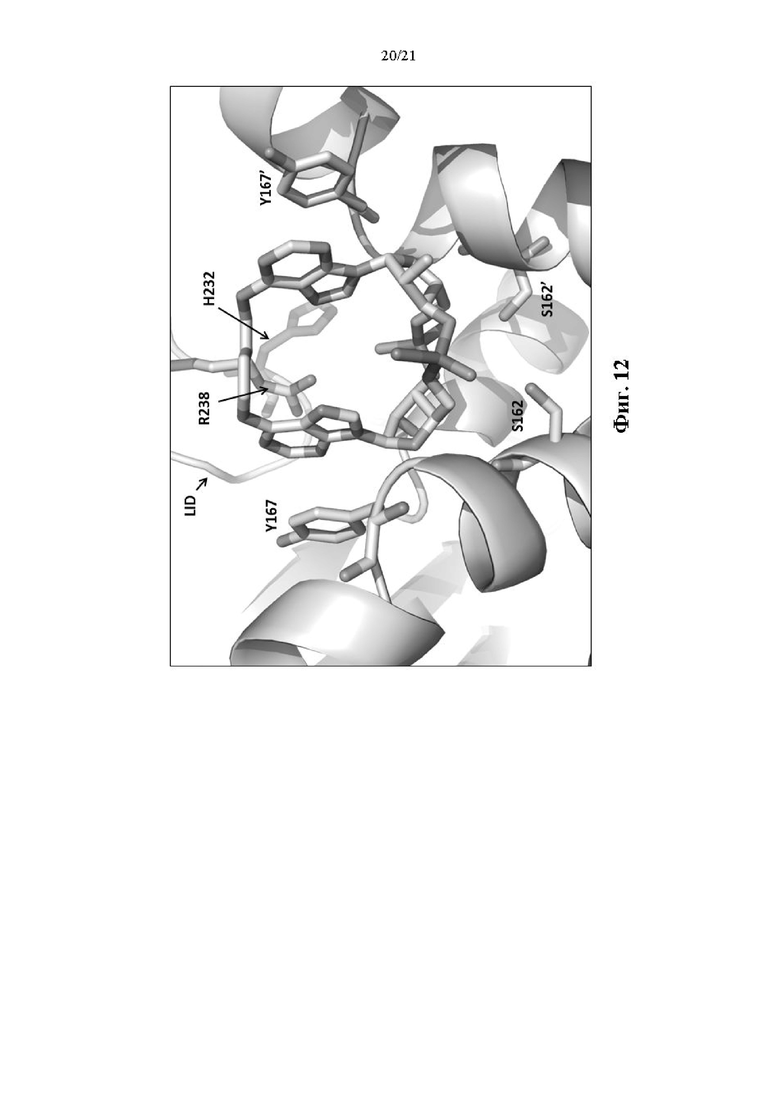
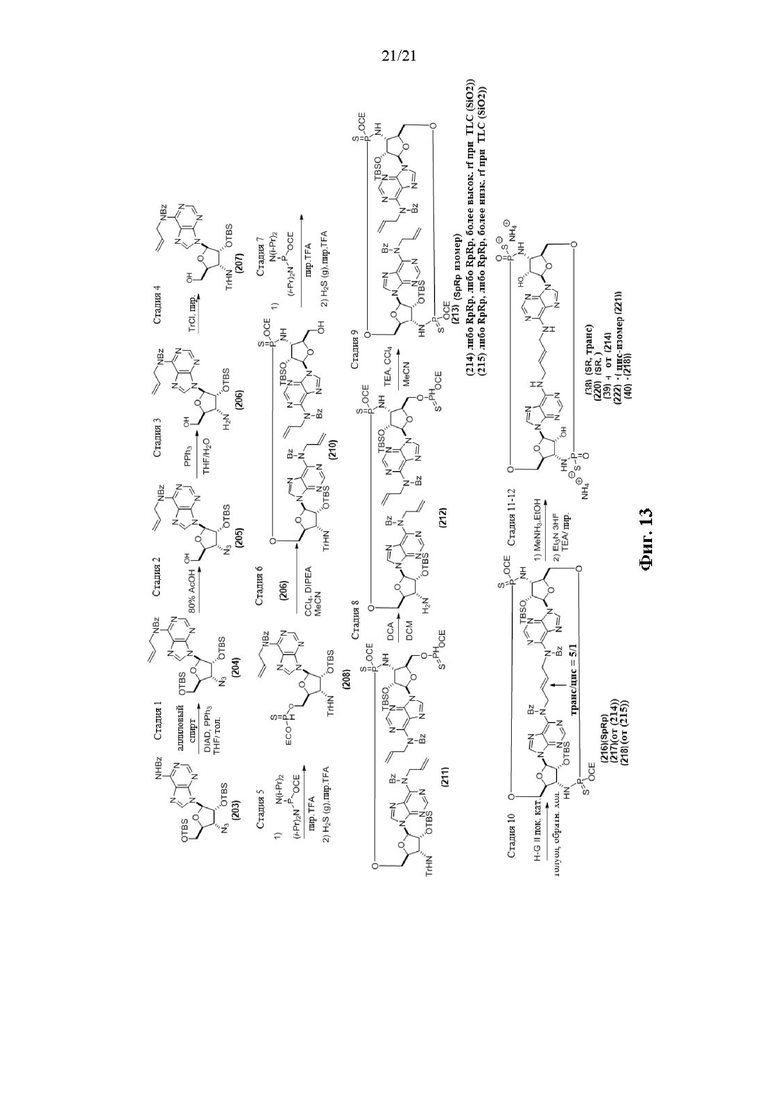
Группа изобретений относится к области органической химии и фармацевтики и направлена на получение новых соединений, которые полезны в лечении рака. Раскрываются соединения формулы (III) или его фармацевтически приемлемая соль, где R1a выбран из группы, состоящей из Н, -ОН и -F; R1b выбран из группы, состоящей из Н, -ОН и F, где по меньшей мере один из R1a и R1b представляет собой -Н; R4a выбран из группы, состоящей из -Н, -ОН и -F; R4b выбран из группы, состоящей из -Н, -OH и -F, где по меньшей мере один из R4a и R4b представляет собой -Н; P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией; Z представляет собой -О- или -NH-; X1a и Х2а такие же или разные и независимо выбраны из =O или =S; X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5; где R5 выбран из Н и -CH2OC(O)ОС1-6алкила; L1 в формуле (III) представляет собой четыре, пять или шесть атомов углерода длиной; где Х10, Х11, Х12, Х13, Х14 и Х15 независимо выбраны из связи, -CH2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН, (iv) -NH2 или (v) -D и, если Х10 или Х15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь; и где любые два смежных члена группы, включая Х10, Х11, Х12, Х13, Х14 и Х15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N атом; где B1 и В2 независимо выбраны из представленных структур в п.1. Кроме того, раскрываются фармацевтическая композиция для лечения рака, содержащая фармацевтически эффективное количество указанных соединений, применение соединений для получения фармацевтической композиции и способы лечения рака, которые предусматривают введение пациенту представленных соединений. Группа изобретений обеспечивает эффективное лечение рака. 21 н. и 16 з.п. ф-лы, 13 ил., 14 табл., 114 пр.
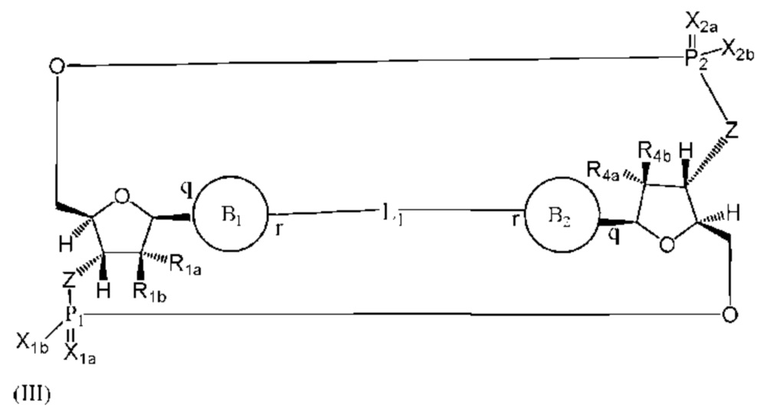
1. Соединение формулы (III)
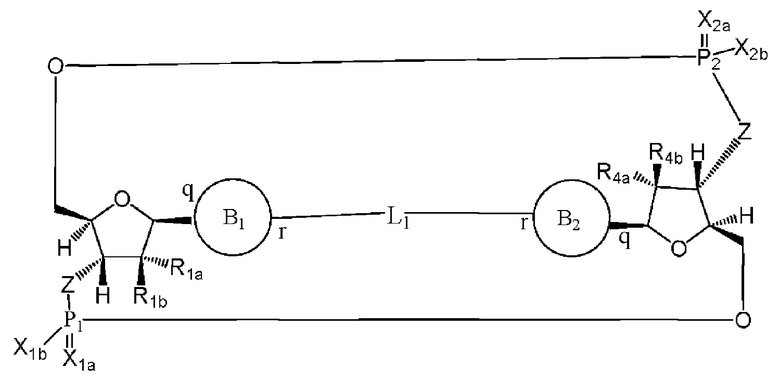
или его фармацевтически приемлемая соль, где
R1a выбран из группы, состоящей из Н, -ОН и -F;
R1b выбран из группы, состоящей из Н, -ОН и F,
где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из -Н, -ОН и -F;
R4b выбран из группы, состоящей из -Н, -OH и -F, где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
Z представляет собой -О- или -NH-;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из Н и -CH2OC(O)ОС1-6алкила;
L1 в формуле (III) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой
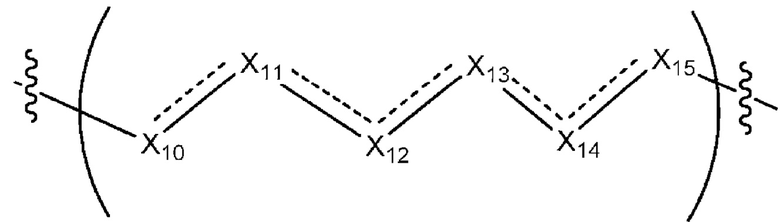 ,
,
где  означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость
означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость  в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости
в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости  в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость
в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость  в L1 означает тройную связь, 0 встречаемостей
в L1 означает тройную связь, 0 встречаемостей  в L1 означают двойную связь; и (iv) где, если 2 встречаемости
в L1 означают двойную связь; и (iv) где, если 2 встречаемости  в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, Х12, Х13, Х14 и Х15 независимо выбраны из связи, -CH2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН, (iv) -NH2 или (v) -D и, если Х10 или Х15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
и где любые два смежных члена группы, включая Х10, Х11, Х12, Х13, Х14 и Х15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N атом;
где B1 и В2 независимо выбраны из:
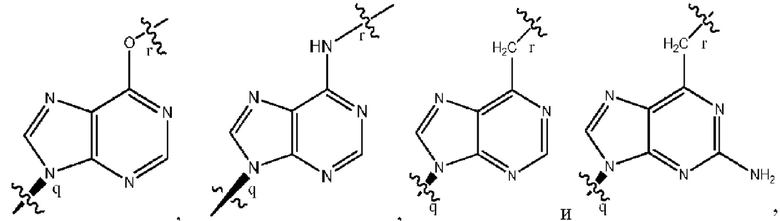
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (III).
2. Соединение или фармацевтически приемлемая соль по п. 1, где
R1a выбран из группы, состоящей из Н и -F;
R1b выбран из группы, состоящей из Н и F, где R1a и R1b оба не могут быть -F;
R4a выбран из группы, состоящей из Н и -F;
R4b выбран из группы, состоящей из -Н и -F, где R4a и R4b оба не могут быть -F;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из группы, состоящей из -Н, C1-6алкила и -С(O)С1-6алкила;
L1 в формуле (III) представляет собой четыре или пять атомов углерода длиной и представляет собой
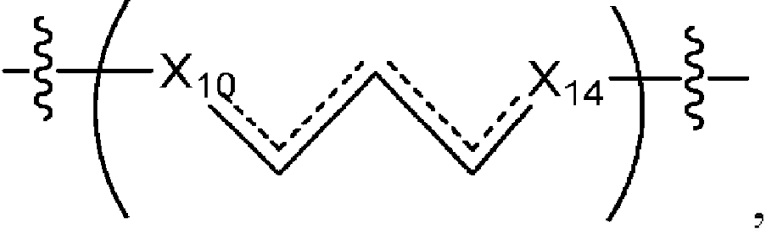
где  означает простую связь или двойную связь и где или 0, или 1 встречаемость
означает простую связь или двойную связь и где или 0, или 1 встречаемость  в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и Х11 независимо выбраны из связи, -СН-, или -СН2- и где, если Х10 или Х14 представляет собой связь, такая связь не представляет собой двойную связь;
где B1 и В2 независимо выбраны из:
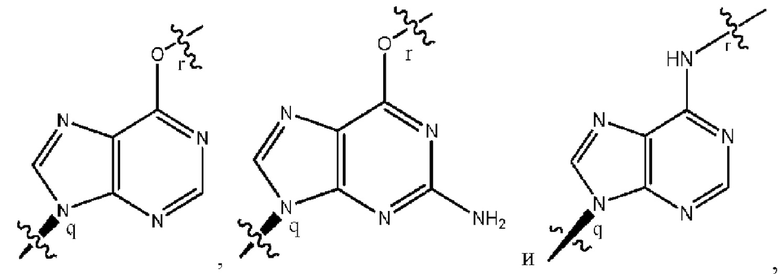
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (III).
3. Соединение формулы (IV)
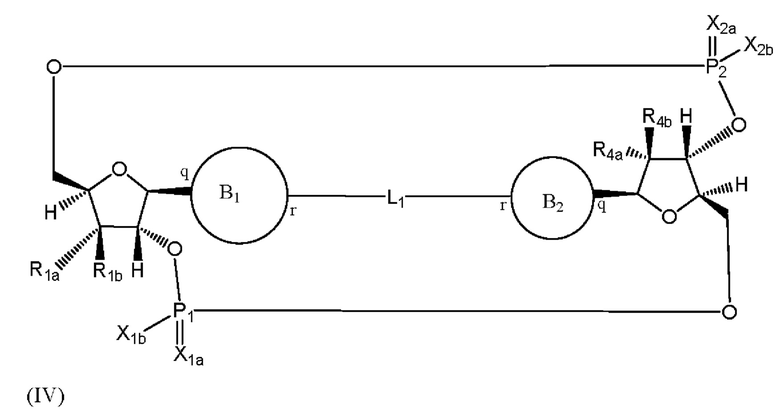
или его фармацевтически приемлемая соль, где R1a выбран из группы, состоящей из -Н, -ОН и -F;
R1b выбран из группы, состоящей из Н, -ОН и -F, где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из -Н, -ОН и -F;
R4b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 выбран из Н и -CH2OC(O)ОС1-6алкила;
L1 в формуле (IV) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой
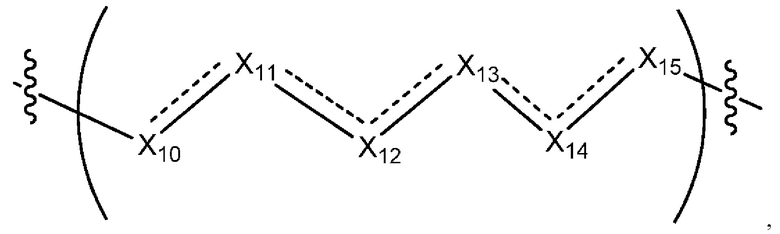
где  означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость
означает простую связь, двойную связь или тройную связь и где (i) или 0, или 1 встречаемость  в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости
в L1 означает тройную связь; или (ii) 0, 1 или 2 встречаемости  в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость
в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость  в L1 означает тройную связь, 0 встречаемостей
в L1 означает тройную связь, 0 встречаемостей  в L1 означают двойную связь; и (iv) где, если 2 встречаемости
в L1 означают двойную связь; и (iv) где, если 2 встречаемости  в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, Х12, Х13, Х14 и Х15 независимо выбраны из связи, -СН2- или -СН-, где -СН2- или -СН- является незамещенным или замещен (i) -ОН или (v) -D и, если Х10 или Х15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
где B1 и В2 независимо выбраны из:
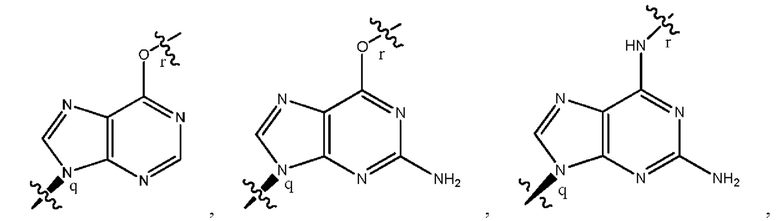
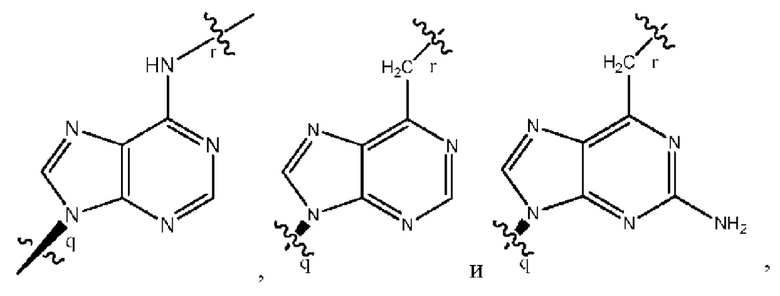
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (IV).
4. Соединение формулы (IV)
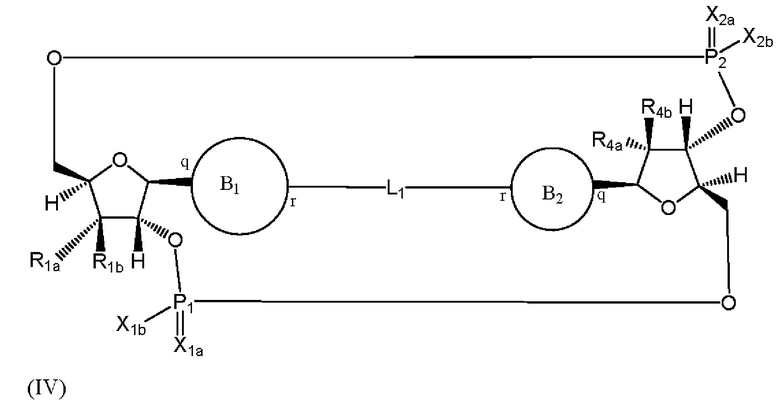
или его фармацевтически приемлемая соль, где R1a выбран из группы, состоящей из Н и -F;
R1b выбран из группы, состоящей из -Н и -F, где R1a и R1b оба не могут быть -F;
R4a выбран из группы, состоящей из -Н и -F;
R4b выбран из группы, состоящей из Н и F,
где R4a и R4b оба не могут быть F;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а такие же или разные и независимо выбраны из =O или =S;
X1b и X2b такие же или разные и независимо выбраны из -OR5 и -SR5;
где R5 представляет собой -Н;
L1 в формуле (IV) представляет собой четыре или пять атомов углерода длиной и представляет собой 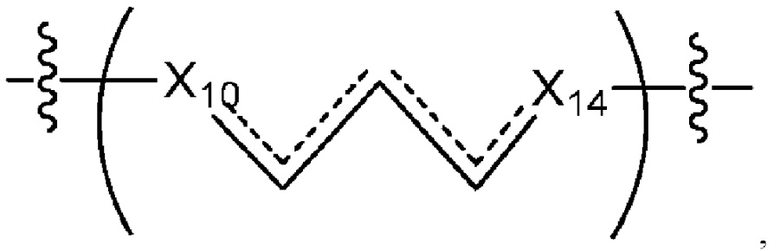
где  означает простую связь или двойную связь и где или 0, или 1 встречаемость
означает простую связь или двойную связь и где или 0, или 1 встречаемость  в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и Х11 независимо выбраны из связи, -СН- или -СН2- и где, если Х10 или X14 представляет собой связь, такая связь не представляет собой двойную связь;
где B1 и В2 независимо выбраны из:
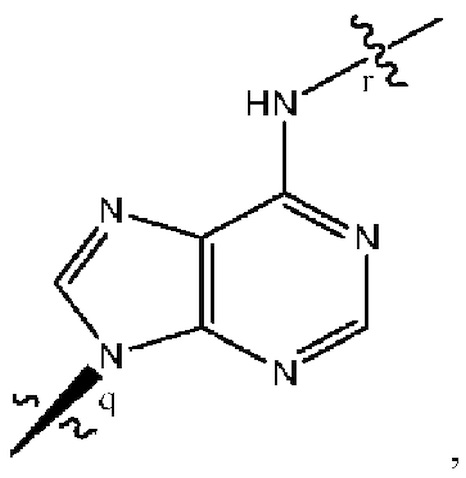
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (IV).
5. Соединение формулы (V)
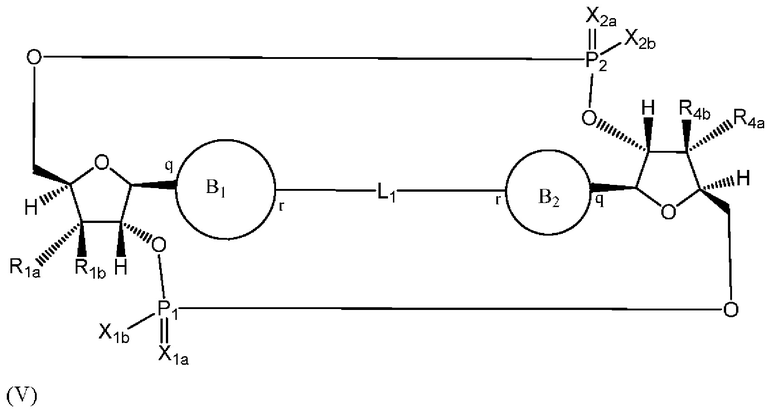
или его фармацевтически приемлемая соль, где R1a выбран из группы, состоящей из -Н, -ОН и -F;
R1b выбран из группы, состоящей из -Н, -ОН и -F, где по меньшей мере один из R1a и R1b представляет собой -Н;
R4a выбран из группы, состоящей из Н, -ОН;
R4b выбран из группы, состоящей из -Н, -ОН и где по меньшей мере один из R4a и R4b представляет собой -Н;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а представляют собой =O;
X1b и X2b представляют собой -SR5;
где R5 представляет собой -Н;
L1 в формуле (V) представляет собой четыре, пять или шесть атомов углерода длиной и представляет собой
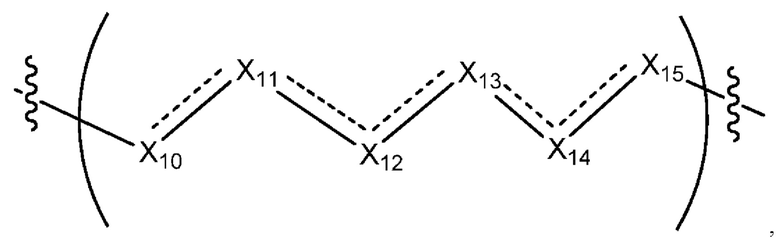
где  означает простую связь или двойную связь и где (i) или 0, или 1 или (ii) 0, 1 или 2 встречаемости
означает простую связь или двойную связь и где (i) или 0, или 1 или (ii) 0, 1 или 2 встречаемости  в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость
в L1 означают двойную связь, где геометрия вокруг каждой двойной связи является цис или транс; и (iii) где, если 1 встречаемость  в L1 означает тройную связь, 0 встречаемостей
в L1 означает тройную связь, 0 встречаемостей  в L1 означают двойную связь; и (iv) где, если 2 встречаемости
в L1 означают двойную связь; и (iv) где, если 2 встречаемости  в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
в L1 означают двойную связь, такие двойные связи являются или смежными связями, или чередующимися связями;
где Х10, Х11, Х12, Х13, Х14 и Х15 независимо выбраны из связи, -СН2- или -СН-, где -СН2- или СН является незамещенным или замещен (i) ОН и, если Х10 или Х15 представляет собой связь, такая связь не представляет собой двойную связь или тройную связь;
и где любые два смежных члена группы, включая Х10, Х11, Х12, Х13, Х14 и Х15, могут необязательно образовывать с дополнительными атомами С3 циклоалкил или С3 гетероциклоалкил, указанный С3 гетероциклоалкил включает в себя N или О атом;
где B1 и В2 независимо выбраны из:
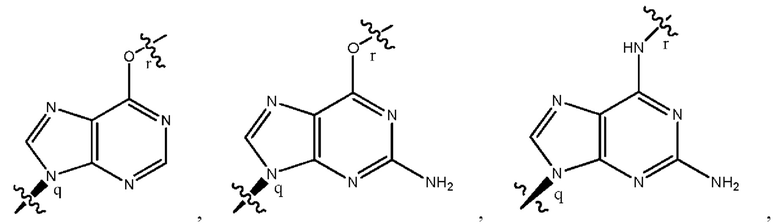
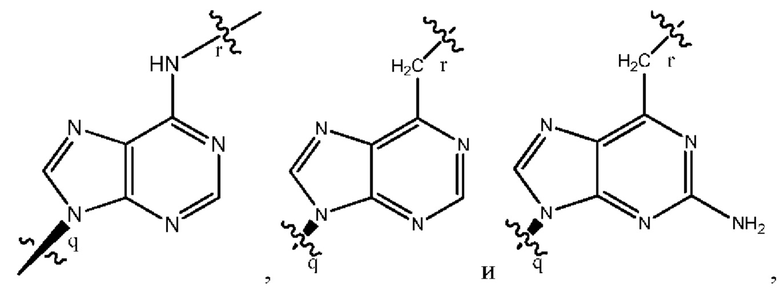
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (V).
6. Соединение формулы (V)
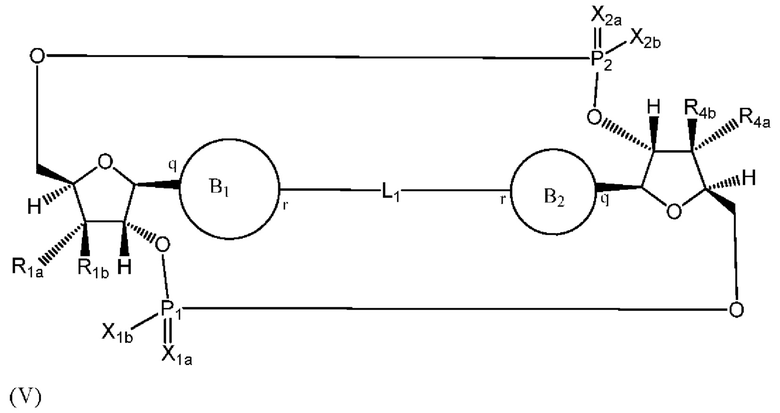
или его фармацевтически приемлемая соль, где
R1a выбран из группы, состоящей из -Н и -F;
R1b выбран из группы, состоящей из -Н и -F, где R1a и R1b оба не могут быть -F;
R4a представляет собой -Н; R4b представляет собой -Н;
P1 и Р2 каждый независимо обладает S или R стереохимической конфигурацией;
X1a и Х2а представляют собой =O
X1b и X2b представляют собой -SR5;
где R5 представляет собой Н;
L1 в формуле (V) представляет собой четыре или пять атомов углерода длиной и представляет собой 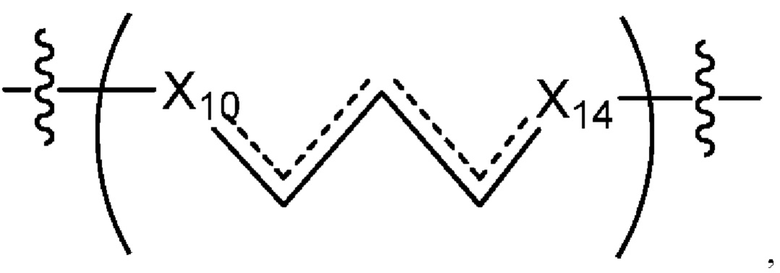
где  означает простую связь или двойную связь и где или 0, или 1 встречаемость
означает простую связь или двойную связь и где или 0, или 1 встречаемость  в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
в L1 означает двойную связь, где геометрия вокруг двойной связи является цис или транс;
где Х10 и Х14 независимо выбраны из связи, -СН- или -СН2- и где, если Х10 или Х14 представляет собой связь, такая связь не представляет собой двойную связь;
где B1 и В2 независимо выбраны из:
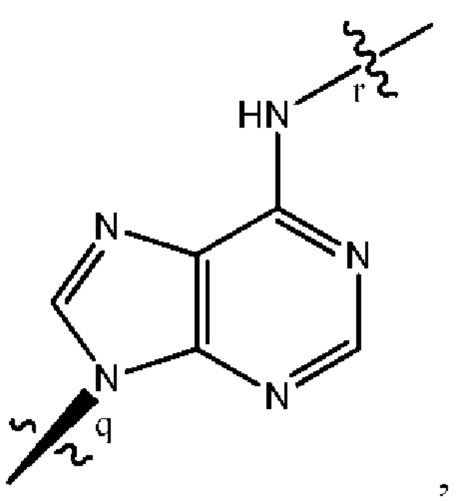
где связи в точках q и r на B1 и В2 присоединены в точках q и r на формуле (V).
7. Соединение или фармацевтически приемлемая соль по любому из пп. 1-6, где
(i) стереохимическая конфигурация P1 и Р2 обоих представляет собой R, стереохимическая конфигурация P1 представляет собой R и Р2 представляет собой S, или стереохимическая конфигурация P1 представляет собой S и Р2 представляет собой R;
(ii) одна встречаемость  в L1 означает двойную связь, где геометрия вокруг двойной связи является транс; и
в L1 означает двойную связь, где геометрия вокруг двойной связи является транс; и
(iii) Z представляет собой -О-.
8. Соединение или фармацевтически приемлемая соль по любому из пп. 1-7, где R1a и R4a каждый представляет собой -F.
9. Соединение или фармацевтически приемлемая соль по любому из пп. 1-7, где R1b и R4b каждый представляет собой -F.
10. Соединение или фармацевтически приемлемая соль по любому из пп. 1-9, где B1 и В2 каждый представляет собой 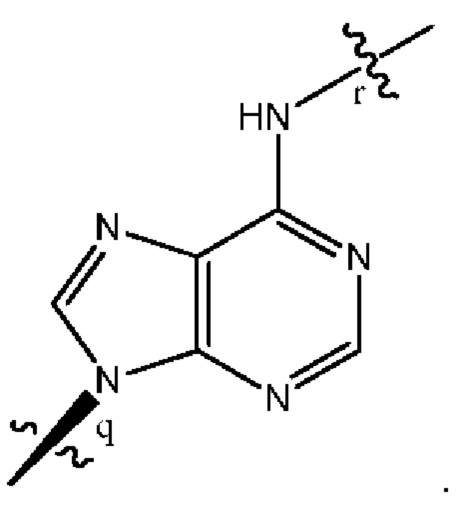
11. Соединение или фармацевтически приемлемая соль по любому из пп. 1-10, где X1a и Х2а оба представляют собой =O и где X1b и X2b оба представляют собой -SH.
12. Соединение или фармацевтически приемлемая соль по любому из пп. 1-11, где L1 представляет собой 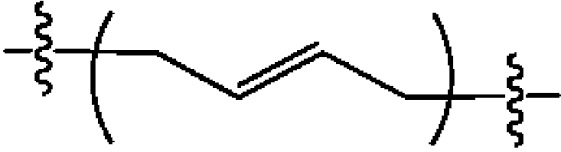 .
.
13. Соединение или фармацевтически приемлемая соль по п. 1 или 2, где L1 представляет собой пять атомов углерода длиной.
14. Соединение или фармацевтически приемлемая соль по любому из пп. 1-11, где L1 представляет собой четыре атома углерода длиной.
15. Соединение или его фармацевтически приемлемая соль, выбранные из группы, состоящей из:
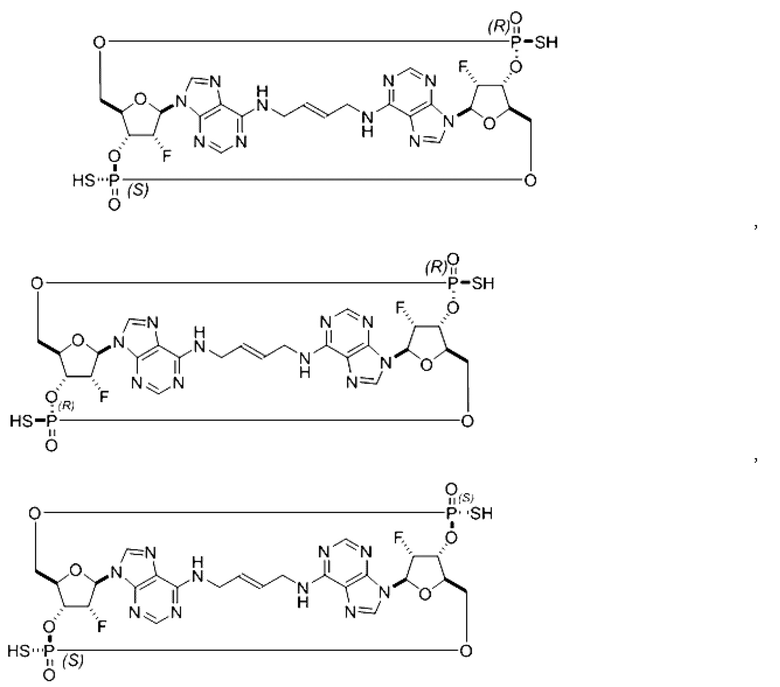
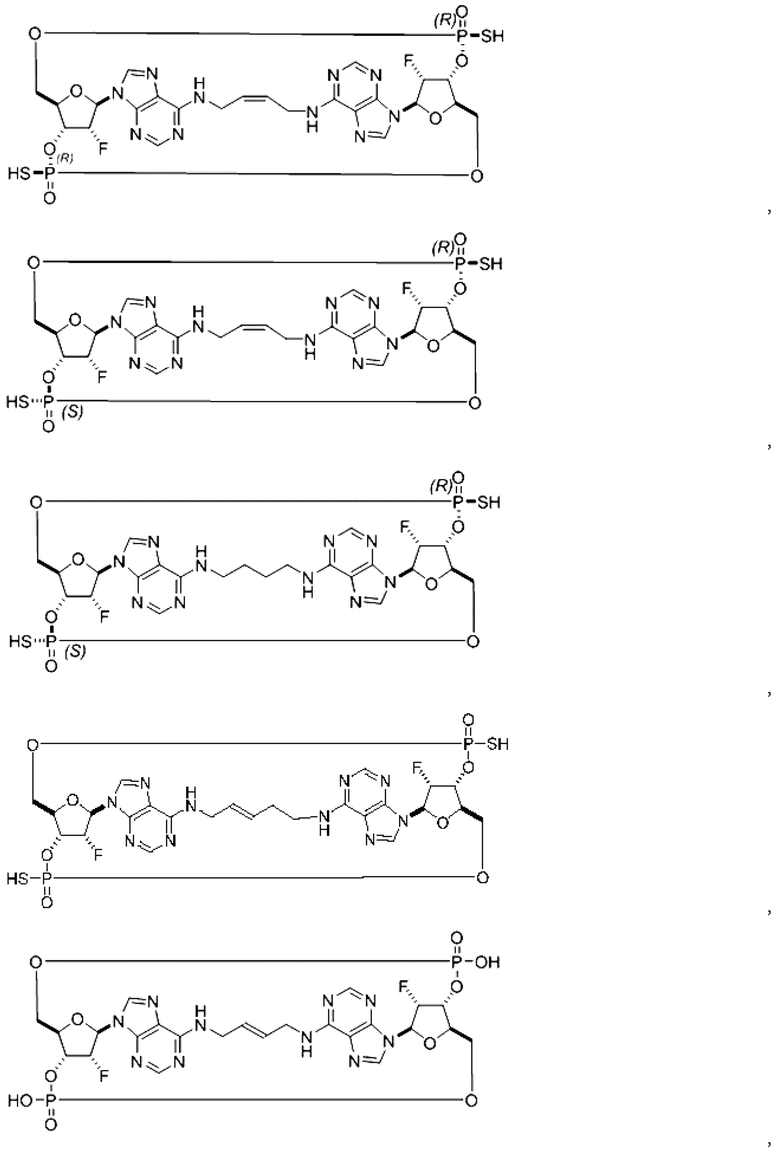
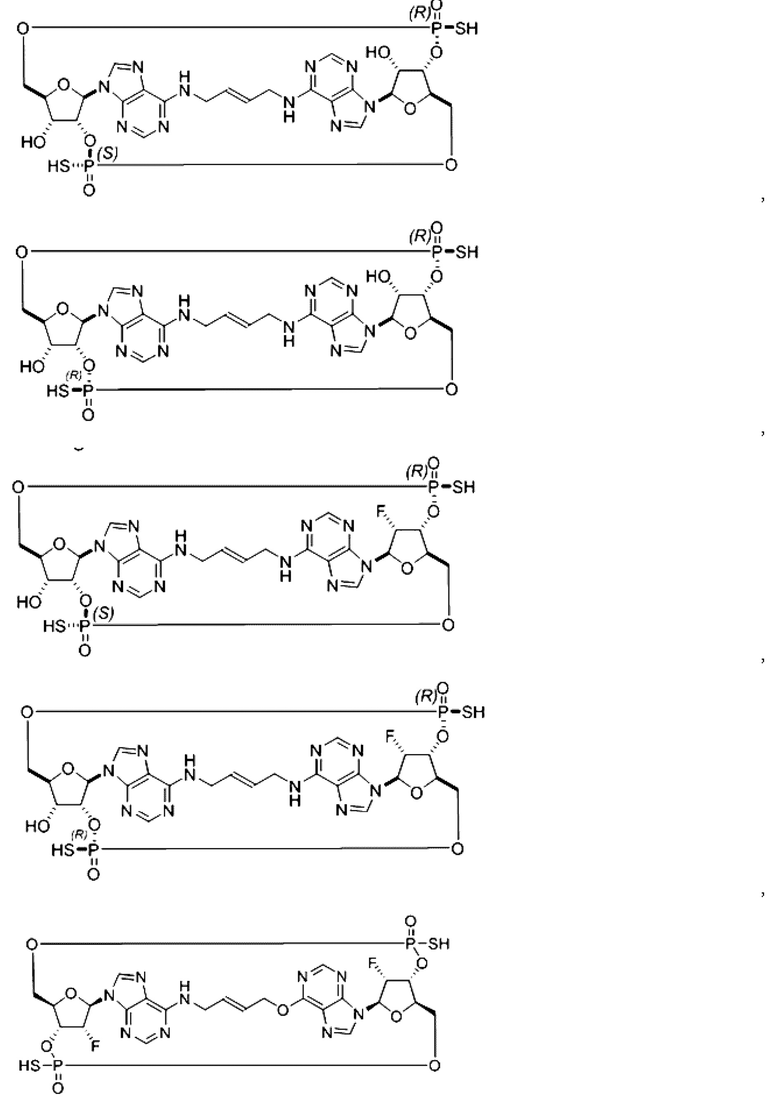
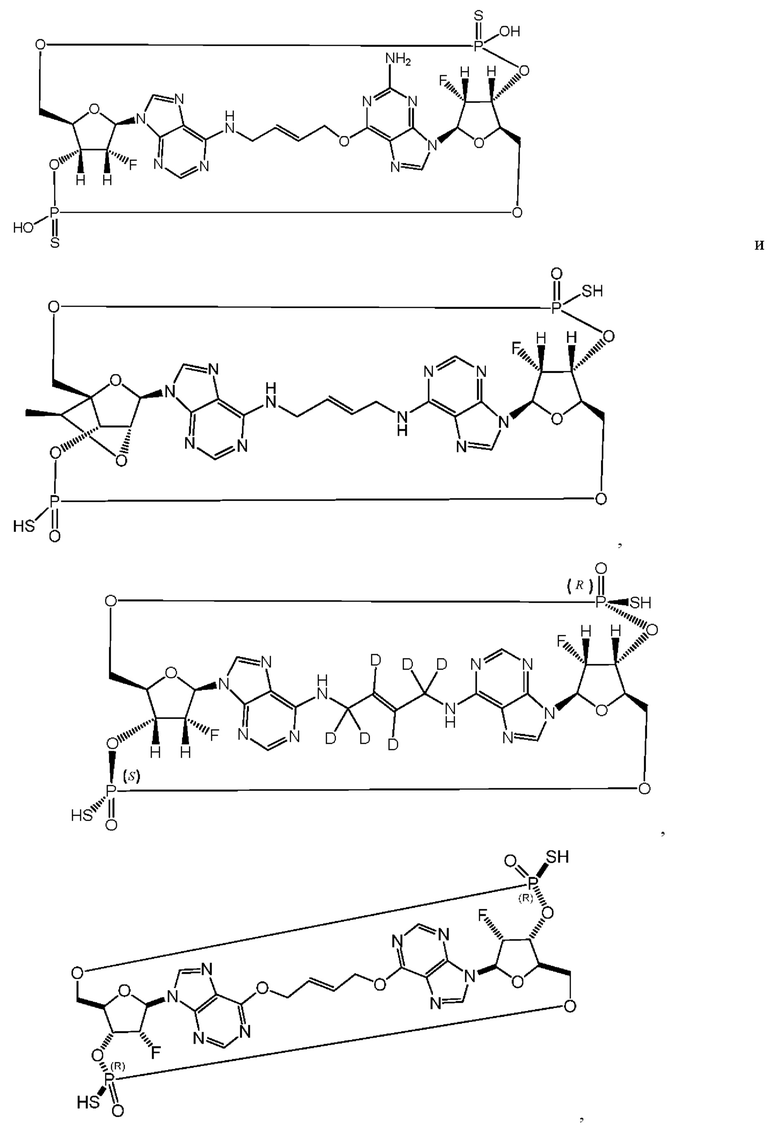
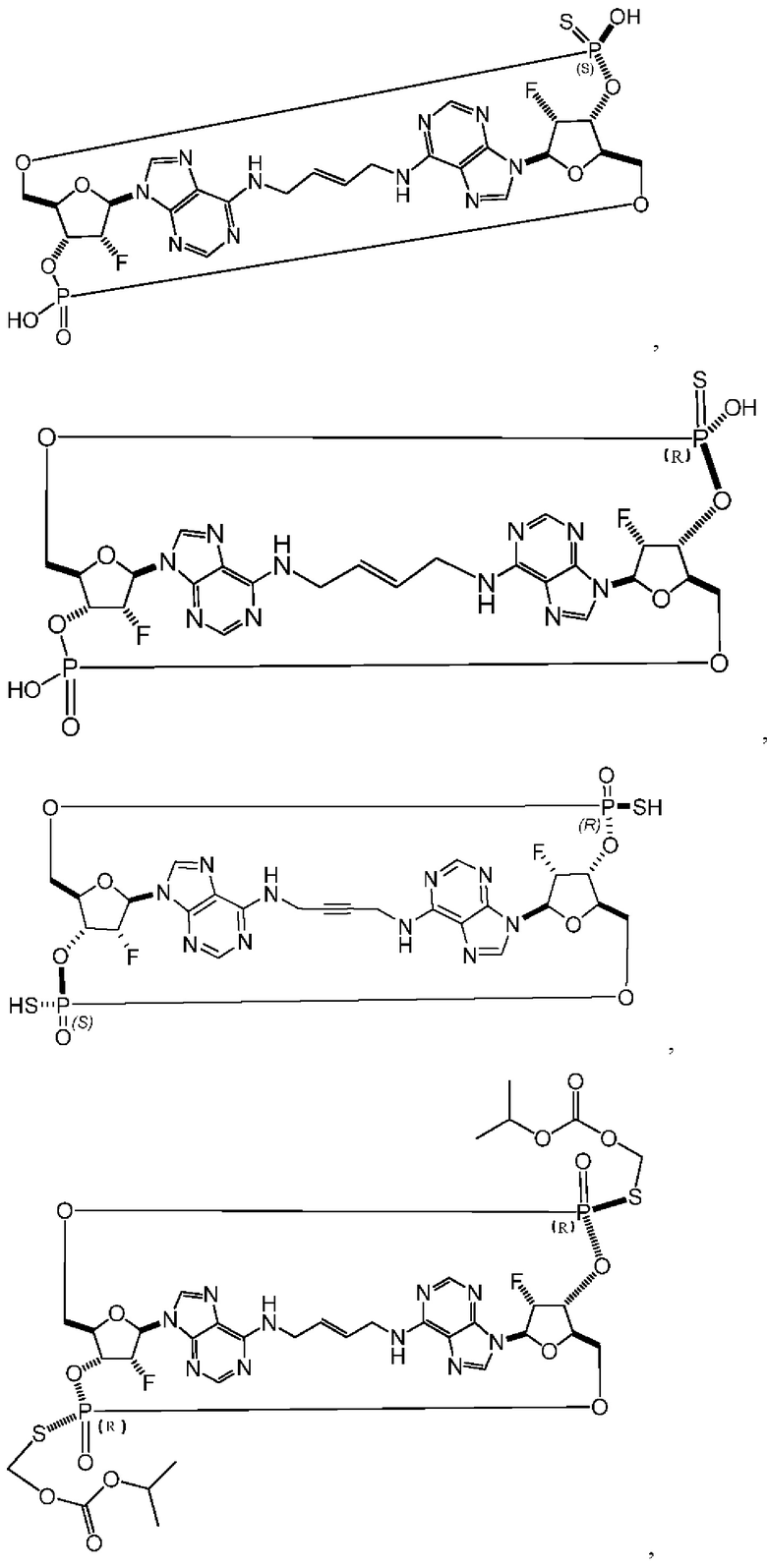
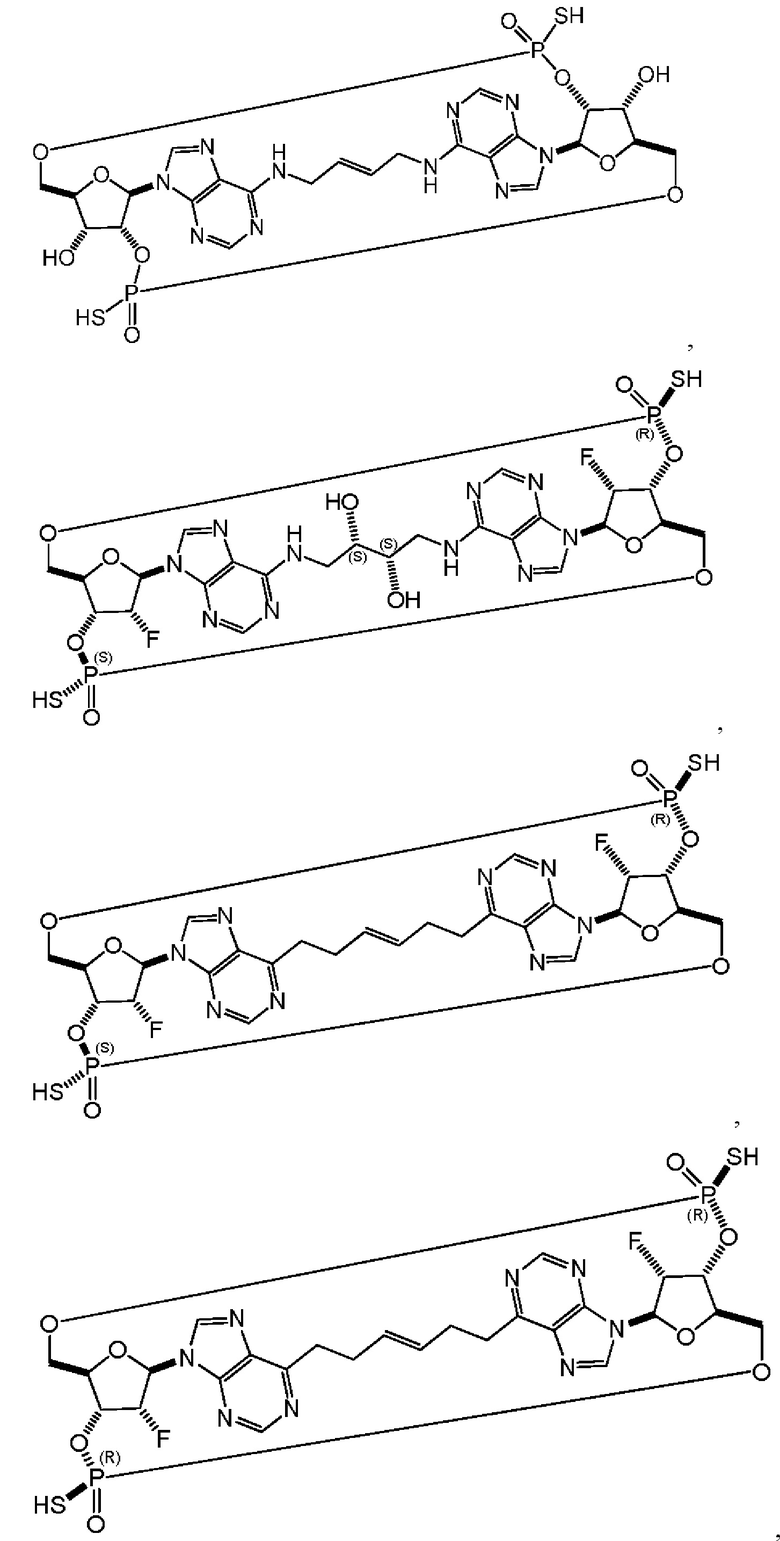
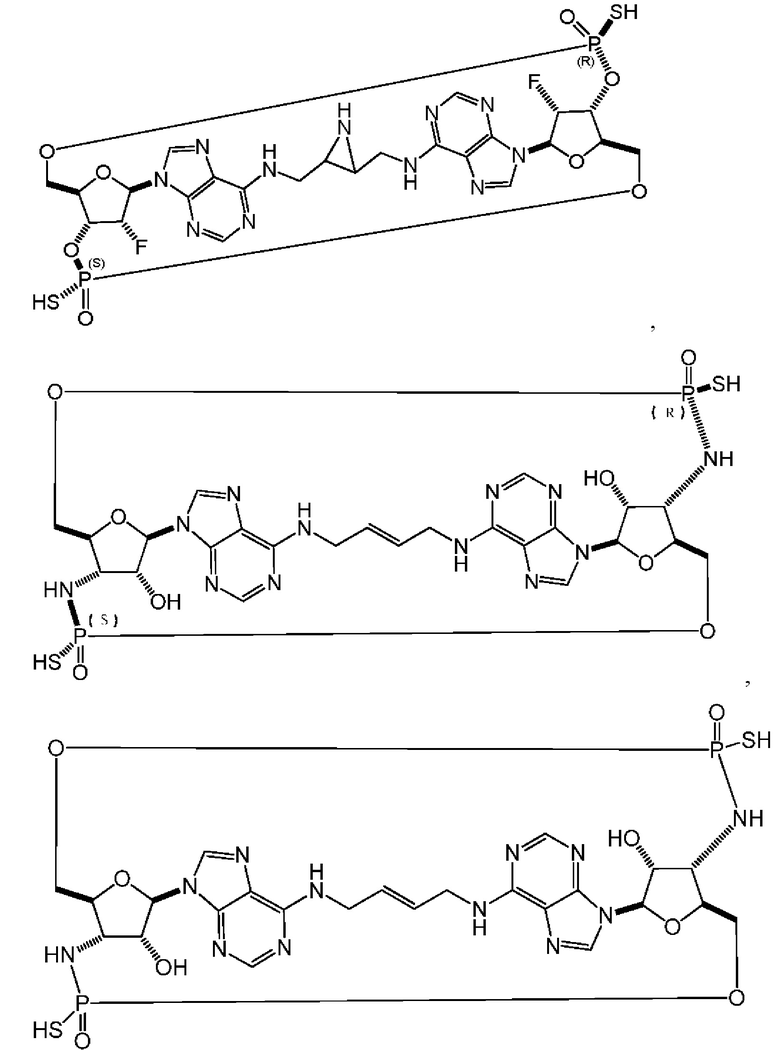
или их фармацевтически приемлемых солей.
16. Соединение или его фармацевтически приемлемая соль по п. 15, выбранные из группы, состоящей из:
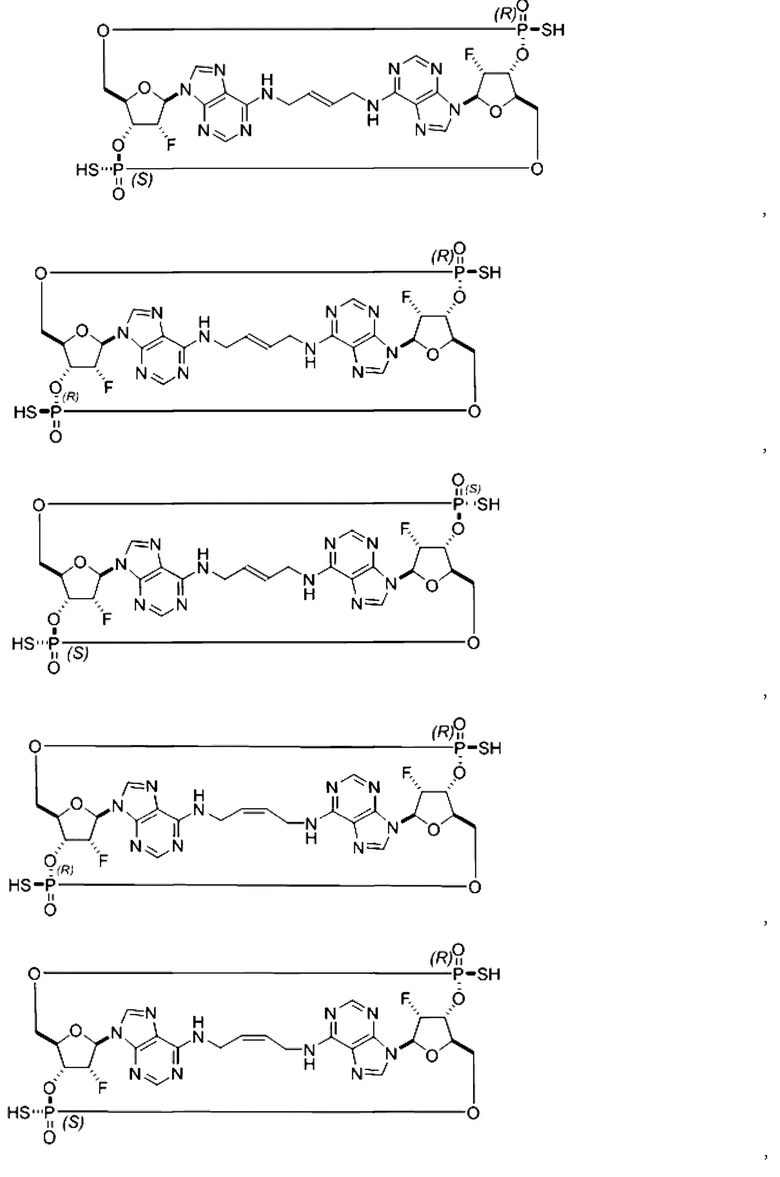
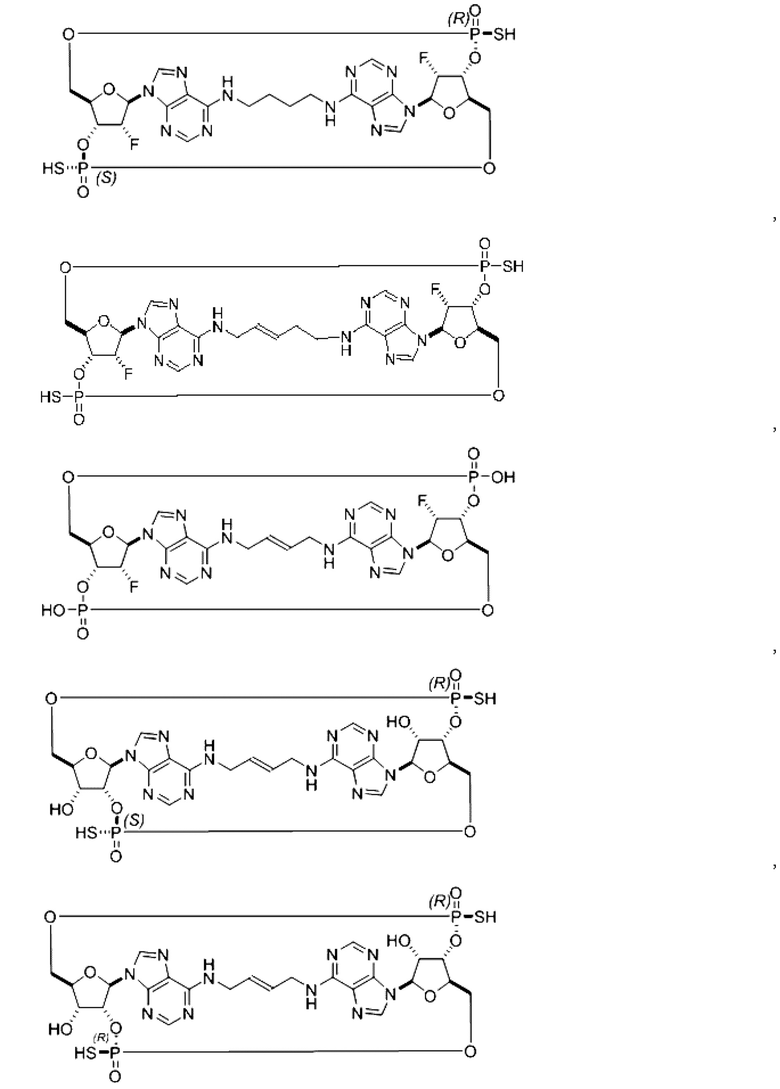
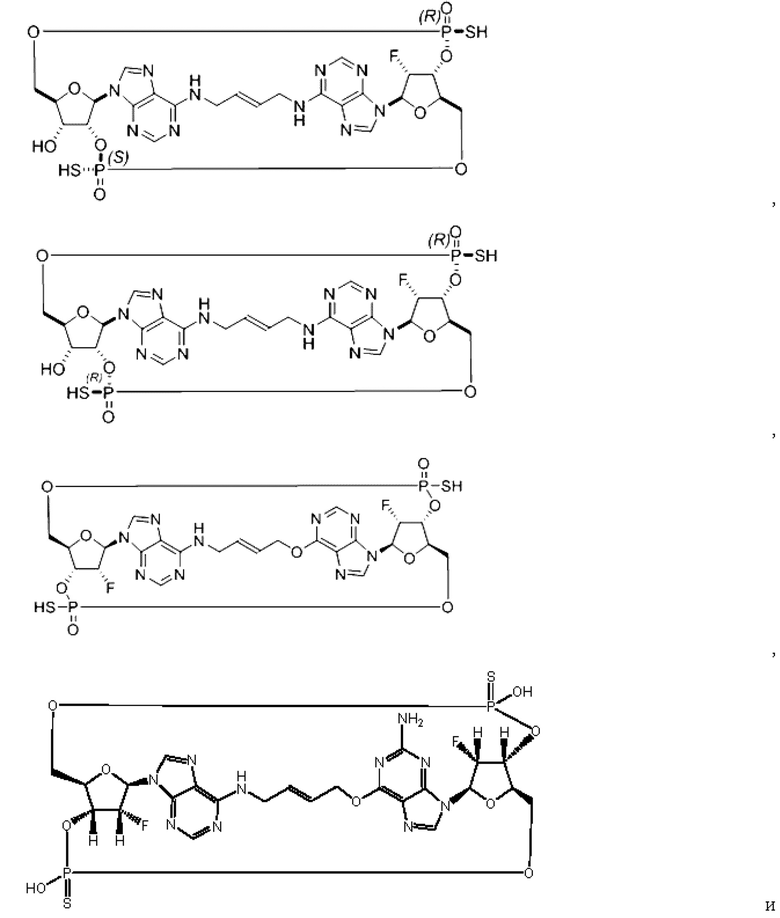
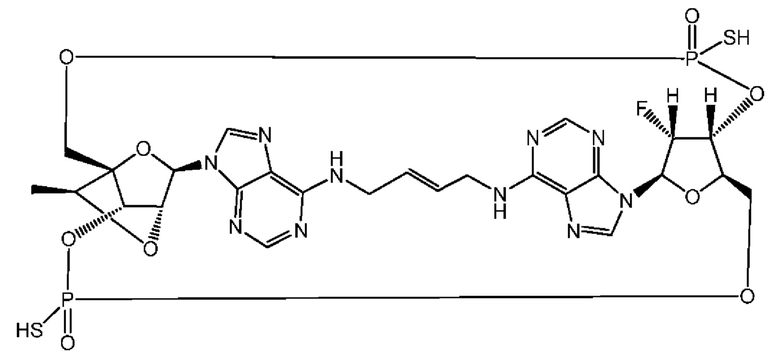
или их фармацевтически приемлемых солей.
17. Соединение или фармацевтически приемлемая соль по любому из пп. 1-16, где соединение или фармацевтически приемлемая соль характеризуется (i) значением ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING HAQ человека; (ii) значением ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING AQ человека; (iii) значением ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING WT человека; или (iv) значением ЕС50 ниже 100 микромоль в репортерных клетках, экспрессирующих вариант STING REF человека.
18. Соединение или фармацевтически приемлемая соль, где соединение представляет собой
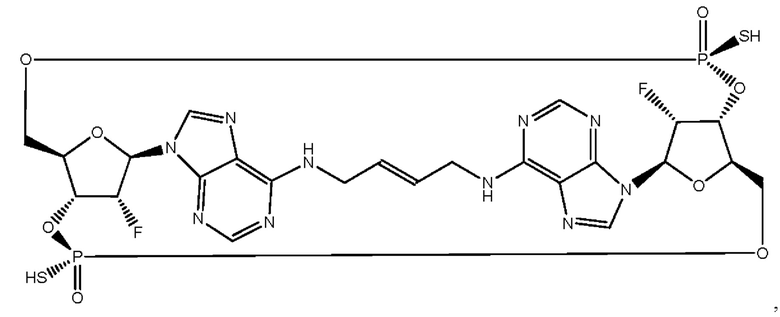
или его фармацевтически приемлемую соль.
19. Соединение или фармацевтически приемлемая соль, где соединение представляет собой
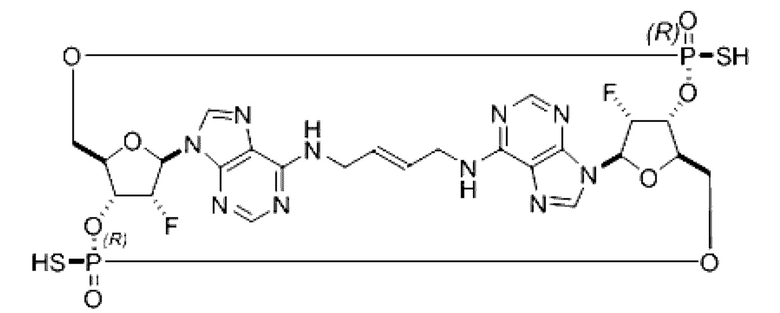
или его фармацевтически приемлемую соль.
20. Соединение или фармацевтически приемлемая соль, где соединение представляет собой
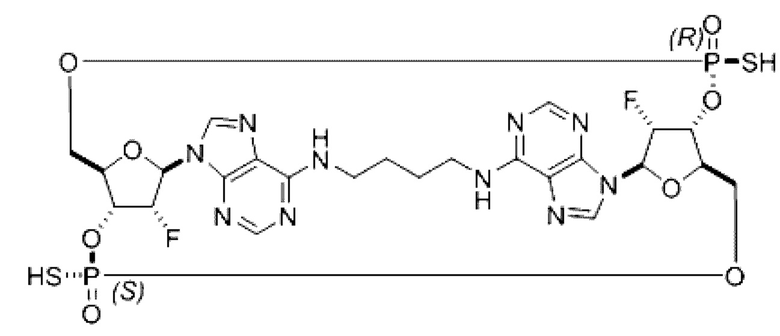
или его фармацевтически приемлемую соль.
21. Соединение или фармацевтически приемлемая соль, где соединение представляет собой
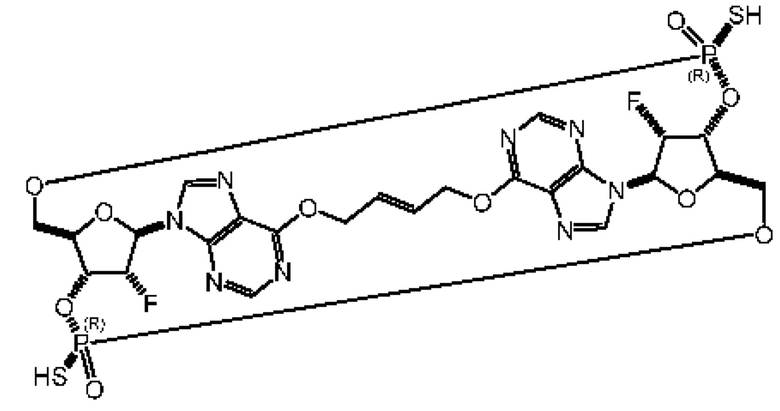
или его фармацевтически приемлемую соль.
22. Соединение или фармацевтически приемлемая соль, где соединение представляет собой
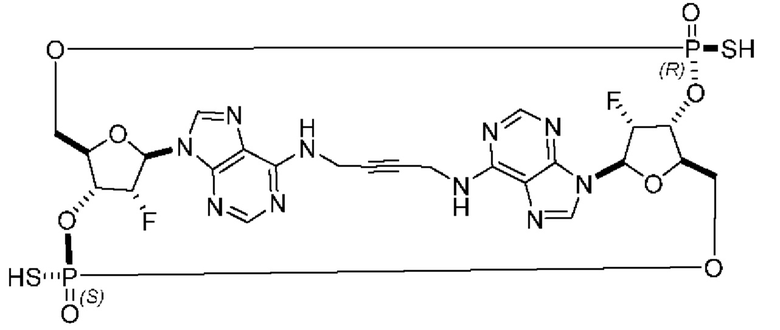
или его фармацевтически приемлемую соль.
23. Фармацевтически приемлемая соль по любому из пп. 1-22, где соль представляет собой диаммонийную соль.
24. Фармацевтическая композиция для лечения рака, содержащая фармацевтически эффективное количество соединения по любому из пп. 1-22 или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель.
25. Способ лечения рака, который предусматривает введение пациенту соединения по любому из пп. 1-22 или фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24.
26. Применение соединения по любому из пп. 1-22 или фармацевтически приемлемой соли по любому из пп. 1-23 для получения фармацевтической композиции для лечения рака.
27. Применение соединения по любому из пп. 1-22 или фармацевтически приемлемой соли по любому из пп. 1-23.
28. Применение фармацевтической композиции по п. 24 для лечения рака.
29. Способ лечения рака, который предусматривает:
идентификацию индивидуума, имеющего рак, подлежащий лечению с помощью соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24; и
введение указанному индивидууму фармацевтически эффективного количества соединения, фармацевтически приемлемой соли или фармацевтической композиции, с помощью которых рак был идентифицирован как подлежащий лечению.
30. Способ по п. 29, при котором указанного индивидуума идентифицируют как имеющего рак, подлежащий лечению с помощью соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24 в результате присутствия аллеля варианта STING REF у пациента.
31. Способ по п. 25 или 29, при котором рак выбран из группы, состоящей из лимфомы, меланомы, колоректального рака, рака молочной железы, острого миелоидного лейкоза, рака толстой кишки, рака печени, рака предстательной железы, рака поджелудочной железы, рака почки и глиомы.
32. Способ лечения рака у пациента, имеющего аллель REF STING, который предусматривает введение указанному пациенту соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24.
33. Способ лечения рака у пациента, имеющего аллель WT STING, который предусматривает введение указанному пациенту соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24.
34. Способ лечения рака у пациента, имеющего аллель AQ STING, который предусматривает введение указанному пациенту соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24.
35. Способ лечения рака у пациента, имеющего аллель HAQ STING, который предусматривает введение указанному пациенту соединения по любому из пп. 1-22, фармацевтически приемлемой соли по любому из пп. 1-23 или фармацевтической композиции по п. 24.
36. Способ по любому из пп. 32-35, при котором рак выбран из группы, состоящей из меланомы, колоректального рака, рака молочной железы, острого миелоидного лейкоза, рака толстой кишки, рака печени и глиомы.
37. Способ по любому из пп. 25 или 32-35, при котором указанный рак является метастатическим.
| WO 2015185565 A1, 10.12.2015 | |||
| THIERRY LIOUX et al | |||
| Design, Synthesis, and Biological Evaluation of Novel Cyclic Adenosine-Inosine Monophosphate (cAIMP) Analogs That Activate Stimulator of Interferon Genes (STING) | |||
| Journal of medicinal chemistry, vol | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Контрольное приспособление к масляным насосам | 1928 |
|
SU10253A1 |
| WO 2014179335 A1, 06.11.2014 | |||
| RU 2013130250 А1, | |||

