Настоящее изобретение относится к области медицины. В частности, настоящее изобретение относится к соединениям, способам и фармацевтическим композициям для лечения бокового амиотрофического склероза (БАС) и других неврологических заболеваний, вызванных изменениями возбудимости двигательных нейронов, включая, помимо прочего, первичный боковой склероз, псевдобульбарный паралич, прогрессирующий бульбарный паралич, прогрессирующую мышечную атрофию и эпилепсию.
БАС (иногда называемый «болезнью Лу Герига») представляет собой фатальное неврологическое заболевание, от которого ежегодно страдают примерно 1,5-3 человека на 100000 человек. Оно характеризуется прогрессирующей потерей двигательных нейронов, что обычно приводит к смерти в течение 2-3 лет с момента постановки диагноза. Хотя исследования продолжаются, большинство случаев БАС, по-видимому, носят спорадический характер без известной причины.
У пациентов с БАС обычно наблюдается повышенная возбудимость периферических и центральных мотонейронов, что приводит к фасцикуляциям, мышечным судорогам и спастичности. Считается, что повышенная возбудимость нейронов приводит к перегрузке кальцием и гибели клеток. Действительно, возбудимость двигательных нейронов отрицательно коррелировала с выживаемостью у пациентов с БАС (см. K. Kanai et. al., J. Neurol. Neurosurg. Psychiatry, 83, 734-738 (2012)).
В исследованиях изучались механизм и причину того, почему двигательные нейроны у пациентов с БАС изменяют возбудимость (по сравнению с пациентами без БАС). (См. K. Kanai et. al., Brain, 129, 953-962 (2006)). Результаты исследования показали, что снижение активности калиевых каналов в нейронах является основным фактором повышенной возбудимости, связанной с заболеванием (см. там же). Кроме того, двигательные нейроны, полученные из плюрипотентных стволовых клеток пациентов с БАС, также обладают повышенной возбудимостью. (См. B.J. Wainger et. al., Cell Rep., 7, 1-11 (2014)). Эти нейроны, полученные из стволовых клеток, показали пониженные калиевые токи замедленного выпрямления, и, фактически, когда агенты ретигабин и флупиртин (известные как потенциаторы калиевых каналов Kv7) добавляют к этим нейронам, полученным из стволовых клеток, повышенная возбудимость мотонейронов нормализовалась и выживаемость клеток in vitro увеличивалась (см. там же). В другом исследовании ретигабин, потенцирующий Kv7, уменьшал симптомы эксайтотоксичности и увеличивал выживаемость в модели БАС in vitro с использованием подъязычных мотонейронов крысы (см. F. Ghezzi et. al., J. Physiol., 596, 2611-2629 (2018)).
В настоящее время единственным лекарственным средством, одобренным для лечения БАС, является рилузол, который, как было показано, увеличивает выживаемость на 2-3 месяца. Совершенно очевидно, что существует потребность в более эффективных, лучших и с более продолжительным действием лекарственных средствах.
Известный ретигабин, усиливающий действие Kv7, действует на калиевые каналы Kv7.2-7.5 (KCNQ2-KCNQ5). Он был одобрен в качестве дополнительной терапии у пациентов с лекарственно-устойчивой эпилепсией. Он был одобрен в Европе и США в 2011 году, но был добровольно отозван в 2017 году. Считалось, что прекращение клинического применения ретигабина связано с рядом проблем с переносимостью, что привело к очень ограниченному применению препарата. Проблемы с переносимостью включают обычное и, предположительно, механическое возникновение сонливости и головокружения, а также менее частые случаи задержки мочи и изменений пигментации сетчатки и кожи. Изменения сетчатки и возможность потери зрения привели к предупреждению в рамке на этикетке ретигабина и, как считается, не связаны с механизмом. Задержка мочи, скорее всего, является результатом потенцирования каналов Kv7.3/7.5 мочевого пузыря. Однако, учитывая его побочные эффекты, существует необходимость в разработке новых потенциаторов Kv7.
Недавнее исследование, сравнивающее эффекты рилузола (например, одобренного лечения БАС) и ретигабина на возбудимость двигательных нейронов у пациентов с БАС, предполагает, что потенциаторы каналов Kv7 могут иметь более высокую эффективность, чем рилузол (см. M. Kovalchuk et. al., Clinical Pharmacology & Therapeutics, 104, 1136-1145 (2018)). Таким образом, для клинического лечения БАС и других расстройств, связанных с повышенной возбудимостью, могут быть использованы улучшенные потенциаторы, которые обладают лучшей переносимостью, чем ретигабин. (См. B. Kalappa, et al., The Journal of Neuroscience, 35(23):8829-8842) (2015). На сегодняшний день никакие агенты, действующие на Kv7, не были одобрены для лечения БАС, и, таким образом, сохраняется потребность в средствах, действующих на Kv7, таких как альтернативные потенциаторы Kv7, которые обеспечивают терапевтический эффект. Кроме того, может быть полезно, чтобы такие потенциаторы были более избирательными в отношении Kv7.2/7.3 по сравнению с другими каналами Kv7. Также существует потребность в новом агенте Kv7, который позволяет избежать нежелательных побочных эффектов и может обеспечить комбинацию улучшенных фармакологических свойств, включая безопасность, действенность, эффективность и переносимость, в частности, для лечения возбудимости периферических и центральных мотонейронов.
В настоящих вариантах осуществления изобретения предложены соединения, которые являются потенциаторами калиевых каналов (такими как потенциаторы Kv7 - также называемые потенциаторами KCNQ), которые могут быть использованы при лечении БАС и других неврологических заболеваний, вызванных изменениями возбудимости двигательных нейронов.
В частности, в качестве потенциаторов Kv7 могут использоваться соединения следующей формулы (которая обозначается как «ФОРМУЛА I»):
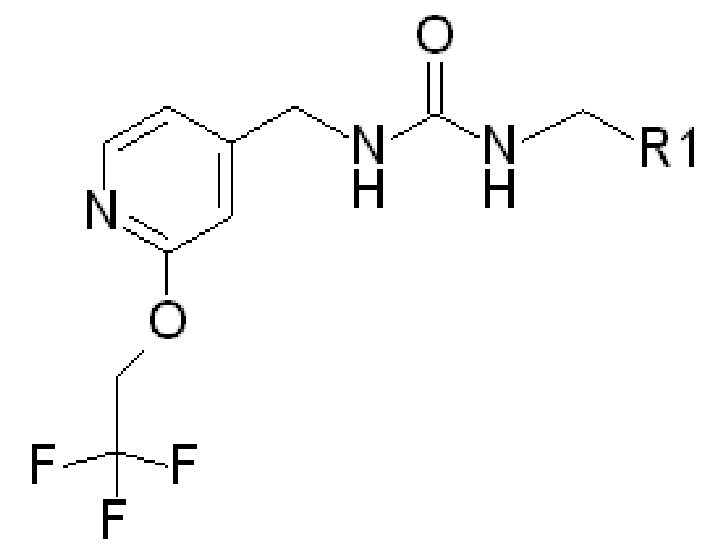
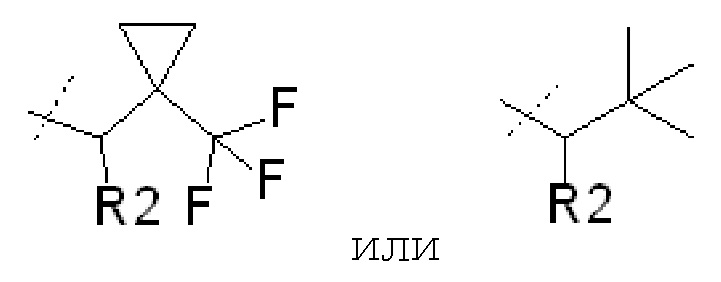
где R1 представляет собой
(ФОРМУЛА I)
где в ФОРМУЛЕ I R2 представляет собой Н или ОН. В дополнение к соединениям ФОРМУЛЫ I из соединений ФОРМУЛЫ I могут быть получены одна или несколько фармацевтически приемлемых солей, и такие фармацевтически приемлемые соли могут быть получены и использованы в качестве потенциаторов Kv7.
В некоторых вариантах осуществления изобретения соединения ФОРМУЛЫ I или их фармацевтически приемлемые соли получают таким образом, что R1 представляет собой
и R2 представляет собой Н.
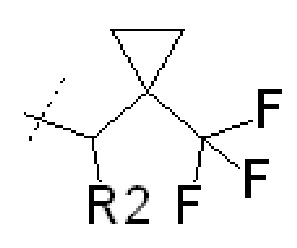
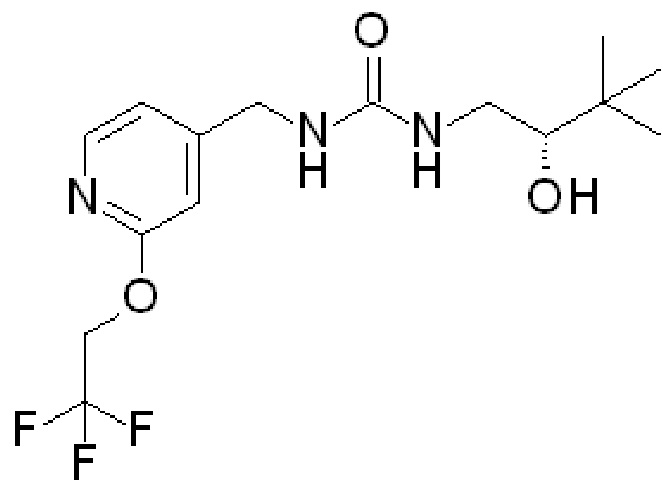
В других вариантах осуществления изобретения соединения ФОРМУЛЫ I (или их фармацевтически приемлемые соли) получают таким образом, что R1 представляет собой
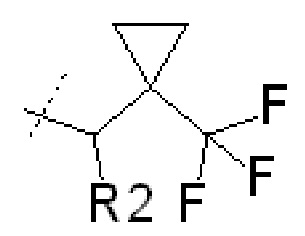
и R2 представляет собой ОН. Более того, в таких вариантах осуществления изобретения, когда R2 представляет собой ОН, углерод, к которому присоединена группа ОН, является стереоцентром. Соответственно, в некоторых вариантах осуществления изобретения соединения (или их фармацевтически приемлемые соли) ФОРМУЛЫ I могут представлять собой рацемическую смесь двух энантиомеров. В других вариантах осуществления изобретения может использоваться конкретный энантиомер, включая любой из следующих энантиомеров:
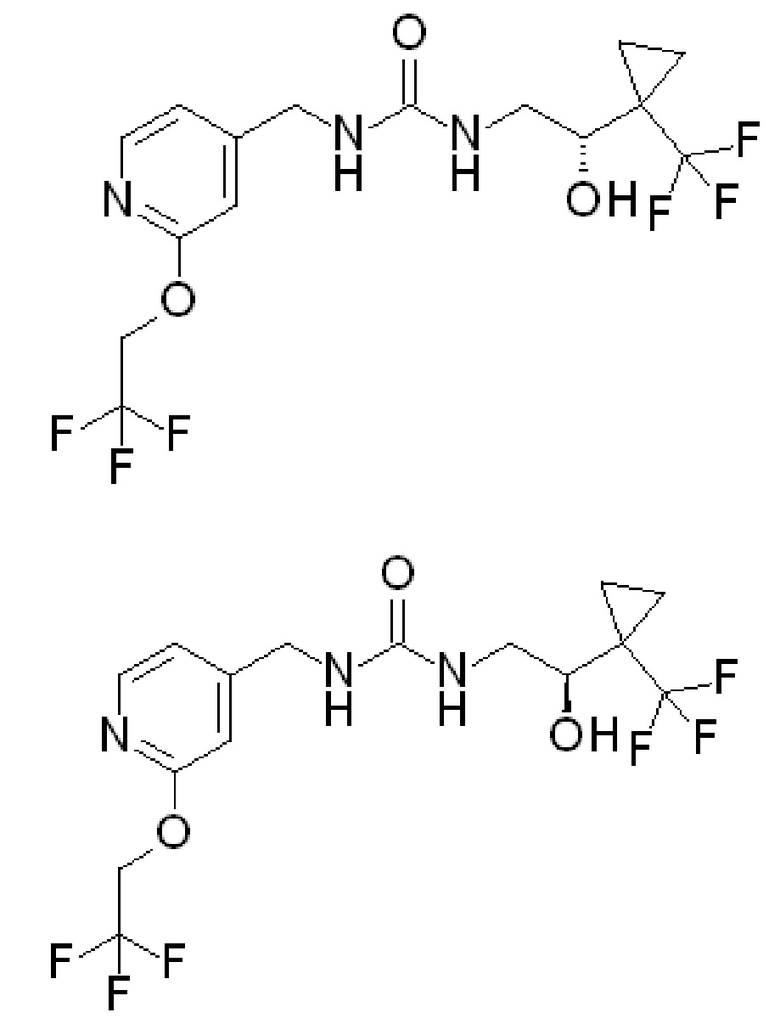
Специалистам в данной области понятно как сконструировать варианты осуществления, в которых используется только один из вышеупомянутых энантиомеров. Другие варианты осуществления изобретения могут быть разработаны со смесями различных энантиомеров, имеющих различное процентное содержание каждого компонента.
В других вариантах осуществления изобретения соединения ФОРМУЛЫ I (или их фармацевтически приемлемые соли) получают таким образом, что
R1 представляет собой
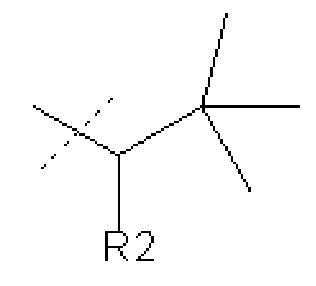
и R2 представляет собой Н.
В еще дополнительных вариантах осуществления изобретения соединения ФОРМУЛЫ I (или их фармацевтически приемлемые соли) получают таким образом, что
R1 представляет собой
и R2 представляет собой ОН. Более того, в таких вариантах осуществления изобретения, когда R2 представляет собой ОН, углерод, к которому присоединена группа ОН, является стереоцентром. Соответственно, в некоторых вариантах осуществления изобретения соединения (или фармацевтически приемлемые соли) ФОРМУЛЫ I могут представлять собой рацемическую смесь двух энантиомеров. В других вариантах осуществления изобретения может использоваться конкретный энантиомер, включая любой из следующих энантиомеров:
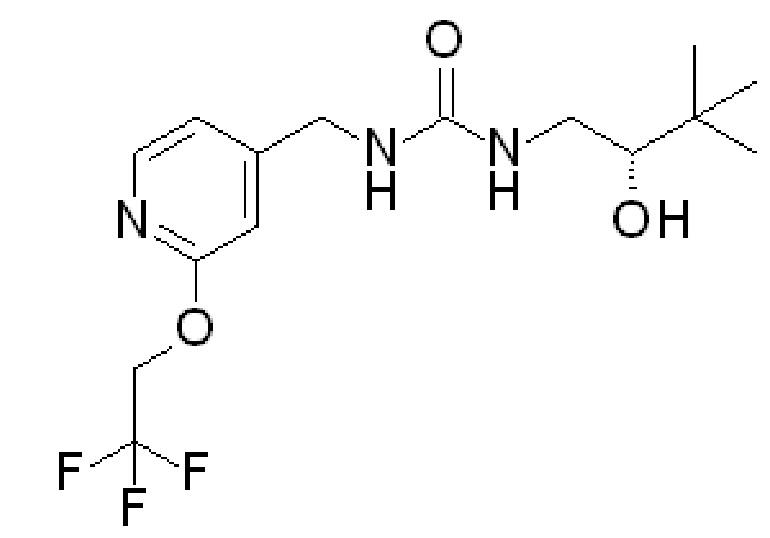
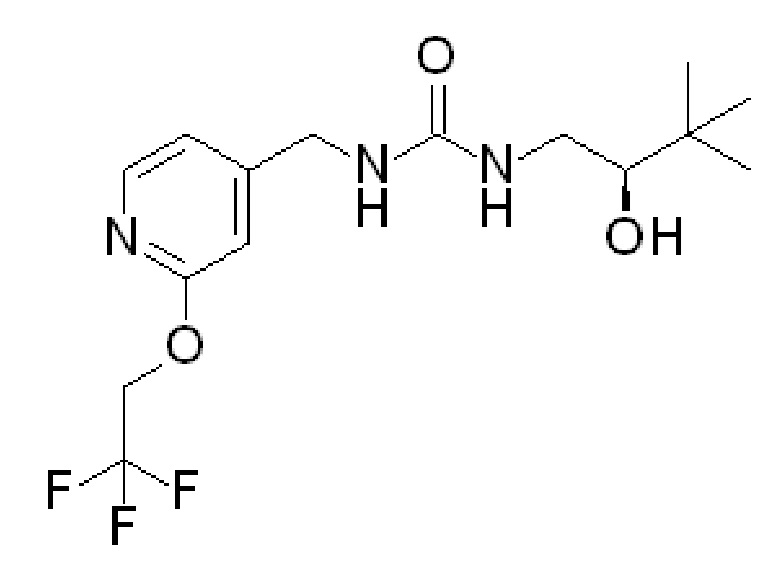
Специалистам в данной области понятно как сконструировать варианты осуществления, в которых используется только один из вышеупомянутых энантионаторов. Другие варианты осуществления могут быть разработаны со смесями различных энантиомеров, имеющих различное процентное содержание каждого компонента.
Настоящие варианты осуществления изобретения дополнительно относятся к фармацевтическим композициям, содержащим соединение или фармацевтически приемлемую соль ФОРМУЛЫ I и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. Такие фармацевтические композиции могут обеспечивать способ лечения заболевания. В частности, настоящие варианты осуществления обеспечивают способ лечения заболевания, вызванного изменениями возбудимости двигательных нейронов, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения ФОРМУЛЫ I (или его фармацевтически приемлемой соли). В некоторых особенно предпочтительных вариантах осуществления изобретения заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой БАС.
В других вариантах осуществления предложен способ лечения заболевания, вызванного изменениями возбудимости двигательных нейронов, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения формулы I (или его фармацевтически приемлемой соли). В некоторых особенно предпочтительных вариантах осуществления изобретения заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой БАС.
Настоящие варианты осуществления также относятся к соединениям ФОРМУЛЫ I (или их фармацевтически приемлемым солям) для использования в терапии. Более конкретно, в настоящих вариантах осуществления изобретения предложено соединение ФОРМУЛЫ I (или его фармацевтически приемлемую соль) для применения при лечении заболевания, вызванного изменениями возбудимости двигательных нейронов. В некоторых вариантах реализации таким заболеванием может быть БАС.
Кроме того, в настоящих вариантах осуществления изобретения предложено применение соединения ФОРМУЛЫ I (или его фармацевтически приемлемых солей) при изготовлении лекарственного средства для лечения заболевания, вызванного изменениями возбудимости двигательных нейронов. В некоторых таких вариантах реализации заболевание представляет собой БАС.
Как используется в настоящем документе, термин «заболевание, связанное с изменениями возбудимости мотонейрона» или «заболевание, вызванное изменениями возбудимости мотонейрона», включает заболевание, выбранное из группы, состоящий из БАС, первичного бокового склероза, псевдобульбарного паралича, прогрессирующего бульбарного паралича, эпилепсии и прогрессирующей мышечной атрофии. Эти термины также включают все заболевания, перечисленные в работе A. Verma, et al., «Atypical Motor Neuron Disease and Related Motor Syndromes», Seminars in Neurology, Volume 21, Number 2, 2001. Такие термины также включают расстройства PNH (гипервозбудимость периферических нервов). Информацию о расстройствах PNH можно найти в работе C.  «Peripheral nerve hyperexcitability syndromes» Rev Neurosci. 2015;26(2):239-51. Соответственно, указанные в настоящем документе соединения и фармацевтически приемлемые соли можно использовать для лечения одного или нескольких из этих заболеваний.
«Peripheral nerve hyperexcitability syndromes» Rev Neurosci. 2015;26(2):239-51. Соответственно, указанные в настоящем документе соединения и фармацевтически приемлемые соли можно использовать для лечения одного или нескольких из этих заболеваний.
Как взаимозаменяемо используется в настоящем документе, термин «пациент», «субъект» и «индивидуум» относится к человеку, более конкретно, пациенту, нуждающемуся в этом. В некоторых вариантах осуществления изобретения пациента дополнительно характеризуется наличием заболевания, расстройства или состояния (например, БАС или другое заболевание), при котором может быть полезно потенцирование Kv7. В другом варианте осуществления изобретения пациент дополнительно охарактеризован как подверженный риску развития состояния, описанного выше, или состояния, которое может быть улучшено от потенцирования Kv7.
Эффективное количество может определить специалист в данной области, используя известные методы и наблюдая за результатами, полученными при аналогичных обстоятельствах. При определении эффективного количества для пациента учитывается ряд факторов, включая, помимо прочего: его размер, возраст и общее состояние здоровья; конкретное заболевание или расстройство; степень или вовлечение, или серьезность заболевания или расстройства; реакция отдельного пациента; конкретное вводимое соединение; режим введения; характеристики биодоступности вводимого препарата; выбранный режим дозирования; прием сопутствующих лекарств; и другие соответствующие обстоятельства. В некоторых вариантах осуществления изобретения эффективное количество обеспечивает клинически значимое снижение возбудимости периферических и центральных двигательных нейронов.
Соединения по настоящему изобретению составляют в виде фармацевтических композиций, вводимых любым путем, который делает соединение биодоступным. Предпочтительно, такие композиции предназначены для перорального введения. Такие фармацевтические композиции и способы их получения хорошо известны в данной области (см., например, Remington: The Science and Practice of Pharmacy, L.V. Allen, Editor, 22nd Edition, Pharmaceutical Press, 2012).
Соединения ФОРМУЛЫ I и их фармацевтически приемлемые соли особенно полезны в способах лечения по изобретению, при этом предпочтительны определенные конфигурации. Такие конфигурации описаны в следующем перечне соединений по настоящему изобретению. Понятно, что эти предпочтения применимы как к способам лечения, так и к соединениям по изобретению.
Соединения по настоящим вариантам осуществления изобретения (в которых R2 представляет собой ОН) включают:
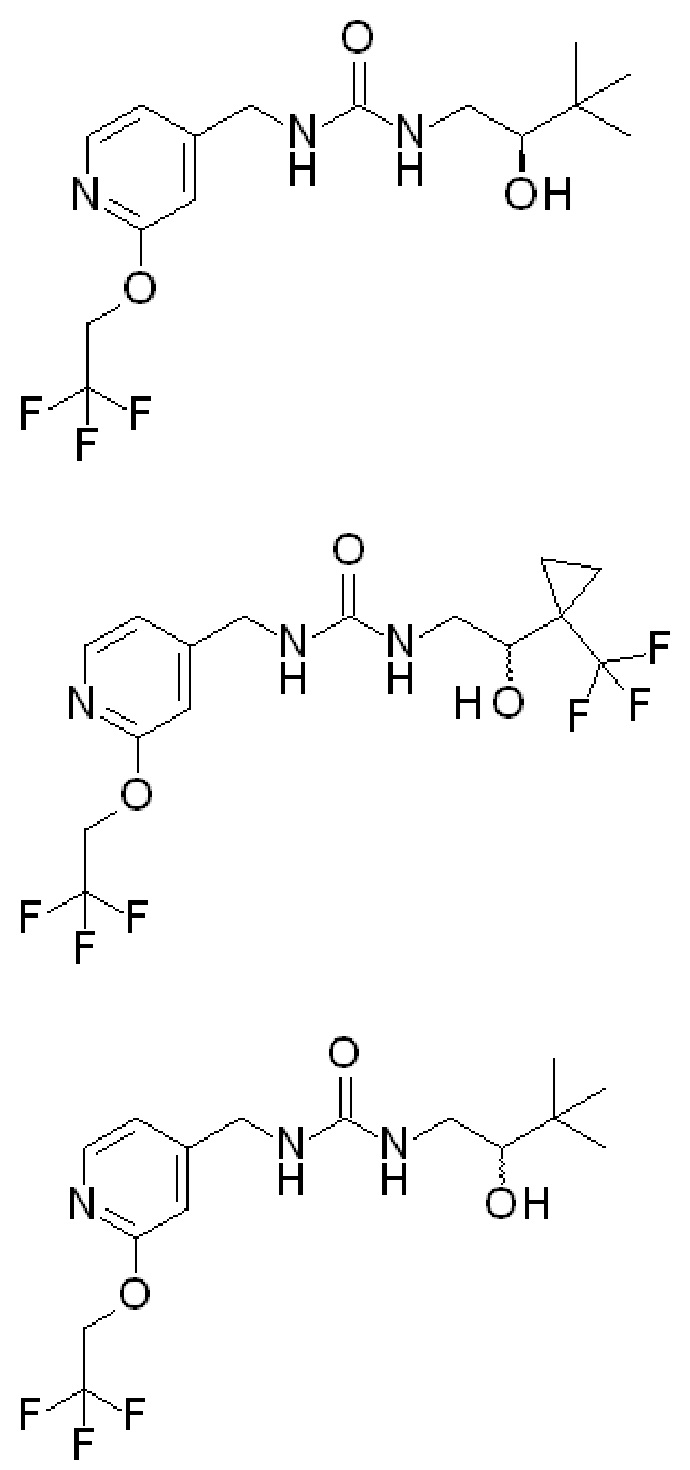
В этих соединениях, в которых R2 представляет собой ОН, соединения с указанной выше «плоской связью» являются рацемическими и/или содержат оба энантиомера (либо в смеси 50/50, либо в каком-либо другом соотношении). Соединения с клиновидной и пунктирной связью выше обозначают конкретный энантиомер. Соединения с «волнистой» связью указывают на то, что это единственный энантиомер, но точная конфигурация этого единственного энантиомера неизвестна, и, таким образом, такие энантиомеры различаются по их оптическому вращению (например, они вращают свет либо в (+), либо в (-) направлении).
Соединения по настоящим вариантам изобретения также включают следующие соединения (в которых R2 представляет собой Н):
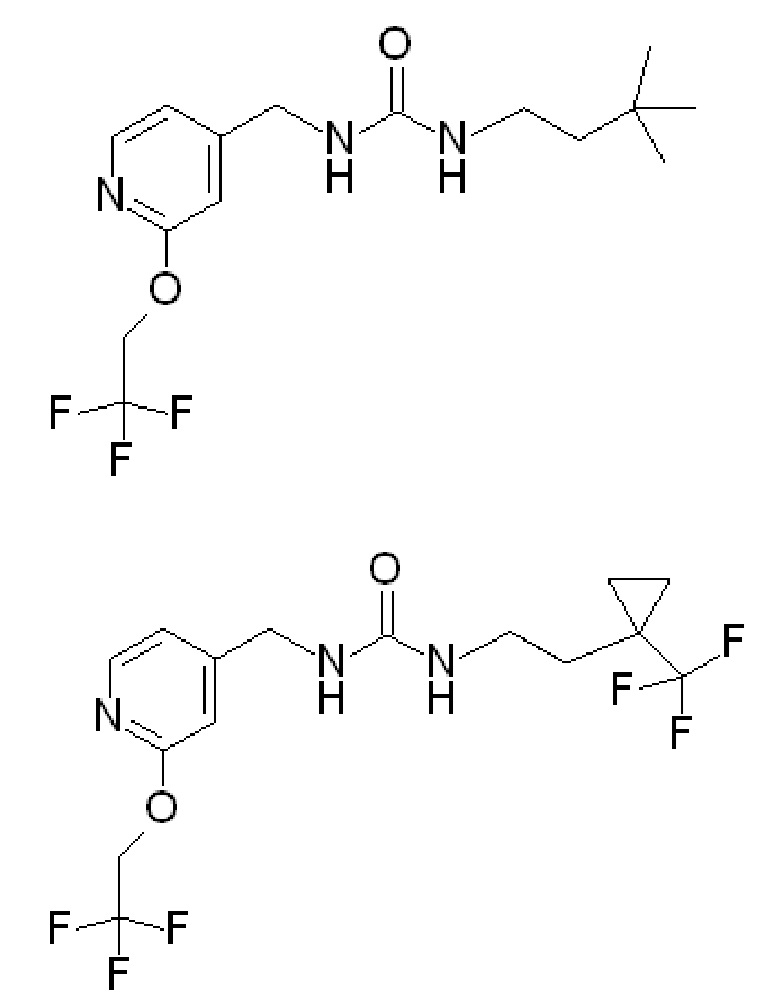
Вышеупомянутые соединения, которые имеют группу ОН в качестве R2, могут быть такими, в которых ОН находится в «верхней» конфигурации (как показано клиновидным выступом). Могут быть разработаны другие варианты, в которых группа ОН находится в «нижней» конфигурации. Специалистам в данной области понятно как получить этот другой энантиомер, например, используя другое исходное вещество и/или используя другие реакции и т. д. Такие энантиомеры имеют следующую структуру и являются частью заявленных в настоящее время вариантов осуществления:
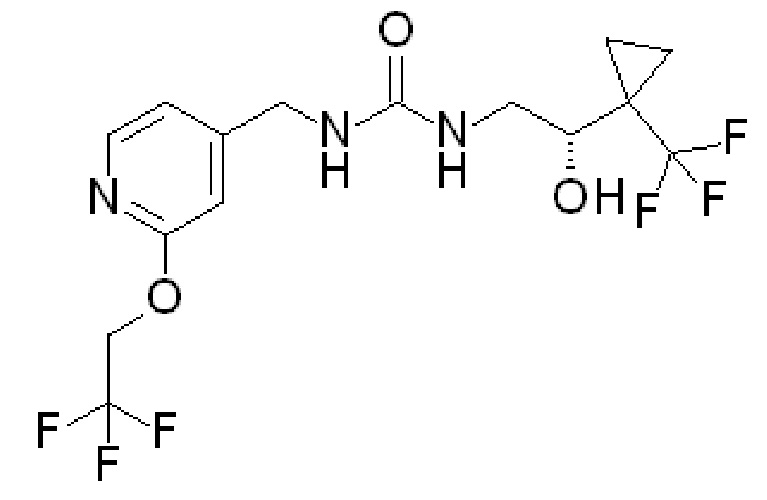
Хотя настоящее изобретение охватывает все отдельные энантиомеры и диастереомеры, а также смеси энантиомеров указанных соединений, включая рацематы, перечисленные выше соединения и их фармацевтически приемлемые соли являются особенно предпочтительными.
Отдельные энантиомеры могут быть выделены или разделены средним специалистом в данной области в любой удобный момент синтеза соединений по изобретению такими способами, как методы селективной кристаллизации, хиральная хроматография (см., например, J. Jacques, et al., «Enantiomers, Racemates, and Resolutions», John Wiley and Sons, Inc., 1981, и E.L. Eliel and S.H. Wilen, «Stereochemistry of Organic Compounds», Wiley-Interscience, 1994), или сверхкритическая жидкостная хроматография (SFC) (см., например, T. A. Berger; «Supercritical Fluid Chromatography Primer», Agilent Technologies, July 2015).
Фармацевтически приемлемая соль соединения по изобретению может быть образована, например, реакцией подходящего свободного основания соединения по изобретению и соответствующей фармацевтически приемлемой кислоты в подходящем растворителе в стандартных условиях, хорошо известных в данной области. См., например, Gould, P.L., «Salt selection for basic drugs», International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., et al. «Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities», Organic Process Research and Development, 4: 427-435 (2000); и Berge, S.M., et al., «Pharmaceutical Salts», Journal of Pharmaceutical Sciences, 66: 1-19, (1977).
Соединения по настоящему изобретению или их соли могут быть получены различными способами, известными среднему специалисту в данной области, некоторые из которых проиллюстрированы схемами, получениями и примерах далее. Продукты каждой стадии на схемах ниже могут быть выделены обычными методами, хорошо известными в данной области, включая экстракцию, выпаривание, осаждение, хроматографию, фильтрацию, растирание и кристаллизацию. На схемах ниже все заместители, если не указано иное, имеют указанные ранее значения. Реагенты и исходные вещества являются легко доступными среднему специалисту в данной области. Не ограничивая объем изобретения, следующие схемы, получения и примеры представлены для дополнительной иллюстрации аспектов изобретения. Кроме того, специалисту в данной области понятно, что соединения ФОРМУЛЫ I могут быть получены с использованием исходного вещества или промежуточного соединения с соответствующей желаемой стереохимической конфигурацией, которая могут быть получена специалистом в данной области.
Далее могут использоваться некоторые сокращения. Эти сокращения означают следующее: «ACN» относится к ацетонитрилу; «Ас» относится к ацетилу; «АсОН» относится к уксусной кислоте; «Ac2O» относится к уксусному ангидриду; «AP5» относится к (2R)-амино-5-фосфонопентаноату; «BDNF» относится к нейротрофическому фактору головного мозга; «ВОС» относится к трет-бутоксикарбонилу; «CAS#» относится к номеру реестра в Chemical Abstracts; «CMAP» относится к потенциалу действия сложной мышцы; «DCM» относится к метиленхлориду или дихлорметану; «DIPEA» относится к N, N-диизопропилэтиламину; «DMEA» относится к диметилэтиламину; «ДМФ» относится к N, N-диметилформамиду; «ДМСО» относится к диметилсульфоксиду; «D-PBS» относится к фосфатно-солевому буферному раствору Дульбекко; «ЭДТА» относится к этилендиаминтетрауксусной кислоте; «EGTA» относится к этиленгликоль-бис(β-аминоэтиловый эфир)-N, N,Nʼ,Nʼ-тетрауксусной кислоте; «ES/MS» относится к масс-спектрометрии с электрораспылением; «Et2O » относится к диэтиловому эфиру; «EtOAc» относится к этилацетату; «EtOH» относится к этанолу или этиловому спирту; «ч» относится к часу или часам; «HEPES» относится к 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоте; «IPA» относится к изопропанолу или изопропиловому спирту; «IPAm» относится к изопропиламину; «IPrOAc» относится к изопропилацетату; «ЖХ/МСМС» относится к жидкостной хроматографии с тандемной масс-спектрометрией; «LiHMDS» относится к бис(триметилсилил)амиду лития; «KOtBu» относится к трет-бутоксиду калия; «Me» относится к метилу; «мсек» относится к миллисекундам или миллисекундам как единице времени; «МТБЭ» относится к метил-трет-бутиловому эфиру; «мин» относится к минуте или минутам; «NaOtBu» относится к трет-бутоксиду натрия; «н-BuLi» относится к н-бутиллитию; «NBQX» относится к (2,3-дигидрокси-6-нитро-7-сульфамоилбензо[f]хиноксалину; «OAc» относится к ацетату или ацетокси; «PBS» относится к физиологическому раствору с фосфатным буфером; «к.т.» относится к комнатной температуре; «SCX» относится к сильному катионному обмену; «SD» относится к стандартному отклонению; «сек» относится к секунде или секундам как единице времени; «SEM» относится к стандартной ошибке среднего; «SFC» относится к сверхкритической жидкостной хроматографии; «TEA» относится к триэтиламину; «ТФУ» относится к трифторуксусной кислоте; «ТГФ» относится к тетрагидрофурану; «ТМА» относится к триметиламину; «TMEDA» относится к тетраметилэтилендиамину; «Трис» относится к трис(гидроксиметил)аминометану или 2-амино-2-(гидроксиметил) пропан-1,3-диолу; «[α]D20» относится к удельному оптическому вращению при 20ºC и 589 нм, где c обозначает концентрацию в г/мл (обычно г/100 мл).
Общие схемы
Схема 1
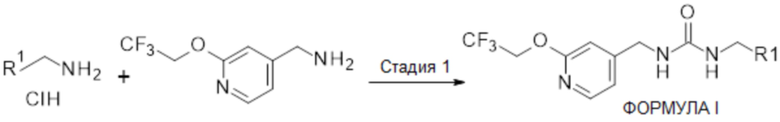
На схеме 1 показано получение соединений ФОРМУЛЫ I, где R1 определен, как описано выше. Образование активированного сложного эфира соответствующим образом замещенного амина (или его соответствующей соли, например, HCl) хорошо описано в данной области техники с использованием, например, карбонилдиимидазола, и селективное N- по сравнению с O-карбонилирование (2-этокси-4-пиридил)метанамина (или его соответствующей солевой формы, например, HCl) указанным активированным промежуточным соединением с подходящим органическим основанием может дать желаемые мочевины по настоящему изобретению (стадия 1). Квалифицированному специалисту понятно, что соединения ФОРМУЛЫ I, имеющие стереохимию, могут быть получены с помощью хирально замещенного амина с получением одного энантиомера, или хиральным разделением соединения ФОРМУЛЫ I с использованием методов хиральной хроматографии, например SFC, или с использованием хирального вспомогательного приема, такого как получение хиральной соли, как хорошо известно в данной области.
Схема 2
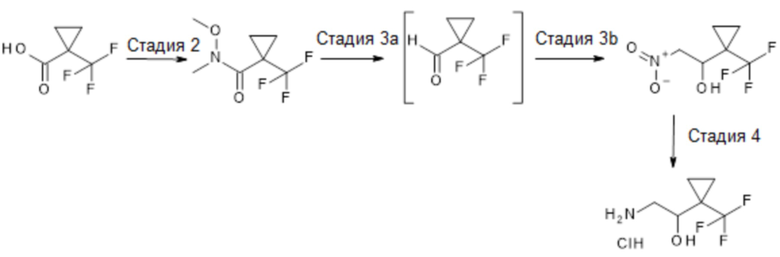
На схеме 2 показано получение гидрохлорида рац-2-амино-1-[1-(трифторметил)циклопропил]этанола. Получение амида типа Вайнреба (стадия 2) из (трифторметил)циклопропанкарбоновой кислоты может осуществляться в различных условиях, хорошо описанных в данной области. Двухстадийный способ получения нитро-1-[1-(трифторметил)циклопропил]этанола путем восстановления амида типа Вейнреба стандартными восстановителями (стадия 3а) с использованием, например, алюмогидрида лития в подходящем органическом растворителе, таком как ТГФ или Et2O, с последующим выделением альдегида и добавлением к соответствующему альдегиду (стадия 3b) аниона нитрометана, образующегося в присутствии сильного основания, такого как NaH или KOtBu, может дать желаемый рацемический 2-нитро-1-[1-(трифторметил)циклопропил]этанол. Последующее восстановление нитрогруппы (стадия 4) до соответствующего амина может быть выполнено в различных условиях, хорошо описанных в данной области, и для простоты использования полученный амин может быть преобразован в подходящую солевую форму, например, HCl. Квалифицированному специалисту понятно, что рацемическая смесь амина может быть разделена на два его хиральных энантиомера с использованием стандартных методик, хорошо известных в данной области, таких как хиральная хроматография или получение с использованием вспомогательной хиральной соли.
Схема 3
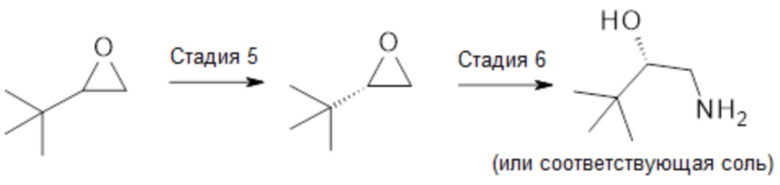
На схеме 3 изображено получение требуемого (2S)-1-амино-3,3-диметилбутан-2-ола или его соответствующей соли. Кинетическое разделение 2-трет-бутилоксирана (стадия 5) с использованием хирального катализатора на основе переходного металла, такого как Co2+, который был активирован до Co3+, может быть достигнуто на основе литературы, представленной в JACS 9 VOL. 124, NO. 7, 2002, 1307. Результирующая стереохимия может быть назначена в зависимости от стереохимии хирального катализатора. Следовательно, (2S)-2-трет-бутилоксиран может быть получен с использованием S, S-(сален)Co3+OAc (см. JACS 9 VOL. 124, NO. 7, 2002, 1307). Кроме того, проверка S-стереохимии может быть выполнена путем сравнения с данными, указанными в Tetrahedron: Asymmetry 13 (2002) 1209-1217. Стереоконтролируемое раскрытие эпоксидного цикла может быть достигнуто с использованием азотного нуклеофила, такого как NH3 в MeOH, в условиях, хорошо известных специалисту в данной области (стадия 6). Полученный аминоспирт можно преобразовать в подходящую солевую форму в условиях, хорошо известных в данной области.
Получение 1
N-метокси-N-метил-1-(трифторметил)циклопропанкарбоксамид
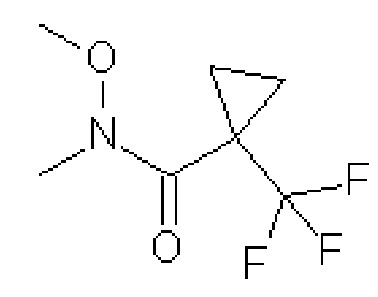
Охлаждают смесь коммерчески доступной 1-(трифторметил) циклопропанкарбоновой кислоты [CAS# 277756-46-4] (4,8 г, 31,4 ммоль) и гидрохлорида N, O-диметилгидроксиламина (4,65 г, 46,7 ммоль) в EtOAc (50 мл) до температуры 0ºC и по каплям через капельную воронку добавляют раствор 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфоран-2,4,6-триоксида (50 мас.% В ДМФ, 28 мл, 47,5 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь охлаждают до температуры 0ºC и гасят, выливая в насыщенный водный раствор NH4Cl. Полученные фазы разделяют и водный слой экстрагируют EtOAc. Органические экстракты объединяют, сушат над MgSO4, фильтруют и упаривают досуха с получением указанного в заголовке соединения (4,4 г, 67%-ный выход). 1H ЯМР (400 МГц, CDCl3) δ 3,74 (с, 3H), 3,29 (с, 3H), 1,33-1,25 (м, 4H). ES/MS: m/z 198 [M+H].
Получение 2
(±)-2-нитро-1-[1-(трифторметил)циклопропил]этанол
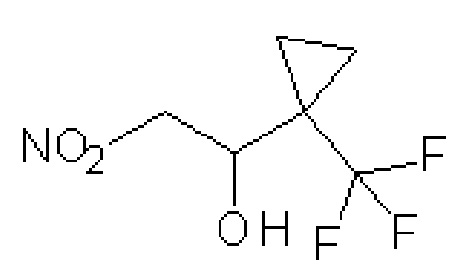
К раствору N-метокси-N-метил-1-(трифторметил)циклопропанкарбоксамида (4,3 г, 20,9 ммоль) в Et2O (50 мл) при температуре 0ºC по каплям добавляют 1M раствор алюмогидрида лития в ТГФ (20 мл, 20 ммоль) и перемешивают полученную смесь 1 ч. Реакцию гасят, добавляя по каплям воду (0,81 мл), затем 2 М водный NaOH (0,81 мл) и затем еще воду (2,43 мл). Добавляют MgSO4 (5 г) и фильтруют полученную смесь через слой диатомовой земли в минимальном вакууме, получая, как предполагается, раствор 1-(трифторметил)циклопропанкарбальдегида в Et2O. Этот раствор при температуре 0ºC по каплям добавляют к быстро перемешиваемому раствору нитрометана (60 мл, 1,1 моль) и KOtBu (360 мг, 3,17 ммоль). Через ~1 ч реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. С коричневой смолы в колбе декантируют растворитель и концентрируют при пониженном давлении. Полученный остаток очищают на силикагеле, элюируя 0-10% MeOH/DCM, с получением указанного в заголовке соединения в виде бесцветного масла (2,9 г, 67%-ный выход). 1H ЯМР (400 МГц, CDCl3) δ 0,97-0,91 (м, 1H), 1,13-0,99 (м, 3H), 2,66 (д, J=5,1 Гц, 1H), 4,41-4,36 (ддд, J=9,8, 5,1, 2,4 Гц, 1H), 4,59-4,53 (дд, J=9,8, 13,8 Гц, 1H), 4,68 (дд, J=2,4, 13,8 Гц, 1H).
Получение 3
гидрохлорид (±)-2-амино-1-[1-(трифторметил)циклопропил]этанола
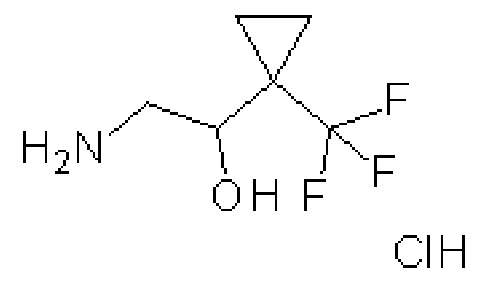
К раствору (±)-2-нитро-1-[1-(трифторметил)циклопропил]этанола (2,9 г, 14,8 ммоль) в Et2O (30 мл) при температуре 0ºC добавляют по каплям 1 M раствор алюмогидрида лития в ТГФ (37 мл, 37 ммоль) и перемешивают полученную реакционную смесь при комнатной температуре в течение ночи. Смесь охлаждают до температуры 0ºC и по каплям добавляют еще 1М раствор алюмогидрида лития в ТГФ (15 мл, 15 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 4 часов. Реакционную смесь гасят при температуре 0ºC, добавляя по каплям воду (1,96 мл), затем 2 M водный NaOH (1,96 мл) и затем воду (5,88 мл). Добавляют MgSO4 (5 г), перемешивают полученную смесь в течение 10 мин и фильтруют через слой диатомитовой земли. Фильтрат обрабатывают 4 M HCl в диоксане (20 мл, 80 ммоль) и концентрируют при пониженном давлении. Полученный остаток суспендируют в Et2O (50 мл) при быстром перемешивании. Полученное белое твердое вещество отделяют фильтрованием с получением указанного в заголовке соединения (1,52 г, 47%-ный выход). ES/MS: m/z=170 [M+H].
Альтернативный способ получения 3
В колбу Парра помещали оксид платины (IV) (242 мг, 1,07 ммоль), раствор (+)-2-нитро-1-[1-(трифторметил)циклопропил]этанола (2,42 г, 11,5 ммоль) в EtOH (24 мл) и уксусную кислоту (4,96 мл). Колбу помещают в атмосферу водорода при давлении 55 фунтов на квадратный дюйм и встряхивают в течение 5 часов при комнатной температуре. Реакционную смесь фильтруют через слой диатомовой земли и концентрируют при пониженном давлении. Остаток суспендируют в 1,4-диоксане (17,2 мл), по каплям добавляют 4 н HCl в 1,4-диоксане (10 мл, 40 ммоль) и перемешивают в течение 2 часов. Смесь фильтруют, промывают слой на фильтре 1,4-диоксаном и сушат в вакууме в течение 15 минут. Полученное твердое вещество сушат в вакуумной печи при 45ºC в течение 3 часов с получением указанного в заголовке соединения (2,21 г, 80%-ный выход).
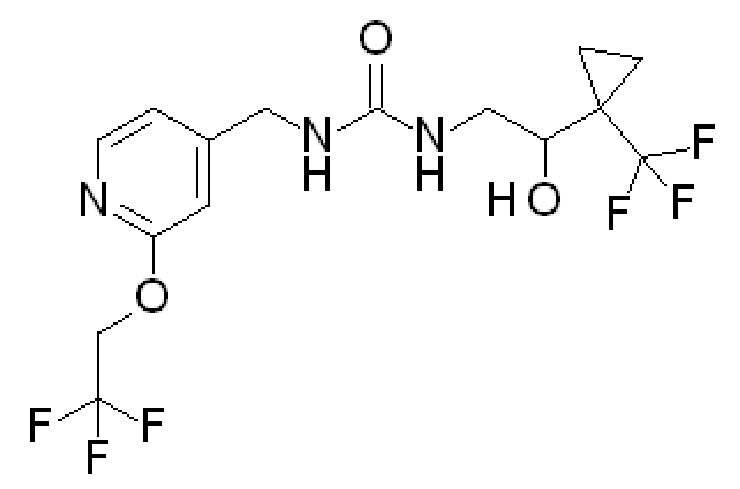
Получение 4 (что дает соединение по примеру 2)
(±)-1-[2-гидрокси-2-[1-(трифторметил)циклопропил]этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевина
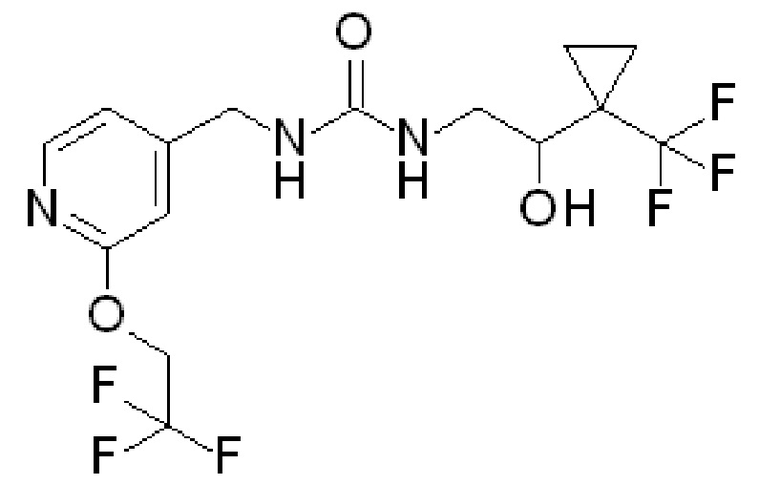
Суспензию гидрохлорида [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина [CAS# 2044704-69-8] (200 мг, 0,8 ммоль) в DCM (4 мл) охлаждают до температуры 0ºC, добавляют DIPEA (580 мкл, 3,3 ммоль) и 1,1ʼ-карбонилдиимидазол (149 мг, 0,9 ммоль). Полученную реакционную смесь энергично перемешивают при температуре 0ºC в течение 15 минут и добавляют гидрохлорид (±)-2-амино-1-[1-(трифторметил)циклопропил]этанола (232 мг, 1,01 ммоль). Полученную смесь перемешивают при комнатной температуре в течение ночи. Гасят добавлением воды, разбавляют DCM и пропускают через гидрофобную фритту. Органические слои упаривают и полученный остаток очищают на силикагеле, элюируя 0-15% MeOH/DCM, с получением указанного в заголовке соединения (198 мг, 57%-ный выход). ES/MS: m/z=402 [M+H].
Альтернативный способ получения 4
Дигидрохлорид [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина (500 мг, 1,79 ммоль) и DIPEA (1,26 мл, 7,16 ммоль) в DCM (10 мл) перемешивают с получением прозрачного раствора. Добавляют 1,1ʼ-карбонилдиимидазол (311 мг, 1,88 ммоль) и перемешивают 30 минут. Добавляют гидрохлорид (±)-2-амино-1-[1-(трифторметил)циклопропил]этанола (465 мг, 2,15 ммоль) и перемешивают реакционную смесь при комнатной температуре в течение 48 часов. Добавляют воду, отделяют органическую фазу и высушивают над сульфатом натрия. Органические слои фильтруют и упаривают и очищали полученный остаток на силикагеле, элюируя 0-15% MeOH/DCM, с получением указанного в заголовке соединения (680 мг, 90%-ный выход).
Хиральная часть этой молекулы может быть синтезирована с использованием кинетического разделения и химии раскрытия цикла.
Получение 5
гидрохлорид (2S)-1-амино-3,3-диметил-бутан-2-ола
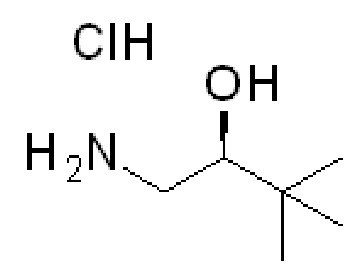
(2S)-2-трет-Бутилоксиран получают коммерчески (CAS# 40102-55-4) или с помощью кинетическое оптического расщепления, как описано в J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 1307, в котором сообщается, что эпоксид с конфигурацией R получали c катализатором RR, следовательно, эпоксид S получали c катализатором SS:

Смесь (2S)-2-трет-бутилоксирана (107 г, 1,01 моль) и NH4OH (1,3 л, 10,7 моль) в EtOH (427 мл) нагревают в закрытом сосуде при 100ºC в течение 4 ч. Охлаждают и концентрируют при пониженном давлении. Полученный остаток растворяют в DCM (100 мл) при 0ºC и медленно в течение 10 минут добавляют 4 M раствор HCl в диоксане (267 мл, 1,1 моль) до образования белого осадка. Полученное твердое вещество фильтруют, промывают холодным DCM и сушат вакуумным отсасыванием с получением указанного в заголовке соединения (103 г; 62,9%-ный выход). 1H ЯМР (300,1 МГц, MeOD): δ 0,94 (с, 9H), 2,76 (дд, J=11,1, 12,6 Гц, 1H), 3,09 (дд, J=2,5, 12,6 Гц, 1H), 3,41 (дд, J=2,5, 11,1 Гц, 1H). [α]D20=+32,38º (c=0,8 г/100 мл, EtOH). Литературные данные представленные в Tetrahedron: Asymmetry 13 (2002) 1209-1217: [α]D20=+25,9º (c=0,47 г/100 мл, EtOH).
Альтернативный способ получения 5
Подготавливают два шприцевых насоса ISCO 2-1000 мл, обозначенных A и B:
Заполняют насос A 7 M раствором NH3 в MeOH. Заполняют насос B раствором (2S)-2-трет-бутилоксирана (25 г, 232 ммоль), растворенного в 7 M растворе NH3 в MeOH (995 мл). Насосы подключают к трубчатому реактору из нержавеющей стали объемом 500 мл (OD=1/8 дюйма) в печи, затем подключают к выходу трубчатый реактор из нержавеющей стали емкостью 7 мл (OD=1/16 дюйма), расположенный вне печи, чтобы он действовал в качестве теплообменника, и подключают регулятор противодавления EQUILIBAR®, установленный на 1200 фунтов на квадратный дюйм после теплообменника.
С помощью насоса A заполняют трубчатый реактор 7 М раствором NH3 в MeOH со скоростью 5 мл/мин. Устанавливают температуру духовки на 200ºC. Когда трубчатые реакторы заполняются, переключают на насос B со скоростью 10 мл/мин в течение 100 минут, а затем переключают на насос A, чтобы доставлять 7 М раствор NH3 в MeOH с той же скоростью потока в течение еще 1 часа.
Собранный раствор концентрируют при пониженном давлении при комнатной температуре с получением сырого (2S)-1-амино-3,3-диметилбутан-2-ола (24,4 г). Сырой продукт растворяют в трет-бутилметиловом эфире (150 мл) и при интенсивном перемешивании добавляют по каплям 5,5 М раствор HCl в IPA (46,4 мл, 255 ммоль) в течение 5 мин. Полученное твердое вещество белого цвета фильтруют, промывают МТВЕ (4×25 мл) и сушат, получая указанное в заголовке соединение (23 г, 72%-ный выход).
Получение 6
дигидрохлорид [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина
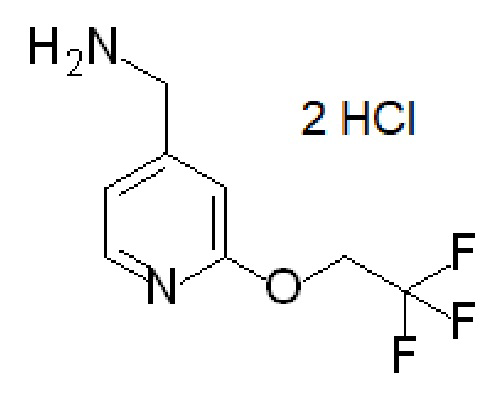
В EtOH (500 мл) перемешивают 2-(2,2,2-трифторэтокси)пиридин-4-карбонитрил [CAS# 618446-30-3] 82,5 г, 367 ммоль), декантируют с нерастворенного твердого вещества раствор и твердое вещество промывают EtOH (3×50 мл), каждый раз декантируя раствор. Растворы в EtOH объединяют, разбавляют дополнительным количеством EtOH (150 мл) и добавляют конц. водный раствор HCl (125 мл). Добавляют суспензию 10% палладия на угле (3,7 г) примерно в 10 мл EtOH. Полученную реакционную смесь встряхивают в атмосфере H2 под давлением 60 фунтов на кв. дюйм при комнатной температуре в течение ночи. Смесь фильтруют через слой диатомовой земли, промывают EtOH и концентририруют фильтрат при пониженном давлении с получением твердого вещества белого цвета. Полученное твердое вещество суспендируют в MTBE при температуре 45ºC в течение 30 мин, смесь охлаждают до комнатной температуры и фильтруют с получением твердого вещества. Твердое вещество растворяют в воде (400 мл) и дважды экстрагируют MTBE (400, затем 200 мл). Водную фазу концентририруют при пониженном давлении с получением твердого вещества кремового цвета, которое суспендируют в ТГФ (100 мл) и фильтруют. Осадок на фильтре промывают ТГФ (2×30 мл) и сушат в вакууме с получением указанного в заголовке соединения (46,16 г, выход 44%). Фильтрат дополнительно концентририруют при пониженном давлении и сушат в вакуумной печи в течение ночи. Полученное твердое вещество суспендируют в ТГФ (50 мл) в течение 30 минут и фильтруют, получая дополнительную порцию указанного в заголовке соединения (34 г, выход 32,5%). ES/MS: m/z 307 [М+Н]. Анализ на хлорид-ион (IC) показал молярное соотношение хлорид-ион:исходный ион 2:1.
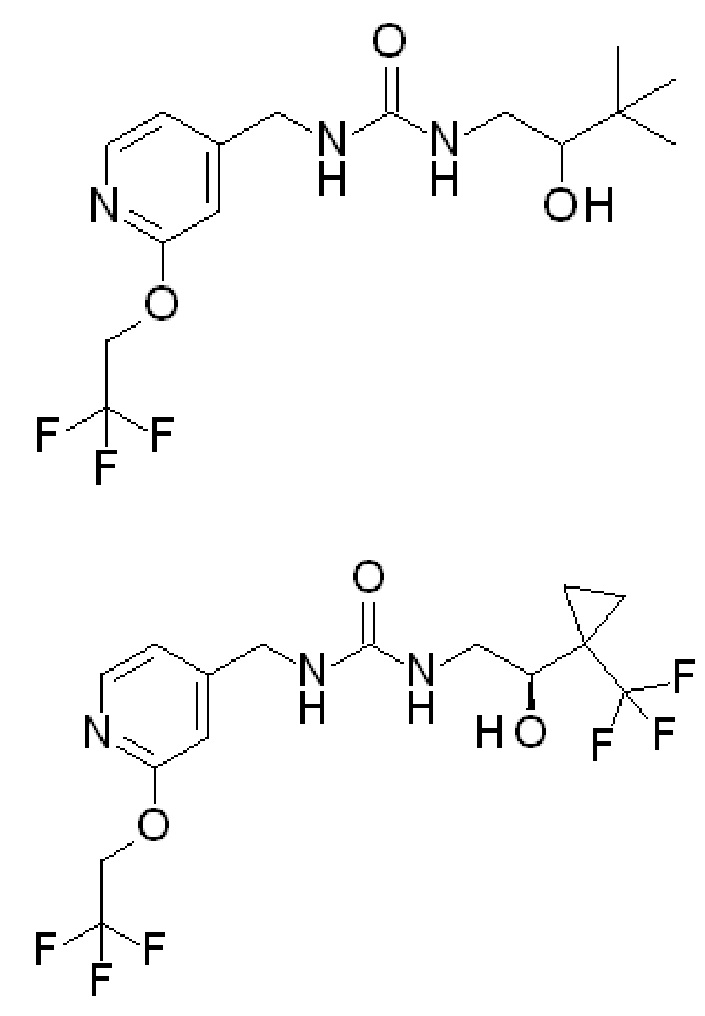
Пример 1
1-[(2S)-2-гидрокси-3,3-диметил-бутил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевина
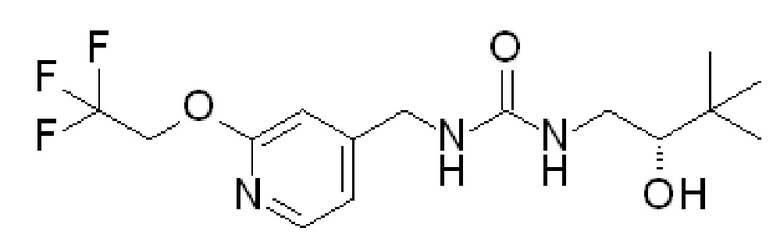
В DCM (50 мл) перемешивают гидрохлорид [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина (8,06 г, 33,2 ммоль) и добавляют DIPEA (29 мл, 166 ммоль). Полученную смесь перемешивают 5 мин и добавляют 1,1ʼ-карбонилдиимидазол (5,7 г, 33,2 ммоль). Смесь перемешивают в течение 10 мин и добавляют гидрохлорид (2S)-1-амино-3,3-диметил-бутан-2-ола (5 г, 32,5 ммоль) и перемешивают полученную реакционную смесь в течение выходных. Промывают реакционную смесь водой, отделяют органическую фазу и концентрируют при пониженном давлении. Полученный остаток очищают колоночной флэш-хроматографией на силикагеле, элюируя 0-10% MeOH в DCM, с получением после выпаривания растворителя указанного в заголовке соединения (8,16 г, 72%-ный выход).
Продукт объединяют с другой партией указанного в заголовке соединения (4,37 г), полученной, как описано выше, и перекристаллизовывают из iPrOAc (45 мл), получая 11,29 г указанного в заголовке соединения. ES/MS: m/z 350 [M+H]. [α]D20=+29,845º (c=0,2 г/100 мл, MeOH).
Пример 2
(+)-1-[2-гидрокси-2-[1-(трифторметил)циклопропил]этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевина и (-)1-[2-гидрокси-2-[1-(трифторметил)циклопропил]этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевина посредством хирального разделения
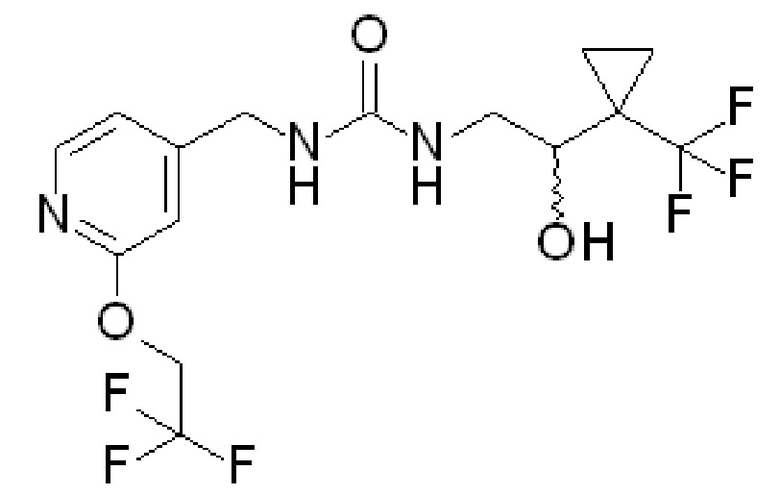
(±)-1-[2-Гидрокси-2-[1(трифторметил)циклопропил]этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевину (680 мг) подвергают очистке хиральной SFC с использованием колонки CHIRALPAK® AD-H (250×30 мм, 5 мк) при температуре 35ºC, 100 бар, элюирование 92:8 CO2/этанол с 0,2% N,N-диметилэтиламина 152 мл/мин и детектирование при 220 нм, фракции упаривают и сушат в вакуумном сушильном шкафу при 45ºC, получая:
энантиомер 1 (1-й пик элюирования, 285,4 мг): (-)-1-[2-гидрокси-2-[1-(трифторметил)циклопропил]этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевина; [α]D20=-21,0º (c=0,20 г/100 мл, MeOH);
энантиомер 2 (2-й пик элюирования, 289,5 мг). Энантиомер 2 подвергают SFC очистке второй раз, используя способ, описанный выше; упаривают фракции и сушат в вакуумной печи при 45ºC с получением (+)-1-[2-гидрокси-2-[1-(трифторметил)циклопропил]-этил]-3-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]мочевины (236 мг); [α]D20=+15,0º (c=0,20 г/100 мл, MeOH).
Пример 3
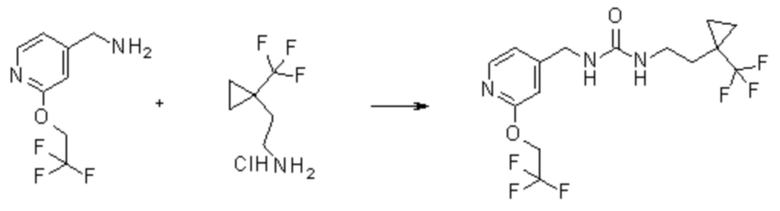
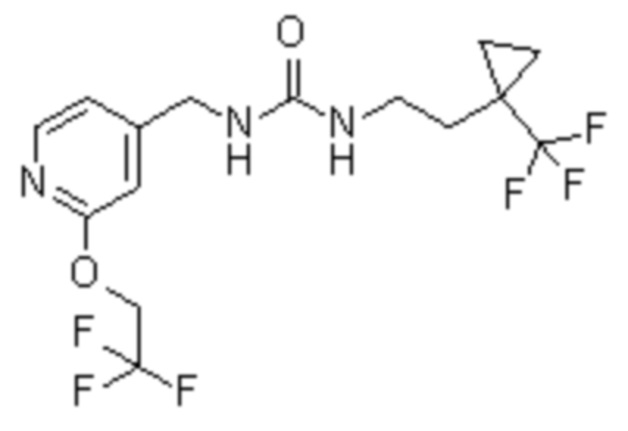
1-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]-3-[2-[1-(трифторметил)циклопропил]этил]мочевина
В круглодонной колбе перемешивают гидрохлорид 2-[1-(трифторметил)циклопропил]этанамина (500 мг, 2,6 ммоль; см. WO 2013/134252, но также имеющийся в продаже [CAS#: 1454690-80-2]) в DCM (5 мл) и добавляют DIPEA (1,4 мл, 8 ммоль). Когда образуется прозрачный раствор, добавляют 1,1ʼ-карбонилдиимидазол (428 мг, 2,6 ммоль) и перемешивают полученную смесь при комнатной температуре в течение 30 мин. Одной порцией добавляют дигидрохлорид [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина (810 мг, 2,9 ммоль) и перемешивают при комнатной температуре в течение 30 мин. Реакционную смесь в течение 3 часов нагревают при температуры 40ºC и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь переносят в сосуд для микроволновой печи и нагревают при температуре 100ºC в течение 30 мин. Полученную смесь очищают хроматографией на силикагеле, элюируя 0-10% MeOH в DCM, с получением указанного в заголовке соединения (892 мг (892 мг, 88%-ный выход). ES/MS: m/z 386 [M+H]
Альтернативный способ для примера 3
В реакционный сосуд для микроволновой печи помещают имеющийся в продаже гидрохлорид 2-[1-(трифторметил)циклопропил]этанамида (758 мг, 0,4 ммоль, CAS# 561297-93-6), DCM (2 мл), DIPEA (698 мкл, 0,4 ммоль) и 1,1ʼ-карбонилдиимидазол (649 мг, 0,4 ммоль). Встряхивают при комнатной температуре в течение 5 часов. Добавляют при комнатной температуре приготовленный 0,5 М раствор коммерчески доступного [2-(2,2,2-трифторэтокси)-4-пиридил]метанамина (CAS# 1454690-80-2 альтернативно, см. WO 2013/134252) в DCM (0,8 мл, 0,4 ммоль). Полученную смесь нагревают в микроволновой печи при температуре 100ºC в течение 30 мин. Реакционную смесь выливают в круглодонную колбу большего размера и разбавляют водой (5 мл) и DCM (5 мл). Перемешивают 15 мин при комнатной температуре и пропускают через гидрофобную фритту для отделения органической фазы. Концентрируют фильтрат при пониженном давлении и очищают полученный остаток обращенно-фазовой хроматографией с высоким pH, используя колонку PHENOMENEX® GEMINI®-NX C18 75×30 мм, размер частиц 5 мкм, 110A, колонку AXIA с GEMINI®-NX C18 15×30 мм защиты, элюируя 23-57% ACN в 10 мМ NH4HCO3 (pH ~10), содержащем 5% MeOH в качестве водной фазы, с получением 1-[[2-(2,2,2-трифторэтокси)-4-пиридил]метил]-3-[2-[1-(трифторметил)циклопропил]этил]мочевины (90,6 мг, 59%-ный выход). ES/MS: m/z 386 [M+H]
Анализы
Данные биологического анализа, показывающие, что соединения по настоящему варианту осуществления активны в потенцировании Kv7, представлены ниже.
Анализ # 1
Анализ возбудимости Optopatch для примера 1 в двигательных нейронах, полученных из линий iPSC пациента с БАС
Измененная возбудимость кортикальных нейронов и нижних мотонейронов является важным фактором патофизиологии БАС (см., например, K. Kanai, et. al., J Neurol Neurosurg Psychiatry, 83, 734-738 (2012); P. Menon, et. al., Eur J Neurol., 24, 816-824 (2017)). Для оценки возбудимости культивируемых мотонейронов, полученных из линий iPSC двух пациентов с БАС с различными патогенными мутациями, была использована полностью оптическая электрофизиология («оптопатч»).
Культура клеток: две линии iPSC получали от одного пациента с патогенной мутацией в TARDBP и одного пациента с мутацией распространения патогенного повтора в C9orf72, соответственно. Моторные нейроны генерировали из этих линий с помощью метода 2D-дифференцировки, адаптированного из протокола формирования нейронального паттерна с двойным ингибированием SMADS. (См S.M. Chambers, et. al., Nat Biotechnol., 27, 275-280 (2009)) путем включения морфогенов моторных нейронов. Дифференциация подтверждалась визуальным осмотром, кариотипированием и окрашиванием на бета-III-тубулин и ядерный маркер мотонейрона ISL1. Нейроны культивировали на монослое мышиной глии в mTeSR (Stem Cell Technologies) с добавлением 10 нг/мл BDNF. За сорок восемь часов до визуализации в среду добавляли 100 нМ трансретиналя.
Трансдукция векторами Optopatch: через 15 дней in vitro культивируемые мотонейроны трансдуцировали лентивирусным вектором для управления совместной экспрессией исполнительного механизма CheRiff-mOrange2 и индикатора напряжения QuasAr3-Citrine (подробнее см. D.R. Hochbaum, et. al., Nat. Methods, 11, 825-833 (2014)). За сорок восемь часов до визуализации в среду добавляли 100 нМ трансретиналя.
Растворы: записи выполняли в буфере для визуализации BrainphysTM с 3 мМ калия. Для устранения электрического взаимодействия между клетками добавляли блокатор щелевых соединений 2-аминоэтоксидифенилборат (50 мкМ), а для блокирования синаптической передачи были использованы NBQX (10 мкМ), габазин (20 мкМ) и AP5 (25 мкМ).
Записи Optopatch: через пять дней после трансдукции на специализированном сверхширокопольном флуоресцентном микроскопе при комнатной температуре выполняли визуализацию Optopatch. На моторные нейроны воздействовали лазерным возбуждением красного диапазона (200 Вт/см2; 635 нм) для отслеживания изменений флуоресценции QuasAr и возбуждением светодиодом синего свечения (0-127 мВт/см2, 470 нм) для деполяризации клеточной мембраны с помощью CheRiff. Использовали настраиваемый протокол синего светового стимула, состоящий из: i) 2-секундного красного освещения только для контроля спонтанной активности, 2) стадий увеличения интенсивности синего света 5×500 мсек и iii) 2×2-секундных линейно увеличивающихся значений синего света, каждый с другой максимальной интенсивностью синего цвета. Данные Optopatch записывали с помощью камеры Hamamatsu ORCA-Flash 4.0 sCMOS с частотой кадров 1 кГц. Размер поля зрения 4 мм×0,5 мм. Программное обеспечение для настраиваемого управления, написанное на MATLAB, использовали для управления протоколами освещения и записи всех картин. Чтобы изучить острые эффекты потенциаторов Kv7.2/7.3, нейроны инкубировали с исследуемым соединением в течение 15 минут перед визуализацией.
Анализ данных: анализ сегментации изображения выполняли с использованием временного анализа основных компонентов и пространственно-временного анализа независимых компонентов для выделения отдельных нейронов. Алгоритм поиска пиков использовали для поиска потенциалов действия, и данные анализировали на предмет сложных эффектов на средний уровень возбуждения, адаптацию, реобазу и форму волны потенциала действия путем сравнения с контролем растворителем (подробности см. C.A. Werley, et. al., Curr. Protoc. Pharmacol., 78, 11,20,1-11,20,24. (2017)).
В соответствии с протоколом, описанным выше, основной эффект соединения по примеру 1 заключался в уровне возбуждения потенциала действия в ответ на освещение синим светом низкой интенсивности. При интенсивности свечения синего светодиода 5,1 мВт/см2 соединение снижало частоту потенциала действия зависимым от концентрации образом.
Подбор 4-параметрического логистического уравнения к данным можно использовать для определения эффективности (ЕС50) воздействия соединения по примеру 1 на частоту потенциала действия. Результаты показаны в таблице 1 для двух отдельных попыток дифференциации каждой из линий, полученных от пациентов, несущих мутации TARDBP и C9orf72. Наблюдаемые эффекты качественно аналогичны, но более сильны, чем эффекты, наблюдаемые с известным потенциатором Kv7.2/7.3 флупиртином, что демонстрирует потенциальную полезность соединения по примеру 1 для лечения БАС за счет снижения возбудимости двигательных нейронов пациента.
Таблица 1: Подавление возбудимости двигательных нейронов, полученных от пациентов с БАС (представлено как среднее значение (95% доверительный
по примеру 1
EC50 (мкМ)
(0,05, 0,25)
(0,06, 0,24)
(0,09, 0,25)
(0,14, 0,25)
EC50 (мкМ)
(0,65, 1,66)
(0,48, 1,21)
(1,31, 2,01)
(0,82, 1,88)
Анализ # 2
Модуляция проводимости Kv7.2/7.3 потенциаторами Kv7 в системе экспрессии млекопитающих
Активность и эффективность потенциаторов Kv7 оценивали с помощью автоматизированной электрофизиологии на платформе IonWorks Barracuda (Molecular Devices) с использованием режима прибора с фиксацией популяции.
Культура клеток: для этих исследований использовали клетки HEK293, стабильно экспрессирующие hKv7.2 (при индукции тетрациклином) и hKv7.3 (Каталог # CT6147, Charles River). Клетки поддерживали в модифицированной Дульбекко среде Игла/питательной смеси Hamʼs F-12 (Sigma-Aldrich) с добавлением 5%-ной фетальной бычьей сыворотки, прошедшей скрининг на тетрациклин (Sigma-Aldrich), 15 мМ HEPES, 500 мкг/мл G418, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина, 29,2 мг/мл L-глутамина, 100 мкг/мл зеоцина и 5 мкг/мл бластицидина. Экспрессию hKv7.2 индуцировали добавлением 1 мкг/мл доксициклина за 24 ч до записи.
Клетки культивировали в колбах Corning T-150 до степени слияния 85%-95%. В начале эксперимента клетки промывали один раз D-PBS без кальция и магния, а затем диссоциировали путем инкубации в 3 мл 0,25% трипсина в течение 8 минут при 37ºC. Клетки ресуспендировали в среде, осторожно растирали и центрифугировали в течение 4 мин при 1000 об/мин. Клетки ресуспендировали во внешнем растворе до конечной концентрации 2,5-3,5 М клеток/мл.
Растворы: Внешний раствор состоял из (в мМ): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 глюкозы, pH 7,4. Внутренний раствор состоял из (в мМ): 90 K-глюконат, 40 KCl, 3,2 EGTA, 3,2 MgCl2, 5 HEPES, pH 7,25. Амфотерицин B, перфорирующий мембрану, готовили ежедневно в виде исходного раствора 27 мг/мл в ДМСО, а затем добавляли к внутреннему раствору до конечной концентрации 0,1 мг/мл. Разведения испытуемых веществ готовили в 384-луночных планшетах из 10 мМ исходных растворов ДМСО и разводили с использованием акустического дозирования (Labcyte ECHO®), так что конечные концентрации ДМСО составляли 0,1%.
Электрофизиологические записи: ресуспендированные клетки помещали в прибор IONWORKS® BARRACUDATM (IWB), внешний раствор добавляли в 384-луночный патч-планшет, и для определения заблокированных лунок и напряжений смещения проводили холл-тест («hole test»). Затем с помощью прибора клетки добавляли в планшет (9 мкл на лунку). Перед введением перфорирующего агента амфотерицина во внутренний раствор проводили два испытания герметичности и давали приблизительно 8 минут для получения электрического доступа, что подтверждалось третьим тестом герметизации. Протокол командного напряжения состоял из семейства шагов напряжения в 1 секунду от -80 мВ до +40 мВ от удерживающего потенциала -80 мВ и применялся до (базового уровня) и через 6 минут после добавления испытуемого образца.
Анализ данных: данные собирали и вычитали утечки с помощью программного обеспечения IWB. Амплитуды тока за последние 10 мс каждого шага напряжения усредняли и экспортировали. Дальнейший анализ выполняли с помощью Microsoft Excel и GraphPad Prism. Амплитуда тока преобразовывали в проводимость (G) по следующей формуле: G=I/(V-Ek), где I=ток, V=ступенчатый потенциал, Ek=обратный потенциал калия (-84 мВ). Электропроводность в присутствии исследуемого образца нормализовали к исходной проводимости при +40 мВ для той же лунки. Кривые проводимости-напряжения (G-V) соответствовали уравнению Больцмана y=снизу+(сверху-снизу)/(1+EXP((Vm-V0,5/k)).
Опосредованный испытуемым образцом сдвиг в средней точке кривой проводимости (V0,5) показан в таблице 2 для соединений по примерам 1-3.
Таблица 2: Отличие от контроля по напряжению при половине максимальной проводимости в присутствии различных концентраций исследуемого соединения.
(SD выше относится к стандартному отклонению).
Подбор 4-параметрического логистического уравнения к данным можно использовать для определения активности (ЕС50) и эффективности (максимального сдвига) для каждого тестируемого соединения. Результаты показаны в таблице 3 для примеров 1-3.
Таблица 3: Активность и эффективность потенциаторов Kv7.2/7.3
Анализ # 3
Отслеживание пороговых значений для измерения действия соединения по примеру 1 при 3,10 и 30 мг/кг IP на возбудимость периферических нервов
Пороговое отслеживание представляет собой неинвазивный метод, который позволяет измерять свойства возбудимости периферических аксонов, предоставляя информацию об их мембранном потенциале и функции ионных каналов.
Метод
Использовали 16 крыс-самцов линии Wistar массой 307-446 г от компании Charles River. Животных размещали в группах со стандартными условиями содержания (по 4 в клетке, с 07:00 до 19:00, фаза освещения, постоянные температура (21ºC) и влажность, свободный доступ к пище и воде, а также обогащение среды).
Крыс анестезировали изофлураном (2-2,5%, O2 при 0,5 л/мин), а затем помещали на спину на грелку, чтобы поддерживать температуру хвоста выше 32ºC. Помечали размещение кольцевых электродов, и отмеченные участки хвоста очищали лезвием для удаления волос и верхнего слоя кожи. Эти участки промывали водой и сушили, чтобы обеспечить хорошую проводимость между кожей и липкими электродами.
Липкий кольцевой стимулирующий электрод (+, анод) оборачивали вокруг стопы. Второй стимулирующий электрод с липким кольцом (-, катод) наматывали на 1,5 см от основания хвоста. Записывающий электрод-игла помещали чуть смещенно от центра на вершине хвоста, на удалении в 6 см от стимулирующего катода у основания хвоста. Эталонный игольчатый электрод помещали чуть не по центру в верхней части хвоста, на расстоянии 2 см от записывающего электрода. Липкий заземляющий электрод оборачивали вокруг хвоста на 2 см проксимальнее записывающего электрода.
Протокол исследования
Запустить программу Multitrack (описание ниже) и Spike.
Записать 15 минут стабильного исходного уровня
Через 15 минут ввести дозу соединения по примеру 1 или HEC.
Взять каплю крови на PK в следующих случаях:
через 10 мин после введения дозы
через 20 минут после введения дозы
через 30 мин после введения дозы
через 40 мин после введения дозы
Через 45 минут после IP (внутрибрюшинного) введения соединения по примеру 1 доза XE-991 (XE-991 представляет собой коммерчески доступное соединение, которое блокирует потенцирующие эффекты KCNQ в формалиновом анализе in vivo - см. Y. Zheng, et al., «Activation of peripheral KCNQ channels relieves gout pain», Pain, 156 (2015) 1025-1035; и R. Zaczek, et al., «Two New Potent Neurotransmitter Release Enhancers, 10,10-Bis(4-Pyridinylmethyl)-9(10H)-Anthracenone and 10,10-Bis(2-Fluoro-4-Pyridinylmethyl)-9(10H)-Anthracenone: Comparison to Linopirdine» The Journal of Pharmacology and Experimental Therapeutics, 285:724-730, 1998.
(XE-991 вводится только после соединения по примеру 1 в дозе 30 мг/кг)
Взять каплю крови на PK в следующих случаях:
через 10 минут после введения дозы (55 минут после введения соединения по примеру 1)
через 20 минут после введения дозы (65 минут после введения соединения по примеру 1)
через 30 минут после введения дозы (75 минут после введения соединения по примеру 1)
через 40 минут после введения дозы (85 минут после введения соединения по примеру 1)
В анализе порогового отслеживания для измерения возбудимости периферических аксонов в исследованиях на крысах соединение вводили внутрибрюшинно в дозе 3, 10 или 30 мг/кг с использованием смеси 1% гидроксиэтилцеллюлоза:0,25% полисорбата 80:0,05% состава пеногасителя:очищенная вода (HEC). Пятна сухой крови (DBS) собирали примерно через 10, 20, 30 и 40 минут после введения дозы. Образцы DBS сушили при комнатной температуре около 2 часов. Образцы головного мозга брали в конечный момент времени и замораживали до анализа. Образцы DBS отправляли и хранили при комнатной температуре.
Готовили маточный раствор соединения по примеру 1 с концентрацией 1 мг/мл, который серийно разбавляли объединенной кровью крыс для получения стандартов в диапазоне от 1 до 10000 нг/мл. Кровь добавляли на пустые карты DBS для создания стандартов. В 96-луночный планшет добавляли один 3-миллиметровый пробойник стандартов или образцов DBS и добавляли 180 мкл внутреннего стандарта (IS) в смеси 1:1 ACN:MeOH. Встряхивали в течение 45 минут, разводили раствор для экстракции водой в два раза и анализировали с помощью ЖХ/МСМС для анализа концентрации лекарственного средства.
Образцы мозга гомогенизировали, используя 1,14 мл смеси MeOH:H2O (2:8). Стандарты готовили путем добавления исходного раствора в серию холостых гомогенатов головного мозга в диапазоне от 5 до 50000 нг/мл. 25 мкл стандарта или образца вносили пипеткой в 96-луночный планшет, добавляли 180 мкл внутреннего стандарта (IS) в смеси 1:1 ACN:MeOH и перемешивали. Образцы центрифугировали при 4000 об/мин при 4°C в течение 10 мин. Супернатант разводили водой в 15 раз и анализировали с помощью ЖХ/МСМС.
Образцы и стандарты анализировали с помощью трехквадрупольного масс-спектрометра Sciex API 4000 (Sciex, Division of MDS Inc., Toronto, Canada), соединенного с системой Shimadzu HPLC (LC-IOAD, Shimadzu Corporation) и автосамплером Gilson 215. Образцы (0,01 мл) вводили в колонку для ВЭЖХ с 5 мкм Betasil C-18, 20×2,1 мм Javelin (Thermo Electron Corp. Cat#70105-022106) и элюировали градиентом. Хроматографические условия включали подвижную фазу A, состоящую из воды/1M NH4HCO3 (2000:10, об./об.), и подвижную фазу B, состоящую из MeOH/1M NH4HCO3 (2000:10, об./об.), которые проходили через 2,5-минутный градиент при скорости потока 1,5 мл/мин. Для масс-спектрометрического обнаружения использовали режим положительных ионов с турбонаддувом и температуру источника ионов 740ºC. Количественное определение выполняли с использованием мониторинга множественных реакций (MRM) на следующих переходах: соединение по примеру 1 (от m/z 350,2 до m/z 233,0) и аналоговый внутренний стандарт (от m/z 263,1 до m/z 148,1). Графики линейной регрессии соединений для соотношений площадей пиков к внутреннему стандарту в зависимости от концентраций лекарственного средства получают с помощью квадратичного коэффициента 1/x2.
Используемый аналог внутреннего стандарта представляет собой 2-(диметиламино)-N-пентил-3-фенилпропанамид, соль 2,2,2-трифторуксусной кислоты (1:1) и имеет следующую структуру:
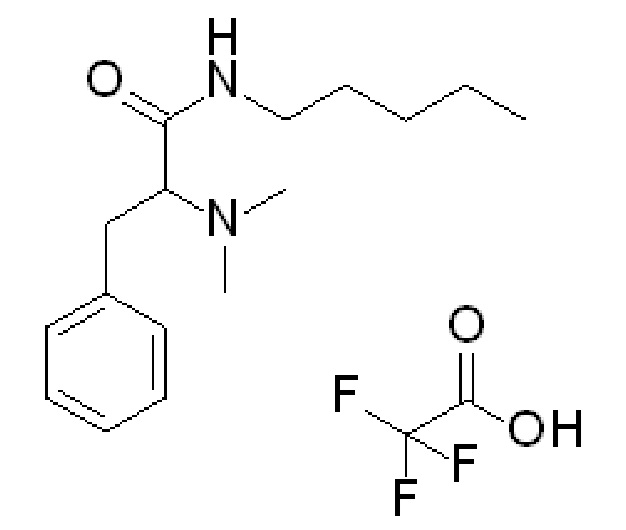
Этот аналоговый внутренний стандарт был приобретен у Syncom, компании из Нидерландов с адресом Kadijk 3, 9747 AT Groningen, Нидерланды.
Связывание лекарственного средства с белками плазмы и гомогенатами мозга крыс определяли с помощью метода диализа in vitro после добавления лекарственного средства в эти матрицы и инкубирования в течение 4,5 часов при 37°C, подвергая орбитальному встряхиванию. Анализ выполняли с использованием устройства для микро-равновесия для диализа и полосок диализной мембраны (MWCO 12-14k). Образец при времени 0 отбирали после белковой матрицы, и образцы отбирали как со стороны белка, так и со стороны буферного раствора мембраны через 4,5 часа инкубации. Исходный продукт количественно определяли с помощью ЖХ-МСМС как во время 0, так и на 45 минуте. Несвязанная фракция рассчитывалась путем деления концентрации на стороне буфера на концентрацию на стороне белка. Процент восстановления также рассчитывали путем деления суммы буферной и белковой камер на концентрацию времени 0 через 4,5 часа. Концентрации несвязанного соединения рассчитывали используя общая концентрация*несвязанная фракция.
Результаты: действие соединения по примеру 1 на абсолютный порог показано в таблице 4.
Таблица 4.
CMAP=потенциал сложного мышечного действия.
(Для получения дополнительной информации об этом анализе см. R. Sittl et al. «The Kv7 potassium channel activator flupirtine affects clinical excitability parameters of myelinated axons in isolated rat sural nerve», Journal of the Peripheral Nervous System 15:63-72 (2010); M. Kovalchuk, et al., «Acute Effects of Riluzole and Retigabine on Axonal Excitability in Patients With Amyotrophic Lateral Sclerosis: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial», Received 7 March 2018; accepted 13 April 2018; предварительная онлайн-публикация 00 месяц 2018 г. doi:10.1002/cpt.1096, CLINICAL PHARMACOLOGY & THERAPEUTICS, VOLUME 00 NUMBER 00, MONTH 2018; and J. Fleckenstein et al., «Activation of axonal Kv7 channels in human peripheral nerve by flupirtine but not placebo - therapeutic potential for peripheral neuropathies: results of a randomised controlled trial», Journal of Translational Medicine 2013, 11:34).
Существует значительная разница во времени и связи время*терапевтический эффект (двусторонний дисперсионный анализ RM; эффект времени F (26, 312)=13,18, p=<0,0001; связь время*терапевтический эффект F (78, 312)=2,888, p<0,0001 Тест множественных сравнений Бонферрони показал, что соединение по примеру 1 в дозе 30 мг/кг значительно увеличивало абсолютный порог (снижение возбудимости) по сравнению с носителем. XE-991 был способен обратить это повышение (повысить возбудимость).
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНИЛА И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2839891C1 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| АМИДОПРОИЗВОДНЫЕ КАК БЛОКАТОРЫ TTX-S | 2013 |
|
RU2632899C2 |
| 6,7-ДИГИДРОПИРАЗОЛО[1,5-a]ПИРАЗИН-4(5H)-ОНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ОТРИЦАТЕЛЬНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ MGLUR2 | 2015 |
|
RU2711382C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
Изобретение относится к низкомолекулярным потенциаторам калиевых каналов (такие как потенциаторы Kv7 - которые также называют потенциаторами KCNQ) формулы
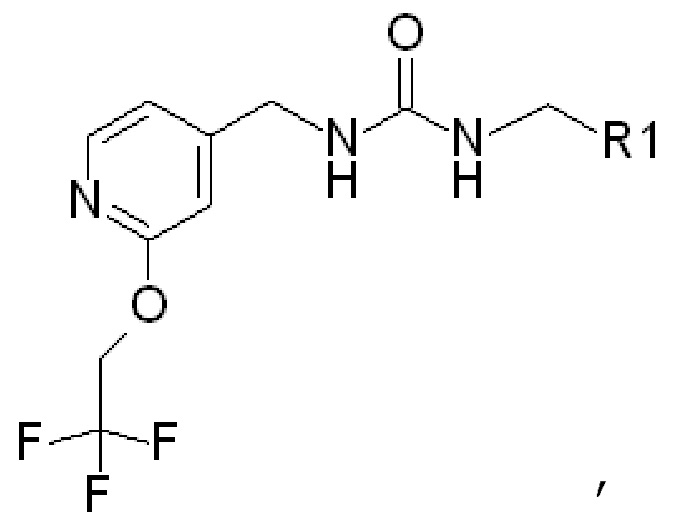
где R1 представляет собой 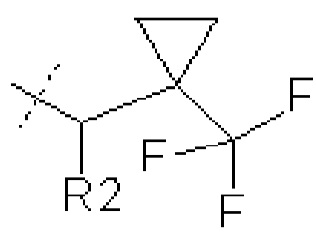 или
или 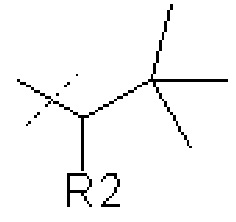 , R2 представляет собой Н или OH, композициям, включающим такие соединения, и способам применения таких соединений для лечения бокового амиотрофического склероза и других неврологических заболеваний, вызванных изменениями возбудимости двигательных нейронов, например, таких как первичный боковой склероз, псевдобульбарный паралич, прогрессирующий бульбарный паралич, прогрессирующую мышечную атрофию и эпилепсию. 9 н. и 16 з.п. ф-лы, 4 табл., 3 пр.
, R2 представляет собой Н или OH, композициям, включающим такие соединения, и способам применения таких соединений для лечения бокового амиотрофического склероза и других неврологических заболеваний, вызванных изменениями возбудимости двигательных нейронов, например, таких как первичный боковой склероз, псевдобульбарный паралич, прогрессирующий бульбарный паралич, прогрессирующую мышечную атрофию и эпилепсию. 9 н. и 16 з.п. ф-лы, 4 табл., 3 пр.
1. Соединение формулы
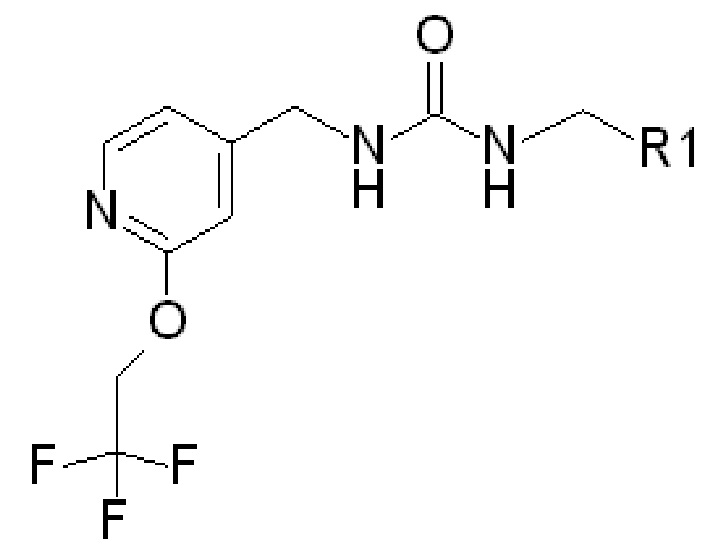
где R1 представляет собой
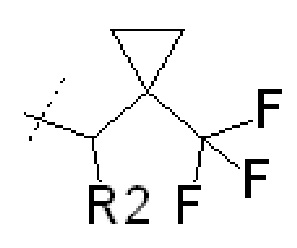
или
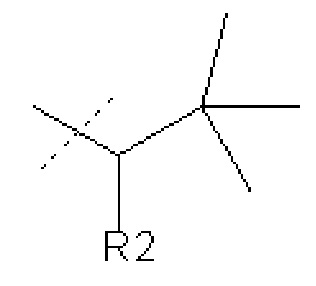
и где R2 представляет собой Н или OH,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой
или его фармацевтически приемлемая соль.
3. Соединение по п.2, где R2 представляет собой ОН, или его фармацевтически приемлемая соль.
4. Соединение по п.3 формулы
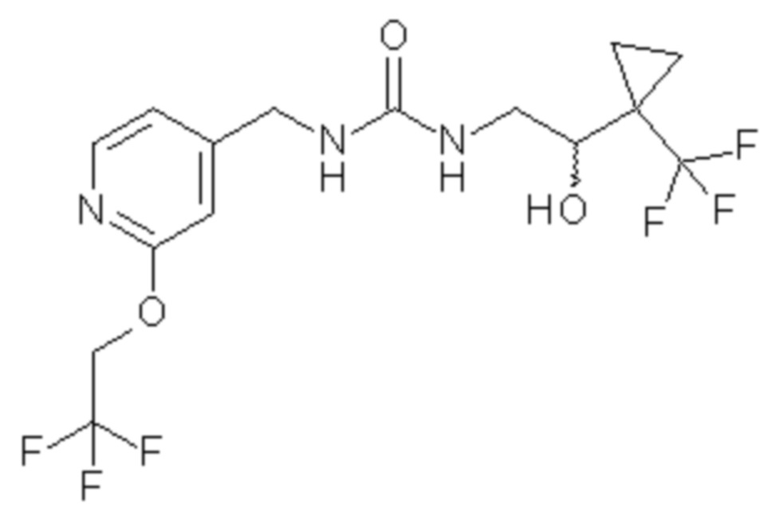
где соединение включает единственный энантиомер, который имеет (+) оптическое вращение в метаноле, или его фармацевтически приемлемую соль.
5. Соединение по п.3 формулы
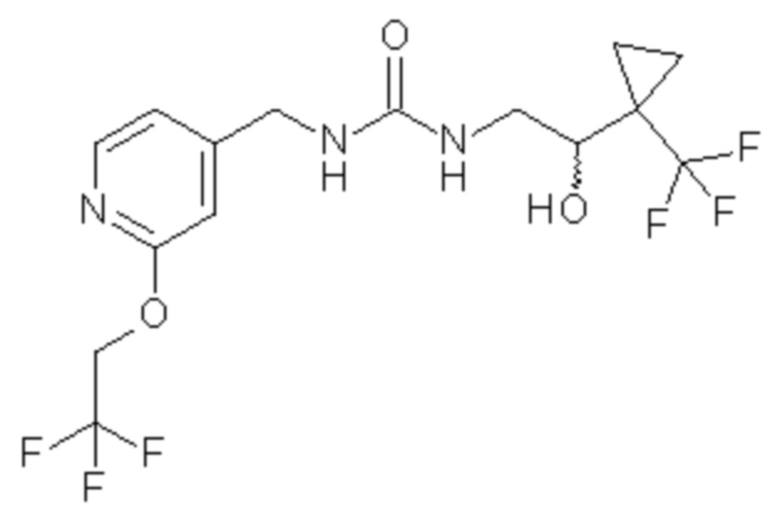
где соединение включает единственный энантиомер, который имеет (-) оптическое вращение в метаноле, или его фармацевтически приемлемая соль.
6. Соединение по п.2, где R2 представляет собой Н, или его фармацевтически приемлемая соль.
7. Соединение по п.1, где R1 представляет собой
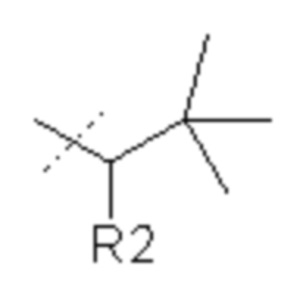
или его фармацевтически приемлемая соль.
8. Соединение по п.7, где R2 представляет собой Н, или его фармацевтически приемлемая соль.
9. Соединение по п.7, где R2 представляет собой ОН, или его фармацевтически приемлемая соль.
10. Соединение по п.9 формулы
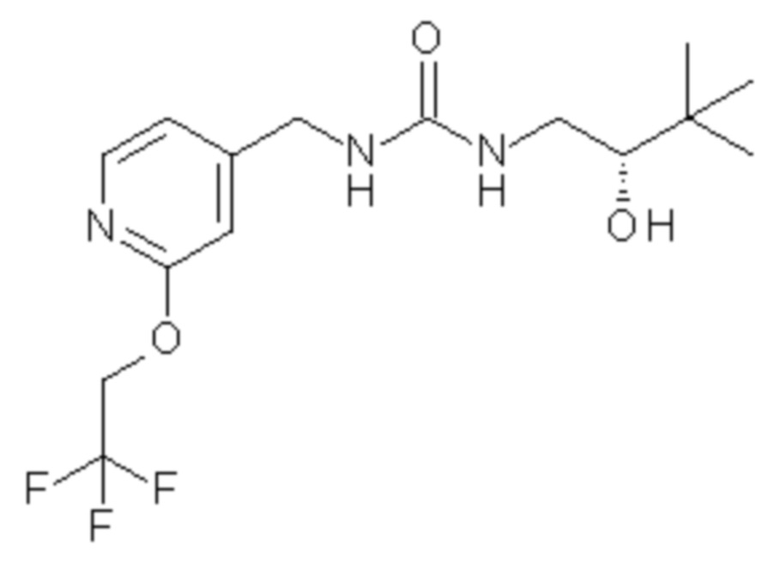
или его фармацевтически приемлемая соль.
11. Соединение по п.9 формулы
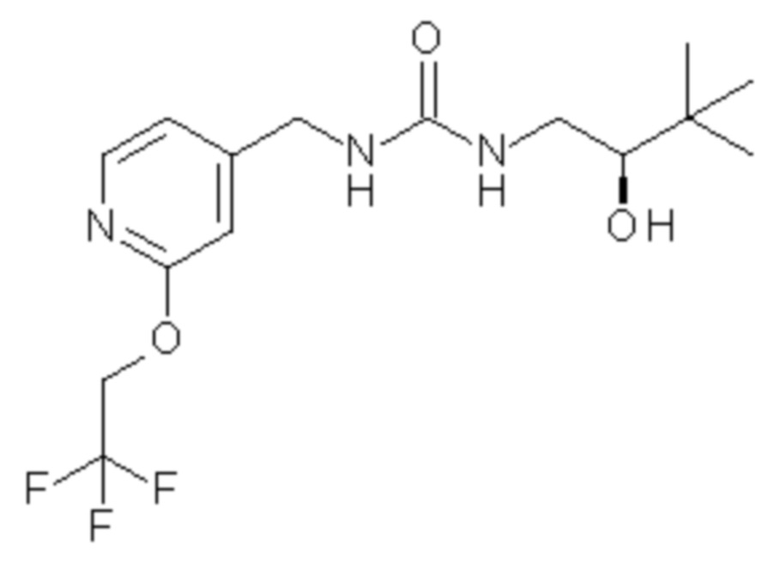
или его фармацевтически приемлемая соль.
12. Фармацевтическая композиция для лечения заболевания, вызванного изменениями возбудимости двигательных нейронов, содержащая эффективное количество соединения по любому из пп.1-11 или его фармацевтически приемлемой соли с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или наполнителями.
13. Способ лечения заболевания, вызванного изменениями возбудимости двигательных нейронов, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-11 или его фармацевтически приемлемой соли.
14. Способ по п.13, в котором заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой боковой амиотрофический склероз.
15. Способ лечения заболевания, вызванного изменениями возбудимости двигательных нейронов, включающий введение пациенту, нуждающемуся в этом, фармацевтической композиции по п.12.
16. Способ по п.15, в котором заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой боковой амиотрофический склероз.
17. Соединение по любому из пп.1-11 или его фармацевтически приемлемая соль для применения в терапии.
18. Соединение по любому из пп.1-11 или его фармацевтически приемлемая соль, предназначенное для лечения заболевания, вызванного изменениями возбудимости двигательных нейронов.
19. Соединение по п.18 или его фармацевтически приемлемая соль, где заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой боковой амиотрофический склероз.
20. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли при изготовлении лекарственного средства для лечения заболевания, вызванного изменениями возбудимости двигательных нейронов.
21. Применение соединения по п.20 или его фармацевтически приемлемой соли, где заболевание, вызванное изменениями возбудимости двигательных нейронов, представляет собой боковой амиотрофический склероз.
22. Способ приготовления фармацевтической композиции, включающий смешивание соединения по любому из пп.1-11 или его фармацевтически приемлемой соли с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или наполнителями.
23. Соединение формулы
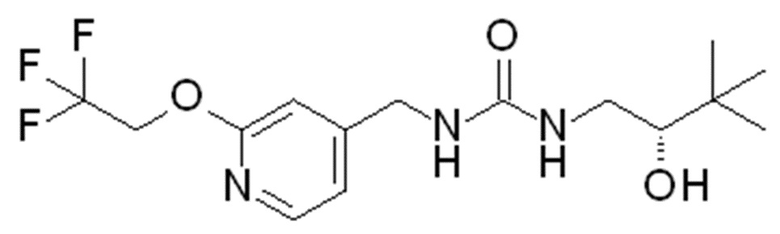
или его фармацевтически приемлемая соль.
24. Соединение формулы
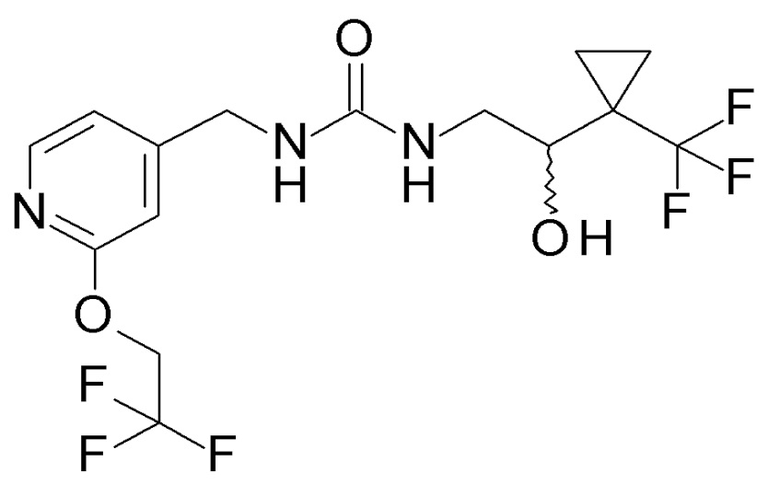
где соединение включает единственный энантиомер, который имеет (+) оптическое вращение в метаноле, или его фармацевтически приемлемую соль.
25. Соединение формулы
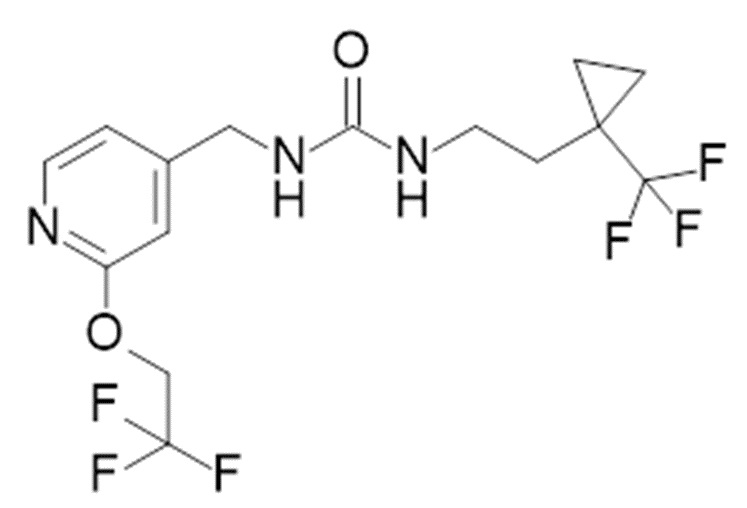
или его фармацевтически приемлемая соль.
| RU 2017117566 A, 26.11.2018 | |||
| RU 2006139933 A, 20.05.2008 | |||
| WO 2015130905 A1, 03.09.2015 | |||
| CN 103709097 A, 09.04.2014. |

