ОБЛАСТЬ ТЕХНИКИ
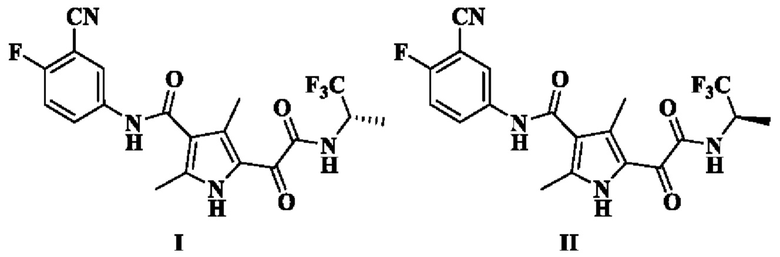
Настоящая заявка относится к соединению формулы I или II, его стереоизомеру, таутомеру, геометрическому изомеру, сольвату, активному метаболиту, гидрату, пролекарству или фармацевтически приемлемой соли, и к способу его получения, к фармацевтической композиции, содержащей его, и его применению для предупреждения или лечения заболевания (такого как заболевание, при котором полезно ингибирование сборки капсидного белка, например, в качестве лекарственного средства для лечения вирусной инфекции гепатита В).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В настоящее время хронический гепатит В невозможно вылечить, а можно только контролировать, и это ограничивается двумя типами лекарственных средств (интерфероны и нуклеозидные аналоги/ингибиторы вирусных полимераз). Более низкая частота излечения от HBV (вирус гепатита В) частично обусловлена присутствием и персистентностью ковалентно замкнутой кольцевой ДНК (кзкДНК (дезоксирибонуклеиновая кислота)) в ядре инфицированных гепатоцитов. Существующие в настоящее время протоколы лечения непригодны для удаления кзкДНК из депо, но, как ожидается, некоторые новые объекты исследования для борьбы с HBV, такие как ингибиторы ядра (такие как ингибиторы образования или сборки вирусного капсидного белка, ингибиторы кзкДНК, активаторы стимулируемых интерфероном генов и так далее) дадут надежду на излечение гепатита В (Mayur Brahmania, et al. New therapeutic agents for chronic hepatitis B).
Капсид HBV собирается из ядерного белка. Необходимо, чтобы обратная транскриптаза HBV и пгРНК (прегеномная РНК (рибонуклеиновая кислота)) были должным образом инкапсулированы перед обратной транскрипцией. Поэтому блокирование сборки капсидного белка или ускорение разрушения капсидного белка будут блокировать процесс сборки капсидного белка и тем самым воздействовать на вирусную репликацию. В последние годы исследователи начали изучать ингибиторы, направленно воздействующие на сборку капсидного белка, например, в WO 2014184350, WO 2015011281, WO 2017156255 и так далее раскрыт ряд родственных соединений. Однако большинство из них находятся на ранней стадии клинических исследований, или исследования были прекращены, и существует потребность в данной области в большем количестве альтернативных эффективных ингибиторов сборки капсидного белка для лечения, ослабления или предупреждения HBV-инфекции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы I, его стереоизомеру, таутомеру, геометрическому изомеру, сольвату, активному метаболиту, гидрату, пролекарству или фармацевтически приемлемой соли,
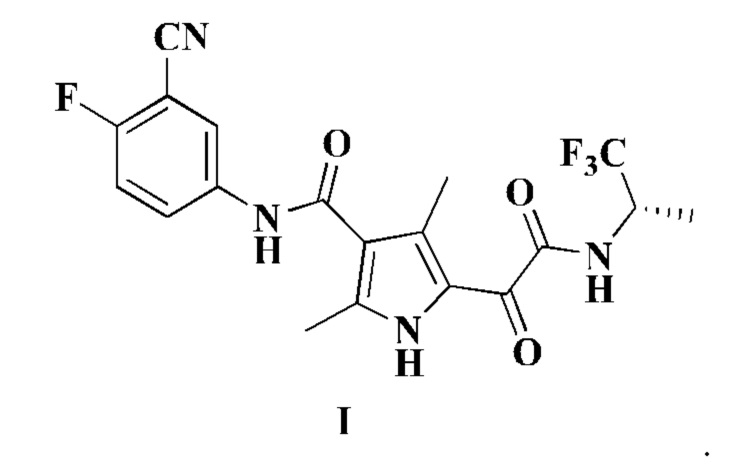
Настоящее изобретение также относится к соединению формулы II, его стереоизомеру, таутомеру, геометрическому изомеру, сольвату, активному метаболиту, гидрату, пролекарству или фармацевтически приемлемой соли,
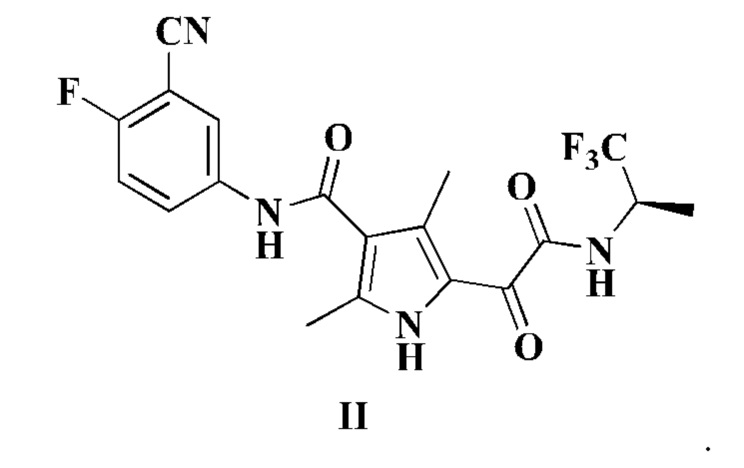
В другом аспекте согласно изобретению предложена фармацевтическая композиция, содержащая соединение формулы I или II или его фармацевтически приемлемую соль по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция по настоящему изобретению дополнительно содержит фармацевтически приемлемый эксципиент.
В другом аспекте согласно настоящему изобретению предложен способ лечения заболевания, при котором полезно ингибирование сборки капсидного белка, включающий введение млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, терапевтически эффективного количества соединения вышеуказанной формулы I или II, его фармацевтически приемлемой соли или его фармацевтической композиции.
В другом аспекте согласно настоящему изобретению также предложено применение соединения вышеуказанной формулы I или II, его фармацевтически приемлемой соли или его фармацевтической композиции в изготовлении лекарственного средства для предупреждения или лечения заболевания, при котором полезно ингибирование сборки капсидного белка.
В другом аспекте согласно настоящему изобретению предложено применение соединения вышеуказанной формулы I или II, его фармацевтически приемлемой соли или его фармацевтической композиции для предупреждения или лечения заболевания, при котором полезно ингибирование сборки капсидного белка.
В другом аспекте настоящего изобретения предложено соединение вышеуказанной формулы I или II, его фармацевтически приемлемой соли или его фармацевтической композиции для применения в предупреждении или лечении заболевания, при котором полезно ингибирование сборки капсидного белка.
В другом аспекте согласно настоящему изобретению предложена фармацевтическая композиция, содержащая соединение формулы I или II, его стереоизомер, таутомер, геометрический изомер, сольват, активный метаболит, гидрат, пролекарство или фармацевтически приемлемую соль по настоящему изобретению. В некоторых воплощениях фармацевтическая композиция по настоящему изобретению дополнительно содержит фармацевтически приемлемый эксципиент.
В другом аспекте согласно настоящему изобретению предложен способ ингибирования сборки капсидного белка, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства, фармацевтически приемлемой соли или его фармацевтической композиции по настоящему изобретению. В некоторых воплощениях указанным субъектом является млекопитающее; в некоторых воплощениях указанным субъектом является человек.
В другом аспекте согласно настоящему изобретению предложен способ предупреждения или лечения заболевания, при котором полезно ингибирование сборки капсидного белка, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства, фармацевтически приемлемой соли или его фармацевтической композиции по настоящему изобретению. В некоторых воплощениях указанным субъектом является млекопитающее; в некоторых воплощениях указанным субъектом является человек.
В другом аспекте согласно настоящему изобретению предложено применение соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства или фармацевтически приемлемой соли или его фармацевтической композиции по настоящему изобретению для ингибирования сборки капсидного белка.
В другом аспекте согласно настоящему изобретению также предложено применение соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства, фармацевтически приемлемой соли или его фармацевтической композиции по настоящему изобретению в изготовлении лекарственного средства для ингибирования сборки капсидного белка.
В другом аспекте согласно настоящему изобретению также предложено применение соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства, фармацевтически приемлемой соли или его фармацевтической композиции по настоящему изобретению в изготовлении лекарственного средства для предупреждения или лечения заболевания, при котором полезно ингибирование сборки капсидного белка.
В другом аспекте согласно настоящему изобретению также предложено применение соединения формулы I или II, его стереоизомера, таутомера, геометрического изомера, сольвата, активного метаболита, гидрата, пролекарства или фармацевтически приемлемой соли, или его фармацевтической композиции для предупреждения или лечения заболевания, при котором полезно ингибирование сборки капсидного белка.
В другом аспекте согласно настоящему изобретению предложено соединение формулы I или II, его стереоизомер, таутомер, геометрический изомер, сольват, активный метаболит, гидрат, пролекарство или фармацевтически приемлемая соль, или его фармацевтическая композиция по настоящему изобретению для применения в ингибировании сборки капсидного белка.
В другом аспекте согласно настоящему изобретению предложено соединение вышеуказанной формулы I или II, его стереоизомер, таутомер, геометрический изомер, сольват, активный метаболит, гидрат, пролекарство, фармацевтически приемлемая соль или его фармацевтическая композиция для применения в предупреждении или лечении заболевания, при котором полезно ингибирование сборки капсидного белка.
В некоторых воплощениях настоящего изобретения заболевание, при котором полезно ингибирование сборки капсидного белка, представляет собой заболевание, вызванное инфицированием вирусом гепатита В (HBV).
В некоторых воплощениях настоящего изобретения заболевание, при котором полезно ингибирование сборки капсидного белка, представляет собой заболевание печени, вызванное инфицированием вирусом гепатита В (HBV).
В некоторых воплощениях настоящего изобретения лечение заболевания, при котором полезно ингибирование сборки капсидного белка, заключается в контролировании, сокращении или устранении HBV для предупреждения, ослабления или излечения заболевания печени у инфицированного пациента.
Определения
Если не оговорено особо, термины и фразы, использованные здесь, имеют нижеследующие значения. Конкретный термин или фразу не следует рассматривать как неопределенную или неясную, когда это конкретно не определено, а следует понимать в соответствии с ее обычным значением. Торговые наименования, использованные здесь, относятся к соответствующим продуктам или их ингредиентам.
Термин "возможный" или "возможно" означает, что описываемое затем событие или ситуация может происходить или нет, и описание включает случаи, когда событие или ситуация имеет место, и случаи, когда событие или ситуация не возникает.Например, этильная группа, "возможно" замещенная галогеном, означает, что этильная группа может быть незамещенной (СН2СН3), монозамещенной (например, CH2CH2F), полизамещенной (например, CHFCH2F, CH2CHF и так далее) или полностью замещенной (CF2CF3). Специалисту в данной области техники будет понятно, что для любой группы, содержащей один или более чем один заместитель, не предусматривается модель незамещения или замещения, которая стерически невозможна и/или которая не может быть синтезирована.
Термин "проведение лечения" или "лечение" относится к введению соединения или препарата, описанного в настоящем изобретения, для ослабления или устранения заболевания или одного или более чем одного симптома, связанного с заболеванием, и включает:
I) подавление заболевания или болезненного состояния, то есть сдерживание его развития;
II) облегчение заболевания или болезненного состояния, то есть регрессирование заболевания или болезненного состояния.
Термин "предупреждение" означает введение соединения или препарата по настоящему изобретению, для предупреждения заболевания или одного или более чем одного симптома, связанного с заболеванием, и включает: предупреждение возникновения заболевания или болезненного состояния у млекопитающего, в частности, когда такое млекопитающее предрасположено к болезненному состоянию, но еще не диагностировано, что страдает болезненным состоянием.
Термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению для (I) лечения или предупреждения конкретного заболевания, состояния или расстройства, (II) облегчения, ослабления или устранения одного или более чем одного симптома конкретного заболевания, состояния или расстройства или (III) предупреждения или задержки возникновения одного или более чем одного симптома заболевания, состояния или расстройства, описанных здесь. "Терапевтически эффективное количество" соединения согласно настоящему изобретению варьирует в зависимости от соединения, болезненного состояния и его тяжести, режима введения и возраста млекопитающего, которого лечат, но может быть определено согласно обычной практике специалистами в данной области техники в соответствии с их собственными знаниями и настоящим описанием изобретения.
Термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые в рамках здравого медицинского суждения являются подходящими для применения в контакте с тканями человека и животного без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, и соизмеримы с разумным соотношением польза/риск.
В качестве фармацевтически приемлемой соли могут быть упомянуты, например, соль металла, соль аммония, соль органического основания, соль неорганической кислоты, соль органической кислоты, соль основной или кислой аминокислотой, или тому подобные.
Термин "фармацевтическая композиция" относится к смеси одного или более чем одного соединения по настоящему изобретению или его соли и фармацевтически приемлемого эксципиента. Назначение фармацевтической композиции заключается в облегчении введения соединений по настоящему изобретению субъекту.
Термин "сольват" относится к веществу, образованному путем объединения соединения по настоящему изобретению с фармацевтически приемлемым растворителем. Фармацевтически приемлемые растворители включают воду, этанол, уксусную кислоту и тому подобное. Сольваты включают стехиометрические сольваты и нестехиометрические сольваты.
Термин "гидрат" относится к сольвату, содержащему раскрытое или заявленное соединение и стехиометрическое или нестехиометрическое количество воды.
Соединения по настоящему изобретению также могут быть получены в виде пролекарств, таких как фармацевтически приемлемые пролекарства. Поскольку известно, что пролекарства улучшают многие желательные свойства лекарственного средства (например, растворимость, биодоступность, приготовление лекарственной формы и так далее), соединения по настоящему изобретению могут быть доставлены в форме пролекарства. Соответственно, предусмотрено, что настоящее изобретение включает пролекарства заявленных здесь соединений, способы их доставки и композиции, содержащие пролекарства.
Термин "пролекарство" включает любое ковалентно связанный носитель, который при введении субъекту-млекопитающему высвобождает активное исходное лекарственное средство по настоящему изобретению in vivo. Пролекарства по настоящему изобретению получают путем модифицирования функциональной группы, присутствующей в соединении, таким образом, что модифицированная группа расщепляется с образованием исходного соединения обычной манипуляцией или in vivo.
В настоящей заявке термин "субъект" включает людей и животных, например, млекопитающих (например приматов, коров, лошадей, свиней, собак, кошек, мышей, крыс, кроликов, коз, овец, птиц и так далее).
Термин "активный метаболит" относится к биологически активному производному соединения, которое образуется, когда соединение метаболизируется.
Термин "фармацевтически приемлемый эксципиент" относится к тем эксципиентам, которые не оказывают значительного раздражающего воздействия на организм и не ухудшают биологическую активность и свойства активного соединения. Специалистам в данной области техники хорошо известны подходящие эксципиенты, такие как углеводы, воски, водорастворимые и/или водонабухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, вода и тому подобное.
Слово "содержать" и его варианты, такие как "содержит" или "содержащий" следует понимать в открытом, неисключающем смысле, то есть "включающий, но не ограничивающийся ими".
Соединения и промежуточные соединения по настоящему изобретению могут также существовать в разных таутомерных формах, и все такие формы охвачены объемом настоящей заявки. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с разными энергиями, которые способны к взаимопревращению через низкий энергетический барьер. Например, протонные таутомеры (также известные как таутомеры протонного переноса) включают взаимопревращения посредством переноса протона, такие как кето-енольная и имин-енаминная изомеризация. Конкретным примером протонного таутомера является имидазольная группировка, в которой протон может мигрировать между двумя кольцевыми атомами азота. Валентные таутомеры включают таутомеры перегруппировки за счет некоторых связеобразующих электронов.
Некоторые соединения по настоящему изобретению могут иметь асимметрические атомы углерода (стереоцентры) или двойные связи. Таким образом, рацематы, диастереомеры, энантиомеры, геометрический изомеры и отдельные изомеры входят в объем настоящего изобретения.
Если не оговорено особо, когда соединения по настоящему изобретению содержат олефиновые двойные связи или другие центры геометрической асимметрии, они включают Е и Z геометрические изомеры.
Соединения по настоящему изобретению могут существовать в конкретной геометрический форме или стереоизомерной форме. Настоящее изобретение предусматривает все такие соединения, включая таутомеры, цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (8)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры и их рацемические смеси, и другие смеси, такие как энантиомерно- или диастереомерно-обогащенные смеси, все из которых входят в объем настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в таких заместителях, как алкил и так далее. Все эти изомеры и их смеси входят в объем настоящего изобретения.
Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры могут быть получены в результате хирального синтеза или с использованием хиральных реагентов, или посредством других обычных способов. Энантиомер определенного соединения по настоящему изобретению может быть получен посредством асимметрического синтеза или дериватизации с использованием хирального вспомогательного вещества, где полученную диастереомерную смесь разделяют, и вспомогательную группу отщепляют с получением чистых требуемых энантиомеров. Альтернативно, когда молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксил), образуют соль диастереомера с подходящей оптически активной кислотой или основанием и затем проводят разделение диастереомеров посредством обычного способа, хорошо известного в данной области техники, затем выделяют чистый энантиомер. Кроме того, разделение энантиомеров и диастереомеров обычно осуществляют с использованием хроматографии с хиральной стационарной фазой и возможно в комбинации с методом химической дериватизации (например образованием карбаматов из аминов).
Настоящее изобретение также включает меченые изотопами соединения по настоящему изобретению, которые идентичны соединениям, описанным здесь, но в которых один или более чем один атом заменен атомами, имеющими атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть введены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, иода и хлора, такие как 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O,31Р, 32Р, 35S, 18F, 123I, 125I, 36Cl и тому подобные, соответственно.
Некоторые меченные изотопами соединения по настоящему изобретению (такие как соединения, меченные 3Н и 14С) могут быть использованы в анализах распределения соединения и/или субстрата в тканях. Изотопы дейтерий (то есть 3Н) и углерод-14 (то есть 14С) особенно предпочтительны благодаря легкости их получения и детектирования. Позитрон-излучающие изотопы, такие как 15O, 13N, 11С и 18F, могут быть использованы в исследованиях методом позитронно-эмиссионной томографии (ПЭТ) для определения занятости субстратом. Меченные изотопами соединения по настоящему изобретению обычно могут быть получены посредством замены немеченого реагента реагентом, меченным изотопом, согласно методикам, аналогичным методикам, раскрытым на схемах и/или в примерах, раскрытых ниже.
Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (то есть 2Н), может давать некоторые терапевтические преимущества, обусловленные более высокой метаболической стабильностью (например увеличенный период полувыведения in vivo или сниженные требования к дозировке), и поэтому может быть предпочтительным в некоторых случаях, причем замещение дейтерием может быть частичным или полным, и частичное замещение дейтерием означает, что по меньшей мере один атом водорода замещен по меньшей мере одним атомом дейтерия, и все такие формы соединений охвачены объемом настоящего изобретения.
Фармацевтическая композиция по настоящему изобретению может быть получена посредством объединения соединения по настоящему изобретению с подходящим фармацевтически приемлемым эксципиентом и может быть приготовлена в виде, например, твердых, полутвердых, жидких или газообразных препаратов, таких как таблетки, пилюли, капсулы, порошки, гранулы, пасты, эмульсии, суспензии, суппозитории, инъекции, средства для ингаляций, гели, микросферы, аэрозоли и тому подобное.
Типичные пути введения соединения по настоящему изобретению или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей их, включают пероральное, ректальное, местное, ингаляционное, парентеральное, сублингвальное, интравагинальное, интраназальное, внутриглазное, интраперитонеальное, внутримышечное, подкожное или внутривенное введение, но не ограничиваются ими.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена посредством общеизвестных в данной области техники способов, таких как традиционный способ смешивания, способ растворения, способ гранулирования, способ изготовления пилюль с сахарным покрытием, способ измельчения, способ эмульгирования и способ сублимационной сушки и так далее.
В некоторых воплощениях фармацевтическая композиция находится в форме для перорального введения. Для перорального введения активное соединение может быть смешано с фармацевтически приемлемыми носителями, общеизвестными в данной области техники, с получением фармацевтической композиции. С этими эксципиентами соединения по настоящему изобретению могут быть приготовлены в виде таблеток, пилюль, облаток, драже, капсул, жидкостей, гелей, сиропа или суспензий и тому подобного для перорального введения пациентам.
Твердая пероральная композиция может быть получена посредством традиционного способа смешивания, заполнения или таблетирования. Например, она может быть получена посредством следующего способа: смешивание активного соединения с твердым эксципиентом; возможно измельчение полученной смеси; добавление других подходящих эксципиентов при необходимости; и затем переработка смеси в гранулы с получением ядер таблеток или драже. Подходящие эксципиенты включают связующие вещества, разбавители, разрыхлители, смазывающие вещества, скользящие агенты, подсластители и/или ароматизаторы и так далее, но не ограничиваются ими.
Фармацевтическая композиция также подходит для парентерального введения, такого как в виде стерильных растворов, суспензий или подвергнутых сублимационной сушке продуктов в соответствующей стандартной лекарственной форме.
Терапевтическая доза соединений по настоящему изобретению может быть определена в соответствии, например, с конкретно применяемым лечением, путем введения соединения, здоровьем и состоянием пациента и заключением лечащего врача. Доля или концентрация соединений по настоящему изобретению в фармацевтической композиции может изменяться в зависимости от множества факторов, включая дозировку, химические свойства (например гидрофобность) и путь введения. Например, соединение по настоящему изобретению может быть представлено в физиологически забуференном водном растворе, содержащем примерно от 0,1 до 10% (мас./об.) соединения, для парентерального введения. Некоторые типичные дозы находятся в диапазоне от примерно 1 мкг/кг до примерно 1 г/кг массы тела в сутки. В некоторых воплощениях диапазон доз составляет от примерно 0,01 мг/кг до примерно 100 мг/кг массы тела в сутки. Дозировка, по всей вероятности, будет зависеть от таких переменных, как тип и степень прогрессирования заболевания или состояния, общее состояние здоровья конкретного пациента, относительной биологической эффективности выбранного соединения, состава эксципиента и пути введения. Эффективная доза может быть получена путем экстраполяции из кривой зависимости «доза-эффект», полученной в результате тестирования in vitro или в системе тестирования на животных.
Соединения по настоящему изобретению могут быть получены посредством различных способов синтеза, хорошо известных специалистам в данной области техники, включая конкретные воплощения, перечисленные ниже, воплощения, полученные путем комбинирования конкретных воплощений с другими способами химического синтеза и эквивалентными альтернативами, известными специалистам в данной области техники. Предпочтительные воплощения включают демонстрационные примеры по настоящему изобретению, но не ограничиваются ими.
Химическое взаимодействие в соответствии с конкретным воплощением по настоящему изобретению проводят в подходящем растворителе, который должен быть подходящим для химических превращений по настоящему изобретению и реагентов и веществ, необходимых в настоящей заявке. Для того чтобы получить соединения по настоящему изобретению, специалисту в данной области техники иногда необходимо модифицировать или выбрать стадию синтеза или реакционный процесс, основываясь на существующих воплощениях.
Важным фактором при разработке путей синтеза в данной области техники является выбор подходящей защитной группы для реакционноспособной функциональной группы (такой как аминогруппа в данной заявке); например, можно сделать ссылку на Greene's Protective Groups in Organic Synthesis (4th Ed). Hoboken, New Jersey: John Wiley & Sons, Inc. Все источники информации, процитированные в настоящей заявке, включены в нее во всей своей полноте.
В настоящей заявке использованы следующие сокращения:
ЕА означает этилацетат; МеОН означает метанол; DMF означает N,N-диметилформамид; HATU означает гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; DIPEA означает N,N-диизопропилэтиламин; РО означает пероральное введение; IV означает внутривенную инъекцию; DMSO означает диметилсульфоксид.
Для ясности настоящая заявка дополнительно проиллюстрирована следующими примерами, но эти примеры не ограничивают объем настоящей заявки. Все реагенты, использованные в данной заявке, имеются в продаже и могут быть использованы без дополнительной очистки.
Примеры
Ядерный магнитный резонанс (ЯМР) по настоящему изобретению регистрировали на спектрометре ядерного магнитного резонанса BRUKER-300 и BRUKER-500, и тетраметилсилан (TMS = δ 0.00) использовали в качестве внутреннего стандарта химического сдвига, и данные ядерного магнитного резонанса записывали в следующем виде: число протонов, тип пика(s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет), константа взаимодействия (в Гц). АВ SCTEX Triple TOF 4600 или АВ SCTEX 3200QTRAP использовали в качестве прибора для масс-спектрометрии.
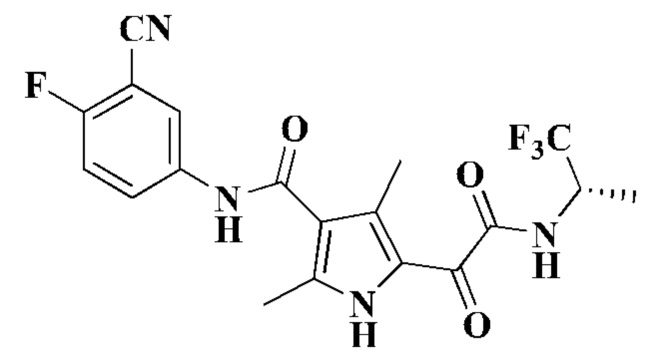
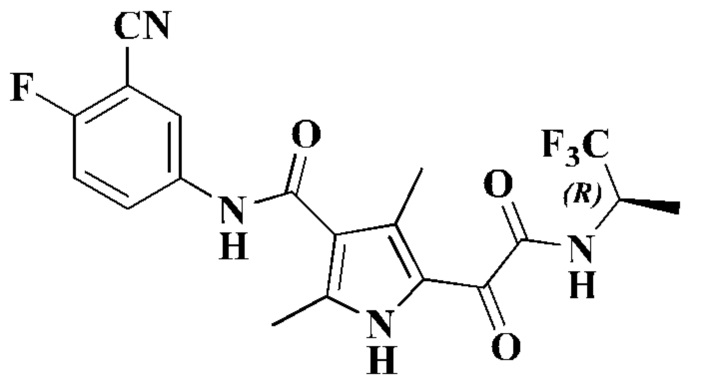
Пример 1
(S)-N-(3-Циано-4-фторфенил)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)-амино)ацетил)-1Н-пиррол-3-карбоксамид
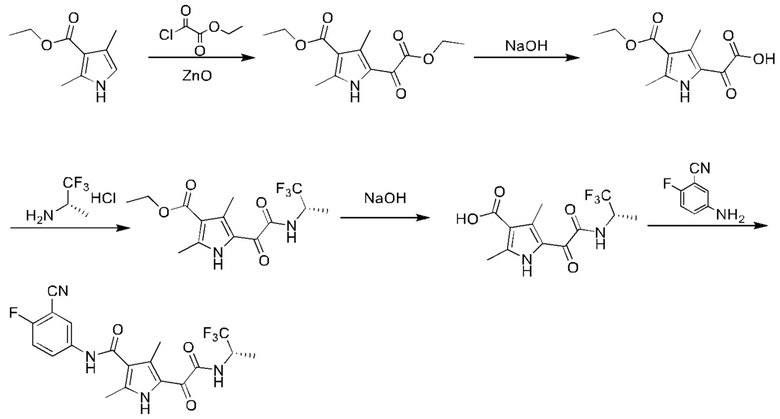
Стадия А: этил-2-хлор-2-оксоацетат (40,8 г) и оксид цинка (1,22 г) последовательно добавляли в реакционную колбу на ледяной бане в защитной атмосфере N2 с последующим добавлением этил-2,4-диметил-1Н-пиррол-3-карбоксилата (5 г). После добавления смесь перемешивали в течение 10 минут на ледяной бане, а затем перемешивали при комнатной температуре после снятия с ледяной бани. После завершения взаимодействия реакционный раствор медленно по каплям добавляли в 200 мл смеси воды со льдом с последующим добавлением ЕА (200 мл). Полученную смесь разделяли. Органическую фазу сушили над безводным сульфатом натрия, концентрировали и подвергали колоночной хроматографии с получением этил-5-(2-этокси-2-оксоацетил)-2,4-диметил-1Н-пиррол-3-карбоксилата (4,5 г). МС (масс-спектрометрия) (ИЭР+ (ионизация электрораспылением), [M+Na]+) m/z: 290,07.
Стадия В: этил-5 -(2-этокси-2-оксоацетил)-2,4-диметил-1Н-пиррол-3-карбоксилат (3,5 г) и МеОН (40 мл) последовательно добавляли в реакционную колбу. Затем по каплям добавляли раствор гидроксида натрия (1,05 г) в воде (20 мл) на ледяной бане, и смесь перемешивали при комнатной температуре. После завершения взаимодействия рН водной фазы доводили 2 н водным раствором соляной кислоты до значения 3-4 и затем экстрагировали ЕА (100 мл × 2). Органическую фазу промывали водой (30 мл) и концентрировали с получением 2-(4-(этоксикарбонил)-3,5-диметил-1Н-пиррол-2-ил)-2-оксоуксусной кислоты (2,7 г). МС (ИЭР-, [М-Н]-) m/z: 238,1.
Стадия С: 2-(4-(этоксикарбонил)-3,5-диметил-1 Н-пиррол-2-ил)-2-оксоуксусную кислоту (1 г), DMF (20 мл), HATU (2,07 г) и DIPEA (1,08 г) последовательно добавляли в реакционную колбу при комнатной температуре. После добавления реакционный раствор перемешивали при комнатной температуре в течение 10 минут с последующим добавлением гидрохлорида (S)-1,1,1-трифторпропан-2-амина (0,63 г). После завершения взаимодействия реакционный раствор выливали в 50 мл воды и затем экстрагировали ЕА (50 мл × 3). Органическую фазу промывали насыщенным водным раствором сульфата натрия (50 мл × 3), сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат собирали, концентрировали и затем очищали посредством колоночной хроматографии с получением этил-(S)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоксилата (0,5 г). МС (ИЭР-, [М-Н]-) m/z: 333,4.
Стадия D: этил-(S)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)-ацетил)-1 Н-пиррол-3-карбоксилат (300 мг) и МеОН (2 мл) добавляли в реакционную колбу с последующим добавлением раствора NaOH (72 мг) в воде (1 мл). После добавления реакционный раствор нагревали до 80°С и подвергали взаимодействию в течение ночи. После завершения взаимодействия полученную смесь концентрировали и затем добавляли в нее воду (20 мл) и ЕА (60 мл). Водный слой разделяли. Органическую фазу промывали водой (30 мл), и слои разделяли. Водные фазы объединяли, рН доводили 2 н соляной кислотой до значения приблизительно 3, экстрагировали посредством добавления ЕА (100 мл × 2), и затем слои разделяли. Органическую фазу концентрировали с получением (S)-2,4-диметил-5- (2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоновой кислоты (230 мг). МС (ИЭР-,[М-Н]-) m/z: 305,4.
Стадия Е: (S)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1H-пиррол-3-карбоновую кислоту (230 мг), DMF (5 мл), HATU (428 мг) и DIPEA (194 мг) последовательно добавляли в реакционную колбу при комнатной температуре. После добавления реакционный раствор перемешивали в течение 10 минут с последующим добавлением 5-амино-2-фторбензонитрила (123 мг). Полученную смесь нагревали до 40°С и перемешивали для проведения взаимодействия в течение 20 часов. После завершения взаимодействия добавляли воду (20 мл) и ЕА (60 мл), и смесь разделяли. Органический слой сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат собирали, подвергали выпариванию на ротационном испарителе досуха, отбирали пробу и очищали посредством колоночной хроматографии с получением (S)-N-(3-циано-4-фторфенил)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоксамида (180 мг). 1Н ЯМР (500 МГц, DMSO-d6) δ 12.05 (s, 1H), 10.20 (s, 1H), 9.49 (d, J=9,0 Гц, 1H), 8.20 (dd, J=6,0, 2,5 Гц, 1H), 7.98-7.91 (m, 1Н),7.53 (t, J=9,0 Гц, 1H), 4.78-4.67 (m, 1Н),2.40 (s, 3Н), 2.32 (s, 3Н), 1.34 (d, J=7,0 Гц, 3Н). МС (ИЭР-, [М-Н]-) m/z: 423,0.
Пример 2
(R)-N-(3-Циано-4-фторфенил)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)-амино)ацетил)-1Н-пиррол-3-карбоксамид
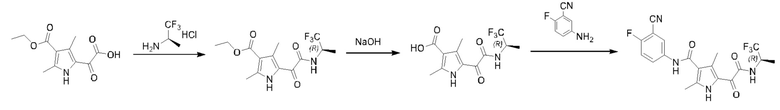
Стадия А: в соответствии с примером 1, гидрохлорид (S)-1,1,1-трифторпропан-2-амина заменяли на гидрохлорид (R)-1,1,1-трифторпропан-2-амина на стадии С с получением этил-(R)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1H-пиррол-3-карбоксилата. МС (ИЭР-, [М-Н]-) m/z: 333,2.
Стадия В: в соответствии с примером 1, этил-(S)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоксилат заменяли на этил-(R)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоксилат на стадии D с получением (R)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)-амино)ацетил)-1Н-пиррол-3-карбоновой кислоты. МС (ИЭР-, [М-Н]-) m/z: 305,4.
Стадия С: в соответствии с примером 1, (S)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоновую кислоту заменяли на (R)-2,4-диметил-5-(2-оксо-2-((1,1,1-трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоновую кислоту на стадии Е с получением (R)-N-(3-циано-4-фторфенил)-2,4-диметил-5-(2-оксо-2-((1,1,1 -трифторпроп-2-ил)амино)ацетил)-1Н-пиррол-3-карбоксамида. 1Н ЯМР (500 МГц, DMSO-d6): δ 12.03 (s, 1H), 10.19 (s, 1H), 9.48 (d, J=8,5 Гц, 1H), 8.20 (dd, J=6,0, 2,5 Гц, 1H), 8.00-7.90 (m, 1H), 7.52 (t, J=9,0 Гц, 1H), 4.90-4.65 (m, 1H), 2.40 (s, 3Н), 2.33 (s, 3Н), 1.34 (d, J=7,5 Гц, 3Н). МС (ИЭР-, [М-Н]-) m/z: 423,2.
Экспериментальный пример 1: исследование активности in vitro
1.1. Ингибирующая активность в отношении ДНК HBV в клетках in vitro Брали флакон с клетками HepG2.2.15 (Wuhan Institute of Virology) или HepAD38 в стадии хорошего экспоненциального роста, однократно промывали посредством добавления 5 мл PBS (забуференный фосфатами физиологический раствор) и затем туда добавляли 3 мл трипсина. После ферментативного расщепления при комнатной температуре в течение 5 минут 2 мл трипсина удаляли, и затем образец помещали в инкубатор для клеточных культур и подвергали ферментативному расщеплению в течение 10 минут. Клетки извлекали и наблюдали под микроскопом (были ли клетки разъединенными и круглыми, и не было ли адгезии между клетками). Добавляли 10 мл полной среды для остановки ферментативного расщепления. После пипетирования в суспензию отдельных клеток 10 мкл клеточной суспензии отбирали для подсчета клеток с использованием счетчика клеток и затем разводили в полной среде и доводили до плотности клеток 1×105 клеток/мл. Затем клеточную суспензию высевали в количестве 1 мл на лунку в 24-луночный планшет с помощью многоканальной пипетки (24-луночный планшет предварительно покрывали раствором коллагена I типа в концентрации 50 мкг/мл) и культивировали при постоянной температуре в СО2 инкубаторе в течение 48 часов.
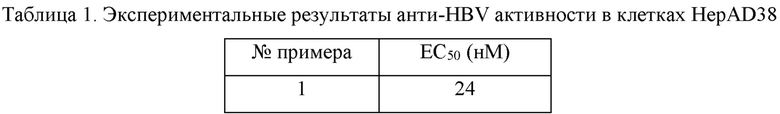
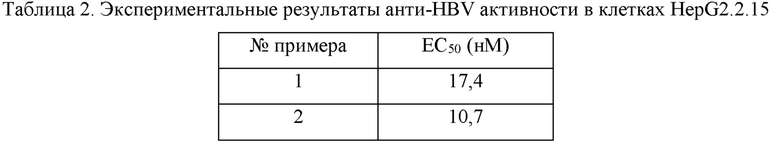
Соединения, растворенные в DMSO, подвергали двукратному серийному разведению (всего 10 концентраций) в полной среде. Добавляли соединение, и каждые 72 часа использовали свежую среду, содержащую соединение, для замены истощенной среды. Клетки обрабатывали соединением в течение 6 суток. После сифонирования надосадочной жидкости 300 мкл лизата (10 мМ трис-HCl, 1 мМ EDTA (этилендиаминтетрауксусная кислота) и 1% NP-40 (детергент)) добавляли в каждую лунку. После лизирования при комнатной температуре в течение 10 минут ДНК экстрагировали, и ДНК HBV во внутриклеточном вирусном капсиде измеряли с помощью метода флуоресцентной количественной ПЦР (полимеразная цепная реакция) в режиме реального времени. Скорость ингибирования вычисляли на основании величины Ct, а величину ЕС50 вычисляли с помощью четырехпараметрического метода. Результаты показаны в таблицах 1 и 2.
1.2. Цитотоксичность in vitro
Брали флакон с клетками HepG2.2.15 (Wuhan Institute of Virology) в стадии хорошего экспоненциального роста, однократно промывали посредством добавления 5 мл PBS и затем туда добавляли 2 мл трипсина. Образец подвергали ферментативному расщеплению в инкубаторе для клеточных культур, и периодически извлекали, и наблюдали под микроскопом. Как только клетки распадались, 1 мл трипсина удаляли. Остаточную жидкость помещали в инкубатор для клеточных культур при 37°С и подвергали ферментативному расщеплению в течение 8-15 минут. Клетки извлекали и наблюдали под микроскопом (были ли клетки разъединенными и круглыми и не было ли адгезии между клетками). 5 мл среды MEM (минимальная поддерживающая среда) добавляли для ресуспендирования клеток. Затем клетки подсчитывали с помощью счетчика клеток, разводили в полной среде и доводили до плотности клеток 2×105 клеток/мл. Затем клетки высевали в количестве 100 мкл на лунку в 96-луночный планшет с помощью многоканальной пипетки (96-луночный планшет предварительно покрывали раствором коллагена I типа в концентрации 50 мкг/мл) и культивировали при постоянной температуре в СО2 инкубаторе в течение 24 часов. Клетки обрабатывали путем введения лекарственного средства, и свежую среду, содержащую соединение, использовали для замены истощенной среды каждые 3 суток. Для контроля в лунки добавляли не содержащую лекарственного средства среду, но содержащую 0,5% DMSO, и создавали контрольную лунку, содержащую обычную среду. Через 6 суток после введения добавляли ССК-8 в количестве 10 мкл на лунку. Через 1-2 часа регистрировали поглощение с помощью ридера для микропланшет при 450 нм и вычисляли степень ингибирования и СС50. Результаты показаны в таблице 3.

1.3. Изучение ингибирования фермента СУР450
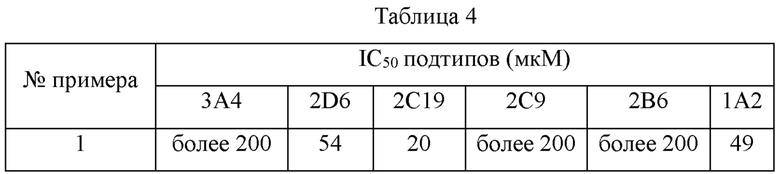
500 мкл конечной инкубационной системы содержат 50 мкл микросом печени человека (концентрация белка: 0,2 мг/мл, Corning), 1 мкл смешанных специфичных субстратов СУР450 (CYP1A2, CYP 2В6, CYP 2С9, CYP2C19, CYP 2D6 и CYP 3А4), 398 мкл PBS буфера (рН 7,4), 1 мкл специфичного положительного ингибитора (положительная контрольная группа) или тестируемого соединения (приготовленного с ацетонитрилом) и 50 мкл NADPH (никотинамидадениндинуклеотидфосфат восстановленный) + MgCl2. Дублированные инкубационные системы по 0,5 мл каждая были приготовлены для каждого подтипа СУР450. Общий объем 450 мкл однородно перемешанного раствора субстрата и фермента был приготовлен в каждой пробирке, и раствор и NADPH предварительно инкубировали при 37°С в течение 5 минут соответственно. Затем 50 мкл смешанного раствора NADPH + MgCl2 добавляли для осуществления взаимодействия. 50 мкл реакционного раствора отбирали через 30 минут, и взаимодействие останавливали с помощью 300 мкл ледяного ацетонитрила, содержащего внутренний стандарт. Дополнительно, две контрольные группы по 500 мкл каждая без NADPH были приготовлены параллельно в качестве отрицательной контрольной группы.
Предварительная обработка образцов: к 50 мкл инкубированного образца добавляли 300 мкл ледяного ацетонитрила, содержащего внутренний стандарт, и затем осаждали. После перемешивания на вортексе в течение 5 минут образец центрифугировали (12000 об/мин, 4°С) в течение 10 минут. Пипетировали 75 мкл надосадочной жидкости, и 75 мкл ультрачистой воды добавляли в нее для разведения и смешивали до однородного состояния. 1 мкл полученного раствора впрыскивали для анализа. Результаты показаны в таблице 4.
1.4. Анализ связывания белков плазмы крови
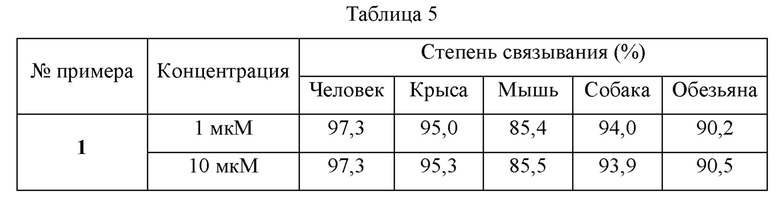
Приготовление образцов плазмы крови: отбирали 495 мкл пустой плазмы соответствующего вида (мыши, крысы, собаки, обезьяны и человека), соответственно, и в них добавляли 5 мкл раствора соответствующего тестируемого соединения или положительного контроля с получением растворов образцов плазмы, имеющих концентрацию в плазме лекарственного соединения 1 мкМ и 10 мкМ соответственно (приготовленного с ацетонитрилом).
Предварительно обработанную диализную мембрану помещали в устройство для равновесного диализа с высокой пропускной способностью, и отбирали по 100 мкл раствора образца плазмы и буферного раствора PBS, и добавляли соответственно с обеих сторон диализной мембраны (сторона образца и сторона буфера) (n=3). После того, как устройство уравновешивания было герметизировано пленкой, его инкубировали при 37°С в течение ночи (100 об/мин). После достижения уравновешивания диализа образцы по 50 мкл отбирали со стороны образца и со стороны буфера соответственно, и взаимодействие останавливали посредством добавления ледяного ацетонитрила, содержащего внутренний стандарт.
Предварительная обработка образцов: К 50 мкл образца со стороны плазмы добавляли 450 мкл ледяного ацетонитрила, содержащего внутренний стандарт, и затем осаждали. После перемешивания на вортексе в течение 5 минут образец центрифугировали (12000 об/мин, 4°С) в течение 10 минут.75 мкл надосадочной жидкости пипетировали, и 75 мкл ультрачистой воды добавляли к ней для разведения и смешивали до однородного состояния. 1 мкл полученного раствора впрыскивали для анализа. К 50 мкл образца со стороны PBS буфера добавляли 250 мкл ледяного ацетонитрила с внутренним стандартом и осаждали. После перемешивания на вортексе в течение 5 минут образец центрифугировали (12000 об/мин, 4°С) в течение 10 мин. 75 мкл надосадочной жидкости пипетировали, и 75 мкл ультрачистой воды добавляли к ней для разведения и смешивали до однородного состояния. 2 мкл полученного раствора впрыскивали для анализа. Результаты показаны в таблице 5.
Экспериментальный пример 2: стабильность микросом печени in vitro
300 мкл конечной инкубационной системы содержат 30 мкл микросом печени (концентрация белка: 0,15 мг/мл, Corning), 30 мкл NADPH + MgCl2, 3 мкл субстрата (приготовленного с ацетонитрилом) и 237 мкл PBS буфера. Дублированные инкубационные системы по 0,3 мл каждая были приготовлены для каждого вида. Общий объем 270 мкл однородно перемешанного раствора субстрата и фермента был приготовлен в каждой пробирке, раствор и NADPH предварительно инкубировали при 37°С в течение 5 минут соответственно. Затем 30 мкл смешанного раствора NADPH + MgCl2 добавляли для осуществления взаимодействия. 50 мкл реакционного раствора отбирали через 0, 10, 30 и 60 минут, и взаимодействие останавливали с помощью 300 мкл ледяного ацетонитрила, содержащего внутренний стандарт.
Предварительная обработка образцов: к 50 мкл инкубированного образца добавляли 300 мкл ледяного ацетонитрила, содержащего диазепам в качестве внутреннего стандарта, и затем осаждали. После перемешивания на вортексе в течение 5 минут образец центрифугировали (12000 об/мин, 4°С) в течение 10 минут.75 мкл надосадочной жидкости пипетировали в 96-луночный планшет, и затем разбавляли с помощью 75 мкл ультрачистой воды и смешивали до однородного состояния. 0,5 мкл полученного раствора впрыскивали и анализировали методом ЖХ-МС/МС (жидкостная хроматография-тандемная масс-спектрометрия). Результаты показаны в таблице 6.


Экспериментальный пример 3: эффективность на животных in vivo
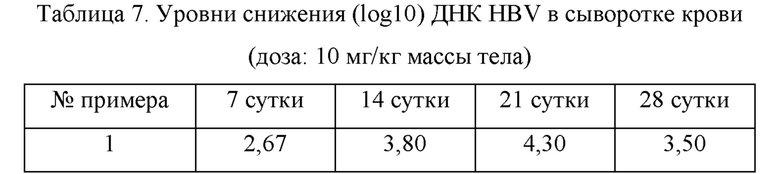
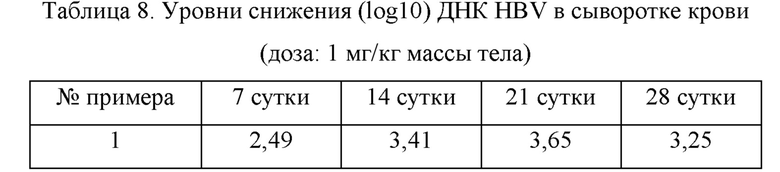
3.1. Оценка противовирусного эффекта в мышиной модели AAV (адено-ассоциированный вирус)
Самцам мышей C57BL/6 (Shanghai Lingchang Biotechnology Co., Ltd.) в возрасте 6-8 недель вводили вирус rAAV8-1.3HBV (Beijing FivePlus Gene Technology Ltd., подтип adr) посредством инъекции в хвостовую вену в дозе 1×1011 гв (геномов вектора). Кровь собирали из глазниц мышей через 2 и 4 недели после введения вируса. Выделяли сыворотку и определяли уровни экспрессии HBeAg и HBsAg в сыворотке и число копий ДНК HBV, чтобы оценить, успешно ли создана модель. Согласно результатам количественного определения серологических HBeAg, HBsAg и ДНК HBV были отобраны мыши, имеющие уровень экспрессии ДНК HBV выше 1x104 МЕ/мл, уровень экспрессии HBeAg выше 1×103 NCU (национальная клиническая единица)/мл и уровень экспрессии HBsAg выше 1×103 нг/мл. Мышей распределяли в группу пустого контроля, группу контроля-носителя и группу тестируемых соединений. Мышам в каждой группе непрерывно вводили внутрижелудочно один раз в сутки в течение 2-3 недель, и затем введение приостанавливали на 1 неделю. Во время эксперимента кровь собирали из глазниц каждые две недели, выделяли сыворотку, и содержание ДНК определяли с помощью метода флуоресцентной количественной ПНР. Результаты показаны в таблицах 7 и 8.
3.2. Экспериментальный метод на модели pAAV/HBV
Использовали самцов мышей C57BL/6 (Shanghai Lingchang Biotechnology Co., Ltd.) в возрасте 6-8 недель, и каждой мыши вводили посредством инъекции очищенную рекомбинантную плазмиду pAAV/HBV1.2 (10 мкг), растворенную в PBS в объеме, эквивалентном примерно 10% от ее массы тела, через хвостовую вену в пределах периода времени 3-8 секунд. Кровь собирали из глазниц мышей через 3 суток после введения плазмиды для определения сывороточной ДНК HBV. Отбирали модельных мышей, имеющих однородную сывороточную ДНК, и распределяли в: модельную контрольную группу, группу контроля-носителя и группу тестируемых соединений. Мышам в каждой группе непрерывно вводили внутрижелудочно один раз в сутки в течение 10 суток в дозе 3 мг/кг. Мышиную сыворотку отбирали на 0, 4, 7 и 10 сутки после введения, и мышей умерщвляли на 10 сутки для взятия образцов ткани печени. Число копий ДНК HBV в сыворотке и печени мышей определяли с помощью метода флуоресцентной количественной ПНР. Результаты показаны в таблице 9.
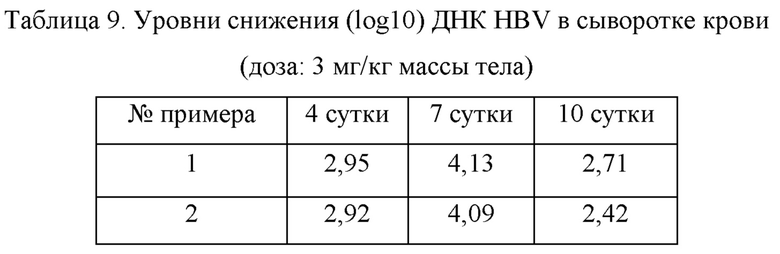
Экспериментальный пример 4: фармакокинетика in vivo
Исследование фармакокинетики (ФК) у крыс in vivo
Крыс линии SD (Shanghai Xipuer-Bikai Laboratory Animal Co., Ltd.) с массой тела 180-220 грамм произвольным образом распределяли в группы по три животных в каждой после 3-5 суток акклиматизации, и серию соединений вводили внутрижелудочно в каждой группе в дозе 20 мг/кг.
Подопытных животных (крысы SD) не кормили в течение 12 часов до введения и кормили через 4 часа после введения. Они имели свободный доступ к воде до и после эксперимента.
После введения собирали приблизительно 0,2 мл крови из глазниц через 0 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов, 24 часа, 30 часов и 48 часов. Не позднее чем через 30 минут после антикоагуляции с использованием EDTA-K2 плазму отделяли посредством центрифугирования при 4°С и 4000 об/мин в течение 10 минут. Сразу после сбора всей плазмы ее хранили при -20°С для тестирования.
50 мкл образца плазмы, подлежащего тестированию, и стандартного образца для калибровочной кривой пипетировали и затем туда добавляли 500 мкл раствора ацетонитрила, содержащего внутренний стандарт (диазепам, 20 мг/мл). Полученную смесь встряхивали и перемешивали до однородного состояния в течение 5 минут и затем центрифугировали при 12000 об/мин в течение 10 мин. 75 мкл надосадочной жидкости пипетировали, и 75 мкл ультрачистой воды добавляли в нее для разведения, и равномерно перемешивали. 1 мкл полученного раствора пипетировали для определения с помощью метода ЖХ-МС/МС. Результаты показаны в таблице 10.
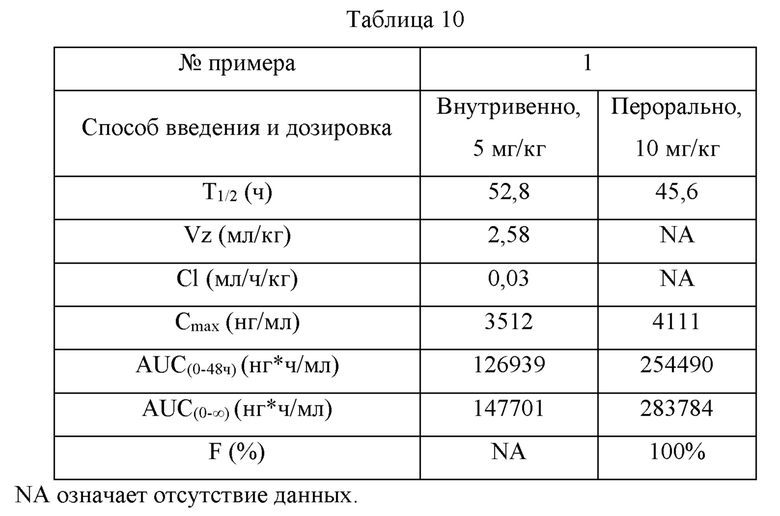
| название | год | авторы | номер документа |
|---|---|---|---|
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| ПРОИЗВОДНОЕ ПИРРОЛОПИРИМИДИНА И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2780254C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2744897C2 |
| ЗАМЕЩЕННОЕ АРОМАТИЧЕСКОЕ ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ И КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ЕГО, И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2811484C1 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ПИПЕРАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2018 |
|
RU2745431C1 |
| СОЕДИНЕНИЕ 3,4-ДИГИДРОИЗОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2825312C1 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ИНГИБИТОРОВ SHP2 | 2018 |
|
RU2776846C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
| СИНТЕЗ НОВОГО АНТАГОНИСТА EP4 И ПРИМЕНЕНИЕ ПРИ РАКЕ И ВОСПАЛЕНИИ | 2021 |
|
RU2804153C1 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
Изобретение относится к соединению формулы I и II или его фармацевтически приемлемой соли, фармацевтической композиции, применению в лечении заболеваний, при которых полезен ингибитор сборки капсидного белка, в частности заболеваний, вызванных инфицированием вирусом гепатита В (HBV). Технический результат: получены новые соединения формул I и II, эффективные ингибиторы сборки капсидного белка, которые могут быть полезны для лечения заболеваний, вызванных инфицированием вирусом гепатита В (HBV). 7 н. и 2 з.п. ф-лы, 10 табл., 6 пр.
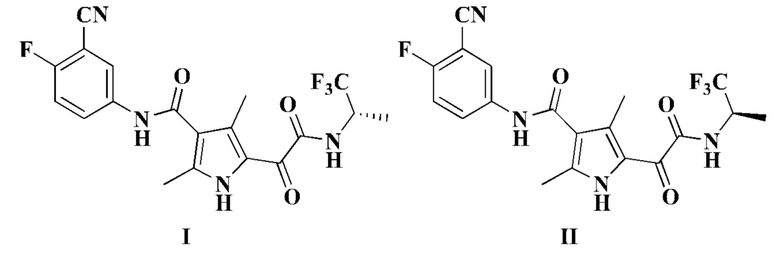
1. Соединение формулы I или II или его фармацевтически приемлемая соль
2. Фармацевтическая композиция, обладающая анти-HBV активностью, содержащая эффективное количество соединения формулы I или II или его фармацевтически приемлемой соли по п. 1; причем фармацевтическая композиция, возможно, дополнительно содержит фармацевтически приемлемый эксципиент.
3. Способ предупреждения или лечения заболевания, вызванного инфицированием вирусом гепатита В (HBV), включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или II или его фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 2.
4. Применение соединения формулы I или II или его фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 2 в изготовлении лекарственного средства для предупреждения или лечения заболевания, вызванного инфицированием вирусом гепатита В (HBV).
5. Применение соединения формулы I или II или его фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 2 для предупреждения или лечения заболевания, вызванного инфицированием вирусом гепатита В (HBV).
6. Соединение формулы I или II или его фармацевтически приемлемая соль по п. 1 или фармацевтическая композиция по п. 2 для применения в предупреждении или лечении заболевания, при котором полезно ингибирование сборки капсидного белка; причем, возможно, заболевание, при котором полезно ингибирование сборки капсидного белка, представляет собой заболевание, вызванное инфицированием вирусом гепатита В (HBV).
7. Способ ингибирования сборки капсидного белка, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или II или его фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 2.
8. Применение соединения формулы I или II или его фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 2 для ингибирования сборки капсидного белка.
9. Соединение формулы I или II или его фармацевтически приемлемая соль по п. 1 или фармацевтическая композиция по п. 2 для применения в ингибировании сборки капсидного белка.
| WO 2017156255 A1, 14.09.2017 | |||
| EA 201690277 A1, 31.05.2016 | |||
| EA 201690760 A1, 29.07.2016 | |||
| ПРОИЗВОДНЫЕ ПИРРОЛА КАК ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА | 2005 |
|
RU2470916C2 |

