Область техники
Изобретение относится к области молекулярной биологии и микробиологии, в частности описывает новые бактериальные нуклеазы системы CRISPR-Cas и новые гибридные формы направляющих CRISPR-РНК. Изобретение может быть использовано в качестве инструмента для строго специфической модификации ДНК в различных организмах.
Изобретение создано при финансовой поддержке Министерства Науки и Высшего образования Российской Федерации в рамках Соглашения № 075-15-2019-1661 от 31.10.2019.
Уровень техники
Изменение последовательности ДНК - одна из актуальных задач биотехнологии на сегодняшний день. Редактирование и изменение геномов эукариотических и прокариотических организмов, а также манипуляции с ДНК in vitro, требуют направленного внесения двунитевых разрывов в последовательности ДНК. Для решения этой задачи в настоящее время используют следующие методики: искусственные нуклеазные системы, содержащей домены типа «цинковые пальцы», TALEN-системы и бактериальные CRISPR-Cas системы. Первые два метода требуют трудозатратой оптимизации аминокислотной последовательности нуклеазы для узнавания конкретной последовательности ДНК. В отличие от них в случае CRISPR-Cas систем структурами, узнающими ДНК мишень, являются не белки, а короткие направляющие РНК. Разрезание конкретной ДНК мишени не требует синтеза нуклеазы или ее гена de novo, а обеспечивается за счет использования направляющих РНК, комплементарных целевой последовательности. Это делает CRISPR-Cas системы удобными и эффективными инструментами разрезания различных ДНК-последовательностей. Методика позволяет осуществлять единовременное разрезание ДНК в нескольких участках при использовании направляющих РНК разной последовательностей. Такой подход используется в том числе для одновременного изменения нескольких генов в эукариотических организмах.
По своей природе CRISPR-Cas системы являются иммунными системами прокариот, способными высоко специфично вносить разрывы в генетический материал вирусов (Mojica et al., 2005). Аббревиатура CRISPR-Cas расшифровывается как “Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR associated genes” (Jansen et al., 2002), что переводе с английского обозначает “короткие палиндромные повторы, регулярно расположенные группами, и aссоциированные с ними гены”. Все CRISPR-Cas системы состоят из CRISPR кассет и генов, кодирующих различные Cas белки (Jansen, 2002). CRISPR кассеты состоят из последовательностей-спейсеров, каждый из которых имеет уникальную нуклеотидную последовательность, и повторяющихся палиндромных повторов (Jansen, 2002). В результате транскрипции CRISPR кассет и их последующего процессинга образуются направляющие крРНК, которые вместе с Cas белками формируют эффекторный комплекс (Brouns et al., 2008). За счет комплементарного спаривания крРНК с целевым участком ДНК, именуемым протоспейсером, Cas-нуклеаза узнает ДНК-мишень и высоко специфично вносит в нее разрыв.
CRISPR-Cas системы, представленные одиночным белком-эффектором, разделяют на шесть различных типов (от I до VI) в зависимости от Cas белков, входящих в состав систем.
Система CRISPR-Cas12d относится к типу V и отличается простотой состава и механизма работы: для ее функционирования необходимо формирование эффекторного комплекса, состоящего лишь из одного белка Cas12d и двух коротких РНК: крРНК (crRNA) и скаутной РНК (scoutRNA). Скаутная РНК комплементарно спаривается с участком крРНК, проиcходящим из CRISPR повтора, образуя вторичную структуру, необходимую для связывания направляющих РНК с Cas эффектором. Несмотря на некоторые аналогии, скаутная РНК и механизм ее связывания с крРНК сильно отличаются от трейсерной РНК, характерной для Cas9 белков, в частности SpCas9 из Streptococcus pyogenes, нуклеазе широко используемой в биотехнологии.
Эффекторный белок Cas12d является РНК-зависимой ДНК эндонуклеазой, образующей двунитевые разрывы в ДНК (Harrington et al., 2020).
Использование CRISPR-Cas нуклеаз для модификации геномов различных организмов, требует воссоздания их активности in vitro и создания гибридной формы направляющих РНК (sgRNA), где крРНК и вторая, дополнительная РНК слиты в единую молекулу. Такие молекулы были созданы для SpCas9 и повышают удобство работы с системой, сокращая количество ее компонентов и снижая число промоторов, необходимых для экспрессии генов в различных организмах. Однако для систем CRISPR-Cas12d такие гибридные РНК не были созданы ранее.
На сегодняшний день известно несколько CRISPR-Cas нуклеаз, работа которых реконструирована in vitro, способных направлено и специфично вносить двунитевые разрывы в ДНК, самой популярной из них является SpCas9 из Streptococcus pyogenes. Одной из основных характеристик, ограничивающих применение CRISPR-Cas систем является PAM последовательность, фланирующая ДНК-мишень с 3'-конца, наличие которой необходимо для правильного узнавания ДНК Cas нуклеазой. Различные CRISPR-Cas белки имеют разные PAM последовательности, которые ограничивают возможности применения нуклеаз на любых участках ДНК. Так, SpCas9 требует 5' -NGG -3' PAM.
Использование CRISPR-Cas белков с новыми разнообразными PAM последовательностями необходимо для обеспечения возможности изменения любого участка ДНК, как in vitro, так и в геноме живых организмов. Изменение эукариотических геномов также требует использования нуклеаз малого размера для обеспечения доставки CRISPR-Cas систем в клетки посредством AAV вирусов. Cas12d нуклеазы, имеющие малый размер, могут решить проблему доставки CRISPR-Cas систем в вирусных капсидах ограниченной емкости.
Несмотря на известность ряда способов разрезания ДНК и изменения последовательности геномной ДНК, на сегодняшний день сохраняется потребность в новых эффективных инструментах для модификации ДНК в различных организмах и в строго определенных местах последовательности ДНК.
Данное изобретение обладает рядом свойств, необходимых для решения этой задачи.
Сущность изобретения
Задачей настоящего изобретения является создание новых инструментов для изменения последовательности геномной ДНК одноклеточных или многоклеточных организмов на основе систем CRISPR-Cas12d.
Существующие в настоящее время CRISPR-Cas системы имеют ограниченное применение из-за специфичной последовательности РАМ, которая должна присутствовать на 3'-конце участка ДНК, подвергающегося модификации. Поиск новых ферментов Cas с другими РАМ последовательностями позволит расширить арсенал имеющихся средств для образования двунитевого разрыва в необходимых, строго определенных местах в молекулах ДНК разных организмов. Поиск новых ферментов Cas малого размера позволит осуществлять их доставку в AAV векторах.
Cas12d белки имеют малый размер и короткие, часто встречающиеся в геномах, PAM последовательности, что расширяет их применение в биотехнологии. Однако на сегодняшний день работа только одной Cas12d нуклеазы из бактерий-симбионтов термитов была реконструирована in vitro. Кроме того, ни для одного представителя Cas12d семейства не создана гибридная направляющая РНК (sgRNA), которая аналогично sgRNA, используемой для белков Cas9, уменьшала бы количество компонентов системы и облегчало бы их экспрессии в клетках организмов.
Задача работы - создание средства разрезания ДНК на основе Cas12d белков и гибридной РНК, полученной путем слияния направляющей CRISPR РНК и scout РНК.
Для решения этой задачи авторами была in vitro реконструирована нуклеазная активность CRISPR-Cas12d системы из Katanobacteria (KbCas12d), чья активность была ранее показана только в бактериальных клетках (US2020255858A1), и впервые для систем Cas12d типа создан новый вид гибридной РНК, представляющий собой слияние направляющей CRISPR РНК и scout РНК.
CRISPR-KbCas12d c направляющей гибридной РНК может быть применена для внесения направленных изменений в геном как этого, так и других организмов. Существенными признаками, отличающими настоящее изобретение, являются: (a) возможность использования одной направляющей РНК (sgRNA) вместо двух (crRNA и scoutRNA) ; (б) малый размер охарактеризованного белка KbCas12d - 1125 аминокислотных остатков (а.о.), что на 243 а.о. меньше, чем размер известного фермента Cas9 из Streptococcus pyogenes (SpCas9); (в) широкий рабочий диапазон температур нуклеазы KbCas12d, которая активна при температурах от 25°С до 45°С с оптимумом при 35°С, что позволит использовать ее в организмах, имеющих различную температуру.
Указанная задача решается путем применения белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, для образования двунитевого разрыва в молекуле ДНК, расположенного непосредственно после нуклеотидной последовательности 5'- TA -3' в указанной молекуле ДНК. В некоторых вариантах изобретения данное применение характеризуется тем, что образование двунитевого разрыва в молекуле ДНК происходит при температуре от 25°С до 45°С.
Указанная задача также решается путем создания способа изменения последовательности геномной ДНК одноклеточного или многоклеточного организма, включающего введение в по меньшей мере одну клетку этого организма эффективного количества: а) либо белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, либо нуклеиновой кислоты, кодирующей ген белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, и б) направляющей гибридной РНК, содержащей последовательность, образующую дуплекс с нуклеотидной последовательностью участка геномной ДНК организма, непосредственно примыкающей к нуклеотидной последовательности 5'-TA -3', и взаимодействующей с указанным белком после образования дуплекса, при этом взаимодействие указанного белка с направляющей РНК и нуклеотидной последовательностью 5'- ТА -3' приводит к образованию двунитевого разрыва в последовательности геномной ДНК, непосредственно примыкающей к последовательности 5'- ТА-3'. В некоторых вариантах изобретения данный способ характеризуется тем, что дополнительно включающий введение экзогенной последовательности ДНК одновременно с направляющей РНК.
В качестве направляющей РНК может быть использована смесь из крРНК (crRNA) и скаутной РНК (scoutRNA), способных образовать комплекс с участком целевой ДНК и белком KbCas12d. В предпочтительных вариантах изобретения в качестве направляющей РНК может быть использована гибридная РНК, разработанная авторами и сконструированная на основе крРНК и скаутной РНК путем слияния их нуклеотидных последовательностей, общей формулой А-В-С-D, где А - последовательность KbCas12d scout РНК, B - последовательность линкера; C - последовательность прямого повтора DR KbCas12d крРНК, D - последовательность, комплементарная ДНК-мишени (спейсерный сегмент); при этом последовательность линкера и спейсерного сегмента может быть любой.
В некоторых вариантах изобретения последовательность KbCas12d scout РНК синтезировна на основе последовательности SEQ ID NO: 6. В некоторых вариантах изобретения последовательность прямого повтора DR KbCas12d крРНК является неизменяемой частью последовательности SEQ ID NO: 3.
Гибридная РНК в свою очередь может быть получена с помощью кассеты для экспрессии гибридной РНК и состоит из последовательности U6 промотора, последовательности гибридной РНК по п.1 и ДНК последовательности, фланкированной с 5' конца PAM последовательностью 5'-TA -3'.
Изобретение может быть использовано как для разрезания целевой ДНК in vitro, так и для модификации генома какого-либо живого организма. Модификация генома может проводиться прямым способом - разрезанием генома в соответствующем сайте, а также вставкой экзогенной последовательности ДНК за счет гомологичной репарации.
В качестве экзогенной последовательности ДНК может быть использован любой участок двунитевой или однонитевой ДНК из генома организма, отличного от организма, используемого при введении (или смесь таких участков между собой и с другими фрагментами ДНК), при этом этот участок (или смесь участков) предназначен для интеграции в место двуцепочечного разрыва в таргетной ДНК, образованного под действием нуклеазы KbCas12d. В некоторых вариантах изобретения в качестве экзогенной последовательности ДНК может быть использован участок двуцепочечной ДНК из генома организма, используемого при введении белка KbCas12d, но при этом измененный мутациями (заменой нуклеотидов), а также вставками или делециями одного или нескольких нуклеотидов.
Техническим результатом настоящего изобретения является повышение универсальности доступных систем CRISPR-Cas12d, позволяющее использовать нуклеазу Cas12d для разрезания геномной или плазмидной ДНК в большем количестве специфических мест и при больших диапазонах температур. Еще одним техническим результатом настоящего изобретения является упрощение редактирования генома системами семейства Cas12d за счет использования гибридной направляющей РНК.
Краткое описание рисунков
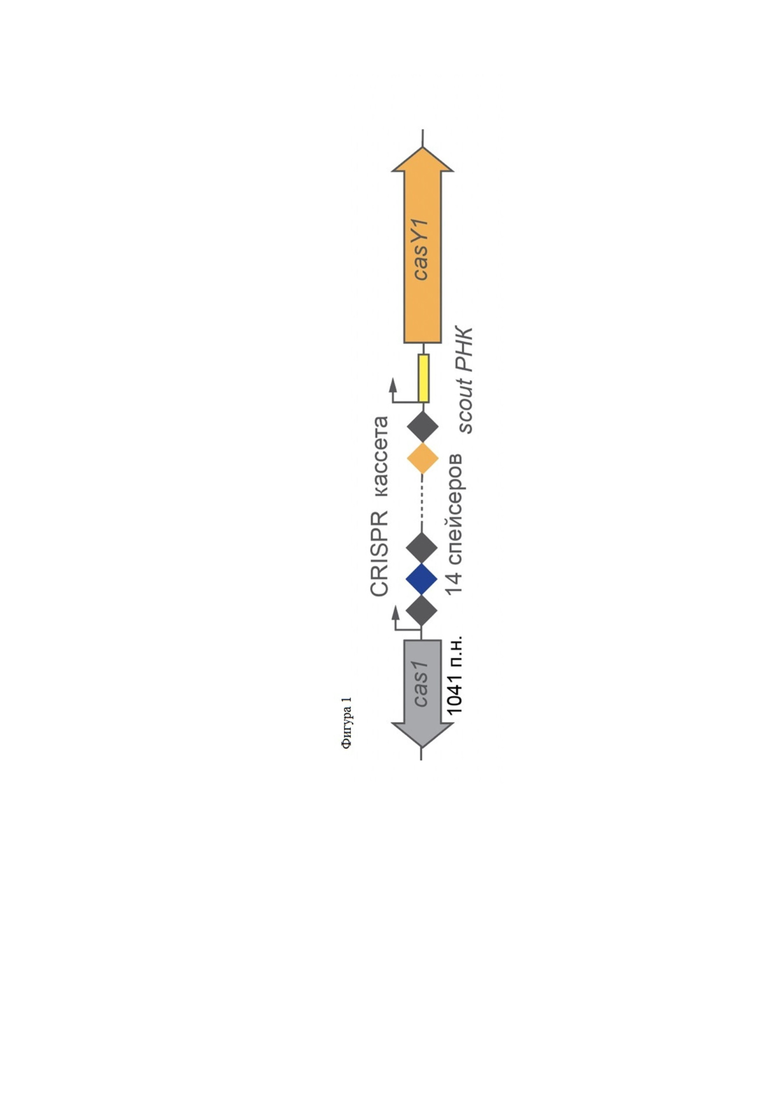
Фиг. 1. Схема устройства CRISPR-KbCas12d локуса.
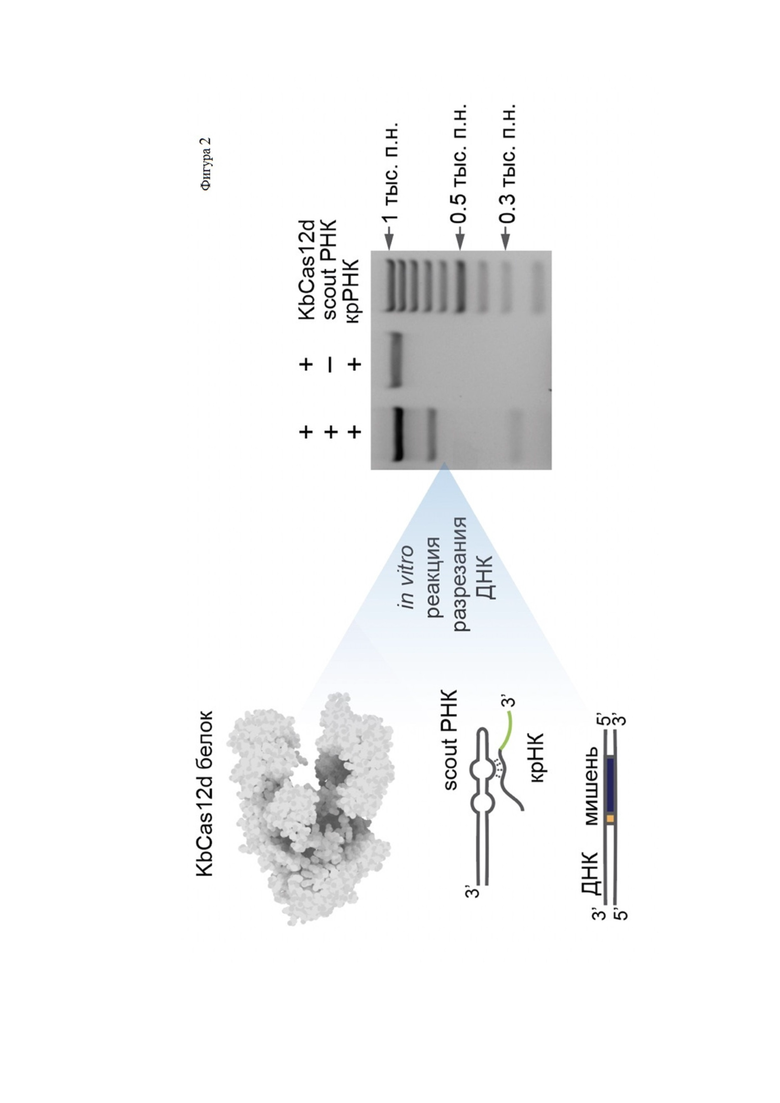
Фиг. 2. KbCas12d в комплексе с скаутной РНК и крРНК разрезает ДНК мишени in vitro.
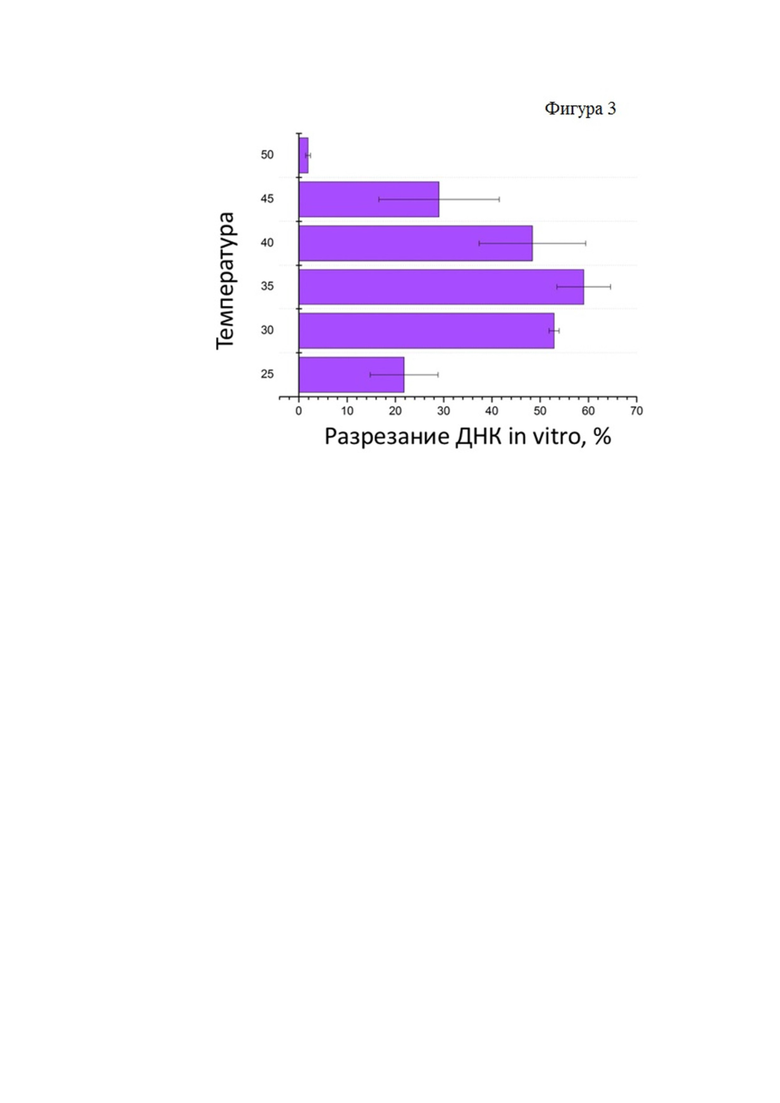
Фиг. 3. Активность белка KbCas12d в разрезании ДНК in vitro в зависимости от температуры.
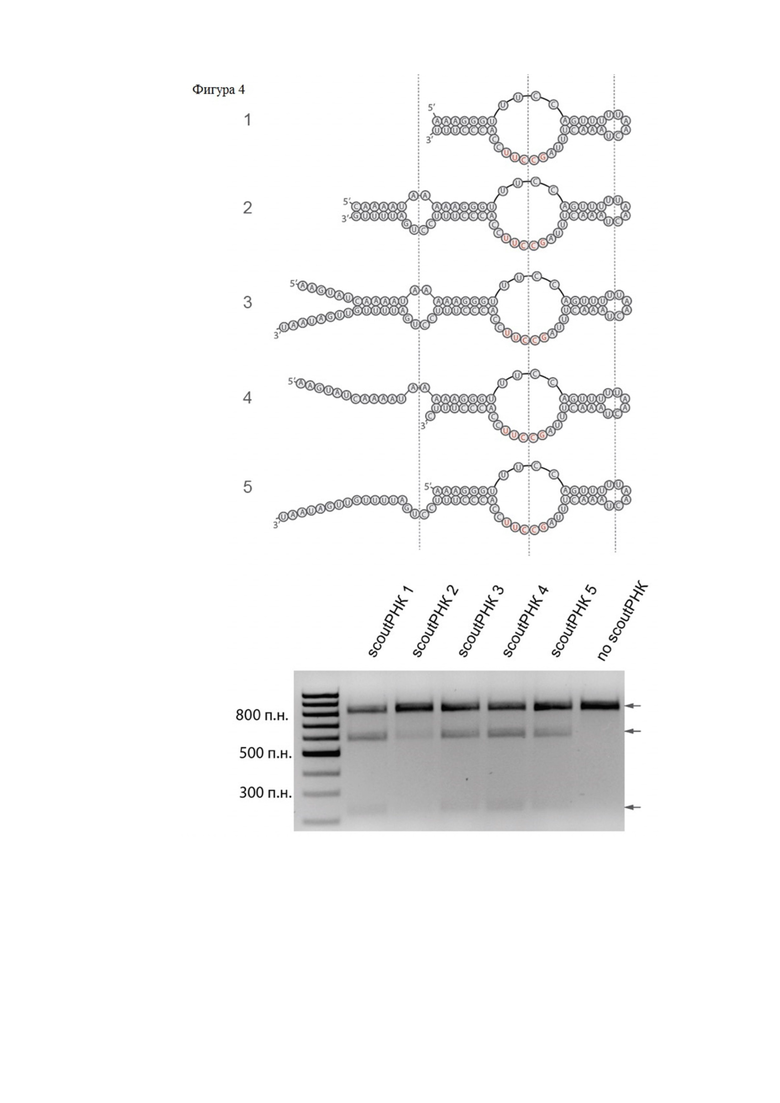
Фиг. 4. Разрезание ДНК белком KbCas12d в комплексе с крРНК и различными формами скаутной РНК, изображенными на рисунке сверху.
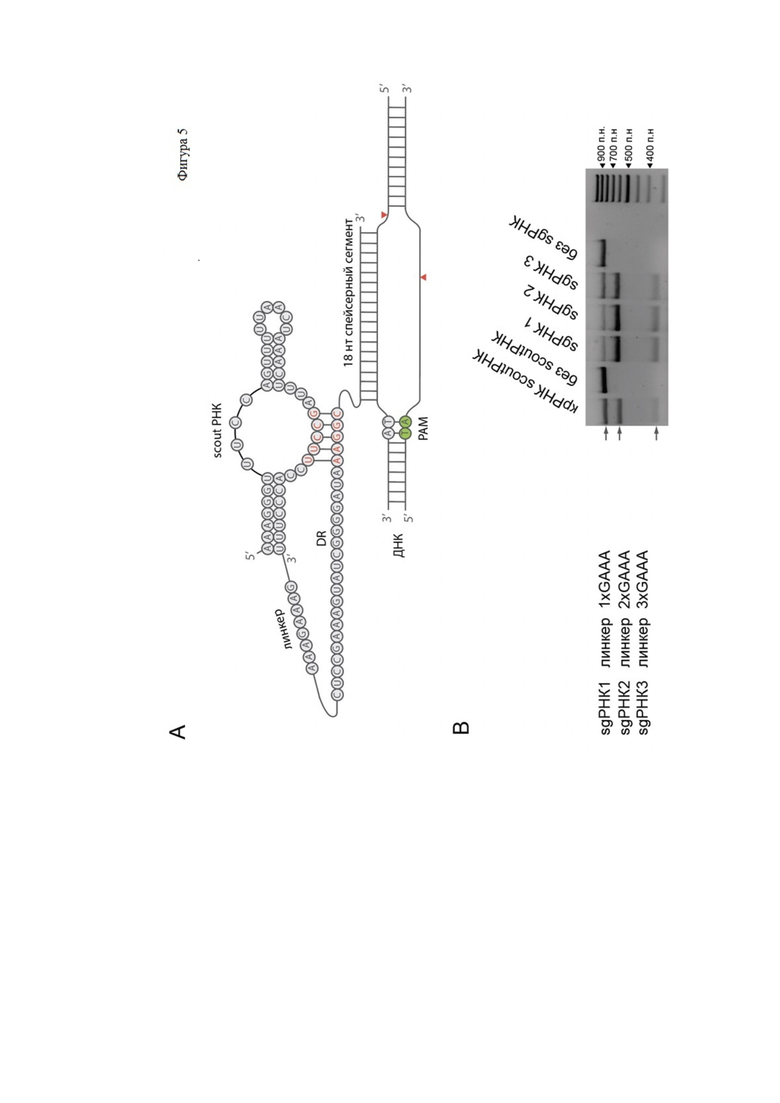
Фиг. 5. Создание гибридной РНК для KbCas12d нуклеазы. (A) схема дизайна гибридной sgРНК. (B) результат разрезания ДНК sgРНК формами, имеющими линкерные последовательности разной длины для соединения крРНК и скаутной РНК.
Фиг. 6. Разрезание KbCas12d различные ДНК мишени in vitro.
Фиг. 7. Выравнивание аминокислотных последовательностей KbCas12d и Cas12d15 из бактерий-симбионтов термитов при помощи программы NCBI BLASTp (default parameters).
Подробное раскрытие изобретения
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из». Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе.
Используемый здесь термин «процент гомологии двух последовательностей» эквивалентен термину «процент идентичности двух последовательностей». Идентичность последовательностей определяется на основании референсной последовательности. Алгоритмы для анализа последовательности известны в данной области, такие как BLAST, описанный Altschul et al. в 1990 году. Для целей настоящего изобретения для определения уровня идентичности и сходства между нуклеотидными последовательностями и аминокислотными последовательностями может быть использовано сравнение нуклеотидных и аминокислотных последовательностей, производимое с помощью пакета программ Blast, предоставляемого National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/blast) с использованием содержащего разрывы выравнивания со стандартными параметрами. Процент идентичности двух последовательностей определяется числом положений идентичных аминокислот в этих двух последовательностях с учетом числа пробелов и длины каждого пробела, которые необходимо ввести для оптимального сопоставления двух последовательностей путем выравнивания. Процент идентичности равен числу идентичных аминокислот в данных положениях с учетом выравнивания последовательностей, разделенному на общее число положений и умноженному на 100.
Термин "специфически гибридизуется" относится к ассоциации между двумя одноцепочечными молекулами нуклеиновых кислот или в достаточной степени комплементарными последовательностями, что разрешает такую гибридизацию в предопределенных условиях, обычно использующихся в данной области.
Фраза "двунитевой разрыв, расположенный непосредственно после нуклеотидной последовательностью РАМ" означает, что двунитевой разрыв в целевой последовательности ДНК будет произведен на расстоянии от 0 до 25 нуклеотидов после нуклеотидной последовательности РАМ.
Под экзогенной последовательностью ДНК, вводимой одновременно с направляющей РНК, следует понимать последовательность ДНК, подготовленную специально для специфической модификации двуцепочечной целевой ДНК в месте разрыва, определяемого специфичностью направляющей РНК. Подобной модификацией может быть, например, вставка или делеция определенных нуклеотидов в месте разрыва целевой ДНК. Экзогенной ДНК может служить как участок ДНК из другого организма, так и участок ДНК из того же организма, что и целевая ДНК.
Под белком, содержащим определенную аминокислотную последовательность следует понимать белок, имеющий аминокислотную последовательность, составленную из указанной аминокислотной последовательности и, возможно, других последовательностей, соединённых пептидными связями с указанной аминокислотной последовательностью. Примером других последовательностей может служить последовательность сигнала ядерной локализации (NLS), или другие последовательности, обеспечивающие повышенную функциональность для указанной аминокислотной последовательности.
Под экзогенной последовательностью ДНК, вводимой одновременно с направляющей РНК, следует понимать последовательность ДНК, подготовленную специально для специфической модификации двуцепочечной целевой ДНК в месте разрыва, определяемого специфичностью направляющей РНК. Подобной модификацией может быть, например, вставка или делеция определенных нуклеотидов в месте разрыва целевой ДНК. Экзогенной ДНК может служить как участок ДНК из другого организма, так и участок ДНК из того же организма, что и целевая ДНК.
Под эффективным количеством вводимых в клетку белка и РНК следует понимать такое количество белка и РНК, которое при попадании в указанную клетку будет способно образовать функциональный комплекс, то есть комплекс, который будет специфически связываться с целевой ДНК и производить в ней двунитевой разрыв в месте, определяемом направляющей РНК и РАМ последовательностью на ДНК. Эффективность этого процесса может быть оценена при помощи анализа целевой ДНК, выделенной из указанной клетки с помощью стандартных методов, известных специалистам.
Доставка белка и РНК в клетку может быть осуществлена различными способами. Например, белок может быть доставлен в виде ДНК-плазмиды, которая кодирует ген этого белка, как мРНК для трансляции этого белка в цитоплазме клетки, или как рибонуклеопротеидный комплекс, включающий этот белок и направляющую РНК. Доставка может быть осуществлена различными методами, известными специалистам.
Нуклеиновая кислота, кодирующая компоненты системы, может быть введена в клетку, непосредственно или опосредованно: за счет трансфекции или трансформации клеток известными специалистам способами, за счет использования рекомбинатного вируса, за счет манипуляций с клеткой, таких как микроинъекция ДНК и т.п.
Доставка рибонуклеинового комплекса, состоящего из нуклеазы и направляющих РНК и экзогенной ДНК (при необходимости) может осуществляться путем трансфекции комплексов в клетку или за счет механического введения комплекса внутрь клетки, например, микроинъекции.
Молекула нуклеиновой кислоты, кодирующая белок, который необходимо ввести в клетку, может быть интегрирована в хромосому или может представлять собой внехромосомно реплицирующуюся ДНК. В некоторых вариантах для обеспечения эффективной экспрессии гена белка с вводимой в клетку ДНК необходимо изменить последовательность этой ДНК в соответствии с типом клетки в целях оптимизации кодонов при экспрессии, обусловленное неравномерностью частот встречаемости синонимичных кодонов в кодирующих областях генома различных организмов. Оптимизация кодонов необходима для увеличения экспрессии в клетках животных, растений, грибов или микроорганизмов.
Для функционирования белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, в эукариотической клетке необходимо, чтобы этот белок оказался в ядре этой клетки. Поэтому, в некоторых вариантах изобретения, для образования двунитевых разрывов в целевой ДНК используют белок, имеющий последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, и который дополнительно модифицирован с одного или с обоих концов добавлением одного или нескольких сигналов ядерной локализации. Например, может быть использован сигнал ядерной локализации из вируса SV40. Для эффективной доставки в ядро сигнал ядерной локализации может быть отделен от основной последовательности белка спейсерной последовательностью, например, описанной Shen et al. в 2013. Также, в других вариантах осуществления, может быть использован другой сигнал ядерной локализации, или альтернативный метод доставки указанного белка в ядро клетки.
Настоящее изобретение охватывает применение белка из организма Katanobateria, гомологичного ранее охарактеризованному in vitro белку Cas12d15 из бактерий симбиотов термита, для внесения двуцепочечных разрывов в молекулы ДНК в строго определенных положениях.
Использование CRISPR нуклеаз для внесения направленных изменений в геном имеет ряд преимуществ. Во-первых, специфичность действия системы определяется последовательностью крРНК, что позволяет использовать один тип нуклеазы для всех локусов-мишеней. Во-вторых, методика позволяет доставить в клетку сразу несколько направляющих РНК, комплементарных разным генам-мишеням, что позволяет осуществлять единовременное изменение сразу нескольких генов.
KbCas9 - Cas нуклеаза, найденная в Katanobacteria. Katanobacteria CRISPR Cas12d1 система (далее CRISPR KbCas12d) относится к V D типу CRISPR Cas систем и состоит из CRISPR кассеты, несущей прямые повторы (direct repeats, DR) последовательностью 5' actccgaaagtatcggggataaaggc 3' разделенных последовательностями уникальных спейсеров. Активность CRISPR-KbCas12d системы в качестве защитной была показана ранее в бактериях (Harrington et al., 2020), в ходе этих же экспериментов были определены требования к PAM, однако in vitro работа системы не была ранее воссоздана. К CRISPR кассете прилегает ген эффекторного Cas12d белка KbCas12d, а также ген белка Cas1, участвующий в адаптации, встраивании новых спейсеров. Рядом с Cas генами находится последовательность, частично комплементарная прямым повторам, складывающаяся в характерную вторичную структуру, - предполагаемая скаутная РНК (scoutRNA) (Фиг. 1) Однако экспериментально функционирование скаутной РНК в KbCas12d показано не было.
Воссоздание ДНК-разрезающего комплекса KbCas12d in vitro
Проведенный ранее биоинформатический анализ (Harrington et al., 2020) предсказал предположительные последовательности крРНК и scout РНК для системы CRISPR-KbCas12d (Таблица 1).
Таблица 1. Последовательности направляющих РНК системы CRISPR-KbCas12d используемые для первичных тестирований активности нуклеазы KbCas12d in vitro. Жирным шрифтом обозначена последовательность прямого повтора DR.
SEQ ID NO: 2
(SEQ ID NO: 3)
Для проверки активности KbCas12d нуклеазы были проведены эксперименты по воссозданию реакции разрезания ДНК in vitro. Для этого необходимо было получить все компоненты эффекторного комплекса KbCas12d: направляющие РНК и нуклеазу в рекомбинантной форме. Определение последовательности направляющих РНК позволило синтезировать in vitro молекулы крРНК и scout РНК. Синтез осуществляли с помощью набора NEB HiScribe T7 RNA synthesis.
В качестве ДНК-мишени использовался линейный ДНК фрагмент длиной 916 п.н. (SEQ ID NO: 4). ДНК-мишень несла целевой сайт 5' GTCATTGGCAGCTACAGG 3', фланкированный с 5' конца PAM последовательностью 5' TA 3'
Для разрезания этой мишени использовали направляющие РНК следующей последовательности: scout РНК (SEQ ID NO: 2; 5' aaguaucaaaauaaaaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuuc -3') и крРНК (SEQ ID NO: 5; 5' cuccgaaaguaucggggauaaaggcGUCAUUGGCAGCUACAGG 3'). Жирным шрифтом выделена последовательность крРНК, комплементарная протоспейсеру (целевой ДНК последовательности).
Для получения рекомбинантного белка KbCas12d его ген был клонирован в плазмиду pET21a вместе с последовательностью, кодирующей MBP (Maltose Binding Protein). В качестве кодирующей ген ДНК, использовалась ДНК с последовательностью, совпадающей с соответствующим геном в геноме бактерии-хозяина Katanobacteria. Клетки Esherichia coli Rosetta были трансформированы полученной плазмидой pET21a-MBP-KbCas12d-6xHis.
5 мл ночной культуры разводили в 500 мл среды LB, и растили клетки при температуре 37°С до достижения оптической плотности 0.6 отн.ед. Синтез целевого белка индуцировали добавлением ИПТГ до концентрации 1 мМ, после чего клетки инкубировали при температуре 16°C в течение 16 часов. Затем проводили центрифугирование клеток на скорости 5000 g в течение 30 минут, полученные осадки клеток замораживали при температуре -20°С.
Осадки размораживали на льду в течение 30 минут, ресуспензировали в 15 мл лизисного буфера (HEPES 50mM pH 7,5 (24°C), 10% глицерина, 500 мМ NaCl, имидазол 10 мМ, 1 мМ бета-меркаптоэтанол) с добавлением 15 мг лизоцима и снова инкубировали на льду в течение 30 минут. Затем клетки разрушали воздействием ультразвука в течение 30 минут и центрифугировали в течение 40 минут на скорости 16000 g. Полученный супернатант пропускали через фильтр 0,2 мкм и наносили на колонку HisTrap HP 1 mL (GE Healthcare) на скорости 0.4 мл/мин.
Хроматографию проводили при помощи FPLC хроматографа AKTA (GE Healthcare) на скорости 0.4 мл/мин. Колонку с нанесенным белком промывали 20 мл лизисного буфера с добавлением 30 мМ имидазола, после чего белок смывали лизисным буфером с добавлением 300 мМ имидазола.
Далее белок концентрировали с помощью концентратора Аmicon (с фильтром на 30 кДа) до объема 500мкл в буфере, не содержащем имидазол. Затем к белку добавляли TEV протеазу (30 мкл TEV из стока концентрацией 2 мг/мл)), чтобы отрезать MBP таг и инкубировали ночь на +4°С.
Затем, фракцию белка, полученную в ходе афинной хроматографии, пропускали через гель-фильтрационную колонку Superdex 200 Increase 10/300 GL (GE Healthcare) уравновешенную буфером, содержащим 50 мМ Hepes-HCl pH=7.5, 500 мМ NaCl, 1 мМ DTT и 10% глицерина. При помощи концентратора Аmicon (с фильтром на 30 кДа) фракции, соответствующие мономерной форме белка KbCas12d, сконцентрировали до 1 мг/мл, после чего очищенный белок хранили при температуре -80 оС в буфере, содержащем 10% глицерин.
In vitro реакцию разрезания линейной ДНК проводили в объёме 20 мкл в следующих условиях. Реакционная смесь состояла из: 1X CutSmart буфера (NEB), 20 нМ ДНК, 4 мкМ scout РНК/крРНК, 400 нМ белка KbCas12d. В качестве контроля аналогичным образом были приготовлены пробы, не содержащие scoutРНК. Пробы инкубировали при температуре 37°С и анализировали методом гель-электрофореза в 1.5 % агарозном геле. В случае правильного узнавания и специфического разрезания ДНК белком KbCas12d должны формироваться два фрагмента ДНК длиной порядка 670 и 246 пар оснований (см. Фиг. 2).
Результаты опыта показали, что воссозданный KbCas12d-крРНК-scoutРНК комплекс активен in vitro и разрезает ДНК, несущую целевую протоспейсерную последовательность.
Градиент температур (Фиг. 3) показал, что белок активен в диапазоне температур 25°С до 45°С. В дальнейшем в работе в качестве рабочей использовалась температура 37°С.
Дизайн гибридной РНК, полученной путем слияния направляющей CRISPR РНК и scout РНК.
Далее проводился подбор оптимальной последовательности скаутной РНК. Для этого in vitro был синтезирован ряд форм скаутной РНК, отличающихся длиной и вторичной структурой (Фиг.4):
Scout РНК 1 aaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuu (SEQ ID NO: 6);
Scout РНК 2 caaaauaaaaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuuccugauuuug (SEQ ID NO: 7);
Scout РНК 3
aaguaucaaaauaaaaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuuccugauuuuguugauaau
(SEQ ID NO: 8);
Scout РНК 4 aaguaucaaaauaaaaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuuc (SEQ ID NO: 9);
Scout РНК 5 auuaucaacaaaaucaggaaaggguggaaggcuaaaguuuaguuaaaaacuggaaacccuuu (SEQ ID NO: 10).
С синтезированными скаутными РНК, крРНК последовательностью SEQ ID NO: 5 и рекомбинатным белком KbCas12d проводилась реакция разрезания ДНК мишени (SEQ ID NO: 4) в объёме 20 мкл на 37°С в течение 30 минут (1X CutSmart буфера (NEB), 20 нМ ДНК, 4 мкМ scoutРНК/крРНК, 400 нМ белка KbCas12d) (Фиг.4)
Результаты эксперимента показали, что оптимальной для разрезания ДНК является scout РНК1 (aaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuu, SEQ ID NO: 6) - самая короткая форма молекулы. В комплексе с ней и крРНК белок KbCas12d активно разрезает двунитевой ДНК фрагмент, несущий протоспейсерную последовательность, фланкированную PAM 5' TA 3'.
Эта молекула была использована как основа для дизайна гибридной РНК - молекулы слияния направляющей крРНК и scout РНК (sgRNA, single guide RNA, sgРНК) (Фиг 5). Для создания sgРНК 3'-конец скаутной РНК был соединен с последовательностью крРНК через линкеры последовательностью “GAAA”. Были проверены sgРНК c различной длиной линкера (Фиг.5):
sgРНК 1 (SEQ ID NO: 11) (aaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuugaaacuccgaaaguaucggggauaaaggcAUCAAUACCAAACUCUGG)
sgРНК 2 (SEQ ID NO: 12)
(aaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuugaaagaaaсuccgaaaguaucggggauaaaggcAUCAAUACCAAACUCUGG)
sgРНК 3 (SEQ ID NO: 13)
(aaaggguuuccaguuuuuaacuaaacuuuagccuuccacccuuugaaagaaagaaaсuccgaaaguaucggggauaaaggcAUCAAUACCAAACUCUGG)
Последовательность sgРНК, узнающая протоспейсер выделена жирным шрифтом; последовательность линкера выделена жирным шрифтом-курсивом.
Белок KbCas12d в комплексе с sgРНК 1, sgРНК 2 или sgРНК 3 инкубировался с ДНК мишенью, использованной в вышеописанных экспериментах, в течение 30 минут на 37°С. Продукты реакции наносили на агарозный электрофорез для оценки эффективности разрезания ДНК фрагмента. В качестве положительного контроля использовалась реакционная смесь, где вместо sgРНК использовался комплекс крРНК и скаутной РНК.
Результат эксперимента показал, что использование sgРНК 1, sgРНК 2 приводят к эффективному разрезанию ДНК мишени, наравне с набором из направляющих РНК двух видов - скаутной и крРНК. Таким образом, удалось найти форму гибридной РНК (sgРНК) для эффективного разрезания ДНК белком KbCas12d. Эти варианты гибридной РНК могут быть использованы для разрезания любой другой целевой ДНК при изменении последовательности, непосредственно спаривающейся с ДНК -мишенью.
Подобная схема соединения скаутной РНК и крРНК может быть использована для любого представителя семейства Cas12d.
Нижеследующие примеры осуществления способа приведены в целях раскрытия характеристик настоящего изобретения и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения.
Пример 1. Тестирование активности белка KbCas12d в разрезании различных ДНК мишеней.
Для того, чтобы проверить способность KbCas12d узнавать различные последовательности ДНК, фланкированные PAM последовательностью, были проведены эксперименты по in vitro разрезанию ДНК-мишеней из последовательности гена grin2b человека (см. Таблицу 2). В качестве целевой ДНК использовался ПЦР-продукт, совпадающий по последовательности с фрагментом гена человека grin2b: SEQ ID NO: 14.
В качестве отрицательного контроля проводилось разрезание ДНК-мишени, фланкированной с 5' -конца нуклеотидами, отличающимися от PAM “TA”.
В реакции разрезания в качестве мишени использовался ПЦР фрагмент гена grin2b, несущий сайты узнавания (Таблица 2), предположительно распознаваемые KbCas12d. Для узнавания этих последовательностей были синтезированы крРНК, направляющие KbCas12d на данные сайты.
Таблица 2. ДНК-мишени гена grin2b человека.
Реакции разрезания проводились в подобранных для KbCas12d условиях, результат представлен на Фиг. 6. Из Фиг. 6 видно, что фермент KbCas12d успешно разрезал все мишени с подходящим PAM и не внес разрывы в мишень 7, используемую в качестве отрицательного контроля.
Таким образом, проведенные исследовательские испытания позволили восстановить активность системы CRISPR-KdCas12d из Katanobacteria in vitro, создать на ее основе средство разрезания ДНК, состоящее из нуклеазы KbCas12d гибридной РНК, полученной путем слияния направляющей CRISPR РНК и scout РНК.
Пример 3. Белки Cas12d из близкородственных организмов, относящихся к Katanobacteria.
На сегодняшний день для систем CRISPR-Cas12d семейства только активность системы CRISPR-Cas12d15 из бактерий-симбионтов термитов была реконструирована in vitro (Harrington et al., 2020). Cas12d15 имеет сравнимый с KbCas12d размер и идентичен KbCas12d на 27 % (Фиг. 7, степень идентичности была рассчитана по программе BLASTp, default parameters).
Таким образом, белок KbCas12d существенно отличается по аминокислотной последовательности от других Cas12d белков, чья активность восстановлена in vitro. KbCas12d - это первый Cas12d белок для которого показана возможность создания гибридной sgРНК.
Специалисту в области генетической инженерии очевидно, что полученный и охарактеризованный в данном Описании вариант последовательности белка KbCas12d может быть изменен без изменения функции самого белка (например, направленным мутагенезом аминокислотных остатков, напрямую не влияющих на функциональную активность (Sambrook et al., 1989). В частности, специалисту известно, что могут быть изменены неконсервативные аминокислотные остатки, не затрагивающие остатки, определяющие функциональность белка (определяющие его функцию или структуру). Примерами таких изменений могут служить замены неконсервативных аминокислотных остатков на гомологичные. В некоторых вариантах осуществления изобретения возможно использование белка, содержащего аминокислотную последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1 и имеет отличия по сравнению с SEQ ID NO: 1 только в неконсервативных аминокислотных остатках, для образования двунитевого разрыва в молекуле ДНК, расположенного непосредственно после нуклеотидной последовательности 5'-TA-3' в указанной молекуле ДНК. Гомологичные белки могут быть получены путем мутагенеза (например, сайт-направленного или ПЦР-опосредуемого мутагенеза) соответствующих молекул нуклеиновых кислот с последующим тестированием кодируемого модифицированного белка Cas12d на сохранение его функций в соответствии с описанными здесь функциональными анализами.
Пример 3. Описанная в настоящем изобретении система KbCas12d в комплексе с направляющими РНК или разработанной в настоящем изобретении гибридной РНК может быть использована для изменения последовательности геномной ДНК многоклеточного организма, в том числе эукариотического. Для введения система KbCas12d в комплексе с направляющими РНК/ sgРНК в клетки этого организма (во все клетки или в часть клеток) могут быть применены различные подходы, известные специалистам. Например, методы доставки CRISPR-Cas систем в клетки организмов раскрыты в источниках (Liu et al., 2017; Lino et al., 2018) и в источниках, раскрытых внутри этих источников.
Для эффективной экспрессии нуклеазы KbCas12d в эукариотических клетках будет желательно провести оптимизацию кодонов для аминокислотной последовательности белка KbCas12d методами, известными специалистам (например, IDT codon optimization tool).
Для эффективной работы нуклеазы KbCas12d в эукариотических клетках необходимо обеспечить импорт этого белка внутрь ядра эукариотической клетки. Для этого можно использовать сигнал ядерной локализации из Т-антигена вируса SV40 (Lanford et al., Cell, 1986, 46: 575-582), соединённый с последовательностью KbCas12d с помощью спейсерной последовательности, описанной в Shen et al., 2013 или без нее. Таким образом, полная аминокислотная последовательность нуклеазы, транспортируемой внутрь ядра эукариотической клетки, будет представлять собой следующую последовательность: MAPKKKRKVGIHGVPAA-KbCas12d-KRPAATKKAGQAKKKK (SEQ ID NO: 15 - KbCas12d - SEQ ID NO: 16; далее KbCas12d NLS). Для доставки белка с приведенной выше аминокислотной последовательностью, могут быть использованы по меньшей мере два подхода.
Доставка в виде гена осуществляется путем создания плазмиды, несущей ген KbCas12d NLS под регуляцией промотора (например, CMV промотора) и последовательности, кодирующей направляющие РНК под регуляцией U6 промотора. В качестве ДНК- мишеней используются ДНК последовательности, фланкированные 5'-TA -3', например, последовательности гена grin2b человека:
5'- AAATAGGATCTACATCAC -3'
Таким образом, кассета для экспрессии sgРНК (SEQ ID NO: 17) выглядит следующим образом:
Жирным шрифтом выделена последовательность U6 промотора, строчными буквами - последовательность, образующая структуру sgRNA, а прописными буквами последовательность, необходимая для узнавания целевой ДНК.
Плазмидную ДНК очищают и трансфицируют в клетки человека HEK293 c помощью реагента Lipofectamine 2000 (Thermo Fisher Scientific). Клетки инкубируют в течение 72 часов, после чего из них выделяется геномная ДНК с помощью колонок для очистки геномной ДНК (Thermo Fisher Scientific). Целевой ДНК сайт анализируется с помощью секвенирования на платформе Illumina с целью определения числа вставок-делеций в ДНК, происходящих в целевом сайте по причине направленного двунитевого разрыва и последующей его репарации.
Для амплификации целевых фрагментов используют праймеры, фланкирующие предположительное место внесения разрыва.
После амплификации пробы готовятся по протоколу реагента Ultra II DNA Library Prep Kit for Illumina (NEB) для подготовки образцов к высокопроизводительному секвенированию. Затем проводится секвенирование на платформе Illumina 300cycles, прямое прочтение. Результаты секвенирования анализируются биоинформатическими методами. В качестве детекции разрезания принимается вставка или делеция нескольких нуклеотидов в целевой последовательности ДНК.
Доставка в виде рибонуклеинового комплекса осуществляется путем инкубации рекомбинантной формы KbCas12d NLS c направляющими РНК в CutSmart буфере (NEB). Рекомбинантный белок получают из бактериальных клеток-продуцентов, очищая его с помощью аффинной хроматографии (NiNTA, Qiagen) разделением по размеру (Superdex 200).
Белок смешивают с РНК в соотношении 1:2 (KbCas9 NLS : sgRNA), инкубируют в течение 10 минут на комнатной температуре, затем смесь трансфицируют в клетки.
Далее проводится анализ экстрагированной из них ДНК на предмет вставок-делеций в целевом ДНК сайте (как описано выше).
Охарактеризованная в настоящем изобретении нуклеаза KbCas12d из Katanobacteria имеет ряд преимуществ относительно ранее охарактеризованных Cas белков.
На сегодняшний день KbCas12d - это второй Cas12d белок, активность которого удалось восстановить in vitro и первый Cas12d белок, для которого была создана гибридная sgРНК. Использование sgРНК вместо комплекса из крРНК и скаутной РНК упрощает систему и является преимуществом в сравнении с Cas12d15 из бактерий симбионтов термитов.
KbCas12d имеет малый размер белка (1125 а.о.), что также является преимуществом по сравнению с другими Cas нуклеазами. Эффективное разрезание ДНК в широком диапазоне температур делает KbCas12d применимым как в клетках рыб и растений, так и в клетках теплокровных животных. Таким образом, KbCas12d в комплексе с созданной гибридной РНК может стать основой нового инструмента геномного редактирования.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные случаи приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть, понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
Список литературы
Altschul S. F. et al., Basic local alignment search tool // J. Mol. Biol., 1990, Oct, 215 (3): 403-410.
Brouns S. J. J. et al., Small CRISPR RNAs guide antiviral defense in prokaryotes // Science, 2008, 321 (5891): 960-964.
Harrington L. B. et al. A scoutRNA is required for some type V CRISPR-Cas systems // Mol. Cell, 2020, Aug, 79 (3): 416-424.
Jansen R. et al., Identification of genes that are associated with DNA repeats in prokaryotes // Molecular microbiology, 2002, Mar, 43 (6): 1565-1575.
Lino C. A. et al., Delivering CRISPR: a review of the challenges and approaches // Drug Deliv., 2018, Nov, 25 (1): 1234-1257.
Liu C. et al., Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications // J Control Release. 2017, Nov, 28 (266): 17-26.
Mojica F. J. M. et al., Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements // Journal of molecular evolution. 2005, 60 (2): 174-182.
Sambrook J. et al., Molecular Cloning: A Laboratory Manual, 1989, CSH Press, pp. 15.3-15.108.
Shen B., et al., Generation of gene-modified mice via Cas9/RNA-mediated gene targeting // Cell Res. 2013 May, 23 (5): 720-3.
--->
<110> Федеральное государственное бюджетное учреждение науки Институт
биологии гена Российской академии наук (Institute of Gene Biology Russian
Academy of Sciences)
<120> Средство разрезания двунитевой ДНК с помощью Cas12d белка из
Katanobacteria и гибридной РНК, полученной путем слияния направляющей
CRISPR РНК и scout РНК
<160> 17
<210> 1
<211> 1125
<212> PRT
<213> Katanobateria
<400> 1
Met Arg Lys Lys Leu Phe Lys Gly Tyr Ile Leu His Asn Lys Arg Leu 16
5 10 15
Val Tyr Thr Gly Lys Ala Ala Ile Arg Ser Ile Lys Tyr Pro Leu Val 32
20 25 30
Ala Pro Asn Lys Thr Ala Leu Asn Asn Leu Ser Glu Lys Ile Ile Tyr 48
35 30 45
Asp Tyr Glu His Leu Phe Gly Pro Leu Asn Val Ala Ser Tyr Ala Arg 64
50 55 60
Asn Ser Asn Arg Tyr Ser Leu Val Asp Phe Trp Ile Asp Ser Leu Arg 80
65 70 75 80
Ala Gly Val Ile Trp Gln Ser Lys Ser Thr Ser Leu Ile Asp Leu Ile 96
85 90 95
Ser Lys Leu Glu Gly Ser Lys Ser Pro Ser Glu Lys Ile Phe Glu Gln 112
100 105 110
Ile Asp Phe Glu Leu Lys Asn Lys Leu Asp Lys Glu Gln Phe Lys Asp 128
115 120 125
Ile Ile Leu Leu Asn Thr Gly Ile Arg Ser Ser Ser Asn Val Arg Ser 144
130 135 140
Leu Arg Gly Arg Phe Leu Lys Cys Phe Lys Glu Glu Phe Arg Asp Thr 160
145 150 155 160
Glu Glu Val Ile Ala Cys Val Asp Lys Trp Ser Lys Asp Leu Ile Val 176
165 170 175
Glu Gly Lys Ser Ile Leu Val Ser Lys Gln Phe Leu Tyr Trp Glu Glu 192
180 185 190
Glu Phe Gly Ile Lys Ile Phe Pro His Phe Lys Asp Asn His Asp Leu 208
195 200 205
Pro Lys Leu Thr Phe Phe Val Glu Pro Ser Leu Glu Phe Ser Pro His 224
210 215 220
Leu Pro Leu Ala Asn Cys Leu Glu Arg Leu Lys Lys Phe Asp Ile Ser 240
225 230 235 240
Arg Glu Ser Leu Leu Gly Leu Asp Asn Asn Phe Ser Ala Phe Ser Asn 256
245 250 255
Tyr Phe Asn Glu Leu Phe Asn Leu Leu Ser Arg Gly Glu Ile Lys Lys 272
260 265 270
Ile Val Thr Ala Val Leu Ala Val Ser Lys Ser Trp Glu Asn Glu Pro 288
275 280 285
Glu Leu Glu Lys Arg Leu His Phe Leu Ser Glu Lys Ala Lys Leu Leu 304
290 295 300
Gly Tyr Pro Lys Leu Thr Ser Ser Trp Ala Asp Tyr Arg Met Ile Ile 320
305 310 315 320
Gly Gly Lys Ile Lys Ser Trp His Ser Asn Tyr Thr Glu Gln Leu Ile 336
325 330 335
Lys Val Arg Glu Asp Leu Lys Lys His Gln Ile Ala Leu Asp Lys Leu 352
340 345 350
Gln Glu Asp Leu Lys Lys Val Val Asp Ser Ser Leu Arg Glu Gln Ile 368
355 360 365
Glu Ala Gln Arg Glu Ala Leu Leu Pro Leu Leu Asp Thr Met Leu Lys 384
370 375 380
Glu Lys Asp Phe Ser Asp Asp Leu Glu Leu Tyr Arg Phe Ile Leu Ser 400
385 390 395 400
Asp Phe Lys Ser Leu Leu Asn Gly Ser Tyr Gln Arg Tyr Ile Gln Thr 416
405 410 415
Glu Glu Glu Arg Lys Glu Asp Arg Asp Val Thr Lys Lys Tyr Lys Asp 432
420 425 430
Leu Tyr Ser Asn Leu Arg Asn Ile Pro Arg Phe Phe Gly Glu Ser Lys 448
435 440 445
Lys Glu Gln Phe Asn Lys Phe Ile Asn Lys Ser Leu Pro Thr Ile Asp 464
450 455 460
Val Gly Leu Lys Ile Leu Glu Asp Ile Arg Asn Ala Leu Glu Thr Val 480
465 470 475 480
Ser Val Arg Lys Pro Pro Ser Ile Thr Glu Glu Tyr Val Thr Lys Gln 496
485 490 495
Leu Glu Lys Leu Ser Arg Lys Tyr Lys Ile Asn Ala Phe Asn Ser Asn 512
500 505 510
Arg Phe Lys Gln Ile Thr Glu Gln Val Leu Arg Lys Tyr Asn Asn Gly 528
515 520 525
Glu Leu Pro Lys Ile Ser Glu Val Phe Tyr Arg Tyr Pro Arg Glu Ser 544
530 535 540
His Val Ala Ile Arg Ile Leu Pro Val Lys Ile Ser Asn Pro Arg Lys 560
545 550 555 560
Asp Ile Ser Tyr Leu Leu Asp Lys Tyr Gln Ile Ser Pro Asp Trp Lys 576
565 570 575
Asn Ser Asn Pro Gly Glu Val Val Asp Leu Ile Glu Ile Tyr Lys Leu 592
580 585 590
Thr Leu Gly Trp Leu Leu Ser Cys Asn Lys Asp Phe Ser Met Asp Phe 608
595 600 605
Ser Ser Tyr Asp Leu Lys Leu Phe Pro Glu Ala Ala Ser Leu Ile Lys 624
610 615 620
Asn Phe Gly Ser Cys Leu Ser Gly Tyr Tyr Leu Ser Lys Met Ile Phe 640
625 630 635 640
Asn Cys Ile Thr Ser Glu Ile Lys Gly Met Ile Thr Leu Tyr Thr Arg 656
645 650 655
Asp Lys Phe Val Val Arg Tyr Val Thr Gln Met Ile Gly Ser Asn Gln 672
660 665 670
Lys Phe Pro Leu Leu Cys Leu Val Gly Glu Lys Gln Thr Lys Asn Phe 688
675 680 685
Ser Arg Asn Trp Gly Val Leu Ile Glu Glu Lys Gly Asp Leu Gly Glu 704
690 695 700
Glu Lys Asn Gln Glu Lys Cys Leu Ile Phe Lys Asp Lys Thr Asp Phe 720
705 710 715 720
Ala Lys Ala Lys Glu Val Glu Ile Phe Lys Asn Asn Ile Trp Arg Ile 736
725 730 735
Arg Thr Ser Lys Tyr Gln Ile Gln Phe Leu Asn Arg Leu Phe Lys Lys 752
740 745 750
Thr Lys Glu Trp Asp Leu Met Asn Leu Val Leu Ser Glu Pro Ser Leu 768
755 760 765
Val Leu Glu Glu Glu Trp Gly Val Ser Trp Asp Lys Asp Lys Leu Leu 784
770 775 780
Pro Leu Leu Lys Lys Glu Lys Ser Cys Glu Glu Arg Leu Tyr Tyr Ser 800
785 790 795 800
Leu Pro Leu Asn Leu Val Pro Ala Thr Asp Tyr Lys Glu Gln Ser Ala 816
805 810 815
Glu Ile Glu Gln Arg Asn Thr Tyr Leu Gly Leu Asp Val Gly Glu Phe 832
820 825 830
Gly Val Ala Tyr Ala Val Val Arg Ile Val Arg Asp Arg Ile Glu Leu 848
835 840 845
Leu Ser Trp Gly Phe Leu Lys Asp Pro Ala Leu Arg Lys Ile Arg Glu 864
850 855 860
Arg Val Gln Asp Met Lys Lys Lys Gln Val Met Ala Val Phe Ser Ser 880
865 870 875 880
Ser Ser Thr Ala Val Ala Arg Val Arg Glu Met Ala Ile His Ser Leu 896
885 890 895
Arg Asn Gln Ile His Ser Ile Ala Leu Ala Tyr Lys Ala Lys Ile Ile 912
900 905 910
Tyr Glu Ile Ser Ile Ser Asn Phe Glu Thr Gly Gly Asn Arg Met Ala 928
915 920 925
Lys Ile Tyr Arg Ser Ile Lys Val Ser Asp Val Tyr Arg Glu Ser Gly 944
930 935 940
Ala Asp Thr Leu Val Ser Glu Met Ile Trp Gly Lys Lys Asn Lys Gln 960
945 950 955 960
Met Gly Asn His Ile Ser Ser Tyr Ala Thr Ser Tyr Thr Cys Cys Asn 976
965 970 975
Cys Ala Arg Thr Pro Phe Glu Leu Val Ile Asp Asn Asp Lys Glu Tyr 992
980 985 990
Glu Lys Gly Gly Asp Glu Phe Ile Phe Asn Val Gly Asp Glu Lys Lys 1008
995 1000 1005
Val Arg Gly Phe Leu Gln Lys Ser Leu Leu Gly Lys Thr Ile Lys Gly 1024
1010 1015 1020
Lys Glu Val Leu Lys Ser Ile Lys Glu Tyr Ala Arg Pro Pro Ile Arg 1040
1025 1030 1035 1040
Glu Val Leu Leu Glu Gly Glu Asp Val Glu Gln Leu Leu Lys Arg Arg 1056
1045 1050 1055
Gly Asn Ser Tyr Ile Tyr Arg Cys Pro Phe Cys Gly Tyr Lys Thr Asp 1072
1060 1065 1070
Ala Asp Ile Gln Ala Ala Leu Asn Ile Ala Cys Arg Gly Tyr Ile Ser 1088
1075 1080 1085
Asp Asn Ala Lys Asp Ala Val Lys Glu Gly Glu Arg Lys Leu Asp Tyr 1104
1090 1095 1100
Ile Leu Glu Val Arg Lys Leu Trp Glu Lys Asn Gly Ala Val Leu Arg 1120
1105 1110 1115 1120
Ser Ala Lys Phe Leu 1125
1125
<210> 2
<211> 59
<212> РНК
<213> artificial sequence
<220>
<223> KbCas12d scout РНК
<400> 2
aaguaucaaa auaaaaaggg uuuccaguuu uuaacuaaac uuuagccuuc cacccuuuc 59
<210> 3
<211> 44
<212> РНК
<213> artificial sequence
<220>
<223> KbCas12d крРНК, где n - любой нуклеотид
<400> 3
acuccgaaag uaucggggau aaaggcnnnn nnnnnnnnnn nnnn 44
<210> 4
<211> 916
<212> ДНК
<213> artificial sequence
<220>
<223> линейный ДНК фрагмент
<400> 4
ccctgcaaac acaaagaaag agcatgttaa aataggatct acatcacgta acctgtctta 60
gaagaggcta gatactgcaa ttcaaggacc ttatctcctt tcattgagca ccaaacccaa 120
ctccatctac cagcctactc tcttatctct ggtatttgct ctgcagaatg agagaaaatg 180
aaactttcaa aagcctcaga aatccttgaa caaggcaata aaaggtgcta ttgctatagt 240
cattggcagc tacaggcaga gacaaaggag gaaaagaggt tgtgagtggt ccaggtagcc 300
atgcgagtat gcatacacaa atctcctggc cctcctgtta cagcccaccc ttgtactgtt 360
cttgggctga aggaaagcaa ggccagacaa agtgagcaga aaaacgtgct cagcagaggt 420
gagcaacaga acatgggctg gataaactgg atgtgggggg ctataagtac acaagccctg 480
cattcttgct gccttcacct tatgtttgcc tcaatgagga caacagccag aaaattcttt 540
agtaaccttg ttagtatctg gctcttaata ttaaactaca agaacaactg atacatgact 600
agtagtttta aaacattgcc tcaattgatc cttacaatga cccagtaggg aataaaataa 660
gaaaaacatt attatcacca tttttataaa tgttgacgcc aaggctcaga gaagctaagt 720
gttctaagac catgaaccaa tcaattaagt gaaacaaacc tgaacccaga tcttctgact 780
tttgtattcc aatatgtgtt ctattacact acgtggaact gcctctcata taaccaatta 840
ttataaggaa cacatattac tccaatctat ttatacacca aaatacagaa acttaaacaa 900
ataccattag cagctg 916
<210> 5
<211> 43
<212> РНК
<213> artificial sequence
<220>
<223> крРНК
<400> 5
cuccgaaagu aucggggaua aaggcgucau uggcagcuac agg 43
<210> 6
<211> 44
<212> РНК
<213> artificial sequence
<220>
<223> scout РНК
<400> 6
aaaggguuuc caguuuuuaa cuaaacuuua gccuuccacc cuuu 44
<210> 7
<211> 62
<212> РНК
<213> artificial sequence
<220>
<223> scout РНК
<400> 7
caaaauaaaa aggguuucca guuuuuaacu aaacuuuagc cuuccacccu uuccugauuu 60
ug 62
<210> 8
<211> 76
<212> РНК
<213> artificial sequence
<220>
<223> scout РНК
<400> 8
aaguaucaaa auaaaaaggg uuuccaguuu uuaacuaaac uuuagccuuc cacccuuucc 60
ugauuuuguu gauaau 76
<210> 9
<211> 59
<212> РНК
<213> artificial sequence
<220>
<223> scout РНК
<400> 9
aaguaucaaa auaaaaaggg uuuccaguuu uuaacuaaac uuuagccuuc cacccuuuc 59
<210> 10
<211> 62
<212> РНК
<213> artificial sequence
<220>
<223> scout РНК
<400> 10
auuaucaaca aaaucaggaa aggguggaag gcuaaaguuu aguuaaaaac uggaaacccu 60
uu 62
<210> 11
<211> 91
<212> РНК
<213> artificial sequence
<220>
<223> sgРНК
<400> 11
aaaggguuuc caguuuuuaa cuaaacuuua gccuuccacc cuuugaaacu ccgaaaguau 60
cggggauaaa ggcaucaaua ccaaacucug g 91
<210> 12
<211> 95
<212> РНК
<213> artificial sequence
<220>
<223> sgРНК, участки спейсерной последовательности (с 78 по 95 п.н.
последовательность aucaauaccaaacucugg) и линкера (с 45 по 52 п.н.
последовательность gaaagaaa) могут быть вариабельными
<400> 12
aaaggguuuc caguuuuuaa cuaaacuuua gccuuccacc cuuugaaaga aacuccgaaa 60
guaucgggga uaaaggcauc aauaccaaac ucugg 95
<210> 13
<211> 99
<212> РНК
<213> artificial sequence
<220>
<223> sgРНК
<400> 13
aaaggguuuc caguuuuuaa cuaaacuuua gccuuccacc cuuugaaaga aagaaacucc 60
gaaaguaucg gggauaaagg caucaauacc aaacucugg 99
<210> 14
<211> 1612
<212> ДНК
<213> H.sapiens
<223> фрагмент гена grin2b
<400> 14
gagagagatg gccaaggctt atattctata gagcattatg tccttagttt gatgcataga 60
ataagattta gggtcatatg tggaagtaaa aaggaaggag ttctttgtag gtaaaaggtg 120
gcaaattata tgaaaatacg gtatcagtca ttttagggaa gtcacgacta taggatggca 180
tcagaccttt tattgccttg ttcagaaaaa aaaaggaaca tttttcaaat gtggctctaa 240
cattacttca gctgctaatg gtatttgttt aagtttctgt attttggtgt ataaatagat 300
tggagtaata tgtgttcctt ataataattg gttatatgag aggcagttcc acgtagtgta 360
atagaacaca tattggaata caaaagtcag aagatctggg ttcaggtttg tttcacttaa 420
ttgattggtt catggtctta gaacacttag cttctctgag ccttggcgtc aacatttata 480
aaaatggtga taataatgtt tttcttattt tattccctac tgggtcattg taaggatcaa 540
ttgaggcaat gttttaaaac tactagtcat gtatcagttg ttcttgtagt ttaatattaa 600
gagccagata ctaacaaggt tactaaagaa ttttctggct gttgtcctca ttgaggcaaa 660
cataaggtga aggcagcaag aatgcagggc ttgtgtactt atagcccccc acatccagtt 720
tatccagccc atgttctgtt gctcacctct gctgagcacg tttttctgct cactttgtct 780
ggccttgctt tccttcagcc caagaacagt acaagggtgg gctgtaacag gagggccagg 840
agatttgtgt atgcatactc gcatggctac ctggaccact cacaacctct tttcctcctt 900
tgtctctgcc tgtagctgcc aatgactata gcaatagcac cttttattgc cttgttcaag 960
gatttctgag gcttttgaaa gtttcatttt ctctcattct gcagagcaaa taccagagat 1020
aagagagtag gctggtagat ggagttgggt ttggtgctca atgaaaggag ataaggtcct 1080
tgaattgcag tatctagcct cttctaagac aggttacgtg atgtagatcc tattttaaca 1140
tgctctttct ttgtgtttgc agggagtcga cgagttgaag atgaagccca gagcggagtg 1200
ctgttctccc aagttctggt tggtgttggc cgtcctggcc gtgtcaggca gcagagctcg 1260
ttctcagaag agccccccca gcattggcat tgctgtcatc ctcgtgggca cttccgacga 1320
ggtggccatc aaggatgccc acgagaaaga tgatttccac catctctccg tggtaccccg 1380
ggtggaactg gtagccatga atgagaccga cccaaagagc atcatcaccc gcatctgtga 1440
tctcatgtct gaccggaaga tccagggggt ggtgtttgct gatgacacag accaggaagc 1500
catcgcccag atcctcgatt tcatttcagc acagactctc acccccatcc tgggcatcca 1560
cgggggctcc tctatgataa tggcagataa ggtaaaaagg ggctgcaggg ag 1612
<210> 15
<211> 17
<212> PRT
<213> artificial sequence
<220>
<223> часть нуклеазы
<400> 15
Met Ala Pro Lys Lys Lys Arg Lys Val Gly Ile His Gly Val Pro Ala 16
1 5 10 15
Ala 17
<210> 16
<211> 16
<212> PRT
<213> artificial sequence
<220>
<223> часть нуклеазы
<400> 16
Lys Arg Pro Ala Ala Thr Lys Lys Ala Gly Gln Ala Lys Lys Lys Lys 16
<210> 17
<211>
<212> ДНК
<213> artificial sequence
<220>
<223> кассета для экспрессии sgРНК
<400> 17
gagggcctat ttcccatgat tccttcatat ttgcatatac gatacaaggc tgttagagag 60
ataattggaa ttaatttgac tgtaaacaca aagatattag tacaaaatac gtgacgtaga 120
aagtaataat ttcttgggta gtttgcagtt ttaaaattat gttttaaaat ggactatcat 180
atgcttaccg taacttgaaa gtatttcgat ttcttggctt tatatatctt gtggaaagga 240
cgaaacaccg aaagggtttc cagtttttaa ctaaacttta gccttccacc ctttgaaaga 300
aactccgaaa gtatcgggga taaaggcaaa taggatctac atcac 345
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| Средство разрезания ДНК на основе Cas9 белка из бактерии Capnocytophaga ochracea | 2021 |
|
RU2778156C1 |
| СРЕДСТВО РАЗРЕЗАНИЯ ДНК НА ОСНОВЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ PASTEURELLA PNEUMOTROPICA | 2019 |
|
RU2722934C1 |
| СРЕДСТВО РАЗРЕЗАНИЯ ДНК НА ОСНОВЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ DEMEQUINA SEDIMINICOLA | 2019 |
|
RU2722933C1 |
| ПРИМЕНЕНИЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ PASTEURELLA PNEUMOTROPICA ДЛЯ МОДИФИКАЦИИ ГЕНОМНОЙ ДНК В КЛЕТКАХ | 2019 |
|
RU2724470C1 |
| СИСТЕМА РЕДАКТИРОВАНИЯ ГЕНОМА CRISPR/CAS9 II ТИПА И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2794774C1 |
| СПОСОБ СОЗДАНИЯ НОВЫХ МУТАЦИЙ В ОРГАНИЗМАХ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2833881C1 |
| ШТАММ ESCHERICHIA COLI BL21(DE3)PLYSS/PET15B-HISCPF1 - ПРОДУЦЕНТ РНК-НАПРАВЛЯЕМОЙ ЭНДОНУКЛЕАЗЫ CRISPR/CPF1 | 2021 |
|
RU2774120C1 |
| КОМПОЗИЦИИ И СПОСОБЫ РЕДАКТИРОВАНИЯ ГЕНОВ | 2019 |
|
RU2804665C2 |
| ЭКСПРЕССИРУЮЩИЕ CAS МЫШИНЫЕ ЭМБРИОНАЛЬНЫЕ СТВОЛОВЫЕ КЛЕТКИ И МЫШИ И ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2782358C2 |
| МЫШИ, СОДЕРЖАЩИЕ МУТАЦИИ, ВСЛЕДСТВИЕ КОТОРЫХ ЭКСПРЕССИРУЕТСЯ УКОРОЧЕННЫЙ НА С-КОНЦЕ ФИБРИЛЛИН-1 | 2017 |
|
RU2721125C1 |


Изобретение относится к области биотехнологии, в частности к гибридной РНК (sgРНК), используемой в качестве направляющей РНК в системе Cas12d для редактирования геномной ДНК, а также к ДНК-кассете для ее экспрессии. Изобретение эффективно для изменения последовательности геномной ДНК одноклеточного или многоклеточного организма. 3 н. и 3 з.п. ф-лы, 7 ил., 2 табл., 3 пр.
1. Гибридная РНК (sgРНК), используемая в качестве направляющей РНК в системе Cas12d для редактирования геномной ДНК, представляющая собой молекулу нуклеиновой кислоты, полученную путем слияния нуклеотидных последовательностей скаутной РНК (scout РНК) и крРНК (crРНК), общей формулой А-В-С-D, где А – последовательность KbCas12d (CRISPR-Cas12d система из Katanobacteria) scout РНК, B – последовательность линкера; C – последовательность прямого повтора DR KbCas12d крРНК, D – последовательность, комплементарная ДНК-мишени (спейсерный сегмент); при этом последовательность KbCas12d scout РНК характеризуется SEQ ID NO: 6, а последовательность линкера и спейсерного сегмента может быть любой.
2. Гибридная РНК по п.1, где последовательность прямого повтора DR KbCas12d крРНК является последовательностью прямого повтора последовательности SEQ ID NO: 3.
3. Гибридная РНК по п.1, представляющая собой последовательности SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13, но не ограничивающаяся ими.
4. ДНК-кассета для экспрессии гибридной РНК по п.1, состоящая из нуклеотидной последовательности U6 промотора, нуклеотидной последовательности, кодирующей гибридную РНК по п.1, и нуклеотидной последовательности, необходимой для узнавания целевой ДНК и фланкированной с 5’ конца PAM последовательностью 5’-TA -3’.
5. Способ изменения последовательности геномной ДНК одноклеточного или многоклеточного организма, включающий введение в по меньшей мере одну клетку этого организма эффективного количества:
а) белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, или нуклеиновой кислоты, кодирующей ген белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1;
б) гибридной РНК по п.1,
при этом взаимодействие указанного белка с гибридной РНК и нуклеотидной последовательностью 5’- TА -3’ приводит к образованию двунитевого разрыва в последовательности геномной ДНК, непосредственно примыкающей к последовательности 5’- TА -3’.
6. Способ по п.8, дополнительно включающий введение экзогенной последовательности ДНК одновременно с гибридной РНК.
| US 2020255858 A1, 13.08.2020 | |||
| WO 2018064352A1 А1, 05.04.2018 | |||
| LUCAS B | |||
| HARRINGTON et al., A scoutRNA Is Required for Some Type V CRISPR-Cas Systems, Molecular Cell, 2020, Vol.79, pp.1-9 | |||
| RUUD | |||
| JANSEN et al., Identification of genes that are associated with DNArepeats in prokaryotes, Molecular Microbiology, 2002, 43(6), pp.1565-1575 | |||
| ЛОМОВ Н | |||
| А | |||
| и |

