ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/675663, поданной 25 июля 2012 г., предварительной заявки на патент США №61/680099, поданной 6 августа 2012 г. и предварительной заявки на патент США №61/729294, поданной 21 ноября 2012 г., полное содержание которых включено в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
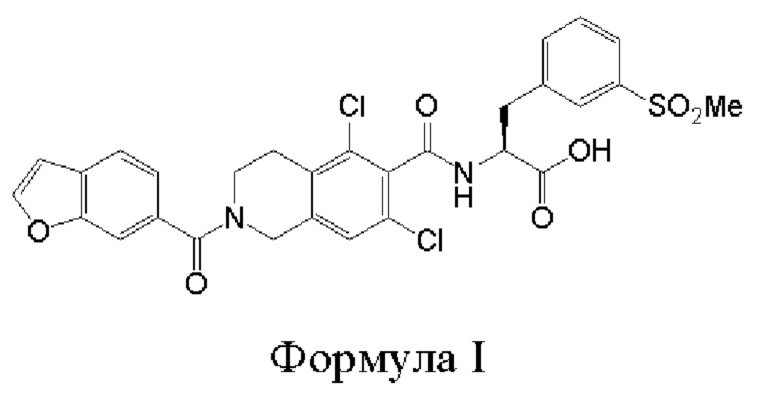
[0002] Было обнаружено, что соединение формулы I:
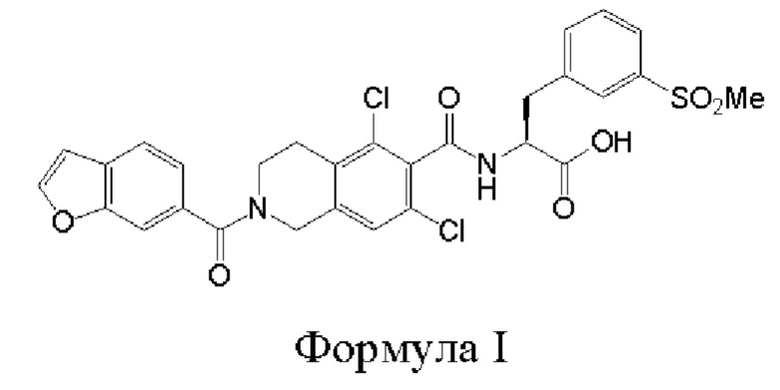
является эффективным ингибитором взаимодействий ассоциированного с функцией лимфоцитов антигена-1 (LFA-1) с семейством молекул межклеточной адгезии (IСAM) и обладает желаемыми фармакокинетическими свойствами, включая быстрый системный клиренс. Тем не менее, целесообразны усовершенствованные способы получения для обеспечения соединения формулы I с повышенной чистотой и/или со сниженным количеством используемых исходных веществ.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0003] Признаки, обеспечивающие новизну изобретения, изложены в прилагаемой формуле изобретения. Лучшее понимание признаков и преимуществ настоящего изобретения будет получено путем ссылки на последующее подробное описание с представленными в нем иллюстративными варианты реализации, в которых используются принципы настоящего изобретения, и прилагаемые графические материалы, на которых:
[0004] ФИГ. 1 представляет собой принципиальную схему, демонстрирующую соотношение между различными формами соединения формулы I.
[0005] ФИГ. 2 представляет собой принципиальную схему, демонстрирующую взаимные превращения между формами I, III и VI соединения формулы I.
[0006] ФИГ. 3 представляет собой диаграмму состояния трехфазной системы соединения формулы I в системе водного раствора ацетона.
[0007] ФИГ. 4 представляет собой графическое отображение дифрактограммы рентгеновской порошковой дифракции кристаллической формы II.
[0008] ФИГ. 5 представляет собой графическое отображение оптической микрофотографии кристаллической формы II.
[0009] ФИГ. 6 представляет собой графическое отображение спектра 1Н ЯМР кристаллической формы II.
[0010] ФИГ. 7 представляет собой графическое отображение термограммы дифференциальной сканирующей калориметрии (ДСК) кристаллической формы II.
[0011] ФИГ. 8 представляет собой графическое отображение термограммы термогравиметрического анализа (ТГА) кристаллической формы II.
[0012] ФИГ. 9 представляет собой графическое отображение кривой гравиметрической сорбции влаги для кристаллической формы II.
[0013] На ФИГ. 10 приведены сводные характеристики форм соединения формулы I.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0014] В первом аспекте настоящего изобретения предложены способы получения соединения формулы I:
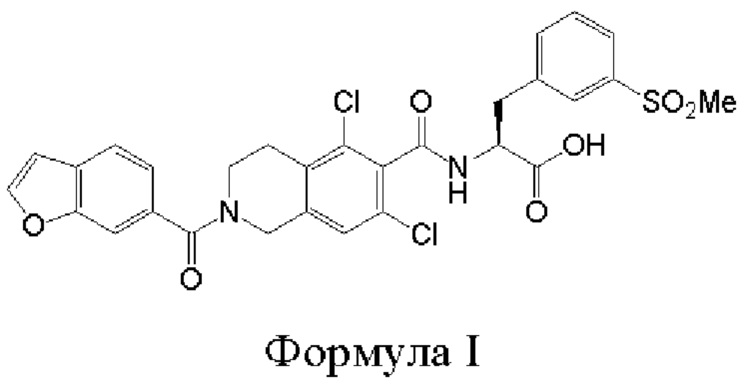
или его соли. Согласно настоящему изобретению, такие способы включают стадии проведения гидролиза сложноэфирного предшественника с основанием в двухфазной среде, где указанная сложноэфирная группа предшественника представляет собой углеродсодержащий фрагмент или силилсодержащий фрагмент; и b) выделения соединения формулы I или его соли. В различных вариантах реализации настоящего изобретения указанная двухфазная среда включает водный раствор ацетона, такой как 30%-й водный раствор ацетона. В различных вариантах реализации настоящего изобретения указанная двухфазная среда меняется со временем, таким образом, что в ходе реакции реакционная смесь, являющаяся двухфазной в начале реакции, становится менее двухфазной или однофазной.
[0015] В различных вариантах реализации настоящего изобретения основание для проведения гидролиза представляет собой гидроксид натрия, например, в количестве, находящемся в диапазоне от примерно 1,0 до примерно 1,5 эквивалента, предпочтительно примерно 1,2 эквивалента.
[0016] В различных вариантах реализации настоящего изобретения указанный сложноэфирный предшественник включает сложноэфирную группу R, которая представляет собой замещенную или незамещенную группу, выбранную из низшей алкильной, низшей алкенильной, низшей алкинильной, цикло(низшей)алкильной, цикло(низшей)алкенильной, арильной, аралкильной, гетероциклильной и гетероарильной групп. Предпочтительно сложноэфирная группа R представляет собой бензильную группу.
[0017] В различных вариантах реализации настоящего изобретения предложены способы получения соединения формулы I, в которых требуется применение катализатора межфазного переноса для проведения катализируемого основанием гидролиза. В различных вариантах реализации настоящего изобретения катализатор межфазного переноса представляет собой соль четвертичного аммония, такую как тетрабутиламмония гидроксид. Такие катализаторы межфазного переноса могут присутствовать в количестве, находящемся в диапазоне от примерно 0,01 эквивалента до примерно 0,5 эквивалента.
[0018] Во втором аспекте настоящего изобретения предложены композиции, которые представляют собой реакционные смеси, соответствующие способам получения соединения формулы I, описанным выше.
[0019] В третьем аспекте настоящего изобретения предложены способы очистки соединения формулы I посредством перекристаллизации. В различных вариантах реализации настоящего изобретения перекристаллизацию проводят с помощью водного раствора ацетона. Соответственно, предложены способы, включающие: а) получение неочищенного соединения формулы I или его соли и перекристаллизацию указанного неочищенного соединения с помощью водного раствора ацетона; и b) выделение соединения формулы I или его соли путем удаления водного раствора ацетона. Предпочтительно указанный водный раствор ацетона представляет собой примерно 30%-й водный раствор ацетона. В различных вариантах реализации настоящего изобретения указанный водный раствор ацетона используют в количестве, составляющем примерно 7 объемов. Предпочтительно указанный способ осуществляют в течение периода времени, составляющего от примерно 1 часа до примерно 48 часов.
[0020] В четвертом аспекте настоящего изобретения предложены композиции, представляющие собой смеси после рекристаллизации, соответствующие способам очистки соединения формулы I, описанным выше.
[0021] В пятом аспекте настоящего изобретения предложено соединение формулы I, синтезированное согласно способам, описанным в настоящем документе, или перекристаллизованное согласно способам, описанным в настоящем документе, или и то, и другое. Предпочтительно указанное соединение по существу не содержит метилэтилкетона. В различных вариантах реализации настоящего изобретения соединение формулы I имеет энантиомерный избыток более чем примерно 96% после выделения его из реакционной смеси катализируемого основанием гидролиза и до перекристаллизации. В различных вариантах реализации настоящего изобретения соединение формулы I, синтезированное и/или перекристаллизованное согласно способам, предложенным в настоящем изобретении, имеет энантиомерный избыток более чем примерно 98%.
[0022] В шестом аспекте настоящего изобретения предложено соединение формулы I, где указанное соединение представляет собой полиморфную форму II, описанную в настоящем документе. В различных вариантах реализации настоящего изобретения полиморфная форма II указанного соединения присутствует в твердой композиции с фармацевтически приемлемым носителем. В различных вариантах реализации настоящего изобретения указанная композиция содержит по меньшей мере примерно 50% по массе формы II или, в качестве альтернативы, менее чем примерно 5% по массе формы II. В различных вариантах реализации настоящего изобретения указанная твердая композиция дополнительно содержит одну или большее количество твердых форм, выбранных из группы, состоящей из аморфной формы, формы I, формы III, формы IV, формы V и формы VI.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
[0023] Все публикации, патенты и заявки на патент, указанные в настоящем описании, включены в настоящее описание посредством ссылки в той же степени, как если бы в отношении каждой отельной публикации, патента или заявки на патент было бы конкретно и отдельно указано, что они включены посредством ссылки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0024] Несмотря не то, что в настоящем описании представлены и описаны избранные варианты реализации настоящего изобретения, специалисту в данной области очевидно, что такие варианты реализации представлены только в качестве примера. Специалист в данной области может, без отклонения от настоящего изобретения, представить многочисленные вариации, изменения и замещения. Следует понимать, что при реализации изобретения на практике могут быть использованы различные альтернативы вариантов реализации настоящего изобретения, описанных в настоящем документе. Предполагается, что объем настоящего изобретения определен прилагаемой формулой изобретения, и что данная формула изобретения охватывает способы и структуры, находящиеся в пределах объема пунктов формулы и их эквиваленты.
[0025] Определения
[0026] Если не указано иное, все технические и научные термины, используемые здесь, имеют такое же значение, как обычно понимается специалистом в области, к которой это изобретение принадлежит.
[0027] В описании и формуле изобретения термины в единственном числе, как в определенной, так и в неопределенной форме, включают данные термины в форме множественного числа, если из контекста явно не следует иное.
[0028] В настоящем описании термин «фармацевтически приемлемая соль» обозначает соли, которые являются подходящими для применения в фармации, предпочтительно для применения в тканях людей и низших животных без ненадлежащего раздражения, аллергических реакций и т.п. Фармацевтически приемлемые соли аминов, карбоновых кислот и других типов соединений хорошо известны в данной области техники. Например, С.М. Бердж (S.М. Berge) и др. подробно описывают фармацевтически приемлемые соли в источнике J Pharmaceutical Sciences, 66: 1-19 (1977), включенном в настоящее описание посредством ссылки. Соли могут быть получены in situ в процессе конечного выделения и очистки соединений согласно настоящему изобретению или отдельно путем взаимодействия функциональной группы свободного основания или свободной кислоты с подходящим реагентом, как в общем описано ниже. Например, функциональная группа свободного основания может быть подвергнута взаимодействию с подходящей кислотой. Кроме того, если соединения согласно настоящему изобретению содержат кислотный фрагмент, подходящие фармацевтически приемлемые соли могут включать соли металлов, такие как соли щелочных металлов, например натриевые или калиевые соли, и соли щелочноземельных металлов, например кальциевые или магниевые соли. Примерами фармацевтически приемлемых нетоксичных кислотно-аддитивных солей являются соли по аминогруппе, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с помощью других методов, применяемых в данной области, таких как ионный обмен. Другие фармацевтически приемлемые соли включают адипатные, альгинатные, аскорбатные, аспартатные, бензоатные, бисульфатные, боратные, бутиратные, камфоратные, камфорсульфонатные, цитратные, циклопентанпропионатные, диглюконатные, додецилсульфатные, формиатные, фумаратные, глюкогептонатные, глицерофосфатные, глюконатные, гемисульфатные, гептаноатные, гексаноатные, гидроиодидные, 2-гидрокси-этансульфонатные, лактобионатные, лактатные, лауратные, лаурилсульфатные, малатные, малеатные, малонатные, метансульфонатные, никотинатные, нитратные, олеатные, оксалатные, пальмитатные, пектинатные, персульфатные, 3-фенилпропионатные, фосфатные, пикратные, пивалатные, пропионатные, стеаратные, сукцинатные, сульфатные, тартратные, тиоцианатные, n-толуолсульфонатные, ундеканоатные, валератные соли и т.п. Примеры солей щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Другие фармацевтически приемлемые соли включают, когда это уместно, нетоксичные аммонийные, четвертичные аммонийные и аминные катионы, образованные путем непосредственного взаимодействия с карбоновой кислотой лекарственного средства или посредством применения противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, сульфонат и арилсульфонат.
[0029] Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемое вспомогательное вещество» включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и задерживающие всасывание агенты и тому подобное. Применение таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. За исключением тех случаев, когда обычные среды или агенты несовместимы с активным ингредиентом, в терапевтических композициях согласно настоящему изобретению предполагается их использование. В композиции также могут быть включены дополнительные активные ингредиенты.
[0030] Термин «пролекарство», как считается, обозначает соединение, которое в физиологических условиях или путем сольволиза может превращаться в биологически активное соединение, описанное в настоящем документе. Таким образом, термин «пролекарство» обозначает предшественник биологически активного соединения, который является фармацевтически приемлемым. Пролекарство может быть неактивным при введении субъекту, то есть быть в форме сложного эфира, но превращается in vivo в активное соединение, например, путем гидролиза с получением свободной карбоновой кислоты. Соединение, представляющее собой пролекарство, имеет преимущества в отношении растворимости, совместимости с тканями или отсроченного высвобождения в организме млекопитающего (см., например, Bundgard, Н., Design of Prodrugs (1985), pp. 7-9, 21-24 (Elsevier, Амстердам). Обсуждение пролекарств приведено в Higuchi, Т., et al., "Pro-drugs as Novel Delivery Systems," A.C.S. Symposium Series, Vol. 14 и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, причем оба из указанных источников полностью включены в настоящее описание посредством ссылки. Также подразумевается, что термин "пролекарство" включает любые ковалентно связанные носители, которые высвобождают активное соединение in vivo, когда такое пролекарство ввели млекопитающему. Пролекарства активного соединения, описанного в настоящем документе, могут быть получены путем модификации функциональных групп, присутствующих в активном соединении, таким образом, что модификации расщепляются либо путем обычной манипуляции, либо in vivo, с получением родительского активного соединения. Пролекарства включают соединения, в которых гидрокси-, амино- или меркаптогруппа связана с любой группой, которая, когда пролекарство активного соединения ввели млекопитающему, расщепляется с образованием свободной гидрокси-, свободной амино- или свободной меркаптогруппы, соответственно. Примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные и бензоатные производные спирта или ацетамидные, формамидные и бензамидные производные функциональной аминогруппы в активном соединении и т.п.
[0031] Термин «субъект» обозначает животное, такое как млекопитающее, например человека. Способы, описанные в настоящем документе, могут быть подходящими как для лечения людей, так и для применения в ветеринарных целях. В некоторых вариантах реализации настоящего изобретения пациент представляет собой млекопитающее, а в некоторых вариантах реализации пациент является человеком. В различных вариантах реализации настоящего изобретения пациент представляет собой животное, не являющееся человеком, такое как собака, кошка, кролик, мышь, крыса, корова, лошадь, свинья или курица.
[0032] Если не указано иное, подразумевается, что структуры, изображенные в настоящем документе, также включают соединения, которые отличаются только присутствием одного или более изотопно обогащенных атомов. Например, соединения, имеющие структуры, где водород заменен на дейтерий или тритий или углерод заменен на 13С- или 14С-обогащенный углерод, находятся в рамках объема настоящего изобретения.
[0033] Соединения согласно настоящему изобретению могут также содержать неестественные пропорции атомных изотопов при одном или нескольких атомах, которые составляют такие соединения. Например, соединения могут быть помечены радиоактивными изотопами, такими как, например, тритий (3Н), иод-125 (125I) или углерод-14 (14С). Все изотопные варианты соединений согласно настоящему изобретению, вне зависимости от того, являются ли они радиоактивными или нет, включены в объем настоящего изобретения.
[0034] В тех случаях, когда для указания характеристик физических свойств, таких как молекулярная масса, или химических свойств, таких как химические формулы, используются диапазоны, подразумевается, что они включают все комбинации и подкомбинации диапазонов и конкретные их варианты. Термин «примерно» в отношении числа или числового диапазона означает, что данное число или числовой диапазон являются приблизительными в пределах экспериментальной изменчивости (или в пределах статистической ошибки эксперимента), и, таким образом, указанное число или числовой диапазон могут варьироваться в диапазоне, например, от 1% до 15% от заявленного числа или числового диапазона. Термин «содержащий» (и родственные термины, такие как «содержит», или «имеющий», или «включающий») включает варианты реализации, например, вариант реализации любого состава вещества, композиции, способа или процесса или тому подобного, при котором они «состоят из» или «по существу состоят из» описанных признаков.
[0035] Используемые в настоящем описании сокращения имеют их обычное для химической и биологической области значение.
[0036] Соединение формулы I
[0037] Было обнаружено, что соединение формулы I:
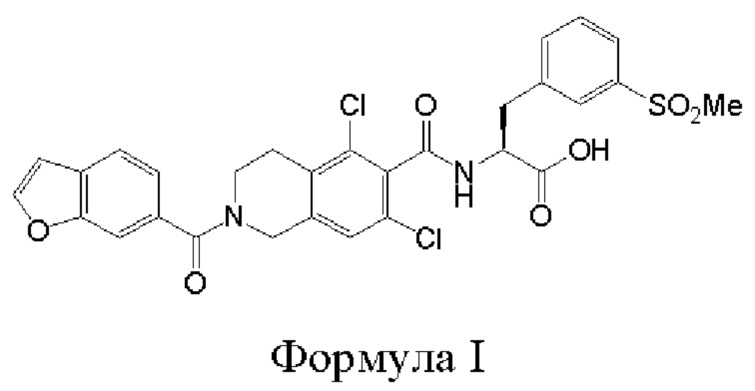
является эффективным ингибитором взаимодействий ассоциированного с функцией лимфоцитов антигена-1 (LFA-1) с молекулой межклеточной адгезии-1 (ICAM-1). Данное соединение является представителем класса прямых конкурентных ингибиторов LFA-1, которые непосредственно связываются с сайтом связывания IСAM на LFA-1 и, таким образом, препятствует связыванию IСAM. Прямые конкурентные ингибиторы LFA-1 могут обеспечить возможность более эффективного модулирования воспалительной и/или иммунологической реакции по сравнению реакцией, которую обеспечивают аллостерические ингибиторы, поскольку прямые конкурентные ингибиторы более эффективно перекрывают сайт связывания. Также в настоящее изобретение включены фармацевтически приемлемые соли соединений формулы I. Дополнительная информация, относящаяся к соединению формулы I, может быть найдена в патенте США 8080562, патентной публикации США 2009/0298869, патентной публикации США 2011/0092707, патенте США 8084047, патентной публикации США 2010/0092542 и патентной публикации США 2006/0281739, причем полное содержание каждого из указанных источников включено в настоящее описание посредством ссылки.
[0038] Для разработки подходящих для клинического применения лекарственных средств лекарственные средства-кандидаты должны быть в достаточной степени химически чистыми для введения субъекту и иметь приемлемую физическую форму, для того чтобы их можно было бы приготовить в виде в фармацевтически приемлемых лекарственных форм. Один из предпочтительных путей достижения более высокой чистоты, воспроизводимости физической формы и стабильности является идентификация одной или более подходящих кристаллических форм. Способность существовать в различных кристаллических формах известна как полиморфизм, и, как известно, имеет место для многих органических молекул. Эти различные кристаллические формы известны как «полиморфные модификации» или «полиморфы». Несмотря на то, что полиморфные модификации имеют одинаковый химический состав, они отличаются по упаковке, геометрическому расположению и другим описательным свойствам кристаллического твердого состояния. Как таковые, эти модификации могут иметь различные физические свойства в твердом состоянии, которые влияют, например, на растворимость, скорость растворения, биодоступность, химическую и физическую стабильность, текучесть, податливость (fractability) и сжимаемость соединения, а также безопасность и эффективность лекарственных продуктов, полученных на основе данных соединений. В процессе получения полиморфа также может быть осуществлена дальнейшая очистка с точки зрения общей физической чистоты или оптической чистоты.
[0039] Был обнаружен ряд различных форм, в том числе кристаллических форм, соединения формулы I, в том числе кристаллические формы А-Е и аморфная форма. Несмотря на то, что для органических соединений кристаллизацию проводят часто, невозможно предсказать заранее, какие условия будут обеспечивать подходящие условия, позволяющие получить определенную кристаллическую форму. Кроме того, невозможно предсказать, какая конкретная кристаллическая форма обеспечит необходимую совокупность физических свойств, неограничивающие примеры которых описаны выше, с получением желаемой лекарственной формы данного лекарственного средства в результате ее приготовления. Дополнительная информация, относящаяся к кристаллическим формам А-Е и аморфной форме соединения формулы I, может быть найдена в патенте США 8080562, патентной публикации США 2009/0298869, патентной публикации США 2011/0092707, патенте США 8084047, патентной публикации США 2010/0092542 и патентной публикации США 2006/0281739, причем полное содержание каждого из указанных источников включено в настоящее описание посредством ссылки.
[0040] Способы получения соединения формулы I
[0041] В одном из вариантов реализации настоящего изобретения соединение формулы I синтезируют, как показано на следующих схемах 1-7. В качестве конечного продукта этого синтеза получают соединение формулы I в виде аморфного твердого вещества или в виде кристаллической формы, такой как формы А-Е, или его фармацевтически приемлемой соли, прямо или косвенно. Варианты этого общего пути синтеза могут обеспечить повышенные выходы, улучшенную стоимость продуктов и/или повышенную хиральную чистоту.
[0042] Защитные группы для амино- и карбоксильных групп известны в данной области техники. Например, см. Greene, Protective Groups in Organic Synthesis, Wiley Interscience, 1981 и последующие издания.
[0043] В различных вариантах реализации настоящего изобретения на последующих схемах HATU используют в качестве реагента в реакциях по образованию амидной связи. В качестве альтернативы, HATU не используют. В различных вариантах реализации настоящего изобретения по меньшей мере одну реакцию по образованию амидной связи осуществляют с использованием в качестве реагента тионихлорида вместо HATU. В различных вариантах реализации настоящего изобретения все реакции по образованию амидной связи осуществляют с использованием в качестве реагента тионилхлорида с получением хлоридов кислот.
[0044] Схема 1
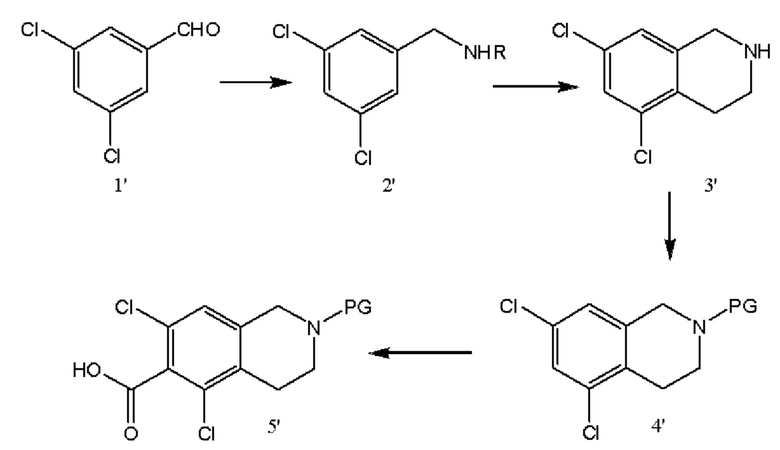
[0045] Согласно первой из альтернативных защитных стратегий, получают соединение 5', защищенные соединения, показанные на схеме 1. Данный синтез начинается с восстановительного аминирования 3,5-дихлорбензальдегида, соединения 1'. Циклизация соединения 2' позволяет получить соединение 3'. Защита свободной аминогруппы соединения 3' с получением защищенного соединения позволяет получить соединение 4'. Функциональную группу карбоновой кислоты вводят путем обработки соединения 4' диоксидом углерода с получением соединения 5'. В различных вариантах реализации настоящего изобретения защитная группа соединения 4' представляет собой бензофуранилкарбонильный фрагмент, полученный из соединения 18'.
[0046] В различных вариантах реализации настоящего изобретения при масштабировании реакции до масштаба в несколько килограмм или большего масштаба обработку соединения 4' сильным основанием (таким как н-бутиллитий (nBuLi) для получения соединений лития или диизопропиламид лития (LDA) для получения соединений лития) осуществляют в проточном режиме, а не путем реакции в периодическом режиме ввиду нестабильности соединений лития не при низких температурах. Скорости потока и время удерживания могут быть откорректированы для получения максимального выхода.
[0047] Схема 1В
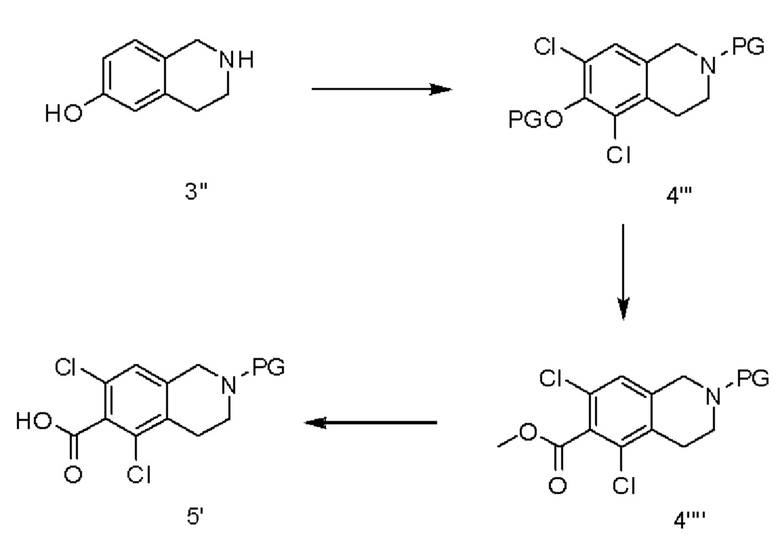
[0048] В различных вариантах реализации настоящего изобретения для получения соединения 5' в качестве исходного вещества используют 6-гидрокси-1,2,3,4-тетрагидроизохинолин (соединение 3''). Данное исходное вещество хлорируют (х2) например, N-хлорсукцинимидом. В различных вариантах реализации настоящего изобретения хлорирование осуществляют в присутствии сульфоновой кислоты. В различных вариантах реализации настоящего изобретения сульфоновая кислота выбрана из n-толуолсульфоновой кислоты и метансульфоновой кислоты. После защиты аминогруппы вводят функциональную группу к гидроксигруппе, например, виде сложного эфира трифлата, который карбонилируют с получением аминозащищенного метилового сложного эфира. Гидролиз данного метилового сложного эфира позволяет получить аминозащищенную карбоновую кислоту.
[0049] Схема 2
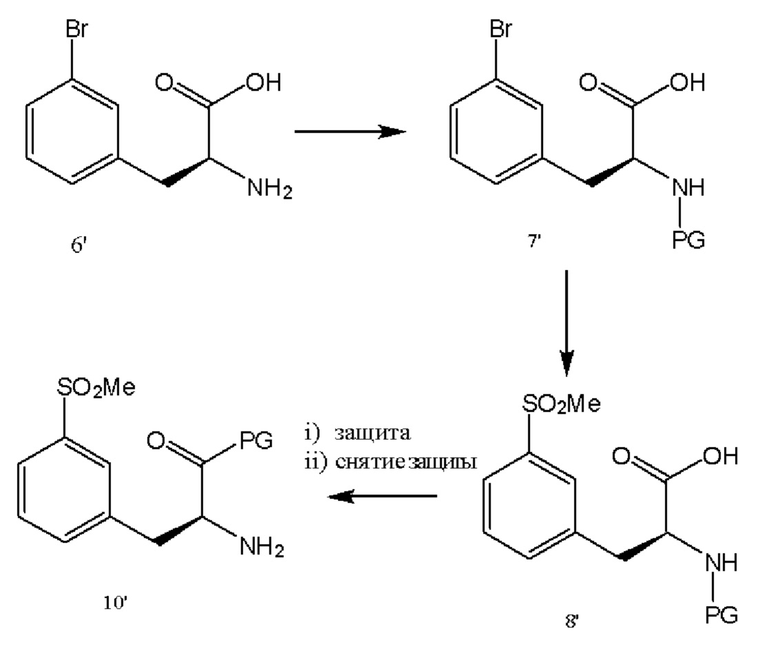
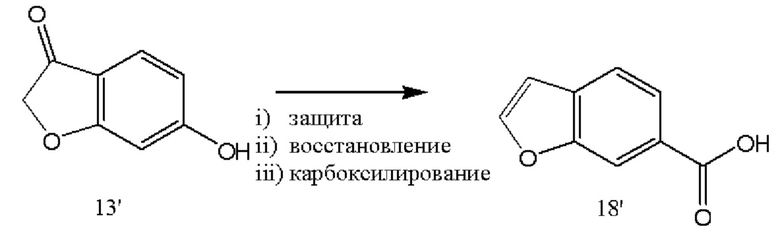
[0050] В различных вариантах реализации настоящего изобретения бромфенилаланин используют в качестве исходного вещества для части конечной молекулы, как показано на схеме 2. Данное исходное вещество защищают аминозащитной группой с обеспечением возможности введения метилсульфоновой функциональной группы в соединение 8'. Защитные группы перегруппировывали путем введения ортогональной защитной группы, защищающей карбоксильный фрагмент, с последующим снятием защиты с аминогруппы с получением соединения 10'. В различных вариантах реализации настоящего изобретения дорогие или малораспространенные основания заменяют карбонатным основанием, таким как карбонат калия или карбонат кальция, в качестве реагентов.
[0051] Схема 2А
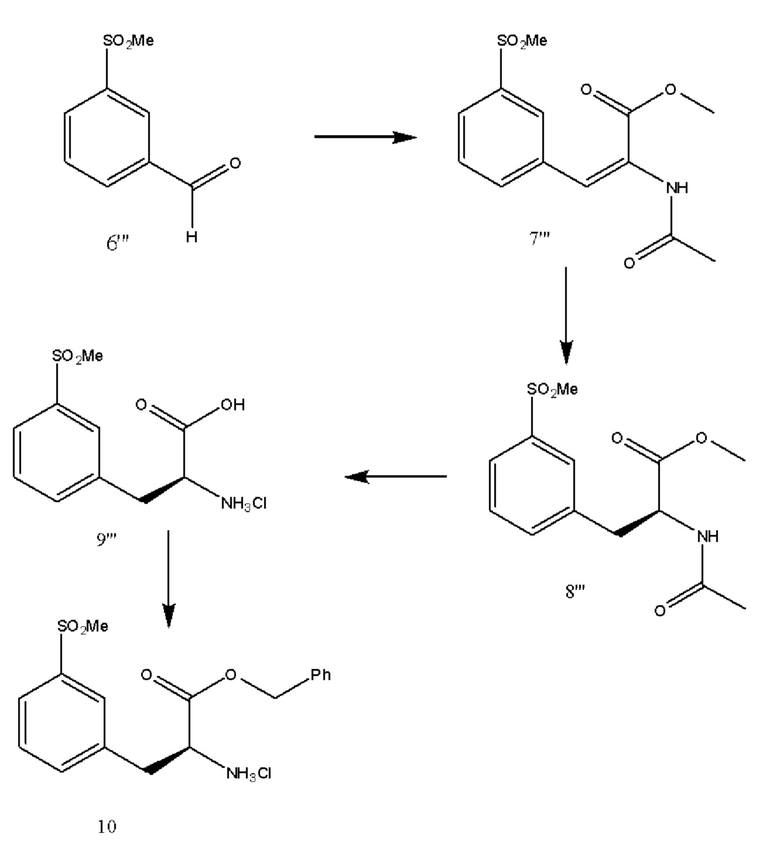
[0052] В различных вариантах реализации настоящего изобретения 3-метилсульфонилбензальденид превращают в 3-метилсульфонилфенилаланиновое производное и вводят функциональную группу с получением соединения 10, как показано выше.
[0053] Схема 3
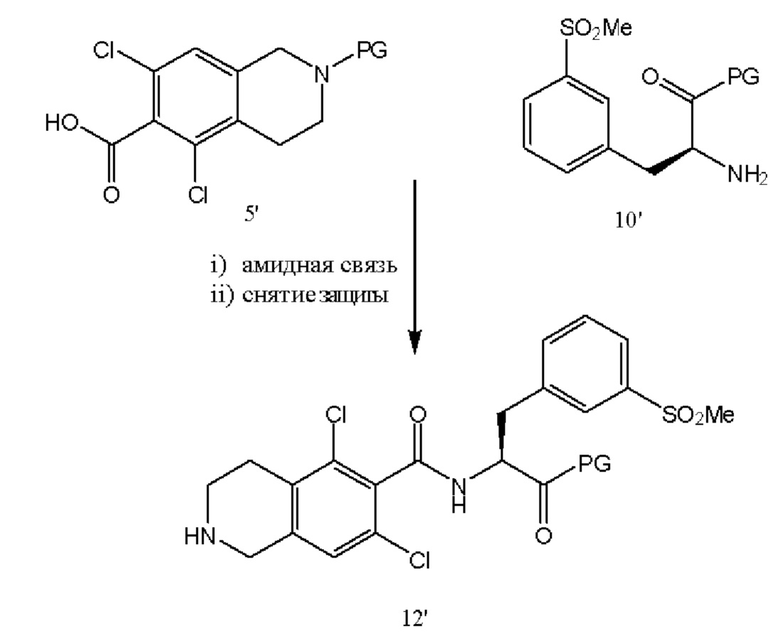
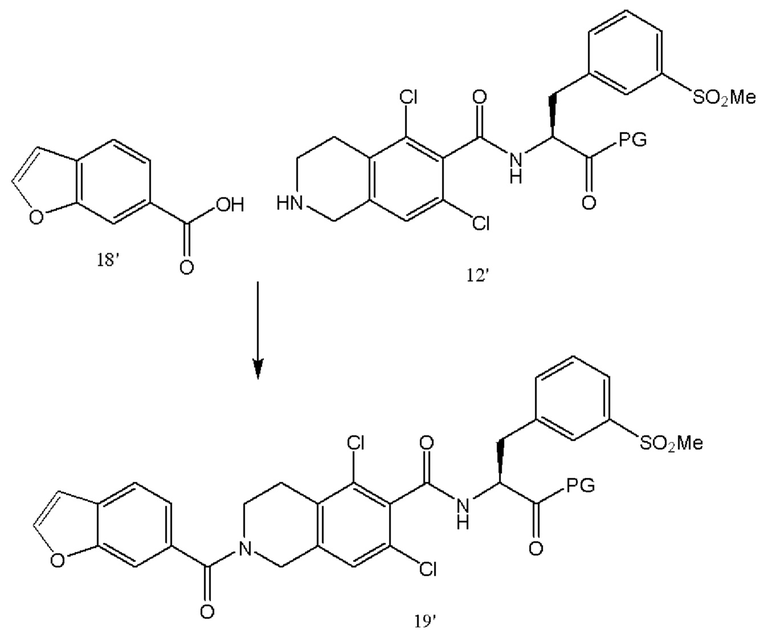
[0054] Соединения 5' и 10' соединяли посредством образования амидной связи с последующим снятием защиты с оставшейся аминогруппы в присутствии карбоксизащитной группы с получением соединения 12' или его соли, такой как соли HCL.
[0055] Схема 3А
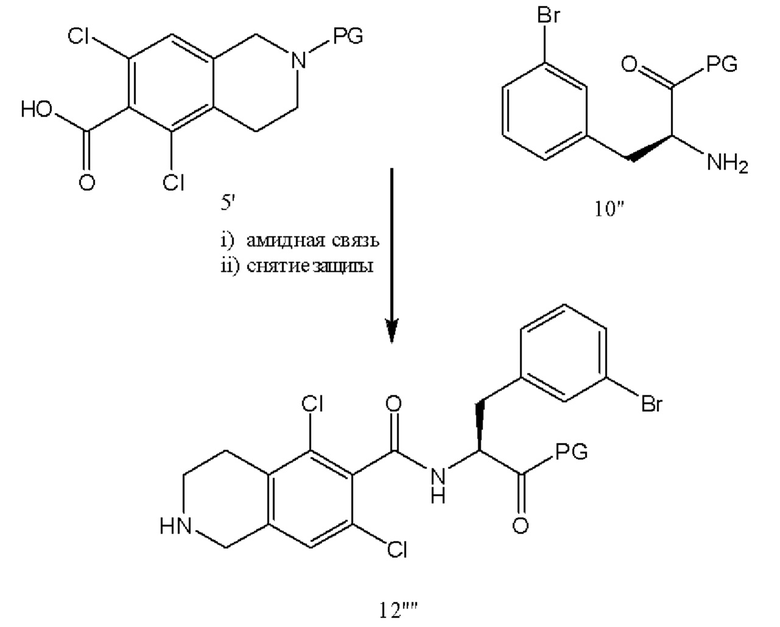
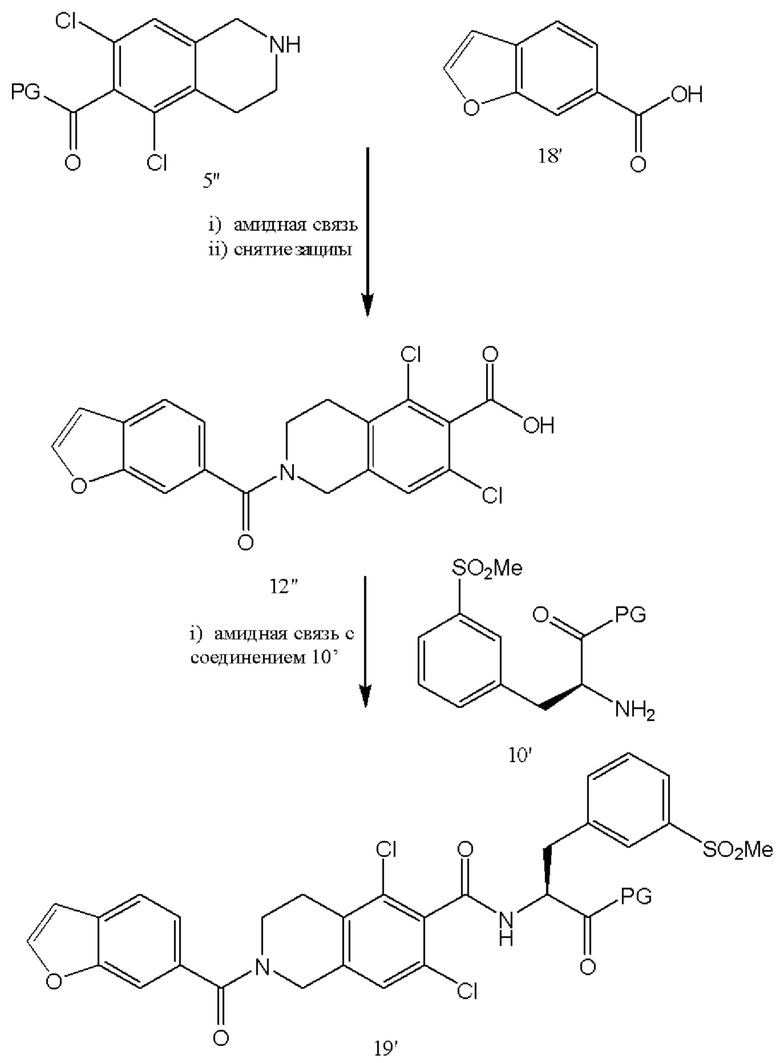
[0056] В качестве альтернативы схеме 3, соединение 10'' подвергают реакции сочетания с соединением 5' с получением бромсодержащего соединения 12'''' с последующим введением на следующей стадии метилсульфоновой функциональной группы на место брома с получением соединения 19'. В качестве альтернативы, вместо брома, соединение 10'' включает X, где X представляет собой любой галогенид (Cl, I, Br, F) или уходящую группу, такую как OTs, OTf или тому подобные.
[0057] Схема 4
[0058] Бензофуранилкарбонильный фрагмент соединения формулы I может быть получен с использованием различных альтернативных схем. В одном из вариантов реализации настоящего изобретения бензофуранилкарбонильный фрагмент получают путем защиты гидроксильной группы соединения 13', восстановления карбонила соединения 13' с получением бензофуранильного фрагмента и последующего карбоксилирования с получением соединения 18'.
[0059] Схема 4А
[0060] В одном из вариантов реализации настоящего изобретения соединение 18' получают из 6-гидроксибензофурана через сложный эфир трифлата и 6-карбоксиметиловый сложный эфир в качестве промежуточных веществ, как показано в примере Пример 4А.
[0061] Схема 5
[0062] Бензофуранкарбоновую кислоту 18' подвергают реакции сочетания с соединением 12' (или его солью) путем образования амидной связи с получением защищенного соединения 19', как показано на схеме 5. Образование амидной связи известно в данной области техники.
[0063] Схема 5А
[0064] В качестве альтернативы схемам 3-5, соединения 18' и 5'' могут быть подвергнуты реакции сочетания с образованием амидной связи с последующим снятием защиты с оставшейся карбоксильной группы с получением соединения 12''. Образование амидной связи между соединениями 12'' и 10' позволяет получить соединение 19' с защищенной карбоксильной группой.
[0065] Схема 5В
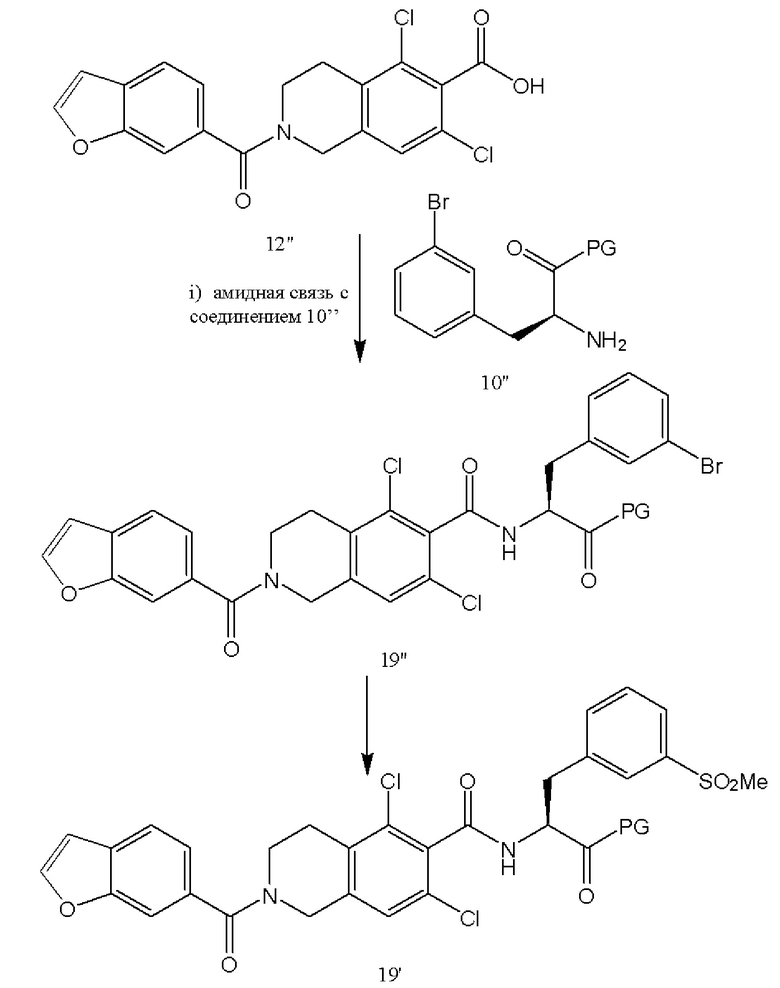
[0066] В качестве альтернативе схемам 1-5, соединения 12'' и 10'' могут быть подвергнуты реакции сочетания с образованием амидной связи с последующим введением метилсульфоновой функциональной группы вместо брома при превращении соединения 19'' в соединение 19' (аналогично схеме 2). В качестве альтернативы, вместо брома, соединение 10'' включает X, где X представляет собой любой галогенид (Cl, I, Br, F) или уходящую группу, такую как OTs, OTf или тому подобное. Соединение 12'' может быть также получено согласно следующей схеме:
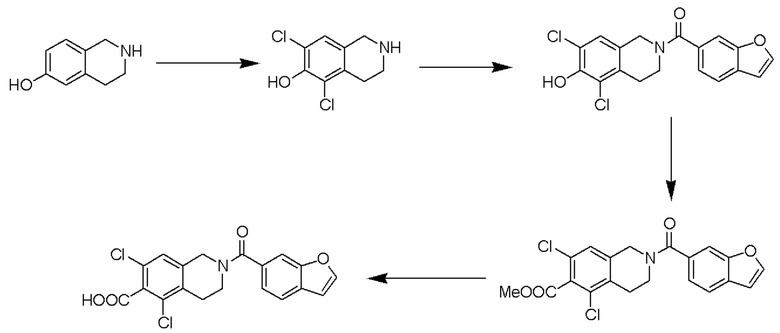
[0067] Схема 6
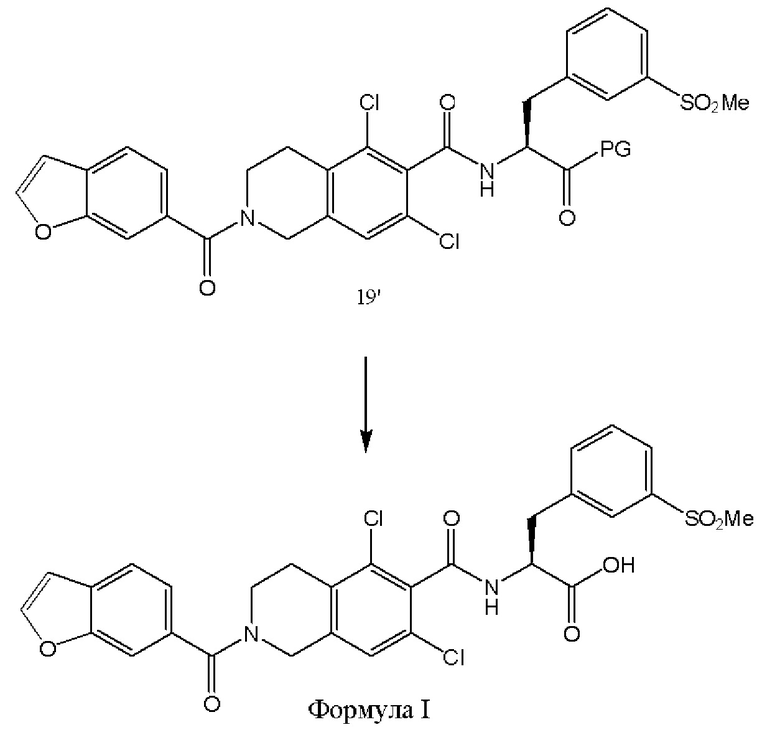
[0068] Конечное снятие защиты с соединения 19' с получением соединения формулы I или его соли осуществляют множеством способов. В различных вариантах реализации настоящего изобретения полученное в результате соединение формулы I имеет более высокую оптическую чистоту, и/или более высокую общую чистоту, и/или более высокий общий выход.
[0069] Согласно одному из подходов, сложноэфирную защитную группу удаляют путем катализируемого кислотой гидролиза. Например, защитную группу в виде метилового эфира удаляют путем катализируемого кислотой гидролиза. В качестве альтернативы, защитную группу в виде бензилового эфира удаляют кислотой, например НСl в диоксане. Растворитель для катализируемого кислотой гидролиза может быть представлять собой любой доступный в промышленности растворитель, такой как апротонный растворитель, протонный растворитель, полярный растворитель, неполярный растворитель, ионный растворитель или сжатый газ, такой как сверхкритический диоксид углерода. В различных вариантах реализации настоящего изобретения растворитель представляет собой апротонный растворитель, такой как диоксан, или тетрагидрофуран, или ацетон. Вариативно растворитель может быть выбран из гексана, бензола, толуола, 1,4-диоксана, хлороформа, диэтилового эфира, дихлорметана, тетрагидрофурана, этилацетата, ацетона, диметилформамида, ацетонитрила, диметилсульфоксида, н-бутанола, изопропанола, н-пропанола, этанола, метанола, воды, муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты и их комбинаций, таких как водный раствор ацетона. Кислота может представлять собой любую кислоту, используемую для реакций гидролиза. В различных вариантах реализации настоящего изобретения кислота представляет собой минеральную кислоту. В различных вариантах реализации настоящего изобретения кислота выбрана из хлористого водорода, серной кислоты, фосфорной кислоты и сульфоновых кислот. В различных вариантах реализации настоящего изобретения кислота представляет собой трифторуксусную кислоту. В одном из вариантов реализации настоящего изобретения сложный эфир может быть удален путем нуклеофильного замещения, например, с использованием иодида натрия в диметилсульфоксиде.
[0070] Согласно одному из подходов, защитную группу в виде бензилового эфира удаляют при помощи палладия на угле. Например, бензиловый эфир соединения 19' удаляют путем гидрогенолиза с использованием 10% палладия на угле, с использованием муравьиной кислоты и триэтиламина в смеси метанол: ТГФ 5:1 с получением соединения формулы I.
[0071] В различных вариантах реализации настоящего изобретения соединение 19' представляет собой соединение формулы АА. Общая стратегия превращения соединения формулы АА обеспечивается основным гидролизом сложного эфира с получением соединения формулы I.
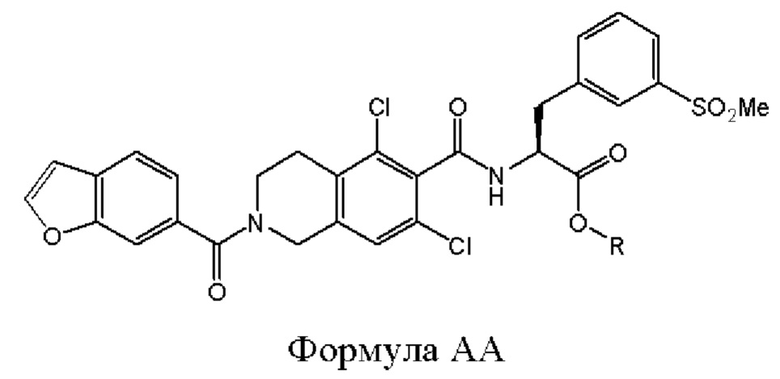
Соединение формулы АА может быть подвергнуто реакции с основанием в растворителе для осуществления катализируемого основанием омыления соединения формулы АА с получением соединения формулы I.
[0072] Растворитель для омыления может представлять собой любой доступный в промышленности растворитель, такой как апротонный растворитель, протонный растворитель, полярный растворитель, неполярный растворитель, ионной растворитель или сжатый газ, такой как сверхкритический диоксид углерода. В различных вариантах реализации настоящего изобретения растворитель представляет собой апротонный растворитель, такой как диоксан или тетрагидрофуран или ацетон. Вариативно растворитель может быть выбран из гексана, бензола, толуола, 1,4-диоксана, хлороформа, диэтилового эфира, дихлорметана, тетрагидрофурана, этилацетата, ацетона, диметилформамида, ацетонитрила, диметилсульфоксида, н-бутанола, изопропанола, н-пропанола, этанола, метанола, воды и их комбинаций. В предпочтительном варианте реализации настоящего изобретения растворитель представляет собой водный раствор ацетона. Основание может представлять собой любое основание, используемое для реакций омыления. В различных вариантах реализации настоящего изобретения основание представляет собой гидроксид, такой как гидроксид калия, или гидроксид натрия, или гидроксид лития.
[0073] В различных вариантах реализации настоящего изобретения группа R представляет собой любой углеродсодержащий фрагмент. Такие соединения могут быть подходящими для применения в качестве синтетических промежуточных веществ для получения соединений формулы I, или в виде пролекарств соединения формулы I. В группе, где R представляет собой любой углеродсодержащий фрагмент, R может быть выбран из низшего алкила, низшего алкенила, низшего алкинила, цикло(низшего)алкила, цикло(низшего)алкенила, арила, аралкила, гетероциклила и гетероарила, любой из которых может быть замещенным или незамещенным. В различных вариантах реализации настоящего изобретения низшая алкильная группа представляет собой метил, этил, пропил, изопропил, бутил, пентил, изобутил, трет-бутил или гексил. В различных вариантах реализации настоящего изобретения группа R соединения формулы АА представляет собой бензильную группу. В различных вариантах реализации соединений формулы АА углеродсодержащий фрагмент R не включает бензильную группу.
[0074] В различных вариантах реализации настоящего изобретения группа R представляет собой силилсодержащий фрагмент, такой что соединение формулы АА представляет собой силиловый эфир.
[0075] В одном из вариантов реализации настоящего изобретения сложноэфирную защитную группу удаляют путем катализируемого основанием гидролиза в гомогенной реакции, такой как реакция в растворе. Например, защитную группу в виде бензилового эфира удаляют с помощью NaOH в водном диоксане. В одном из вариантов реализации настоящего изобретения защитную группу в виде бензилового эфира удаляют с помощью NaOH в водном растворе ацетона. В различных вариантах реализации гомогенной жидкостной реакции концентрация NaOH может находиться в диапазоне от примерно 0,1 н. до примерно 2 н., в частности может составлять примерно 0,5 н., 0,6 н., 0,7 н., 0,8 н., 0,9 н., 1,0 н., 1,1 н., 1,2 н., 1,3 н., 1,4 н. или 1,5 н., где все указанные концентрации следует понимать как «примерные».
[0076] В одном варианте реализации настоящего изобретения сложноэфирную защитную группу удаляют из соединения 19' или соединения формулы АА путем катализируемого основанием гидролиза в гетерогенной реакции в присутствии катализатора межфазного переноса. Например, соединение 19' или соединение формулы АА приводят в контакт с катализатором межфазного переноса в водном растворе ацетона. В различных вариантах реализации настоящего изобретения реакцию проводят при наличии границы раздела фаз твердое-жидкость. В различных вариантах реализации настоящего изобретения реакцию проводят в суспензии растворителя и кристаллического материала. В различных вариантах реализации настоящего изобретения реакция является двухфазной. В различных вариантах реализации настоящего изобретения реакция начинается как двухфазная реакция в периодическом режиме и становится все более гомогенной по мере протекания реакции и превращения исходного вещества в продукт, который остается в растворе. В различных вариантах реализации настоящего изобретения рацемизация исходного материала сведена к минимуму благодаря снижению воздействия непрореагировавшего исходного вещества на основание при использовании двухфазной среды.
[0077] В различных вариантах реализации настоящего изобретения ход реакции контролируют путем оценки уровня оставшегося твердого вещества. В различных вариантах реализации настоящего изобретения реакцию считают по существу завершенной, когда реакционная смесь является по существу монофазной (то есть все твердые вещества растворены в растворе).
[0078] В различных вариантах реализации настоящего изобретения щелочной гидролиз проводят при количестве основания, находящемся в диапазоне от примерно 0,9 эквивалента до примерно 3 эквивалентов, таком как примерно 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,1, 2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9 или 3,0 эквивалента, где все количества являются примерными. В различных вариантах реализации настоящего изобретения количество основания находится в диапазоне от примерно 1,0 до примерно 1,5 эквивалента, в частности составляет примерно 1,2 эквивалента. В различных вариантах реализации настоящего изобретения основание представляет собой NaOH. В различных вариантах реализации настоящего изобретения основной гидролиз проводят с использованием NaOH в качестве основания в присутствии менее чем стехиометрического количества тетрабутиламмония гидроксида.
[0079] В различных вариантах реализации настоящего изобретения реакцию проводят в периодическом режиме при времени до завершения более 0 часов и менее чем примерно 24 часа, менее чем примерно 12 часов, менее чем примерно 8 часов, менее чем примерно 6 часов или менее чем примерно 4 часа.
[0080] В различных вариантах реализации настоящего изобретения катализируемый основанием гидролиз соединения 19' или соединения формулы АА проводят в присутствии катализатора межфазного переноса. В различных вариантах реализации настоящего изобретения катализатор межфазного переноса представляет собой соль четвертичного аммония, соль фосфония или краун-эфир. В различных вариантах реализации настоящего изобретения катализатор межфазного переноса выбран из бензилтриметиламмония хлорида, гексадецилтрибутилфосфония бромида, тетрабутиламмония гидроксида, тетрабутиламмония бромида, метилтриоктиламмония хлорида и тетрабутиламмония хлорида. В различных вариантах реализации настоящего изобретения катализатор межфазного переноса представляет собой тетрабутиламмония гидроксид. В одном из вариантов реализации настоящего изобретения количество катализатора межфазного переноса составляет менее чем стехиометрического. Например, количество катализатора межфазного переноса составляет примерно 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8 или 0,9 эквивалента, где все количества являются примерными.
[0081] В других вариантах реализации настоящего изобретения сложноэфирная защитная группа может быть удалена другими способами, известными в литературе, включая применение слабокислых или слабощелочных сред. Сложноэфирные защитные группы также могут быть удалены путем обработки ферментами, гидролизующими сложные эфиры, такими как эстераза печени свиньи, холестеринэстераза, аминоэстераза и т.д. Удаление сложноэфирной защитной группы из соединения формулы АА может быть также осуществлено с применением сильнокислых смол, слабокислых смол, сильноосновных смол или слабоосновных смол.
[0082] После получения соединения формулы I в виде неочищенного соединения, доступны различные способы выделения и/или очистки. Соединение формулы I может быть выделено в виде неочищенного продукта путем отгонки или выпаривания растворителя с конечной стадии снятия защиты. Удаление растворителя может быть произведено посредством удаления досуха, или посредством удаления части растворителя с получением твердой/жидкой смеси, которую фильтруют и/или промывают. Неочищенное соединение может быть очищено путем суспендирования в растворителе, таком как метилэтилкетон (МЭК), ацетонитрил, метиленхлорид или ацетон, где указанные растворители могут быть водными или неводными. Соединение формулы I может быть выделено/очищено путем перекристаллизации и/или промывки дополнительными растворителями. Методики перекристаллизации в малых и больших масштабах известны в данной области техники.
[0083] Частичный список растворителей, подходящих для получения и очистки соединения формулы I включает, например, воду, алифатические растворители, такие как пентан, петролейный эфир и гексан; ароматические растворители, такие как толуол и ксилол, алифатические кетоны и сложные эфиры, такие как метилэтилкетон, ацетон, этилацетат, изопропилацетат и бутилацетат, спирты, такие как этиловый спирт, пропиловый спирт и метиловый спирт, ацетонитрил, простые эфиры, такие как этиловый простой эфир, трет-бутилметиловый эфир (МТБЭ), и тетрагидрофуран, алкены и алкины, алкениловые сложные эфиры и спирты, алкиниловые сложные эфиры и спирты и ароматические сложные эфиры и спирты. В одном из вариантов реализации настоящего изобретения рекристаллизацию проводят в фармацевтически приемлемом(ых) растворителе(ях). В одном из вариантов реализации настоящего изобретения подходящий растворитель представляет собой водный раствор ацетона.
[0084] В различных вариантах реализации настоящего изобретения перекристаллизацию проводят с применением от примерно 0,5 объемов до примерно 15 объемов растворителя для перекристаллизации, например от примерно 5 объемов до примерно 15 объемов или, например, примерно 1, 2, 3, 4, 5, 6, 7, 8 или 9 объемов. В различных вариантах реализации настоящего изобретения перекристаллизацию проводят с применением по меньшей мере примерно 10 объемов растворителя для перекристаллизации. В различных вариантах реализации настоящего изобретения в результате перекристаллизации получают одну или более партий кристаллов, например 1 партию, 2 партии, 3 партии или более. В различных вариантах реализации настоящего изобретения перекристаллизация обеспечивает выход, составляющий по меньшей мере 50%, или по меньшей мере 60%, или по меньшей мере 70%, или по меньшей мере 80%, или по меньшей мере 90% в результате первого фильтрования и/или комбинации фильтрований.
[0085] В различных вариантах реализации настоящего изобретения конечное снятие защиты и/или перекристаллизацию проводят в водном растворе ацетона. Вода и ацетон являются смешиваемыми, что позволяет получить диапазон от 100%/0% вода/ацетон до 0%/100% вода/ацетон. В различных вариантах реализации настоящего изобретения отношение вода/ацетон составляет примерно 10/90, 20/80, 30/70, 40/60, 50/50, 60/40, 70/30, 80/20 или 90/10, где все количества являются «примерными». Предпочтительно растворитель для конечного снятия защиты и/или перекристаллизации представляет собой примерно 30%-й водный раствор ацетона. В различных вариантах реализации настоящего изобретения перекристаллизация водным раствором ацетона обеспечивает выход, составляющий по меньшей мере 50%, или по меньшей мере 60%, или по меньшей мере 70%, или по меньшей мере 80%, или по меньшей мере 90% в результате первого фильтрования и/или комбинации фильтрований. В различных вариантах реализации настоящего изобретения водный раствор ацетона используют в количестве от примерно 0,5 объема до примерно 15 объемов, таком как примерно 7 объемов, как представлено выше.
[0086] В различных вариантах реализации настоящего изобретения в процессе выделения и/или очистки соединения формулы I используют модификатор рН. Не желая ограничиваться теорией, полагают, что растворимость соединения формулы I изменяют, подвергая соль соединения формулы I воздействию кислой среды, таким образом, что фрагмент карбоновой кислоты соединения формулы I протонируется, что делает соединение формулы I более растворимым в органических растворителях. В различных вариантах реализации настоящего изобретения модификатор рН добавляют в композицию неочищенного соединения формулы I с получением рН менее чем примерно 7. В различных вариантах реализации настоящего изобретения рН снижен до менее чем примерно 5, менее чем примерно 4 или менее чем примерно 3. В различных вариантах реализации настоящего изобретения рН находится в диапазоне от примерно 1 до примерно 5. В различных вариантах реализации настоящего изобретения рН составляет примерно 2. Модификатор рН может представлять собой кислоту, такую как органическая или минеральная кислота. В различных вариантах реализации настоящего изобретения модификатор рН представляет собой хлористоводородную кислоту. В различных вариантах реализации настоящего изобретения модификатор рН представляет собой разбавленный раствор НСl, такой как 4 н. раствор НСl, 1 н. раствор НСl, 0,1 н. раствор НСl или 0,01 н. раствор НСl. В различных вариантах реализации настоящего изобретения локального рН менее чем примерно 1 избегают, чтобы уменьшить рацемизацию и/или гидролиз.
[0087] В различных вариантах реализации настоящего изобретения перекристаллизацию проводят при температуре выше комнатной температуры. В различных вариантах реализации настоящего изобретения перекристаллизацию проводят при температуре от примерно 50°С до примерно 90°С. В различных вариантах реализации настоящего изобретения соединение формулы I растворяют в растворителе для перекристаллизации при температуре выше комнатной температуры, фильтруют для удаления твердых частиц, охлаждают до комнатной температуры или температуры ниже комнатной с обеспечением тем самым прохождения кристаллизация и фильтруют для разделения кристаллов и маточного раствора.
[0088] В различных вариантах реализации настоящего изобретения перекристаллизацию проводят в виде периодического процесса в течение времени до завершения более 0 часов и менее чем примерно 3 суток, менее чем примерно 2 суток, менее чем примерно 36 часов, менее чем примерно 24 часов, менее чем примерно 12 часов, менее чем примерно 8 часов, менее чем примерно 6 часов или менее чем примерно 4 часов.
[0089] В различных вариантах реализации настоящего изобретения перекристаллизацию проводят в ходе периодического процесса при масштабе более чем примерно 10 килограмм, 100 килограмм, одной метрической тонны или 10 метрических тонн, где все количества являются примерными. В различных вариантах реализации настоящего изобретения конечное снятие защиты и/или перекристаллизацию проводят с выходом, составляющим по меньшей мере 60%, или по меньшей мере 70%, или по меньшей мере 80%, или по меньшей мере 90% в результате первого фильтрования и/или комбинации фильтрований.
[0090] В других вариантах реализации настоящего изобретения соединение формулы I может быть очищено посредством других методик, известных в литературе, включающих, но не ограничивающихся ими, выпадение (crashing out) из раствора, сублимационную сушку или лиофилизацию, диализ или т.п.
[0091] В некоторых вариантах реализации способов получения согласно настоящему изобретению хиральная чистота соединения формулы I, измеренная посредством хиральной хроматографии при 260 нм, составляет более чем примерно 75%, примерно 75,5%, примерно 76%, примерно 76,5%, примерно 77%, примерно 77,5%, примерно 78%, примерно 78,5%, примерно 79%, примерно 79,5%, примерно 80%, примерно 80,5%, примерно 81%, примерно 81,5%, примерно 82%, примерно 82,5%, примерно 83%, примерно 83,5%, примерно 84%, примерно 84,5%, примерно 85%, примерно 85,5%, примерно 86%, примерно 86,5%, примерно 87%, примерно 87,5%, примерно 88%, примерно 88,5%, примерно 89%, примерно 89,5%, примерно 90%, примерно 90,5%, примерно 91,0%, примерно 91,5%, примерно 92,0%, примерно 92,5%, примерно 93,0%, примерно 93,5%, примерно 94,0%, примерно 94,5%, примерно 95,0%, примерно 95,5%, примерно 96,0%, примерно 96,5%, примерно 97,0%, примерно 97,5%, примерно 98,0%, примерно 98,5%, примерно 99,0%, примерно 99,5% или примерно 99,9% S-энантиомера. В различных вариантах реализации настоящего изобретения хиральная чистота соединения формулы I, измеренная посредством хиральной хроматографии, составляет более чем примерно 99%. В некоторых вариантах реализации настоящего изобретения хиральная чистота соединения формулы I, измеренная посредством хиральной хроматографии при 260 нм, составляет примерно 100%.
[0092] В некоторых вариантах реализации способов получения согласно настоящему изобретению соединение формулы I имеет менее чем примерно 2,0%, примерно 1,9%, примерно 1,8%, примерно 1,7%, примерно 1,6%, примерно 1,5%, примерно 1,4%, примерно 1,3%, примерно 1,2%, примерно 1,1%, примерно 1,0%, примерно 0,9%, примерно 0,8%, примерно 0,7%, примерно 0,6%, примерно 0,5%, примерно 0,4%, примерно 0,3%, примерно 0,2%, примерно 0,1%, примерно 0,09%, примерно 0,08%, примерно 0,07%, примерно 0,06%, примерно 0,05%, примерно 0,04%, примерно 0,03%, примерно 0,02%, примерно 0,01% или примерно 0,009% любой одной примеси, введенной, полученной или произведенной в результате химического синтеза, по результатам измерения посредством хроматографии при 220 нм. В некоторых вариантах реализации настоящего изобретения указанная примесь представляет собой побочный продукт синтеза. В различных вариантах реализации настоящего изобретения указанная примесь представляет собой бромсодержащее соединение. В различных вариантах реализации настоящего изобретения указанная примесь представляет собой монохлорсодержащее соединение.
[0093] В некоторых вариантах реализации способа получения согласно настоящему изобретению соединение формулы I содержит менее примерно 3,0%, примерно 2,8%, примерно 2,6%, примерно 2,4%, примерно 2,2%, примерно 2,1%, примерно 2,0%, примерно 1,9%, примерно 1,8%, примерно 1,7%, примерно 1,6%, примерно 1,5%, примерно 1,4%, примерно 1,3%, примерно 1,2%, примерно 1,1%, примерно 1,0%, примерно 0,9%, примерно 0,8%, примерно 0,7%, примерно 0,6%, примерно 0,5%, примерно 0,4%, примерно 0,3%, примерно 0,2%, примерно 0,1% или примерно 0,09% общего количества примесей, введенных, полученных или произведенных в результате химического синтеза, по результатам измерения посредством хроматографии при 220 нм. В некоторых вариантах реализации настоящего изобретения указанные примеси включают побочный продукт химического синтеза.
[0094] В одном из вариантов реализации настоящего изобретения продукт перекристаллизации содержит менее чем 0,5%, 0,4%, 0,3%, 0,2% или 0,1% не являющегося фармацевтически приемлемым растворителя. В различных вариантах реализации настоящего изобретения продукт перекристаллизации по существу не содержит не являющихся фармацевтически приемлемых растворителей. В одном из вариантов реализации настоящего изобретения продукт перекристаллизации содержит менее чем 0,5%, 0,4%, 0,3%, 0,2% или 0,1% метилэтилкетона.
[0095] В различных вариантах реализации настоящего изобретения соединения, синтезированные согласно настоящему изобретению, могут иметь различные преимущества, такие как простота очистки, сниженная стоимость, сниженное число стадий синтеза, более высокие общие выходы, сниженное количество примесей, отличающиеся профили примесей и уменьшенная рацемизация хирального центра. В одном из вариантов реализации настоящего изобретения соединение, синтезированное согласно настоящему изобретению, имеет энантиомерный избыток (э. и.), выбранный из более чем примерно 95% э. и., примерно 96%, примерно 97%, примерно 98%, примерно 99% и примерно 99,9%. В различных вариантах реализации настоящего изобретения соединение, синтезированное согласно настоящему изобретению, имеет сниженные уровни химического катализатора в качестве примеси, по сравнению с соединением формулы I, полученным с применением палладия в качестве катализатора для удаления сложноэфирной группы с получением карбоновой кислоты. Например, в различных вариантах реализации настоящего изобретения указанное соединение имеет менее чем 100 ppm загрязнения палладием, или менее чем 50 ppm, или менее чем 10 ppm, или менее чем 1 ppm загрязнения палладием. В различных вариантах реализации настоящего изобретения указанное соединение по существу не содержит химического катализатора.
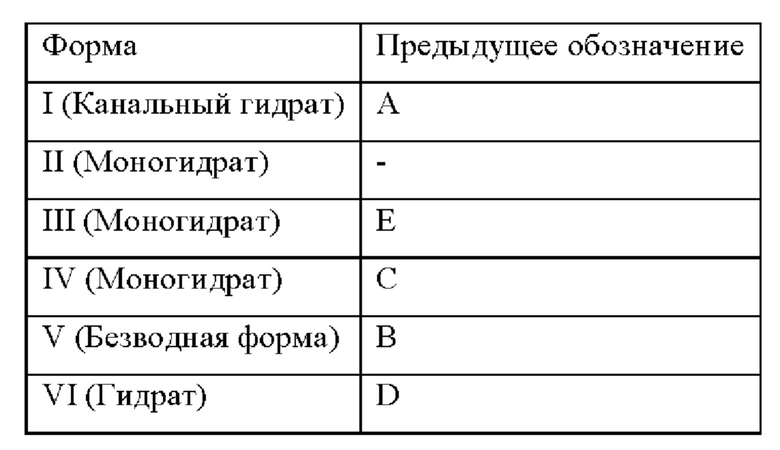
[0096] Безводную форму соединения формулы I и пять полиморфов, формы А, В, С, D и Е были ранее выделены и охарактеризованы. См. патент США 8080562. В настоящем изобретении был идентифицирован, выделен и полностью охарактеризован новый полиморф соединения формулы I. Указанные шесть форм в настоящем описании обозначены как формы I-VI, представленные в таблице 1, в которой приведены взаимоотношения между ранее принятой номенклатурой и номенклатурой в настоящем описании.
[0097] Таблица 1
[0098] Фармацевтические композиции, составы и наборы
[0099] В различных вариантах реализации настоящего изобретения аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I вводят в виде фармацевтических композиций. Фармацевтические композиции согласно настоящему изобретению содержат фармацевтически приемлемые носители и вспомогательные вещества, а также аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I с получением композиции для соответствующего введения субъекту.
[00100] В некоторых вариантах реализации настоящего изобретения кристаллическая форма остается в кристаллической форме в фармацевтической композиции. В других вариантах реализации настоящего изобретения аморфная форма и/или кристаллическая форма солюбилизируется и перестает быть кристаллической. В последнем случае, однако, повышенная чистота или другие физико-химические свойства аморфной формы и/или кристаллической формы способствуют, например, легкости обработки данной формы соединение формулы I с получением композиции, улучшенным возможностям хранения кристаллической формы до ее введения в составы, обеспечению лучшего терапевтического индекса, переносимости соединения формулы I субъектом или снижению побочных эффектов, вызываемых соединением формулы I. Аморфная форма или кристаллические формы А, В, С, D или Е могут быть измельчены с обеспечением желаемых свойств для введения в состав.
[00101] Фармацевтические композиции согласно настоящему изобретению могут быть приготовлены в виде геля, крема, лосьона, раствора, суспензии, эмульсии, мази, порошка, кристаллических форм, спрея, аэрозоля, пены, бальзама (salve), пасты, пластыря, краски, микрочастицы, наночастицы или биоадгезива, и могут быть приготовлены таким образом, чтобы содержать липосомы, мицеллы и/или микросферы. Пероральные композиции могут представлять собой таблетки, капсулы, пастилки, пилюли, облатки, жевательные резинки, лепешки, водные растворы или суспензии, масляные суспензии, сиропы, эликсиры, или диспергируемые порошки или гранулы и т.п. и могут быть приготовлены любым способом, известным в данной области техники. Пероральные композиции также могут содержать подслащающие, ароматизирующие, красящие и консервирующие агенты.
[00102] Аморфная форма или любая из кристаллических форм соединения формулы I, или их комбинация, могут быть приготовлены в виде стерильного раствора или суспензии в подходящих носителях, хорошо известных в данной области техники. Подходящие составы и дополнительные носители и вспомогательные вещества описаны в источнике Remington "The Science and Practice of Pharmacy" (20th Ed., Lippincott Williams & Wilkins, Baltimore MD), содержание которого полностью включено в настоящее описание посредством ссылки.
[00103] Составы согласно настоящему изобретению могут дополнительно включать другие фармакологически активные ингредиенты, если они не противоречат цели настоящего изобретения. В комбинации множества активных ингредиентов их соответствующие содержания может быть соответствующим образом увеличены или уменьшены с учетом их воздействия и безопасности.
[00104] Настоящее изобретение также относится к наборам. Наборы включают соединения согласно настоящему изобретению в подходящей упаковке, а также письменные материалы, которые могут включать инструкции по применению, обсуждение клинических исследований, перечень побочных эффектов и тому подобное. Набор может дополнительно содержать другой терапевтический агент, который вводят совместно с соединением формулы I, в том числе в аморфной форме, или в любой из кристаллических форм соединения формулы I, или их комбинации. В некоторых вариантах реализации настоящего изобретения указанный терапевтический агент и аморфная форма, или любая из кристаллических форм соединения формулы I, или их комбинация, представлены в наборе в виде отдельных композиций в отдельных емкостях. В некоторых вариантах реализации настоящего изобретения указанный терапевтический агент и аморфная форма, или любая из кристаллических форм соединения формулы I, или их комбинация представлены в наборе в виде единой композиции, находящейся в емкости. Подходящая упаковка и дополнительные продукты для применения (например, мерный стакан для жидких препаратов, оберточная фольга для сведения к минимуму воздействия воздуха, распылители и т.п.) известны в данной области техники и могут быть включены в набор.
[00105] Дополнительная информация, относящаяся к фармацевтическим композициям, составам и наборам, может быть найдена в патенте США 8080562, патентной публикации США 2009/0298869, патентной публикации США 2011/0092707, патенте США 8084047, патентной публикации США 2010/0092542 и патентной публикации США 2006/0281739, причем полное содержание каждого из указанных источников включено в настоящее описание посредством ссылки.
[00106] Способы применения
[00107] Не предполагая ограничение способов применения единственным механизмом действия, предложенные способы включают ингибирование начала и прогрессирования связанного с воспалением заболевания путем ингибирования взаимодействия LFA-1 и ICAM-1 путем введения соединения формулы I, включая аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I. В некоторых вариантах реализации настоящего изобретения такие способы обеспечивают противовоспалительное действие in-vitro и in-vivo и являются подходящими для применения для лечения опосредованных воспалением заболеваний и/или исследования механизмов заболевания.
[00108] В частности, аморфная форма, или любая из кристаллических форм А, В, С, D или Е, или комбинация указанных форм соединения формулы I может обеспечивать модулирование воспаления, опосредованного лейкоцитами. Аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I можно применять в качестве терапевтического агента при любом расстройстве, при котором показана эффективность антител к LFA-1. В одном из вариантов реализации настоящего изобретения субъекту вводят аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I для модулирования воспаления, связанного с воспалением глаз. В другом варианте реализации указанных способов субъекту с воспалением, связанным с синдромом сухого глаза, вводят аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I.
[00109] Введение фармацевтический композиции, содержащей соединение формулы I, включая аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I может быть произведено любыми подходящими средствами. В некоторых вариантах реализации настоящего изобретения фармацевтическую композицию, содержащую аморфную форму, или любую из кристаллических форм А, В, С, D или Е, или комбинацию указанных форм соединения формулы I, вводят путем перорального, трансдермального введения, путем инъекции, путем внутриглазной имплантации с медленным высвобождением или путем введения посредством аэрозоля.
[00110] Дополнительная информация, относящаяся к применению соединения формулы I, может быть найдена в патенте США 8080562, патентной публикации США 2009/0298869, патентной публикации США 2011/0092707, патенте США 8084047, патентной публикации США 2010/0092542 и патентной публикации США 2006/0281739, причем полное содержание каждого из указанных источников включено в настоящее описание посредством ссылки. Дополнительная информация, относящаяся к введению соединения формулы I, может быть найдена в патенте США 8080562, патентной публикации США 2009/0298869, патентной публикации США 2011/0092707, патенте США 8084047, патентной публикации США 2010/0092542 и патентной публикации США 2006/0281739, причем полное содержание каждого из указанных источников включено в настоящее описание посредством ссылки.
ПРИМЕРЫ
[00111] Пример 1
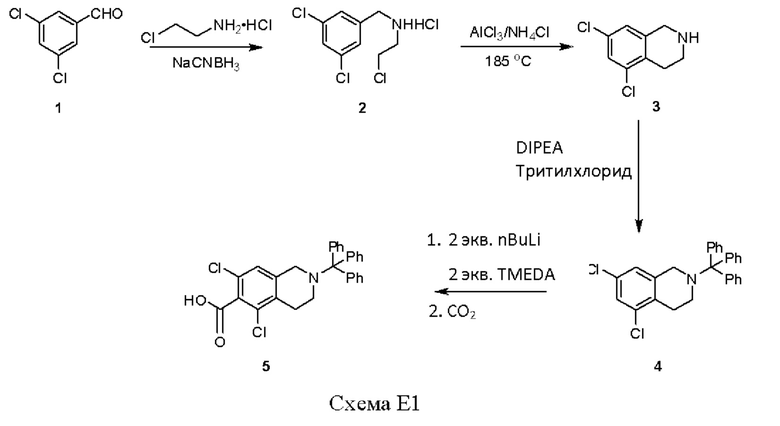
[00112] Восстановительное аминирование 3,5-дихлорбензальдегида, соединения 1, с 1-хлор-2-аминоэтаном и цианоборгидридом натрия позволило получить 35%-й выход соединения 2. Циклизация соединения 2 с применением катализа, где в качестве катализатора использовали хлорид алюминия, и хлорида аммония при 185°С позволила получить соединение 3 с выходом 91%. Защита свободной аминогруппы соединения 3 в виде тритилзащищенного соединения позволила получить соединение 4 с выходом 89%. Функциональную группу карбоновой кислоты вводили путем обработки соединения 4 н-бутиллитием (nBuLi) и тетраметилэтилендиамином (TMEDA) с последующим введением диоксида углерода с получением тритилзащищенного соединения 5 с выходом 75%.
[00113] Пример 1А
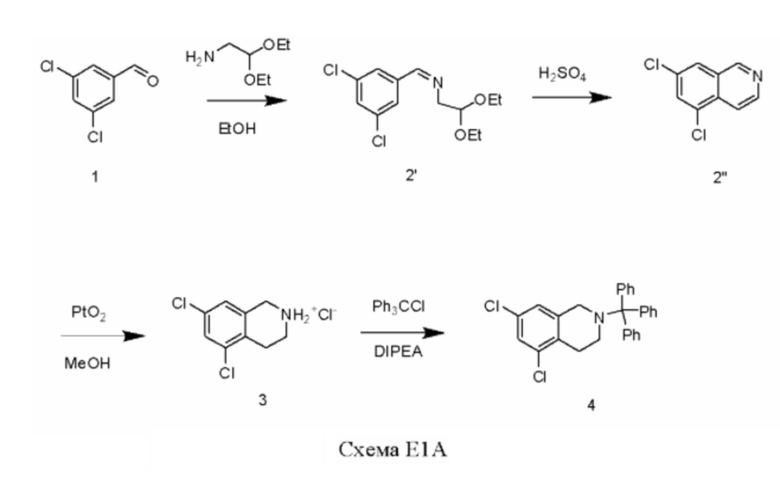
[00114] В стеклянный реактор помещали 3,5-дихлорбензальдегид. К содержимому реактора медленно добавляли абсолютный этанол (данное добавление является умеренно экзотермическим), и начинали перемешивание. К содержимому реактора медленно добавляли 2,2-диэтоксиэтиламин (1,03 экв.), поддерживая температуру в реакторе в диапазоне 20-78°С. Содержимое реактора затем нагревали до 76-78°С в течение 2 ч. Анализ посредством газовой хроматографии-масс-спектрометрии (ГХ-МС) показал окончание реакции (количество исходного материала <1%). Содержимое реактора охлаждали до температуры окружающей среды для обработки. Содержимое реактора концентрировали под вакуумом до получения остатка, и подвергали азеотропной перегонке с гептаном (х2). Полученный остаток охлаждали и выдерживали при 0-5°С в течение 12 ч с получением суспензии. Твердые вещества собирали путем фильтрования, остаток на фильтре промывали холодным (0-5°С) гептаном и сушили в теплом азоте (45-50°С) с получением соединения 2' в виде твердого вещества белого цвета (выход 94%).
[00115] В стеклянный реактор помещали концентрированную 95-98%-ю серную кислоту (25,9 эквив.). Содержимое реактора нагревали до 120-125°С, и медленно добавляли раствор соединения 2' в CH2Cl2 в течение 1 ч, поддерживая температуру внутри реактора в диапазоне 120-125°С. Содержимое реактора далее суспендировали при 120-125°С в течение 6 ч. Содержимое реактора охлаждали до температуры <50°С. В стеклянный реактор помещали деионизованную (ДИ) воду, и температуру внутри реактора доводили до 0-5°С. Реакционную смесь медленно переносили, поддерживая температуру внутри реактора в диапазоне 0-50°С. Для облегчения переноса использовали ДИ воду. К содержимому реактора добавляли Дикалит 4200 (Dicalite 4200). Содержимое реактора фильтровали через слой Дикалита 4200. К фильтрату добавляли 50%-й водный раствор гидроксида натрия медленно в течение 3 ч, поддерживая температуру внутри реактора в диапазоне 0-50°С, с обеспечением доведения рН до 12. Полученную суспензию перемешивали при 45-50°С в течение 2 ч, и собирали твердые вещества посредством фильтрования. Остаток на фильтре суспендировали в ДИ воде при 30-35°С в течение 1 ч. Содержимое реактора фильтровали. Остаток на фильтре промывали гептаном и сушили в вакуумной печи при 45-50°С в течение 22 ч с получением неочищенного соединения 2'' в виде твердого вещества коричневого (tan) цвета (выход 75%), которое далее очищали путем перекристаллизации.
[00116] В реактор добавляли диоксид платины (0,012 эквив.), соединение 2'' и МеОН (10 об.), и полученную суспензию перемешивали при комнатной температуре в атмосфере аргона в течение 10 минут. Создавали инертную атмосферу реакционной смеси путем ее обработки аргоном три раза, и далее реакционную смесь перемешивали в атмосфере водорода при давлении примерно 862 кПа (125 psi) при комнатной температуре в течение 25 часов. Анализ посредством ВЭЖХ показал завершение реакции с остатком исходного вещества менее 1%. После выдерживания надосадочную жидкость отделяли от твердых веществ (катализатора) путем декантации под вакуумом. К полученным твердым веществам добавляли метанол, и суспензию перемешивали в атмосфере азота. Твердым веществам давали осесть на дне в течение нескольких часов. Надосадочную жидкость отделяли от твердых веществ путем декантации под вакуумом. Объединенные надосадочные жидкости фильтровали через целит в атмосфере азота, и фильтрующий слой промывали МеОН (х2). Объединенные фильтрат и жидкость, полученную в результате промывки, концентрировали досуха. Остаток суспендировали в МТБЭ. Полученную смесь обрабатывали 3 М НСl при поддержании температуры <40°С с получением тяжелого осадка. Смесь перемешивали при 35-40°С в течение 60-90 минут. Содержимое реактора охлаждали до 0-5°С, перемешивали в течение 60-90 минут и далее фильтровали. Остаток на фильтре промывали холодной ДИ водой (х2), а после этого проводили промывку вытеснением посредством МТВЕ (х2). Остаток на фильтре сушили при пониженном давлении с получением гидрохлоридной соли соединения 3 (выход 86%). Катализатор гидрогенирования может быть восстановлен и использован повторно.
[00117] В реакционную колбу помещали соединение 3 и тритилхлорид. В реактор добавляли ДХМ (10 об.), и начинали перемешивание для получения суспензии. Реакционную смесь охлаждали до 10-15°С. К реакционной смеси медленно добавляли N,N-диизопропилэтиламин (2,5 эквив.), поддерживая температуру в диапазоне 15-25°С в процессе добавления. После завершения добавления содержимое реактора перемешивали при 15-25°С в течение минимум 60 минут. Проводили анализ реакционной смеси посредством ВЭЖХ путем разбавления образца ацетонитрилом и введения в прибор для ВЭЖХ. Первый анализ через 30 минут показал, что реакция была завершена, и, по данным ВЭЖХ-анализа, наблюдалось <1% исходного вещества. Реакционную смесь разбавляли ДИ водой (5 об.). Реакционную смесь перемешивали в течение 5 минут, после чего ее переносили на делительную воронку, и обеспечивали разделение фаз. ДХМ-слой промывали ДИ водой (5 об.) при перемешивании в течение 5 минут, и далее обеспечивали разделение фаз. ДХМ-слой промывали солевым раствором (5 об.) при перемешивании в течение 5 минут, и далее обеспечивали разделение фаз. ДХМ-слой сушили над сульфатом магния, фильтровали, и остаток на фильтре промывали ДХМ (х2). Объединенные фильтрат и жидкость, полученную в результате промывки, концентрировали с получением остатка, который подвергали азеотропной перегонке с EtOAc (х2). Остаток суспендировали в EtOAC и перемешивали в течение 1 часа в водяной бане с температурой 40°С. Полученную суспензию охлаждали до 0-5°С в течение 1 часа и далее фильтровали. Остаток на фильтре дважды промывали EtOAc и далее сушили при пониженном давлении с получением соединения 4.
[00118] Пример 1В
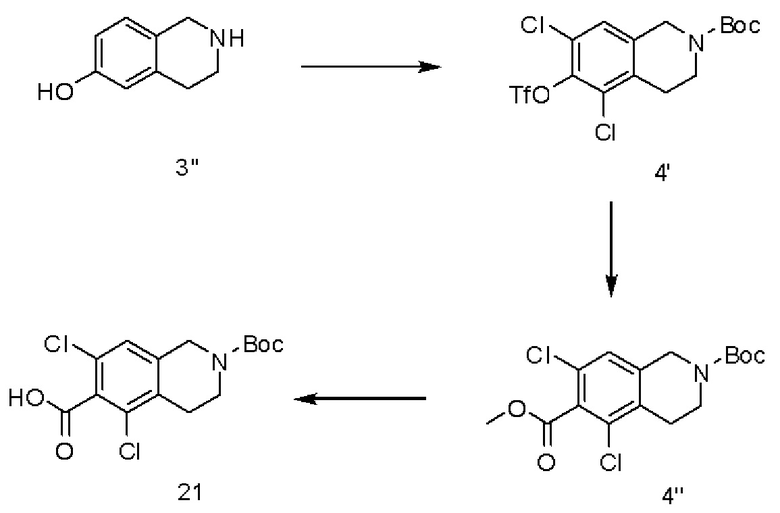
[00119] К 1,2,3,4-тетрагидро-6-гидроксиизохинолину в ацетонитриле добавляли n-толуолсульфоновую кислоту и N-хлорсукцинимид. Суспензию охлаждали до температуры окружающей среды, и выделяли продукт путем фильтрования с выходом приблизительно 61% с чистотой более 95%. Выделенную соль TsOH перекристаллизовывали до достижения чистоты более 99,7%. К одному эквиваленту соли TsOH, суспендированной в метаноле, добавляли 2М карбонат натрия (0,55 экв.) и 1,2 экв. ангидрида Boc. Суспензию перемешивали при комнатной температуре в течение ночи. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции смесь охлаждали до температуры менее 10°С, добавляли воду, и выделяли Вос-защищенное дихлорсоединение путем фильтрования. Полученный продукт промывали и сушили при 40°С с достижением выхода 95% и чистоты >97%. Полученное Вос-защищенное дихлорсоединение суспендировали в дихлорметане (10 объемов), и добавляли пиридина (5 объемов). Полученную смесь охлаждали до температуры менее 2°С, и добавляли трифторметансульфоновый ангидрид (1,25 экв.). Смесь перемешивали при 0-2°С в течение 10 минут и далее приливали к 10 объемам 6% водного раствора гидрокарбоната натрия. После промывки дихлорметаном органические фазы объединяли и сушили над сульфатом магния. Проводили очистку, после которой получали продукт (соединение 4') с выходом 90% и чистотой >98%. Соединение 4' растворяли в диметилформамиде и метаноле при комнатной температуре. Добавляли диизопропиламин (4 экв.). В атмосфере СО добавляли 1,3-бис(дифенилфосфино)пропан (0,1 экв.) и ацетат палладия (0,1 экв.). Реакционную смесь нагревали до кипения, и контролировали ход реакции посредством ВЭЖХ. Когда реакция была близка к завершению, смесь охлаждали до температуры окружающей среды. В результате обработки водой, этилацетатом и солевым раствором получали соединение 4'', которое использовали без дополнительной очистки. Соединение 4'' растворяли в метаноле и 2,4 М гидроксиде натрия (10 объемов каждого) и кипятили с обратным холодильником. Полученную смесь охлаждали до температуры окружающей среды, и добавляли толуол. Проводили обработку водой, после чего доводили значение рН до 2,3 посредством 3М хлористоводородной кислоты, и выделяли неочищенный продукт путем фильтрования с достижением выхода 53% и чистоты более 80%.
[00120] Пример 2
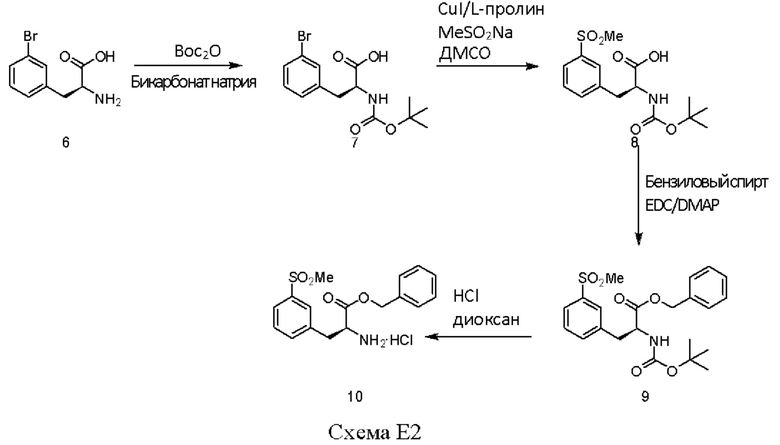
[00121] Защиту аминогруппы бромфенилаланина m-бутилкарбаматной группой (Воc) осуществляли с использованием бикарбоната натрия (3 эквивалента), m-бутилдикарбоната (Вос2О, 1,1 эквивалента) в диоксане и воде с получением соединения 7 с выходом 98%. Метилсульфоновую функциональную группу вводили путем обработки бромсодержащего соединения 7 иодидом меди (0,4 эквивалента), карбонатом цезия (0,5 эквивалента), L-пролином (0,8 эквивалента) и натриевой солью метансульфиновой кислоты (3,9 эквивалента) в диметилсульфоксиде (ДМСО) при 95-100°С в общем в течение 9 часов, при этом в течение указанного периода проводили два дополнительных введения иодида меди (0,2 эквивалента) и L-пролина (0,4 эквивалента). Соединение 8 выделяли с выходом 96%. Соединение 8 в виде карбоновой кислоты превращали в его бензиловый эфир, соединение 9, с выходом 99% с использованием бензилового спирта (1,1 эквивалента), диметиламинопиридина (DMAP, 0,1 эквивалента) и N-(3-диметиламинопропил)-N-этилкарбодиимида (EDC, 1,0 эквивалента). С аминогруппы соединения 9 снимали защиту путем добавления 4 н. раствора НСl в диоксане к соединению 9 при 0°С в метиленхлориде. Соль НСl соединения со свободной аминогруппой, соединение 10, выделяли с выходом 94%.
[00122] Пример 2А
[00123] Повторяли пример 2 с карбонатом калия вместо карбоната цезия.
[00124] Пример 2В
[00125] Вос-защищенный бромфенилаланин (соединение 7) (100 г) растворяли в ДМСО (400 мл) при перемешивании и дегазации аргоном. Добавляли метансульфинат натрия (98 г), иодид меди (28,7 г), карбонат калия (40 г) и L-пролин (26,75 г) при 28-30°С. Реакционную смесь нагревали до примерно 87°С в течение примерно 17-19 часов. Реакционную смесь охлаждали и гасили дробленым льдом, перемешивали в течение 30-40 минут, и доводили рН до значения от примерно 12 до примерно 3-4 лимонной кислотой (350 г). Реакционную смесь после гашения реакции фильтровали, подвергали экстракции дихлорметаном х3, промывали раствором хлорида аммония, промывали раствором бисульфита натрия и потом промывали солевым раствором. Неочищенный продукт в дихлорметане концентрировали под вакуумом до достижения влажности менее чем примерно 0,5%, и использовали на следующей стадии без дополнительного выделения. К неочищенному соединению 8 в дихлорметане добавляли бензиловый спирт и DMPA при перемешивании в атмосфере азота. Реакционную смесь охлаждали до 0-5°С. EDC-HCL (1,03 эквив.) добавляли при перемешивании в течение 30 минут. После завершения реакции по данным ТСХ и ВЭЖХ реакцию гасили раствором бикарбоната натрия, органический слой отделяли, и водный слой подвергали экстракции дихлорметаном. Органический слой промывали раствором лимонной кислоты, и объединенные органические слои промывали солевым раствором. Удаляли дихлорметан при 45-50°С, и полученный концентрат использовали на следующей стадии без дополнительного выделения. С аминогруппы соединения 9 снимали защиту путем добавления 4 н. раствора НСl в диоксане к соединению 9 при 10-15°С в метиленхлориде. Соль НСl соединения со свободной аминогруппой, соединение 10, выделяли посредством фильтрования из диэтилового эфира. Выделение соединения 10 осуществляли путем перекристаллизации с использованием системы растворителей диметилформамид/дихлорметан.
[00126] Пример 3
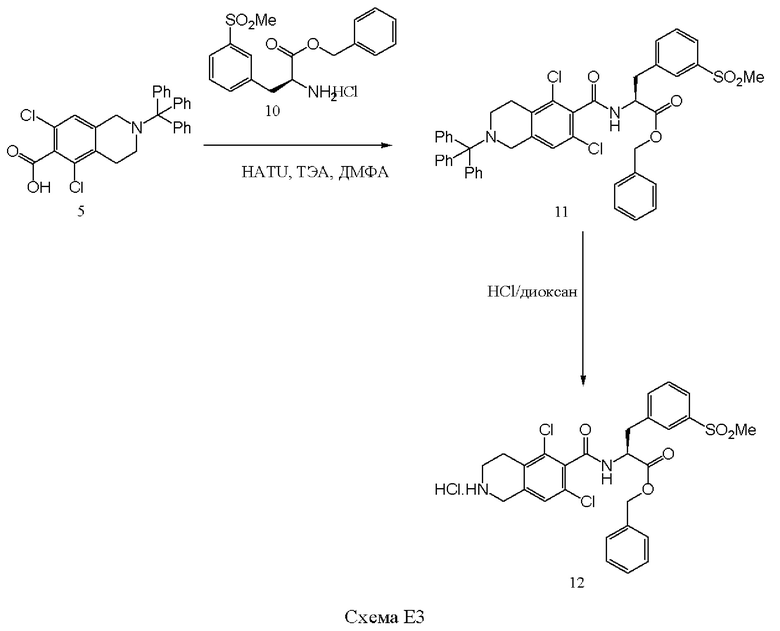
[00127] Соединение 5 обрабатывали триэтиламином (ТЭА, 5 эквивалента) и 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфатом (HATU, 1,25 эквивалента) в течение 10 минут в диметилформамиде (ДМФА), и далее к раствору добавляли соединение 10. После перемешивания при комнатной температуре в течение 18 часов, полученный продукт, соединение 11 выделяли с выходом 70%. Удаление тритильной защитной группы осуществляли путем обработки соединения 11 НСl в диоксане (4 н., избыток) при комнатной температуре в течение 2 часов, добавляли диэтиловый эфир, и полученный твердый продукт, соединение 12, выделяли посредством фильтрования с выходом 95%. Соединение 12 существует как в аморфной, так и в кристаллической форме, и может быть выделено в любой форме.
[00128] Пример 3А
[00129] Соединение 5 растворяли в изопропилацетате и охлаждали до 20-25°С. Добавляли тионилхлорид при охлаждении до 10-15°С, и медленно добавляли N-метилморфолин. Ход реакции контролировали посредством ВЭЖХ. Соединение 10, воду и изопропилацетат перемешивали при 15-20°С до получения раствора. Добавляли N-метилморфолин с последующим добавлением реакционной смеси соединения 5 (хлорангидрида соединения 5). Ход реакции контролировали посредством ВЭЖХ. После завершения реакции двухфазным слоям давали осесть, и водный слой удаляли. Верхний органический слой экстрагировали водой, а оставшийся органический слой подвергали дистилляции под вакуумом. Добавляли диоксан и IpAc, и проводили следующую дистилляцию. После того как остаток становился сухим, добавляли 4 н. безводную НСl в диоксане. Смесь перемешивали при температуре 20-25°С в течение 12 часов, и окончание снятия защиты контролировали посредством ВЭЖХ. После завершения снятия защиты полученную густую суспензию фильтровали, промывали IPАс и сушили под вакуумом при 45-55°С. Выход соединения 12 составил 88%.
[00130] Пример 4
[00131] Бензофуранилкарбонильный фрагмент соединения формулы I получали с применением различных схем (схем Е4, Е4А и Е4В).
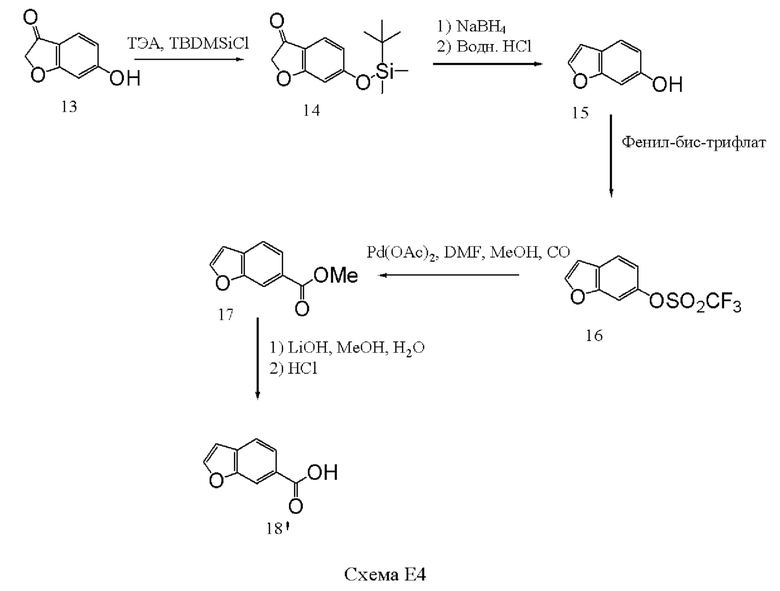
[00132] Бензофуранилкарбонильный фрагмент получали путем защиты гидроксильной группы соединения 13 путем проведения реакции с трет-бутилдиметилсилилхлоридом (1,0 эквивалента) и триэтиламином (ТЭА, 1,1 эквивалента) в ацетоне с получением соединения 14 с выходом 79%. Раствор соединения 14 в метаноле далее обрабатывали боргидридом натрия (1,0 эквивалента) при комнатной температуре в течение ночи. Реакцию гасили путем добавления ацетона, перемешивали при комнатной температуре еще в течение 2,5 часов, добавляли водный НСl (4 н.) при контролируемой температуре менее 28°С, добавляли тетрагидрофуран (ТГФ), и раствор перемешивали в течение ночи в атмосфере аргона и в отсутствие света. Полученный продукт, соединение 15, выделяли количественно путем экстракции в метиленхлорид, концентрировали при небольшом нагревании и использовали без дополнительной очистки. Трифлатный сложный эфир, соединение 16, получали с выходом 69% из соединения 15 путем проведения реакции соединения 15 с N-фенил-бис(трифторметансульфонимидом) (1,0 эквивалента) в метиленхлориде в течение 72 часов. Соединение 16 в смеси ДМФА, метанола и триэтиламина добавляли к подготовленному раствору палладия ацетата, 1,3-бис(дифенилфосфино)пропана (dppp), ДМФА и метанола в автоклаве. В автоклав добавляли монооксид углерода до давления 8 бар, и реакционную смесь нагревали при 70°С в течение 6 часов. После обработки соединение 17 выделяли с выходом 91%. Для гидролиза сложного эфира использовали гидроксид лития (4 эквивалента) в метаноле и воду, и проводили выделение соединения 18' с выходом 97%.
[00133] Пример 4А
[00134] Повторяли пример 4 с ангидридом трифторметансульфоновой кислоты и гидроксидом натрия в качестве реагентов для гидролиза сложного эфира.
[00135] Соединение 15 (6-гидроксибензофуран) перемешивали в дихлорметане и диизопропилэтиламине. Добавляли ангидрид трифторметансульфоновой кислоты (1,2 экв.), поддерживая температуру ниже 20°С. Ход реакции контролировали посредством ВЭЖХ. Реакцию гасили метанолом, удаляли растворитель под вакуумом, и неочищенный остаток, содержащий соединение 16, использовали без дополнительной очистки. Соединение 16 в виде неочищенного остатка растворяли в 4 объемах диметилформамида и 2 объемах метанола. К полученному раствору добавляли 0,02 экв. ацетата палладия, 0,02 экв. dppp и СО под давлением. Ход реакции контролировали посредством ВЭЖХ. После обработки соединение 17 выделяли в виде неочищенного маслянистого остатка без дальнейшей очистки. Остаток соединения 17 растворяли в метаноле (5 объемов), и добавляли 1 объем гидроксида натрия (27,65%). Смесь нагревали до 40°С до полного превращения по данным ВЭЖХ. Смесь охлаждали до температуры окружающей среды, и добавляли 3 объема воды. Доводили рН примерно до 2 посредством 3М хлористоводородной кислоты. Суспензию фильтровали, промывали водой и сушили с получением соединения 18' с общим выходом примерно 75% при чистоте >99,5%.
[00136] Пример 4В
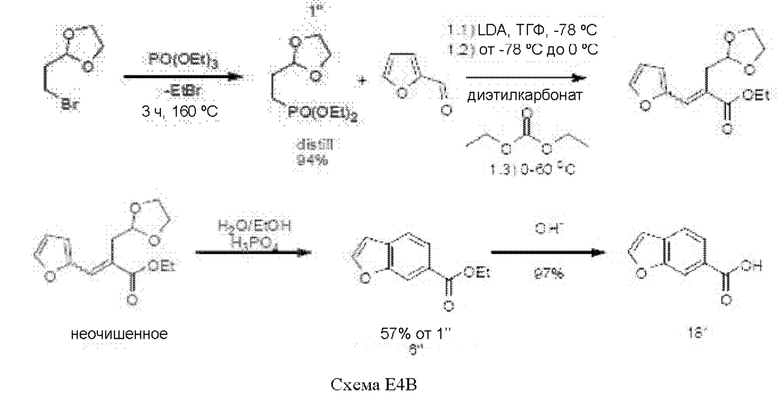
[00137] Диэтил-2-(1,3-диоксолан-2-ил)этилфосфонат, соединение 1'', получали из 2-(2-бромэтил)-1,3-диоксолана путем добавления триэтилфосфата. После удаления этилбромида путем дистилляции при 210°С неочищенную реакционную смесь охлаждали, и далее путем вакуумной дистилляции собирали соединение 1'' в виде бесцветного масла с выходом 94%.
[00138] На следующей стадии н-бутиллитий (2,15 эквивалента) в гексане охлаждали до -70°С, и добавляли диизопропиламин (2,25 эквивалента) при поддержании температуры ниже -60°С. Добавляли соединение 1'' (1 эквивалент), растворенное в тетрагидрофуране (ТГФ), в течение 30 мин при -70°С. Через 10 мин добавляли диэтилкарбонат (1,05 эквивалента), растворенный в ТГФ, в течение 30 мин, поддерживая температуру реакционной смеси ниже -60°С. После перемешивания в течение одного часа при -60°С, реакционной смеси давали нагреться до 15°С, и добавляли фуран-2-карбальдегид (1,3 эквивалента), растворенный в ТГФ. После перемешивания в течение 20 ч при комнатной температуре реакционную смесь упаривали на роторном испарителе досуха с получением этил-2-((1,3-диоксолан2-ил)метил-3-(фуран-2-ил)акрилат, который непосредственно использовали в следующей реакции.
[00139] Полученное неочищенное соединение (1 эквивалент) растворяли в этаноле и добавляли к смеси воды и фосфорной кислоты (85%, 15 эквивалента) в течение 30 мин при поддержании температуры ниже 50°С. После перемешивания в течение 20 ч при комнатной температуре добавляли дополнительные 200 мл фосфорной кислоты (85%) добавляли, и нагревали смесь до 50°С в течение еще двух часов. После удаления этанола путем выпаривания на роторном испарителе полученное вещество подвергали экстракции толуолом, промывали водой, сушили над сульфатом натрия, обрабатывали углем, фильтровали и сушили с получением масла. Данное масло подвергали дистилляции с получением этил-бензофуран-6-карбоксилата, соединения 6'', (температура кипения 111-114,5°С), которое кристаллизовалось при выдерживании. Соединение 6'' выделяли с выходом 57% отсоединения 1''.
[00140] Соединение 6'' (875 ммоль) растворяли в метаноле и тетрагидрофуране (ТГФ). Добавляли гидроксид натрия (4 М, 3 эквивалента) добавляли, и реакционную смесь перемешивали в течение ночи. После концентрирования путем упаривания на роторном испарителе, водный раствор подвергали экстракции метил-трет-бутиловым эфиром (МТБЭ), подкисленным до рН 2 путем добавления хлористоводородной кислоты (НСl), и охлаждали с получением мелких кристаллов бензофуран-6-карбоновой кислоты, то есть соединения 18'. Соединение 18' выделяли, промывали водой и сушили с достижением конечного выхода 97%.
[00141] Пример 5
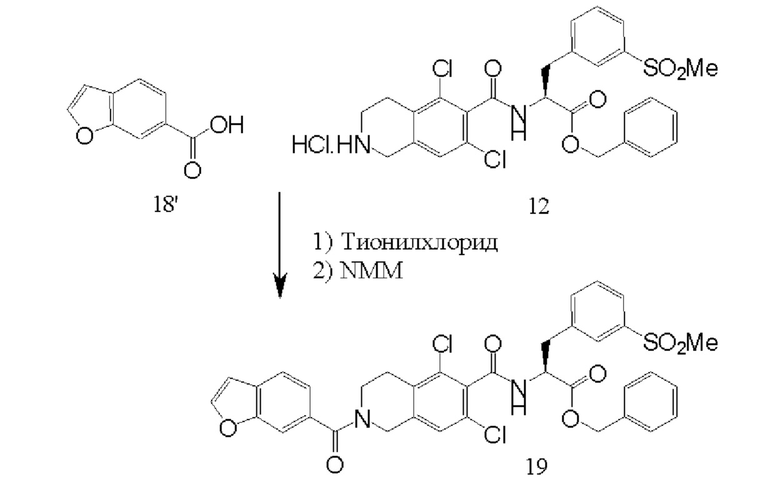
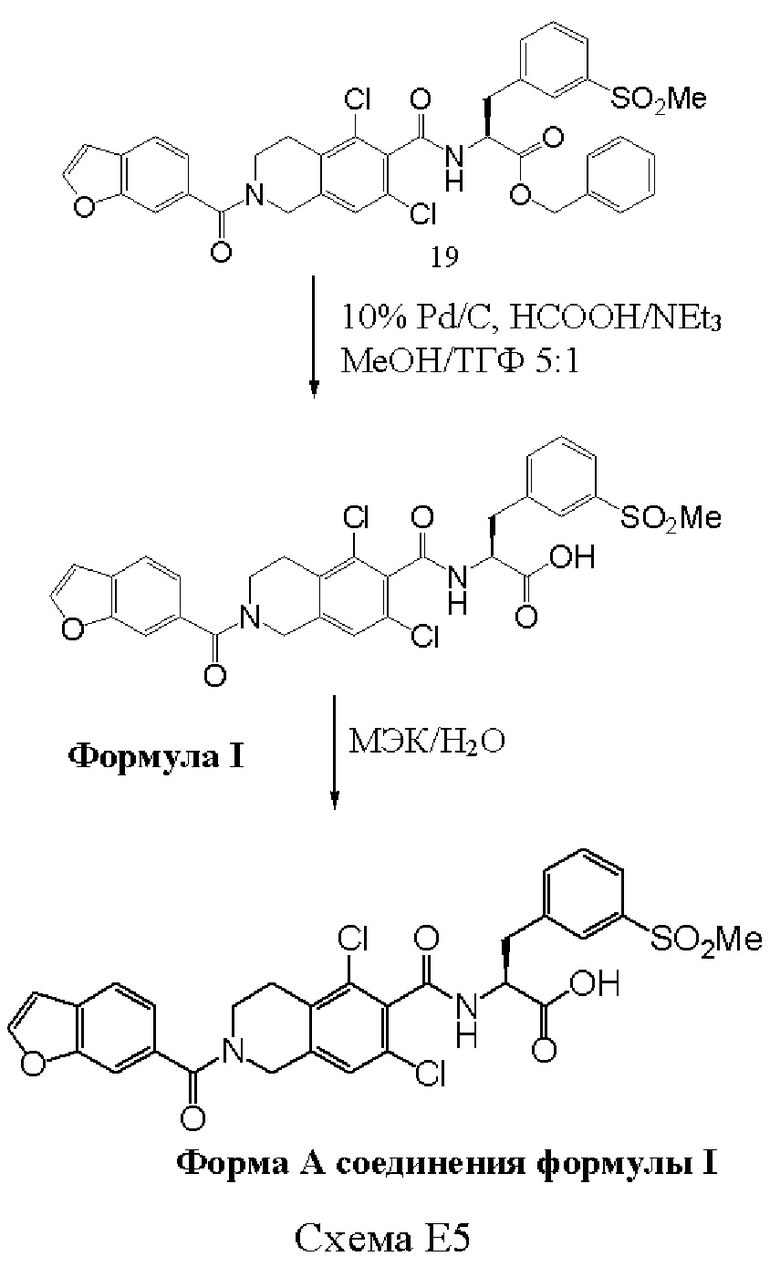
[00142] Бензофуранкарбоновую кислоту 18' обрабатывали оксалилхлоридом (1,2 эквивалента) и каталитическим количеством ДМФА, перемешивая в течение 5,5 часов до получения прозрачного раствора. Растворитель удаляли при пониженном давлении, и полученный хлорангидрид соединения 18' хранили в атмосфере аргона до применения на следующий день. Указанный хлорангидрид в метиленхлориде медленно добавляли к метиленхлоридному раствору соединения формулы 12 и диизопропилэтиламина (DIPEA), который охлаждали до 0-5°С. Температуре реакционной смеси не давали подняться выше 5°С, и после окончания добавления смесь перемешивали при 5°С еще 0,5 часа. После обработки водой и экстракции метиленхлоридом полученный продукт, соединение 19, выделяли с количественным выходом.
[00143] Бензилововую сложноэфирную группу соединения 19 удаляли путем гидрогенолиза с переносом (transfer hydrogenolysis) с применением 10% палладия на угле, муравьиной кислоты и триэтиламина в смеси метанол: ТГФ 5:1 с получением соединения формулы I с выходом 95%.
[00144] На заключительной стадии суспендирования в метилэтилкетоне (МЭК) получали форму А соединения формулы I. Полученный продукт промывали водой для удаления остаточного МЭК. В качестве альтернативы, продукт на стадии гидрогенолиза суспендировали в ацетонитриле с получением формы А соединения формулы I.
[00145] При использовании соединения формулы I непосредственно в виде неочищенного продукта реакции после гидрогенолиза с переносом и концентрирования из раствора в метиленхлориде получали аморфную форму соединения формулы I с чистотой 97%.
[00146] Пример 6
[00147] Альтернативная стратегия защиты приведена на схеме Е6.
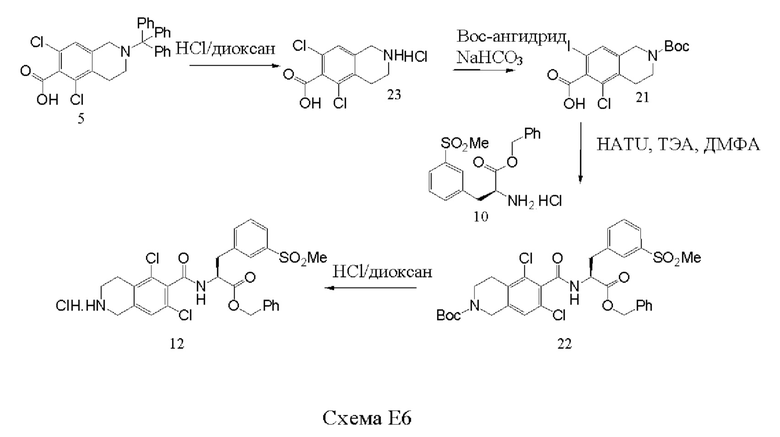
[00148] Защиту с помощью использовали для азота в кольце промежуточных соединений 21 и 22. С соединения 5 снимали защиту посредством НСl в диоксане с получением соединения 23 с выходом более 97%. Вводили защитную группу Воc с применением ди-трет-бутилдикарбоната (1,1 эквивалента), и получали соединение 21 с выходом более 95%. Соединение 10 подвергали реакции сочетания с соединением 21 с применением HATU и триэтиламина в ДМФА, в результате чего получали соединение 22. Полученный продукт, соединение 22, получали с количественным выходом и чистотой более 90%. После снятия защиты посредством НСl получали соединение формулы 12 с выходом 97,4%.
[00149] В результате гидрогенолиза с переносом, проведенного в отношении соединения 19, получали соединение формулы I с оптической чистотой (S)-энантиомера 98,5% по сравнению с оптической чистотой (S)-энантиомера 79-94,5%, полученной при гидролизе соответствующего метилового эфира.
[00150] Пример 6А
[00151] Повторяли пример 6 при использовании тионилхлорида вместо HATU с получением хлорангидрида для реакции сочетания с образованием амидной связи.
[00152] Пример 7
[00153] Реализовывали альтернативную стратегию по превращению соединения 19 в соединение формулы I путем щелочного гидролиза бензилового эфира (соединения 19).
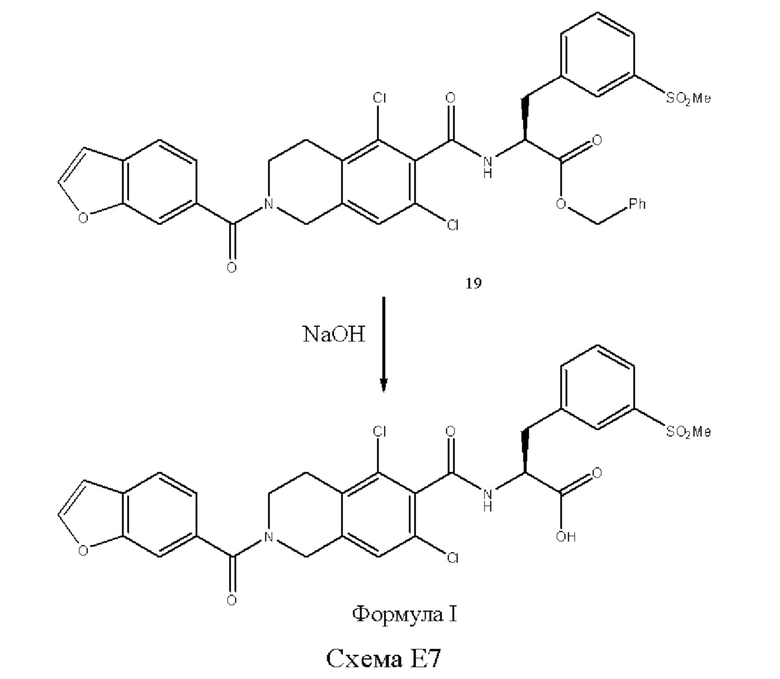
[00154] Соединение 19, представляющее собой ((S)-бензил-2-(2-(бензофуран-6-карбонил)-5,7-дихлор-1,2,3,4-тетрагидроизохинолин-6-карбоксамидо)-3-(3-метилсульфонил)фенил)пропаноат), (70,9 моль) растворяли в диоксане, и добавляли воду. Данный раствор охлаждали до 8°С. В течение 45 минут к 68,0 ммоль добавляли NaOH (0,5 М). После перемешивания в течение 2 ч диоксан удаляли путем выпаривания на роторном испарителе. Водный раствор дважды подвергали экстракции толуолом для удаления непрореагировавших исходных веществ. К водному слою добавляли этилацетат, и при интенсивном перемешивании указанный водный слой подкисляли до рН 2 посредством НСl (4 М водный раствор). После перемешивания слои разделяли, и водный слой подвергали экстракции этилацетатом. Объединенные этилацетатные фракции промывали солевым раствором, сушили над сульфатом натрия и упаривали досуха с получением пены, которая имела чистоту 95% по данным ВЭЖХ и имела э. и. 94,8%. Данную пену растворяли в метилэтилкетоне (МЭК), и к ней добавляли затравочные кристаллы (чистота 99%, э. и. 99%), что привело к интенсивной кристаллизации. После перемешивания в течение 24 ч суспензию фильтровали, промывали водой и сушили под вакуумом. Выход соединения формулы I составил 77% при чистоте 98,9% и оптической чистоте с э. и. 97,9%. При концентрировании маточного раствора была получена дополнительная партия соединения формулы I (чистота >98%).
[00155] Пример 8
[00156] Были проведены альтернативная реакция сочетания, снятие защиты и процесс очистки.
[00157] Соединение 18' растворяли в изопропилацетате, и доводили температуру до 20-25°С. Добавляли тионилхлорид, и доводили температуру до 10-15°С. Добавляли N-метилморфолин. Ход реакции контролировали посредством ВЭЖХ. Соединение 12 растворяли в воде, метилэтилкетоне и N-метилморфолине, и данную смесь охлаждали до 15-20°С. Медленно добавляли раствор хлорангидрида соединения 18' и перемешивали в течение 30 минут. Ход реакции контролировали посредством ВЭЖХ. Добавляли затравку соединения 19, и полученную смесь перемешивали в течение 1 часа, и далее часть органических растворителей отгоняли под вакуумом. Смесь охлаждали до 20-25°С, перемешивали в течение 2 часов и фильтровали. Остаток на фильтре промывали изопропилацетатом, и далее остаток на фильтре суспендировали в воде, фильтровали, промывали водой и сушили при 40°С под вакуумом с получением выхода 90%.
[00158] Соединение 19 смешивали с ацетоном, водой и тетрабутиламмония гидроксидом (ТВАН) и перемешивали при температуре 18-22°С до образования раствора. В течение 1 часа медленно добавляли 2 н. гидроксид натрия, и проводили перемешивание при 25°С до окончания реакции по данным ВЭЖХ. Смесь подвергали дистилляции под вакуумом при 30-35°С для удаления ацетона. Полученный раствор охлаждали до 10°С, и добавляли 4 н. водн. НСl, поддерживая температуру менее 15°С, до достижения рН ~2. Полученную суспензию перемешивали примерно в течение часа, фильтровали, а остаток на фильтре промывали водой. Влажный остаток на фильтре суспендировали в ацетоне и воде (примерно 2/1) и нагревали до 40-45°С для улучшения растворения. Полученный раствор фильтровали через 10-микронный фильтр. Смесь охлаждали до 18-22°С. Добавляли затравки, и полученную смесь перемешивали в течение 12 часов. Продукт собирали посредством фильтрования, промывали 30%-м водным раствором ацетона и сушили под вакуумом при 45-55°С с выходом 88%.
[00159] Пример 9
[00160] Неочищенное соединение формулы I перекристаллизовывали из 10 объемов метилэтилкетона при перемешивании в течение 3 дней с получением очищенного соединения формулы I с выходом 60-65%.
[00161] Пример 10
[00162] Неочищенное соединение формулы I перекристаллизовывали из 30%-го водного раствора ацетона и далее одного объема воды в течение 24-36 часов с получением очищенного соединения формулы I с выходом 73-77%.
[00163] Пример 10А
[00164] Неочищенное соединение формулы I перекристаллизовывали из 30%-го водного раствора ацетона и далее одного объема воды в течение 24-36 часов с получением очищенного соединения формулы I с выходом 80-90% после нескольких фильтрований. Полученное соединение формулы I не содержало определяемого остатка метилэтилкетона.
[00165] Пример 11
[00166] Кристаллическая форма II
[00167] Маломасштабный синтез
[00168] Приблизительно 50 мг кристаллической формы I растворяли в ацетоне (2,5 мл) при 50°С. Раствор фильтровали через фильтр тонкой очистки в предварительно нагретую емкость. Добавляли агент, препятствующий растворению, н-гептан, и смесь помещали в холодильник с температурой примерно 5°С. Полученное твердое вещество отфильтровывали и сушили под вакуумом.
[00169] Синтез в большем масштабе
[00170] Приблизительно 320 мг формы I растворяли в ацетоне (15 мл). Раствор фильтровали через фильтр тонкой очистки в предварительно нагретый флакон. Далее добавляли н-гептан (10 мл), и смесь охлаждали в холодильнике в течение 30 минут. В охлажденный раствор добавляли затравку вещества формы II, и давали смеси прийти в равновесие в течение 12 ч при 5°С. Полученное твердое вещество отфильтровывали и сушили в течение ночи под вакуумом.
[00171] Спектр ядерного магнитного резонанса 1Н кристаллической формы II, представленный на фигуре 6, соответствует структуре соединения и содержит 0,2 масс. % ацетона. Кристаллическая форма II характеризуется дифрактограммой рентгеновской порошковой дифракции, представленную на фигуре 4, и имеет игольчатую морфологию, как показано на фигуре 5. Анализ формы II посредством дифференциальной сканирующей калориметрии (ДСК) демонстрирует наличие небольшой эндотермы при температуре 37,8°С, вероятно связанное с потерей ацетона и/или воды, и переход в расплавленное состояние при температуре 155,5°С, как представлено на фигуре 7.
[00172] График термогравиметрического анализа кристаллической формы II представлен на фигуре 8. Наблюдается потеря массы, составляющая 1,5 масс. %, связанная с выделением воды в процессе плавления, и последующее начало разложения при температуре 260°С. Гравиметрический анализ сорбции влаги показал, что форма II является умеренно гигроскопичной, впитывая 3,0 масс. % воды при относительной влажности 60% и 3,4 масс. % воды при относительной влажности 90%. Содержание воды в форме II при влажности менее 40% составляет 2,8 масс. %, что близко к теоретическому содержанию воды в моногидрате данного соединения (2,9 масс. %). Содержание воды в форме II, определенное титрованием по методу Карла Фишера, составляет 3,2 масс. %, что также соответствует моногидрату данного соединения.
[00173] Несмотря на то, что в настоящем описании были показаны и описаны отдельные варианты реализации настоящего изобретения, специалисту в данной области техники очевидно, что такие варианты реализации представлены только для примера. Многочисленные вариации, изменения и замены, которые могут быть сделаны, не выходя за рамки настоящего изобретения, теперь будут очевидны специалистам в данной области техники. Следует понимать, что для реализации изобретения на практике могут быть использованы различные альтернативы вариантов реализации настоящего изобретения, приведенных в настоящем описании. Предполагается, что следующая формула изобретения определяет объем настоящего изобретения и что данная формула изобретения охватывает способы и структуры, входящие в объем пунктов формулы и их эквивалентов.
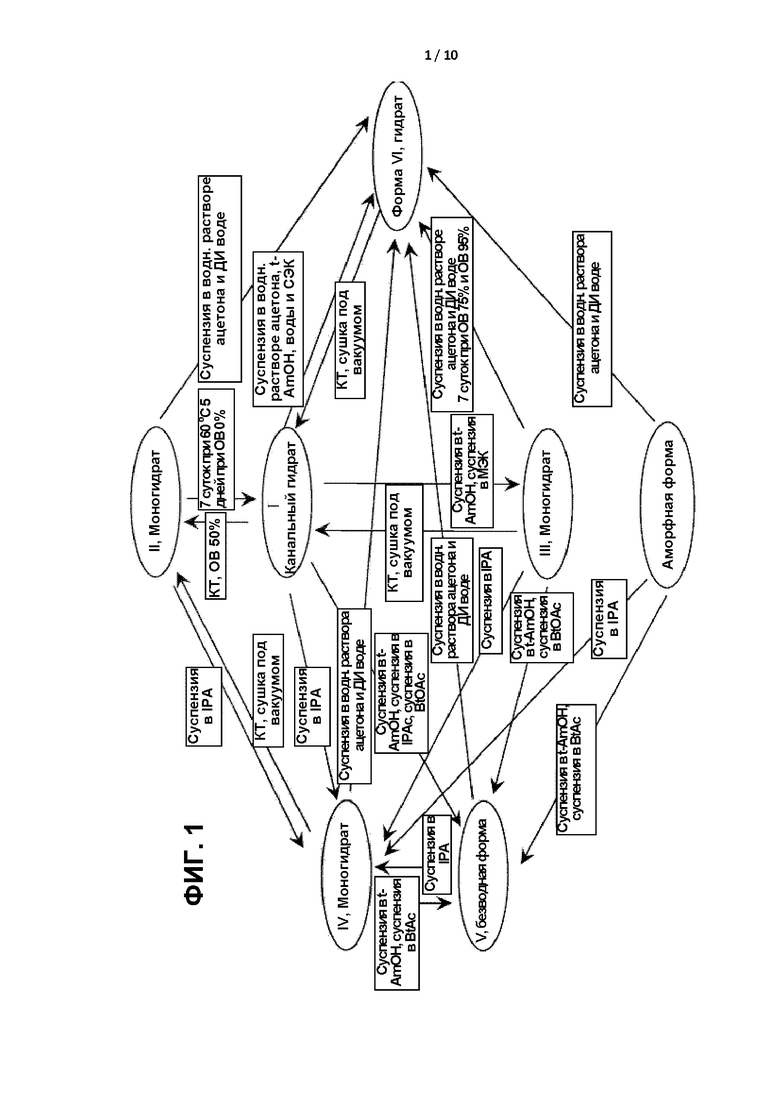
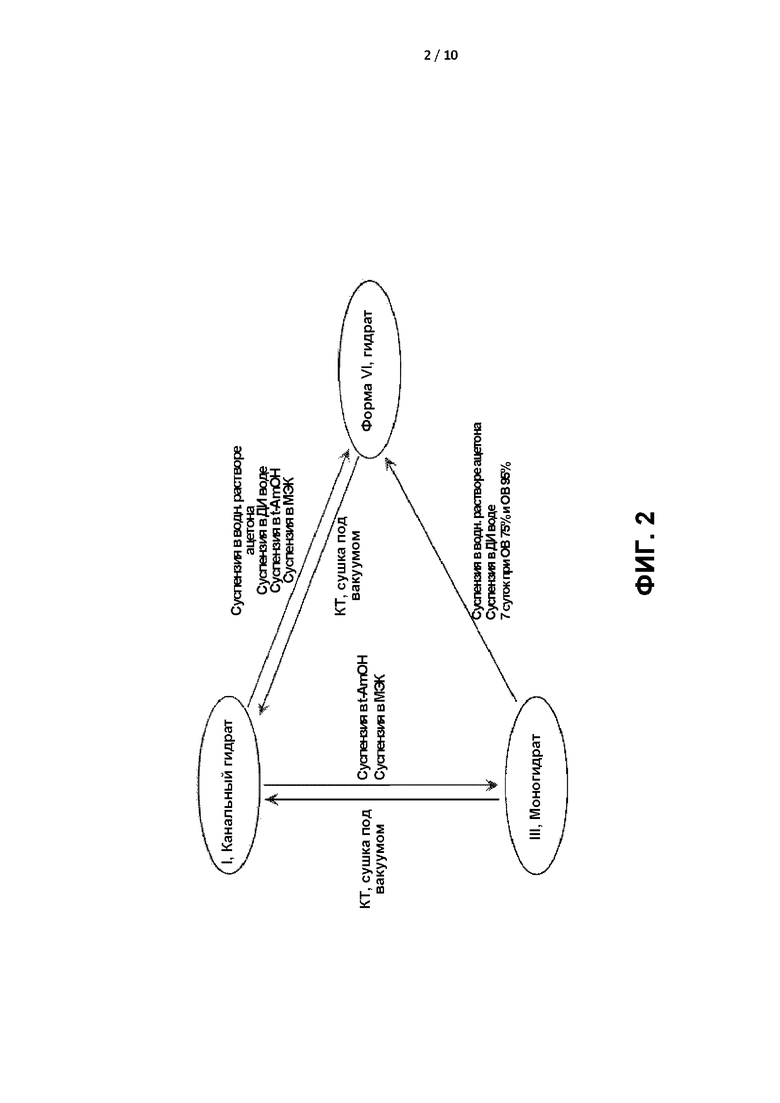

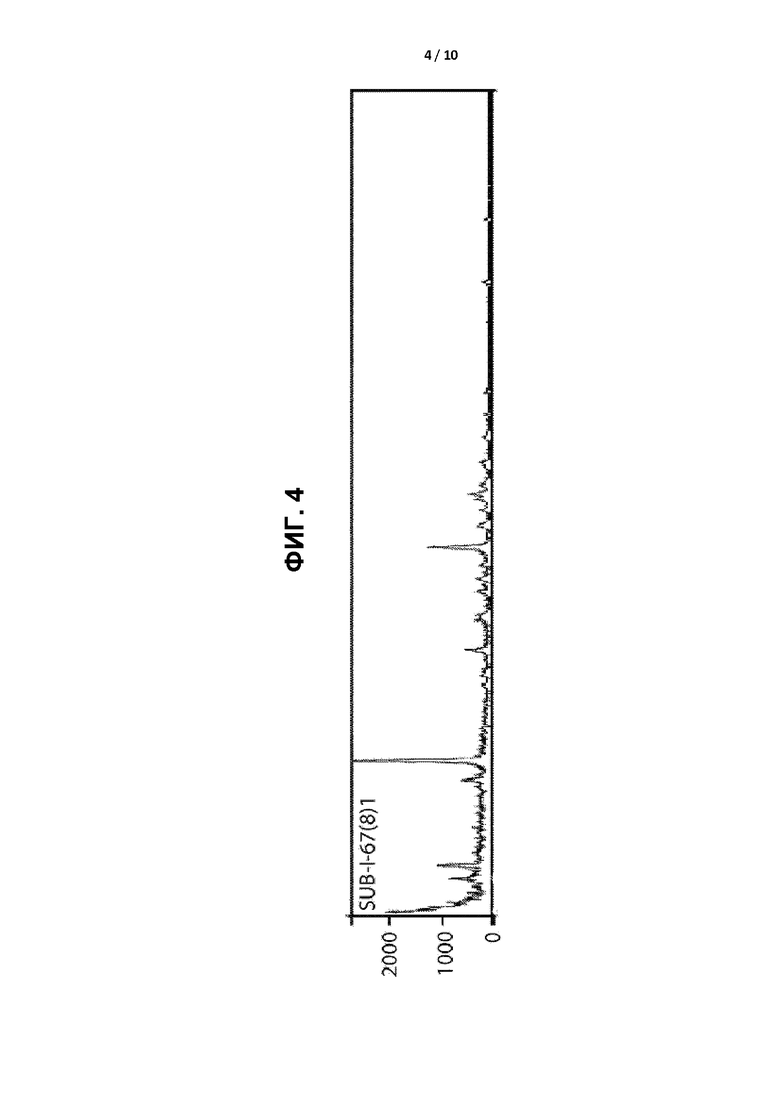

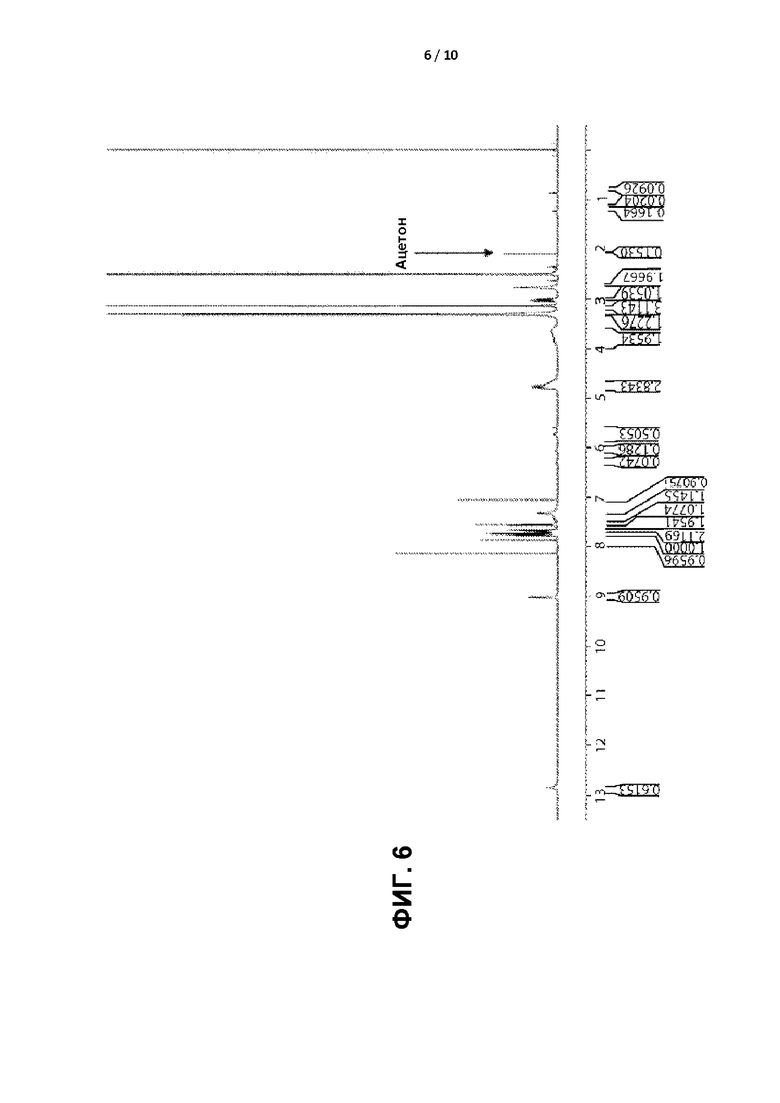
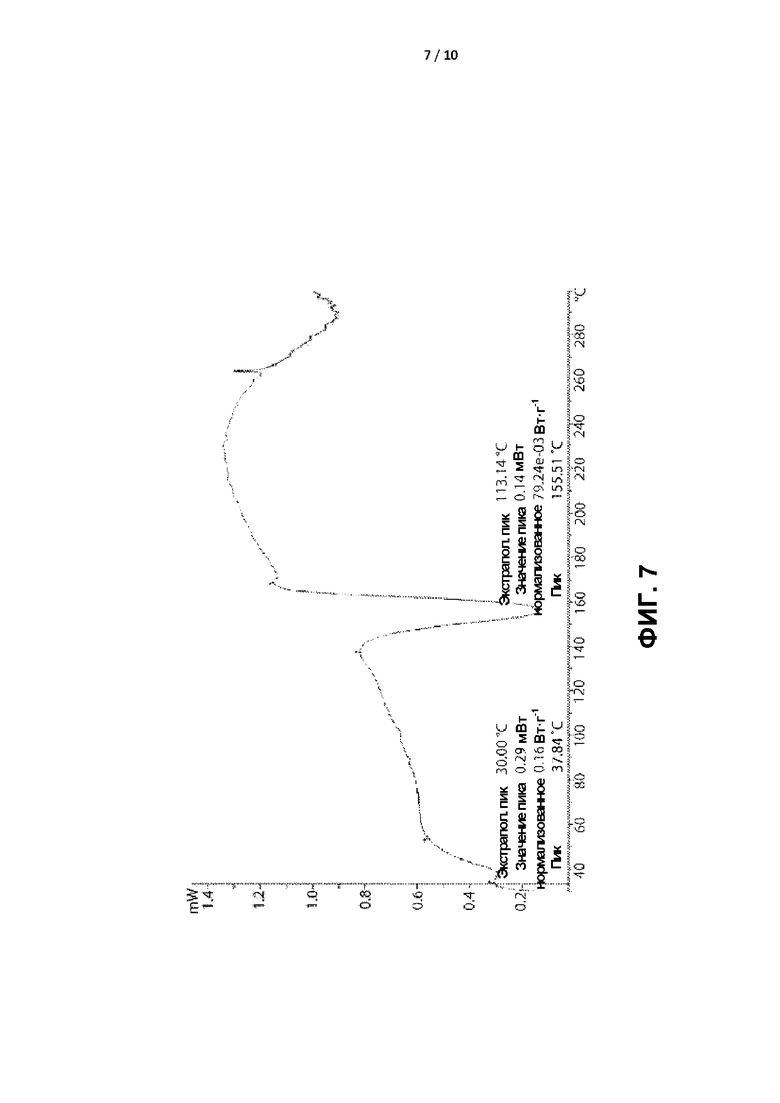
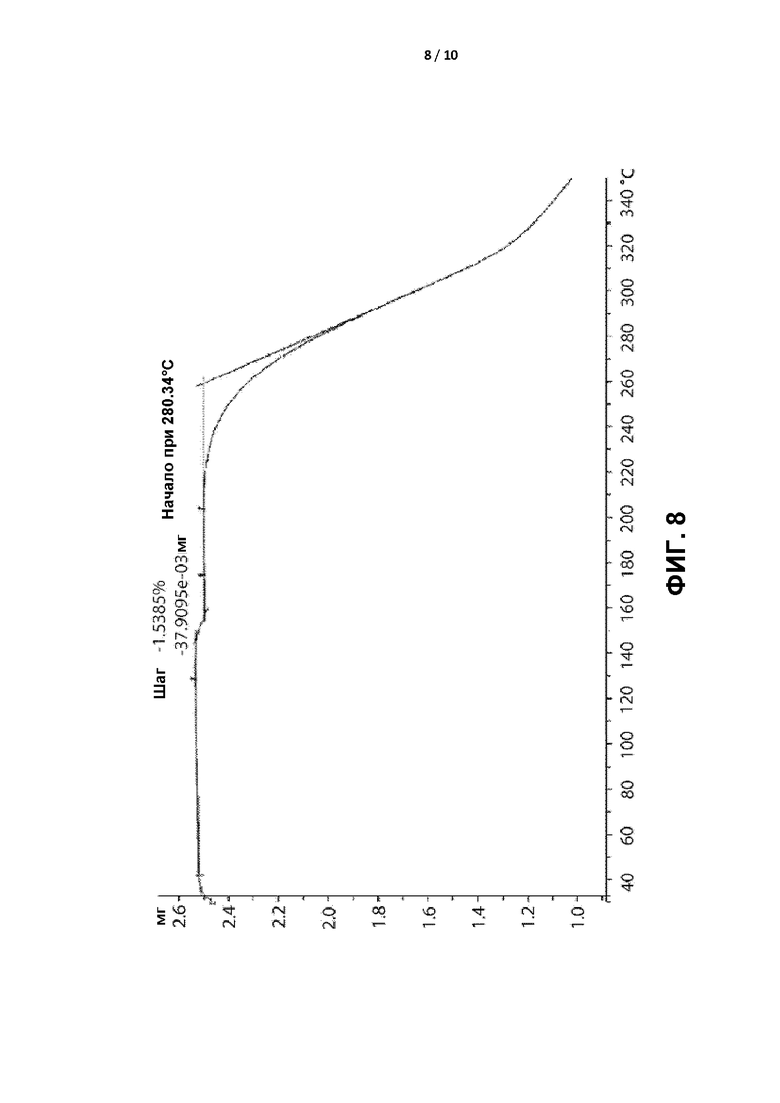
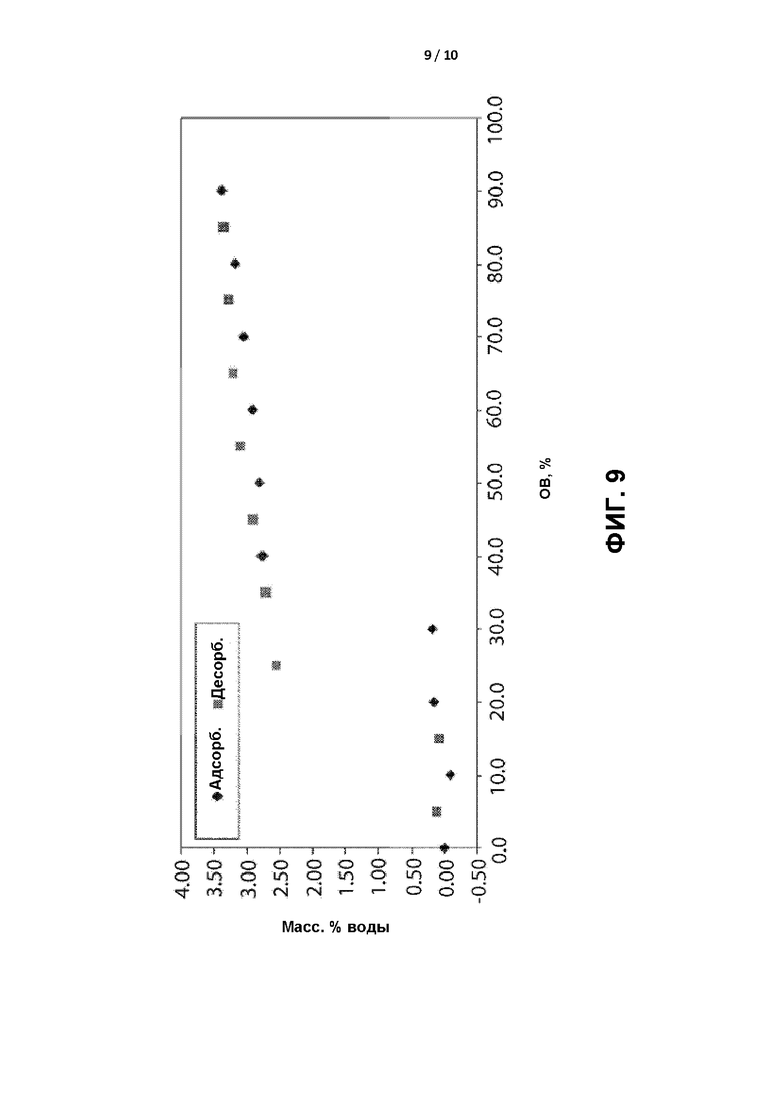
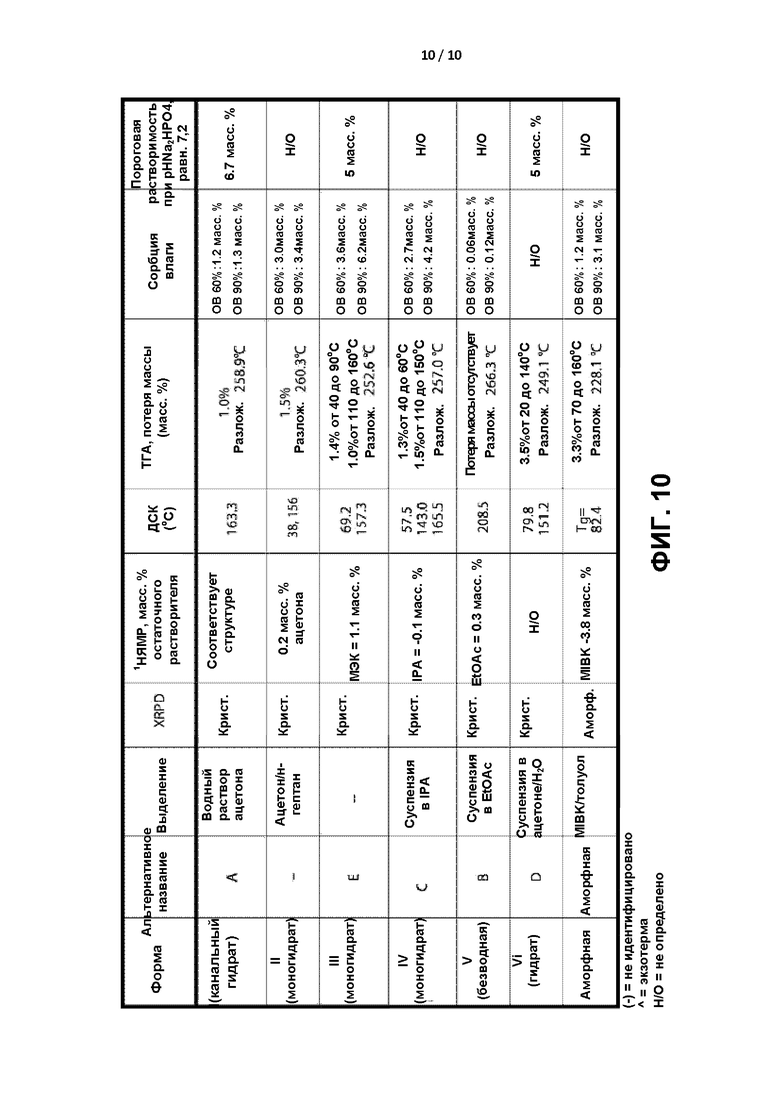
Изобретение относится к способу очистки соединения формулы I, включающему стадии: a) получения неочищенного соединения формулы I или его соли и перекристаллизации указанного неочищенного соединения с помощью водного раствора ацетона путём растворения неочищенного соединения Формулы I в 30% водном растворе ацетона с подогревом до 40-45°С, фильтрования полученного раствора и охлаждения указанного раствора до 18-22°С; и b) выделения указанного соединения формулы I или его соли путем удаления водного раствора ацетона. Технический результат: разработан новый способ очистки соединения формулы I, который позволяет получить соединение формулы I с повышенной чистотой. 7 з.п. ф-лы, 10 ил., 11 пр.
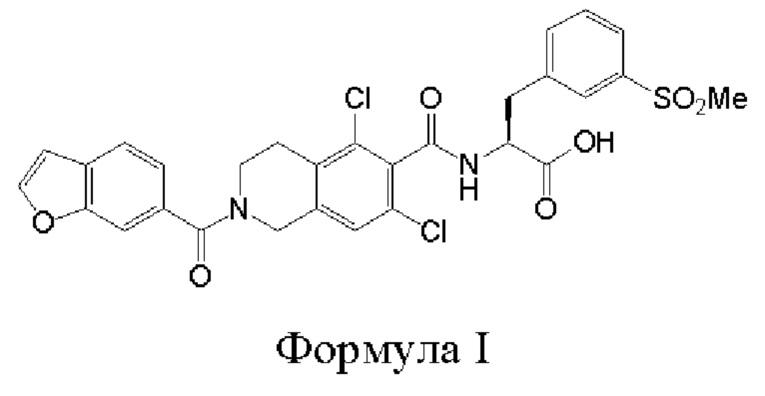
1. Способ очистки соединения формулы I:
Формула I
или его соли, включающий стадии:
a) получения неочищенного соединения формулы I или его соли и перекристаллизации указанного неочищенного соединения с помощью водного раствора ацетона путём растворения неочищенного соединения Формулы I в 30%-ном водном растворе ацетона с подогревом до 40-45°С, фильтрования полученного раствора и охлаждения указанного раствора до 18-22°С; и
b) выделения указанного соединения формулы I или его соли путем удаления водного раствора ацетона.
2. Способ по п. 1, в котором указанный водный раствор ацетона используют в количестве, составляющем примерно 7 объемов.
3. Способ по п. 1, дополнительно включающий добавление модификатора pH с получением pH менее примерно 5.
4. Способ по п. 3, где в указанном способе получают pH от примерно 1 до примерно 5.
5. Способ по п. 3, в котором указанный модификатор pH выбран из органической или минеральной кислоты.
6. Способ по п. 3, в котором указанный модификатор pH представляет собой хлористоводородную кислоту.
7. Способ по п. 1, в котором указанное удаление водного раствора ацетона осуществляют путем фильтрования.
8. Способ по п. 1, где указанный способ осуществляют в течение периода времени, составляющего от примерно 1 часа до примерно 48 часов.
| US 2011092707A1, 21.04.2011 | |||
| WO 2006125119A1, 23.11.2006 | |||
| ABDEL-MAGID AHMED F ET AL, "Hydrolysis of polypeptide esters with tetrabutylammonium hydroxide", TETRAHEDRON LETTERS, vol | |||
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| RU 2005117343 A, 27.01.2006. | |||

