Настоящее изобретение относится к соединениям общей формулы (I), представляющим собой метаболически устойчивые аналоги биоактивных липидных медиаторов, полученных из полиненасыщенных жирных кислот омега-3 (n-3 ПНЖК). Настоящее изобретение также относится к композициям, содержащим одно или более указанных соединений, и к применению указанных соединений или композиций для лечения или предотвращения сердечно-сосудистых заболеваний.
УРОВЕНЬ ТЕХНИКИ
Омега-6 и омега-3 полиненасыщенные жирные кислоты (n-6 и n-3 ПНЖК) являются важными компонентами рациона млекопитающих. Биологически наиболее важными n-3 ПНЖК являются эйкозапентаеновая кислота (ЕРА, 20:5 n-3) и докозагексаеновая кислота (DHA, 22:6 n-3). Поступающие с пищей n-3 ПНЖК оказывают влияние на различные физиологические процессы, влияющие на состояние здоровья и хронические заболевания, например, на регуляцию уровней липидов в плазме крови, сердечно-сосудистую и иммунную функции, воспаление, действие инсулина, нейрональное развитие и зрительную функцию.
При употреблении внутрь n-3 ПНЖК распределяются практически по всем клеткам тела и оказывают влияние на состав и функцию мембран, синтез эйкозаноидов, сигнализацию, а также на регуляцию экспрессии генов.
Эпидемиологические, клинические и экспериментальные исследования показали, что употребление рыбьего жира n-3 ПНЖК (ЕРА и DHA) защищает от сердечно-сосудистых заболеваний. n-3 ПНЖК снижают смертность от ишемической болезни сердца и частоту случаев внезапной сердечной смерти.
Вероятно, основным фактором, обеспечивающим предотвращение внезапной смерти от сердечной недостаточности при употреблении n-3 ПНЖК пациентами после инфаркта миокарда и пациентами с сердечной недостаточностью, является защита от желудочковой аритмии. Значительное антиаритмическое действие n-3 ПНЖК также наблюдалось в исследованиях фибрилляции предсердий человека. Потенциальное благоприятное влияние n-3 ПНЖК на сердце также выражается в профилактике и лечении застойной сердечной недостаточности и атеросклероза, а также в снижении общих факторов риска, таких как высокие уровни триглицеридов и провоспалительных цитокинов в плазме крови.
Кроме того, эпидемиологические и экспериментальные исследования показали, что с употреблением n-3 ПНЖК связано уменьшение риска макулярной дегенерации и более низкая частота возникновения рака толстой кишки, молочной железы, предстательной железы и других видов рака. Основным распространенным механизмом защиты от макулярной дегенерации и рака является способность n-3 ПНЖК ингибировать патологический ангиогенез. ЕРА и DHA ингибируют проницаемость сосудов и воспаление. Ангиогенез является важной стадией роста опухоли и метастазов, при этом n-6 ПНЖК и ее метаболиты способствуют ангиогенезу, а n-3 ПНЖК и ее метаболиты ингибируют ангиогенез.
Кроме того, одной из наиболее важных биологических функций ПНЖК является обеспечение исходных веществ для получения биоактивных метаболитов жирных кислот, способных модулировать многие функции. Например, арахидоновая кислота (АА, 20:4, n-6) метаболизируется ферментами цитохром Р450 (CYP) в несколько классов окисленных метаболитов, обладающих мощной биологической активностью. Основные метаболиты включают 20-гидроксиэйкозотетраеновую кислоту (20-НЕТЕ) и ряд регио- и стереоизомерных эпоксиэйкозатриеновых кислот (ЕЕТ). Изоформы CYP4A и CYP4F продуцируют 20ЕТ-НЕТЕ и CYP2C и CYP2J изоформы ЕЕТ.
Как известно, ЕРА (20:5, n-3) и DHA (22:6, n-3) могут служить альтернативными субстратами для АА-метаболизирующих изоформ CYP (Arnold С. et al., J Biol Chem. 2010 Oct 22; 285(43):32720-33.; Fischer R. et al., J Lipid Res. 2014 Mar 16; 55(6):1150-1164.). Члены подсемейства CYP2C и CYP2J, которые эпоксидируют АА до ЕЕТ, метаболизируют ЕРА до эпоксиейкозатетраеновых кислот (EEQ) и DHA до эпоксидокозапентаеновых кислот (EDP). Двойная связь ω-3, отличающая ЕРА и DHA от АА, является предпочтительным местом атаки большинства эпоксигеназ, что приводит к образованию 17, 18-EEQ и 19,20-EDP в качестве основных метаболитов. Изоформы CYP4A и CYP4F, гидроксилирующие АА до 20-НЕТЕ, метаболизируют ЕРА до 20-гидроксиэйкозапентаеновой кислоты (20-НЕРЕ) и DHA до 22-гидроксидокозагексаеновой кислоты (22-HDHA). CYP1A1, CYP2E1 и другие изоформы, превращающие АА преимущественно в 19-НЕТЕ, демонстрируют выраженную эпоксигеназную активность ω-3 с ЕРА и DHA. Варианты CYP1A1 человека приводят к дифференцированным схемам метаболизма эйкозапентаеновой кислоты. Метаболиты эйкозапентаеновой кислоты, зависящие от цитохрома Р450, являются новыми активаторами ВК-канала. Примечательной особенностью CYP-зависимого метаболизма n-3 ПНЖК является предпочтительное эпоксидирование двойной связи n-3, что отличает ЕРА и DHA от АА. Полученные метаболиты - 17,18-EEQ из ЕРА и 19,20-EDP из DHA - уникальны в том, что у них отсутствует гомолог в серии продуктов АА. В соответствии с субстратной специфичностью изоформ CYP поступление EPA/DHA с пищей вызывает глубокий сдвиг эпоксидных и ω-гидрокси-метаболитов от АА- до ЕРА- и DHA во всех основных органах и тканях у крыс и, предположительно, также и у человека.
ЕЕТ и 20-НЕТЕ играют важную роль в регуляции различных сердечно-сосудистых функций (Roman RJ., Physiol Rev., 2002; 82:131-85). Было показано, что гипертензия, индуцированная Ang II, связана с пониженной регуляцией CYP-зависимого метаболизма АА (Kaergel et l., Hypertension. 2002; 40:273-9) в модели Ang II-индуцированной гипертензии и циркуляторно-ишемического поражения органов на двойных трансгенных крысах (dTGR) (Luft et al., Hypertension. 1999; 33:212-8). Трансгенные крысы несут гены ренина и ангиотензиногена человека, локально продуцируют Ang II, и у них развивается значительная гипертензия, инфаркт миокарда и альбуминурия. Животные умирают от сердечной и почечной недостаточности до достижения ими возраста восьми недель. Модель демонстрирует серьезные признаки воспаления, вызванного Ang II. Вырабатываются реакционноспособные формы кислорода, активируются факторы транскрипции NF-κВ и АР-1 и активируются гены, несущие места связывания для указанных факторов транскрипции.
Недавно было показано, что добавка эйкозапентаеновой кислоты (ЕРА) значительно снижает смертность у dTGR (Theuer et al., Kidney Int. 2005; 67:248-58). Кроме того, было показано, что у dTGR развиваются желудочковые аритмии на основе индуцированного Ang II электрического ремоделирования (Fischer et al., Am J Physiol Heart Circ Physiol. 2007; 293:H1242-1253). Обработка крыс dTGR активатором PPAR-альфа в большой степени индуцировала CYP2C23-зависимое продуцирование ЕЕТ и защищала от гипертонии и циркуляторно-ишемического поражения органов (Muller et al., Am J Pathol. 2004; 164:521-32).
Долгосрочная кормежка dTGR (с возраста 4 недели по 7 недель) смесью чистых ЕРА- и DHA-этиловых эфиров (Omacor от Solvay Arzneimittel, Ганновер, Германия) улучшало электрическое ремоделирование сердца в модели гипертонии, индуцированной ангиотензином II. В частности, ЕРА и DHA уменьшали смертность, подавляли возникновение сердечных аритмий и защищали от ремоделирования щелевых контактов коннексина 43 (Fischer et al., Hypertension. 2008 Feb; 51(2):540-6). В целом CYP-зависимые эйкозаноиды следует рассматривать как второстепенные мессенджеры: ЕЕТ и 20-НЕТЕ производятся ферментами CYP после индуцированного внеклеточным сигналом высвобождения АА из мембранных фосфолипидов (фосфолипазой А2), и выполняют свою функцию в отношении сигнальных путей, модулируя перенос ионов, пролиферацию клеток и воспаление. В зависимости от рациона n-3 ПНЖК частично заменяют АА в положении sn2 фосфолипидов и могут, таким образом, участвовать в качестве альтернативных молекул в последующих сигнальных путях.
Ряд исследований биологической активности CYP-зависимых эйкозаноидов в сердце указывают на важную роль ЕЕТ и 20-НЕТЕ в регуляции L-типа Са2+ и сарколеммальных и митохондриальных АТФ-чувствительных калиевых каналов (KАТФ). В кардиомиоцитах токи L-типа Са2+ и короткое замыкание клеток снижаются при ингибировании образования ЕЕТ, и эти эффекты можно обратить вспять путем добавления 11,12-ЕЕТ (Xiao et al., J Physiol. 1998; 508 (Pt 3):777-92). Также было показано, что ЕЕТ активируют сердечные каналы KАТФ. Указанное влияние является высоко стереоселективным: эффективен только S,R-энантиомер 11,12-ЕЕТ, но не R,S-энантиомер (Lu et al., Mol Pharmacol. 2002; 62:1076-83). Сверхэкспрессия ЕЕТ-продуцирующего CYP2J2 у человека приводит к улучшенному постишемическому функциональному восстановлению сердца у трансгенных мышей за счет активации каналов KАТФ (Seubert et al., Circ Res., 2004; 95: 506-14). 20-НЕТЕ играет противоположную роль, выступая в качестве эндогенного блокатора каналов KАТФ (Gross et al., J Mol Cell Cardiol. 2004; 37:1245-9; Nithipatikom et al., Circ Res. 2004; 95:e65-71).
Известная в настоящее время биологическая активность метаболитов CYP, полученных из ЕРА и DHA, в некотором роде похожа на активность их аналогов, полученных из АА, а частично представляется уникальной или может даже иметь противоположный эффект (Westphal et al., Prostaglandins Other Lipid Mediat. 2011; 96:99-108). Эпокси-метаболиты всех трех ПНЖК обладают сосудорасширяющими свойствами, в результате чего действенность EEQ и EDP может превышать действенность ЕЕТ в некоторых сетях сосудов (Lauterbach et al. Hypertension. 2002; 39:609-13). Противовоспалительные эффекты были впервые обнаружены для 11,12- и 14,15-ЕЕТ, но также их проявляют эпоксиды ЕРА, примером которых является 17,18-EEQ (Morin et al., Am J Respir Cell Mol Biol. 2010; 43:564-575). 17,18-EEQ и 19,20-EDP ингибируют индуцированную Ca2+ и изопротеренолом повышенную сократимость неонатальных кардиомиоцитов, что указывает на то, что эти метаболиты могут действовать как эндогенные медиаторы антиаритмических эффектов ЕРА и DHA, как описано выше (Arnold et al., J Biol Chem. 2010 Oct 22; 285(43):32720-33). В недавнее время были описаны химически синтезированные соединения, которые обладают антиаритмическим действием 17,18-EEQ на неонатальные кардиомиоциты и уменьшают желудочковую тахиаритмию в модели инфаркта миокарда у крыс (Falck et al., J Med Chem. 2011 Jun 23; 54(12):4109-18; WO 2010/081683 A1, также опубликованный в виде патентной публикации США 2012/0122972). Образование 17,18-EEQ и 19,20-EDP может дополнительно способствовать антитромботическому действию n-3 ПНЖК (Jung et al., Clin Hemorheol Microcirc. 2012; 52(2-4):403-16). Кроме того, имеются свидетельства того, что CYP-зависимые эпоксиметаболиты играют важную роль в опосредовании противоположных эффектов n-6 ПНЖК и n-3 ПНЖК в описанных выше процессах патологического ангиогенеза, и, таким образом, ЕЕТ, полученные из АА, способствуют ангиогенезу опухоли и метастазированию (Panigrahy et al., J Clin Invest. 2012; 122:178-191). Напротив, 19,20-EDP и другие региоизомерные DHA-эпоксиды ингибируют эти важные для канцерогенеза события (Zhang et al., Proc Natl AcadSci USA. 2013; 110:6530-6535).
Несмотря на то, что метаболиты CYP, полученные из n-3 ПНЖК, такие как 17,18-EEQ и 19,20-EDP, играют важную роль в опосредовании положительных эффектов n-3 ПНЖК в организме млекопитающих, их не применяют в качестве терапевтических средств из-за их ограниченной биодоступности, а также химической и метаболической нестабильности. Указанные эпоксиметаболиты n-3 ПНЖК подвержены автоокислению, быстрой инактивации растворимой эпоксидгидролазой и деградации путем β-окисления. Наконец, новые агенты для лечения или предотвращения состояний и заболеваний, связанных с пролиферацией, патологическим ангиогенезом, гипертонией, коагуляцией, иммунной функцией, сердечной недостаточностью и сердечными аритмиями, представляют значительный интерес, поскольку эти состояния обуславливают значительное количество смертей пациентов, а с приемом многих применяемых в настоящее время лекарственных средств связаны комплексные взаимодействия между лекарственными средствами и различные неблагоприятные побочные эффекты.
Поэтому задача, лежащая в основе настоящего изобретения, заключается в создании новых соединений, предпочтительно новых и улучшенных аналогов метаболитов n-3 ПНЖК. Одной из задач, лежащих в основе настоящего изобретения, является обеспечение улучшенных соединений, стабильных в отношении дезактивации эпоксидными гидролазами, менее подверженных автоокислению и предпочтительно обладающих активностью против фибрилляции предсердий, желудочковой аритмии или сердечной недостаточности.
В первом аспекте вышеуказанная задача решается путем обеспечения соединений общей формулы (I):

или их фармацевтически приемлемых солей, где:
Р представляет собой группу, представленную общей формулой (II):

где
n представляет собой 0 или целое число от 3 до 8; и
k представляет собой 0, 1 или 2; предпочтительно с условием, что в случае, когда n равно 0, k равно 1, более предпочтительно k равно 1;
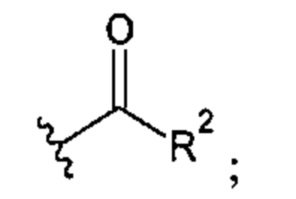
X представляет собой CH2OH, СН2ОАс, СН(О) или группу, выбранную из группы, состоящей из:

 и
и 
предпочтительно X представляет собой 
где
каждый из R и R' независимо представляет собой атом водорода; или C1-С6 алкильную группу, которая может быть замещена одним или более атомами фтора или хлора или гидроксильными группами;
R1 представляет собой гидроксильную группу, C1-С6 алкокси, -NHCN, -NH(C1-C6 алкил), -NH(C3-C6 циклоалкил), -NH(арил) или -О(С1-С6алкилдиил)О(С=О)R11; R11 представляет собой C1-С6 алкильную группу, необязательно замещенную одним или более атомами фтора или хлора; или С3-С6 циклоалкильную группу, необязательно замещенную одним или более атомами фтора или хлора или гидроксильными группами;
R2 представляет собой -NHR3; -NR20R21; -OR22; -(OCH2-CH2)i-R23; -С3-С10-гетероциклил, необязательно замещенный одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из гидроксильной группы, C1-С6 алкокси, C1-С6алкила и оксо; -(Хаа)о; моно- или дисахарида или его производного, присоединенных к -С(О) посредством сложноэфирной связи по положению 1-О-, 3-О- или 6-О- указанного сахарида;
или выбран из группы, состоящей из:

 и
и 
где
R3 представляет собой (SO2R30); (OR31); -C1-C6aлкaндиил(SO2R32); -С1-С6алкандиил(CO2H), арильную группу, предпочтительно фенил, гетероарильную группу, предпочтительно содержащую одно кольцо и 5 или 6 атомов, циклоалкильную группу, предпочтительно С3-С10циклоалкил, или гетероциклоалкильную группу, предпочтительно содержащую одну или две системы колец, где указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6 алкила, C1-С6 алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-C6 алкил), -N(C1-С6)диалкила и -C(=O)OR51; где указанная гетероарильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из С1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкил), -N(С1-С6)диалкила и -C(=O)OR51; где указанная циклоалкильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6 алкила, C1-С6 алкокси, C1-С6 алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-C6 алкил), -N(С1-С6)диалкила и -C(=O)OR51; и где указанная циклоалкильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6 алкила, C1-С6 алкокси, C1-С6 акилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-C6 алкил), -Н(C1-С6)диалкила и -C(=O)OR51;
R30 представляет собой C1-С6 алкильную или арильную группу, где указанная C1-С6алкильная группа необязательно замещена -NH2, -NH(C1-С6)алкилом, -N(C1-С6)диалкилом, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(С1-С6)алкиламинокарбонил-, одним, двумя или тремя атомами фтора или хлора или гидроксильными группами; и где указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, С1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкил) и -N(С1-С6)диалкила;
R31 представляет собой C1-С6алкильную группу, необязательно замещенную одним или более атомами фтора или хлора или гидроксильными группами; или С3-С6циклоалкильную группу, необязательно замещенную одним или более атомами фтора или хлора или гидроксильными группами;
R32 представляет собой C1-С6алкильную группу, необязательно замещенную одним или более атомами фтора или хлора или гидроксильными группами; или С3-С6циклоалкильную группу, необязательно замещенную одним или более атомами фтора или хлора или гидроксильными группами;
каждый из R20 и R21 независимо представляет собой атом водорода; C1-С6алкильную группу, которая может быть замещена одним или более атомами фтора или хлора или гидроксильными группами; С3-С6циклоалкильную группу, которая может быть замещена одним или более атомами фтора или хлора или гидроксильными группами; или -C1-С6-алкилдиил(СО2Н) или совместно образуют С3-С10гетероциклоалкил, предпочтительно С5-С6гетероциклоалкил, где указанный С3-С10-гетероциклоалкил может быть замещен одной или более C1-С6алкильными группами, C1-С6алкоксигруппами, атомами фтора или хлора или гидроксильными группами;
R22 представляет собой атом водорода, C1-С6алкильную группу; или С3-С6циклоалкильную группу; где указанная C1-С6алкильная группа или С3-С6циклоалкильная группа необязательно замещена -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидроксилами или C1-С6алкокси, аралкильной группой, гетероалкильной группой или гетероалкилциклоалкильной группой;
R23 представляет собой -ОН, -O(С1-С3)алкил или -N(С1-С3)диалкил;
i представляет собой целое число от 1 до 10, т.е. 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10, предпочтительно от 2 до 4;
каждый из R24, R25 и R26 независимо представляют собой атом водорода; -С(=O)С11-С21алкил; или -С(=O)С11-С21алкенил;
R27 представляет собой -ОН; -O(CH2)2NH2, -OCH2-[CH(NH2)(CO2H)], -О(CH2)2N(CH3)3; или 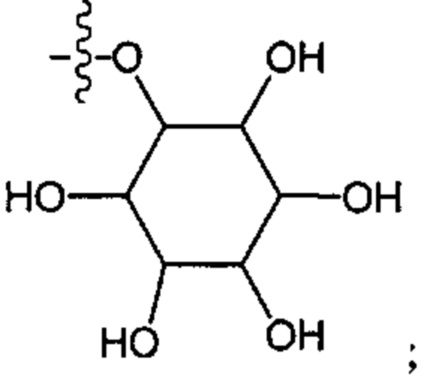
Xaa представляет собой Gly, природную D,L-, D- или L-аминокислоту, неприродную D,L-, D- или L-аминокислоту, или от 2- до 10-мерный пептид; и присоединена к -С(=О) амидной связью;
о представляет собой целое число от 1 до 10, т.е. 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;
R4 выбран из группы, состоящей из:

 и
и 
h представляет собой 0, 1 или 2;
R5 представляет собой атом водорода; атом фтора или хлора; -CF3; -C(=O)OR51; -NHC(=O)R52; -C(=O)NR53R54; или -S(O2)OH;
R51 представляет собой атом водорода, C1-С6алкильную группу; или С3-С6циклоалкильную группу; где указанная C1-С6алкильная группа или С3-С6циклоалкильная группа необязательно замещена -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидроксилами или C1-С6алкокси;
каждый из R52, R53 и R54 независимо представляет собой C1-С6алкильную группу, необязательно замещенную одним или более атомами фтора или хлора; С3-С6циклоалкильную группу, необязательно замещенную одним или более атомами фтора или хлора; или арильную группу, необязательно замещенную одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила, и оксозаместителя;
каждый из R6 и R7 независимо представляет собой гидроксильную группу; -O(C1-С6)алкильную группу, -O(С2-С6)алкенильную группу, -O(С1-С6)алкилдиилО(С=О)(С1-С6)алкильную группу или -О(С1-С6)алкилдиилО(С=О)(С2-С6)алкенильную группу; где указанная C1-С6алкильная группа и указанная С2-С6алкенильная группа могут быть замещены NH2, -NH(C1-С6)алкилом, -N(С1-С6)диалкилом, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(С1-С6)алкиламинокарбонил-, или одним, двумя или тремя атомами фтора или хлора; или
R6 представляет собой гидроксильную группу и R7 представляет собой группу:

R9 представляет собой C1-С6алкил или арил; где указанный C1-С6алкил необязательно замещен -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(С1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси, C1-С6алкокси, арилом, арилокси, -С(=O)-арилом, -С(=O)С1-С6алкокси; и где указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкил),
-N(С1-С6)диалкила и оксозаместителя;
g представляет собой 1 или 2;
X1 представляет собой атом кислорода; атом серы или NH;
X2 представляет собой атом кислорода; атом серы, NH или N(СН3);
X3 представляет собой атом кислорода; атом серы; атом азота; атом углерода или С-ОН; и
прерывистая линия представляет собой связь углерод-углерод или двойную связь углерод-углерод;
Е представляет собой группу, представленную общей формулой (III) или (IV):


где R12 и R13 предпочтительно находятся в цис-конфигурации, и где
кольцо А в формуле (III) представляет собой 5-членное или 6-членное карбоциклическое или гетероциклическое кольцо, содержащее по меньшей мере одну двойную связь, включая ароматическое карбоциклическое или гетероциклическое кольцо, которое может быть замещено от 1 до 3 или от 1 до 4 заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкил) и -N(С1-С6)диалкила; и каждый из L и Т независимо представляет собой атом кольца, где L и Т смежны друг с другом;
каждый из R12 и R13 независимо представляет собой атом водорода, атом фтора, гидроксил, -NH2, C1-С6алкил, C1-С6алкокси, -С(=O)-арил, -С(=O)С1-С6-алкил или -SO2(C1-С6-алкил); или -SO2арил; где любой из указанных С1-С6алкила, C1-С6алкокси или арила необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(C1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(C1-С6)алкиламинокарбонил-, атома фтора или хлора и гидроксила; или R12 и R13 совместно образуют 5-членное или 6-членное кольцо, причем указанное кольцо необязательно замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(C1-С6)алкиламинокарбонил-, атома фтора или хлора и гидроксила;
I представляет собой -(CH2)m-Y, где
m представляет собой целое число от 3 до 6, при условии, что m представляет собой целое число от 3 до 5, когда Е представляет собой группу общей формулы (III);
Y представляет собой -U-V-W-(CH2)p-(CH3)q, где р представляет собой целое число от 0 до 6; q равно 0 или 1; U отсутствует или выбран из группы, состоящей из СН, СН2 и NR40, при условии, что U представляет собой только СН, если он образует эпоксидную группу совместно с V и W; V выбран из группы, состоящей из -С(О)-, -С(O)-С(O)-, -О- и -S-; W выбран из группы, состоящей из СН, CH2 и NR40, при условии, что W представляет собой только СН, если он образует эпоксидную группу совместно с U и V;
или Y представляет собой группу, выбранную из группы, состоящей из:

где
каждый из R40, R41, R43, R44, R46, R48 и R49 независимо представляет собой атом водорода, -C1-С6алкил, -С3-С6циклоалкил, -C1-С6алкокси, -С(=O)арил или -С(=O)С1-С6алкил, где любой из указанных C1-С6алкила, С3-С6циклоалкила, C1-С6алкокси или арила необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(С1-С6)алкиламинокарбонил-, атома фтора или хлора, и гидроксила; или R40 и R41, или R43 и R44 совместно образуют 5-членное или 6-членное кольцо, причем указанное кольцо может быть необязательно замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(C1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(С1-С6)алкиламинокарбонил-, атома фтора или хлора, и гидроксила;
каждый из R42, R45, R47 и R50 независимо представляет собой -C1-С3алкил, где указанный C1-С3алкил может быть необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С3)алкила, -N(C1-С3)диалкила, C1-С3алкилкарбонилокси-, C1-С3алкоксикарбонилокси-, C1-С3алкилкарбонилтио-, C1-С3алкиламинокарбонил-, ди(С1-С3)алкиламинокарбонил-, атома фтора или хлора, и гидроксила; или R40 и R41, R43 и R44; R49 и R50 совместно образуют 5-членное или 6-членное кольцо, причем указанное кольцо может быть необязательно замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонил-, ди(С1-С6)алкиламинокарбонил-, атома фтора или хлора, и гидроксила;
f представляет собой целое число от 0 до 2;
при условии, что
когда X не содержит фрагмент -С(=О)О с карбонильным углеродом в альфа- или бета-положении к атому кислорода общей формулы (II), Y представляет собой оксамид, карбамат или карбамид, предпочтительно Y представляет собой оксамид, как определено выше.
В предпочтительном варианте реализации соединения согласно настоящему изобретению представляют собой соединения, как описано выше, с дополнительным условием, что когда n равно 3, 5, 6, 7 или 8, k равно 1 и Е представляет собой группу общей формулы (III) или общей формулы (IV), где каждый из R12 и R13 представляет собой атом водорода;
Р представляет собой группу:
-(СН2)3-O-(СН2)-Х81; -(СН2)5-O-(СН2)-Х81;
где
X81 представляет собой группу, выбранную из группы, состоящей из:
 ;и
;и
R1' определен так же, как R1 выше;
R2' представляет собой -NHR3'; -OR22'; -(OCH2-CH2)i-R23; моно- или дисахарид или его производное, присоединенные к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-O-положение указанного сахарида;
или где R2 выбран из группы, состоящей из:

 и
и 
где
R3' представляет собой (SO2R30); (OR31); -C1-C6aлкaндиил(SO2R32); или -С2-С6алкандиил(СО2Н);
R22' представляет собой водород или С3-С6циклоалкильную группу, необязательно замещенную -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(С1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси или C1-С6алкокси;
R23 и i являются такими, как определено выше;
R24, R25, R26 и R27 являются такими, как определено выше;
R4' определен так же, как R4 выше; и h является таким, как определено выше;
R6' и R7' определены так же, как R6 и R7 выше;
R8 и R8'' определены так же, как R8 и R8' выше;
R9' определен так же, как R9 выше; R9'' представляет собой арил, необязательно замещенный одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкил), -N(С1-С6)диалкила и оксозаместителя.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой
где R2 представляет собой -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или его производное, присоединенные к -С(=О) сложноэфирной связью через 1-О-, 3-О- или 6-О-положение указанного сахарида;
или где R2 выбран из группы, состоящей из:

 и
и 
где R23 и i являются такими, как определено выше, предпочтительно i равно 3;
где R22 и с R23 по R27 такие, определено в п. 1, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода.
В одном варианте реализации настоящего изобретения R3 не представляет собой арильную группу, гетероарильную группу, циклоалкильную группу или гетероциклоалкильную группу.
В одном варианте реализации настоящего изобретения R20 и R21 совместно не образуют С3-С10-гетероциклоалкил.
В одном варианте реализации настоящего изобретения X не представляет собой 
В одном варианте реализации настоящего изобретения R2 не представляет собой -С3-С10-гетероциклил.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой  более предпочтительно
более предпочтительно 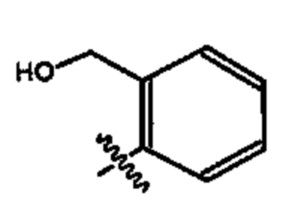 . В указанном варианте реализации дополнительно предпочтительно, чтобы Y представлял собой один из указанных выше оксамидов.
. В указанном варианте реализации дополнительно предпочтительно, чтобы Y представлял собой один из указанных выше оксамидов.
В еще более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, где X представляет собой -С(=O)ОН или подходящую соль карбоновой кислоты, предпочтительно свободную карбоновую кислоту.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой  . В указанном варианте реализации особенно предпочтительно, чтобы g составляло 2. В другом предпочтительно варианте реализации настоящего изобретения R9 представляет собой C1-С6алкил, т.е. метил, этил, пропил, бутил, пентил или гексил, предпочтительно метил, или арил, предпочтительно фенил; где указанный C1-С6алкил необязательно замещен -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-C6)алкилдиил-C1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси, C1-С6алкокси, арилом, арилокси, -С(=O)-арилом, -С(=O)С1-С6алкокси; и где указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH-(С1-С6алкил),
. В указанном варианте реализации особенно предпочтительно, чтобы g составляло 2. В другом предпочтительно варианте реализации настоящего изобретения R9 представляет собой C1-С6алкил, т.е. метил, этил, пропил, бутил, пентил или гексил, предпочтительно метил, или арил, предпочтительно фенил; где указанный C1-С6алкил необязательно замещен -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-C6)алкилдиил-C1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси, C1-С6алкокси, арилом, арилокси, -С(=O)-арилом, -С(=O)С1-С6алкокси; и где указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH-(С1-С6алкил),
-N(С1-С6)диалкила и оксозаместителя. В указанном варианте реализации настоящего изобретения k предпочтительно равно 1 или 2; более предпочтительно k равно 1. В указанном варианте реализации предпочтительно, чтобы n составляло 0. В указанном варианте реализации дополнительно предпочтительно, чтобы Y представлял собой один из указанных выше оксамидов.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой  . В указанном варианте реализации указанная гидроксильная группа может быть в пара-, мета- или орто-положении, предпочтительно в пара-положении. В указанном предпочтительном варианте также предпочтительно, чтобы R5 представлял собой водород. В указанном варианте реализации дополнительно предпочтительно, чтобы Y представлял собой один из указанных выше оксамидов.
. В указанном варианте реализации указанная гидроксильная группа может быть в пара-, мета- или орто-положении, предпочтительно в пара-положении. В указанном предпочтительном варианте также предпочтительно, чтобы R5 представлял собой водород. В указанном варианте реализации дополнительно предпочтительно, чтобы Y представлял собой один из указанных выше оксамидов.
В другом предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором Y представляет собой один из оксамидов, как описано выше.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой
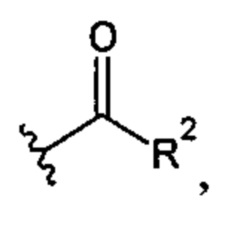
где R2 представляет собой -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или его производное, присоединенные к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-O-положение указанного сахарида; или где R2 выбран из группы, состоящей из:

 и
и 
где R22, с R23 по R27 и i определены выше, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода, предпочтительно i составляет от 2 до 4, более предпочтительно 3, и где Y предпочтительно представляет собой один из описанных выше оксамидов.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой

и где R2 представляет собой -С3-С10-гетероциклил, необязательно замещенный одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из гидроксильной группы, C1-С6алкокси, C1-С6алкила и оксо.
В другом предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, в котором X представляет собой С(=O)ОН, предпочтительно свободную карбоновую кислоту, и Y представляет собой один из оксамидов, как описано выше.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение следующей формулы (V):

где
R55 представляет собой -ОН; -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или его производное, присоединенные к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-O-положение указанного сахарида;
R22, R23 и i являются такими, как определено выше, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода, и i предпочтительно составляет от 2 до 4, более предпочтительно 3;
Y представляет собой группу, выбранную из группы, состоящей из:


где с R40 по R50 такие, определено выше, предпочтительно R40 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода, R57 и R58 представляют собой водород, или совместно образуют 5- или 6-членное кольцо, предпочтительно ароматическое кольцо, необязательно замещенное от 1 до 3 или от 1 до 4 заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С3алкил), -N(С1-С6)диалкила и оксозаместителя;
s равно 0, 1 или 2, при условии, что s равно 0, если R57 и R58 совместно образуют пяти- или шестичленное кольцо;
двойная связь в формуле (V) представляет собой двойную углерод-углеродную связь в цис-конфигурации, если R57 и R58 представляют собой водород, или указанная двойная связь является частью пяти- или шестичленного кольца, образованного R57 и R58 совместно.
В другом наиболее предпочтительном варианте реализации настоящего изобретения соединения формулы (V) представляют собой соединения, в которых
R55 представляет собой -ОН или - (OCH2-CH2)i-R23; i принимает значения от 2 до 4, предпочтительно i равно 3; R23 предпочтительно представляет собой ОН;
Y представляет собой оксамид, карбамид или карбамат, предпочтительно C1-С6алкилзамещенный оксамид, карбамид или карбамат;
R57 и R58 оба представляют собой Н или совместно образуют замещенное или незамещенное пяти- или шестичленное ароматическое кольцо, предпочтительно образуют замещенное или незамещенное бензильное кольцо; и
s равно 1 или s равно 0, если R57 и R58 совместно образуют замещенное или незамещенное пяти- или шестичленное ароматическое кольцо.
Наиболее предпочтительными определенными соединениями по настоящему изобретению являются те, которые выбраны из группы, состоящей из:



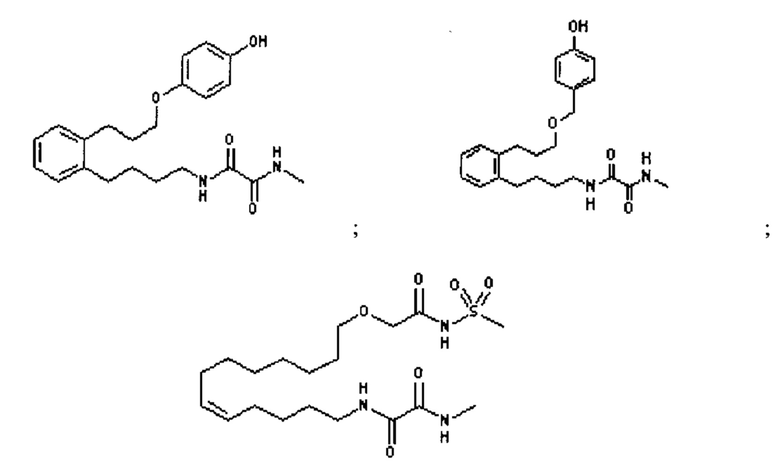
или их фармацевтически приемлемая соль.
Среди вышеуказанных соединений соединение следующей формулы (VI)

или его фармацевтически приемлемая соль являются наиболее предпочтительными.
Соединения согласно настоящему изобретению имеют следующее преимущество, как показано ниже в разделе, описывающем эксперименты: они эффективны для лечения сердечных заболеваний. В то же время указанные соединения метаболически устойчивы для фармацевтической композиции и для введения пациентам, нуждающимся в таком введении.
Соединения, описанные в настоящем изобретении, в целом описаны с использованием стандартной номенклатуры. Понимают, что для соединений с асимметричными центрами включены все оптические изомеры и их смеси, если не указано иное. Соединения с двумя или более асимметричными элементами могут также находиться в виде смесей диастереомеров. Кроме того, соединения с двойными связями углерод-углерод могут встречаться в Z- и Е-формах, причем все изомерные формы указанных соединений включены в настоящее изобретение, если не указано иное. В тех случаях, когда соединение существует в различных таутомерных формах, указанное соединение не ограничено каким-либо одним конкретным таутомером, а скорее охватывает все таутомерные формы. Описанные в настоящем изобретении соединения также включают соединения, в которых один или более атомов замещены изотопом, то есть атомом, имеющим тот же атомный номер, но с другим массовым числом. В качестве общего примера, но без ограничения, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают 11С, 13С и 14С.
Соединения формул, приведенных в настоящем изобретении, с одним или более стереогенными центрами, имеют энантиомерный избыток по меньшей мере 50%. Например, указанные соединения могут иметь энантиомерный избыток по меньшей мере 60%, 70%, 80%, 85%, 90%, 95% или 98%. Некоторые варианты реализации указанных соединений имеют энантиомерный избыток по меньшей мере 99%. Очевидно, что отдельные энантиомеры (оптически активные формы) можно получать при помощи асимметричного синтеза, синтеза из оптически чистых предшественников, биосинтеза, например, с использованием модифицированного CYP102 (CYP ВМ-3) или путем разделения рацематов, например, ферментативного разделения или обычными способами, такими как кристаллизация в присутствии разделяющего агента или хроматография, с использованием, например, хиральной колонки ВЭЖХ.
Некоторые соединения описаны в настоящем изобретении общей формулой, которая содержит такие переменные, как, например, Р, Е, I, R1-R50, Х-Х81 и Y. Если не указано иное, каждая переменная в такой формуле определена независимо от любой другой переменной, и любая переменная, которая встречается более одного раза в формуле, определена в каждом случае независимо. Так, например, если указано, что группа замещена при помощи 0-2 R*, то указанная группа может быть незамещенной или замещена при помощи вплоть до двух групп R*, и R* в каждом случае выбран независимо из определения R*. Кроме того, комбинации заместителей и/или переменных допустимы только в том случае, если такие комбинации приводят к образованию стабильных соединений, т.е. соединений, которые могут быть выделены, охарактеризованы и испытаны на биологическую активность.
«Фармацевтически приемлемая соль» соединения, как описано в настоящем изобретении, представляет собой кислую или основную соль, которая обычно считается в данной области техники подходящей для применения в контакте с тканями людей или животных без чрезмерной токсичности или канцерогенности и предпочтительно без раздражения, аллергической реакции или других проблем или осложнений. Такие соли включают соли минеральных и органических кислот основных остатков, таких как амины, а также щелочные или органические соли кислотных остатков, таких как карбоновые кислоты.
Подходящие фармацевтические соли включают, но не ограничиваются ими, соли кислот, таких как соляной, фосфорной, бромистоводородной, яблочной, гликолевой, фумаровой, серной, сульфаминовой, сульфанильной, муравьиной, толуолсульфоновой, метансульфоновой, бензолсульфоновой, этандисульфоновой, 2-гидроксиэтилсульфоновой, азотной, бензойной, 2-ацетоксибензойной, лимонной, винной, молочной, стеариновой, салициловой, глутаминовой, аскорбиновой, памовой, янтарной, фумаровой, малеиновой, пропионовой, гидроксималеиновой, йодистоводородной, фенилуксусной, алкановой, такой как уксусная, НООС-(CH2)n-СООН, где n представляет собой любое целое число от 0 до 6, т.е. 0, 1, 2, 3, 4, 5 или 6 и т.п. Аналогичным образом фармацевтически приемлемые катионы включают, но не ограничиваются ими: натрий, калий, кальций, алюминий, литий и аммоний. Специалисты в данной области техники смогут определить дополнительные фармацевтически приемлемые соли для соединений, представленных в настоящем изобретении. В целом фармацевтически приемлемая соль кислоты или основания может быть синтезирована из исходного соединения, которое содержит основную или кислотную часть, любым обычным химическим способом. В целом указанные соли можно получать путем взаимодействия свободных кислых или основных форм указанных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе, или в их смеси. Обычно предпочтительным является использование неводных сред, таких как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Очевидно, что каждое соединение формулы (I) может присутствовать в виде гидратного, сольватного или нековалентного комплекса, но это не обязательно. Кроме того, различные кристаллические формы и полиморфы входят в объем настоящего изобретения, а также пролекарства соединений формулы (I), представленных в настоящем изобретении.
«Пролекарство» представляет собой соединение, которое может не полностью удовлетворять структурным требованиям для соединений, представленных в настоящем изобретении, но которое модифицируется in vivo после введения субъекту или пациенту с получением соединения формулы (I), представленного в настоящем изобретении. Например, пролекарство может представлять собой ацилированное производное соединения, представленного в настоящем изобретении. Пролекарства включают соединения, в которых гидрокси-, карбокси-, амино- или сульфгидрильные группы связаны с любой группой, которая при введении субъекту, представляющему собой млекопитающее, отщепляется с образованием свободной гидрокси-, карбокси-, амино- или сульфгидрильной группы, соответственно. Примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные, фосфатные и бензоатные производные спиртовых и аминофункциональных групп в представленных в настоящем изобретении соединениях. Пролекарства соединений, представленных в настоящем изобретении, могут быть получены путем модификации функциональных групп, присутствующих в указанных соединениях, таким образом, что модифицированные части отщепляются in vivo с получением исходных соединений.
Термин «заместитель», используемый в настоящем изобретении, относится к молекулярному фрагменту, ковалентно связанному с атомом в пределах интересующей молекулы. Например, «заместитель в кольце» может представлять собой фрагмент, такой как галоген, алкильную группу, галогеналкильную группу или другой заместитель, описанный в настоящем изобретении, который ковалентно связан с атомом, предпочтительно атомом углерода или азота, который является членом кольца. Используемый в настоящем изобретении термин «замещенный» означает, что любой один или более атомов водорода на обозначенном атоме заменен на указанный заместитель по выбору при условии, что нормальная валентность указанного атома не будет превышена и что указанная замена приводит к получению стабильного соединения, т.е. соединения, которое может быть выделено, охарактеризовано и проверено на биологическую активность. Если заместитель представляет собой оксо, т.е. =О, то на атоме заменены 2 атома водорода. Оксигруппа, являющаяся заместителем ароматического атома углерода, приводит к превращению -СН- в -С(=O)- и к утрате ароматичности. Например, пиридильная группа, замещенная оксо, представляет собой пиридон.
Выражение «необязательно замещенный» относится к группе, в которой один, два, три или более атомов водорода могут быть независимо друг от друга заменены на соответствующие заместители.
Используемый в настоящем изобретении термин «аминокислота» относится к любой органической кислоте, содержащей один или более аминозаместителей, например α-, β- или γ-амино-производным алифатических карбоновых кислот. В используемых в настоящем изобретении обозначениях полипептидов, например, Xaa5, т.е. Хаа1Хаа2Хаа3Хаа4Хаа5, где каждый из от Xaa1 до Xaa5 независимо выбран из аминокислот, как определено в настоящем изобретении, левое направление представляет собой аминоконцевое направление и правое направление представляет собой карбоксиконцевое направление в соответствии со стандартными использованием и условными обозначениями.
Термин «природная аминокислота» относится к двадцати встречающимся в природе аминокислотам и охватывает все стереоизомерные изоформы, то есть D,L-, D- и L-аминокислоты. Такие природные аминокислоты могут также быть упомянуты в виде обычных трехбуквенных или однобуквенных аббревиатур, и такие сокращения соответствуют обычно использующимся (см., например, Immunology - A Synthesis, 2nd Edition, Е.S. Golub and D.R. Gren, Eds., Sinauer Associates, Sunderland Mass. (1991)).
Термин «неприродная аминокислота» относится к аминокислотам не природного происхождения или к химическим аналогам аминокислот, например, к α,α-дизамещенным аминокислотам, N-алкиламинокислотам, гомоаминокислотам, дегидроаминокислотам, ароматическим аминокислотам (отличным от фенилаланина, тирозина и триптофана) и орто-, мета- или пара-аминобензойной кислоте. Неприродные аминокислоты также включают соединения, которые содержат аминовую и карбоксильную функциональные группы, разделенные по типу замещения 1,3 или более, такие как β-аланин, γ-аминомасляная кислота, лактам Фрейдингера, бициклический дипептид (BTD), аминометилбензойную кислоту и другие, хорошо известные в данной области. Также можно применять статиноподобные изостеры, изостеры гидроксиэтилена, изостеры с восстановленными амидными связями, изостеры тиоамида, изостеры мочевины, изостеры карбамата, изостеры тиоэфира, изостеры винила и другие изостеры с амидными связями, известные в данной области. Применение аналогов аминокислот или неприродных аминокислот может улучшить стабильность и биологический период полураспада добавленного пептида, поскольку они более устойчивы к разрушению в физиологических условиях. Специалисту в данной области известны подобные типы замещения, которые могут быть сделаны. Неограничивающий список неприродных аминокислот, которые можно применять в качестве подходящих структурных единиц для пептида, а также их стандартных сокращений (в скобках) выглядит следующим образом: α-аминомасляная кислота (Abu), L-N-метилаланин (Nmala), α-амино-α-метилбутират (Mgabu), L-N-метиларгинин (Nmarg), аминоциклопропан (Cpro), L-N-метиласпарагин (Nmasn), карбоксилат L-N-метиласпарагиновой кислоты (Nmasp), анииноизомасляная кислота (Aib), L-N-метилцистеин (Nmcys), аминонорборнил (Norb), L-N-метилглутамин (Nmgln), карбоксилат L-N-метилглутаминовой кислоты (Nmglu), циклогексилаланин (Chexa), L-N-метилгистидин (Nmhis), циклопентилаланин (Cpen), L-N-метилизоллейцин (Nmile), L-N-метиллейцин (Nmleu), L-N-метиллизин (Nmlys), L-N-метилметионин (Nmmet), L-N-метилнорлейцин (Nmnle), L-N-метилнорвалин (Nmnva), L-N-метилорнитин (Nmorn), L-N-метилфенилаланин (Nmphe), L-N-метилпролин (Nmpro), L-N-метилстерин (Nmser), L-N-метилтреонин (Nmthr), L-N-метилтриптофан (Nmtrp), D-орнитин (Dorn), L-N-метилтирозин (Nmtyr), L-N-метилвалин (Nmval), L-N-метилтиглицин (Nmetg), L-N-метил-t-бутилглицин (Nmtbug), L-норлейцин (NIe), L-норвалин (Nva), α-метиламиноизобутират (Maib), α-метил-γ-аминобутират (Mgabu), D-α-метилаланин (Dmala), α-метилциклогексилаланин (Mchexa), D-α-метиларгинин (Dmarg), α-метилциклопентилаланин (Mcpen), D-α-метиласпарагин (Dmasn), α-метил-α-нафтилаланин (Manap), D-α-метиласпартат (Dmasp), α-метилпеницилламин (Mpen), D-α-метилцистеин (Dmcys), N-(4-аминобутил)глицин (NgIu), D-α-метилглутамин (Dmgln), N-(2-аминоэтил)глицин (Naeg), D-α-метилгистидин (Dmhis), N-(3-аминопропил)глицин (Norn), D-α-метилизолейцин (Dmile), N-амино-α-метилбутират (Nmaabu), D-α-метиллейцин (Dmleu), α-нафтилаланин (Anap), D-α-метиллизин (Dmlys), N-бензилглицин (Nphe), D-α-метилметионин (Dmmet), N-(2-карбамилэтил)глицин (NgIn), D-α-метилорнитин (Dmorn), N-(карбамилметил)глицин (Nasn), D-α-метилфенилаланин (Dmphe), N-(2-карбоксиэтил)глицин (NgIu), D-α-метилпролин (Dmpro), N-(карбоксиметил)глицин (Nasp), D-α-метилсерин (Dmser), N-циклобутилглицин (Ncbut), D-α-метилтреонин (Dmthr), N-циклогептилглицин (Nchep), D-α-метилтриптофан (Dmtrp), N-циклогексилглицин (Nchex), D-α-метилтирозин (Dmty), N-циклодецилглицин (Ncdec), D-α-метилвалин (Dmval), N-циклододецилглицин (Ncdod), D-N-метилаланин (Dnmala), N-циклооктилглицин (Ncoct), D-N-метиларгинин (Dnmarg), N-циклопропилглицин (Ncpro), D-N-метиласпарагин (Dnmasn), N-циклоундецилглицин (Ncund), D-N-метиласпартат (Dnmasp), N-(2,2-дифенилэтил)глицин (Nbhm), D-N-метилцистеин (Dnmcys), N-(3,3-дифенилпропил)глицин (Nbhe), D-N-метилглютамин (Dnmgln), N-(3-гуанидинопропил)глицин (Narg), D-N-метилглутамат (Dnmglu), N-(1-гидроксиэтил)глицин (Ntbx), D-N-метилгистидин (Dnmhis), N-(гидроксиэтил))глицин (Nser), D-N-метилизолейцин (Dnmile), N-(имидазолилэтил))глицин (Nhis), D-N-метиллейцин (Dnmleu), N-(3-индолилэтил)глицин (Nhtrp), D-N-метиллизин (Dnnilys), N-метил-γ-аминобутират (Nmgabu), N-метилциклогексилаланин (Nmchexa), D-N-метилметионин (Dnmmet), D-N-метилорнитин (Dnmorn), N-метилциклопентилаланин (Nmcpen), N-метилглицин (NaIa), D-N-метилфенилаланин (Dnmphe), N-метиламиноизобутират (Nmaib), D-N-метилпролин (Dnmpro), N-(1-метилпропил)глицин (Nile), D-N-метилсерин (Dnmser), N-(2-метилпропил)глицин (Nleu), D-N-метилтреонин (Dnmthr), D-N-метилтриптофан (Dnmtrp), N-(1-метилэтил)глицин (Nval), D-N-метилтирозин (Dnmtyr), N-метила-нафтилаланин (Nmanap), D-N-метилвалин (Dnmval), N-метилпеницилламин (Nmpen), γ-аминомасляная кислота (Gabu), N-(р-гидроксифенил)глицин (Nhtyr), L-/-бутилглицин (Tbug), N-(тиометил)глицин (Ncys), L-этилглицин (Etg), пеницилламин (Pen), L-гомофенилаланин (Hphe), L-α-метилаланин (Mala), L-α-метиларгинин (Marg), L-α-метиласпарагин (Masn), L-α-метиласпартат (Masp), L-α-метил-t-бутилглицин (Mtbug), L-α-метилцистеин (Mcys), L-метилэтилглицин (Metg), L-α-метилглютамин (MgIn), L-α-метилглутамат (MgIu), L-α-метилгистидин (Mhis), L-α-метилгомофенилаланин (Mhphe), L-α-метилизолейцин (Mile), N-(2-метилтиоэтил)глицин (Nmet), L-α-метиллейцин (Mleu), L-α-метиллизин (Mlys), L-α-метилметионин (Mmet), L-α-метилнорлейцин (MnIe), L-α-метилнорвалин (Mnva), L-α-метилорнитин (Morn), L-α-метилфенилаланин (Mphe), L-α-метилпролин (Mpro), L-α-метилсерин (Mser), L-α-метилтреонин (Mthr), L-α-метилтриптофан (Mtrp), L-α-метилтирозин (Mtyr), L-α-метилвалин (Mval), L-N-метилгомофенилаланин (Nmhphe), N-(N-(2,2-дифенилэтил)карбамилметил)глицин (Nnbhm), N-(N-(3,3-дифенилпропил)карбамилметил)глицин (Nnbhe), 1-карбокси-1-(2,2-дифенилэтиламино)циклопропан (Nmbc), L-O-метилсерин (Omser), L-O-метилгомосерин (Omhser).
Термин «алкил» относится к насыщенной углеводородной группе с неразветвленной или разветвленной цепью, которая содержит от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, например н-октильной группе, особенно от 1 до 6 атомов углерода, то есть 1, 2, 3, 4, 5 или 6, например, метилу, этилу, пропилу, изопропилу, н-бутилу, изо-бутилу, втор-бутилу, трет-бутилу, н-пентилу, изо-пентилу, н-гексилу или 2,2-диметилбутилу.
Термин «алкенил» относится к по меньшей мере частично ненасыщенной углеводородной группе с неразветвленной или разветвленной цепью, которая содержит от 2 до 21 атомов углерода, предпочтительно от 2 до 6 атомов углерода, то есть 2, 3, 4, 5 или 6 атомов углерода, например, к этенилу (винилу), пропенилу (аллилу), изопропенилу, бутенилу, изопренилу или гекс-2-енильной группе, или от 11 до 21 атомов углерода, то есть 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или 21 атомов углерода, например, к углеводородной группе, содержащей метиленовую цепь, прерванную одной двойной связью, как, например, в мононенасыщенных жирных кислотах, или к углеводородной группе, содержащей прерванные метиленом полиены, например, к углеводородным группам, содержащим два или более следующих структурных единиц -[СН=СН-CH2]-, как, например, в полиненасыщенных жирных кислотах. Алкенильные группы содержат одну или более двойных связей, предпочтительно 1, 2, 3, 4, 5 или 6.
Термин «алкинил» относится к по меньшей мере частично ненасыщенным углеводородным группам с неразветвленной или разветвленной цепью, которые содержат от 2 до 20 атомов углерода, предпочтительно от 2 до 10 атомов углерода, особенно от 2 до 6, то есть 2, 3, 4, 5 или 6 атомов углерода, например, к этинилу, пропинилу, бутинилу, ацетиленилу или пропаргилу. Предпочтительно алкинильные группы содержат одну или две (особенно предпочтительно одну) тройную связь.
Кроме того, термины «алкил», «алкенил» и «алкинил» относятся к группам, в которых один или более атомов водорода были заменены, например, на атом галогена, предпочтительно F или Cl, таким как, например, 2,2,2-трихлорэтильная или трифторметильная группа.
Термин «гетероалкил» относится к алкильной, алкенильной или алкинильной группе, в которой один или более атомов углерода, предпочтительно 1, 2 или 3, были заменены независимо друг от друга на атом кислорода, азота, фосфора, бора, селена, кремния или серы, предпочтительно на атом кислорода, серы или азота. Термин «гетероалкил» может также относиться к карбоновой кислоте или к группе, полученной из карбоновой кислоты, такой как, например, ацил, ацилалкил, алкоксикарбонил, ацилокси, ацилоксиалкил, карбоксиалкиламид или алкоксикарбонилокси.
Предпочтительно гетероалкильная группа содержит от 1 до 10 атомов углерода и от 1 до 4 гетероатомов, выбранных из кислорода, азота и серы (особенно кислорода и азота). Особенно предпочтительно гетероалкильная группа содержит от 1 до 6 атомов углерода, то есть, 1, 2, 3, 4, 5 или 6 атомов углерода и 1, 2 или 3 гетероатомов, особенно 1 или 2, выбранных из кислорода, азота и серы, особенно кислорода и азота.
Примерами гетероалкильных групп являются группы формул: Ra-O-Ya-, Ra-S-Ya-, Ra-N(Rb)-Ya-, Ra-CO-Ya-, Ra-O-CO-Ya-, Ra-CO-O-Ya-, Ra-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-Ya-, Ra-O-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-O-Ya-, Ra-N(Rb)-CO-N(Rc)-Ya-, Ra-O-CO-O-Ya-, Ra-N(Rb)-C(=NRd)-N(Rc)-Ya-, Ra-CS-Ya-, Ra-O-CS-Ya-, Ra-CS-O-Ya-, Ra-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-Ya-, Ra-O-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-O-Ya-, Ra-N(Rb)-CS-N(Rc)-Ya-, Ra-O-CS-O-Ya, Ra-S-CO-Ya-, Ra-CO-S-Ya, Ra-S-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-S-Ya-, Ra-S-CO-O-Ya-, Ra-O-CO-S-Ya-, Ra-S-CO-S-Ya-, Ra-S-CS-Ya-, Ra-CS-S-Ya-, Ra-S-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-S-Ya-, Ra-S-CS-O-Ya-, Ra-O-CS-S-Ya-, где Ra представляет собой атом водорода, C1-С6 алкильную, C2-C6 алкенильную или С2-С6 алкинильную группу; Rb представляет собой атом водорода, C1-С6 алкильную, C2-С6 алкенильную или C2-С6 алкинильную группу; Rc представляет собой атом водорода, C1-С6 алкильную, C2-С6 алкенильную или C2-С6 алкинильную группу; Rd представляет собой атом водорода, C1-С6 алкильную, C2-С6 алкенильную или C2-С6 алкинильную группу, и Ya представляет собой прямую связь, C1-С6 алкиленовую, C2-С6 алкениленовую или C2-С6 алкиниленовую группу, при этом каждая гетероалкильная группа содержит по меньшей мере один атом углерода и один или более атомов водорода может быть заменен на атом фтора или атом хлора.
Конкретными примерами гетероалкильных групп являются метокси, трифторметокси, этокси, н-пропилокси, изопропилокси, бутокси, трет-бутилокси, метоксиметил, этоксиметил, -CH2CH2OH, -CH2OH, метоксиэтил, 1-метоксиэтил, 1-этоксиэтил, 2-метоксиэтил или 2-этоксиэтил, метиламино, этиламино, пропиламино, изопропиламино, диметиламино, диэтиламино, изопропилэтиламино, метиламинометил, этиламинометил, диизопропиламиноэтил, метилтио, этилтио, изопропилтио, енольный эфир, диметиламинометил, диметиламиноэтил, ацетил, пропионил, бутирилокси, ацетилокси, метоксикарбонил, этоксикарбонил, пропионилокси, ацетиламино или пропиониламино, карбоксиметил, карбоксиэтил или карбоксипропил, N-этил-N-метилкарбамоил или N-метилкарбамоил. Другими примерами гетероалкильных групп являются нитрильные, изонитрильные, цианатные, тиоцианатные, изоцианатные, изотиоцианатные и алкилнитрильные группы.
Термин «алкокси» обозначает алкильную группу, связанную с кислородом.
Термин «алкилтио» обозначает алкильную группу, связанную с серой.
Термины циклоалкильное и карбоциклическое кольцо относятся к насыщенной циклической углеводородной группе, которая содержит одно или более колец, предпочтительно 1 или 2, и содержит от 3 до 14 атомов углерода кольца, предпочтительно от 3 до 10, особенно 3, 4, 5, 6 или 7 атомов углерода кольца, например, циклопропил, циклобутил, циклопентил, спиро[4,5]деканил, норборнил, циклогексил, декалинил, бицикло[4.3.0]нонил, тетралин или циклопентилциклогексил. Термин «циклоалкил» относится, кроме того, к группам, в которых один или более атомов водорода были заменены на атомы фтора, хлора, брома или йода или группами ОН, =O, SH, NH2, NH, N3 или NO2, например, к циклическим кетонам, таким как, например, циклогексанон, 2-циклогексенон или циклопентанон. Другими конкретными примерами циклоалкильных групп являются циклопропил, циклобутил, циклопентил, спиро[4,5]деканил, норборнил, циклогексил, циклопентенил, циклогексадиенил, декалинил, бицикло[4.3.0]нонил, тетралин, циклопентилциклогексил, фторциклогексил или циклогекс-2-енил.
Термин «арил» относится к ароматической группе, которая содержит одно или более колец, содержащих от 6 до 14 атомов углерода кольца, предпочтительно от 6 до 10, особенно 6, атомов углерода кольца.
Термин «гетероарил» относится к ароматической группе, которая содержит одно или более колец, содержащих от 5 до 14 атомов кольца, предпочтительно от 5 до 10, особенно 5 или 6 атомов кольца и содержит один или более, предпочтительно 1, 2, 3 или 4 атомов кислорода, азота, фосфора или серы кольца, предпочтительно О, S или N. Примерами являются пиридил (например, 4-пиридил), имидазолил (например, 2-имидазолил), фенилпирролил (например, 3-фенилпирролил), тиазолил, изотиазолил, 2,3-триазолил, 1,2,4-триазол, оксадиазолил, тиадиазолил, индолил, индазолил, тетразолил, пиразинил, пиримидинил, пиридазинил, оксазолил, изоксазолил, триазолил, тетразолил, изоксазолил, индазолил, индолил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, пиридазинил, хинолинил, изохинолинил, пирролил, пуринил, карбазолил, акридинил, пиримидил, 2,3-бифурил, пиразолил (например, 3-пиразолил) и изохинолинил. Термин «гетероциклоалкил» относится к циклоалкильной группе, как определено выше, в которой один или более (предпочтительно 1,2 или 3) атомов углерода в кольце, каждый независимо, заменены на атом кислорода, азота, кремния, селена, фосфора или серы (предпочтительно на атом кислорода, серы или азота). Гетероциклоалкильная группа содержит предпочтительно 1 или 2 кольца, содержащих от 3 до 10 (особенно 3, 4, 5, 6 или 7) атомов кольца (предпочтительно выбранных из С, О, N и S). Термин «гетероциклоалкил» относится, кроме того, к группам, в которых один или более атомов водорода были заменены на атомы фтора, хлора, брома или йода или группами ОН, =O, SH, =S, NH2, =NH, N3 или NO2. Примерами являются пиперидил, пролинил, имидазолидинил, пиперазинил, морфолинил, уротропинил, пирролидинил, тетрагидротиофенил, тетрагидропиранил, тетрагидрофурил или 2-пиразолинил, а также лактамы, лактоны, циклические имиды и циклические ангидриды.
Термин «алкилциклоалкил» относится к группе, которая содержит как циклоалкильную группу, так и алкильную, алкенильную или алкинильную группу в соответствии с приведенными выше определениями, например, алкилциклоалкильная, циклоалкилалкильная, алкилциклоалкенильная, алкенилциклоалкильная и алкинилциклоалкильная группы. Алкилциклоалкильная группа предпочтительно содержит циклоалкильную группу, которая содержит одну или две кольцевые системы, содержащие от 3 до 10 (особенно 3, 4, 5, 6 или 7) атомов углерода кольца, и одну или две алкильные, алкенильные или алкинильные группы, содержащие от 1 или 2 до 6 атомов углерода. Термин «аралкил» относится к группе, содержащей как арильную группу, так и алкильную, алкенильную, алкинильную и/или циклоалкильную группы в соответствии с приведенными выше определениями, такой как, например, арилалкильная, арилалкенильная, арилалкинильная, арилциклоалкильная, арилциклоалкенильная, алкиларилциклоалкильная и алкиларилциклоалкенильная группы. Конкретными примерами аралкилов являются толуол, ксилол, мезитилен, стирол, бензилхлорид, о-фтортолуол, 1Н-инден, тетралин, дигидронафталин, инданон, фенилциклопентил, кумен, циклогексилфенил, флуорен и индан. Аралкильная группа предпочтительно содержит одну или две ароматические кольцевые системы (1 или 2 кольца), содержащие от 6 до 10 атомов углерода и одну или две алкильные, алкенильные и/или алкинильные группы, содержащие от 1 или 2 до 6 атомов углерода и/или циклоалкильных групп, содержащих 5 или 6 атомов углерода кольца.
Термин «гетероалкилциклоалкил» относится к алкилциклоалкильным группам, как определено выше, в которых один или более, предпочтительно 1, 2 или 3, атомов углерода были заменены независимо друг от друга на атомы кислорода, азота, кремния, селена, фосфора или серы (предпочтительно на атом кислорода, серы или азота). Гетероалкилциклоалкильная группа предпочтительно содержит 1 или 2 кольцевые системы, содержащие от 3 до 10 (особенно 3, 4, 5, 6 или 7) атомов кольца и одну или две алкильные, алкенильные, алкинильные или гетероалкильные группы, содержащие от 1 или 2 до 6 атомов углерода. Примерами таких групп являются алкилгетероциклоалкил, алкилгетероциклоалкенил, алкенилгетероциклоалкил, алкинилгетероциклоалкил, гетероалкилциклоалкил, гетероалкилгетероциклоалкил и гетероалкилгетероциклоалкенил, при этом циклические группы являются насыщенными или моно-, ди- или триненасыщенными.
Термин «гетероциклическое кольцо» относится к гетероарильной группе, как определено выше, а также к циклоалкильной группе или карбоциклическому кольцу, как определено выше, в которых один или более (предпочтительно 1, 2 или 3) атомов углерода кольца, каждый независимо, заменены на атом кислорода, азота, кремния, селена, фосфора или серы, предпочтительно на атом кислорода, серы или азота. Гетероциклическое кольцо содержит предпочтительно 1 или 2 кольца, содержащих от 3 до 10, особенно 3, 4, 5, 6 или 7 атомов кольца, предпочтительно выбранных из С, О, N и S. Примерами являются азиридинил, оксиранил, тииранил, оксазиридинил, диоксиранил, ацетидинил, оксетанил, тиетанил, диазетидинил, диоксетанил, дитиетанил, пирролидинил, тетрагидрофуранил, тиоланил, фосфоланил, силоланил, азолил, тиазолил, изотиазолил, имидазолидинил, пиразолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, изотиазолидинил, диоксоланил, дитиоланил, пиперазинил, морфолинил, тиоморфолинил, триоксанил, азепанил, оксепанил, типанил, гомопиперазинил или уротропинил.
Термин «гетероаралкил» относится к аралкильной группе, как определено выше, в которой один или более (предпочтительно 1, 2, 3 или 4) атомов углерода, каждый независимо, заменены на атом кислорода, азота, кремния, селена, фосфора, бора или серы (предпочтительно атомом кислорода, серы или азота), то есть к группе, содержащей как арил, так и гетероарил, соответственно, а также алкильную, алкенильную, алкинильную и/или гетероалкильную и/или циклоалкильную и/или гетероциклоалкильную группы в соответствии с приведенными выше определениями. Гетероаралкильная группа предпочтительно содержит одну или две ароматические кольцевые системы (1 или 2 кольца), содержащие от 5 или 6 до 10 атомов углерода и одну или две алкильные, алкенильные и/или алкинильные группы, содержащие от 1 или 2 до 6 атомов углерода и/или циклоалкильных групп, содержащих 5 или 6 атомов углерода кольца, при этом 1, 2, 3 или 4 из указанных атомов углерода заменены на атом кислорода, серы или азота.
Примерами являются арилгетероалкил, арилгетероциклоалкил, арилгетероциклоалкенильная, арилалкилгетероциклоалкил, арилалкенилгетероциклоалкил, арилалкинилгетероциклоалкил, арилалкилгетероциклоалкенил, гетероарилалкил, гетероарилалкенил, гетероарилалкинил, гетероарилгетероалкил, гетероарилциклоалкил, гетероарилциклоалкенил, гетероарилгетероциклоалкил, гетероарилгетероциклоалкенил, гетероарилалкилциклоалкил, гетероарилалкилгетероциклоалкенил, гетероарилгетероалкилциклоалкил, гетероарилгетероалкилциклоалкенил и гетероарилгетероалкилгетероциклоалкил, при этом циклические группы являются насыщенными или моно-, ди- или триненасыщенными. Конкретными примерами являются тетрагидроизохинолинил, бензоил, 2- или 3-этилиндолил, 4-метилпиридино, 2-, 3- или 4-метоксифенил, 4-этоксифенил, 2-, 3- или 4-карбоксифенилалкил.
Как уже указывалось выше, термины циклоалкил, гетероциклоалкил, алкилциклоалкил, гетероалкилциклоалкил, арил, гетероарил, аралкил и гетероаралкил также относятся к группам, в которых один или более атомов водорода таких групп были заменены независимо друг от друга на атом фтора, хлора, брома или йода или группами ОН, =O, SH, =S, NH2, NH, N3 или NO2.
Используемый в настоящем изобретении общий термин «кольцо», если не указано иное, включает циклоалкильные группы или карбоциклические кольца, гетероциклические кольца, арильные группы и гетероарильные группы.
Используемые в настоящем изобретении термины «галоген», «гало-» или «атом галогена», обозначают фтор, хлор, бром или йод, предпочтительно фтор и/или хлор.
Используемое в настоящем изобретении выражение «моно- или дисахарид и их производные» обозначают углевод или сахар, принадлежащий к группе моносахаридов или дисахаридов или полученный из них.
Примеры моно-, дисахаридов и их производных включают глюкозу, 3-O-метил-глюкозу, 1-деокси-глюкозу, 6-деокси-глюкозу, галактозу, маннозу, фруктозу, ксилозу, рибозу, целлобиозу, мальтозу, лактозу, гентиобиозу, сахарозу, трегалозу и маннит, сорбит и рибитол. Предпочтительно сахариды представляют собой сахариды D-формы, например D-глюкозу, 3-О-метил-D-глюкозу, 1-деокси-D-глюкозу или 6-деокси-D-глюкозу, D-галактозу, D-маннозу.
Используемая в настоящем изобретении формулировка, определяющая пределы диапазона длины, такие как, например, «от 1 до 5», означает любое целое число от 1 до 5, то есть 1, 2, 3, 4 и 5. Другими словами, любой диапазон, определенный двумя явно упомянутыми целыми числами, должен содержать и описывать любое целое число, определяющее указанные пределы, и любое целое число, содержащееся в указанном диапазоне.
Термин «-С(=O)O-фрагмент» используется в настоящем изобретении для четкого определения группы, содержащей sp2-гибридированный карбонильный углерод, присоединенный (i) к любому атому углерода или гетероатому, и (ii) к кислороду, который в свою очередь может быть присоединен к атому водорода или любому другому атому химического элемента. Термин «карбоксильная группа» исключен из описания «-С(=O)O-фрагмента», поскольку его ошибочно можно принять за описание только карбоновой кислоты.
Термин «в альфа-положении» используется для описания непосредственно смежного положения, тогда как термин «в бета-положении» указывает на соседствующее положение атома или группы А и атома или группы В, отличающееся тем, что между А и В расположен еще один атом или группа.
Используемый в настоящем тексте термин «оксамид» относится к произвольно замещенному органическому соединению, содержащему 2 карбонильных атома углерода и два атома азота, причем указанное соединение представляет собой произвольно замещенный диамид, полученный из любой щавелевой кислоты.
Специалисты в данной области легко поймут, что некоторые из аналогов n-3 ПНЖК общей формулы (I) согласно настоящему изобретению представляют собой «биоизостеры» встречающихся в природе эпоксиметаболитов, продуцируемых ферментами цитохром Р450 (CYP) из омега-3 (n-3) полиненасыщенных жирных кислот (ПНЖК). Биоизостер представляет собой соединение, образующееся в результате замены атома или группы атомов на альтернативный в целом схожий атом или группу атомов, тем самым создается новое соединение, обладающее аналогичными биологическими свойствами, что и исходное соединение. Например, химики-фармацевты используют биоизостеризм для улучшения желаемых биологических или физических свойств соединения, например, для ослабления токсичности, изменения активности, изменения фармакокинетики и/или метаболизма соединения. Например, замена атома водорода на атом фтора в месте метаболического окисления в соединении может препятствовать прохождению такого метаболизма. Поскольку фтор имеет сходство по размеру с атомом водорода, на общую топологию молекулы не оказывается существенного влияния, и желаемая биологическая активность оказывается не затронута. Однако указанное соединение, имея заблокированный путь метаболизма, может иметь более длительный период полувыведения. Другим примером является биоизостерическая замена групп карбоновой кислоты, что привело к получению аналогов, демонстрирующих улучшенную биодоступность, усиленное проникновение гематоэнцефалического барьера, повышенную активность, улучшенную химическую стабильность и/или селективность по отношению к мишени (см., например, учебник „The practice of medicinal chemistry”, под редакцией Camille Georges Wermuth, 3rd edition, Academic Press, 2008, e.g. p. 303-310; Ballatore C. et al. “Carboxylic Acid (Bio)Isosteres in Drug Design”, ChemMedChem 8, 385-395 (2013)). Кроме того, биоизостеризм можно также использовать для получения «пролекарства» соединения, то есть соединения, которое первоначально вводят субъекту или пациенту в неактивной (или менее активной) форме, и которое затем модифицируется in vivo в активную форму при помощи нормальных метаболических процессов в организме. Например, конъюгирование соединения с липидными и/или сахарными единицами приводит к получению аналогов (пролекарств), демонстрирующим увеличение доставки лекарственного средства по сравнению с исходным соединением (см., например, Wong A. and Toth I. "Lipid, Sugar and Liposaccharide Based Delivery Systems", Current Medicinal Chemistry 8, 1123-1136 (2001)).
Аналоги n-3 ПНЖК общей формулы (I) согласно настоящему изобретению можно получить несколькими способами, хорошо известными специалисту в области органического синтеза. Например, соединения согласно настоящему изобретению можно синтезировать в соответствии с общими схемами реакций, показанными ниже, с использованием синтетических методов, известных в области синтетической органической химии, или их вариаций, которые сможет оценить специалист в данной области техники. Если не указано иное, все переменные, например, n, k, R2 (также может быть обозначен как R2), R6, R7, R8, R41, R42, R44 и R45, имеют такое значение, как определено выше. В качестве исходных материалов можно использовать реагенты стандартного коммерческого сорта без дополнительной очистки или их можно легко получить из таких материалов обычными методами. Специалисты в области органического синтеза поймут, что исходные вещества и условия реакции могут варьироваться, включая дополнительные этапы, используемые для получения соединений, охватываемых настоящим изобретением.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) в комбинации с физиологически приемлемым вспомогательным веществом.
Особенно предпочтительно комбинировать предпочтительные варианты реализации отдельных общих групп формулы (I) любыми возможными способами.
В следующем аспекте настоящее изобретение относится к соединению формулы (I) или к фармацевтической композиции, содержащей соединение формулы (I), для применения для лечения сердечно-сосудистых заболеваний, предпочтительно выбранных из группы, состоящей из фибрилляции предсердий, желудочковой аритмии и сердечной недостаточности.
В следующем аспекте настоящее изобретение относится к соединению формулы (I) или фармацевтической композиции, содержащей соединение формулы (I), для применения для лечения фибрилляции предсердий, желудочковой аритмии, сердечной недостаточности, заболевания коронарной артерии, инфаркта миокарда, неадаптивной сердечной гипертрофии и сердечных аритмий, включая желудочковые экстрасистолы, желудочковую тахикардию, злокачественную желудочковую тахикардию, предсердную тахикардию, трепетание предсердий и фибрилляцию предсердий, дилатационную кардиомиопатию и гипертоническую болезнь сердца, предпочтительно выбранных из группы, состоящей из фибрилляции предсердий, предсердной тахикардии, желудочковой аритмии, сердечной недостаточности, предпочтительно выбранных из группы, состоящей из фибрилляции предсердий, предсердной тахикардии, желудочковой аритмии и сердечной недостаточности.
В предпочтительном варианте реализации соединение или композицию для применения согласно настоящему изобретению вводят перорально, местно, подкожно, внутримышечно, внутрибрюшинно, внутривенно или интраназально, предпочтительно перорально или внутривенно, более предпочтительно перорально.
Дополнительно предпочтительно соединение или композиция для применения согласно настоящему изобретению представляет собой лекарственную форму, выбранную из группы, состоящей из спрея, аэрозоля, пены, лекарственной формы для ингалятора, порошка, таблетки, капсулы, мягкой желатиновой капсулы, чая, сиропа, гранулы, жевательной таблетки, мази, крема, геля, суппозитория, пастилки для рассасывания, липосомальной композиции и раствора, подходящего для инъекций.
Фармацевтические композиции в соответствии с настоящим изобретением содержат по меньшей мере одно соединение формулы (I) и, необязательно, одно или более веществ-носителей, например, циклодекстринов, таких как гидроксипропил β-циклодекстрин, мицеллы или липосомы, вспомогательные вещества и/или адъюванты. Фармацевтические композиции могут дополнительно содержать, например, одно или более из воды, буферов, таких как, например, нейтральный забуференный солевой раствор или забуференный фосфатом физиологический раствор, этанол, минеральное масло, растительное масло, диметилсульфоксид, углеводы, такие как, например, глюкоза, манноза, сахароза или декстраны, маннит, белки, адъюванты, полипептиды или аминокислоты, такие как глицин, антиоксиданты, хелатирующие агенты, такие как ЭДТА или глутатион, и/или консерванты. Кроме того, один или более других активных ингредиентов могут, но не обязательно, входить в состав описанных в настоящем изобретении фармацевтических композиций. Например, соединения согласно настоящему изобретению можно преимущественно применять в комбинации с антибиотиком, противогрибковым или противовирусным агентом, антигистаминным, нестероидным противовоспалительным лекарственным средством, модифицирующим течение заболевания противоревматическим лекарственным средством, противовоспалительным лекарственным средством для лечения аутоиммунного заболевания, цитостатическим лекарственным средством, лекарственным средством, модулирующим активность гладкой мускулатуры, антигипертензивным лекарственным средством, бетаблокатора, антиаритмического лекарственного средства, лекарственного средства для лечения сердечной недостаточности, антитромботического лекарственного средства, антиагрегантного лекарственного средства или их смесей.
Предпочтительно настоящее изобретение относится к комбинированному препарату или состоящему из частей набору, которые содержат по меньшей мере одно соединение согласно настоящему изобретению и по меньшей мере одно лекарственное средство из группы, включающей антигипертензивное лекарственное средство, бетаблокатор, антиаритмическое лекарственное средство, лекарственное средство для лечения сердечной недостаточности, антитромботическое лекарственное средство, антитромботическое лекарственное средство, противоревматическое лекарственное средство и/или противовоспалительное лекарственное средство для лечения аутоиммунного заболевания.
Фармацевтические композиции могут быть составлены для любого подходящего способа введения, включая, например, местное, такое как, например, трансдермальное или глазное, пероральное, буккальное, назальное, вагинальное, ректальное или парентеральное введение. Используемый в настоящем изобретении термин «парентеральный» включает подкожную, внутрикожную, внутрисосудистую, такую как, например, внутривенная, внутримышечную, спинальную, внутричерепную, интратекальную, внутриглазную, подглазничную, внутриглазничную, внутрисуставную и внутрибрюшинную инъекцию, а также любой подобный метод инъекции или инфузии. В некоторых вариантах реализации предпочтительны композиции в форме, пригодной для перорального применения. Такие формы включают, например, таблетки, формованные пастилки, пастилки для рассасывания, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсию, твердые или мягкие капсулы, сиропы или эликсиры. В других вариантах реализации описанные в настоящем изобретении композиции могут быть составлены в виде лиофилизата. Композиция для местного введения может быть предпочтительной при определенных состояниях, таких как, например, при лечении кожных состояний, таких как ожоги или зуд.
Композиции, предназначенные для перорального применения, могут дополнительно содержать один или более компонентов, таких как подслащивающие агенты, ароматизаторы, красители и/или консерванты для обеспечения привлекательности и вкуса составов. Таблетки содержат активный ингредиент в смеси с физиологически приемлемыми вспомогательными веществами, подходящими для изготовления таблеток. Такие вспомогательные вещества включают, например, инертные разбавители, такие как, например, карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия, гранулирующие и дезинтегрирующие агенты, такие как, например, кукурузный крахмал или альгиновая кислота, связывающие агенты, такие как, например, крахмал, желатин или аравийская камедь и смазывающие агенты, такие как, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми или могут быть покрыты известными способами, чтобы задержать дезинтеграцию и абсорбцию в желудочно-кишечном тракте и тем самым обеспечить пролонгированное действие в течение более длительного периода. Например, можно использовать материал, обеспечивающий задержку по времени, такой как глицерилмоностеарат или глицерилдистеарат. Способы получения таких композиций известны (см., например, Н.С. Ansel and N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5th ed., Lea and Febiger (1990)).
Композиции для перорального применения могут быть также представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, таким как, например, карбонат кальция, фосфат кальция или каолин, или в виде мягких желатиновых капсул, в которых указанный активный ингредиент смешан с водой или масляной средой, например, с арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии согласно настоящему изобретению содержат активное(ые) вещество(а) в смеси со вспомогательными веществами, пригодными для изготовления водных суспензий. Такие вспомогательные вещества включают суспендирующие агенты, такие как, например, натрий карбоксиметилцеллюлозу, метилцеллюлозу, гидропропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь и камедь акации; и диспергирующие или смачивающие агенты, такие как, например, природные фосфатиды, такие как лецитин, продукты конденсации алкиленоксида с жирными кислотами, как полиоксиэтиленстеарат, продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, как гептадекаэтиленоксицетанол, продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот, и гекситолом, как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными эфирами, полученными из жирных кислот и ангидридов гексита, например, моноолеат полиэтиленсорбитана. Водные суспензии могут также содержать один или более консервант, например, этил или н-пропил-п-гидроксибензоат, один или более красителей, один или более ароматизирующих агентов, и один или более подслащивающих агентов, таких как сахароза или сахарин.
Масляные суспензии могут быть приготовлены суспендированием активных ингредиентов в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например, пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подсластители, такие, как описано в настоящем изобретении, и ароматизаторы, чтобы обеспечить вкус перорального препарата. Для сохранения этих суспензии может быть добавлен антиоксидант, такой как аскорбиновая кислота.
Диспергируемые порошки и гранулы, пригодные для получения водной суспензии при добавлении воды, обеспечивают активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или более консервантами. Примеры подходящих диспергирующих или смачивающих агентов и суспендирующих агентов уже приведены выше. Также могут присутствовать дополнительные вспомогательные вещества, например, подслащивающие, ароматизирующие и окрашивающие агенты.
Фармацевтические композиции согласно настоящему изобретению могут также быть в виде эмульсий масло-в-воде. Масляная фаза может представлять собой растительное масло, например, оливковое масло или арахисовое масло, минеральное масло, например, жидкий парафин, или их смесь. Подходящие эмульгаторы включают природные смолы, такие как, например, аравийская камедь или камедь трагаканта, природные фосфатиды, такие как, например, лецитин соевых бобов, и сложные эфиры или неполные эфиры, полученные из жирных кислот и гексита, ангидриды, такие как, например, сорбитанмоноолеат, и продукты конденсации неполных эфиров, полученных из жирных кислот и гексита с этиленоксидом, например, моноолеат полиоксиэтиленсорбитана. Эмульсия может также содержать один или несколько подслащивающих и/или ароматизирующих агентов.
Сиропы и эликсиры могут быть составлены с подслащивающими агентами, таких как глицерин, пропиленгликоль, сорбит или сахароза. Такие составы могут также содержать одно или более из средств, уменьшающих раздражение, консервантов, ароматизаторов и/или красителей.
Соединения могут быть составлены для локального или местного введения, например, для местного нанесения на кожу или слизистые оболочки, например, в глаз. Препараты для местного введения обычно содержат местный носитель в сочетании с активным агентом (агентами) с дополнительными необязательными компонентами или без них. Подходящие местные носители и дополнительные компоненты хорошо известны в данной области техники, и будет очевидно, что выбор носителя будет зависеть от конкретной физической формы и способа доставки. Местные носители включают воду; органические растворители, такие как спирты, такие как, например, этаноловый или изопропиловый спирт или глицерин; гликоли, такие как, например, бутилен-, изопрен- или пропиленгликоль; алифатические спирты, такие как, например, ланолин; смеси воды и органических растворителей и смеси органических растворителей, таких как спирт и глицерин; липидные материалы, такие как жирные кислоты, ацилглицерины, включая масла, такие как, например, минеральное масло и жиры природного или синтетического происхождения, фосфоглицериды, сфинголипиды и воски; белковые материалы, такие как коллаген и желатин; материалы на основе силикона, как нелетучие, так и летучие; и материалы на основе углеводородов, такие как микрогубки и полимерные матрицы. Композиция может дополнительно содержать один или более компонентов, предназначенных для улучшения стабильности или эффективности нанесенного состава, такие как стабилизирующие агенты, суспендирующие агенты, эмульгаторы, регуляторы вязкости, гелеобразующие агенты, консерванты, антиоксиданты, усилители проникновения в кожу, увлажнители и материалы пролонгированного высвобождения. Примеры таких компонентов описаны в Martindale The Extra Pharmacopoeia (Pharmaceutical Press, London 1993) и в Martin (ред.), Remington's Pharmaceutical Sciences. Составы могут содержать микрокапсулы, такие как гидроксиметилцеллюлозные или желатиновые микрокапсулы, липосомы, микросферы альбумина, микроэмульсии, наночастицы или нанокапсулы.
Композиция для местного применения может быть приготовлена во множестве физических форм, включая, например, твердые вещества, пасты, кремы, пены, лосьоны, гели, порошки, жидкости на водной основе, эмульсии, спреи, глазные капли и пластыри для кожи. Внешний вид и вязкость таких форм могут определяться наличием и количеством эмульгатора(ов) и регулятора(ов) вязкости, присутствующих в составе. Твердые вещества обычно твердые и не текучие, и обычно изготавливаются в виде стержней или палочек или в виде частиц; твердые вещества могут быть непрозрачными или прозрачными и необязательно могут содержать растворители, эмульгаторы, увлажнители, смягчающие вещества, ароматизаторы, красители/окрашивающих вещества, консерванты и другие активные ингредиенты, которые повышают или улучшают эффективность конечного продукта. Кремы и лосьоны часто похожи друг на друга, отличаются в основном своей вязкостью; как лосьоны, так и кремы могут быть непрозрачными, полупрозрачными или прозрачными и часто содержат эмульгаторы, растворители и средства для регулирования вязкости, а также увлажняющие средства, смягчающие вещества, ароматизаторы, красители/окрашивающие вещества, консерванты и другие активные ингредиенты, которые повышают или улучшают эффективность конечного продукта. Гели могут быть получены с диапазоном вязкостей, от густого геля или высокой вязкости до жидкого или низкой вязкости. Указанные составы, такие как составы лосьонов и кремов, также могут содержать растворители, эмульгаторы, увлажнители, смягчающие вещества, ароматизаторы, красители/окрашивающие вещества, консерванты и другие активные ингредиенты, которые повышают или улучшают эффективность конечного продукта. Жидкости жиже, чем крема, лосьоны или гели и часто не содержат эмульгаторов. Жидкие продукты для наружного применения часто содержат растворители, эмульгаторы, увлажнители, смягчающие вещества, ароматизаторы, красители/окрашивающие вещества, консерванты и другие активные ингредиенты, которые повышают или улучшают эффективность конечного продукта.
Подходящие эмульгаторы для применения в композициях для местного применения включают, но не ограничиваются ими, ионные эмульгаторы, цетеариловый спирт, неионогенные эмульгаторы, такие как полиоксиэтиленовый олеиловый эфир, стеарат ПЭГ-40, цетеарет-12 (ceteareth-12), цетеарет-20 (ceteareth-20), цетеарет-30 (ceteareth-30), цетиловый спирт, стеарат ПЭГ-100 и глицерилстеарат. Подходящие средства для регулирования вязкости включают, но не ограничиваются ими, защитные коллоиды или неионные камеди, такие как гидроксиэтилцеллюлоза, ксантановая камедь, алюмосиликат магния, оксид кремния, микрокристаллический воск, пчелиный воск, парафин и цетилпальмитат. Гелевую композицию можно получить путем добавления гелеобразующего агента, такого как хитозан, метилцеллюлоза, этилцеллюлоза, поливиниловый спирт, поликватерниумы, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбомер или аммонизированный глицирризинат. Подходящие поверхностно-активные вещества включают, но не ограничиваются ими, неионные, амфотерные, ионные и анионные поверхностно-активные вещества. Например, в композициях для местного применения можно применять один или более из сополиола диметикона, полисорбата 20, полисорбата 40, полисорбата 60, полисорбата 80, лаурамида DEA, кокамида DEA и кокамида МЕА, олеилбетаина, кокамидопропил хлорида PG-димониум хлорида и лауретсульфата аммония.
Подходящие консерванты включают, но не ограничиваются ими, противомикробные средства, такие как метилпарабен, пропилпарабен, сорбиновую кислоту, бензойную кислоту и формальдегид, а также стабилизаторы и антиоксиданты, такие как витамин Е, аскорбат натрия/аскорбиновая кислота и пропилгаллат. Подходящие увлажнители включают, но не ограничиваются ими, молочную кислоту и другие гидроксикислоты и их соли, глицерин, пропиленгликоль и бутиленгликоль. Подходящие смягчающие средства включают ланолиновый спирт, ланолин, производные ланолина, холестерин, вазелин, изостеарил неопентаноат и минеральные масла. Подходящие ароматы и красители включают, но не ограничиваются ими, FD&C Красный №40 и FD&C Желтый №5. Другие подходящие дополнительные ингредиенты, которые можно включать в состав композиции для местного применения, включают, но не ограничиваются ими, абразивы, абсорбенты, анти-слеживающие добавки, противопенные агенты, антистатические агенты, вяжущие вещества, такие как, например, гамамелис, алкогольные и травяные экстракты, такие как экстракт ромашки, связующие вещества/вспомогательные вещества, буферные агенты, хелатирующие агенты, пленкообразующие агенты, кондиционирующие агенты, пропелленты, замутняющие агенты, регуляторы рН и защитные вещества.
Примером подходящего носителя для местного применения для состава геля является гидроксипропилцеллюлоза (2,1%); 70/30 изопропиловый спирт/вода (90,9%); пропиленгликоль (5,1%); и полисорбат 80 (1,9%). Примером подходящего носителя для местного применения для состава в виде пены является цетиловый спирт (1,1%); стеариловый спирт (0,5%); кватерниум 52 (1,0%); пропиленгликоль (2,0%); этанол 95 PGF3 (61,05%); деионизированная вода (30,05%); Р75 углеводородный пропеллент (4,30%). Все проценты приведены по массе.
Типичные способы доставки для местных композиций включают нанесение пальцами; нанесение с использованием физического аппликатора, такого как материя, ткань, тампон, палочка или щетка; распыление, включая мелкодисперсный аэрозоль (англ. mist), аэрозоль или распыление пены; нанесение пипеткой; разбрызгивание; замачивание; полоскание. Также можно применять носители с контролируемым высвобождением, и композиции могут быть составлены для трансдермального введения в виде трансдермального пластыря.
Фармацевтическая композиция может быть составлена в виде ингаляционных составов, включая спреи, мелкодисперсные аэрозоли (англ. mist) или аэрозоли. Такие составы особенно полезны для лечения астмы или других респираторных состояний. Соединения, описанные в настоящем изобретении, в виде ингаляционных составов можно вводить с помощью любых способов ингаляции, известных специалистам в данной области. Такие способы ингаляции и устройства для ингаляции включают, но не ограничиваются ими, дозированные ингаляторы с пропеллентами, такими как ХФУ (хлорфторуглерод) или ГФА (гидрофторалкан), или пропеллентами, которые являются физиологически и экологически приемлемыми. Другими подходящими устройствами являются активизируемые вдохом ингаляторы, ингаляторы с несколькими дозами сухого порошка и аэрозольные небулайзеры. Аэрозольные композиции для применения в способе согласно настоящему изобретению обычно включают пропелленты, поверхностно-активные вещества и сорастворители и ими можно заполнять обычные аэрозольные контейнеры, которые закрываются подходящим дозирующим клапаном.
Композиции для ингаляции могут содержать жидкие или порошкообразные композиции, содержащие активный ингредиент и пригодные для распыления и внутрибронхиального применения, или аэрозольные композиции, вводимые через аэрозольную установку, высвобождающую отмеренные дозы. Подходящие жидкие композиции содержат активный ингредиент в водном фармацевтически приемлемом растворителе для ингаляций, например, в изотоническом солевом растворе или бактериостатической воде. Указанные растворы вводят с помощью распылителя с насосом или сжимаемым приводом или любым другим обычным способом для обеспечения требуемого количества для дозировки жидкой композиции для вдыхания в легкие пациента. Подходящие композиции, в которых носитель представляет собой жидкость, для введения, например, в виде назального спрея или в виде назальных капель, включают водные или масляные растворы активного ингредиента.
Составы или композиции, подходящие для назального введения, где носитель представляет собой твердое вещество, включают грубый порошок с размером частиц, например, в диапазоне от 20 до 500 микрон, который вводят тем же способом, которым потребляют нюхательный табак, т.е. порошок, находящийся в контейнере, удерживают близко к носу и быстро вдыхают через носовой проход. Подходящие порошкообразные композиции включают, в качестве иллюстрации, порошкообразные составы активного ингредиента, тщательно перемешанные с лактозой или другими инертными порошками, приемлемыми для внутрибронхиального введения. Порошкообразные композиции можно вводить при помощи аэрозольного распылителя или заключать в хрупкую капсулу, которую пациент может вставить в устройство, которое прокалывает капсулу и выдувает порошок в устойчивом потоке, подходящем для ингаляции.
Фармацевтические композиции также могут быть получены в виде суппозиториев, таких как, например, для ректального введения. Такие композиции можно получать путем смешивания лекарственного средства с подходящим не раздражающим вспомогательным веществом, которое является твердым при обычных температурах, но жидким при ректальной температуре и, следовательно, расплавляется в прямой кишке с высвобождением лекарственного средства. Подходящие вспомогательные вещества включают, например, масло какао и полиэтиленгликоли.
Фармацевтические композиции могут быть составлены в виде композиций с замедленным высвобождением, таких как, например, состав, такой как капсула, который обеспечивает медленное высвобождение модулятора после введения. Указанные составы в целом можно получать с использованием хорошо известной технологии и вводить, например, при помощи перорального, ректального или подкожного имплантата или при помощи имплантата в желаемый целевой участок. Носители для применения в таких составах являются биосовместимыми и могут также быть биоразлагаемыми; предпочтительно состав обеспечивает относительно постоянный уровень высвобождения модулятора. Количество модулятора, содержащегося в составе с замедленным высвобождением, зависит от, например, места имплантации, скорости и ожидаемой продолжительности высвобождения и характера состояния, подлежащего лечению или предотвращению.
Для лечения сердечных повреждений, особенно сердечных аритмий, доза биологически активного соединения согласно настоящему изобретению может варьироваться в широких пределах и может быть адаптирована к индивидуальным требованиям. Активные соединения согласно настоящему изобретению обычно вводят в эффективном количестве, например, в терапевтически эффективном количестве. Предпочтительные дозы варьируются от примерно 0,1 мг до примерно 140 мг на килограмм массы тела в сутки, от примерно 0,5 мг до примерно 7 г на пациента в сутки. Суточную дозу можно вводить в виде разовой дозы или во множестве доз. Количество активного ингредиента, которое можно комбинировать с материалами носителями для получения единичной лекарственной формы, будет варьироваться в зависимости от пациента и конкретного способа введения. Единичные дозы обычно содержат от примерно 1 до примерно 500 мг активного ингредиента.
Однако следует понимать, что конкретный уровень дозы для любого отдельного пациента будет зависеть от множества факторов, включая активность определенного применяемого соединения, возраст пациента, массу тела, общее состояние здоровья, пол, диету, время введения, маршрут введения и скорость экскреции, комбинации лекарственных средств, то есть, других лекарственных средств, применяемых для лечения указанного пациента, и от степени тяжести конкретного заболевания, подлежащего терапии.
Предпочтительные соединения согласно настоящему изобретению будут обладать определенными фармакологическими свойствами. Такие свойства включают, но не ограничиваются ими, пероральную биодоступность, так что предпочтительные пероральные лекарственные формы, описанные выше, могут обеспечить терапевтически эффективные уровни соединения in vivo.
Примеры состояний и заболеваний, связанных с сердечно-сосудистыми заболеваниями и которые можно лечить при помощи соединений для применения согласно настоящему изобретению, включают фибрилляцию предсердий, желудочковую аритмию, сердечную недостаточность, заболевания коронарной артерии, инфаркт миокарда, неадаптивную сердечную гипертрофию и сердечные аритмий, включая желудочковые экстрасистолы, желудочковую тахикардию, злокачественную желудочковую тахикардию, предсердную тахикардию, трепетание предсердий и фибрилляцию предсердий, дилатационную кардиомиопатию и гипертоническую болезнь сердца, предпочтительно выбранные из группы, состоящей из фибрилляции предсердий, предсердной тахикардии, желудочковой аритмии, сердечной недостаточности.
Описанные в настоящем изобретении производные n-3 ПНЖК предпочтительно вводят пациенту, такому как, например, человек, перорально или парентерально, и они присутствуют по меньшей мере в одной жидкости или ткани организма пациента.
В следующем аспекте настоящее изобретение относится к способу лечения сердечно- сосудистого заболевания, предпочтительно сердечно-сосудистого заболевания, указанного выше, включающему стадию введения соединения или композиции согласно настоящему изобретению субъекту, нуждающемуся в этом, предпочтительно млекопитающему, более предпочтительно человеку, в эффективном количестве.
Используемый в настоящем изобретении термин «лечение» охватывает любой тип лечения, модифицирующего заболевание, и включает симптоматическое лечение, то есть, лечение после появления симптомов; при этом любое лечение может быть профилактическим. Однако лечение, модифицирующее заболевание, может включать введение лекарственного средства до появления симптомов, чтобы предотвратить, по меньшей мере задержать или уменьшить тяжесть симптомов после их начала. Лечение, модифицирующее заболевание, также может быть терапевтическим, т.е. происходить после появления симптомов, чтобы уменьшить их тяжесть и/или продолжительность. Лечение после появления симптомов может также просто включать остановку прогрессирования заболевания (стабильное заболевание). В определенном варианте реализации настоящего изобретения производные n-3 ПНЖК, описанные в настоящем изобретении, вводят профилактически, то есть, до наступления заболевания и/или симптомов, в идеале, но необязательно, для того, чтобы фактически предотвратить заболевания и/или симптомы. Следует понимать, что термин «профилактика» и «предотвращение» в контексте настоящего изобретения просто описывает, что соединение (соединения) согласно настоящему изобретению вводят до появления симптомов. Профилактическое введение может представлять собой введение до появления симптомов, которые явно связаны с заболеванием, обсуждаемым в настоящем изобретении: производные n-3 ПНЖК, представленные в настоящем изобретении, можно, например, вводить субъекту профилактически, когда у него или нее присутствуют определенные состояния, которые могут указывать на склонность к развитию одного из состояний или заболеваний, которые можно лечить одним из производных n-3 ПНЖК согласно настоящему изобретению. Такими индикативными условиями являются, например, повышенное кровяное давление или диабет. Такое профилактическое лечение называется первичной профилактикой. В другом варианте реализации настоящего изобретения производные n-3 ПНЖК, описанные в настоящем изобретении, можно вводить субъекту профилактически в случаях, когда он или она ранее страдал(а) состоянием или заболеванием, которое можно лечить с помощью производных n-3 ПНЖК согласно настоящему изобретению, но в настоящее время пациент не проявляет каких-либо симптомов. Такое профилактическое лечение называется вторичной профилактикой. Предполагается, что пациенты, получающие производные n-3 ПНЖК в целях первичной или вторичной профилактики, нуждаются в таком лечении. Пациенты могут включать, но не ограничиваются ими, млекопитающих, особенно людей, домашних животных-компаньонов, таких как собаки, кошки, лошади, и скот, такой как крупный рогатый скот, свиньи, овцы, с дозировками, описанными в настоящем изобретении.
Как будет понятно специалисту в данной области, введение производных n-3 ПНЖК согласно настоящему изобретению благотворно влияет на множество состояний и заболеваний, при этом наиболее значимыми среди них являются сердечно-сосудистые заболевания.
Активность аналогов n-3 ПНЖК согласно настоящему изобретению может быть, например, определена в соответствующих анализах in vitro и/или in vivo. Например, биологическая активность аналогов n-3 ПНЖК согласно настоящему изобретению может быть определена с использованием устоявшейся клеточной модели Канга и Лифа (Kang and Leaf Proc Natl Acad Sci USA, 1994. 91(21): p. 9886-90.), известной специалистам в данной области техники.
Следующие фигуры и примеры служат для иллюстрации настоящего изобретения и не предназначены для того, чтобы ограничивать объем изобретения, описанный в прилагаемой формуле изобретения.
Фигуры
Фиг. 1 представляет собой столбчатую диаграмму, на которой представлены соединения 1-5 (Соед.-01 - Соед.-05), которые являются примерами настоящего изобретения, и другие родственные структуры (Соед.-06 - Соед.-13), а также их потенциал в отношении уменьшения спонтанного биения NRCM (подробнее см. пример 2 ниже).
На фиг. 2 приведена столбчатая диаграмма, на которой показана обработка соединением 2 (Соед.-02), синтетическим агонистом 17,18-EEQ, облегчающую выраженность AF-бремени (А) и тяжести (В) фибрилляции предсердий в мышиной модели умеренной сердечной гипертрофии.
Фиг. 3 представляет собой столбчатую диаграмму, показывающую обработку соединением 3 (Соед.-03), синтетическим агонистом 17,18-EEQ, снижающую продолжительность (А) и тяжесть (В) сердечных аритмий в крысиной модели инфаркта миокарда.
Фиг. 4 представляет собой столбчатую диаграмму, показывающую обработку соединением 3 (Соед.-03), синтетическим агонистом 17,18-EEQ, снижающую продолжительность (А) и тяжесть (В) пост-ишемического функционального восстановления изолированных перфузированных сердец мышей.
Фиг. 5 представляет собой столбчатую диаграмму, показывающую ингибирование растворимой эпоксидгидролазы (sEH) соединениями согласно настоящему изобретению (Соед.-01 - Соед.-03) по сравнению с родственными аналогами (Соед.-07, Соед.-08 и Соед.-10).
Фиг. 6 представляет собой столбчатую диаграмму, показывающую потенциал проницаемости метаболически устойчивых аналогов CYP-эйкозаноидов, относящихся к настоящему изобретению (Соед.-01 - Соед.-04), протестированный на клетках Сасо-2.
Фиг. 7 представляет собой таблицу, суммирующую данные о включении соединений согласно настоящему изобретению (Соед.-02 и Соед.-04) и других родственных соединений в мембранные фосфолипиды.
На фиг. 8 показано, что непрерывная инфузия 100 нм Соед.-02 не вызывала никаких очевидных отрицательных побочных эффектов. После глобальной ишемии резко уменьшилась сократительная способность контрольных сердец (n=5).
На фиг. 9А-9В показано, что Соед.-02 частично защищало от повреждения, вызванного OGD, в первичных кардиомиоцитах. На Фиг. 9С показано высвобождение LDH от Соед.-02 и 17,18-EEQ.
Пример 1: Синтез соединений
Ниже проиллюстрирован синтез некоторых соединений согласно настоящему изобретению.
Соединение 1 (Соед.-01)
Синтез соединения 1 (Соед.-01) был аналогичен синтезу соединения 3 (Соед.-03), в то время как мочевину вводили в соответствии со способом синтеза, описанным в заявке на патент WO 02010/081683 (пример 13).
Соединение 2 (Соед.-02)
Краткое описание синтеза

Общий способ
Спектры ЯМР получали на Bruker Avance 400 МГц для 1Н ЯМР и 100 МГц для 13С ЯМР. ЖХМС проводили на квадрупольном масс-спектрометре Shimadzu LCMS-2010 (колонка: Sepax ODS 50×2,0 мм, 5 мкм) или Agilent 1200 HPLC, 1956 MSD (колонка: Shimpack XR-ODS 30×3,0 мм, 2,2 мкм), работающем в ES (+) ионизационном режиме. Хроматографическое очищение проводили с помощью флэш-хроматографии с использованием силикагеля 100~200 меш. Безводные растворители перед использованием предварительно обрабатывали на колонке 3А MS. Все коммерчески доступные реагенты использовали в том виде, в котором они были получены, если не указано иное.
Общая схема получения соединения 2
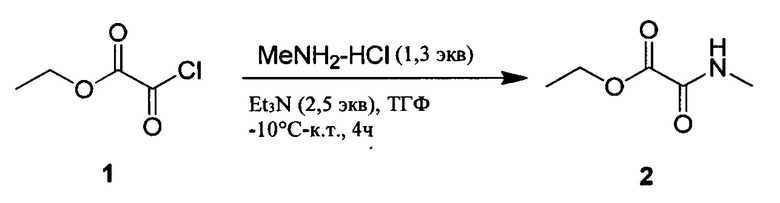

К метанамину (64,29 г, 952,17 ммоль, 1,30 экв.) в 500 мл ТГФ добавляли Et3N (75 г, 732,44 ммоль), полученный раствор добавляли к соединению 1 (100,00 г, 732.44 ммоль, 1,00 экв), Et3N (111 г, 1,1 моль) в ТГФ (1,5 л) при температуре -10°C. Указанную смесь перемешивали при температуре 25°C в течение 16 часов. Затем смесь фильтровали, фильтрат промывали 2 н. HCl (500 мл), экстрагировали ЕА (300 мл × 4), концентрировали и очищали силикагелем (РЕ : ЕА = от 3:1 до 1:1) с получением соединения 2 (70,00 г, 533,82 ммоль, выход 72,88%) в виде желтого масла.
Данные ТСХ (РЕ : EtOAc = 2:1); Rf (Соед.-02) = 0,39; ЖХМС: ЕТ2662-1-Р1А (М+Н+): 131,7; 1Н ЯМР (CDCl3, 400 МГц) 4,36~4,24 (q, J=8 Гц, 2Н), 2,93~2,85 (d, J=4 Гц, 3Н), 1,38~1,30 (t, J=8 Гц, 3Н)
Общая схема получения соединения 4



Раствор соединения 3 (47,50 г, 484,00 ммоль, 1,00 экв.) и DIAD (107,66 г, 532,40 ммоль, 1,10 экв.) в сухом ТГФ (50 мл) медленно добавляли через канюлю к раствору соединения 4 (78,33 г, 532,40 ммоль, 1,10 экв.) и PPh3 (133,30 г, 508,20 ммоль, 1,05 экв.) в сухом ТГФ (100 мл), находящемуся при температуре 0°C. Колбу и канюлю промывали дополнительной порцией сухого ТГФ (30 мл), чтобы обеспечить полное добавление. Реакционной смеси давали постепенно нагреться до температуры 25°C и перемешивали в течение 18 часов. Затем добавляли H2O (1000 мл), экстрагировали ЕА (500 мл × 2), концентрировали и очищали силикагелем (РЕ : ЕА = 0-10:1) с получением соединения 5 (42,5 г, 374,02 ммоль, 77,28%) в виде белого твердого вещества.
Данные ТСХ (РЕ : EtOAc = 5:1); Rf (Соед.-03) = 0,2; Rf (Соед.-05) = 0,5; 1Н ЯМР (CDCl3, 400 МГц) 7,86~7,79 (m, 2Н), 7,72~7,67 (m, 2Н), 3,73~3,66 (t, J=8 Гц, 2Н), 2,27~2,20 (m, 2Н), 1,95~1,91 (t, J=4 Гц, 1Н), 1,85~1,75 (m, 2Н), 1,61~1,52 (m, 2Н)
Общая схема получения соединения 6



NIS (130,68 г, 580,83 ммоль, 1,50 экв.) добавляли одной порцией к раствору соединения 5 (88,00 г, 387,22 ммоль, 1,00 экв.) и AgNO3 (16,44 г, 96,81 ммоль, 0,25 экв.) в сухом ТГФ (1600 мл) при температуре 25°C. Свободное пространство над реакционной смесью промывали N2, реакционную смесь защищали от света, обернув алюминиевой фольгой, и перемешивали в течение 16 часов. Смесь выливали в воду (1000 мл), экстрагировали ЕА (600 мл × 3), концентрировали и очищали на силикагеле (РЕ : ЕА = от 10:1 до 2:1) с получением соединения 6 (118,6 г, 1,01 моль, выход 86,78%) в виде белого твердого вещества.
Данные ТСХ (РЕ : EtOAc = 20:1); Rf (Соед.-05) = 0,22; Rf (Соед 6) = 0,21; 1Н ЯМР (CDCl3, 400 МГц) 7,87~7,82 (m, 2Н), 7,74~7,69 (m, 2Н), 3,74~3,67 (t, J=8 Гц, 2Н), 2,45~2,39 (t, J=8 Гц, 2Н), 1,84~1,74 (m, 2Н), 1,61~1,52 (m, 2Н)
Общая схема получения соединения 7


2-метилбут-2-ен (87,30 г, 1,24 моль, 2,80 экв.) добавляли в течение 30 мин к раствору BH3.Me2S (43,91 г, 577,94 ммоль, 1,30 экв.) в ТГФ (300 мл), находящемуся при температуре 0°C. После 1 ч указанную реакционную смесь нагревали до температуры 25°C и перемешивали в течение 90 минут. После повторного охлаждения до температуры 0°C медленно добавляли раствор соединения 6 (157,00 г, 444,57 ммоль, 1,00 экв.) в ТГФ (900 мл) в течение 1 часа. После полного добавления удаляли холодную баню и указанную реакционную смесь перемешивали при температуре 25°C. Спустя 2 ч указанную реакционную смесь снова охлаждали до температуры 0°C, после чего медленно добавляли ледяной АсОН (260 мл) в течение 30 мин (осторожно: выделение газа) и перемешивали при температуре 25°С в течение 16 часов. ТСХ (РЕ : ЕА = 10:1) показала завершение реакции, смесь выливали в воду (1 л), экстрагировали ЕА (300 мл × 2), концентрировали и очищали силикагелем (РЕ : ЕА = 0-10:1) с получением соединения 7 (135 г, 380,1 ммоль, выход 85,50%) в виде желтого масла.
Данные ТСХ (РЕ : EtOAc = 10:1); Rf (Соед.6) = 0,5; Rf (Соед.7) = 0,55; 1Н ЯМР: (CDCl3, 400 МГц) 7,88~7,80 (m, 2Н), 7,75~7,67 (m, 2Н), 6,24~6,11 (m, 2Н), 3,74~3,66 (t, J=8 Гц, 2Н), 2,24~2,15 (q, J=8 Гц, 2Н), 1,78~1,67 (m, 2Н), 1,55~1,44 (m, 2Н)
Общая схема получения соединения 8


N2H4⋅H2O (97,25 г, 1,94 моль, 5,00 экв.) добавляли к раствору соединения 7 (138,00 г, 388,55 ммоль, 1,00 экв.) в сухом МеОН (2,00 л) при температуре 0°C и перемешивали температуре при 25°C в течение 18 часов, ТСХ (РЕ : ЕА = 10:1) показала завершение реакции, затем указанную реакционную смесь концентрировали, остаток выливали в ДХМ (5000 мл) и перемешивают в течение 30 минут. Отфильтровывали и осадок на фильтре промывали ДХМ (1 л × 2), фильтрат концентрировали с получением соединения 8 (162,00 г, неочищенный) в виде желтого масла.
Данные ТСХ (РЕ : EtOAc = 10:1); Rf (Соед.7) = 0,5; Rf (Соед.8) = 0; Данные ТСХ (ДХМ : МеОН = 10:1); Rf (Соед.7) = 1; Rf (Соед.8) = 0,2; 1Н ЯМР: (CDCl3, 400 МГц) 6,19~6,07 (m, 2Н), 2,73~2,59 (m, 2Н), 2,20~2,05 (m, 2Н), 1,75~1,55 (m, 2Н), 1,51~1,36 (m, 4Н)
Общая схема получения соединения 9


Соединение 8 (92,00 г, 408,76 ммоль, 1,00 экв.), соединение 2 (53,60 г, 408,76 ммоль, 1,00 экв.) и Et3N (49,64 г, 490,51 ммоль, 1,20 экв.) в сухом этаноле (1,5 л) нагревали при температуре 60°C в течение 20 часов. ТСХ (ДХМ : МеОН = 10:1) показала завершение реакции, затем указанную смесь концентрировали до примерно 300 мл. Отфильтровывали и концентрировали с получением соединения 9 (90 г, 232,16 ммоль, выход 57%) в виде белого твердого вещества.
Данные ТСХ (ДХМ : МеОН = 10:1); Rf (Соед.8) = 0,2; Rf (Соед.9) = 0,5; 1Н ЯМР: (CDCl3, 400 МГц) 7,57~7,37 (s, 2Н), 6,25-6,20 (d, J=8 Гц, 1Н), 6,18~6,11 (q, J=8 Гц, 1Н), 3,37~3,30 (q, J=8 Гц, 2Н), 2,93~2,88 (d, J=4 Гц, 3Н), 2,21~2,13 (m, 2Н), 1,66~1,56 (m, 2Н), 1,53-1,43 (m, 2Н)
Общая схема получения соединения 12
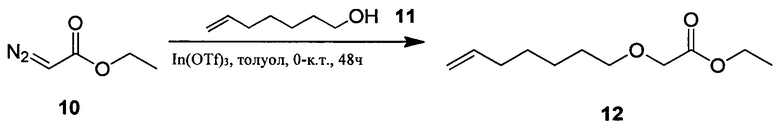
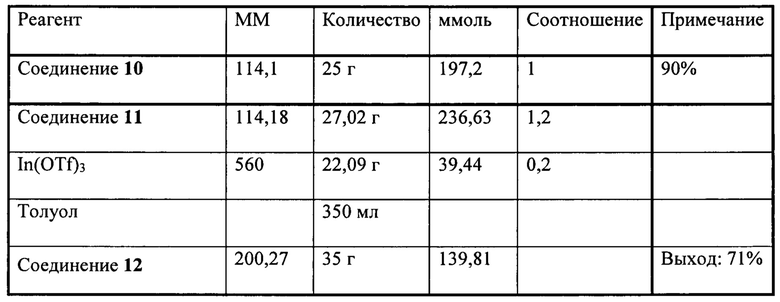
Соединение 10 (25,00 г, 197,20 ммоль, 1,00 экв.) в толуоле (75 мл) добавляли к раствору соединения 11 (27,02 г, 236,63 ммоль, 1,20 экв.) In(OTf)3 (22,09 г, 39,44 ммоль, 0,20 экв.) в толуоле (275 мл) в течение 20 мин. Затем смесь перемешивали при температуре 25°C в течение 48 часов. Смесь концентрировали и очищали силикагелем (РЕ : ЕА = 20:1) с получением этилового соединения 12 (35,00 г, 139,81 ммоль, выход 70,90%, чистота 80%) в виде желтого масла.
Данные ТСХ (РЕ : EtOAc = 10:1); Rf (Соед.11) = 0,21; Rf (Соед.12) = 0,55; 1Н ЯМР: (CDCl3, 400 МГц) 5,86~5,72 (m, 1Н), 5,03~5,86 (m, 2Н), 4,24~4,17 (q, J=8 Гц, 2Н), 4,07~4,01 (s, 2Н), 3,54~3,47 (t, J=8 Гц, 2Н), 2,09~1,98 (m, 2Н), 1,68~1,55 (m, 2Н), 1,45~1,32 (m, 4Н), 1,30~1,25 (t, J=8 Гц, 3Н)
Общая схема получения соединения 13
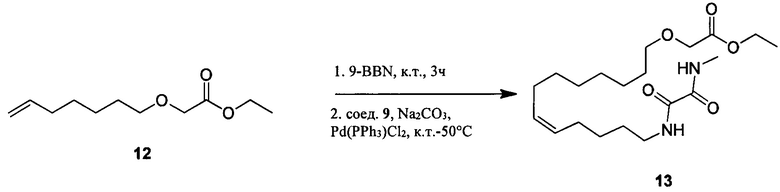


В высушенную в печи колбу, содержащую 9-BBN (17,53 г, 100,60 ммоль, 2,40 экв.) в ТГФ (540 мл), добавляли раствор соединения 12 (10,07 г, 50,30 ммоль, 1,20 экв.) в ТГФ (60 мл) при температуре 0°C. После перемешивания при температуре 25°C в течение 16 ч добавляли водный раствор Na2CO3 (200 мл 2 М раствора, полученного из H2O, барботированной аргоном). Через 2 часа добавляли Pd(PPh3)2Cl2 (1,47 г, 2,10 моль, 0,05 экв.), а затем соединение 9 (13,00 г, 41,92 ммоль, 1,00 экв.), растворенное в ТГФ (200 мл). Полученный раствор красного цвета защищали от света. Указанную смесь перемешивали при температуре 50°C в течение 5 часов. ТСХ показала завершение реакции. После охлаждения до температуры 25°C указанную реакционную смесь концентрировали в вакууме и остаток очищали на силикагеле (РЕ : ЕА = от 10:1 до 3:1) с получением соединения 13 (6,5 г, 16,06 ммоль, выход 38,31%, чистота 95%) в виде желтого твердого вещества.
Данные ТСХ (РЕ : EtOAc = 2:1); Rf (Соед.12) = 0,3; Rf (Соед.13) = 0,3; ЖХМС: ET2662-38-P1D (М+Н+): 385,1; 1Н ЯМР: (CDCl3, 400 МГц) 7,57~7,38 (s, 1Н), 5,41~5,25 (m, 2Н), 4,25~4,17 (q, J=8 Гц, 2Н), 4,07~4,02 (s, 2Н), 3,54~3,47 (t, J=8 Гц, 2Н), 3,34~3,26 (q, J=8 Гц, 2Н), 2,92~2,87 (d, J=8 Гц, 3Н), 2,08~1,94 (m, 4Н), 1,65~1,51 (m, 4Н), 1,43~1,23 (m, 13Н)
Общая схема получения соед.-02


К раствору соединения 13 (7,50 г, 19,51 ммоль, 1,00 экв.) в ТГФ (70,00 мл) добавляли LiOH (934,31 мг, 39,02 ммоль, 2,00 экв.) в H2O (40,00 мл) при температуре 0°C и затем указанную реакционную смесь перемешивали при температуре 0-25°C в течение 1 часа. ЖХМС показала завершение реакции. Затем к указанной реакционной смеси добавляли H2O (60 мл), водную фазу обрабатывали 3 н. HCl (10 мл) до рН 3-4, экстрагировали ЕА (100 мл * 3), сушили, органическую фазу концентрировали с получением неочищенного продукта. Остаток очищали с помощью колоночной хроматографии на силикагеле (РЕ : ЕА = 5:1 до ЕА) с получением Соед.-02 (4,00 г, 10,72 ммоль, выход 54,95%, чистота 95,51%)
Данные ТСХ (ДХМ : МеОН = 10:1); Rf (Соед.13) = 0,9; Rf (Соед.-02) = 0,4; МС: ЕТ2662-43-Р1С (M+Na+): 379,2; 1Н ЯМР (CDCl3, 400 МГц) 7,84 (s, 1Н), 7,74 (s, 1Н), 5,40~5,32 (m, 2Н), 4,11 (s, 2Н), 3,59~3,55 (t, J=6,4 Гц, 2Н), 3,35~3,32 (t, J=6,8 Гц, 2Н), 2,92~2,91 (d, J=5,2 Гц, 3Н), 2,07~2,00 (m, 4Н), 1,64-1,59 (m, 4Н), 1,42~1,32 (m, 10Н); 13С ЯМР (CDCl3, 100 МГц) δ 173,7, 160,7, 159,8, 130,5, 129,0, 72,0, 67,8, 39,7, 29,4, 29,3, 29,0, 29,0, 28,6, 27,1,26,8, 26,7, 25,8
Соединение 3 (Соед.-03)
Краткое описание синтеза


Синтез 2-(гекс-5-ин-1-ил)изоиндолин-1,3-диона (2): Следуя описанной в литературе методике1, раствор 5-гексин-1-ола (1) (5 г, 1 экв.) и диизопропилазодикарбоксилата (DIAD, 10,5 г, 1,02 экв.) в сухом ТГФ (30 мл) медленно добавляли через канюлю к раствору фталимида (7,5 г, 51 ммоль) и трифенилфосфина (ТРР, 13,4 г, 1 экв.) в сухом ТГФ (50 мл), находящемуся при температуре 0°C. Колбу и канюлю промывали дополнительной порцией сухого ТГФ (20 мл), чтобы обеспечить полное добавление. Затем смесь оставляли постепенно нагреваться до комнатной температуры в течение ночи. Через 18 часов все летучие вещества выпаривали и остаток очищали с помощью хроматографической системы Teledyne Isco Combiflash® RF (80 г колонка SiO2, элюировали гексанами, 2 мин; 0-20% EtOAc/гексанами, 12 мин; 20% EtOAc/гексанами, 6 мин) с получением 2 (8,3 г, 72%) в виде белого твердого вещества, спектральные данные которого были идентичны указанным2.

Синтез 2-(6-йодгекс-5-ин-1-ил)изоиндолин-1,3-диона (3): Следуя описанному в литературе прецеденту3, N-йодсукцинимид (NIS, 7,4 г, 1,5 экв.) добавляли одной порцией к раствору алкина 2 (5,0 г, 22 ммоль) и AgNO3 (0,93 мг, 0,25 экв.) в сухом ТГФ (120 мл), находящемуся при комнатной температуре. Свободное пространство над реакционной смесью промывали аргоном, и реакционную смесь защищали от света, обернув алюминиевой фольгой. Через 4 часа указанную реакционную смесь выливали в Н2О (200 мл) и экстрагировали Et2O (2×50 мл). Эфирные экстракты промывали солевым раствором (3×60 мл) (Примечание: двухфазная смесь становилась коричневой). Объединенные водные фазы повторно экстрагировали Et2O (2×50 мл). Объединенные эфирные экстракты сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Остаток очищали с применением хроматографической системы Teledyne Isco Combiflash® RF (80 г колонка SiO2, элюировали гексанами, 2 мин; 0-40% EtOAc/гексанами, 8 мин; 40% EtOAc/гексанами, 10 мин; 40-100%) EtOAc/гексанами, 5 мин; 100%, EtOAc, 3 мин) с получением 3 (97%) в виде белого твердого вещества, т.пл. 132,5-132,7°C. 1Н ЯМР (500 МГЦ, CDCl3) δ 7,85 (ddd, J=5,4, 3,0, 1,0 Гц, 2Н), 7,72 (ddd, J=5,5, 3,0, 1,0 Гц, 2Н), 3,71 (t, J=7,1 Гц, 2Н), 2,42 (t, J=7,0 Гц, 2Н), 1,83-1,73 (m, 2Н), 1,61-1,51 (m, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 168,62, 134,14, 132,30, 123,44, 94,04, 37,60, 27,91, 25,89, 20,60, -6,27.

Синтез 2-(6-йодгекс-5(Z)-ен-1-ил)изоиндолин-1,3-диона (4): Следуя литературному прецеденту4, чистый 2-метил-2-бутен (4,2 мл, 2,8 экв.) в течение 5 мин добавляли к раствору ВН3⋅Me2S (2,0 М в ТГФ, 9,2 мл, 1,3 экв.) в ТГФ (3 мл), находящемуся при температуре 0°C. После 1 ч указанную реакционную смесь нагревали до комнатной температуры и перемешивали в течение 90 минут. После повторного охлаждения до температуры 0°C медленно добавляли раствор йодоалкина 3 (5 г, 1 экв.) в ТГФ (30 мл) в течение 5 мин. После полного добавления удаляли холодную баню и реакционную смесь перемешивали при комнатной температуре. Спустя 2 ч реакционную смесь снова охлаждали до температуры 0°C, после чего медленно добавляли ледяной АсОН (8,5 мл) в течение 5 мин (Осторожно: выделение газа). После перемешивания в течение ночи (14 ч) указанную реакционную смесь разбавляли Н2О (20 мл), затем осторожно выливали в перемешиваемый насыщенный раствор бикарбоната натрия (40 мл). Двухфазную смесь экстрагировали эфиром (2×40 мл) и объединенные эфирные экстракты промывали водой, солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с применением хроматографической системы Teledyne Isco Combiflash® RF (40 г колонка SiO2, элюировали 0-20% EtOAc/гексанами, 8 мин; 20% EtOAc/гексанами, 6 мин) с получением смеси (4,52 г) 4 и побочного продукта борана. Дальнейшую очистку откладывали до следующего шага.

Синтез 6-йодгекс-5(Z)-ен-1-амина (5): Следуя литературному прецеденту5, 40% мас. MeNH2 в H2O (15 мл) добавляли к раствору неочищенного 4 (4,52 г) в сухом этаноле (20 мл), находящемуся при комнатной температуре. После перемешивания в течение ночи (18 часов) указанную реакционную смесь выливали в ледяную воду (100 мл) и экстрагировали Et2O (30 мл × 2). Объединенные эфирные экстракты промывали холодным 1 н. раствором HCl (20 мл × 2). Объединенные водные смывы доводили до значения рН 8 разбавлением водн. NaOH. Раствор экстрагировали Et2O (30 мл × 2), сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного 5 (1,12 г) в виде коричневого масла, которое использовали на следующей стадии без дальнейшего очистки. 1Н ЯМР (500 МГц, CDCl3) δ 6,29-6,08 (m, 2Н), 2,71 (tt, J=7,0, 1,8 Гц, 2Н), 2,16 (прим. q, J=6,5 Гц, 2Н), 1,78-1,52 (m, 2Н).
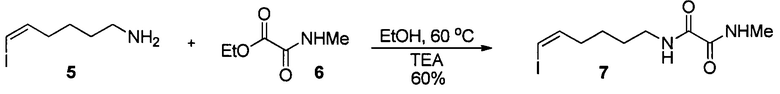
Синтез N1-(6-йодгекс-5(Z)-ен-1-ил)-N2-метилоксаламида (7): Следуя литературному прецеденту6, раствор йодалкена 5 (1,12 г, 4,98 ммоль), этил 2-(метиламино)-2-оксоацетата (6) (0,62 г, 1,2 экв.) и триэтиламина (0,83 мл, 1,2 экв.) в сухом этаноле (10 мл) нагревали при температуре 60°C. Через 20 ч коричневый раствор охлаждали до комнатной температуры и концентрировали в вакууме. Проводили очистку остатка с применением хроматографической системы Teledyne Isco Combiflash® RF (25 г колонка SiO2, элюировали 0-50% EtOAc/гексанами, 10 мин; 50% EtOAc/гексанами, 10 мин) с получением 7 (0,93 г, 60%) в виде белого твердого вещества, 99,7-99,8°C. 1Н ЯМР (500 МГц, CDCl3) δ 7,46 (шир. s, 2H), 6,32-6,02 (m, 2H), 3,34 (прим. q, J=6,9 Гц, 2H), 2,91 (d, J=5,3 Гц, 3Н), 2,18 (dt, J=7,5, 7,0 Гц, 2H), 1,68-1,59 (m, 2Н), 1,54-1,42 (m, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 160,47, 159,70, 140,43, 83,07, 39,40, 34,11, 28,61, 26,15, 25,11.
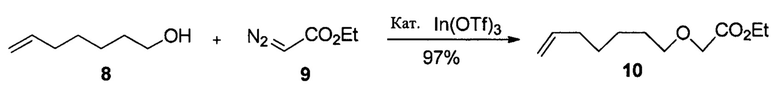
Синтез этил-2-(окт-7-ен-1-илокси)ацетата (10): В соответствии с литературным прецедентом7 к суспензии In(OTf)3 (1,57 г, 20 мол.%) в сухом толуоле (20 мл) комнатной температуры добавляли чистое соединение 8 (1,92 г, 1,2 экв.). Этилдиазоацетат (9) (1,60 г, 14 ммоль) медленно добавляли в атмосфере аргона в течение 5 мин (осторожно: экзотермическая реакция) с получением раствора желтого цвета. Через 2 дня указанную реакционную смесь концентрировали под вакуумом и остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash® RF (колонка 25 г SiO2, элюировали 0-10% EtOAc/гексанами, 5 мин; 10% EtOAc/гексанами, 8 мин) с получением 10 (2,72 г, 97%) в виде бесцветного масла. 1Н ЯМР (500 МГц, CDCl3) δ 5,80 (ddt, J=16,9, 10,2, 6,6 Гц, 1Н), 5,08-4,84 (m, 2H), 4,22 (q, J=7,1 Гц, 2H), 4,06 (s, 2H), 3,52 (t, J=6,7 Гц, 2H), 2,13-1,96 (m, 2H), 1,72-1,52 (m, 2H), 1,48-1,33 (m, 4H), 1,28 (t, J=7,1 Гц, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 170,70, 138,99, 114,48, 71,97, 68,48, 60,86, 33,84, 29,55, 28,84, 25,63, 14,34.

Синтез этил-2-((13-(2-(метиламино)-2-оксоацетамидо)тридец-8(Z)-ен-1-ил)окси)ацетата (11): В высушенную в печи колбу, содержащую этил 2-(окт-7-ен-1-илокси)ацетат (10) (220 мг, 1,2 экв.), добавляли раствор 9-BBN (0,5 М в ТГФ, 2,4 экв., 4,40 мл). После перемешивания при комнатной температуре в течение 3 ч добавляли водный раствор Na2CO3 (1,5 мл 2 М раствора, полученного из H2O, барботированной аргоном). Через 5 мин добавляли Pd(PPh3)2Cl2 (33 мг, 5 мол.%), а затем 7 (284 мг, 0,92 ммоль), растворенный в ТГФ (4 мл). Полученный раствор красного цвета был защищен от света, в то время как добавляли другую порцию водн. Na2CO3 (0,5 мл 2 М раствора). Реакция продолжалась в течение ночи (14 часов) при комнатной температуре, затем при температуре 50°C в течение 4 часов. После охлаждения до комнатной температуры указанную реакционную смесь концентрировали под вакуумом и остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash® RF (колонка 24 г SiO2, элюировали 0-40% EtOAc/гексанами, 5 мин; 40% EtOAc/гексанами, 8 мин; 40-100% EtOAc/гексанами, 4 мин) с получением эфира 11 (330 мг, 90%) в виде беловатого твердого вещества. Аналитический образец очищали при помощи препаративной ТСХ с получением 11 в виде белого низко плавкого твердого вещества.
ТСХ: 50% EtOAc/гексаны, Rf ~ 0,49. 1Н ЯМР (500 МГц, CDCl3) δ 7,45 (шир. s, 2Н), 5,42-5,26 (m, 2Н), 4,22 (q, J=7,2 Гц, 2Н), 4,06 (s, 2Н), 3,52 (t, J=6,7 Гц, 2Н), 3,31 (dt, J=7,0, 6,5 Гц, 2Н), 2,91 (d, J=5,1 Гц, 3Н), 2,15-1,91 (m, 4Н), 1,70-1,50 (m, 2Н), 1,44-1,31 (m, 12Н), 1,29 (t, J=7,1 Гц, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 170,62, 160,55, 159,66, 130,58, 128,86, 71,96, 68,64, 39,55, 29,61, 29,51, 29,30, 29,19, 27,20, 26,83, 26,67, 26,15, 25,93, 14,21.
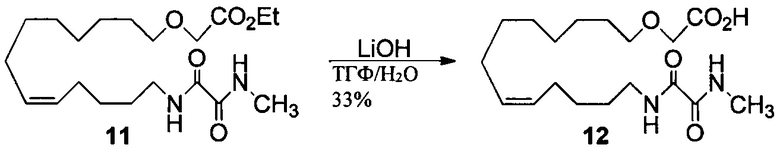
Синтез 2-((13-(2-(метиламино)-2-оксоацетамидо)тридец-8(Z)-ен-1-ил)окси)уксусной кислоты (12): К раствору 11 (720 мг, 1,87 ммоль) в ТГФ (44 мл), находящемуся при комнатной температуре, добавляли LiOH (9 мл 1,0 М водного раствора). Через 48 часов указанную реакционную смесь охлаждали до температуры 4°C и подкисляли до значения рН 4 с помощью водного раствора 2 н. HCl. Реакционную смесь разбавляли Н2О (10 мл) и экстрагировали EtOAc (15 мл × 3). Объединенные органические фазы сушили над Na2SO4, фильтровали через воронку из фриттованного стекла и концентрировали в вакууме. Неочищенный материал очищали с применением хроматографической системы Teledyne Isco Combiflash® RF (12 г колонка SiO2, элюировали 0-80% EtOAc/гексанами, 15 мин; 80% EtOAc/гексанами, 5 мин) с получением 12 (232 мг, 33%) в виде белого твердого вещества, т.пл. 94,6-94,7°C.
1Н ЯМР (500 МГц, CDCl3) δ 7,90 (s, 1H), 7,66 (s, 1H), 5,48-5,22 (m, 2H), 4,10 (s, 2H), 3,58 (t, J=6,5 Гц, 2H), 3,32 (dt, J=7,0, 6,5 Гц, 2H), 2,91 (d, J=5,2 Гц, 3Н), 2,16-1,90 (m, 4H), 1,71-1,48 (m, 4H), 1,45-1,18 (m, 10Н); 13C ЯМР (75 МГц, CD3OD) δ 176,96, 160,32, 160,12, 130,65, 129,99, 72,51, 69,84, 39,45, 29,82, 29,58, 29,15, 27,71, 27,38, 27,24, 27,08, 26,83, 25,84, 25,03.

Синтез Соед.-03: Смесь EDCI (275 мг, 1,3 экв.) и триэтиленгликоля (1,5 мл, 10 экв.) сушили в высоком вакууме в течение 90 мин. Реакционную колбу промывали аргоном и DMAP (175 мг, 1,3 экв.), ацетонитрилом (50 мл) и добавляли кислоту 12 (395 мг, 1,1 ммоль), растворенную в CH2Cl2 (20 мл). Через 3 дня указанную реакционную смесь концентрировали в вакууме, неочищенный остаток растворяли в EtOAc (20 мл) и промывали 1 н. HCl (20 мл) и солевым раствором (20 мл). Водные смывы повторно экстрагировали EtOAc (20 мл × 2). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с применением хроматографической системы Teledyne Isco Combiflash® RF (12 г колонка SiO2, элюировали 0-80% EtOAc/гексанами, 8 мин; 80%) EtOAc/гексанами, 4 мин; 80-100% EtOAc/гексанами, 3 мин; 100% EtOAc, 15 мин; 10% МеОН/CH2Cl2, 5 мин) с получением аналога 13 (174 мг, 32%) в виде белого твердого вещества, т.пл. 65,3-65,8°C.
1Н ЯМР (500 МГц, CDCl3) δ 7,46 (s, 2Н), 5,41-5,27 (m, 2Н), 4,33 (t, J=4,7 Гц, 2Н), 4,11 (s, 2Н), 3,77-3,70 (m, 4Н), 3,70-3,64 (m, 4Н), 3,61 (прим. t, J=4,5 Гц, 2Н), 3,52 (t, J=6,7 Гц, 2Н), 3,42 (t, J=6,1 Гц, ОН), 3,31 (dt, J=7,0, 6,5 Гц, 2Н), 2,91 (d, J=5,2 Гц, 3Н), 2,44 (s, 1Н), 2,05 (dt, J=7,5, 7,0 Гц, 2Н), 2,00 (dt, J=7,0, 6,5 Гц, 2Н), 1,62-1,50 (m, 4Н), 1,45-1,21 (m, 10Н); 13С ЯМР (125 МГц, CDCl3) δ 170,86, 160,83, 159,95, 130,76, 129,12, 72,76, 72,21, 70,77, 70,52, 69,19, 68,34, 63,84, 61,92, 39,78, 29,84, 29,73, 29,54, 29,42, 29,02, 27,44, 27,09, 26,92, 26,42, 26,15.
Соединение 4 (Соед.-04)
Синтез соединения 4 (Соед.-04) был аналогичен синтезу соединения 2 (Соед.-02), в то время как мочевинную группу вводили в соответствии со способом синтетеза, описанным в заявке на патент WO 02010/081683 (пример 6).
Соединение 5 (Соед.-05)
Краткое описание синтеза

Общая схема получения соединения 4-2


Смесь Соед.4-1 (30,0 г, 137 ммоль, 1,0 экв.) в TEA (480 мл) добавляли соед.1 (13,4 г, 137 ммоль, 1,0 экв.), CuI (522 мг, 2,74 ммоль, 0,02 экв.), Pd(PPh3)4 (1,58 г, 1,37 ммоль, 0,01 экв.) под N2 при температуре 25°C и перемешивали при температуре 25°C в течение 16 часов. ТСХ (петролейный эфир/этилацетат = 1/1, Rf=0,5) показала завершение реакции. Раствор выливали в водн. NH4Cl (1,0 л), экстрагировали ДХМ (200 мл × 5), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя петролейным эфиром : EtOAc (10:1, 1:1) с получением Соед.4-2 (21,0 г, выход 73%) в виде желтого масла.
1Н ЯМР: ЕТ5008-6-P1b1 400 МГц CDCl3; 7,30-7,24 (m, 1Н), 7,10 (t, J=7,6 Гц, 1Н), 6,73-6,65 (m, 2Н), 4,19 (шир., 2Н), 3,74 (m, 2Н), 2,54 (t, J=6,0 Гц, 2Н), 1,87-1,68 (m, 4Н), 1,50-1,45 (m, 1Н).
Общая схема получения соединения 4-3
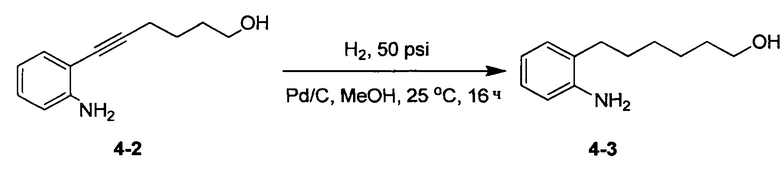

Смесь Соед.4-2 (21,0 г, 111 ммоль, 1,0 экв.) в МеОН (500 мл) добавляли Pd/C (500 мг) и перемешивали при температуре 25°C при 50 фунтах на квадратный дюйм (psi) (~344,74 кПа) Н2 в течение 16 часов. ЖХМС (ЕТ5008-10-Р1А5, продукт: RT = 1,10 мин) показала завершение реакции. Затем раствор фильтровали и концентрировали с получением Соед.4-3 (17,0 г, выход 75%) в виде желтого масла.
1Н ЯМР: ЕТ5008-10-P1b1 400 МГц CDCl3; 7,08-7,03 (m, 2Н), 6,78-6,69 (m, 2Н), 3,69-3,62 (m, 4Н), 2,52 (t, J=8,0 Гц, 2Н), 1,68-1,59 (m, 4Н), 1,47-1,42 (m, 4Н), 1,31-1,27 (m, 1Н).
Общая схема получения соединения 4-4
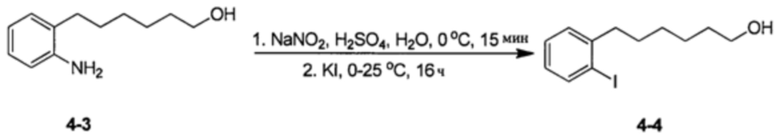

Конц. NH2SO4 (30,2 г, 308 ммоль, 3,5 экв.) добавляли к соед.4-3 (17,0 г, 88,0 ммоль, 1,0 экв.) в Н2О (500 мл) при температуре 0°C в атмосфере N2. К указанному раствору при температуре 0°C добавляли раствор NaNO2 (6,07 г, 88,0 ммоль, 1,0 экв.) в Н2О (30,0 мл) и перемешивали при температуре 0°C в течение 15 минут. Добавляли раствор KI (43,8 г, 264 ммоль, 3,0 экв.) в Н2О (30,0 мл) при температуре 0°C и полученную суспензию нагревали до температуры 25°C и перемешивали в течение 45 минут. ТСХ (петролейный эфир/этилацетат = 1/1, Rf=1) показала, что реакция завершена. Добавляли Н2О (400 мл), экстрагировали EtOAc (350 мл × 3), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя петролейным эфиром : EtOAc (100:1, 10:1) с получением соед.4-4 (17,0 г, выход 57%) в виде коричневого масла.
1Н ЯМР: ЕТ5008-22-P1b1 400 МГц CDCl3; 7,80 (d, J=7,2 Гц, 1Н), 7,28-7,23 (m, 1H), 7,21-7,18 (m, 1H), 6,89-6,85 (m, 1H), 3,65 (t, J=6,8 Гц, 2H), 2,71 (t, J=8,0 Гц, 2H), 1,61-1,50 (m, 4H), 1,45-1,40 (m, 4H), 1,31-1,28 (m, 1H).
Общая схема получения соединения 4-5'


Смесь BrCH2CO2tBu (7,70 г, 39,5 ммоль, 1,2 экв.) и соед.4-4 (10,0 г, 32,9 ммоль, 1,0 экв.) в толуоле (50,0 мл) добавляли Bu4NHSO4 (5,58 г, 16,4 ммоль, 0,50 экв.), KOH (33,0 г, 588 ммоль, 17,9 экв.) в H2O (50,0 мл) при температуре 0°C, затем смесь перемешивали при температуре 25°C в течение 16 часов. ТСХ (петролейный эфир/этилацетат = 10/1, Rf=0,62) показала, что оставалось 40% SM. Добавляли H2O (200 мл), экстрагировали ДХМ (200 мл × 2), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя петролейным эфиром : EtOAc (40:1) с получением Соед.4-5' (5,40 г, выход 37%) в виде желтого масла.
1Н ЯМР: ЕТ5008-26-P1b1 400 МГц CDCl3;  7,82 (d, J=7,2 Гц, 1Н), 7,30-7,26 (m, 1H), 7,23-7,20 (m, 1H), 6,91-6,88 (m, 1H), 3,97 (s, 2H), 3,53 (t, J=6,8 Гц, 2H), 2,72 (t, J=8,0 Гц, 2H), 1,69-1,59 (m, 4H), 1,58-1,43 (m, 13H).
7,82 (d, J=7,2 Гц, 1Н), 7,30-7,26 (m, 1H), 7,23-7,20 (m, 1H), 6,91-6,88 (m, 1H), 3,97 (s, 2H), 3,53 (t, J=6,8 Гц, 2H), 2,72 (t, J=8,0 Гц, 2H), 1,69-1,59 (m, 4H), 1,58-1,43 (m, 13H).
Общая схема получения соединения 4-6


Смесь Соед.4-5' (5,40 г, 12,9 ммоль, 1,0 экв.) и Соед.2 (2,18 г, 12,9 ммоль, 1,0 экв.) в Et3N (110 мл) добавляли CuI (49,2 мг, 258 мкмоль, 0,02 экв.), PdCl2(PPh3)2 (181 мг, 258 мкмоль, 0,02 экв.), при температуре 25°C в атмосфере N2 и перемешивали при температуре 25°C в течение 16 часов. ТСХ (петролейный эфир/этилацетат = 1/1, Rf=0,3) показала завершение реакции. Добавляли водн. NH4Cl (200 мл), экстрагировали EtOAc (200 мл × 3), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя петролейным эфиром: EtOAc (10:1, 1:1) с получением Соед.4-6 (3,00 г, выход 48%) в виде желтого масла.
1Н ЯМР: ЕТ5008-32-P1b1 400 МГц CDCl3; 7,41-7,34 (m, 1Н), 7,23-7,06 (m, 3Н), 4,97-4,87 (m, 1Н), 3,97 (s, 2Н), 3,53 (t, J=6,8 Гц, 2Н), 3,43-3,33 (m, 2Н), 2,72 (t, J=8,0 Гц, 2Н), 2,64 (J=8,0 Гц, 2Н), 1,69-1,59 (m, 4Н), 1,55-1,43 (m, 22Н).
Общая схема получения соединения 4-7


Смесь Соед.4-6 (3,00 г, 6,53 ммоль, 1,0 экв.) в МеОН (20,0 мл) добавляли Pd/C (200 мг) и перемешивали при температуре 25°C при 50 фунтах на квадратный дюйм (~344,74 кПа) H2 в течение 5 часов. ЖХМС (ЕТ5008-33-Р1А4, продукт: RT = 1,04 мин) показала завершение реакции. Затем раствор фильтровали и концентрировали с получением Соед.4-7 (2,50 г, выход 75%) в виде желтого масла.
1Н ЯМР: ЕТ5008-33-P1b1 400 МГц CDCl3; 7,13 (s, 4Н), 4,54 (s, 1Н), 3,96 (s, 2H), 3,52 (t, J=6,8 Гц, 2H), 3,18-3,14 (m, 2H), 2,65-2,57 (m, 4H), 1,75-1,54 (m, 10Н), 1,53-1,37 (m, 20H).
Общая схема получения соединения 4-10
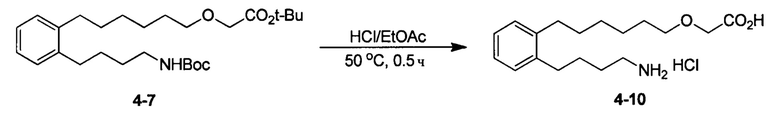

Смесь Соед.4-7 (1,00 г, 2,16 ммоль, 1,0 экв.) в HCl/EtOAc (30,0 мл) при температуре 50°C и перемешивали при температуре 50°C в течение 0,5 часа. ЖХМС (ЕТ5008-34-Р1А4, продукт: RT = 0,698 мин) показала завершение реакции. Смесь концентрировали с получением неочищенного продукта Соед.4-10 (800 мг) в виде желтого твердого вещества.
Общая схема получения Соед.-05
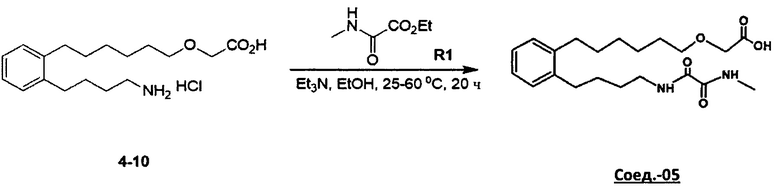

Смесь Соед.4-10 (800 мг, 2,33 ммоль, 1,0 экв.) в EtOH (40,0 мл) добавляли Et3N (2,36 г, 23,3 ммоль, 10,0 экв.) и Соед. R1 (611 мг, 4,66 ммоль, 2,0 экв.) при температуре 25°C. Затем указанный раствор перемешивали при температуре 60°C в течение 20 часов. ЖХМС (ЕТ5008-35-Р1А1, продукт: RT = 0,81 мин) показала завершение реакции. Раствор концентрировали. Остаток очищали с помощью предварительной ВЭЖХ (условия TFA) с получением Соед.-05 (370 мг, выход 40%) в виде белого твердого вещества.
Метод выделения ВЭЖХ:

1Н ЯМР: ЕТ5008-35-P1b1 400 МГц CDCl3; 10,46 (шир., 1Н), 8,35 (s, 1Н), 7,74 (s, 1Н), 7,12 (s, 4Н), 4,12 (s, 2Н), 3,59 (t, J=6,0 Гц, 2Н 2Н), 3,35 (q, J=7,2 Гц, 2Н), 2,92 (d, J=5,2 Гц, 3Н), 2,65-2,57 (m, 4Н), 1,68-1,44 (m, 14Н).
Для синтеза соединений с Соед.-14 по Соед.-34 были предварительно синтезированы общие структурные единицы:
Структурная единица 1 (ВВ-1)
N'-[(5Z)-6-иодогекс-5-ен-1-ил]-Nметилэтандиамид
Стадия 1:
PPh3 (140 г) и фталимид (82,5 г) суспендировали в сухом ТГФ (500,0 мл) и охлаждали до температуры 0°C. Затем по каплям в течение 45 мин добавляли раствор 5-гексин-1-ола (50,0 г) и диизопропилазодикарбоксилата (110 мл) в сухом ТГФ (100 мл). Полученную смесь перемешивали при температуре 0°C в течение 1 часа и затем при комнатной температуре в течение ночи.
ТГФ удаляли в вакууме, насколько это возможно. Остаток суспендировали в РЕ/EtOAc = 9:1 (700 мл) и интенсивно перемешивали. Растворитель декантировали из осажденного OPPh3. Во время этого процесса в декантированном растворителе образовывались белые иглы (продукт), которые были отфильтрованы и отложены (F1).
Затем осадок OPPh3 дополнительно промывали РЕ/EtOAc = 9:1 несколько раз. Затем все фильтраты объединяли и выпаривали в вакууме (F2). F1 в виде игл растворяли в EtOAc (200 мл) и промывали 1 н. NaOH (2×75 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток фильтровали через слой SiO2 (элюент CH2Cl2). Растворитель удаляли в вакууме и маслянистый остаток оставляли стоять в холодильнике в течение выходных, после чего образовались белые иглы. Смесь разбавляли РЕ, затем продукт отфильтровывали, промывали РЕ и сушили в вакууме с получением F1 в виде белых игл. Маточный раствор объединяли с F2.
F2 в виде желтого масла растворяли в EtOAc (400 мл) и промывали 1 н. NaOH (3×150 мл) и солевым раствором (50 мл). Органический слой сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент CH2Cl2). Фракции, содержащие продукт, объединяли и выпаривали. РЕ добавляли к желтому маслянистому остатку, после чего образовался осадок. Смесь охлаждали до температуры 0°C, затем твердое вещество отфильтровывали, промывали РЕ и сушили в вакууме с получением F2 в виде белого твердого вещества. Маточный раствор выпаривали. РЕ добавляли к маслянистому остатку, после чего образовался осадок. Смесь оставляли стоять в холодильнике в течение 2 часов, затем осадок отфильтровывали, промывали РЕ и сушили в вакууме с получением F3 в виде бледно-желтого твердого вещества.
Стадия 2:
2-(гекс-5-ин-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион (46,3 г), AgNO3 (8,65 г) и NIS (68,8 г) помещали в колбу объемом 1 л. Добавляли сухой ТГФ (500 мл), колбу промывали аргоном и обворачивали алюминиевой фольгой, чтобы защитить реакцию от света. Затем смесь перемешивали в атмосфере Ar при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь декантировали от образовавшегося осадка, разбавляли водой (400 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали водой (100 мл), насыщ. Na2SO3 (3×100 мл) и солевым раствором (100 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток перекристаллизовывали из EtOH с получением F1 в виде белого твердого вещества. Маточный раствор выпаривали и снова перекристаллизовывали из EtOH с получением F2 в виде желтого твердого вещества.
Стадия 3:
2-метил-2-бутен (29,4 мл) добавляли по каплям к холодному раствору BH3*SMe2 (2,00 М в ТГФ, 64,4 мл), находящемуся при температуре 0°C, и перемешивали при температуре 0°C в течение 1 часа, а затем при комнатной температуре в течение 1 часа. Затем указанную смесь добавляли по каплям к холодной суспензии 2-(6-йодгекс-5-ин-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона (17,5 г) в ТГФ (200 мл), находящейся при температуре 0°C. После добавления полученную смесь перемешивали при комнатной температуре в течение 1 часа. Контроль по ЖХМС показал, что исходный материал был полностью израсходован. Реакционную смесь охлаждали до температуры 0°C, затем добавляли по каплям НОАс (30,0 мл), перемешивали в течение 30 мин при температуре 0°C и затем при комнатной температуре в течение ночи. Контроль с помощью ЖХМС показал продукт.
ТГФ удаляли в вакууме, насколько это возможно. Остаток медленно выливали в раствор NaOH (15,0 г) в H2O (200 мл) и экстрагировали CH2Cl2 (3×100 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток использовали для дальнейшей трансформации как есть.
Стадия 4:
2-[(5Z)-6-иодгекс-5-ен-1-ил]-2,3-дигидро-1Н-изоиндол-1,3-дион (17,6 г, неочищенный IK-0353/4) растворяли в МеОН (150 мл). Добавляли гидразингидрат (6,00 мл) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
МеОН удаляли в вакууме. Остаток суспендировали в CH2Cl2 (300 мл). Твердое вещество отфильтровывали и промывали CH2Cl2 (2×100 мл). Затем объединенные фильтраты промывали водой (2×100 мл), сушили над Na2SO4 и концентрировали в вакууме с получением неочищенного продукта в виде оранжевого масла, который использовали для дальнейшей трансформации как есть.
Стадия 5:
Этилхлорформилформиат (10,0 г) растворяли в ТГФ (50 мл) и охлаждали до температуры 0°C.Добавляли по каплям пиридин (7,70 мл) и указанную смесь перемешивали при температуре 0°C в течение 30 мин. Затем по каплям добавляли метиламин (2,0 М в ТГФ, 47,6 мл). Перемешивание продолжали при температуре 0°C в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:3) показал продукт.
Осажденную соль отфильтровывали и фильтрат выпаривали. Остаток растворяли в EtOAc (200 мл), промывали 1 н. HCl (2×50 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта с достаточной степенью чистоты в виде коричневого масла.
Стадия 6:
(5Z)-6-йодгекс-5-ен-1-амин (11,15 г) растворяли в EtOH (200 мл). Добавляли этил (метилкарбамоил)формиат (6,50 г) и NEt3 (8,26 мл) и полученную смесь перемешивали при температуре 50°C в течение 24 часов. Контроль с помощью ЖХМС показал неполное преобразование. Добавляли дополнительный (метилкарбамоил)формиат (1,00 г) и NEt3 (4,00 мл) и перемешивание продолжали при температуре 50°C в течение 24 часов. Контроль с помощью ЖХМС показал продукт.
EtOH удаляли в вакууме. Остаток очищали колоночной хроматографией на SiO2 (CH2Cl2 -> CH2Cl2/МеОН = 50:1 -> CH2Cl2/МеОН = 20:1). Фракции, содержащие продукт, объединяли и выпаривали. EtOAc (30 мл) добавляли к частично твердому остатку, обрабатывали ультразвуком и оставляли стоять в холодильнике в течение выходных. Затем осадок отфильтровывали, промывали небольшим количеством ледяного EtOAc и сушили в вакууме.
Выход: 10,3 г (67%) в виде твердого вещества бледно-желтого цвета.
Структурная единица 2 (ВВ-2)
N'-[4-(2-йодфенил)бутил]-Nметилэтандиамид
Стадия 1:
PPh3 (95,5 г), фталимид (56,1 г) и 3-бутен-1-ол (25,0 г) суспендировали в сухом ТГФ (250 мл) и охлаждали до температуры 0°C. Затем добавляли по каплям диизопропилазодикарбоксилат (75,1 мл) в течение 20 мин. Полученную смесь перемешивали при температуре 0°C в течение 30 минут и затем при комнатной температуре в течение ночи. Контроль с помощью ЖХМС показал продукт.
ТГФ удаляли в вакууме, насколько это возможно. Маслянистый остаток разбавляли РЕ/EtOAc = 9:1 (400 мл) и энергично перемешивали до тех пор, пока не образовался осадок. Осажденный OPPh3 отфильтровывали и интенсивно промывали РЕ/EtOAc = 9:1. Объединенные фильтраты фильтровали через слой SiO2 и затем выпаривали. Остаток разбавляли РЕ (200 мл), тщательно перемешивали и помещали в ледяную баню. Затем осажденный продукт отфильтровывали и промывали при помощи РЕ с получением продукта достаточной чистоты в виде бледно-желтого твердого вещества.
Стадия 2:
2-(бут-3-ен-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион (22,1 г) помещали в колбу объемом 1 л под Ar атмосферу. 9-BBN (0,5 М в ТГФ, 273 мл) добавляли по каплям при температуре 0°C и полученную смесь перемешивали при температуре 0°C в течение 30 мин, а затем при комнатной температуре в течение ночи. Затем добавляли раствор Na2CO3 (48,4 г) в воде (250 мл) и перемешивание продолжали при комнатной температуре в течение 30 мин. Затем добавляли 2-йодфениламин (20,0 г) и PdCl2(PPh3)2 (2,80 г) и смесь нагревали до температуры 50°C в течение 4 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь разбавляли EtOAc (200 мл) и слои разделяли. Водный слой экстрагировали EtOAc (300 мл) и объединенные органические слои промывали солевым раствором (200 мл) и сушили над Na2SO4. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/ EtOAc = 6:4).
Стадия 3:
2-[4-(2-аминофенил)бутил]-2,3-дигидро-1Н-изоиндол-1,3-дион (22,0) растворяли в ацетоне (100 мл). Затем добавляли воду (200 мл) и конц. H2SO4 (13,9 мл) и полученную суспензию охлаждали до температуры 0°C. По каплям добавляли раствор NaNO2 (5,23 г) в воде (50 мл) и указанную смесь перемешивали при температуре 0°C в течение 30 мин. Затем по каплям добавляли раствор KI (37,2 г) в воде (50 мл), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 20 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь разбавляли насыщ. Na2SO3 (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали солевым раствором (150 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/ EtOAc = 8:2).
Стадия 4:
2-[4-(2-иодфенил)бутил]-2,3-дигидро-1Н-изоиндол-1,3-дион (21,2 г) суспендировали в МеОН (300 мл). Добавляли гидразингидрат (5,10 мл) и полученную смесь перемешивали при комнатной температуре в течение 3 дней. Контроль с помощью ЖХМС показал продукт.
МеОН удаляли в вакууме. Остаток суспендировали в CH2Cl2 (200 мл). Твердое вещество отфильтровывали и промывали CH2Cl2 (100 мл). Затем объединенные фильтраты промывали водой (2×100 мл). Объединенные водные слои повторно экстрагировали CH2Cl2 (50 мл) и затем объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта с достаточной степенью чистоты в виде желтого масла.
Стадия 5:
Этилхлорформилформиат (10,0 г) растворяли в ТГФ (50 мл) и охлаждали до температуры 0°C. Добавляли по каплям пиридин (7,70 мл) и указанную смесь перемешивали при температуре 0°C в течение 30 мин. Затем по каплям добавляли метиламин (2,0 М в ТГФ, 47,6 мл). Перемешивание продолжали при температуре 0°C в течение 3 часов. Контроль с помощью ТСХ (PE/EtOAc = 1:3) показал продукт.
Осажденную соль отфильтровывали и фильтрат выпаривали. Остаток растворяли в EtOAc (200 мл), промывали 1 н. HCl (2×50 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта с достаточной степенью чистоты в виде коричневого масла.
Стадия 6:
4-(2-йодфенил)бутан-1-амин (11,0 г, неочищенный IK-0355710) растворяли в EtOH (100 мл). Добавляли этил (метилкарбамоил)формиат (5,76 г) и NEt3 (6,67 мл) и полученную смесь перемешивали при температуре 50°С в течение 18 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь охлаждали до комнатной температуры и EtOH удаляли в вакууме. Остаток фильтровали через слой SiO2 (CH2Cl2/МеОН = 98:2). Дальнейшую очистку проводили путем перекристаллизации из EtOAc.
Выход: 7,76 г (54%) бежевого твердого вещества.
Структурная единица 4 (ВВ-4)
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусная кислота
Стадия 1
NaH (60% в минеральном масле, 771 мг) суспендировали в сухом ТГФ (20,0 мл). Смесь охлаждали до температуры 0°С, затем добавляли 6-гептен-1-ол (1,18 мл). Перемешивание продолжали при температуре 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (1,34 г) в ТГФ (10,0 мл). После полного добавления ледяную баню удаляли и перемешивали в течение 15 мин, затем смесь нагревали до температуры 70°С в течение 1,5 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) показал продукт.
Указанную реакционную смесь выливали в 1 н. NaOH (50 мл) и экстрагировали EtOAc (2×30 мл). Объединенные органические слои не содержали продукта и были отброшены. Водные слои осторожно подкисляли конц. HCl и затем снова экстрагировали EtOAc (3×30 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта с достаточной чистотой в виде бесцветного масла.
Стадия 2
1,1'-карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при комнатной температуре в течение 6 часов. Затем ТГФ удаляли в вакууме и к остатку добавляли МеОН (200 мл). Смесь перемешивали при комнатной температуре в течение 12 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 9:1) показал продукт.
МеОН удаляли в вакууме. К остатку добавляли РЕ (200 мл) и перемешивали в течение 5 мин. Затем из густого маслянистого остатка декантировали растворитель, остаток далее промывали РЕ (2×100 мл) и затем отбрасывали. Объединенные фракции РЕ промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде бесцветной жидкости.
Стадия 3
Метил-2-(гепт-6-ен-1-илокси)ацетат (2,88 г) помещали в колбу объемом 100 мл и охлаждали до температуры 0°С в Ar атмосфере. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 38,7 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (6,84 г) в воде (30,0 мл) и перемешивание продолжали при комнатной температуре в течение 30 мин. Затем добавляли N'-[(5Z)-6-йодгекс-5-ен-1-ил]-N-метилэтандиамид (ВВ-1, 4,00 г) и PdCl2(PPh3)2 (453 мг) и указанную смесь нагревали до температуры 50°С в течение 1,5 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь охлаждали до комнатной температуры и разделяли слои. Водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/ EtOAc 1:1).
Стадия 4
Метил 2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}ацетат (400 мг) суспендировали в МеОН (20,0 мл). Добавляли NaOH (3N, 5,00 мл) и полученную смесь перемешивали при комнатной температуре в течение 15 минут. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь выливали в 1 н. HCl (30 мл). Осажденный продукт отфильтровывали, промывали водой и сушили в вакууме.
Выход: 869 мг (86%) бежевого твердого вещества.
Структурная единица 6 (ВВ-6)
2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусная кислота
Стадия 1:
NaH (60% в минеральном масле, 15,2 г) суспендировали в сухом ТГФ (250 мл). Смесь охлаждали до температуры 0°С, затем добавляли аллиловый спирт (11,8 мл). Перемешивание продолжали при температуре 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (25,3 г) в ТГФ (50,0 мл). После полного добавления ледяную баню удаляли и перемешивали в течение 15 мин, смесь затем нагревали до температуры 70°С в течение 3 часов и перемешивали при комнатной температуре в течение ночи.
Указанную реакционную смесь выливали в воду (250 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические слои не содержали продукта и были отброшены. Водные слои осторожно подкисляли конц. HCl и затем снова экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде жидкости светло-коричневого цвета.
Стадия 2:
1,1'-карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли 2-(проп-2-ен-1-илокси)уксусную кислоту (неочищенный IK-0352/9) и полученную смесь перемешивали при комнатной температуре в течение 7 часов. Затем ТГФ удаляли в вакууме и к остатку добавляли МеОН (200 мл). Смесь перемешивали при комнатной температуре в течение ночи. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) показал продукт.
МеОН удаляли в вакууме. К остатку добавляли РЕ (200 мл) и перемешивали в течение 5 мин. Затем из густого маслянистого остатка декантировали растворитель, остаток далее промывали РЕ (2×100 мл). Контроль с помощью ТСХ показал, что большая часть продукта оставалась в маслянистом остатке, который в этой связи промывали МТБЭ (4×100 мл). Слои РЕ и МТБЭ объединяли и промывали 1 н. HCl (3×100 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной степени чистоты в виде бледно-желтой жидкости.
Стадия 3:
Метил-2-(проп-2-ен-1-илокси)ацетат (1,30 г) помещали в колбу объемом 100 мл и охлаждали до температуры 0°С в Ar атмосфере. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 25,0 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (4,41 г) в воде (25,0 мл) и перемешивание продолжали при комнатной температуре в течение 30 мин. Затем добавляли N'-[4-(2-йодфенил)бутил]-N-метилэтандиамид (ВВ-2, 3,00 г) и PdCl2(PPh3)2 (292 мг) и указанную смесь нагревали до температуры 50°С в течение 4 часов, затем перемешивали при комнатной температуре в течение ночи. Контроль с помощью ЖХМС показал неполное преобразование. Дополнительный метил 2-(проп-2-ен-1-илокси)ацетат (650 мг) помещали в отдельную колбу под Ar-атмосферу. 9-BBN (0,5 М в ТГФ, 12,5 мл) добавляли при комнатной температуре и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли насыщ. раствор Na2CO3 (10 мл) и перемешивание продолжали при комнатной температуре в течение 30 мин. Затем смесь добавляли к указанной реакционной смеси. После добавления свежего PdCl2(PPh3)2 (200 мг) указанную смесь перемешивали при температуре 50°С в течение 2 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь охлаждали до комнатной температуры и разделяли слои. Водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc 3:7).
Стадия 4:
Метил-2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]ацетат (2,04 г) растворяли в ТГФ (30 мл). Добавляли NaOH (3 н., 30 мл) и МеОН (20 мл) и полученную смесь перемешивали при комнатной температуре в течение 5 мин. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь подкисляли 6 н. HCl и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали при помощи короткой колонки на SiO2 (CH2Cl2/МеОН = 9:1).
Выход: 1,56 г (80%) бежевого твердого вещества.
Структурная единица 8 (ВВ-8)
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-Nметилэтандиамид
Стадия 1:
6-гептен-1-ол (3,00 г) и имидазол (3,57 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCl (6,18 мл) и полученную смесь перемешивали при температуре 60°С в течение 6 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) показал почти полное преобразование.
Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. HCl (2×50 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 95:5).
Стадия 2:
(Гепт-6-ен-1-илокси)трис(пропан-2-ил)силан (1,57 г) помещали в колбу объемом 100 мл и охлаждали до температуры 0°С в Ar-атмосфере. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 14,5 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (2,56 г) в воде (15,00 мл) и перемешивание продолжали при комнатной температуре в течение 30 минут, затем добавляли N'-[(5Z)-6-йодгекс-5-ен-1-ил]-N-метилэтандиамид (ВВ-1, 1,50 г) и PdCl2(PPh3)2 (170 мг) и указанную смесь нагревали до температуры 50°С в течение 2 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь охлаждали до комнатной температуры и разделяли слои. Водный слой экстрагировали EtOAc (2×50 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток фильтровали через слой SiO2 (PE/EtOAc = 4:6). Полученный таким образом неочищенный продукт использовали для дальнейшей трансформации как есть.
Стадия 3:
(гепт-6-ен-1-илокси)трис(пропан-2-ил)силан (2,20 г, неочищенный IK-0357/16) растворяли в ТГФ (50 мл) и охлаждали до температуры 0°С. Добавляли TBAF*3H2O (2,29 г) и полученную смесь перемешивали при температуре 0°С в течение 30 минут, а затем при комнатной температуре в течение 6 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) и ЖХМС показал полное преобразование.
Указанную реакционную смесь выливали в воду (100 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток пропускали через короткую колонку на SiO2 (РЕ/EtOAc = 1:1 → EtOAc).
Выход: 1,11 г (77%) бежевого твердого вещества.
Структурная единица 9 (ВВ-9)
N'-{4-[2-(3-(гидроксипропил)фенил]бутил}-Nметилэтандиамид
Стадия 1:
2-пропен-1-ол (3,00 г) и имидазол (7,03 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCl (14,4 мл) и полученную смесь перемешивали при температуре 60°С в течение 6 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) показал почти полное преобразование. Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. HCl (2×50 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 95:5).
Стадия 2:
(проп-2-ен-1-илокси)трис(пропан-2-ил)силан (1,33 г) помещали в колбу объемом 100 мл и охлаждали до температуры 0°С и атмосфере Ar. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 14,2 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов.
Затем добавляли раствор Na2CO3 (2,21 г) в воде (15,0 мл) и перемешивание продолжали при комнатной температуре в течение 30 мин. Затем добавляли N'-[4-(2-йодфенил)бутил]-N-метилэтандиамид (1,50 г) и PdCl2(PPh3)2 (146 мг) и смесь нагревали до температуры 50°С в течение 3 часов. Контроль с помощью ЖХМС показал продукт. Реакционную смесь охлаждали до комнатной температуры и разделяли слои. Водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток пропускали через короткую колонку на SiO2 (РЕ/EtOAc 1:1). Неочищенный продукт использовали для дальнейшей трансформации как есть.
Стадия 3:
N-метил-N'-{4-[2-(3-{[трис(пропан-2-ил)силил]окси}пропил)фенил]бутил}этандиамид (1,87 г, неочищенный IK-0357/17) растворяли в ТГФ (50 мл) и охлаждали до температуры 0°С. Добавляли TBAF*3H2O (1,97 г) и полученную смесь перемешивали при температуре 0°С в течение 30 минут, а затем при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал полное преобразование. Указанную реакционную смесь выливали в воду (100 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток пропускали через короткую колонку на SiO2 (РЕ/EtOAc = 1:1 → EtOAc).
Выход: 911 мг (75%) бежевого твердого вещества.
Структурная единица 11 (ВВ-11)
N'-[(5Z)-13-(2-аминоэтокси)тридец-5-ен-1-ил]-Nметилэтандиамид
Стадия 1:
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до температуры 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Перемешивание продолжали при температуре 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После полного добавления ледяную баню удаляли и перемешивали в течение 15 мин, затем смесь нагревали до температуры 70°С в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) показал продукт.
Указанную реакционную смесь выливали в 1 н. NaOH (300 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические слои не содержали продукта и были отброшены. Водные слои осторожно подкисляли конц. HCl и затем снова экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде жидкости светло-коричневого цвета.
Стадия 2:
1,1'-карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при комнатной температуре в течение 6 часов. Затем ТГФ удаляли в вакууме и к остатку добавляли МеОН (200 мл). Смесь перемешивали при комнатной температуре в течение 12 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 9:1) показал продукт.
МеОН удаляли в вакууме. К остатку добавляли РЕ (200 мл) и перемешивали в течение 5 мин. Затем из густого маслянистого остатка декантировали растворитель, остаток далее промывали РЕ (2×100 мл) и затем отбрасывали. Объединенные фракции РЕ промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде бесцветной жидкости.
Стадия 3:
Метил-2-(гепт-6-ен-1-илокси)ацетат (5,00 г) растворяли в CH2Cl2 (100 мл) и охлаждали до температуры 0°С. По каплям добавляли DIBALH (1,00 М в CH2Cl2, 61,7 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение ночи. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) показал полное преобразование.
Реакционную смесь охлаждали до температуры 0°С и осторожно гасили насыщенным водным раствором Na2SO4. Затем указанную смесь разбавляли CH2Cl2 (100 мл), перемешивали в течение 20 мин и далее фильтровали через целит. Фильтровальный осадок несколько раз промывали CH2Cl2. Объединенные фильтраты концентрировали в вакууме с получением продукта достаточной чистоты в виде бесцветной жидкости.
Стадия 4:
PPh3 (7,17 г), фталимид (4,21 г) и 2-(гепт-6-ен-1-илокси)этан-1-ол (4,12 г) суспендировали в сухом ТГФ (100 мл) и охлаждали до температуры 0°С. Затем по каплям добавляли диизопропилазодикарбоксилат (5,79 мл) в течение 20 мин. Полученную смесь перемешивали при температуре 0°С в течение 30 мин и затем при комнатной температуре в течение ночи.
ТГФ удаляли в вакууме, насколько это возможно. Маслянистый остаток разбавляли РЕ/EtOAc = 9:1 (200 мл) и энергично перемешивали до тех пор, пока не образовался осадок. Осажденный OPPh3 отфильтровывали и интенсивно промывали PE/EtOAc = 9:1. Объединенные фильтраты фильтровали через слой SiO2 (элюент РЕ/EtOAc = 9:1) и выпаривали. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 8:2).
Стадия 5:
2-[2-(гепт-6-ен-1-илокси)]-2,3-дигидро-1Н-изоиндол-1,3-дион (2,22 г) помещали в колбу объемом 1 мл и охлаждали до температуры 0°С в Ar атмосфере. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 19,3 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (3,42 г) в воде (20,0 мл) и перемешивание продолжали при комнатной температуре в течение 30 минут, затем добавляли N'-[(5Z)-6-йодгекс-5-ен-1-ил]-N-метилэтандиамид (ВВ-1, 2,00 г) и PdC12(PPh3)2 (226 мг) и указанную смесь нагревали до температуры 50°С в течение 1,5 часов. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь охлаждали до комнатной температуры и разделяли слои. Водный слой экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/ EtOAc = 4:6).
Стадия 6:
N'-[(5Z)-13-[2-(1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-ил)этокси]тридец-5-ен-1-ил]-N-метилэтандиамид (2,36 г) суспендировали в МеОН (100 мл). Добавляли гидразингидрат (486 мкл) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
МеОН удаляли в вакууме. Остаток суспендировали в CH2Cl2/7 н. NH3 в МеОН = 9:1 (100 мл), фильтровали через слой SiO2 и дополнительно элюировали CH2Cl2/7 н. NH3 в МеОН = 9:1 (300 мл). Фильтрат концентрировали в вакууме с получением 1,68 г неочищенного продукта. 60 мг продукта подвергали очистке с помощью препаративной ТСХ (CH2Cl2/7 н. NH3 в МеОН = 9:1). Полученный таким образом неочищенный материал использовали для дальнейшей трансформации как есть.
Выход: 41 мг (2%) бледно-желтого твердого вещества (очищенного).
Соединение 14 (Соед.-14)
N-метил-N'-[(5Z)-13-[(1Н-1,2,3,4-тетразол-5-ил)метокси]тридец-5-ен-1-ил]этандиамид
Стадия 1
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до температуры 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Перемешивание продолжали при температуре 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После полного добавления ледяную баню удаляли и перемешивали в течение 15 мин, затем смесь нагревали до температуры 70°С в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) показал продукт. Указанную реакционную смесь выливали в 1 н. NaOH (300 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические слои не содержали продукта и были отброшены. Водные слои осторожно подкисляли конц. HCl и затем снова экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде жидкости светло-коричневого цвета.
Выход: 14,1 г (93%) бледно-коричневого масла
Стадия 2
Смесь 2-(гепт-6-ен-1-илокси)уксусной кислоты (3,00 г) и SOCl2 (15,00 мл) нагревали до температуры 70°С в течение 1 часа. Затем избыток SOCl2 удаляли в вакууме и остаток растворяли в дихлорэтане (15,0 мл). Затем через указанный раствор медленно пропускали аммиак в течение 5 мин. Реакционную смесь разбавляли водой (50 мл) и экстрагировали CH2Cl2 (3×30 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (30 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде белого твердого вещества, m=2,16 г (выход=62%). Аналог IK-0367/1 по ТСХ.
Стадия 3
2-(гепт-6-ен-1-илокси)ацетамид (2,61 г) растворяли в CH2Cl2 (50 мл). Добавляли NEt3 (6,35 мл) и указанную смесь охлаждали до температуры 0°С. Медленно добавляли раствор POCl3 (1,54 мл) в CH2Cl2 (4 мл). Затем перемешивание продолжали при температуре 0°С в течение 15 минут. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) показал продукт.
Добавляли насыщ. NaHCO3 (5,00 мл) при температуре 0°С и перемешивали при этой температуре в течение 30 минут. Указанную смесь доводили до комнатной температуры, разбавляли водой (15,0 мл) и экстрагировали CH2Cl2 (3×20 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (10,0 мл) и солевым раствором (10,0 мл), сушили над Na2SO4 и затем фильтровали через слой SiO2 (элюент CH2Cl2). После выпаривания получали продукт достаточной степени чистоты в виде бесцветного масла. m=2,08 г, выход = 89%.
Стадия 4
2-(гепт-6-ен-1-илокси)ацетонитрил помещали в колбу объемом 10 мл и охлаждали до температуры 0°С в Ar атмосфере. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 1,63 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (288 мг) в дегазированной воде (1 мл) и перемешивание продолжали при комнатной температуре в течение 30 минут. Затем добавляли N'-[(5Z)-6-йодгекс-5-ен-1-ил]-М-метилэтандиамид (168 мг) и PdCl2(PPh3)2 (19 мг) и указанную смесь нагревали до температуры 50°С в течение ночи. Добавляли воду и смесь экстрагировали ДХМ. Органический слой сушили над MgSO4, фильтровали и выпаривали растворитель. Смесь очищали при помощи препаративной ТСХ (ДХМ/МеОН 95/5). m = 70 мг, выход = 38%.
Стадия 5
N'-[(5Z)-13-(цианометокси)тридец-5-ен-1-ил]-N-метилэтандиамид, азид натрия и триэтиламина гидрохлорид растворяли в ТГФ и реакционную смесь перемешивали при температуре 70°С в течение ночи.
Затем добавляли воду и этилацетат. Смесь подкисляли 3 н. HCl. Затем водный слой (кислотный рН) экстрагировали этилацетатом (х3) и объединенные органические слои промывали рассолом. Органический слой сушили над MgSO4, фильтровали и растворитель удаляли под вакуумом, m = 82 мг. Продукт очищали препаративной ТСХ (ДХМ/МеОН 95/5)
Выход: 8 мг (10%) в виде белого порошка.
Соединение 15 (Соед. - 15)
N'-(4-{2-[3-(карбамоилметокси)пропил]фенил}бутил)-N-метилэтандиамид
250 мг (0,72 ол) 2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусной кислоты (ВВ-6) и 164,1 мг (0,86 ммоль) EDCI растворяли в 20 мл ДХМ. Добавляли 36,5 мг (2,14 ммоль, 5,35 мл) аммиака (0,4 М в ТГФ) и указанную смесь перемешивали при комнатной температуре в течение выходных.
Смесь выливали в 50 мл воды и экстрагировали ДХМ (3×50 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали.
Выход: 50 мг (20%), твердое вещество белого цвета.
Соединение 16 (Соед. - 16)
N-метил-N'-[(5Z)-13-[(5-оксо-4,5-дигидро-1Н-1,2,4-триазол-3-ил)метокси]тридец-5-ен-1-ил]этандиамид
Стадия 1:
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до температуры 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Перемешивание продолжали при температуре 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После полного добавления ледяную баню удаляли и перемешивали в течение 15 мин, затем смесь нагревали до температуры 70°С в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) показал продукт. Указанную реакционную смесь выливали в 1 н. NaOH (300 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические слои не содержали продукта и были отброшены. Водные слои осторожно подкисляли конц. HCl и затем снова экстрагировали EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде жидкости светло-коричневого цвета.
Стадия 2:
1,1'-карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при комнатной температуре в течение 6 часов. Затем ТГФ удаляли в вакууме и к остатку добавляли МеОН (200 мл). Смесь перемешивали при комнатной температуре в течение 12 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 9:1) показал продукт. МеОН удаляли в вакууме. К остатку добавляли РЕ (200 мл) и перемешивали в течение 5 мин. Затем из густого маслянистого остатка декантировали растворитель, остаток далее промывали РЕ (2×100 мл) и затем отбрасывали. Объединенные фракции РЕ промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали в вакууме с получением продукта достаточной чистоты в виде бесцветной жидкости.
Стадия 3:
500 мг (2,68 ммоль) и 1,34 г (26,9 ммоль, 1,30 мл) гидразингидрата растворяли в 5 мл EtOH и перемешивали при температуре 70°С в течение 4,5 часов (→ прозрачный раствор).
→ ЖХ/MC: GH-0513/1-1
→ ТСХ (ЕА/РЕ 1:1): Полное потребление исходного материала
Указанную смесь выпаривали досуха
Стадия 4:
500 мг (2,68 ммоль) 2-(гепт-6-ен-1-илокси)ацетогидразида (GH-0513/1) растворяли в 3 мл АсОН. Добавляли 653,3 мг (8,05 ммоль) цианата калия, растворенного в 3 мл воды, и указанную смесь перемешивали при комнатной температуре в течение 1,5 ч (желтый раствор). Указанную смесь выпаривали досуха.
Стадия 5:
Маслянистый остаток растворяли в 10 мл 2 М NaOH и нагревали с обратным холодильником в течение 2 часов. → ЖХМС: GH-0515/1-2: Полное потребление промежуточного продукта 1. Смесь подкисляли конц. HCl и экстрагировали ЕА (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали досуха. Неочищенное твердое вещество перекристаллизовывали из ACN.
Стадия 6:
В атмосфере аргона 92,0 мг (0,44 ммоль) 3-[(гепт-6-ен-1-илокси)метил]-4,5-дигидро-1Н-1,2,4-триазол-5-он (GH-0515/1), растворенного в 2 мл безводного ТГФ, добавляли к раствору 88,5 мг (0,73 ммоль, 1,45 мл) 9BBN (0,5 М в ТГФ) и указанную смесь перемешивали при комнатной температуре в течение ночи. Добавляли раствор 153,8 мг (1,45 ммоль) Na2CO3 в 1 мл воды и продолжали перемешивание при комнатной температуре в течение 15 мин. Затем добавляли 90,0 мг (0,29 ммоль) N'-[(5Z)-6-йодгекс-5-ен-1-ил]-метилэтандиамида (IK-0356/2), растворенного в 2 мл ТГФ, и 10,2 мг (14,5 мкмоль) PdCl2(PPh3)2, и указанную смесь нагревали до температуры 50°С в течение 4 часов (желтая двухфазная смесь).
→ ЖХ/МС: GH-0516/1-1: Был обнаружен продукт
Органический слой отделяли пипеткой и выпаривали досуха. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (ДХМ/МеОН 20:1:9:1, Rf возможного продукта: 0,62). Перекристаллизация из CAN.
Выход: 51 мг (0,13 ммоль, 45%).
Соединение 17 (Соед. - 17)
N-метил-N'-[(5Z)-13-[(фенилкарбамоил)метокси]тридец-5-ен-1-ил]этандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг, 140,3 мкмоль), анилин (26 мкл, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (20 мл) и экстрагировали Et2O (3×20 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Продукт лиофилизировали.
Выход: 52 мг (87%), твердое вещество белого цвета.
Соединение 18 (Соед. - 18)
N-метил-N'-[(5Z)-13-{[(оксан-4-ил)карбамоил]метокси}тридец-5-ен-1-ил]этандиамид
2-{[(82)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг, 140,3 мкмоль), 4-аминотетрагидропиран (29 мкл, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt 3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт. Указанную реакционную смесь разбавляли водой (20 мл) и экстрагировали Et2O (3×20 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Продукт лиофилизировали.
Выход: m = 58 мг (94%) белого твердого вещества.
Соединение 19 (Соед. - 19)
N-метил-N'-[(5Z)-13-{[(1,3-оксазол-2-ил)карбамоил]метокси}тридец-5-ен-l-ил]этандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг, 140,3 мкмоль), 1,3-оксазол-2-амин (24 мг, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (20 мл) и экстрагировали Et2O (3×20 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (ДХМ/МеОН = 95:5).
Выход: 11 мг (19%), твердое вещество белого цвета.
Соединение 20 (Соед. - 20)
N'-[(5Z)-13-{[(4-метоксифенил)карбамоил]метокси}тридец-5-ен-1-ил]-Nметилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг, 140,3 мкмоль), п-анизидин (35 мг, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (20 мл) и экстрагировали Et2O (3×20 мл). Объединенные органические фазы промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (ДХМ/МеОН = 98:2).
Выход: 18 мг (28%) бежевого твердого вещества.
Соединение 21 (Соед. - 21)
N-метил-N'-[4-(2-{3-[(фенилкарбамоил)метокси]пропил}фенил)бутил]этандиамид 40 мг (0,11 ммоль)
2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусную кислоту (ВВ-6), 26,3 мг (0,14 ммоль) EDCI и 11,7 мг (0,13 ммоль, 11,5 мкл) анилина растворяли в 3 мл ДХМ и перемешивали в течение выходных (прозрачный раствор).
Смесь выпаривали досуха и очищали при помощи пТСХ (1 мм, ДХМ/МеОН 20:1, Rf возможного продукта: 0,54).
Выход: 24 мг (51%), твердое вещество белого цвета.
Соединение 22 (Соед. - 22)
N-метил-N'-[4-(2-{3-[(2-оксо-2-(пирролидин-1-ил)этокси]пропил}фенил)бутил]этандиамид 50 мг (0,14 ммоль)
2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусной кислоты (ВВ-6), 32,8 мг (0,17 ммоль) EDCI и 20,3 мг (0,29 ммоль, 23,4 мкл) пирролидина растворяли в 3 мл ДХМ и перемешивали при комнатной температуре в течение 1,5 часов (прозрачный раствор).
Смесь выпаривали досуха и очищали при помощи пТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,46).
Выход: 20 мг (35%), твердое вещество белого цвета.
Соединение 23 (Соед. - 23)
N-метил-N'-[4-(2-{3-[2-(морфолин-4-ил)-2-оксоэтокси]пропил}фенил)бутил]этандиамид 50 мг (0,14 ммоль)
2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусной кислоты (ВВ-6, IK-0558/6), 32,8 мг (0,17 ммоль) EDCI и 24,9 мг (0,29 ммоль, 24,9 мкл) морфолина растворяли в 3 мл ДХМ и перемешивали при комнатной температуре в течение выходных (прозрачный раствор).
Смесь выпаривали досуха и очищали при помощи пТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,48).
Выход: 34 мг (58%), твердое вещество белого цвета.
Соединение 24 (Соед. - 24)
N'-[(5Z)-13-{[(бензолсульфонил)карбамоил]метокси}тридец-5-ен-1-ил]-Nметилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг) растворяли в ТГФ (2,00 мл). Добавляли 1,1'-карбонилдиимидазол (29,6 мг) и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли DBU (52,9 мкл) и бензолсульфонамид (33,1 мг) и перемешивание продолжали при комнатной температуре в течение 25 часов. Контроль с помощью ЖХМС показал ОМТ-121.
Указанную реакционную смесь разбавляли водой (30 мл) и экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои промывали 1 н. HCl (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 44 мг (63%), твердое вещество белого цвета.
Соединение 25 (Соед. - 25)
4-{2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]ацетамидо}бензойная кислота
Стадия 1:
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусной кислоты (ВВ-6, IK-0358/6), 32,8 мг (0,17 ммоль) EDCI и 43,1 мг (0,29 ммоль) метил-4-аминобензоата растворяли в 3 мл ДХМ и перемешивали при комнатной температуре в течение 1,5 часов (прозрачный раствор).
Смесь выпаривали досуха и очищали при помощи пТСХ (1 мм, ЕА/РЕ 4:1, Rf возможного продукта: 0,31).
Стадия 2:
К раствору 48 мг (0,10 ммоль) N-метил-N'-{4-[2-(3-{[(пиридин-2-ил)карбамоил]метокси}пропил)фенил]бутил}этандиамида (GH-0498/1) в 2 мл ТГФ добавляли 16,7 мг (0,40 ммоль) моногидрата LiOH, растворенного в 0,5 мл воды, и смесь перемешивали при комнатной температуре в течение ночи (двухфазная смесь).
Смесь выливали в 1 н. раствор HCl (10 мл) и экстрагировали ДХМ (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали досуха. Неочищенный продукт очищали пТСХ (ДХМ/MeOH/FA 100:10:1, Rf возможного продукта: 0,43).
Выход: 10 мг (21%), твердое вещество белого цвета.
Соединение 26 (Соед. - 26)
N'-[(5Z)-13-[2-(4-гидрокси-2-оксо-2,5-дигидрофуран-3-ил)-2-оксоэтокси]тридец-5-ен-1-ил]-Nметилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50 мг), DCC (34,7 мг), DMAP (22,3 мг) и 2,4(3Н,5Н)-фурандион (15,4 мг) помещали во флакон G16. Добавляли CH2Cl2 (3,00 мл) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (30 мл) и экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 9:1).
Выход: 20 мг (33%) бежевого твердого вещества.
Соединение 27 (Соед. - 27)
N'-[4-(2-(3-[2-(гидроксиметил)феноксил]пропил)фенил)бутил]-N-метилэтандиамид
Стадия 1:
Салицилальдегид (2,00 г) и имидазол (2,79 г) растворяли в ДМФ (20,0 мл). Добавляли TIPSCl (5,96 мл) и полученную смесь перемешивали при температуре 60°С в течение 2 дней. Контроль с помощью ТСХ (РЕ/EtOAc = 95:5) и ЖХМС показал неполное преобразование. Добавляли дополнительную порцию TIPSCl (2,00 мл) и перемешивание продолжали при температуре 60°С в течение 3 дней. Контроль с помощью ТСХ (РЕ/EtOAc = 95:5) и ЖХМС показал почти полное преобразование. Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. NaOH (30 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 95:5).
Выход: 3,54 г (78%) жидкости бледно-желтого цвета.
Стадия 2:
2-{[трис(пропан-2-ил)силил]окси}бензальдегид (3,54 г) растворяли в EtOH (30,0 мл) и охлаждали до температуры 0°С. Добавляли NaBH4 (481 мг) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 18 ч. Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) и ЖХМС показал продукт. Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 8:2).
Выход: 2,73 г (77%) масла желтого цвета.
Стадия 3:
(2-{[трис(пропан-2-ил)силил]окси}фенил)метанол (400 мг) растворяли в сухом ТГФ (15 мл). Добавляли NaH (60% в минеральном масле, 85,6 мг) и указанную смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли аллилбромид (309 мкл) и полученную смесь перемешивали при комнатной температуре в течение ночи. Контроль с помощью ЖХМС показал продукт. Указанную реакционную смесь разбавляли водой (50 мл) и экстрагировали CH2Cl2 (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Полученный таким образом неочищенный продукт использовали для дальнейшей трансформации как есть.
Выход: 480 мг (неочищенный), масло желтого цвета.
Стадия 4:
{2-[(проп-2-ен-1-илокси)метил]фенокси}трис(пропан-2-ил)силан (178 мг) помещали в колбу объемом 10 мл и охлаждали до температуры 0°С в атмосфере Ar. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 1,38 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов. Затем добавляли раствор Na2CO3 (147 мг) в воде (1,50 мл) и перемешивание продолжали при комнатной температуре в течение 30 минут. Затем добавляли N'-[4-(2-йодфенил)бутил]-N-метилэтандиамид (ВВ-2, 100 мг) и PdCl2(PPh3)2 (9,7 мг) и смесь нагревали до температуры 50°С в течение 3 часов. Контроль с помощью ЖХМС показал продукт. Реакционную смесь охлаждали до комнатной температуры, разводили водой (20 мл) и разделяли слои. Водный слой экстрагировали EtOAc (15 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток пропускали через короткую колонку на SiO2 (РЕ/EtOAc 1:1). Неочищенный продукт использовали для дальнейшей трансформации как есть.
Выход: 235 мг (неочищенный), масло желтого цвета.
Стадия 5:
N-метил-N'-[4-(2-{3-[(2-{[трис(пропан-2-ил)силил]окси}фенил)метокси]пропил}фенил)бутил]этандиамид (154 мг, неочищенный IK-0357/19) растворяли в ТГФ (5,00 мл). Добавляли TBAF*3H2O (87,0 мг) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Контроль с помощью ЖХМС показал полное преобразование. Указанную реакционную смесь выливали в воду (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 75 мг (66%), твердое вещество белого цвета.
Соединение 28 (Соед. - 28)
N'-[(5Z)-13-(2-бензолсульфонамидоэтокси)тридец-5-ен-1-ил]-Nметилэтандиамид
N'-[(5Z)-13-(2-аминоэтокси)тридец-5-ен-1-ил]-N-метилэтандиамид (ВВ-11, 50,0 мг, неочищенный IK-0355/8) суспендировали в CH2Cl2 (3,00 мл). Добавляли бензолсульфонилхлорид (37,5 мкл) и NEt3 (61,1 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Контроль с помощью ЖХМС показал продукт. Контроль с помощью ЖХМС показал продукт. Контроль с помощью ЖХМС показал продукт.
Реакционную смесь разбавляли водой (20 мл) и насыщ. NaHCO3 (20 мл) и затем экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои промывали 1 н. HCl (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 42 мг (60%), твердое вещество белого цвета.
Соединение 29 (Соед. - 29)
N'-[(5Z)-13-(2-гидроксифенокси)тридец-5-ен-1-ил]-Nметилэтандиамид
Стадия 1:
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-N-метилэтандиамид (ВВ-8, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (598 мг) и CBr4 (756 мг) и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) и ЖХМС показал полное преобразование.
Реакционную смесь концентрировали в вакууме и остаток очищали при помощи препаративной ТСХ (РЕ/EtOAc = 1:1).
Стадия 2:
N'-[(5Z)-13-бромтридец-5-ен-1-ил]-N-метилэтандиамид (50 мг), пирокатехин (76,2 мг) и K2СО3 (57,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при температуре 60°С в течение 2,5 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (40 мл) и экстрагировали МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (СН2С12/МеОН = 95:5).
Выход: 43 мг (80%), твердое вещество белого цвета.
Соединение 30 (Соед. - 30)
N-метил-N'-[4-(2-{3-[2-(морфолин-4-ил)-2-оксоэтокси]пропил}фенил)бутил]этандиамид
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)формамидо]бутил}фенил)пропокси]уксусной кислоты (ВВ-8, IK-0558/6), 32,8 мг (0,17 ммоль) EDCI и 24,9 мг (0,29 ммоль, 24,9 мкл) морфолина растворяли в 3 мл ДХМ и перемешивали при комнатной температуре в течение выходных (прозрачный раствор).
Смесь выпаривали досуха и очищали при помощи пТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,48).
Выход: 34 мг (58%), твердое вещество белого цвета.
Соединение 31 (Соед. - 31)
N'-(4-{2-[3-(3-гидроксифенокси)пропил]фенил}бутил)-Nметилэтандиамид
Стадия 1:
N'-{4-[2-(3-гидроксипропил)фенил]бутил}-N-метилэтандиамид (ВВ-9, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (610 мг) и CBr4 (771 мг) и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) и ЖХМС показал полное преобразование. Реакционную смесь концентрировали в вакууме и остаток очищали при помощи препаративной ТСХ (РЕ/EtOAc = 1:1).
Стадия 2:
N'-{4-[2-(3-бромпропил)фенил]бутил}-N-метилэтандиамид (50 мг), 1,3-бензодиол (77,5 мг) и K2СО3 (58,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при температуре 60°С в течение 1 часа. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (40 мл) и экстрагировали МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 40 мг (74%), твердое вещество белого цвета.
Соединение 32 (Соед. - 32)
N'-[(5Z)-13-(4-гидроксифенокси)тридец-5-ен-1-ил]-Nметилэтандиамид
Стадия 1:
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-N-метилэтандиамид (ВВ-8, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (598 мг) и CBr4 (756 мг) и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) и ЖХМС показал полное преобразование.
Реакционную смесь концентрировали в вакууме и остаток очищали при помощи препаративной ТСХ (РЕ/EtOAc = 1:1).
Стадия 2:
N'-[(5Z)-13-бромтридец-5-ен-1-ил]-N-метилэтандиамид (50 мг), гидрохинон (76,2 мг) и K2СО3 (57,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при температуре 60°С в течение 2,5 часов. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (40 мл) и экстрагировали МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 43 мг (80%), твердое вещество белого цвета
Соединение 33 (Соед. - 33)
N'-(4-{2-[3-(4-гидроксифенокси)пропил]фенил}бутил)-Nметилэтандиамид
Стадия 1:
N'-{4-[2-(3-гидроксипропил)фенил]бутил}-N-метилэтандиамид (ВВ-9, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (610 мг) и CBr4 (771 мг) и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Контроль с помощью ТСХ (РЕ/EtOAc = 1:1) и ЖХМС показал полное преобразование.
Реакционную смесь концентрировали в вакууме и остаток очищали при помощи препаративной ТСХ (РЕ/EtOAc = 1:1).
Стадия 2:
N'-{4-[2-(3-бромпропил)фенил]бутил}-N-метилэтандиамид (50 мг), гидрохинон (77,5 мг) и K2СО3 (58,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при температуре 60°С в течение 1 часа. Контроль с помощью ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (40 мл) и экстрагировали МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 39 мг (72%), твердое вещество белого цвета
Соединение 34 (Соед. - 34)
N'-[4-(2-{3-[(4-гидроксифенил)метокси]пропил}фенил)бутил]-N-метилэтандиамид
Стадия 1
4-гидроксибензальдегид (2,00 г) и имидазол (2,79 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCl (5,96 мл) и полученную смесь перемешивали при температуре 60°С в течение 2 дней.
Контроль с помощью ТСХ (РЕ/EtOAc = 95:5) и ЖХМС показал полное преобразование.
Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. NaOH (30 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 95:5).
Выход: 3,94 г (86%), масло бледно-желтого цвета.
Стадия 2
4-{[трис(пропан-2-ил)силил]окси}бензальдегид (3,94 г) растворяли в EtOH (30,0 мл) и охлаждали до температуры 0°С. Добавляли NaHB4 (535 мг) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 18 ч.
Контроль с помощью ТСХ (РЕ/EtOAc = 8:2) и ЖХМС показал продукт.
Указанную реакционную смесь разбавляли водой (100 мл) и экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (РЕ/EtOAc = 8:2).
Стадия 3
(4-{[трис(пропан-2-ил)силил]окси}фенил)метанол (300 мг) растворяли в сухом ТГФ (5,00 мл). Добавляли NaH (60% в минеральном масле, 64,2 мг) и указанную смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли аллилбромид (231 мкл) и полученную смесь перемешивали при комнатной температуре в течение 2,5 часов. Контроль с помощью ЖХМС показал полное преобразование.
Указанную реакционную смесь выливали в воду (30 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток использовали для дальнейшей трансформации как есть.
Выход: 372 мг (неочищенный), масло желтого цвета.
Стадия 4
{4-[(проп-2-ен-1-илокси)метил]фенокси}трис(пропан-2-ил)силан (356 мг) помещали в колбу объемом 10 мл и охлаждали до температуры 0°С в атмосфере Ar. Затем добавляли по каплям 9-BBN (0,5 М в ТГФ, 3,33 мл) и полученную смесь перемешивали при температуре 0°С в течение 30 мин, а затем при комнатной температуре в течение 2 часов.
Затем добавляли раствор Na2CO3 (147 мг) в воде (3,00 мл) и перемешивание продолжали при комнатной температуре в течение 30 минут. Затем добавляли N'-[4-(2-йодфенил)бутил]-N-метилэтандиамид (ВВ-2, 100 мг) и PdCl2(PPh3)2 (9,7 мг) и смесь нагревали до температуры 50°С в течение 2 часов. Контроль с помощью ЖХМС показал продукт. Реакционную смесь охлаждали до комнатной температуры, разводили водой (20 мл) и разделяли слои. Водный слой экстрагировали EtOAc (15 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток пропускали через короткую колонку на SiO2 (РЕ/EtOAc 1:1). Неочищенный продукт использовали для дальнейшей трансформации как есть.
Выход: 248 мг (неочищенный), масло желтого цвета.
Стадия 5
N-метил-N'-[4-(2-{3-[(4-{[трис(пропан-2-ил)силил]окси}фенил)метокси]пропил}фенил)бутил]этандиамид (154 мг, неочищенный IK-0357/20) растворяли в ТГФ (5,00 мл). Добавляли TBAF*3H2O (131 мг) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Контроль с помощью ЖХМС показал полное преобразование.
Указанную реакционную смесь выливали в воду (40 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 76 мг (68%), твердое вещество белого цвета.
Соединение 35 (Соед. - 35)
N'-[(5Z)-13-[(метансульфонилкарбамоил)метокси]тридец-5-ен-1-ил]-Nметилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)формамидо]тридец-8-ен-1-ил]окси}уксусную кислоту (ВВ-4, 50,0 мг) растворяли в ТГФ (2,00 мл). Добавляли 1,1'-карбонилдиимидазол (25,0 мг) и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов. Затем добавляли DBU (50,0 мкл) и метансульфонамид (16,0 мг) и перемешивание продолжали при комнатной температуре в течение 18 часов. Контроль с помощью ЖХМС показал продукт. Указанную реакционную смесь разбавляли водой (30 мл) и экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои промывали 1 н. HCl (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали препаративной ТСХ (CH2Cl2/МеОН = 95:5).
Выход: 27 мг (44%), твердое вещество белого цвета
Аналитические устройства, используемые для анализа Соед. - 14 - Соед. - 34
Аналит. ЖХ/ESI-MC: Waters 2700 автосэмплер. Система подачи растворителей Waters 1525. 5 мкл пробоотборная петля. Колонка, Phenomenex Onyx Monolythic С18 50×2 мм, с фильтром предварительной очистки из нержавеющей стали 2 мкм. Элюент А, H2O+0,1% НСООН; элюент В, MeCN. Градиент, от 5% В до 100% В в течение 3,80 мин, затем изократический в течение 0,20 мин, затем обратно до 5% В в пределах 0,07 мин, затем изократический в течение 0,23 мин; 0,6 мл/мин и 1,2 мл/мин.
Waters Micromass ZQ 4000 с одним квадрупольным масс-спектрометром с источником электрораспыления. Метод МС, MS4_15минРМ-80-800-35V; сканирование в положительном/отрицательном ионном режиме, m/z 80-800 за 0,5 с; капиллярное напряжение 3,50 кВ; конусное напряжение, 50 В; напряжение умножителя, 650 В; температура исходного блока и газа десольватации, 120°С и 300°С, соответственно. Двухволновой детектор поглощения Waters 2487, установленный на 254 нм. Программное обеспечение, Waters Masslynx V 4.0.
Waters Micromass LCZ Platform 4000 с одним квадрупольным масс-спектрометром с источником электрораспыления. Метод МС, MS4_15минРМ-80-800-35V; сканирование в положительном/отрицательном ионном режиме, m/z 80-800 за 1 с; капиллярное напряжение 4,0 кВ; конусное напряжение, 30 В; напряжение умножителя, 900 В; температура исходного блока и газа десольватации, 120°С и 300°С, соответственно. Детектор с фотодиодной матрицей Waters 996, установленный на от 200 до 400 нм. Программное обеспечение, Waters Masslynx V4.0.
Значения для [М+Н]+, приведенные в примерах, представляют собой значения, указанные в соответствующей хроматограмме ЖХ/МС для определенного соединения. Эти значения были получены в допустимых пределах +/- 0,3 ед. по сравнению с рассчитанной точной массой при протонировании соединения.
Препаративная тонкослойная хроматография (препаративная ТСХ): Пластины для ТСХ Merck, силикагель 60 F254, 0,5 мм, 1,0 мм или 2,0 мм.
Колоночная хроматография: Acros силикагель 60А, 0,035-0,070 мм.
Препаративная ЖХМС-МС: Автосэмплер Waters 2767, система подачи растворителей Waters 600 с насосом с аналитическими головками (100 мкл); контроллер Waters 600; бинарный градиентный модуль Waters 2525 с насосом с препаративными головками (500 мкл). Раствор на колонке: растворитель 1, MeCN: Н20 70:30 (об./об.), растворитель 2, MeCN:MeOH:ДМФА 80:15:5 (об./об./об.); скорость потока 5 мл/мин. Автосамплер 2767 со шприцем 10 мл и пробоотборной петлей 10 мл. 6-позиционный клапан для колонки Flom 401 с Waters X-Terra RP18, 5 мкм, 19×150 мм с защитным картриджем X-Terra RP18 5 мкм, 19×10 мм, использовался при скорости потока 20 мл/мин; Waters SunFire Prep OBD 5 мкм, 30×50 мм с защитным картриджем SunFire RP18 5 мкм, 19×10 мм, использовался со скоростью потока 25 мл/мин; Waters Atlantis Prep Т3 OBD 5 мкм, 30×50 мм с защитным картриджем Atlantis, использовался со скоростью потока 50 мл/мин; Waters X-Bridge Prep OBD 5 мкм, 19×150 мм с защитным картриджем X-Bridge RP18 5 мкм, 19×10 мм использовался при скорости потока 20 мл/мин; Waters Atlantis Prep Т3 OBD 5 мкм, 19×50 мм с защитным картриджем Atlantis, использовался со скоростью потока 25 мл/мин и YMC-Actus Hydrosphere C18 5 мкм, 20×50 мм с защитным картриджем Actus, использовался со скоростью потока 20 мл/мин. Элюент А, Н2О с содержанием 0,1% (об./об.) НСО2Н или Н2О с содержанием 0,1% (об./об.) NEt3; элюент В, MeCN. Различные линейные градиенты, индивидуально адаптированные для образца. Объем впрыска 9 мл, в зависимости от образца. Составной растворитель MeOH-MeCN-H2O-HCO2H 80:15:4,95:0,05 (об./об./об./об.). Насос подпитки Waters Reagent Manager, скорость потока 0,5 мл/мин. Waters ZQ с одним квадрупольным масс-спектрометром с источником электрораспыления. Сканирование в положительном/отрицательном ионном режиме m/z 105-950 за 1 с; капиллярное напряжение 3,6 кВ; конусное напряжение, 45 В; напряжение умножителя, 700 В; температура исходного блока и газа десольватации, 120°С и 250°С, соответственно. Коллектор фракций Waters 2767, сбор фракций по массе или запускается ультрафиолетовым излучением. Двухволновой детектор поглощения Waters 2487, установленный на 254 нм. Программное обеспечение, Waters Masslynx V 4.0 SP4.
Спектры 1Н-ЯМР регистрировали при комнатной температуре на спектрометре Bruker Supraleitendes Fourier NMR Spektrometer, AvanceTM 300 МГц. Химические сдвиги 8 представлены в ppm. Множественность определенного сигнала (синглет, дублет, триплет, квартет, мультиплет) обозначается соответствующей аббревиатурой (s, d, t, q, m соответственно). «Шир. s» обозначает широкий синглет, «mC» - центрированный мультиплет. Остаточные сигналы растворителя использовали в качестве внутренних стандартов: δ(CDCl3)=7,26, δ(d6-ДМСО)=2,50, δ(CD3OD)=3,31, δ(d6-ацетон)=2,05.



Пример 2: Антиаритмический эффект метаболически устойчивых аналогов 17,18-EEQ на NRCM
Материалы и способы
Структуры всех тестируемых соединений приведены на фиг. 1. Соединения включают аналоги, являющиеся частью настоящего изобретения (Соед. - 01 - Соед. - 05), которые были синтезированы, как описано в примере 1, и дополнительные родственные соединения (Соед. - 06 - Соед. - 13). Перед применением готовили 1000-кратные исходные растворы тестируемых соединений в этаноле.
Для измерения биологической активности метаболически устойчивых аналогов CYP-эйкозаноидов использовали установленная клеточная модель (Kang, J.X. et al., Proc Natl Acad Sci USA, 1994. 91(21): p. 9886-90). Спонтанно бьющиеся кардиомиоциты новорожденных крыс (NRCM) являются модельной системой для исследования антиаритмических эффектов тестируемых соединений. Тестируемые соединения снижали скорость спонтанного биения этих клеток и предотвращали их нерегулярное и асинхронное сокращение как ответ на аритмические вещества, что указывает на их антиаритмический потенциал.
Выделение и культивирование NRCM проводили так, как описано ранее (Wallukat, G et al., J Clin Invest. 1999;103: 945-952; Falck, JR et al., JMedChem. 2011 Jun 23;54(12):4109-18). Выделенные клетки культивировали в виде монослоев на дне (12,5 см2) колб Falcon в 2,5 мл среды Halle SM 20-1, находящейся в равновесии с увлажненным воздухом. Среда содержала 10% инактивированной нагреванием FCS и 2 мкмоль/л фтордезоксиуридина (Serva, Хайдельберг, Германия), которая предотвращала пролиферацию немышечных клеток. NRCM (2,4×106 клеток/колбу) культивировали при температуре 37°С в инкубаторе. Через 5-7 дней NRCM образовывали спонтанно бьющиеся клеточные кластеры. Клетки в каждом кластере показали синхронизированное сжатие с частотой биения от 120 до 140 ударов в минуту. В день эксперимента культуральную среду заменяли на 2,0 мл свежей среды, содержащей сыворотку. После четырех часов инкубации клетки адаптировали в течение 10 мин к температуре 31°С, а биение записывали с использованием инвертированного микроскопа (Leica DM IRB), оборудованного камерой CCD и соединенного с IonOptix (программное обеспечение: IonWizard6, IonOptix). Для определения фоновой частоты отбирали от 6 до 8 отдельных кластеров и подсчитывали количество биений в течение 15 секунд. После этого к культуре добавляли тестируемое соединение, и частоту биения тех же самых кластеров снова контролировали через 5 минут. Исходя из разницы между фоновой и индуцированной соединениями частотой биения отдельных кластеров, вычисляли хронотропный эффект (Δ биения/мин) и приводили их как средние значения +/- SE. N относится к числу контролируемых кластеров, которые возникли в целом по меньшей мере из трех независимых культур NRCM. Готовили исходные растворы соединений в этаноле и использовали их для получения конечной концентрации 20 нМ или 30 нМ на NRCM (n=6 на соединение). Контроль носителем (0,1%) не оказывал влияния на фоновую частоту биения.
Результаты
Результаты приведены на фиг. 1. Потенциал аналогов 17,18-EEQ по снижению скорости спонтанного биения варьировался от -1,3±1,0 дельта bpm до -38,0+/- 3,3 дельта bpm в соответствии с различными структурными особенностями. Производные свободной карбоновой кислоты показали большее снижение, чем аналоги, где карбоксигруппа была этерифицирована до полиалкоксиалкила или аминокислоты (Соед. - 10 по сравнению с Соед. - 07 и Соед. - 08 и Соед. - 06 по сравнению с Соед. - 09). Результаты также показали, что можно заменить двойную связь в 11,12-положении на фенильное кольцо без значительной потери активности (Соед. - 06 и Соед. - 05). Однако сдвиг 11,12-двойной связи на 14,15-позицию сильно уменьшил отрицательный хронотропный эффект на NRCM (Соед. - 06 по сравнению с Соед. - 11). Кроме того, аналоги, содержащие 3-окса группу, показали равную эффективность в снижении частоты биения (Соед. - 06 по сравнению с Соед. - 02 и Соед. - 03 или Соед. - 10 по сравнению с Соед. - 01) или увеличивали отрицательный хронотропный эффект (Соед. - 10 по сравнению с Соед. - 04). Для оксидамидной группы было показано, что она необходима для эффективности in vitro, поскольку два продукта разложения оксидамидной группы были неактивны (Соед. - 06 по сравнению с Соед. - 12 и Соед. - 13). Как показано на фиг. 1, дальнейшие соединения, содержащие как 3-окса, так и оксамидную группу, продемонстрировали хорошую активность (Соед. - 14 - Соед. - 35).
Пример 3: Антиаритмический эффект агонистов 17,18-EEQ Соед. - 02 при фибрилляции предсердий
Этот пример показывает, что агонистический аналог Соед.-02 уменьшает выраженность фибрилляции предсердий.
Материалы и способы
Дизайн исследования: Чтобы получить представление об эффекте in vivo синтетических 17,18-EEQ-агонистов, проводили исследования фибрилляции предсердий у самцов мышей Bl6, как описано в Westphal, С. et al., PLoS ONE. 2013, 8(8): e73490. Вкратце, умеренная сердечная гипертрофия индуцировалась непрерывной инфузией изопротеренола посредством подкожно имплантированных осмотических мининасосов (Alzet) со скоростью 40 мг/кг/сут в течение двух недель. Через две недели лечения регистрировали ЭКГ и электрофизиологические данные. Программируемую электрическую стимуляцию (PES) проводили в правом предсердии или в правом желудочке с использованием цифровой электрофизиологической лаборатории (ЕР Tracer, CardioTek) для определения рефракторных фаз и индуктивности аритмии. Предсердные аритмии определялись как быстрая (>800 уд/мин) электрическая активность на электрограммах правых предсердий, с Р-зубцами ЭКГ, отличными от нормального синусового ритма, и последующая быстрая, но физиологическая активация желудочков (зубец R ЭКГ и электрограммы правых желудочков, подобные нормальному синусовому ритму). Фибрилляция предсердий определялась как быстрая, нерегулярная активность на электрограммах правых предсердий с нерегулярной проводимостью к желудочкам (высокая изменчивость интервалов R-R). Желудочковые аритмии определялись как быстрая (>800 уд/мин) активность, происходящая из желудочкового миокарда (изменение морфологии волн ЭКГ и локальных электрограмм правого желудочка по сравнению с нормальным синусовым ритмом). Во время ингаляционной анестезии изофлураном (2% с потоком воздуха 360 мл/мин, аппарат для наркоза Univentor 400) температуру тела животного поддерживали постоянной при 37°С с использованием гомеотермического одеяла с блоком управления (Hugo Sachs Elektronik, Harvard Apparatus) с ректальным контролем температуры. После приготовления правой яремной вены в сердце справа, включая предсердие и желудочек, помещали восьмиполярный электрофизиологический катетер 2 French (CIB'ER mouse cath, NuMed). PES проводили с использованием стандартизованного протокола, который включал череду из 10 базовых стимулов (S1), а затем до 3 дополнительных стимулов (S2-S4), доставляемых с интервалом сцепления, уменьшающимся с шагом 5 мс до достижения рефрактерности желудочков или предсердий. Процедуры стимуляции повторяли с тремя различными базальными циклами (100 мс, 90 мс, 80 мс) с каждым животным. Документировали возникновение и продолжительность индуцируемых аритмий. В качестве положительных рассматривали только протоколы стимуляции с воспроизводимыми аритмиями дольше, чем пять последовательных ударов в желудочке и эпизоды дольше 350 мс в предсердиях. «Индуктивность аритмии» была рассчитана как процент эффективных (положительных) протоколов из общего числа применяемых протоколов. Соответственно, индуцируемость аритмии у отдельных животных может принимать значение 0, 33, 66 или 100%. Для статистической оценки данные, полученные для отдельных животных в данной группе, усредняли и приводили как среднее ± SEM. Для оценки тяжести индуцированных аритмий были определены три категории ответа: устойчивый (≥10 последовательных желудочковых экстрасистол, VES или эпизоды фибрилляции предсердий ≥30 секунд по меньшей мере в одном протоколе), неустойчивый (<10 VES или эпизоды фибрилляции предсердий <30 сек по меньшей мере в одном протоколе) и без аритмий во всех трех протоколах. Данные приведены в процентах от животных в данной группе, относящейся к этим категориям.
Результаты
Результаты приведены на фиг. 2. Болюсные инъекции синтетического агониста 17,18-EEQ (Соед. - 02) не вызывали каких-либо негативных побочных эффектов. Программируемая электрическая стимуляция индуцировала фибрилляцию предсердий у большинства обработанных носителем мышей (n=12). Однократная внутривенная инъекция синтетического агониста 17,18-EEQ Соед. - 02 (2 мг/кг массы тела) значительно уменьшала количество эпизодов полностью индуцированной фибрилляции предсердий (бремя фибрилляции предсердий) (n=14); Фиг. 2А. Более того, тяжесть эпизодов индуцированной фибрилляции предсердий значительно снижалась. В частности, индуцируемость эпизодов устойчивой аритмии была значительно снижена на 62%; Фиг. 2В.
Пример 4: Антиаритмический эффект агонистов 17,18-EEQ на спонтанные желудочковые тахикардии при остром инфаркте миокарда
Этот пример показывает, что агонистический аналог Соед.-03 уменьшает выраженность аритмии, вызванной инфарктом миокарда.
Материалы и способы
Дизайн исследования: Чтобы получить представление о эффектах in vivo синтетических агонистов 17,18-EEQ, проводили исследования инфаркта миокарда у самцов крыс Wistar. Вкратце, крысы весом 220-250 г были рандомизированы для получения внутривенного болюса Соед. - 03 (100 мкг в 300 мкл 0,9% NaCl) или только 300 мкл 0,9% NaCl в качестве контроля носителем за десять минут до того, как у крыс вызывали инфаркт миокарда. Для безопасного применения болюса животных слегка анестезировали с использованием изофларана. Начинали непрерывный мониторинг поверхностной ЭКГ (EPTracer, Нидерланды) и продолжали до конца исследования (через 45 минут после того, как был вызван инфаркт миокарда). После регистрации базальной ЭКГ у крыс вызывали инфаркт миокарда лигированием левой передней нисходящей артерии (LAD). Через 45 минут после инфаркта миокарда животных умерщвляли и отбирали органы. Для дальнейшего анализа хранили образцы мочи, крови, печени, почек и сердца.
Метод анализа аритмии: Бремя желудочковой тахикардии рассчитывалось как сумма всех аритмических событий, возникших в желудочковом миокарде, которые наблюдались в течение первого часа после того, как был вызван инфаркт миокарда. Для количественной оценки не только частоты, но и тяжести желудочковых аритмий рассчитывали показатель тяжести аритмии. Этот показатель рассчитывался как сумма количеств различных случаев аритмии (ПЖС, дублет, триплет, ЖТ<1,5 с, ЖТ>=1,5 с), каждый класс факторизовали увеличением индекса тяжести 1-5 (например, ПЖС x 1, дублеты x 2, триплеты 1,5 с х3, ЖТ>=1,5 с х 5).
Результаты
Результаты приведены на фиг. 3. Болюсные инъекции синтетического агониста 17,18-EEQ (Соед. - 03) не вызвали каких-либо негативных побочных эффектов. Желудочковые аритмии возникали после лигирования коронарной артерии и наблюдались как единичные преждевременные желудочковые сокращения (ПЖС), короткие периоды неустойчивой желудочковой тахикардии (ЖТ) и желудочковой тахикардии/фибрилляции. У крыс, обработанных синтетическим агонистом 17,18-EEQ (n=10), наблюдали значительно сокращенную продолжительность желудочковой тахикардии по сравнению с контролем (n=9); Фиг. 3А. Кроме того, показатель тяжести аритмии значительно снижался. Фиг. 3В.
Пример 5: Кардиопротекторное действие 17,18-EEQ при ишемическом/реперфузионном повреждении сердца
Этот пример показывает, что агонистический аналог Соед. - 03 улучшает пост-ишемическое восстановление после определенного периода ишемии.
Материалы и способы
Дизайн исследования: Чтобы получить представление о кардиопротекторном действии синтетических агонистов 17,18-EEQ, проводили исследования ишемии-реперфузии в изолированных сердцах мышей. Вкратце, сердца были перфузированы по методике Лангендорфа, как описано в Seubert et al., Circ Res. 2004; 95: 506-514. Сердца перфузировали буфером в течение 10-минутного периода стабилизации, затем перфузировали либо агонистическим аналогом Соед.-03 (1 мкМ), либо носителем в течение 10 минут, затем подвергали 35-минутной глобальной ишемии без потока с последующей реперфузией в течение 40 минут. Во время ишемии и реперфузии поддерживали постоянную инфузию агонистическим аналогом Соед.-03 или носителя. Восстановление сократительной функции измеряли как давление, развиваемое в левом желудочке (LVDP) в конце реперфузии, выраженное в процентах от предишемического LVDP.
Результаты
Результаты приведены на фиг. 4. Непрерывная инфузия 1 мкМ синтетического агониста 17,18-EEQ (Соед-03) не вызывала каких-либо очевидных негативных побочных эффектов. Сразу же после 35-минутного эпизода глобальной ишемии сократительная способность контрольных сердец (n=14), выраженная как давление, развиваемое в левом желудочке (LVdP), сильно снижалось и возвращалось затем постепенно в фазу реперфузии примерно до 50% доишемических значений. Сердца, обработанные синтетическим агонистом 17,18-EEQ Соед.-03 (n=14), показали значительно улучшенное восстановление сократительной способности.
Пример 6: Ингибирование in vitro рекомбинантной растворимой эпоксигидролазы человека
Материалы и способы
Набор соединений тестировали на способность ингибировать растворимую эпоксигидролазу человека (sEH). Метаболически устойчивые аналоги 17,18-EEQ сами по себе не склонны к метаболизму с помощью sEH, но могут выступать в качестве ингибиторов этого фермента. Вкратце, анализ проводили при температуре 37°C в течение 20 мин в конечном объеме 100 мкл калий-фосфатного буфера (0,1 М, pH 7,2), содержащего 50 мкМ 14,15-ЕЕТ в качестве субстрата. Реакции начинали с добавления фермента (107,5 нг на реакцию, активность 12,06 ед/мл, Cayman Chemicals) и заканчивали этилацетатом 300 мкл. Оставшийся субстрат и его продукт (14,15-DHET) экстрагировали и анализировали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (RP-HPLC) (Muller, D.N. et al., Biochem J 2007. 403(1): p. 109-118). Метаболически устойчивые аналоги, показанные на фиг. 5, испытывали при конечной концентрации 10 мкМ на эффективность ингибирования sEH по сравнению с контролем носителем (1% ДМСО), n=2-4.
Результаты
На фиг. 5 показано, что некоторые из тестируемых аналогов 17,18-EEQ ингибировали sEH человека до 76,6%. Эти соединения имеют один структурный признак, они содержат группу мочевины (Соед.-01, -07, -08 и Соед.-10). Напротив, оба тестируемых соединения, содержащие оксидамидную группу, не проявляли ингибирование sEH (Соед.-02 и Соед.-03).
Пример 7: Потенциал проницаемости метаболически устойчивых аналогов CYP-эйкозаноидов, тестируемый в клетках Сасо-2.
Материалы и способы
Чтобы предсказать проницаемость кишечника человека и исследовать отток лекарственного средства, т.е. параметры, влияющие на биодоступность соединения (van Breemen RB & Li Y., Expert Opin Drug Metab Toxicol. 2005 Aug; 1(2):175-85.), несколько метаболически устойчивых аналогов 17,18-EEQ были протестированы на предмет их способности проникать через конфлюэнтный монослой клеток Сасо-2 (клеточная линия аденокарциномы толстой кишки человека). Исходные растворы соединений получали в ДМСО (1 мМ), и соединения испытывали при конечной концентрации 1 мкМ. Для оценки проницаемости монослоя раствор тестируемого соединения помещали на апикальную сторону монослоя Сасо-2 (время инкубации 2 ч), и измеряли скорости появления тестируемых соединений на базолатеральной стороне клеток, эти данные приводятся как проницаемость от А до В в см/с для тестируемых соединений (фиг. 6).
Результаты
Результаты, представленные на фиг. 6, показывают, что соединения с группой мочевины показали меньшую проницаемость (Соед.-01 и Соед.-04), чем соединения, содержащие оксидамидную группу (Соед.-02 и Соед.-03). Дополнительный полиалкоксиалкил, этерифицированный к группе карбоновой кислоты, дополнительно улучшает проницаемость (Соед.-02 по сравнению с Соед.-03).
Пример 8: Способность метаболически устойчивых аналогов CYP-эйкозаноидов внедряться в фосфолипидные мембраны
Материалы и способы
Чтобы понять, могут ли модифицированные структуры (выбранные метаболически устойчивые аналоги CYP-эйкозаноидов, таблица 1) внедряться в фосфолипидные мембраны, клеточную линию кардиомиоцитов (Н9с2, крыса) инкубировали с тестируемыми соединениями в течение четырех часов при конечной концентрации 1 мкМ (n=2). После инкубации клеточные липиды экстрагировали смесью хлороформ/метанол 1:2. Для того, чтобы отличить соединения в свободной форме от внедренных соединений аликвоту образцов подвергали щелочному гидролизу. Оба экстракта анализировали на испытуемые соединения с использованием ЖХ-МС/МС. 17,18-EEQ и 20-НЕТЕ (Kaduce TL et al., J Biol Chem. 2004 Jan 23; 279(4):2648-56.) служили положительным контролем для внедрения в мембрану.
Результаты
На фиг. 7 показано, что, в отличие от положительных контролей (17,18-EEQ и 20-НЕТЕ), соединения согласно настоящему изобретению (Соед.-02 и Соед.-04), а также соединение, содержащее оксидамид (Соед.-06), не показали внедрение в фосфолипидные мембраны. Содержащее мочевину Соед.-10, лишенное 3-оха-группы, показало слабое внедрение.
Пример 9: Оценка растворимости метаболически устойчивых аналогов 17,18-EEQ
Материалы и способы
Водную растворимость Соед.-06 и Соед.-02 определяли в деионизированной воде при температуре 37°C. Известные количества соединений суспендировали при перемешивании в течение 24 часов. Фильтраты изолировали и с помощью ВЭЖХ-анализа определяли содержание соответствующих соединений.
Результаты
В таблице 1 показано, что введение в структуру 3-оксагруппы улучшает растворимость. При сравнении растворимости в деионизированной воде растворимость Соед.-02 была примерно в 10 раз больше, чем у Соед.-06.

Пример 10: Оценка растворимости метаболически устойчивых аналогов 17,18-EEQ
Материалы и способы
Дизайн исследования: Чтобы получить представление о фармакологических свойствах синтетических агонистов 17,18-EEQ, с двумя выбранными соединениями (Соед.-02 и Соед.-06) проводили фармакокинетические исследования. Указанные соединения были фармакологически оценены в плазме после однократного внутривенного и перорального введения. Таким образом, мышам C57BL/6 (Janvier Labs (Франция), n=12 на группу) вводили тестируемое соединение в дозе 2 мг/кг (внутривенно) и 8 мг/кг (перорально), соответственно. Внутривенное введение Соед.-02 (натриевая соль в изотоническом солевом растворе) и Соед.-06 (свободная кислота в ДМСО/PEG400 20:80) выполнялось через хвостовую вену, а пероральное введение через желудочный зонд. Для обеспечения равной растворимости Соед.-06 приготовили в ДМСО/PEG400 20:80, поскольку Соед.-06 в виде натриевой соли показало нарушение растворимости. Отбор образцов крови по 100 мкл проводили два раза в разное время для каждой мыши (0,083/2 ч, 0,25/4 ч, 0,5/8 ч и 1/24 ч). Биологические образцы подвергали процедуре экстракции (40 мкл ацетонитрила + 22,4 мкл образца, встряхивание, центрифугирование 10 мин при 6000 g при комнатной температуре, разбавление 50 мкл супернатанта 1:1 водой). Для анализа 20 мкл образца вводили в ЖХМС, система УВЭЖХ Accela 1250, открытый аутосэмплер Accela, Q-Exactive масс-спектрометр (Thermo Fisher Scientific). В качестве контролей использовались соответствующие образцы: нулевой образец, калибровочный стандарт и образец QC. Фармакокинетический анализ проводили с использованием некомпартментной модели с использованием программного обеспечения Kinetica 5.0 (Thermo Scientific, Waltham, США). Все данные параметры были получены путем расчета площади трапеции.
Cmax (нг/мл) максимальная наблюдаемая концентрация в плазме
tmax (ч) время максимальной наблюдаемой концентрации в плазме
AUC0-∞ (нг*ч/мл) площадь под кривой концентрация-время, экстраполированная до бесконечности
F (%) пероральная биодоступность, выраженная в процентах
Результаты
Результаты, представленные в таблице 2, показывают улучшенные свойства в фармакокинетических параметрах Соед.-02 по сравнению с Соед.-06. Соединение согласно настоящему изобретению (Соед.-02) отличается только дополнительной 3-оксагруппой, которая отсутствует в Соед.-06. Эта структурная особенность значительно улучшала оральную биодоступность (87% по сравнению с 5,3%). Более того, максимальная наблюдаемая концентрация была намного выше и достигалась быстрее для Соед.-02 (903,3 нг/мл и 0,3 часа), чем для Соед.-06 (24,4 нг/мл и 0,5 часа). Кроме того, AUC0-∞, как мера общего содержания соединения во времени, была выше для Соед.-02 (1818 нг*ч/мл) по сравнению с Соед.-06 (31 нг*ч/мл).


Пример 11: Кардиопротекторное действие синтетического агониста 17,18-EEQ при ишемии/реперфузии сердца
В дополнение к примеру 5 этот пример показывает, что другой агонистический аналог согласно настоящему изобретению, а именно Соед.-02 улучшает пост-ишемическое восстановление после определенного периода ишемии.
Материалы и способы
Дизайн исследования: Чтобы получить представление о кардиопротекторном действии синтетических агонистов 17,18-EEQ, проводили исследования ишемии-реперфузии в изолированных сердцах мышей, 12-14-недельных самцов C57BL6/n. Вкратце, мышей анестезировали инъекцией пентобарбитала/гепарина внутривенно, быстро удаляли сердца и перфузировали в аппарате Лангендорфа. Вырезанные сердца помещали в ледяной буфер Кребса-Хенселейта и канюлировали аорты. Модифицированный буфер Кребса-Хенселейта для ретроградной перфузии, содержащий 118 мМ NaCl, 3,5 мМ KCl, 1,3 мМ MgSO4, 2,5 мМ CaCl2, 24,7 мМ NaHCO3, 1,4 мМ KH2PO4 и 11 мМ глюкозы, аэрировали 95% O2 - 5% CO2 и выдерживали при постоянной температуре 37°C и pH 7,3. Перфузионное давление доводили до 80 мм рт.ст.; латексный шарик, соединенный с измерительным датчиком давления, вводили в левый желудочек через левое предсердие. Сердца перфузировали буфером в течение периода стабилизации 15 минут с последующей 10-минутной перфузией агонистическим аналогом Соед.-02 (100 нМ) или носителем (0,9% NaCl в воде). После этого сердца подвергали 35-минутной глобальной ишемии без потока, а затем 40-минутной реперфузии. Перфузия соединением или носителем поддерживалась постоянной во время реперфузии. Для анализа восстановления сократительной функции измеряли давление, развиваемое в левом желудочке (LVdP) в разные моменты времени во время реперфузии и выражалось в процентах от предишемического LVdP. Критериями для исключения сердец были: (i) LVdP ниже 60 мм рт.ст. в конце фазы стабилизации (базовое измерение), (ii) частота сердечных сокращений ниже 260 уд/мин, (iii) коронарный поток ниже 1,5 или более 4 мл/мин и, наконец, (iv) тяжелая устойчивая аритмии во время фазы стабилизации. Результаты представлены на фиг. 1 и выражены как среднее ± SEM в процентах от доишемического LVdP. Данные анализировали с помощью двухстороннего непарного теста Стьюдента и считались значимыми, если p<0,05*.
Результаты
На фиг. 8 показано, что непрерывная инфузия 100 нм Соед.-02 не вызывала никаких очевидных отрицательных побочных эффектов. После глобальной ишемии резко уменьшилась сократительная способность контрольных сердец (n=5). Через 10 мин реперфузионного времени доишемические значения LVdP снизились до 1%, но постепенно возвращались к примерно 18% после 40 минут реперфузии. Напротив, сердца, обработанные Соед.-02 (n=5), показали значительно улучшенное пост-ишемическое восстановление функции сократимости по сравнению с контрольной группой. Для ранних (10 минут), а также для поздних (40 минут) моментов времени во время фазы реперфузии показано улучшение функционального восстановления со значениями 18% и 59% от доишемических значений LVdP, соответственно.
Пример 12: Кардиопротекторное действие устойчивых аналогов 17,18-EEQ на изолированные первичные кардиомиоциты
Материалы и способы
Ишемическое повреждение in vitro индуцировали в культуре первичных кардиомиоцитов новорожденных мышей посредством кислородно-глюкозной депривации в течение 18 часов и 24-часового периода реоксигенации в увлажненном инкубаторе с 5% CO2/95% воздух. Кардиомиоциты инкубировали с испытуемыми соединениями в течение 4 часов до индукции кислородно-глюкозной депривацией (OGD). Тестируемые соединения также присутствовали во время OGD и в течение 24-часового периода реоксигенации. После периода реоксигенации количество клеток оценивали по окрашиванию Hoechst 33342, измеряли апоптоз, определяя активацию каспазы 3/7, и целостность клеточной мембраны или некроз измеряли как высвобождение LDH.
Данные выражали как среднее ± SD трех отдельных лунок, и индивидуальные сравнения проводились с t-критерием Стьюдента (программа SigmaPlot 9.0). Результаты были нормализованы для клеток, обработанных при нормоксии. Статистическая значимость (*) p<0,05 по сравнению с клетками, обработанными нормоксией/ДМСО, и  с клетками, обработанными гипоксией/ДМСО.
с клетками, обработанными гипоксией/ДМСО.
Результаты
На фиг. 9А-9В показано, что Соед.-02 частично защищало от повреждения, вызванного OGD, в первичных кардиомиоцитах. Оно снижало количество потерь клеток (на 35% при 30 нМ или на 44% при 10 мкМ) и уменьшало OGD-индуцированные апоптоз и некроз (на 43% и 29% соответственно). Естественный предшественник соединения согласно настоящему изобретению 17,18-EEQ оказался менее эффективным, так как он существенно не влиял на число клеток (при 30 нМ и 10 мкМ) и уменьшал апоптоз только при высокой концентрации (10 мкМ, 43%), но не при низкой концентрации (30 нМ). Однако при предотвращении вызванного OGD-реоксигенацией клеточного некроза Соед.-02 и 17,18-EEQ казались одинаково эффективными, что было проверено при высвобождении LDH. Соед.-02 уменьшало клеточный некроз на 43% (30 нМ) и на 45% (10 мкМ) соответственно. Сравнительно, 17,18-EEQ уменьшало высвобождение LDH на 41% (30 нМ) и на 37% (10 мкМ) соответственно, см. фиг. 9С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| Новые производные CYP-эйкозаноидов | 2015 |
|
RU2730512C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2021 |
|
RU2819642C1 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2020 |
|
RU2834484C2 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2773346C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И СПОСОБ ИДЕНТИФИКАЦИИ АГЕНТА НА ЕГО ОСНОВЕ | 1999 |
|
RU2245335C2 |
| ИНГИБИТОРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2019 |
|
RU2806033C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
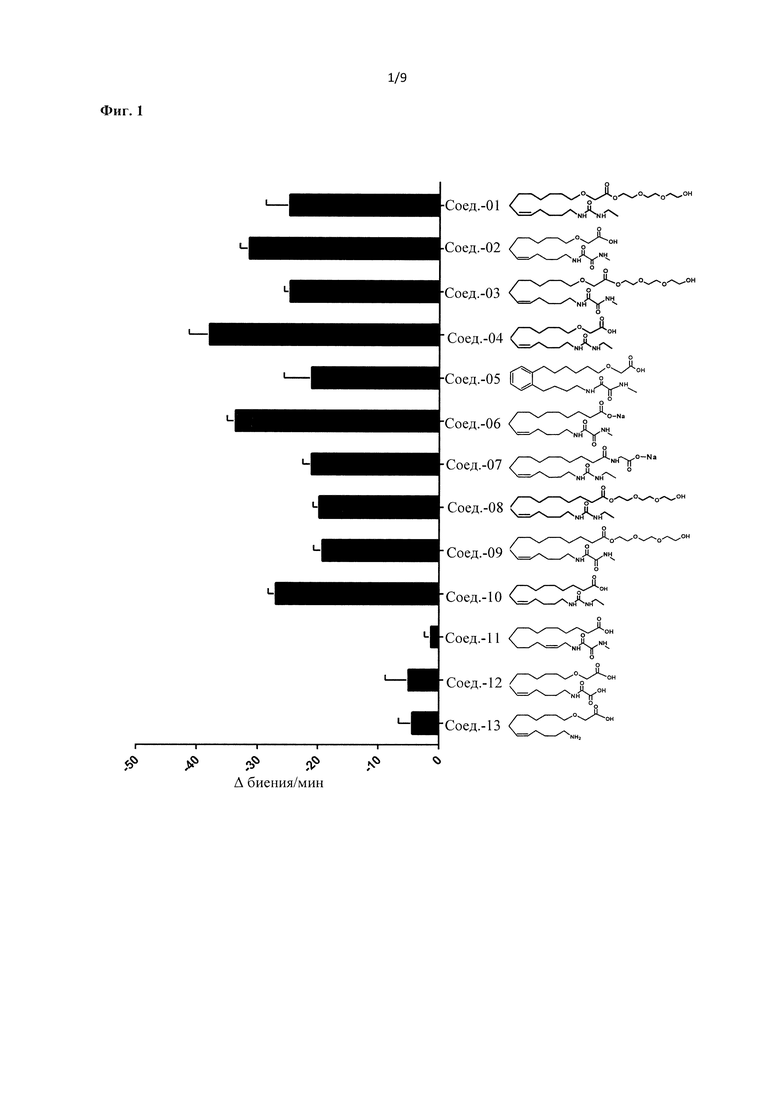








Изобретение относится к соединению общей формулы P-E-I или его фармацевтически приемлемым солям, которые представляют собой метаболически устойчивые аналоги биоактивных липидных медиаторов, полученных из полиненасыщенных жирных кислот омега-3 (n-3 ПНЖК), фармацевтическим композициям на их основе, их применению и способу лечения сердечно-сосудистых заболеваний с их использованием. В общей формуле P-E-I P представляет собой -(CH2)n-O-(CH2)k-X, n представляет собой целое число от 3 до 8; и k представляет собой 0, 1 или 2; X представляет собой CH2OH, CH2OAc, CH(O) или группу, выбранную из

 и
и 
R2 представляет собой -NHR3; -NR20R21; -OR22; -(OCH2-CH2)i-R23; -C3-C10-гетероциклил, содержащий один атом кислорода; R3 представляет собой (SO2R30); C6-арильную группу, необязательно замещенную -O-CH3 или COOH, гетероарильную группу, содержащую 3 атома углерода и два гетероатома, представляющих собой O и N; или 6-членный гетероциклоалкил, содержащий 1 гетероатом, представляющий собой O; R30 представляет собой C1-C6 алкильную или С6-арильную группу; каждый из R20 и R21 независимо представляет собой Н, C1-C6алкильную группу, которая может быть замещена одной или более гидроксильными группами; C3-C6циклоалкильную группу, или совместно образуют 5-6-членный гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из O и N; R22 представляет собой Н или C1-C6алкильную группу; R23 представляет собой -OH; i представляет собой целое число от 1 до 10; R5 представляет собой Н; атом фтора или хлора; -CF3; -C(=O)OR51; -NHC(=O)R52; -C(=O)NR53R54; или -S(O2)OH; R51 представляет собой Н или C1-C6алкильную группу; каждый из R52, R53 и R54 независимо представляет собой C1-C6алкильную группу; R9 представляет собой C1-C6алкил или С6-арил; g представляет собой 1 или 2; X1 представляет собой атом кислорода; атом серы или NH; E представляет собой группу, представленную общей формулой (III) или (IV), и кольцо A в формуле (III) представляет собой 6-членное карбоциклическое кольцо, содержащее по меньшей мере одну двойную связь, включая 6-членное ароматическое карбоциклическое кольцо; и каждый из L и T независимо представляет собой атом кольца, L и T смежны друг с другом; каждый из R12 и R13 независимо представляет собой Н, атом фтора или гидроксил; I представляет собой -(CH2)m-Y, m представляет собой целое число от 3 до 6, при условии, что m представляет собой целое число от 3 до 5, когда E представляет собой группу общей формулы (III); Y представляет собой группу, выбранную из

каждый из R40, R41, R43 и R44 независимо представляет собой Н или -C1-C6алкил; каждый из R42 или R45 независимо представляет собой -C1-C3алкил. 5 н. и 12 з.п. ф-лы, 9 ил., 3 табл., 12 пр.

1. Соединение общей формулы (I)
P-E-I (I),
или его фармацевтически приемлемая соль, где
P представляет собой группу, представленную общей формулой (II)
-(CH2)n-O-(CH2)k-X (II),
где
n представляет собой целое число от 3 до 8; и
k представляет собой 0, 1 или 2; наиболее предпочтительно k равно 1;
X представляет собой CH2OH, CH2OAc, CH(O) или группу, выбранную из группы, состоящей из:

 и
и  ; предпочтительно X представляет собой
; предпочтительно X представляет собой 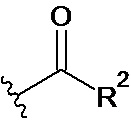 ;
;
где
R2 представляет собой -NHR3; -NR20R21; -OR22; -(OCH2-CH2)i-R23; -C3-C10-гетероциклил, содержащий один гетероатом, представляющий собой кислород;
где
R3 представляет собой (SO2R30); C6-арильную группу, необязательно замещенную -O-CH3 или COOH, гетероарильную группу, содержащую 3 атома углерода и два гетероатома, представляющих собой O и N; или 6-членный гетероциклоалкил, содержащий 1 гетероатом, представляющий собой O;
R30 представляет собой C1-C6 алкильную или С6-арильную группу;
каждый из R20 и R21 независимо представляет собой атом водорода, C1-C6алкильную группу, которая может быть замещена одной или более гидроксильными группами; C3-C6циклоалкильную группу, или совместно образуют 5-6-членный гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из O и N;
R22 представляет собой атом водорода или C1-C6алкильную группу;
R23 представляет собой -OH;
i представляет собой целое число от 1 до 10;
R5 представляет собой атом водорода; атом фтора или хлора; -CF3; -C(=O)OR51; -NHC(=O)R52; -C(=O)NR53R54; или -S(O2)OH;
R51 представляет собой атом водорода или C1-C6алкильную группу;
каждый из R52, R53 и R54 независимо представляет собой C1-C6алкильную группу;
R9 представляет собой C1-C6алкил или С6-арил;
g представляет собой 1 или 2;
X1 представляет собой атом кислорода; атом серы или NH;
E представляет собой группу, представленную общей формулой (III) или (IV)
 ,
,
где R12 и R13 предпочтительно находятся в цис-конфигурации, и
где кольцо A в формуле (III) представляет собой 6-членное карбоциклическое кольцо, содержащее по меньшей мере одну двойную связь, включая 6-членное ароматическое карбоциклическое кольцо; и каждый из L и T независимо представляет собой атом кольца, где L и T смежны друг с другом;
каждый из R12 и R13 независимо представляет собой атом водорода, атом фтора или гидроксил;
I представляет собой -(CH2)m-Y, где
m представляет собой целое число от 3 до 6, при условии, что m представляет собой целое число от 3 до 5, когда E представляет собой группу общей формулы (III);
Y представляет собой группу, выбранную из группы, состоящей из:
(оксамид)
где
каждый из R40, R41, R43 и R44 независимо представляет собой атом водорода или -C1-C6алкил;
каждый из R42 или R45 независимо представляет собой -C1-C3алкил;
при условии, что
когда X не содержит фрагмент -C(=O)O с карбонильным углеродом в альфа- или бета-положении по отношению к атому кислорода общей формулы (II), Y представляет собой оксамид, карбамат или карбамид, предпочтительно Y представляет собой оксамид, как определено выше.
2. Соединение по п. 1,
при дополнительном условии, что
когда n равно 3 или 5, k равно 1 и E представляет собой группу общей формулы (III) или общей формулы (IV), где каждый из R12 и R13 представляет собой атом водорода;
P представляет собой группу:
-(CH2)3-O-(CH2)-X81; -(CH2)5-O-(CH2)-X81;
где
X81 представляет собой группу, выбранную из группы, состоящей из:
 ,
,
R2' представляет собой -NHR3'; -OR22'; -(OCH2-CH2)i-R23;
где
R3' представляет собой (SO2R30);
R22' представляет собой водород;
R23 и i являются такими, как определено выше;
R9' определен как R9 выше; R9'' представляет собой С6-арил.
3. Соединение по п. 1 или 2, где Х представляет собой
 ;
;
где R2 представляет собой -ОR22; -(OCH2-CH2)i-R23;
где R23 и i являются такими, как определено выше; и
где R22 и R23 являются такими, как определено в п. 1.
4. Соединение по любому из пп. 1-3, отличающееся тем, что X представляет собой -C(=O)OH или подходящую соль карбоновой кислоты, предпочтительно свободную карбоновую кислоту.
5. Соединение по любому из пп. 1-4, отличающееся тем, что Y представляет собой один из оксамидов, определенных в п. 1.
6. Соединение по любому из пп. 1-3 и 5, отличающееся тем, что Х представляет собой
 ,
,
где R2 представляет собой -ОR22; -(OCH2-CH2)i-R23;
где R22, R23 и i являются такими, как определено в п. 1, и где Y представляет собой один из оксамидов, определенных в п. 1.
7. Соединение по любому из пп. 1-6, отличающееся тем, что Х представляет собой C(=O)OH, предпочтительно свободную карбоновую кислоту, и Y представляет собой один из оксамидов, определенных в п. 1.
8. Соединение по п. 1 формулы (V)
 (V),
(V),
где
R55 представляет собой -OH; -OR22; -(OCH2-CH2)i-R23;
R22, R23 и i являются такими, как определено в п. 1, предпочтительно R22 представляет собой атом водорода или C1-C6алкильную группу, более предпочтительно атом водорода, и i предпочтительно составляет от 2 до 4, более предпочтительно 3;
Y представляет собой группу, выбранную из группы, состоящей из:
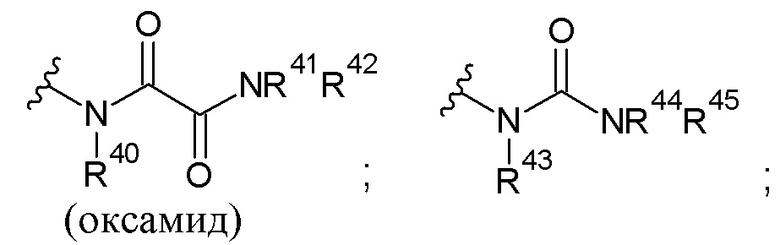
где R40-R50 являются такими, как определено в п. 1, предпочтительно R40 представляет собой атом водорода или C1-C6алкильную группу, более предпочтительно атом водорода,
R57 и R58 представляют собой водород;
s равно 1 или 2;
двойная связь в формуле (V) представляет собой двойную углерод-углеродную связь в цис-конфигурации, если R57 и R58 представляют собой водород.
9. Соединение по п. 8, где
R55 представляет собой -ОН или -(OCH2-CH2)i-R23; i принимает значения от 2 до 4, предпочтительно i равно 3; R23 предпочтительно представляет собой ОН;
Y является таким, как определено в п. 1;
R57 и R58 оба представляют собой H; и
s равно 1.
10. Соединения по любому из пп. 1-3, отличающиеся тем, что указанное соединение выбрано из группы, состоящей из:

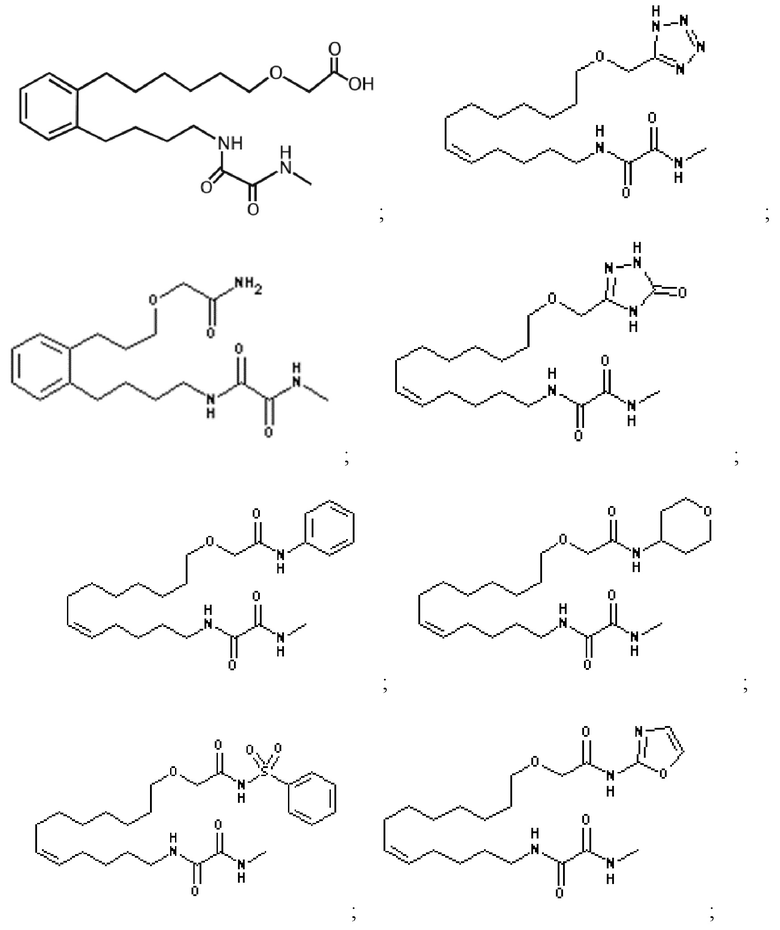
 ;
;  ;
;  ;
;  ;
;  ;
;  ;
; ;
;  ;
;  ;
;  ;
;  ;
;  ;
;
 ;
;  ;
; 
или их фармацевтически приемлемая соль.
11. Соединение по любому из пп. 1-10 формулы (VI)
 (VI)
(VI)
или его фармацевтически приемлемая соль.
12. Фармацевтическая композиция для лечения или предотвращения сердечно-сосудистого заболевания, содержащая эффективное количество соединения по любому из пп. 1-11 совместно с физиологически приемлемым вспомогательным веществом.
13. Применение соединения по любому из пп. 1-11 для лечения сердечно-сосудистых заболеваний, предпочтительно выбранных из группы, состоящей из фибрилляции предсердий, желудочковой аритмии, сердечной недостаточности, заболевания коронарной артерии, инфаркта миокарда, неадаптивной сердечной гипертрофии и сердечных аритмий, включая желудочковые экстрасистолы, желудочковую тахикардию, злокачественную желудочковую тахикардию, предсердную тахикардию, трепетание предсердий и фибрилляцию предсердий, дилатационную кардиомиопатию и гипертоническую болезнь сердца, предпочтительно выбранные из группы, состоящей из фибрилляции предсердий, предсердной тахикардии, желудочковой аритмии, сердечной недостаточности.
14. Применение фармацевтической композиции по п. 12 для лечения сердечно-сосудистых заболеваний, предпочтительно выбранных из группы, состоящей из фибрилляции предсердий, желудочковой аритмии, сердечной недостаточности, заболевания коронарной артерии, инфаркта миокарда, неадаптивной сердечной гипертрофии и сердечных аритмий, включая желудочковые экстрасистолы, желудочковую тахикардию, злокачественную желудочковую тахикардию, предсердную тахикардию, трепетание предсердий и фибрилляцию предсердий, дилатационную кардиомиопатию и гипертоническую болезнь сердца, предпочтительно выбранные из группы, состоящей из фибрилляции предсердий, предсердной тахикардии, желудочковой аритмии, сердечной недостаточности.
15. Применение по п. 13 или 14, отличающееся тем, что указанные соединение или композицию вводят перорально, местно, подкожно, внутримышечно, внутрибрюшинно, внутривенно, интраназально, предпочтительно перорально или внутривенно, более предпочтительно перорально.
16. Применение по любому из пп. 13-15, отличающееся тем, что указанные соединение или композиция представляют собой лекарственную форму, выбранную из группы, состоящей из спрея, аэрозоля, пены, лекарственной формы для ингалятора, порошка, таблетки, капсулы, мягкой желатиновой капсулы, чая, сиропа, гранулы, жевательной таблетки, мази, крема, геля, суппозитория, пастилки для рассасывания, липосомальной композиции и раствора, подходящего для инъекций.
17. Способ лечения сердечно-сосудистого заболевания, включающий стадию введения соединения или композиции на его основе по любому из пп. 1-12 в эффективном количестве нуждающемуся в этом субъекту, предпочтительно млекопитающему, более предпочтительно человеку.
| Компенсированный рычаг управления рулем высоты | 1929 |
|
SU23104A1 |
| WO 2010081683 A1 22.07.2010 | |||
| Falck, J | |||
| R., Kodela, R., Manne, R., Atcha, K | |||
| R., Puli, N., Dubasi, N | |||
| Campbell, W | |||
| B | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Journal of Medicinal | |||

