Заявляемое изобретение относится к области химической технологии азотсодержащих соединений кремния, в частности, к способам получения прекерамических волокнообразующих олигоорганосилазанов взаимодействием органогалогенсиланов с аммиаком, дисилазанами с последующей обработкой продукта аммонолиза основным катализатором, депротонирующим NH-группы. Керамические волокна, благодаря своим эксплуатационным характеристикам: легкому весу, огнестойкости, высокой прочности, износостойкости, стабильности при высоких температурах в агрессивных и окислительных средах, низкому коэффициенту термического расширения (КТР), обеспечивают при использовании их в качестве армирующих компонентов керамоматричных композиционных материалов (ККМ) повышение эксплуатационных характеристик. Наряду с волокнами, олигоорганосилазаны позволяют реализовать различные способы получения керамических материалов, которые не могут быть получены традиционными методами, а, именно: керамических покрытий на различных подложках или ККМ с керамической матрицей на основе карбонитридо-, нитридокремния.
Авторами патента (US №4,543,344, МПК С04В 35/58, 1985 г.) сообщается о способе получения прекерамических волокнообразующих полиорганосилазанов, содержащих в своей структуре от одного до нескольких циклосилазановых звеньев. Полисилазаны получают взаимодействием трихлорсилана и органодисилазана с органическими заместителями различной природы при кремнии с образованием сополимера, который далее обрабатывают основным катализатором, депротонирующим группы NH и SiH, находящиеся рядом. Перегруппировки, происходящие при этом в реакционной смеси, сопровождаются увеличением молекулярной массы полиорганосилазанов.
К недостаткам способа можно отнести то, что полученный полимер не рассматривается как коммерческий продукт.
В патенте (US №4,395,460, МПК С04В 35/52, 1983 г.) описан способ получения прекерамических полисилазанов аммонолизом смеси органохлордисиланов с различными заместителями при кремнии в инертном растворителе с последующей обработкой продукта аммонолиза основным катализатором, способным депротонировать группы NH. Преимущество этого процесса заключается в возможности остановить реакцию в любой момент путем охлаждения реакционной массы, что позволяет управлять вязкостью полимеров, а, следовательно, и их молекулярной массой. По внешнему виду полисилазаны могут быть: жидкостями с различной вязкостью или твердыми стеклообразными материалами. В изобретении описано получение полисилазанов с высоким содержанием кремния, гидролитически стабильных и простых в использовании. После пиролиза полисилазанов при 1200°C в инертной атмосфере выход неорганического остатка составил 46,3 мас. % и имел следующий состав: углерод - 29 мас. %; водород - 7-8 мас. %; кремний - 45 мас. % и азот - 8,1 мас. %.
К недостаткам метода можно отнести то, что в процессе полимеризации не удается достичь высоких молекулярных масс, поэтому для обеспечения формования волокон необходимо использовать мелкодисперсные наполнители. Также недостатком приведенного способа является получение низкого выхода неорганического остатка после пиролиза. В патенте не сообщается о стабильности волокнообразующих полисилазанов в виде гранул, особенно, для составов с содержанием нелетучих веществ более 97 мас. %, что является необходимым условием для формования волокон из расплава.
Наиболее близким по технической сущности и принятым нами в качестве прототипа, является предложенный авторами в патенте (US №4,482,669, МПК С08К 3/34, 1984) способ получения полиорганосилазанов в растворителе в инертной атмосфере взаимодействием органодигалогенсилана с аммиаком с образованием продукта аммонолиза, для которого после обработки основным катализатором, способным депротонировать рядом находящиеся группы NH и SiH, наблюдается повышение молекулярной массы полисилазана. Прекерамический полимер характеризуется циклолинейной структурой, поскольку содержит множество циклических и/или линейных звеньев, связанных между собой мостиками состава Si2N2. Такие полимеры образуют новые лестничные или плоские решетчатые структуры и могут находиться в различных физических состояниях: в форме порошков, растворимых в органических растворителях; в виде воскообразных твердых или вязких материалов. Пиролиз полимеров приводит к высокому выходу керамики до 83 мас. %, содержащей нитрид кремния, карбид кремния и углерод в мольном соотношении 0,88:1,27:0,75, соответственно. Прекерамические олигосилазаны используют для получения керамических покрытий, волокон и керамических изделий на их основе, различного применения.
Недостатком данного метода является отсутствие исследований полисилазанов с содержанием нелетучих веществ более 97 мас. %, а содержание нелетучих веществ более 97 мас. % является необходимым условием для формования волокон из расплава. Кроме того, не приведены данные о возможности получения полисилазанов в виде гранул с высокой стабильностью при хранении. Стабильность полисилазанов в виде гранул существенно сокращается время контакта с воздухом при дозировке, что препятствует появлению кислорода в полимере.
Задачей настоящего изобретения является разработка универсального способа получения прекерамических волокнообразующих олигоорганосилазанов с высоким выходом из легко доступных и относительно дешевых отечественных исходных материалов с содержанием нелетучих веществ более 97 мас. % с выходом неорганического остатка не менее 75 мас. % после пиролиза до 1200°C как в инертной атмосфере, так и на воздухе, со стабильностью в виде гранул до 6 месяцев, обеспечивающих формование волокон из расплава в непрерывном режиме с высокими физико-механическими характеристиками. Причем, способ может быть эффективно осуществлен в промышленном производстве.
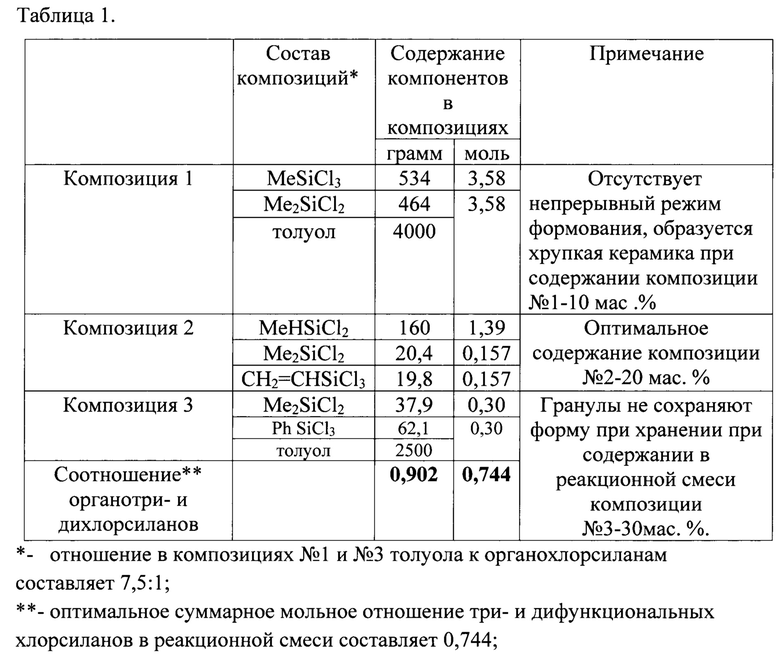
Поставленная задача достигается тем, что предложен способ получения прекерамических волокнообразующих олигоорганосилазанов в инертной атмосфере в растворителе путем прямого аммонолиза органодихлорсиланов с последующей полимеризацией продукта аммонолиза при повышенной температуре в присутствии катализатора, заключающийся в том, что аммонолизу подвергают реакционную смесь, состоящая из смеси органотри- и органодихлорсиланов в различных комбинациях при их суммарном мольном отношении в интервале более 0,66, но менее 0,85, причем аммонолиз осуществляют ступенчато путем последовательного ввода первой эквимолярной композиции, состоящей из диметилдихлорсилана и метилтрихлорсилана с концентрацией 3,42 моль в толуоле; по завершению процесса - второй композиции, состоящей из метилдихлорсилана, винилтрихлорсилана, диметилдихлорсилана с концентрациями 1,39 моль, 0,138 моль, 0,138 моль, соответственно; по завершению процесса - третьей эквимолярной композиции с концентрацией 0,294 моль в толуоле, состоящей из метилдихлорсилана, фенилтрихлорсилана с отделением образующегося осадка хлористого аммония и удалением растворителя известными методами после введения третьей композиции и завершении процесса, при последующей полимеризации продукта аммонолиза от 3 до 6 часов при атмосферном и пониженном давлении при температурах более 149°C, но менее до 171°C в присутствии полиалкоксифосфортитаноксана в качестве катализатора с концентрацией не менее 0,2 мас. % и не более 2 мас. %, приводящей к образованию полисилазанов с содержанием нелетучих веществ более 97 мас. %, с выходом неорганического остатка не менее 75 мас. % после пиролиза до 1200°C как на воздухе, так и инертной атмосфере, со стабильностью полисилазана в виде гранул до 6 месяцев.
Органические заместители при кремнии в органохлорсиланах могут быть выбраны из предложенного списка как однотипные, так и в различных комбинациях: водород, низшие алкильные группы: метил, этил, н-пропил, изопропил и т.д.; винил, аллил, бензил; диметил-, метилэтил-, диизопропиламино и т.д.; триметил-, диметил-, триэтилсилил и др. группы; низшие арильные группы: фенил, толил, ксилил и т.д., предпочтительно: водород, метил, фенил, винил.
Растворители, используемые в процессе аммонолиза, депротонирования /полимеризации, могут быть выбраны из: простых эфиров; циклических простых эфиров; алифатических углеводородов; ароматических углеводородов, предпочтительно толуола.
Присутствие толуола в реакционной смеси не оказывает существенного влияния на свойства конечного продукта. Расчетное количество толуола должно обеспечивать достижение однородной смеси на всех стадиях аммонолиза, при соблюдении предпочтительного отношения толуола к сумме органохлорсиланов равного 6:1.
Содержание в реакционной смеси органотри- и оргноадихлорсиланов в различных комбинациях составляет при суммарном мольном отношении более 0,66, но менее 0,85.
Продолжительность термообработки от 3 до 6 часов в зависимости от суммарного мольного отношения органотри- и органодихлорсиланов при проведении процесса при атмосферном и пониженном давлении при температурах более 149°C, но менее 171°C. В качестве катализатора полимеризации используют полиалкоксифосфортитаноксан следующей формулы: [1-Rn TiO 2R(4-n)-], где 1R=Р; n=1-3; 2R= низшие алкилы (С1-С5), с концентрацией более 0,2 мас. %, но не менее 2 мас. %. Поскольку природа радикала (2R) не оказывает существенного влияния на свойства получаемых полисилазанов, в данном процессе использовали бутокси - производный титаноксан, так как бутиловый спирт является самым дешевым.
Качество полученных волокнообразующих полимеров на стадии первичного тестирования оценивали по их способности обеспечивать формование волокон, которые должны сохранять форму после окисления на воздухе в течение 30 минут при 60°C.
Сущность изобретения иллюстрируют следующими примерами, которые должны рассматриваться только для иллюстрации, а не как ограничение. Условия синтеза и свойства волокнообразующих полисилазанов приведены в таблицах:
таблица 1 - «Составы композиций органохлорсиланов в толуоле для получения волокнообразующих полисилазанов»;
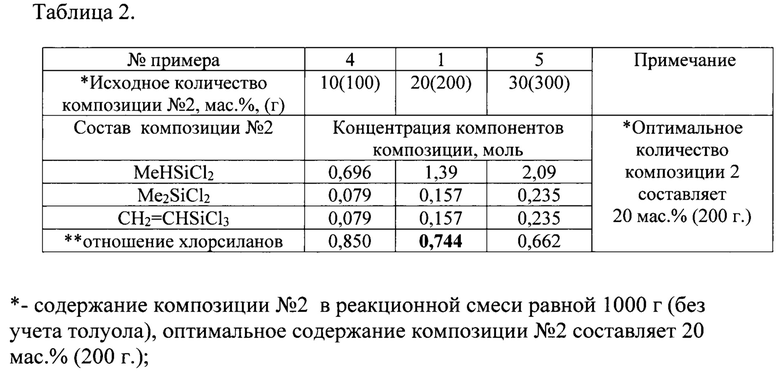
таблица 2 - «Концентрация исходных органохлорсиланов композиции №2»;
таблица 3 - «Режимы получения волокнообразующих полисилазанов и их свойства».
Пример 1.
В четырехгорлый реактор с рубашкой, снабженный мешалкой, обратным холодильником и капельной воронкой, в азоте при температуре 0°C загружают эквимолярную композицию №1 с концентрацией 3,58 моль в 4 кг толуола (Таблицы 1, 2). Далее, после завершения процесса аммонолиза вводят композицию №2 в количестве 200 г, состоящую из метилдихлорсилана, винилтрихлорсилана, диметилдихлорсилана с концентрациями 1,39 моль, 0,138 моль, 0,138 моль, соответственно. Затем, после завершения процесса аммонолиза вводят эквимолярную композицию №3 в 3,5 кг толуола с концентрацией 0,294 моль, состоящую из фенилтрихлорсилана, метилдихлорсилана. Суммарное мольное отношение органотри- и органодихлорсиланов при аммонолизе составляет 0,744 (Таблицы 1, 2, 3). После удаления осадка хлористого аммония и дистилляции растворителя известными методами к продукту аммонолиза добавляют катализатор полимеризации, в качестве которого использовали полиалкоксифосфортитаноксана с концентрацией 1,5 мас. %. При температуре 160°C при перемешивании проводят полимеризацию в течение 6 часов. Для волокнообразующего полисилазана получены следующие характеристики: сухой остаток 97 мас. %; стабильность в виде гранул достигает 6 месяцев (Таблицы 1, 2, 3); выход неорганического остатка около 75 мас. % как на воздухе, так и в аргоне; характеристики молекулярно-массового распределения равны Mw=4275; Mn=988; D=4,32.
Пример 2. В примере 2 использована другая последовательность введения композиций органохлорсиланов: первой вводят композицию №2, затем №1 и далее композицию №3 (Таблицы 1, 2, 3).
Пример 3. В примере 3 использована следующая последовательность ввода композиций органохлорсиланов: первой вводят композицию №3, затем композицию №2 и далее композицию №1 (Таблицы 1, 2, 3).
Пример 4. В примере 4 композицию №2 вводят в количестве 100 г (Таблицы 1, 2, 3).
Пример 5. В примере 5 композиций №2 вводят в количестве 300 г (Таблицы 1, 2, 3).
При сохранении всех других параметров примера №1 в примерах 2-10, проведен выбор оптимальных параметров: последовательность ввода композиций (примеры 1-3); количество вводимой в реакционную смесь композиции №2 (примеры 1, 4, 5), температуры полимеризации (примеры 1, 6, 7) и продолжительности полимеризации (примеры 1, 8), концентрации катализатора (пример 9-10).
Последовательность ввода композиций органохлорсиланов определяет свойства получаемых полисилазанов, а, следовательно, характеристики и режимы проведения процесса их получения. Первоначально проведенное тестирование волокнообразующей способности полисилазанов, позволило выбрать для дальнейших испытаний образцы полисилазанов, полученные в примерах 1,4-9. Используемая в примере 1 последовательность ввода композиций хлорсиланов обеспечивает получение полисилазана с повышенной термоокислительной стабильностью, поскольку выход неорганического осадка составляет около 75 мас. % как на воздухе, так и в аргоне. Используемая последовательность ввода композиций в примерах 2 и 3 не обеспечивает получение полиорганосилазана, соответствующего предлагаемому изобретению, поскольку сформованные на их основе волокна, не прошли первоначальное тестирование, поскольку расплавились при термообработке при 60°C в течение 30 минут.
В примерах 1, 3, 4 количество вводимой композиции №2 влияет на мольное отношение органотри- и органодифункциональных хлорсиланов в реакционной смеси в интервале 0,662-0,850. Так, в примере 4 композицию №2 вводят в количестве 100 г (10 мас. %). ри этом мольное отношение три- и дифункциональных хлорсиланов составляет 0,850. Полученные при этом полисилазаны не обеспечивают непрерывный режим формования волокна из расплава, а керамика, полученная после пиролиза, характеризуется повышенной хрупкостью (Таблицы 1,2,3). В примере 5 композицию №2 вводят в количестве 300 г (30 мас. %) с мольным отношением три- и дифункциональных хлорсиланов равном 0,662 (Таблица 1, 2, 3). При этом снижается жизнеспособность волокнообразующего полисилазана в форме гранул при хранении. Наблюдается снижение выхода неорганического остатка (ВНО) при пиролизе и снижение их термоокислительной стабильности, поскольку появляются существенные различия ВНО после пиролиза в аргоне и на воздухе.
В предлагаемом способе получение волокнообразующих олигоорганосилазанов с молекулярными массами в широком интервале значений достигают путем контроля продолжительности полимеризации реакционной массы от 3 до 6 часов при изменении температуры от 149°C до 171°C. Выбор температуры полимеризации при сохранении остальных условий синтеза примера 1 проводили в примерах 6 и 7. Так, в примере 6 при проведении полимеризации при 149°C, получен полисилазан с содержанием нелетучих веществ менее 97 мас. %, а при полимеризации при 171°C в примере 7 полисилазан не обеспечил формование волокна из расплава.
Установлено, что при проведении полимеризации 2,8 часов, форма волокна не сохраняется при выдержке при 60°C в течение 30 мин, поэтому время полимеризации менее 3 является недостаточным. (Таблица 3, пример 8). Термообработка в течение 6 часов (Таблица 3, примеры 1-7,9) обеспечивает сохранение формы волокна при выдержке в течение 30 мин. при 60°C, поэтому дальнейшее увеличение длительности процесса термообработки не требуется.
Проведение процесса при различной концентрации катализатора показало, что концентрация катализатора более 0,2 мас. %, но не менее 2 мас. % являет оптимальной. При введении катализатора в количестве более 2,1 мас. % (Таблица 3, пример 9), полученные полисилазаны не обеспечивают непрерывный режим формования волокна из расплава, а керамика, полученная после пиролиза, характеризуется повышенной хрупкостью. Концентрация катализатора менее 0,2 мас. % (Таблица 3, пример 10) не обеспечивает получение полиорганосилазана, соответствующего предлагаемому изобретению, поскольку сформованные на их основе волокна, не прошли первоначальное тестирование, так как расплавились при 60°C в течение 30 минут.
Использование каждой смеси по отдельности не приводит к получению полисилазана, позволяющего выполнить задачу изобретения. По-видимому, при использовании трех смесей в соответствии с предложенным изобретением наблюдается синергетический эффект, приводящий к получению полисилазана, согласно настоящему изобретению.
Таким образом, при проведении процесса в контролируемом режиме путем последовательного ввода трех композиций органохлорсилазанов в последовательности, приведенной в примере 1, с мольным отношением органотри- и органодихлорсиланов более 0,66, но менее 0,85, после отделения образующегося осадка хлористого аммония и удаления растворителя известными методами после введения третьей композиции и завершении процесса с последующей полимеризацией продукта аммонолиза от 3 до 6 часов при атмосферном и пониженном давлении при температурах более 149°C, но менее 171°C в присутствии полиалкоксифосфортитаноксана с концентрацией более 0,2 мас. %, но не менее 2 мас. %, взятого в качестве катализатора являются необходимыми для получения полисилазанов, соответствующих предложенному изобретению.
За пределами вышеуказанных диапазонов параметров процесса, полученные полимеры не обеспечивают заданных свойств.
В примере 1 установлены оптимальные условия проведения синтеза полиорганосилазана со свойствами в соответствии с предлагаемым изобретением: последовательность ввода композиций органохлорсиланов их состав; содержание композиции №2 в реакционной смеси в количестве 200 г (20 мас. %) при мольном отношении три - и дифункциональных хлорсиланов равном 0,744; температуры полимеризации, составляющей 160°C; при продолжительности полимеризации равной 6 часов.
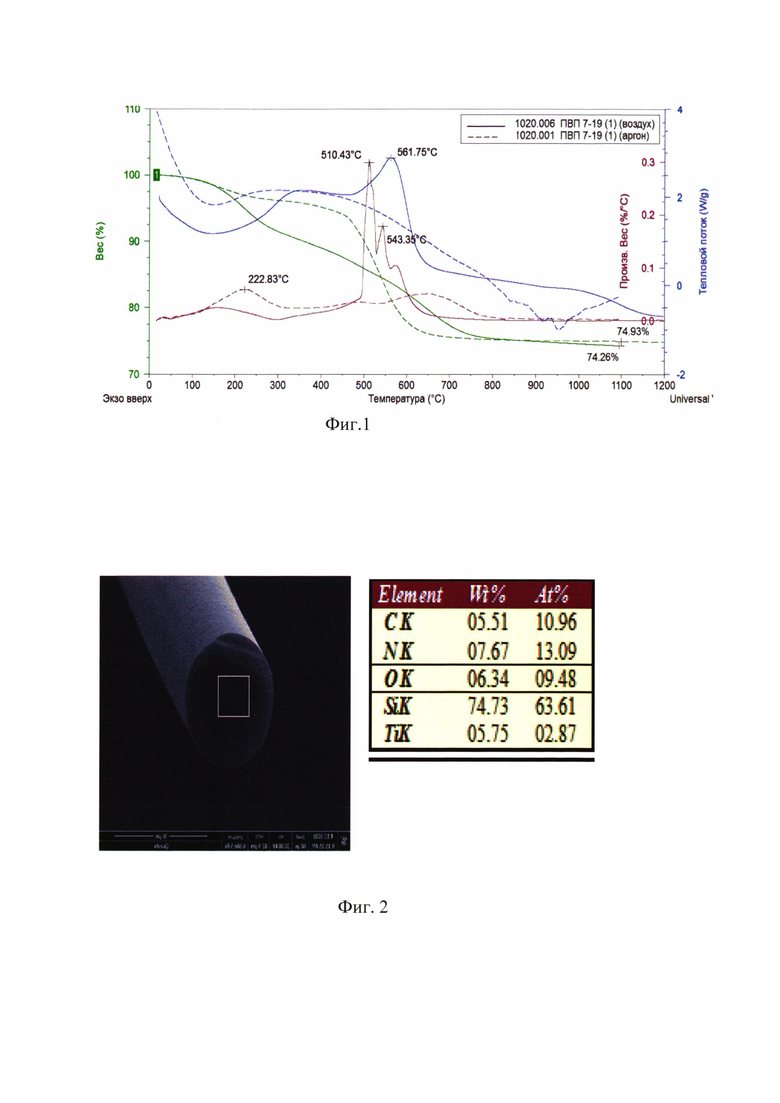
Техническим результатом предлагаемого изобретения является получение олигоорганосилазанов с содержанием нелетучих веществ более 97 мас. % с молекулярной массой, обеспечивающей получение высокотермостойких, окислительностойких олигоорганосилазанов с высоким выходом керамики около 75 мас. % после пиролиза до 1200°C как на воздухе, так и инертной атмосфере, состоящей преимущественно из кремния, азота, углерода, что подтверждается данными, представленными на фиг. 1 «Термогравиметрические кривые волокнообразующего полисилазана на воздухе и в аргоне (оптимальный образец)».
Стабильность полученных полисилазанов в виде гранул достигает шесть месяцев. Волокнообразующий полисилазан, согласно настоящему изобретению, имеет структуру, отличную от структуры полисилазанового полимера по прототипу, за счет наличия различных типов повторяющихся сесквисилазановых звеньев, соединенных различными мостиками, что обеспечивает повышение характеристик и появление новых свойств.
Последовательность ввода композиций органохлорсиланов, выбранная в примере 1, обеспечивает формирование структуры полимера, при которой после формования из расплава и проведения окислительного отверждения волокон на воздухе в течение 1,5 часа наблюдается сохранение состава и формы волокна в процессе последующего пиролиза в азоте до 1200°C. Морфологию поверхности керамических волокон, их состав исследовали методом SEM и рентгеновского микроанализа. На фиг. 2 представлена «Микрофотография волокна, изготовленного на основе полученного волокнообразующего полисилазана и результаты элементного анализа».
Несмотря на то, что разработка методов получения прекерамических волокнообразующих полимеров для волокон состава SiCN проводится зарубежными и отечественными исследователями с 1970 г («Ceramic Fibers Based on SiC and SiCN Systems: Current Research, Development, and Commercial Status», Flores O., Bordia R., Advanced engineering materials, 2014, 16, No. 6, p. 621-635; «Processing and characterization of large diameter ceramic SiCN monofilaments from commercial oligosilazanes», Flores O., Bordia R., et al., RSC Adv. 2015. Vol. 5. P. 107001-107011; «Selective cross-linking of oligosilazanes to tailored meltable polysilazanes for the processing of ceramic SiCN fibres», Flores O., Schmalz Т., Krenkel W. et al., J. Mater. Chem. A. 2013) на данном этапе отсутствуют коммерчески доступные волокна, что связано с их низкой жизнеспособностью. Отсутствие на рынке коммерчески доступных волокнообразующих прекерамических полимеров является одной из основных причин, препятствующих появлению коммерчески доступных волокон. Следует заметить, что достигнутые нами характеристики волокон состава SiCN (прочности на разрыв (1,1-1,3) ГПа) на основе олигоорганосилазанов, с использованием окислительного отверждения находятся в интервале максимальных значений, полученных зарубежными исследователями («Ceramic Fibers Based on SiC and SiCN Systems: Current Research, Development, and Commercial Status)), Flores O., Bordia R., Advanced engineering materials, 2014, 16, No. 6, p. 621-635; «Processing and characterization of large diameter ceramic SiCN monofilaments from commercial oligosilazanes», Flores O., Bordia R., et al., RSC Adv. 2015. Vol. 5. P. 107001-107011). Причем, максимальные прочностные характеристики (1,3 ГПа) наблюдаются у волокон, отвержденных только облучением электронным пучком.
Волокнообразующие олигоорганосилазаны, полученные на базе отечественного сырья, являются коммерческими продуктами для керамических волокон состава SiCN.
Разработанная технология на основе доступного отечественного сырья может быть осуществлена в промышленных масштабах с выпуском объемов, обеспечивающих все потребности промышленности. Технологические характеристики волокнообразующих полисилазанов для волокон состава SiCN соответствуют ТУ 20.16.57-250-00209013-2018 (таблица 4 - «Характеристики предкерамического волокнообразующего полисилазана»).
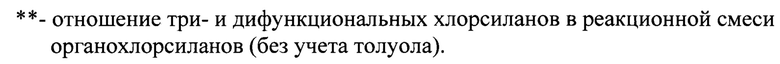

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОБОРСИЛАЗАНОВ | 2016 |
|
RU2624442C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОФУНКЦИОНАЛЬНЫХ КЕРАМОМАТРИЧНЫХ КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ (ВАРИАНТЫ) | 2015 |
|
RU2603330C2 |
| Материал на основе карбида кремния для 3D-печати | 2021 |
|
RU2774467C1 |
| ВЫСОКОТЕМПЕРАТУРНЫЙ АДГЕЗИВ ДЛЯ СОЕДИНЕНИЯ КЕРАМИЧЕСКИХ И КОМПОЗИЦИОННЫХ МАТЕРИАЛОВ | 1992 |
|
RU2034892C1 |
| ВЫСОКОТЕМПЕРАТУРНЫЙ АДГЕЗИВ ДЛЯ СОЕДИНЕНИЯ КОНСТРУКЦИОННЫХ МАТЕРИАЛОВ | 1992 |
|
RU2034890C1 |
| Способ получения раствора неорганического полисилазана в дибутиловом эфире | 2020 |
|
RU2745823C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОРГАНОСИЛОКСАНОВ НА ОСНОВЕ ОРГАНОАЛКОКСИСИЛАНОВ | 2009 |
|
RU2428438C2 |
| ВОЛОКНООБРАЗУЮЩИЕ ОРГАНОИТТРИЙОКСАНАЛЮМОКСАНЫ | 2014 |
|
RU2551431C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОБОРСИЛАЗАНОВ | 2013 |
|
RU2546664C1 |
| СПОСОБ ОТВЕРЖДЕНИЯ ТЕРМОРЕАКТИВНЫХ ПОЛИОРГАНОСИЛОКСАНОВЫХ СМОЛ | 1992 |
|
RU2038359C1 |
Изобретение относится к способу получения прекерамических волокнообразующих олигоорганосилазанов для получения керамических волокон состава SiCN. Реакционную смесь три- и дифункциональных органохлорсиланов при их суммарном мольном соотношении более 0,66, но менее 0,85 подвергают аммонолизу. Аммонолиз осуществляют ступенчато путём последовательного ввода первой, второй и третьей композиций, каждая из которой состоит из смеси три- и дифункциональных органохлорсиланов в толуоле. Продукт аммонолиза обрабатывают катализатором и полимеризуют от 3 до 6 часов при атмосферном и пониженном давлении при температурах более 149°С и менее 171°С с получением полисилазанов с содержанием нелетучих веществ более 97 мас.% с выходом неорганического остатка около 75 мас.% после пиролиза до 1200°C как в инертной атмосфере, так и на воздухе. Данный способ может быть эффективно осуществлен в промышленном производстве прекерамических волокнообразующих олигоорганосилазанов, получаемых с высоким выходом и стабильностью в виде гранул до 6 месяцев. Волокнообразующие полисилазаны обеспечивают формование из расплава в непрерывном режиме и керамизацию волокон состава SiCN с заданными характеристиками с высокими физико-механическими свойствами. 1 з.п. ф-лы, 4 табл., 10 пр., 2 ил.
1. Способ получения в инертной атмосфере предкерамических волокнообразующих олигоорганосилазанов в растворителе путем прямого аммонолиза органодихлорсиланов с последующей полимеризацией продукта аммонолиза в присутствии катализатора, отличающийся тем, что аммонолизу подвергают реакционную смесь, состоящую из смеси органотри- и органодихлорсиланов в различных комбинациях при их суммарном мольном отношении в интервале более 0,66, но менее 0,85, причем аммонолиз осуществляют ступенчато путем последовательного ввода первой эквимолярной композиции, состоящей из диметилдихлорсилана и метилтрихлорсилана с концентрацией 3,42 моль в толуоле; по завершению процесса - второй композиции, состоящей из метилдихлорсилана, винилтрихлорсилана, диметилдихлорсилана с концентрациями 1,39 моль, 0,138 моль, 0,138 моль, соответственно; по завершении процесса - третьей эквимолярной композиции в толуоле с концентрацией 0,294 моль, состоящей из диметилдихлорсилана, фенилтрихлорсилана, с отделением образующегося осадка хлористого аммония и удалением растворителя известными методами после введения третьей композиции и завершении процесса, с последующей полимеризацией продукта аммонолиза от 3 до 6 часов при атмосферном и пониженном давлении при температурах более 149°C, но менее 171°C в присутствии полиалкоксифосфортитаноксана с концентрацией более 0,2 мас.%, но менее 2 мас.%, взятого в качестве катализатора, приводящей к образованию полисилазанов с содержанием нелетучих веществ более 97 мас.%, с выходом неорганического остатка не менее 75 мас.% после пиролиза как на воздухе, так и в инертной атмосфере до 1200°C со стабильностью полисилазана в виде гранул до 6 месяцев.
2. Способ по п. 1, отличающийся тем, что в качестве катализатора полимеризации использовали полиалкоксифосфортитаноксан следующей формулы: [1Rn TiO 2R(4-n)-], где 1R=Р; n=1-3; 2R= низшие алкилы (С1-С5).
| US 4482669 A1, 13.11.1984 | |||
| ПРИМЕНЕНИЕ ПОЛИСИЛАЗАНОВ ДЛЯ ПОКРЫТИЯ МЕТАЛЛИЧЕСКИХ ПОЛОС | 2005 |
|
RU2388777C2 |
| 1971 |
|
SU433182A1 | |
| Способ получения полиорганосилазанов | 1984 |
|
SU1235877A1 |
| КЕРАМИКООБРАЗУЮЩАЯ КОМПОЗИЦИЯ, КЕРАМИЧЕСКИЙ КОМПОЗИЦИОННЫЙ МАТЕРИАЛ НА ЕЕ ОСНОВЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2190582C2 |
| WO 1994003529 A1, 17.02.1994. | |||

