Изобретение относится к области биотехнологии и может быть использовано в молекулярно-генетической диагностике онкологических заболеваний, а именно неходжкинской лимфомы, и для секвенирования и проведения надежного картирования полученных последовательностей на нуклеотидную последовательность референсного генома.
Основным эпигенетическим механизмом, обеспечивающим регуляцию генной активности, служит метилирование ДНК, широко распространенное в живых организмах. Определение степени модификации тех или иных участков генома в норме и патологии позволяет выявлять нарушения в регуляции экспрессии генов и имеет большой диагностический потенциал (Caseres L., Cairus P. Methylated DNA sequences for early cancer detection, molecular classification and chemotherapy response prediction // Clin. Transi. Oncol. - 2007. - Vol.9. - P. 429-437). Изучение метилирования ДНК также важно для понимания молекулярных механизмов онтогенеза, старения, приспосабливаемости организмов к изменениям среды обитания.
Следует разделять методы определения метилирования отдельных участков в протяженной ДНК и поиск максимально возможного числа метилированных участков (т.н. эпигенетическое профилирование или картирование) во всей геномной ДНК.
Процедура пробоподготовки в данном случае включает в себя: а) расщепление геномной ДНК эндонуклеазой рестрикции, например EcoRI; б) раздельную обработку полученного гидролизата метил-чувствительной и метил-нечувствительной эндонуклеазами; в) сшивку полученных фрагментов с олигонуклеотидными адаптерами, подобранными в соответствии с образуемыми при ферментативном расщеплении концами фрагментов.
Первая группа методов, таких как GLAD-ПЦР-анализ (RU2525710, C12Q 1/68, 20.08.2014), применяется, в основном, для диагностических целей. Эти методы позволяют достаточно просто определить состояние метилирования одного или нескольких участков геномной ДНК.
При этом данные методы не предусматривают поиск этих участков, поскольку в процессе поиска необходимо проведение отдельного эксперимента для каждого кандидатного участка, что существенно увеличивает трудоемкость такого поиска.
Вторая группа методов (эпигенетического картирования или профилирования) нацелена на поиск и выявление аномально метилированных сайтов во всей протяженной ДНК и последующий анализ связи метилирования отдельных участков геномной ДНК с различными патологиями, изучение механизмов онтогенеза, старения, приспосабливаемости организмов к изменениям среды обитания. (Grigg G.W. Sequencing 5-methylcytosine residues by the bisulphite method // DNA Seq. (1996) 6, 189-198).
Существующие способы эпигенетического профилирования геномов включают в себя методы, основанные на секвенировании бисульфитно- обработанной ДНК (например, WGBS и RRBS), и методы, использующие ферментативное расщепление ДНК эндонуклеазами, чувствительными к метилированию цитозиновых остатков ДНК (Grigg G.W. Sequencing 5-methylcytosine residues by the bisulphite method // DNA Seq. (1996) 6, 189-198). Метод WGBS (Whole Genome Bisulfite Sequencing, полногеномное бисульфитное секвенирование) позволяет определить статус метилирования цитозиновых остатков во всем геноме. Метод RRBS (Reduced Representation Bisulfite Sequencing, бисульфитное секвенирование с пониженной представленностью) представляет собой упрощенный вариант метода WGBS и отличается тем, что для проведения секвенирования отбираются короткие MspI - фрагменты генома (40-220 п. н.), включающие в себя большую часть регуляторных CpG-островков. Для проведения анализа с помощью методов геномного бисульфитного секвенирования требуется сложная пробоподготовка образцов ДНК, включающая в себя: а) случайную фрагментацию ДНК ультразвуком или неспецифической нуклеазой; б) восстановление концов фрагментов ДНК-полимеразой и добавление адениновых нуклеотидов к их концам для сшивки с адаптерами; в) сшивку фрагментов с олигонуклеотидными адаптерами, содержащими метилированные цитозиновые основания; г) очистку и отделение фракции фрагментов нужной длины; д) проведение бисульфитной конверсии; е) ПЦР-амплификацию продуктов конверсии; ж) очистку амплифицированных фрагментов
Следует отметить, что методы, основанные на секвенировании бисульфитно-обработанной ДНК, не нашли широкого применения в связи со значительной степенью деградации ДНК при обработке бисульфитом (до 90% от всей обрабатываемой ДНК) и трудностью получения полностью конвертированной ДНК (Smith ZD, Gu Н, Bock С, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009; 48: 226-232).
Ферментативный способ требует использования пары сайт-специфических эндонуклеаз, отличающихся по чувствительности к метилированию одного и того же сайта узнавания, например HpaII/MspI (Xia Z, Zou M, Zhang S, Feng B, Wang W. AFSM sequencing approach: a simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci Rep.2014; 4: 7300).
Однако известно лишь несколько таких ферментов, что резко сужает возможности этого метода. Кроме того, необходимость использования двух гидролизатов ДНК для проведения геномного секвенирования также усложняет и повышает стоимость исследований (Booth MJ, Raiber ЕА, Balasubramanian S. Chemical methods for decoding cytosine modifications in DNA. Chem Rev. 2015; 115(6): 2240-2254).
Наиболее близким к заявленному способу является способ обнаружения и анализа метилирования геномной ДНК, включающий расщепление образца геномной ДНК с помощью метилзависимой эндонуклеазы для получения фрагментов, секвенирование фрагментов для получения прочтений, выравнивание прочтений с эталонной последовательностью для выбора однозначно выровненного прочтения, определение сайта в эталонной последовательности, который метилируется в однозначно выровненном прочтении (US2014364321, C12Q 1/68, 11.12.2014). Сайт эталонной последовательности соответствует сайту С, по меньшей мере, в одном из YNCGNR, YCNGR, CNNG, GNNC, CYNRG, CNYRNG, YNNGCNNR, YNNGNCNNR, CNNR, YNNG и их комплементарной цепи, где Y представляет собой С или Т, R представляет собой А или G, N представляет собой А, С, Т или G, и Н представляет собой С, А или Т. Далее проводится вычисление типового распределения CG, CHG или СНН в метилированном С-сайте, где Н представляет собой С, А или Т; аннотирование следующего одного или нескольких видов информации на карте всего генома для получения карты метилирования всего генома, включающей глубина секвенирования каждого метилированного С-сайта; с получением информации, включающей аннотацию метилированного одиночного нуклеотида, номера хромосомы, в которой находится каждый определенный метилированный С-сайт, положение С-сайта, прямую или обратную цепь, а также глубину покрытия. расщепленный и распознанный сайт, типы цитозина и общее количество, и покрытие положения метилированного цитозина. В данном способе в качестве метилзависимой эндонуклеазы используется MspJI, которая представляет собой рестрикцию, зависящую от модификации. Данный фермент узнает последовательность 5'-(5mC)NNPu-3' и гидролизует ДНК преимущественно в позициях 9/13 (верхняя/нижняя цепи ДНК) в 3'-направлении от сайта узнавания. При расщеплении геномной ДНК ферментом MspJI образуются короткие фрагменты длиной 32 п. н., содержащие модифицированные CG-динуклеотиды в составе последовательности 5'-PyN(5mC)GNPu-3'. Определение нуклеотидного состава коротких MspJI-фрагментов методами NGS-секвенирования и установление их позиций в референсном геноме позволяет выявить значительную часть модифицированных CG-динуклеотидов.
Данный способ обнаружения и анализа метилированных геномных участков ДНК обладает недостаточной надежностью, в связи со значительной вариабельность позиций расщепления эндонуклеазы MspJI и необходимость достройки выступающих концов фрагментов, образуемых при расщеплении двуцепочечной ДНК ферментом MspJI. Кроме того показана сниженная степень достоверности картирования коротких фрагментов ДНК на референсный геном (Cohen-Karni D, Xu D, Apone L, Fomenkov A, Sun Z, Davis PJ, Kinney SR, Yamada-Mabuchi M, Xu SY, Davis T, Pradhan S, Roberts RJ, Zheng Y. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci USA. 2011; 108(27): 11040-11045).
Задачей заявляемого изобретения является повышение надежности и достоверности способа обнаружения и анализа метилирования геномных участков ДНК посредством более надежного и достоверного определения позиций (картирования) большого числа метилированных сайтов в геноме, представленных последовательностью Pu(5mC)GPy.
Поставленная задача решается тем, что в способе обнаружения и анализа метилирования геномной ДНК в биологических образцах периферической крови больных неходжкинской лимфомой, включающем расщепление образца геномной ДНК с помощью метилзависимой эндонуклеазы для получения фрагментов, секвенирование фрагментов для получения прочтений, выравнивание прочтений с эталонной последовательностью для выбора однозначно выровненного прочтения, определение сайта в эталонной последовательности, который метилируется в однозначно выровненном прочтении, а в качестве метилзависимой эндонуклеазы применяют GlaI, с помощью которого проводят анализ последовательности Pu(5mC)GPy по меньшей мере в одном из генов RARB, SIPA, SEPT9, SYNE1, FGF12, GAD2, HAND1, CACNA1G, CNRIP1, TLX2, LOX, LAMA1, GAS7, SLC6A15, BOLL, TFPI2, SOCS3, TRH, MDFI, Clorf70, RUNX3, TMEFF2, SFRP2, WNT5A, IGFBP7, TFPI2, FOXE1, NPY, WIF1, CIDEB, RIC3, KCNQ5, COL4A1, SPG20, SND1, PENK, CDO1, THBD, EDIL3, DUSP26, UCHL1, STOX2, ELMO1, SLC30A10, IGF2, ESR1, EYA4, NALCN, FL11, RXRg, RYR2, PRKAR1B, BEND5, ITGA4, ADHFE1, NGFR, GATA4, NEYROG1, FBN1, что позволяет оценить количество фрагментов, имеющих одинаковую начальную координату, свидетельствует о высокой частоте метилирования сайта 5’- Pu(5mC)GPy -3’ в данной позиции генома, и указывает на расположение координаты нуклеотида G в метилированном CG-динуклеотиде и количестве метилированных фрагментов Pu(5mC)GPy в исследуемом геноме больных неходжкинской лимфомой.
Способ осуществляется следующим образом. Для определения положений ряда метилированных нуклеотидных последовательностей Pu(5mC)GPy в протяженной ДНК для построения эпигенетического профиля и выявления аномально метилированных участков ДНК больных неходжкинской лимфомой, выделяют геномную ДНК из легкой фракции клеток крови, которую гидролизуют в реакционном буфере метилзависимой сайт-специфической эндонуклеазой Glal в течение времени и при температуре, достаточных для полного гидролиза продукта с получением смеси фрагментов геномной ДНК; выделяют указанную смесь фрагментов ДНК из раствора осаждением, а для дальнейшего анализа отбирают GlaI-фрагменты ДНК размером 150-500 п. н., наиболее оптимально подходящие для NGS-секвенирования, определяют нуклеотидный состав концов образовавшихся коротких GlaI фрагментов геномной ДНК методами NGS-секвенирования; программно фильтруют полученные данные для исключения фрагментов с низким качеством чтения, с концами, неспецифичными для GlaI-фрагментов; и по признаку присутствия динуклеотида GPy на 5'-концах последовательностей и определяют положения в референсном геноме нуклеотидных последовательностей Pu(5mC)GPy отфильтрованных GlaI-фрагментов с использованием специализированного любого программного обеспечения. Далее определяют нуклеотидный состав концов образовавшихся коротких GlaI-фрагментов геномной ДНК методами NGS-секвенирования путем парноконцевого чтения при длине чтения 75 п. н. с каждого конца фрагмента ДНК, программно фильтруют полученные данные для исключения фрагментов с низким качеством чтения, с концами, неспецифичными для GlaI-фрагментов, и по признаку присутствия динуклеотида GРу на 5'-концах последовательностей и определяют положения в референсном геноме нуклеотидных последовательностей Pu(5mC)GPy отфильтрованных GlaI-фрагментов с использованием любого программного обеспечения.
Заявляемый способ определения положений ряда метилированных нуклеотидных последовательностей Pu(5mC)GPy в протяженной ДНК для построения эпигенетического профиля и выявления аномально метилированных участков ДНК включает следующие шаги:
1) получение образцов высокоочищенной геномной ДНК, пригодной для секвенирования следующего поколения;
2) обработка ДНК метилзависимой сайт-специфической эндонуклеазой GlaI и получение GlaI-фрагментов;
3) определение нуклеотидного состава концов образовавшихся GlaI-фрагментов методами NGS-секвенирования;
4) фильтрация полученных данных для исключения фрагментов с низким качеством чтения и с концами, неспецифичными для GlaI-фрагментов;
5) установление положения секвенированных последовательностей ДНК в референсном геноме для определения положения метилированных участков.
GlaI-фрагменты получают при расщеплении ДНК путем добавления метилзависимой сайт-специфической ДНК-эндонуклеазы GlаI (2-3 е.а. на 1 мкг ДНК) в реакционном буфере А (20 мМ Tris-S04, рН 8.0, 4 мМ MgCl2, 1 мМ β-меркаптоэтанол, 100 нг/мкл BSA) в течение 1 часа при 30°С с последующей инактивацией при 65°С в течение 20 минут. Подготовку и проверку библиотеки ДНК-фрагментов для секвенирования проводят в соответствии с рекомендациями производителя секвенирующей машины NGS. Определяют нуклеотидный состав фрагментов из полученной библиотеки с использованием секвенирующей машины NGS.
По итогам секвенирования проводят фильтрацию суммарного массива данных для исключения из него фрагментов с низким качеством чтения, определяемым аппаратно при проведении секвенирования. Проводят дополнительную фильтрацию массива данных, исключая из него фрагменты, не несущие на 5'-конце динуклеотид GPy и, следовательно, образовавшиеся при неспецифической деструкции ДНК при ее выделении или в процессе случайной сшивки во время пробоподготовки. Определение положения отфильтрованных Glal-фрагментов на референсный геном проводят с помощью любого программного обеспечения, предназначенного для этой цели, например, с помощью программ CLC Genomics Workbench, Bowtie, GMAP и т.д. Начальные координаты положения картированных фрагментов в геноме соответствуют координатам нуклеотида G в метилированном CG-динуклеотиде. Значительное количество фрагментов, имеющих одинаковую начальную координату, свидетельствует о высокой частоте метилирования сайта 5'-Pu(5mC)GPy-3' в данной позиции генома. Результатом картирования являются данные о расположении (координаты нуклеотида G в метилированном CG-динуклеотиде) и количестве метилированных фрагментов Pu(5mC)GPy в исследуемом геноме.
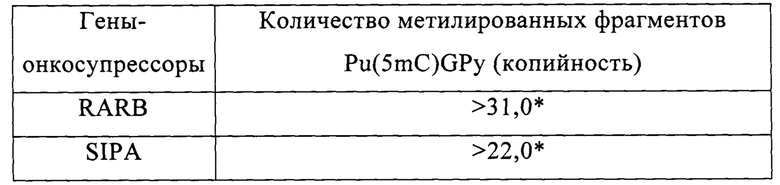
В таблице показано количество метилированных фрагментов сайта Pu(5mC)GPy в исследуемом геноме (RARB и SIPA) в пробах ДНК больных неходжкинской лимфомой (n=7). Показано достоверное различие по сравнению с условно-здоровыми донорами (*р<0,05), где р - это уровень значимости полученных данных.
Использование заявляемого способа позволяет повысить надежность и достоверность способа обнаружения и анализа метилирования геномных участков ДНК в биологических образцах периферической крови больных неходжкинской лимфомой посредством более надежного и достоверного определения позиций (картирования) большого числа метилированных сайтов Pu(5mC)GPy в геноме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КАРТИРОВАНИЯ ПОЛОЖЕНИЙ РЯДА МЕТИЛИРОВАННЫХ НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ Pu(5mC)GPy В ПРОТЯЖЕННОЙ ДНК ДЛЯ ПОСТРОЕНИЯ ЭПИГЕНЕТИЧЕСКОГО ПРОФИЛЯ И ВЫЯВЛЕНИЯ АНОМАЛЬНО МЕТИЛИРОВАННЫХ УЧАСТКОВ ДНК | 2015 |
|
RU2586502C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ Pu(5mC)GPy В ЗАДАННОМ ПОЛОЖЕНИИ ПРОТЯЖЕННОЙ ДНК | 2013 |
|
RU2525710C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТИЛИРОВАНИЯ САЙТОВ PuCGPy РЕГУЛЯТОРНЫХ ОБЛАСТЕЙ ГЕНОВ-ОНКОМАРКЕРОВ КОЛОРЕКТАЛЬНОГО РАКА МЕТОДОМ GLAD-ПЦР-АНАЛИЗА И ОЛИГОНУКЛЕОТИДНЫЕ ПРАЙМЕРЫ И ФЛУОРЕСЦЕНТНО-МЕЧЕНЫЕ ЗОНДЫ ДЛЯ ОСУЩЕСТВЛЕНИЯ УКАЗАННОГО СПОСОБА | 2015 |
|
RU2596404C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ R(5mC)GY В ЗАДАННОМ ПОЛОЖЕНИИ ПРОТЯЖЕННОЙ ДНК | 2015 |
|
RU2587631C1 |
| Способ определения метилирования сайтов PuCGPy регуляторных областей генов-онкомаркеров колоректального рака методом GLAD-ПЦР-анализа и набор олигонуклеотидных праймеров и флуоресцентно-меченых зондов для осуществления указанного способа | 2016 |
|
RU2630669C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ГИПЕРМЕТИЛИРОВАННЫХ CpG ОСТРОВКОВ В ОБЛАСТИ ГЕНОВ-СУПРЕССОРОВ ОПУХОЛЕВОГО РОСТА В ДНК ЧЕЛОВЕКА | 2009 |
|
RU2413773C1 |
| Набор олигонуклеотидных праймеров и флуоресцентно-меченых зондов для выявления эпигенетических маркеров рака шейки матки | 2021 |
|
RU2760573C1 |
| Набор синтетических олигонуклеотидов для проведения метилчувствительной амплификации ДНК | 2017 |
|
RU2662664C1 |
| Штамм бактерий Micrococcus luteus 805 - продуцент сайт-специфической метилзависимой эндонуклеазы MluVI | 2016 |
|
RU2614262C1 |
| ШТАММ БАКТЕРИЙ Plantibacter flavus 3Kz - ПРОДУЦЕНТ МЕТИЛЗАВИСИМОЙ САЙТ-СПЕЦИФИЧЕСКОЙ ЭНДОНУКЛЕАЗЫ PfsI | 2015 |
|
RU2593723C1 |
Изобретение относится к области биотехнологии. Предложен способ обнаружения и анализа метилирования геномных участков ДНК в биологических образцах периферической крови больных неходжкинской лимфомой. Способ включает расщепление образца геномной ДНК с помощью метилзависимой эндонуклеазы для получения фрагментов. Он также включает секвенирование фрагментов для получения прочтений, выравнивание прочтений с эталонной последовательностью для выбора однозначно выровненного прочтения, определение сайта в эталонной последовательности, который метилируется в однозначно выровненном прочтении. В качестве метилзависимой эндонуклеазы применяют GlaI, с помощью которого проводят анализ последовательности Pu(5mC)GPy, по меньшей мере, в одном из генов. Техническим результатом является повышение надежности и достоверности способа обнаружения и анализа метилирования геномных участков ДНК посредством более надежного и достоверного определения позиций (картирования) большого числа метилированных сайтов в геноме, представленных последовательностью Pu(5mC)GPy. 1 табл.
Способ обнаружения и анализа метилирования геномных участков ДНК в биологических образцах периферической крови больных неходжкинской лимфомой, включающий расщепление образца геномной ДНК с помощью метилзависимой эндонуклеазы для получения фрагментов, секвенирование фрагментов для получения прочтений, выравнивание прочтений с эталонной последовательностью для выбора однозначно выровненного прочтения, определение сайта в эталонной последовательности, который метилируется в однозначно выровненном прочтении, отличающийся тем, что в качестве метилзависимой эндонуклеазы применяют GlaI, с помощью которого проводят анализ последовательности Pu(5mC)GPy, по меньшей мере, в одном из генов RARB, SIPA, SEPT9, SYNE1, FGF12, GAD2, HAND1, CACNA1G, CNRIP1, TLX2, LOX, LAMA1, GAS7, SLC6A15, BOLL, TFPI2, SOCS3, TRH, MDFI, Clorf70, RUNX3, TMEFF2, SFRP2, WNT5A, IGFBP7, TFPI2, FOXE1, NPY, WIF1, CIDEB, RIC3, KCNQ5, COL4A1, SPG20, SND1, PENK, CDOI, THBD, EDIL3, DUSP26, UCHL1, STOX2, ELMOI, SLC30A10, IGF2, ESR1, EYA4, NALCN, FL11, RXRg, RYR2, PRKAR1B, BEND5, ITGA4, ADHFE1, NGFR, GATA4, NEYROG1, FBN1, что позволяет оценить количество фрагментов, имеющих одинаковую начальную координату, свидетельствует о высокой частоте метилирования сайта 5’- Pu(5mC)GPy -3’ в данной позиции генома, и указывает на расположение координаты нуклеотида G в метилированном CG-динуклеотиде и количестве метилированных фрагментов Pu(5mC)GPy в исследуемом геноме больных неходжкинской лимфомой.
| СПОСОБ КАРТИРОВАНИЯ ПОЛОЖЕНИЙ РЯДА МЕТИЛИРОВАННЫХ НУКЛЕОТИДНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ Pu(5mC)GPy В ПРОТЯЖЕННОЙ ДНК ДЛЯ ПОСТРОЕНИЯ ЭПИГЕНЕТИЧЕСКОГО ПРОФИЛЯ И ВЫЯВЛЕНИЯ АНОМАЛЬНО МЕТИЛИРОВАННЫХ УЧАСТКОВ ДНК | 2015 |
|
RU2586502C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ГИПЕРМЕТИЛИРОВАННЫХ CpG ОСТРОВКОВ В ОБЛАСТИ ГЕНОВ-СУПРЕССОРОВ ОПУХОЛЕВОГО РОСТА В ДНК ЧЕЛОВЕКА | 2009 |
|
RU2413773C1 |
| Niu Y | |||
| et al | |||
| Oxidative stress alters global histone modification and DNA methylation //Free Radical Biology and Medicine | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| - Т | |||
| Машина для разделения сыпучих материалов и размещения их в приемники | 0 |
|
SU82A1 |
| - С | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Abdurashitov M | |||
| A | |||
| et al | |||
| GlaI digestion of mouse γ-satellite DNA: study of primary structure and ACGT sites methylation //BMC genomics | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
