В настоящей заявке испрашивается приоритет по патентной заявке КНР No. 201910117530.7, озаглавленной «КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА FGFR И СПОСОБ ЕЕ ПОЛУЧЕНИЯ», поданной 15 февраля 2019 в Патентное ведомство КНР, которая во всей полноте включена в данную заявку посредством ссылки.
Область изобретения
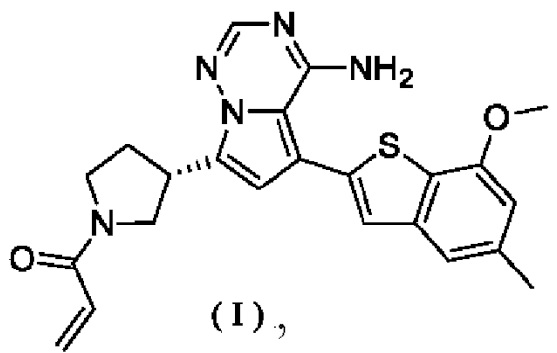
Настоящее изобретение относится к твердой форме, кристаллической форме, кристаллической форме А и кристаллической форме В соединения формулы (I), а также к способу получения кристаллической формы А и кристаллической формы В, а также относится к применению твердой формы, кристаллической формы, кристаллической формы А, кристаллической формы В в изготовлении лекарственного средства для лечения заболеваний, связанных с FGFR (рецептором фактора роста фибробластов).
Уровень изобретения
Рецептор фактора роста фибробластов (FGFR) является рецептором фактора роста фибробластов (FGF) для передачи сигнала, который относится к семейству, состоящему из четырех представителей (FGFR1, FGFR2, FGFR3, FGFR4), и представляет собой гликопротеин, состоящий из внеклеточного иммуноглобулино(Ig)-подобного домена, гидрофобного трансмембранного домена и внутриклеточной части, содержащей тирозинкиназный домен. С помощью рецептора (FGFR) фактор роста фибробластов (FGF) оказывает свое важное влияние на многие физиологические регуляторные процессы, такие как пролиферация клеток, дифференциация клеток, миграция клеток и ангиогенез. Кроме того, с помощью многих доказательств было показано, что нарушение FGF сигнальных путей (например, высокая экспрессия, амплификация гена, мутация гена, хромосомная рекомбинация) напрямую связано со многими патологическими процессами, такими как пролиферация, миграция, инвазия и ангиогенез опухолевых клеток. Таким образом, FGFR стал важной терапевтической мишенью, вызывающей широкий исследовательский интерес.
В WO 2015008844 заявлена серия соединений, обладающих ингибирующей способностью в отношении FGFR, включая соединения сравнения 1 и 2. В WO 2013124316, WO 2013087647 и US 20130158000 также сообщается о серии соединений с ингибирующей способностью в отношении FGFR, включая бензотиофеновую структуру из настоящей заявки и соединение сравнения 3.
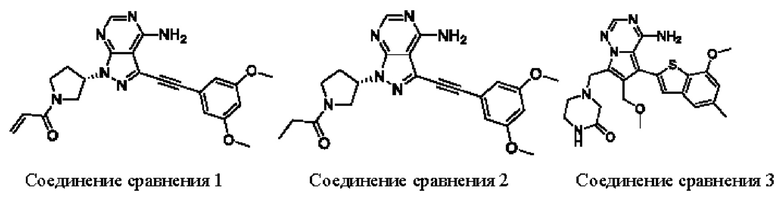
В WO 2019034076 раскрыт класс соединений с ингибирующей способностью в отношении FGFR, включая соединение WX001 (включая пару WX001A и WX001B в виде оптических изомеров) с хорошей активностью, но не удалось получить его продукт в твердой форме.
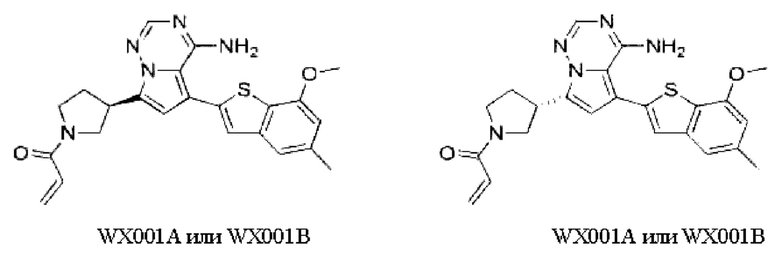
Авторы настоящего изобретения также обнаружили, что трудно получить твердую форму соединения WX001 традиционными способами, такими как применение метанола, этанола, тетрагидрофуранав качестве растворителя в обычных условиях обработки.
Таким образом, проблема, которую необходимо решить, состоит в том, чтобы получить соединение WX001A или WX001B в твердой форме, чтобы обеспечить продукты, удобные при производстве, очистке, хранении и применении.
Сущность изобретения
Авторы настоящего изобретения неожиданно обнаружили способ, позволяющий получать соединение WX001A или WX001B, описанное выше, в твердой форме, а также полученный соответствующий продукт в твердой форме, а также в кристаллической форме.
На основании указанного выше, в первом аспекте настоящего изобретения предложено соединение, представленное формулой (I), в твердой форме:
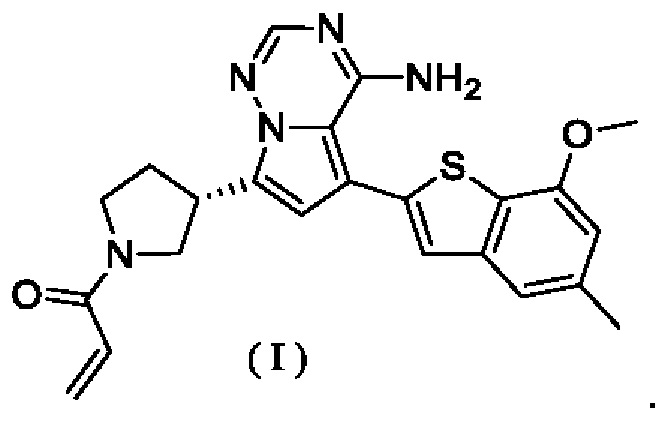
Во втором аспекте настоящего изобретения предложено соединение, представленное формулой (I), в кристаллической форме.
В предпочтительном аспекте настоящего изобретения предложена кристаллическая форма А соединения, представленного формулой (I), с дифракционными пиками при угле 2θ 6,37±0,2°, 9,90±0,2° и 19,07±0,2° на ее порошковой рентгенодифрактограмме.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет дифракционные пики при угле 2θ 6,37±0,2°, 9,90±0,2°, 12,74±0,2°, 13,35±0,2°, 14,26±0,2°, 16,31±0,2°, 19,07±0,2° и 21,83±0,2° на ее порошковой рентгенодифрактограмме.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет порошковую рентгенодифрактограмму, показанную на фиг. 1.
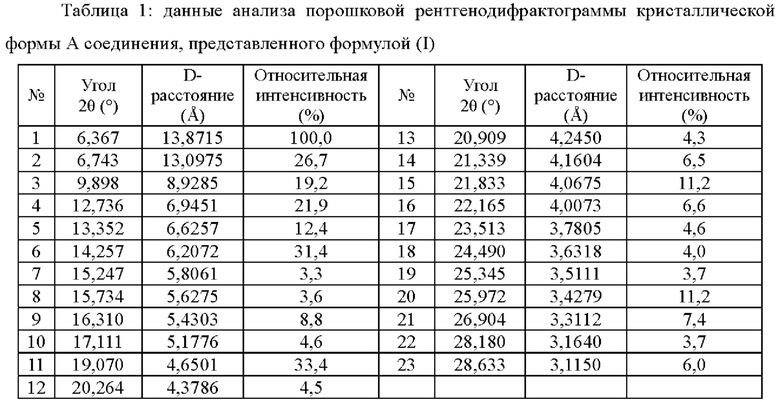
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет данные анализа порошковой рентгенодифрактограммы, показанные в таблице 1.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет на ее кривой дифференциальной сканирующей калориметрии (ДСК) эндотермический пик при температуре 141,05°С±5°С.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет кривую ДСК, показанную на фиг. 2.
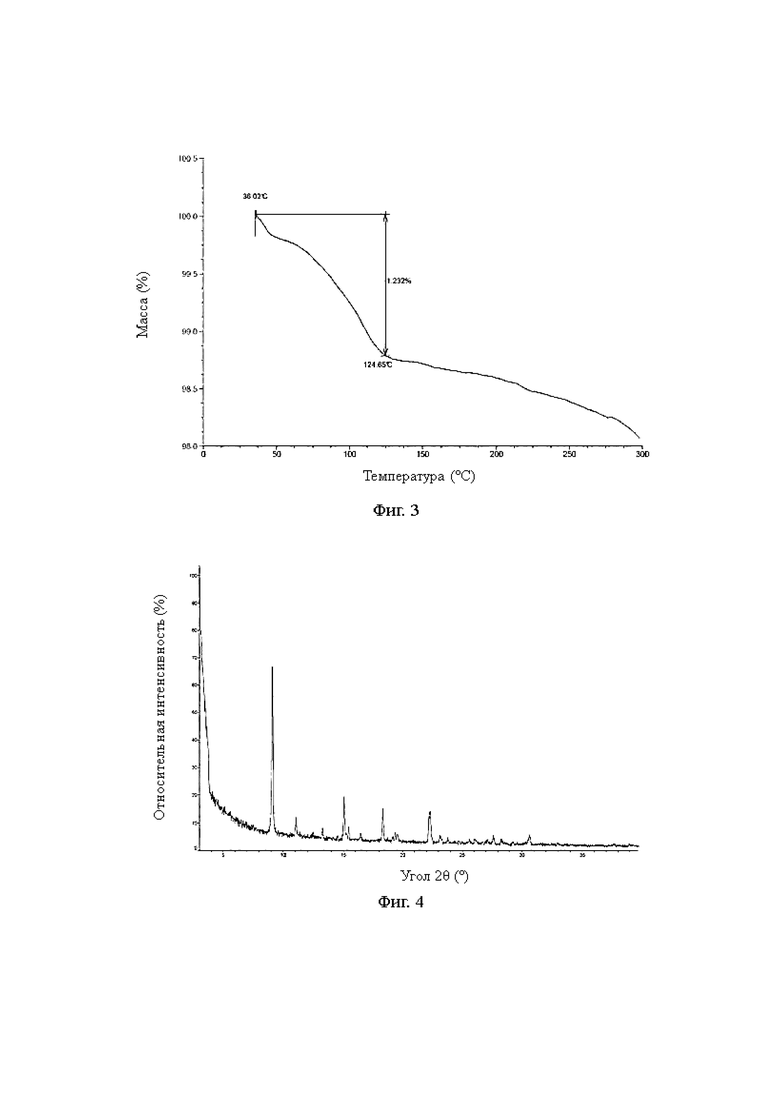
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет потерю массы 1,232% при температуре 124,65±3°С на ее кривой термогравиметрического анализа (ТГА).
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма А соединения, представленного формулой (I), имеет кривую ТГА, показанную на фиг. 3.
В настоящем изобретении также предложен способ получения описанной кристаллической формы А соединения, представленного формулой (I), включающий:
(a) добавление соединения, представленного формулой (I), в нитрильный растворитель или сложноэфирный растворитель;
(b) перемешивание при температуре 30-50°С в течение 40-55 часов и
(c) отделение кристаллической формы А соединения, представленного формулой (I).
В указанном выше способе стадия (с) отделения может быть осуществлена путем центрифугирования, фильтрации и т.д. и предпочтительно путем центрифугирования; при этом необязательно за стадией (с) отделения может быть осуществлена сушка.
В некоторых воплощениях настоящего изобретения нитрильный растворитель, описанный выше, выбирают из ацетонитрила, пропионитрила и бутиронитрила.
В некоторых воплощениях настоящего изобретения сложноэфирный растворитель, описанный выше, выбирают из этилацетата, метилацетата, изопропилацетата и этилформиата.
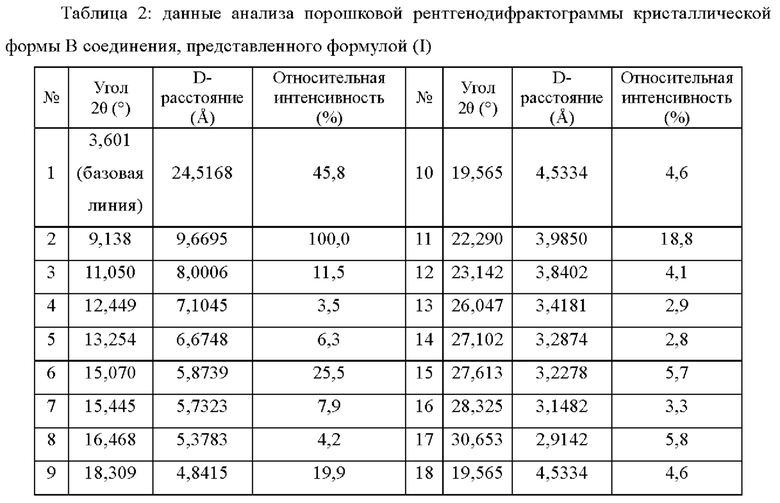
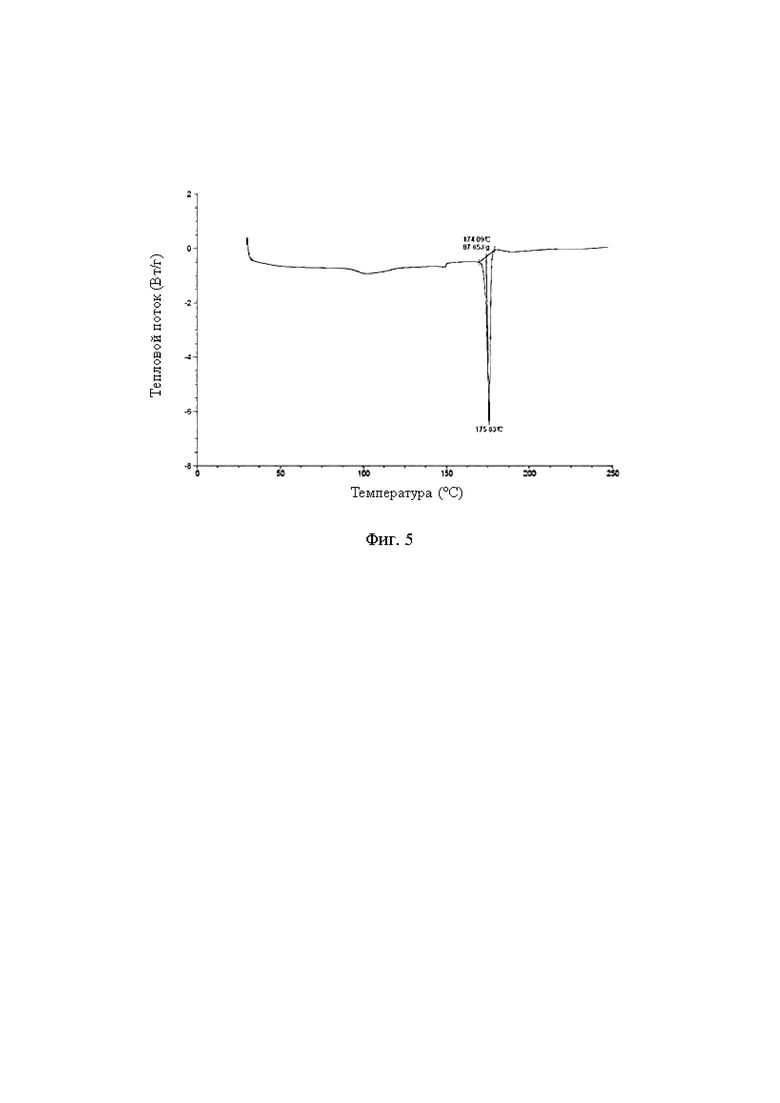
В другом аспекте настоящего изобретения предложена также кристаллическая форма В соединения, представленного формулой (I), с дифракционными пиками при угле 20 3,60±0,2°, 9,14±0,2° и 15,07±0,2° на ее порошковой рентгенодифрактограмме.
Кристаллическая форма В соединения, представленного формулой (I), предложенная в настоящем изобретении, имеет дифракционные пики при угле 20 9,14±0,2° и 15,07±0,2° на ее порошковой рентгенодифрактограмме.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет дифракционные пики при угле 2θ 3,60±0,2°, 9,14±0,2°, 11,05±0,2°, 13,25±0,2°, 15,07±0,2°, 16,47±0,2°, 18,31±0,2° и 22,29±0,2° на ее порошковой рентгенодифрактограмме.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет дифракционные пики при угле 2θ 9,14±9,2°, 11,05±0,2°, 13,25±0,2°, 15,97±9,2°, 16,47±0,2°, 18,31±0,2° и 22,29±0,2° на ее порошковой рентгенодифрактограмме.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет порошковую рентгенодифрактограмму, показанную на фиг. 4.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет данные анализа порошковой рентгенодифрактограммы, показанные в таблице 2.
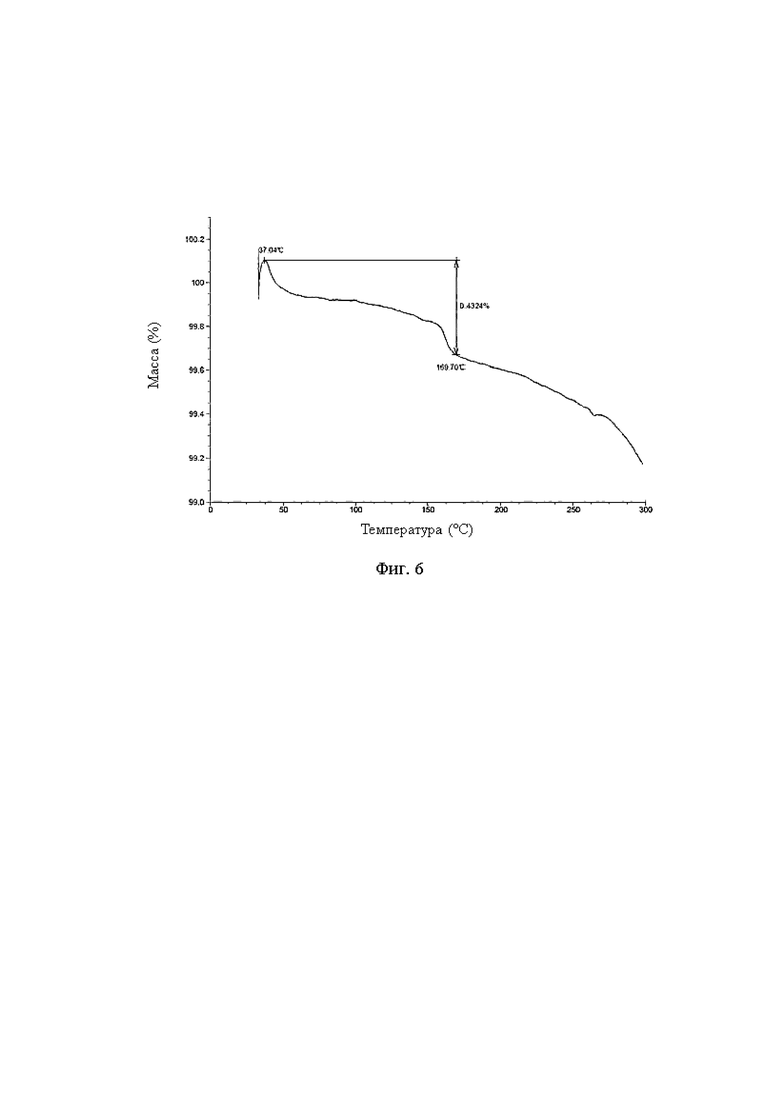
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет на ее кривой дифференциальной сканирующей калориметрии эндотермический пик с начальной точкой при температуре 174,09°С±5°С.
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет кривую ДСК, показанную на фиг. 5.
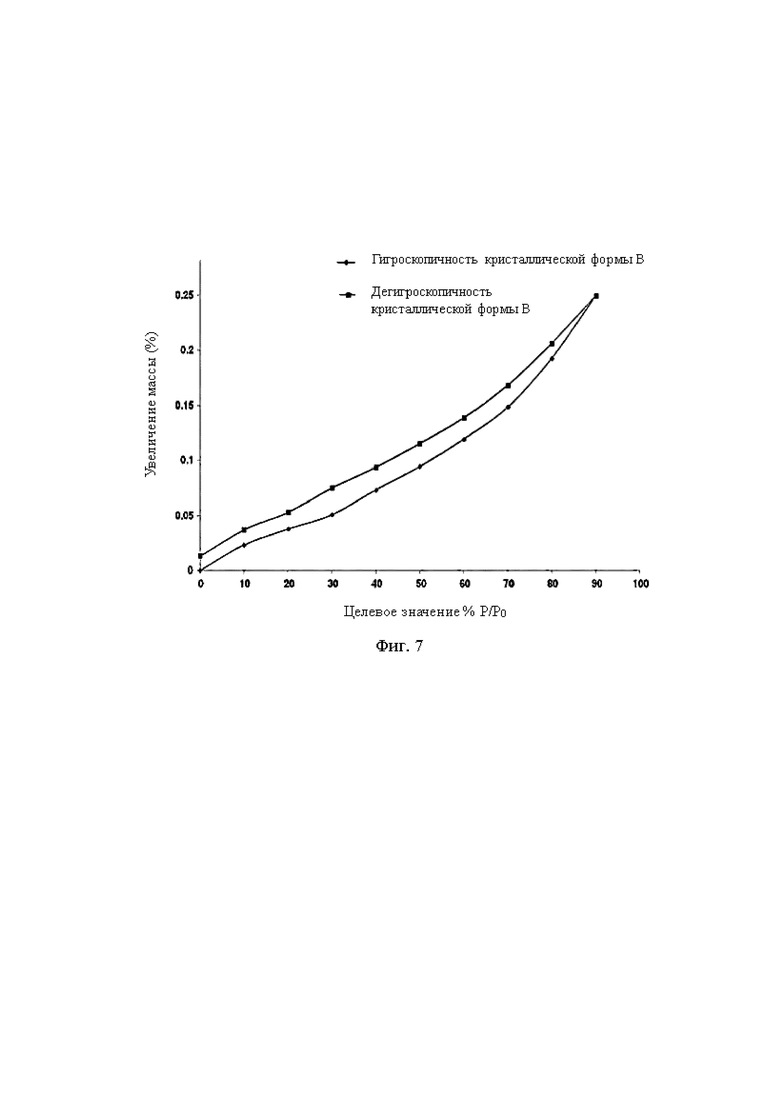
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет потерю массы 0,4324% при температуре 169,70±3°С на ее кривой термогравиметрического анализа (ТГА).
В некоторых воплощениях настоящего изобретения описанная кристаллическая форма В соединения, представленного формулой (I), имеет кривую ТГА, показанную на фиг. 6.
В настоящем изобретении также предложен способ получения описанной кристаллической формы В соединения, представленного формулой (I), включающий:
(a) добавление кристаллической формы А соединения, представленного формулой (I), в спиртовый растворитель или смешанный растворитель из спиртового растворителя и воды;
(b) перемешивание при температуре 30-50°С в течение 5-30 часов;
(c) выстаивание при температуре 10-20°С в течение 3-10 часов и
(d) отделение кристаллической формы В соединения, представленного формулой (I).
В указанном выше способе стадия (d) отделения может быть осуществлена путем центрифугирования, фильтрации и т.д. и предпочтительно путем центрифугирования; при этом необязательно за стадией (d) отделения может быть осуществлена сушка.
В некоторых воплощениях настоящего изобретения описанный спиртовый растворитель выбирают из метанола, этанола и изопропанола.
В некоторых воплощениях настоящего изобретения описанный смешанный растворитель из спиртового растворителя и воды выбирают из смешанного растворителя метанола и воды, смешанного растворителя этанола и воды и смешанного растворителя изопропанола и воды.
В настоящем описании также предложено применение соединения, представленного формулой (I), в твердой форме, описанной выше, соединения, представленного формулой (I), в кристаллической форме, кристаллической формы А соединения, представленного формулой (I), кристаллической формы В соединения, представленного формулой (I), в производстве лекарственного средства для лечения заболеваний, связанных с FGFR.
В некоторых воплощениях настоящего изобретения описанное заболевание, связанное с FGFR, представляет собой твердую опухоль.
Технический результат
Кристаллическая форма В соединения формулы (I) имеет увеличение массы, вызванное адсорбцией влаги, составляющее 0,1928% при температуре 25°С и 80% ОВ (относительной влажности), не проявляя или почти не проявляя гигроскопичности и показывая хорошие перспективы для изготовления лекарственных средств. Соединение формулы (I) проявляло хорошую ингибирующую способность в отношении FGFR дикого типа, а также хорошую ингибирующую способность в отношении FGFR мутантного типа и имело хорошие фармакокинетические параметры.
Определения и описание
Если не указано иное, следующие термины и выражения, используемые в данной заявке, имеют значения, указанные ниже. Конкретный термин или конкретное выражение, не определенное конкретно, следует понимать в соответствии с его обычным значением, а не считать его неопределенным или неясным. Когда в данной заявке указывается торговое наименование, оно относится к соответствующему товару или его активному ингредиенту.
Промежуточные соединения в настоящем изобретении можно получить разными способами синтеза, хорошо известными специалистам в данной области техники, включая конкретные воплощения, приведенные ниже, воплощения, образованные с помощью этих конкретных воплощений в сочетании с другими способами химического синтеза, а также эквивалентными им альтернативными способами, хорошо известными специалистам в данной области техники. Предпочтительные воплощения включают, не ограничиваясь указанными, примеры по настоящему изобретению.
Растворители, используемые в описании настоящей заявки, являются коммерчески доступными и могут использоваться без дальнейшей очистки.
В настоящем описании используют следующие сокращения: ДМФ обозначает диметилформамид; MsOH обозначает метансульфоновую килоту; EtOH обозначает этанол; NaOH обозначает гидроксид натрия; ДМСО обозначает диметилсульфоксид.
Названия соединениям даны без использования специальных программ или с помощью программного комплекса ChemDraw®, а для коммперчески доступных соединений используют название по каталогу поставщика.
Способ рентгеновской порошковой дифракции (XRPD) в описании настоящей заявки
Модель прибора: рентгеновский дифрактометр Bruker D8 advance
Способ испытания: Приблизительно 10-20 мг образца использовали для измерения методом рентгеновской порошковой дифракции (XRPD).
Подробные параметры XRPD были следующими:
Световая трубка: Cu, kα, 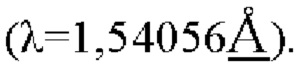
Напряжение световой трубки: 40 кВ, ток световой трубки: 40 мА
Щель расходимости: 0,60 мм
Щель детектора: 10,50 мм
Антирассеивающая щель: 7,10 мм
Диапазон сканирования: 3-40 град
Диаметр шага: 0,02 град
Длина шага: 0,12 секунд
Скорость вращения лотка для образцов: 15 об/мин
Способ дифференциальной сканирующей калориметрии (ДСК) в описании настоящей заявки
Модель прибора: дифференциальный сканирующий калориметр ТА Q2000
Способ испытания: Образец (0,5-1 мг) помещали в алюминиевую чашку ДСК прибора и испытывали в интервале 25°С-300°С при скорости 10°С/мин. Скорость нагревания m-ДСК составляла 2°С/мин.
Способ термического гравиметрического анализа (ТГА) в описании настоящей заявки
Модель прибора: анализатор термического гравиметрического анализа ТА Q5000
Способ испытания: Образец (2-5 мг) помещали в платиновую чашку ТГА анализатора для испытания в условиях 25 мл/мин N2 и нагревания со скоростью нагревания 10°С/мин от комнатной температуры до 350°С или до потери массы 20%.
Динамическая сорбция паров (DVS)
Условия испытания: Приблизительно 10-15 мг образца использовали для определения DVS.
Уравновешивание dm/dt: 0,01%/мин: (время: 10 мин - 180 мин (максимум))
Сушка: 0% ОВ (относительной влажности), 120 мин
Градиент измерения ОВ (%): 10%
Диапазон градиента измерения ОВ (%): 0% - 90% - 0%

Краткое описание чертежей
На фиг. 1 показана порошковая рентгенодифрактограмма кристаллической формы А соединения формулы (I).
На фиг. 2 показана ДОС кривая кристаллической формы А соединения формулы (I).
На фиг. 3 показан ТГА спектр кристаллической формы А соединения формулы (I).
На фиг. 4 показана порошковая рентгенодифрактограмма кристаллической формы В соединения формулы (I).
На фиг. 5 показана ДОС кривая кристаллической формы В соединения формулы (I).
На фиг. 6 показан ТГА спектр кристаллической формы В соединения формулы (I); и
На фиг. 7 показан ДВС спектр кристаллической формы В соединения формулы (I).
Подробное описание изобретения
Далее в заявке, чтобы лучше понять содержание настоящей заявки, приводятся конкретные примеры для дополнительной иллюстрации содержания настоящего изобретения, но они не предназначены для ограничения содержания настоящего изобретения.
Примеры
Промежуточное соединение А1:
Путь синтеза:
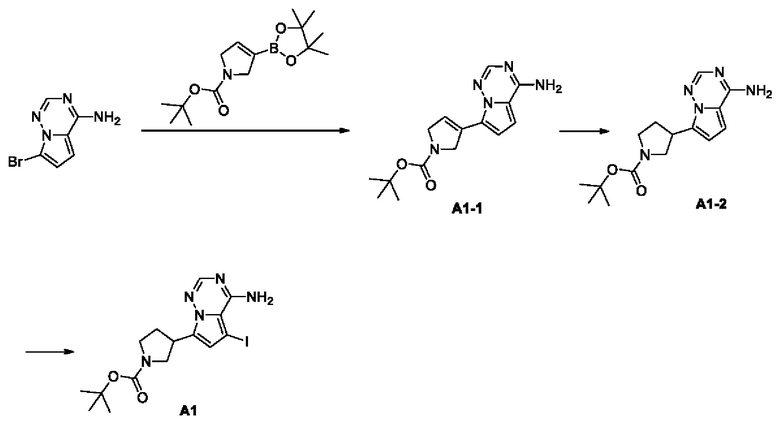
Стадия 1: синтез соединения А1-1
При комнатной температуре сначала 4-амино-7-бромопирроло[1,2-f][1,2,4]триазин (3,00 г, 14,1 ммоль, 1,00 экв.) растворяли в смешанном растворе 1,4-диоксана (40 мл) и воды (8 мл) и затем в тот же смешанный раствор последовательно добавляли сложный пинаколиновый эфир 1-Вос-2,5-дигидро-1Н-пиррол-3-бороновой кислоты (4,36 г, 14,8 ммоль, 1,05 экв.), фосфат калия (8,97 г, 42,2 ммоль, 3,00 экв.) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (1,03 г, 1,41 ммоль, 0,10 экв.). Под защитой газообразным азотом реакционный раствор нагревали до 80°С и перемешивали в течение 2 часов. После завершения реакции реакционный раствор охлаждали до 25°С и выливали в 20 мл воды. Черное твердое вещество получали и собирали путем фильтрации, а затем растворяли в смешанном растворе дихлорметана и метанола (100 мл, 5:1) и снова отфильтровывали. Фильтрат сушили с помощью безводного сульфата натрия и упаривали при пониженном давлении с помощью роторного испарителя для удаления органического растворителя и получения неочищенного продукта. Неочищенный продукт суспендировали в этилацетате (30 мл) и отфильтровывали с получением соединения А1-1. ЖХ-МС (ESI) m/z: 302,1 [М+Н]+, 1Н ЯМР (400 МГц, дейтерированный ДМСО) δ=7,95 (s, 1H), 7,79 (шир. s, 2Н), 6,92 (s, 1H), 6,80-6,66 (m, 2Н), 4,47 (s, 2Н), 4,24 (s, 2Н), 1,44 (s, 9Н).
Стадия 2: синтез соединения А1-2
При комнатной температуре гидроксид палладия (615 мг, 438 мкмоль) добавляли в раствор А1-1 (1,20 г, 3,98 ммоль, 1,00 экв.) в метаноле (30 мл). После замены газа с помощью газообразного водорода 3 раза реакционный раствор нагревали до 50°С и перемешивали при 50 psi давления водорода в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и отфильтровывали для удаления катализатора. Фильтрат упаривали при пониженном давлении с помощью роторного испарителя для удаления растворителя с получением А1-2. 1Н ЯМР (400 МГц, дейтерированный метанол) δ: 7,80 (s, 1Н), 6,86 (d, J=4,4 Гц, 1Н), 6,53 (d, J=4,4 Гц, 1H), 3,96-3,79 (m. 2Н), 3,60-3,51 (m, 1H), 3,49-3,38 (m, 2Н), 2,39-2,36 (m, 1H), 2,19-2,13 (m, 1H), 1,49 (d, J=3,6 Гц, 9Н).
Стадия 3: синтез соединения А1
При комнатной температуре йодосукцинимид (26,7 г, 119 ммоль, 3,00 экв.) добавляли порциями в раствор А1-2 (12,0 г, 39,6 ммоль, 1,00 экв.) в N,N-диметилформамиде (150 мл). Затем реакционный раствор перемешивали при комнатной температуре в течение 1 часа, его медленно добавляли в ледяную баню (200 мл) и высаживали твердое вещество. После фильтрации растворитель удаляли и фильтровальный кек сушили с помощью роторного испарителя при пониженном давлении с получением соединения A1. 1Н ЯМР (400 МГц, дейтерированный ДМСО) δ=7,88 (s, 1Н), 6,75 (s, 1H), 3,77-3,68 (m, 2Н), 3,42-3,38 (m, 1H), 3,28-3,23 (m, 2Н), 2,31-2,22 (m, 1H), 2,05-1,98 (m, 1Н), 1,39 (d, J=5,2 Гц, 9Н).
Промежуточное соединение В1:
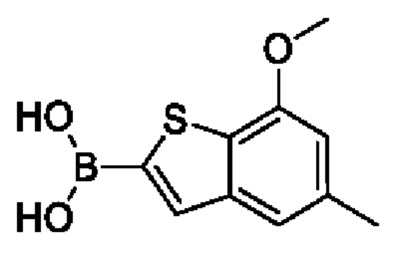
Путь синтеза:

Раствор В1-1 (2,00 г, 11,22 ммоль, 1,00 экв.) в тетрагидрофуране (20,00 мл) охлаждали до -70°С. В охлажденный раствор медленно по каплям добавляли раствор бутиллития в н-гексане (2,5 моль/л, 8,98 мл, 2,00 экв.) и перемешивали в течение 1 часа после добавления. Затем добавляли триизопропилборат (2,11 г, 11,22 ммоль, 1,00 экв.) и перемешивали в течение 1 часа после добавления. Реакцию останавливали путем добавления по каплям воды (10 мл). Дезактивированную реакционную смесь концентрировали для удаления тетрагидрофурана. Остаток промывали петролейным эфиром (50 мл) и затем доводили до рН 5 разбавленной соляной кислотой для получения белого твердого вещества. После фильтрации фильтровальный кек промывали водой (50 мл) и затем сушили под вакуумом с получением промежуточного соединения В1. 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ=7,72 (s, 1H), 7,28 (s, 1H), 6,67 (s, 1H), 4,01 (s, 3Н), 2,50 (s, 3Н).
Пример 1 Синтез соединения формулы (I)
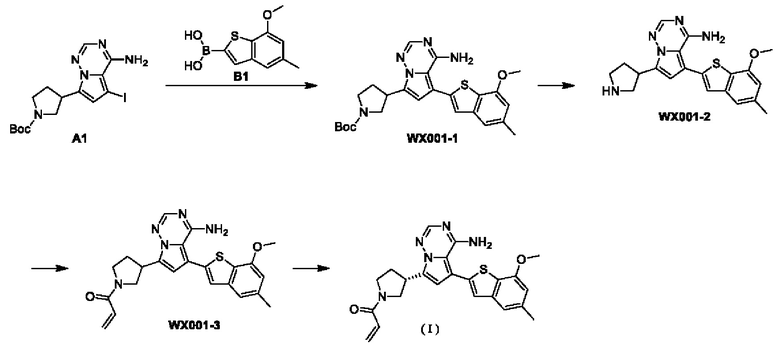
Стадия 1: Синтез соединения WX001-1
При комнатной температуре соединение В1 (777,25 мг, 3,50 ммоль, 2,50 экв.), карбонат натрия (296,77 мг, 2,80 ммоль, 2,00 экв.) и тетракис(трифенилфосфин)палладий (161,78 мг, 140,00 мкмоль, 0,10 экв.) последовательно добавляли в смешанный раствор соединения А1 (600,00 мг, 1,40 ммоль, 1,00 экв.) в диметиловом эфире этиленгликоля (9 мл), этанола (3 мл) и воды (0,5 мл). После замены газа газообразным азотом 3 раза смесь нагревали до 90°С. После этого смесь перемешивали в течение 5 часов, охлаждали ее до комнатной температуры и выливали в 30 мл воды и потом экстрагировали дихлорметаном (10 мл) 5 раз. Органические фазы объединяли и сушили безводным сульфатом натрия. После фильтрации фильтрат подвергали упариванию с помощью роторного испарителя при пониженном давлении для удаления растворителя с получением неочищенного продукта. Неочищенный продукт очищали методом колоночной хроматографии (петролейный эфир / этилацетат = от 10/1 до 1/3) с получением WX001-1. ЖХ-МС (ESI) m/z: 480,2 [М+Н]+, 502,2 [M+Na]+, 1H ЯМР (400 МГц, дейтерированный метанол) δ=7,91 (s, 1Н), 7,27 (s, 2Н), 6,77 (s, 1H), 6,70 (s, 1H), 4,00 (s, 3Н), 3,96-3,90 (m, 2Н), 3,64-3,50 (m, 3Н), 2,49 (s, 3Н), 2,44-2,36 (m, 2Н), 1,50 (s, 9Н).
Стадия 2: Синтез соединения WX001-2
При комнатной температуре раствор соляной кислоты в этилацетате (4 моль/л, 2,00 мл, 9,51 экв.) медленно добавляли по каплям в раствор WX001-1 (350,00 мг, 729,79 мкмоль, 1,00 экв.) в этилацетате (2 мл) и перемешивали в течение 1 часа. После фильтрации твердое вещество получали и сушили при пониженном давлении с получением гидрохлоридной соли соединения WX001-2. ЖХ-МС (ESI) m/z: 380,1 [М+Н]+, 1Н ЯМР (400 МГц, дейтерированный метанол) δ=8,17 (s, 1Н), 7,46 (s, 1H), 7,33 (s, 1H), 7,12-7,06 (m, 1H), 6,84 (s, 1H), 4,12-4,06 (m, 1H), 4,02 (s, 3H), 3,92-3,82 (m, 2H), 3,67-3,58 (m, 2H), 2,66-2,60 (m, 1H), 2,51 (s, 3H), 2,39-2,32 (m, 1H).
Стадия 3: Синтез соединения формулы (I)
При 0°С диизопропилэтиламин (258,56 мг, 2,00 ммоль, 349,41 мкл, 4,00 экв.) и раствор акрилоилхлорида в дихлорметане (0,25 М, 1,80 мл, 0,90 экв.) добавляли последовательно в раствор гидрохлоридной соли WX001-2 (200,00 мг, 500,16 мкмоль, 1,00 экв.) в дихлорметане (4,00 мл) и перемешивали в течение 5 минут. Затем реакционную жидкость выливали в 2 мл воды. После разделения слоев водную фазу экстрагировали дихлорметаном (1 мл) 3 раза. Органические фазы объединяли и сушили безводным сульфатом натрия. После фильтрации фильтрат подвергали упариванию с помощью роторного испарителя при пониженном давлении для удаления растворителя и с получением неочищенного продукта. Неочищенный продукт очищали методом тонкослойной препаративной хроматографии (дихлорметан/метанол=10/1) с получением соединения WX001-3. Соединение WX001-3 подвергали хиральному разделению (колонка: AS (250 мм × 30 мм, 5 мкм), подвижная фаза: [0,1% гидроксида аммония, этанол], диоксид углерода: 40%-40%) с получением соединения формулы (I) (время удерживания: 6,98 минут). Время удерживания определяли с помощью аналитической колонки Chiralpak AS-3 150×4,6 мм 3 мкм, подвижная фаза А: диоксид углерода В: метанол (0,05% диэтиламина), 40% диоксида углерода, со скоростью потока 2,5 мл/мин и температурой колонки 35°С. ЖХ-МС (ESI) m/z: 434,2 [М+Н]+, 456,1 [M+Na]+, 1Н ЯМР (400 МГц, дейтерированный метанол) δ=7,75 (d, J=2,8 Гц, 1H), 7,08 (s, 2Н), 6,61 (s, 1H), 6,54 (d, J=6,4 Гц, 1H), 6,41-6,51 (m, 1H), 6,20-6,16 (m, 1H), 5,66-5,42 (m, 1Н), 4,09-3,96 (m, 1Н), 3,85 (s, 3Н), 3,80-3,38 (m, 4Н), 2,44-2,25 (m, 4Н), 2,21-1,99 (m, 1H).
Пример 2: Получение кристаллической формы А
500 мг соединения формулы (I), полученного в Примере 1, взвешивали добавляли в стеклянную колбу на 40 мл, затем добавляли 8 мл ацетонитрила и магнитную палочку для перемешивания. Смесь перемешивали, пока она не стала образцом-суспензией. Указанный выше образец еще перемешивали в течение 2 дней (с защитой от света) на нагревательном столике с магнитным перемешиванием (40°С). Образец быстро центрифугировали и оставшееся твердое вещество сушили под вакуумом в вакуумном сушильном шкафу при температуре 30°С в течение ночи для удаления остатков растворителя и с получением кристаллической формы А соединения формулы (I).
Пример 3: Получение кристаллической формы В
600 мг кристаллической формы А соединения формулы (I), полученного в Примере 2, взвешивали и добавляли в стеклянную колбу на 40 мл, затем добавляли 12 мл этанола в качестве растворителя и магнитную палочку для перемешивания. Смесь перемешивали, пока она не стала образцом-суспензией. Указанный выше образец еще перемешивали в течение ночи (с защитой от света) на нагревательном столике с магнитным перемешиванием (40°С) и оставляли при стоянии в течение 5 часов при комнатной температуре (примерно 15°С). Образец быстро центрифугировали и удаляли надосадочную жидкость. Твердое вещество, полученное путем центрифугирования, сушили под вакуумом в вакуумном сушильном шкафу сначала при температуре 40°С в течение 2 часов и потом при температуре 30°С в течение 60 часов с получением кристаллической формы В соединения формулы (I).
Пример 4: Исследование гигроскопичности кристаллической формы В соединения формулы (I)
Экспериментальные материалы:
Прибор для динамической адсорбции паров SMS DVS Advantage
Экспериментальный метод:
Брали 10-15 мг кристаллической формы В соединения формулы (I) и помещали на панель для DVS образца для испытания.
Результат эксперимента:
Кристаллическая форма В соединения формулы (I) имела DVS спектр, показанный на фиг. 7. При температуре 25°С и 80% влажности увеличение массы составляло ΔW=0,1928%.
Вывод:
Кристаллическая форма В соединения формулы (I) имела увеличение массы, вызванное адсорбцией влаги 0,1928% при 25°С и 80% ОВ, что является показателем того, что кристаллическая форма В соединения формулы (I) не имела или практически не имела гигроскопичности.
Экспериментальный пример 1: Оценка ингибирующей способности киназы дикого типа in vitro
IC50 определяли, используя количественное определение активности киназы, меченной изотопом 33Р (Reaction Biology Corp), для оценки ингибирующей способности испытуемого соединения против FGFR1 и FGFR4 человека.
Буфер: 20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ ЭГТК (этиленгликоль тетрауксусной кислоты), 0,02% Brij35, 0,02 мг/мл БСА (бычьего сывороточного альбумина), 0,1 мМ Na3VO4, 2 мМ ДТТ (дитиотреитола) и 1% ДМСО.
Методика испытания: При комнатной температуре соединение формулы (I) растворяли в ДМСО для получения 10 мМ раствора. Субстрат растворяли в свежеприготовленном буфере и добавляли в испытуемую киназу и хорошо перемешивали. С помощью акустической технологии (Echo 550) раствор испытуемого соединения в ДМСО добавляли в указанный выше хорошо перемешиваемый реакционный раствор, чтобы концентрация соединения в реакционном растворе составляла 10 мкМ, 3,33 мкМ, 1,11 мкМ, 0,370 мкМ, 0,123 мкМ, 41,2 нМ, 13,7 нМ, 4,57 нМ, 1,52 нМ и 0,508 нМ или составляла 10 мкМ, 2,50 мкМ, 0,62 мкМ, 0,156 мкМ, 39,1 нМ, 9,8 нМ, 2,4 нМ, 0,61 нМ, 0,15 нМ и 0,038 нМ. Через 15 минут инкубации добавляли 33Р-АТР (имеющую активность 0,01 мкСi/мкл, соответствующие концентрации приведены в таблице 3) для начала реакции. Информация о FGFR1, FGFR4 и поставщиках их субстратов, номера по каталогу и номер партии, а также их концентрации в реакционном растворе перечислены в таблице 3. После проведения реакции при комнатной температуре в течение 120 минут реакционный раствор наносили пятнами на ионообменную фильтровальную бумагу Р81 (номер Whatman 3698-915). После многократной промывки фильтровальной бумаги 0,75% раствором фосфорной кислоты определяли радиоактивность фосфорилированного субстрата, оставшегося на фильтровальной бумаге. Данные по активности киназы выражали путем сравнения активности киназы испытуемого соединения с активностью контрольной группы (содержащей только ДМСО), a IC50 определяли путем аппроксимации кривой с помощью программного обеспечения Prism4 (GraphPad). Результаты эксперимента представлены в таблице 4.

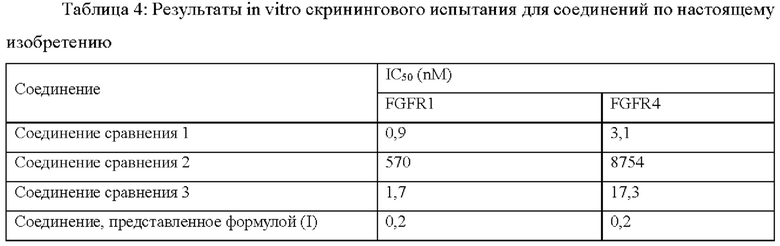
Вывод: Соединение формулы (I) в настоящем изобретении проявляет более хорошую ингибирующую способность против FGFR дикого типа.
Экспериментальный пример 2: Оценка ингибирующей способности киназы мутантного типа in vitro
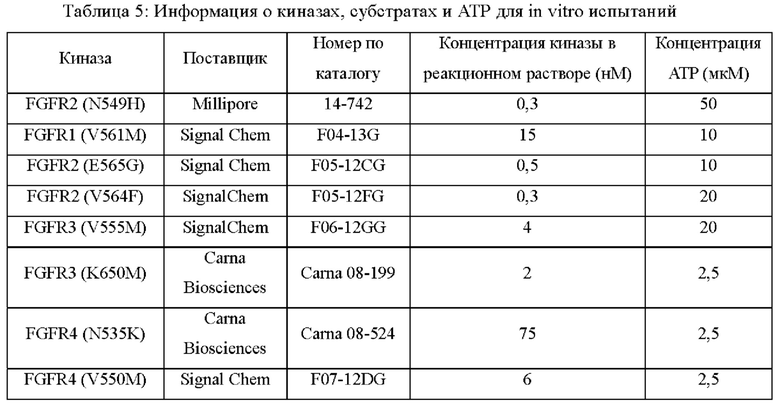
IC50 определяли, используя количественное определение активности киназы, меченной изотопом 33Р (Reaction Biology Corp), для оценки ингибирующей способности испытуемого соединения против FGFR мутантного типа. Релевантная информация о киназе, субстрате и АТР для in vitro испытания показана в таблице 5.

Буфер: 20 мМ Hepes (рН 7,5), 10 мМ MgCl2, 1 мМ ЭГТК, 0,02% Brij35, 0,02 мг/мл БСА, 0,1 мМ Na3VO4, 2 мМ ДТТ и 1% ДМСО.
Методика испытания: При комнатной температуре соединение формулы (I) растворяли в ДМСО для получения 10 мМ раствора. Субстрат растворяли в свежеприготовленном буфере и добавляли в испытуемую киназу и хорошо перемешивали. С помощью акустической технологии (Echo 550) раствор испытуемого соединения в ДМСО добавляли в указанный выше хорошо перемешиваемый реакционный раствор, чтобы концентрация соединения в реакционном растворе составляла 10 мкМ, 3,33 мкМ, 1,11 мкМ, 0,370 мкМ, 0,123 мкМ, 41,2 нМ, 13,7 нМ, 4,57 нМ, 1,52 нМ и 0,508 нМ или составляла 10 мкМ, 2,50 мкМ, 0,62 мкМ, 0,156 мкМ, 39,1 нМ, 9,8 нМ, 2,4 нМ, 0,61 нМ, 0,15 нМ и 0,038 нМ. Через 15 минут инкубации добавляли 33Р-АТР (имеющую активность 0,01 мкCi/мкл, соответствующие концентрации приведены в таблице 5) для начала реакции. Информация о FGFR1, FGFR4 и поставщиках их субстратов, номера по каталогу и номер партии, а также их концентрации в реакционном растворе перечислены в таблице 5. После проведения реакции при комнатной температуре в течение 120 минут реакционный раствор наносили пятнами на ионообменную фильтровальную бумагу Р81 (номер Whatman 3698-915). После многократной промывки фильтровальной бумаги 0,75% раствором фосфорной кислоты определяли радиоактивность фосфорилированного субстрата, оставшегося на фильтровальной бумаге. Данные по активности киназы выражали путем сравнения активности киназы испытуемого соединения с активностью контрольной группы (содержащей только ДМСО), a IC50 определяли путем аппроксимации кривой с помощью программного обеспечения Prism4 (GraphPad). Результаты эксперимента представлены в таблице 6.
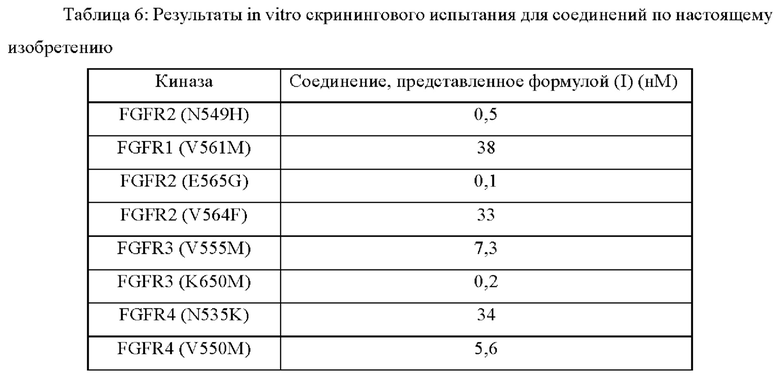
Вывод: Соединение формулы (I) в настоящем изобретении проявляет более хорошую ингибирующую способность против FGFR мутантного типа.
Экспериментальный пример 3: фармакокинетические исследования на собаках
Цель экспериментов
Целью экспериментов является испытание фармакокинетики испытуемого соединения на собаках породы бигль.
Экспериментальные материалы:
Собаки породы бигль (кобели)
Экспериментальный метод:
Две собаки породы бигль были отобраны в одну группу. Соединение вводили в состав смеси препарат с определенным назначением. В качестве носителя для внутривенного введения использовали смесь ДМСО: полиэтиленликоль 1400 (PEG400): водный раствор хлорида натрия=10:40:50 (объемное отношение) или смесь 10% ДМСО/10% солютола/80% воды. В качестве носителя для перорального введения была смесь 0,5% метилцеллюлозы (МЦ) + 0,2% Tween. Каждому животному вводили препарат внутрижелудочно в заранее заданной дозе.
Образцы цельной крови, каждый примерно по 500 мкл, собирали из головной вены или подкожной вены в 12 моментов времени, а именно: 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа после введения животным.
Образцы плазмы помещали в пробирку для центрифуги, содержащую антикоагулянт, и центрифугировали при 3000 g в течение 10 мин при 4°С. Супернатант плазмы быстро замораживали на сухом льду и после этого хранили в холодильнике при -70±10°С до проведения ЖХ-МС/МС анализа.
Обработка данных:
Концентрацию соединения в плазме обрабатывали с использованием некомпартментной модели с помощью фармакокинетического программного обеспечения WinNonlin™ Version 6,3,0 (Pharsight, Mountain View, CA). Пиковую концентрацию (Смакс), время достижения пиковой концентрации (Тмакс) и время последней определяемой концентрации определяли напрямую по кривой зависимости концентрации в плазме от времени.
Следующие фармакокинетические параметры: период полувыведения (Т1/2), среднее время пребывания лекарственного средства в организме от момента времени 0 до конечного момента времени (MRTO-last), среднее время пребывания лекарственного средства в организме от момента времени 0 до бесконечного времени (MRTO-inf), площадь под кривой зависимости концентрации плазмы от времени от момента времени 0 до конечного момента времени (AUC0-last), площадь под кривой зависимости концентрации плазмы от времени от момента времени 0 до бесконечного времени (AUCO-inf), вычисляли линейно-логарифмическим методом трапеций.
Для отдельных концентраций в плазме ниже предела обнаружения, если она появлялась до Тмакс, учитывалась при расчетах как 0; если появлялась после Тмакс, ее просто исключали. Все параметры и соотношения записывались в виде трех значащих цифр.
В этих экспериментах фармакокинетические параметры рассчитывали на основе теоретического времени забора крови и теоретической вводимой концентрации в воплощениях изобретения. Отклонение между фактической вводимой концентрацией и теоретической концентрацией находилось в пределах ±20%. Отклонение между фактическим временем забора крови и теоретическим временем забора крови соответствовало релевантным стандартным рабочим методикам (SOP) (моменты времени в пределах 1 часа после введения находились в пределах ±1 минуты, а другие - в пределах 5% от теоретического времени).
Результаты эксперимента:
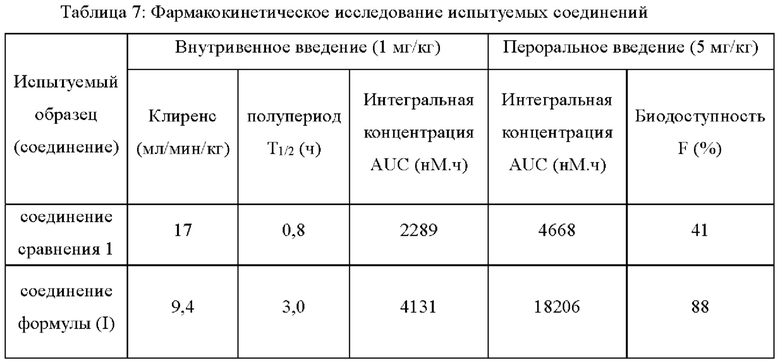
Результаты эксперимента с испытуемыми соединениями показаны в Таблице 7.
Вывод эксперимента:
Соединение формулы (I) имеет хорошие фармакокинетические параметры в опытах на собаках.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЬ АРИЛАМИНОХИНАЗОЛИН-СОДЕРЖАЩЕГО СОЕДИНЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2833198C1 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЕВАЯ ФОРМА ИНГИБИТОРА TGF-βRI И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2750702C1 |
| Тиазололактамные соединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2805569C1 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
| Спиросоединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2800042C1 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИАЗАБЕНЗОФЛУОРАНТРЕНОВЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2762189C2 |
| ТОЗИЛАТНАЯ СОЛЬ ПРОИЗВОДНОГО 5-ПИРАЗОЛИЛ-2-ПИРИДОНА, ПОЛЕЗНАЯ В ЛЕЧЕНИИ COPD | 2010 |
|
RU2526038C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| Соединение-ингибитор мультикиназ и его кристаллическая форма и применение | 2017 |
|
RU2723985C1 |
Изобретение относится к соединению, представленному формулой (I), в твердой форме, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), с дифракционными пиками при угле 2θ 6,37±0,2°, 9,90±0,2°, 12,74±0,2°, 13,35±0,2°, 14,26±0,2°, 16,31±0,2°, 19,07±0,2° и 21,83±0,2° на порошковой рентгенодифрактограмме или представляет собой кристаллическую форму В соединения, представленного формулой (I), с дифракционными пиками при угле 2θ 9,14±0,2°, 11,05±0,2°, 13,25±0,2°, 15,07±0,2°, 16,47±0,2°, 18,31±0,2° и 22,29±0,2° на порошковой рентгенодифрактограмме. Изобретение относится к способу получения кристаллической формы А соединения, представленного формулой (I), включающему (а) добавление соединения, представленного формулой (I), в ацетонитрил; (b) перемешивание при температуре 30-50°С в течение 40-55 ч и (c) отделение кристаллической формы А соединения, представленного формулой (I). Изобретение относится к способу получения кристаллической формы В соединения, представленного формулой (I), включающему (a) добавление кристаллической формы А соединения, представленного формулой (I), в этанол; (b) перемешивание при температуре 30-50°С в течение 5-30 ч; (c) выстаивание при температуре 10-20°С в течение 3-10 ч и (d) отделение кристаллической формы В соединения, представленного формулой (I). Изобретение также относится к применению соединения, представленного формулой (I), в твердой форме, которое представляет собой кристаллическую форму А или кристаллическую форму В для производства лекарственного средства для лечения заболевания, связанного с FGFR. Технический результат - соединение формулы (I) проявляло хорошую ингибирующую способность в отношении FGFR дикого типа, а также кристаллическая форма В соединения формулы (I) не проявляет гигроскопичности и показывает хорошие перспективы для изготовления лекарственных средств. 4 н. и 11 з.п. ф-лы, 7 ил., 7 табл., 4 пр.
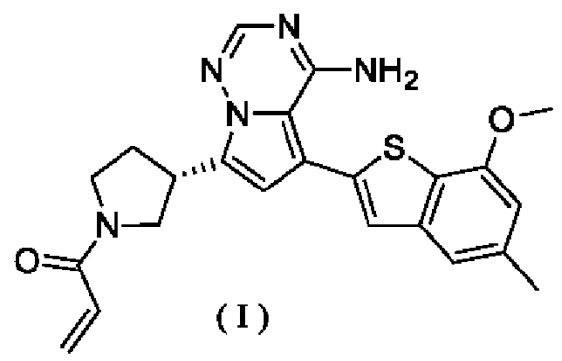
1. Соединение, представленное формулой (I), в твердой форме
которое представляет собой кристаллическую форму А соединения, представленного формулой (I), с дифракционными пиками при угле 2θ 6,37±0,2°, 9,90±0,2°, 12,74±0,2°, 13,35±0,2°, 14,26±0,2°, 16,31±0,2°, 19,07±0,2° и 21,83±0,2° на ее порошковой рентгенодифрактограмме или
которое представляет собой кристаллическую форму В соединения, представленного формулой (I), с дифракционными пиками при угле 2θ 9,14±0,2°, 11,05±0,2°, 13,25±0,2°, 15,07±0,2°, 16,47±0,2°, 18,31±0,2° и 22,29±0,2° на ее порошковой рентгенодифрактограмме.
2. Соединение, представленное формулой (I), в твердой форме по п. 1, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), которая имеет порошковую рентгенодифрактограмму, показанную на фиг. 1.
3. Соединение, представленное формулой (I), в твердой форме по п. 1 или 2, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), которая имеет эндотермический пик при температуре 141,05±5°С на кривой дифференциальной сканирующей калориметрии.
4. Соединение, представленное формулой (I), в твердой форме по п. 3, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), которая имеет кривую ДСК, показанную на фиг. 2.
5. Соединение, представленное формулой (I), в твердой форме по любому из пп. 1-4, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), которая имеет потерю массы 1,232% при температуре 124,65±3°С на кривой термогравиметрического анализа (ТГА).
6. Соединение, представленное формулой (I), в твердой форме по п. 5, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), которая имеет кривую ТГА, показанную на фиг. 3.
7. Соединение, представленное формулой (I), в твердой форме по п. 1, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), которая имеет порошковую рентгенодифрактограмму, показанную на фиг. 4.
8. Соединение, представленное формулой (I), в твердой форме по п. 1 или 7, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), которая имеет эндотермический пик с начальной точкой при температуре 174,09±5°С на кривой дифференциальной сканирующей калориметрии.
9. Соединение, представленное формулой (I), в твердой форме по п. 8, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), которая имеет кривую ДСК, показанную на фиг. 5.
10. Соединение, представленное формулой (I), в твердой форме по любому из пп. 1, 8 и 9, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), которая имеет потерю массы 0,432% при температуре 169,70±3°С на спектре термогравиметрического анализа (ТГА).
11. Соединение, представленное формулой (I), в твердой форме по п. 10, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), которая имеет спектр ТГА, показанный на фиг. 6.
12. Способ получения соединения, представленного формулой (I), в твердой форме по любому из пп. 1-6, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), включающий:
(а) добавление соединения, представленного формулой (I), в ацетонитрил;
(b) перемешивание при температуре 30-50°С в течение 40-55 ч и
(c) отделение кристаллической формы А соединения, представленного формулой (I).
13. Способ получения соединения, представленного формулой (I), в твердой форме по любому из пп. 1 и 7-11, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), включающий:
(a) добавление кристаллической формы А соединения, представленного формулой (I), в этанол;
(b) перемешивание при температуре 30-50°С в течение 5-30 ч;
(c) выстаивание при температуре 10-20°С в течение 3-10 ч и
(d) отделение кристаллической формы В соединения, представленного формулой (I).
14. Применение соединения, представленного формулой (I), в твердой форме по любому из пп. 1-6, которое представляет собой кристаллическую форму А соединения, представленного формулой (I), или соединения, представленного формулой (I), в твердой форме по любому из пп. 1 и 7-11, которое представляет собой кристаллическую форму В соединения, представленного формулой (I), в производстве лекарственного средства для лечения заболевания, связанного с FGFR.
15. Применение по п. 14, в котором заболевание, связанное с FGFR, представляет собой твердую опухоль.
| Механизм для автоматической подачи электродной проволоки при дуговой электрической сварке | 1930 |
|
SU29556A1 |
| WO 2013087647 A1, 20.06.2013 | |||
| WO 2013124316 A1, 29.08.2013 | |||
| MINO R | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208 | |||
| Дж | |||
| Бернштейн "Полиморфизм молекулярных кристаллов" Москва, Наука, 2007, гл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Биодоступность, с.324-330. | |||

