ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области фармацевтических составов и, в частности, относится к твердому фармацевтическому составу, содержащему соединение, являющееся антагонистом орексинового рецептора, к способу ее получения и применению для изготовления лекарственного средства для лечения заболевания, связанного с орексином.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Орексин (гипокретин) включает два нейропептида, синтезируемых в гипоталамусе: орексин А (ОХ-А) (пептид, содержащий 33 аминокислоты) и орексин В (ОХ-В) (пептид, содержащий 28 аминокислот) (Sakurai Т. et al, Cell, 1998, 92, 573-585). Было установлено, что орексин стимулирует потребление пищи у крыс, что дает основания полагать, что данные пептиды играют физиологическую роль медиаторов в центральных механизмах обратной связи, регулирующих пищевое поведение (Sakurai Т. et al., Cell, 1998, 92, 573-585). Орексин может регулировать состояние сна и бессонницы, что потенциально предполагает новый подход к лечению пациентов с нарколепсией или бессонницей (Chemelli R.M. et al., Cell, 1999, 98, 437-451). Орексин также играет роль в реакциях пробуждения, мотивации, обучении и памяти (Harris, et al, Trends Neurosci., 2006, 29(10), 571-577). У млекопитающих были клонированы и охарактеризованы два орексиновых рецептора: рецептор орексина 1 и рецептор орексина 2. Они принадлежат к суперсемейству сопряженных с G-белком рецепторов (Sakurai Т. et al., Cell, 1998, 92, 573-585), причем рецептор орексина 1 (ОХ или OX1R) является селективным в отношении ОХ-А, а рецептор орексина 2 (ОХ2 или OX2R) может связываться как с ОХ-А, так и с ОХ-В. Можно предположить, что физиологические роли орексина достигаются за счет экспрессии одного или обоих рецепторов ОХ1 и ОХ2 (представляющих собой два подтипа орексиновых рецепторов).
Орексиновые рецепторы находятся в головном мозге теплокровных животных и связаны с такими расстройствами, как: депрессия; тревожность; зависимость; обессивно-компульсивное расстройство; аффективный невроз; депрессивный невроз; невроз страха и тревоги; психотическое депрессивное расстройство; нарушение поведения; расстройство настроения; сексуальная дисфункция; психосексуальное расстройство; тендерное расстройство; шизофрения; маниакальная депрессия; психоз; деменция; умственная отсталость тяжелой степени и дискинезия, как, например, болезнь Хантингтона и синдром Туретта; расстройство пищевого поведения, такое как анорексия, булимия, истощение и ожирение; зависимое пищевое поведение; компульсивное переедание; сердечно-сосудистые заболевания; диабет; расстройство аппетита/вкуса; рвотные позывы, тошнота, рвота; астма; рак; болезнь Паркинсона; синдром/болезнь Кушинга; базофильные аденомы; пролактиномы; гиперпролактинемия; опухоль/аденомы гипофиза; гипоталамическое расстройство; воспалительное заболевание кишечника; дисфункция желудка; язва желудка; адипозогенитальная дистрофия; заболевание передней доли гипофиза; болезнь гипофиза; пониженная функция передней доли гипофиза; повышенная функция передней доли гипофиза; гипоталамический гипогонадизм; синдром Калльманна (отсутствие обоняния, пониженное обоняние); функциональная или психогенная аменорея; гипопитуитаризм (гипофункция гипофиза); гипоталамический гипотиреоз; гипоталамо-надпочечниковая дисфункция; внезапная гиперпролактинемия; гипоталамический дефицит гормона роста; внезапный дефицит роста; карликовость; гигантизм; акромегалия; нарушения биологического и циркадного ритмов; нарушения сна, связанные с такими заболеваниями, как психоз, невропатическая боль и синдром беспокойных ног; заболевания сердца и легких, острая и хроническая сердечная недостаточность; гипотензия; гипертензия; задержка мочеиспускания; остеопороз; стенокардия; острый инфаркт миокарда; ишемический или геморрагический инсульт; арахноидальное кровоизлияние; язвы; аллергия; доброкачественная гиперплазия предстательной железы; хроническая почечная недостаточность; болезнь почек; нарушенная толерантность к глюкозе; мигрень; гипералгезия; боль; повышенная или преувеличенная чувствительность к боли, такая как гипералгезия, жгучая боль и аллодиния; острая боль; жгучая боль; атипичная лицевая боль; невропатическая боль; боль в спине; комплексные региональные болевые синдромы I и II типа; боль при артрите; боль при спортивных травмах; боль, связанная с инфекциями, такими как ВИЧ (англ. HIV, Human Immunodeficiency Virus - вирус иммунодефицита человека), боль после химиотерапии; постинсультная боль; послеоперационная боль; невралгия; рвотные позывы, рвота, тошнота; состояния, связанные с висцеральной болью, такие как синдром раздраженного кишечника и стенокардические боли; мигрень; недержание мочевого пузыря, такое как императивное недержание; толерантность к наркотическим средствам или синдром отмены наркотических средств; расстройство сна; апноэ во сне; нарколепсия; бессонница; парасомния; синдром смены часового пояса; и нейродегенеративное расстройство, включая объекты классификации заболеваний, такие как синдром расторможенности-деменции-болезни Паркинсона-мышечной дистрофии; эпилепсия; судорожные расстройства и другие заболевания, связанные с общей дисфункцией орексиновой системы.
В патенте CN 106414439 A раскрыт класс производных пиперидина в качестве антагонистов орексиновых рецепторов, обладающих значительным ингибирующим действием на рецепторы ОХ1 и ОХ2 GPCR (англ. G-protein coupled receptor - рецептор, сопряженный с G-белком). В ходе исследований было установлено, что при получении их в виде твердых фармацевтических композиций имеют место недостатки, заключающиеся в более низких скорости растворения и уровне растворения, а также более низкой биодоступности и более длительном периоде полувыведения in vivo, поскольку длительный период полувыведения лекарственных средств для лечения бессонницы имеет неблагоприятные последствия, такие как остаточное воздействие на вторые сутки, что может негативно сказаться на здоровье пациентов. Следовательно, необходимо разработать твердую лекарственную форму с более высокими скоростью растворения и уровнем растворения, а также с более высокой биодоступностью и более низким периодом полувыведения для удовлетворения более широкого спектра потребностей клинического применения.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение имеет целью предложить твердый фармацевтический состав, содержащий соединение формулы I, обладающую хорошей стабильностью, хорошей растворимостью и высокой биодоступностью.
Первый аспект настоящего изобретения относится к твердому фармацевтическому составу, где твердый фармацевтический состав содержит активный ингредиент, при этом активный ингредиент представляет собой соединение, имеющее формулу (I), или его фармацевтически приемлемую соль либо их смесь;

где Ra представляет собой водород, фтор, хлор, метил, этил, пропил, изопропил, метокси, этокси, пропокси или изопропокси;
Z представляет собой N или CR0; R0 представляет собой водород, галоген или С1-3 алкил;
n равно 0, 1 или 2;
а размер частиц D90 активного ингредиента составляет 50 мкм или менее.
Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 35 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 30 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 20 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 10 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 30 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому предпочтительному примеру, соединение, имеющее формулу (I), представляет собой соединение формулы (II):
 ;
;
((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон. Как описано в патенте CN 106414439 A, ингибирующее действие соединения формулы (II) на рецепторы ОХ1 и ОХ2 GPCR оценивали, измеряя при помощи FLIPR (англ. Fluorescence Imaging Plate Reader - устройство для чтения планшетов для визуализации флуоресценции) изменение внутриклеточного кальциевого сигнала, и определяли значение IC50 соединения, при этом его активность составляла 20 нМ (hOX1R) и 36 нМ (hOX2R), соответственно.
Фармацевтически приемлемая соль по настоящему изобретению включает фармацевтически приемлемую соль присоединения кислоты и фармацевтически приемлемую соль присоединения основания. Фармацевтически приемлемая соль присоединения кислоты относится к соли, образованной неорганической или органической кислотой, способной сохранять биологическую эффективность свободного основания при отсутствии других побочных эффектов. Соль неорганической кислоты включает, не ограничиваясь перечнем, гидрохлорид, гидробромид, сульфат, фосфат и т.п. Соль органической кислоты включает, не ограничиваясь перечнем, формиат, ацетат, пропионат, гликолят, глюконат, лактат, оксалат, малеат, сукцинат, фумарат, тартрат, цитрат, глутамат, аспартат, бензоат, мезилат, п-толуолсульфонат, салицилат и т.п. Эти соли могут быть получены известными способами. Фармацевтически приемлемая соль присоединения основания включает, не ограничиваясь перечнем, соль неорганического основания, такую как соль натрия, соль калия, соль кальция, соль магния и т.п., и включает, не ограничиваясь перечнем, соль органического основания, такую как соль аммония, соль триэтиламина, соль лизина, соль аргинина и т.п. Эти соли могут быть получены известными способами.
Соединение, представленное формулой (I) и формулой (II) по настоящему изобретению, или его фармацевтически приемлемая соль могут иметь любую форму, при этом конкретная форма включает, не ограничиваясь перечнем, аморфную форму, любую кристаллическую форму, гидрат, сольват и т.п. Согласно некоторым вариантам осуществления, соединение, представленное формулой (II), существует в виде кристаллической формы А, и его спектр XRPD (англ. X-ray powder diffraction - рентгеновская порошковая дифракция), полученный в Cu-Kα излучении, имеет следующее разрешение, а его структура изображена на Фиг. 6.
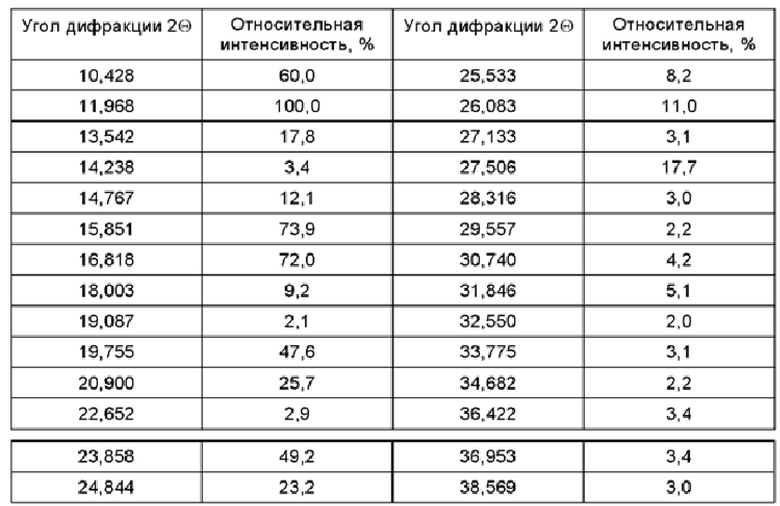
Согласно некоторым вариантам осуществления, соединение, представленное формулой (II), существует в виде кристаллической формы В, и его спектр XRPD, полученный в Cu-Kα излучении, имеет следующее разрешение, а его структура изображена на Фиг. 7.
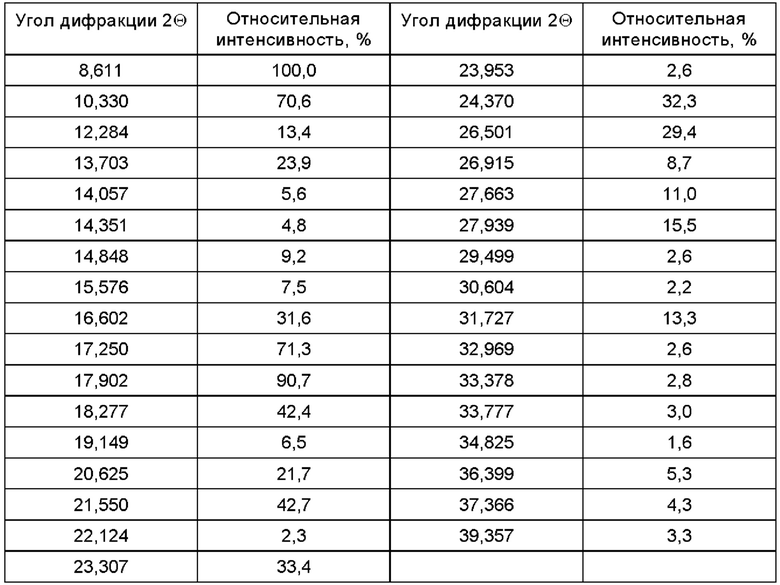
Кристаллическая форма А и кристаллическая форма В, упоминаемые в контексте настоящего изобретения, могут быть получены и охарактеризованы в соответствии со способом, описанным в патенте CN 107709318 А. Согласно некоторым вариантам осуществления, соединение формулы (II) существует в виде кристаллической формы, при этом рентгеновский дифракционный спектр его монокристалла показывает, что соединение формулы (II) имеет трехмерную эллипсоидную структуру, как изображено на Фиг. 10.
Согласно другому предпочтительному примеру, содержание активного ингредиента составляет от 1% до 15%, более предпочтительно, от 4% до 10%, более предпочтительно, от 9,5% до 10%, в расчете на общую сухую массу твердого фармацевтического состава.
Согласно другому предпочтительному примеру, твердый фармацевтический состав дополнительно содержит связующее, выбранное из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации. Содержание связующего составляет от 0,5% до 10%, более предпочтительно, от 1,5% до 3%, в расчете на общую сухую массу твердого фармацевтического состава.
Согласно другому предпочтительному примеру, связующее выбрано из группы, состоящей из гипромеллозы-Е5, гипромеллозы-К4М, гипромеллозы-Е50, карбопола, поливинилового спирта и их комбинации.
Согласно другому предпочтительному примеру, связующее представляет собой гипромеллозу-Е5.
Согласно другому предпочтительному примеру, твердый фармацевтический состав дополнительно содержит наполнитель, выбранный из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлозы-лактозы, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации. Содержание наполнителя составляет от 60% до 90%, более предпочтительно, от 73% до 85%, более предпочтительно, от 73% до 82,5%, в расчете на общую сухую массу твердого фармацевтического состава.
Согласно другому предпочтительному примеру, наполнитель представляет собой микрокристаллическую целлюлозу и лактозу.
Согласно другому предпочтительному примеру, твердый фармацевтический состав дополнительно содержит разрыхлитель, выбранный из группы, состоящей из кроскармеллозы натрия, гипромеллозы-К4М, кросповидона, карбоксиметилкрахмала натрия и их комбинации. Содержание разрыхлителя составляет от 5% до 15% в расчете на общую сухую массу твердого фармацевтического состава.
Согласно другому предпочтительному примеру, разрыхлитель представляет собой кроскармеллозу натрия или гипромеллозу-К4М.
Согласно другому предпочтительному примеру, твердый фармацевтический состав дополнительно содержит смазывающее вещество, выбранное из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации. Содержание смазывающего вещества составляет от 0,1% до 1%, более предпочтительно, от 0,4% до 0,5%, более предпочтительно, от 0,48% до 0,5%, в расчете на общую сухую массу твердого фармацевтического состава.
Согласно другому предпочтительному примеру, твердый фармацевтический состав представляет собой таблетку, капсулу, порошок, гранулу, капельную пилюлю или пленку, предпочтительно, таблетку.
Согласно другому предпочтительному примеру, твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 1% до 15%, более предпочтительно, от 4% до 10%, более предпочтительно, от 9,5% до 10%;
b) наполнитель, выбранный из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлозы-лактозы, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации, где содержание наполнителя составляет от 60% до 90%, более предпочтительно, от 73% до 85%, более предпочтительно, от 73% до 82,5%;
c) связующее, выбранное из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации, где содержание связующего составляет от 0,5% до 10%, более предпочтительно, от 1,5% до 3%;
d) разрыхлитель, выбранный из группы, состоящей из кроскармеллозы натрия, гипромеллозы-К4М, кросповидона, карбоксиметилкрахмала натрия и их комбинации, где содержание разрыхлителя составляет от 5% до 15%; и
e) смазывающее вещество, выбранное из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации, где содержание смазывающего вещества составляет от 0,1% до 1%, более предпочтительно, от 0,4% до 0,5%, более предпочтительно, от 0,48% до 0,5%;
где размер частиц D90 активного ингредиента составляет 35 мкм или менее.
Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 30 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 20 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 10 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 30 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому предпочтительному примеру, твердый фармацевтический состав представляет собой таблетку.
Согласно другому предпочтительному примеру, содержание активного ингредиента составляет от 5 мг до 100 мг. Согласно другому предпочтительному примеру, содержание активного ингредиента составляет от 10 мг до 50 мг. Согласно другому предпочтительному примеру, содержание активного ингредиента составляет 10 мг, 20 мг или 40 мг.
Согласно другому предпочтительному примеру, связующее выбрано из группы, состоящей из гипромеллозы-Е5, гипромеллозы-К4М, гипромеллозы-Е50, карбопола, поливинилового спирта и их комбинации.
Согласно другому предпочтительному примеру, связующее представляет собой гипромеллозу-Е5.
Согласно другому предпочтительному примеру, наполнитель представляет собой микрокристаллическую целлюлозу и лактозу.
Согласно другому предпочтительному примеру, разрыхлитель представляет собой кроскармеллозу натрия или гипромеллозу-К4М.
Согласно другому предпочтительному примеру, твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 4% до 10%;
b) от 24% до 27,5% микрокристаллической целлюлозы;
c) от 48,5% до 56,5% лактозы;
d) от 1,5% до 3% гипромеллозы-Е5;
e) от 5% до 15% кроскармеллозы натрия или гипромеллозы-К4М; и
f) от 0,4% до 0,5% стеарата магния;
где размер частиц D90 активного ингредиента составляет 35 мкм или менее, или D90 составляет 30 мкм или менее, или D90 составляет 20 мкм или менее, или D90 составляет 10 мкм или менее, или D90 составляет от 1 мкм до 30 мкм, или D90 составляет от 1 мкм до 20 мкм, или D90 составляет от 1 мкм до 10 мкм, или D90 составляет от 10 мкм до 20 мкм.
Согласно другому предпочтительному примеру, твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 9,5% до 10%;
b) от 24% до 27,5% микрокристаллической целлюлозы;
c) от 48,5% до 56,5% лактозы;
d) от 1,5% до 3% гипромеллозы-Е5;
e) от 5% до 15% кроскармеллозы натрия или гипромеллозы-К4М; и
f) от 0,48% до 0,5% стеарата магния;
где размер частиц D90 активного ингредиента составляет от 1 мкм до 30 мкм, или D90 составляет от 1 мкм до 20 мкм, или D90 составляет от 1 мкм до 10 мкм, или D90 составляет от 10 мкм до 20 мкм, при этом твердый фармацевтический состав представляет собой таблетку.
Согласно другому предпочтительному примеру, лактоза имеет размер частиц 200 меш, 100 меш, 50 меш и т.п. Более предпочтительно, лактоза имеет размер частиц 200 меш.
Согласно другому предпочтительному примеру, наполнитель представляет собой микрокристаллическую целлюлозу и лактозу. Массовое соотношение микрокристаллической целлюлозы и лактозы составляет (24-27,5):(48,5-56,5).
Согласно другому предпочтительному примеру, твердый фармацевтический состав включает любую таблетку из примера получения.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: примерно 10 мг, примерно 20 мг или примерно 40 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); примерно от 25 до 150 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); примерно от 50 до 300 мг лактозы (например, Granulac 200); примерно от 1 до 15 мг гипромеллозы (например, гипромеллозы Е5); примерно от 10 до 50 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711) и гипромеллозы (например, НРМС-К4М); и примерно от 0,5 до 3 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: примерно 10 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); примерно от 25 до 26 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); примерно 51 мг лактозы (например, Granulac 200); примерно 3 мг гипромеллозы (например, гипромеллозы Е5); примерно 10 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и примерно 0,5 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: примерно 20 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); примерно 51 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); примерно 102 мг лактозы (например, Granulac 200); примерно 6 мг гипромеллозы (например, гипромеллозы Е5); примерно 20 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и примерно 1 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: примерно 40 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); примерно 102 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); примерно 204 мг лактозы (например, Granulac 200); примерно 12 мг гипромеллозы (например, гипромеллозы Е5); примерно 40 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и примерно 2 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит от 8 мг до 12 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); от 20 мг до 30 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); от 46 мг до 56 мг лактозы (например, Granulac 200); от 2 мг до 4 мг гипромеллозы (например, гипромеллозы Е5); от 5 мг до 15 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и от 0,3 мг до 0,7 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: от 18 мг до 22 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); от 46 мг до 56 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); от 97 мг до 107 мг лактозы (например, Granulac 200); от 5 мг до 7 мг гипромеллозы (например, гипромеллозы Е5); от 15 мг до 25 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и от 0,8 мг до 1,2 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно другому аспекту, в настоящем изобретении предложена дозированная лекарственная форма, при этом в расчете на общую массу дозированной лекарственной формы дозированная лекарственная форма содержит: от 38 мг до 42 мг активного ингредиента, где активный ингредиент представляет собой соединение формулы (II); от 97 мг до 107 мг микрокристаллической целлюлозы (например, микрокристаллической целлюлозы РН101); от 199 мг до 209 мг лактозы (например, Granulac 200); от 11 мг до 13 мг гипромеллозы (например, гипромеллозы Е5); от 35 мг до 45 мг кроскармеллозы натрия (например, кроскармеллозы натрия SD711); и от 1,8 мг до 2,2 мг стеарата магния, где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно некоторым вариантам осуществления, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм или D90 составляет от 10 мкм до 20 мкм.
Согласно некоторым вариантам осуществления, в любой из упомянутых выше дозированных лекарственных форм по настоящему изобретению соединение формулы (II), используемое в качестве активного ингредиента, может существовать в виде любой формы, включая аморфную форму, любую кристаллическую форму, гидрат, сольват и т.п. Согласно некоторым вариантам осуществления, соединение формулы (II), используемое в качестве активного ингредиента, существует в виде аморфной формы, кристаллической формы А или кристаллической формы В. Согласно некоторым вариантам осуществления, соединение формулы (II), используемое в качестве активного ингредиента, существует в виде кристаллической формы А.
Согласно некоторым вариантам осуществления, упомянутая выше дозированная лекарственная форма представляет собой таблетку или капсулу. Согласно некоторым вариантам осуществления, упомянутая выше дозированная лекарственная форма представляет собой таблетку. Согласно некоторым вариантам осуществления, упомянутая выше дозированная лекарственная форма представляет собой таблетку и дополнительно содержит покрытие.
Таблетка может быть получена при помощи способов, включающих обычное прессование, влажное гранулирование или сухое гранулирование. Согласно некоторым вариантам осуществления, лекарственная форма по настоящему изобретению представляет собой таблетку, полученную способом влажного гранулирования. Таблетка также может содержать одно или более поверхностных покрытий, таких как прозрачное покрытие и/или окрашенное покрытие. В данной области техники известны различные покрытия и способы их нанесения, включая способы, раскрытые в работе Remington's Pharmaceutical Sciences (17-е издание, Mack Publishing Company, Easton, Pa., 1985). При наличии соответствующего количества покрытия масса таблетки обычно увеличивается на от 2% до 3%, так что масса таблетки обычно может составлять примерно от 50 мг до 1000 мг. Согласно некоторым вариантам осуществления, масса таблетки составляет примерно 100 мг, примерно 150 мг, примерно 200 мг, примерно 250 мг, примерно 400 мг, примерно 500 мг, примерно 600 мг, примерно 700 мг, примерно 800 мг, примерно 900 мг и т.п., в зависимости от дозы, необходимой для терапевтического применения.
Выражение "примерно", использованное перед значением массы упомянутых выше дозированных лекарственных форм по настоящему изобретению, означает диапазон значений ±10 мг, или ±5 мг, или ±2 мг, или ±1 мг, или ±0,5 мг, или ±0,2 мг, или ±0,1 мг.
Размер частиц D90 активного ингредиента в контексте настоящего изобретения относится к размеру частиц, соответствующему моменту, когда выраженное в процентах кумулятивное распределение частиц по размерам в образце достигает 90%.
Пленочные покрытия, которые могут быть использованы в составах по настоящему изобретению, известны в данной области техники и обычно включают полимеры (как правило, полимеры на основе целлюлозы), красители и пластификаторы. Для придания пленочному покрытию определенных свойств в его состав могут быть включены дополнительные ингредиенты, такие как сахара, вкусо-ароматические агенты, масла и смазывающие вещества. Композиции и составы по настоящему изобретению также могут быть объединены и преобразованы в твердые вещества, которые затем помещают в капсульные формы, такие как желатиновые капсулы.
Следует понимать, что некоторые компоненты составов по настоящему изобретению могут выполнять несколько функций. Например, определенный компонент может быть использован как в качестве наполнителя, так и в качестве разрыхлителя. В некоторых таких случаях функцию этого компонента можно считать единственной, хотя его свойства могут допускать многофункциональность.
Согласно настоящему изобретению, лактоза может быть выбрана из коммерчески доступной лактозы, подходящей для фармацевтического применения, включая Flow100, Granulac 200, Tableffose 100, Spherolac 100 и т.п. Микрокристаллическая целлюлоза может быть выбрана из коммерчески доступной микрокристаллической целлюлозы, подходящей для фармацевтического применения, включая рН101, рН102, рН301, рН302, KG1000, KG802, UF702, UF711 и т.п. Прежелатинизированный крахмал (также известный как модифицированный крахмал) может быть выбран из коммерчески доступного прежелатинизированного крахмала, подходящего для фармацевтического применения, включая Starch 1500, РС10 и т.п. Гипромеллоза может быть выбрана из коммерчески доступной гипромеллозы, подходящей для фармацевтического применения, включая НРМС Е3, НРМС Е5, НРМС К4М, НРМС Е15 и т.п. Повидон может быть выбран из коммерчески доступного повидона, подходящего для фармацевтического применения, включая повидон К30 и повидон К90. Кросповидон может быть выбран из коммерчески доступного кросповидона, подходящего для фармацевтического применения, включая PVPP XL-10, PVPP VL-10 и PVPP XL. Кроскармеллоза натрия может быть выбрана из коммерчески доступной кроскармеллозы натрия, подходящей для фармацевтического применения, включая RC-A591NF, SD-711 и т.п.
Второй аспект настоящего изобретения относится к способу получения таблетки, при этом способ включает следующие стадии, на которых:
(a) проводят влажное гранулирование после смешивания частиц активного ингредиента, наполнителя, связующего и первого разрыхлителя;
(b) сушат продукт, полученный на стадии (а);
(c) проводят сухое смешивание продукта, полученного на стадии (b), второго разрыхлителя и смазывающего вещества; и
(d) проводят прессование продукта, полученного на стадии (с), в таблетку;
где активный ингредиент представляет собой ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь;
размер частиц D90 активного ингредиента составляет 50 мкм или менее.
Первый разрыхлитель и второй разрыхлитель могут быть одинаковыми или разными.
Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 35 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 30 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 20 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет 10 мкм или менее. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 30 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 1 мкм до 10 мкм. Согласно другому предпочтительному примеру, размер частиц D90 активного ингредиента составляет от 10 мкм до 20 мкм.
Согласно другому предпочтительному примеру, наполнитель выбирают из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлозы-лактозы, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации.
Согласно другому предпочтительному примеру, связующее выбирают из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации.
Согласно другому предпочтительному примеру, каждый из первого разрыхлителя и второго разрыхлителя независимо выбирают из группы, состоящей из кроскармеллозы натрия, гипромеллозы-К4М, кросповидона, карбоксиметилкрахмала натрия и их комбинации.
Согласно другому предпочтительному примеру, смазывающее вещество выбирают из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации.
Согласно другому предпочтительному примеру, содержание активного ингредиента составляет от 4% до 10% в расчете на общую сухую массу смеси, полученной на стадии (с).
Согласно другому предпочтительному примеру, содержание наполнителя составляет от 73% до 82,5% в расчете на общую сухую массу смеси, полученной на стадии (с).
Согласно другому предпочтительному примеру, содержание связующего составляет от 1,5% до 3% в расчете на общую сухую массу смеси, полученной на стадии (с).
Согласно другому предпочтительному примеру, содержание первого разрыхлителя составляет от 2% до 10%, более предпочтительно, от 5% до 6%, в расчете на сухую массу всех компонентов на стадии (а); содержание второго разрыхлителя составляет от 5% до 10% в расчете на сухую массу всех компонентов на стадии (с).
Согласно другому предпочтительному примеру, содержание смазывающего вещества составляет от 0,1% до 1%, более предпочтительно, от 0,4% до 0,5%, более предпочтительно, от 0,48% до 0,5%, в расчете на общую сухую массу всех компонентов на стадии (с).
Согласно другому предпочтительному примеру, способ дополнительно включает стадию (е), на которой: наносят покрытие на продукт, полученный на стадии (d).
Согласно другому предпочтительному примеру, материал покрытия на стадии (е) представляет собой премикс растворимого в желудке пленочного покрытия, а концентрация материала покрытия составляет от 10% до 20%.
Согласно другому аспекту, в настоящем изобретении также предложен продукт, полученный способом, описанным в настоящем документе.
Третий аспект настоящего изобретения относится к применению твердого фармацевтического состава, описанного в соответствии с первым аспектом настоящего изобретения, для изготовления лекарственного средства для лечения заболевания, связанного с орексином.
Более предпочтительно, заболевания, связанные с орексином, включают бессонницу, хроническую обструктивную болезнь легких, синдром обструктивного апноэ во сне, сонливость, тревожность, обессивно-компульсивное расстройство, состояние паники, никотиновую зависимость или расстройство пищевого поведения.
Материалы, способы и примеры, представленные в контексте настоящего документа, предназначены для иллюстрации и не предназначены для ограничения объема настоящего изобретения. Все публикации, заявки на патенты, патенты и другие ссылочные материалы, упоминаемые в контексте настоящего документа, полностью включены в него посредством ссылки.
Следует понимать, что в пределах объема настоящего изобретения упомянутые выше технические признаки настоящего изобретения и технические признаки, конкретно описанные ниже (например, примеры), могут быть объединены друг с другом с образованием новых или предпочтительных технических решений. Для краткости изложения в рамках настоящего документа они не повторяются.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
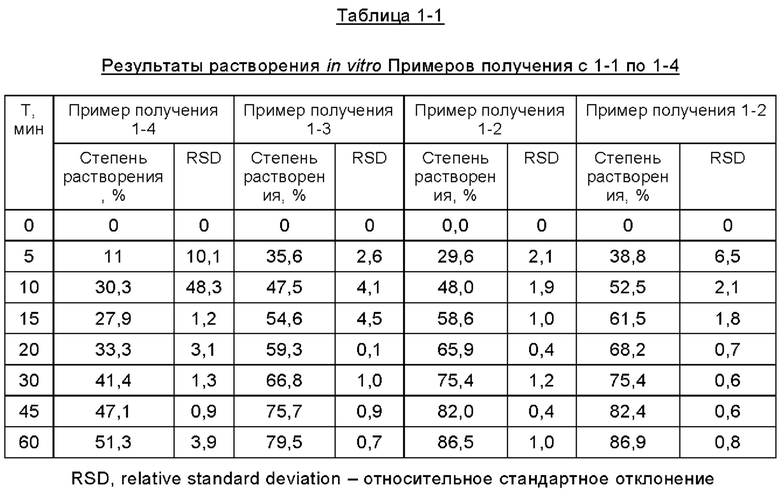
Фиг. 1 представляет собой график, изображающий кривые растворения составов Примеров получения 1-1, 1-2, 1-3 и 1-4.
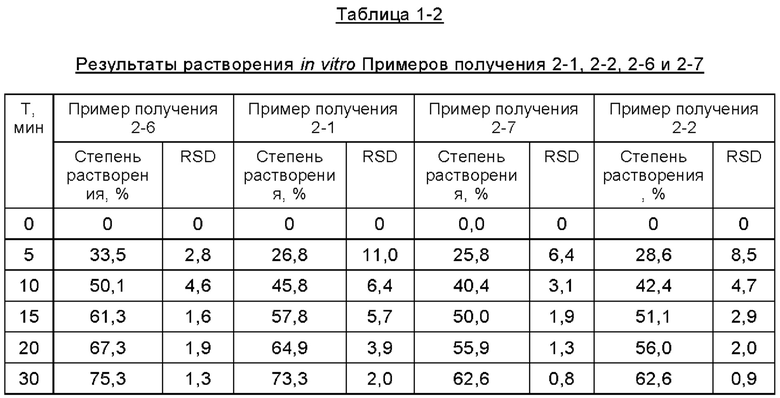
Фиг. 2 представляет собой график, изображающий кривые растворения составов Примеров получения 2-1, 2-2, 2-6 и 2-7.
Фиг. 3 представляет собой график, изображающий кривые растворения составов Примеров получения 2-1, 2-2, 2-3, 2-4 и 2-5.
Фиг. 4 представляет собой график, изображающий кривые растворения составов Примеров получения 3, 2-1 и 2-4.
Фиг. 5 представляет собой график, изображающий кривые растворения составов Примеров получения 4-1, 4-2 и 4-3.
Фиг. 6 представляет собой спектр XRPD, полученный в Cu-Kα излучении, соединения формулы (II) в кристаллической форме А.
Фиг. 7 представляет собой спектр XRPD, полученный в Cu-Kα излучении, соединения формулы (II) в кристаллической форме В.
Фиг. 8 представляет собой график, изображающий кривые растворения in vitro составов Примера получения 2-8 и Примера получения 6, при использовании в качестве среды растворения фосфатного буфера при величине рН 6,8.
Фиг. 9 представляет собой график, изображающий кривые растворения in vitro составов Примера получения 2-8 и Примера получения 6, при использовании в качестве среды растворения 0,1 N HCl.
На Фиг. 10 изображена трехмерная эллипсоидная структура монокристалла соединения формулы (II).
СПОСОБ ПОЛУЧЕНИЯ
Твердый фармацевтический состав по настоящему изобретению может быть получена способами, известными в данной области техники. Например, в случае гранул соединение, представленное формулой (I) или формулой (II), или его фармацевтически приемлемая соль и вспомогательное вещество, связующее, разрыхлитель, увлажняющее средство и т.п. могут быть смешаны и подвергнуты гранулированию при перемешивании, экструзионному гранулированию, роторному гранулированию, одностадийному гранулированию распылением и т.п., или прямому сухому гранулированию, как требуется для получения гранул. Кроме того, гранула также может быть получена путем нанесения лекарственного средства на пеллеты. Кроме того, по мере необходимости можно также проводить гранулирование и измельчение. При этом к описанным выше гранулам при таблетировании также могут быть дополнительно добавлены вспомогательные вещества, разрыхлители, связующие, антиокислители, красители и т.п.
В качестве дополнительной иллюстрации, таблетка без покрытия (плоская таблетка) или таблетка с покрытием по настоящему изобретению может быть получена при помощи следующего способа путем изменения добавляемого количества или соответствующего компонента в соответствии с различными рекомендациями. Способ получения включает: измельчение активного фармацевтического ингредиента, просеивание вспомогательного материала, взвешивание, смешивание и гранулирование, влажную сортировку гранул по размерам, сушку, сухую сортировку гранул по размерам, окончательное смешивание, таблетирование и нанесение покрытия (требуется при получении таблеток с покрытием).
Измельчение активного фармацевтического ингредиента: размер частиц отвечающего требованиям активного фармацевтического ингредиента (соединения формулы (I) или формулы (II), его фармацевтически приемлемой соли или их смеси) должен составлять от 120 до 150 мкм, что может быть достигнуто за счет регулирования различных параметров оборудования для измельчения. (1) Контроль размера D90 частиц на уровне 10 мкм или менее, 20 мкм или менее или 30 мкм или менее: отвечающий требованиям активный фармацевтический ингредиент (соединение формулы (I) или формулы (II), его фармацевтически приемлемую соль или их смесь) пропускают через сито, затем пересыпают в струйную мельницу для измельчения, размер частиц измельченного активного фармацевтического ингредиента контролируют, регулируя скорость вращения подающего механизма в диапазоне от 100 до 500 об/мин, устанавливая давление подачи на уровне от 3 до 7 бар и давление при измельчении на уровне от 2 до 7 бар, и затем измеряют распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, где распределение частиц по размерам должно соответствовать размеру D90 частиц 10 мкм или менее, размеру D90 20 мкм или менее или размеру D90 30 мкм или менее; или (2) контроль размера D90 частиц на уровне 50 мкм или менее: используют отвечающий требованиям активный фармацевтический ингредиент (формулы (I) или формулы (II)), измельчают его при помощи лабораторной ножевой мельницы для тонкого размола, измельчение проводят в течение 3 минут с интервалом 5 минут, после измельчения в течение определенного периода времени измеряют размер частиц активного фармацевтического ингредиента, при этом распределение частиц по размерам, определенное при помощи лазерного анализатора распределения частиц по размерам, должно соответствовать размеру D90 50 мкм или менее.
Просеивание вспомогательного материала: получают отвечающий требованиям вспомогательный материал - лактозу (Granulac 200 меш).
Взвешивание: взвешивают активный фармацевтический ингредиент (измельченный), микрокристаллическую целлюлозу (РН101), лактозу (Granulac 200 меш) (просеянную), гипромеллозу-Е5, кроскармеллозу натрия или гипромеллозу-К4М (с добавлением разрыхлителя).
Смешивание и гранулирование: суспендирование, т.е. приготовление связующего: взвешивают 300 г очищенной воды, при перемешивании добавляют 30 г гипромеллозы-Е5, продолжают перемешивание до растворения и получают 10% водный раствор гипромеллозы-Е5, пропускают его через сито 60 меш и откладывают. Смешивание: в гранулятор для влажного смешивания последовательно добавляют активный фармацевтический ингредиент (измельченный) и лактозу (просеянную) и начинают перемешивание и смешивание при скорости перемешивания от 300 до 500 об/мин (например, 300 об/мин или 400 об/мин), скорости дробления от 350 до 400 об/мин или от 400 до 500 об/мин (например, 400 об/мин) и времени перемешивания 300 с; открывают крышку резервуара, в резервуар последовательно добавляют кроскармеллозу натрия или гипромеллозу-К4М (с добавлением разрыхлителя), микрокристаллическую целлюлозу (РН101) и начинают перемешивание и смешивание при скорости перемешивания от 300 до 500 об/мин (например, 350 об/мин или 400 об/мин), скорости дробления от 400 до 500 об/мин (предпочтительно 400 об/мин) и времени перемешивания 600 с. Приготовление влажной массы, при этом весь процесс приготовления влажной массы разделен на две стадии, где на первой стадии: открывают гранулятор для влажного смешивания с заданными параметрами, устанавливают скорость перемешивания на уровне от 350 до 500 об/мин (например, 350 об/мин) и скорость резки на уровне от 1000 до 1500 об/мин (например, 1000 об/мин), включают на 10 с, после чего в гранулятор для влажного смешивания медленно добавляют всю массу 10% (мас./мас.) водного раствора связующего гипромеллозы-Е5, после добавления раствора связующего гомогенизируют полученные влажные гранулы с помощью лопастной мешалки и измельчителя до исчезновения заметных агломератов, время гранулирования (время добавления суспензии) составляет 300 с или менее (например, 60 с); и на второй стадии: устанавливают скорость перемешивания на уровне 500 об/мин или 350 об/мин и скорость резки на уровне 1500 об/мин или 1000 об/мин, одновременно начинают перемешивание и резку и для получения подходящих влажных гранул продолжают перемешивание и резку в течение 60 с.
Влажная сортировка гранул по размерам: полученные влажные гранулы сортируют по размерам, пропуская через сито 18 меш из нержавеющей стали в качающемся устройстве для сортировки гранул по размерам, или подвергают ручной влажной сортировке гранул по размерам, пропуская через сито 20 меш.
Сушка: влажные гранулы после сортировки по размерам равномерно распределяют на поддоне, причем толщина распределенных влажных гранулы на поддоне должна составлять 1,5 см ±0,5 см, поддон с распределенными по нему влажными гранулами помещают в печь и начинают сушку при температуре сушки 65,0°С ±5,0°С. Переворачивают распределенные влажные гранулы на поддоне и измеряют влажность гранул каждые 30 минут сушки до конечной точки сушки, когда содержание влаги в гранулах после сушки составит 2,0% или менее.
Сухая сортировка гранул по размерам: высушенные гранулы сортируют по размерам, пропуская их через сито 20 меш из нержавеющей стали в качающемся устройстве для сортировки гранул по размерам.
Окончательное смешивание: в смеситель одновременно помещают кроскармеллозу натрия или гипромеллозу-К4М (с добавлением разрыхлителя), гранулы после сухой сортировки гранул по размерам и стеарат магния и перемешивают в течение 300 с при скорости перемешивания 16 об/мин. После окончательного смешивания общая влажность гранул должна составлять 3,0% или менее.
Таблетирование: получают конечные смешанные гранулы активного фармацевтического ингредиента и таблетируют их при помощи роторной таблеточной машины.
Нанесение покрытия: на плоские таблетки, полученные таблетированием, наносят покрытие до получения требуемого увеличение массы покрытия в диапазоне от 2% до 3%. В качестве материала покрытия выбирают премикс растворимого в желудке пленочного покрытия, при этом концентрация раствора для нанесения покрытия составляет 15%. Конкретный способ получения включает следующие этапы: используют 30 г порошка для нанесения покрытия, добавляют в 200 г очищенной воды, перемешивают до однородного диспергирования и получают раствор для нанесения покрытия (приготовленный из расчета 200% с учетом увеличения массы на 3%), после чего наносят покрытие на таблетки при помощи высокоэффективного устройства для нанесения покрытия. Нанесение покрытия можно проводить при использовании следующих технологических параметров.

ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно описано ниже применительно к конкретным примерам. Следует понимать, что эти примеры использованы исключительно для иллюстрации настоящего изобретения и не предназначены для ограничения объема настоящего изобретения. В следующих примерах методики проведения экспериментов, для которых не указаны конкретные условия, обычно соответствуют общепринятым условиям или условиям, рекомендованным производителем. Проценты и части означают массовые проценты и массовые части, если не указано иное. Экспериментальные материалы и реагенты, использованные в следующих примерах, могут быть получены из коммерческих источников, если не указано иное.
Структуру монокристалла исследовали при помощи рентгеновского монокристалльного дифрактометра D8 Venture, источник излучения: мишень Cu, рентгеновские лучи: Cu-Kα (=1,54178 Å), детектор: поверхностный детектор CMOS (англ. complementary metal-oxide-semiconductor - комплементарный металл-оксидный проводник), разрешение: 0,8 Å, ток и напряжение: 50 кВ, 1,2 мА, время экспозиции: 10 с, расстояние от поверхности детектора до образца: 40 мм, температура испытания: 150(2)К.
Если не предусмотрено иное, массовые проценты, указанные для активного ингредиента, компонента наполнителя, компонента связующего, компонента разрыхлителя и компонента смазывающего вещества твердого фармацевтического состава, раскрытые в контексте настоящего документа, означают процентное содержание каждого из компонентов в конечного твердого фармацевтического состава без учета каких-либо поверхностных оболочек, таких как покрытие таблетки (например, любое прозрачное покрытие или окрашенное покрытие) или капсула. Расчет массовых процентов активного ингредиента, компонента наполнителя, компонента связующего, компонента разрыхлителя и компонента смазывающего вещества может незначительно измениться, поскольку таблетка с покрытием в следующих конкретных примерах включает массу покрытия. Однако следующие примеры использованы исключительно для иллюстрации настоящего изобретения и не предназначены для ограничения объема настоящего изобретения. Если не указано иное, в конкретных композициях следующих примеров получения активный ингредиент соединения формулы (II) существует в виде кристаллической формы А, микрокристаллическая целлюлоза представляет собой микрокристаллическую целлюлозу РН101, лактоза представляет собой лактозу Granulac 200 меш, а кроскармеллоза натрия представляет собой кроскармеллозу натрия SD711.
Пример получения 1-1
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 6 до 7 бар, давлении на входе от 7 до 8 бар, давлении подачи от 6 до 7 бар и скорости вращения при подаче от 100 до 150 об/мин. После завершения измельчения отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 10 мкм или менее (D90=7,461 мкм). Соединение формулы (II) с размером D90 частиц 10 мкм или менее (D90=7,461 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали плоские таблетки без покрытия со спецификацией 500 мг.
Пример получения 1-2
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали плоские таблетки без покрытия со спецификацией 500 мг.
Пример получения 1-3
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 2 до 4 бар, давлении на входе от 4 до 5 бар, давлении подачи от 3 до 4 бар и скорости вращения при подаче от 300 до 400 об/мин. После завершения измельчения отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 35 мкм или менее (D90=30,7 мкм). Соединение формулы (II) с D90 35 мкм или менее (D90=30,7 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали плоские таблетки без покрытия со спецификацией 500 мг.
Пример получения 1-4
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
После просеивания соединения формулы (II) через сито 20 меш измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 150 мкм или менее (D90=144,5 мкм). Соединение формулы (II) с размером D90 частиц 150 мкм или менее (D90=144,5 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали плоские таблетки без покрытия со спецификацией 500 мг.
Пример получения 2-1
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с размером D90 частиц 20 мкм или менее (D90=17,89 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. Затем проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример получения 2-2
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 410 мг.
Пример получения 2-3
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 6 до 7 бар, давлении на входе от 7 до 8 бар, давлении подачи от 6 до 7 бар и скорости вращения при подаче от 100 до 150 об/мин. После завершения измельчения отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 10 мкм или менее (D90=7,461 мкм). Соединение формулы (II) с размером D90 частиц 10 мкм или менее (D90=7,461 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример получения 2-4
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш и измельчали при помощи лабораторной ножевой мельницы для тонкого размола, измельчая в течение 3 минут с интервалом 5 минут. После измельчения в течение 10 минут измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 50 мкм или менее (D90=45,71 мкм). Соединение формулы (II) с размером D90 частиц 50 мкм или менее (D90=45,71 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример получения 2-5
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
После просеивания соединения формулы (II) через сито 20 меш измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 150 мкм или менее (D90=144,5 мкм). Соединение формулы (II) с размером D90 частиц 150 мкм или менее (D90=144,5 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример получения 2-6
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с размером D90 частиц 20 мкм или менее (D90=17,89 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали таблетки без покрытия со спецификацией 200 мг.
Пример получения 2-7
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Добавляли соединение формулы (II) с размером D90 частиц 20 мкм или менее (D90=17,89 мкм) и лактозу, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали плоские таблетки без покрытия со спецификацией 400 мг.
Пример получения 2-8
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 6 до 7 бар, давлении на входе от 7 до 8 бар, давлении подачи от 6 до 7 бар и скорости вращения при подаче от 100 до 150 об/мин. После завершения измельчения отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 10 мкм или менее (D90=8,93 мкм). Соединение формулы (II) с размером D90 частиц 10 мкм или менее (D90=8,93 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример получения 3
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 6 до 7 бар, давлении на входе от 7 до 8 бар, давлении подачи от 6 до 7 бар и скорости вращения при подаче от 100 до 150 об/мин. После завершения измельчения отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 10 мкм или менее (D90=7,461 мкм). Соединение формулы (II) с размером D90 частиц 10 мкм или менее (D90=7,461 мкм), лактозу и микрокристаллическую целлюлозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали. Затем медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали таблетки со спецификацией 500 мг.
Пример получения 4-1
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм), лактозу, микрокристаллическую целлюлозу и гипромеллозу-К4М помещали в гранулятор для влажного смешивания, перемешивали и смешивали. Затем медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, гипромеллозу-К4М и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали таблетки со спецификацией 200 мг.
Пример получения 4-2
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм), лактозу, микрокристаллическую целлюлозу и гипромеллозу-К4М помещали в гранулятор для влажного смешивания, перемешивали и смешивали. Затем медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам, и гранулы, полученные после сухой сортировки по размерам, гипромеллозу-К4М и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали таблетки со спецификацией 200 мг.
Пример получения 4-3
Состав плоской таблетки без покрытия
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм), лактозу, микрокристаллическую целлюлозу и гипромеллозу-К4М помещали в гранулятор для влажного смешивания, перемешивали и смешивали. Затем медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам, и гранулы, полученные после сухой сортировки по размерам, гипромеллозу-К4М и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали и получали таблетки со спецификацией 200 мг.
Пример получения 5
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 4 до 6 бар, давлении на входе от 6 до 7 бар, давлении подачи от 5 до 6 бар и скорости вращения при подаче от 200 до 300 об/мин. Отбирали образцы для тестирования, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=17,89 мкм). Соединение формулы (II) с D90 20 мкм или менее (D90=17,89 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Полученные сухие гранулы, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе. Суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 102 мг.
Пример получения 6
Состав таблетки с покрытием
Состав таблетки, включающий соединение формулы (II) (мас. %):
Соединение формулы (II) (в виде кристаллической формы В) пропускали через сито 20 меш, просеянный активный фармацевтический ингредиент помещали в струйную мельницу и измельчали при давлении измельчения от 6 до 7 бар, давлении на входе от 7 до 8 бар, давлении подачи от 6 до 7 бар и скорости вращения при подаче от 100 до 150 об/мин. Отбирали образцы для испытаний, измеряли распределение частиц по размерам при помощи лазерного анализатора распределения частиц по размерам, показывающего размер частиц D90 20 мкм или менее (D90=10,09 мкм). Соединение формулы (II) с размером D90 частиц 20 мкм или менее (D90=10,09 мкм) и лактозу помещали в гранулятор для влажного смешивания, перемешивали и смешивали, затем добавляли микрокристаллическую целлюлозу и кроскармеллозу натрия, перемешивали и смешивали. После этого в гранулятор для влажного смешивания медленно добавляли 10% (мас. %) водный раствор гипромеллозы-Е5 для проведения влажного гранулирования. Полученную смесь подвергали влажной сортировке гранул по размерам и сушили в печи. Содержание влаги в высушенных гранулах составляло 2,0% или менее. После этого проводили сухую сортировку гранул по размерам. Гранулы, полученные после сухой сортировки по размерам, кроскармеллозу натрия и стеарат магния окончательно смешивали в смесителе, суммарная влажность гранул после окончательного смешивания составляла 3,0% или менее. Гранулы таблетировали, покрывали премиксом растворимого в желудке пленочного покрытия и получали таблетки со спецификацией 205 мг.
Пример испытаний 1
Испытание на растворение in vitro
Согласно второму методу раздела 0931 общих принципов 4-ой редакции Китайской Фармакопеи 2015, составы таблеток, полученные в описанных выше примерах получения, подвергали испытанию на растворение in vitro для определения их уровней растворения в фосфатном буфере при величине рН 6,8, где в устройстве для растворения использовали лопастную мешалку, среда растворения представляла собой буфер с величиной рН 6,8, температура водяной бани составляла 37,0±0,5°С, объем растворения составлял 900 мл, скорость вращения составляла 50 об/мин, время отбора проб соответствовало 5 мин, 10 мин, 15 мин, 20 мин, 30 мин, 45 мин, 60 мин, объем отбора проб составлял 5 мл, а фильтрующая мембрана представляла собой фильтр из полиэфирсульфона. Результаты испытаний представлены ниже в Таблицах с 1-1 по 1-5 и на Фиг. с 1 по 5.

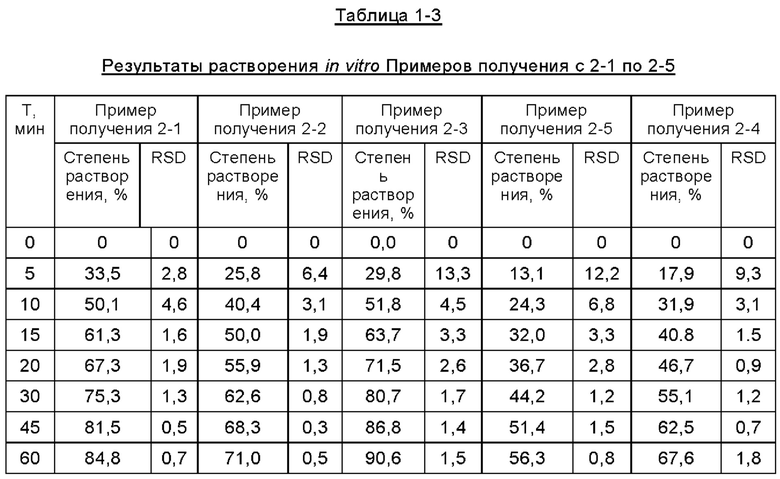
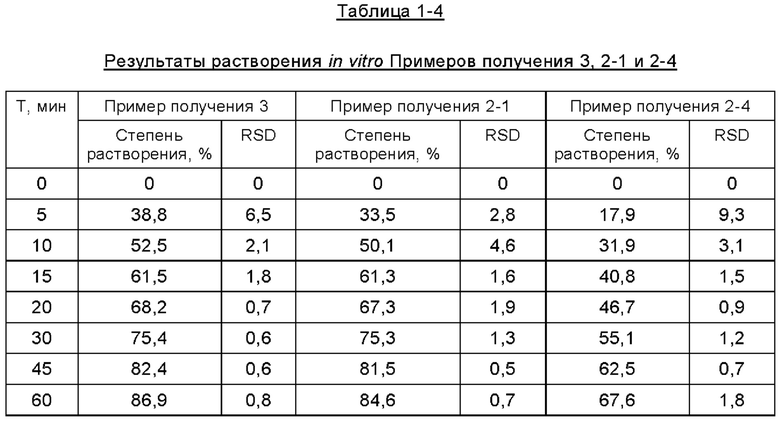

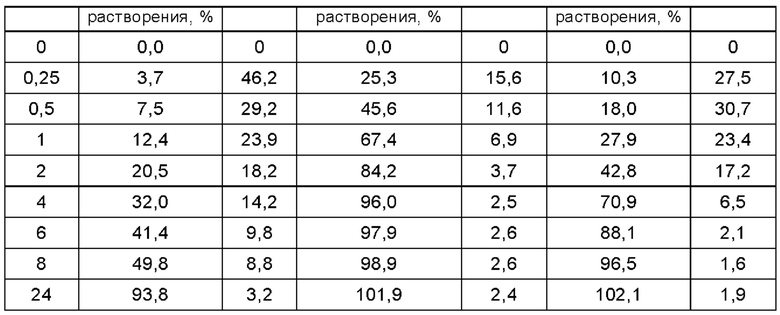
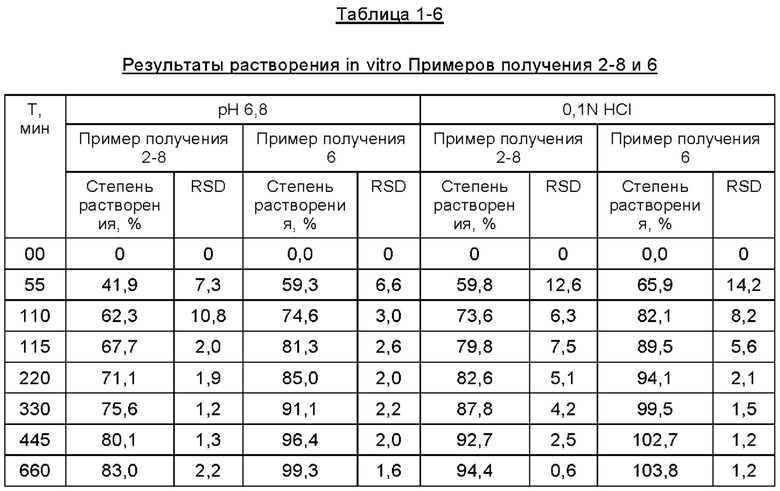
Пример испытаний 2
Испытание стабильности
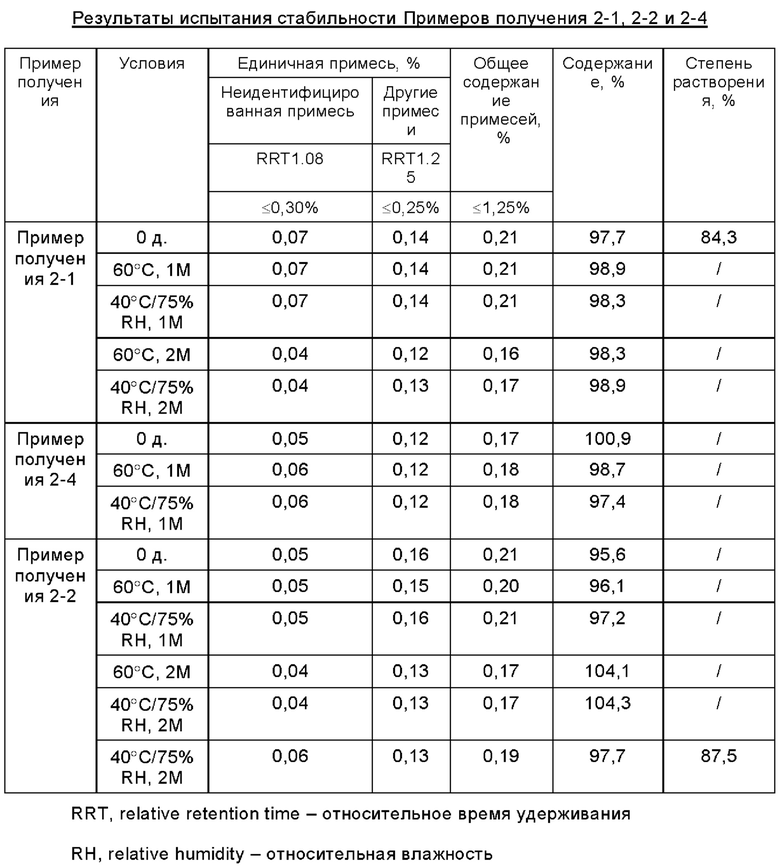
Подготовленные образцы помещали в условия высокой температуры (60°С) и условия ускоренного старения (40°C/75%RH) на определенный период времени и оценивали стабильность образцов, исследуя их состав, проверяя на сопутствующие примеси и степень растворения в соответствии со вторым методом разделов 0512 и 0931 общих принципов 4-ой редакции Китайской Фармакопеи 2015. Результаты испытаний представлены в Таблице 2-1.
Пример испытаний 3
Испытание биодоступности при пероральном введении у собак
Изучали пероральную биодоступность приготовленных образцов у собак. Исследование было разработано в соответствии с техническими рекомендациями по доклиническим фармакокинетическим исследованиям прежнего Управления по контролю качества пищевых продуктов и лекарственных средств Китая (англ. CFDA, China Food and Drug Administration) и ICH M3(R2) (англ. International Conference on Harmonisation - Международная конференция по гармонизации). Таблетка имела спецификацию 20 мг, 1 таблетка/1 собаку. Образцы отбирали в соответствии с расчетным моментом времени. Концентрации активных ингредиентов в образцах определяли методом ВЭЖХ-МС/МС (англ. HPLC-MS/MS, High Pressure Liquid Chromatography-Mass Spectrometry/Mass Spectrometry - высокоэффективная хроматография-тандемная масс-спектрометрия) и рассчитывали фармакокинетические параметры. Суть испытания заключалась в следующем.
Животные и введение: использовали собак породы бигль общего класса в возрасте от 7 до 14 месяцев весом от 9,51 до 11,14 кг. Пероральное введение проводили однократно в каждый день введения при времени введения с 8:00 до 12:00 ч. Все животные голодали в течение ночи перед каждым введением и продолжали голодание в течение от 2 до 3 часов после введения, но общее время голодания не превышало 24 часов.
Забор крови: проводили отбор венозной крови из передней конечности. Время забора крови - каждый день введения (т.е. 1 день, 5 день, 8 день, 12 день и 15 день). Моменты времени забора крови - перед введением (0 ч), 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения. Отбирали примерно 1 мл крови в вакуумированную пробирку для забора крови с антикоагулянтом калиевой солью ЭДТА (англ. EDTA, Ethylene Diamine Tetraacetic Acid - этилендиаминтетрауксусная кислота). После забора крови пробирку с кровью осторожно встряхивали для тщательного перемешивания крови и антикоагулянта. После забора кровь помещали в дробленый лед и центрифугировали (4°С, 2000 г, 10 мин) в течение 1 ч после забора. После центрифугирования отбирали примерно 400 мкл плазмы в пластиковую пробирку коричневого цвета и сразу же помещали ее в контейнер с сухим льдом, затем переносили в сверхнизкотемпературную холодильную камеру (≤-65°С) и хранили в темноте.
Анализ образцов: для определения концентрации активных ингредиентов в образце использовали метод ВЭЖХ-МС/МС, а для обработки данных и расчета использовали Microsoft Excel 2013. После анализа и определения концентрации образцов в плазме с помощью программного обеспечения WinNonlin6.3 строили кривую зависимости концентрация-время и рассчитывали фармакокинетические параметры в соответствии с некомпартаментной моделью. Результаты фармакокинетических параметров соответствующих составов представлены ниже в Таблице 3-1, Таблице 3-2 и Таблице 3-3, соответственно. Результаты в одной и той же таблице получены при тестировании одной и той же партии, а результаты в разных таблицах получены при тестировании разных партий.

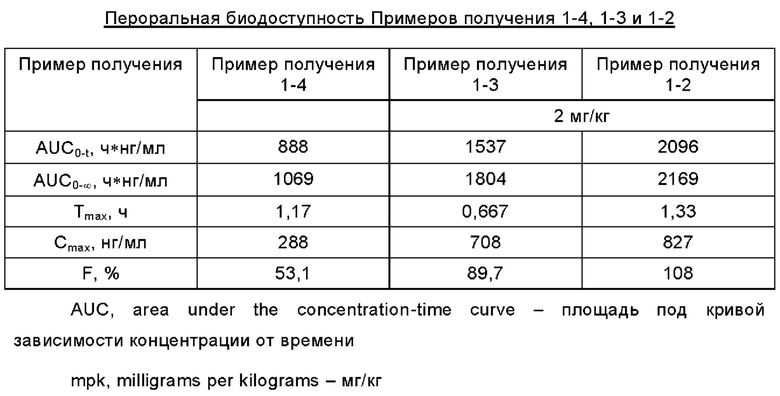

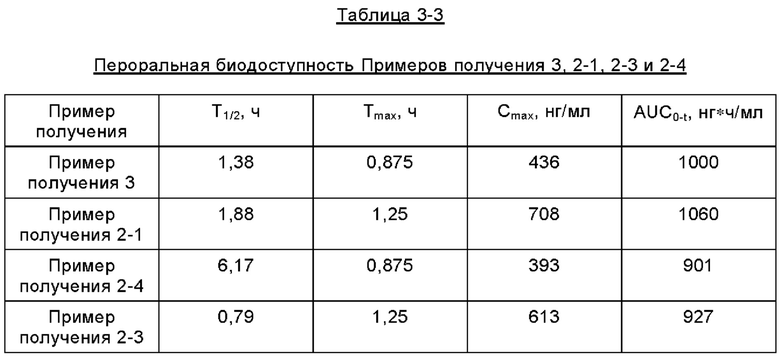
Из представленной выше таблицы видно, что разные размеры частиц активного ингредиента в композиции по-разному влияют на фармакокинетические параметры, такие как пролонгированный период полувыведения или снижение экспозиции у собак породы бигль.
Все документы, упоминаемые в контексте настоящего изобретения, включены в настоящий документ посредством ссылки, как если бы каждый из документов был отдельно включен посредством ссылки. Кроме того, следует понимать, что после ознакомления с приведенным выше описанием настоящего изобретения специалисты в данной области техники могут вносить в настоящее изобретение различные изменения или модификации, и эти эквивалентные формы также подпадают под объем, определяемый прилагаемой формулы изобретения настоящей заявки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТАБЛЕТКА И СПОСОБ ЕЕ ПРИГОТОВЛЕНИЯ | 2019 |
|
RU2796301C2 |
| ТАБЛЕТКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2016 |
|
RU2697660C2 |
| ЛЕЧЕБНОЕ ПРИМЕНЕНИЕ ПИРРОЛОПИРИМИДИНОВОГО СОЕДИНЕНИЯ И ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПИРРОЛОПИРИМИДИНОВОГО СОЕДИНЕНИЯ | 2019 |
|
RU2810112C2 |
| ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АГОНИСТ TLR7 | 2020 |
|
RU2822480C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 2019 |
|
RU2756452C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2828311C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ФИМАСАРТАН И ГИДРОХЛОРТИАЗИД | 2013 |
|
RU2613900C2 |
| ФАРМАЦЕВТИЧЕСКОЕ КОМБИНИРОВАННОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2014 |
|
RU2639818C2 |
| КОМБИНИРОВАННАЯ КОМПОЗИЦИЯ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ, СОДЕРЖАЩАЯ ЭЗЕТИМИБ И РОЗУВАСТАТИН | 2014 |
|
RU2683937C2 |
| НОВАЯ КОМПОЗИЦИЯ ЛАПАТИНИБА В ВИДЕ ТВЕРДОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ | 2019 |
|
RU2821950C2 |
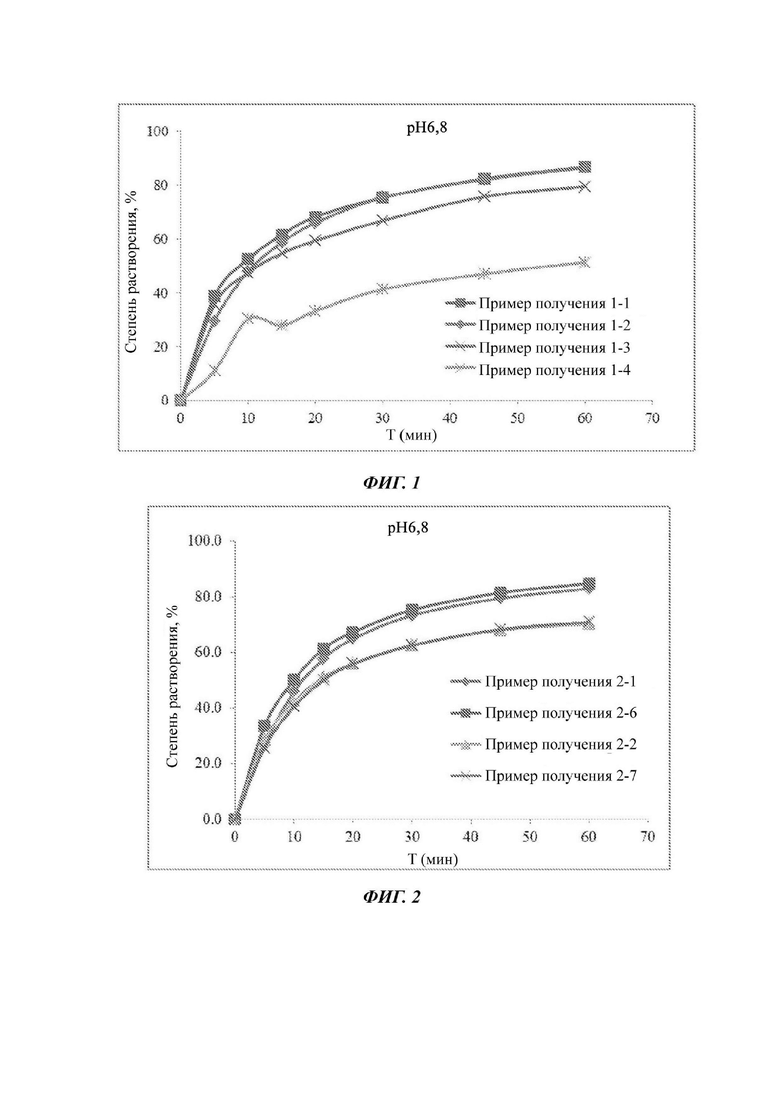

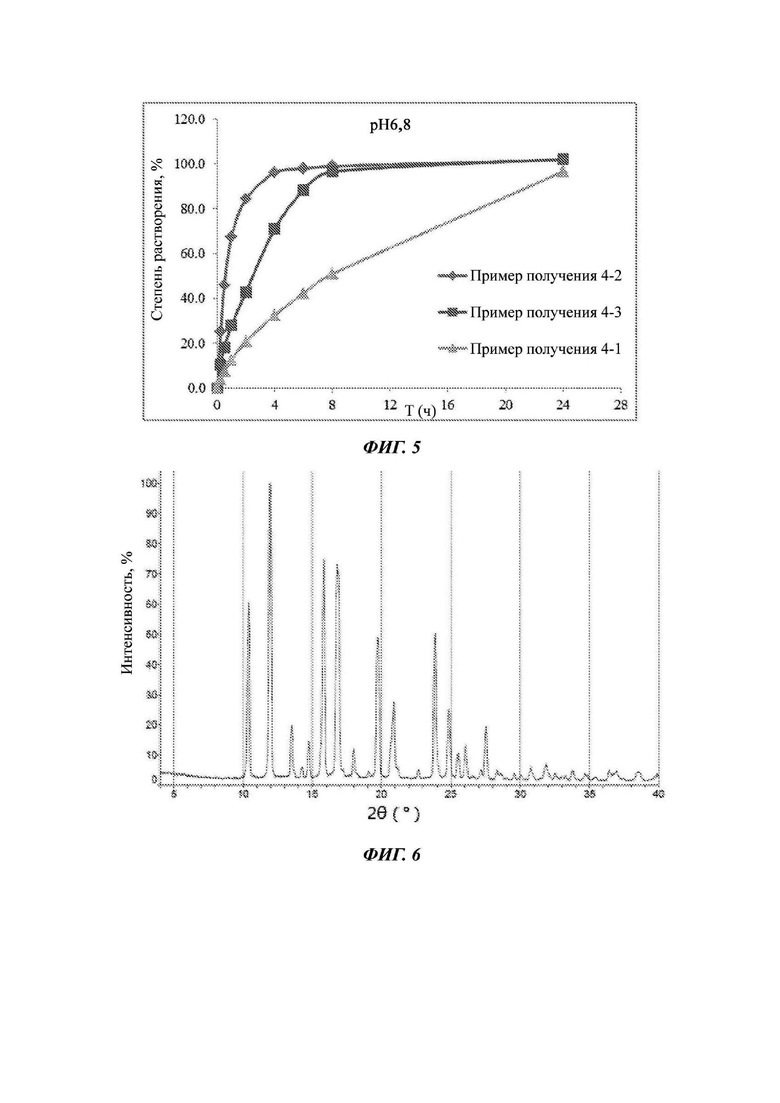
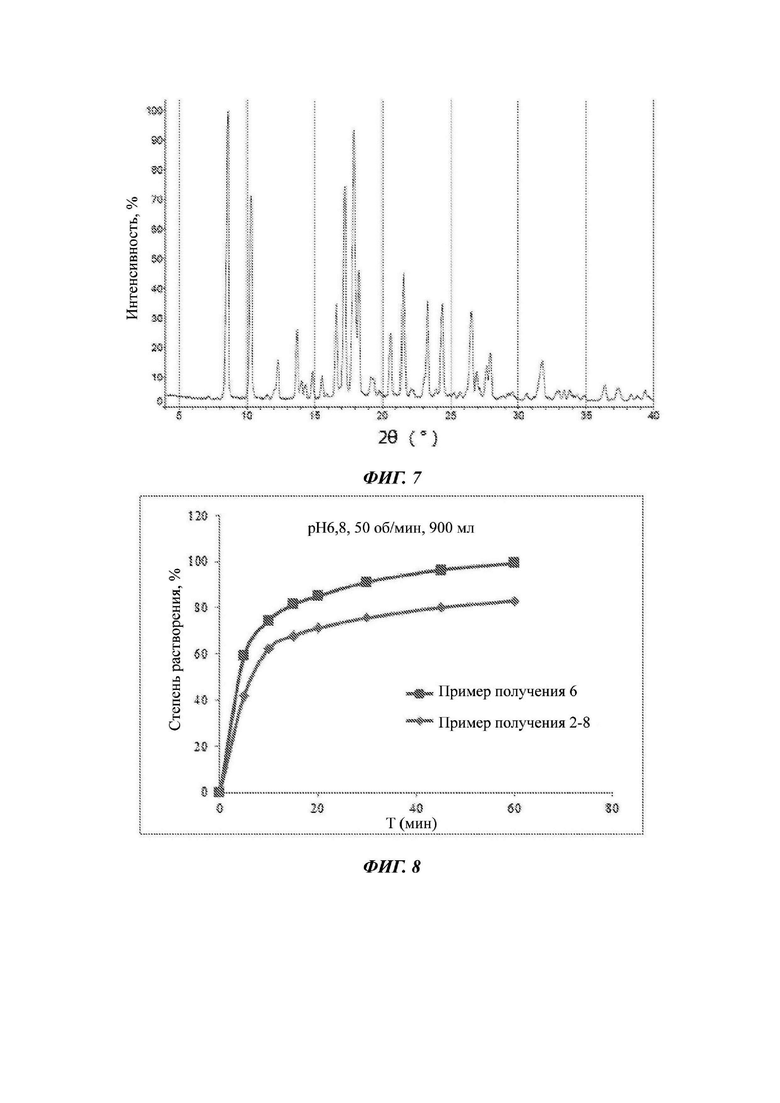

Группа изобретений относится к области медицины и раскрывает твердый фармацевтический состав для лечения заболевания, связанного с орексином, где указанный твердый фармацевтический состав содержит активный ингредиент, при этом указанный активный ингредиент представляет собой соединение формулы (II) или его фармацевтически приемлемую соль либо их смесь; и размер частиц D90 активного ингредиента составляет 35 мкм или менее. Также группа изобретений раскрывает способ получения таблетки, дозированную лекарственную форму для лечения заболевания, связанного с орексином и способ лечения заболевания, связанного с орексином. Техническим результатом группы изобретений является обеспечение хорошей стабильности, хорошей растворимости и высокой биодоступности твердого фармацевтического состава, содержащего соединение формулы (II). 4 н. и 16 з.п. ф-лы, 10 ил., 10 табл., 21 пр.

1. Твердый фармацевтический состав для лечения заболевания, связанного с орексином, где указанный твердый фармацевтический состав содержит активный ингредиент, при этом указанный активный ингредиент представляет собой соединение формулы (II) или его фармацевтически приемлемую соль либо их смесь

и размер частиц D90 активного ингредиента составляет 35 мкм или менее.
2. Твердый фармацевтический состав по п. 1, где размер частиц D90 активного ингредиента составляет 20 мкм или менее.
3. Твердый фармацевтический состав по п. 1, где содержание активного ингредиента составляет от 1% до 15% в расчете на общую сухую массу твердого фармацевтического состава.
4. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав дополнительно содержит связующее, выбранное из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации, при этом содержание связующего составляет от 0,5% до 10% в расчете на общую сухую массу твердого фармацевтического состава; и/или
где указанный твердый фармацевтический состав дополнительно содержит наполнитель, выбранный из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлоза–лактоза, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации, при этом содержание наполнителя составляет от 60% до 90% в расчете на общую сухую массу твердого фармацевтического состава.
5. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав дополнительно содержит разрыхлитель, выбранный из группы, состоящей из кроскармеллозы натрия, гипромеллозы-K4M, кросповидона, карбоксиметилкрахмала натрия и их комбинации, при этом содержание разрыхлителя составляет от 5% до 15% в расчете на общую сухую массу твердого фармацевтического состава; и/или
где указанный твердый фармацевтический состав дополнительно содержит смазывающее вещество, выбранное из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации, при этом содержание смазывающего вещества составляет от 0,1% до 1% в расчете на общую сухую массу твердого фармацевтического состава.
6. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав представляет собой таблетку, капсулу, порошок, гранулу, драже или пленку.
7. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 1% до 15%;
b) наполнитель, выбранный из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлоза–лактоза, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации, где содержание наполнителя составляет от 60% до 90%;
c) связующее, выбранное из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации, где содержание связующего составляет от 0,5% до 10%;
d) разрыхлитель, выбранный из группы, состоящей из кроскармеллозы натрия, гипромеллозы-K4M, кросповидона, карбоксиметилкрахмала натрия и их комбинации, где содержание разрыхлителя составляет от 5% до 15%; и
e) смазывающее вещество, выбранное из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации, где содержание смазывающего вещества составляет от 0,1% до 1%;
где размер частиц D90 активного ингредиента составляет 35 мкм или менее, или D90 составляет 30 мкм или менее, или D90 составляет 20 мкм или менее, или D90 составляет 10 мкм или менее, или D90 составляет от 1 мкм до 30 мкм, или D90 составляет от 1 мкм до 20 мкм, или D90 составляет от 1 мкм до 10 мкм, или D90 составляет от 10 мкм до 20 мкм.
8. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 4% до 10%;
b) от 24% до 27,5% микрокристаллической целлюлозы;
c) от 48,5% до 56,5% лактозы;
d) от 1,5% до 3% гипромеллозы-E5;
e) от 5% до15% кроскармеллозы натрия или гипромеллозы-K4M; и
f) от 0,4% до 0,5% стеарата магния;
где размер частиц D90 активного ингредиента составляет 35 мкм или менее, или D90 составляет 30 мкм или менее, или D90 составляет 20 мкм или менее, или D90 составляет 10 мкм или менее, или D90 составляет от 1 мкм до 30 мкм, или D90 составляет от 1 мкм до 20 мкм, или D90 составляет от 1 мкм до 10 мкм, или D90 составляет от 10 мкм до 20 мкм.
9. Твердый фармацевтический состав по п. 1, где указанный твердый фармацевтический состав содержит следующие компоненты в расчете на общую сухую массу твердого фармацевтического состава:
a) активный ингредиент: ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь, где содержание активного ингредиента составляет от 9,5% до 10%;
b) от 24% до 27,5% микрокристаллической целлюлозы;
c) от 48,5% до 56,5% лактозы;
d) от 1,5% до 3% гипромеллозы-E5;
e) от 5% до15% кроскармеллозы натрия или гипромеллозы-K4M; и
f) от 0,48% до 0,5% стеарата магния;
где размер частиц D90 активного ингредиента составляет от 1 мкм до 30 мкм, или D90 составляет от 1 мкм до 20 мкм, или D90 составляет от 1 мкм до 10 мкм, или D90 составляет от 10 мкм до 20 мкм, при этом указанный твердый фармацевтический состав представляет собой таблетку.
10. Способ получения таблетки, где способ включает следующие стадии, на которых:
a) проводят влажное гранулирование после смешивания частиц активного ингредиента, наполнителя, связующего и первого разрыхлителя;
b) сушат продукт, полученный на стадии (a);
c) проводят сухое смешивание продукта, полученного на стадии (b), второго разрыхлителя и смазывающего вещества; и
d) прессуют продукт, полученный на стадии (c), в таблетку;
при этом указанный активный ингредиент представляет собой ((1S,2R,5S)-2-(((5-фторпиридин-2-ил)окси)метил)-8-азабицикло[3.2.1]октан-8-ил)(5-метил-2-(пиримидин-2-ил)фенил)метанон или его фармацевтически приемлемую соль либо их смесь;
размер частиц D90 активного ингредиента составляет 35 мкм или менее;
наполнитель выбран из группы, состоящей из микрокристаллической целлюлозы, лактозы, комплекса целлюлоза–лактоза, прежелатинизированного крахмала, двузамещенного фосфата кальция, карбоната кальция и их комбинации;
связующее выбрано из группы, состоящей из гипромеллозы, гидроксипропилцеллюлозы, повидона, альгината натрия, карбопола, поливинилового спирта и их комбинации;
первый разрыхлитель и второй разрыхлитель каждый независимо выбран из группы, состоящей из кроскармеллозы натрия, гипромеллозы-K4M, кросповидона, карбоксиметилкрахмала натрия и их комбинации; и
смазывающее вещество выбрано из группы, состоящей из стеарата магния, порошка талька, глицеринмоностеарата, стеарилфумарата натрия и их комбинации.
11. Способ по п. 10, где содержание активного ингредиента составляет от 4% до 10% в расчете на общую сухую массу смеси, полученной на стадии (c).
12. Способ по п. 10, где содержание наполнителя составляет от 73% до 82,5% в расчете на общую сухую массу смеси, полученной на стадии (c); и/или
где содержание связующего составляет от 1,5% до 3% в расчете на общую сухую массу смеси, полученной на стадии (c).
13. Способ по п. 10, где содержание первого разрыхлителя составляет от 2% до 10% в расчете на общую сухую массу всех компонентов на стадии (a); и где содержание второго разрыхлителя составляет от 5% до 10% в расчете на общую сухую массу всех компонентов на стадии (c).
14. Способ по п. 10, где содержание смазывающего вещества составляет от 0,1% до 1% в расчете на общую сухую массу всех компонентов на стадии (c).
15. Способ по п. 10, где указанный способ дополнительно включает:
e) нанесение покрытия на продукт, полученный на стадии (d).
16. Дозированная лекарственная форма для лечения заболевания, связанного с орексином, где в расчете на общую массу дозированной лекарственной формы указанная дозированная лекарственная форма содержит:
10 мг, 20 мг или 40 мг активного ингредиента, где указанный активный ингредиент представляет собой соединение формулы (II)
от 25 до 150 мг микрокристаллической целлюлозы;
от 50 до 300 мг лактозы;
от 1 до 15 мг гипромеллозы;
от 10 до 50 мг кроскармеллозы натрия; и
от 0,5 до 3 мг стеарата магния,
где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм; или
где в расчете на общую массу дозированной лекарственной формы указанная дозированная лекарственная форма содержит:
от 8 мг до 12 мг активного ингредиента, где указанный активный ингредиент представляет собой соединение формулы (II)
от 20 мг до 30 мг микрокристаллической целлюлозы;
от 46 мг до 56 мг лактозы;
от 2 мг до 4 мг гипромеллозы;
от 5 мг до 15 мг кроскармеллозы натрия; и
от 0,3 мг до 0,7 мг стеарата магния,
где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм; или
где в расчете на общую массу дозированной лекарственной формы указанная дозированная лекарственная форма содержит:
от 18 мг до 22 мг активного ингредиента, где указанный активный ингредиент представляет собой соединение формулы (II)
от 46 мг до 56 мг микрокристаллической целлюлозы;
от 97 мг до 107 мг лактозы;
от 5 мг до 7 мг гипромеллозы;
от 15 мг до 25 мг кроскармеллозы натрия; и
от 0,8 мг до 1,2 мг стеарата магния,
где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм; или
где в расчете на общую массу дозированной лекарственной формы указанная дозированная лекарственная форма содержит:
от 38 мг до 42 мг активного ингредиента, где указанный активный ингредиент представляет собой соединение формулы (II)
от 97 мг до 107 мг микрокристаллической целлюлозы;
от 199 мг до 209 мг лактозы;
от 11 мг до 13 мг гипромеллозы;
от 35 мг до 45 мг кроскармеллозы натрия; и
от 1,8 мг до 2,2 мг стеарата магния,
где размер частиц D90 активного ингредиента составляет от 1 мкм до 20 мкм.
17. Твердый фармацевтический состав по п. 1, где соединение формулы (II) в качестве активного ингредиента существует в виде кристаллической формы A или кристаллической формы B, где кристаллическая форма A имеет спектр XRPD, полученный в Cu-Kα излучении:
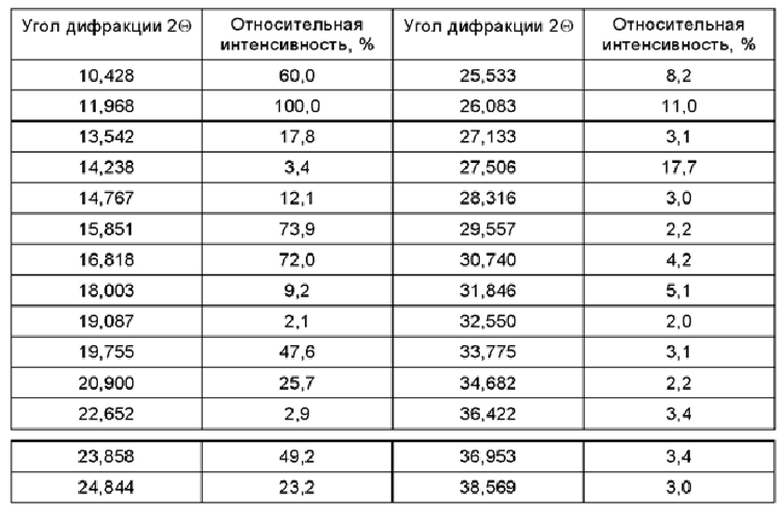
и где кристаллическая форма В имеет спектр XRPD, полученный в Cu-Kα излучении:
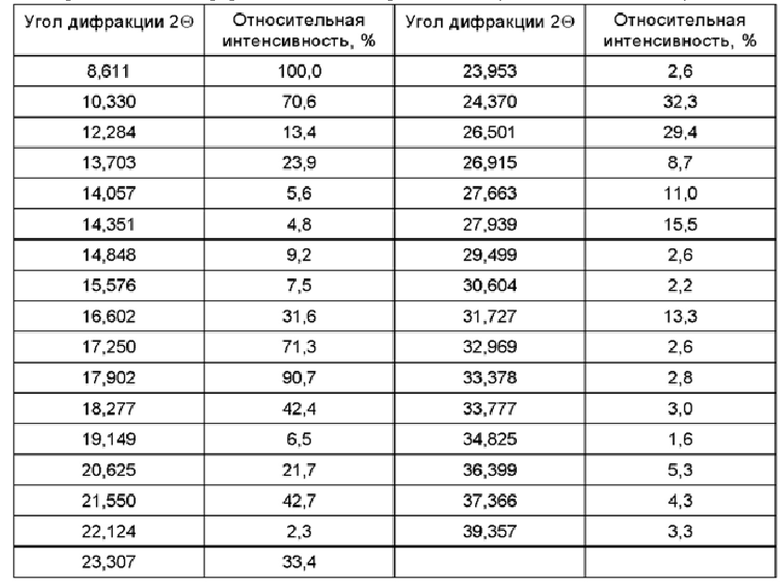
18. Дозированная лекарственная форма по п. 16, где указанная дозированная лекарственная форма представляет собой таблетку или капсулу.
19. Способ лечения заболевания, связанного с орексином, включающий введение твердого фармацевтического состава по п. 1.
20. Способ по п. 19, где заболевание, связанное с орексином, включает бессонницу, хроническую обструктивную болезнь легких, синдром обструктивного апноэ во сне, сонливость, тревожность, обессивно-компульсивное расстройство, состояние паники, никотиновую зависимость или расстройство пищевого поведения.
| WO 2014072961 A2, 15.05.2014 | |||
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ОРЕКСИНА | 2015 |
|
RU2669701C2 |
| US 20180235965 A1, 23.08.2018 | |||
| WO 2007014124 A2, 01.02.2007 | |||
| Станок для сборки цепей Галля | 1931 |
|
SU28386A1 |
| EP 1531805 B1, 30.04.2008 | |||
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ БЕССОННИЦЫ | 2015 |
|
RU2703297C2 |
