Изобретение относится к области масштабируемого синтеза углеродных структур, в частности - к способу синтеза оксида графена, получившего широкое применение в электронике, медицине, фармакологии и строительстве.
Графен - новый 20-наноматериал толщиной в один атом, который представляет собой двумерную аллотропную модификацию углерода. Атомы углерода в графене находятся в состоянии sp2-гибридизации и соединены посредством σ- и π-связей в гексагональную двумерную кристаллическую решетку. Впервые графен был получен при помощи метода механической эксфолиации, а в дальнейшем были разработаны такие методы синтеза графена как химическое расслоение, эпитаксия и химическое осаждение из газовой фазы. Оксид графена представляет собой окисленную форму графена, которая содержит кислородные функциональные группы на поверхности графенового листа, (карбонильные, карбоксильные, лактольные, гидроксильные и эпоксидные группы). Использование окисленной формы графена имеет следующие преимущества: возможность получения стабильных водных дисперсий; возможность дальнейшей функционализации поверхности оксида графена с использованием различных функциональных групп, расположенных на поверхности оксида графена.
Оксид графена может применяться в различных областях биомедицины в качестве носителя для адресной доставки лекарств и биологически активных молекул, в качестве основы для создания противовирусных, антибактериальных и противогрибковых средств, в тканевой инженерии, как компонент биосенсоров и биомаркеров, для диагностики и биоимиджинга. Кроме того, он может найти применение в энергетике, для создания солнечных элементов, суперконденсаторов, топливных элементов и транзисторов.
В настоящее время для получения оксида графена используют различные методы. В работе L. Sun, В. Fugetsu, Mass production of graphene oxide from expanded graphite, Mater. Lett. 109 (2013) 207-210 описан масштабируемый метод получения оксида графена. Авторы перемешивали графит (5 г) и перманганат калия (15 г) до получения гомогенной смеси. Затем к смеси добавляли концентрированную серную кислоту (98%, 100 мл) при охлаждении с помощью льда и непрерывном перемешивании до образования однородной жидкой пасты. Далее продолжали перемешивание в течение 30 мин без охлаждения до образования пенообразного промежуточного продукта. Затем к реакционной смеси добавляли деионизированную воду (400 мл) и возобновляли перемешивание для предотвращения вскипания. Далее колбу помещали в водяную баню и выдерживали при 90°С в течение 1 ч до получения гомогенной суспензии темно-желтого цвета. После этого суспензию фильтровали, многократно промывали водой и центрифугировали (10000 об/мин, 2 ч на цикл) для удаления примесей. Заключительной стадией описанной методики являлась ультразвуковая обработка дисперсии оксида графена (мощность 100 Вт) для деагломерации частиц оксида графена.
Выход оксида графена в описанном методе составляет около 75%. Конечный продукт (оксид графена) содержит карбоксильные, карбонильные, гидроксильные, эпоксидные группы. Процентное содержание функциональных групп и их соотношение не изучалось. Для идентификации оксида графена авторы использовали следующие физико-химические методы анализа: высокоразрешающая электронная микроскопия (ВРЭМ), спектроскопия комбинационного рассеяния (Рамановская спектроскопия), инфракрасная (ПК) спектроскопия, сканирующая электронная микроскопия (СЭМ), рентгеновская фотоэлектронная спектроскопия, термогравиметрический анализ (ТГА), рентгеноструктурный анализ, оптическая спектроскопия, энергодисперсионная рентгеновская спектроскопия.
Недостатками вышеупомянутого метода являются:
- низкий выход конечного продукта (70-75%);
- невозможность масштабирования методики синтеза, поскольку при увеличении исходного количества графита процесс окисления затруднен, что, в свою очередь, приводит к снижению выхода и степени функционализации оксида графена;
- приведенный метод позволяют получить оксид графена с низким содержанием поверхностных функциональных групп. Анализ литературы показывает, что содержание функциональных групп в оксиде графена составляет 30-35% [N. Morimoto, Т. Kubo, Y. Nishina, Tailoring the oxygen content of graphite and reduced graphene oxide for specific applications, Sci. Rep. 6 (2016). https://doi.org/10.1038/srep21715]. Данный факт приводит к низкой стабильности дисперсий и необходимости использования поверхностно-активных веществ (ПАВ) для их стабилизации. При этом использование ПАВ ограничивает области применения таких дисперсий, например, в биомедицине;
- длительность процесса синтеза (1-3 дня);
- содержание примесей в конечном продукте, образующихся из непрореагировавшего графита (особенно при использовании большой исходной массы последнего).
Известен способ получения оксида графена [Патент РФ на изобретение №RU 2709594 C1]. В реактор при комнатной температуре добавляли 6 л 100% серной кислоты и загружали 200 г графита. Далее закрывали реактор крышкой с присоединенной мешалкой и включали перемешивание. На первой стадии при непрерывно работающей мешалке в реактор тонкой струей через капиллярную трубку добавляли 60 мл 60% пероксида водорода (плотность 1,2505 г/см3, 60 мл=75,03 г) и проводили перемешивание в течение 1 ч. На второй стадии в реакционную смесь добавляли 166 мл воды с помощью дозатора тонкой струей через капиллярную трубку и перемешивали еще в течение 30 мин. На третьей стадии в реактор загружали 600 г перманганата калия на протяжении 3 ч, поддерживая температуру при 20°С. После загрузки перманганата калия перемешивание при 20°С продолжали еще 1 ч. На четвертой стадии к реакционной смеси при непрерывном перемешивании в течение 40 мин добавляли 331 мл дистиллированной воды. В таком виде реакционную смесь оставляли на ночь. Выполнение всех перечисленных выше технологических операций занимает полную рабочую смену. На следующий день включали перемешивание и медленно приливали в реактор 24 л воды со скоростью, исключающей превышение температуры реакционной смеси выше 60°С. После этого в реактор при непрерывном перемешивании добавляли 1 л 60% пероксида водорода, при этом цвет реакционной смеси переходил из коричневого в золотисто-желтый, что связано с восстановлением диоксида марганца перекисью водорода. Полученная реакционная смесь представляла собой суспензию объемом приблизительно 32 л, содержащую блестящие чешуйки золотистого цвета. Эту суспензию переносили на полипропиленовый фильтр, фильтровали и промывали 1 М раствором серной кислоты (9,2%). Промывку проводили до полного удаления сульфата марганца из суспензии. Для тестирования полноты отмывки к пробе фильтрата добавляли водный раствор аммиака или раствор щелочи до щелочной реакции. Образование черного или бурого осадка, или помутнение свидетельствует о наличии сульфата марганца. Когда промывка доведена до конца, проба фильтрата после подщелачивания остается совершенно прозрачной и бесцветной. Количество 1 М серной кислоты, необходимое для полной промывки, зависит от режима фильтрации, но ориентировочно составляет 30-35 л. Полученный продукт, содержащий некоторое количество серной кислоты, отмывали от кислоты с применением диализатора барабанного типа, после чего добавлением воды концентрацию оксида графена доводили концентрации 0,5% в расчете на исходный графит.
Недостатками данного метода являются:
- длительность синтеза (более одного дня);
- в патенте отсутствует информация о выходе продукта;
- отсутствует информация о концентрации полученного оксида графена в водной дисперсии;
- для характеризации образца авторы использовали только рентгенофазовый анализ, который не позволяет судить о химическом составе оксида графена.
В патенте [№RU 2720780 C2 Способ синтеза оксида графена] для получения оксида графена на первой стадии смешивали 75 мл концентрированной серной кислоты с 90 г сухого льда с размером частиц 1-5 мм, затвердевшую смесь загружали в автоклав вместимостью 500 мл, выполненный из нержавеющей стали. Затем помещали в автоклав смесь, состоящую из 12 г измельченного природного графита (размер частиц составляет D50=15 мкм и D95 - 70 мкм), 72 г перманганата калия и 150 г сухого льда с размером частиц 1-5 мм. Автоклав закрывали, герметизировали и встряхивали на шейкере со скоростью вращения 12 об/мин в течение 3 ч. С началом вращения включали обдув горячим воздухом, направленный на внешнюю поверхность стенок автоклава. В течение 1,5 ч внешние стенки автоклава достигают температуры 40°С, которую поддерживали до конца вращения. Затем автоклав переносили на вибрационный стол, закрепляли горизонтально на столе и подвергали воздействию вибрации с частотой 50 Гц и амплитудой 2 мм в течение 1 ч, поддерживая температуру внешних стенок автоклава около 40°С. Затем давление в автоклаве медленно снижали в результате стравливания углекислого газа через бак с водой. Затем открывали автоклав и выгружали интеркалированный графит. Количество выгруженного интеркалированного графита составляло 330 г. Полученный материал смешивали с 2 л воды при перемешивании на магнитной мешалке. Затем во время перемешивания в реакционную смесь загружали 100 г дигидрата щавелевой кислоты порциями по 10 г, суспензия при этом меняла цвет с фиолетового на желтый. Затем суспензию подвергали ультразвуковой обработке в течение 15 мин в ванне с использованием источника ультразвуковых волн мощностью 100 Вт. После этого суспензию переносили в восемь диализных мешков, изготовленных из фторопласта-4 с размером пор 0,6 мкм, мешки помещали в бак с водой емкостью 24 л на магнитной мешалке. Перемешивание останавливали через 24 ч, когда внутри и снаружи мешков величина рН составляет 1. Затем переносили материал из диализных мешков в полиэтиленовый пакет и после добавляли в пакет 170 мл 30% соляной кислоты, проводя обработку ультразвуком в течение 15 мин. После материал переносят в восемь новых диализных мешков, помещенных в бак с водой (24 л) при перемешивании на магнитной мешалке в течение 24 ч. Диализные мешки закрепляли на баке с водой таким образом, чтобы вначале уровень суспензии в диализных мешках был выше уровня воды в баке. За счет перетекания жидкости через стенки мешков разница уровней исчезает во время диализа. Объем суспензии после диализа примерно соответствует объему суспензии перед добавлением 30% соляной кислоты. Первоначальная разница уровней необходима для поддержания высокой концентрации оксида графена в водной суспензии после добавления соляной кислоты и диализа. Процедура добавления 170 мл соляной кислоты и воздействие ультразвуком в течение 15 мин с последующим диализом в течение 24 ч (время необходимое для достижения выравнивания рН внутри и снаружи диализных мешков) в новых мешках с новой водой в баке повторяется пять раз. Затем аналогичный процесс диализа повторяется еще шесть раз, но исключает добавление соляной кислоты. В результате получили 2,3 л коричневой суспензии оксида графена с рН=7,0. Затем 20 мл суспензии смешивали с 40 мл воды в полиэтиленовом пакете, помещенном в ультразвуковую ванну на 15 мин. Разбавленная и концентрированная суспензии оксида графена стабильны, и выпадение осадка в процессе хранения в течение одного месяца не наблюдалось. Концентрированную суспензию подвергали сублимационной сушке, в результате которой получили 15,2 г порошкообразного оксида графена. Полученный оксид графена исследовали методом рентгеновской фотоэлектронной спектроскопии для определения содержания кислорода (42 весовых процента).
Недостатками данного метода являются:
- длительность синтеза (более одного дня);
- необходимость выполнения трудоемких процедур выделения и очистки конечного продукта;
- в патенте отсутствует информация о выходе продукта;
- отсутствует информация о квалификации использованных реактивов;
- приведенных методов идентификации полученного продукта недостаточно для полноценной характеризации оксида графена. В частности, не указана информация о химическом составе оксида графена;
- низкое содержание кислородсодержащих функциональных групп (42%).
Наиболее близким техническим решением является способ производства окисленного графена [Toward Large-Scale Production of Oxidized Graphene. Nanomaterials (Basel) 2020 Italy], который выбран нами в качестве прототипа.
В предложенном способе-прототипе 3,0 г графитового порошка (99,99%) добавляли к 70 мл концентрированной серной кислоты (H2SO4, 95,0-98,0%) при перемешивании и охлаждении при помощи льда. Затем постепенно добавляли 9,0 г KMnO4 (99,0%), поддерживая температуру суспензии ниже 20°С. Полученную смесь переносили на масляную баню и интенсивно перемешивали при 50°С в течение 30 мин. Затем добавляли дистиллированную воду (150 мл), и раствор перемешивали в течение 20 минут при 90°С. После чего добавляли дополнительное количество дистиллированной воды (500 мл) с последующим постепенным добавлением 15 мл перекиси водорода (Н2О2, 30%) и перемешивали до изменения цвета раствора от темно-коричневого до бледно-желтого. Полученный продукт многократно промывали водным раствором соляной кислоты (HCl, соотношение кислоты к воде составляло 1:10; исходная концентрация кислоты - 37%) и дистиллированной водой (каждый раз по 10 мин) до значения рН ~ 6. Отделение осадка от раствора осуществлялось при помощи центрифугирования при 3000 об/мин. Затем проводили высушивание при 80°С в течение 24 ч. На заключительном этапе ля дополнительной эксфолиации полученный продукт повторно диспергировали в дистиллированной воде в ультразвуковой бане в течение 1 ч.
Несомненно, преимуществами известного способа-прототипа являются отсутствие выделения токсичных газов при его реализации, экспрессность синтеза и отсутствие длительной ультразвуковой обработки и трудоемких этапов очистки конечного продукта.
Однако, на наш взгляд, данный способ не лишен ряда существенных недостатков.
1. Отсутствие данных по масштабируемости методики синтеза оксида графена. В прототипе указано, что предложенная методика может быть масштабируемой, однако в работе отсутствуют доказательства масштабируемости методики и не указаны пределы масштабирования.
2. Низкое содержание кислородсодержащих функциональных групп. По данным энергодисперсионной рентгеновской спектроскопии содержание кислорода составляет 50,3%.
3. Отсутствуют данные количественного определения кислородсодержащих функциональных групп. По данным ИК-спектроскопии полученный образец содержит меньшее количество функциональных групп, что является ограничением для дальнейшей модификации поверхности оксида графена.
4. Отсутствуют данные по стабильности полученных водных дисперсий оксида графена.
5. В способе-прототипе не приводятся данные о выходе конечного продукта.
Технический результат настоящего изобретения состоит в разработке масштабируемой методики синтеза оксида графена с увеличенным содержанием кислородсодержащих функциональных групп (до 85%) путем использования дополнительных окислителей и выбором технологических режимов, позволяющих проводить дальнейшее модифицирование поверхности оксида графена и получать стабильные водные дисперсии.
Этот результат достигается тем, что в известном способе масштабирования синтеза оксида графена, включающем диспергирование графитового порошка в концентрированной серной кислоте, добавление к полученной смеси перманганата калия, разбавление ее дистиллированной водой, фильтрацию осадка и промывку его до нейтрального значения рН с последующим высушиванием, согласно изобретению, дополнительно при диспергировании графитового порошка в концентрированной серной кислоте добавляют оксид фосфора(V), затем полученную смесь обрабатывают ультразвуком в течение 10 мин и после добавления перманганата калия к смеси добавляют нитрат натрия, а после разбавления ее дистиллированной водой, температуру смеси повышают до 95°С в течение 1,5 ч с последующим добавлением дополнительного количества дистиллированной воды, а фильтрацию осадка осуществляют на фильтре Шотта, затем полученный осадок промывают дистиллированной водой и разбавленной HCl (концентрация - 5 мас. %) с последующим высушиванием при температуре 65°С в течение 5 ч и диспергируют в дистиллированной воде в ультразвуковой бане в течение 1 ч.
Использование в предложенном способе синтеза дополнительных окислителей оксид фосфора(V) Р2О5 и нитрат натрия NaNO3, приводит к значительно более глубокому окислению графена и увеличению процентного содержания кислородсодержащих функциональных групп. Кроме того, после добавления нитрата натрия, происходит выделение оксида азота(IV), что приводит к большой степени эксфолиации оксида графена и увеличению его площади поверхности.
Обработка смеси ультразвуком в течение 10 минут позволяет усилить интеркаляцию серной кислоты и оксида фосфора(V) в межслоевое пространство графита, что в дальнейшем приводит к его более полной конверсии в оксид графена.
Введение в схему синтеза стадии, связанной с температурной обработкой реакционной смеси при 95°С в течение 1,5 ч, позволяет регулировать размер полученных наночастиц (размер частиц уменьшается с увеличением продолжительности обработки) и тем самым регулировать стабильность водных дисперсий.
Фильтрация осадка на фильтре Шотта позволяет очистить оксид графена от примесей минеральных солей и кислот, а последующее высушивание оксида графена при температуре 65°С в течение 5 ч позволяет избавиться от присутствия воды в образце. Дополнительное диспергирование в дистиллированной воде в ультразвуковой бане в течение 1 ч проводилось для более полной эксфолиации оксида графена.
Для лучшего понимания заявленного изобретения приводим методику синтеза:
Графит и оксид фосфора(V) (Р2О5) диспергировали в концентрированной серной кислоте (H2SO4, 98%). Полученную смесь обрабатывали ультразвуком в течение 10 мин и перемешивали в течение 15 мин при комнатной температуре. После этого к реакционной смеси постепенно добавляли нитрат натрия (NaNO3) при перемешивании и охлаждении при помощи льда в течение 30 мин. Далее к полученной смеси постепенно добавляли перманганата калия (KMnO4) при непрерывном перемешивании в течение 30 мин (температура системы поддерживалась при 5°С), а затем постепенно повышали температуру до 40°С в течение 1 ч при непрерывном перемешивании с последующим постепенным добавлением дистиллированной воды Н2О (1). Далее температуру реакционной повышали до 95°С в течение 1,5 ч с последующим добавлением дополнительного количества дистиллированной воды Н2О (2) и 30% Н2О2. Полученный осадок отфильтровывали на фильтре Шотта (пористость - 10), промывали дистиллированной водой и разбавленной HCl (концентрация - 5 мас. %) до нейтрального значения рН, высушивали при 65°С и повторно диспергировали в дистиллированной воде в ультразвуковой бане в течение 1 ч для эксфолиации.
Пятикратный избыток H2SO4 был взят для приготовления более однородной дисперсии графита и других окислителей. Более того, было установлено, что именно такое соотношение между исходным графитом и H2SO4, а также другими окислителями (Р2О5, NaNO3, KMnO4) приводит к повышенному содержанию кислородсодержащих функциональных групп и более полной конверсии графита в оксид графена. Десятикратный избыток графита по сравнению с NaNO3 обусловлен мерами предосторожности в связи с сильным тепловым эффектом, выделяющимся при взаимодействии компонентов, и, соответственно, взрывоопасностью реакционной смеси. Добавление воды к реакционной смеси, содержащей графит и окислители в соотношении графит (кг): вода (л) = 1:2,5, обусловлено следующими причинами: необходимостью активации химической реакции при 95°С и взрывоопасностью полученной смеси (без разбавления водой).
Дополнительное добавление воды к реакционной смеси, содержащей графит и окислители в соотношении графит (кг): вода (л) = 1:0,1, а также Н2О2 в соотношении графит (кг): Н2О2 (л)=1:5 необходимо для завершения реакции окисления графита.
В Таблице 1 представлено соотношение реагентов для проведения синтеза.

Для лучшего понимания приводим краткое описание чертежей.
Фиг. 1 - Дисперсии оксида графена в диапазоне концентраций С=1,8-9,0 г⋅л-1.
Фиг. 2 - Спектр 13С ЯМР оксида графена, полученный при прямом возбуждении (время контакта - 2 мс), где:
1 - химический сдвиг (м. д.);
2 - структурный фрагмент С-О-С, расположенный на плоскостях графенового листа;
3 - структурный фрагмент С-ОН, расположенный на плоскостях и краях графенового листа;
4 - структурный фрагмент 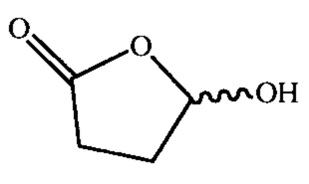 на краях графенового листа;
на краях графенового листа;
5 - структурный фрагмент С=С в плоскости графена;
6 - структурный фрагмент С=O на краях графенового листа;
7 - структурный фрагмент -СООН на краях графенового листа.
Фиг. 3 - Спектры рентгеноструктурного анализа графита (синяя линия) и оксида графена (голубая линия), где:
8 - интенсивность (пр. е.);
9 - угол 2θ.
Фиг. 4 - Комплексный термический анализ оксида графена (ТГ - сплошная линия, ДТГ - пунктирная линия),
где: 10 - ТГ (%), 11 - 1 (°С), 12 - ДТГ (%⋅мин-1).
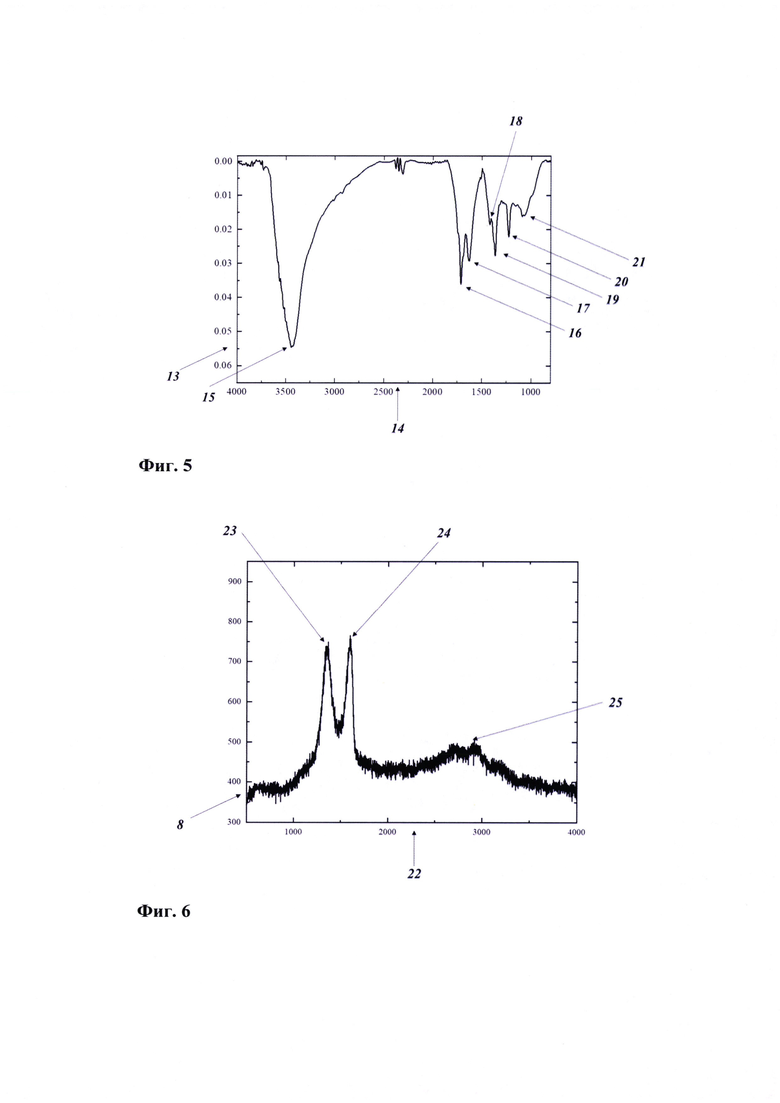
Фиг. 5 - ИК-спектр оксида графена, где:
13 - Т(%);
14 - v (см-1);
15 - валентные колебания О-Н фрагмента карбоксильной группы (3440 см-1);
16 - валентные колебания С=0 фрагмента карбоксильных групп (1718 см-1);
17 - колебания ароматических доменов графена (1630 см-1);
18 - колебания С-ОН фрагмента карбоксильных групп (1418 см-1);
19 - колебания С-ОН фрагмента спиртовых групп (1363 см-1);
20 - валентные колебания С-ОН в составе лактольной группы (1226 см-1);
21 - валентные колебания С-О, в составе эпоксидных и спиртовых групп (1085 см-1).
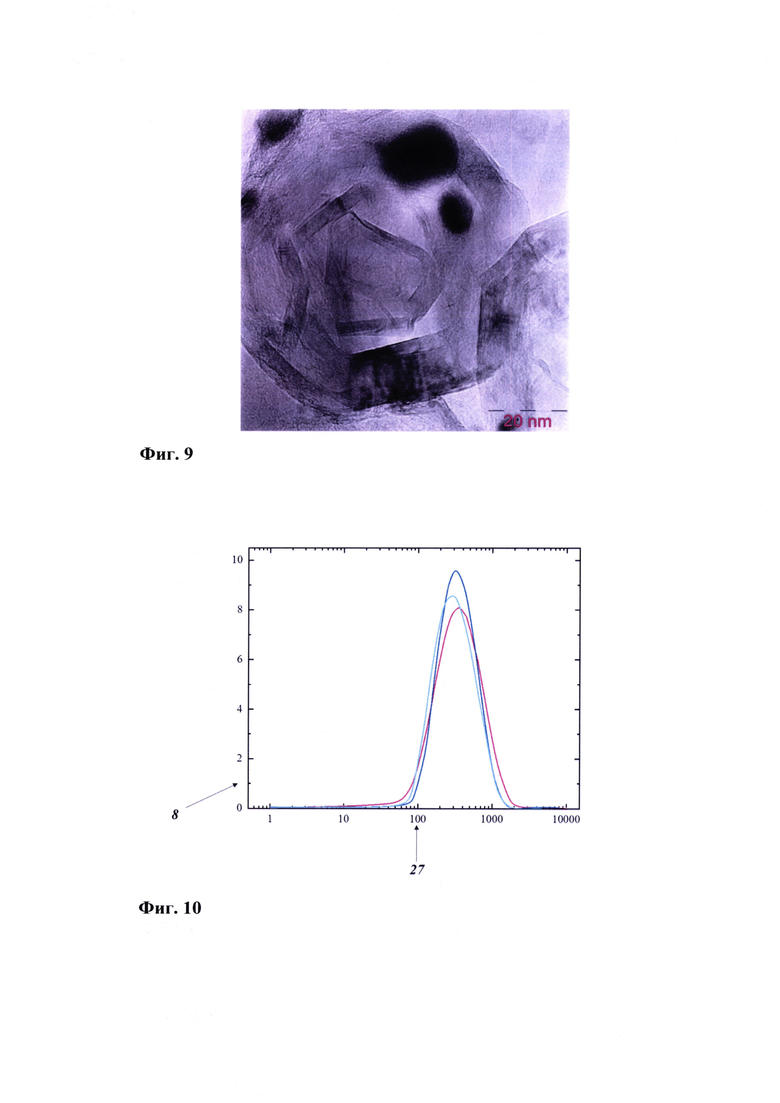
Фиг. 6 - Рамановский спектр оксида графена, где:
8 - интенсивность (пр. е.);
22 - Рамановское смещение (см-1);
23 - полоса D;
24 - полоса G;
25 - полоса 2D.
Фиг. 7 - Спектр оптического возбуждения оксида графена (С=1,8 г⋅л-1), где:
8 - интенсивность (пр. е.);
26 - λ (нм).
Фиг. 8 - СЭМ-изображение оксида графена.
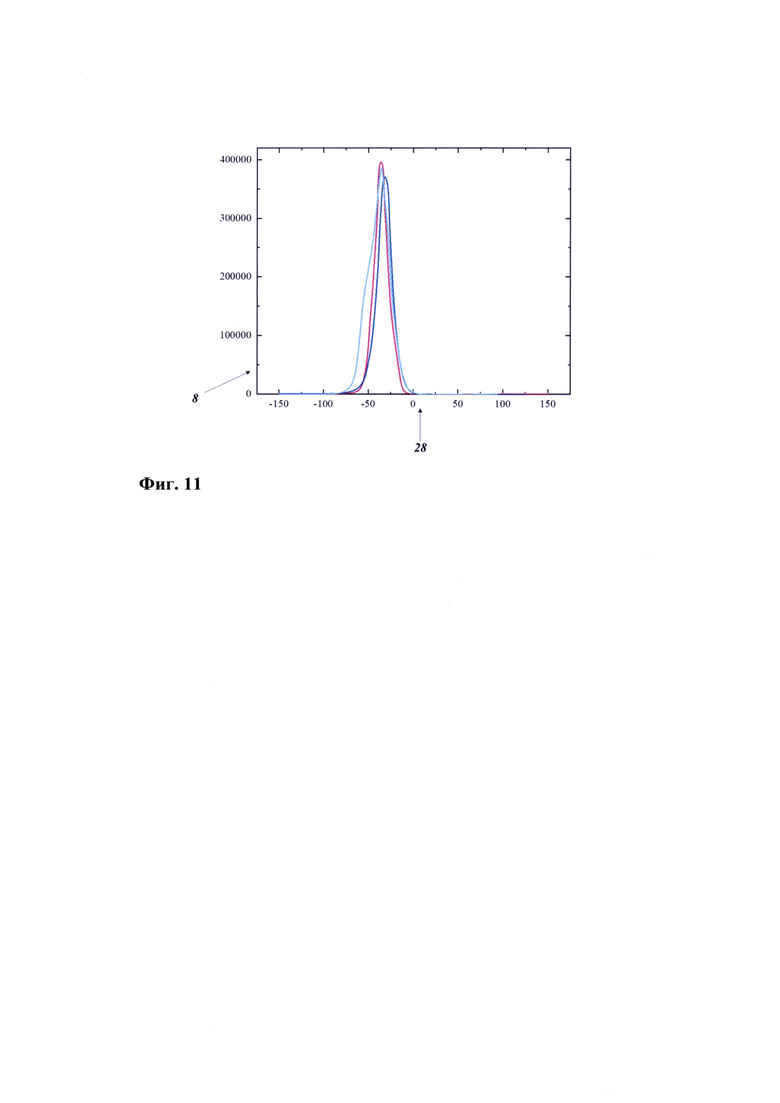
Фиг.9 - ВРЭМ-изображение оксида графена
Фиг. 10 - Распределение наночастиц оксида графена по размерам в водной дисперсии (С=9 г⋅л-1), где:
8 - интенсивность (пр. е.);
27 - размер (нм).
Фиг. 11 - Распределение ξ-потенциалов наночастиц оксида графена в водной дисперсии (С=9 г⋅л-1), где:
8 - интенсивность (пр. е.);
28 - ξ(мВ).
Сущность способа поясняется примерами.
Пример 1.
В круглодонной колбе смешивали 500 г графита, 50 г Р2О5 и 2,5 л H2SO4 и обрабатывали в ультразвуковой бане (мощность ультразвука - 160 Вт) в течение 10 мин, затем полученную смесь перемешивали в течение 15 мин при комнатной температуре. После этого к реакционной смеси постепенно добавляли 50 г NaNO3 при охлаждении с помощью льда до 5°С при непрерывном перемешивании в течение 30 мин. Далее добавляли 500 г KMnO4 при непрерывном перемешивании в течение 30 мин (температура системы поддерживалась с помощью льда при 5°С), а затем повышали температуру до 40°С в течение 1 ч при непрерывном перемешивании с последующим постепенным добавлением 1,25 л дистиллированной воды. После этого температуру реакционной смеси повышали до 95°С на 1,5 ч, затем добавляли 2,5 л дистиллированной воды и 50 мл 30% Н2О2. Полученный осадок отделяли от раствора с использованием фильтра Шотта (пористость - 10), промывали дистиллированной водой и разбавленной HCl (концентрация - 5 мас. %) до нейтрального значения рН, высушивали при 65°С и повторно диспергировали в дистиллированной воде в ультразвуковой бане в течение 1 ч для эксфолиации. Полученные дисперсии оксида графена в концентрационном диапазоне С = 1,8-9,0 г⋅л-1 по сравнению с дистиллированной водой представлены на Фигура (Фиг.) 1.
Для характеризации полученного материала использовались следующие физико-химические методы анализа:
13С спектроскопия ядерного магнитного резонанса (ЯМР).
На Фиг. 2 представлен 13С ЯМР-спектр с использованием твердотельной ЯМР-спектроскопии в режиме прямого возбуждения (время контакта - 2 мс). Полученные данные позволяют охарактеризовать образец оксида графена, где слабовыраженный пик в области 60 м.д. соответствует эпоксидным группам (а); интенсивный пик при 69 м.д. относится к гидроксильным группам (б); малоинтенсивный пик в области 100 м.д. отвечает атому углерода в составе лактольной группы (в); пик при 129 м.д. относится к -С=С- структурному фрагменту плоскости графена (г); слабовыраженный пик в области 165 м.д. соответствует карбонильным группам (д); широкий пик в районе 191 м.д. отвечает карбоксильным группам (е).
Из представленных данных было определено процентное содержание кислородсодержащих функциональных групп на поверхности оксида графена:
- гидроксильные группы (55%). Структурные фрагменты С-ОН, расположенные на обеих базальных плоскостях и краях графенового листа;
- эпоксидные группы (22%). Структурные фрагменты С-О-С, расположенные на базальных плоскостях;
- лактольные группы (4%). Структурные фрагменты 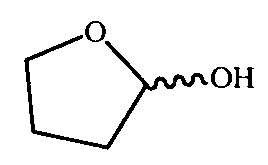 на краях графенового листа;
на краях графенового листа;
- карбоксильные группы (3%). Структурные фрагменты -СООН на краях графенового листа;
- карбонильные группы (1%). Структурные фрагменты С=O на краях графенового листа.
Рентгеноструктурный анализ.
На Фиг. 3 представлены результаты рентгеноструктурного анализа графита (синяя линия) и оксида графена (голубая линия). Анализ спектров показывает, что в случае оксида графена пик, отвечающий плоскости 002, соответствует 2θ=12,2°, а в случае графита 2θ=26,5°. Данный факт подтверждает образование оксида графена. ТГА.
На Фиг. 4 представлены данные ТГА (кривые термогравиметрии (ТГ, сплошная линия) и дифференциальной термогравиметрии (ДТГ, пунктирная линия)) оксида графена в интервале температур 30-1020°С, скорость нагрева - 5°С⋅мин-1 в инертной атмосфере азота. Хорошо видно, что оксид графена термически стабилен до 90°С, дальнейшее повышение температуры приводит к постепенному разложению оксида графена, сопровождающемуся деградацией функциональных групп, различными типами перегруппировок и, наконец, дальнейшей деградацией поверхности графенового остова.
ИК-спектроскопия.
На Фиг. 5 представлен ИК-спектр оксида графена. Широкий пик при 3440 см-1 (а) относится к валентным колебаниям О-Н фрагмента карбоксильной группы; интенсивный пик при 1718 см-1 (б) соответствует валентным колебаниям С=0 фрагмента карбоксильных групп; пик при 1630 см-1 (в) соответствует колебаниям ароматических доменов графена; пики при 1418 (г) и 1363 см-1 (д) относятся колебаниям С-ОН фрагмента карбоксильных и спиртовых групп, соответственно; пик при 1226 см-1 (е) относится к валентным колебаниям С-ОН в составе лактольной группы; пик при 1085 см-1 (ж) соответствует валентным колебаниям С-О, в составе эпоксидных и спиртовых групп.
Рамановская спектроскопия.
На Фиг. 6 представлен Рамановский спектр образца оксида графена. Спектр содержит D-, G- и 20-полосы. Полоса D (а) описывает дефектную моду, связанную с функционализацией поверхности, приводящей к переходу атомов углерода в sp3-гибридизованное состояние и разупорядочению л-системы. Полоса G (б) характерна для всех зр2-гибридизованных графитоподобных материалов; она показывает, что синтезированный образец содержит фрагмент С-С, формирующий л-структуру. Полоса 2D (в) описывает количество слоев графена и определяет, являются ли данные структуры однослойными, двухслойными или многослойными. В частности, в случае однослойного оксида графена отношение I2D/IG=2; уменьшение этого отношения свидетельствует об увеличении количества слоев. Отношение ID/IG позволяет судить о степени функционализации поверхности оксида графена. Анализ Фиг. 6 показывает наличие D- и G-полос при 1360 см-1 и 1592 см-1, имеющих соотношение ID/ID=0,97, указывающее на функционализацию графена с образованием оксида графена. Присутствие 2D-полосы (отношение I2D/IG=0,65) свидетельствует о многослойной структуре оксида графена.
Оптическая спектроскопия.
На Фиг. 7 представлен спектр оптического возбуждения образца оксида графена (С=1,8 г⋅л-1) в диапазоне длин волн λ=250-800 нм. Полученный спектр не имеет видимых полос поглощения и является относительно малоинформативным.
СЭМ и ВРЭМ.
На Фиг. 8 (СЭМ) и на Фиг. 9 (ВРЭМ) представлены микрофотографии оксида графена. Видно, что синтезированный образец состоит из чешуйчатых слоев оксида графена с острыми краями. В то же время, анализ ВРЭМ показывает, что образовавшиеся слои являются скрученными, а структура образца представляет собой однородную поверхность, состоящую из слоев графена.
Распределение частиц оксида графена по размеру и ξ-потенциалы.
Распределение частиц оксида графена по размеру в водной дисперсии (С=9,0 г⋅л-1), представленное на Фиг. 10, показывает, что средний размер ассоциатов составляет 300-350 нм. В то же время значения ξ-потенциалов (-(35-30) мВ, С=9,0 г⋅л-1) доказывают, что полученные дисперсии электрокинетически устойчивы (Фиг. 11).
Предлагаемый способ получения оксида графена имеет следующие преимущества перед аналогами:
- приводит к получению оксида графена с увеличенным содержанием кислородсодержащих функциональных групп (карбоксильных, гидроксильных, карбонильных, эпоксидных и лактольных). Содержание кислорода в конечном продукте составляет более 80%, тогда как в ранее описанных методиках содержание варьируется в интервале 30-35%;
- позволяет получать стабильные водные дисперсии оксида графена без добавления поверхностно-активных веществ, в то время как в описанных в литературе применяются дополнительные поверхностно-активные вещества для повышения стабильности дисперсий;
- разработанный способ синтеза оксида графена является экспрессным и проводится в течение 4 ч. При этом ранее предложенные литературные методики выполняются несколько дней;
- разработанный способ может применяться для масштабирования процесса получения оксида графена до 1 кг.
Способ масштабирования синтеза оксида графена разработан на кафедре общей и биоорганической химии и в лаборатории биомедицинского материаловедения в ФГБОУ ВО «ПСПбГМУ им. академика И.П. Павлова» Минздрава РФ совместно с ФГБУ «РНЦРХТ им. академика А. М. Гранова» Минздрава РФ. Характеризация полученного оксида графена была выполнена на базе следующих ресурсных центров Научного парка ФГБОУ ВО СПбГУ: Геомодель, Центр диагностики функциональных материалов для медицины, фармакологии и наноэлектроники, Междисциплинарный ресурсный центр по направлению «Нанотехнологии», Центр магнитно-резонансных методов исследования, Центр физических методов исследования поверхности, Центр методов анализа состава вещества, Центр термогравиметрических и калориметрических методов исследования.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ создания конъюгатов на основе углеродных наноструктур | 2023 |
|
RU2829735C1 |
| Способ получения малослойных форм восстановленного оксида графена из графита однореакторным методом | 2024 |
|
RU2829356C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА ГРАФЕНА | 2018 |
|
RU2709594C1 |
| Способ получения наноструктурированного порошкового композита на основе графена и диоксида циркония с использованием уротропина | 2023 |
|
RU2812131C1 |
| ПРОВОДЯЩИЙ КОМПОЗИТНЫЙ МАТЕРИАЛ НА ОСНОВЕ СОПРЯЖЕННЫХ ПОЛИМЕРОВ С ГРАФЕНОМ | 2022 |
|
RU2804778C1 |
| Способ получения композитных наноструктурированных порошков на основе графена и оксидов Al, Ce и Zr | 2018 |
|
RU2706652C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНА | 2016 |
|
RU2657504C2 |
| ПРОЦЕСС КАТИОННОЙ ПОЛИМЕРИЗАЦИИ ДЛЯ СИНТЕЗА НАНОСТРУКТУРНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩИХ ГРАФЕН | 2014 |
|
RU2652115C2 |
| Способ получения оксида графена | 2022 |
|
RU2796672C2 |
| Способ получения наноструктурированного композита на основе бескислородного графена и ZrO | 2022 |
|
RU2788977C1 |
Изобретение относится к способу масштабирования синтеза оксида графена, включающему диспергирование графитового порошка в концентрированной серной кислоте. Далее добавление к полученной смеси перманганата калия, разбавление ее дистиллированной водой, фильтрацию осадка и промывку его до нейтрального значения рН с последующим высушиванием. Способ характеризуется тем, что дополнительно, при диспергировании графитового порошка в концентрированной серной кислоте, добавляют оксид фосфора(V), затем полученную смесь обрабатывают ультразвуком в течение 10 мин и после добавления перманганата калия к смеси добавляют нитрат натрия. После разбавления смеси дистиллированной водой температуру смеси повышают до 95 °С в течение 1,5 ч с последующим добавлением дополнительного количества дистиллированной воды и 30 % Н2О2, фильтрацию осадка осуществляют на фильтре Шотта. Затем полученный осадок промывают дистиллированной водой с последующим высушиванием при температуре 65 °С в течение 5 ч и диспергируют в дистиллированной воде в ультразвуковой бане в течение 1 ч. При этом количество реагентов рассчитывают таким образом, что количество серной кислоты в 5 раз превышает исходное количество графитового порошка, при этом его исходное количество в 10 раз превышает количество оксида фосфора(V), нитрата натрия и перекиси водорода, разбавление смеси перед повышением ее температуры до 95 °С дистиллированной водой выполняют в количестве, в 2,5 раза превышающем исходное количество графитового порошка. После указанного диспергирование смеси осуществляют в дистиллированной воде в количестве, превышающем исходное количество графитового порошка в 5 раз. Технический результат настоящего изобретения состоит в разработке масштабируемой методики синтеза оксида графена с увеличенным содержанием кислородсодержащих функциональных групп (до 85 %) путем использования дополнительных окислителей и выбором технологических режимов, позволяющих проводить дальнейшее модифицирование поверхности оксида графена и получать стабильные водные дисперсии. 11 ил., 1 табл., 1 пр.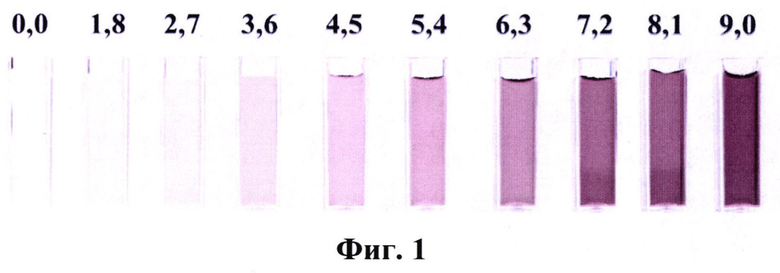
Способ масштабирования синтеза оксида графена, включающий диспергирование графитового порошка в концентрированной серной кислоте, добавление к полученной смеси перманганата калия, разбавление ее дистиллированной водой, фильтрацию осадка и промывку его до нейтрального значения рН с последующим высушиванием, отличающийся тем, что дополнительно, при диспергировании графитового порошка в концентрированной серной кислоте, добавляют оксид фосфора(V), затем полученную смесь обрабатывают ультразвуком в течение 10 мин и после добавления перманганата калия к смеси добавляют нитрат натрия, а после разбавления смеси дистиллированной водой температуру смеси повышают до 95 °С в течение 1,5 ч с последующим добавлением дополнительного количества дистиллированной воды и 30 % Н2О2, фильтрацию осадка осуществляют на фильтре Шотта, затем полученный осадок промывают дистиллированной водой с последующим высушиванием при температуре 65 °С в течение 5 ч и диспергируют в дистиллированной воде в ультразвуковой бане в течение 1 ч, при этом количество реагентов рассчитывают таким образом, что количество серной кислоты в 5 раз превышает исходное количество графитового порошка, при этом его исходное количество в 10 раз превышает количество оксида фосфора(V), нитрата натрия и перекиси водорода, разбавление смеси перед повышением ее температуры до 95 °С дистиллированной водой выполняют в количестве, в 2,5 раза превышающем исходное количество графитового порошка, после чего диспергирование смеси осуществляют в дистиллированной воде в количестве, превышающем исходное количество графитового порошка в 5 раз.
| ПОЛУЧЕНИЕ ОКСИДА ГРАФЕНА | 2014 |
|
RU2691365C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОССТАНОВЛЕННОГО ОКСИДА ГРАФЕНА ИЗ КИШ-ГРАФИТА | 2018 |
|
RU2741045C1 |
| СПОСОБ СИНТЕЗА ОКСИДА ГРАФЕНА | 2018 |
|
RU2720780C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА ГРАФЕНА | 2018 |
|
RU2709594C1 |
| CN 106477572 B, 28.08.2018 | |||
| CN 107352535 A, 17.11.2017 | |||
| CN 109368631 A, 22.02.2019 | |||
| CN 103241983 A, 14.08.2013 | |||
| CN 107673344 A, 09.02.2018 | |||
| CN 112010298 A, 01.12.2020 | |||
| CN 109504012 A, 22.03.2019. | |||

