Настоящее изобретение относится к способу получения C-H-кислотных (мет)акрилатов и к вариантам их применения.
В предшествующем уровне техники описан способ получения производных метакрилатных соединений на основе ацетоацетамидов и ацетоацетатов.
Синтез C-H-кислотных мономеров на основе производных цианоуксусной кислоты не является возможным в соответствии со способом, описанным в документе EP 0013147, хотя он включен в описание формулы изобретения. В документе EP 0013417 различные аминоспирты или диамины и их метариклатные производные вводят в реакцию с дикетеном и, таким образом, получают соответствующие производные ацетоуксусной кислоты. Они также представляют собой C-H-кислотные соединения. Однако дикетен не подходит в качестве исходного материала для синтеза цианоуксусной кислоты и ее производных карбоновой кислоты (сложных эфиров, амидов и т.д.) уже по той причине, что углеродный скелет представляет собой C4-структуру, а цианоуксусная кислота представляет C3-структуру. Для синтеза несимметрично замещенных диаминов в документе EP 0013147 также задействуют химию защитных групп для временного блокирования амина с целью защиты его от реакции. Это является неэкономичным и из-за дополнительных стадий реакции является дорогостоящим с точки зрения технологии способа.
Производные карбоновой кислоты ацетоуксусной кислоты легко образуют окрашенные комплексы, которые являются неподходящими для широкого разнообразия применений покровного лака.
Таким образом, целью являлось обеспечение способа получения C-H-кислотных (мет)акрилатов на основе цианоуксусной кислоты. В частности, целью было получение C-H-кислотного мономера без 1,3-дикетоновой структуры, как в ацетоуксусной кислоте.
Дополнительная цель состояла в обеспечении устойчивых в отношении гидролиза (мет)акрилатов, чтобы обеспечить особенно надлежащую стабильность при хранении в продукте.
Цели достигались с помощью способа получения C-H-кислотных (мет)акрилатов путем осуществления реакции диамина (B) формулы R2 CnHmOxNy,
где
n = 2-15,
m = 4-30,
x = 0-4, и
y = 0-4,
со сложным эфиром цианоуксусной кислоты (A) для получения промежуточного продукта (IP). В данном случае отдается особое предпочтение применению метиловых и этиловых сложных эфиров (Z = CH3, C2H5) цианоуксусной кислоты, поскольку они являются общедоступными на рынке, однако высшие сложные эфиры также являются вполне подходящими для реакции. В качестве побочного продукта в данной реакции (как будет предполагаться специалистами в данной области) обе аминогруппы диамина вступают в реакцию с цианоуксусной кислотой с получением бис-цианоацетамида (BP). Данная реакция является нежелательной, поскольку для отделения BP будут требоваться дополнительные стадии, что имеет отрицательное влияние на экономическую целесообразность и стоимость способа.
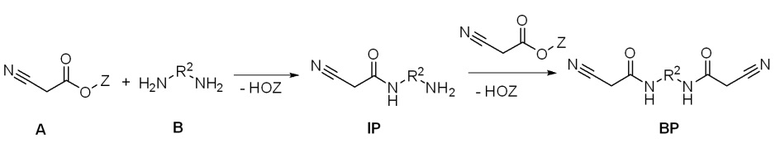
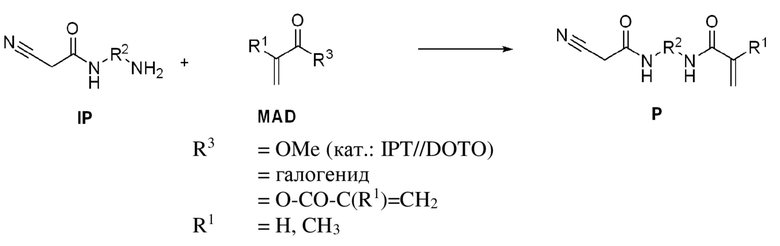
Проще работать с избытком диамина, который также характеризоваться более низкой температурой кипения, чем IP, и может, таким образом, удаляться легче, чем BP, который является таким же термолабильным, как и IP. Неожиданно образование BP очень сильно подавляется даже небольшим избытком B, например, избытком 1,001:1 и избытком 10:1, особенно предпочтительно 4:1 B, неочищенный продукт с >99% IP получают после удаления диамина B. Отделение B можно в данном случае проводить посредством экстракции или кристаллизации и, в частности, дистилляции, предпочтительно при пониженном давлении. Затем IP, полученный после удаления B, можно вводить в реакцию в виде неочищенного продукта с (мет)акрилатным производным (MAD) без дальнейшего выделения. В данном случае реакцию можно продолжать с галогенангидридами (c2) (предпочтительно с применением оснований для поглощения образующихся галогеноводородов), а также путем проведения реакции со сложными эфирами (мет)акриловой кислоты (c1). При этом смесь оксида диоктилолова (DOTO) и изопропилтитаната (IPT), как было доказано, является особенно подходящей в качестве катализатора. Однако реакция BP с кислотными ангидридами (мет)акриловой кислоты (c2), как было показано, является особенно эффективной.
Неожиданно было обнаружено, что с помощью способа в соответствии с настоящим изобретением достигаются высокие значения степени превращения, а количество побочных продуктов значительно снижается.
Выражение «(мет)акрилат» в данном документе означает как метакрилат, например, метилметакрилат, этилметакрилат и т.д., так и акрилат, например, метилакрилат, этилакрилат и т.д., а также смеси обоих из них.
Диамины
Подходящие диамины выбраны из группы алифатических, линейных, или разветвленных, или циклических замещенных и незамещенных диаминов и ароматических (орто-, мета- или пара-) замещенных диаминов.
Особое предпочтение отдают диаминам, выбранным из группы, состоящей из этилендиамина, 1,3-диаминопропана, 1,4-диаминобутана, 1,5-диаминопентана, 1,6-гександиамина, 1,7-диаминогептана, 1,8-диаминооктана, 1,10-диаминодекана, 1,2-диаминопропана, выделенных цис- и транс-изомеров 1,2-диаминоциклогексана и их смесей, выделенных цис- и транс-изомеров 1,3-диаминоциклогексана и их смесей, выделенных цис- и транс-изомеров 1,4-диаминоциклогексана и их смесей, 1,3-диамино-2-гидроксипропана, 2,2-диметил-1,3-пропандиамина, выделенных цис- и транс-изомеров изофорондиамина и их смесей, выделенных цис- и транс-изомеров 1,3-циклогексанбис(метиламина) и их смесей, 4,4'-метиленбис(циклогексиламина), выделенных цис- и транс-изомеров 4,4'-метиленбис(2-метилциклогексиламина) и их смесей.
Соотношение ангидрида (мет)акриловой кислоты или галогенида (мет)акриловой кислоты и промежуточного продукта в (c2) составляет от 0,2:1 до 5:1.
Особое предпочтение отдают проведению реакции при соотношении 0,5:1, поскольку в этом случае 1 экв. промежуточного продукта амина нейтрализует кислоту, образованную из ангидрида или галогенангидрида.
Особое предпочтение отдают соотношению 1:1, поскольку в этом случае большие порции амина, до сих пор характеризующегося дорогостоящим получением, дополнительно вступают в реакцию с получением продукта, несмотря на более раннюю реакцию с карбоновой кислотой ангидрида.
Аналогично, может быть предпочтительно работать с избытком ангидрида, составляющим >1:1, поскольку это обеспечивает более быстрое достижение полного превращения.
(Мет)акриловый ангидрид
Промежуточный продукт (IP) на стадии c2) вводят в реакцию с (мет)акриловыми ангидридами, выбранными из группы, состоящей из метакрилового ангидрида и акрилового ангидрида.
Условия реакции
Реакцию проводят при значениях температуры от 0°C до 120°C, предпочтительно от 10°C до 40°C, во время дозирования и при значениях температуры, составляющих не более 100°C, во время фазы после реакции и при подготовке к выделению. На стадии c1 - в диапазоне от 60 до 140°C, предпочтительно от 100 до 120°C. На стадии c2 - от 0 до 40°C, после реакции возможно не более 100°C.
Время реакции составляет от 15 мин до 10 ч.
Чтобы избежать образования нежелательных побочных продуктов, температуру поддерживают насколько возможно низкой и на стадии a применяют избыток амина.
Нейтрализация и выделение
Промежуточный продукт (IP) можно применять на следующей стадии без выделения.
Предпочтительные варианты способа
Промежуточный продукт (IP) со стадии b поглощают растворителем в горячем состоянии, поскольку в другом случае он будет затвердевать с образованием стекловидного вещества, а также за счет этого он вступает в реакцию со значительно большей легкостью. Подходящие растворители представляют собой H2O, MTBE, THF, ацетонитрил, диоксан, MAD и спирты. Выбор является очевидными для специалиста в данной области техники, исходя из соответствующей цели реакции.
На стадии c2 применяют растворитель, выбранный из группы, состоящей из H2O, MTBE, THF, ацетонитрила, диоксана, MAD и спиртов.
Экстракция или кристаллизация
Продукт (P) можно применять на следующей стадии без выделения. При необходимости из него также можно удалять низкокипящие соединения при пониженном давлении, его можно подвергать перекристаллизации путем добавления полярного растворителя или экстрагировать путем добавления несмешивающегося растворителя.
Обнаружили, что заявленные C-H-кислотные (мет)акрилаты являются устойчивыми в отношении гидролиза и, таким образом, являются устойчивыми при хранении в течение длительного периода времени.
C-H-кислотные (мет)акрилаты имеют множество областей применения. Предпочтение отдают применению в покрытиях и красках, в частности, в покровных лаках. Аналогично, их можно применять в качестве способного к полимеризации мономера для получения полимеров, которые могут сшиваться с кетонами, альдегидами, изоцианатами и активированными двойными связями при комнатной температуре.
Приведенные ниже примеры лучше иллюстрируют настоящее изобретение, но без ограничения настоящего изобретения признаками, раскрытыми в них.
ПРИМЕРЫ
Пример 1. Получение N-(2-этиламино)-2-цианоацетамида
Изначально загружали 600 г (10,0 моль) этилендиамина в четырехгорлую круглодонную колбу объемом 2 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл. В нее дозировали по каплям 248 г (2,5 моль) метилцианоацетата в течение 60 минут так, чтобы температура реакции не превышала 30°C. В течение данного времени четырехгорлую круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в розовый цвет, и затем - в сиреневый. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при комнатной температуре.
Затем избыток этилендиамина удаляли при пониженном давлении. Для данной цели реакционную смесь нагревали до 100°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 2 часа, при давлении, составляющем не более 5 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей 97,9% площади (определяли с применением GC-RV). Выход продукта составил 309 г (95%).
Сравнительный пример 1. Получение N,N’-этилен-бис-метакриламида
Изначально загружали 40% водный раствор этилендиамина (25,5 г, 0,17 моль) в четырехгорлую круглодонную колбу объемом 250 мл с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 100 мл. В нее дозировали 26 г (0,17 моль) метакрилового ангидрида в течение 60 минут так, чтобы температура реакции не превышала 30°C. В течение данного времени четырехгорлую круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метакрилового ангидрида образовывалось белое твердое вещество.
Белое твердое вещество отделяли посредством фильтрации и высушивали. Оно представляло собой N,N´-этилен-бис-метакриламид с чистотой 74,8% площади (определяли с применением GC-RV). Выход продукта составил 20 г (60%).
Пример 2. Получение N-(2-цианоэтиламидоэтил)метакриламида
Изначально загружали смесь 147 г (1,2 моль) N-(2-этиламино)-2-цианоацетамида и 600 г (6,0 моль) метилметакрилата в четырехгорлую круглодонную колбу объемом 1 л со впуском для воздуха, сабельной мешалкой, электродвигателем для мешалки и колонкой из зеркального материала длиной 50 см и толщиной 29 мм с неупорядоченной насадкой, заполненной кольцами Рашига 6×6. В нее добавляли 7 мг (10 ppm) 4-гидрокси-2,2,6,6-тетраметилпиперидинооксила и 0,15 г (200 ppm) гидрохинон-монометилового простого эфира с последующим добавлением 7,4 г смеси, содержащей 65,6 вес.% оксида диоктилолова и 34,4 вес.% тетраизопропилтитаната.
Реакционную смесь нагревали с обратным холодильником, при этом образованный метанол отгоняли в виде азеотропной смеси с помощью колонки с неупорядоченной насадкой. Через примерно 3,5 часа степень превращения, определенная с помощью GC, составляла 58%.
Пример 3. Получение N-(2-цианоэтиламидоэтил)метакриламида
Растворяли 312 г (2,4 моль) N-(2-этиламино)-2-цианоацетамида из примера 1 в 468 г воды в трехгорлой круглодонной колбе объемом 2 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл. В нее медленно добавляли по каплям 370 г (2,4 моль) метакрилового ангидрида, при этом образовывался светло-коричневый осадок. Затем реакционную смесь перемешивали дополнительно в течение 1,5 часа при 80°C.
Из полученной прозрачной темно-красной реакционной смеси удаляли низкокипящие вещества при пониженном давлении, ее концентрировали до 646 г и к ней добавляли 400 г изопропанола. Это приводило к образованию осадка, который отделяли посредством фильтрации.
Продукт получали в виде коричневого кристаллического твердого вещества с чистотой 77,0% площади (определяли с применением GC-RV). Выход продукта составил 346 г (73,9%).
Пример 4. Получение N-(2-бутиламино)-2-цианоацетамида
Плавили 353 г (4,0 моль) 1,4-диаминобутана при примерно 30°C в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 250 мл. В нее дозировали по каплям 99 г (1,0 моль) метилцианоацетата в течение 30 минут так, чтобы температура реакции оставалась на уровне от примерно 30°C до 40°C. В течение данного времени четырехгорлую круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в интенсивный желтый цвет. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при комнатной температуре, при этом реакционная смесь окрашивалась в красный цвет.
Затем удаляли избыток 1,4-диаминобутана при пониженном давлении. Для данной цели реакционную смесь нагревали до 100°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 2,5 часа, при давлении, составляющем не более 2 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей 89,1% площади (определяли с применением GC-RV). Выход продукта составил 146 г (84%).
Пример 5. Получение N-(2-цианоэтиламидобутил)метакриламида
Растворяли 360 г (0,93 моль) N-(2-бутиламино)-2-цианоацетамида из примера 4 в 540 г воды в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл, и охлаждали до 0°C на бане с ледяной водой. В нее медленно добавляли по каплям 143 г (0,93 моль) метакрилового ангидрида, растворенного в 300 мл метанола. Затем реакционную смесь перемешивали в течение ночи при комнатной температуре, при этом реакционная смесь окрашивалась в зеленый цвет.
Реакционную смесь концентрировали при пониженном давлении при 80°C и 35 мбар до 263 г. Остаток растворяли в 160 г изопропанола и полученный раствор хранили при комнатной температуре. Это приводило к образованию осадка, который отделяли посредством фильтрации.
Продукт получали в виде желтого кристаллического твердого вещества. Чистота представляла собой примерно 94,0% площади (определяли с применением GC-RV).
Пример 6. Получение N-(2-гексиламино)-2-цианоацетамида
Плавили 465 г (4,0 моль) 1,6-диаминогексана при примерно 41°C в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 250 мл. В нее дозировали по каплям 99 г (1,0 моль) метилцианоацетата в течение 30 минут так, чтобы температура реакции оставалась на уровне от примерно 50°C до 75°C. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в интенсивный желтый цвет. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при температуре от примерно 50°C до 75°C, при этом реакционная смесь окрашивалась в красный цвет.
Затем удаляли избыток 1,6-диаминогексана при пониженном давлении. Для данной цели реакционную смесь нагревали до 120°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 4 часа, при давлении, составляющем не более 2 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей примерно 100% площади (определяли с применением GC-RV). Выход продукта составил 172 г (94%).
Пример 7. Получение N-(2-цианоэтиламидобутил)метакриламида
Изначально загружали 31 г (0,2 моль) метакрилового ангидрида и 150 г воды в четырехгорлую круглодонную колбу объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл и охлаждали до 0°C на бане с ледяной водой.
Растворяли 360 г (0,93 моль) N-(2-гексиламино)-2-цианоацетамида из примера 6 в 3240 г метанола при 60°C и его охлаждали до комнатной температуры. Данный раствор добавляли в течение периода времени, составляющего 30 минут, с помощью капельной воронки к метакриловому ангидриду. Температуру реакции поддерживали на уровне ниже 20°C. Затем реакционную смесь перемешивали дополнительно в течение 3 часов при комнатной температуре.
Образованный продукт обнаруживали в реакционной смеси с применением GC-RV, и его можно выделять посредством кристаллизации из изопропанола.
Анализ
Газовая хроматография (GC)
Прибор: 7820A от Agilent Technologies
Колонка: DB5, 30 м, диаметр 0,250 мм, пленка 0,25 мкм
Температурный режим
Введение при 60°C, затем удерживание в течение 2 мин. Затем нагревание до 240°C при 20°C/мин и после достижения данной температуры удерживание в течение 240°C в течение 8 мин.
| название | год | авторы | номер документа |
|---|---|---|---|
| CH-КИСЛОТНЫЕ СЛОЖНЫЕ МЕТАКРИЛОВЫЕ ЭФИРЫ ДЛЯ ПОЛУЧЕНИЯ ВОДНЫХ ПОЛИМЕРНЫХ ДИСПЕРСИЙ | 2019 |
|
RU2777540C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЕТО-ФУНКЦИОНАЛИЗИРОВАННЫХ АРОМАТИЧЕСКИХ (МЕТ)АКРИЛАТОВ | 2019 |
|
RU2777539C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2515985C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛИРОВАННЫХ БЕНЗОФЕНОНОВ | 2009 |
|
RU2536471C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАКИЛЕНГЛИКОЛЬДИ(МЕТ)АКРИЛАТОВ | 2009 |
|
RU2522453C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ НОРБОРНИЛОВЫХ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ | 2018 |
|
RU2730856C1 |
| ПОЛУЧЕНИЕ СЛОЖНЫХ ДИЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ ИЗ ЭПОКСИДОВ | 2019 |
|
RU2797811C2 |
| ФОТОАКТИВИРУЕМАЯ КРОЮЩАЯ КОМПОЗИЦИЯ НА ВОДНОЙ ОСНОВЕ | 2001 |
|
RU2275403C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИМЕТИЛАМИНОАЛКИЛ(МЕТ)АКРИЛАТОВ | 2018 |
|
RU2749072C1 |
| МАКРОМОНОМЕРЫ, СОДЕРЖАЩИЕ ПОЛИИЗОБУТЕНОВЫЕ ГРУППЫ, И ИХ ГОМО- И СОПОЛИМЕРЫ | 2017 |
|
RU2745788C2 |
Изобретение относится к способу получения C-H-кислотных (мет)акрилатов формулы CH2=CR1-CO-NH-R2-NH-CO-CH2-CN (I), (P), где R1: H или метил, R2: CnHm, где n = 2-15 и m = 4-30. Способ характеризуется тем, что a) сложный эфир цианоуксусной кислоты (A) вводят в реакцию с избытком диамина (B), представляющего собой соединение формулы H2N-R2-NH2; b) непрореагировавший диамин (B) удаляют из реакционного раствора; c) промежуточный продукт (IP), полученный на стадии b), функционализированный аминогруппой цианоацетамид: c1) вводят в реакцию со сложными эфирами (мет)акриловой кислоты (MAD) или c2) вводят в реакцию с (мет)акриловым ангидридом (MAD) или (мет)акрилоилгалогенидом (MAD); d) продукт (P), полученный на стадии c), необязательно выделяют посредством экстракции или кристаллизации. Предлагаемый способ позволяет достичь высоких значений степени превращения, а также снизить количество побочных продуктов. 10 з.п. ф-лы, 7 пр.
1. Способ получения C-H-кислотных (мет)акрилатов формулы (I)
CH2=CR1-CO-NH-R2-NH-CO-CH2-CN (I),
(P),
где
R1: H или метил,
R2: CnHm,
где
n = 2-15 и
m = 4-30,
отличающийся тем, что
a) сложный эфир цианоуксусной кислоты (A) вводят в реакцию с избытком диамина (B), представляющего собой соединение формулы H2N-R2-NH2,
b) непрореагировавший диамин (B) удаляют из реакционного раствора,
c) промежуточный продукт (IP), полученный на стадии b), функционализированный аминогруппой цианоацетамид:
c1) вводят в реакцию со сложными эфирами (мет)акриловой кислоты (MAD) или
c2) вводят в реакцию с (мет)акриловым ангидридом (MAD) или (мет)акрилоилгалогенидом (MAD),
d) продукт (P), полученный на стадии c), необязательно выделяют посредством экстракции или кристаллизации.
2. Способ по п. 1, отличающийся тем, что сложные эфиры цианоуксусной кислоты выбраны из группы, состоящей из метилцианоацетата и этилцианоацетата.
3. Способ по п. 1, отличающийся тем, что диамины выбраны из группы алифатических, линейных, или разветвленных, или циклических замещенных и незамещенных диаминов и ароматических (орто-, мета- или пара-) замещенных диаминов.
4. Способ по п. 1, отличающийся тем, что избыток диаминов относительно сложных эфиров цианоуксусной кислоты составляет от 10:1 до 1,001:1.
5. Способ по п. 1, отличающийся тем, что непрореагировавший диамин на стадии b) удаляют посредством дистилляции, экстракции или кристаллизации.
6. Способ по п. 1, отличающийся тем, что реакцию промежуточного продукта на стадии c1) проводят со сложными эфирами, выбранными из группы, состоящей из метил(мет)акрилата, этил(мет)акрилата, бутил(мет)акрилата, этилакрилата, бутилакрилата, а также с высшими спиртами (мет)акрилата.
7. Способ по п. 1, отличающийся тем, что промежуточный продукт на стадии c2) вводят в реакцию с галогенидами (мет)акриловой кислоты, выбранными из группы, состоящей из (мет)акрилоилбромидов и (мет)акрилоилхлоридов.
8. Способ по п. 1, отличающийся тем, что промежуточный продукт на стадии c2) вводят в реакцию с (мет)акриловыми ангидридами, выбранными из группы метакрилового ангидрида и акрилового ангидрида.
9. Способ по п. 1, отличающийся тем, что реакция сложного эфира цианоуксусной кислоты с диамином проходит при значениях температуры от 0 до 120°C, предпочтительно от 0 до 30°C.
10. Способ по п. 1, отличающийся тем, что соотношение сложных эфиров (мет)акриловой кислоты и промежуточного продукта на стадии c1) составляет от 1,01:1 до 20:1.
11. Способ по п. 1, отличающийся тем, что соотношение ангидрида (мет)акриловой кислоты или галогенида (мет)акриловой кислоты и промежуточного продукта на стадии c2) составляет от 0,2:1 до 5:1.
| Масленка, действующая сжатым воздухом | 1926 |
|
SU13147A1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛОВ И ЭФИРОВ ДИКАРБОНОВЫХ КИСЛОТ | 0 |
|
SU234254A1 |

