Область техники, к которой относится изобретение
Изобретение относится к области медицины, фармации, биотехнологии, а именно к количественному определению стабилизаторов углеводной природы в биологически активных лекарственных препаратах. Заявленное изобретение может быть использовано в производственной биотехнологии и фармацевтической промышленности, а также в смежных областях.
Уровень техники
Вещества углеводной природы, такие как сорбитол, маннитол, трегалоза, глюкоза, лактоза, сахароза и мальтоза как индивидуально так и в различных комбинациях друг с другом входят в состав большого количества биологически активных лекарственных препаратов (БЛП): препараты интерферонов, соматропинов, генно-инженерные препараты крови, вакцины, препараты моноклональных антител, иммуностимулирующие препараты бактериального происхождения, препараты аллергенов, препараты иммуноглобулина человека нормального, иммунодепрессивные препараты. Перечисленные углеводы выполняют функции стабилизаторов как крио- и лиопротекторов в лиофилизированных формах БЛП, а также роль модификаторов тоничности в восстановленных и жидких формах БЛП.
В соответствии с фармакопейными требованиями при оценке качества БЛП необходимо количественно определять вносимые в лекарственный препарат вспомогательные вещества [1].
Из уровня техники известны физические способы определения углеводов - поляриметрический, основанный на измерении величины угла вращения плоскости поляризации света при прохождении через оптически активные вещества, к которым относятся углеводы [2, 3] и рефрактометрический, основанный на определении величины коэффициента преломления. Основной недостаток этих способов определения углеводов - недостаточная специфичность, поэтому данные способы не применимы для индивидуального определения сахаров в смеси, кроме того поляриметрический способ не применим для определения в присутствии других оптически активных соединений.
Из уровня техники известен спектрофотометрический способ определения глюкозы, лактозы, сахарозы и мальтозы, основанный на измерении оптической плотности окрашенных комплексов, полученных при взаимодействии углеводов с антроновым реактивом [4]. В способе раскрыты условия реакции и спектрофотометрической детекции продукта реакции при длине волны 625 нм. Так же известен способ определения, основанный на взаимодействии моносахаридов с пикриновой кислотой с образованием пикраминовой кислоты и спектрофотометрическим детектированием при максимуме поглощения в области от 440 до 460 нм [4]. Данные способы не применимы для определения сорбитола и маннитола, так как эти вещества не обладают свойствами восстановителей. Кроме того перечисленные способы позволяют определить только суммарное содержание сахаров в смеси без достоверной информации о присутствии отдельных компонентов.
Из уровня техники известен другой способ определения соединений углеводной природы, основанный на реакции окисления моносахаридов реактивом Фелинга. Определение проводят методом спектрофотомерии при длине волны 670 нм или методом титриметриии с индикатором. Недостатком данного способа является его применимость только для определения восстанавливающих сахаров (глюкоза, лактоза и мальтоза) и сахарозы, предварительно подвергнутой гидролизу с кислотой или с ферментом инвертазой. Кроме того способ не обладает достаточной селективностью для определения индивидуальных соединений углеводной природы в смеси [5].
Из уровня техники известны другие способы определения, основанные на окислительно-восстановительных реакциях: йодометрический, феррицианидный, метод Бертрана. Количество сахаров определяется по количеству реактива, пошедшего на их окисление. Указанные методы являются титриметрическими и отличаются друг от друга применяемыми окислителями: йод в йодометрическом, железосинеродистый калий в феррицианидном, сульфат меди II - в методе Бертрана [2, 3]. Перечисленные способы не обладают достаточной селективностью для определения индивидуальных соединений углеводной природы в смеси. Кроме того сахароза не окисляется железосинеродистым калием, а метод Бертрана не применим для определения сорбитола и маннитола, так как они не восстанавливают медь.
Из уровня техники известен спектрофотометрический способ определения маннитола периодатом натрия [6]. Данный способ используют для количественного определения маннитола в биологических лекарственных препаратах [7].
Из уровня техники известны ферментативные способы определения полиолов, маннитола, трегалозы, глюкозы, лактозы, сахарозы и мальтозы [6, 8, 9]. Разнообразные ферментативные методики отличаются высокой специфичностью, но при этом требуют применения для каждого углевода своего уникального фермента. Кроме того в большинстве случаев для воспроизведения ферментативного анализа предусмотрен набор или тест-система, коммерческая доступность которых в настоящее время затруднена.
Из уровня техники известен способ определения углеводов, основанный на переводе углеводов в летучие триметилсилильные производные с последующим разделением и идентификацией на газовом хроматографе с помощью пламенноионизационного или масс-селективного детекторов [10]. Трудоемкость подготовки проб является существенным недостатком способа.
Из уровня техники известен способ определения соединений углеводной природы на основе гидрофильной или ион-эксклюзионной ВЭЖХ с рефрактометрическим детектированием [11, 12]. В способах раскрыты условия хроматографического разделения и детектирования анализируемых соединений. Как правило, данные способы не применимы для одновременного определения полиолов, моно - и дисахаридов.
Из области техники известно, что в щелочных условиях соединения углеводной природы ведут себя как слабые кислоты [13]. Внедрение стационарных фаз на основе полимеров, обеспечивающих стабильность в широком диапазоне pH, способствовало разработке универсального и эффективного метода разделения углеводов и родственных соединений на основе ионообменной ВЭЖХ. Кроме того современная конструкция амперометрических детекторов позволяет автоматически чередовать напряжение на рабочем электроде (импульсный режим), что позволяет получать воспроизводимые результаты анализа.
Прототипом заявляемого изобретения являются известные из уровня техники способы определения лактозы [14]; лактозы, сахарозы и глюкозы [15]; полиолов (глицерин, эритритол, арабитол, сорбитол, маннитол), глюкозы и трегалозы [16]; ксилитола, сорбитола, маннитола, лактитола, изомальтитола, глюкозы, мальтитола, фруктозы, лактозы, сахарозы и мальтозы [17]; полиолов (глицерин, эритритол, арабитол, сорбитол, маннитол) и трегалозы [18] ионообменной высокоэффективной хроматографией с импульсным амперометрическим детектированием. В способах раскрыты условия хроматографического разделения и детектирования анализируемых соединений.
Недостатками вышеизложенных способов, является, то что, не представляется возможным применить их, в случае если углеводные стабилизаторы (сорбитол или маннитол или трегалоза или глюкоза или лактоза или сахароза или мальтоза) находятся между собой в определенной комбинации. Кроме того, анализируемыми объектами являются растения и пищевые продукты, отличающиеся матрицей от биологически активных лекарственных препаратов.
Авторами была выявлена потребность в разработке способа определения стабилизаторов углеводной природы, выбранных из сорбитола, маннитола, трегалозы, глюкозы, лактозы, сахарозы, мальтозы их комбинаций в смеси ионообменной высокоэффективной жидкостной хроматографией (ионообменная ВЭЖХ) с импульсным амперометрическим детектированием в биологически активных лекарственных препаратах.
Раскрытие сущности изобретения
Технической задачей является разработка способа определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографией (ионообменная ВЭЖХ) с импульсным амперометрическим детектированием.
Техническим результатом является разработанный способ определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографией (ионообменная ВЭЖХ) с импульсным амперометрическим детектированием.
Достижение технического результата обеспечивается благодаря:
- использованию ионообменной высокоэффективной хроматографии с импульсным амперометрическим детектированием;
- проведению процедуры пробоподготовки;
- использованию стандартного образца.
Способ определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографии с импульсным амперометрическим детектированием, включающий разведение содержащих по меньшей мере один углеводный стабилизатор в биологически активном лекарственном препарате и исходном стандартном растворе до концентрации углеводных стабилизаторов от 10 мкг/мл до 50 мкг/мл и от 1 мкг/мл до 50 мкг/мл соответственно, либо от 25 мкг/мл до 120 мкг/мл и от 2 до 120 мкг/мл соответственно, хроматографируют полученные растворы в изократическом режиме элюирования, а затем проводят расчеты.
Способ, дополнительно характеризующийся тем, что биологически активными лекарственными препаратами могут быть вакцины или аллергены или препараты содержащие плазму крови или препараты фактора крови VIII или иммуноглобулины человека нормального
3.Способ, дополнительно характеризующийся тем, что стабилизаторы углеводной природы выбраны по меньшей мере из мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы.
Способ, дополнительно характеризующийся тем, что для хроматографирования используют подвижную фазу в виде гидроксида натрия или гидроксида калия с концентрацией 160 ммоль/л.
Способ, дополнительно характеризующийся тем, что исходный стандартный раствор с концентрацией 1,0 мг/л.
Способ, дополнительно характеризующийся тем, что для детектирования используют импульсный амперометрический детектор с золотым рабочим электродом
Способ, дополнительно характеризующийся тем, что хроматографируют при скорости потока 1,0 мл/мин.
Способ, дополнительно характеризующийся тем, что для хроматографирования используют неподвижную фазу в виде колонки размером 250х4 мм.
Способ, дополнительно характеризующийся тем, что на колонку наносят последовательно воду, стандартные растворы, раствор биологически активного лекарственного препарата.
Краткое описание чертежей и иных материалов (Приложение 1-16)
Фиг. 1 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации сорбитола в воде при объеме вкола 10 мкл.
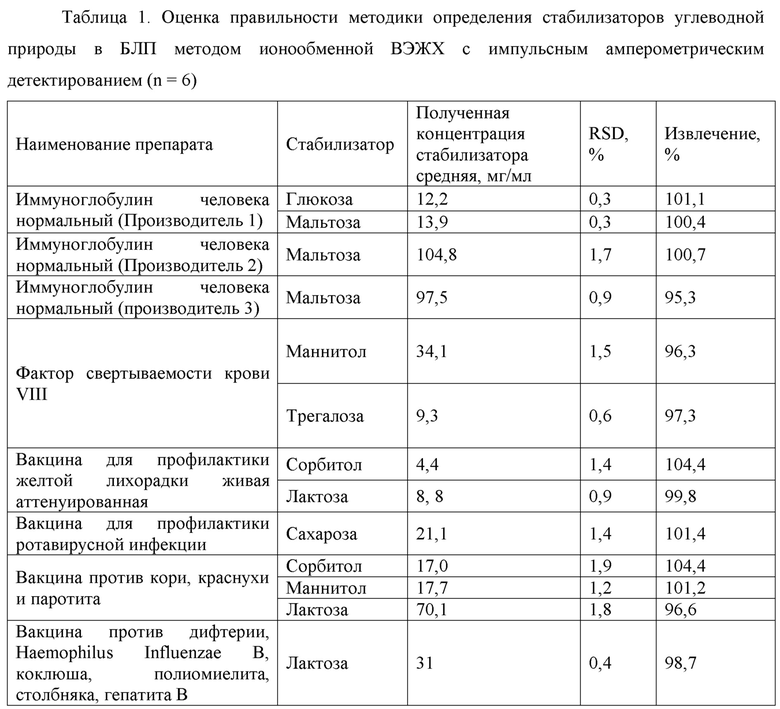
Фиг. 2 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации маннитола в воде при объеме вкола 10 мкл.
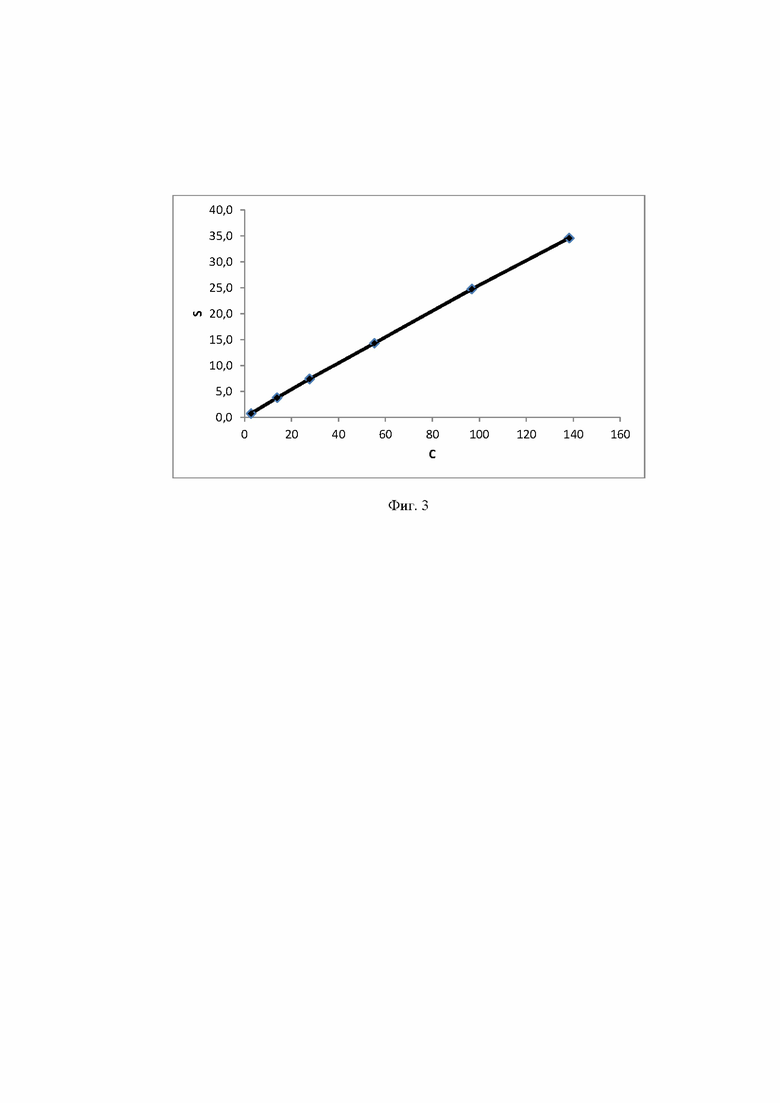
Фиг. 3 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации трегалозы в воде при объеме вкола 10 мкл.
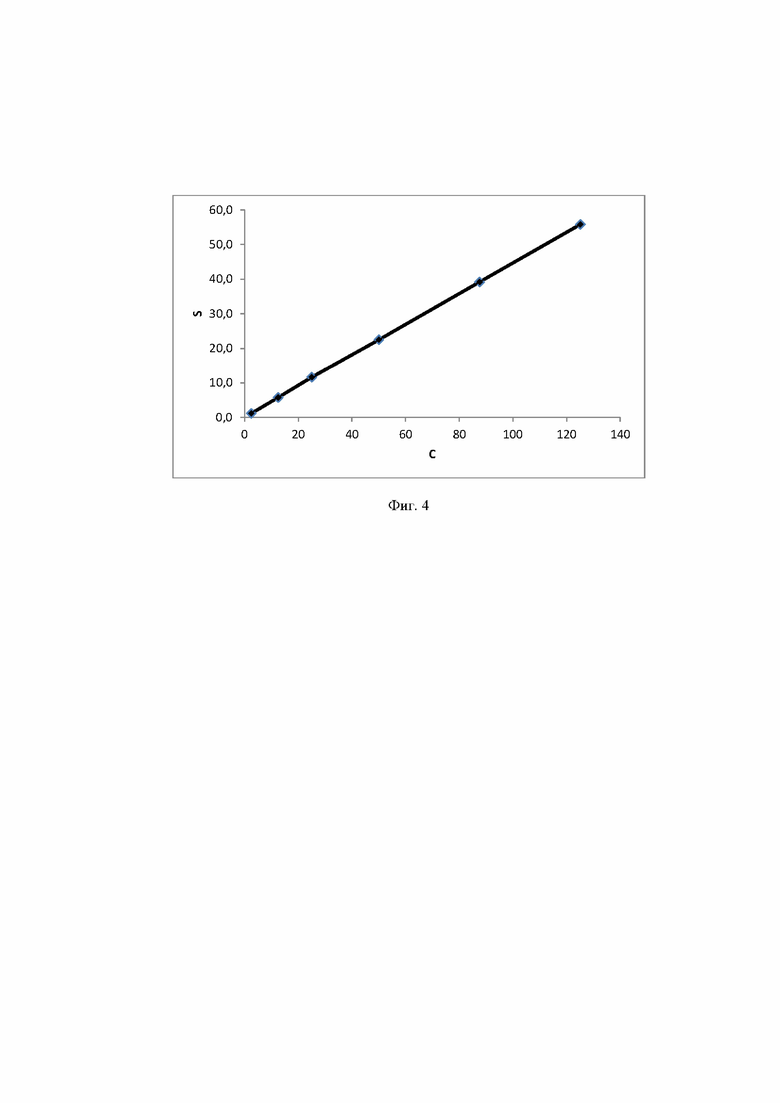
Фиг. 4 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации глюкозы в воде при объеме вкола 10 мкл.
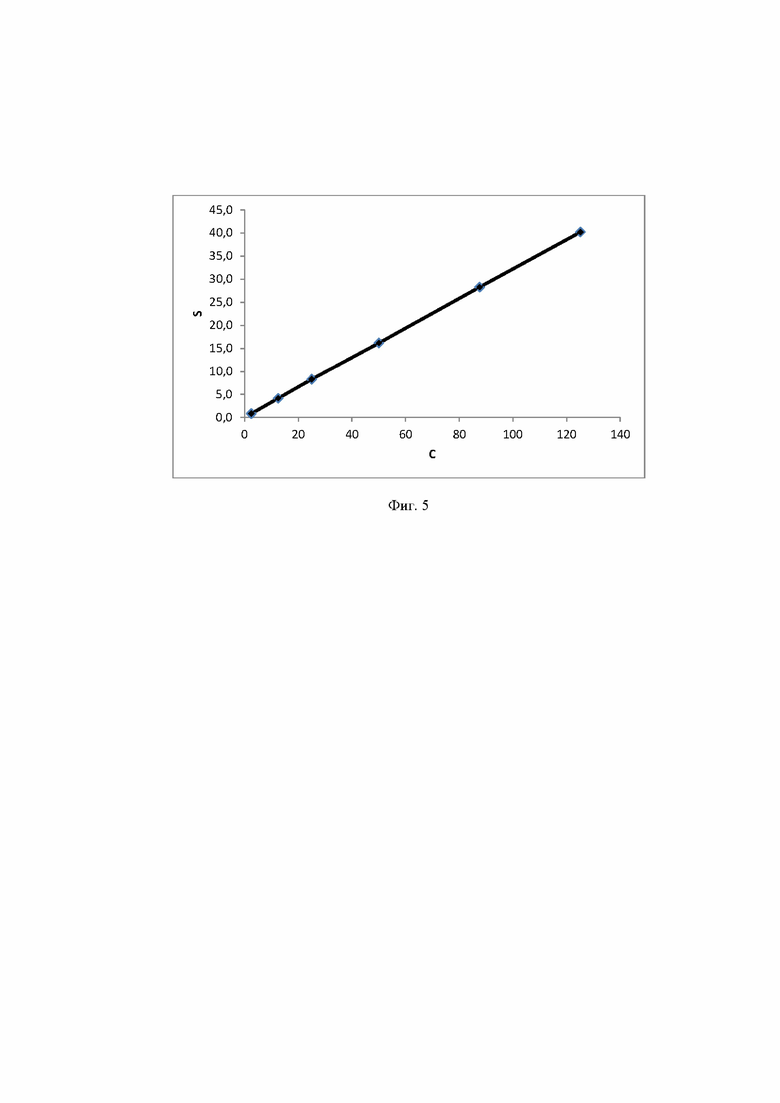
Фиг. 5 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации лактозы в воде при объеме вкола 10 мкл.
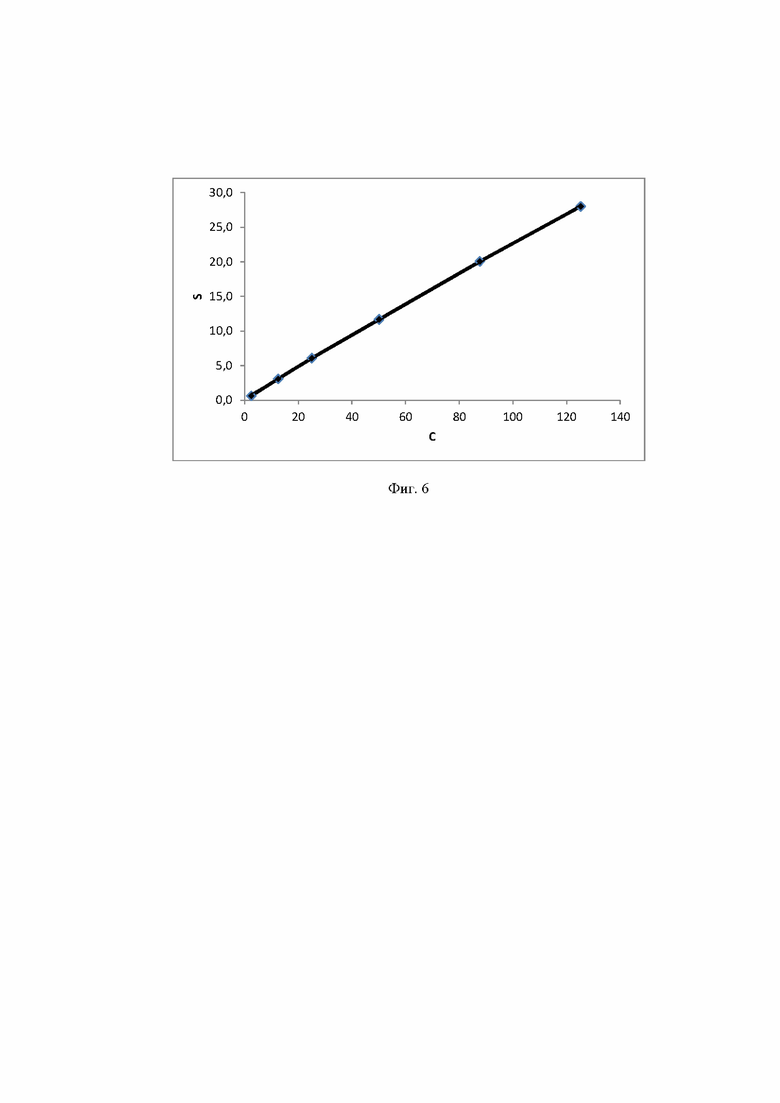
Фиг. 6 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации сахарозы в воде при объеме вкола 10 мкл.
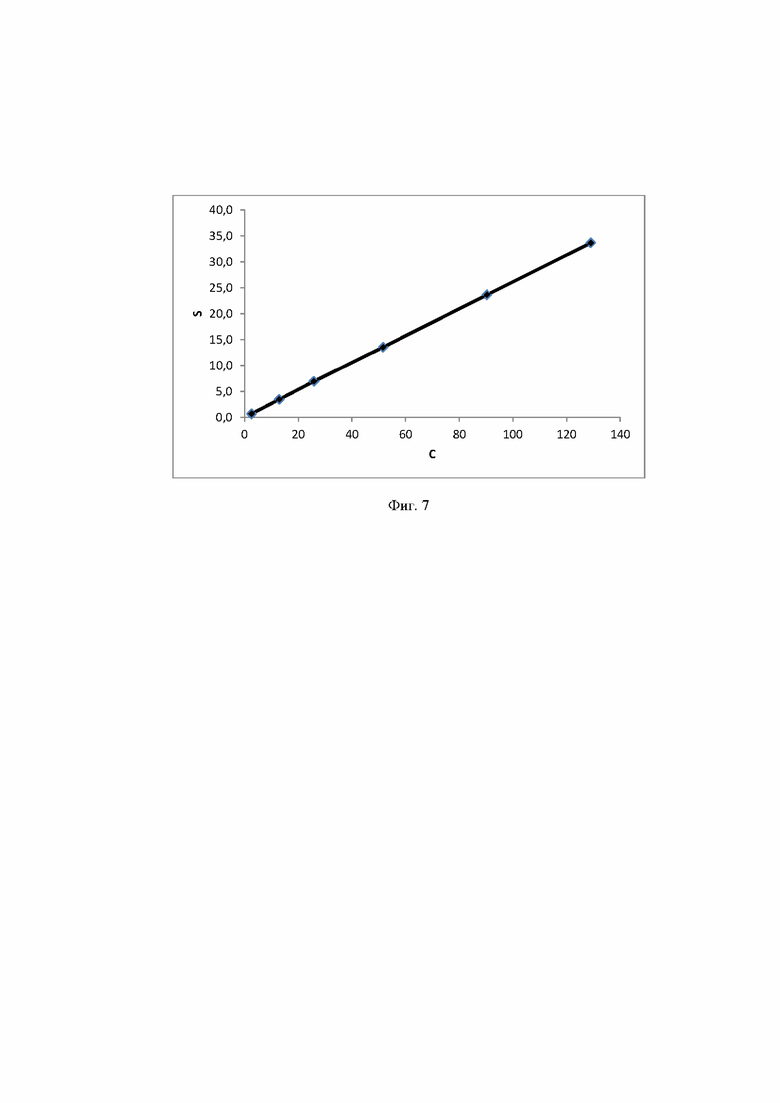
Фиг. 7 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации мальтозы в воде при объеме вкола 10 мкл.
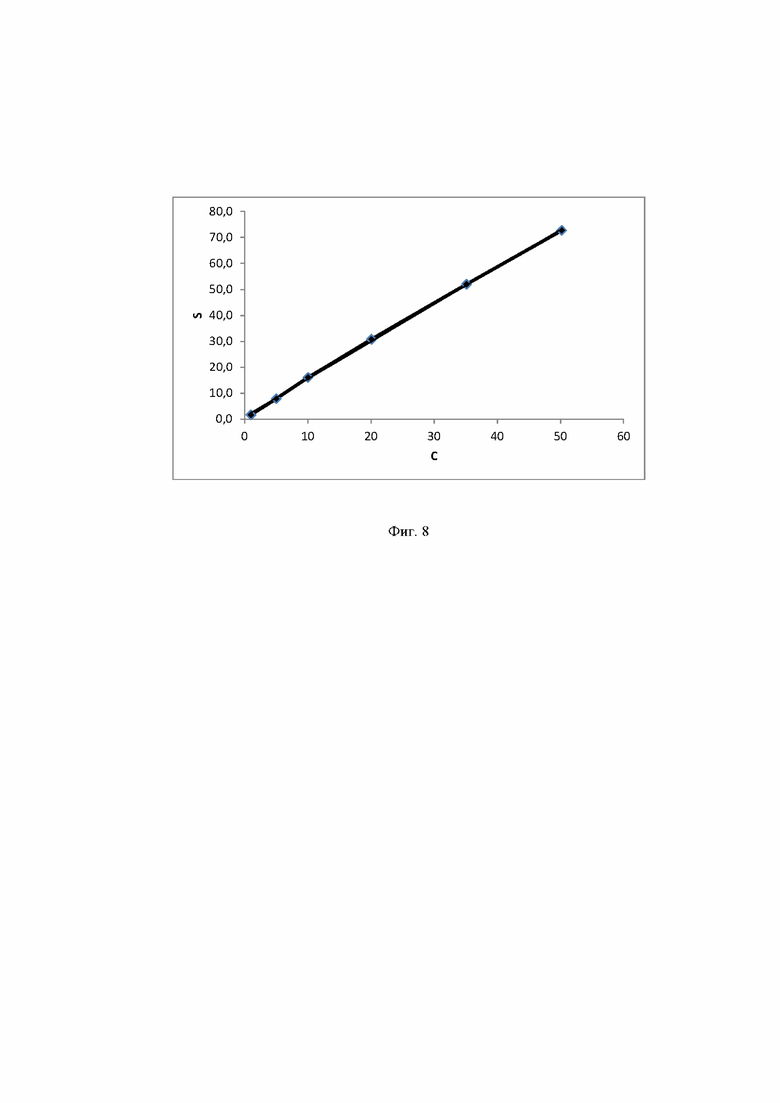
Фиг. 8 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации сорбитола в воде при объеме вкола 25 мкл.
Фиг. 9 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации маннитола в воде при объеме вкола 25 мкл.
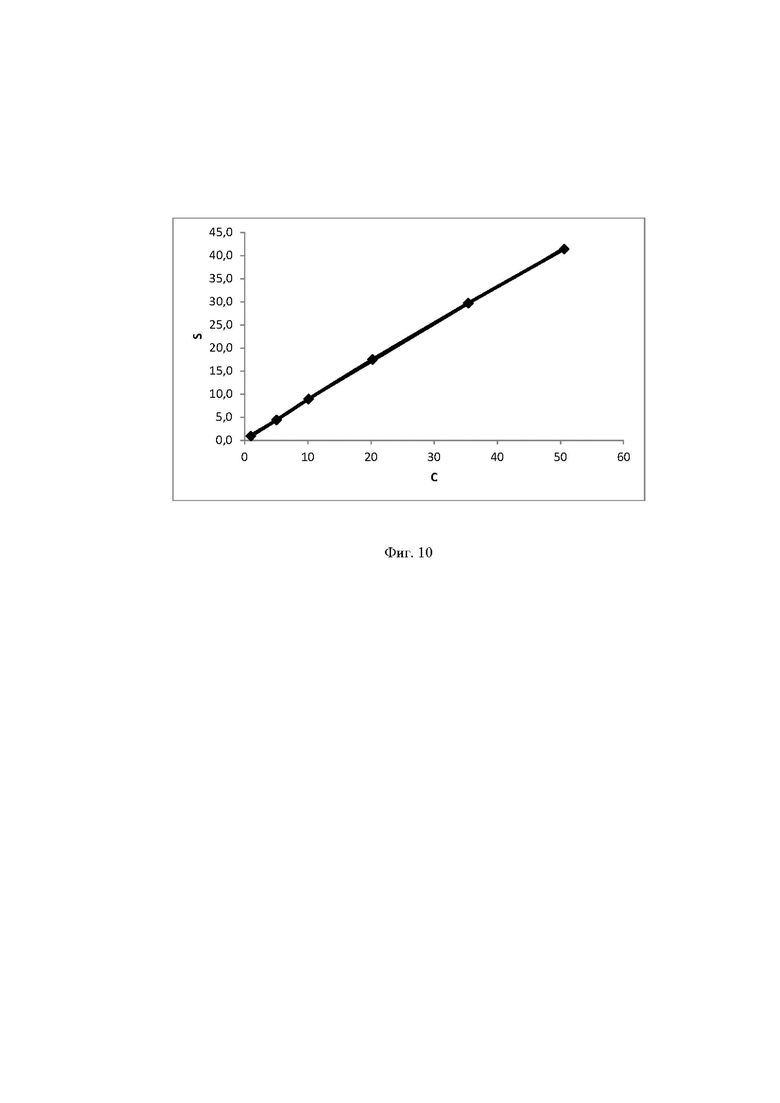
Фиг. 10 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации трегалозы в воде при объеме вкола 25 мкл.
Фиг. 11 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации глюкозы в воде при объеме вкола 25 мкл.
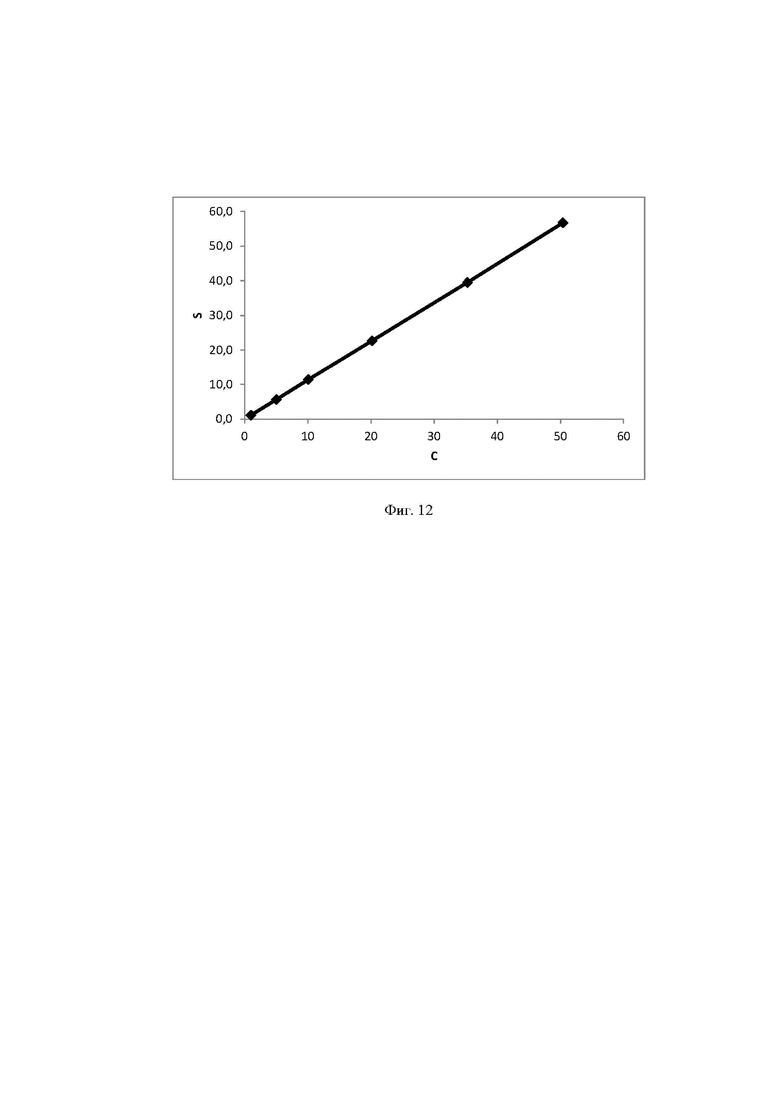
Фиг. 12 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации лактозы в воде при объеме вкола 25 мкл.
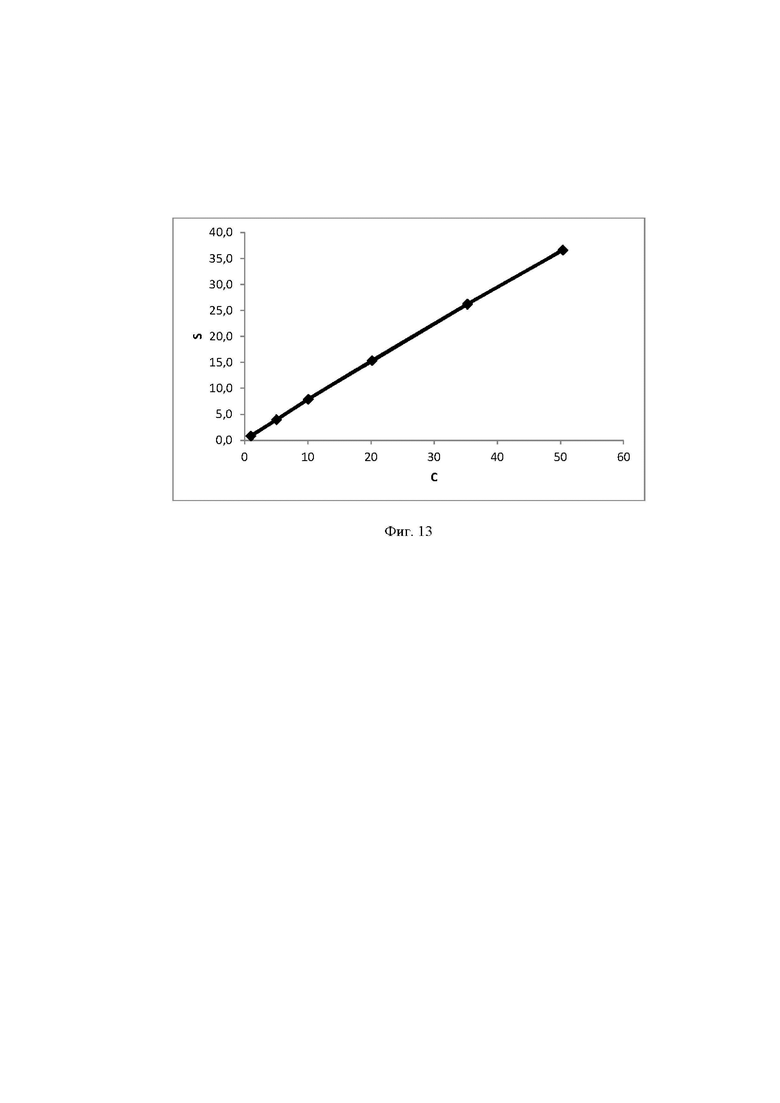
Фиг. 13 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации сахарозы в воде при объеме вкола 25 мкл.
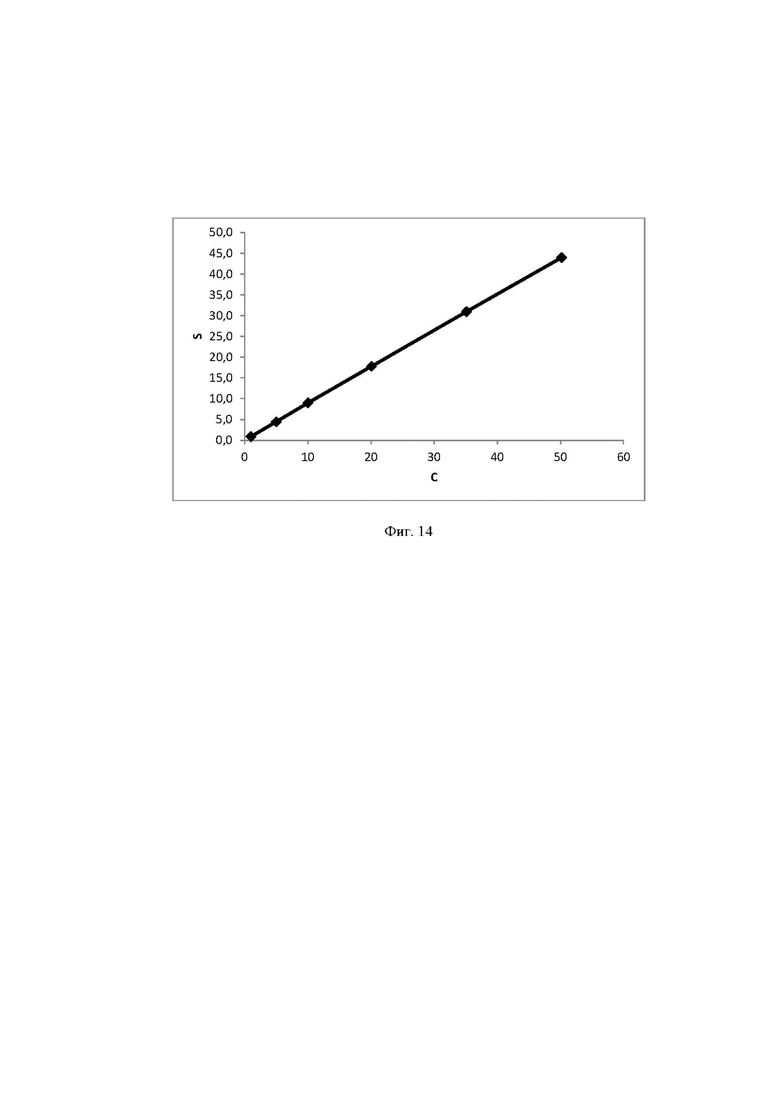
Фиг. 14 Калибровочная прямая, характеризующая зависимость аналитического сигнала от концентрации мальтозы в воде при объеме вкола 25 мкл.
Фиг. 15 График зависимости потенциала относительно времени. В интервале 1 сек напряжение на электроде меняется от 0,05 В до - 0,15 В с максимумом 0,75 В.
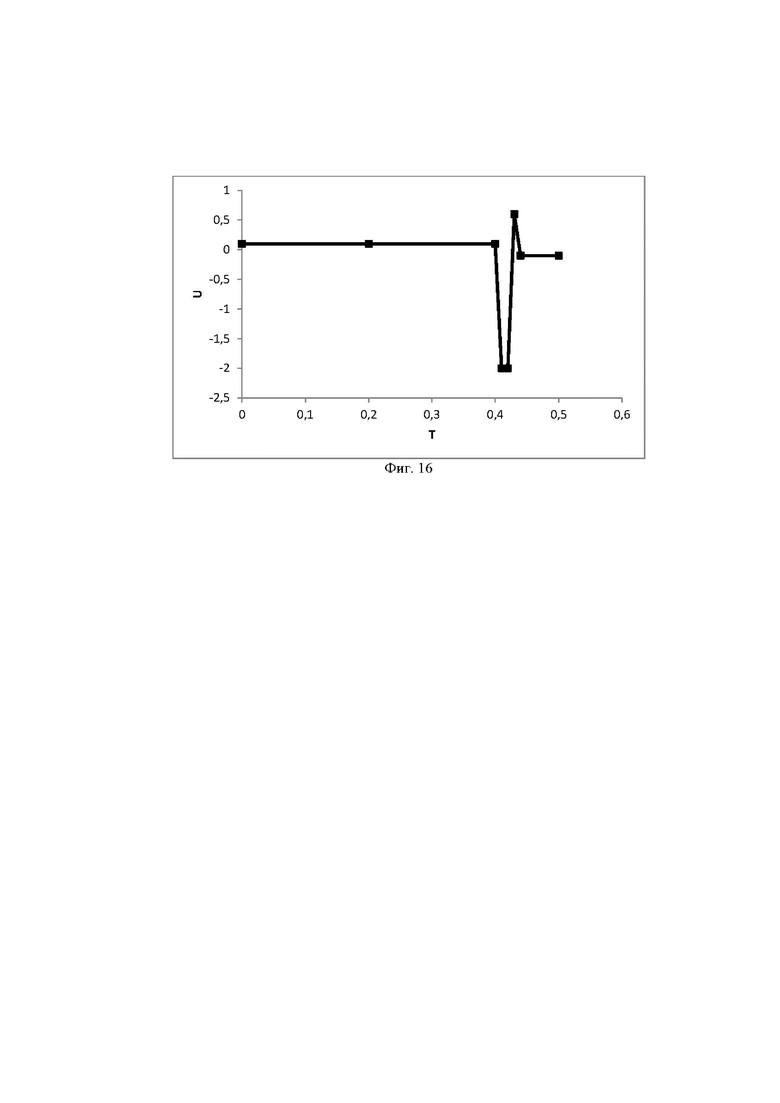
Фиг. 16 График зависимости потенциала относительно времени. В интервале 0,5 сек напряжение на электроде меняется от 0,1 В до -0,1 В с максимумом -2,0 В.
Стандартный образец - это материал, достаточно однородный и стабильный по отношению к одному или нескольким определенным свойствам, которые были установлены для того, чтобы использовать его по назначению в измерительном процессе. По уровню утверждения и области применения стандартные образцы разделяют на следующие категории: государственный стандартный образец (ГСО), международный стандартный образец (МСО) фармакопейный стандартный образец (ФСО), и др. [19].
Биологически активные лекарственные препараты (далее - БЛП) - это лекарственные препараты, действующее вещество которых произведено или выделено из биологического источника и для определения свойств и качества которых необходима комбинация биологических и физико-химических методов. К биологическим лекарственным препаратам относятся иммунобиологические лекарственные препараты, лекарственные препараты, полученные из крови, плазмы крови человека и животных (за исключением цельной крови), биотехнологические лекарственные препараты, генотерапевтические лекарственные препараты [20].
Импульсное амперометрическое детектирование-режим детектирования, при котором повторяются серии изменений потенциала на рабочем электроде. За счет повторяющихся импульсов для перехода между высоким положительным и отрицательным потенциалами поверхность электрода постоянно регенерируется. Значение потенциалов и продолжительность импульсов задаются во время анализа и представляют собой серию шагов, определенных как точки на графике зависимости потенциала относительно времени [21].
Метод стандартных добавок - метод, основанный на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества [22].
Осуществление изобретения
Заявленный способ характеризуется тем, что используют ионообменную высокоэффективную жидкостную хроматографию с импульсным амперометрическим детектированием. Проводят пробоподготовку, путем разведения водой биологически активного лекарственного препарата содержащего по меньшей мере один стабилизатор углеводной природы и исходного стандартного раствора с концентрацией стабилизатора/(ов) углеводной природы 1,0 мг/мл до концентрации стабилизатора/(ов) углеводной природы в диапазоне от 10 мкг/мл до 50 мкг/мл или от 25 до 120 мкг/мл (в зависимости от объем вкола) для биологически активных лекарственных препаратов и концентрации стабилизатора/(ов) углеводной природы в диапазоне от 1 мкг/мл до 50 мкг/мл или от 2 мкг/мл до 120 мкг/мл (в зависимости от объем вкола) для исходного стандартного раствора, хроматографируют полученные растворы, а затем проводят расчеты.
Стабилизаторы углеводной природы, содержащиеся в исходном стандартном растворе и приготовленных из него стандартных растворах выбраны по меньшей мере из мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы. В исходном стандартном растворе и в приготовленных из него стандартных растворах, содержится по меньшей мере один стабилизатор углеводной природы.
Для осуществления заявленного способа биологически активные лекарственные препараты, содержащие по меньшей мере один углеводный стабилизатор, выбирают из: иммуноглобулина человека нормальный, фактора свертываемости крови VIII, вакцин для профилактики различных инфекций. Выбранный биологически активный лекарственный препарат, разводят водой до концентрации в диапазоне от 10 мкг/мл до 50 мкг/мл или от 25 мкг/мл до 120 мкг/мл в зависимости от объема вкола.
Углеводные стабилизаторы, содержащиеся в биологически активных лекарственных препаратах выбраны по меньшей мере из мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы.
При проведении ионообменной высокоэффективной жидкостной хроматографии используют подвижную фазу, состоящую из водного раствора натрия гидроксида или калия гидроксида с концентрацией 160 ммоль/л.
Для регистрации аналитического сигнала анализируемых соединений используют импульсный амперометрический детектор с золотым рабочим электродом.
После хроматографического разделения определяют количественное содержание стабилизатора/(ов) углеводной природы в биологически активных лекарственных препаратах в сравнении со стандартными растворами, содержащими стабилизатор/(ы) углеводной природы в диапазоне концентраций либо от 1 до 50 мкг/мл, либо от 2 до 120 мкг/мл в зависимости от объема вкола.
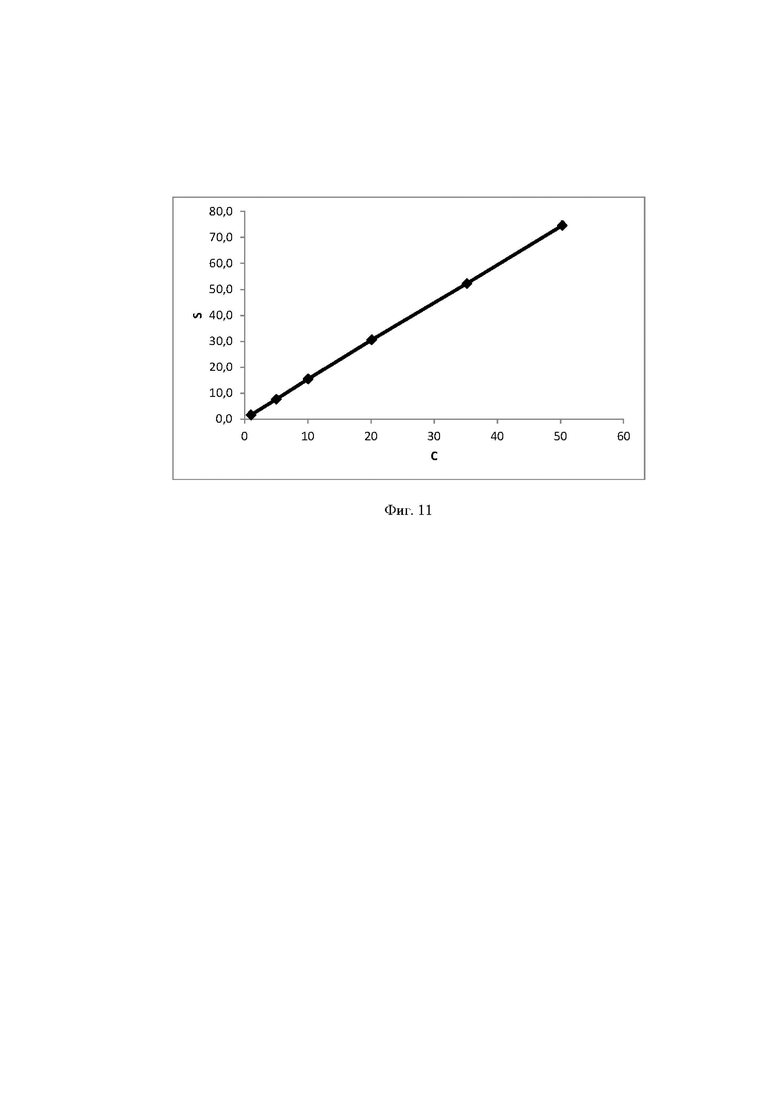
Концентрация стабилизатора/(ов) углеводной природы в биологически активных лекарственных препаратах определялась с использованием калибровочной кривой для соответствующего углеводного стабилизатора (фиг. 1-14).
Условия проведения ионообменной высокоэффективной жидкостной хроматографии (ионообменная ВЭЖХ):
- хроматографическая колонка размером 250 мм × 4,0 мм;
- защитная предколонка размером 50 мм × 4,0 мм;
- изократический режим элюирования;
- подвижная фаза состава: 160 мМ раствор натрия/калия гидроксида;
- скорость потока - 1,0 мл/мин;
- импульсный амперометрический детектор;
- объем нанесения на колонку - 25 мкл или 10 мкл;
- температура хроматографической колонки: 20°С.
При нанесении проб на колонку используют последовательность: вода, стандартные растворы, испытуемые растворы.
Расчет содержания стабилизатора/(ов) углеводной природы производят, используя график регрессионной зависимости площадей пиков стабилизатора/(ов) углеводной природы от их концентрации в стандартных растворах (1).
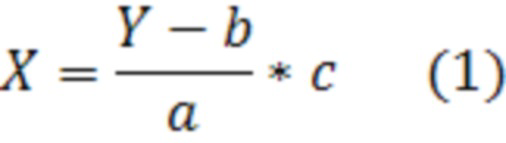
где:
Y - средняя площадь пика соответствующего стабилизатора углеводной природы на хроматограммах испытуемого раствора;
a - угловой коэффициент графика регрессионной зависимости;
b - свободный член графика регрессионной зависимости;
c - разведение испытуемого образца.
Возможность осуществления заявляемого изобретения раскрыта в следующих примерах.
Пример 1. Приготовление растворов для построения калибровочного графика.
Готовят исходный стандартный раствор с концентрацией 1 мг/мл каждого стабилизатора, выбранных из мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы. Путем разведения исходного стандартного раствора с концентрацией 1 мг/мл готовят 6 стандартных растворов углеводных стабилизаторов с концентрацией каждого углевода (мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы) в диапазоне: от 1 мкг/мл до 50 мкг/мл: 1 мкг/мл, 5 мкг/мл, 10 мкг/мл, 20 мкг/мл, 35 мкг/мл и 50 мкг/мл соответственно; или от 2 мкг/мл до 120 мкг/мл: 2 мкг/мл, 12 мкг/мл, 25 мкг/мл, 50 мкг/мл, 90 мкг/мл, 120 мкг/мл соответственно.
Стандартные растворы углеводных стабилизаторов являются калибровочными растворами углеводных стабилизаторов.
Анализируют полученные растворы на ионном хроматографе, фиксируя аналитический сигнал каждого углеводного стабилизатора.
Выводы. Полученная линейная зависимость аналитического сигнала от концентрации каждого стабилизатора (фиг. 1 - 14) может быть использована в качестве калибровочной при анализе проб с неизвестной концентрацией стабилизаторов углеводной природы.
Пример 2. Определение количественного содержания глюкозы и мальтозы в препарате иммуноглобулин человека нормальный (производитель 1) заявленным способом.
Образец восстановили в 25 мл воды (в соответствии с инструкцией производителя). Полученный раствор развели водой до содержания стабилизаторов углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводородных стабилизаторов выбранных из глюкозы и мальтозы разводят водой согласно примеру 1.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал мальтозы и глюкозы при следующих условиях:
- хроматографическая колонка размером 250 мм × 4,0 мм;
- защитная предколонка размером 50 мм × 4,0 мм;
- изократический режим элюирования;
- подвижная фаза состава: 160 мМ раствор натрия гидроксида;
- скорость потока - 1,0 мл/мин;
- импульсный амперометрический детектор, где интервал 1 сек напряжение на электроде меняется от 0,05 В до - 0,15 В с максимумом 0,75В (фиг. 15);
- объем нанесения на колонку - 25 мкл;
- температура хроматографической колонки: 20°С.
Полученное значение аналитического сигнала пересчитывают по калибровочному графику с учетом степени разведения.
Вывод. Полученные значения, 12,2 мг/мл глюкозы и 13,9 мг/мл мальтозы находятся в диапазоне, определенном нормативной документацией на препарат - от 10,0 мг/мл до 16,6 мг/мл.
Пример 3. Определение количественного содержания мальтозы в препарате иммуноглобулин человека нормальный (Производитель 2) заявленным способом.
Лекарственный препарат развели водой до содержания стабилизатора углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из мальтозы разводят водой согласно примеру 1.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал мальтозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала пересчитывают по калибровочному графику с учетом степени разведения.
Вывод. Полученное значение 104,8 мг/мл мальтозы находится в диапазоне, определенном нормативной документацией на препарат - от 90,0 мг/мл до 110 мг/мл.
Пример 4. Определение количественного содержания мальтозы в препарате иммуноглобулина человека нормальный (производитель 3) заявленным способом.
Лекарственный препарат развели водой до содержания стабилизатора углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из мальтозы разводят водой согласно примеру 1.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал мальтозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала пересчитывают по калибровочному графику с учетом степени разведения.
Вывод. Полученное значение 97,5 мг/мл мальтозы находится в диапазоне, определенном нормативной документацией на препарат - от 90 мг/мл до 110 мг/мл мальтозы.
Пример 5. Определение количественного содержания маннитола и трегалозы в препарате фактора свертывания крови VIII заявленным способом.
Образец восстановили в 5 мл воды (в соответствии с инструкцией производителя). Полученный раствор развели водой до содержания стабилизаторов углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из маннитола и трегалозы разводят водой согласно примеру 1.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал маннитола и трегалозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала пересчитывают по калибровочному графику с учетом степени разведения.
Вывод. Полученные значения (с учетом пересчета содержания на флакон) 170,5 мг/флакон маннитола и 46,5 мг/флакон трегалозы дигидрата находятся в диапазоне, определенном нормативной документацией на препарат - от 128 мг/флакон до 192 мг/флакон маннитола и от 32 мг/флакон до 48 мг/флакон трегалозы дигидрата.
Пример 6. Определение количественного содержания сорбитола и лактозы в препарате вакцины для профилактики желтой лихорадки живой аттенуированной заявленным способом.
Образец восстановили в 5 мл воды (в соответствии с инструкцией производителя). Полученный раствор развели водой до содержания стабилизаторов углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из сорбитола и лактозы разводят водой согласно примеру 1.
Для определения правильности готовят дополнительные растворы: раствор сорбитола и лактозы с концентрацией 1 мг/мл в воде и испытуемый раствор с добавкой раствора сорбитола и лактозы.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал сорбитола и лактозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала для испытуемого раствора и испытуемого раствора с добавкой пересчитывают по калибровочному графику с учетом степени разведения. По разности содержания стабилизатора углеводной природы в испытуемом растворе с добавкой и испытуемом растворе без добавки находят количество введенного в препарат стабилизатора. Сравнивают количество введенного стабилизатора (известного количества вещества, которое добавлено) и найденного
Вывод. Полученные значения: 4,4 мг/мл сорбитола и 8,8 мг/мл лактозы. Правильность полученных данных подтверждена методом стандартных добавок и составила 104,4 % и 99,8 % соответственно (табл. 1).
Пример 7. Определение количественного содержания сахарозы в препарате вакцины для профилактики ротавирусной инфекции заявленным способом.
Образец восстановили в 2,5 мл воды (в соответствии с инструкцией производителя). Полученный раствор развели водой до содержания стабилизатора углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из сахарозы разводят водой согласно примеру 1.
Для определения правильности готовят дополнительные растворы: раствор сахарозы с концентрацией 1 мг/мл в воде и испытуемый раствор с добавкой раствора сахарозы.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал сахарозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала для испытуемого раствора и испытуемого раствора с добавкой пересчитывают по калибровочному графику с учетом степени разведения. По разности содержания стабилизатора углеводной природы в испытуемом растворе с добавкой и испытуемом растворе без добавки находят количество введенного в препарат стабилизатора. Сравнивают количество введенного стабилизатора (известного количества вещества, которое добавлено) и найденного
Вывод. Полученное значение: 21,1 мг/мл сахарозы. Правильность полученных данных подтверждена методом стандартной добавки и составила 101,4 % (табл. 1).
Пример 8. Определение количественного содержания сорбитола, маннитола и лактозы в препарате вакцины для профилактики кори, краснухи и паротита заявленным способом.
Препарат развели водой до содержания стабилизаторов углеводной природы, соответствующей диапазону от 10 мкг/мл до 50 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из сорбитола, маннитола и лактозы разводят водой согласно примеру 1.
Для определения правильности готовят дополнительные растворы: раствор сорбитола, маннитола и лактозы с концентрацией 1 мг/мл в воде и испытуемый раствор с добавкой раствора сорбитола, маннитола и лактозы.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал сорбитола, маннитола, лактозы при условиях раскрытых в примере 2.
Полученное значение аналитического сигнала для испытуемого раствора и испытуемого раствора с добавкой пересчитывают по калибровочному графику с учетом степени разведения. По разности содержания стабилизатора углеводной природы в испытуемом растворе с добавкой и испытуемом растворе без добавки находят количество введенного в препарат стабилизатора. Сравнивают количество введенного стабилизатора (известного количества вещества, которое добавлено) и найденного
Вывод. Полученные значения: 17,0 мг/мл сорбитола, 17,7 мг/мл маннитола и 70,1 мг/мл лактозы. Правильность полученных данных подтверждена методом стандартной добавки и составила 104,4 %, 101,2 % и 96,6 % соответственно (табл. 1).
Пример 9. Определение количественного содержания лактозы в препарате вакцины против дифтерии, Haemophilus Influenzae B, коклюша, полиомиелита, столбняка, гепатита B заявленным способом.
Образец восстановили в 0,5 мл воды (в соответствии с инструкцией производителя). Полученный раствор развели водой до содержания стабилизатора углеводной природы, соответствующей диапазону от 25 мкг/мл до 120 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из лактозы разводят водой согласно примеру 1.
Для определения правильности готовят дополнительные растворы: раствор лактозы с концентрацией 1 мг/мл в воде и испытуемый раствор с добавкой раствора лактозы.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал лактозы при условиях раскрытых в примере 2, и где объем нанесения на колонку 10 мкл.
Полученное значение аналитического сигнала для испытуемого раствора и испытуемого раствора с добавкой пересчитывают по калибровочному графику с учетом степени разведения. По разности содержания стабилизатора углеводной природы в испытуемом растворе с добавкой и испытуемом растворе без добавки находят количество введенного в препарат стабилизатора. Сравнивают количество введенного стабилизатора (известного количества вещества, которое добавлено) и найденного
Вывод. Полученные значения: 31,0 мг/мл лактозы. Правильность полученных данных подтверждена методом стандартной добавки и составила 96,6 % (табл. 1).
Пример 10. Определение количественного содержания мальтозы в препарате иммуноглобулина человека нормальный (производитель 3) заявленным способом.
Лекарственный препарат развели водой до содержания стабилизатора углеводной природы, соответствующей диапазону 25 мкг/мл до 120 мкг/мл.
Исходный стандартный раствор с концентрацией 1,0 мг/мл углеводных стабилизаторов выбранных из мальтозы разводят водой согласно примеру 1.
Анализируют растворы на ионном хроматографе, фиксируя аналитический сигнал мальтозы при условиях раскрытых в примере 2, и где объем нанесения на колонку 10 мкл.
Полученное значение аналитического сигнала пересчитывают по калибровочному графику с учетом степени разведения.
Вывод. Полученное значение 100,6 мг/мл мальтозы находится в диапазоне, определенном нормативной документацией на препарат - от 90 мг/мл до 110 мг/мл мальтозы.
Пример 11. Робастность заявленного способа оценивали при изменении состава подвижной фазы, графика изменения потенциалов (фиг. 16), скорости потока подвижной фазы и температуры колонки.
Выводы. Заявленный способ обладает робастностью в исследованных условиях (таблица 2).
6652 - маннитол
6105 - трегалоза
6858 - глюкоза
5381 - лактоза
5540 - сахароза
5264 - мальтоза
1,2 - маннитол
1,1 - трегалоза
1,0 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,7 - маннитол
6,3 - трегалоза
10,8 - глюкоза
4,4 - лактоза
11, 3- сахароза
0,2 - маннитол
1,5 - трегалоза
0,3 - глюкоза
0,7 - лактоза
1,3 - сахароза
1,6 - мальтоза
0,5 - маннитол
0,6 - трегалоза
0,6 - глюкоза
0,9 - лактоза
1,1 - сахароза
1,1 - мальтоза
6041 - маннитол
5515 - трегалоза
6249- глюкоза
4917 - лактоза
4973 - сахароза
4709 - мальтоза
1,2 - маннитол
1,1 - трегалоза
1,1 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,6 - маннитол
6,1 - трегалоза
10,3 - глюкоза
4,2 - лактоза
10,7 - сахароза
1,0 - маннитол
1,1 - трегалоза
1,7 - глюкоза
0,8 - лактоза
1,3 - сахароза
1,0 - мальтоза
0,09- маннитол
0,08 - трегалоза
0,06 - глюкоза
0,05 - лактоза
0,05 - сахароза
0,08 - мальтоза
4453 - маннитол
4057 - трегалоза
4475 - глюкоза
3794 - лактоза
3798 - сахароза
3616 - мальтоза
1,0 - маннитол
1,0 - трегалоза
1,0 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,2 - маннитол
5,1 - трегалоза
8,8 - глюкоза
3,6 - лактоза
9,4 - сахароза
0,03 - маннитол
0,4 - трегалоза
0,4 - глюкоза
0,1 - лактоза
0,3 - сахароза
0,4 - мальтоза
0,08 - маннитол
0,04 - трегалоза
0,07 - глюкоза
0,04 - лактоза
0,04 - сахароза
0,04 - мальтоза
6935 - маннитол
5057 - трегалоза
5548 - глюкоза
4507- лактоза
4450 - сахароза
4119 - мальтоза
1,1 - маннитол
1,0 - трегалоза
1,0 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
3,1 - маннитол
5,1 - трегалоза
10,1 - глюкоза
4,7 - лактоза
9,7 - сахароза
1,1 - маннитол
1,7 - трегалоза
1,2 - глюкоза
1,3 - лактоза
1,6 - сахароза
1,4 - мальтоза
0,24 - маннитол
0,24 - трегалоза
0,17 - глюкоза
0,07 - лактоза
0,05 - сахароза
0,06 - мальтоза
6041 - маннитол
5515- трегалоза
6249 - глюкоза
4917 - лактоза
4973 - сахароза
4709 - мальтоза
1,2 - маннитол
1,1 - трегалоза
1,1 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,6 - маннитол
6,1 - трегалоза
10,3 - глюкоза
4,2 - лактоза
10,7 - сахароза
1,0 - маннитол
1,1 - трегалоза
1,7 - глюкоза
0,8 - лактоза
1,3 - сахароза
1,0 - мальтоза
0,09 - маннитол
0,08 - трегалоза
0,06 - глюкоза
0,05 - лактоза
0,05 - сахароза
0,08 - мальтоза
6343 - маннитол
5820 - трегалоза
6545 - глюкоза
5299 - лактоза
5306 - сахароза
5037 - мальтоза
1,1 - маннитол
1,1 - трегалоза
1,1 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,1 - маннитол
6,8 - трегалоза
10,2 - глюкоза
3,4 - лактоза
11,4 - сахароза
0,8 - маннитол
0,2 - трегалоза
0,3 - глюкоза
0,3 - лактоза
0,4 - сахароза
0,5 - мальтоза
0,04 - маннитол
0,08 - трегалоза
0,04 - глюкоза
0,01 - лактоза
0,01 - сахароза
0,05 - мальтоза
6772 - маннитол
5144 - трегалоза
5874 - глюкоза
4818 - лактоза
4559 - сахароза
4426 - мальтоза
1,2 - маннитол
1,0 - трегалоза
1,1 - глюкоза
1,1 - лактоза
1,0 - сахароза
1,0 - мальтоза
2,0 - маннитол
6,1 - трегалоза
8,6 - глюкоза
3,4 - лактоза
10,3 - сахароза
1,9 - маннитол
1,0 - трегалоза
0,6 - глюкоза
0,7 - лактоза
1,3 - сахароза
0,8 - мальтоза
0,2 - маннитол
0,3 - трегалоза
0,2 - глюкоза
0,1 - лактоза
0,1 - сахароза
0,2 - мальтоза
6987 - маннитол
6317 - трегалоза
7162 - глюкоза
5728 - лактоза
5750 - сахароза
5410 - мальтоза
1,2 - маннитол
1,1 - трегалоза
1,1 - глюкоза
1,0 - лактоза
1,0 - сахароза
1,0 - мальтоза
1,7 - маннитол
7,4 - трегалоза
10,0 - глюкоза
2,8 - лактоза
11,7 - сахароза
0,3 - маннитол
0,6 - трегалоза
0,1 - глюкоза
0,4 - лактоза
0,1 - сахароза
1,2 - мальтоза
0,01 - маннитол
0,14 - трегалоза
0,02 - глюкоза
0,01- лактоза
0,02- сахароза
0,04 - мальтоза
Пример 12. Повторяемость и правильность заявленного способа оценивали на трех уровнях концентраций, охватывающих диапазон применения 10 - 50 мкг/мл: 10, 25 и 50 мкг/мл, и 25 - 120 мкг/мл: 25, 60, 120 мкг/мл с использованием 6 модельных растворов для каждого уровня, приготовленных не зависимо друг от друга. Прецизионность заявленного способа оценивали на двух уровнях концентраций для каждого диапазона: 10 и 50 мкг/мл и 25 и 120 мкг/мл с использованием 6 модельных растворов для каждого уровня, приготовленных не зависимо друг от друга [23, 24].
Выводы. Заявленный способ обладает прецизионностью и правильностью, соответствующей требованиям Государственной фармакопеи Российской Федерации (таблица 4, 5).
Таблица 3. Валидационные параметры методики определения стабилизаторов углеводной природы в БЛП методом ионообменной ВЭЖХ с импульсным амперометрическим детектированием (объем вкола 25 мкл).
0,9998 - маннитол
0,9994 - трегалоза
0,9999 - глюкоза
1,0000 - лактоза
0,9996 - сахароза
1,0000 - мальтоза
10 мкг/мл
1,1 % - маннитол
0,9 % - трегалоза
0,9 % - глюкоза
1,6 % - лактоза
1,1 % - сахароза
1,0 % - мальтоза
25 мкг/мл
0,7 % - маннитол
1,0 % - трегалоза
0,4 % - глюкоза
0,5 % - лактоза
0,6 % - сахароза
0,7 % - мальтоза
50 мкг/мл
0,6 % - маннитол
0,8 % - трегалоза
0,4 % - глюкоза
0,3 % - лактоза
0,2 % - сахароза
0,4 % - мальтоза
(n = 12)
0,0 % - маннитол
0,4 % - трегалоза
0,7 % - глюкоза
1,4 % - лактоза
0,0 % - сахароза
1,2 % - мальтоза
(n = 12)
1,5 % - маннитол
1,1 % - трегалоза
0,8 % - глюкоза
0,0 % - лактоза
0,6 % - сахароза
0,5 % - мальтоза
(n = 6)
102,8 % - маннитол
102,3 % - трегалоза
100,7 % - глюкоза
100,1 % - лактоза
101,6 % - сахароза
100,7 % - мальтоза
(n = 6)
99,6 % - маннитол
101,2 % - трегалоза
100,1 % - глюкоза
99,4 % - лактоза
101,2 % - сахароза
99,4 % - мальтоза
(n = 6)
98,0 % - маннитол
97,9 % - трегалоза
97,9 % - глюкоза
98,2 % - лактоза
97,9 % - сахароза
98,3 % - мальтоза
Таблица 4. Валидационные параметры методики определения стабилизаторов углеводной природы в БЛП методом ионообменной ВЭЖХ с импульсным амперометрическим детектированием (объем вкола 10 мкл)
0,9995 - маннитол
0,9995 - трегалоза
0,9998 - глюкоза
1,0000 - лактоза
0,9996 - сахароза
1,0000 - мальтоза
25 мкг/мл
0,4 % - маннитол
0,5 % - трегалоза
0,8 % - глюкоза
0,5 % - лактоза
0,5 % - сахароза
0,4 % - мальтоза
60 мкг/мл
1,0 % - маннитол
1,2 % - трегалоза
0,5 % - глюкоза
1,2 % - лактоза
0,9 % - сахароза
1,1 % - мальтоза
120 мкг/мл
1,5 % - маннитол
1,7 % - трегалоза
1,2 % - глюкоза
1,9 % - лактоза
1,3 % - сахароза
2,0 % - мальтоза
(n = 12)
3,0 % - маннитол
2,1 % - трегалоза
2,9 % - глюкоза
3,6 % - лактоза
1,8 % - сахароза
3,1 % - мальтоза
(n = 12)
1,9 % - маннитол
2,3 % - трегалоза
2,1 % - глюкоза
2,6 % - лактоза
1,9 % - сахароза
2,8 % - мальтоза
(n = 6)
100,4 % - маннитол
98,7 % - трегалоза
98,1 % - глюкоза
97,8 % - лактоза
98,9 % - сахароза
97,7 % - мальтоза
(n = 6)
99,8 % - маннитол
98,8 % - трегалоза
97,2 % - глюкоза
96,7 % - лактоза
98,0 % - сахароза
96,2 % - мальтоза
(n = 6)
97,5 % - маннитол
95,3 % - трегалоза
95,4 % - глюкоза
96,5 % - лактоза
95,0 % - сахароза
95,4 % - мальтоза
Вышеизложенное техническое решение раскрыто довольно подробно, с целью ясности понимания, и не должно рассматриваться, как ограничивающее объем притязаний.
Представленные примеры не ограничивают объем притязаний настоящего изобретения и служат только для цели иллюстрации и раскрытия заявленного способа.
Промышленная применимость
Все представленные примеры подтверждают возможность применения заявленного способа в фармации, биотехнологии, биологии, химии, медицине, а также в производственной биотехнологии, фармацевтической промышленности и смежных отраслях. Таким образом, поставленная техническая задача, а именно, разработка способа определения стабилизаторов углеводной природы в биологически активных препаратах выполнена.
Список литературы:
1. Государственная фармакопея РФ, Биологические лекарственные препараты ОФС.1.7.1.0010.18 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/v14/vol2/807/(дата обращения 12.07.2023).
2. ГОСТ Р 54667-2011. Молоко и продукты переработки молока. Методы определения массовой доли сахаров.
3. Молочная промышленность. «Методы определения массовой доли сахарозы в сладких молочных продуктах», 2020 г.
4. Государственная фармакопея РФ, Определение сахаров спектрофотометрическим методом ОФС.1.2.3.0019.15 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/v14/vol1/1087/ (дата обращения 12.07.2023).
5. Ralph Daniels, Clyde C. Rush, Ludwig Bauer. The Fehling and Benedict tests. J. Chem. Educ. 1960, 37(4), 205
6. Juan Sanchez. Colorimetric Assay of Alditols in Complex Biological Samples J. Agric. Food Chem. 1998, 46, 157-160.
7. Государственная фармакопея РФ, Определение маннита (маннитола) в биологических лекарственных препаратах ОФС 1.7.2.0037.18 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/v14/vol2/1247/ (дата обращения 12.07.2023).
8. Jane Blood, A R Ingle, N Allison, G R Davies P G Hill. Rapid enzymatic method for the measurement of mannitol in urine. Ann Clin Biochem. 1991, 28, 401-406.
9. [Электронный ресурс] URL: https://www.brenda -enzymes.org/enzyme.php?ecno=1.1.1.118 (дата обращения 12.07.2023).
10. Beligh Mechri, Meriem Tekaya, Hechmi Cheheb, Mohamed Hammami. Determination of Mannitol Sorbitol and Myo-Inositol in Olive Tree Roots and Rhizospheric Soil by Gas Chromatography and Effect of Severe Drought Conditions on Their Profiles. Journal of Chromatographic Science, 2015, Vol. 53, No. 10, 1631-1638.
11. Veena Nagaraj, Neelam Upadhyay, Battula Surendra Nath, Ashish Kumar Sinh. Advances in Fractionation and Analysis of Milk Carbohydrates. Technological Approaches for Novel Applications in Dairy Processing.
12. Ting Fang, Yaming Cai, Qiurui yang, Collins O Ogutu, Liao Liao, Yuepeng Han. Analysis of sorbitol content variation in wild and cultivated apples. Journal of the Science of Food and Agriculture. DOI 10.1002/jsfa.10005.
13. Rendleman JA Jr. In, Isbell HS. Carbohydrates in solution. Advances in Chemistry Ser ACS, Washington, 1973;117;51-68.
14. Wibo B.van Scheppingen, Piet H. van Hilten, Marieke P. Vijverberg, Alexander L.L. Duchateau. Selective and sensitive determination of lactose in low-lactose dairy products with HPAEC-PAD. Journal of Chromatography B. Volume 1060, 15 August 2017, Pages 395-399.
15. Sensitive Analysis of the Lactose Content of Lactose-Free Labelled Products Using HPAEC-PAD. The Application Notebook-12-01-2015, Volume 0, Issue 0. https://www.chromatographyonline.com/
16. Christiane Grunewald, Stefan Bohnert, Stefan Jacob. The Determination of Carbohydrates by High-Performance Anion-Exchange Chromatography Coupled with Pulsed Amperometric Detection (HPAEC-PAD). Methods in Molecular biology 2356/ Springer protocols.
17. Rikke Andersen, Annemarine Sorensen. Separation and determination of alditols and sugars by high-pH anion exchange chromatograph with pulsed amperometric detection. Journal of Chromatography A. Volume 897, Issue 1-2, 2000, pages 195- 204.
18. John E Hallsworth, Naresh Magan. A rapid HPLC protocol for detection of polyols and trehalose. Journal of Microbiological methods. Volume 29, Issue 1, 1997, pages 7-13.
19. Межгосударственный стандарт ГОСТ 8.315-2019 "Государственная система обеспечения единства измерений. Стандартные образцы состава и свойств веществ и материалов. Основные положения" (введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 30 октября 2019 г. N 1059 -ст). [Электронный ресурс] URL: https://dokipedia.ru/document/5347908 (дата обращения 12.07.2023).
20. Государственная фармакопея РФ, Биолонические лекарственые препараты ОФС.1.7.1.0010.18 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/v14/vol2/807/ (дата обращения 12.07.2023).
21. Dionex ICS-5000/ICS-5000+, руководство по эксплуатации.
22. Государственная фармакопея РФ, Хроматография ОФС.1.2.1.2.0001.15 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/v14/vol1/863/ (дата обращения 12.07.2023)
23. Государственная фармакопея РФ, Статистическая обработка результатов химического эксперимента ОФС.1.1.0013.15 [Электронный ресурс] URL: https: // docs.rucml.ru / feml / pharma / v14 / vol1 / 289 /(дата обращения 12.07.2023).
24. Государственная фармакопея РФ, Валидация аналитических методик ОФС.1.1.0012.15 [Электронный ресурс] URL: https://docs.rucml.ru/feml/pharma/ v14 /vol1/275/ (дата обращения 12.07.2023).
| название | год | авторы | номер документа |
|---|---|---|---|
| Определение полисорбата 80 в биологических лекарственных препаратах | 2023 |
|
RU2812788C1 |
| Способ определения арбутина в листьях толокнянки | 2023 |
|
RU2802173C1 |
| Способ количественного определения ионов алюминия атомно-абсорбционной спектрометрией с электротермической атомизацией | 2022 |
|
RU2799235C1 |
| Способ определения алкалоидов в экстракте термопсиса | 2022 |
|
RU2796599C1 |
| Способ количественного определения аскорбиновой кислоты в лекарственных растительных препаратах | 2023 |
|
RU2801885C1 |
| Способ определения летучих компонентов в лекарственных препаратах | 2022 |
|
RU2790000C1 |
| Способ количественного определения анти-D-антител IgG в препаратах иммуноглобулина человека антирезус Rh0(D) | 2021 |
|
RU2777845C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| Способ определения лозартана, его основного метаболита лозартан карбоновой кислоты и глибенкламида в сыворотке крови и моче человека | 2020 |
|
RU2749567C1 |
| Способ количественного определения фтивазида | 2024 |
|
RU2828350C1 |


Изобретение относится к области аналитической химии. Раскрыт способ определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографией с импульсным амперометрическим детектированием, включающий разведение содержащих по меньшей мере один углеводный стабилизатор в биологически активном лекарственном препарате и исходном стандартном растворе с концентрацией 1,0 мг/л до концентрации углеводных стабилизаторов от 10 до 50 мкг/мл и от 1 до 50 мкг/мл соответственно, либо от 25 до 120 мкг/мл и от 2 до 120 мкг/мл соответственно, хроматографируют полученные растворы в изократическом режиме элюирования, где подвижная фаза в виде гидроксида натрия или гидроксида калия с концентрацией 160 ммоль/л, а затем проводят расчеты. Изобретение обеспечивает разработку способа определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографией (ионообменная ВЭЖХ) с импульсным амперометрическим детектированием. 6 з.п. ф-лы, 16 ил., 4 табл., 12 пр.
1. Способ определения стабилизаторов углеводной природы в биологически активных лекарственных препаратах ионообменной высокоэффективной жидкостной хроматографией с импульсным амперометрическим детектированием, включающий разведение содержащих по меньшей мере один углеводный стабилизатор в биологически активном лекарственном препарате и исходном стандартном растворе с концентрацией 1,0 мг/л до концентрации углеводных стабилизаторов от 10 до 50 мкг/мл и от 1 до 50 мкг/мл соответственно, либо от 25 до 120 мкг/мл и от 2 до 120 мкг/мл соответственно, хроматографируют полученные растворы в изократическом режиме элюирования, где подвижная фаза в виде гидроксида натрия или гидроксида калия с концентрацией 160 ммоль/л, а затем проводят расчеты.
2. Способ по п. 1, дополнительно характеризующийся тем, что биологически активными лекарственными препаратами могут быть вакцины, или аллергены, или препараты, содержащие плазму крови, или препараты фактора крови VIII, или иммуноглобулины человека нормального
3. Способ по п. 1 или 2, дополнительно характеризующийся тем, что стабилизаторы углеводной природы выбраны по меньшей мере из мальтозы, сахарозы, маннитола, трегалозы, сорбитола, лактозы, глюкозы.
4. Способ по п. 1, дополнительно характеризующийся тем, что для детектирования используют импульсный амперометрический детектор с золотым рабочим электродом
5. Способ по п. 1, дополнительно характеризующийся тем, что хроматографируют при скорости потока 1,0 мл/мин.
6. Способ по п. 1, дополнительно характеризующийся тем, что для хроматографирования используют неподвижную фазу в виде колонки размером 250×4 мм.
7. Способ по п. 6, дополнительно характеризующийся тем, что на колонку наносят последовательно воду, стандартные растворы, раствор биологически активного лекарственного препарата.
| BAZHENOVA A | |||
| et al | |||
| Glycoconjugate vaccines against Salmonella enterica serovars and Shigella species: existing and emerging methods for their analysis // Biophysical Reviews, 2021, V | |||
| Насос | 1917 |
|
SU13A1 |
| Способ изготовления замочных ключей с отверстием для замочного шпенька из одной болванки с помощью штамповки и протяжки | 1922 |
|
SU221A1 |
| СИСТЕМА ПОДАЧИ БЕЗ ЕСТЕСТВЕННОГО БАЛАНСА ДЛЯ УСТРОЙСТВА ИНЖЕКЦИОННОГО ФОРМОВАНИЯ | 2012 |
|
RU2565176C2 |
| CN 103323562 B, 05.08.2015 | |||
| СПОСОБ ОПРЕДЕЛЕНИЯ IN VITRO ГЛИКЕМИЧЕСКОГО ИНДЕКСА ПИЩЕВЫХ ПРОДУКТОВ | 2008 |
|
RU2451938C2 |
| MARK R | |||
| et al | |||
| Monosaccharide Analysis of Glycoconjugates by | |||

