ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к дипептидилкетоамид м-метоксифенилпроизводным и к их применению для лечения заболеваний и состояний, ассоциированных с повышенной активностью кальпаина, таких как повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее, нейродегенеративные нарушения, малярия, диабетическая нефропатия, нейротоксичность, индуцированная вирусом ВИЧ, злокачественная опухоль и фиброзные заболевания.
УРОВЕНЬ ТЕХНИКИ
Кальпаины представляют собой внутриклеточные белки, принадлежащие семейству кальций-зависимых нелизосомальных цистеиновых протеаз (протеолитические ферменты), широко экспрессируемых у млекопитающих и многих других организмов. Описаны две основные формы, кальпаин 1 и кальпаин 2, также известные как µ-кальпаин и m-кальпаин, однако также допускается существование других изоферментов кальпаина (M.E. Saez et al.; Drug Discovery Today 2006, 11 (19/20), pp. 917-923).
Кальпаины играют важную роль в различных физиологических процессах, которые включают расщепление различных регуляторных белков (K.K. Wang et al., Trends in Pharmacol.Sci. 1994, 15, pp. 412-419).
Повышенные уровни кальпаина были измерены при различных патофизиологических процессах, например: ишемии сердца, почек, легких, печени или центральной нервной системы, воспалениях, мышечных дистрофиях, катарактах глаз, диабете, ВИЧ-опосредуемых нарушениях, повреждениях центральной нервной системы (например, травма головного мозга), болезни Альцгеймера, болезни Гентингтона, болезни Паркинсона, рассеянном склерозе и т.д. (см. K.K. Wang, выше) и инфекционных заболеваниях, таких как малярия (IM Medana et al., Neuropath and Appl. Neurobiol. 2007, 33, pp.179-192). Полагают, что существует связь между этими заболеваниями и часто или постоянно повышенными внутриклеточными уровнями кальция. Это приводит к гиперактивации кальций-зависимых процессов и тому, что они перестают поддаваться нормальному физиологическому контролю. Соответствующая гиперактивация кальпаинов также может запускать патофизиологические процессы. По этой причине утверждалось, что ингибиторы кальпаина можно использовать для лечения этих заболеваний.
Yoshida, Ken Ischi et al. (Jap. Circ. J. 1995, 59 (1), pp. 40-48) описали, что ингибиторы кальпаина имели благоприятные эффекты после повреждения сердца, вызванного ишемией или реперфузией. Недавно было описано, что кальпаины активируются в ходе ишемии-реперфузии миокарда и участвуют в реперфузионном повреждении, а также вовлечение кальпаинов в ремоделирование после инфаркта и сердечную недостаточность. После острого инфаркта миокарда сердце в целом претерпевает серию структурных изменений, называемых постинфарктным ремоделированием миокарда, что ведет к возникновению сердечной недостаточности. Ремоделирование желудочков включает расширение, гипертрофию и образование обособленного коллагенового рубца. Нарушение регуляции активности кальпаина играет важную роль в реперфузионном повреждении и ремоделировании миокарда, что указывает на то, что ингибирование кальпаина является потенциальной терапевтической стратегией (Ye et al., PLoS ONE, 2015, 10(3), e0120178; Kudo-Sakamoto et al., Journal Biological Chemistry, 2014, 289(28), pp. 19408-19419). Ингибиторы кальпаина описаны Neuhof et al. (World J Cardiol, 2014, 6(7), 638-652) в качестве новой профилактической и терапевтической возможности для пациентов с инфарктом миокарда, реваскуляризацией и хирургической операцией на коронарных сосудах. Исследование, описанное Poncelas et al. (Cardiovascular Research, 2017, 113(8), pp. 950-961), подтверждает эту роль кальпаинов и демонстрирует, что фармакологический ингибитор кальпаинов длительного действия является перспективной терапевтической стратегией против неблагоприятного постинфарктного ремоделирования.
Также было показано, что ингибиторы кальпаина обеспечивают нейропротективный эффект при острых нейродегенеративных нарушениях или ишемиях, таких как те, которые возникают после церебрального инсульта (Seung-Chyul Hong et al., Stroke 1994, 25 (3), pp. 663-669, и R. T. Bartus et al., Neurological Res. 1995, 17, pp. 249-258).
Также было показано, что после экспериментальной травмы головного мозга ингибиторы кальпаина улучшают восстановление после нарушений памяти и нейро-двигательных нарушений (K. E. Saa™an et al., Proc. Natl. Acad. Sci. USA, 1996, 93, pp. 3428-3433), и что ингибиторы кальпаина обладают защитным эффектом на поврежденные гипоксией почки (C. L. Edelstein et al., Proc. Natl. Acad. Sci. USA, 1995, 92, pp. 7662-6).
Более поздние исследования показали, что кальпастатин (натуральный ингибитор кальпаина) значительно ослабляет патофизиологические эффекты активированного кальпаина при ряде заболеваний, таких как a) экспериментальный гломерулонефрит (J. Peltier et al., J A, Soc Nephrol. 2006, 17, pp. 3415-3423), b) сердечно-сосудистое ремоделирование при индуцированной ангиотензином II гипертензии, c) нарушение синаптической передачи при врожденном миастеническом синдроме медленных каналов (Groshong JS et al., J Clin Invest. 2007, 117 (10), pp 2903-2912), d) эксайтотоксическая фрагментация ДНК посредством митохондриальных каскадов (J Takano et al., J Biol Chem. 2005, 280 (16) pp. 16175-16184), и e) некротические процессы в дистрофичных мышцах (M J Spencer et al., Hum Mol Gen, 2002, 11(21), pp 2645-2655).
Также известно, что кальпаины связаны с болезнью Альцгеймера (AD) (Nixon R.A., "The calpains in aging and aging-related заболевания", Ageing Res Rev. 2003 Oct; 2(4):407-18). Кальпаин 1 аномально активируется в головном мозге при AD (Saito K, et al. "Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration", Proc Natl Acad Sci USA. 1993 Apr 1; 90(7):2628-32). Уровень кальпастатина, эндогенного ингибитора кальпаинов, значительно снижен при этом же нейродегенеративном расстройстве (Nixon R.A., "The calpains in aging and aging-related diseases", Ageing Res Rev. 2003 Oct; 2(4):407-18). Сверхактивация кальпаина посредством аномально высоких уровней кальция и истощения кльпастатина приводит к ограниченному расщеплению или деградации ключевых нейрональных белков при AD (Wang KK, "Calpain and caspase: can you tell the difference?", Trends Neurosci. 2000 Jan; 23(1):20-6). Кальпаины непрямо модулируют протеолитический процессинг белка-предшественника амилоида (APP) - полипептида, предположительно играющего фундаментальную роль при AD (Siman R. at al. "Proteolytic processing of beta-amyloid precursor by calpain I", J Neurosci. 1990 Jul; 10(7):2400-11).
Другие субстраты кальпаина, изменяющиеся при AD, включают CaM-киназу IIα (CaMK-IIα) и PKC - 2 фермента, которые регулируют фосфорилирование APP и влияют на его метаболизм (Wang KK et al., "Development and therapeutic potential of calpain inhibitors" Adv Pharmacol. 1997; 37():117-52); связанные со вторичными посредниками ферменты, такие как фосфолипаза C-1, -2, -β3 (Banno Y. et al, "Endogenous cleavage of phospholipase C-beta 3 by agonist-induced activation of calpain in human platelets", J Biol Chem. 1995 Mar 3; 270(9):4318-24), и циклин-зависимую киназу 5 (Cdk-5) (Lee MS. et al., "Neurotoxicity induces cleavage of p35 to p25 by calpain", Nature. 2000 May 18; 405(6784):360-4); факторы транскрипции, такие как c-Jun, c-Fos, and IκB (Carillo S, "Differential sensitivity of FOS and JUN family members to calpains", Oncogene. 1994 Jun; 9(6):1679-89 и Lin YC, "Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B", Proc Natl Acad Sci U S A. 1995 Jan 17; 92(2):552-6); и связанный с памятью ген связывающего регуляторный элемент cAMP белка (CREB) (Mbebi C, "Amyloid precursor protein family-induced neuronal death is mediated by impairment of the neuroprotective calcium/calmodulin protein kinase IV-dependent signalling pathway", J Biol Chem. 2002 Jun 7; 277(23):20979-90). Недавно было показано, что действие кальпаина на субъединицу GluR1 рецепторов AMPA (24), амфифизин I (25) и белок циркадных ритмов супрахиазмального ядра (26) модулирует синаптическую активность и формирование воспоминаний.
Накапливающиеся данные указывают на то, что когнитивное нарушение при AD начинается задолго до нейрональной смерти, и что передача сигнала между нейронами прерывается на ранних стадиях заболевания. Важность синаптических изменений при AD была подтверждена исследованиями с моделями AD на трансгенных мышах (Sant'Angelo A, "Usefulness of behavioral and electrophysiological studies in transgenic models of Alzheimer's disease", Neurochem Res. 2003 Jul; 28(7):1009-15) и в отношении индуцируемого пептидом амилоида β (индуцируемым Aβ) нарушения долговременной потенциации (LTP), широко исследованной клеточной модели обучения и памяти (Bliss TV. et al., "A synaptic model of memory: long-term potentiation in the hippocampus"; Collingridge GL Nature. 1993 Jan 7; 361(6407):31-9).
Более того, кальпаины влияют на фосфорилирование и протеолиз тау, другого белка, ассоциированного с AD (Wang KK, "Calpain and caspase: can you tell the difference?", Trends Neurosci. 2000 Jan; 23(1):20-6). Кроме того, накопление фосфорилированного тау приводит к образованию так называемых нейрофибриллярных клубков (NFT), которые вместе с широко известными амилоидными бляшками являются патологическим признаком болезни Альцгеймера. Сходные изменения белка тау, обычно считающиеся важным признаком таупатий, также наблюдают при других (нейро)дегенеративных нарушениях, например, таких как нарушения после инсульта, воспаление головного мозга, паркинсонизм, гидроцефалия нормального давления и болезнь Крейтцфельда-Якоба.
Вовлечение кальпаина в нейродегенеративные процессы было продемонстрировано у трансгенных мышей с помощью кальпастатина, специфического и натурального ингибитора кальпаинов (Higuchi et al.; J. Biol. Chem. 2005, 280 (15), pp. 15229-15237). Было возможно с помощью ингибитора кальпаина значительно снизить клинические признаки аутоиммунного энцефаломиелита в модели рассеянного склероза на мышах (F. Mokhtarian et al.; J. Neuroimmunology 2006, Vol. 180, pp. 135-146). Кроме того, было показано, что ингибиторы кальпаина с одной стороны блокируют индуцируемую A@ дегенерацию нейронов (Park et al.; J. Neurosci. 2005, 25, pp. 5365-5375), и, кроме того, снижают высвобождение белка-предшественника β-амилоида (β-APP) (J. Higaki et al., Нейрон, 1995, I4, pp. 651-659).
В связи с этим, ингибиторы кальпаина представляют собой новый терапевтический подход для лечения нейродегенеративных нарушений в общем и, в частности, болезни Альцгеймера, болезни Паркинсона, рассеянного склероза, острого аутоиммунного энцефалита и болезни Крейтцфельда-Якоба.
Было продемонстрировано, что ВИЧ-индуцируемая нейротоксичность опосредуется кальпаином (O'Donnell et al.; J. Neurosci. 2006, 26 (3), pp. 981-990) и было показано вовлечение кальпаина в репликацию ВИЧ (Teranishi et al.; Biochem. Biophys. Res. Comm. 2003, 303 (3), pp. 940-946).
Также недавно было продемонстрировано вовлечение кальпаина в развитие заболеваний почек, таких как хронические заболевания почек, например, диабетическая нефропатия. Таким образом, Y. Shi et al. продемонстрировали в моделях на животных, что натуральный ингибитор кальпаина кальпастатин подавляется в ходе ишемии-реперфузии почек (Am. J. Physiol. Renal Physiol. 2000, 279, pp. 509-517). Более того, A Dnyanmote et al., Toxicology and Applied Pharmacology 2006, 215, pp. 146-157 показали, что ингибирование кальпаина посредством сверхэкспрессии кальпастатина снижает прогрессирование индуцируемого DCVC повреждения почек в модели острой почечной недостаточности. Кроме того, Peltier et al. продемонстрировали, что активация и секреция кальпаина способствуют гломерулярному повреждению при экспериментальном гломерулонефрите (J. Am. Soc. Nephrol. 2006, 17, pp. 3415-3423). Также было показано, что ингибиторы кальпаина снижают дисфункцию и повреждение почек, вызываемые ишемией-реперфузией почек, и, таким образом, могут быть пригодными для повышения устойчивости почек против повреждения почек, ассоциированного с аортально-сосудистой хирургической операцией или трансплантацией почек (P. Chatterjee et al., Biochem. Pharmacal. 2005, 7, pp. 1121-1131). Исходя из этого, ингибирование кальпаина можно считать терапевтическим подходом, пригодным для лечения заболеваний почек, таких как хронические заболевания почек, например, диабетическая нефропатия.
Кальпаин также был идентифицирован в качестве центрального медиатора, необходимого для паразитарной активности. Паразиты, такие как Plasmodium falciparum и Toxoplasma gondii используют кальпаины клетки-хозяина для способствования выходу из внутриклеточной паразитофорной вакуоли и/или мембраны клетки-хозяина. Ингибирование кальпаина-1 в гипотонически лизированных и повторно запечатанных эритроцитах препятствовало выходу паразитов P. falciparum, которое восстанавливалось посредством добавления очищенного кальпаина-1. Аналогично, эффективный выход T. gondii из фибробластов млекопитающих блокировался либо посредством опосредуемой малой интерферирующей РНК супрессии, либо посредством генетическим устранением активности кальпаина, и мог быть восстановлен посредством генетической комплементации (D. Greenbaum et al., Science 324, 794 (2009)). Поскольку паразиты, которые не выходят из клеток-хозяев, не способны пролиферировать, это предполагает стратегию для антипаразитарных терапевтических средств. Было показано, что фармакологическое ингибирование кальпаина демонстрирует антималярийную активность и, таким образом, обеспечивает новую антипаразитарную стратегию для лечения таких заболеваний, которые вызываются инфекциями простейшими, такими как малярия или токсоплазмоз (Li et al., Mol Biochem Parasitol. 2007; 155(1): 26-32; Jung et al. Archives of Pharmacal Research (2009), 32(6), 899-906, Chandramohanadas et al. Science (2009), 324, 794).
Также было описано (Leloup and Wells, Expert Opin Ther Targets., 2011, 15(3), 309-323; Storr et al., Nat Rev Cancer., 2011, 11(5), 364-374; Storr et al., Pathobiology., 2015, 82(3-4), 133-141; Selvakumar and Sharma, Experimental Therapeutic Medicine, 2010, 1, 413-417; Storr et al., Oncotarget, 2016, 30(7), 47927-47937; и Guan et al., Proc Amer Assoc Cancer Res., 2005, 46), что кальпаины, в частности, кальпаин 1 и кальпаин 2, вовлечены в широкое множество часто встречающихся злокачественных опухолей, таких как рак молочной железы, рак ободочной и прямой кишки и лейкоз. Кальпаины 1 и 2 вовлечены в развитие и прогрессирование злокачественной опухоли путем обеспечения возможности трансформации, миграции и ускользания опухолевых клеток, и неоваскуляризации опухоли. В этих сообщениях также упоминается, что многочисленные опухолевые клетки демонстрируют аномально высокую активность этих кальпаинов. Таким образом, ингибирование активности кальпаина может быть эффективным способом блокирования развития опухоли посредством блокирования трансформации и пролиферации клеток, а также васкуляризации опухоли.
Фиброз относится к накоплению молекул внеклеточного матрикса, которые формируют рубцовую ткань (богатый коллагеном внеклеточный матрикс), и является частым признаком фиброзных заболеваний. Фиброзные заболевания могут поражать любой орган, такой как почка, печень, легкие, кожа, сердце и глаз, вызывая дисфункцию и недостаточность органа и потенциально смерть. В эпителиальных органах, особенно в легком, печени, коже и почке, замещение нормальных функциональных элементов клеток богатой коллагеном рубцовой тканью и нарушение архитектоники, вызванное стягиванием рубцами, являются основными факторами прогрессирующего снижения функции органа и в итоге его недостаточности [Friedman et al. Science Translational Medicine, 2013, 5, 167]. Было описано, что активность кальпаина является необходимой для заживления ран и формирования рубца [Nassar et al., PLoS ONE, 2012, 7(5), e37084]. Также было описано, что кальпаины вовлечены в фиброзные заболевания [WO 2017/156074 A1; Buckman et al., Am J Respir Crit Care Med, 2018, 197, A5747; Potz et al., J Nat Sci 2016, 2(9), e218; Ono et al., Nature Reviews Drug Discovery, 2016, 15, 854-876], такие как фиброз печени (алкогольный, вирусный, аутоиммунный, метаболический и наследственное хроническое заболевание), фиброз почек (например, в результате хронического воспаления, инфекций или диабета типа II), фиброз легких (идиопатический или в результате воздействия факторов внешней среды, включая токсические частицы, саркоидоз, асбестоз, обусловленный гиперчувствительностью пневмонит, бактериальные инфекции, включая туберкулез, среди прочих), интерстициальный фиброз, системная склеродермия (аутоиммунное заболевание, при котором во многих органах развивается фиброз), макулярная дегенерация (фиброзное заболевание глаза), панкреатический фиброз (в результате, например, злоупотребления алкоголем и хронического воспалительного заболевания поджелудочной железы), фиброз селезенки (в результате серповидноклеточной анемии и других нарушений крови), фиброз сердца (в результате инфекции, воспаления и гипертрофии), медиастинальный фиброз, миелофиброз, эндомиокардиальный фиброз, ретроперитонеальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные осложнения хирургических операций, особенно хирургических имплантатов, инъекционный фиброз и вторичные состояния и болезненные состояния с фиброзом, такие как цирроз, диффузное паренхиматозное заболевание легких, болевой синдром после вазектомии и артрофиброз, среди прочих. Конкретный ингибитор кальпаина BLD-2660 в настоящее время находится в фазе 1 клинических испытаний против фиброза. Таким образом, ингибирование активности кальпаина может быть эффективной стратегией управления течением фиброзных заболеваний.
В WO 2004/078908 A2 и WO 2005/056519 A1 описаны ингибиторы кальпаина с потенциалом в отношении ингибирования кальпаина 1 для лечения различных заболеваний, включая болезнь Альцгеймера. В частности, соединение примера 17 WO 2005/056519 A1, которому было присвоено кодовое наименование SNJ-1945, продемонстрировало эффективность в отношении уменьшения клинических показателей экспериментального аутоиммунного энцефаломиелита (EAE) in vivo (Trager N. et al., "Effects of a novel orally administered calpain inhibitor SNJ-1945 on immunomodulation and neurodegeneration in a murine model of multiples sclerosis", J. Neurochem. 2014 July; 130(2): 268-279).
Остается потребность в новых стратегиях для управления течением заболеваний или состояний, ассоциированных с повышенной активностью кальпаина, таких как повреждение сердца, вызванное инфарктом, ишемией с реперфузий или без нее; нейродегенеративными нарушениями; малярией; диабетической нефропатией; нейротоксичностью, индуцированной вирусом ВИЧ; злокачественной опухолью; и фиброзными заболеваниями.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения неожиданно открыли, что соединения формулы (I), которые имеют метоксигруппу в мета-положении фенильного кольца, являются мощными ингибиторами кальпаина-1. Положение этой группы имеет первостепенную важность для достижения высокой эффективности ингибирования кальпаина-1, как показано в сравнительных примерах. Таким образом, эти соединения формулы (I) являются пригодными для лечения и/или предупреждения заболеваний или состояний, ассоциированных с повышенной активностью кальпаина, таких как повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее; нейродегенеративные нарушения; малярия; диабетическая нефропатия; нейротоксичность, индуцированная вирусом ВИЧ; и злокачественная опухоль.
В первом аспекте, настоящее изобретение относится к соединению формулы (I):
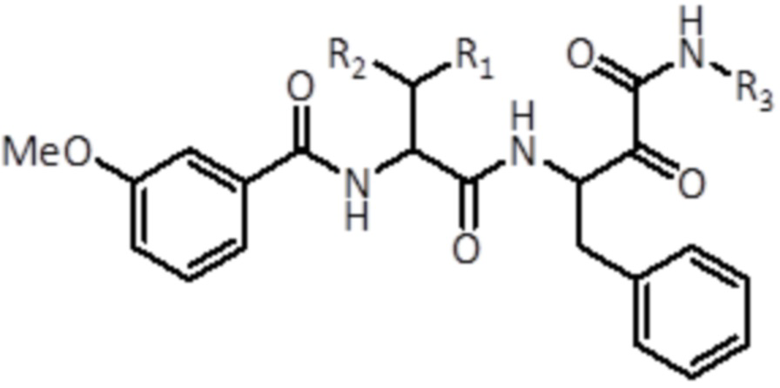
(I)
где
R1 выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила,
R2 выбран из группы, состоящей из H и C1-C6 алкила,
R3 выбран из группы, состоящей из C1-C6 алкокси и C3-C6 циклоалкила,
или его фармацевтически приемлемой соли или стереоизомеру, для применения для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), как определено в первом аспекте, и фармацевтически приемлемый эксципиент, для применения для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
В другом аспекте настоящее изобретение относится к применению соединения формулы (I), как определено в первом аспекте, или фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый эксципиент, для производства лекарственного средства для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
В другом аспекте настоящее изобретение относится к способу лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли и фиброзных заболеваний, причем способ включает введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), как определено в первом аспекте, или фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый эксципиент.
В следующем аспекте настоящее изобретение относится к соединению формулы (II):
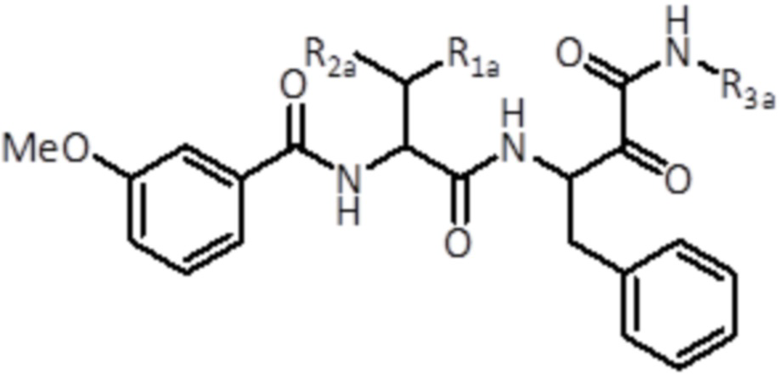
(I)
где
R1a выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила,
R2a выбран из группы, состоящей из H и C1-C6 алкила,
R3a представляет собой C3-C6 циклоалкила,
или его фармацевтически приемлемой соли или их стереоизомеру.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (II), как определено в третьем аспекте, и фармацевтически приемлемый эксципиент.
В другом аспекте настоящее изобретение относится к соединению формулы (II), как определено в третьем аспекте, или к фармацевтической композиции, как определено в четвертом аспекте, для применения в медицине, в частности, для лечения и/или предупреждения заболевания или состояния, ассоциированного с повышенной активностью кальпаина; предпочтительно выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
С целью облегчения понимания изобретения приводятся некоторые из определений.
Термин "алкил", как используют в настоящем описании отдельно или в качестве части другой группы, означает линейную или разветвленную насыщенную одновалентную углеводородную цепь, содержащую количество атомов углерода, указанное в каждом случае, которое обычно составляет от одного до шести атомов углерода, и предпочтительно от одного до трех. Примерами алкилов являются метил, этил, пропил, 2-пропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, втор-пентил, трет-пентил, неопентил и т.п.
Термин "алкокси", как используют в настоящем описании отдельно или в качестве части другой группы, означает алкильную группу, как определено выше, связанную через кислород, т.е. алкил-O-. Примеры алкокси включают метокси, этокси, изопропокси, трет-бутокси и т.п.
Термин "циклоалкил", как используют в настоящем описании отдельно или в качестве части другой группы, означает моноциклический радикал, который является насыщенным или частично насыщенным, предпочтительно насыщенным, и который состоит только из атомов углерода и водорода, и содержит количество атомов углерода, указанное в каждом случае, которое обычно составляет от 3 до 6 и предпочтительно от 3 до 5. Примерами циклоалкилов являются циклопропил, циклобутил, циклопентил, циклогексил.
Термин "соль" необходимо понимать как любую форму соединения в соответствии с настоящим изобретением, где указанное соединение находится в ионной форме, или является заряженным или сопряженным с противоионом (катионом или анионом), или находится в растворе. Это определение также включает четвертичные соли аммония. В частности, определение включает фармацевтически приемлемые соли.
Термин "фармацевтически приемлемая соль" охватывает соли с фармацевтически приемлемой кислотой или основанием, которые синтезированы из исходного соединения, которое содержит кислотную часть, посредством добавления фармацевтически приемлемого основания, или которые синтезированы из исходного соединения, которое содержит основную часть, посредством добавления фармацевтически приемлемой кислоты. Фармацевтически приемлемые кислоты включают как неорганические кислоты, например, хлористоводородную, серную, фосфорную, дифосфорную, бромистоводородную, йодистоводородную и азотистую кислоты, так и органические кислоты, например, лимонную, фумаровую, малеиновую, яблочную, миндальную, аскорбиновую, щавелевую, янтарную, виннокаменную, бензойную, уксусную, метансульфоновую (мезилат), этансульфоновую, бензолсульфоновую (безилат) или п-толуолсульфоновую (тозилат) кислоту. Фармацевтически приемлемые основания включают гидроксиды щелочных металлов (например, натрия или калия) и щелочноземельных металлов (например, кальция или магния), и органические основания, такие как алкиламины, арилалкиламины и гетероциклические амины. Например, фармацевтически приемлемые соли соединений, описанных в настоящем описании, синтезируют из исходного соединения, которое содержит основную или кислотную часть, общепринятыми химическими способами. Как правило, такие соли получают, например, посредством реакции форм свободного основания или свободной кислоты этих соединений со стехиометрическим количеством соответствующей кислоты или основания, соответственно, в воде или в органическом растворителе, или в их смеси.
Предусматриваются все стереоизомеры соединений по настоящему изобретению, либо отдельно, либо в качестве их смесей. В процессе получения в качестве исходных материалов могут использоваться рацематы, энантиомеры или диастереомеры. Когда получают диастереомерные или энантиомерные продукты, их можно разделять общепринятыми способами, например, хроматографией или функциональной кристаллизацией.
Если нет иных указаний, соединения по изобретению также включают соединения, которые отличаются только присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие настоящие структуры, за исключением замены атома водорода дейтерием или тритием, или замены углерода 13C- или 14C-обогащенным углеродом или 15N-обогащенным азотом, входят в объем настоящего изобретения.
Определенные соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. Как правило, сольватированные формы эквивалентны несольватированным формам, и охватываются объемом настоящего изобретения.
Определенные соединения по настоящему изобретению могут существовать в множестве кристаллических или аморфных форм. Как правило, все физические формы эквивалентны для применений, предусматриваемых настоящим изобретением, и подразумевается, что они входят в объем настоящего изобретения.
Термин "предупреждение", как используют в рамках изобретения, относится к введению соединения по изобретению на начальной или ранней стадии заболевания или состояния, или также для предупреждения его возникновения.
Термин "лечение" используют для обозначения введения соединения по изобретению для контроля прогрессирования нарушения до или после появления клинических признаков. Под контролем прогрессирования нарушения подразумевают определение благоприятных или желаемых клинических результатов, включая, но не ограничиваясь ими, уменьшение симптомов, уменьшение длительности нарушения, стабилизацию патологического состояния (в частности, предотвращение дальнейшего ухудшения), замедление прогрессирования нарушения, улучшение патологического состояния и ремиссию (как частичную, так и общую).
Термин "индивидуум", как используют в рамках изобретения, относится к любому животному или человеку, которые страдают от одного из заболеваний или состояний, описанных в настоящем описании. Предпочтительно, индивидуум является млекопитающее. Термин "млекопитающее", как используют в рамках изобретения, относится к любому виду млекопитающих, включая, но не ограничиваясь ими, домашних и сельскохозяйственных животных (коровы, лошади, свиньи, овцы, козы, собаки, кошки или грызуны), приматов и людей. Предпочтительно, млекопитающим является человек.
Термин "повреждение сердца", как используют в рамках изобретения, относится к любому повреждению сердечной мышцы, вызванному инфарктом, ишемией с реперфузией или без нее, такой как ремоделирование.
Термин "ремоделирование", как используют в рамках изобретения, относится к группе молекулярных, клеточных и интерстициальных изменений, которые клинически проявляются изменениями размера, формы и функции сердца в результате повреждений сердца, таких как изменения диаметра полости, массы (гипертрофия и атрофия), геометрии (толщина и стенка стенок сердца), площади рубца, фиброз и воспалительный инфильтрат. Сердечная дисфункция является основным следствием ремоделирования сердца, которое составляет патофизиологическую основу для начала и прогрессирования дисфункции желудочков. Это взаимодействие начинается с генетических изменений в ответ на повреждение сердца с реэкспрессией фетальных генов. Следовательно, возникают клеточные и молекулярные изменения, которые приводят к постепенному ухудшению функции желудочков, сначала бессимптомному, вслед за которым возникают признаки и симптомы сердечной недостаточности. Ремоделирование сердца ассоциировано со злокачественными желудочковыми аритмиями, включая устойчивую желудочковую тахикардию и фибрилляцию желудочков. В ходе первых часов после окклюзии коронарной артерии может произойти дезинтеграция межфибриллярного коллагена одновременно с некрозом миофибрилл. Потеря поддерживающей ткани делает эту область более чувствительной к растяжению и деформации. Вследствие потери некротических мышечных клеток и перестройки миоцитов в претерпевшей инфаркт стенке возникает истончение претерпевшей инфаркт области и расширение полости. Это острое расширение желудочков, характеризующееся истончением и вытягиванием области инфаркта, называют экспансией инфаркта. Экспансия инфаркта повышает вероятность разрыва миокарда и является анатомической основой для аневризм.
Термин "нейродегенеративные нарушения", как используют в рамках изобретения, относится к нарушениям, которые приводят к прогрессирующей дегенерации и/или гибели нейронов. Примерами нейродегенеративных нарушений являются болезнь Альцгеймера, болезнь Паркинсона, рассеянный склероз, острый аутоиммунный энцефалит и болезнь Крейтцфельда-Якоба.
Термин "злокачественная опухоль" или "опухоль", как используют в рамках изобретения, относится к широкой группе заболеваний, вовлекающих нерегулируемый рост клеток, которые также называют злокачественными новообразованиями. Этот термин обычно применяют в отношении заболевания, характеризующегося неконтролируемым делением клеток (или повышением выживаемости или резистентности к апоптозу) и способностью указанных клеток вторгаться в другие соседние ткани (инвазия) и распространяться в другие области организма, где эти клетки обычно не присутствуют (метастаз), через лимфатические и кровеносные сосуды, циркулировать в кровотоке, а затем вторгаться в нормальные ткани в других частях тела. В зависимости от того, могут ли они распространяться посредством инвазии и метастазирования, опухоли классифицируют на доброкачественные или злокачественные: доброкачественные опухоли представляют собой опухоли, которые не могут распространяться посредством инвазии или метастазирования, т.е. они растут только локально; в то время как злокачественные опухоли представляют собой опухоли, которые способны распространяться посредством инвазии и метастазирования. Известные биологические процессы, связанные со злокачественной опухолью, включают ангиогенез, инфильтрацию иммунных клеток, миграцию клеток и метастазирование. Обычно злокачественные опухоли имеют некоторые из следующих общих характеристик: поддержание пролиферативной передачи сигнала, ускользание от супрессоров роста, сопротивление клеточной смерти, возможность репликативного бессмертия, индукция ангиогенеза и активация инвазии и в конечном итоге метастазирования. Злокачественные опухоли вторгаются в близлежащие части организма, а также могут распространяться в более отдаленные части организма через лимфатическую систему или кровоток. Злокачественные опухоли классифицируют по типу клеток, с которыми сходны опухолевые клетки, которые, таким образом, считаются источником опухоли.
Злокачественные опухоли, которые можно лечить или предупреждать посредством медицинских применений по настоящему изобретению, представляют собой солидные опухоли, например, рак ободочной и прямой кишки, рак молочной железы, рак легкого, рак поджелудочной железы, рак гортани, рак языка, рак яичника, рак предстательной железы, рак печени, рак головы и шеи, рак пищевода, аденокарциному, карциному потовых желез, карциному сальных желез, папиллярную карциному, папиллярные аденокарциномы, цистаденокарциному, медуллярную карциному, бронхогенную карциному, почечноклеточный рак, гепатому, карциному желчного протока, хориокарциному, семиному, дисгерминому, эмбриональную карциному, опухоль Вильмса, рак шейки матки, опухоль яичка, карциному яичника, эпителиальную карциному, глиому, астроцитому, медуллобластому, краниофарингиому, эпендимому, пинеалому, геманиобластому, невриному слухового нерва, олигоденроглиому, менингиому, нейробластому, ретинобластому и лейкоз; предпочтительно рак молочной железы, рак ободочной и прямой кишки и лейкоз.
Термин "лейкоз", как используют в рамках изобретения, относится к типу злокачественной опухоли крови или костного мозга, характеризующемуся аномальным повышением уровня незрелых лейкоцитов, называемых бластами, и возникающему в кровеобразующей ткани. Лейкоз начинается в костном мозге, где развивающиеся клетки крови, обычно развивающиеся лейкоциты, претерпевают злокачественное (раковое) изменение. Это означает, что они увеличиваются в количестве неконтролируемым образом, заполняя костный мозг и препятствуя нормальному продуцированию клеток крови. Увеличившиеся в количестве аномальные клетки, называемые бластными клетками или лейкобластами, в конечном итоге выходят из костного мозга и перемещаются по организму через кровоток. В некоторых случаях эти аномальные клетка накапливаются в различных органах, включая лимфатические узлы, селезенку, печень и центральную нервную систему (головной и спинной мозг). Существует четыре основных типа лейкоза: острый лимфобластный лейкоз, или ALL; острый миелоидный лейкоз, или AML; хронический лимфоцитарный лейкоз, или CLL; хронический миелогенный лейкоз, или CML.
Термин "фиброзное заболевание", как используют в рамках изобретения, относится к группе заболеваний, которые вовлекают образование чрезмерной фиброзной соединительной ткани в органе или ткани. Фиброз приводит к рубцеванию и утолщению пораженной ткани. Фиброз может происходить во многих тканях организма, вызывая различные фиброзные заболевания, такие как фиброз сердца, фиброз легких, фиброз печени, фиброз почек, ретроперитонеальный фиброз, обусловленный гиперчувствительностью пневмонит, интерстициальный фиброз, системная склеродермия, макулярная дегенерация, фиброз поджелудочной железы, фиброз селезенки, медиастинальный фиброз, миелофиброз, эндомиокардиальный фиброз, прогрессирующий массивный фиброз, нефрогенный системный фиброз, фиброзные осложнения хирургической операции, хроническую васкулопатию аллотрансплантата и/или хроническое отторжение трансплантированных органов, фиброз, ассоциированный с ишемией-реперфузией, инъекционный фиброз, цирроз, диффузное паренхиматозное заболевание легких, синдром боли после вазэктомии и артрофиброз. Предпочтительно фиброзное заболевание относится к фиброзу сердца, фиброзу легких, фиброзу печени, фиброзу почек или ретроперитонеальному фиброзу.
Соединения формулы (I)
В первом аспекте настоящее изобретение относится к соединению формулы (I)
(I)
где
R1 выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила,
R2 выбран из группы, состоящей из H и C1-C6 алкила,
R3 выбран из группы, состоящей из C1-C6 алкокси и C3-C6 циклоалкила,
или его фармацевтически приемлемой соли или стереоизомеру, для применения для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
В конкретном варианте осуществления R1 выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила; предпочтительно из группы, состоящей из метила, этила, пропила, изопропила, циклопропила, циклобутила и циклопентила; более предпочтительно из группы, состоящей из метила, изопропила и циклопропила; еще более предпочтительно из группы, состоящей из изопропила и циклопропила; наиболее предпочтительным является циклопропил.
В другом конкретном варианте осуществления R2 выбран из группы, состоящей из H и C1-C3 алкила; предпочтительно из группы, состоящей из H, метила, этила, пропила и изопропила; более предпочтительно из группы, состоящей из H и метила; наиболее предпочтительным является H.
В другом конкретном варианте осуществления R3 выбран из группы, состоящей из C1-C3 алкокси и C3-C5 циклоалкила; предпочтительно из группы, состоящей из метокси, этокси, пропокси, изопропокси, циклопропила, циклобутила и циклопентила; еще более предпочтительно из группы, состоящей из метокси и циклопропила; наиболее предпочтительным является циклопропил. В другом конкретном варианте осуществления R3 представляет собой C3-C6 циклоалкил; предпочтительно C3-C5 циклоалкил; предпочтительно циклопропил, циклобутил или циклопентил; наиболее предпочтительным является циклопропил.
В конкретном варианте осуществления R1 выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила; R2 выбран из группы, состоящей из H и C1-C3 алкила; и R3 выбран из группы, состоящей из C1-C3 алкокси и C3-C5 циклоалкила.
В другом конкретном варианте осуществления R1 выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила; R2 выбран из группы, состоящей из H и C1-C3 алкила; и R3 представляет собой C3-C5 циклоалкил.
В другом конкретном варианте осуществления R1 выбран из группы, состоящей из изопропила и циклопропила; R2 представляет собой H; и R3 представляет собой циклопропил.
В конкретном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ia):
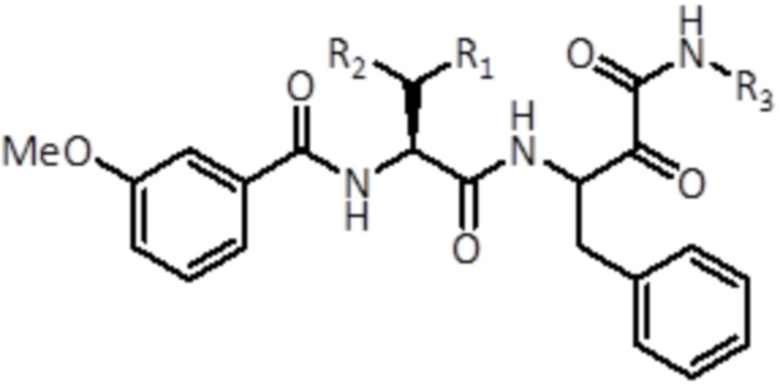
(Ia)
где R1, R2 и R3 имеют то же значение, как и в соединении формулы (I), или его стереоизомер или фармацевтически приемлемую соль.
В конкретном варианте осуществления конкретный стереоизомер соединения формулы (I) представляет собой соединение формулы (Ib):
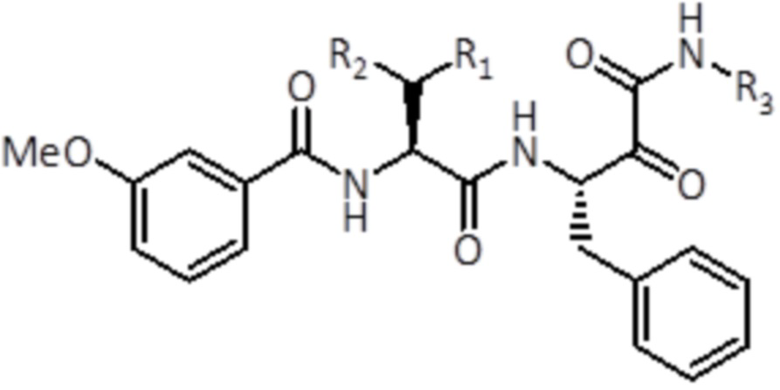
(Ib)
где R1, R2 и R3 имеют то же значение, как и в соединении формулы (I), или его фармацевтически приемлемую соль.
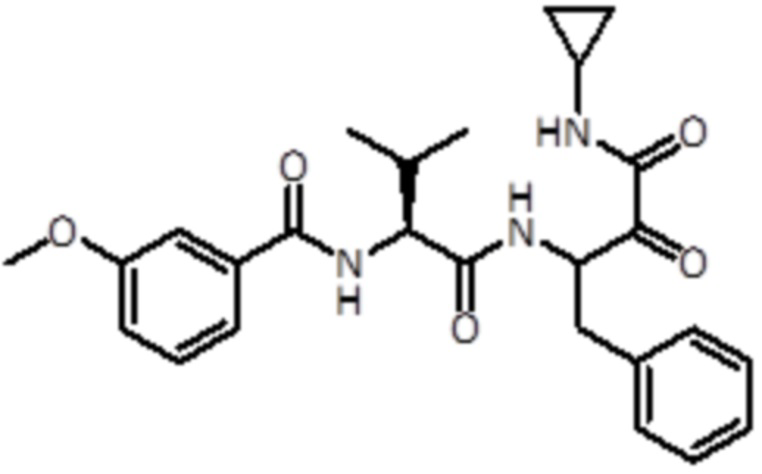
В предпочтительном варианте осуществления соединение формулы (I) выбран из группы, состоящей из:
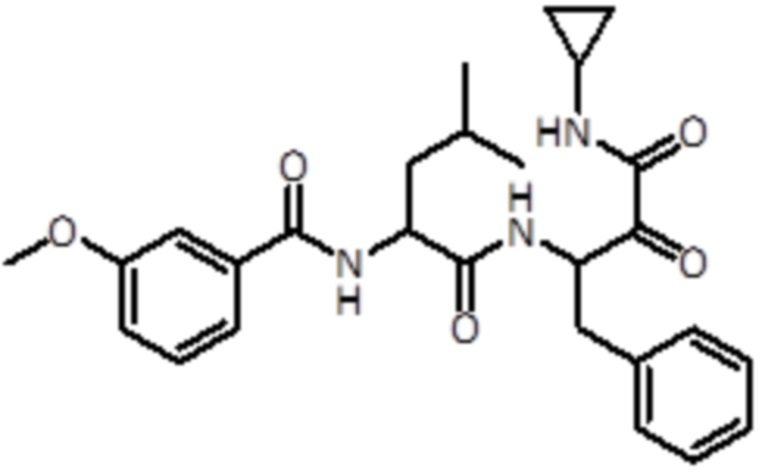 ,
, 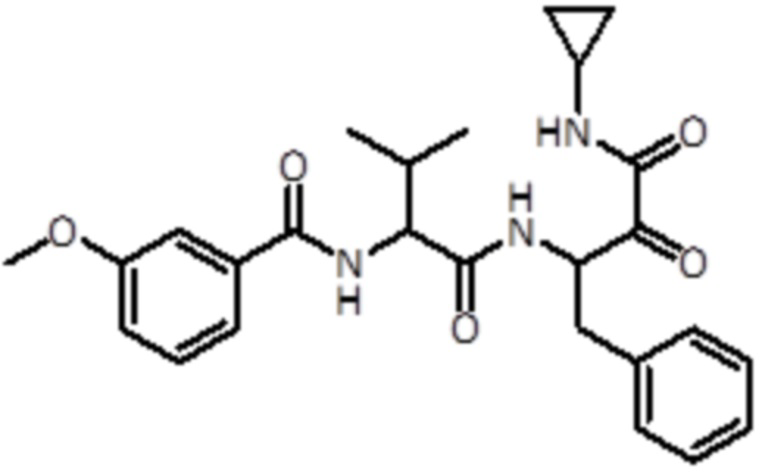 ,
, 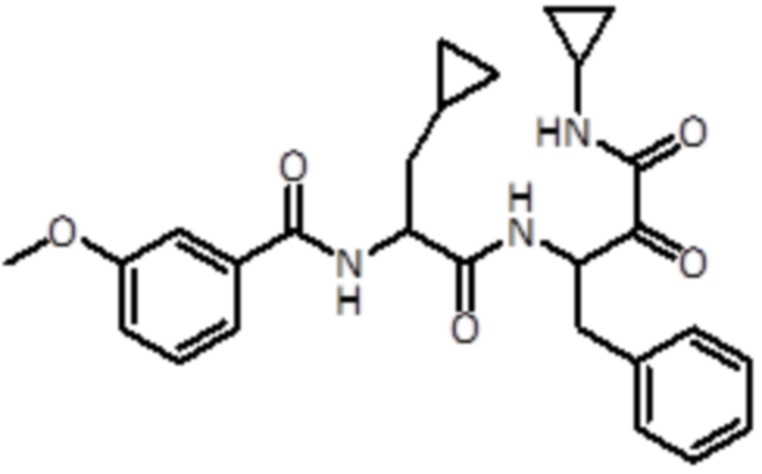 и
и  ,
,
или их стереоизомера или фармацевтически приемлемой соли.
В конкретном варианте осуществления соединение формулы (Ia) выбран из группы, состоящей из:
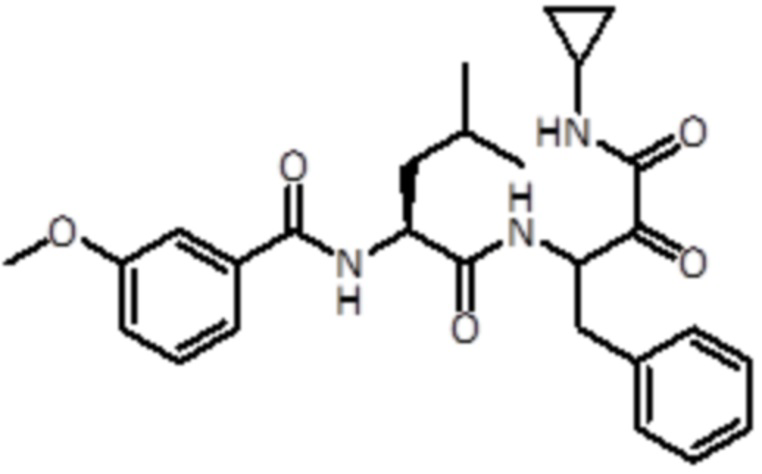 ,
,  ,
, 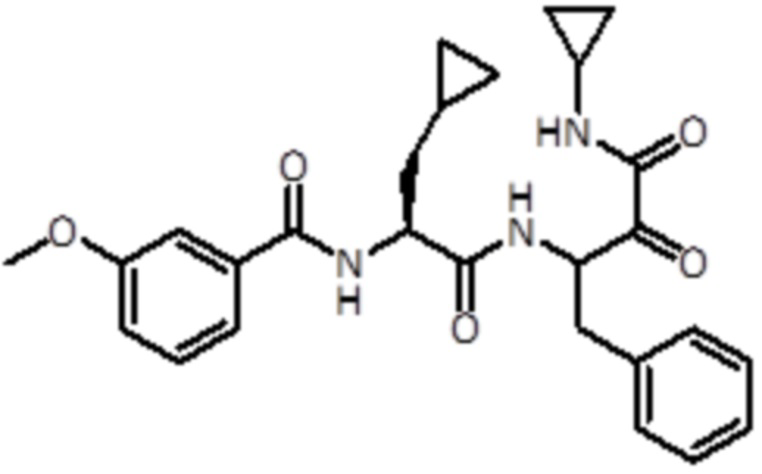 и
и 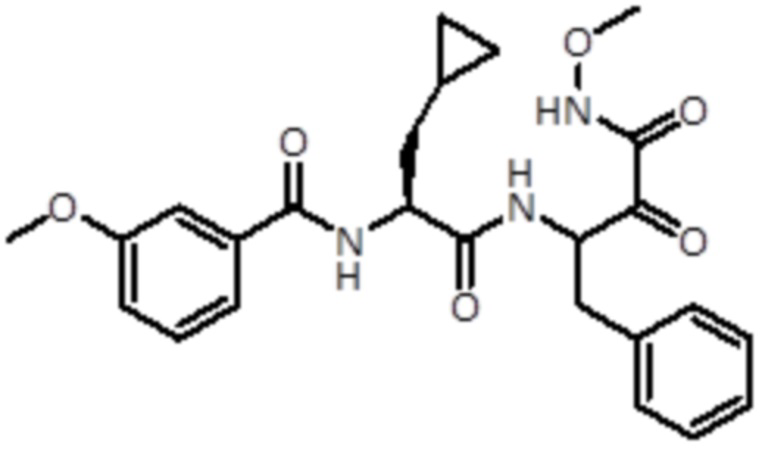 ,
,
или их стереоизомера или фармацевтически приемлемой соли.
В конкретном варианте осуществления соединение формулы (Ib) выбрано из группы, состоящей из:
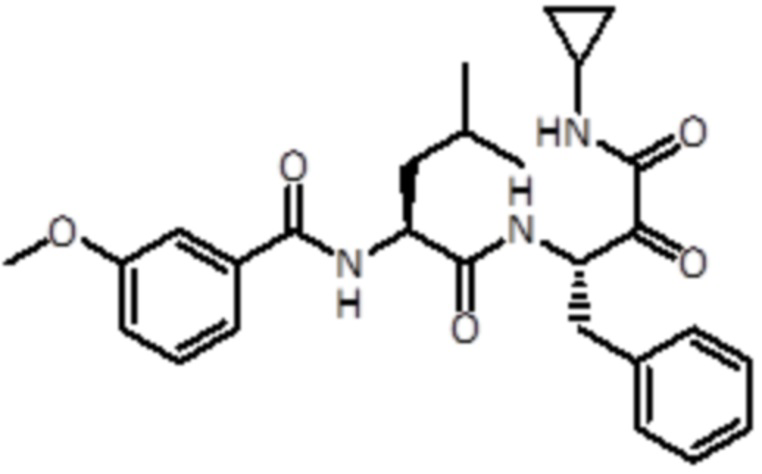 ,
, 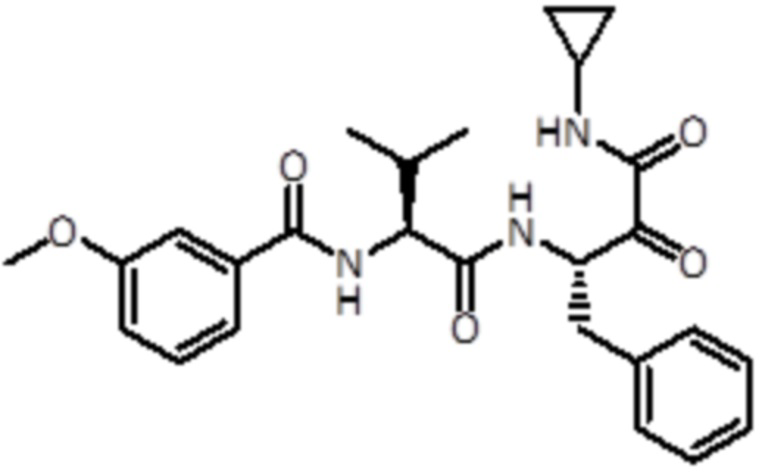 ,
, 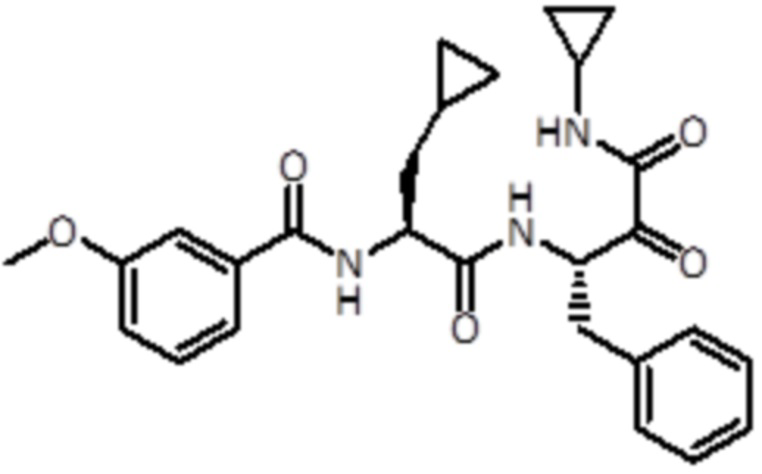 и
и 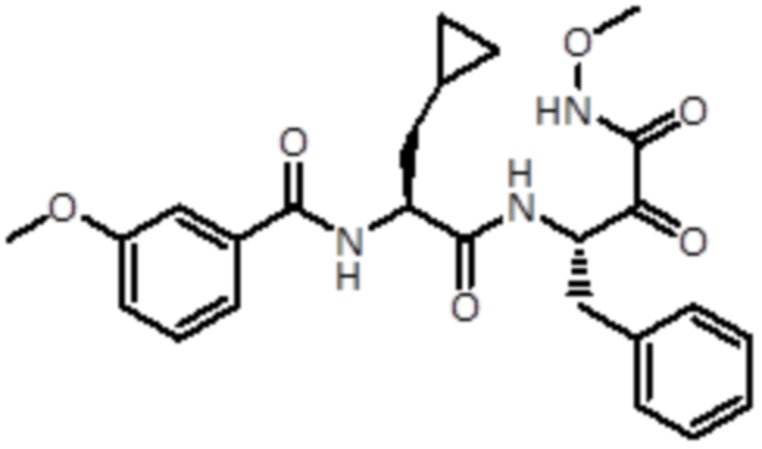 ,
,
или их фармацевтически приемлемой соли.
В конкретном варианте осуществления соединение формулы (I) предназначено для применения для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; и злокачественной опухоли.
В предпочтительном варианте осуществления соединение формулы (I) предназначено для применения для лечения и/или предупреждения повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее, предпочтительно ремоделирования после инфаркта миокарда.
В конкретном варианте осуществления соединение формулы (I) предназначено для применения для лечения и/или предупреждения нейродегенеративного нарушения, выбранного из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, рассеянного склероза, острого аутоиммунного энцефалита и болезни Крейтцфельда-Якоба.
В другом конкретном варианте осуществления соединение формулы (I) предназначено для применения для лечения и/или предупреждения злокачественной опухоли, выбранной из группы, состоящей из рака молочной железы, рака ободочной и прямой кишки и лейкоза.
В другом конкретном варианте осуществления соединение формулы (I) предназначено для применения для лечения и/или предупреждения фиброзного заболевания, выбранного из группы, состоящей из фиброза сердца, фиброза легких, фиброза печени, фиброза почек и ретроперитонеального фиброза.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), как определено в первом аспекте, и фармацевтически приемлемый эксципиент, для применения для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
В другом аспекте изобретение относится к применению соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли или их стереоизомера, для производства лекарственного средства для лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний.
В следующем аспекте настоящее изобретение относится к способу лечения и/или предупреждения заболевания или состояния, выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний, который включает введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), как определено выше, или его фармацевтически приемлемой соли или стереоизомера.
В конкретных вариантах осуществления применений и способов лечения, определенных выше, заболевание или состояние выбрано из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; и злокачественной опухоли.
В других конкретных вариантах осуществления применений и способов лечения, определенных выше, предпочтительным вариантом осуществления заболевания или состояния является повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее, предпочтительно ремоделирование после инфаркта миокарда.
В других конкретных вариантах осуществления применений и способов лечения, определенных выше, нейродегенеративное нарушение выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, рассеянного склероза, острого аутоиммунного энцефалита и болезни Крейтцфельда-Якоба.
В других конкретных вариантах осуществления применений и способов лечения, определенных выше, злокачественная опухоль выбрана из группы, состоящей из рака молочной железы, рака ободочной и прямой кишки и лейкоза.
В других конкретных вариантах осуществления применений и способов лечения, определенных выше, фиброзное заболевание выбрано из группы, состоящей из фиброза сердца, фиброза легких, фиброза печени, фиброза почек и ретроперитонеального фиброза.
Соединения формулы (II)
В третьем аспекте настоящее изобретение относится к соединению формулы (II):
(II)
где
R1a выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила,
R2a выбран из группы, состоящей из H и C1-C6 алкила,
R3a представляет собой C3-C6 циклоалкил,
или его фармацевтически приемлемой соли или стереоизомеру.
Соединения формулы (II) представляют собой подгруппу соединений формулы (I).
В конкретном варианте осуществления R1a выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила; предпочтительно из группы, состоящей из метила, этила, пропила, изопропила, циклопропила, циклобутила и циклопентила; более предпочтительно из группы, состоящей из метила, изопропила и циклопропила; еще более предпочтительно из группы, состоящей из изопропила и циклопропила; наиболее предпочтительным является циклопропил.
В другом конкретном варианте осуществления R2a выбран из группы, состоящей из H и C1-C3 алкила; предпочтительно из группы, состоящей из H, метила, этила, пропила и изопропила; более предпочтительно из группы, состоящей из H и метила; наиболее предпочтительным является H.
В другом конкретном варианте осуществления R3a представляет собой C3-C6 циклоалкил; предпочтительно C3-C5 циклоалкил; предпочтительно циклопропил, циклобутил или циклопентил; наиболее предпочтительным является циклопропил.
В конкретном варианте осуществления R1a выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила; R2a выбран из группы, состоящей из H и C1-C3 алкила; и R3a представляет собой C3-C5 циклоалкил.
В другом конкретном варианте осуществления R1a выбран из группы, состоящей из изопропила и циклопропила; R2a представляет собой H; и R3a представляет собой циклопропил.
В конкретном варианте осуществления соединение формулы (II) представляет собой соединение формулы (IIa):

(IIa)
где R1a, R2a и R3a имеют то же значение, как и в соединениях формулы (II), или его стереоизомер или фармацевтически приемлемую соль.
В конкретном варианте осуществления конкретный стереоизомер соединения формулы (II) представляет собой соединение формулы (IIb):
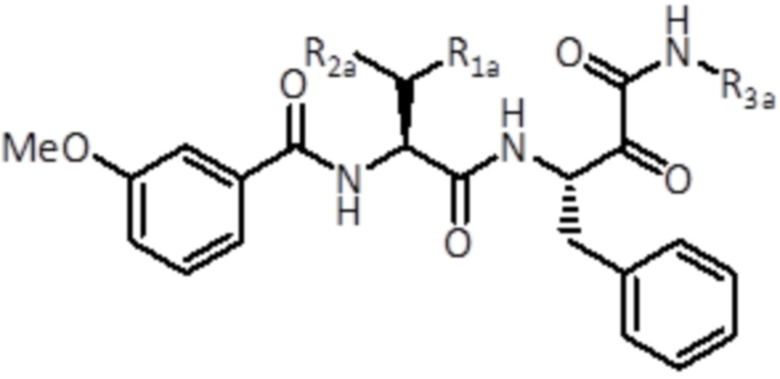
(IIb)
где R1a, R2a и R3a имеют те же значения, как определено для соединений формулы (II), или его фармацевтически приемлемую соль.
В предпочтительном варианте осуществления соединение формулы (II) выбрано из группы, состоящей из:
, и ,
или их стереоизомера или фармацевтически приемлемой соли.
В конкретном варианте осуществления соединение формулы (IIa) выбрано из группы, состоящей из:
, и ,
или их стереоизомера или фармацевтически приемлемой соли.
В конкретном варианте осуществления соединение формулы (IIb) выбрано из группы, состоящей из:
, и ,
или их фармацевтически приемлемой соли.
Фармацевтические композиции
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (II), как определено выше, или его стереоизомер или фармацевтически приемлемую соль, и один или несколько фармацевтически приемлемых эксципиентов.
Термин "фармацевтически приемлемый эксципиент" относится к носителю, разбавителю или адъюванту, который вводят с активным ингредиентом. Такие фармацевтические эксципиенты могут представлять собой стерильные жидкости, такие как вода и масла, включая масла из нефти, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и сходные. В качестве носителей предпочтительно используют воду или водные солевые растворы и водные растворы декстрозы и глицерина, в частности, для инъецируемых растворов. Подходящие фармацевтические носители описаны в "Remington's Pharmaceutical Sciences", E.W. Martin, 21st Edition, 2005.
Соединения формулы (II) по изобретению можно вводить пероральным, сублингвальным, парентеральным, подкожным, внутримышечным, внутривенным, трансдермальным, интраназальным, внутриглазным и/или ректальным путями. Соединения можно вводить отдельно или в комбинации с одним или несколькими другими соединениями по изобретению или одним или несколькими другими лекарственными средствами.
Фармацевтические композиции по настоящему изобретению могут содержать соединения формулы (II) в липосомах или микровезикулах, и они могут иметь форму дисперсий, растворов, лосьонов, гелей и т.п., включая местные препараты.
Описанные выше определения также применимы к фармацевтическим композициям, содержащим соединение формулы (I).
Применение соединений формулы (II)
Как объяснено выше, соединения формулы (II) представляют собой подгруппу соединений формулы (I). Таким образом, соединения формулы (II) также являются ингибиторами кальпаина-1.
Таким образом, в четвертом аспекте настоящее изобретение относится к соединениям формулы (II) для применения в медицине, в частности, для лечения и/или предупреждения заболеваний или состояний, ассоциированных с повышенной активностью кальпаина. Предпочтительные заболевания или состояния представляют собой заболевания или состояния, выбранные из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; и злокачественной опухоли, таких как повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее; нейродегенеративные нарушения; малярия; диабетическая нефропатия; нейротоксичность, индуцированная вирусом ВИЧ; злокачественная опухоль и фиброзные заболевания; более предпочтительным заболеванием или состоянием является повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее, предпочтительно ремоделирование после инфаркта миокарда.
В другом аспекте изобретение относится к применению соединения формулы (II), как определено выше, или его фармацевтически приемлемой соли или стереоизомера, для производства лекарственного средства для лечения и/или профилактики заболевания или состояния, ассоциированного с повышенной активностью кальпаина; предпочтительно выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний; более предпочтительно выбранного из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее, еще более предпочтительно ремоделирования после инфаркта миокарда.
В следующем аспекте настоящее изобретение относится к способу лечения и/или предупреждения заболевания или состояния, ассоциированного с повышенной активностью кальпаина, который включает введение индивидууму, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (II), как определено выше, или его фармацевтически приемлемой соли или стереоизомера. Предпочтительно заболевание или состояние, ассоциированное с повышенной активностью кальпаина, выбрано из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; злокачественной опухоли; и фиброзных заболеваний; более предпочтительно выбрано из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее, еще более предпочтительно ремоделирования после инфаркта миокарда.
В конкретных вариантах осуществления заболевание или состояние выбрано из группы, состоящей из повреждения сердца, вызванного инфарктом, ишемией с реперфузией или без нее; нейродегенеративных нарушений; малярии; диабетической нефропатии; нейротоксичности, индуцированной вирусом ВИЧ; и злокачественной опухоли.
В других конкретных вариантах осуществления заболевание или состояние представляет собой повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее, предпочтительно ремоделирование после инфаркта миокарда.
В других конкретных вариантах осуществления нейродегенеративное нарушение выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, рассеянного склероза, острого аутоиммунного энцефалита и болезни Крейтцфельда-Якоба.
В других конкретных вариантах осуществления злокачественная опухоль выбрана из группы, состоящей из рака молочной железы, рака ободочной и прямой кишки и лейкоза.
В других конкретных вариантах осуществления фиброзное заболевание выбрано из группы, состоящей из фиброза сердца, фиброза легких, фиброза печени, фиброза почек и ретроперитонеального фиброза.
Способ получения соединений формулы (I) и (II)
Соединения формулы (I) и (II), предпочтительно где R3 представляет собой циклоалкильную группу, можно получать, начиная с гидрохлоридов N-замещенных 3-амино-2-гидроксиамидов формулы (VIII), по схеме синтеза, представленной ниже:
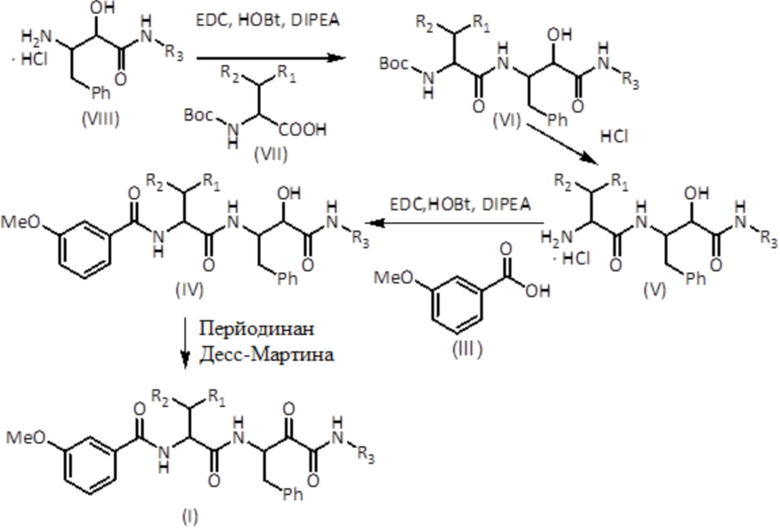
На первой стадии защищенную аминокислоту формулы (VII), HOBt (гидроксибензотриазол), EDC (1-этил-3-(3-диметиламинопропил)карбодиимид) и гидрохлориды N-замещенных 3-амино-2-гидроксиамидов формулы (VIII) растворяют в растворителе, таком как дихлорметан (DCM). Затем добавляют DIPEA (N, N-диизопропилэтиламин) и смесь оставляют реагировать с получением соединения формулы (VI). Другие агенты для присоединения амидов являются в равной степени эффективными, такие как HATU (гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиния) в присутствии DIPEA в диметилформамиде (DMF) или T3P (пропилфосфоновый ангидрид) в присутствии NEt3 в DMF. Соединение формулы (VI) подвергают реакции в растворителе, таком как 1,4-диоксан, с сильной кислотой, такой как хлористоводородная кислота, или в DCM, и обрабатывают трифторуксусной кислотой (TFA) с получением соединения формулы (V). Соединение формулы (III), HOBt, EDC и соединение формулы (V) растворяют в растворителе, таком как DCM. Затем добавляют DIPEA и смесь оставляют реагировать с получением соединения формулы (IV). Другие агенты присоединения амидов являются в равной степени эффективными, такие как HATU в присутствии DIPEA в DMF или T3P в присутствии NEt3 в DMF. Наконец, соединение формулы (IV) растворяют в растворителях, таких как DCM, DMF или их смеси, и добавляют перйодинан Десс-Мартина с получением соединения формулы (I). Другие окислители, такие как DCC (N, N'-дициклогексилкарбодиимид) в DMSO (диметилсульфоксид) являются в равной степени пригодными.
Гидрохлориды N-замещенные 3-амино-2-гидроксиамида формулы (VIII) можно получать, начиная с защищенных аминоальдегидов формулы (XIII), согласно схеме синтеза, показанной ниже:
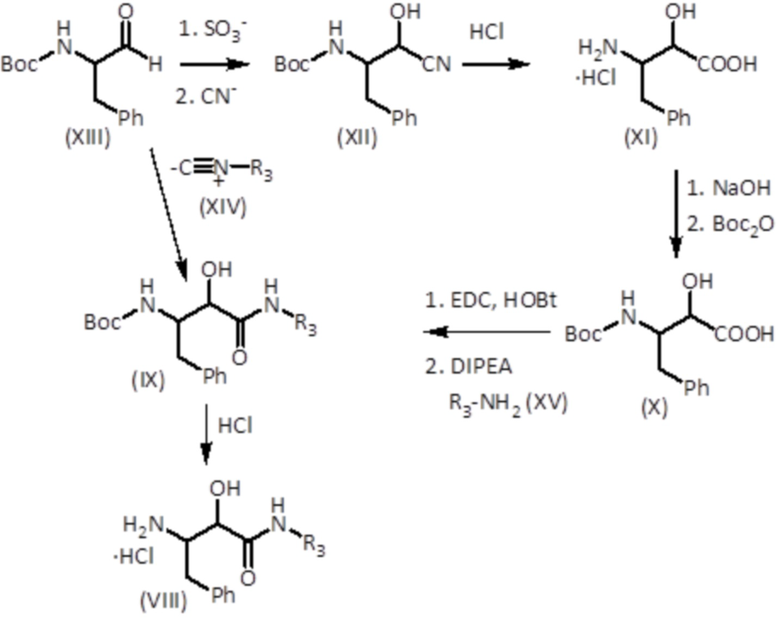
На первой стадии защищенные аминоальдегиды формулы (XIII) растворяют в растворителе, таком как 1,4-диоксан, и добавляют бисульфит натрия, а затем добавляют водный раствор цианида калия с получением соединения формулы (XII). Соединение формулы (XII) растворяют в концентрированном водном растворе кислоты, такой как концентрированная хлористоводородная кислота, и кипятят с обратным холодильником с получением соединения формулы (XI). Водный раствор соединения формулы (XI) доводят до щелочного pH (предпочтительно в диапазоне 10-12), например, гидроксидом натрия, и добавляют Boc2O (ди-трет-бутилдикарбонат). После полного конвертирования смесь подкисляют, например, KHSO4, и соединение формулы (X) экстрагируют с использованием не смешивающегося с водой растворителя, такого как этилацетат. Соединение формулы (X), HOBt и EDC растворяют в растворителе, таком как безводный DCM. Затем добавляют DIPEA и амин формулы (XV), и смесь оставляют реагировать в течение 8-24 часов с получением соединения формулы (VIII). Также можно использовать T3P или HATU вместо EDC и HOBt с хорошими результатами. Соединение формулы (IX) подвергают реакции в растворителе, таком как 1,4-диоксан, с сильной кислотой, такой как хлористоводородная кислота, с получением соединения формулы (VIII).
В альтернативном синтетическом пути, которой также проиллюстрирован на предыдущей схеме, защищенные аминоальдегиды формулы (XIII) растворяют в растворителе, таком как безводный DCM, и добавляют кислоту, такую как уксусная кислота, и соединение изоцианида формулы (XIV) и оставляют реагировать в инертной атмосфере, такой как атмосфера аргона, при комнатной температуре. Затем растворитель удаляют и полученное соединение экстрагируют этилацетатом и промывают насыщенным водным раствором бикарбоната натрия. Затем продукт растворяют в смеси THF (тетрагидрофуран) и MeOH (метанол) и обрабатывают водным раствором гидроксида лития с получением соединения формулы (IX). Затем, соединение формулы (IX) подвергают реакции, как описано выше, в растворителе, таком как 1,4-диоксан, с сильной кислотой, такой как хлористоводородная кислота, с получением соединения формулы (VIII).
Соединения формулы (I), где R3 представляет собой алкоксигруппу, можно получать, начиная с гидрохлоридов N-замещенных 3-амино-2-гидроксиамидов формулы (VIII), по схеме синтеза, представленной ниже:
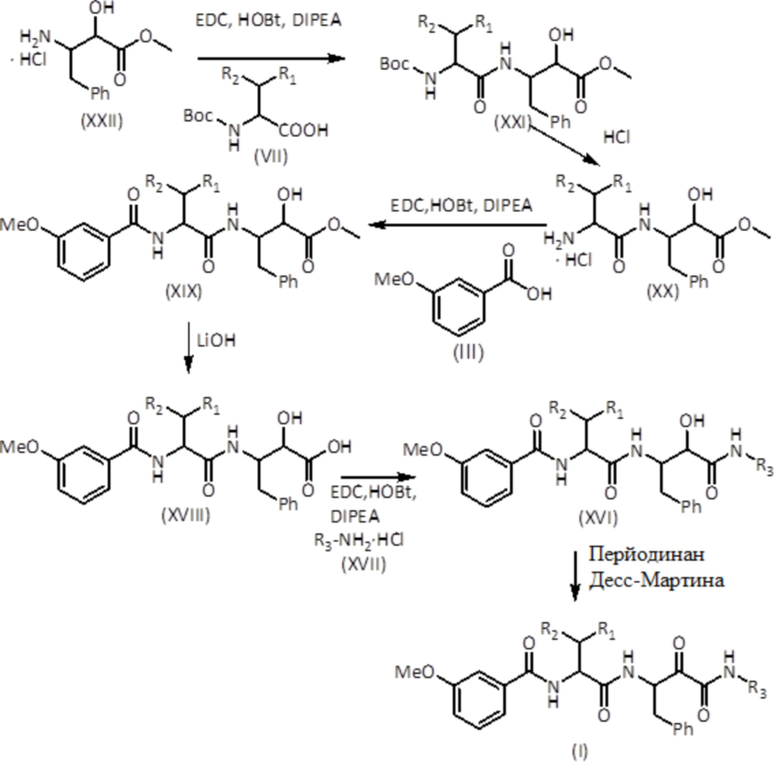
На первой стадии защищенную аминокислоту формулы (VII), HOBt и соединение формулы (XXII) растворяют в растворителе, таком как дихлорметан (DCM). Затем добавляют DIPEA и смесь оставляют реагировать с получением соединения формулы (XXI). Другие агенты присоединения амидов являются в равной степени эффективными, такие как HATU в присутствии DIPEA в DMF или T3P в присутствии NEt3 в DMF. Соединение формулы (XXI) подвергают реакции в растворителе, таком как 1,4-диоксан, с сильной кислотой, такой как хлористоводородная кислота, или в DCM, и обрабатывают трифторуксусной кислотой (TFA) с получением соединения формулы (XX). Соединение формулы (III), HOBt, EDC и соединение формулы (XX) растворяют в растворителе, таком как DCM. Затем добавляют DIPEA и смесь оставляют реагировать с получением соединения формулы (XIX). Другие агенты присоединения амидов являются в равной степени эффективными, такие как HATU в присутствии DIPEA в DMF или T3P в присутствии NEt3 в DMF. Затем соединение формулы (XIX) обрабатывают LiOH в смеси THF/MeOH/вода, а затем обрабатывают водным раствором кислоты, такой как HCl, с получением соединения формулы (XVIII). Соединение формулы (XVII), HOBt, EDC и соединение формулы (XVIII) растворяют в растворителе, таком как DCM. Затем добавляют DIPEA и смесь оставляют реагировать с получением соединения формулы (XVI). Другие агенты присоединения амидов являются в равной степени эффективными, такие как HATU в присутствии DIPEA в DMF или T3P в присутствии NEt3 в DMF. Наконец, соединение формулы (XVI) растворяют в растворителе, таком как DCM, DMF или их смесь, и добавляют перйодинан Десс-Мартина с получением соединения формулы (I). Другие окислители, такие как DCC (N, N'-дициклогексилкарбодиимид) в DMSO (диметилсульфоксид), являются в равной степени пригодными.
Соединение формулы (XXII) можно получать из соединения формулы (X), синтез которого описан выше, посредством обработки оксалилхлоридом в метаноле, как показано на схеме синтеза ниже:
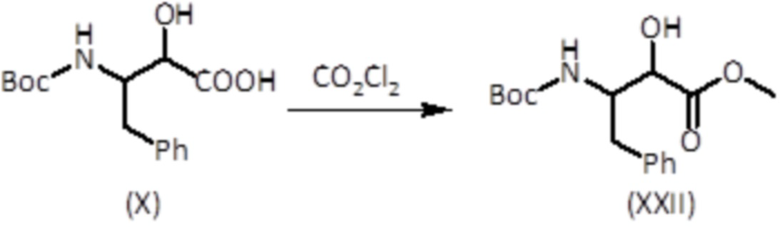
Исходные соединения формул (XVII), (XIII), (XIV), (XV), (VII) и (III) либо коммерчески доступны, либо могут быть получены способами, описанными в литературе.
Следующие примеры являются только иллюстративными для определенных вариантов осуществления изобретения и не могут считаться ограничивающими его каким-либо образом.
Примеры
Сокращенные обозначения
В примерах используют следующие сокращенные обозначения:
Boc: трет-бутоксикарбонил
конц.: концентрат
Boc2O: ди-трет-бутилдикарбонат
DCM: дихлорметан
DIPEA: N, N-диизопропилэтиламин
DMSO: диметилсульфоксид
EDC·HCl: гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида
EtOAc: этилацетат
HBTU: гексафторфосфат N, N,N′,N′-тетраметил-O-(1H-бензотриазол-1-ил)урония
HCl: хлористоводородная кислота
HEPES: 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота
HOBt: гидроксибензотриазол
LC-MS: жидкостная хроматография-масс-спектрометрия
Leu-OH: лейцин
MeOH: метанол
Мин: минуты
Phe-OH: фенилаланин
Насыщ.: насыщенный
T3P: пропилфосфоновый ангидрид
TBME: трет-бутилметиловый эфир
THF: тетрагидрофуран
tR: время удержания
Val-OH: валин
Материалы и способы
LC-MS: Продукты согласно примерам охарактеризовывали с использованием жидкостной хроматографии, сопряженной с масс-спектрометрией (LC-MS). Анализ посредством ВЭЖХ-MS проводили следующим образом: в хроматографе Alliance HT 2795 (Waters), оборудованном детектором с фотодиодной матрицей 2996 и сопряженном с детектором масс LC/MS 3100. Разделение осуществляли с использованием колонки XBridge C18 (50×4,6 мм, S-3,5 мкм) и с использованием смесей 10 мМ водного раствора NH4CO3 с pH=9 (A) и ацетонитрила (B) в качестве элюентов при 50°C и скорости потока 1,6 мл/мин со следующими условиями элюирования: от 5% до 100% B за 3,5 мин. Детектор был установлен в позитивный режим электрораспыления (ESI+) в диапазоне масс 100-700. Напряжение на конусе 10 В. Источник T: 120°C. Десольватация T: 350°C.
Реагенты: 2-метоксибензойная кислота (Sigma Aldrich); 3-метоксибензойная кислота (Sigma Aldrich); 4-метоксибензойная кислота (Sigma Aldrich); уксусная кислота (VWR); Boc-L-Phe-OH (Fluorochem); Boc-L-Leu-OH (Fluorochem); Boc-L-Val-OH (Fluorochem); циклопропиламин (Sigma Aldrich); циклопропилизоцианид (Fluorochem); 15% раствор перйодинана Десс-Мартина в DCM (Acros); EDC (Iris Biotech); HBTU (Iris Biotech); 4 Н HCl в 1,4-диоксане (TCI Europe Organic Chem); HOBt (Carbosynth); LiOH·H2O (Sigma Aldrich); 1,0 M раствор алюмогидрида лития в тетрагидрофуране (Sigma Aldrich); гидрохлорид N, O-диметилгидроксиламина (Fluorochem); (S)-2-(Boc-амино)-3-циклопропилпропионовая кислота (Fluorochem).
Растворители: DCM (Scharlab); этанол (Panreac); EtOAc (Scharlab); гексан (Scharlab); MeOH (Scharlab); TBME (SDS); THF (Panreac).
Синтез промежуточных соединений
Промежуточное соединение 1: (S)-трет-бутил(1-(метокси(метил)амино)-1-оксо-3-фенилпропан-2-ил)карбамат
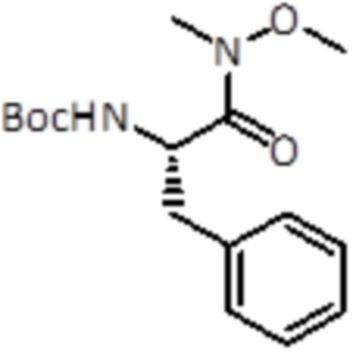
К раствору Boc-L-Phe-OH (10,0 г, 37,7 ммоль, 1,0 экв.) в DCM (150 мл), добавляли гидрохлорид N, O-диметилгидроксиламина (4,0 г, 41,1 ммоль, 1,1 экв.), HBTU (III) (15,7 г, 41,1 ммоль, 1,1 экв.) и DIPEA (20 мл, 113,0 ммоль, 3,0 экв.). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Затем летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали насыщенным раствором NaHCO3 (2×200 мл). Неочищенный материал очищали флэш-хроматографией (ISCO Rf) с использованием гексана/EtOAc в качестве элюентов, от 0% до 40% в EtOAc, продукт элюировался при 35%. Было получено 11,6 г (37,7 ммоль, выход 100%).
LC-MS: tR=2,58 мин; m/z=309
Промежуточное соединение 2: Синтез (S)-трет-бутил (1-оксо-3-фенилпропан-2-ил)карбамата
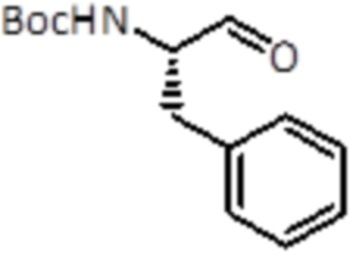
Раствор алюмогидрида лития 1,0 M в тетрагидрофуране (37,7 мл, 37,7 ммоль, 1,0 экв.) добавляли к раствору промежуточного соединения 1 (11,6 г, 37,7 ммоль, 1,0 экв.) в THF (250 мл), охлажденному до 0°C. Смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли EtOAc и растворитель выпаривали. Неочищенный материал очищали хроматографией на силикагеле с использованием гексана/EtOAc в качестве растворителей, от 0% до 60% в EtOAc, продукт элюировался при 25%. Было получено 6,7 г желаемого продукта (26,9 ммоль, выход 68%).
LC-MS: tR=2,48 мин; m/z=250
Промежуточное соединение 3: Синтез трет-бутил ((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)карбамата
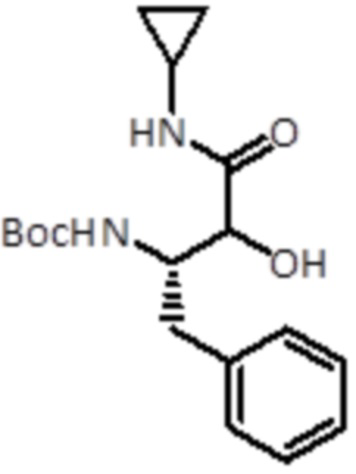
Уксусную кислоту (0,35 мл, 8,0 ммоль, 2,0 экв.) и циклопропилизоцианид (0,30 мл, 4,4 ммоль, 1,1 экв.) добавляли к раствору промежуточного соединения 2 (1,0 г, 4,0 ммоль, 1,0 экв.) в DCM (20 мл), и смесь перемешивали при к.т. Через 15 мин к смеси добавляли смесь THF/MeOH/H2O (7/5/3), а затем LiOH·H2O (0,67 г, 16,0 ммоль, 4 экв.). Через 15 мин растворители удаляли в вакууме. Продукт экстрагировали в EtOAc, и раствор промывали насыщ. раствором NaHCO3 и очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве растворителей, от 0% до 50% в смеси гексан:этанол (8:2), продут элюировался при 25%. Было получено 0,31 г продукта (выход 23%).
LC-MS: tR=2,32 мин; m/z=335
Промежуточное соединение 4: Синтез гидрохлорида (3S)-3-амино-N-циклопропил-2-гидрокси-4-фенилбутанамида
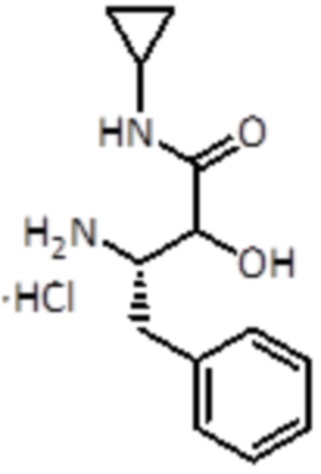
Раствор 4 Н HCl в 1,4-диоксане (1 мл, 4,1 ммоль, 4 экв.) добавляли к промежуточному соединению 3 (0,31 г, 0,9 ммоль, 1,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали до сухого состояния. Этот продукт использовали для следующей стадии без дальнейшей очистки.
LC-MS: tR=1,50 мин; m/z=235
Промежуточное соединение 5: трет-бутил ((2S)-1-(((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)карбамат
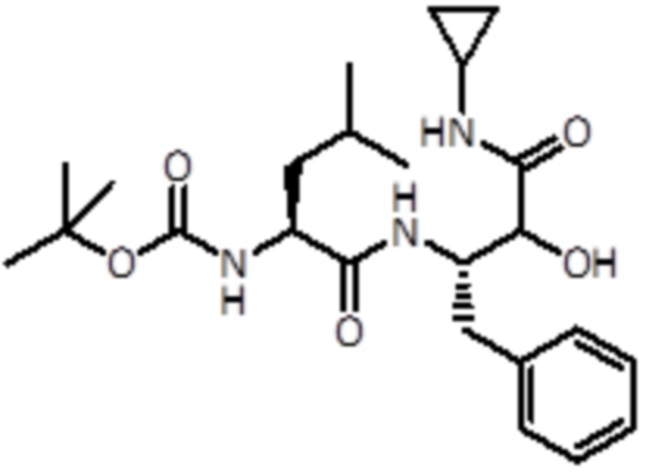
Раствор промежуточного соединения 4 (251 мг, 0,9 ммоль, 1,0 экв.) и DIPEA (0,6 мл, 3,7 ммоль, 4 экв.) в DCM (5 мл) добавляли к раствору Boc-L-Leu-OH (300 мг, 1,2 ммоль, 1,2 экв.), EDC (249 мг, 1,3 ммоль, 1,5 экв.) и HOBt (199 мг, 1,3 ммоль, 1,5 экв.) в DCM (5 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (2×10 мл) и очищали флэш-хроматографией (ISCO Rf) с использованием гексана/TBME в качестве элюентов, от 0% до 80% в TBME, продукт элюировался при 60%. Было получено 298 мг продукта (0,7 ммоль, выход 72%).
LC-MS (Способ A): tR =2,60 мин; m/z=448.
Промежуточное соединение 6: гидрохлорид (2S)-2-амино-N-((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)-4-метилпентанамида
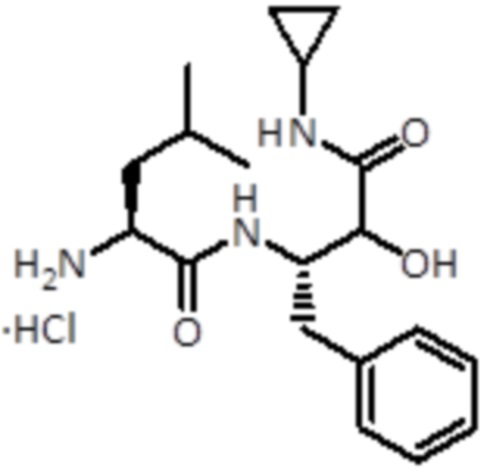
Раствор HCl (4 N) в 1,4-диоксане (3,3 мл,13,2 ммоль, 20 экв.) добавляли к промежуточному соединению 5 (298 мг, 0,6 ммоль, 1,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали до сухого состояния. Продукт использовали для следующей стадии без дальнейшей очистки.
LC-MS: tR =1,92-2,03 мин; m/z=348.
Промежуточное соединение 7: N-((2S)-1-(((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-3-метоксибензамид
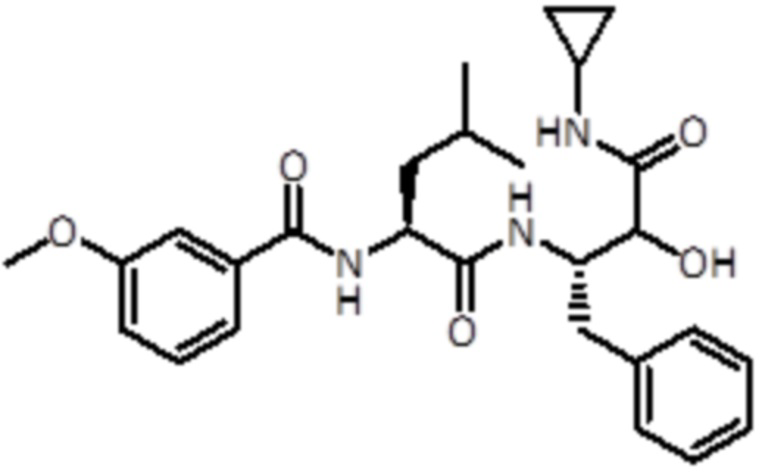
Раствор промежуточного соединения 6 (115 мг, 0,3 ммоль, 1,0 экв.) и DIPEA (231 мкл, 1,3 ммоль, 4,0 экв.) в DCM (5 мл) добавляли к раствору 3-метоксибензойной кислоты (60,4 мг, 0,4 ммоль, 1,2 экв.), EDC (108 мг, 0,6 ммоль, 1,7 экв.) и HOBt (86 мг, 0,6 ммоль, 1,5 экв.) в DCM (4 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (2×20 мл). Материал очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве элюентов, от 0% до 60% в гексане:этаноле (8:2), продукт элюировался при 40%. Получали 100 мг продукта (0,2 ммоль, 100%).
LC-MS: tR =2,47 мин; m/z=482.
Промежуточное соединение 8: Синтез трет-бутил ((2S)-1-((4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-3-метил-1-оксобутан-2-ил)карбамата
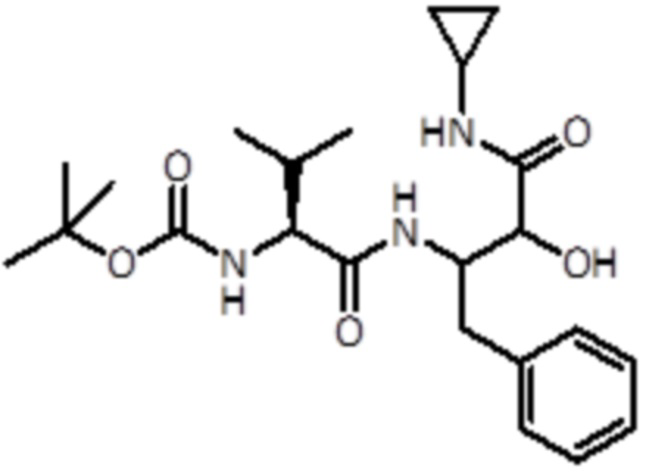
Раствор промежуточного соединения 4 (1,06 г, 3,91 ммоль, 1,0 экв.) и DIPEA (2,74 мл, 15,66 ммоль, 4 экв.) в DCM (5 мл) добавляли к раствору Boc-L-Val-OH (1,28 г, 5,87 ммоль, 1,5 экв.), EDC (1,13 г, 5,87 ммоль, 1,5 экв.) и HOBt (0,9 г, 5,87 ммоль, 1,5 экв.) в DCM (5 мл). Реакционную смесь перемешивали в течение 45 минут при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенный материал очищали флэш-хроматографией (ISCO Rf) с использованием DCM/ DCM:метанола (8:2) в качестве элюентов, от 0% до 50% в DCM:метаноле (8:2), продукт элюировался при 15%. Было получено 600 мг продукта (1,384 ммоль, выход 35%).
LC-MS: tR=2,47 мин; m/z=434
Промежуточное соединение 9: Синтез гидрохлорида 3-((S)-2-амино-3-метилбутанамидо)-N-циклопропил-2-гидрокси-4-фенилбутанамида
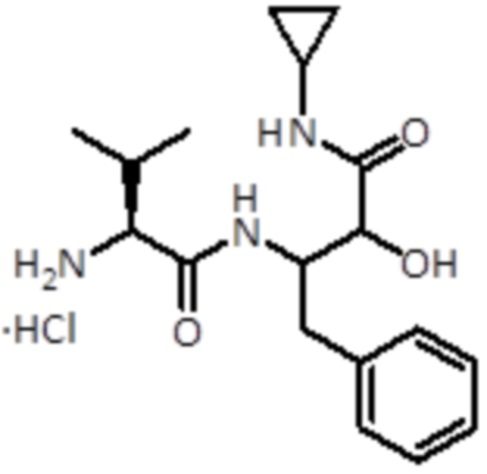
Раствор 4 Н HCl в 1,4-диоксане (6,9 мл,27,7 ммоль, 20 экв.) добавляли к промежуточному соединению 8 (600 мг, 1,384 ммоль, 1,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали до сухого состояния. Продукт использовали для следующей стадии без дальнейшей очистки.
LC-MS: tR=1,77-1,88 мин; m/z=334
Промежуточное соединение 10: Синтез N-((2S)-1-((4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-3-метил-1-оксобутан-2-ил)-3-метоксибензамида
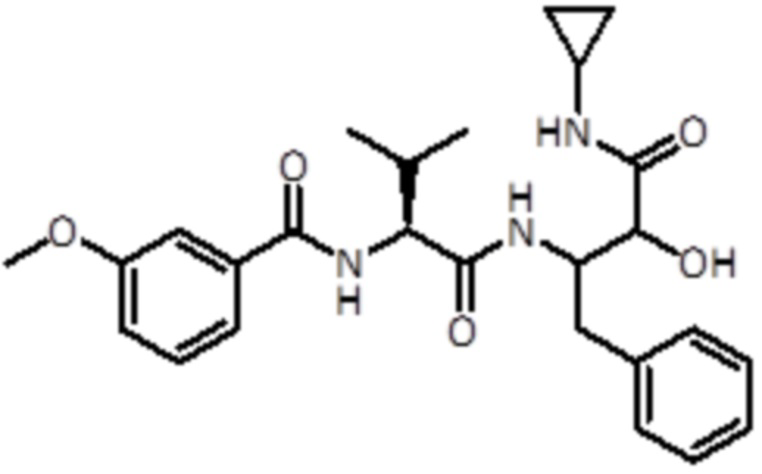
Раствор промежуточного соединения 9 (461 мг, 0,3 ммоль, 1,0 экв.) и DIPEA (0,97 мл, 5,53 ммоль, 4,0 экв.) в DCM (9 мл) добавляли к раствору 3-метоксибензойной кислоты (316 мг, 2,07 ммоль, 1,5 экв.), EDC (398 мг, 2,07 ммоль, 1,5 экв.) и HOBt (318 мг, 2,07 ммоль, 1,5 экв.) в DCM (9 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенный материал очищали флэш-хроматографией (ISCO Rf) с использованием DCM/ DCM:метанола (8:2) в качестве элюентов, от 0% до 20% в DCM:метаноле (8:2), продукт элюировался при 15%. Было получено 470 мг продукта (1 ммоль, 73%).
LC-MS: tR=2,32 мин; m/z=468.
Промежуточное соединение 11: Синтез трет-бутил ((2S)-3-циклопропил-1-((4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-1-оксопропан-2-ил)карбамата
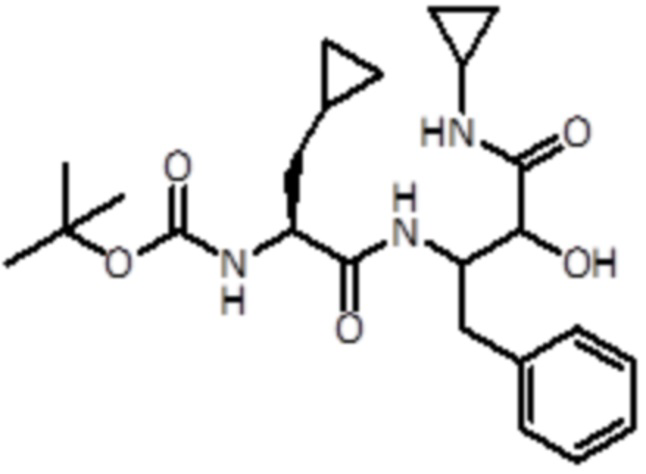
Раствор промежуточного соединения 4 (530 мг, 1,96 ммоль, 1,0 экв.) и DIPEA (1,37 мл, 7,83 ммоль, 4 экв.) в DCM (3 мл) добавляли к раствору (S)-2-(Boc-амино)-3-циклопропилпропионовой кислоты (673 мг, 2,94 ммоль, 1,5 экв.), EDC (563 мг, 2,94 ммоль, 1,5 экв.) и HOBt (450 мг, 2,94 ммоль, 1,5 экв.) в DCM (2 мл). Реакционную смесь перемешивали в течение полутора часов при комнатной температуре. Летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенную смесь очищали флэш-хроматографией (ISCO Rf) с использованием DCM/DCM:метанола (8:2) в качестве элюентов, от 0% до 20% в DCM:метаноле (8:2), продукт элюировался при 12%. Было получено 430 мг продукта (0,96 ммоль, выход 49%).
LC-MS: tR=2,5 мин; m/z=446
Промежуточное соединение 12: Синтез гидрохлорида 3-((S)-2-амино-3-циклопропилпропанамидо)-N-циклопропил-2-гидрокси-4-фенилбутанамида
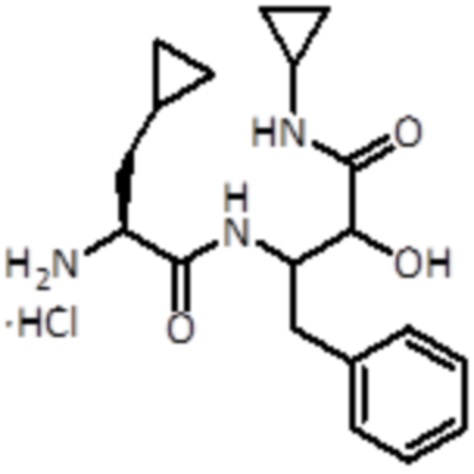
Раствор 4 Н HCl в 1,4-диоксане (4,83 мл,19,3 ммоль, 20 экв.) добавляли к промежуточному соединению 11 (430 мг, 0,96 ммоль, 1,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали до сухого состояния. Продукт использовали для следующей стадии без дальнейшей очистки.
LC-MS (Способ A): tR=1,82-1,92 мин; m/z=346
Промежуточное соединение 13: Синтез N-((2S)-3-циклопропил-1-((4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-1-оксопропан-2-ил)-3-метоксибензамида
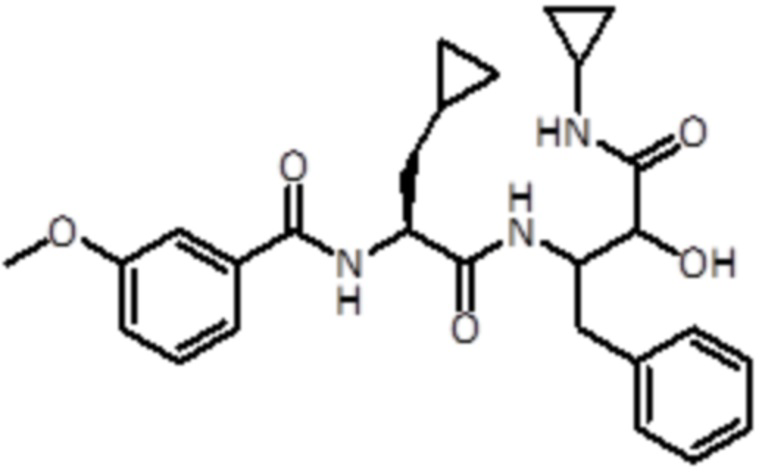
Раствор промежуточного соединения 12 (333 мг, 0,3 ммоль, 1,0 экв.) и DIPEA (0,67 мл, 3,86 ммоль, 4,0 экв.) в DCM (6 мл) добавляли к раствору 3-метоксибензойной кислоты (220 мг, 1,45 ммоль, 1,5 экв.), EDC (277 мг, 1,45 ммоль, 1,5 экв.) и HOBt (221 мг, 1,45 ммоль, 1,5 экв.) в DCM (6 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенный материал очищали флэш-хроматографией (ISCO Rf) с использованием DCM/DCM:метанола (8:2) в качестве элюентов, от 0% до 50% в DCM:метаноле (8:2), продукт элюировался при 38%. Было получено 440 мг продукта (0,88 ммоль, 91%).
LC-MS: tR=2,37 мин; m/z=480
Промежуточное соединение 14: Синтез трет-бутил ((2S)-1-циано-1-гидрокси-3-фенилпропан-2-ил)карбамата

Бисульфат натрия (11,2 г, 107,0 ммоль, 4 экв.) добавляли к раствору промежуточного соединения 2 (6,7 г, 26,9 ммоль, 1,0 экв.) в 1,4-диоксане (150 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 минут и добавляли цианид калия (7,0 г, 107,0 ммоль, 4 экв.), растворенный в воде (45 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Органический растворитель выпаривали, добавляли воду и EtOAc, и слои разделяли. Объединенные органические слои промывали насыщ. водн. NaHCO3, сушили и концентрировали при пониженном давлении. Было получено 7,0 г продукта (23,5 ммоль, выход 94%).
LC-MS: tR=2,83 мин; m/z=277
Промежуточное соединение 15: Синтез (3S)-3-((трет-бутоксикарбонил)амино)-2-гидрокси-4-фенилбутановой кислоты
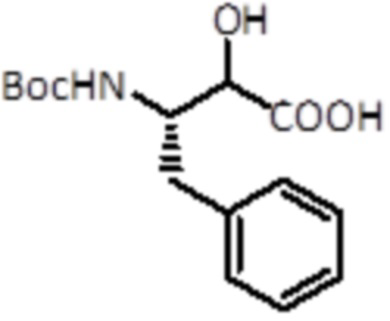
Промежуточное соединение 14 (6,5 г, 23,5 ммоль, 1,0 экв.) растворяли в 20 мл раствора конц. HCl и кипятили в течение 1 часа. Затем неочищенную смесь охлаждали и промывали DCM. К водному слою добавляли 10 Н NaOH до pH 11 и промывали DCM. К водному слою добавляли Boc2O (5,6 г, 25,8 ммоль, 1,1 экв.). Смесь перемешивали при комнатной температуре в течение ночи. После полного конвертирования смесь подкисляли KHSO4 до pH 2 и экстрагировали EtOAc. Органический слой выпаривали и продукт использовали для следующей стадии без дальнейшей очистки. Было получено 5,1 г (17,2 ммоль, выход 73%, 2 стадии).
LC-MS: tR=1,62 мин; m/z=296
Промежуточное соединение 16: Синтез гидрохлорида (3S)-метил 3-амино-2-гидрокси-4-фенилбутаноата
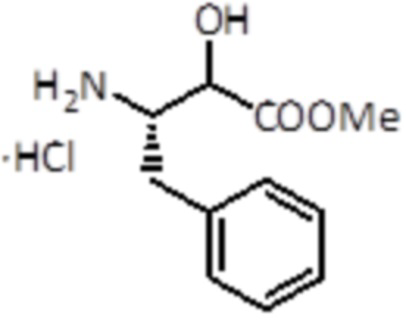
Оксалилхлорид (6 мл, 69,9 ммоль, 4,0 экв.) медленно добавляли к раствору промежуточного соединения 15 (5,1 г, 17,26 ммоль, 1,0 экв.) в MeOH (300 мл) при 0°C. Смесь перемешивали в течение ночи при комнатной температуре. Неочищенную смесь упаривали, а затем совместно упаривали с MeOH, и продукт использовали для следующей стадии без дальнейшей очистки. Было получено 4,2 г (17,26 ммоль, количественный выход).
LC-MS: tR=1,58-1,65 мин; m/z=210.
Промежуточное соединение 17: Синтез метил 3-((S)-2-((трет-бутоксикарбонил)амино)-3-циклопропилпропанамидо)-2-гидрокси-4-фенилбутаноата
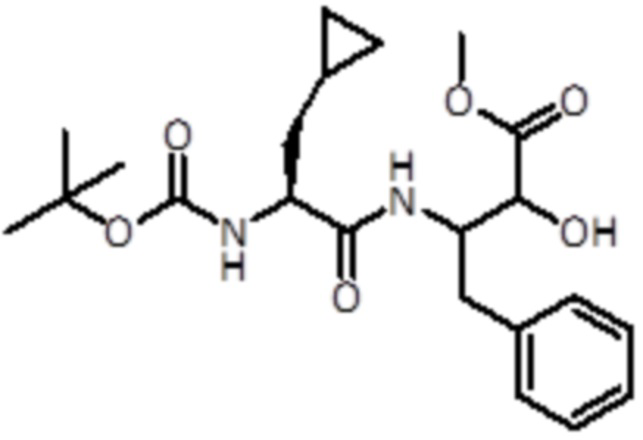
Раствор промежуточного соединения 16 (3 г, 12,21 ммоль, 1,0 экв.) и DIPEA (8,53 мл, 48,8 ммоль, 4 экв.) в DCM (15 мл) добавляли к раствору (S)-2-(Boc-амино)-3-циклопропилпропионовой кислоты (2,8 г, 12,21 ммоль, 1 экв.), EDC (3,51 г, 18,31 ммоль, 1,5 экв.) и HOBt (2,8 г, 18,31 ммоль, 1,5 экв.) в DCM (10 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенный продукт очищали флэш-хроматографией (Biotage) с использованием DCM/DCM:метанола (8:2) в качестве элюентов, от 0% до 30% в DCM:метаноле (8:2), продукт элюировался при 20%. Было получено 2,2 г продукта (5,23 ммоль, выход 43%).
LC-MS: tR=2,64 мин; m/z=421.
Промежуточное соединение 18: Синтез гидрохлорида метил 3-((S)-2-амино-3-циклопропилпропанамидо)-2-гидрокси-4-фенилбутаноата

Раствор 4 Н HCl в 1,4-диоксане (26,2 мл, 105 ммоль, 20 экв.) добавляли к промежуточному соединению 17 (2,2 г, 5,23 ммоль, 1,0 экв.). Смесь перемешивали при комнатной температуре в течение 1 ч, а затем упаривали до сухого состояния. Продукт использовали для следующей стадии без дальнейшей очистки.
LC-MS: tR=1,78 мин; m/z=321.
Промежуточное соединение 19: Синтез метил 3-((S)-3-циклопропил-2-(3-метоксибензамидо)пропанамидо)-2-гидрокси-4-фенилбутаноат
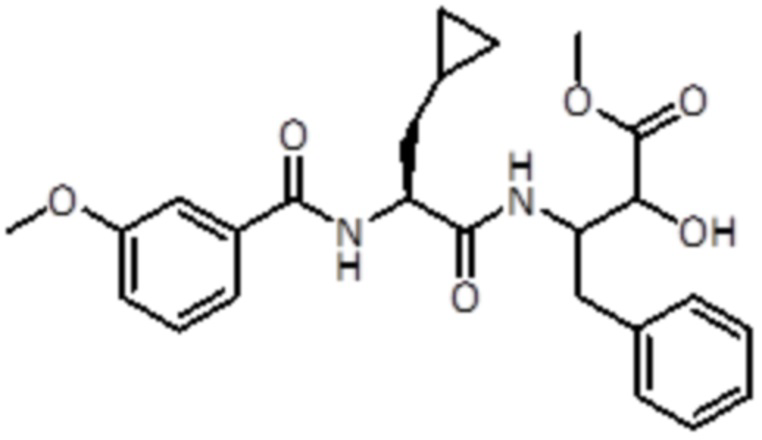
Раствор промежуточного соединения 18 (1,87 г, 5,23 ммоль, 1,0 экв.) и DIPEA (3,66 мл, 3,86 ммоль, 4,0 экв.) в DCM (10 мл) добавляли к раствору 3-метоксибензойной кислоты (0,955 г, 6,28 ммоль, 1,2 экв.), EDC (1,5 г, 7,85 ммоль, 1,5 экв.) и HOBt (1,2 г, 7,85 ммоль, 1,5 экв.) в DCM (10 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3. Неочищенный продукт очищали флэш-хроматографией (Biotage) с использованием DCM/ DCM:метанола (8:2) в качестве элюентов, от 0% до 50% в DCM:метаноле (8:2), продукт элюировался при 25%. Было получено 1 г продукта (2,2 ммоль, 42%).
LC-MS: tR=2,47 мин; m/z=455.
Промежуточное соединение 20: Синтез 3-((S)-3-циклопропил-2-(3-метоксибензамидо)пропанамидо)-2-гидрокси-4-фенилбутановой кислоты
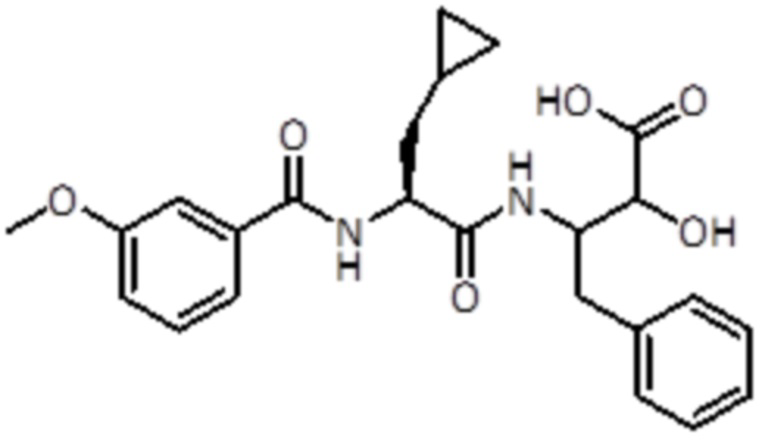
LiOH·H2O (111 мг, 2,64 ммоль, 3,0 экв.) добавляли к раствору промежуточного соединения 19 (400 мг, 0,88 ммоль, 1,0 экв.), растворенного в 13,5 мл смеси THF/MeOH/H2O (5/3/1). После полного конвертирования растворители удаляли в вакууме. Неочищенный материал растворяли в 1 M HCl и экстрагировали DCM до тех пор, пока все промежуточное соединение 20 не экстрагировалось в органический слой. Органические слои упаривали до сухого состояния. Промежуточное соединение 20 использовали для следующей стадии без дальнейшей очистки.
LC-MS: tR=1,85 мин; m/z=441.
Промежуточное соединение 21: Синтез N-((2S)-3-циклопропил-1-((3-гидрокси-4-(метоксиамино)-4-оксо-1-фенилбутан-2-ил)амино)-1-оксопропан-2-ил)-3-метоксибензамида

К раствору промежуточного соединения 20 (470 мг, 1,07 ммоль, 1,0 экв.) и гидрохлорида O-метилгидроксиламина (134 мг, 1,6 ммоль, 1,5 экв.) в DCM (20 мл) добавляли EDC (307 мг, 1,6 ммоль, 1,5 экв.) и HOBt (245 мг, 1,6 ммоль, 1,5 экв.) и DIPEA (0,75 мл, 4,27 ммоль, 4,0 экв.). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. После этого добавляли 0,1 мл DIPEA и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли 0,5 экв. как EDC, так и HOBt, и перемешивание продолжали в течение 2 часов при комнатной температуре. Летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 и 5% водным раствором лимонной кислоты. Неочищенный продукт очищали флэш-хроматографией (Biotage) с использованием DCM/DCM:метанола (8:2) в качестве элюентов, от 0% до 50% в DCM:метаноле (8:2), продукт элюировался при 20%. Было получено 170 мг продукта (0,37 ммоль, выход 35%).
LC-MS: tR=2,18 мин; m/z=470
Промежуточное соединение 22: N-((2S)-1-(((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-4-метоксибензамид

Раствор промежуточного соединения 6 (200 мг, 0,6 ммоль, 1,0 экв.) и DIPEA (392 мкл, 2,3 ммоль, 4,0 экв.) в DCM (6 мл) добавляли к раствору 4-метоксибензойной кислоты (105 мг, 0,7 ммоль, 1,2 экв.), EDC (188 мг, 1,0 ммоль, 1,7 экв.) и HOBt (150 мг, 1,0 ммоль, 1,7 экв.) в DCM (6 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и летучие вещества удаляли в вакууме. Неочищенный материал экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (3×10 мл) и 5% водным раствором лимонной кислоты (3×10 мл). Неочищенную смесь очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве элюентов, от 0% до 100% в гексане:этаноле (8:2), продукт элюировался при 35%. Было получено 115 мг продукта (0,2 ммоль, 41%).
LC-MS: tR=2,35 мин; m/z=482
Промежуточное соединение 23: N-((2S)-1-(((2S)-4-(циклопропиламино)-3-гидрокси-4-оксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-2-метоксибензамид
Раствор промежуточного соединения 6 (200 мг, 0,5 ммоль, 1,0 экв.) и DIPEA (350 мкл, 2,0 ммоль, 4,0 экв.) в DCM (6 мл) добавляли к раствору 2-метоксибензойной кислоты (95 мг, 0,6 ммоль, 1,2 экв.), EDC (169 мг, 0,9 ммоль, 1,7 экв.) и HOBt (135 мг, 0,9 ммоль, 1,7 экв.) в DCM (5 мл). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (3×10 мл) и 5% водным раствором лимонной кислоты (3×10 мл). Неочищенный продукт очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве элюентов, от 0% до 100% в гексане:этаноле (8:2), продукт элюировался при 35%. Было получено 250 мг (0,5 ммоль, 100%).
LC-MS: tR=2,47 мин; m/z=482.
Получение соединений примеров и сравнительных примеров
Пример 1: N-((S)-1-(((S)-4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-3-метоксибензамид
5 мкл воды добавляли к 15% раствору перйодинана Десс-Мартина в DCM (640 мкл, 0,3 ммоль, 1,5 экв.). Затем смесь добавляли к раствору промежуточного соединения 7 (100 мг, 0,2 ммоль, 1,0 ммоль) в DCM (4 мл). Смесь перемешивали в течение 15 минут при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (2×10 мл). Неочищенный продукт очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве растворителей, от 0% до 60% в гексане:этаноле (8:2), продукт элюировался при 30% в гексане:этаноле (8:2). Было получено 47,6 мг продукта (0,1 ммоль, выход 47%).
LC-MS: tR =2,63 мин; m/z=480.
1H-ЯМР (400 МГц, DMSO-d6) δ 8,75 (дд, J=26,3, 5,1 Гц, 1H), 8,38 (дд, J=18,9, 8,1 Гц, 1H), 8,33 (д, J=7,7 Гц, 1H), 7,49-7,31 (м, 3H), 7,27-7,15 (м, 5H), 7,09 (м, 1H), 5,22-5,13 (м, 1H), 4,54 (м, 1H), 3,80 (с, 3H), 3,13 (м, 1H), 2,88-2,77 (м, 1H), 2,76-2,70 (м, 1H), 1,67-1,55 (м, 1H), 1,53-1,43 (м, 1H), 1,29 (м, 1H), 0,91-0,78 (м, 6H), 0,65 (м, 2H), 0,61-0,53 (м, 2H).
Пример 2: Синтез N-((S)-1-((4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-3-метил-1-оксобутан-2-ил)-3-метоксибензамида
15% раствор перйодинана Десс-Мартина в DCM (3,13 мл, 1,51 ммоль, 1,5 экв.) добавляли к раствору промежуточного соединения 10 (470 мг, 1 ммоль, 1,0 экв.), растворенному в предварительно полученном растворе DCM (10 мл) и воды (5 мкл). Смесь перемешивали в течение 15 минут при комнатной температуре. После этого добавляли 1,5 экв. 15% раствора перйодинана Десс-Мартина и смесь перемешивали в течение дополнительных 15 минут при комнатной температуре. Летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали 1 M раствором NaOH. Неочищенный продукт растирали с использованием гексана и фильтровали. Было получено 352 мг продукта (0,76 ммоль, выход 76%).
LC-MS: tR =2,52 мин; m/z=466
1H-ЯМР (400 МГц, DMSO-d6) δ 0,61 (с, 2H), 0,61-0,68 (м, 2H), 0,73 (дд, J=23,0, 6,7 Гц, 3H), 0,87 (дд, J=6,7, 1,8 Гц, 3H), 1,92-2,10 (м, 1H), 2,68-2,85 (м, 2H), 3,07-3,18 (м, 1H), 3,81 (д, J=1,1 Гц, 3H), 4,32 (тд, J=8,3, 5,6 Гц, 1H), 5,18-5,28 (м, 1H), 7,10 (дддд, J=8,0, 2,8, 1,9, 1,1 Гц, 1H), 7,15-7,25 (м, 5H), 7,34-7,46 (м, 3H), 8,15 (дд, J=11,8, 9,0 Гц, 1H), 8,45 (дд, J=14,4, 7,2 Гц, 1H), 8,76 (дд, J=21,8, 5,1 Гц, 1H).
Пример 3: Синтез N-((S)-3-циклопропил-1-((4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-1-оксопропан-2-ил)-3-метоксибензамида
15% раствор перйодинана Десс-Мартина в DCM (2,73 мл, 1,31 ммоль, 1,5 экв.) добавляли к раствору промежуточного соединения 13 (420 мг, 0,88 ммоль, 1,0 экв.), растворенного в 8 мл предварительно полученного раствора DCM (10 мл) и 5 мкл воды. Смесь перемешивали в течение 15 минут при комнатной температуре. После этого добавляли 1,5 экв. 15% раствора перйодинана Десс-Мартина и смесь перемешивали в течение дополнительных 15 минут при комнатной температуре. Летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали 1 M раствором NaOH. Неочищенный продукт растирали с использованием гексан и фильтровали. Было получено 274 мг продукта (0,57 ммоль, выход 65%).
LC-MS: tR =2,57 мин; m/z=478
1H-ЯМР (400 МГц, DMSO-d6) δ -0,01-0,20 (м, 2H), 0,25-0,43 (м, 1H), 0,54-0,70 (м, 4H), 0,70-0,79 (м, 1H), 1,18-1,35 (м, 1H), 1,47-1,68 (м, 2H), 2,69-2,88 (м, 2H), 3,12 (тд, J=13,5, 4,2 Гц, 1H), 3,80 (д, J=1,2 Гц, 3H), 4,55 (тд, J=8,7, 5,1 Гц, 1H), 5,20 (дтд, J=9,0, 7,8, 4,1 Гц, 1H), 7,08-7,12 (м, 1H), 7,16-7,25 (м, 5H), 7,34-7,46 (м, 3H), 8,29-8,45 (м, 2H), 8,75 (дд, J=27,7, 5,1 Гц, 1H).
Пример 4: Синтез N-((S)-3-циклопропил-1-((4-(метоксиамино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-1-оксопропан-2-ил)-3-метоксибензамида
15% раствор перйодинана Десс-Мартина в DCM (0,827 мл, 0,4 ммоль, 1,5 экв.) добавляли 4 порциями к раствору промежуточного соединения 21 (170 мг, 0,36 ммоль, 1,0 экв.) в DMSO (2,5 мл), перемешивая смесь в течение 10 минут между добавлениями. Смесь перемешивали в течение 2 часов при комнатной температуре, а затем летучие вещества удаляли в вакууме. Неочищенную смесь экстрагировали EtOAc и промывали 1 M раствором NaOH. Летучие вещества выпаривали и неочищенную смесь очищали обращено-фазовой хроматографией (Biotage) с использованием H2O/ацетонитрила в качестве растворителей, от 0% до 100% в ацетонитриле, продукт элюировался при 20%. Было получено 14,2 мг продукта (0,03 ммоль, выход 8%).
LC-MS: tR =2,03 мин; m/z=468
1H-ЯМР (400 МГц, DMSO-d6) δ -0,04-0,23 (м, 1H), 0,34 (ддд, J=17,7, 9,1, 5,5 Гц, 1H), 1,43-1,68 (м, 1H), 2,52 (д, J=1,9 Гц, 1H), 2,67-2,83 (м, 1H), 3,10-3,22 (м, 2H), 3,65 (д, J=13,1 Гц, 2H), 3,79 (д, J=2,2 Гц, 3H), 4,53 (тт, J=9,0, 4,6 Гц, 1H), 5,32 (с, 1H), 6,95-7,33 (м, 6H), 7,33-7,46 (м, 3H), 8,09 (д, J=72,5 Гц, 1H), 8,34 (дд, J=26,3, 8,3 Гц, 1H).
Сравнительный пример 5: N-((S)-1-(((S)-4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-4-метоксибензамид
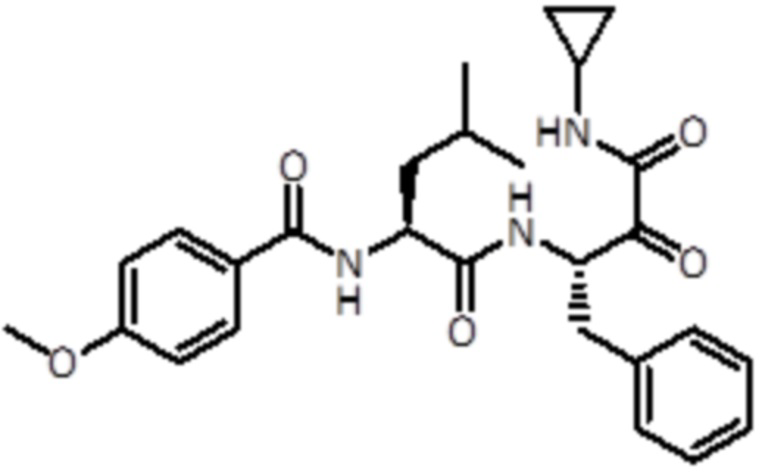
5 мкл воды добавляли к 15% раствору перйодинана Десс-Мартина в DCM (744 мкл, 0,3 ммоль, 1,5 экв.). Затем смесь добавляли к раствору промежуточного соединения 22 (115 мг, 0,2 ммоль, 1,0 экв.) в DCM (5 мл). Смесь перемешивали в течение 15 минут при комнатной температуре. Летучие вещества удаляли в вакууме. Неочищенный материал экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (2×10 мл). Неочищенный материал очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве растворителей, от 0% до 30% в гексане:этаноле (8:2), продукт элюировался при 20%. Было получено 31,2 мг (0,2 ммоль, 27%).
LC-MS: tR =2,60 мин; m/z=480
1H-ЯМР (400 МГц, DMSO-d6) δ 8,74 (дд, J=25,5, 5,1 Гц, 1H), 8,33 (дд, J=34,8, 7,4 Гц, 1H), 8,19 (дд, J=14,2, 8,4 Гц, 1H), 7,88-7,82 (м, 2H), 7,24-7,14 (м, 5H), 7,03-6,95 (м, 2H), 5,17 (м, 1H), 4,52 (м, 1H), 3,81 (с, 3H), 3,12 (м, 1H), 2,91-2,74 (м, 1H), 2,74-2,65 (м, 1H), 1,60 (м, 1H), 1,48 (м, 1H), 1,30 (м, 1H), 0,92-0,78 (м, 6H), 0,68-0,61 (м, 2H), 0,58 (м, 2H).
Сравнительный пример 6: N-((S)-1-(((S)-4-(циклопропиламино)-3,4-диоксо-1-фенилбутан-2-ил)амино)-4-метил-1-оксопентан-2-ил)-2-метоксибензамид

5 мкл воды добавляли к 15% раствору перйодинана Десс-Мартина в DCM (1,6 мл, 0,7 ммоль, 1,5 экв.). Затем смесь добавляли к раствору промежуточного соединения 23 (250 мг, 0,5 ммоль, 1,0 экв.) в DCM (10 мл). Смесь перемешивали в течение 15 минут при комнатной температуре. Летучие вещества удаляли в вакууме и неочищенную смесь экстрагировали EtOAc и промывали насыщ. раствором NaHCO3 (2×10 мл). Неочищенный продукт очищали флэш-хроматографией (ISCO Rf) с использованием гексана/гексана:этанола (8:2) в качестве растворителей, от 0% до 30% в гексане:этаноле (8:2), продукт элюировался при 20%. Было получено 89,2 мг продукта (0,2 ммоль, 35%).
LC-MS: tR =2,68 мин; m/z=480
1H-ЯМР (400 МГц, DMSO-d6) δ 8,78 (дд, J=19,7, 5,1 Гц, 1H), 8,51 (дд, J=32,3, 7,5 Гц, 1H), 8,26 (дд, J=8,2, 6,1 Гц, 1H), 7,79 (м, 1H), 7,49 (м, 1H), 7,25-7,21 (м, 4H), 7,20-7,13 (м, 2H), 7,05 (м, 1H), 5,24-5,17 (м, 1H), 4,63-4,55 (м, 1H), 3,86 (с, 3H), 3,15 (м, 1H), 2,85-2,75 (м, 1H), 2,75-2,70 (м, 1H), 1,66-1,46 (м, 2H), 1,36 (м, 1H), 0,91-0,77 (м, 6H), 0,67-0,62 (м, 2H), 0,61-0,55 (м, 2H).
Биологические данные
Анализ для определения способности ингибировать кальпаин-1
Активность кальпаина-1 измеряли с использованием набора Calpain-Glo Protease kit от Promega. Анализ проводили в 384-луночных черных планшетах с прозрачным дном. 5 мкл соединения в 10 мМ HEPES и 1 мМ водном растворе EDTA инкубировали с 15 мкл раствора 16 нг/мл кальпаина-1 (Sigma Aldrich) в течение 2 минут, а затем в каждую лунку добавляли 20 мкл реагента Calpain-GloT™ Reagent, предоставляемого в наборе. Люминесценцию, индуцированную посредством индуцированного кальпаином-1 лизиса субстрата, измеряли на устройстве для считывания Hamamatsu FDSS 7000, и данные вычисляли из максимального пика люминесценции. Ингибирование кальпаина вычисляли по формуле:
% ингибирование = (LT-LB)*100/(LC-LB);
где LT представляет собой люминесценцию, наблюдаемую для тестируемого соединения, LB представляет собой люминесценцию, полученную для пустой лунки без кальпаина-1, и LC представляет собой люминесценцию, наблюдаемую в контрольных лунках, включающих кальпаин 1, в отсутствии ингибитора.
Все соединения тестировали с использованием по меньшей мере 6 концентраций для обеспечения возможности вычисления IC50 (концентрация, которая ингибирует активность кальпаина-1 на 50%).
В каждом случае в анализе включали кальпастатин и ALLN в качестве контрольных ингибиторов.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ СУЛЬФОНАМИДА, ОБЛАДАЮЩИЕ СЕЛЕКТИВНОЙ NOX-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2019 |
|
RU2809030C2 |
| Замещенные цианопирролидины, обладающие активностью ингибиторов USP30 | 2020 |
|
RU2822680C1 |
| ПРОИЗВОДНЫЕ ИМИНОМОЧЕВИНЫ | 2018 |
|
RU2780118C2 |
| ПРОИЗВОДНЫЕ РАПАМИЦИНА | 2018 |
|
RU2807188C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2016 |
|
RU2718915C2 |
| ПУТИ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2020 |
|
RU2795503C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| ПРОИЗВОДНЫЕ N-ГИДРОКСИЛСУЛЬФОНАМИДА КАК НОВЫЕ ФИЗИОЛОГИЧЕСКИ ПРИМЕНИМЫЕ ДОНОРЫ НИТРОКСИЛА | 2008 |
|
RU2485097C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ИНГИБИРУЮЩИЕ ASK1 ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА И ПИРРОЛОПИРИДИНА | 2018 |
|
RU2772422C2 |
Изобретение относится к производным дипептидилкетоамид м-метоксифенила формулы II, где R1a выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила, R2a выбран из группы, состоящей из H и C1-C6 алкила, R3a представляет собой C3-C6 циклоалкил или метокси, при условии, что когда R3a представляет собой метокси, R1a представляет собой циклопропил, и R2a представляет собой H, или его фармацевтически приемлемой соли или стереоизомеру, которые могут быть использованы для лечения заболеваний и состояний, ассоциированных с повышенной активностью кальпаина, таких как повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее, нейродегенеративные нарушения, малярия, диабетическая нефропатия, нейротоксичность, индуцированная вирусом ВИЧ, злокачественная опухоль и фиброзные заболевания. 3 н. и 13 з.п. ф-лы, 6 пр.
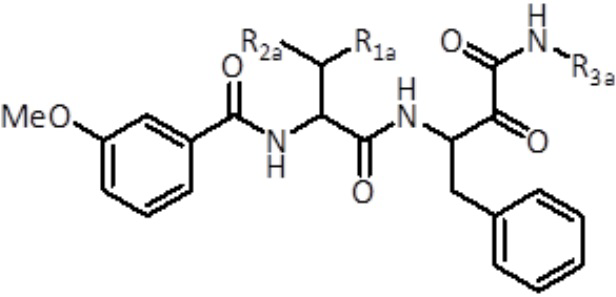 II
II
1. Соединение формулы (II):
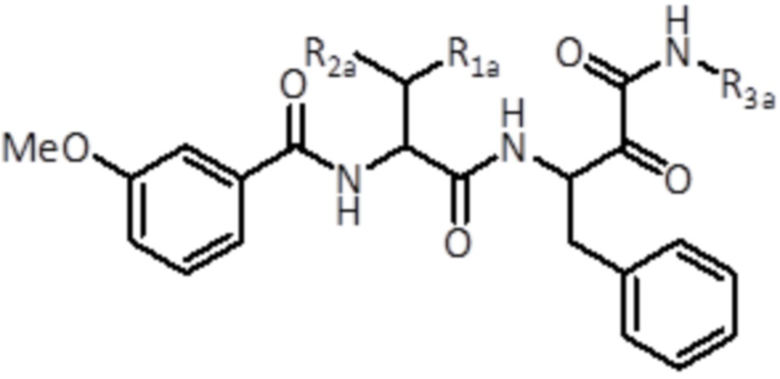
(II),
где
R1a выбран из группы, состоящей из C1-C6 алкила и C3-C6 циклоалкила,
R2a выбран из группы, состоящей из H и C1-C6 алкила,
R3a представляет собой C3-C6 циклоалкил или метокси,
при условии, что когда R3a представляет собой метокси, R1a представляет собой циклопропил, и R2a представляет собой H,
или его фармацевтически приемлемая соль или стереоизомер.
2. Соединение формулы (II) по п.1, где
R1a выбран из группы, состоящей из C1-C3 алкила и C3-C5 циклоалкила,
R2a выбран из группы, состоящей из H и C1-C3 алкила,
R3a представляет собой C3-C5 циклоалкил или метокси,
при условии, что когда R3a представляет собой метокси, R1a представляет собой циклопропил, и R2a представляет собой H.
3. Соединение формулы (II) по любому из пп.1 или 2, где R1a выбран из группы, состоящей из метила, изопропила и циклопропила.
4. Соединение формулы (II) по любому из пп.1 или 2, где R2a выбран из группы, состоящей из H и метила.
5. Соединение формулы (II) по любому из пп.1 или 2, где R3a представляет собой циклопропил.
6. Соединение формулы (II) по любому из пп.1 или 2, которое выбрано из группы, состоящей из:
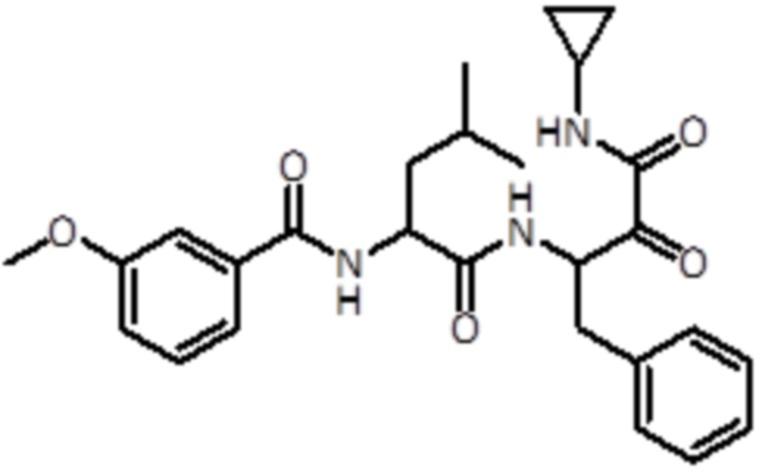 ,
, 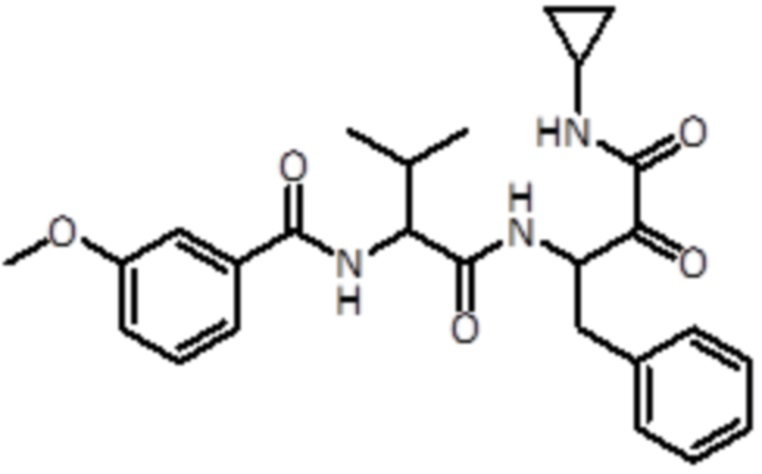 ,
, 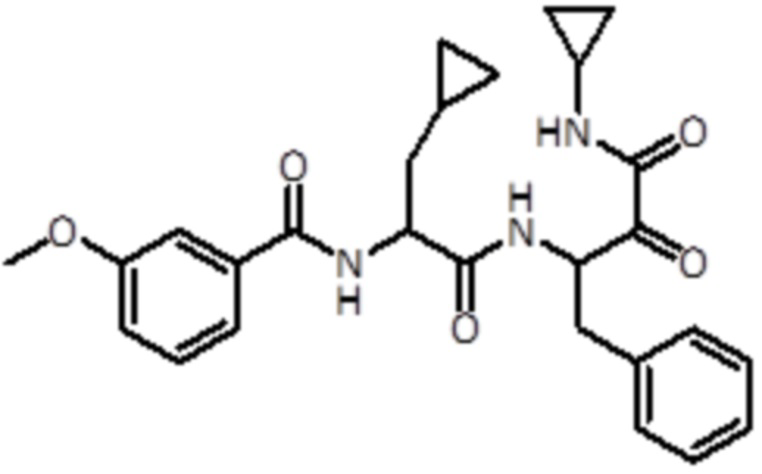 и
и 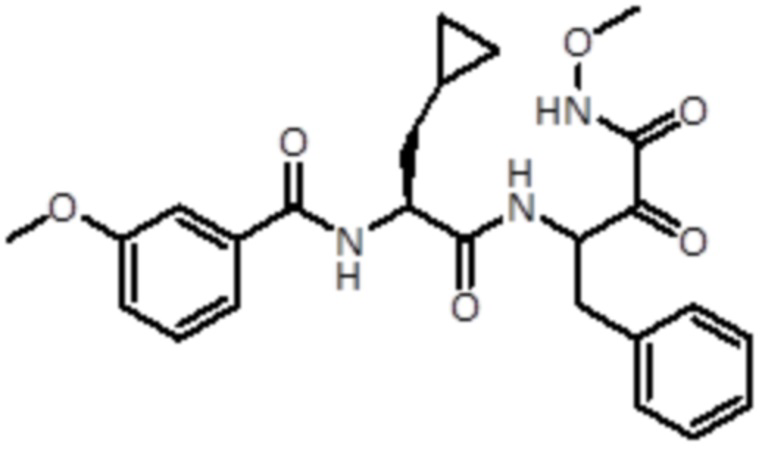 ,
,
или их стереоизомера или фармацевтически приемлемой соли.
7. Соединение формулы (II) по любому из пп.1 или 2, которое выбрано из группы, состоящей из:
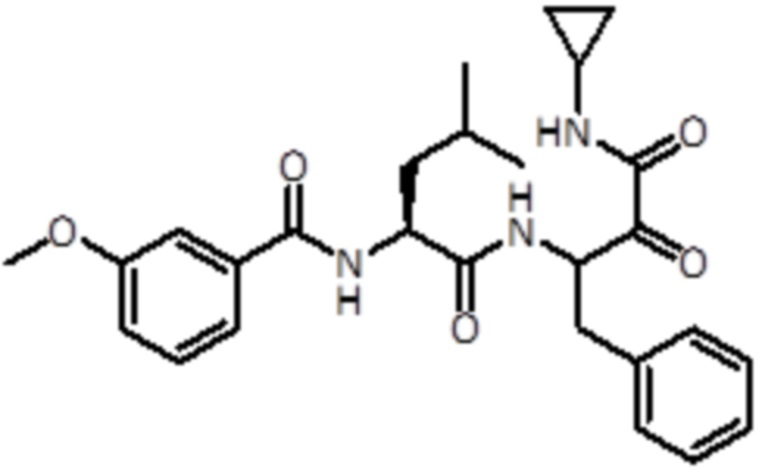 ,
, 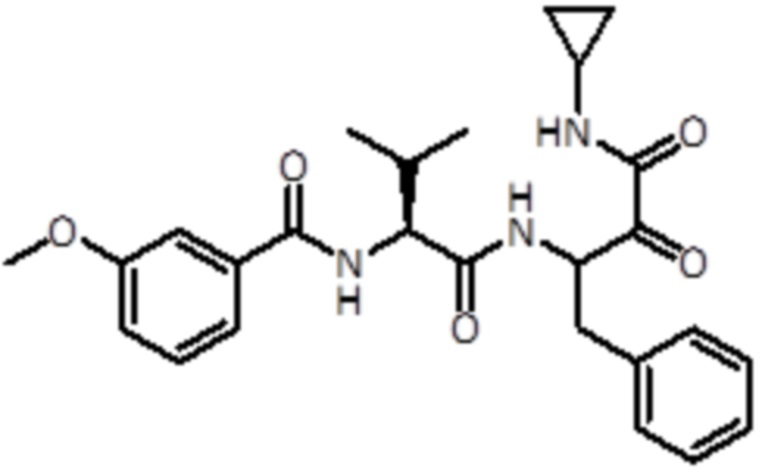 ,
, 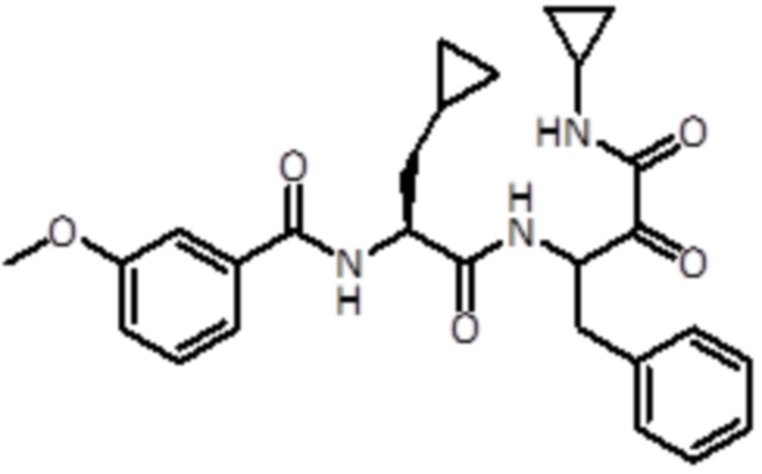 и ,
и ,
или их стереоизомера или фармацевтически приемлемой соли.
8. Соединение формулы (II) по любому из пп.1 или 2, которое выбрано из группы, состоящей из:
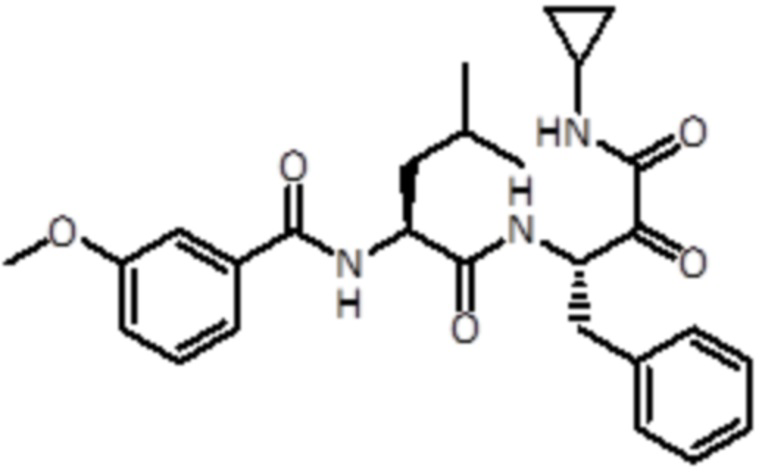 ,
, 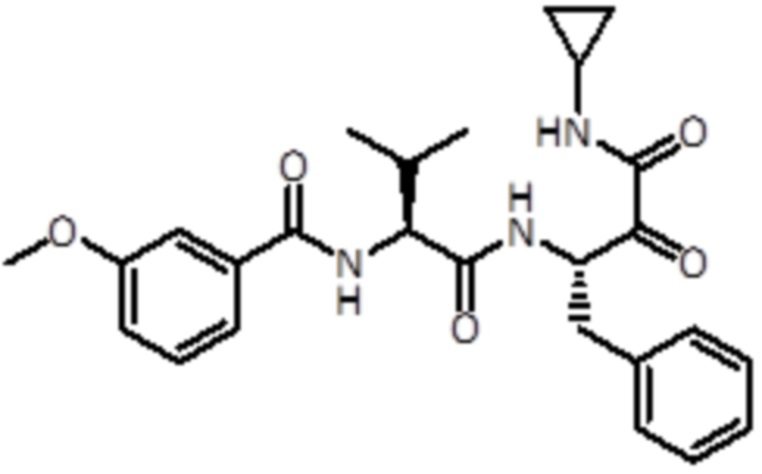 ,
, 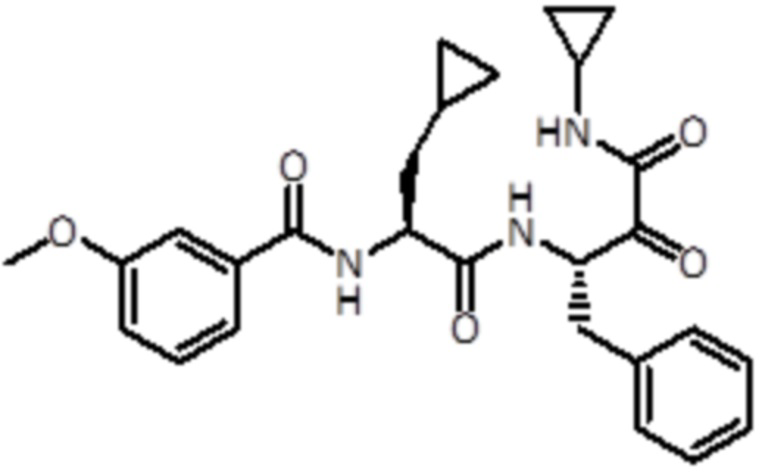 и
и 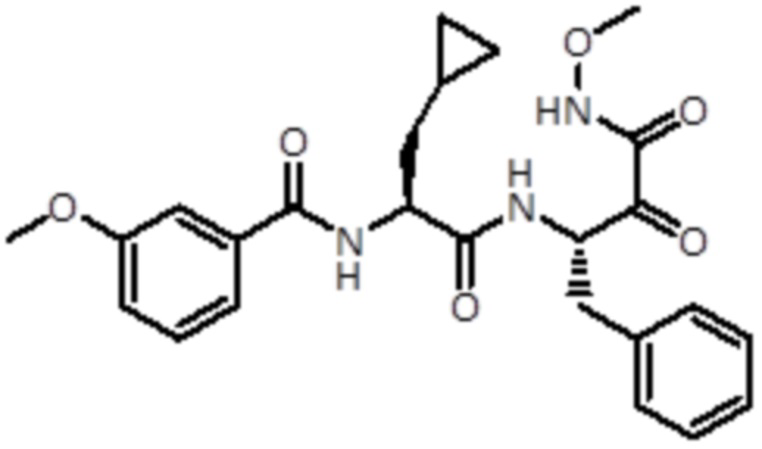 ,
,
или их фармацевтически приемлемой соли.
9. Фармацевтическая композиция для лечения и/или предупреждения заболеваний или состояний, восприимчивых к лечению ингибированием кальпаина 1, содержащая соединение формулы (II) по любому из пп.1-8, и фармацевтически приемлемый эксципиент.
10. Способ лечения и/или предупреждения заболевания или состояния, восприимчивого к лечению ингибированием кальпаина 1; где способ включает введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (II) по любому из пп.1-8.
11. Способ по п.10, где заболевание или состояние представляет собой повреждение сердца, вызванное инфарктом, ишемией с реперфузией или без нее.
12. Способ по п.11, где повреждение сердца представляет собой ремоделирование после инфаркта миокарда.
13. Способ по п.10, где заболевание или состояние выбрано из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, рассеянного склероза, острого аутоиммунного энцефалита и болезни Крейтцфельда-Якоба.
14. Способ по п.10, где заболевание или состояние выбрано из группы, состоящей из рака молочной железы, рака ободочной и прямой кишки и лейкоза.
15. Способ по п.10, где заболевание или состояние выбрано из группы, состоящей из фиброза сердца, фиброза легких, фиброза печени, фиброза почек и ретроперитонеального фиброза.
16. Способ по любому из пп.10-15 лечения и/или предупреждения заболевания или состояния, восприимчивого к лечению ингибированием кальпаина 1; где способ включает введение нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.9.
| WO 2016038040 A2, 17.03.2016 | |||
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНА | 2010 |
|
RU2567392C2 |
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНА | 2010 |
|
RU2540856C2 |
| LESCOP C ET AL, "Novel cell-penetrating alpha-keto-amide calpain inhibitors as potential treatment for muscular dystrophy", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, 2005, vol | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |

