Перекрестная ссылка на родственные заявки
[0001] Эта заявка основана и испрашивает преимущество приоритета по китайской патентной заявке № 202010144983.1, поданной 4 марта 2020 года, которая полностью включена в данный документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
[0002] Настоящее изобретение относится к области химии и медицины и, в частности, к пиразольному производному и его применению.
УРОВЕНЬ ТЕХНИКИ
[0003] Простагландин E2 (ПГE2) представляет собой эндогенный биоактивный липид. ПГE2 активирует простагландиновые рецепторы, вызывая широкий восходящий и нисходящий зависимый биологический ответ (Legler, D. F. et al., hit. J Biochem. Cell Biol. 2010, 42, p. 198-201), вовлеченный в регуляцию многочисленных физиологических и патологических процессов, включая воспаление, боль, функцию почек, сердечно-сосудистую систему, функцию легких и рак. Сообщается, что ПГE2 в высокой степени экспрессируется в раковых тканях различных видов рака, и было подтверждено, что ПГE2 связан с возникновением, ростом и развитием рака и болезненных состояний у пациентов. Общепринято считается, что ПГE2 связан с активацией клеточной пролиферации и гибелью клеток (апоптозом) и играет важную роль в процессах пролиферации раковых клеток, прогрессирования заболевания и метастаза при раке.
[0004] Рецепторы ПГE2 подразделяются на 4 подтипа, т.е., EP1, EP2, EP3 и EP4, которые широко распространены в различных тканях. Среди этих подтипов ПГE2 воздействует на рецептор EP4, препятствуя воспалительным реакциям (включая иммуновоспалительные реакции), расслаблению гладких мышц, болям, дифференцировке лимфоцитов, гипертрофии или пролиферации сосудистых мезангиальных клеток, секреции желудочно-кишечной слизи и т.п. Таким образом, можно считать, что антагонисты рецептора ЕР4 перспективны в качестве противовоспалительных и/или обезболивающих средств для лечения заболеваний, связанных с путем ПГE2-ЕР4, таких как воспалительные заболевания, заболевания, сопровождающиеся различными болями, и т.п.
[0005] EP4 является первичным рецептором, вовлеченным при артритной боли в моделях ревматоидного артрита и остеоартрита у грызунов (например, смотрите J. Pharmacol. Exp. Ther., 325, 425 (2008)), который при активации приводит к накоплению внутриклеточной сигнальной молекулы цАМФ. Были проведены исследования по обнаружению экспрессии рецептора EP4 на периферических нервных окончаниях болевых рецепторов, макрофагов и нейтрофилов, и было подтверждено, что эти типы клеток чрезвычайно важны для эндометриоза. В исследованиях сообщалось, что пероральные антагонисты EP4 могут снижать протеинурию у мышей с диабетом II типа, подавляя прогрессирование диабетической нефропатии. В дополнительных исследованиях сообщалось, что активация EP4 и повышенная выработка ПГE2 в слизистой оболочке мочевого пузыря могут быть важными причинами гиперактивности мочевого пузыря при простатите, а внутрипузырное введение антагонистов EP4 может быть эффективным для облегчения гиперактивности мочевого пузыря вследствие простатита. Таким образом, селективные антагонисты ЕР4 могут быть полезны при лечении артрита, включая артритную боль, а также эндометриоз, диабетическую нефропатию, гиперактивность мочевого пузыря. Существующие средства лечения артрита в основном представляют собой традиционные нестероидные противовоспалительные лекарственные препараты (НПВП) или селективные ингибиторы ЦОГ-2, которые могут вызывать побочные эффекты со стороны сердечно-сосудистой системы и/или желудочно-кишечного тракта. Однако селективные антагонисты EP4 с меньшей вероятностью вызывают побочные эффекты со стороны сердечно-сосудистой системы.
[0006] ПГE2 непрерывно активирует рецепторы EP (обильно продуцируемые опухолевыми клетками) в микроокружении опухоли (Ochs et al., J Neurochem. 2016, 136, p. 1142-1154; Zelenay, S. et al., Cell 2015, 162, p. 1257-1270), что способствует накоплению различных иммуносупрессивных клеток и усиливает их активность. Иммуносупрессивные клетки включают опухолеассоциированные макрофаги (ОАМ) II типа, Treg-клетки и супрессорные клетки миелоидного происхождения (МСК). Одной из основных особенностей иммуносупрессивного микроокружения опухоли является наличие значительного количества МСК и ОАМ, что, в свою очередь, тесно связано с низкой общей выживаемостью у пациентов с карциномой желудка, яичника, молочной железы, мочевого пузыря, гепатоцеллюлярной карциномой (ГЦК), раком головы и шеи и другими видами рака. Кроме того, сообщалось, что ПГE2 может индуцировать иммунную толерантность путем подавления накопления антигенпрезентирующих дендритных клеток (ДК) в опухолях, а также подавления активации проникающих в опухоль ДК (Wang et al., Trends in Molecular Medicine 2016, 22, p. 1-3). Все эти ПГE2-опосредованные эффекты вместе могут помочь опухолевым клеткам избежать иммунного надзора. ПГE2 играет важную роль в содействии развитию онкогенеза. Повышенные уровни экспрессии ПГE2 и родственных ему рецепторов EP2 и EP4 были обнаружены при различных типах злокачественных новообразований, включая рак толстой кишки, рак легкого, рак молочной железы и рак головы и шеи, и они часто тесно связаны с неблагоприятным прогнозом (Bhooshan, N. et al., Lung Cancer 101, 88-91). Таким образом, селективная блокада сигнальных путей EP2 и EP4 может подавлять онкогенез путем изменения микроокружения опухоли и модуляции опухолевых иммунных клеток.
[0007] Существующие данные доклинических исследований показывают, что EP2- и EP4-специфичные антагонисты могут предотвращать или ингибировать рост опухоли в различной степени на животных моделях, таких как рак толстой кишки, пищевода, легкого и молочной железы. Среди лекарственных препаратов на основе рецептора ПГE2, введенных в клиническую практику, Грапипрант, антагонист EP4, разработанный Pfizer, был одобрен FDA для лечения артрита у собак, а тем временем в 2015 году он вошел в клинические испытания фазы II противоопухолевого препарата для лечения нескольких типов солидных опухолей, таких как рак предстательной железы, немелкоклеточный рак легкого и рак молочной железы (De Vito, V. et al. J Pharm Biomed Anal 118, 251-258). В 2015 году для E7046, антагониста EP4, разработанного Eisai, также начались клинические исследования фазы I, а клинические испытания фазы Iб в комбинации с лучевой терапией или химиотерапией рака прямой кишки были начаты в 2017 году. ONO-4578, разработанный Ono Pharmaceutical, в 2017 году вошел в клинические испытания фазы I для распространенных или метастатических солидных опухолей, и в клинические испытания фазы I/II для лечения распространенных солидных опухолей отдельно или в комбинации с ниволумабом в 2018 году.
[0008] В настоящее время в отношении антагонистов EP4 достигнут определенный прогресс в лечении воспалительных заболеваний, боли, рака и др. Однако по-прежнему актуальной является разработка новых лекарственных препаратов в качестве усовершенствований или замен существующих лекарственных препаратов.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0009] В настоящем изобретении представлено соединение, способное эффективно антагонизировать EP4, которое можно применять в качестве усовершенствования или замены существующих лекарственных препаратов или антагонистов EP4.
[0010] С этой целью в первом аспекте в настоящем изобретении представлено соединение, которое представляет собой соединение, представленное формулой V, или таутомер, стереоизомер, гидрат, сольват, соль или пролекарство соединения, представленного формулой V:
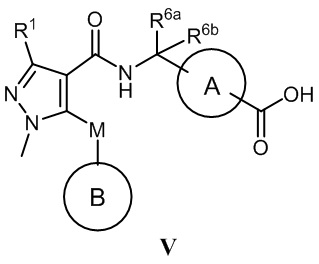 , в которой
, в которой
кольцо A выбрано из  ,
,  ,
,  и
и 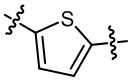 ;
;
кольцо B выбрано из 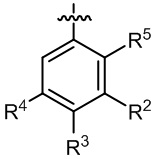 и
и 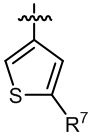 ;
;
R1 выбран из -CH3, -CHF2 и -CF3;
R2 выбран из C2-C6алкила, C3-C6циклоалкила, фенила, трифторметила, C2-C6галогензамещенного алкила, C3-C6галогензамещенного циклоалкила, C2-C6гидроксизамещенного алкила, C2-C6цианозамещенного алкила, -SF5 и -X-R2a, где X выбран из кислорода, серы, -CO-, -SO2- и SO-, и R2a выбран из C1-C6алкила и C1-C6галогензамещенного алкила;
R3 выбран из водорода, галогена, C1-C2алкила, C1-C2фторзамещенного алкила и фенила;
R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила;
R5 выбран из водорода и галогена;
один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил; или R6a и R6b вместе образуют циклобутил;
R7 выбран из -CH3, -CHF2 и -CF3; и
M выбран из кислорода, серы и метилена;
при условии, что:
когда R2 представляет собой трифторметил, M представляет собой кислород, один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил, кольцо A выбрано из и ; и
когда R2 представляет собой трифторметил, M представляет собой кислород, а кольцо A представляет собой , R6a и R6b вместе образуют циклобутил.
[0011] В соответствии с вариантами осуществления настоящего изобретения вышеуказанное соединение дополнительно может включать по меньшей мере один из следующих дополнительных технических признаков.
[0012] В соответствии с вариантами осуществления настоящего изобретения соединение представляет собой соединение, представленное формулой III, или соединение представляет собой таутомер, стереоизомер, гидрат, сольват, соль или пролекарство соединения, представленного формулой III:
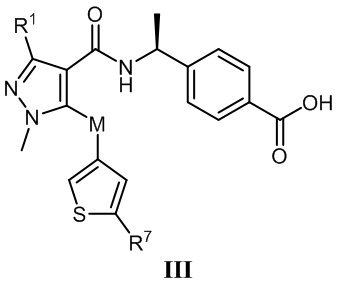 , в которой R1 выбран из -CH3, -CHF2 и -CF3; R7 выбран из -CH3, -CHF2 и -CF3; и M выбран из кислорода, серы и метилена.
, в которой R1 выбран из -CH3, -CHF2 и -CF3; R7 выбран из -CH3, -CHF2 и -CF3; и M выбран из кислорода, серы и метилена.
[0013] В соответствии с вариантами осуществления настоящего изобретения соединение представляет собой соединение, представленное формулой II, или соединение представляет собой таутомер, стереоизомер, гидрат, сольват, соль или пролекарство соединения, представленного формулой II:
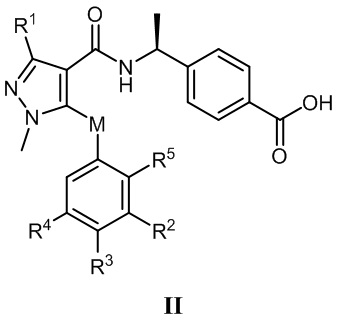 , в которой R1 выбран из -CH3, -CHF2 и -CF3, и предпочтительно R1 представляет собой -CHF2; R2 выбран из этила, пропила, изопропила, н-бутила, изобутила, трет-бутила, фторэтила, фторпропила, фторизопропила, фторбутила, фторизобутила, гидроксиэтила, гидроксиизопропила, цианометила, цианоэтила, фенила, -SF5 и -X-R2a, где X выбран из кислорода, серы и -CO-, и R2a выбран из метила, этила, фторметила и фторэтила; R3 выбран из водорода, фтора, хлора, метила, этила, фторметила, фторэтила и фенила; R4 выбран из водорода, фтора, хлора, метила, этила, фторметила и фторэтила; R5 выбран из водорода, фтора и хлора; и M выбран из кислорода, серы и метилена.
, в которой R1 выбран из -CH3, -CHF2 и -CF3, и предпочтительно R1 представляет собой -CHF2; R2 выбран из этила, пропила, изопропила, н-бутила, изобутила, трет-бутила, фторэтила, фторпропила, фторизопропила, фторбутила, фторизобутила, гидроксиэтила, гидроксиизопропила, цианометила, цианоэтила, фенила, -SF5 и -X-R2a, где X выбран из кислорода, серы и -CO-, и R2a выбран из метила, этила, фторметила и фторэтила; R3 выбран из водорода, фтора, хлора, метила, этила, фторметила, фторэтила и фенила; R4 выбран из водорода, фтора, хлора, метила, этила, фторметила и фторэтила; R5 выбран из водорода, фтора и хлора; и M выбран из кислорода, серы и метилена.
[0014] В соответствии с вариантами осуществления настоящего изобретения соединение представляет собой соединение, представленное формулой I (также называемое соединение I), или соединение представляет собой таутомер, стереоизомер, гидрат, сольват, соль или пролекарство соединения, представленного формулой I:
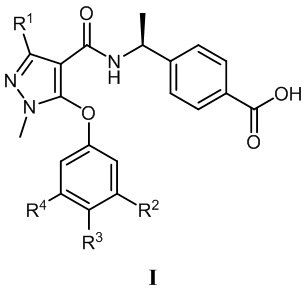 , в которой R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, C2-C6галогензамещенного алкила и C3-C6галогензамещенного циклоалкила; R3 выбран из водорода, галогена, C1-C2алкила и C1-C2фторзамещенного алкила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила.
, в которой R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, C2-C6галогензамещенного алкила и C3-C6галогензамещенного циклоалкила; R3 выбран из водорода, галогена, C1-C2алкила и C1-C2фторзамещенного алкила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила.
[0015] В соответствии с вариантами осуществления настоящего изобретения вышеуказанное соединение дополнительно может включать по меньшей мере один из следующих дополнительных технических признаков.
[0016] В соответствии с вариантами осуществления настоящего изобретения R2 выбран из C2-C3алкила, C3-C6циклоалкила, C2-C3фторзамещенного алкила и C3-C6фторзамещенного циклоалкила.
[0017] В соответствии с вариантами осуществления настоящего изобретения R2 предпочтительно выбран из -CH2CH3, -CH(CH3)2, циклопропила, -CF2CH3 и -CH2CF3.
[0018] В соответствии с вариантами осуществления настоящего изобретения R3 выбран из водорода, фтора и хлора.
[0019] В соответствии с вариантами осуществления настоящего изобретения R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила.
[0020] В соответствии с некоторыми вариантами осуществления настоящего изобретения R4 выбран из водорода, фтора, хлора, C1-C4алкила, C1-C4алкоксила, C1-C4фтор- или хлорзамещенного алкила, C1-C4фтор- или хлорзамещенного алкоксила; и предпочтительно R4 выбран из водорода, фтора и хлора.
[0021] В соответствии с вариантами осуществления настоящего изобретения соединение представляет собой любое из следующих соединений, или соединение представляет собой таутомер, стереоизомер, гидрат, сольват, фармацевтически приемлемую соль или пролекарство любого из следующих соединений:
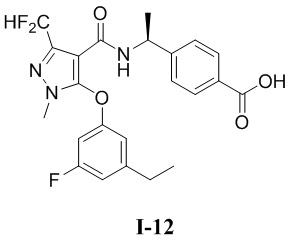
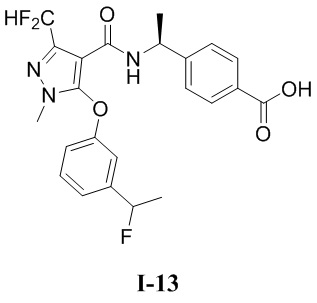
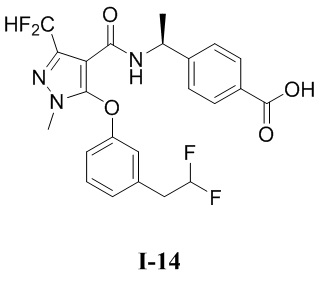
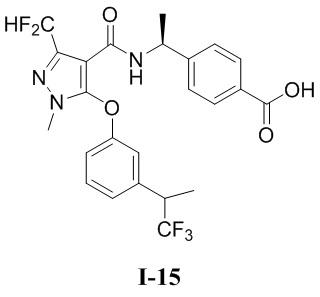
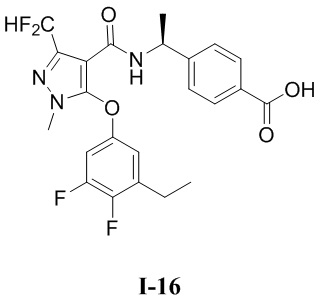
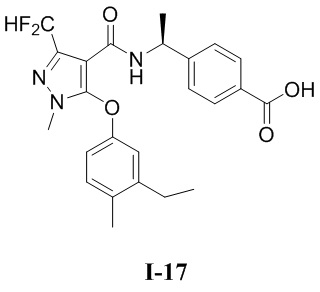
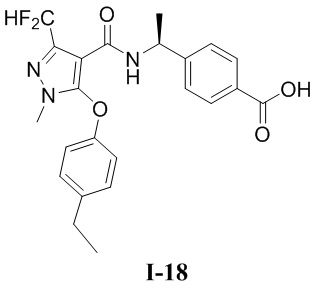
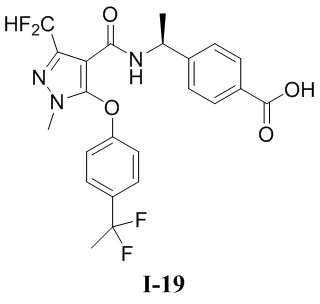
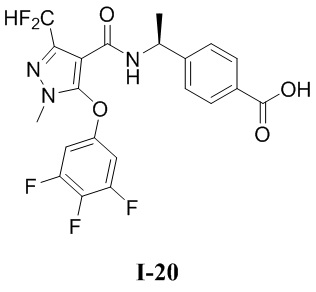
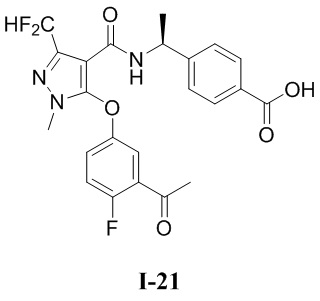
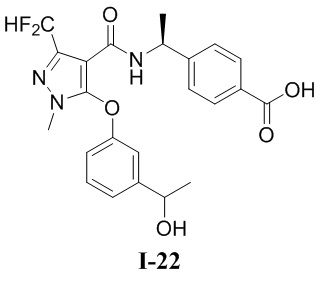
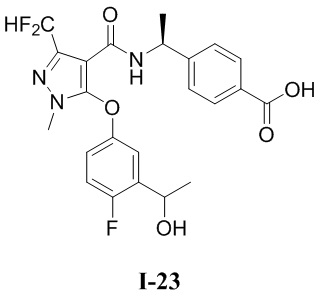
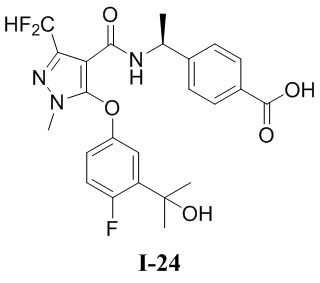
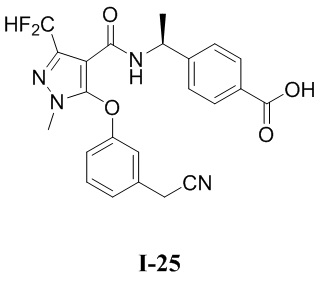
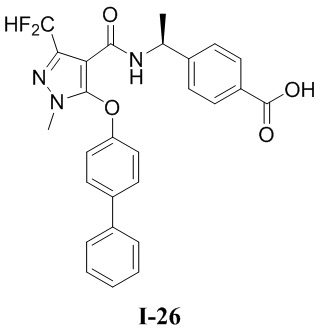
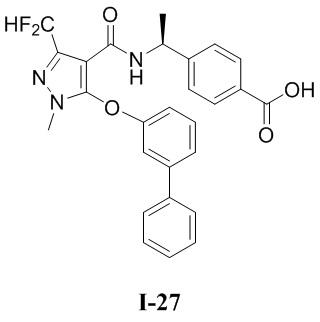
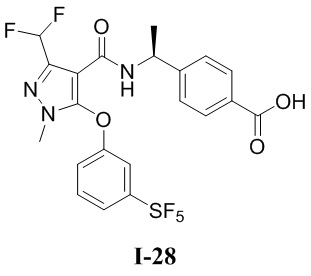
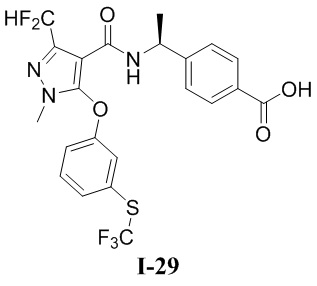
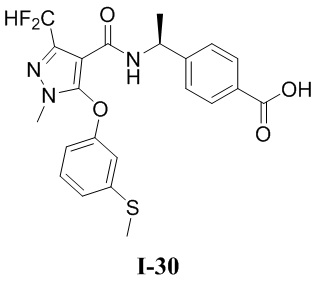
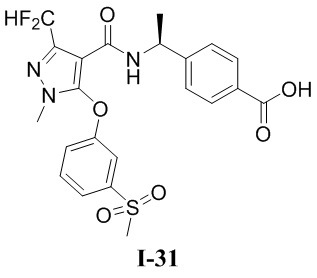
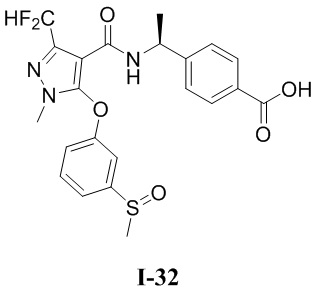
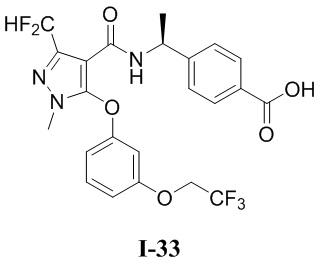
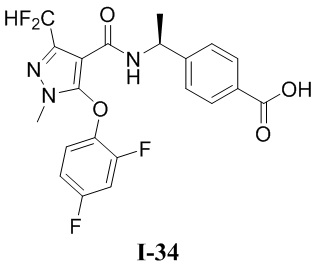
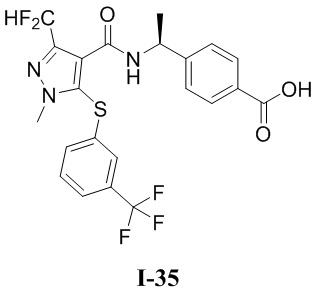
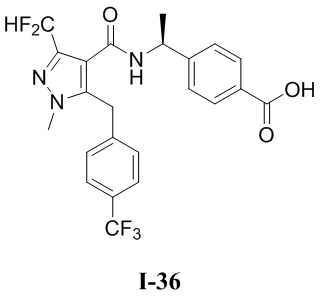
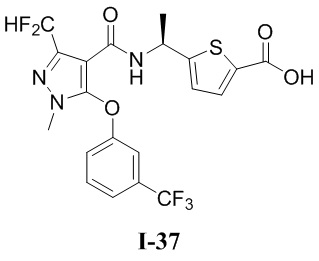
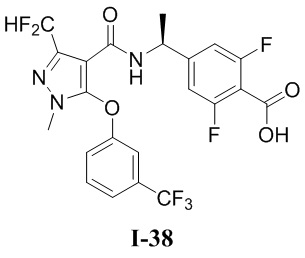
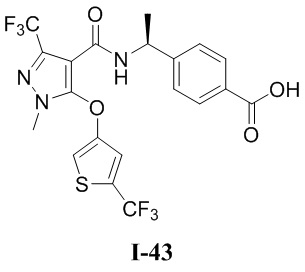 .
.
[0022] В соответствии с вариантами осуществления настоящего изобретения соль включает фармацевтически приемлемую соль и представляет собой по меньшей мере одну, выбранную из серной кислоты, фосфорной кислоты, азотной кислоты, бромистоводородной кислоты, хлористоводородной кислоты, муравьиной кислоты, уксусной кислоты, пропионовой кислоты, бензолсульфоновой кислоты, бензойной кислоты, фенилуксусной кислоты, салициловой кислоты, альгиновой кислоты, антраниловой кислоты, камфорной кислоты, лимонной кислоты, винилсульфоновой кислоты, муравьиной кислоты, фумаровой кислоты, фурановой кислоты, глюконовой кислоты, глюкуроновой кислоты, глутаминовой кислоты, гликолевой кислоты, изэтионовой кислоты, молочной кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, слизевой кислоты, памовой кислоты, пантотеновой кислоты, стеариновой кислоты, янтарной кислоты, сульфаниловой кислоты, винной кислоты, п-толуолсульфоновой кислоты, малоновой кислоты, 2-гидроксипропионовой кислоты, щавелевой кислоты, гликолевой кислоты, глюкуроновой кислоты, галактуроновой кислоты, лимонной кислоты, лизина, аргинина, аспарагиновой кислоты, коричной кислоты, п-толуолсульфоновой кислоты, метансульфоновой кислоты, этансульфоновой кислоты или трифторметансульфоновой кислоты. Специалистам в данной области техники понятно, что, кроме фармацевтически приемлемых солей, в настоящем изобретении также могут быть использованы другие соли, выступающие в качестве промежуточных веществ в очистке соединений или в получении других фармацевтически приемлемых солей, или для идентификации, характеристики или очистки соединений по настоящему изобретению.
[0023] Во втором аспекте настоящего изобретения в настоящем изобретении представлена фармацевтическая композиция. В соответствии с вариантами осуществления настоящего изобретения фармацевтическая композиция включает фармацевтически приемлемый эксципиент и вышеописанное соединение.
[0024] В третьем аспекте настоящего изобретения в настоящем изобретении представлено применение вышеописанного соединения или вышеописанной фармацевтической композиции в получении лекарственного средства для лечения или предупреждения связанного с EP4 заболевания.
[0025] В соответствии с вариантами осуществления настоящего изобретения применение может дополнительно включать по меньшей мере один из следующих дополнительных технических признаков.
[0026] В соответствии с вариантом осуществления настоящего изобретения связанное с EP4 заболевание включает по меньшей мере одно выбранное из группы, состоящей из воспалительного заболевания, боли, рака, метаболического заболевания и заболевания мочевыделительной системы.
[0027] В соответствии с вариантом осуществления настоящего изобретения воспалительное заболевание включает по меньшей мере одно выбранное из группы, состоящей из артрита и ревматоидного артрита.
[0028] В соответствии с вариантом осуществления настоящего изобретения боль включает остеоартритическую боль и вызванную эндометриозом боль.
[0029] В соответствии с вариантом осуществления настоящего изобретения вышеописанные соединение или фармацевтическая композиция могут быть введены в комбинации с лучевой терапией и/или терапией антителами. Терапия антителами выбрана из группы, состоящей из терапии антителами CTLA4, терапии антителами PDL1, терапии антителами PD1 и их комбинаций.
[0030] В соответствии с вариантом осуществления настоящего изобретения рак включает солидный рак.
[0031] В соответствии с вариантами осуществления настоящего изобретения рак включает рак молочной железы, рак шейки матки, колоректальный рак, рак эндометрия, глиобластому, рак головы и шеи, рак почки, рак печени, рак легкого, медуллобластому, рак яичника, рак поджелудочной железы, рак предстательной железы, рак кожи и рак уретры.
[0032] В соответствии с вариантом осуществления настоящего изобретения метаболическое заболевание включает диабет, а заболевание мочевыделительной системы включает гиперактивность мочевого пузыря.
[0033] В соответствии с вариантом осуществления настоящего изобретения при помощи соединения или фармацевтической композиции по настоящему изобретению пациенту, который в этом нуждается, могут быть обеспечены более оптимальные, более эффективные препарат или схема клинического лечения. В соответствии с вариантами осуществления настоящего изобретения в настоящем изобретении представлен ряд антагонистов EP4, характеризующихся новыми структурами, лучшими фармакокинетическими свойствами, лучшим действием лекарственного препарата и хорошими лечебными свойствами, которые могут эффективно лечить связанные с EP4 заболевания или нарушения.
[0034] Настоящее изобретение также относится к способу лечения заболевания, связанного с EP4, при этом способ включает введение пациенту терапевтически эффективного количества фармацевтического состава, содержащего описанное в настоящем документе соединение или его фармацевтически приемлемую соль.
[0035] В настоящем изобретении дополнительно представлен способ лечения воспалительных заболеваний, боли, рака, метаболических заболеваний, заболеваний мочевыделительной системы. Способ включает: введение пациенту терапевтически эффективного количества фармацевтического состава, содержащего вышеописанное соединение или его фармацевтически приемлемую соль. В настоящем изобретении дополнительно представлен способ лечения заболевания путем введения соединения или фармацевтической композиции в комбинации с лучевой терапией и/или терапией антителами, в котором терапия антителами выбрана из группы, состоящей из терапии антителами CTLA4, терапии антителами PDL1, терапии антителами PD1 и их комбинаций.
[0036] Определения и пояснения терминов
[0037] Если не указано иное, определения групп и терминов, описанных в описании и формуле изобретения включают фактические определения, примерные определения, предпочтительные определения, определения, записанные в таблицах, и определения конкретных соединений в примерах и т.д., которые можно произвольно комбинировать и интегрировать друг с другом. Определения групп и структуры соединений, которые объединены и интегрированы, должны подпадать под объем настоящего изобретения.
[0038] Если не определено иное, все технические и научные термины в настоящем документе имеют такое же значение, которое обычно понимается специалистом средней квалификации в области техники, к которой принадлежит заявляемый объект. Патенты, патентные заявки, публикации, цитируемые в настоящем документе, в полном объеме включены в настоящий документ посредством ссылки, если не указано иное. Если термин имеет несколько определений, то определение, данное в этой главе, имеет преимущественную силу.
[0039] Если не указано иное, используются традиционные методы в соответствующей области техники, такие как масс-спектроскопия, ЯМР, ИК- и УФ-спектроскопия/спектроскопия в видимой области света, а также фармакологические методы. Если не указаны конкретные определения, термины в соответствующем описании аналитической химии, органической синтетической химии, а также медицины и медицинской химии являются известными из уровня техники. Стандартные методики могут быть использованы в химическом синтезе, химическом анализе, фармацевтическом получении, составлении и доставке, а также при лечении пациентов. Например, реакции и очистки можно проводить, используя инструкции производителя набора, или способом, хорошо известным в соответствующей области техники, или как описано в настоящем документе. Вышеописанные методики и процедуры, как правило, можно проводить традиционными способами, хорошо известными в данной области техники, в соответствии с описанием в ряде общих и более конкретных документов, цитируемых и обсуждаемых в настоящем описании. Во всем описании группы и их заместители могут быть выбраны специалистом в данной области техники для обеспечения устойчивых фрагментов и соединений. Если группы заместителей изображены обычными химическими формулами, написанными слева направо, группы заместителей также включают химически эквивалентные заместители, которые возникли бы при написании структурной формулы справа налево. Например, CH2O является эквивалентным OCH2.
[0040] Как используется в настоящем документе, в описании и формуле изобретения указываются числовые диапазоны, прочитанные как «целые числа», которые следует понимать как указанные как конечные точки диапазона и каждое целое число в пределах диапазона. Например, «целое число от 1 до 6» следует понимать как перечисление всех без исключения целых чисел от 0, 1, 2, 3, 4, 5 и 6. Когда числовой диапазон понимается как «число», подразумевается, что он указывает обе конечные точки диапазона и каждое целое число в пределах диапазона и каждое десятичное число в пределах диапазона. Например, «число от 1 до 10» следует понимать как перечисление не только каждого из целых чисел 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10, но также по меньшей мере суммы каждого из целых чисел с 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 соответственно.
[0041] Термин «фармацевтически приемлемый» означает соединения, материалы, композиции и/или лекарственные формы, которые подходят для применения в контакте с тканями человека и животных без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений в рамках надежного медицинского заключения и соизмеримы с разумным соотношением польза/риск.
[0042] Термин «фармацевтически приемлемая соль» или «их фармацевтически приемлемая соль» относится к солям фармацевтически приемлемых нетоксичных кислот или оснований, включая соли неорганических кислот и оснований, органических кислот и оснований.
[0043] Помимо фармацевтически приемлемых солей в настоящем изобретении могут быть приняты другие соли, и они могут служить как промежуточные соединения в очистке соединений или в получении других фармацевтически приемлемых солей или могут использоваться для идентификации, характеристики или очистки соединений по настоящему изобретению.
[0044] Термин «стереоизомер» относится к изомеру, получаемому другим пространственным расположением атомов в молекуле. Определения и правила стереохимии, используемые в настоящем изобретении, в целом соответствуют “McGraw-Hill Dictionary of Chemical Terms (1984)”, S. P. Parker, Ed., McGraw-Hill Book Company, New York; и “Stereochemistry of Organic Compounds”, Eliel, E. and Wilen, S., John Wiley & Sons, Inc., New York, 1994. Соединение по настоящему изобретению может содержать асимметричный центр или хиральный центр и, таким образом, могут существовать различные стереоизомерные формы. Все стереоизомерные формы соединения по настоящему изобретению, в том числе без ограничения диастереоизомеры, энантиомеры, атропоизомеры, геометрические (или конформационные) изомеры, и их смеси, такие как рацемические смеси, должны подпадать под объем настоящего изобретения.
[0045] Многие органические соединения существуют в оптически активных формах, т.е. они способны вращать плоскость плоскополяризованного света. При описании оптически активных соединений приставки D и L или R и S используются для обозначения абсолютных конфигураций молекулы по отношению к одному или более хиральным центрам. Приставки D и L или (+) и (-) представляют собой символы, используемые для обозначения вращения плоскополяризованного света, вызванного соединением, где (-) или L указывает, что соединение является левовращающим, а приставка (+) или D указывает, что соединение является правовращающим. Для данной химической структуры эти стереоизомеры идентичны, за исключением того, что эти стереоизомеры являются зеркальными отображениями друг друга. Конкретные стереоизомеры можно назвать энантиомерами, а смесь таких изомеров называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называется рацемической смесью или рацематом, что может иметь место, когда в химической реакции или процессе отсутствует стереоселективность или стереоспецифичность.
[0046] В соответствии с выбором исходных материалов и методов соединение по настоящему изобретению может существовать в форме одного из возможных изомеров или их смеси, например, в виде оптически чистого изомера или в виде смеси изомеров, такой как рацемический изомер и диастереоизомерная смесь, в зависимости от количества асимметрических атомов углерода. Оптически активный (R)- или (S)-изомер может быть получен с помощью хиральных синтонов или хиральных препаратов или отделен с использованием традиционных методик. Если соединение содержит двойную связь, то заместители могут быть в E- или Z-конфигурациях; если соединение содержит двузамещенный циклоалкил, то заместитель циклоалкила может иметь цис- или транс-конформацию.
[0047] Если связь с хиральным атомом углерода в формуле по настоящему изобретению изображена прямой линией, следует понимать, что две конфигурации (R) и (S) хирального атома углерода и полученные в результате энантиомерно чистое соединение и смесь включены в объем, определенный общей формулой. Схематическое представление рацемата или энантиомерно чистого соединения в настоящем документе взято из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, клиновидная связь и связь, показанная пунктирной линией, используются для представления абсолютной конфигурации стереоцентра.
[0048] Соединения по настоящему изобретению, содержащие асимметрически замещенные атомы углерода, могут быть разделены в оптически активной форме или рацемической форме. Разделение рацемической смеси соединения может проводиться любым из множества способов, известных из уровня техники. Например, способы включают фракционную рекристаллизацию с использованием кислот для хирального разделения, которые представляют собой оптически активные солеобразующие органические кислоты. Например, подходящие разделяющие агенты для фракционной рекристаллизации представляют собой оптически активные кислоты, такие как винная кислота, диацетилвинная кислота, дибензоилвинная кислота, миндальная кислота, яблочная кислота, молочная кислота или различные оптически активные камфорсульфоновые кислоты, такие как D- и L-формы β-камфорсульфоновой кислоты. Другие разделяющие агенты, подходящие для фракционной рекристаллизации, включают α-метилбензиламин в стереоизомерно чистой форме (например, S- и R-формы или диастереомерно чистая форма), 2-фенилглицинол, норэфедрин, эфедрин, N-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и др. Разделение рацемической смеси также может проводиться элюированием колонки, заполненной оптически активным разделяющим агентом (например, динитробензоилфенилглицин). Также можно использовать высокоэффективную жидкостную хроматографию (ВЭЖХ) или сверхкритическую флюидную хроматографию (СФХ). Конкретный метод, условия элюирования и хроматографические колонки могут быть выбраны специалистом в данной области техники в соответствии со структурами соединений и экспериментальными результатами. Кроме того, оптически чистые активные исходные материалы или реагенты известной конфигурации также можно использовать для получения любых энантиомеров или диастереомеров соединений, описанных в настоящем изобретении, посредством стереоорганического синтеза.
[0049] Многие геометрические изомеры олефинов, двойные связи C=N или т.п. также могут присутствовать в соединениях, описанных в настоящем документе, и все эти стабильные изомеры рассматриваются в настоящем изобретении. Если соединение, описанное в настоящем документе, содержит этиленовую двойную связь, такая двойная связь включает геометрические E- и Z-изомеры, если не указано иное.
[0050] Термин «таутомер» относится к изомеру функциональной группы, образующемуся в результате быстрого перемещения атома между двумя положениями в молекуле. Соединение по настоящему изобретению может демонстрировать таутомерию. Таутомерные соединения могут присутствовать в двух или более взаимопревращающихся формах. Прототропный таутомер образуется в результате переноса ковалентно связанных атомов водорода между двумя атомами. Таутомер обычно существует в равновесной форме. При попытке отделить один таутомер, обычно получают смесь, физические и химические свойства которой соответствуют смеси соединений. Положение равновесия зависит от внутримолекулярных химических свойств. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладающим является кетоновый тип; а для фенолов преобладает енольный тип. Все таутомерные формы соединений включены в настоящее изобретение.
[0051] Термин «фармацевтическая композиция» относится к смеси одного или более соединений, описанных в настоящем документе, или их физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов. Другие химические компоненты могут представлять собой, например, физиологически/фармацевтически приемлемые носители и эксципиенты. Фармацевтическая композиция направлена на облегчение введения соединения в организм.
[0052] Термин «сольват» относится к соединению по настоящему изобретению или его соли, включающим стехиометрический или нестехиометрический растворитель, связанный посредством межмолекулярной нековалентной силы. Если растворитель представляет собой воду, сольват представляет собой гидрат.
[0053] Термин «пролекарство» может быть превращен в соединение по настоящему изобретению, характеризующееся биологической активностью, при физиологических условиях или посредством сольволиза. Пролекарство по настоящему изобретению получают путем модификации функциональных групп в соединении, и модифицированный фрагмент может быть удален стандартными операциями или in vivo так, чтобы получить исходное соединение. Пролекарство включает соединение, которое образуется присоединением фрагмента к гидроксильной группе или аминогруппе в соединении по настоящему изобретению. Когда пролекарство соединения по настоящему изобретению вводится млекопитающему, пролекарство диссоциирует с образованием свободной гидроксильной или аминогруппы.
[0054] Соединение по настоящему изобретению может содержать неприродное соотношение атомных изотопов в одном или более атомах, составляющих соединение. Например, соединение может быть меченным радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). Превращение всех изотопных составов соединений по настоящему изобретению, независимо от того, радиоактивны они или нет, включено в объем настоящего изобретения.
[0055] Термин «эксципиент» относится к фармацевтически приемлемому инертному ингредиенту. Примеры «эксципиента» включают без ограничения связующие, разрыхлители, смазывающие вещества, вещества, способствующие скольжению, стабилизаторы, наполнители, разбавители и т.п.
[0056] Термин «C1-C6 алкил» относится к насыщенной одновалентной углеводородной группе с линейной или разветвленной цепью, содержащей 1, 2, 3, 4, 5 или 6 атомов углерода. Указанный алкил представляет собой, например, метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изоамил, 2-метилбутил, 1-метилбутил, 1-этилпропил, 1,2-диметилпропил, неопентил, 1,1-диметилпропил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 2-этилбутил, 1-этилбутил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 2,3-диметилбутил, 1,3-диметилбутил, 1,2-диметилбутил и др. или их изомер. Конкретно, указанная группа содержит 1, 2 или 3 атома углерода («C1-C3 алкил»), например, метил, этил, н-пропил или изопропил.
[0057] Термин «C3-C6 циклоалкил» относится к насыщенному одновалентному моно- или бициклическому углеводородному кольцу, содержащему 3-6 атомов углерода, включая конденсированные или мостиковые полициклические кольцевые системы, например, циклопропил, циклобутил, циклопентил и циклогексил.
[0058] Термин «C1-C6 алкоксил» следует понимать как -O-(C1-6 алкил), в котором «C1-6 алкил» имеет вышеуказанное определение.
[0059] Термин «галогеногруппа» или «галоген» представляет собой фтор, хлор, бром или йод.
[0060] «Галогензамещенный алкил» относится к насыщенной алифатической углеводородной группе с разветвленной и прямой цепью, содержащей конкретное число атомов углерода и замещенной одним или более атомами галогена (например, -CvFw, где v = 1-3, w = 1-(2v + l)). Примеры галогензамещенного алкила включают без ограничения трифторметил, трихлорметил, пентафторэтил, пентахлорэтил, 2,2,2-трифторэтил, гептафторпропил и гептахлорпропил.
[0061] Благоприятные эффекты
[0062] В соответствии с вариантами осуществления настоящего изобретения соединения и/или их композиции, описанные в настоящем документе, обладают активностью в отношении эффективного антагонизирования рецептора EP4, и они имеют преимущества превосходной метаболической стабильности в печени и кардиологической безопасности, а также характеризуются лучшими фармакокинетическими свойствами, более высоким воздействием in vivo, более низким дозированием и лучшей приверженностью. Следовательно, они имеют хорошие перспективы применения в получении лекарственных средств для лечения связанных с EP4 заболеваний.
[0063] Дополнительные аспекты и преимущества настоящего изобретения будут частично изложены в последующем описании, а частично будут очевидны из описания или могут быть изучены при применении настоящего изобретения на практике.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0064] Фиг. 1 представляет собой результат ингибирования опухоли соединениями в соответствии с вариантом осуществления настоящего изобретения.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0065] Решения в соответствии с настоящим изобретением будут пояснены в связи с примерами. Специалистам в данной области техники будет понятно, что следующие примеры являются только иллюстрацией настоящего изобретения и не должны рассматриваться как ограничивающие объем настоящего изобретения. Методики или условия, которые не указаны в примерах, должны выполняться в соответствии с методиками или условиями, описанными в литературных источниках в данной области техники, или в соответствии с инструкцией к продукту. Реагенты или инструменты без указания производителя и являются коммерчески доступными.
[0066] Соединения по настоящей заявке идентифицированы с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС), если не указано иное. Единица сдвига ЯМР составляет 10-6 (ч./млн.). Растворители для ЯМР представляли собой дейтерированный диметилсульфоксид, дейтерированный хлороформ, дейтерированный метанол и др., а внутренним стандартом являлся тетраметилсилан (ТМС).
[0067] Сокращения в настоящем изобретении определены следующим образом:
[0068] BAST: бис(2-метоксиэтил)аминосеры трифторид
[0069] m-CPBA: м-хлорпероксибензойная кислота
[0070] L-селектрид: три-втор-бутилборгидрид лития
[0071] Pd (dppf) Cl2: 1,1-бис(дифенилфосфино)ферроценпалладия хлорид
[0072] ДХМ: дихлорметан
[0073] ГАТУ: гексафторфосфат O-(7-азабензотриазол-1-ил)-N, N, N', N'-тетраметилурония
[0074] ДИПЭА: диизопропилэтиламин т.е., N, N-диизопропилэтиламин
[0075] ДМФА: N, N-диметилформамид
[0076] н.: нормальность, например, 1 н. хлористоводородная кислота означает раствор хлористоводородной кислоты концентрацией 1 моль/л
[0077] ТГФ: тетрагидрофуран
[0078] ДМА: N, N-диметилацетамид
[0079] ДМСО: диметилсульфоксид
[0080] ЭА: этилацетат
[0081] IC50: концентрация полумаксимального ингибирования, указывающая на концентрацию, при которой достигается половина максимального ингибирующего эффекта.
[0082] CHO: клетки яичника китайского хомяка
[0083] HBSS: сбалансированный солевой раствор Хэнкса
[0084] БСА: альбумин из бычьей сыворотки
[0085] ГЭПЭС: гидроксиэтилпиперазинэтантиосульфоновая кислота
[0086] IBMX: 3-изобутил-1-метил-7H-ксантин
[0087] FLIPR: спектрофотометр для чтения планшетов для визуализации флуоресценции
[0088] EC80: концентрация, при которой достигается 80% максимального эффекта
[0089] Если не указано иное, соединения, приведенные в качестве примеров в настоящем документе, названы и пронумерованы с помощью ChemBioDraw Ultra 13.0.
[0090] Контрольный пример 1: получение контрольного соединения
[0091] 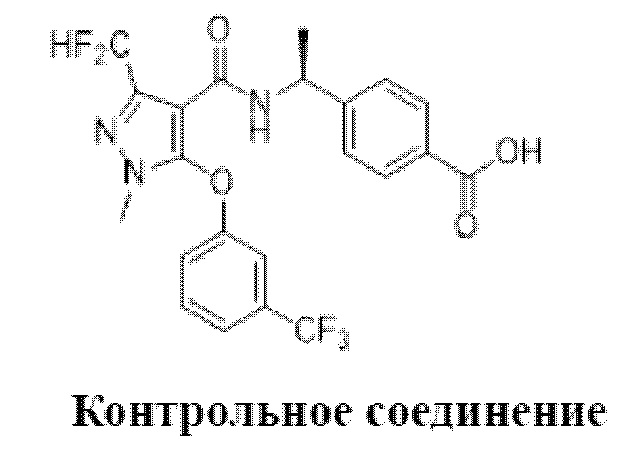
[0092] Контрольное соединение синтезировали в соответствии с патентной заявкой WO2012039972A1.
[0093] Контрольные соединения в следующих тестовых примерах обозначаются как соединение, описанное в контрольном примере 1.
[0094] Подготовительный пример 1: Получение промежуточного соединения А
[0095] Метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А)
[0096] 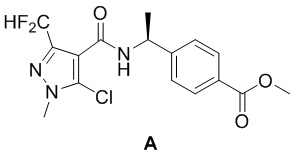
[0097] Схема синтеза для промежуточного соединения А показана ниже:
[0098] 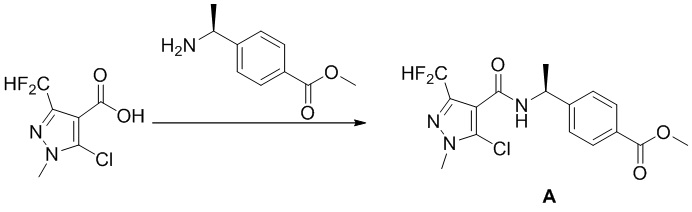
[0099] Исходный материал 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоновую кислоту (5 г, 23,8 ммоль), которую синтезировали в соответствии с патентной заявкой WO2011151369A1, добавляли к ДХМ (200 мл). Добавляли метил (S)-4-(l-аминоэтил)бензоат (5,1 г, 28,6 ммоль), ГАТУ (10,9 г, 28,6 ммоль) и ДИПЭА (4,6 г, 35,7 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; добавляли воду (200 мл) и экстрагировали смесь с помощью ДХМ (50 мл ×3) и разделяли, и органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата.
[00100] ЖХМС (ИЭР) m/z: 372,5 [M+H]+
[00101] Подготовительный пример 2: Кислотный способ получения А
[00102] Настоящий пример является примером очистки продукта, в котором очистку выполняют с использованием высокоэффективной жидкостной хроматографии со следующими условиями очистки: колонка Welch, Ultimate C18, 10 мкм, 21,2 мм × 250 мм.
[00103] Подвижная фаза А представляла собой 1‰ трифторуксусную кислоту в чистой воде, подвижная фаза Б представляла собой ацетонитрил. Условия градиента: в пределах 0-3 мин, подвижную фазу А выдерживали при 90%; после элюирования градиентом от 3 мин до 18 мин подвижная фаза А изменялась от 90% до 5%, и подвижную фазу А выдерживали при 5% от 18 мин до 22 мин.
[00104] «Кислотный способ получения А», описанный в следующем разделе «Примеры», полностью относится к кислотному способу получения А подготовительного примера 2.
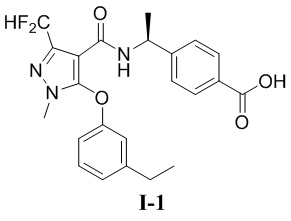
[00105] Пример 1: получение соединения I-1
[00106] (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-1)
[00107] 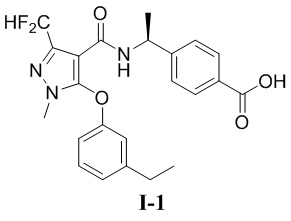
[00108] Схема синтеза для соединения I-1 показана ниже:
[00109] 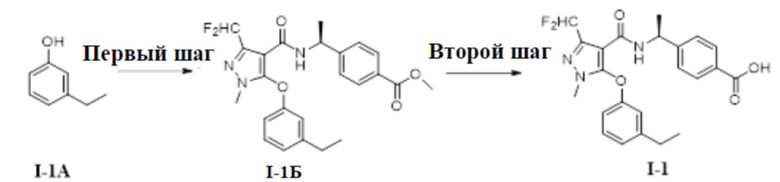
[00110] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-1Б)
[00111] 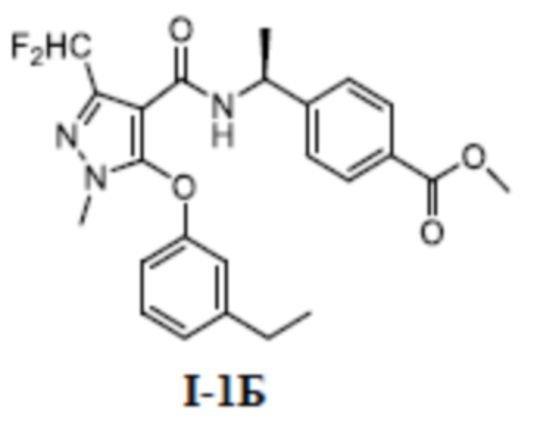
[00112] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (370 мг, 1,0 ммоль) добавляли к ДМФА (10 мл) при комнатной температуре, и добавляли 3-этилфенол (I-1А) (183 мг, 1,5 ммоль) и KOH (168 мг, 3,0 ммоль); смесь нагревали до 120°C и перемешивали в течение 6 ч, а затем охлаждали до комнатной температуры и разбавляли водой (40 мл); pH доводили до 7 с помощью 1 н. хлористоводородной кислоты; раствор экстрагировали этилацетатом (20 мл ×3) и отделенные органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-1Б) (220 мг, выход 48,1%).
[00113] ЖХМС (ИЭР) m/z: 458,1 [M+H]+
[00114] Второй шаг: (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-1)
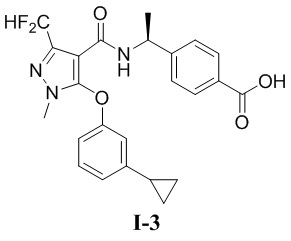
[00115] 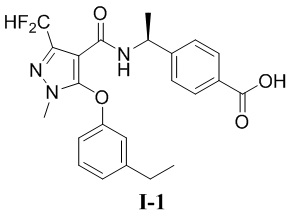
[00116] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-1Б) (220 мг, 0,48 ммоль) добавляли к ТГФ (4 мл) при комнатной температуре; добавляли воду (2 мл) и гидроксид лития моногидрат (42 мг, 1,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(3-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-1) (80 мг, выход 37,5%).
[00117] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 7,90 (д, 1H), 7,71 (д, 2H), 7,32 (т, 1H), 7,25 (т, 1H), 7,12 (д, 2H), 7,07 (д, 1H), 6,90 (с, 1H), 6,76 (дд, 1H), 4,90 (т, 1H), 3,72 (с, 3H), 2,61 (к, 2H), 1,22 (д, 3H), 1,14 (т, 3H).
[00118] ЖХМС (ИЭР) m/z: 444,1 [M+H]+
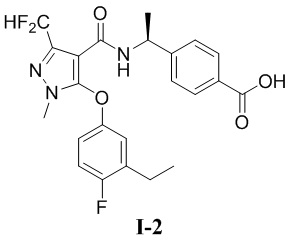
[00119] Пример 2: получение соединения I-2
(S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-2)
[00120] 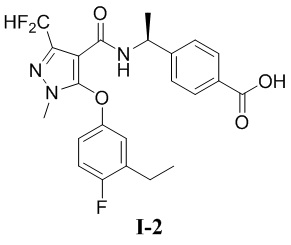
[00121] Схема синтеза для соединения I-2 показана ниже:
[00122] 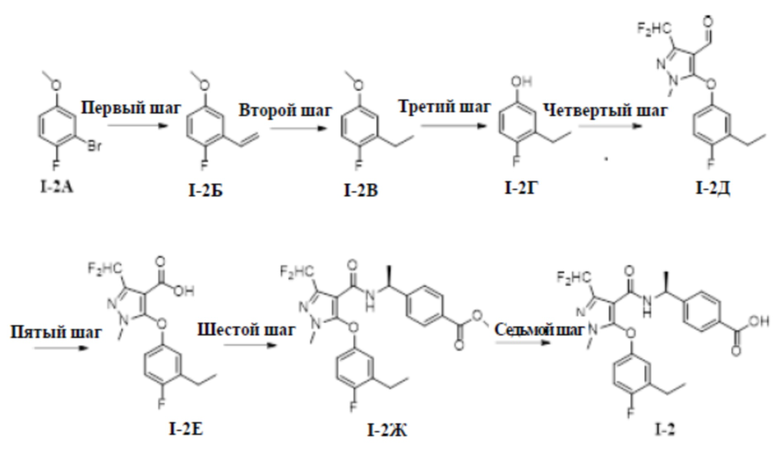
[00123] Первый шаг: 1-фтор-4-метокси-2-винилбензол (соединение I-2Б)
[00124] 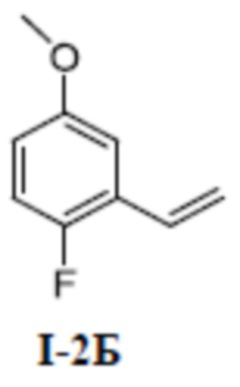
[00125] 2-бром-1-фтор-4-метоксибензол (соединение I-2А) (1,02 г, 5,0 ммоль) добавляли к 1, 4-диоксану (20 мл) при комнатной температуре; добавляли винилфторборат калия (740 мг, 5,52 ммоль), и [l, l-бис(дифенилфосфино)ферроцен]дихлорпалладий (430 мг, 0,50 ммоль), и карбонат калия (1,52 г, 11,0 ммоль). Смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 14 часов, а затем охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали с помощью ДХМ (80 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (чистый петролейный эфир) с получением бесцветного жидкого неочищенного продукта 1-фтор-4-метокси-2-винилбензола (соединение I-2Б) (680 мг, выход 89,9%).
[00126] Второй шаг: 2-этил-1-фтор-4-метоксибензол (соединение I-2В)
[00127] 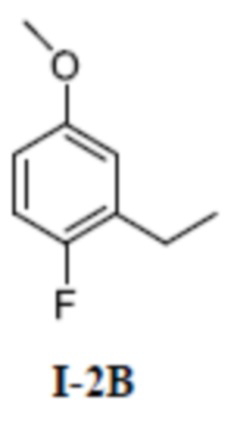
[00128] 1-фтор-4-метокси-2-винилбензол (3,60 г, 23,7 ммоль) добавляли к метанолу (50 мл) при комнатной температуре; добавляли 10% палладия на угле (200 мг); продували H2, а затем раствор перемешивали в течение 16 часов при комнатной температуре. Раствор фильтровали и фильтрат промывали метанолом (30 мл×3), а органические фазы объединяли и концентрировали с получением бесцветного жидкого неочищенного продукта 2-этил-1-фтор-4-метоксибензола (соединение I-2В) (2,90 г, выход 79,5%).
[00129] Третий шаг: 3-этил-4-фторфенол (соединение I-2Г)
[00130] 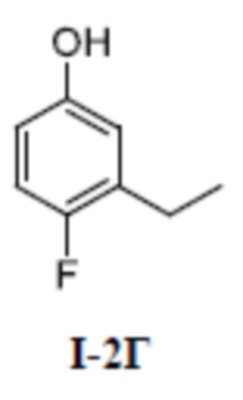
[00131] 2-этил-1-фтор-4-метоксибензол (100 мг, 0,65 ммоль) добавляли к ДХМ (3 мл) при комнатной температуре, а затем смесь охлаждали до -60°C. Добавляли 1 моль/л раствор BBr3 ДХМ (2 мл), смесь подогревали естественным путем до комнатной температуры и перемешивали при комнатной температуре в течение 4 ч. Остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением бесцветной жидкости 3-этил-4-фторфенола (соединение I-2Г) (60 мг, выход 89,9%).
[00132] Четвертый шаг: 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбальдегид (соединение I-2Д)
[00133] 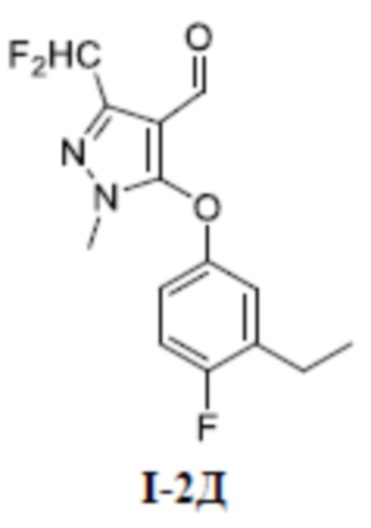
[00134] Соединение 5-хлор-3-(дифторметил)-1-метил-lH-пиразол-4-карбальдегид (350 мг, 1,18 ммоль) добавляли к ДМФА (5 мл) при комнатной температуре; добавляли 3-этил-4-фторфенол (379 мг, 2,70 ммоль) и карбонат калия (546 мг, 3,95 ммоль); и смесь нагревали до 100°C и перемешивали в течение 1,5 ч. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (15 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением бесцветного жидкого неочищенного продукта 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбальдегида (соединение I-2Д) (600 мг, выход 100%).
[00135] ЖХМС (ИЭР) m/z: 299,1 [M+H]+
[00136] Пятый шаг: 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоновая кислота (соединение I-2Е)
[00137] 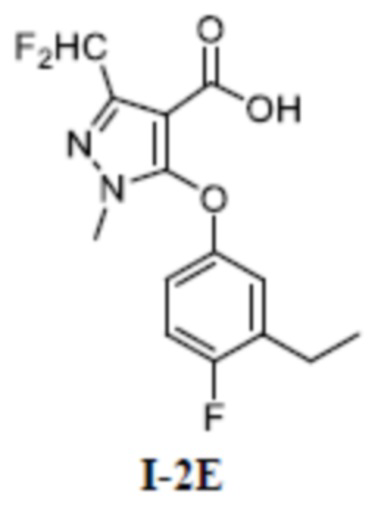
[00138] Соединение 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбальдегид (350 мг, 1,80 ммоль) добавляли к трет-бутанолу (10 мл) и воде (2 мл) при комнатной температуре; добавляли 2-метил-2-бутен (246 мг, 3,52 ммоль), хлорит натрия (316 мг, 3,52 ммоль) и дигидрофосфат натрия (281 мг, 2,34 ммоль). Смесь перемешивали в течение 4 часов при комнатной температуре. Раствор разбавляли водой (5 мл), экстрагировали этилацетатом (10 мл× 3) и разделяли, а органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоновой кислоты (соединение I-2Е) (350 мг, выход 94,9%).
[00139] ЖХМС (ИЭР) m/z: 315,1 [M+H]+
[00140] Шестой шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-2Ж)
[00141] 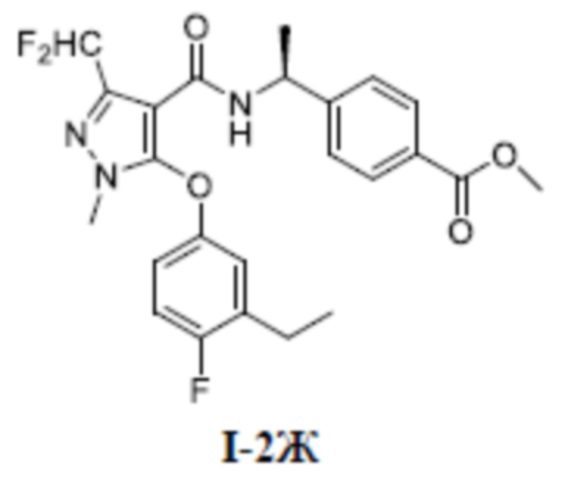
[00142] Соединение 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоновую кислоту (350 мг, 1,11 ммоль) добавляли к ДМФА (5 мл) при комнатной температуре, и добавляли метил (S)-4-(l-аминоэтил)бензоат (220 мг, 1,23 ммоль), ГАТУ (467 мг, 1,23 ммоль) и ДИПЭА (301 мг, 2,33 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли водой (20 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли, а органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 4: 1) с получением бесцветной жидкости метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-2Ж) (400 мг, выход 75,5%).
[00143] ЖХМС (ИЭР) m/z: 476,2 [M+H]+
[00144] Седьмой шаг: (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-2)
[00145]
[00146] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (400 мг, 0,84 ммоль) добавляли к ТГФ (5 мл), воде (5 мл) и метанолу (5 мл) при комнатной температуре, и добавляли гидроксид лития моногидрат (141 мг, 3,36 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, и реакционную смесь концентрировали с получением белого твердого вещества ((S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (300 мг, выход 77,2%).
[00147] 1H ЯМР (400 МГц, ДМСО-d6) 12,8 (с, 1H), 8,03 (д, 1H), 7,74 (д, 2H), 7,17 (т, 1H), 7,14-7,10 (м, 3H), 7,01-6,97 (м, 1H), 6,82-6,79 (м, 1H), 4,92 (т, 1H), 3,73 (с, 3H), 2,59 (к, 2H), 1,25 (д, 3H), 1,11 (т, 3H).
[00148] ЖХМС (ИЭР) m/z: 462,2 [M+H]+
[00149] Пример 3: получение соединения I-3
[00150] (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-3)
[00151] 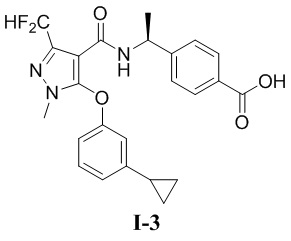
[00152] Схема синтеза для соединения I-3 показана ниже:
[00153] 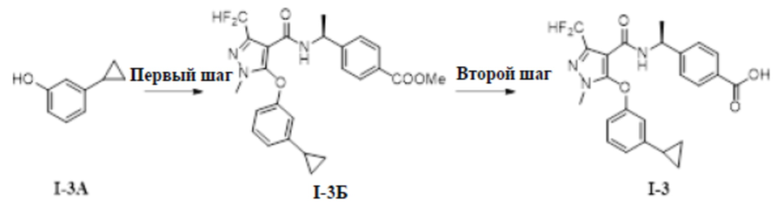
[00154] Первый шаг: метил (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-3Б)
[00155] 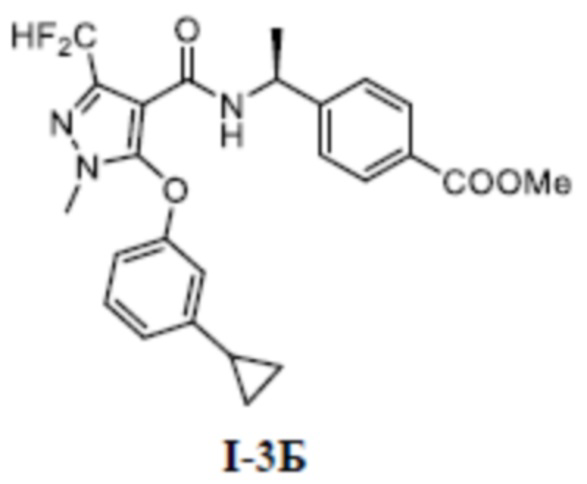
[00156] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (145 мг, 0,39 ммоль) добавляли к ДМА (2 мл) при комнатной температуре, добавляли 3-циклопропилфенол (80 мг, 0,59 ммоль) и KOH (34 мг, 0,61 ммоль), и смесь нагревали до 120°C, перемешивали в течение 2 часов, а затем охлаждали до комнатной температуры, разбавляли водой (100 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-3Б) (20 мг, выход 10,9%).
[00157] ЖХМС (ИЭР) m/z: 470,6 [M+H]+
[00158] Второй шаг: (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-3)
[00159]
[00160] Исходный материал метил (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-3Б) (20 мг, 0,04 ммоль) добавляли к ТГФ (1 мл) при комнатной температуре, и добавляли воду (1 мл) и гидроксид лития моногидрат (2 мг, 0,048 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(3-циклопропилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-3) (1,5 мг, выход 7,7%).
[00161] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,8 (с, 1H), 7,92 (д, 1H), 7,72 (д, 2H), 7,27 (т, 1H), 7,11 (т, 1H), 7,10 (д, 2H), 6,90 (д, 1H), 6,80 (т, 1H), 6,70 (дд, 1H), 4,90 (т, 1H), 3,72 (с, 3H), 1,92-1,98 (м, 1H), 1,23 (д, 3H), 0,96-0,92 (м, 2H), 0,66-0,27 (м, 2H).
[00162] ЖХМС (ИЭР) m/z: 456,6 [M+H]+
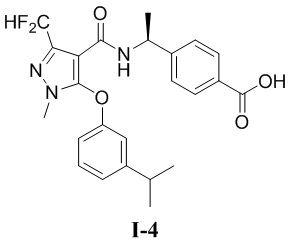
[00163] Пример 4: получение соединения I-4
[00164] (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-4)
[00165] 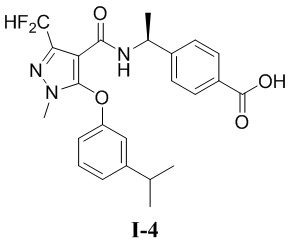
[00166] Схема синтеза для соединения I-4 показана ниже:
[00167] 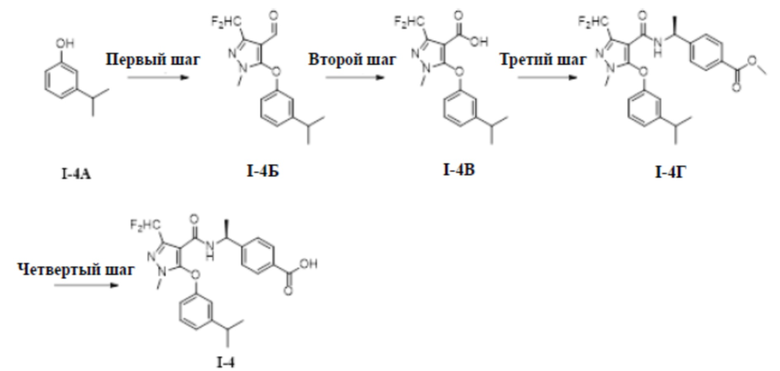
[00168] Первый шаг: 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбальдегид (соединение I-4Б)
[00169] 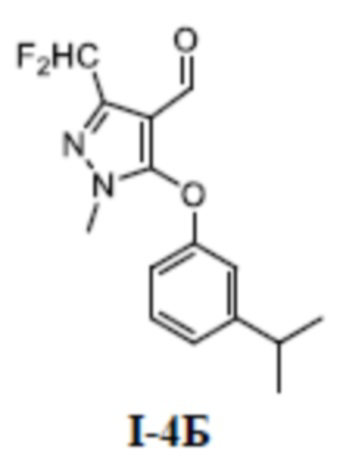
[00170] Соединение 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (500 мг, 2,57 ммоль) добавляли к ДМФА (5 мл) при комнатной температуре; добавляли 3-изопропилфенол (386 мг, 2,80 ммоль) и KOH (216 мг, 3,85 ммоль); и смесь нагревали до 150°C и перемешивали в течение 4 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (15 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтого жидкого неочищенного продукта 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбальдегида (соединение I-4Б) (750 мг, выход 98,9%).
[00171] ЖХМС (ИЭР) m/z: 295,1 [M+H]+
[00172] Второй шаг: 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоновая кислота (соединение I-4В)
[00173] 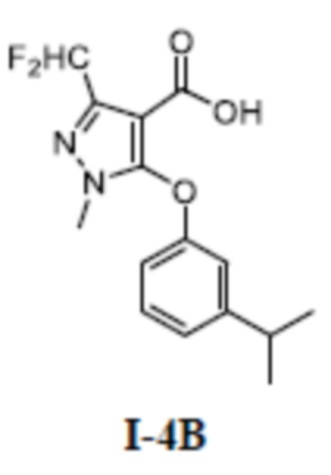
[00174] Соединение 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбальдегид (750 мг, 2,55 ммоль) добавляли к трет-бутанолу (6 мл) и воде (7 мл) при комнатной температуре, добавляли 2-метил-2-бутен (355 мг, 5,07 ммоль), хлорит натрия (456 мг, 5,07 ммоль) и дигидрофосфат натрия (669 мг, 5,57 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре. Смесь разбавляли водой (15 мл) и экстрагировали этилацетатом (30 мл ×3) и разделяли, а органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого твердого неочищенного продукта 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоновой кислоты (соединение I-4В) (800 мг, выход 100%).
[00175] ЖХМС (ИЭР) m/z: 311,1 [M+H]+
[00176] Третий шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-4Г)
[00177] 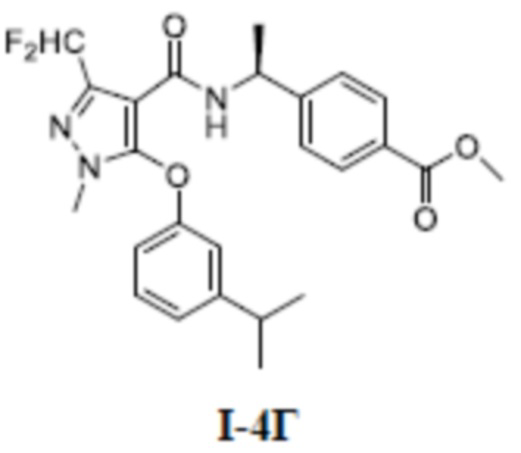
[00178] Соединение 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоновую кислоту (800 мг, 2,58 ммоль) добавляли к ДХМ (20 мл); добавляли (S)-метил 4-(l-аминоэтил)бензоат (459 мг, 2,56 ммоль), ГАТУ (1,40 г, 3,68 ммоль) и ДИПЭА (991 мг, 7,68 ммоль); и смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли ДХМ (40 мл), промывали водой (20 мл ×3) и разделяли. Органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтой жидкости метил (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-4Г) (720 мг, выход 59,2%).
[00179] ЖХМС (ИЭР) m/z: 472,2 [M+H]+
[00180] Четвертый шаг: (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-4)
[00181]
[00182] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-4Г) (440 мг, 0,93 ммоль) добавляли к метанолу (10 мл) и воде (1 мл) при комнатной температуре, и добавляли гидроксид натрия (93 мг, 2,32 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (93 мг, выход 21,7%).
[00183] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,7 (с, 1H), 7,89 (д, 1H), 7,70 (д, 2H), 7,32 (т, 1H), 7,12 (т, 1H), 7,10 (с, 1H), 7,06 (д, 2H), 6,99 (с, 1H), 6,72-6,70 (м, 1H), 4,91 (т, 1H), 3,73 (с, 3H), 2,91-2,85 (м, 1H), 1,21 (д, 3H), 1,17 (д, 6H).
[00184] ЖХМС (ИЭР) m/z: 458,3 [M+H]+
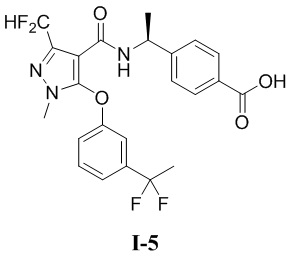
[00185] Пример 5: получение соединения I-5
[00186] (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-5)
[00187] 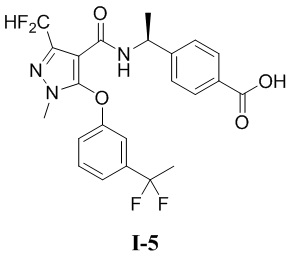
[00188] Схема синтеза для соединения I-5 показана ниже:
[00189] 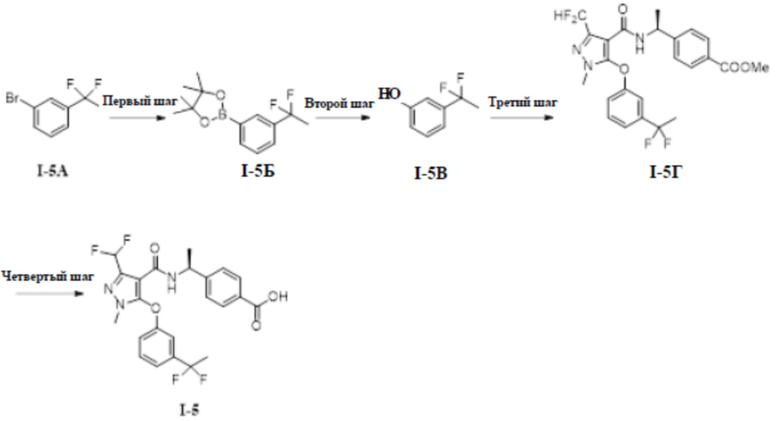
[00190] Первый шаг: 2-(3-(1,1-дифторэтил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (соединение I-5Б)
[00191] 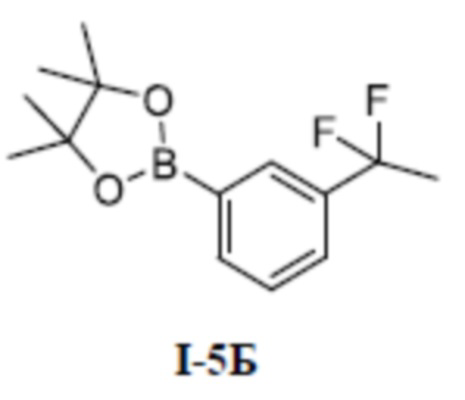
[00192] 1-бром-3-(1, 1-дифторэтил)бензол (800 мг, 3,62 ммоль) добавляли к 1, 4-диоксану (30 мл) при комнатной температуре, и добавляли бис(пинаколато)диборон (17,0 г, 156,3 ммоль), йодид меди (II) (2,5 г, 13,0 ммоль), L-пролин (2,76 г, 10,86 ммоль), ацетат калия (710 мг, 7,24 ммоль) и [1, 1-бис(дифенилфосфино)ферроцен]дихлорпалладий (295 мг, 0,36 ммоль). Смесь нагревали до 90°C в защитной атмосфере азота и перемешивали в течение 16 часов, а затем смесь охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали дихлорметаном (80 мл ×3) и разделяли. Органические фазы объединяли и сушили с помощью безводного сульфата натрия, фильтровали и концентрировали, а остаток очищали путем разделения на колонке с силикагелем (чистый петролейный эфир) с получением бесцветного жидкого неочищенного продукта 2-(3-(1,1-дифторэтил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (соединение I-5Б) (900 мг, выход 92,7%).
[00193] Второй шаг: 3-(1,1-дифторэтил)фенол (соединение I-5В)
[00194] 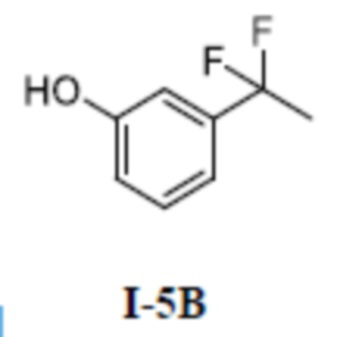
[00195] 2-(3-(1,1-дифторэтил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (соединение I-5Б) (900 мг, 3,36 ммоль) добавляли к ТГФ (15 мл) и воде (15 мл) при комнатной температуре, и добавляли перборат натрия моногидрат (1,01 г, 10,07 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре, и смесь разбавляли водой (200 мл), экстрагировали с помощью ДХМ (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 8:1) с получением бесцветной жидкости 3-(1,1-дифторэтил)фенол (соединение I-5В) (280 мг, выход 52,7%).
[00196] Третий шаг: метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-5Г)
[00197] 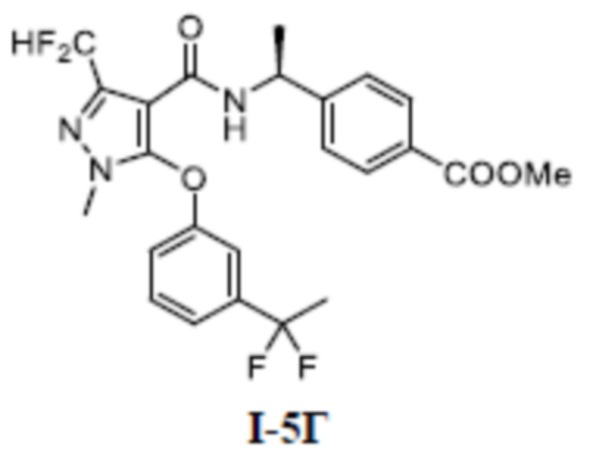
[00198] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (650 мг, 1,75 ммоль) добавляли к ДМФА (12 мл) при комнатной температуре; добавляли 3-(l, l-дифторэтил)фенол (360 мг, 2,27 ммоль) и гидроксид калия (147 мг, 2,62 ммоль); и смесь нагревали до 120°C и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали этилацетатом (80 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-5Г) (1,2 г, неочищенный продукт).
[00199] ЖХМС (ИЭР) m/z: 494,6 [M+H]+.
[00200] Четвертый шаг: (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-5)
[00201]
[00202] Исходный материал метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-5Г) (1,0 г, 2,03 ммоль) добавляли к ТГФ (5 мл) при комнатной температуре, и добавляли воду (4 мл) и гидроксид лития моногидрат (340 мг, 8,11 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-5) (88 мг, выход 7,9%).
[00203] ЖХМС (ИЭР) m/z: 480,5 [M+H]+
[00204] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,10 (д, 1H), 7,71 (д, 2H), 7,53 (т, 1H), 7,47 (д, 1H), 7,27 (д, 1H), 7,11 (т, 1H), 7,11 (д, 2H), 7,07 (дд, 1H), 4,88 (т, 1H), 3,74 (с, 3H), 1,96 (т, 3H), 1,96 (д, 3H).
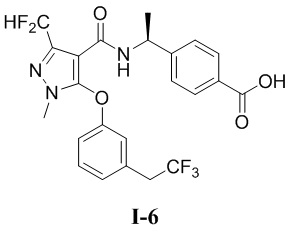
[00205] Пример 6: получение соединения I-6
[00206] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-6)
[00207] 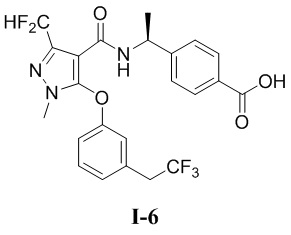
[00208] Схема синтеза для соединения I-6 показана ниже:
[00209] 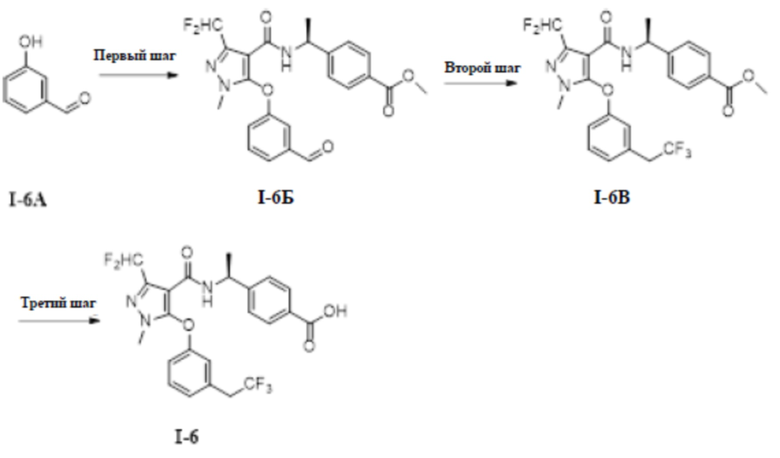
[00210] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-6Б)
[00211] 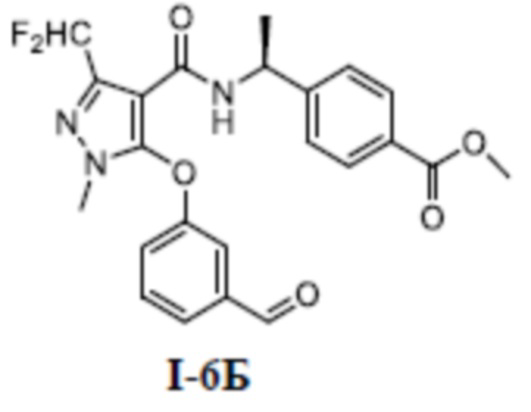
[00212] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (371 мг, 1,0 ммоль) добавляли к ДМСО (5 мл) при комнатной температуре, и добавляли 3-гидроксибензальдегид (122 мг, 1,0 ммоль), K2CO3 (270 мг, 2,0 ммоль) и йодид меди (I) (76 мг, 0,4 ммоль), фенантролин (72 мг, 0,4 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 8:1) с получением бесцветной жидкости метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-6Б) (180 мг, выход 39,3%).
[00213] ЖХМС (ИЭР) m/z: 458,1 [M+H]+
[00214] Второй шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-6В)
[00215] 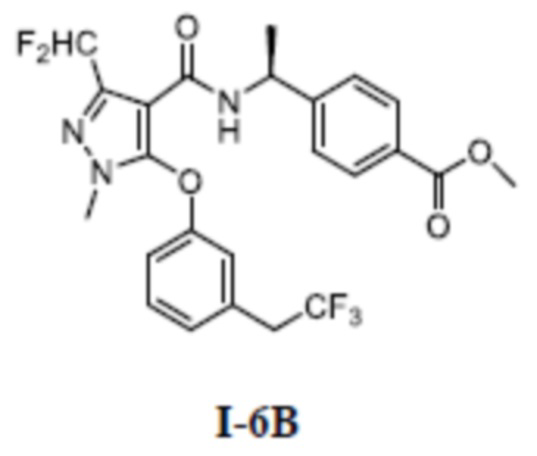
[00216] Метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-6Б) (41,4 мг, 0,09 ммоль) добавляли к ДМФА (2 мл) и добавляли 2,2-дифтор-2-трифенилфосфаниумилацетат (64 мг, 0,18 ммоль) при комнатной температуре, и смесь нагревали до 60°C и перемешивали в течение 2 часов. Добавляли раствор тетрабутиламмония фторида в тетрагидрофуране (0,3 мл, 0,30 ммоль) и перемешивание продолжали в течение 4 часов. Затем смесь охлаждали до комнатной температуры, реакционную смесь концентрировали и остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 8:1) с получением бесцветного твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-6В) (40 мг, выход 91,2%).
[00217] ЖХМС (ИЭР) m/z: 512,1 [M+H]+
[00218] Третий шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-6)
[00219]
[00220] Исходный материал (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-6В) (40 мг, 0,082 ммоль) добавляли к метанолу (2 мл) при комнатной температуре, и добавляли воду (2 мл) и гидроксид лития моногидрат (12 мг, 0,3 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-6) (4,8 мг, выход 12,3%).
[00221] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,7 (с, 1H), 7,95 (д, 1H), 7,73 (д, 2H), 7,43 (т, 1H), 7,22 (д, 1H), 7,13 (т, 1H), 7,12 (д, 2H), 7,10 (с, 1H), 6,97 (дд, 1H), 4,89 (т, 1H), 3,74 (с, 3H), 3,72-3,63 (м, 2H), 1,24 (д, 3H).
[00222] ЖХМС (ИЭР) m/z: 498,5 [M+H]+
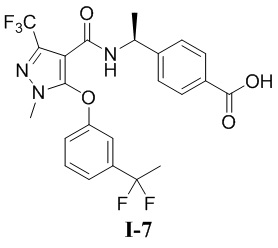
[00223] Пример 7: получение соединения I-7
[00224] (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-7)
[00225]
[00226] Схема синтеза для соединения I-7 показана ниже:
[00227] 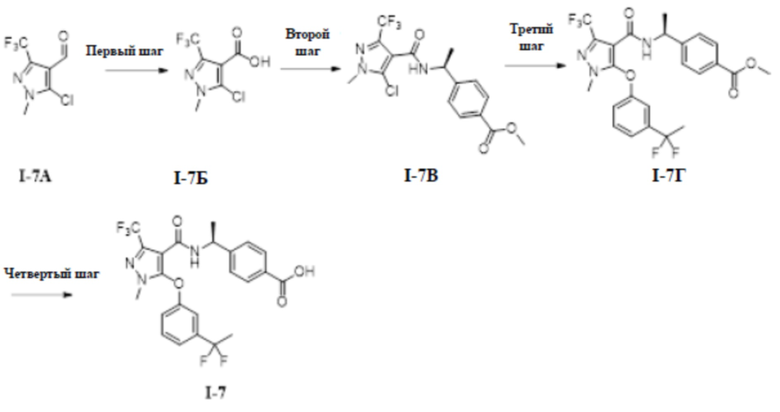
[00228] Первый шаг: 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновая кислота (соединение I-7Б)
[00229] 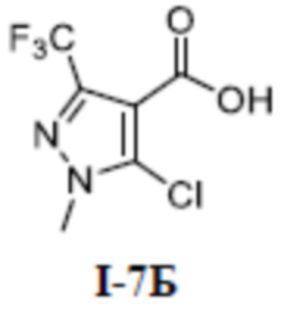
[00230] Соединение 5-хлор-1-метил-3-(трифторметил)-lH-пиразол-4-карбальдегид (700 мг, 3,30 ммоль) добавляли к трет-бутанолу (20 мл) и воде (5 мл) при комнатной температуре, и добавляли 2-метил-2-бутен (1,80 г, 25,7 ммоль), хлорит натрия (1,48 г, 16,4 ммоль) и дигидрофосфат натрия (3,10 г, 25,8 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре, а затем смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного остатка. Затем неочищенный остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого вещества 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновой кислоты (соединение I-7Б) (680 мг, выход 90,3%).
[00231] ЖХМС (ИЭР) m/z: 229,6 [M+H]+
[00232] Второй шаг: метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-7В)
[00233] 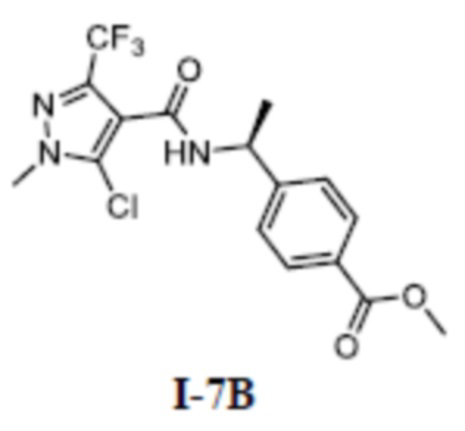
[00234] Соединение 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновую кислоту (соединение I-7Б) (680 мг, 2,98 ммоль) добавляли к ДМФА (20 мл), и добавляли метил (S)-4-(l-аминоэтил)бензоат (537 мг, 3,00 ммоль), ГАТУ (1,70 г, 4,47 ммоль) и ДИПЭА (1,90 г, 14,7 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь разбавляли водой (200 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого вещества метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-7В) (700 мг, выход 60,3%).
[00235] ЖХМС (ИЭР) m/z: 390,5 [M+H]+
[00236] Третий шаг: метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-7Г)
[00237] 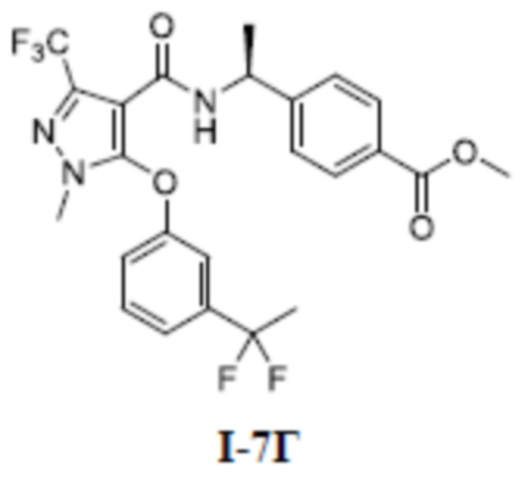
[00238] Соединение метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-7В) (700 мг, 1,00 ммоль) добавляли к ДМФА (10 мл) при комнатной температуре, и добавляли 3-(l, l-дифторэтил)фенол (284 мг, 1,00 ммоль) и KOH (264 мг, 4,63 ммоль). Смесь нагревали до 120°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-7Г) (100 мг, выход 10,8%).
[00239] ЖХМС (ИЭР) m/z: 512,3 [M+H]+
[00240] Четвертый шаг: (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-7)
[00241]
[00242] Исходный материал метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-7Г) (100 мг, 0,19 ммоль) добавляли к ТГФ (5 мл) при комнатной температуре, и добавляли воду (4 мл) и гидроксид лития моногидрат (10 мг, 0,24 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-7) (47,2 мг, выход 48,5%).
[00243] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,8 (с, 1H), 8,58 (д, 1H), 7,73 (д, 2H), 7,53 (т, 1H), 7,40 (д, 1H), 7,27 (с, 1H), 7,15 (д, 2H), 7,11 (д, 1H), 4,85 (т, 1H), 3,29 (с, 3H), 1,96 (т, 3H), 1,17 (д, 3H).
[00244] ЖХМС (ИЭР) m/z: 498,3 [M+H]+
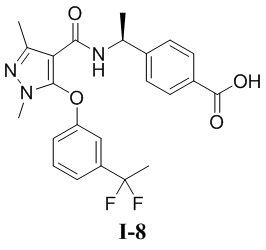
[00345] Пример 8: получение соединения I-8
[00246] (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-8)
[00247] 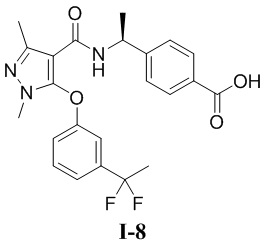
[00248] Схема синтеза для соединения I-8 показана ниже:
[00249] 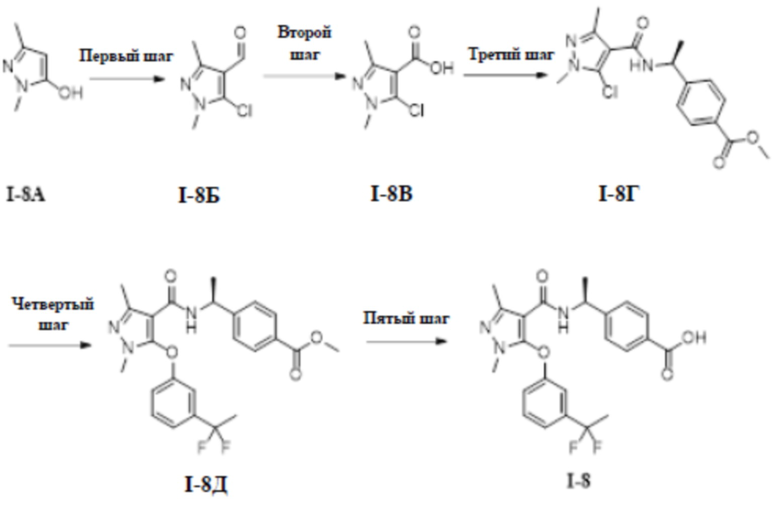
[00250] Первый шаг: 5-хлор-1,3-диметил-1H-пиразол-4-карбальдегид (соединение I-8Б)
[00251] 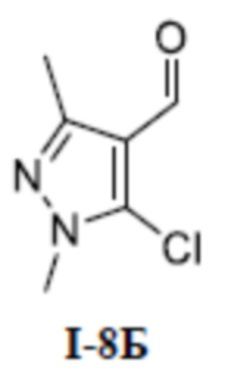
[00252] 1, 3-диметил-5-гидроксипиразол (5,5 г, 49,1 ммоль) добавляли к ДМФА (10,9 г) при комнатной температуре, и смесь охлаждали до 0°C, и добавляли POCl3 (53,0 г, 346,4 ммоль), а затем подогревали естественным путем до комнатной температуры, нагревали до 120°C и перемешивали в течение 1 ч. Затем смесь охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали с помощью ЭА (200 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветной жидкости 5-хлор-1,3-диметил-1H-пиразол-4-карбальдегида (соединение I-8Б) (4,3 г, выход 55,4%).
[00253] ЖХМС (ИЭР) m/z: 159,6 [M+H]+
[00254] Второй шаг: 5-хлор-1,3-диметил-1H-пиразол-4-карбоновая кислота (соединение I-8В)
[00255] 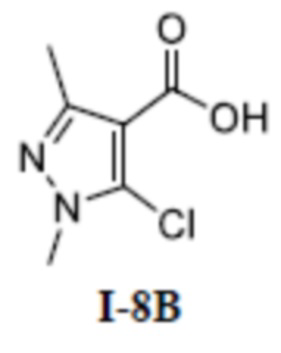
[00256] Соединение 5-хлор-1,3-диметил-1H-пиразол-4-карбальдегид (соединение I-8Б) (3,20 г, 20,2 ммоль) добавляли к трет-бутанолу (50 мл) и воде (15 мл) при комнатной температуре; и добавляли 2-метил-2-бутен (11,3 г, 161,4 ммоль), хлорит натрия (9,10 г, 101,1 ммоль) и дигидрофосфат натрия (19,4 г, 161,6 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре; а затем смесь разбавляли водой (50 мл), экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного бесцветного жидкого остатка указанного соединения. Остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого вещества 5-хлор-1,3-диметил-1H-пиразол-4-карбоновой кислоты (соединение I-8В) (3,10 г, выход 87,9%).
[00257] ЖХМС (ИЭР) m/z: 175,6 [M+H]+
[00258] Третий шаг: метил (S)-4-(1-(5-хлор-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-8Г)
[00259] 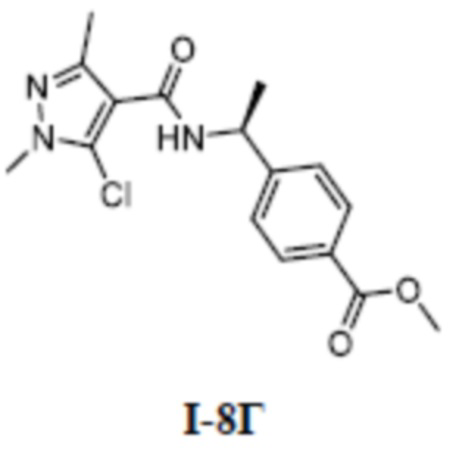
[00260] Соединение 5-хлор-1,3-диметил-1H-пиразол-4-карбоновую кислоту (соединение I-8В) (1,20 г, 6,89 ммоль) добавляли к ДМФА (30 мл); и добавляли (S)-метил 4-(l-аминоэтил)бензоат (1,23 г, 6,87 ммоль), ГАТУ (3,90 г, 10,1 ммоль) и ДИПЭА (4,50 г, 34,8 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли водой (200 мл), экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого вещества метил (S)-4-(1-(5-хлор-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-8Г) (1,90 г, выход 82,2%).
[00261] ЖХМС (ИЭР) m/z: 336,6 [M+H]+
[00262] Четвертый шаг: метил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-8Д)
[00263] 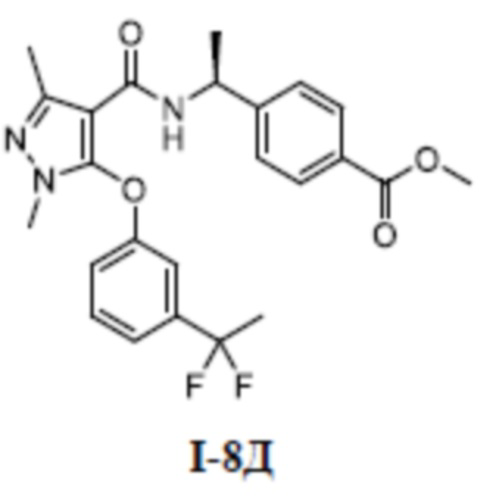
[00264] Соединение метил (S)-4-(1-(5-хлор-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-8Г) (335 мг, 1,00 ммоль) добавляли к ДМФА (10 мл) при комнатной температуре, и добавляли 3-(l, l-дифторэтил)фенол (158 мг, 1,00 ммоль) и KOH (150 мг, 2,67 ммоль). Смесь нагревали до 120°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл ×3) и разделяли. Органические фазы объединяли и сушили с помощью безводного сульфата натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта этил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-8Д) (170 мг, выход 37,1%).
[00265] ЖХМС (ИЭР) m/z: 458,5 [M+H]+
[00266] Пятый шаг: (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-8)
[00267]
[00268] Исходный материал этил (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-8Д) (500 мг, 1,09 ммоль) добавляли к ТГФ (5 мл) при комнатной температуре, и добавляли воду (4 мл) и гидроксид лития моногидрат (340 мг, 8,11 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(3-(1,1-дифторэтил)фенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-8) (53 мг, выход 10,9%).
[00269] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,8 (с, 1H), 7,73 (д, 2H), 7,71 (д, 1H), 7,52 (т, 1H), 7,37 (д, 1H), 7,21 (с, 1H), 7,14 (д, 2H), 7,01 (дд, 1H), 4,91 (т, 1H), 3,59 (с, 3H), 2,27 (с, 3H), 1,96 (т, 3H), 1,22 (д, 3H).
[00270] ЖХМС (ИЭР) m/z: 444,5 [M+H]+
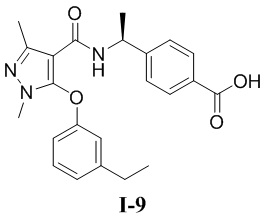
[00271] Пример 9: получение соединения I-9
[00272] (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-9)
[00273] Схема синтеза для соединения I-9 показана ниже:
[00274] 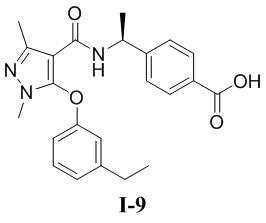
[00275] Схема синтеза для соединения I-9 показана ниже:
[00276] 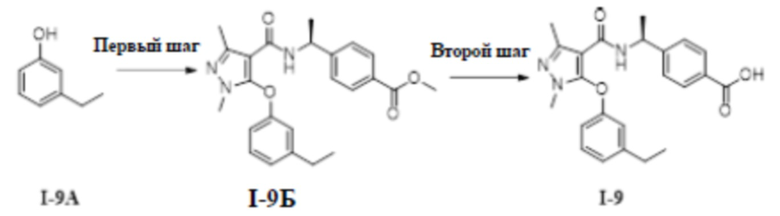
[00277] Первый шаг: метил (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-9Б)
[00278] 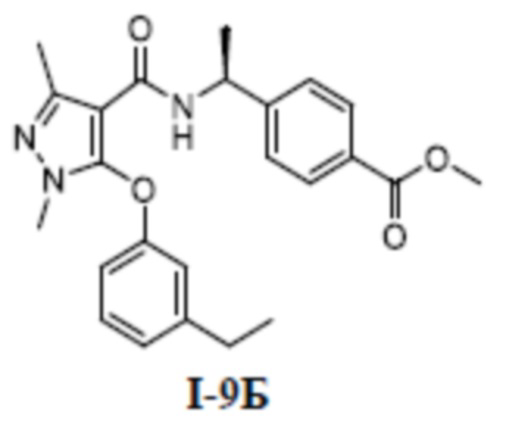
[00279] Соединение метил (S)-4-(1-(5-хлор-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-8Г) (100 мг, 0,30 ммоль) добавляли к ДМФА (3 мл) при комнатной температуре; добавляли 3-этилфенол (36 мг, 0,30 ммоль) и KOH (49 мг, 0,86 ммоль); и смесь нагревали до 120°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (20 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-9Б) (60 мг, выход 47,7%).
[00280] ЖХМС (ИЭР) m/z: 422,6 [M+H]+
[00281] Второй шаг: (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-9)
[00282]
[00283] Исходный материал метил (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-9Б) (60 мг, 0,14 ммоль) добавляли к ТГФ (2 мл) при комнатной температуре, и добавляли воду (2 мл) и гидроксид лития моногидрат (6 мг, 0,14 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(3-этилфенокси)-1,3-диметил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-9) (10 мг, выход 17,2%).
[00284] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,6 (с, 1H), 7,72 (д, 2H), 7,50 (д, 1H), 7,31 (т, 1H), 7,12 (д, 2H), 7,04 (д, 1H), 6,85 (с, 1H), 6,71 (дд, 1H), 4,92 (т, 1H), 3,56 (с, 3H), 2,67 (к, 2H), 2,27 (с, 3H), 1,24 (д, 3H), 1,14 (т, 3H).
[00285] [00207] ЖХМС (ИЭР) m/z: 408,6 [M+H]+
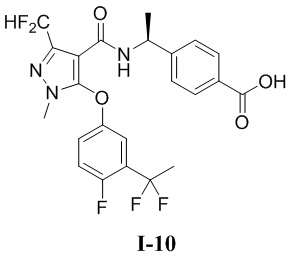
[00286] Пример 10: получение соединения I-10
[00287] (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-10)
[00288] 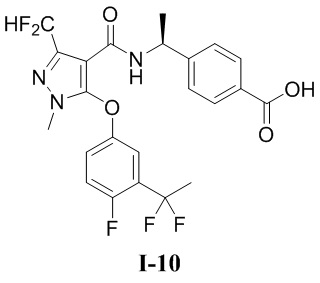
[00289] Схема синтеза для соединения I-10 показана ниже:
[00290] 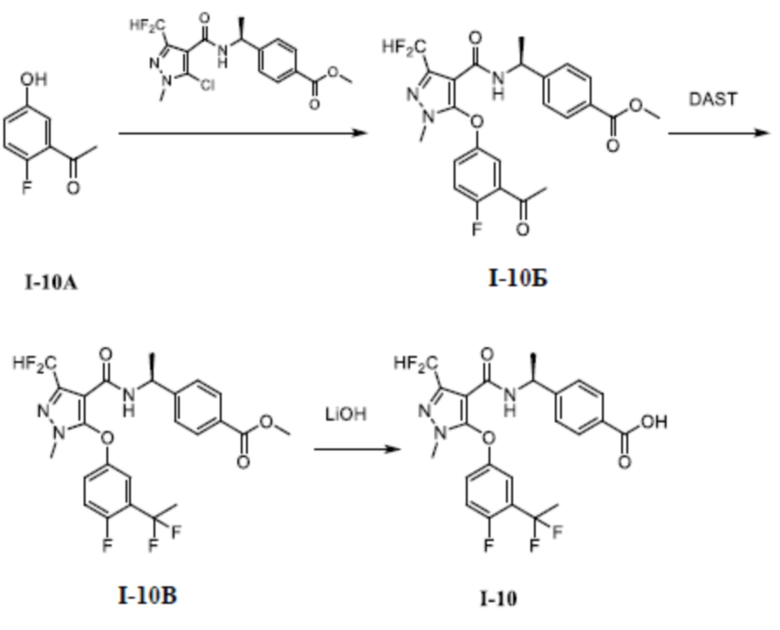
[00291] Первый шаг: метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-10Б)
[00292] 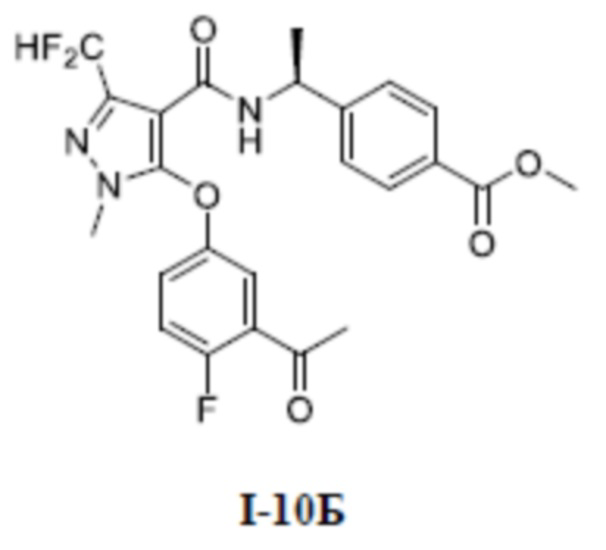
[00293] Соединение 1-(2-фтор-5-гидроксифенил)этан-1-он (1,58 г, 10,8 ммоль) добавляли к N, N-диметилформамиду (30 мл) при комнатной температуре; добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (2,0 г, 5,4 ммоль) и гидроксид калия (450 мг, 8,1 ммоль); и смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Смесь охлаждали до комнатной температуры и добавляли воду (30 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением белого твердого вещества метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-10Б) (346 мг, выход 13%).
[00294] ЖХ-МС, M/Z (ИЭР): 490,2 [M+H]+
[00295] Второй шаг: метил (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-10В)
[00296] 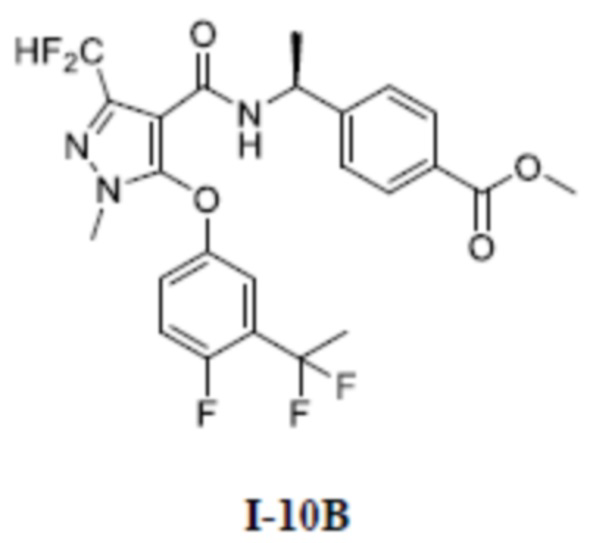
[00297] Соединение метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-10Б) (346 мг, 0,71 ммоль) добавляли к (диэтиламино)серы трифториду (10 мл) при комнатной температуре. Смесь нагревали до 50°C и перемешивали в течение 16 часов. Затем смесь охлаждали до 0°C, разбавляли водой (50 мл), экстрагировали с помощью ЭА (100 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого неочищенного продукта метил (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-10Б) (390 мг, выход 100%).
[00298] ЖХ-МС, M/Z (ИЭР): 512,2 [M+H]+
[00299] Третий шаг 3: (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-10)
[002300]
[00301] Соединение метил (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-10В) (390 мг, 0,76 ммоль) добавляли к тетрагидрофурану (10 мл), метанолу (10 мл) и воде (3 мл) при комнатной температуре, и добавляли гидроксид лития (64 мг, 1,52 ммоль). Смесь перемешивали при комнатной температуре в течение 36 часов, а затем pH доводили до 7 с помощью 1 н. хлористоводородной кислоты. Смесь концентрировали с получением, посредством кислотного способа получения, белого твердого вещества (S)-4-(1-(5-(3-(1,1-дифторэтил)-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-10) (116 мг, выход 31%).
[00302] ЖХ-МС, M/Z (ИЭР):498,1 [M+H]+
[00303] 1H ЯМР (400 МГц, ДМСО-d6) δ12,10 (с, 1H), 8,19 (д, 1H), 7,75 (д, 2H), 7,39 (т, 1H), 7,26 (т, 1H), 7,15 (д, 2H), 7,13 (д, 1H), 7,12 (т, 1H), 4,90 (т, 1H), 3,75 (с, 3H), 2,01 (т, 3H), 1,22 (д, 3H).
[00304] Пример 11: получение соединения I-11
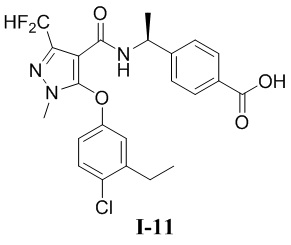
[00305] (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-11)
[00306] 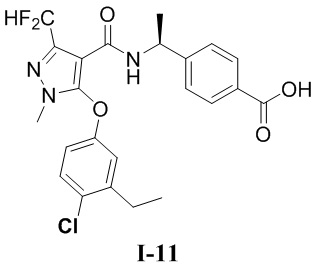
[00307] Схема синтеза для соединения I-11 показана ниже:
[00308] 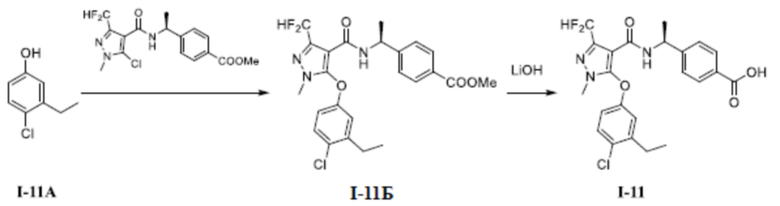
[00309] Первый шаг: метил (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-11Б)
[00310] 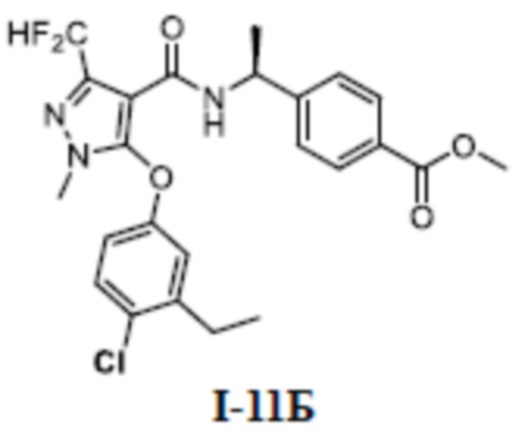
[00311] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (371 мг, 1,00 ммоль) добавляли к диметилсульфоксиду (3 мл) при комнатной температуре; добавляли 4-хлор-3-этилфенол (156 мг, 1,00 ммоль) и гидроксид калия (112 мг, 2,00 ммоль); и смесь нагревали до 120°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (10 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта метил (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-11Б) (80 мг, выход 16%).
[00312] ЖХ-МС, M/Z (ИЭР): 492,3 (M+1).
[00313] Второй шаг: (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-11)
[00314]
[00315]
[00316] Исходный материал метил (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-11Б) (80 мг, 0,16 ммоль) добавляли к тетрагидрофурану (1 мл) при комнатной температуре; и добавляли воду (1 мл) и гидроксид лития моногидрат (21 мг, 0,48 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(5-(4-хлор-3-этилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-11) (13,3 мг, выход 17%).
[00317] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,04 (д, 1H), 7,73 (д, 2H), 7,41 (д, 1H), 7,11 (т, 1H), 7,10 (д, 2H), 7,08 (д, 1H), 6,82 (д, 1H), 4,91 (т, 1H), 3,73 (с, 3H), 2,67 (к, 2H), 1,24 (д, 3H),1,11 (т, 3H).
[00318] ЖХ-МС, M/Z (ИЭР): 478,3 (M+1)
[00319] Пример 12: получение соединения I-12
[00320] (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-12)
[00321] 
[00322] Схема синтеза для соединения I-12 показана ниже:
[00323] 
[00324] Первый шаг: 1-фтор-3-метокси-5-винилбензол (соединение I-12Б)
[00325] 
[00326] 1-бром-3-фтор-5-метоксибензол (2,0 г, 9,7 ммоль) добавляли к 1, 4-диоксану (50 мл) при комнатной температуре; добавляли винилфторборат калия (1,6 г, 11,7 ммоль); [1, 1-бис(дифенилфосфино)ферроцен]дихлорпалладий (1,4 г, 1,96 ммоль) и карбонат калия (2,7 г, 19,6 ммоль); и смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали дихлорметаном (80 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (чистый петролейный эфир) с получением 1-фтор-3-метокси-5-винилбензола (соединение I-12Б) (600 мг, выход 40%).
[00327] Второй шаг: 1-этил-3-фтор-5-метоксибензол (соединение I-12В)
[00328] 
[00329] 1-фтор-3-метокси-5-винилбензол (300 мг, 1,97 ммоль) добавляли к метанолу (20 мл) при комнатной температуре и добавляли 10% палладий на угле (200 мг). При введении H2 смесь перемешивали при комнатной температуре в течение 16 часов. Смесь фильтровали и промывали метанолом (30 мл ×3), а органические фазы объединяли и концентрировали с получением бесцветного жидкого неочищенного продукта 1-этил-3-фтор-5-метоксибензола (соединение I-12В) (200 мг, выход 66%).
[00330] Третий шаг: 3-этил-5-фторфенол (соединение I-12Г)
[00331] 
[00332] l-этил-3-фтор-5-метоксибензол (200 мг, 1,3 ммоль) добавляли к дихлорметану (2 мл) при комнатной температуре. Смесь охлаждали до 0°C, и добавляли BBr3 (1 моль/л в дихлорметане, 2 мл). Смесь подогревали естественным путем до комнатной температуры и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили добавлением воды (3 мл), и смесь экстрагировали дихлорметаном (5 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта 3-этил-5-фторфенола (соединение I-12Г) (180 мг, выход 99%).
[00333] Четвертый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-12Д)
[00334] 
[00335] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (250 мг, 0,67 ммоль) добавляли к N, N-диметилформамиду (15 мл) и нагревали до 120°C; добавляли 3-этил-5-фторфенол (180 мг, 1,28 ммоль), карбонат калия (270 мг, 1,96 ммоль), йодид меди (I) (50 мг, 0,26 ммоль) и 1, 10-фенантролин (90 мг, 0,50 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре. Затем смесь охлаждали до комнатной температуры, разбавляли водой (30 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением бесцветной жидкости метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-12Д) (200 мг, выход 62%).
[00336] ЖХ-МС, M/Z (ИЭР): 476,2 (M+1).
[00337] Пятый шаг: (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-12)
[00338] 
[00339] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (200 мг, 0,42 ммоль) добавляли к тетрагидрофурану (10 мл) и воде (5 мл) при комнатной температуре, и добавляли гидроксид лития моногидрат (53 мг, 1,26 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением белого твердого вещества соединения (S)-4-(1-(3-(дифторметил)-5-(3-этил-5-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (130 мг, выход 67%).
[00340] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,10 (д, 1H), 7,74 (д, 2H), 7,16 (д, 2H), 7,11 (т, 1H), 6,93 (д, 1H), 6,72 (с, 1H), 6,69 (д, 1H), 4,92 (т, 1H), 3,73 (с, 3H), 2,58-2,54 (м, 2H), 1,25 (д, 3H), 1,13 (т, 3H).
[00341] ЖХ-МС, M/Z (ИЭР): 462,1 (M+1)
[00342] Пример 13: получение соединения I-13
[00343] 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-13)
[00344]
[00345] Схема синтеза для соединения I-13 показана ниже:
[00346] 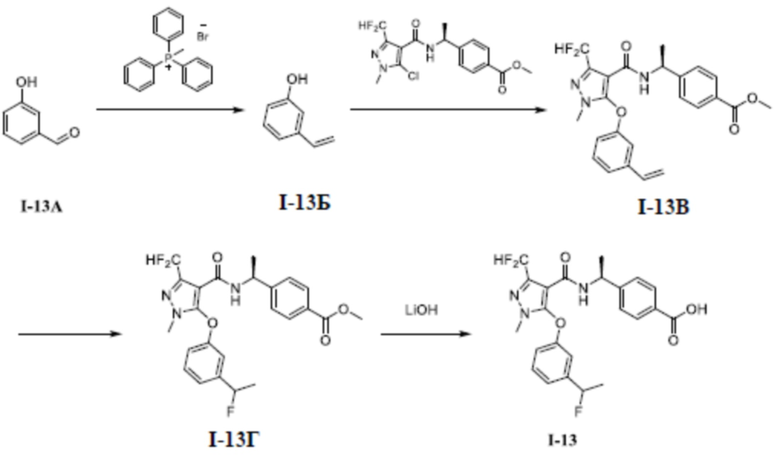
[00347] Первый шаг: 3-винилфенол (соединение I-13Б)
[00348] 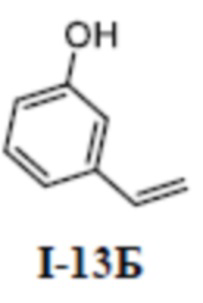
[00349] Соединение метилтрифенилфосфина бромид (7,30 г, 20,5 ммоль) добавляли к безводному тетрагидрофурану (40 мл) при 0°C, и добавляли трет-бутоксид калия (2,3 г, 20,5 ммоль). Смесь перемешивали при низкой температуре в течение 2 часов. Раствор 3-гидроксибензальдегида (1,0 г, 8,20 ммоль) и тетрагидрофурана (15 мл) добавляли по каплям, и, после завершения добавления, смесь подогревали естественным путем до комнатной температуры и перемешивали в течение 16 часов. Смесь разбавляли водой (100 мл), экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением желтой жидкости 3-винилфенола (соединение I-13Б) (500 мг, выход 49%).
[00350] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-13В)
[00351] 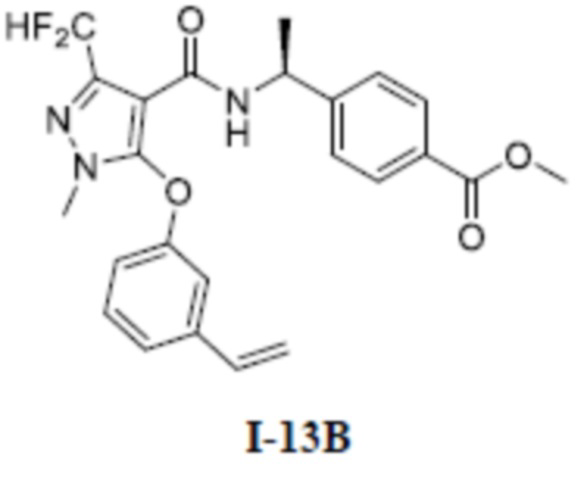
[00352] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (600 мг, 1,62 ммоль) добавляли к N, N-диметилформамиду (18 мл) при комнатной температуре; и добавляли 3-винилфенол (соединение I-13Б) (388 мг, 3,23 ммоль) и гидроксид калия (181 мг, 3,23 ммоль). Смесь нагревали до 120°C в микроволновой печи и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (60 мл), экстрагировали этилацетатом (60 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-13В) (200 мг, выход 27%).
[00353] ЖХ-МС, M/Z (ИЭР): 456,2 (M+1).
[00354] Третий шаг: метил 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-13Г)
[00355] 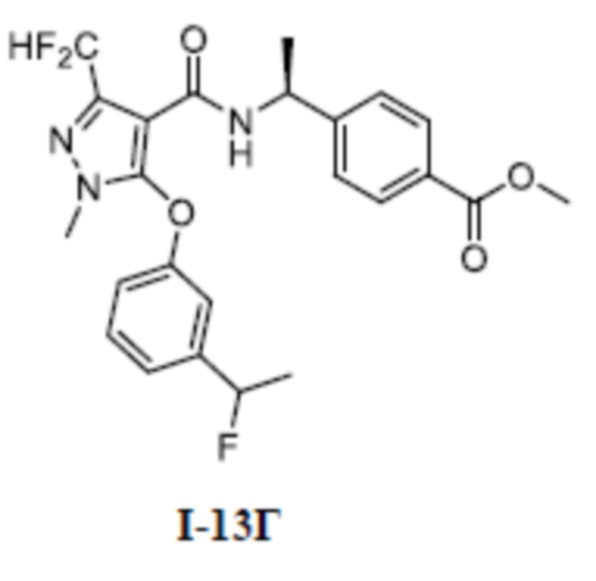
[00356] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-13В) (100 мг, 0,22 ммоль) добавляли к ацетонитрилу (25 мл); и селективный фторирующий реагент (233 мг, 0,66 ммоль), палладий тетракистрифенилфосфоний (25 мг, 0,02 ммоль) и триэтилсилан (40,0 мг, 0,33 ммоль) добавляли при комнатной температуре. Смесь перемешивали в течение 16 часов в защитной атмосфере азота при комнатной температуре. Затем смесь разбавляли водой (60 мл), экстрагировали этилацетатом (60 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 5:1) с получением белого твердого вещества метил 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-13Г) (65,0 мг, выход 62%).
[00357] ЖХ-МС, M/Z (ИЭР): 476,3 (M+1)
[00358] Четвертый шаг: 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-13)
[00359]
[00360] Исходный материал метил 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-13Г) (150 мг, 0,19 ммоль) добавляли к тетрагидрофурану (24 мл); и добавляли воду (12 мл) и метанол (12 мл), гидроксид лития (15,0 мг, 0,36 ммоль) при комнатной температуре. Смесь перемешивали в течение 4 часов при комнатной температуре. Реакционную смесь концентрировали с получением, посредством кислотного способа получения A, белого твердого вещества 4-((1S)-1-(3-(дифторметил)-5-(3-(1-фторэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-13) (50,0 мг, выход 32%).
[00361] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,03 (д, 1H), 7,72 (д, 2H), 7,45 (т, 1H), 7,25 (т, 1H), 7,11 (д, 2H), 6,98 (т, 1H), 6,94 (т, 1H), 5,78 (дд, 1H),4,89 (м, 1H), 3,73 (с, 3H), 3,60 (с, 1H), 1,55 (дд, 3H), 1,24 (д, 3H).
[00362] ЖХ-МС, M/Z (ИЭР): 462,3 (M+1)
[00363] Пример 14: получение соединения I-14
[00364] (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-14)
[00365] 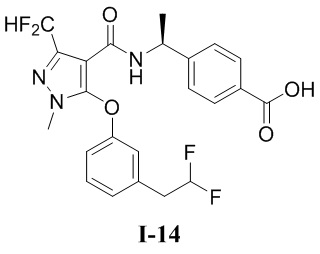
[00366] Схема синтеза для соединения I-14 показана ниже:
[00367] 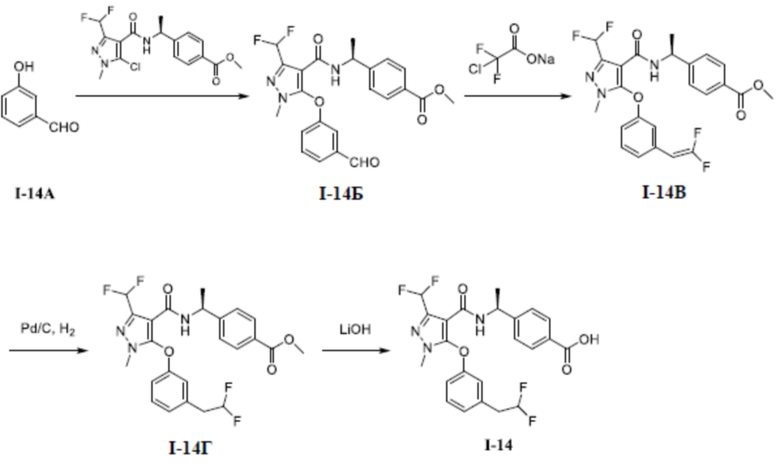
[00368] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14Б)
[00369] 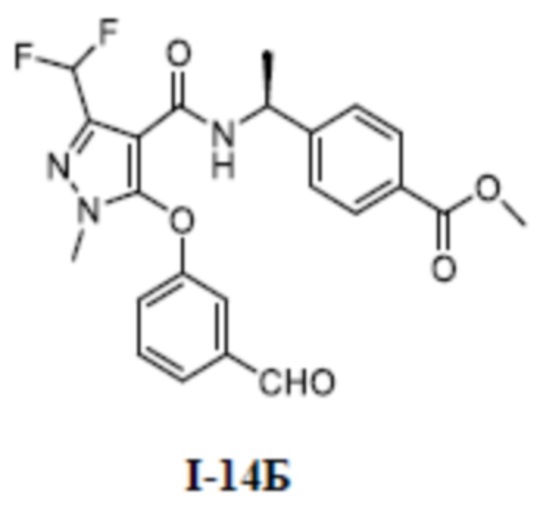
[00370] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (574 мг, 1,54 ммоль) добавляли к N, N-диметилформамиду (15 мл) при комнатной температуре; и добавляли 3-гидроксибензальдегид (378 мг, 3,09 ммоль), йодид меди (II) (60 мг, 0,31 ммоль), 1, 10-фенантролин (114 мг, 0,62 ммоль) и карбонат цезия (1,50 г, 4,62 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты. Смесь экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением светло-желтого твердого вещества метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-14Б) (280 мг, выход 40%).
[00371] ЖХ-МС, M/Z (ИЭР): 458,3 [M+H]+.
[00372] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-(2,2-дифторвинил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14В)
[00373] 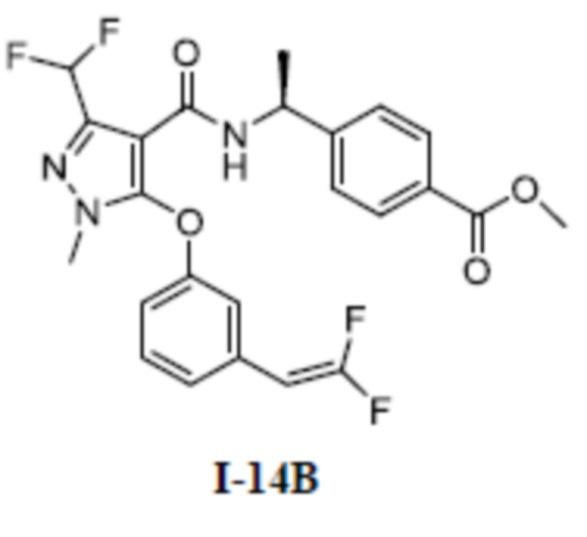
[00374] Соединение метил (S)-4-(1-(3-(дифторметил)-5-(3-формилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14Б) (280 мг, 0,61 ммоль) добавляли к N, N-диметилформамиду (15 мл) при комнатной температуре, и добавляли трифенилфосфин (200 мг, 0,76 ммоль) и дифторхлорацетат натрия (140 мг, 0,92 ммоль), и смесь нагревали до 100°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 1 ч. Смесь охлаждали до комнатной температуры, разбавляли посредством добавления воды (50 мл), и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, а смесь экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтого твердого вещества метил (S)-4-(1-(3-(дифторметил)-5-(3-(2,2-дифторвинил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-14В) (120 мг, выход 40%).
[00375] ЖХ-МС, M/Z (ИЭР): 492,4 [M+H]+.
[00376] Третий шаг: метил (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14Г)
[00377] 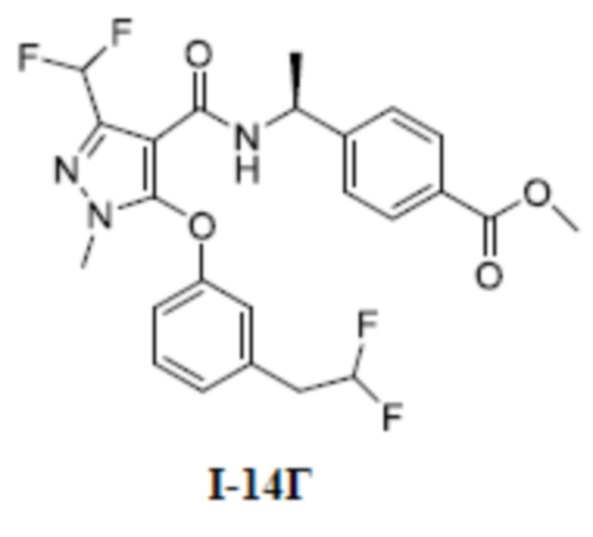
[00378] Соединение метил (S)-4-(1-(3-(дифторметил)-5-(3-(2,2-дифторвинил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14В) (120 мг, 0,24 ммоль) добавляли к метанолу (20 мл) при комнатной температуре; добавляли 10% палладий на угле (20 мг); и вводили водород. Смесь перемешивали при комнатной температуре в течение 16 часов. Смесь фильтровали и концентрировали с получением светло-желтого твердого неочищенного продукта метил (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-14Г) (120 мг, выход 99%).
[00379] ЖХ-МС, M/Z (ИЭР): 494,4 [M+H]+.
[00380] Четвертый шаг: (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-14)
[00381] 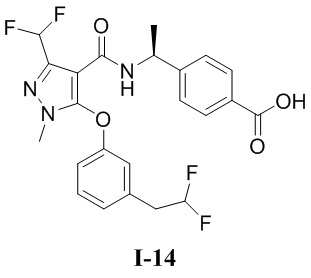
[00382] Исходный материал метил (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-14Г) (120 мг, 0,24 ммоль) добавляли к тетрагидрофурану (5 мл) и воде (5 мл) при комнатной температуре, а затем добавляли гидроксид лития (31 мг, 0,72 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-(3-(2,2-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-14) (90 мг, выход 77%).
[00383] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 7,95 (д, 1H), 7,74 (д, 2H), 7,39 (т, 1H), 7,25 (т, 1H), 7,16 (д, 2H), 7,13 (д, 1H), 7,04 (д, 1H), 6,91 (т, 1H), 6,34 (дт, 1H), 4,91 (т, 1H), 3,71 (с, 3H), 3,22 (к, 2H), 1,22 (д, 3H).
[00384] ЖХ-МС, M/Z (ИЭР): 480,4 [M+H]+
[00385] Пример 15: получение соединения I-15
[00386] 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-15)
[00387] 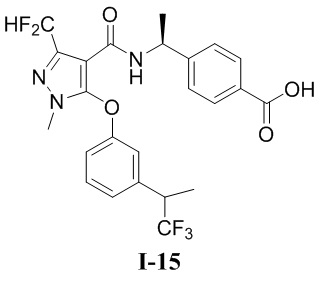
[00388] Схема синтеза для соединения I-15 показана ниже:
[00389] 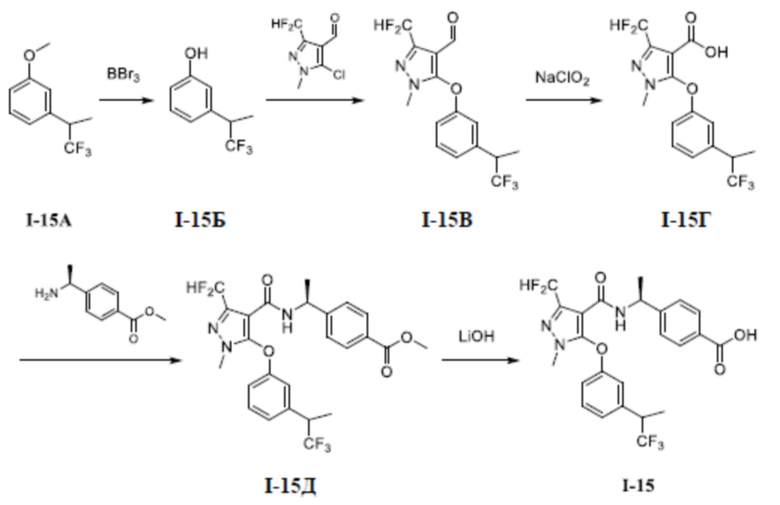
[00390] Первый шаг: 3-(1,1,1-трифторпропан-2-ил)фенол (соединение I-15Б)
[00391] 
[00392] Соединение l-метокси-3-(l, l, l-трифторпропан-2-ил)бензол (800 мг, 0,72 ммоль) добавляли к дихлорметану (1 мл) при комнатной температуре, и затем смесь охлаждали до -78°C, и затем добавляли трибромид бора (1,50 г, 5,88 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов; а затем смесь разбавляли водой (50 мл), экстрагировали дихлорметаном (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 5:1) с получением светло-желтой жидкости 3-(1,1,1-трифторпропан-2-ил)фенола (соединение I-15Б) (640 мг, выход 86%).
[00393] Второй шаг: 3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбальдегид (соединение I-15В)
[00394] 
[00395] Соединение 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (660 мг, 3,37 ммоль) добавляли к N, N-диметилформамиду (6 мл) при комнатной температуре, а затем добавляли 3-(1, 1, 1-трифторпропан-2-ил)фенол (640 мг, 3,37 ммоль) и карбонат калия (930 мг, 6,72 ммоль). Смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 8 ч; а затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 5:1) с получением светло-желтой жидкости 33-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбальдегида (соединение I-15В) (1,0 г, выход 18%).
[00396] ЖХ-МС, M/Z (ИЭР): 349,2 [M+H]+.
[00397] Третий шаг: 3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоновая кислота (соединение I-15Г)
[00398] 
[00399] Соединение 3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбальдегид (800 мг, 2,30 ммоль) добавляли к трет-бутанолу (8 мл) и воде (8 мл) при комнатной температуре. Добавляли 2-метил-2-бутен (322 мг, 4,60 ммоль), хлорит натрия (414 мг, 4,60 ммоль) и дигидрофосфат натрия (617 мг, 5,06 ммоль). Смесь перемешивали при комнатной температуре в течение 8 ч; а затем смесь разбавляли водой (15 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого твердого неочищенного вещества 3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоновой кислоты (соединение I-15Г) (850 мг, выход 100%).
[00340] ЖХ-МС, M/Z (ИЭР): 365,1 [M+H]+.
[00341] Четвертый шаг: метил 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-15Д)
[00342] 
[00343] Соединение 3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоновую кислоту (850 мг, 2,34 ммоль) добавляли к N, N-диметилформамиду (10 мл); и добавляли метил (S)-4-(1-аминоэтил)бензоат (418 мг, 2,34 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (1,3 г, 3,50 ммоль) и N, N-диизопропилэтиламин (900 мг, 7,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли водой (40 мл), экстрагировали этилацетатом (40 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением светло-желтой жидкости метил 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-15Д) (1,0 г, выход 81%).
[00344] ЖХ-МС, M/Z (ИЭР): 526,3 [M+H]+.
[00345] Пятый шаг: 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-15)
[00346] 
[00347] Исходный материал метил 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-15Д) (1,0 г, 1,90 ммоль) добавляли к тетрагидрофурану (10 мл); и воде (2 мл) при комнатной температуре; и добавляли гидроксид лития (200 мг, 4,76 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(1,1,1-трифторпропан-2-ил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-15) (356 мг, выход 37%).
[00348] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 7,97 (дд, 1H), 7,73 (д, 2H), 7,43 (дд, 1H), 7,28 (д, 1H), 7,25 (т, 1H), 7,13 (д, 2H), 7,11 (д, 1H), 6,99 (т, 1H), 4,90 (т, 1H), 3,89-3,85 (м, 1H), 3,73 (с, 3H), 1,41 (д, 3H), 1,21 (т, 3H).
[00349] ЖХ-МС, M/Z (ИЭР): 512,3 [M+H]+
[00350] Пример 16: получение соединения I-16
[00351] (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-16)
[00352]
[00353] Схема синтеза для соединения I-16 показана ниже:
[00354] 
[00355] Первый шаг: 1-(бензилокси)-2,3-дифтор-5-метоксибензол (соединение I-16Б)
[00356] 
[00357] Соединение 1, 2, 3-трифтор-5-метоксибензол (8,5 г, 52,4 ммоль) добавляли к толуолу (100 мл) при комнатной температуре; добавляли бензиловый спирт (11,3 г, 104 ммоль) и гидроксид калия (5,86 г, 104 ммоль); и смесь нагревали до 120°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (500 мл) и pH доводили до 7-8 с помощью 1 н. хлористоводородной кислоты. Смесь экстрагировали дихлорметаном (100 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.), 20:1) с получением бесцветной жидкости 1-(бензилокси)-2,3-дифтор-5-метоксибензола (соединение I-16Б) (5,2 г, выход 40%).
[00358] Второй шаг: 2,3-дифтор-5-метоксифенол (соединение I-16В)
[00359] 
[00360] Соединение 1-(бензилокси)-2,3-дифтор-5-метоксибензол (5,2 г, 20,8 ммоль) добавляли к тетрагидрофурану (100 мл) при комнатной температуре; добавляли 10% палладий на угле (520 мг); и вводили водород. Смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь фильтровали, концентрировали и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 5:1) с получением бесцветного жидкого неочищенного продукта 2,3-дифтор-5-метоксифенола (соединение I-16В) (3,8 г, выход 100%).
[00361] Третий шаг: 2,3-дифтор-5-метоксифенилтрифторметансульфонат (соединение I-16Г)
[00362] 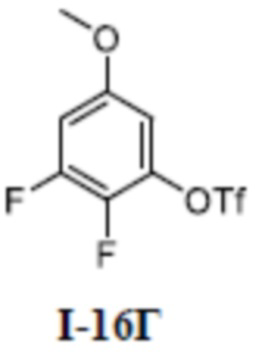
[00363] Соединение 2,3-дифтор-5-метоксифенол (500 мг, 3,12 ммоль) добавляли к дихлорметану (20 мл) при комнатной температуре; и добавляли трифторметансульфоновый ангидрид (881 мг, 3,12 ммоль) и пиридин (493 мг, 6,25 ммоль). Смесь перемешивали в течение 4 часов при комнатной температуре; добавляли насыщенный бикарбонат натрия (20 мл), а затем смесь разделяли и органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта 2,3-дифтор-5-метоксифенилтрифторметансульфоната (соединение I-16Г) (600 мг, выход 66%).
[00364] Четвертый шаг: 1,2-дифтор-5-метокси-3-винилбензол (соединение I-16Д)
[00365] 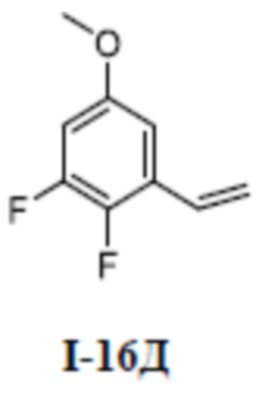
[00366] Соединение 2,3-дифтор-5-метоксифенилтрифторметансульфонат (600 мг, 2,05 ммоль) добавляли к 1, 4-диоксану (50 мл) при комнатной температуре; добавляли винилтрифторборат калия (1,25 г, 9,37 ммоль); карбонат калия (1,30 г, 9,37 ммоль) и дихлор[1, 1’-бис(дифенилфосфино)ферроцен]палладий (254 мг, 0,31 ммоль); и смесь нагревали до 90°C в защитной атмосфере азота и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (200 мл), экстрагировали дихлорметаном (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 100:1) с получением светло-желтой жидкости 1,2-дифтор-5-метокси-3-винилбензола (соединение I-16Д) (200 мг, выход 57%).
[00367] Пятый шаг: 1-этил-2,3-дифтор-5-метоксибензол (соединение I-16Е)
[00368] 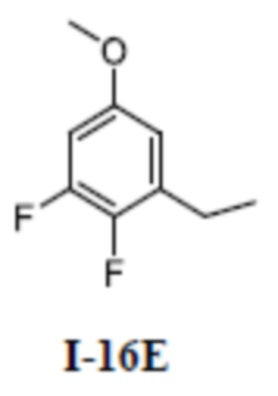
[00369] Соединение 1,2-дифтор-5-метокси-3-винилбензол (130 мг, 0,82 ммоль) добавляли к метанолу (15 мл) при комнатной температуре; добавляли 10% палладий на угле (20 мг); и вводили водород при перемешивании при комнатной температуре в течение 16 часов. Смесь фильтровали и концентрировали с получением 1-этил-2,3-дифтор-5-метоксибензола (соединение I-16Е) (100 мг, выход 76%), светло-желтой жидкости.
[00370] Шестой шаг: 3-этил-4,5-дифторфенол (соединение I-16Ж)
[00371] 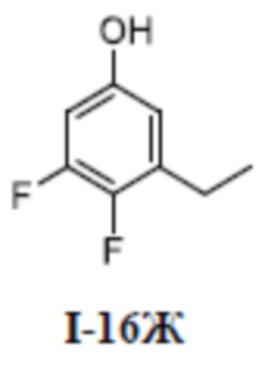
[00372] Соединение 1-этил-2,3-дифтор-5-метоксибензол (100 мг, 0,58 ммоль) добавляли к дихлорметану (10 мл) при комнатной температуре; добавляли трибромид бора (726 мг, 2,90 ммоль) при 0°C при перемешивании в течение 3 ч; и смесь разбавляли посредством добавления воды (20 мл), экстрагировали дихлорметаном (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого жидкого неочищенного продукта 3-этил-4,5-дифторфенола (соединение I-16Ж) (60 мг, выход 66%).
[00373] Седьмой шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-16И)
[00374] 
[00375] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (600 мг, 1,61 ммоль) добавляли к N, N-диметилформамиду (15 мл) при комнатной температуре; и добавляли 3-этил-4, 5-дифторфенол (450 мг, 3,0 ммоль) и гидроксид калия (210 мг, 3,9 ммоль). Смесь нагревали до 120°C в микроволновой печи и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты. Смесь экстрагировали этилацетатом (60 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтого твердого вещества метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-16И) (350 мг, выход 44%).
[00376] ЖХ-МС, M/Z (ИЭР): 494,4 [M+H]+.
[00377] Восьмой шаг: (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-16)
[00378] 
[00379] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (325 мг, 0,66 ммоль) добавляли к тетрагидрофурану (15 мл), метанолу (3 мл) и воде (3 мл) при комнатной температуре; и добавляли гидроксид лития (32 мг, 1,32 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(3-этил-4,5-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (170 мг, выход 54%).
[00380] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,15 (д, 1H), 7,76 (д, 2H), 7,24 (т, 1H), 7,18 (д, 2H), 7,01 (т, 1H), 6,80 (д, 1H), 4,94 (т, 1H), 3,74 (с, 3H), 2,62 (к, 2H), 1,27 (д, 3H), 1,11 (т, 3H).
[00381] ЖХ-МС, M/Z (ИЭР): 480,4 [M+H]+
[00382] Пример 17: получение соединения I-17
[00383] (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-17)
[00384] 
[00385] Схема синтеза для соединения I-17 показана ниже:
[00386] 
[00387] Первый шаг: метил (S)-4-(1-(5-(3-бром-4-метилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-17Б)
[00388] 
[00389] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (500 мг, 1,35 ммоль) добавляли к N, N-диметилформамиду (10 мл) при комнатной температуре; добавляли 3-бром-4-метилфенол (380 мг, 2,02 ммоль), йодид меди (I) (100 мг, 0,52 ммоль), 1, 10-фенантролин (180 мг, 1,0 ммоль) и карбонат цезия (1,30 г, 4,05 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты. Смесь экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением светло-желтого твердого вещества метил (S)-4-(1-(5-(3-бром-4-метилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-17Б) (270 мг, выход 38%).
[00390] ЖХ-МС, M/Z (ИЭР): 522,3 [M+H]+.
[00391] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-метил-3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-17В)
[00392] 
[00393] Соединение метил (S)-4-(1-(5-(3-бром-4-метилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-17Б) (260 мг, 0,50 ммоль) добавляли к 1, 4-диоксану (5 мл) при комнатной температуре; добавляли дихлор[l, l’-бис(дифенилфосфино)ферроцен]палладий (73 мг, 0,10 ммоль), винилтрифторборат калия (100 мг, 0,75 ммоль) и карбонат калия (140 мг, 1,0 ммоль); и смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-метил-3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-17В) (160 мг, выход 68%).
[00394] ЖХ-МС, M/Z (ИЭР): 470,3 [M+H]+.
[00395] Третий шаг: метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-17Г)
[00396] 
[00397] Соединение метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-метил-3-винилфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (160 мг, 0,34 ммоль) добавляли к метанолу (10 мл) при комнатной температуре и добавляли 10% палладий на угле (20 мг); и вводили водород при перемешивании в течение 16 часов при комнатной температуре; и смесь фильтровали и концентрировали с получением светло-желтого твердого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-17Г) (150 мг, выход 93%).
[00398] ЖХ-МС, M/Z (ИЭР): 472,3 [M+H]+
[00399] Четвертый шаг: (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-17)
[00400]
[00401] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-17Г) (150 мг, 0,32 ммоль) добавляли к тетрагидрофурану (5 мл) и воде (5 мл) при комнатной температуре, и добавляли гидроксид лития (24 мг, 1,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью I н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-метилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (85 мг, выход 58%).
[00402] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 7,90 (д, 1H), 7,69 (д, 2H), 7,25 (т, 1H), 7,13 (д, 1H), 7,11 (д, 2H), 6,85 (д, 1H), 6,64 (дд, 1H), 4,93 (т, 1H), 3,71 (с, 3H), 2,55 (к, 2H), 2,23 (с, 3H), 1,24 (д, 3H), 1,07 (т, 3H).
[00403] ЖХ-МС, M/Z (ИЭР): 458,3 [M+H]+
[00404] Пример 18: получение соединения I-18
[00405] (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-18)
[00406]
[00407] Схема синтеза для соединения I-18 показана ниже:
[00408] 
[00409] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-18Б)
[00410] 
[00411] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (371 мг, 1,0 ммоль) добавляли к диметилсульфоксиду (3 мл) при комнатной температуре; добавляли 4-этилфенол (122 мг, 1,0 ммоль) и гидроксид калия (114 мг, 2,0 ммоль); и смесь нагревали до 120°C в микроволновой печи и перемешивали в течение 20 мин. Затем смесь охлаждали до комнатной температуры и разбавляли водой (20 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (20 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-18Б) (175 мг, выход 38%).
[00412] ЖХ-МС, M/Z (ИЭР): 458,3 [M+H]+
[00413] Второй шаг: (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-18)
[00414]
[00415] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-18Б) (175 мг, 0,38 ммоль) добавляли к тетрагидрофурану (2 мл), метанолу (2 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития (3 мг, 0,16 ммоль). Смесь перемешивали в течение 12 часов при комнатной температуре; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(4-этилфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-18) (40 мг, выход 24%).
[00416] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 7,95 (д, 1H), 7,71 (д, 2H), 7,24 (д, 2H), 7,21 (т, 1H), 7,11 (д, 2H), 6,92 (д, 2H), 4,91 (т, 1H), 3,72 (с, 3H), 2,62 (к, 2H), 1,24 (д, 3H), 1,19 (т, 3H).
[00417] ЖХ-МС, M/Z (ИЭР): 444,3 [M+H]+
[00418] Пример 19: получение соединения I-19
[00419] (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-19)
[00420] 
[00421] Схема синтеза для соединения I-19 показана ниже:
[00422] 
[00423] Первый шаг: 5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (соединение I-19Б)
[00424] 
[00425] Соединение 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (1,0 г, 5,15 ммоль) добавляли к N, N-диметилформамиду (15 мл) при комнатной температуре; добавляли 4-ацетилфенол (1,05 г, 7,73 ммоль), йодид меди (I) (918 мг, 5,15 ммоль), 1, 10-фенантролин (1,05 г, 7,73 ммоль) и карбонат калия (1,42 г, 10,3 ммоль); и смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением светло-желтого твердого неочищенного продукта 5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегида (соединение I-19Б) (270 мг, выход 18%).
[00426] ЖХ-МС, M/Z (ИЭР): 295,1 [M+H]+
[00427] Второй шаг: 5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоновая кислота (соединение I-19В)
[00428] 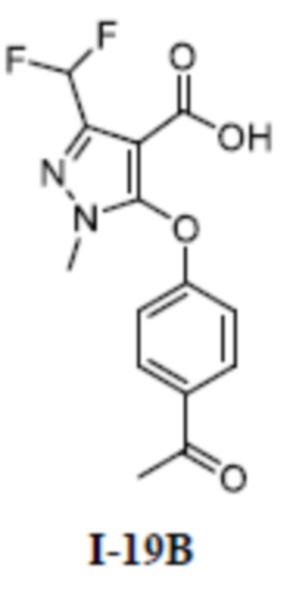
[00429] Соединение 5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (соединение I-19Б) (400 мг, 1,36 ммоль) добавляли к трет-бутанолу (9 мл) и воде (3 мл) при комнатной температуре; и добавляли 2-метил-2-бутен (476 мг, 6,80 ммоль), хлорит натрия (369 мг, 4,08 ммоль) и дигидрофосфат натрия (408 мг, 3,40 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре; смесь разбавляли посредством добавления воды (15 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенной 5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоновой кислоты (соединение I-19C) (350 мг, выход 100%), светло-желтого твердого вещества.
[00430] ЖХ-МС, M/Z (ИЭР): 311,1 [M+H]+.
[00431] Третий шаг: метил (S)-4-(1-(5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-19Г)
[00432] 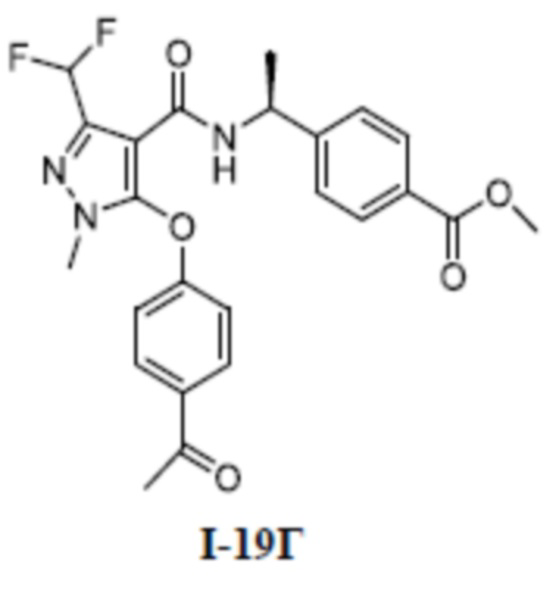
[00433] Соединение 3-(дифторметил)-5-(3-изопропилфенокси)-1-метил-1H-пиразол-4-карбоновую кислоту (350 мг, 1,13 ммоль) добавляли к N, N-диметилформамиду (7 мл); добавляли метил (S)-4-(l-аминоэтил)бензоат (242 мг, 1,36 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (650 мг, 1,71 ммоль) и N, N-диизопропилэтиламин (437 мг, 3,67 ммоль); и смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли водой (40 мл), экстрагировали этилацетатом (40 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтой жидкости метил (S)-4-(1-(5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-19Г) (500 мг, выход 94%).
[00434] ЖХ-МС, M/Z (ИЭР): 472,2 [M+H]+
[00435] Четвертый шаг: метил (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-19Д)
[00436] 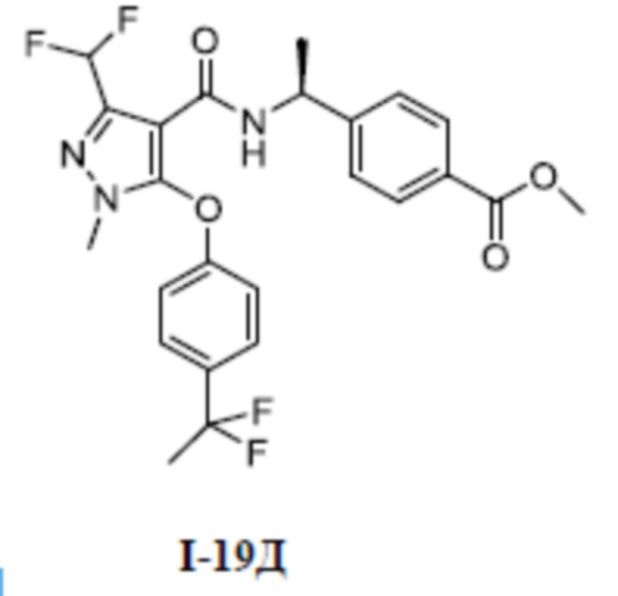
[00437] Соединение метил (S)-4-(1-(5-(4-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-19Г) (50 мг, 0,53 ммоль) добавляли к бис(2-метоксиэтил)аминосеры трифториду (5 мл) при комнатной температуре. Смесь нагревали до 50°C и перемешивали в течение 48 ч; затем смесь охлаждали до комнатной температуры, и добавляли по каплям к насыщенному раствору бикарбоната натрия (50 мл) и экстрагировали этилацетатом (40 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого твердого неочищенного продукта метил (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-19Д) (250 мг, выход 96%).
[00438] ЖХ-МС, M/Z (ИЭР): 494,5 [M+H]+.
[00439] Пятый шаг: (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-19)
[00440] 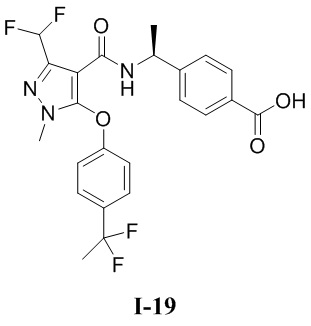
[00441] Исходный материал метил (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-19Д) (250 мг, 0,51 ммоль) добавляли к тетрагидрофурану (4 мл); и добавляли метанол (2 мл) и воду (2 мл) при комнатной температуре и гидроксид лития (61 мг, 2,55 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-(4-(1,1-дифторэтил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-19) (19 мг, выход 8%).
[00442] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,11 (д, 1H), 7,71 (д, 2H), 7,60 (д, 2H), 7,25 (т, 1H), 7,12 (д, 2H), 7,08 (д, 2H), 4,90 (т, 1H), 3,73 (с, 3H), 2,01 (т, 3H), 1,23 (д, 3H).
[00443] ЖХ-МС, M/Z (ИЭР): 480,5 [M+H]+
[00444] Пример 20: получение соединения I-20
[00445] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-20)
[00446] 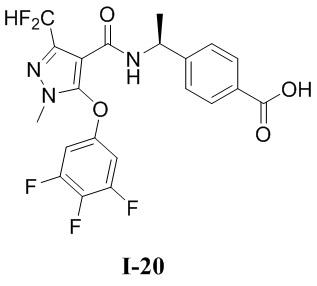
[00447] Схема синтеза для соединения I-20 показана ниже:
[00448] 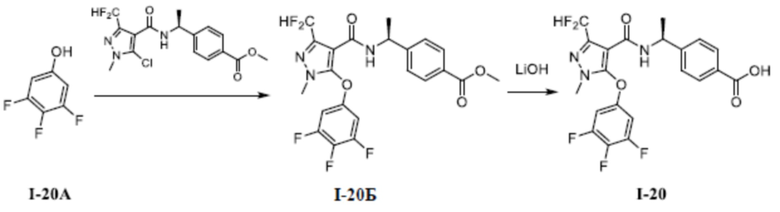
[00449] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-20Б)
[00450] 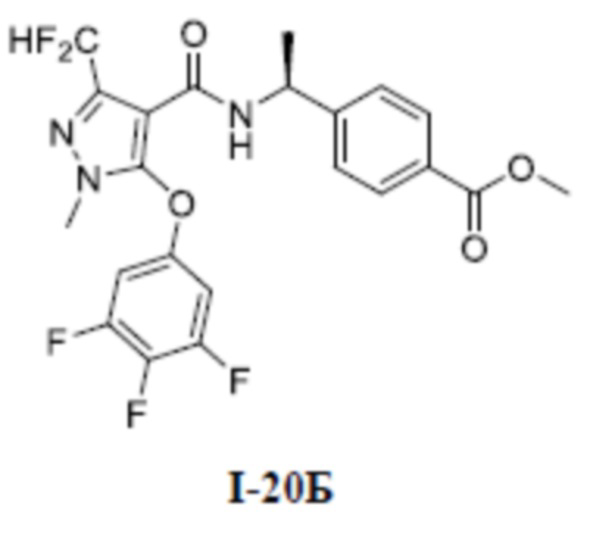
[00451] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (370 мг, 1,0 ммоль) добавляли к N, N диметилформамиду (10 мл) при комнатной температуре; добавляли 3, 4, 5-трифторфенол (222 мг, 1,5 ммоль) и гидроксид калия (168 мг, 3,0 ммоль); и смесь нагревали до 120°C и перемешивали в течение 6 часов. Смесь охлаждали до комнатной температуры и разбавляли водой (50 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (60 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-20Б) (80 мг, выход 17%).
[00452] ЖХ-МС, M/Z (ИЭР): 484,1 [M+H]+.
[00453] Второй шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-20)
[00454]
[00455] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (80 мг, 0,16 ммоль) добавляли к тетрагидрофурану (2 мл) и воде (1 мл) при комнатной температуре; добавляли гидроксид лития (17 мг, 0,40 ммоль); и смесь нагревали до 50°C и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3,4,5-трифторфенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-20) (7 мг, выход 9,0%).
[00456] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,24 (д, 1H), 7,78 (д, 2H), 7,24 (д, 2H), 7,22 (т, 1H), 7,09 (д, 1H), 7,02 (д, 1H), 4,96 (т, 1H), 3,73 (с, 3H), 1,30 (д, 3H).
[00457] ЖХ-МС, M/Z (ИЭР): 470,1 [M+H]+
[00458] Пример 21: получение соединения I-21
[00459] (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-21)
[00460]
[00461] Схема синтеза для соединения I-21 показана ниже:
[00462] 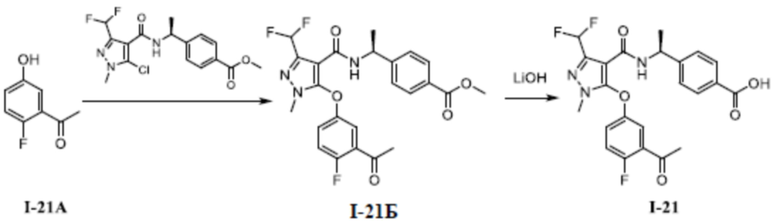
[00463] Первый шаг: метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-21Б)
[00464] 
[00465] Соединение 1-(2-фтор-5-гидроксифенил)этан-1-он (1,58 г, 10,8 ммоль) добавляли к N, N-диметилформамиду (30 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (2,0 г, 5,4 ммоль) и гидроксид калия (450 мг, 8,1 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов; смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением белого твердого вещества метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-21Б) (346 мг, выход 13%).
[00467] ЖХ-МС, M/Z (ИЭР): 490,2 [M+H]+
[00468] Второй шаг: (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-21)
[00469] 
[00470] Исходный материал метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-21Б) (65 мг, 0,13 ммоль) добавляли к тетрагидрофурану (5 мл), метанолу (5 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития (20 мг, 0,47 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-21) (30 мг, выход 66%).
[00471] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,18 (д, 1H), 7,72 (д, 2H), 7,38-7,32 (м, 2H), 7,29-7,24 (м, 1H), 7,26 (т, 1H), 7,16 (д, 2H), 4,91 (т, 1H), 3,73 (с, 3H), 2,54 (д,3H), 1,24 (д, 3H).
[00472] ЖХ-МС, M/Z (ИЭР): 476,1 [M+H]+
[00473] Пример 22: получение соединения I-22
[00474] 4-((1S)-1-(3-(дифторметил)-5-(3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-22)
[00475]
[00476] Схема синтеза для соединения I-22 показана ниже:
[00477] 
[00478] Первый шаг: метил (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-22Б)
[00479] 
[00480] Соединение 1-(3-гидроксифенил)этан-1-он (275 мг, 2,02 ммоль) добавляли к N, N-диметилформамиду (6 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (500 мг, 1,35 ммоль), йодид меди (II) (100 мг, 0,52 ммоль), 1, 10-фенантролин (180 мг, 1,0 ммоль) и карбонат цезия (1,30 г, 4,05 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-22Б) (300 мг, выход 47%).
[00481] ЖХ-МС, M/Z (ИЭР): 472,3 [M+H]+
[00482] Второй шаг: (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-22В)
[00483] 
[00484] Исходный материал метил (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-22Б) (290 мг, 0,62 ммоль) добавляли к тетрагидрофурану (5 мл) и воде (5 мл) при комнатной температуре; и добавляли гидроксид лития (45 мг, 1,87 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-22В) (180 мг, выход 64%).
[00485] ЖХ-МС, M/Z (ИЭР): 458,3 [M+H]+
[00486] Третий шаг: 4-((1S)-1-(3-(дифторметил)-5-(3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-22)
[00487] 
[00488] Соединение (S)-4-(1-(5-(3-ацетилфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойную кислоту (соединение I-22В) (170 мг, 0,37 ммоль) добавляли к метанолу (15 мл) и воде (15 мл) при комнатной температуре; и добавляли боргидрид натрия (15 мг, 0,39 ммоль). Смесь перемешивали в течение 2 часов при комнатной температуре; и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты и концентрировали с получением 4-((1S)-1-(3-(дифторметил)-5-(3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4 карбоксамидо)этил)бензойной кислоты (соединение I-22) (168 мг, выход 98%), белого твердого вещества, посредством кислотного способа получения A.
[00489] ЖХ-МС, M/Z (ИЭР): 460,3 [M+H]+
[00490] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 7,92 (д, 1H), 7,73 (д, 2H), 7,36 (т, 1H), 7,26 (т, 1H), 7,18 (д, 1H), 7,13 (д, 2H), 7,11 (т, 1H), 6,83 (дд, 1H), 5,26 (д, 1H), 4,92 (т, 1H), 4,71 (с, 1H), 3,72 (с, 3H), 1,28 (дд, 3H), 1,22 (д, 3H).
[00491] ЖХ-МС, M/Z (ИЭР): 478,1 [M+H]+
[00492] Пример 23: получение соединения I-23
[00493] 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-23)
[00494]
[00495] Схема синтеза для соединения I-23 показана ниже:
[00496] 
[00497] Первый шаг: метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-23Б)
[00498] 
[00499] Соединение 1-(2-фтор-5-гидроксифенил)этан-1-он (1,58 г, 10,8 ммоль) добавляли к N, N-диметилформамиду (30 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (2,0 г, 5,4 ммоль) и гидроксид калия (450 мг, 8,1 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов; смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением белого твердого вещества метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-23Б) (346 мг, выход 13%).
[00500] ЖХ-МС, M/Z (ИЭР): 490,2 [M+H]+
[00501] Второй шаг: метил 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-23В)
[00502] 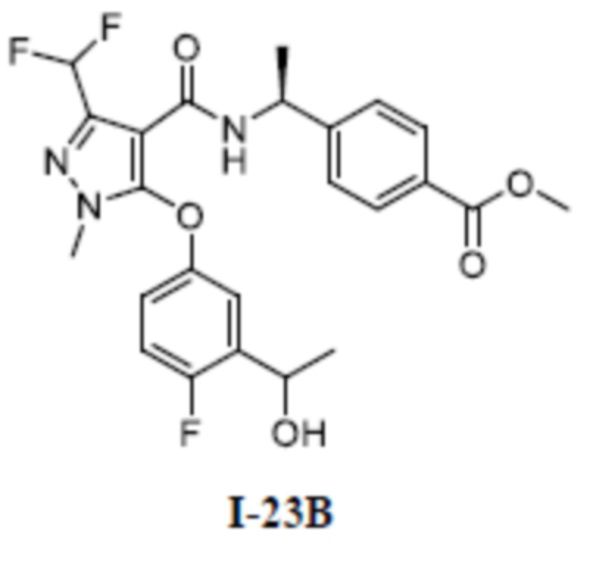
[00503] Соединение метил (S)-4-(1-(5-(3-ацетил-4-фторфенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-23Б) (100 мг, 0,20 ммоль) добавляли к тетрагидрофурану (10 мл) при комнатной температуре, охлаждали до 0°C; и добавляли боргидрид натрия (23 мг, 0,60 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч; смесь разбавляли посредством добавления воды (100 мл), экстрагировали этилацетатом (80 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого неочищенного продукта метил 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-23C) (75 мг, выход 75%).
[00504] ЖХ-МС, M/Z (ИЭР): 492,1 [M+H]+.
[00505] 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-23)
[00506] 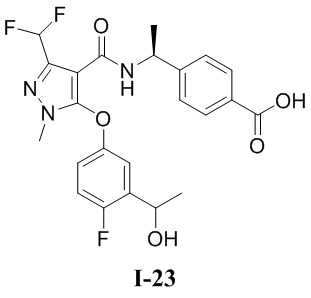
[00507] Исходный материал метил 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-23В) (75 мг, 0,15 ммоль) добавляли к тетрагидрофурану (5 мл), метанолу (5 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития моногидрат (20 мг, 0,47 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества 4-((1S)-1-(3-(дифторметил)-5-(4-фтор-3-(1-гидроксиэтил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-23) (48 мг, выход 66%).
[00508] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,02 (д, 1H), 7,74 (д, 2H), 7,25 (т, 1H), 7,21-7,10 (м, 4 H), 6,90-6,81 (м, 1H), 4,96-4,86 (м, 2H), 3,72 (с, 3H), 3,67 (с,1H), 1,28 (д, 3H), 1,24 (д, 3H).
[00509] ЖХ-МС, M/Z (ИЭР): 478,1 [M+H]+
[00510] Пример 24: получение соединения I-24
[00511] (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-24)
[00512]
[00513] Схема синтеза для соединения I-24 показана ниже:
[00514] 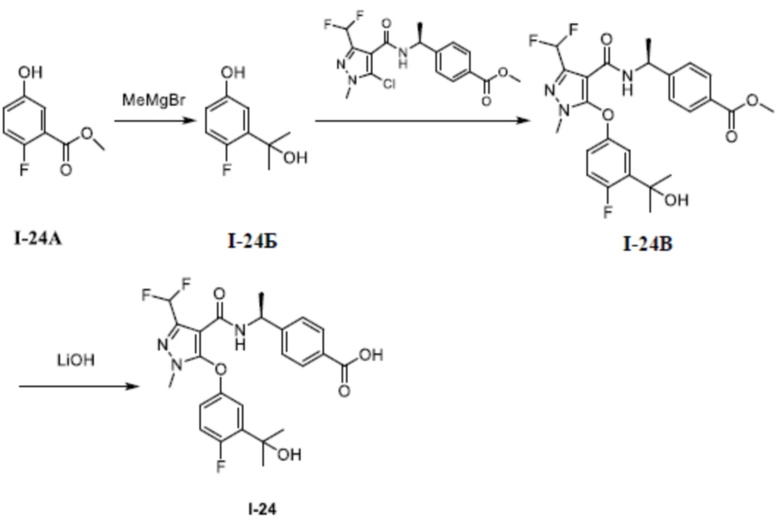
[00515] Первый шаг: 4-фтор-3-(2-гидроксипропан-2-ил)фенол (соединение I-24Б)
[00516] 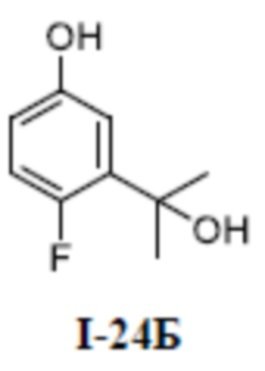
[00517] Соединение метил-2-фтор-5-гидроксибензоат (340 мг, 2,0 ммоль) добавляли к раствору метилмагния бромида (1 моль/л) в тетрагидрофуране (5 мл) при комнатной температуре и перемешивали в течение 16 часов; добавляли насыщенный раствор хлорида аммония (10 мл) и экстрагировали дихлорметаном (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта 4-фтор-3-(2-гидроксипропан-2-ил)фенола (соединение I-24Б) (340 мг, выход 100%).
[00518] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-24В)
[00519] 
[00520] Соединение 4-фтор-3-(2-гидроксипропан-2-ил)фенол (соединение I-24Б) (340 мг, 2,0 ммоль) добавляли к N, N-диметилформамиду (9 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (495 мг, 1,34 ммоль), йодид меди (I) (100 мг, 0,52 ммоль), 1, 10-фенантролин (180 мг, 1,0 ммоль) и карбонат цезия (1,30 г, 4,05 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-24В) (120 мг, выход 32%).
[00521] ЖХ-МС, M/Z (ИЭР): 506,3 [M+1]+
[00522] Третий шаг: (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-24)
[00523] 
[00524] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-24В) (120 мг, 0,24 ммоль) добавляли к тетрагидрофурану (5 мл), и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития (20 мг, 0,47 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(4-фтор-3-(2-гидроксипропан-2-ил)фенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-24) (22 мг, выход 19%).
[00525] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 7,96 (д, 1H), 7,73 (д, 2H), 7,33 (т, 1H), 7,26 (т, 1H), 7,17 (д, 2H), 7,14 (т, 1H), 6,85 (т, 1H), 4,92 (т, 1H), 3,73 (с, 3H), 3,72 (с, 1H), 1,44 (с, 6H), 1,25 (д, 3H).
[00526] ЖХ-МС, M/Z (ИЭР): 492,3 [M+1]+
[00527] Пример 25: получение соединения I-25
[00528] (S)-4-(1-(5-(3-(цианометил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-25)
[00529] 
[00530] Схема синтеза для соединения I-25 показана ниже:
[00531] 
[00532] Первый шаг: (S)-4-(1-(5-(3-(цианометил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-25)
[00533] 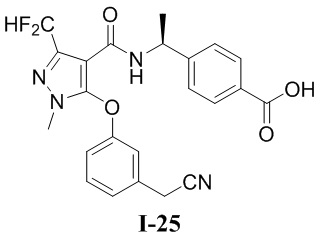
[00534] Соединение 2-(3-гидроксифенил)ацетонитрил (72 мг, 0,40 ммоль) добавляли к N, N-диметилформамиду (2 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (100 мг, 0,27 ммоль) и гидроксид калия (30 мг, 0,53 ммоль). Смесь нагревали до 170°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 5 мин; а затем смесь охлаждали до комнатной температуры и добавляли воду (30 мл). Смесь перемешивали в течение 0,5 часа; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-(3-(цианометил)фенокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (10 мг, выход 8%).
[00535] 1H ЯМР (400 МГц, CDCl3) δ12,7 (с, 1H), 7,93 (д, 2H), 7,38 (т, 1H), 7,25 (т, 1H), 7,19 (д, 1H), 7,16 (д, 2H), 6,94 (с, 1H), 6,82 (т, 1H), 6,35 (д, 1H), 5,18 (т, 1H), 3,73 (с, 2H), 3,70 (с, 3H), 1,41 (д, 3H).
[00536] ЖХ-МС, M/Z (ИЭР): 455,5 [M+H]+
[00537] Пример 26: получение соединения I-26
[00538] (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-26)
[00539]
[00540] Схема синтеза для соединения I-26 показана ниже:
[00541] 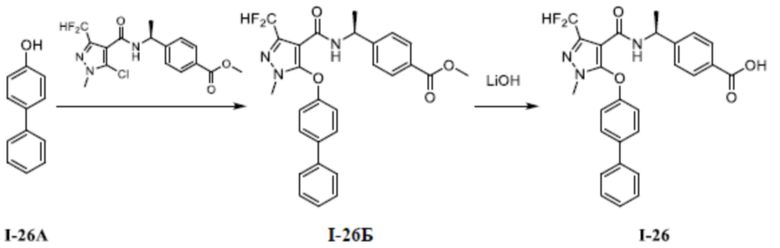
[00542] Первый шаг: метил (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-26Б)
[00543] 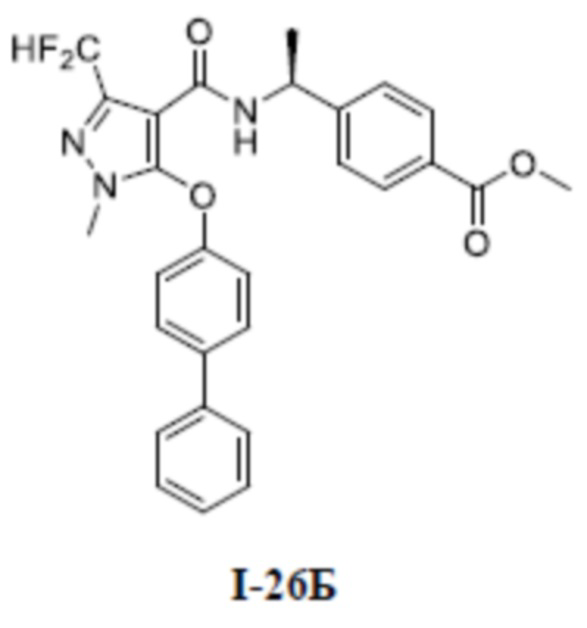
[00544] Соединение [l, l’-бифенил]-4-ол (458 мг, 2,70 ммоль) добавляли к N, N-диметилформамиду (10 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (500 мг, 1,35 ммоль) и гидроксид калия (227 мг, 4,05 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 1 часа. Затем смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением белого твердого неочищенного продукта метил (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-26Б) (500 мг, выход 73%).
[00545] ЖХ-МС, M/Z (ИЭР): 506,4 [M+H]+
[00546] Второй шаг: (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-26)
[00547] 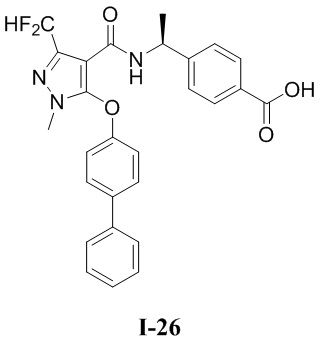
[00548] Исходный материал метил (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-26Б) (450 мг, 0,89 ммоль) добавляли к тетрагидрофурану (10 мл), метанолу (5 мл) и воде (4 мл) при комнатной температуре; и добавляли гидроксид лития (108 мг, 4,5 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-([1,1'-бифенил]-4-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-26) (70 мг, выход 16%).
[00549] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,08 (д, 1H), 7,71 (д, 2H), 7,69 (д, 2H), 7,63 (д, 2H), 7,48 (т, 2H), 7,38 (т, 1H), 7,26 (т, 1H), 7,15 (д, 2H), 7,12 (д, 2H), 4,92 (т, 1H), 3,76 (с, 3H), 1,25 (д, 3H).
[00550] ЖХ-МС, M/Z (ИЭР): 492,4 [M+H]+
[00551] Пример 27: получение соединения I-27
[00552] (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-27)
[00553]
[00554] Схема синтеза для соединения I-27 показана ниже:
[00555] 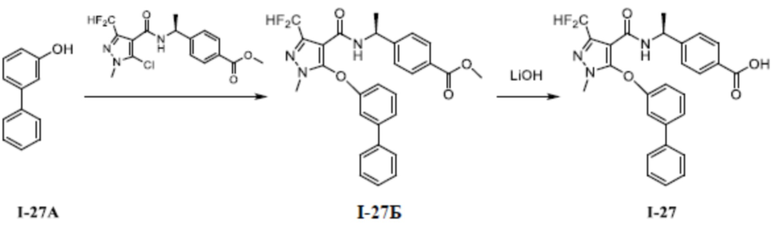
[00556] Первый шаг: метил (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-27Б)
[00557] 
[00558] Соединение [l, l’-бифенил]-3-ол (458 мг, 2,70 ммоль) добавляли к N, N-диметилформамиду (10 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (500 мг, 1,35 ммоль) и гидроксид калия (227 мг, 4,05 ммоль). Смесь нагревали до 120°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 1 часа. Затем смесь охлаждали до комнатной температуры и добавляли воду (30 мл); pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением белого твердого неочищенного продукта метил (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-27Б) (450 мг, выход 66%).
[00559] ЖХ-МС, M/Z (ИЭР): 506,4 [M+H]+
[00560] Второй шаг: (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-27)
[00561] 
[00562] Исходный материал метил (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-27Б) (400 мг, 0,79 ммоль) добавляли к тетрагидрофурану (10 мл), метанолу (5 мл) и воде (5 мл) при комнатной температуре; и добавляли гидроксид лития (96 мг, 4,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(5-([1,1'-бифенил]-3-илокси)-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-27) (115 мг, выход 30%).
[00563] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,07 (д, 1H), 7,68 (д, 2H), 7,62 (д, 2H), 7,50-7,44 (м, 4 H), 7,41 (д, 1H), 7,31 (с, 1H), 7,25-6,96 (м, 4 H), 4,89 (т, 1H), 3,77 (с, 3H), 1,19 (д, 3H).
[00564] ЖХ-МС, M/Z (ИЭР): 492,4 [M+H]+
[00565] Пример 28: получение соединения I-28
[00566] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-28)
[00567]
[00568] Схема синтеза для соединения I-28 показана ниже:
[00569] 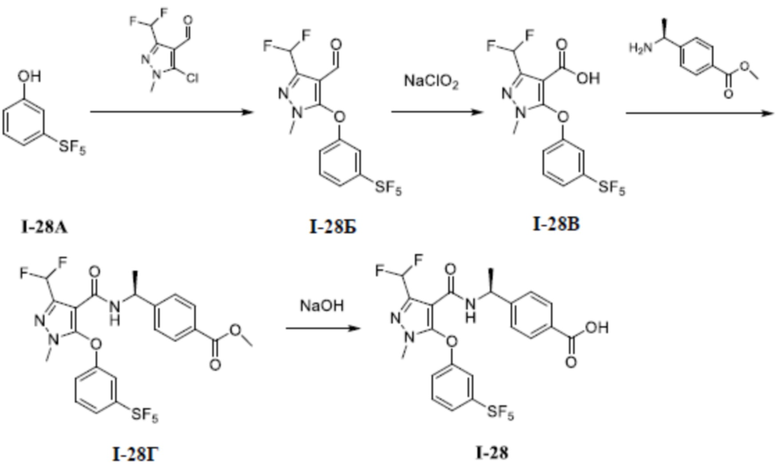
[00570] Первый шаг: 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбальдегид (соединение I-28Б)
[00571] 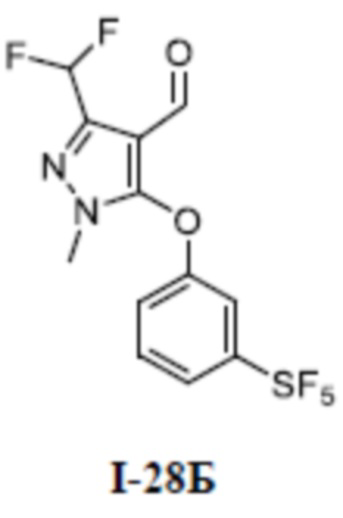
[00572] Соединение 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (500 мг, 2,57 ммоль) добавляли к N, N-диметилформамиду (5 мл) при комнатной температуре; добавляли 3-(пентафтор-λ6-сульфанил)фенол (386 мг, 2,80 ммоль) и гидроксид калия (216 мг, 3,85 ммоль); и смесь нагревали до 150°C и перемешивали в течение 4 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (15 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтого жидкого неочищенного продукта 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбальдегида (соединение I-28Б) (750 мг, выход 98%).
[00573] ЖХ-МС, M/Z (ИЭР): 379,1 [M+H]+
[00574] Второй шаг: 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоновая кислота (соединение I-28В)
[00575] 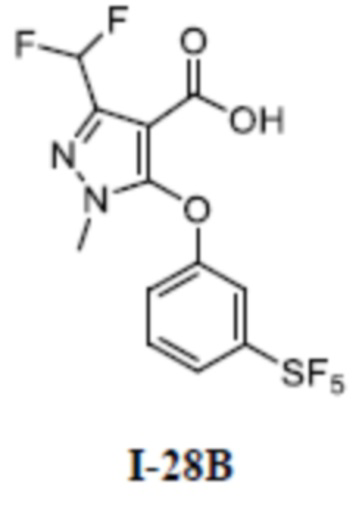
[00576] Соединение 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбальдегид (соединение I-28Б) (750 мг, 2,55 ммоль) добавляли к трет-бутанолу (6 мл) и воде (7 мл) при комнатной температуре; и добавляли 2-метил-2-бутен (355 мг, 5,07 ммоль), хлорит натрия (456 мг, 5,07 ммоль) и дигидрофосфат натрия (669 мг, 5,57 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре; смесь разбавляли водой (15 мл) и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением светло-желтого твердого неочищенного продукта 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоновой кислоты (соединение I-28В) (800 мг, выход 100%).
[00577] ЖХ-МС, M/Z (ИЭР): 395,1 [M+H]+
[00578] Третий шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-28Г)
[00579] 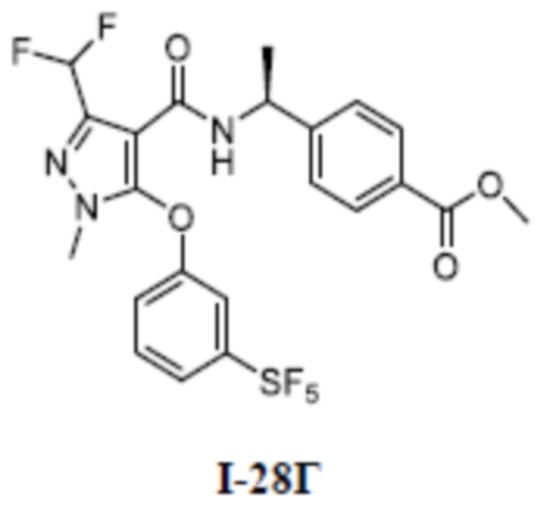
[00580] Соединение 3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоновую кислоту (соединение I-28В) (800 мг, 2,58 ммоль) добавляли к дихлорметану (20 мл); и добавляли метил (S)-4-(l-аминоэтил)бензоат (459 мг, 2,56 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (1,40 г, 3,68 ммоль) и N, N-диизопропилэтиламин (991 мг, 7,68 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли дихлорметаном (40 мл), промывали водой (20 мл ×3) и разделяли. Органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением светло-желтой жидкости (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-28Г) (720 мг, выход 59%).
[00581] ЖХ-МС, M/Z (ИЭР): 556,1 [M+H]+
[00582] Четвертый шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-28)
[00583] 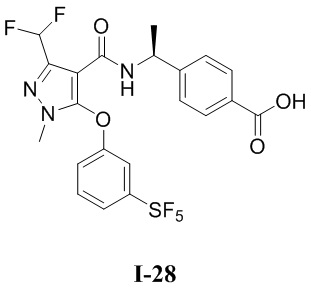
[00584] Исходный материал (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-28Г) (440 мг, 0,93 ммоль) добавляли к метанолу (10 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид натрия (93 мг, 2,32 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(пентафтор-λ6-сульфанил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-28) (93 мг, выход 22%).
[00585] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 8,18 (д, 1H), 7,74 (д, 2H), 7,73 (д, 1H), 7,71 (д, 1H), 7,65 (т, 1H), 7,26 (д, 1H), 7,23 (т, 1H), 7,21 (д, 2H), 4,89 (т, 1H), 3,78 (с, 3H), 1,11 (д, 3H).
[00586] ЖХ-МС, M/Z (ИЭР): 542,1 [M+H]+
[00587] Пример 29: получение соединения I-29
[00588] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-29)
[00589]
[00590] Схема синтеза для соединения I-29 показана ниже:
[00591] 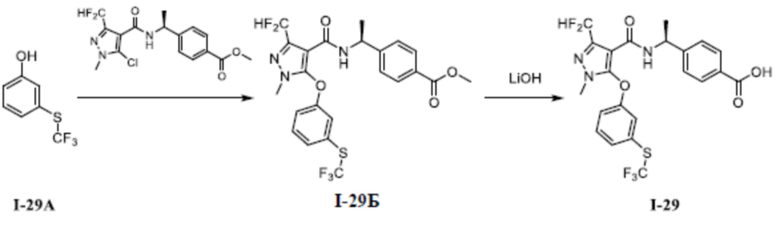
[00592] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-29Б)
[00593] 
[00594] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (186 мг, 0,50 ммоль) добавляли к N, N-диметилформамиду (5 мл) при комнатной температуре; добавляли 3-((трифторметил)сульфанил)фенол (120 мг, 0,60 ммоль) и гидроксид калия (45 мг, 0,80 ммоль); и смесь нагревали до 120°C и перемешивали в течение 12 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (15 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-29Б) (20 мг, выход 7,5%).
[00595] ЖХ-МС, M/Z (ИЭР): 530,2 [M+H]+
[00596] Второй шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-29)
[00597] 
[00598] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-29Б) (20 мг, 0,04 ммоль) добавляли к тетрагидрофурану (2 мл), метанолу (2 мл) и воде (1 мл) при комнатной температуре; добавляли гидроксид лития (3 мг, 0,16 ммоль); и смесь нагревали до 50°C и перемешивали в течение 4 часов. Затем смесь охлаждали до комнатной температуры и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-((трифторметил)тио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-29) (14 мг, выход 72%).
[00599] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,16 (д, 1H), 7,74 (д, 2H), 7,57 (т, 2H), 7,39 (с, 1H), 7,22-6,69 (м, 4 H), 4,91-4,84 (м, 1H), 3,74 (с, 3H), 1,21 (д, 3H).
[00600] ЖХ-МС, M/Z (ИЭР): 516,2 [M+H]+
[00601] Пример 30: получение соединения I-30
[00602] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-30)
[00603]
[00604] Схема синтеза для соединения I-30 показана ниже:
[00605] 
[00606] Первый шаг: 3-(метилтио)фенол (соединение I-30Б)
[00607] 
[00608] Соединение (3-метоксифенил)(метил)сульфид (4,0 г, 26 ммоль) добавляли к раствору 30% бромистоводородной кислоты в уксусной кислоте (12 мл) и раствору 48% бромистоводородной кислоты в воде (3 мл) при комнатной температуре; смесь нагревали с обратным холодильником в защитной атмосфере N2 и перемешивали в течение 6 часов; а затем смесь охлаждали до комнатной температуры, разбавляли водой (100 мл), экстрагировали диэтиловым эфиром (100 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бесцветного жидкого неочищенного продукта 3-(метилтио)фенола (соединение I-30Б) (3,8 г, выход 100%).
[00609] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-30В)
[00610] 
[00611] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (700 мг, 1,89 ммоль) добавляли к N, N-диметилформамиду (5 мл) при комнатной температуре; и добавляли 3-(метилтио)фенол (528 мг, 3,77 ммоль) и гидроксид калия (318 мг, 5,67 ммоль). Смесь нагревали до 150°C в микроволновой печи и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры и разбавляли водой (20 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-30В) (800 мг, выход 89%).
[00612] ЖХ-МС, M/Z (ИЭР): 476,5 [M+H]+
[00613] Третий шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-30)
[00614] 
[00615] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-30В) (800 мг, 1,68 ммоль) добавляли к тетрагидрофурану (10 мл), метанолу (10 мл) и воде (2 мл) при комнатной температуре; добавляли гидроксид лития (121 мг, 5,0 ммоль); и смесь нагревали до 50°C и перемешивали в течение 4 часов. Затем смесь охлаждали до комнатной температуры и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-30) (165 мг, выход 21%).
[00616] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,03 (д, 1H), 7,74 (д, 2H), 7,34 (т, 2H), 7,14 (д, 2H), 7,11 (т, 1H), 6,97 (т, 1H), 6,70 (д, 1H), 4,91 (т, 1H), 3,73 (с, 3H), 2,45 (с, 3H), 1,24 (д, 3H).
[00617] ЖХ-МС, M/Z (ИЭР): 462,5 [M+H]+
[00618] Пример 31: получение соединения I-31
[00619] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилсульфонил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-31)
[00620]
[00621] Схема синтеза для соединения I-31 показана ниже:
[00622] 
[00623] Исходный материал (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-30) (80 мг, 0,17 ммоль) добавляли к дихлорметану (5 мл); и добавляли м-хлорпероксибензойную кислоту (83 мг, 0,48 ммоль) при комнатной температуре и перемешивали в течение 3 часов при комнатной температуре. Реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилсульфонил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-31) (64 мг, выход 75%).
[00624] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,21 (д, 1H), 7,77 (д, 2H), 7,75 (д, 1H), 7,70 (т, 1H), 7,59 (т, 1H),7,37 (д, 1H), 7,25 (т, 1H), 7,15 (д, 2H), 4,87 (т, 1H), 3,77 (с, 3H), 3,19 (с, 3H), 1,19 (д, 3H).
[00625] ЖХ-МС, M/Z (ИЭР): 494,1 [M+H]+
[00626] Пример 32: получение соединения I-32
[00627] 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(метилсульфинил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-32)
[00628]
[00629] Схема синтеза для соединения I-32 показана ниже:
[00630] 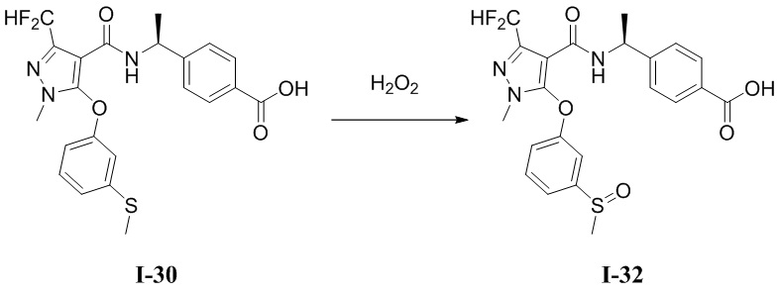
[00631] Исходный материал (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(метилтио)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойную кислоту (соединение I-30) (60 мг, 0,13 ммоль) добавляли к тетрагидрофурану (5 мл); добавляли 30% пероксид водорода (1 мл); и смесь нагревали до 60°C и перемешивали в течение 7 ч при комнатной температуре. Затем смесь охлаждали до комнатной температуры и концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества 4-((1S)-1-(3-(дифторметил)-1-метил-5-(3-(метилсульфинил)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-32) (35 мг, выход 75%).
[00632] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,15 (д, 1H), 7,73 (д, 2H), 7,61 (д, 1H), 7,50 (т, 1H), 7,36 (с, 1H), 7,25 (т, 1H), 7,15 (д, 1H), 7,13 (д, 2H), 4,87 (т, 1H), 3,75 (с, 3H), 2,69 (с, 3H), 1,20 (д, 3H).
[00633] ЖХ-МС, M/Z (ИЭР): 478,2 [M+H]+
[00634] Пример 33: получение соединения I-33
[00635] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-33)
[00636] 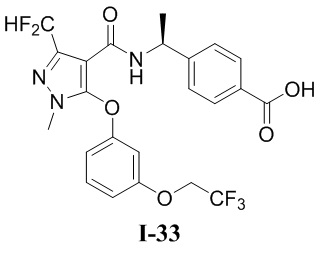
[00637] Схема синтеза для соединения I-33 показана ниже:
[00638] 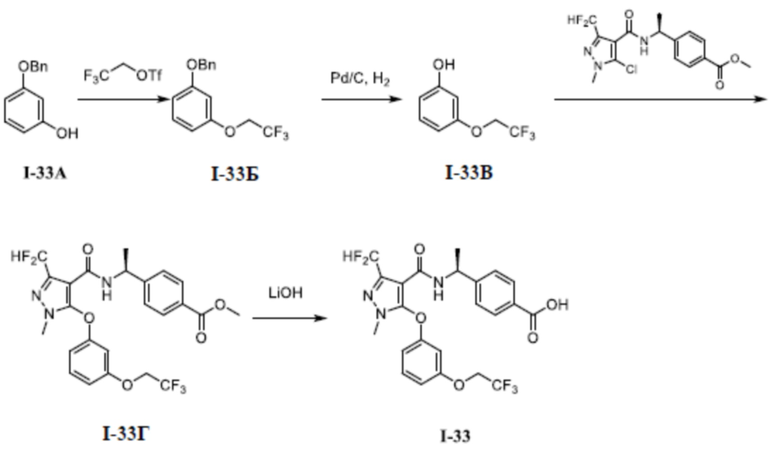
[00639] Первый шаг: 1-(бензилокси)-3-(2,2,2-трифторэтокси)бензол (соединение I-33Б)
[00670] 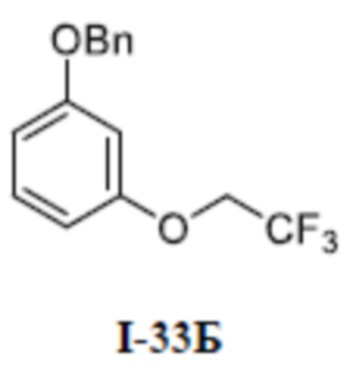
[00671] Соединение 3-(бензилокси)фенол (600 мг, 3,0 ммоль) добавляли к N, N-диметилформамиду (10 мл) при комнатной температуре; добавляли трифтор-2, 2, 2-трифторэтан-1-сульфонат (840 мг, 3,6 ммоль) и карбонат калия (1,24 г, 9,0 ммоль); и смесь нагревали до 60°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (30 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали; остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 10:1) с получением бесцветной жидкости 1-(бензилокси)-3-(2,2,2-трифторэтокси)бензола (соединение I-33Б) (380 мг, выход 45%).
[00672] Второй шаг: 3-(2,2,2-трифторэтокси)фенол (соединение I-33В)
[00673] 
[00674] Соединение 1-(бензилокси)-3-(2,2,2-трифторэтокси)бензол (соединение I-33Б) (520 мг, 1,84 ммоль) добавляли к этанолу (10 мл) при комнатной температуре; добавляли 10% палладий на угле (50 мг); и добавляли каплю муравьиной кислоты. Затем вводили водород и смесь перемешивали в течение 12 часов; и смесь разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого вещества указанного соединения, т.е., 3-(2,2,2-трифторэтокси)фенола (соединение I-33В) (210 мг, выход 59%).
[00675] Третий шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-33Г)
[00676] 
[00677] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (371 мг, 1,0 ммоль) добавляли к N, N-диметилформамиду (5 мл) при комнатной температуре; добавляли 3-(2,2,2-трифторэтокси)фенол (соединение I-33В) (210 мг, 1,1 ммоль), йодид меди (II) (191 мг, 1,0 ммоль), карбонат цезия (1,0 г, 3,1 ммоль) и 1, 10-фенантролин (72 мг, 0,40 ммоль); и смесь нагревали до 120°C в микроволновой печи и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-33Г) (120 мг, выход 8%).
[00678] ЖХ-МС, M/Z (ИЭР): 528,2 [M+H]+
[00679] Четвертый шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-33)
[00680] 
[00681] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-33Г) (120 мг, 0,23 ммоль) добавляли к тетрагидрофурану (5 мл), метанолу (5 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития (20 мг, 0,83 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(2,2,2-трифторэтокси)фенокси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-33) (45 мг, выход 72%).
[00682] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,02 (д, 1H), 7,74 (д, 2H), 7,38 (т, 1H), 7,16 (т, 1H), 7,14 (д, 2H), 6,93 (д, 1H), 6,76 (т, 1H), 6,65 (д, 1H), 4,92 (т, 1H), 4,78 (дд, 2H), 3,72 (с, 3H), 1,25 (д, 3H).
[00683] ЖХ-МС, M/Z (ИЭР): 514,2 [M+H]+
[00684] Пример 34: получение соединения I-34
[00685] (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-34)
[00686]
[00687] Схема синтеза для соединения I-34 показана ниже:
[00688] 
[00689] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-34Б)
[00690] 
[00691] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (370 мг, 1,0 ммоль) добавляли к N, N-диметилформамиду (10 мл) при комнатной температуре; добавляли 2, 4-дифторфенол (195 мг, 1,5 ммоль) и гидроксид калия (168 мг, 3,0 ммоль); и смесь нагревали до 120°C и перемешивали в течение 6 часов. Затем смесь охлаждали до комнатной температуры и разбавляли водой (50 мл), pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь экстрагировали этилацетатом (60 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта метил (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-34Б) (160 мг, выход 34%).
[00692] ЖХ-МС, M/Z (ИЭР): 466,1 [M+H]+
[00693] Второй шаг: (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-34)
[00694]
[00695] Исходный материал метил (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-34Б) (160 мг, 0,34 ммоль) добавляли к тетрагидрофурану (4 мл) и воде (2 мл) при комнатной температуре; добавляли гидроксид лития (32 мг, 0,76 ммоль); и смесь нагревали до 50°C и перемешивали в течение 2 часов. Затем смесь охлаждали до комнатной температуры и pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(2,4-дифторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-34) (11 мг, выход 7,0%).
[00696] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 8,28 (д, 1H), 7,78 (д, 2H), 7,45 (т, 1H), 7,21 (д, 2H), 7,17 (т, 1H), 7,04 (д, 1H), 7,02 (д, 1H), 4,88 (т, 1H), 3,76 (с, 3H), 1,26 (д, 3H).
[00697] ЖХ-МС, M/Z (ИЭР): 452,1 [M+H]+
[00698] Пример 35: получение соединения I-35
[00699] (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-
[00700]
[00701] Схема синтеза для соединения I-35 показана ниже:
[00702] 
[00703] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-35Б)
[00704] 
[00705] Соединение 3-(трифторметил)тиофенол (215 мг, 1,20 ммоль) добавляли к N, N-диметилформамиду (20 мл) при комнатной температуре; и добавляли гидрид натрия (80 мг, 2,00 ммоль). Смесь перемешивали в течение 0,5 часа; добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (375 мг, 1,00 ммоль); и смесь нагревали до 120°C и перемешивали в течение 8 ч. Затем смесь охлаждали до комнатной температуры, разбавляли водой (100 мл), экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-35Б) (350 мг, выход 67%).
[00706] ЖХ-МС, M/Z (ИЭР): 514,2 (M+1).
[00707] Второй шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-35)
[00708]
[00709] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-35Б) (220 мг, 0,43 ммоль) добавляли к тетрагидрофурану (5 мл) при комнатной температуре; добавляли воду (5 мл), метанол (5 мл) и гидроксид лития (30 мг, 1,25 ммоль); и смесь нагревали до 50°C и перемешивали в течение 18 часов. Реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-((3-(трифторметил)фенил)тио)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-35) (105 мг, выход 49%).
[00710] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,68 (д, 1H), 7,79 (д, 2H), 7,63 (д, 1H), 7,58 (т, 2H), 7,39 (д, 2H), 7,27 (т, 1H), 7,14 (т, 1H), 5,10-5,03 (м, 1H), 3,89 (с, 3H), 1,38 (д, 3H).
[00711] ЖХ-МС, M/Z (ИЭР): 500,2 (M+1)
[00712] Пример 36: получение соединения I-36
[00713] (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-36)
[00714]
[00715] Схема синтеза для соединения I-36 показана ниже:
[00716] 
[00717] Первый шаг: (4-(трифторметил)бензил)цинк(II) бромид (соединение I-36Б)
[00718] 
[00719] Порошок цинка (2,60 г, 40,0 ммоль) добавляли к безводному тетрагидрофурану (15 мл) при комнатной температуре; добавляли 1, 2-дибромэтан (0,02 мл) в защитной атмосфере азота; а смесь нагревали до 60°C. Затем добавляли хлортриметилсилан (0,02 мл), и смесь перемешивали в течение 15 мин и охлаждали до 0°C. Добавляли по каплям раствор 4-(трифторметил)бензилбромида (4,80 г, 20,0 ммоль) в тетрагидрофуране (5 мл); а затем смесь нагревали до 60°C и перемешивали в течение 1 часа. Смесь охлаждали до комнатной температуры с получением раствора (1 моль/л) (4-(трифторметил)бензил)цинк(II) бромида (соединение I-36Б) в тетрагидрофуране (20 мл).
[00720] Второй шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-36В)
[00721] 
[00722] Соединение (4-(трифторметил)бензил)цинк бромид в тетрагидрофуране (соединение I-36Б) (2,70 мл, 2,70 ммоль) добавляли к N, N-диметилформамиду (15 мл) при комнатной температуре; и добавляли метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (500 мг, 1,35 ммоль), 4, 4’-ди-трет-бутил-2, 2'-бипиридин (35 мг, 0,13 ммоль) и комплекс хлорида никеля и диметоксиэтана (30 мг, 0,13 ммоль). Смесь нагревали до 100°C в микроволновой печи в защитной атмосфере азота и перемешивали в течение 1 часа; смесь охлаждали до комнатной температуры и добавляли воду (50 мл); а смесь экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-36В) (180 мг, выход 27%).
[00723] ЖХ-МС, M/Z (ИЭР): 496,3 [M+1]+.
[00724] Третий шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-36)
[00725]
[00726] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-36В) (150 мг, 0,30 ммоль) добавляли к 48% раствору бромгидрированной уксусной кислоты (3 мл) при комнатной температуре, и смесь нагревали до 65°C и перемешивали в течение 16 часов. Затем смесь охлаждали до комнатной температуры, и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(4-(трифторметил)бензил)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-36) (90 мг, выход 62%).
[00727] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,47 (д, 1H), 7,86 (д, 2H), 7,62 (д, 2H), 7,41 (д, 2H), 7,33 (д, 2H), 7,23 (т, 1H), 5,12 (т, 1H),4,37 (дд, 2H), 3,77 (с, 3H), 1,41 (с, 3H).
[00728] ЖХ-МС, M/Z (ИЭР): 482,3 [M+1]+
[00729] Пример 37: получение соединения I-37
[00730] (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоновая кислота (соединение I-37)
[00731]
[00732] Схема синтеза для соединения I-37 показана ниже:
[00733] 
[00734] Первый шаг: (R, E)-N-(1-(5-бромтиофен-2-ил)этилиден)-2-метилпропан-2-сульфинамид (соединение I-37Б)
[00735] 
[00736] Соединение l-(5-бромтиофен-2-ил)этан-1-он (2,05 г, 10,0 ммоль) добавляли к дихлорметану (50 мл) при комнатной температуре; добавляли (R)-трет-бутилсульфинил (1,8 г, 15,0 ммоль) и тетраэтилтитанат (5,70 г, 25,0 ммоль); и смесь нагревали до 50°C, перемешивали в течение 20 часов и добавляли воду (80 мл). Смесь экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением белого твердого вещества (R, E)-N-(1-(5-бромтиофен-2-ил)этилиден)-2-метилпропан-2-сульфинамида (соединение I-37Б) (500 мг, выход 16%).
[00737] Второй шаг: (R)-N-((S)-1-(5-бромтиофен-2-ил)этил)-2-метилпропан-2-сульфинамид (соединение I-37В)
[00738] 
[00739] Соединение (R, E)-N-(1-(5-бромтиофен-2-ил)этилиден)-2-метилпропан-2-сульфинамид (соединение I-37Б) (500 мг, 1,62 ммоль) добавляли к тетрагидрофурану (10 мл) при комнатной температуре; смесь охлаждали до -78°C, и добавляли 1 моль/л раствор три-втор-бутилборгидрид лития в тетрагидрофуране (2,4 мл, 2,4 ммоль); а затем смесь подогревали естественным путем до комнатной температуры и перемешивали в течение 16 часов. pH доводили до 4 с помощью 1 н. хлористоводородной кислоты, и смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого вещества (R)-N-((S)-1-(5-бромтиофен-2-ил)этил)-2-метилпропан-2-сульфинамида (соединение I-37В) (320 мг, выход 64%).
[00740] ЖХ-МС, M/Z (ИЭР): 310,2 [M+H]+.
[00741] Третий шаг: (S)-1-(5-бромтиофен-2-ил)этан-1-амина гидрохлорид (соединение I-37Г)
[00742] 
[00743] Исходный материал (R)-N-((S)-1-(5-бромтиофен-2-ил)этил)-2-метилпропан-2-сульфинамид (соединение I-37В) (310 мг, 1,0 ммоль) добавляли к метанолу (10 мл) при комнатной температуре, и добавляли 4 моль/л раствор диоксана гидрохлорида (0,5 мл, 2,0 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; реакционную смесь концентрировали и добавляли диэтиловый эфир (10 мл); смесь фильтровали с получением (S)-1-(5-бромтиофен-2-ил)этан-1-амина гидрохлорида (60097D) (190 мг, выход 78%) в виде белого твердого вещества.
[00744] Четвертый шаг: (S)-N-(1-(5-бромтиофен-2-ил)этил)-3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамид (соединение I-37Д)
[00745] 
[00746] Соединение 3-(дифторметил)-1-метил-5-(3-(трифторметил) фенокси)-lH-пиразол-4-карбоновую кислоту (140 мг, 0,41 ммоль) добавляли к дихлорметану (5 мл) и N, N-диметилформамиду (5 мл) при комнатной температуре; добавляли (S)-1-(5-бромтиофен-2-ил)этан-1-амина гидрохлорид (соединение I-37Г) (118 мг, 0,49 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (239 мг, 0,63 ммоль) и N, N-диизопропилэтиламин (135 мг, 1,05 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре. Затем смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; а остаток очищали путем разделения с помощью тонкослойной хроматографии на силикагеле (петролейный эфир:этилацетат (об./об.) = 4:1) с получением белого твердого неочищенного продукта (S)-N-(1-(5-бромтиофен-2-ил)этил)-3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамида (соединение I-37Д) (170 мг, выход 78%).
[00747] ЖХ-МС, M/Z (ИЭР): 524,2 [M+H]+
[00748] Пятый шаг: метил (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоксилат (соединение I-37Е)
[00749] 
[00750] Соединение (S)-N-(1-(5-бромтиофен-2-ил)этил)-3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамид (соединение I-37Д) (170 мг, 0,32 ммоль) добавляли к метанолу (5 мл) при комнатной температуре; добавляли триэтиламин (320 мг, 3,20 ммоль) и [l, l’-бис(дифенилфосфино) ферроцен]дихлорпалладий (53 мг, 0,06 ммоль); вводили монооксид углерода; а смесь нагревали до 120°C и перемешивали в течение 48 ч. Затем смесь разбавляли водой (20 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоксилата (соединение I-37Е) (140 мг, выход 86%).
[00751] ЖХ-МС, M/Z (ИЭР): 504,5 [M+H]+
[00752] Шестой шаг: (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоновая кислота (соединение I-37)
[00753]
[00754] Исходный материал метил (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоксилат (соединение I-37Е) (140 мг, 0,28 ммоль) добавляли к тетрагидрофурану (5 мл) и воде (5 мл) при комнатной температуре, и добавляли гидроксид лития (20 мг, 0,47 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения A, (S)-5-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)тиофен-2-карбоновой кислоты (соединение I-37) (110 мг, выход 81%) в виде белого твердого вещества.
[00755] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 8,32 (д, 1H), 7,62 (т, 1H), 7,54 (т, 1H), 7,42 (д, 2H), 7,28 (т, 1H), 7,23 (т, 1H), 6,75 (д, 1H), 5,51-5,04 (м, 1H), 3,76 (с, 3H), 1,27 (д, 3H).
[00756] ЖХ-МС, M/Z (ИЭР): 490,5 [M+H]+
[00757] Пример 38: получение соединения I-38
[00758] (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензойная кислота (соединение I-38)
[00759]
[00760] Схема синтеза для соединения I-38 показана ниже:
[00761] 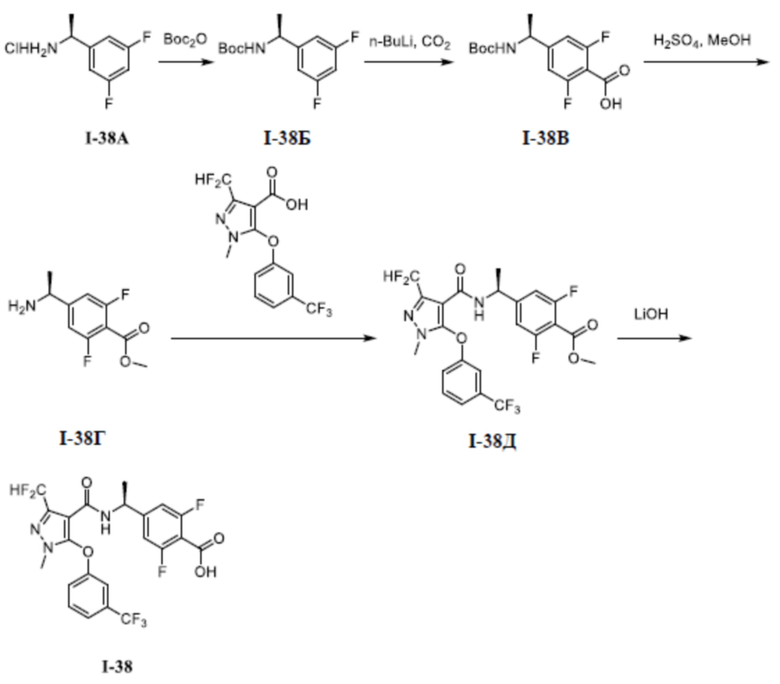
[00762] Первый шаг: трет-бутил (S)-(1-(3,5-дифторфенил)этил)карбамат (соединение I-38Б)
[00763] 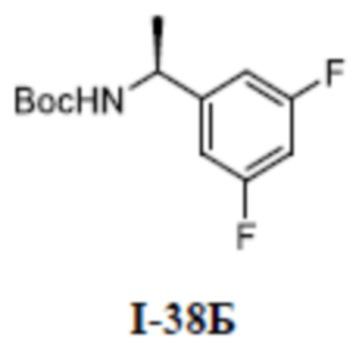
[00764] Соединение (S)-1-(3, 5-дифторфенил)этан-1-аминогидрохлорид (2,0 г, 10,3 ммоль) добавляли к дихлорметану (50 мл) при комнатной температуре; добавляли ди-трет-бутилдикарбонат (4,5 г, 20,7 ммоль) и триэтиламин (3,1 г, 31,0 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре; затем реакционную смесь концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 10:1) с получением трет-бутил (S)-(1-(3,5-дифторфенил)этил)карбамата (соединение I-38Б) (2,2 г, выход 72%) в виде белого твердого вещества.
[00765] Второй шаг: S)-4-(1-((трет-бутоксикарбонил)амино)этил)-2,6-дифторбензойная кислота (соединение I-38В)
[00766] 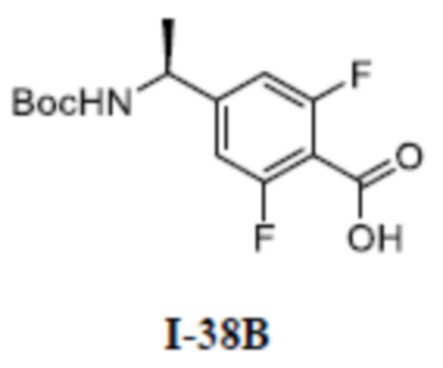
[00767] Соединение (S)-трет-бутил (1-(3, 5-дифторфенил)этил)карбамат (500 мг, 2,50 ммоль) добавляли к тетрагидрофурану (10 мл) при комнатной температуре. Смесь охлаждали до -78°C и добавляли 2,5 моль/л раствор бутиллития в ТГФ (2,0 мл, 5,0 ммоль). Смесь перемешивали в течение 2 часов; вводили диоксид углерода при низкой температуре. Смесь подогревали естественным путем до комнатной температуры и перемешивали в течение 16 часов; pH доводили до 2 с помощью 1 н. хлористоводородной кислоты; и смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта (S)-4-(1-((трет-бутоксикарбонил)амино)этил)-2,6-дифторбензойной кислоты (соединение I-38В) (550 мг, выход 90%).
[00768] Третий шаг: метил (S)-4-(1-аминоэтил)-2,6-дифторбензоат (соединение I-38Г)
[00769] 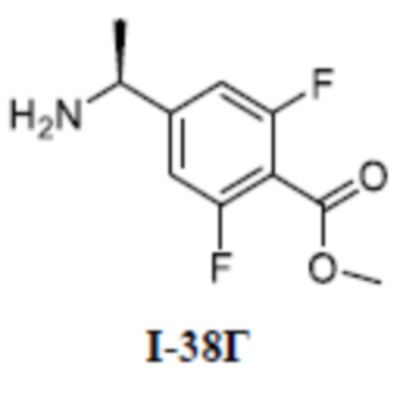
[00770] Исходный материал (S)-4-(1-((трет-бутоксикарбонил)амино)этил)-2,6-дифторбензойная кислота (соединение I-38В) (500 мг, 2,0 ммоль) добавляли к метанолу (100 мл) при комнатной температуре и добавляли концентрированную серную кислоту (20 мл). Смесь нагревали до 80°C и перемешивали в течение 6 часов; а затем смесь разбавляли посредством добавления воды (100 мл), экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта метил (S)-4-(1-аминоэтил)-2,6-дифторбензоата (соединение I-38Г) (1,2 г, выход 100%).
[00771] Четвертый шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензоат (соединение I-38Д)
[00772] 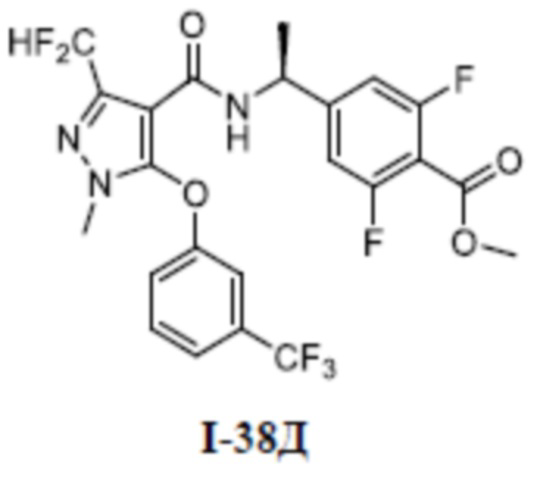
[00773] Соединение 3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоновую кислоту (100 мг, 0,30 ммоль) добавляли к дихлорметану (8 мл) и N, N-диметилформамиду (2 мл) при комнатной температуре; добавляли метил (S)-4-(1-аминоэтил)-2,6-дифторбензоат (соединение I-38Г) (78 мг, 0,36 ммоль); а затем добавляли O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (171 мг, 0,45 ммоль) и N, N-диизопропилэтиламин (58 мг, 0,45 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре, и смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали; остаток отделяли и очищали с помощью пластинки со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 3:1) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензоата (соединение I-38Д) (82 мг, выход 52%).
[00774] ЖХ-МС, M/Z (ИЭР): 534,3 [M+H]+
[00775] Пятый шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензойная кислота (соединение I-38)
[00776]
[00777] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензоат (82 мг, 0,15 ммоль) добавляли к тетрагидрофурану (2 мл) и воде (4 мл) при комнатной температуре, и добавляли гидроксид лития (18,9 мг, 0,45 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 4 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)этил)-2,6-дифторбензойной кислоты (соединение I-38) (16 мг, выход 20%).
[00778] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,12 (д, 1H), 7,61 (т, 1H), 7,56 (т, 1H), 7,45 (с, 1H), 7,27 (т, 1H), 7,25 (т, 1H), 6,62 (д, 2H),4,76 (т, 1H), 3,76 (с, 3H), 1,13 (д, 3H).
[00779] ЖХ-МС, M/Z (ИЭР): 520,3 [M+H]+
[00780] Пример 39: получение соединения I-39
[00781] (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензойная кислота (соединение I-39)
[00782]
[00783] Схема синтеза для соединения I-39 показана ниже:
[00784] 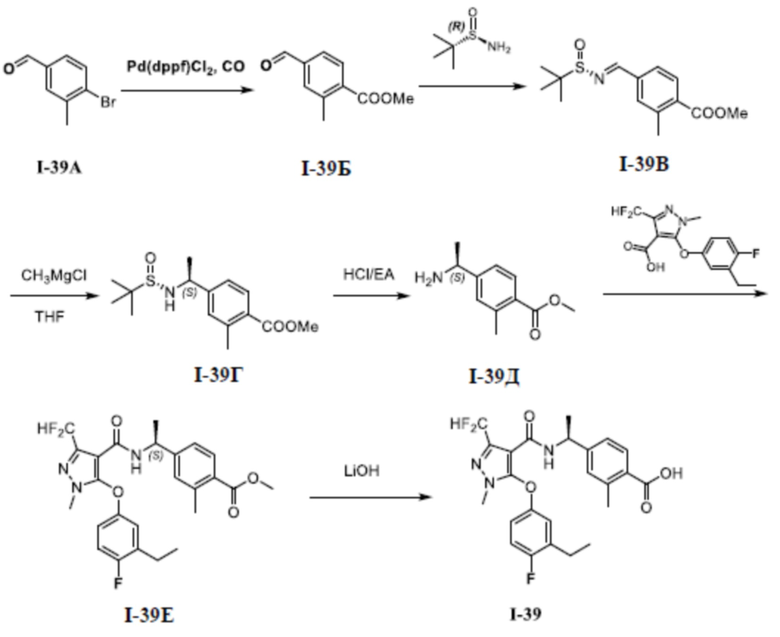
[00785] Первый шаг: синтез метил-4-формил-2-метилбензоата (соединение I-39Б)
[00786] 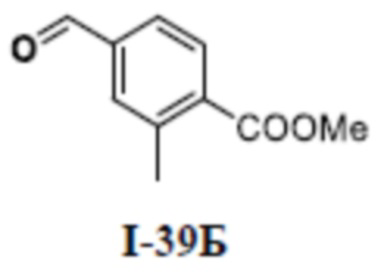
[00787] 4-бром-3-метилбензальдегид (соединение I-39А) (10,0 г, 50,2 ммоль) и триэтиламин (15,3 г, 151 ммоль) добавляли к метанолу (200 мл); и добавляли 1, 1-бис(дифенилфосфоний)ферроценпалладия хлорид (2,94 г, 4,02 ммоль) при комнатной температуре. Смесь заменяли монооксидом углерода 3 раза, и затем осуществляли реакцию в течение 15 часов при 65° под давлением 50 фунтов/кв. дюйм; и затем смесь охлаждали до комнатной температуры и фильтровали через целит. Осадок на фильтре промывали этилацетатом 3 раза и фильтрат концентрировали с получением неочищенного продукта, который отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 100:1-5:1) с получением соединения метил 4-формил-2-метилбензоата (соединение I-39Б) (8,0 г, выход 89%).
[00788] Второй шаг: синтез метил (S,E)-4-(((трет-бутилсульфинил)имино)метил)-2-метилбензоата (соединение I-39В)
[00789] 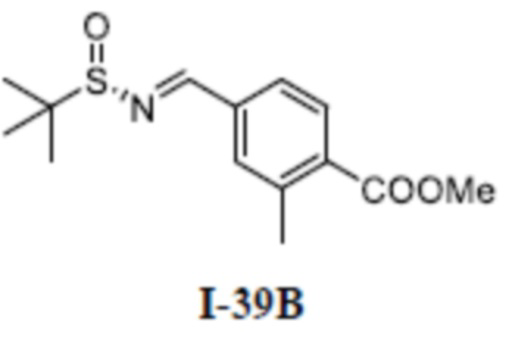
[00790] Метил-4-формил-2-метилбензоат (соединение I-39Б) (8,0 г, 44,9 ммоль) и R-(+)-трет-бутилсульфинамид (6,53 г, 53,9 ммоль) и карбонат цезия (17,6 г, 53,9 ммоль) добавляли к дихлорметану (200 мл). Осуществляли реакцию смеси при 50°C в течение 15 ч, добавляли воду (200 мл) и дихлорметан (200 мл), и водную фазу экстрагировали дихлорметаном (200 мл ×3), объединяли и промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и сушили в центробежной сушилке с получением неочищенного продукта. Неочищенный продукт разделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об) = 100:1-1:1) с получением соединения метил (S, E)-4-(((трет-бутилсульфинил)имино)метил)-2-метилбензоата (соединение I-39В) (10,0 г, выход 79%).
[00791] Третий шаг: синтез метил 4-((S)-1-(((S)-трет-бутилсульфинил)амино)этил)-2-метилбензоата (соединение I-39Г)
[00792] 
[00793] Метил (S, E)-4-(((трет-бутилсульфинил)имино)метил)-2-метилбензоат (соединение I-39В) (1,0 г, 3,55 ммоль) добавляли к тетрагидрофурану (20 мл); и смесь охлаждали до -10°C. Затем медленно по каплям добавляли метилмагния хлорид (7,1 мл, 21,3 ммоль, 3 моль/л) и осуществляли реакцию при комнатной температуре в течение 15 часов; к реакционной смеси добавляли воду (50 мл) и этилацетат (100 мл). Водную фазу экстрагировали этилацетатом (100 мл ×3); объединенный экстрагированный раствор промывали солевым раствором (100 мл), сушили над сульфатом натрия, фильтровали и сушили в центробежной сушилке с получением неочищенного продукта. Неочищенный продукт разделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об) = 20:1-1:1) с получением соединения метил 4-((S)-1-(((S)-трет-бутилсульфинил)амино)этил)-2-метилбензоата (соединение I-39Г) (0,7 г, выход 66%).
[00794] Четвертый шаг: синтез метил (S)-4-(1-аминоэтил)-2-метилбензоата (соединение I-39Д)
[00795] 
[00796] Метил 4-((S)-1-(((S)-трет-бутилсульфинил)амино)этил)-2-метилбензоат (соединение I-39Г) (0,7 г, 2,35 ммоль) добавляли к этилацетату (10 мл); раствор (10 мл) этилацетата/хлористого водорода добавляли при комнатной температуре. Осуществляли реакцию реакционной смеси при комнатной температуре в течение 2 часов, и сушили в центробежной сушилке с получением метил (S)-4-(1-аминоэтил)-2-метилбензоата (соединение I-39Д) (0,4 г, выход 88%).
[00797] Пятый шаг: синтез метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензоата (соединение I-39Е)
[00798] 
[00799] Метил (S)-4-(1-аминоэтил)-2-метилбензоат (соединение I-39Д) (0,1 г, 0,52 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (0,24 г, 0,62 ммоль), N, N-диизопропилэтиламин (0,20 г, 1,55 ммоль) и 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоновую кислоту (0,20 г, 0,62 ммоль) добавляли к N, N-диметилформамиду (5 мл) и осуществляли реакцию при комнатной температуре в течение 12 часов. Затем в реакционную смесь добавляли воду (50 мл) и этилацетат (100 мл), и экстрагировали этилацетатом (100 мл x 4). Объединенный экстрагированный раствор промывали солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и сушили в центробежной сушилке с получением неочищенного продукта. Неочищенный продукт разделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об) = 20:1-1:1) с получением метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензоата (соединение I-39Е) (0,15 г, выход 59%).
[00800] Шестой шаг: синтез (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензойной кислоты (соединение I-39)
[00801]
[00802] Метил (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензоат (соединение I-39Е) (0,15 г, 0,31 ммоль) добавляли к тетрагидрофурану (5 мл), метанолу (5 мл) и воде (1 мл), и добавляли гидроксид лития (0,03 г, 1,23 ммоль) при комнатной температуре. Осуществляли реакцию реакционной смеси при комнатной температуре в течение 2 часов, затем смесь сушили в центробежной сушилке. К неочищенному продукту добавляли воду (50 мл) и этилацетат (100 мл), затем pH доводили до 7 с помощью 1 н. хлористоводородной кислоты и смесь экстрагировали этилацетатом (100 мл x 2). Органические слои объединяли и промывали солевым раствором (50 мл), и органическую фазу сушили над сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт разделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об) = 1:1) с получением белого твердого вещества (S)-4-(1-(3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоксамидо)этил)-2-метилбензойной кислоты (соединение I-39) (83 мг, выход 57%).
[00803] 1H ЯМР (400 МГц, ДМСО) δ 7,94 (д, J = 7,6 Гц, 1H), 7,59 (д, J = 8,0 Гц, 1H), 7,10 (дд, J = 7,6, 6,4 Гц, 2H), 6,99 - 6,94 (м, 2H), 6,89 (д, J = 8,2 Гц, 1H), 6,80 - 6,75 (м, 1H), 4,84 - 4,79 (м, 1H), 3,69 (с, 3H), 2,36 (с, 3H), 2,53 (дд, J = 14,8, 7,4 Гц, 2H) 1,19 (д, J = 7,0 Гц, 3H), 1,06 (т, J = 7,6 Гц, 3H).
[00804] ЖХ-МС, M/Z (ИЭР): 476,3 [M+H]+
[00805] Пример 40: получение соединения I-40
[00806] 4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензойная кислота (соединение I-40)
[00807]
[00808] Схема синтеза для соединения I-40 показана ниже:
[00809] 
[00810] Первый шаг: N-циклобутилиден-2-метилпропан-2-сульфинамид (соединение I-40Б)
[00811] 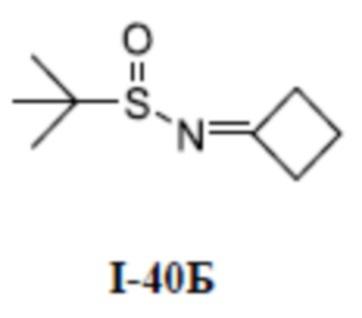
[00812] Соединение циклобутанон (5,0 г, 71,4 ммоль) добавляли к дихлорметану (100 мл) при комнатной температуре; добавляли трет-бутилсульфинамид (10,3 г, 85,6 ммоль) и тетраэтилтитанат (32,6 г, 143 ммоль); и смесь нагревали до 40°C и перемешивали в течение 24 часов. Смесь охлаждали до комнатной температуры и добавляли воду (150 мл), и смесь экстрагировали этилацетатом (100 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением светло-желтой жидкости N-циклобутилиден-2-метилпропан-2-сульфинамида (соединение I-40Б) (6,5 г, выход 53%).
[00813] Второй шаг: N-(1-(4-бромфенил)циклобутил)-2-метилпропан-2-сульфинамид (соединение I-40В)
[00814] 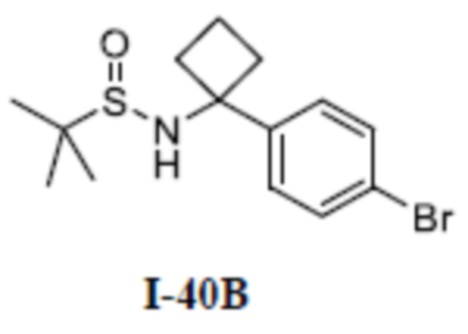
[00815] Соединение 1, 4-дибромбензол (14,0 г, 59,8 ммоль) добавляли к тетрагидрофурану (150 мл) при комнатной температуре; смесь охлаждали до -78°C; и добавляли 2,5 моль/л раствор бутиллития тетрагидрофурана (24,0 мл, 60,0 ммоль). Смесь перемешивали в течение 40 мин, а затем раствор N-циклобутилиден-2-метилпропан-2-сульфинамида (соединение I-40Б) (7,0 г, 40,4 ммоль) в тетрагидрофуране (150 мл) добавляли по каплям в защитной атмосфере азота. Затем смесь подогревали естественным путем до комнатной температуры и перемешивали в течение 2,5 часа, а затем разбавляли насыщенным водным хлоридом аммония (200 мл), экстрагировали этилацетатом (200 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением желтого твердого вещества N-(1-(4-бромфенил)циклобутил)-2-метилпропан-2-сульфинамида (соединение I-40В) (1,8 г, выход 64%).
[00816] ЖХ-МС, M/Z (ИЭР): 330,0 [M+H]+
[00817] Третий шаг: метил 4-(1-((трет-бутилсульфинил)амино)циклобутил)бензоат (соединение I-40Г)
[00818] 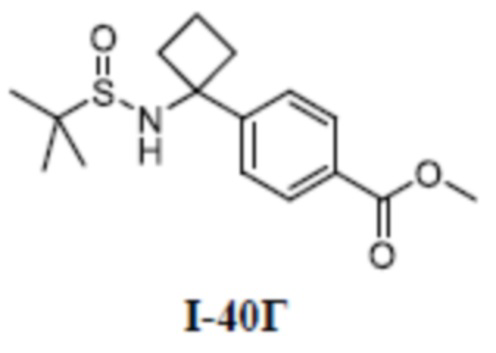
[00819] Исходный материал N-(1-(4-бромфенил)циклобутил)-2-метилпропан-2-сульфинамид (соединение I-40В) (1,80 г, 5,47 ммоль) добавляли к метанолу (30 мл) при комнатной температуре; добавляли триэтиламин (2,76 г, 27,4 ммоль) и l, l’-бисдифенилфосфиноферроценпалладия дихлорид (450 мг, 0,55 ммоль) и добавляли монооксид углерода. Смесь нагревали до 85°C и перемешивали в течение 24 часов. Смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, и остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (дихлорметан: метанол (об./об.) = 10:1) с получением желтой жидкости метил 4-(1-((трет-бутилсульфинил)амино)циклобутил)бензоата (соединение I-40Г) (1,30 г, выход 77%).
[00820] Четвертый шаг: метил 4-(1-аминоциклобутил)бензоат (соединение I-40Д)
[00821] 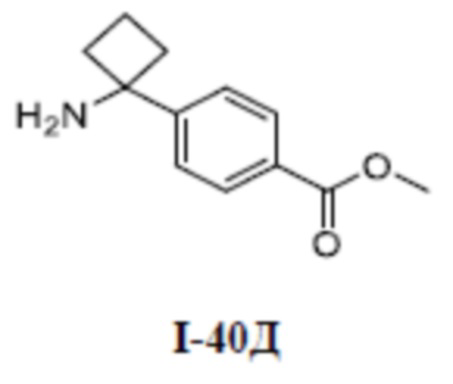
[00822] Соединение метил 4-(1-((трет-бутилсульфинил)амино)циклобутил)бензоат (соединение I-40Г) (600 мг, 1,94 ммоль) добавляли к дихлорметану (10 мл), и добавляли 4 моль/л раствор хлористоводородной кислоты в диоксане (2,0 мл, 8,0 ммоль); смесь перемешивали при комнатной температуре в течение 4 часов; добавляли насыщенный раствор бикарбоната натрия (30 мл), и смесь экстрагировали дихлорметаном (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и разделяли с получением бесцветного жидкого неочищенного продукта метил 4-(1-аминоциклобутил)бензоата (соединение I-40Д) (350 мг, выход 88%).
[00823] ЖХ-МС, M/Z (ИЭР): 206,5 [M+H]+
[00824] Пятый шаг: метил 4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензоат (соединение I-40Е)
[00825] 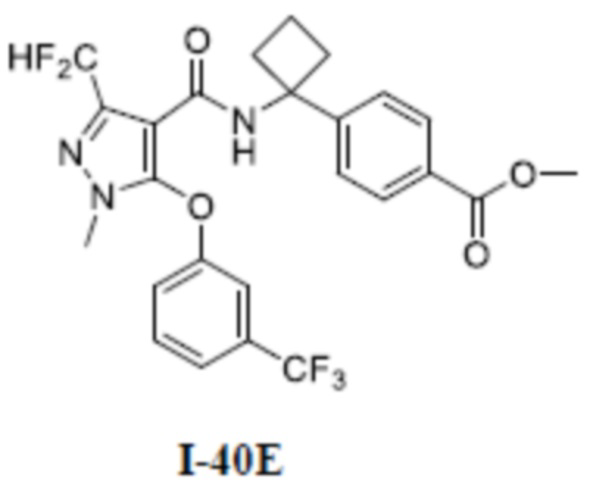
[00826] Соединение 3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-lH-пиразол-4-карбоновую кислоту (100 мг, 0,30 ммоль) добавляли к N, N-диметилформамиду (3 мл); добавляли метил-4-(1-аминоциклобутил)бензоат (соединение I-40Д) (74 мг, 0,36 ммоль), и O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (125 мг, 0,33 ммоль), и N, N-диизопропилэтиламин (85 мг, 0,66 ммоль) при комнатной температуре. Смесь перемешивали в течение 16 часов при комнатной температуре; и смесь разбавляли посредством добавления воды (10 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали на пластинке со слоем силикагеля для тонкослойной хроматографии (петролейный эфир:этилацетат (об./об.) = 1:1) с получением белого твердого вещества метил-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензоата (соединение I-40Е) (60 мг, выход 39%).
[00827] ЖХ-МС, M/Z (ИЭР): 524,5 [M+H]+
[00828] Шестой шаг: 4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензойная кислота (соединение I-40)
[00829]
[00830] Исходный материал метил-4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензоат (соединение I-40Е) (60 мг, 0,12 ммоль) добавляли к тетрагидрофурану (1 мл), метанолу (1 мл) и воде (1 мл) при комнатной температуре; и добавляли гидроксид лития (20 мг, 0,47 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов; pH доводили до 2 с помощью 1 н. хлористоводородной кислоты; и реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества 4-(1-(3-(дифторметил)-1-метил-5-(3-(трифторметил)фенокси)-1H-пиразол-4-карбоксамидо)циклобутил)бензойной кислоты (соединение I-40) (16 мг, выход 27%).
[00831] 1H ЯМР (400 МГц, ДМСО-d6) δ12,7 (с, 1H), 8,52 (с, 1H), 7,74 (д, 2H), 7,68 (д, 1H), 7,62 (д, 1H), 7,50 (с, 1H), 7,34 (т, 1H), 7,25 (д, 2H), 7,18 (т, 1H), 3,78 (с, 3H), 2,33-2,28 (м, 2H), 2,16-2,13 (м, 2H), 1,70-1,65 (м, 1H), 1,59-1,54 (м, 1H).
[00832] ЖХ-МС, M/Z (ИЭР): 510,5 [M+H]+
[00833] Пример 41: получение соединения I-41
[00834] (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-метилтиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-41)
[00835]
[00836] Схема синтеза для соединения I-41 показана ниже:
[00837] 
[00838] Первый шаг: метил-3-((3-(дифторметил)-4-формил-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилат (соединение I-41Б)
[00839] 
[00840] Соединение 5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбальдегид (120 мг, 0,62 ммоль) добавляли к N, N-диметилформамиду (5 мл); добавляли метил-3-гидрокси-5-метилтиофен-2-карбоксилат (143 мг, 0,74 ммоль), карбонат цезия (403 мг, 1,24 ммоль), йодид меди (I) (24,0 мг, 0,12 ммоль) и 1, 10-фенантролин (45,0 мг, 0,25 ммоль) при комнатной температуре и нагревали до 130°C в микроволновой печи и перемешивали в течение 1 ч; затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (15 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением бесцветной жидкости метил-3-((3-(дифторметил)-4-формил-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилата (соединение I-41Б) (32,0 мг, выход 16%).
[00841] ЖХ-МС, M/Z (ИЭР): 331,4 (M+1).
[00842] Второй шаг: 3-(дифторметил)-5-((2-(метоксикарбонил)-5-метилтиофен-3-ил)окси)-1-метил-1H-пиразол-4-карбоновая кислота (соединение I-41В)
[00843] 
[00844] Соединение метил-3-((3-(дифторметил)-4-формил-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилат (соединение I-41Б) (30,0 мг, 0,09 ммоль) добавляли к трет-бутанолу (6 мл) и воде (3 мл) при комнатной температуре; добавляли 2-метил-2-бутен (18,9 мг, 0,27 ммоль), хлорит натрия (24,0 мг, 0,27 ммоль) и дигидрофосфат натрия (27,0 мг, 2,34 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре; а затем смесь разбавляли водой (5 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого вещества 3-(дифторметил)-5-((2-(метоксикарбонил)-5-метилтиофен-3-ил)окси)-1-метил-1H-пиразол-4-карбоновой кислоты (соединение I-41В) (32,0 мг, выход 100%).
[00845] ЖХ-МС, M/Z (ИЭР): 347,4 (M+1)
[00846] Третий шаг: метил (S)-3-((3-(дифторметил)-4-((1-(4-(метоксикарбонил)фенил)этил)карбамоил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилат (соединение I-41Г)
[00847] 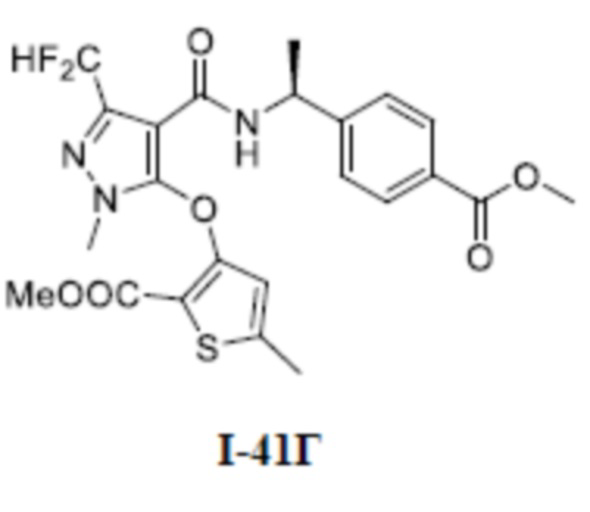
[00848] Соединение 3-(дифторметил)-5-(3-этил-4-фторфенокси)-1-метил-1H-пиразол-4-карбоновую кислоту (24,0 мг, 0,07 ммоль) добавляли к дихлорметану (5 мл); добавляли (S)-метил-4-(l-аминоэтил)бензоат (15,0 мг, 0,08 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (40,0 мг, 0,10 ммоль) и N, N-диизопропилэтиламин (14,0 мг, 0,10 ммоль); и смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли водой (20 мл), экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и остаток очищали путем разделения на колонке с силикагелем (петролейный эфир:этилацетат (об./об.) = 3:1) с получением бесцветной жидкости метил (S)-3-((3-(дифторметил)-4-((1-(4-(метоксикарбонил)фенил)этил)карбамоил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилата (соединение I-41Г) (25,0 мг, выход 71,0%).
[00849] ЖХ-МС, M/Z (ИЭР): 508,6 (M+1).
[00850] Четвертый шаг: (S)-3-((4-((1-(4-карбоксифенил)этил)карбамоил)-3-(дифторметил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоновая кислота (соединение I-41Д)
[00851] 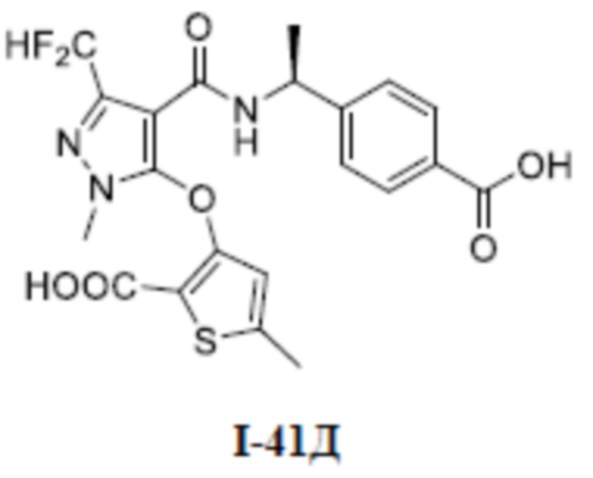
[00852] Исходный материал метил (S)-3-((3-(дифторметил)-4-((1-(4-(метоксикарбонил)фенил)этил)карбамоил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоксилат (38,0 мг, 0,08 ммоль) добавляли к тетрагидрофурану (5 мл) и воде (3 мл) при комнатной температуре, и добавляли гидроксид лития (10,0 мг, 0,22 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов; pH доводили до 6 с помощью 1 н. хлористоводородной кислоты; и смесь экстрагировали этилацетатом (10 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого неочищенного продукта (S)-3-((4-((1-(4-карбоксифенил)этил)карбамоил)-3-(дифторметил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоновой кислоты (соединение I-41Д) (33,0 мг, выход 92%).
[00853] ЖХ-МС, M/Z (ИЭР): 480,3 (M+1)
[00854] Пятый шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-метилтиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-41)
[00855]
[00856] Исходный материал (S)-3-((4-((1-(4-карбоксифенил)этил)карбамоил)-3-(дифторметил)-1-метил-1H-пиразол-5-ил)окси)-5-метилтиофен-2-карбоновую кислоту (30,0 мг, 0,06 ммоль) добавляли к N-метилпирролидону (6 мл) при комнатной температуре; добавляли ацетат серебра (10,2 мг, 0,06 ммоль); и смесь нагревали до 100°C в защитной атмосфере азота и перемешивали в течение 0,5 часа. Реакционную смесь фильтровали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-метилтиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-41) (6,7 мг, выход 25%).
[00857] 1H ЯМР (400 МГц, CDCl3) δ12,8 (с, 1H), 7,99 (д, 2H), 7,20 (т, 1H), 7,20 (д, 2H), 6,51 (с, 1H), 6,39 (д, 1H), 6,20 (с, 1H), 5,22 (т, 1H), 3,75 (с, 3H), 2,45 (с, 3H), 1,43 (д, 3H).
[00858] ЖХ-МС, M/Z (ИЭР): 436,1 (M+1)
[00859] Пример 42: получение соединения I-42
[00860] (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-42)
[00861]
[00862] Схема синтеза для соединения I-42 показана ниже:
[00863] 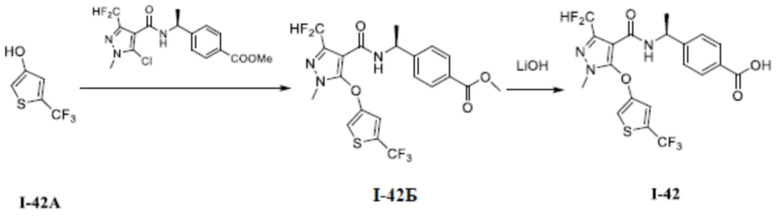
[00864] Первый шаг: метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-42Б)
[00865] 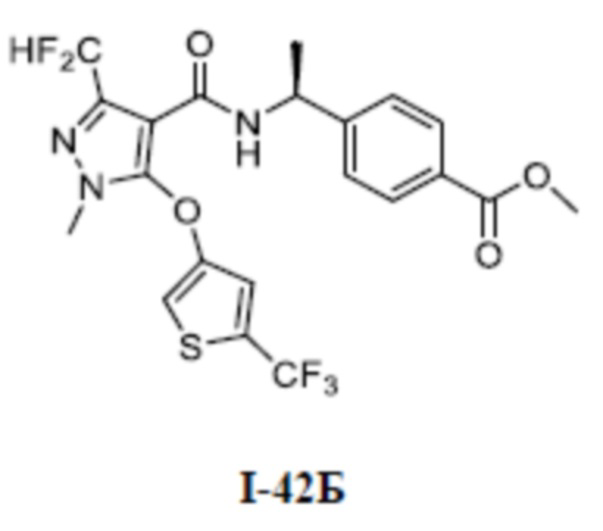
[00866] Соединение метил (S)-4-(1-(5-хлор-3-(дифторметил)-1-метил-1H-пиразол-4-карбоксамидо)этил)бензоат (промежуточное соединение А) (1,0 г, 2,69 ммоль) добавляли к диметилсульфоксиду (5 мл); добавляли 5-(трифторметил)тиофен-3-ол (908 мг, 5,40 ммоль), карбонат калия (1,13 г, 8,18 ммоль), йодид меди (I) (206 мг, 1,08 ммоль) и 1, 10-фенантролин (388 мг, 2,16 ммоль) при комнатной температуре; и смесь нагревали до 120°C в микроволновой печи и перемешивали в течение 4 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:3) с получением белого твердого вещества метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-42Б) (980 мг, выход 72%).
[00867] ЖХ-МС, M/Z (ИЭР): 504,2 (M+1)
[00868] Второй шаг: (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-42)
[00869] 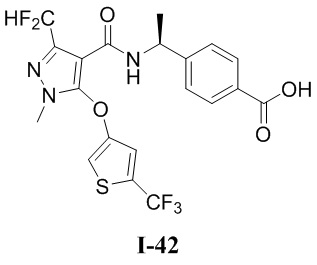
[00870] Исходный материал метил (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-42Б) (980 мг, 1,89 ммоль) добавляли к тетрагидрофурану (5 мл); добавляли воду (3 мл) и гидроксид лития (238 мг, 5,67 ммоль) при комнатной температуре; и смесь перемешивали в течение 3 часов при комнатной температуре. Затем реакционную смесь концентрировали с получением, посредством кислотного способа получения А, белого твердого вещества (S)-4-(1-(3-(дифторметил)-1-метил-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-42) (345 мг, выход 36%).
[00871] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,20 (д, 1H), 7,79 (д, 2H), 7,67 (с, 1H), 7,25 (д, 2H), 7,23 (д, 1H), 7,09 (т, 1H), 4,99-4,92 (м, 1H), 3,77 (с, 3H), 1,29 (д, 3H).
[00872] ЖХ-МС, M/Z (ИЭР): 490,2 (M+1)
[00873] Пример 43: получение соединения I-43
[00874] (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-43)
[00875]
[00876] Схема синтеза для соединения I-43 показана ниже:
[00877] 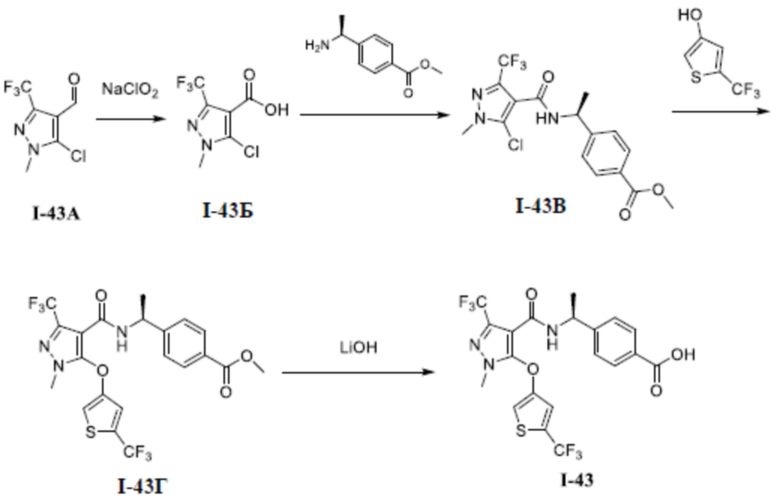
[00878] Первый шаг: 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновая кислота (соединение I-43Б)
[00879] 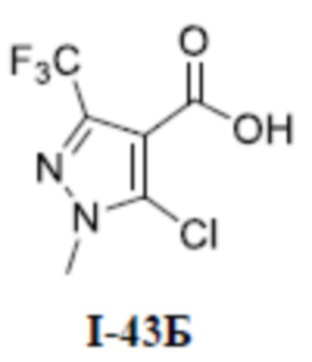
[00880] Соединение 5-хлор-1-метил-3-(трифторметил)-lH-пиразол-4-карбальдегид (700 мг, 3,30 ммоль) добавляли к трет-бутанолу (20 мл) и воде (5 мл) при комнатной температуре; добавляли 2-метил-2-бутен (1,80 г, 25,7 ммоль), хлорит натрия (1,48 г, 16,4 ммоль) и дигидрофосфат натрия (3,10 г, 25,8 ммоль). Смесь перемешивали в течение 14 часов при комнатной температуре; а затем смесь разбавляли водой (50 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого неочищенного продукта 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновой кислоты (соединение I-43Б) (680 мг, выход 90%).
[00881] ЖХ-МС, M/Z (ИЭР): 229,6 (M+1).
[00882] Второй шаг: метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-43В)
[00883] 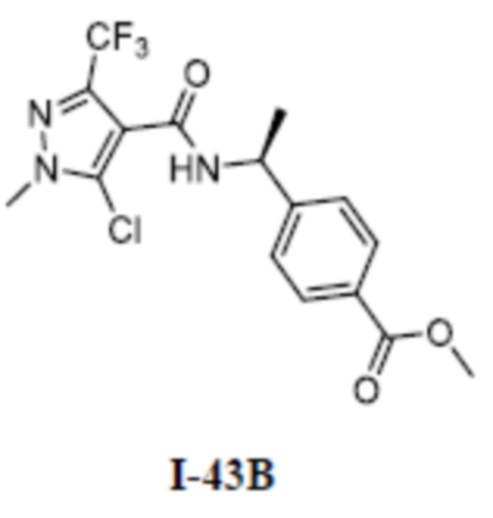
[00884] Соединение 5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоновую кислоту (соединение I-43Б) (680 мг, 2,98 ммоль) добавляли к N, N-диметилформамиду (20 мл); добавляли метил (S)-4-(l-аминоэтил)бензоат (537 мг, 3,00 ммоль), O-(7-азабензотриазол-1-ил)-N, N, N, N-тетраметилурония гексафторфосфат (1,70 г, 4,47 ммоль) и N, N-диизопропилэтиламин (1,90 г, 14,7 ммоль) и перемешивали при комнатной температуре в течение 16 часов; смесь разбавляли водой (200 мл), экстрагировали этилацетатом (30 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением бесцветного твердого вещества метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-43В) (700 мг, выход 60%).
[00885] ЖХ-МС, M/Z (ИЭР): 390,5 (M+1).
[00886] Третий шаг: метил (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-43Г)
[00887] 
[00888] Соединение метил (S)-4-(1-(5-хлор-1-метил-3-(трифторметил)-1H-пиразол-4-карбоксамидо)этил)бензоат (500 мг, 1,3 ммоль) добавляли к диметилсульфоксиду (10 мл); добавляли 5-(трифторметил)тиофен-3-ол (216 мг, 1,3 ммоль), карбонат калия (360 мг, 2,6 ммоль), йодид меди (I) (100 мг, 0,5 ммоль) и 1, 10-фенантролин (100 мг, 0,5 ммоль) при комнатной температуре; и смесь нагревали до 95°C в микроволновой печи и перемешивали в течение 5 часов. Затем смесь охлаждали до комнатной температуры, разбавляли водой (50 мл), экстрагировали этилацетатом (20 мл ×3) и разделяли. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, а остаток отделяли и очищали с помощью колонки с силикагелем (петролейный эфир:этилацетат (об./об.) = 1:1) с получением желтого твердого вещества метил (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоата (соединение I-43Г) (200 мг, выход 30%).
[00889] ЖХ-МС, M/Z (ИЭР): 522,1 (M+1).
[00890] Четвертый шаг: (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойная кислота (соединение I-43)
[00891] 
[00892] Исходный материал метил (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензоат (соединение I-43Г) (100 мг, 0,19 ммоль) добавляли к тетрагидрофурану (30 мл); добавляли воду(10 мл) и гидроксид лития (24 мг, 0,57 ммоль) при комнатной температуре; и смесь нагревали до 50°C и перемешивали в течение 2 часов. Реакционную смесь концентрировали с получением, посредством кислотного способа получения А, желтого твердого вещества (S)-4-(1-(1-метил-3-(трифторметил)-5-((5-(трифторметил)тиофен-3-ил)окси)-1H-пиразол-4-карбоксамидо)этил)бензойной кислоты (соединение I-43) (54 мг, выход 56%).
[00893] 1H ЯМР (400 МГц, ДМСО-d6) δ12,8 (с, 1H), 8,64 (д, 1H), 7,80 (д, 2H), 7,68 (с, 1H), 7,31 (д, 1H), 7,24 (д, 2H), 4,92 (т, 1H), 3,80 (с, 3H), 1,26 (д, 3H).
[00894] ЖХ-МС, M/Z (ИЭР): 508,1 (M+1)
[00895] Аналитические примеры для оценки биологической активности и соответствующих свойств
[00896] Аналитический пример 1: оценка антагонистического эффекта в отношении EP4
[00897] Контрольное соединение и соединения, полученные в примерах 1-43, тестировали отдельно для оценки антагонистического эффекта в отношении EP4, и анализ осуществляли в стабильной клеточной линии CHO, которая в высокой степени экспрессирует рецептор ЕР4 человека.
[00898] После трипсинизации клетки ресуспендировали в буфере (l× HBSS, 0,1% БСА, 20 мМ ГЭПЭС и 500 мкм IBMX) и 8000 клеток на лунку высевали в 384-луночные планшеты в посевной объем 15 мкл. Рабочий раствор соединения с 8X концентрацией готовили с использованием экспериментального буфера, а затем 2,5 мкл рабочего раствора соединения с 8X концентрацией добавляли в 384-луночный планшет, соответственно, и инкубировали при 37°C в течение 30 мин. Рабочий раствор агониста ПГE2 с 8X концентрацией (4 нМ) готовили с использованием экспериментального буфера и 2,5 мкл на лунку добавляли в 384-луночный планшет (конечная концентрация ПГE2 составляла 0,5 нМ) и инкубировали при 37°C в течение 30 мин. После завершения реакции количество цАМФ в клетках количественно определяли в соответствии с инструкциями тестового набора цАМФ (Perkin Elmer, № по кат. TRF0263). Вычисляли антагонистические эффекты (значение IC50) в отношении тестируемых соединений.
[00899]
[00900] Экспериментальные результаты свидетельствуют о том, что соединения по настоящей заявке характеризуются хорошим антагонистическим эффектом в отношении EP4; в сравнении с контрольным соединением, антагонистический эффект большинства соединений в более чем 5 раз выше, чем у контрольного соединения; и соединения по настоящему изобретению проявляют лучший антагонистический эффект в отношении рецептора ЕР4.
[00901] Аналитический пример 2: Анализ ингибирования кальциевого тока в отношении рецептора EP4
[00902] Контрольное соединение и соединения, полученные в примерах 1-43, соответственно тестировали в отношении их ингибирующего эффекта на ток кальция EP4, и анализ выполняли на клетках 293, которые сверхэкспрессируют рецептор EP4 человека.
[00903] Клетки быстро размораживали на водяной бане при 37°C, центрифугировали, ресуспендировали и подсчитывали. Суспензии клеток высевали при 20 мкл/лунка в два 384-луночных планшета (20000 клеток/лунка) и помещали на ночь в термостат (37°C, 5% CO2). Приготовление загрузочного буфера 2X Fluo-4 Direct™ (Invitrogen, № по кат. F10471): 77 мг пробенецида добавляли к 1 мл буфера для FLIPR, с концентрацией 250 мМ. 10 мл буфера для FLIPR и 0,2 мл пробенецида (250 мМ) добавляли в каждую пробирку кристаллов Fluo-4 Direct™ (F10471).
[00904] Один из из планшетов с клетками извлекали из термостата и среду удаляли. 20 мкл аналитического буфера и непромывной загрузочный буфер 2X Fluo-4 Direct™ добавляли в 384-луночный планшет для культуры клеток до конечного объема 40 мкл, и затем планшет с культурой клеток инкубировали в термостате (37°C, 5% CO2) в течение 50 минут, и затем при комнатной температуре в течение 10 минут, и затем планшет помещали в устройство FLIPR. 10 мкл буфера переносили в планшет с клетками и считывали сигнал флуоресценции. Агонист ПГE2 был составлен в виде 10 мМ исходного раствора в растворителе ДМСО и осуществляли серийное разбавление 10 точек концентрации 6X рабочего раствора с использованием буфера. 10 мкл агониста ПГE2 переносили в планшет с клетками, считывали сигнал флуоресценции для вычисления значения EC80.
[00905] Агонист ПГE2 готовили при концентрации 6X EC80 и тестируемое соединение составляли в виде 10 мМ исходного раствора в растворителе ДМСО и осуществляли серийное разбавление 10 точек концентрации 6X рабочего раствора соединения с использованием буфера.
[00906] Другой планшет с клетками извлекали для удаления среды и 20 мкл аналитического буфера и добавляли непромывной загрузочный буфер 2X Fluo-4 Direct™; планшет с клетками в термостате при 37°C, 5% CO2 в течение 50 минут и при комнатной температуре в течение 10 минут, а затем помещали в устройство FLIPR. 10 мкл рабочего раствора соединения, ДМСО и полный антагонист EP4 переносили в планшет с клетками и считывали сигнал флуоресценции. 10 мкл агониста ПГE2 с концентрацией 6X EC80 переносили в планшет с клетками, считывали сигнал флуоресценции для вычисления степени ингибирования.
[00907] Ингибирование (%) = 100-(тестируемая группа-группа полного антагониста Ep4)/(группа ДМСО-группа полного антагониста EP4) * 100
[00908] Значение IC50 ингибирования тока кальция EP4 соединения вычисляли на основе степени ингибирования при различных концентрациях соединения.
[00909]
[00910] Экспериментальные результаты свидетельствуют о том, что соединения по настоящей заявке демонстрируют лучшие ингибирующие эффекты в отношении тока кальция EP4, и они значительно выше, чем у контрольного соединения, особенно соединение I-4; и ингибирующие эффекты соединений в отношении тока кальция EP4 повышаются в более чем 5 раз в сравнении с контрольным соединением. Соединения по настоящему изобретению демонстрируют лучшие ингибирующие эффекты в отношении тока кальция EP4.
[00911] Аналитический пример 3: Анализ связывания радиолиганда в отношении рецептора EP4
[00912] Связывания с радиолигандом ЕР4 контрольного соединения и соединений, полученных в примерах 1-43, тестировали с использованием рекомбинантного мембранного белка рецептора EP4 человека (полученного из клеток 293, сверхэкспрессирующих рецептор EP4 человека). Тестируемые соединения и ПГE2 были составлены в виде 10 мМ исходных растворов в растворителе ДМСО, а затем осуществляли серийное разбавление 8 точек концентрации 4×рабочего раствора с использованием буфера (50 мМ HBSS, 0,1% БСА, 500 мМ NaCl). 1 мкл рабочего раствора соединения, ДМСО и рабочего раствора ПГE2 добавляли в аналитический планшет, соответственно; добавляли 100 мкл рекомбинантного мембранного белка рецептора EP4 человека (20 мкг /лунка) и 100 мкл радиолиганда [3H]-ПГE2 (PerkinElmer, кат.: NET428250UC, парт.: 2469552) (конечная концентрация 1,5 нМ) и инкубировали в запечатанном состоянии при комнатной температуре в течение 1 часа. Фильтрационный планшет Unifilter-96 GF/C (Perkin Elmer) пропитывали 0,5% БСА, 50 мкл на лунку, в течение по меньшей мере 30 мин при комнатной температуре. После завершения связывания реакционную смесь собирали с помощью планшета GF/C с использованием устройства для сбора Perkin Elmer Filtermate с последующим промыванием фильтрационного планшета и высушиванием фильтрационного планшета в течение 1 часа при 50°C. После высушивания дно лунок фильтрационного планшета запечатывали с использованием герметизирующей ленты Perkin Elmer Unifilter-96 и добавляли 50 мкл смеси MicroScint ™-20 (Perkin Elmer), чтобы запечатать верхнюю часть фильтрационного планшета. Подсчеты 3H, захваченные на фильтре, считывали с использованием устройства для считывания Perkin Elmer MicroBeta2.
[00913] Данные анализировали с использованием GraphPad Prism 5 и вычисляли значения степени ингибирования в соответствии со следующей формулой:
[00914] Ингибирование (%) = 100-(тестируемая группа-группа ПГE2)/(группа ДМСО-группа ПГE2) * 100
[00915] Значения IC50 и Ki соединений, определенных с помощью анализа радиолигандного связывания в отношении рецептора EP4 вычисляли на основе степени ингибирования различных концентраций соединений.
[00916]
[00917] Экспериментальные результаты свидетельствуют о том, что в сравнении с контрольным соединением соединения по настоящему изобретению характеризуются лучшей афинностью к рецептору ЕР4, что превосходит контрольное соединение, особенно соединение I-2 и соединение I-5; а их афинность к рецептору EP4 увеличивается в более чем 5 раз в сравнении с контрольным соединением. Соединения по настоящему изобретению демонстрируют более высокую афинность к рецептору EP4.
[00918] Аналитический пример 4: Фармакокинетический анализ
[00919] В данном анализе фармакокинетические параметры контрольного соединения и соединений, полученных в примерах 1-43, тестировали на субъектах, представляющих собой мышей, крыс и собак, соответственно.
[00920] В фармакокинетическом анализе на мышах отбирали самцов мышей ICR (от 20 г до 25 г) и не кормили в течение ночи. Отбирали трех мышей и внутрижелудочно вводили 5 мг/кг соответствующего соединения. Кровь мышей собирали до введения и через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч, 24 ч после введения. Еще 3 мышам внутривенно вводили 1 мг/кг соответствующего соединения и собирали их кровь до введения и через 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 8 ч, 24 ч после введения. Образцы крови центрифугировали при 6800 g при температуре от 2°C до 8°C в течение 6 минут, собирали плазму и хранили при -80°C. Плазму брали в соответствующий момент времени, смешивали с 3-5-кратным количеством ацетонитрила, содержащего внутренний стандарт, встряхивали в течение 1 минуты, центрифугировали при 4°C в течение 10 минут при 13000 об/мин, в супернатант добавляли в 3 раза больше воды, и соответствующую смесь брали для анализа ЖХ-МС/МС. Первичные фармакокинетические параметры анализировали с помощью программного обеспечения WinNonlin 7.0 с некомпартментной моделью.
[00921] В фармакокинетическом анализе на крысах отбирали самцов крыс SD (от 180 г до 240 г) и не кормили в течение ночи. Отбирали трех крыс, внутрижелудочно вводили 5 мг/кг соответствующего соединения. Другим 3 крысам внутривенно вводили 1 мг/кг соответствующего соединения. Следующие операции были такими же, как и при фармакокинетическом анализе на мышах.
[00922] В фармакокинетическом анализе на собаках отбирали самцов собак породы бигль (от 8 кг до 10 кг) и не кормили в течение ночи. Отбирали трех собак породы бигль и внутрижелудочно вводили 3 мг/кг соответствующего соединения. Другим 3 собакам породы бигль внутривенно вводили 1 мг/кг соответствующего соединения. Следующие операции были такими же, как и при фармакокинетическом анализе на мышах.
[00923]
[00924]
[00925]
[00926]
[00927]
[00928]
[00929] Экспериментальные результаты свидетельствуют о том, что в сравнении с контрольным соединением соединения по настоящему изобретению имеют более низкий клиренс при внутривенном введении и более высокое воздействие при пероральном введении, в частности соединение I-5; воздействие после перорального введения у крыс и собак примерно в 2 раза выше, чем у контрольного соединения; и соединения по настоящему изобретению проявляют превосходные фармакокинетические свойства и обладают хорошими лекарственными свойствами.
[00930] Аналитический пример 5: Противоопухолевый эффект тестируемых соединений на мышиной модели CT-26 опухоли рака толстой кишки, в комбинации с лучевой терапией
[00931] В этом анализе противоопухолевые эффекты контрольного соединения и соединений, полученных в примерах 1-43, в комбинации с лучевой терапией тестировали на мышиной модели CT-26 опухоли рака толстой кишки.
[00932] После одной недели адаптивного кормления мышей клетки CT-26 в логарифмической фазе ресуспендировали в ФСБ, 5 x 105 клеток CT-26 инокулировали подкожно в правый бок в количестве 100 мкл на мышь, и регулярно наблюдали рост опухоли. Когда опухоль выростала до среднего объема от 60 до 80 мм3, мышей с опухолью случайным образом разделили на 5 групп в зависимости от размера опухоли, каждая группа содержала 10 мышей из 10. Эти группы представляли собой группу контрольного соединения (150 мг/кг) в комбинации с лучевой терапией (3Gry), группу соединения I-2 (150 мг/кг) в комбинации с лучевой терапией, группу соединения I-5 (150 мг/кг) в комбинации с лучевой терапией, группу лучевой терапии (3Gry) отдельно и группу контроля среды-носителя. Мышам внутрижелудочно вводили соответствующее соединение один раз в сутки в целом в течение 23 дней; лучевую терапию проводили однократно в первый день введения. Объемы опухолей и массу тела мышей измеряли два раза в неделю, а массу опухолей взвешивали в конце эксперимента. Оценки эффективности проводились на основе относительного ингибирования роста опухоли (TGI), оценки безопасности проводились на основе изменений массы тела животных и гибели. Объем опухоли и относительное ингибирование роста опухоли вычисляли следующим образом:
[00933] Объем опухоли (TV) = 1/2 × a × b2, где a и b представляют собой длину и ширину измерения опухоли, соответственно.
[00934] Степень относительного ингибирования роста опухоли TGI (%) = (TWc-TWt/TWc) x 100%, где TWc представляет собой среднее значение массы опухоли группы контроля среды-носителя, а TW представляет собой среднее значение массы опухоли в группе лечения.
[00935] Экспериментальные результаты проиллюстрированы на фиг. 1. На фиг. 1 показано, что контрольное соединение, тестируемые соединения 1-2 и 1-5 в комбинации с лучевой терапией проявляли значительный эффект ингибирования опухоли на 23-й день после начала введения, а значения степени относительного ингибирования роста опухоли TGI (%) составляли 54. %, 63% и 79% соответственно, все из которых статистически значимо отличаются от контрольной группы среды-носителя (среднее значение p меньше 0,05); группа соединения I-5 в комбинации с лучевой терапией статистически значимо отличалась от группы только лучевой терапии (p меньше 0,05) и была лучше, чем группа контрольного соединения в комбинации с лучевой терапией. Ни в одной из групп в комбинации с лучевой терапией не было ни гибели животных, ни проявления выраженной лекарственной токсичности, демонстрируя хорошую переносимость во время лечения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕКОТОРЫХ ЛЕЙКОЗОВ | 2019 |
|
RU2804709C2 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА КАК ИНГИБИТОРЫ RSV | 2016 |
|
RU2738232C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРАЗОЛ-3-КАРБОКСАМИДА, ОБЛАДАЮЩЕЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА 5-НТ | 2009 |
|
RU2528406C2 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
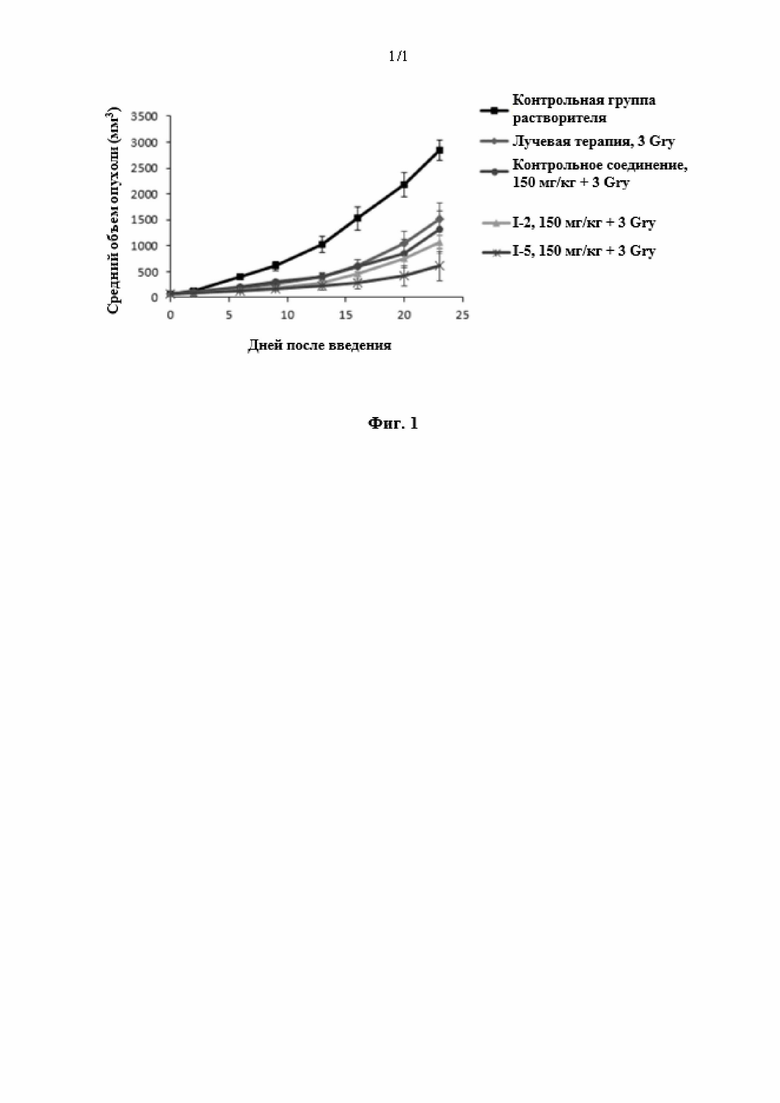
Настоящее изобретение относится к новому пиразольному производному, а именно к новому соединению, эффективному в антагонизировании EP4. Соединение представлено формулой V, где кольцо A выбрано из 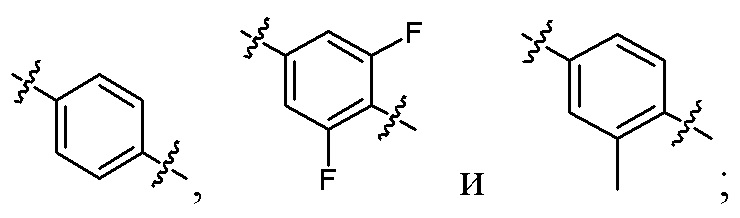 кольцо B выбрано из
кольцо B выбрано из 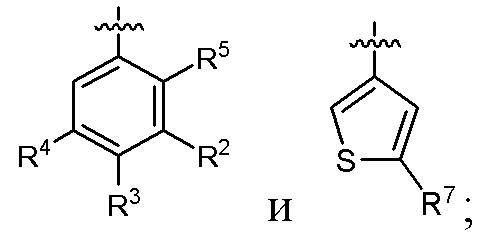 R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, фенила, трифторметила, C2-C6галогензамещенного алкила, C3-C6галогензамещенного циклоалкила, C2-C6гидроксизамещенного алкила, C2-C6цианозамещенного алкила и -X-R2a, где X выбран из серы, -CO-, -SO2- и SO-, и R2a выбран из C1-C6алкила и C1-C6галогензамещенного алкила; R3 выбран из водорода, галогена, C1-C2алкила, C1-C2фторзамещенного алкила и фенила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C2-C6галогензамещенного алкоксила; R5 выбран из водорода и галогена; один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил; или R6a и R6b вместе образуют циклобутил; R7 выбран из -CH3, -CHF2 и -CF3; и M выбран из кислорода, серы и метилена; при условии, что когда R2 представляет собой трифторметил, M представляет собой кислород, один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил, кольцо A выбрано из
R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, фенила, трифторметила, C2-C6галогензамещенного алкила, C3-C6галогензамещенного циклоалкила, C2-C6гидроксизамещенного алкила, C2-C6цианозамещенного алкила и -X-R2a, где X выбран из серы, -CO-, -SO2- и SO-, и R2a выбран из C1-C6алкила и C1-C6галогензамещенного алкила; R3 выбран из водорода, галогена, C1-C2алкила, C1-C2фторзамещенного алкила и фенила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C2-C6галогензамещенного алкоксила; R5 выбран из водорода и галогена; один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил; или R6a и R6b вместе образуют циклобутил; R7 выбран из -CH3, -CHF2 и -CF3; и M выбран из кислорода, серы и метилена; при условии, что когда R2 представляет собой трифторметил, M представляет собой кислород, один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил, кольцо A выбрано из 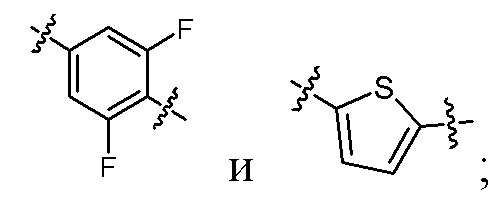 и когда R2 представляет собой трифторметил, M представляет собой кислород, а кольцо A представляет собой
и когда R2 представляет собой трифторметил, M представляет собой кислород, а кольцо A представляет собой  R6a и R6b вместе образуют циклобутил. Также предложено соединение, представленное формулой I, где R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, C2-C6галогензамещенного алкила и C3-C6галогензамещенного циклоалкила; R3 выбран из водорода, галогена, C1-C2алкила и C1-C2фторзамещенного алкила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила. Кроме того, изобретение относится к соединению, выбранному из группы индивидуальных соединений, применению указанных соединений в получении лекарственного средства для лечения или предупреждения связанного с EP4 заболевания и способу лечения связанного с EP4 заболевания. Технический результат изобретения заключается в разработке новых соединений, являющихся антагонистами EP4. 7 н. и 6 з.п. ф-лы, 1 ил., 9 табл., 43 пр.
R6a и R6b вместе образуют циклобутил. Также предложено соединение, представленное формулой I, где R1 выбран из -CH3, -CHF2 и -CF3; R2 выбран из C2-C6алкила, C3-C6циклоалкила, C2-C6галогензамещенного алкила и C3-C6галогензамещенного циклоалкила; R3 выбран из водорода, галогена, C1-C2алкила и C1-C2фторзамещенного алкила; R4 выбран из водорода, галогена, C1-C6алкила, C1-C6алкоксила, C1-C6галогензамещенного алкила и C1-C6галогензамещенного алкоксила. Кроме того, изобретение относится к соединению, выбранному из группы индивидуальных соединений, применению указанных соединений в получении лекарственного средства для лечения или предупреждения связанного с EP4 заболевания и способу лечения связанного с EP4 заболевания. Технический результат изобретения заключается в разработке новых соединений, являющихся антагонистами EP4. 7 н. и 6 з.п. ф-лы, 1 ил., 9 табл., 43 пр.
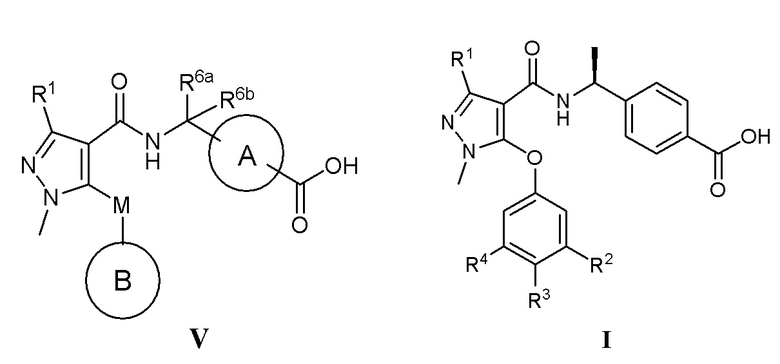
1. Соединение, представляющее собой соединение, представленное формулой V:
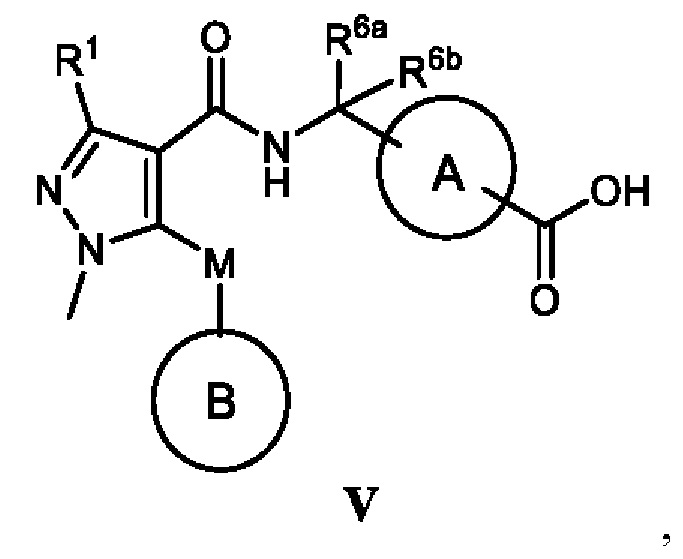 где
где
кольцо А выбрано из 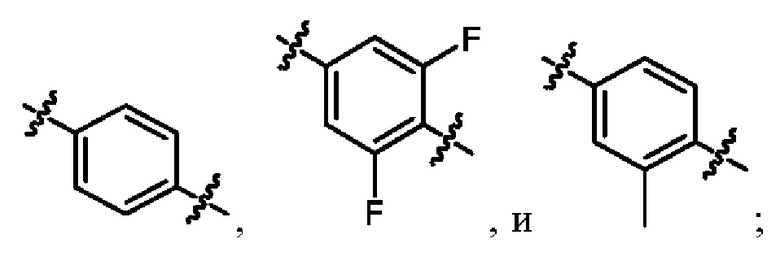
кольцо В выбрано из 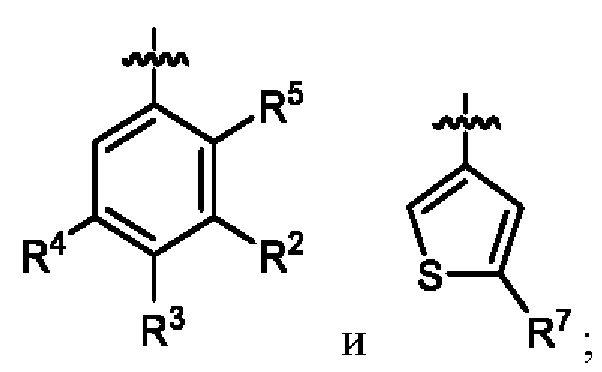 ;
;
R1 выбран из -CH3, -CHF2 и -CF3;
R2 выбран из C2-С6алкила, C3-С6циклоалкила, фенила, трифторметила, C2-С6галогензамещенного алкила, C3-С6галогензамещенного циклоалкила, С2-С6гидроксизамещенного алкила, С2-С6цианозамещенного алкила, и -X-R2a, где X выбран из серы, -СО-, -SO2- и SO-, и R2a выбран из C1-С6алкила и C1-С6галогензамещенного алкила;
R3 выбран из водорода, галогена, С1-С2алкила, С1-С2фторзамещенного алкила и фенила;
R4 выбран из водорода, галогена, C1-С6алкила, C2-С6алкоксила, C1-С6галогензамещенного алкила и C2-С6галогензамещенного алкоксила;
R5 выбран из водорода и галогена;
один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил; или R6a и R6b вместе образуют циклобутил;
R7 выбран из -CH3, -CHF2 и -CF3; и
М выбран из кислорода, серы и метилена;
при условии, что:
когда R2 представляет собой трифторметил, М представляет собой кислород, один из R6a и R6b представляет собой водород, а другой один из R6a и R6b представляет собой метил, кольцо А выбрано из 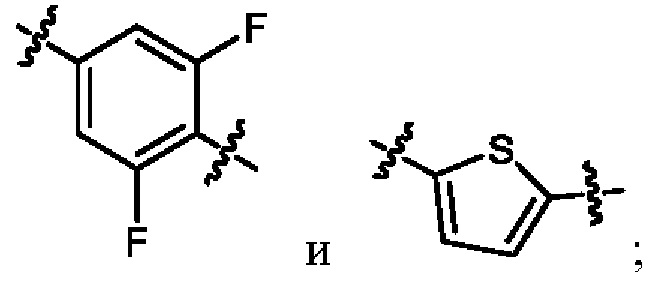 и
и
когда R2 представляет собой трифторметил, М представляет собой кислород, а кольцо А представляет собой  R6a и R6b вместе образуют циклобутил.
R6a и R6b вместе образуют циклобутил.
2. Соединение по п. 1, представляющее собой соединение, представленное формулой III:
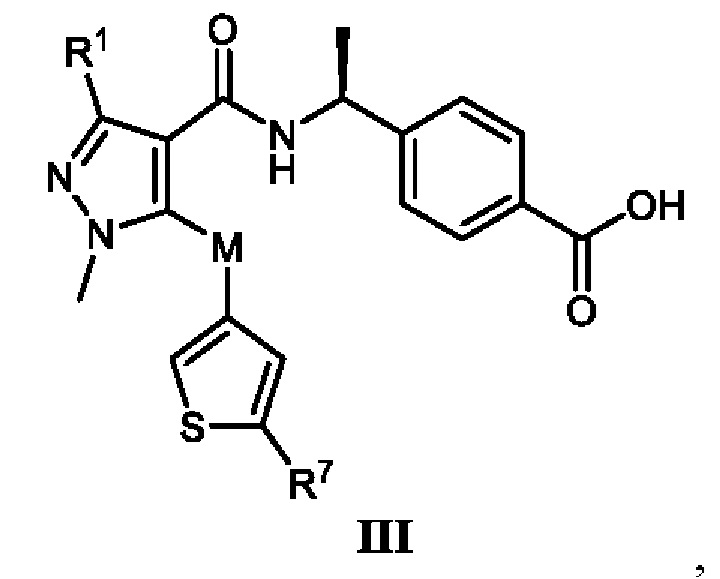 где
где
R1 выбран из -CH3, -CHF2 и -CF3;
R7 выбран из -CH3, -CHF2 и -CF3; и
М выбран из кислорода, серы и метилена.
3. Соединение по п. 1, представляющее собой соединение, представленное формулой II:
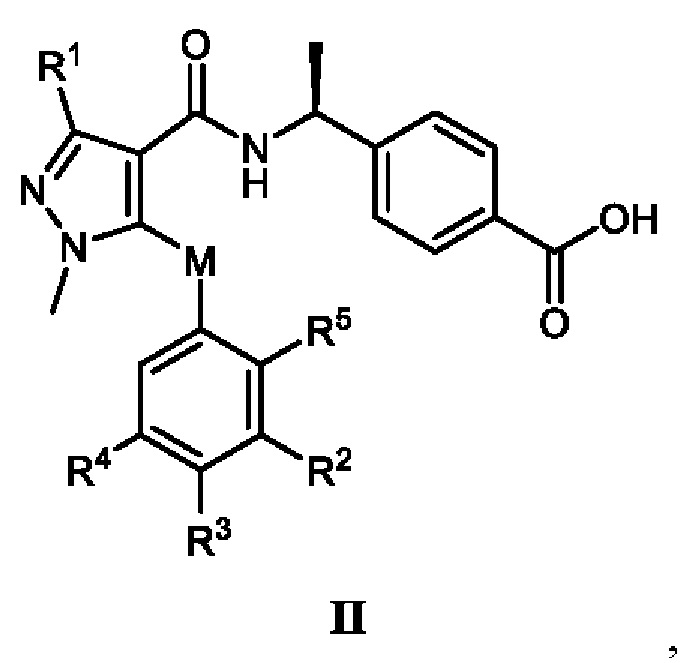 где
где
R1 выбран из -CH3, -CHF2 и -CF3, и предпочтительно R1 представляет собой -CHF2;
R2 выбран из этила, пропила, изопропила, н-бутила, изобутила, трет-бутила, фторэтила, фторпропила, фторизопропила, фторбутила, фторизобутила, гидроксиэтила, гидроксиизопропила, цианометила, цианоэтила, фенила, и -X-R2a, где X выбран из серы и -СО-, и R2a выбран из метила, этила, фторметила и фторэтила;
R3 выбран из водорода, фтора, хлора, метила, этила, фторметила, фторэтила и фенила;
R4 выбран из водорода, фтора, хлора, метила, этила, фторметила и фторэтила;
R5 выбран из водорода, фтора и хлора; и
М выбран из кислорода, серы и метилена.
4. Соединение, представляющее собой соединение, представленное формулой I:
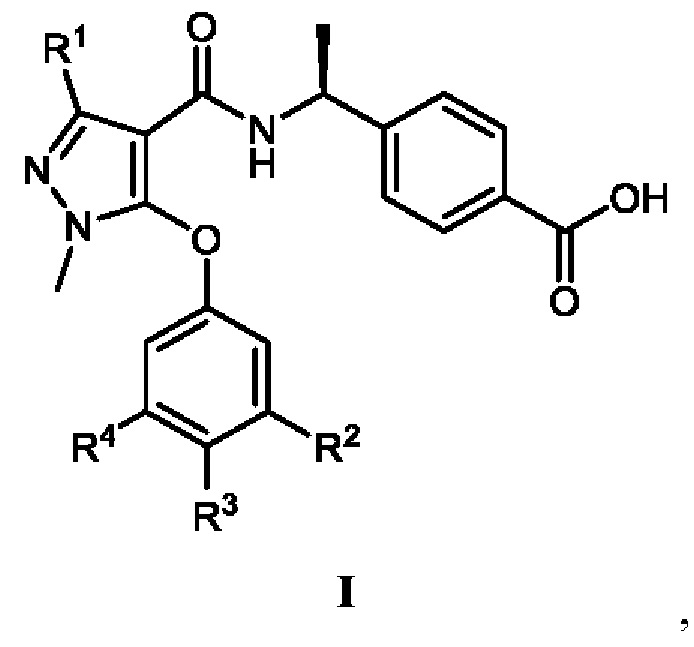 где
где
R1 выбран из -CH3, -CHF2 и -CF3;
R2 выбран из C2-С6алкила, C3-С6циклоалкила, С2-С6галогензамещенного алкила и C3-С6галогензамещенного циклоалкила;
R3 выбран из водорода, галогена, С1-С2алкила и C1-С2фторзамещенного алкила;
R4 выбран из водорода, галогена, C1-С6алкила, C1-С6алкоксила, C1-С6галогензамещенного алкила и C1-С6галогензамещенного алкоксила.
5. Соединение по п. 4, где
R2 выбран из C2-С3алкила, C3-С6циклоалкила, C2-С3фторзамещенного алкила и C3-С6фторзамещенного циклоалкила, и предпочтительно R2 выбран из -CH2CH3, -СН(CH3)2, циклопропила, -CF2CH3 и -CH2CF3;
R3 выбран из водорода, фтора и хлора;
R4 выбран из водорода, фтора, хлора, С1-С4алкила, С1-С4алкоксила, С1-С4фтор- или хлорзамещенного алкила и С1-С4фтор- или хлорзамещенного алкоксила, и предпочтительно R4 выбран из водорода, фтора и хлора.
6. Соединение, представляющее собой любое из следующих соединений:
7. Применение соединения по любому из пп. 1-6 в получении лекарственного средства для лечения или предупреждения связанного с ЕР4 заболевания.
8. Применение соединения по любому из пп. 1-6 в получении лекарственного средства для лечения или предупреждения рака.
9. Применение по п. 8, где рак выбран из видов солидного рака, предпочтительно из рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, глиобластомы, рака головы и шеи, рака почки, рака печени, рака легкого, медуллобластомы, рака яичника, рака поджелудочной железы, рака предстательной железы, рака кожи и рака уретры.
10. Способ лечения или предупреждения связанного с ЕР4 заболевания, включающий:
введение пациенту соединения по любому из пп. 1-6.
11. Способ лечения рака, при этом способ включает: введение пациенту соединения по любому из пп. 1-6.
12. Способ по п. 11, при этом рак выбран из видов солидного рака, предпочтительно из рака молочной железы, рака шейки матки, колоректального рака, рака эндометрия, глиобластомы, рака головы и шеи, рака почки, рака печени, рака легкого, медуллобластомы, рака яичника, рака поджелудочной железы, рака предстательной железы, рака кожи и рака уретры.
13. Способ по п. 11, в котором соединение вводят в комбинации с лучевой терапией и/или терапией антителами, при этом терапия антителами выбрана из группы, состоящей из терапии антителами CTLA4, терапии антителами PDL1, терапии антителами PD1 и их комбинаций.
| WO 2012039972 A1, 29.03.2012 | |||
| WO 2015179615 A1, 26.11.2015 | |||
| BENDER A | |||
| et al, Evaluation of a candidate anti-arthritic drug using the mouse collagen antibody induced arthritis model and clinically relevant biomarkers, Am J Transl Res, 2013, vol.5, no.1, p.92-102 | |||
| YANG J | |||
| et al, Discovery and Characterization of 1H-1,2,3-Triazole Derivatives as |
