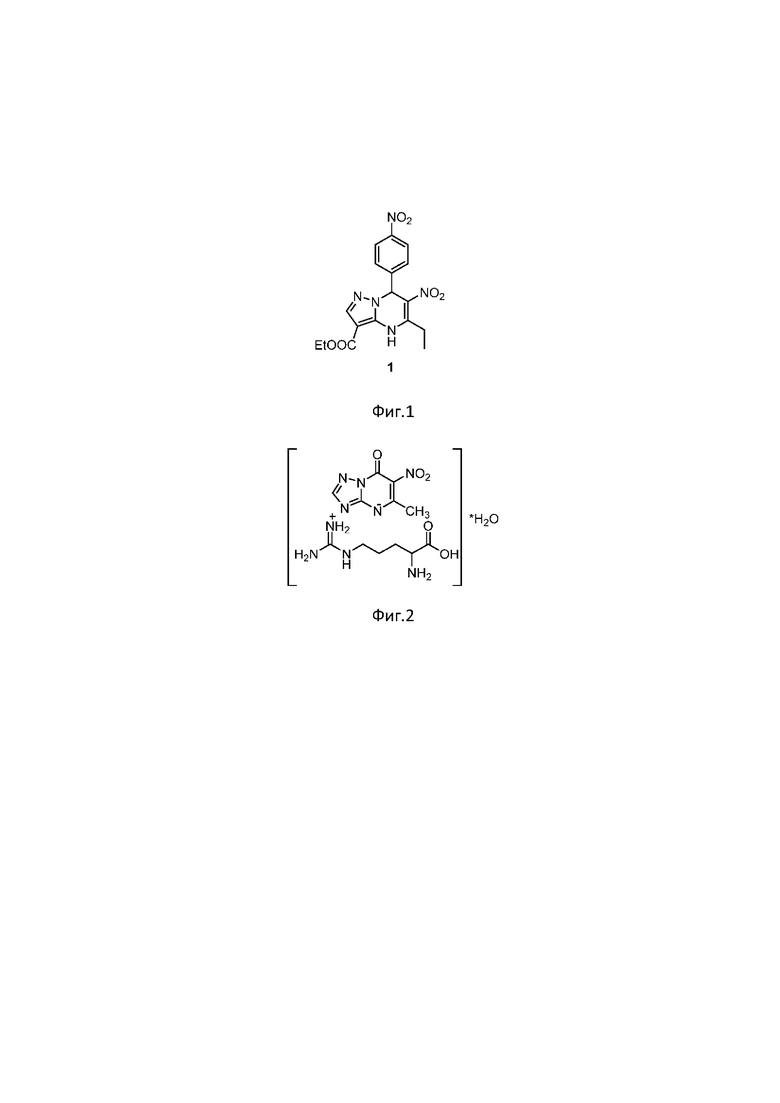
Изобретение относится к области фармацевтической и аналитической химии, в частности к вольтамперометрическому способу определения нового нитросоединения из ряда пиразолопиримидинов - этил 6-нитро-7-(4’-нитрофенил)-5-этил-4,7-дигидропиразоло[1,5-а]пиримидин-3-карбоксилата (соединение 1), фиг.1 - химическая структура), проявляющего биологическую активность в отношении казеин киназы 2, которая является многообещающей мишенью химиотерапии опухолевых заболеваний и отвечает за рост и пролиферацию раковых клеток.
В настоящее время из патентной и научно-технической литературы неизвестны сведения о способах количественного определения соединения 1.
Задача разработки чувствительного и экспрессного способа количественного определения соединения 1 в стандартных образцах является актуальной и важной для будущего обеспечения контроля качества лекарственного средства на предмет возможных фальсификаций и деструкции в процессе хранения.
Известны способы количественного определения органических лекарственных веществ, заключающиеся в применении высокоэффективной жидкостной хроматографии (ВЭЖХ) и спектрофотометрии. Эти методы имеют ряд недостатков: ВЭЖХ отличает длительность и трудоемкость анализа, необходимость использования большого количества реактивов, включая токсичные органические растворители, высокая стоимость оборудования, расходных материалов и обслуживающего персонала, а спектрофотометрия в некоторых случаях не обладает необходимой селективностью и чувствительностью.
Известны способы определения биологически активных нитросоединений вольтамперометрическими методами [Сагликоглу Г., Йилмаз С. Высокочувствительное вольтамперометрическое определение метронидазола на стеклоуглеродном электроде, модифицированном поли(р-аминобензолсульфокислотой) // Электрохимия. 2015. 51: 977-981], [Шайдарова Л.Г., Гедмина А.В., Жалдак Э.Р., Челнокова А.И., Будников Г.К. Вольтамперометрическое определение Ацикловира в лекарственных средствах на электроде, модифицированном пленкой из гексахлороплатината или гексацианокобальтата рутения // Химико-фармацевтический журнал. 2014. 48: 37-43]. Область применения данных способов ограничена ввиду их специфичности.
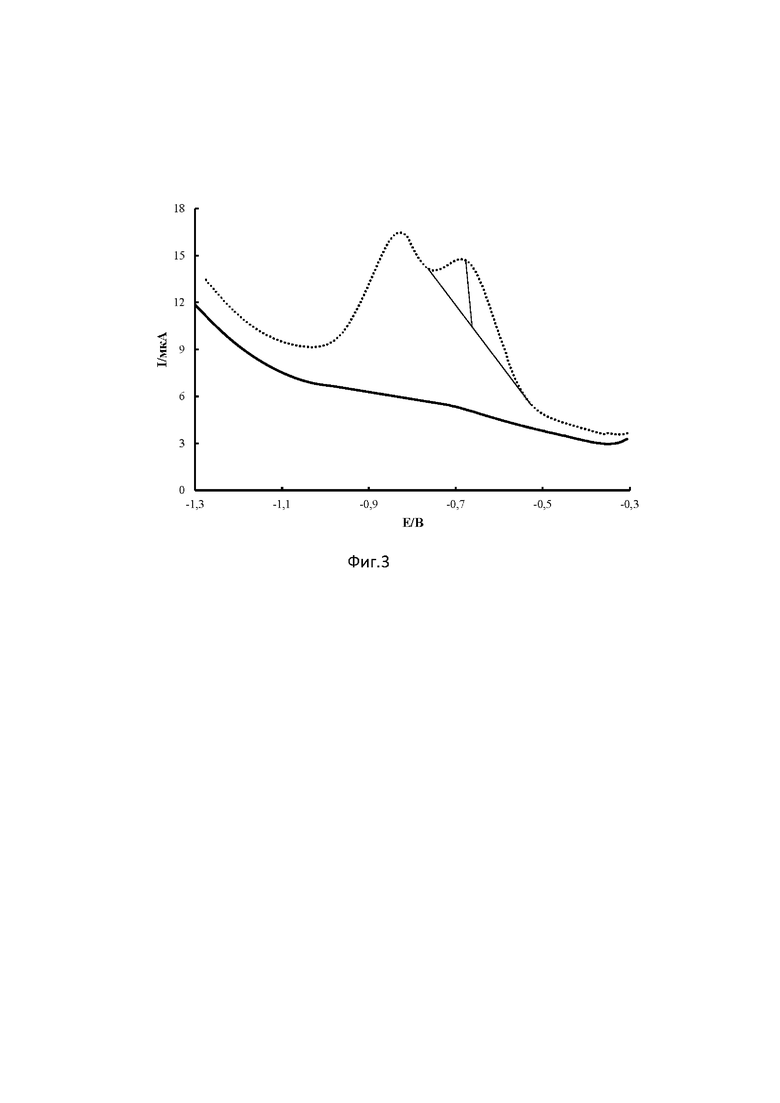
Также известен способ определения противовирусного лекарственного соединения с похожей структурой Триазида[(5-метил-6-нитро-7-оксо-4,7-дигидро-1,2,4-триазоло[1,5-α] пиримидинида l-аргининия моногидрата] из класса азолоазинов (фиг.2 - химическая структура), методом вольтамперометрии. Сущность его заключается в электровосстановлении триазида на толстопленочном углеродсодержащем электроде (ТУЭ). на фоне раствора Бриттона-Роббинсона при рН 7.0 в присутствии 0.01 М сульфита натрия в диапазоне потенциалов от от (-0.40) до (-1.4) В при скорости развертки потенциала 150 мВ/с, амплитуде импульса 50 мВ. [Малахова Н.А., Ивойлова А.В., Цмокалюк А.Н., Козицина А.Н, Иванова А.В., Русинов В.Л.,. способ количественного определения триазида методом вольтамперометрии // Патент РФ на изобретение № 2733397, опубл. 01.10.2020]. Наличие в структуре молекуле двух нитрогрупп может существенно влиять на электрохимическую активность соединения [Vyskočil V., Barek J.Polarographic and voltammetric study of genotoxic 2,7-dinitrofluoren-9-one and its determination using mercury electrodes// Collection of Czechoslovak Chemical Communications. 2009, 74, 1675-1696], в связи с чем использование перечисленных выше условий в способе определения триазида делает невозможным его применение для определения соединения 1 ввиду того, что в структуре соединения 1 есть 2 нитрогруппы. На фиг.3 представлены катодные квадратно-волновые (КвВ) вольтамперограммы, зарегистрированные на стеклоуглеродном электроде (СУЭ) с физическим удалением кислорода в смешанном растворе трис-НСl и этанола (1:1) рН 7.5 со скоростью развертки 0.15 В/с без добавления (сплошная кривая) и после добавления 50 мг/л соединения 1 (точечная кривая). В качестве аналитического сигнала (АС) берут первый пик восстановления. Пример обработки АС представлен на фиг.3.
Задачей, решаемой данным изобретением, является создание чувствительного и экспрессного способа количественного вольтампероаметрического определения соединения 1 в стандартном образце для будущего обеспечения контроля качества лекарственного средства на производстве.
Поставленная задача достигается тем, что способ количественного определения соединения 1 (этил 6-нитро-7-(4’-нитрофенил)-5-этил-4,7-дигидропиразоло[1,5-а]пиримидин-3-карбоксилата) в стандартном образце, заключается в растворении навески вещества в органическом растворителе - диметилсульфоксиде до достижения концентрации - и внесении аликвоты подготовленного раствора анализируемой пробы в электрохимическую ячейку с фоновым электролитом смешанного раствора трис-НСl и этанола (1:1) рН 7.5 после чего осуществляют регистрацию катодных вольтамперных кривых в квадратно-волновом режиме на поверхности стеклоуглеродного электрода в интервале от (-0.30) до (-1.3) В при скорости развертки потенциала 150 мВ/с, амплитуде импульса 80 мВ, а концентрацию соединения 1 определяют по площади пика под первым пиком восстановления в диапазоне потенциалов от (-0.55) до (-0.75) В относительно хлоридсеребряного электрода из выражения:
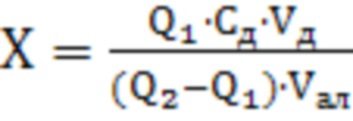 , где
, где
X - содержание массовой концентрации соединения 1, г/л,
Q1 -среднее значение величины площади пика соединения 1 для пробы, мкКл,
Q2 - среднее значение величины площади пика соединения 1 для пробы c добавленным стандартным раствором соединения 1, мкКл,
Cд - концентрация стандартного раствора соединения 1, из которого делают добавку в пробу, г/л,
Vд - объем стандартного раствора пробы, помещенная в электролизер, мл,
Vал - объем аликвотной части пробы, помещенный в электролизер, мл.
Отличительные признаками является то, что ранее не был разработана технология количественного определения потенциального противоопухолевого средства на стеклоуглеродном электроде в смешанном растворе трис-HCl и этанола при рН 7.5 с физическим удалением кислородом - продувкой инертным газом. В качестве фонового электролита использовать буферные растворы, содержащие ионы натрия, нежелательно, поскольку ионы влияют на растворимость вещества и как следствие на истинность раствора. [Serajuddin, Abu T.M., Salt formation to improve drug solubility// Advanced Drug Delivery Reviews, 2007,59,7,603-616] Также использовать ранее запатентовый метод удаления кислорода сульфитом натрия не рекомендуется [Малахова Н.А., Ивойлова А.В., Цмокалюк А.Н., Козицина А.Н, Иванова А.В., Русинов В.Л., Способ количественного определения триазида методом вольтамперометрии // Патент РФ на изобретение № 2733397, опубл. 01.10.2020].
Предлагаемое техническое решение может быть использовано как в лабораториях фармацевтического контроля для определения соединения 1 в стандартных образцах, так и в фармацевтической промышленности для контроля технологических процессов и качества фармпрепаратов.
Все условия определения соединения 1 подобраны экспериментально. В процессе поиска оптимальных условий вольтамперометрического определения соединения 1 было изучено влияние ряда факторов: кислотность фонового электролита, частота импульса, амплитуда импульса и скорость квадратно-волновой (КвВ) развертки потенциала на высоту пика соединения 1.
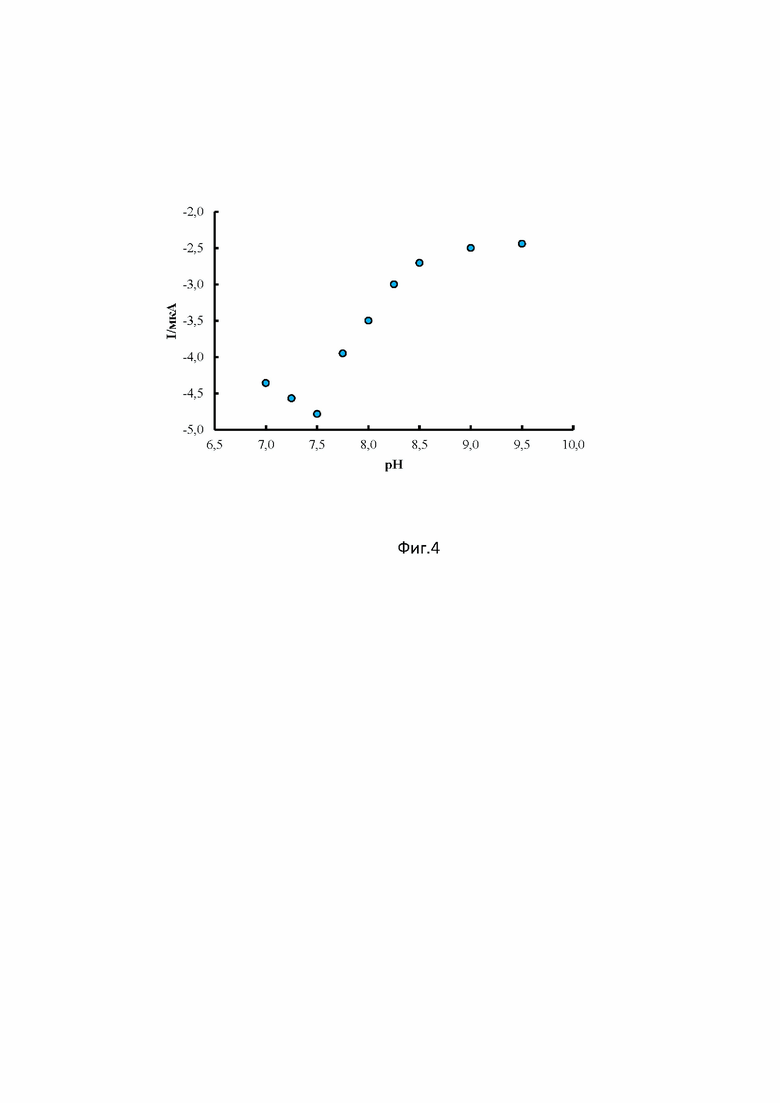
Влияние кислотности растворов трис-HCl и этанола (1:1) на ток восстановления соединения 1 изучали в диапазоне рН 6.75-9.5. (фиг.4) При более низких рН раствор имел мутность (неистинный), что не позволяло получать корректные результаты. Ток восстановления соединения 1 достигает максимального значения при рН 7.5. Снижение величины пика соединения 1 в более щелочной среде может быть обусловлено затруднением восстановления из-за нехватки протонов. Полученные результаты свидетельствуют об участии ионов водорода в процессе электровосстановления соединения 1. Исходя из полученных результатов в качестве фонового электролита был выбран раствор трис-HCl и этанола (1:1) с рН 7.5, так как на его фоне наблюдалась четкая волна восстановления соединения 1 максимальной величины, кроме того, данный раствор обеспечивал хорошую электропроводность, широкую рабочую область и необходимую площадь для обработки сигнала.
Величина АС в КвВ режиме зависит от инструментальных параметров таких как: частота амплитуды, шаг импульса и амплитуда импульса.
Линейная зависимость тока восстановления соединения 1 от частоты импульсов при амплитуде импульса 0.08 В и шаге импульса 0.004 В наблюдается в области от 5 до 50 Гц. После увеличения частоты величина тока не возрастает. При этом увеличение сигнала по абсолютной величине в диапазоне частот от 25 до 50 Гц незначительно (не превышает 5%). При сравнении базовых линии остаточного тока при 25 и 50 Гц наблюдается увеличение тока при 50 Гц почти в 2 раза. Таким образом, отношение полезный сигнал/остаточный ток значительно снижается, что усложняет регистрацию пика соединения 1 и ухудшает воспроизводимость измерения полученных результатов. Поэтому для аналитических целей была выбрана частота 25 Гц.
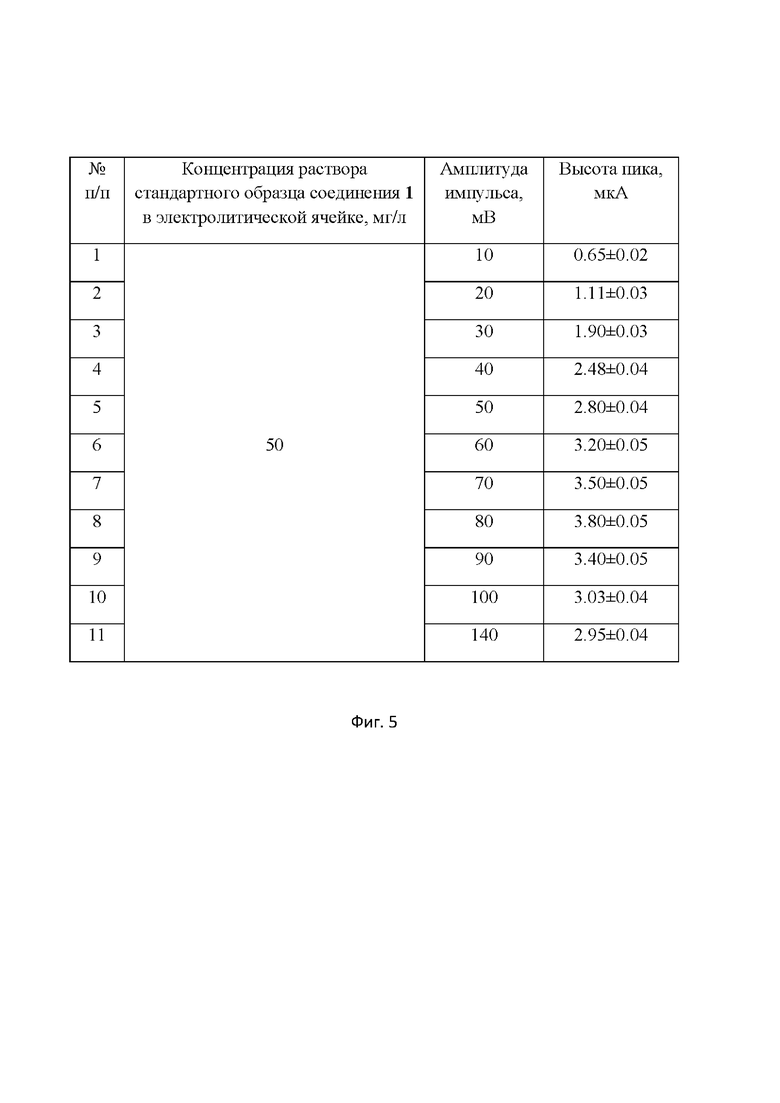
Оптимальная амплитуда импульса составила 80 мВ. При значениях амплитуды импульса менее 80 мВ высота пика не достигает максимального значения, что снижает чувствительность определения соединения 1. При дальнейшем увеличении значения амплитуды импульса рост пика соединения 1 существенно замедляется. (табл.1 влияние амплитуды импульса на ток пика соединения 1 на фиг.5 (Примечание: шаг развертки потенциала 4 мВ; границы развертки потенциала от (-0.3) до (-1.3) В; скорость развертки потенциала 150 мВ/с, частота 25 Гц)). Кроме того, регистрация пика соединения 1 затрудняется в диапазоне амплитуд от 10 до 60 и от 100 до 140мВ вследствие асимметричности сигнала и увеличения его ширины.
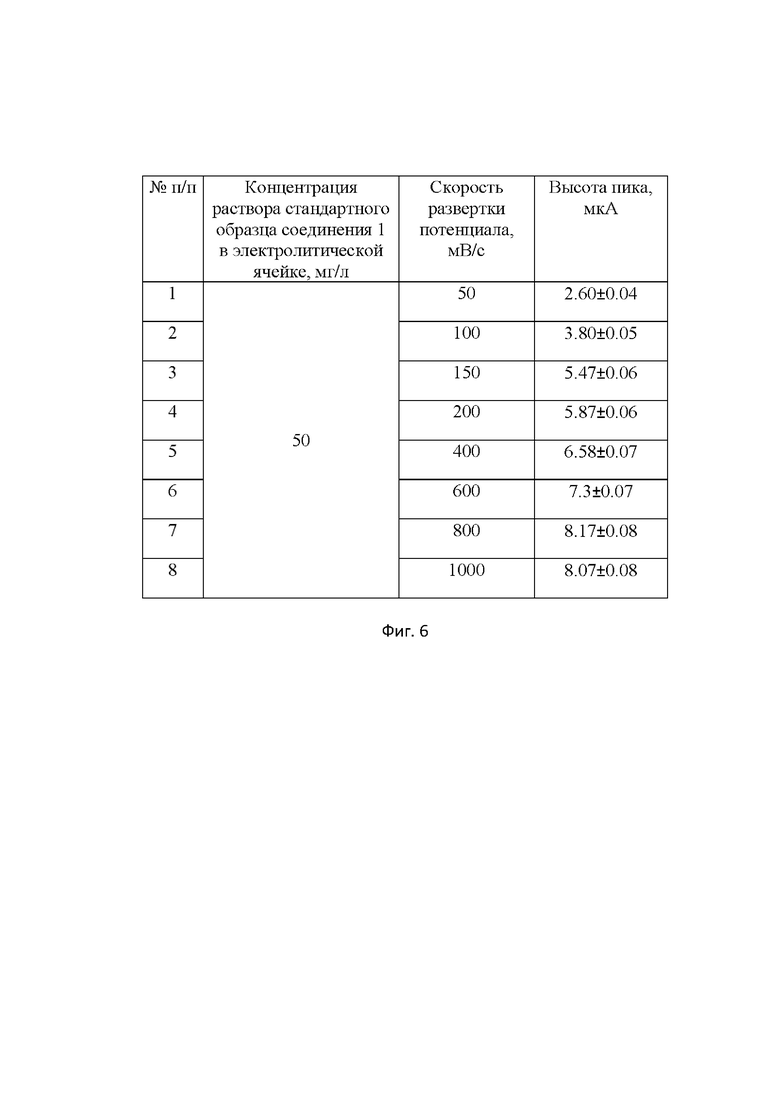
Важным для определения соединения 1 методом КвВ ВА является выбор скорости развертки потенциала. Экспериментально установлено, что оптимальной является скорость развертки 150 мВ/с. Изменение скорости развертки потенциала в сторону уменьшения увеличивает время анализа и понижает высоту пика соединения 1. При увеличении скорости развертки рост пика соединения 1 существенно замедляется, базовая линия остаточного тока возрастает в 3 раза, что уменьшает соотношение полезный сигнал/остаточный ток и ухудшает воспроизводимость результатов измерения (табл.2 Влияние скорости развертки потенциала на высоту пика соединения 1 (Примечание: шаг развертки потенциала 5 мВ; амплитуда импульса 80 мВ; частота 25 Гц,границы развертки потенциала от (-0.3) до (-1.3) В) на фиг. 6). Кроме того, регистрация пика соединения 1 затрудняется вследствие асимметричности сигнала и увеличения его ширины.
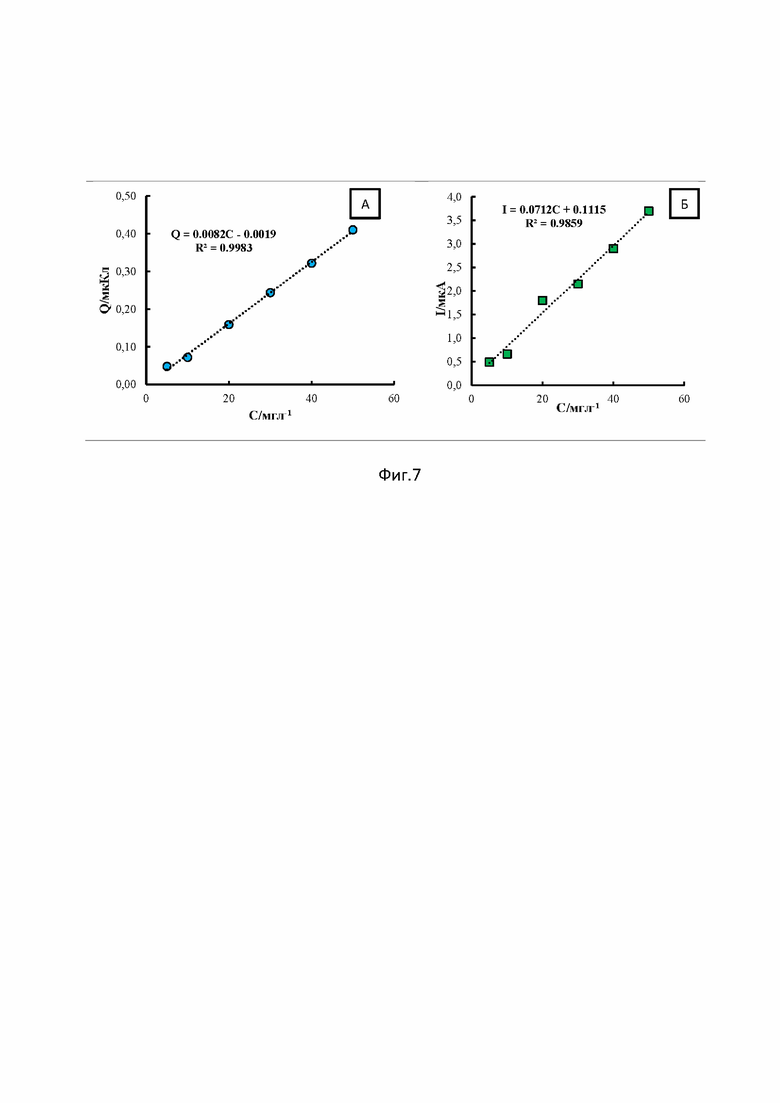
При оптимальных условиях регистрации соединения 1 площадь пика линейно растет в интервале 5 - 50 мг/л, в то время как для тока пика восстановления линейность градуировочного графика снижается, на что указывает коэффициент корреляции (0.998 и 0.986, соответственно) (фиг 7А и Б). В диапазоне концентраций от 50 до 500 мг/л тенденция сохраняется: коэффициенты корреляции составляют 0.96 для площади пика и 0.79 для тока пика. Предпочтительнее использовать в качестве аналитического сигнала использовать площадь пика. Уравнение регрессии имеет для диапазона концентрации 5 - 500 мг/л имеет вид Q(мкКл) = (0.0065C±0.0001) + (0.120±0.027) с коэффициентом корреляции 0.987.
Предложенный способ количественного определения соединения 1 отличается простотой и экспрессностью. Время единичного анализа не превышает 20 мин. Способ не требует больших трудозатрат, исключает использование большого количества реактивов, дорогостоящих электродов и и может быть применен в любой химической лаборатории, имеющей вольтамперометрический анализатор как отечественного, так и зарубежного производства.
Метрологические характеристики данного способа: предел обнаружения составляет 2.0 10-6 моль/дм3, предел количественного определения 6.2 10-6 Область определяемых содержаний соединения 1 от 1.3·10-5 до 1.3·10-3 моль/дм3. Относительная ошибка определения не превышает 1 %. Способ позволяет измерять в выбранных условиях пик восстановления соединения 1 с хорошей воспроизводимостью. Относительное стандартное отклонение (RSD) измеряемого тока восстановления соединения 1 не превышает 1.5 % при последовательной регистрации 3 катодных сигналов для концентрации соединения 1 на уровне его определяемых содержаний.
Способ иллюстрируется следующим примером.
Пример 1. Определение соединения 1 в стандартном образце методом КвВ вольтамперометрии
В электролитическую ячейку вольтамперометрического анализатора вносят помещают 3 см3 трис-НСl буферного раствора Бриттона-Робинсона (рН 7.5 ± 0,2), 3 см3 этанола медицинского 95%. Через раствор в течение 5-7 мин с включенным перемешиванием пропускают инертный газ - аргон для удаление кислорода. Опускают в раствор электроды: индикаторный - СУЭ, вспомогательный стеклоуглеродный и электрод сравнения - насыщенный хлоридсеребряный (нас. х.с.э.). Затем регистрируют 3 вольтамперограммы фонового раствора со скоростью 150 мВ/с в интервале от (-0.3) до (-1.3) В для контроля удаления растворенного кислорода путем продувки инертным газом. Отсутствие пиков на вольтамперограмме свидетельствует о полноте удаления растворенного кислорода. Затем в ячейку с фоновым раствором вносят аликвоту 0,5 мл подготовленного органического раствора анализируемой пробы, включают продувку и перемешивание раствора в течение 5-7 мин и вновь регистрируют вольтамперограмму в тех же условиях. Пик для указанной концентрации вещества регистрируют в диапазоне потенциалов от (-0.55) до (-0.75) В (отн. нас. х.с.э.). Содержание соединения 1 оценивают методом добавки стандартного раствора соединения 1, измеряя площадь пика под первым пиком восстановления в диапазоне потенциалов от (-0.55) до (-0.75) В относительно хлоридсеребряного электрода, по следующей формуле:
, где
X - содержание массовой концентрации соединения 1, г/л;
Q1 -среднее значение величины площади пика соединения 1 для пробы, мкКл;
Q2 - среднее значение величины площади пика соединения 1 для пробы c добавленным стандартным раствором соединения 1, мкКл;
Cд - концентрация стандартного раствора соединения 1, из которого делают добавку в пробу, г/л;
Vд - объем стандартного раствора пробы, помещенная в электролизер, мл;
Vал - объем аликвотной части пробы, помещенный в электролизер, мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ТРИАЗИДА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ | 2019 |
|
RU2733397C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ТРИАЗАВИРИНА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ (ВАРИАНТЫ) | 2015 |
|
RU2614022C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РТУТИ КАТОДНО-АНОДНОЙ ВОЛЬТАМПЕРОМЕТРИЕЙ | 2013 |
|
RU2533337C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В РУДАХ И РУДНЫХ КОНЦЕНТРАТАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ ПО ПИКАМ СЕЛЕКТИВНОГО ЭЛЕКТРООКИСЛЕНИЯ ВИСМУТА ИЗ ИНТЕРМЕТАЛЛИЧЕСКОГО СОЕДИНЕНИЯ PtBi | 2011 |
|
RU2479837C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БУТОПРОФИДА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2289127C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЕРЕБРА КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИЕЙ | 2014 |
|
RU2580635C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПЛАТИНЫ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ ПО ПИКАМ СЕЛЕКТИВНОГО ЭЛЕКТРООКИСЛЕНИЯ ВИСМУТА ИЗ ИНТЕРМЕТАЛЛИЧЕСКОГО СОЕДИНЕНИЯ PtBi | 2009 |
|
RU2390011C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ МОЛОЧНОЙ КИСЛОТЫ МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ НА СТЕКЛОУГЛЕРОДНОМ ЭЛЕКТРОДЕ | 2013 |
|
RU2526821C1 |
| Вольтамперометрический способ определения дифениламина в продуктах выстрела | 2017 |
|
RU2657552C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МОЛИБДЕНА КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИЕЙ | 2013 |
|
RU2533333C1 |
Изобретение относится к области фармацевтической и аналитической химии. Раскрыт способ количественного определения потенциального противоопухолевого средства этил 6-нитро-7-(4’– нитрофенил) – 5 -этил-4,7-дигидропиразоло[1,5-a]пиримидин-3-карбоксилата (соединения 1) методом вольтамперометрии, заключающийся в переводе соединения 1 из пробы в неводный раствор и внесении аликвоты подготовленного раствора анализируемой пробы в электрохимическую ячейку с фоновым электролитом с последующей регистрацией катодных вольтамперных кривых в квадратно-волновом режиме на стеклоуглеродном электроде, на фоне смешанного раствора трис-НСl и этанола (1:1) рН 7.5±0.1 с физическим удалением растворенного кислорода путем пропускания инертного газа аргона в интервале от (-0.30) до (-1.3) В при скорости развертки потенциала 150 мВ/с, амплитуде импульса 80 мВ, а концентрацию соединения 1 оценивают добавлением стандартного раствора соединения 1, измеряя площадь пика под первым пиком восстановления в диапазоне потенциалов от (-0.55) до (-0.75) В относительно хлоридсеребряного электрода. Изобретение обеспечивает создание чувствительного и экспрессного способа количественного вольтамперометрического определения соединения 1 для будущего обеспечения контроля качества лекарственного средства на производстве. 7 ил., 1 пр.
Способ количественного определения потенциального противоопухолевого средства этил 6-нитро-7-(4’– нитрофенил) – 5 -этил-4,7-дигидропиразоло[1,5-a]пиримидин-3-карбоксилата (соединения 1) методом вольтамперометрии, заключающийся в переводе соединения 1 из пробы в неводный раствор и внесении аликвоты подготовленного раствора анализируемой пробы в электрохимическую ячейку с фоновым электролитом с последующей регистрацией катодных вольтамперных кривых в квадратно-волновом режиме на стеклоуглеродном электроде, на фоне смешанного раствора трис-НСl и этанола (1:1) рН 7.5±0.1 с физическим удалением растворенного кислорода путем пропускания инертного газа аргона в интервале от (-0.30) до (-1.3) В при скорости развертки потенциала 150 мВ/с, амплитуде импульса 80 мВ, а концентрацию соединения 1 оценивают добавлением стандартного раствора соединения 1, измеряя площадь пика под первым пиком восстановления в диапазоне потенциалов от (-0.55) до (-0.75) В относительно хлоридсеребряного электрода, по следующей формуле:
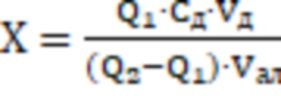 , где
, где
X - концентрация соединения 1, г/л;
Q1 -среднее значение величины площади пика соединения 1 для пробы, мкКл;
Q2 - среднее значение величины площади пика соединения 1 для пробы c добавленным стандартным раствором соединения 1, мкКл;
Cд - концентрация стандартного раствора соединения 1, из которого делают добавку в пробу, г/л;
Vд - объем стандартного раствора пробы, помещенный в электролизер, мл;
Vал - объем аликвотной части пробы, помещенный в электролизёр, мл.
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ТРИАЗИДА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ | 2019 |
|
RU2733397C2 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ НИБЕНТАНА | 2003 |
|
RU2247368C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ТРИАЗАВИРИНА МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ (ВАРИАНТЫ) | 2015 |
|
RU2614022C1 |
| CN 109781813 A, 21.05.2019. | |||
