Область техники
Настоящее изобретение относится к производному пиримидин-2,4-диамина, способу его получения и фармацевтической композиции для профилактики или лечения рака, содержащей его в качестве активного ингредиента.
Уровень техники
Развитие рака связано с различными факторами окружающей среды, включая химические вещества, радиацию и вирусы, а также с изменениями в онкогенах, генах-супрессорах опухолей и генах, связанных с апоптозом и репарацией ДНК. Недавно, благодаря пониманию этих молекулярных механизмов рака, стал возможен новый способ лечения, таргетная противораковая терапия.
Таргетные терапевтические агенты обычно создаются для оказания их воздействия путем таргетирования молекул, которые характерны для раковых клеток. Молекулярные мишени включают гены, связанные с путем передачи сигнала, ангиогенезом, матриксом, регулятором клеточного цикла и апоптозом раковых клеток. В настоящее время важные таргетные терапевтические агенты, используемые в лечении, включают «ингибиторы пути трансдукции сигнала» и «ингибиторы ангиогенеза», включая ингибиторы тирозинкиназы.
Было обнаружено, что протеинтирозинкиназы играют важную роль во многих злокачественных опухолях. В частности, известно, что рецептор эпидермального фактора роста (EG FR), рецепторная тирозинкиназа семейства erbB, аномально активируется при многих опухолях эпителиальных клеток, включая немелкоклеточную карциному легких (NSCLC), рак молочной железы, глиому, плоскоклеточную карциному легких головы и шеи, рак толстого кишечника, аденокарциному прямой кишки, рак головы и шеи, рак желудка и рак простаты, и активация EGFR-тирозинкиназы вызывает непрерывную пролиферацию клеток, инвазию в окружающие ткани, отдаленные метастазы, образование кровеносных сосудов, и увеличивает выживаемость клеток.
В частности, EGFR является одним из семейства рецепторов тирозинкиназы ErbB (EGFR, HER2, ErbB3, ErbB4) и представляет собой трансмембранную тирозинкиназу, которая имеет внутриклеточный домен, включающий внеклеточный лигандсвязывающий домен и тирозинкиназный домен. Когда лиганд связывается с гомодимерным или гетеродимерным рецептором, внутриклеточная тирозинкиназа активируется, и сигнал, стимулируемый EGFR, активирует путь трансдукции сигнала фосфатидилинозит 3-киназы (PI3K)/AKT/mTOR, RAS/RAF/MAPK и JAK/STAT (Nat Rev Cancer 2007;7:169-81).
В частности, EGFR сверхэкспрессируется более чем в половине случаев немелкоклеточного рака легких (NSCLC), и было проведено множество исследований в качестве мишени для лечения. Были разработаны EGFR TKI (ингибиторы тирозинкиназы), которые ингибируют активность тирозинкиназы EGFR, и типовые лекарственные средства включают гефитиниб (IRESSA™), эрлотиниб (TARCEVA™) и лапатиниб (TYKERB™, TYVERB™).
Между тем, в 2004 году сообщалось, что активирующие мутации в EGFR коррелируют с ответом на терапию гефитинибом при немелкоклеточном раке легких (NSCLC) (Science [2004] Vol.304, 1497-500 и New England Journal of Medicine [2004] Vol. 350, 2129-2139).
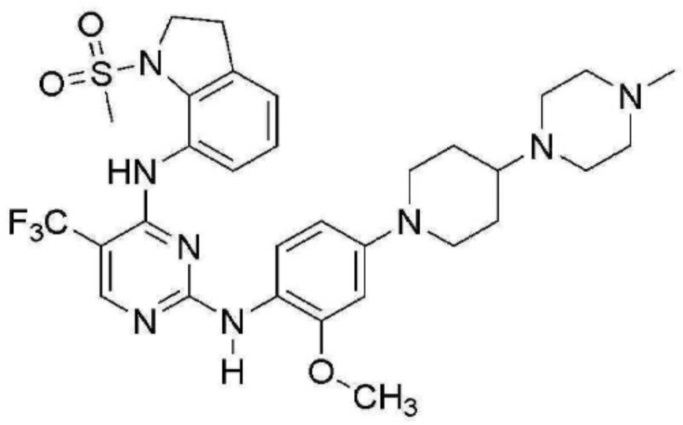
В частности, мутации EGFR в основном делятся на сенсибилизирующие мутации и резистентные мутации. Делеция экзона 19 и точечная мутация L858R в экзоне 21 являются наиболее важными сенсибилизирующими мутациями, на их долю приходится приблизительно 85-90%, и del мутация экзона 19, как известно, обладает большей сенсибилизирующей способностью к TKI. С другой стороны, точечная мутация T790M в экзоне 20 является наиболее важной резистентной мутацией и, как известно, обнаруживается более чем у 50% пациентов с приобретенной резистентностью (Clin Cancer Res 2006;12:6494-6501).
Соматические мутации, идентифицированные на сегодняшний день, включают точечные мутации (например, L858R, G719S, G719C, G719A, L861Q), при которых один нуклеотидный остаток модифицируется в экспрессируемом белке, а также делеции в рамке считывания в экзоне 19 или вставки в экзоне 20 (Fukuoka et al. JCO 2003; Kris et al JAMA 2003 and Shepherd et al NEJM 2004).
Несмотря на первоначальную клиническую эффективность гефитиниба/эрлотиниба у пациентов с NSCLC с мутациями EGFR, у большинства пациентов на фоне терапии этими агентами в конечном итоге развивается прогрессирование рака. Первоначальные исследования образцов с рецидивами выявили вторичную мутацию EGFR, T790M, которая делает гефитиниб и эрлотиниб неэффективными ингибиторами киназной активности EGFR (Kobayashi et al., NEJM 2005 и Pao et al., PLOS Medicine 2005). Последующие исследования показали, что мутация EGFR T790M была обнаружена приблизительно в 50% (24/48) опухолей, полученных от пациентов, которые приобрели резистентность к гефитинибу или эрлотинибу (Kosaka et al CCR 2006; Balak et al CCR 2006 и Engelman et al Science 2007). Эти вторичные генетические модификации возникают в положениях, аналогичных остаткам «привратника» и связанным с ними аллелям вторичной резистентности у пациентов, получающих ингибиторы киназы (например, T315I в ABL при CML с резистентностью к иматинибу).
Давно известно, что мутации EGFR, EGFR_del19 или EGFR_L858R, являются основной причиной немелкоклеточного рака легких и рака головы и шеи, и были разработаны лекарственные средства для их лечения, Иресса и Тарцева, и в настоящее время используются клинически. Однако при применении этих лекарственных средств у пациентов наблюдалась приобретенная резистентность, при которой происходили вторичные мутации EGFR, обусловленные структурой лекарственного средства, а также было выявлено, что это является основной причиной фактической лекарственной резистентности. Когда ингибиторы EGFR первого поколения используют в среднем в течение 10 месяцев, возникает приобретенная резистентность, называемая мутацией T790M, расположенной в привратнике киназы EGFR, что делает ингибиторы EGFR первого поколения неэффективными. Другими словами, происходит двойная мутация EGFR_del19_T790M или EGFR_L858R_T790M, что делает традиционные терапевтические агенты неэффективными.
На основании этих фактов возникла необходимость разработки лекарственных средств второго и третьего поколений с превосходной эффективностью и новой структурой.
За последние 10 лет, различные лекарственные средства третьего поколения, показавшие эффективность против двойных мутаций EGFR T790M, включают AZD9291 (осимертиниб, Тагриссо) от транснациональной фармацевтической компании AstraZeneca. Однако сообщалось, что резистентность к AZD9291 снова развивается примерно через 10 месяцев, и эффективность AZD9291 теряется. В частности, сообщалось, что резистентность возникает из-за тройной мутации, включая C797S (Thress et al., Nature Medicine 2015).
Соответственно, существует потребность в разработке ингибиторов, которые демонстрируют ингибирование относительно низкого WT EGFR и, в то же время, более высокое ингибирование специфических активированных или различных резистентных мутантных форм EGFR.
TKI EGFR (ингибиторы тирозинкиназы) гефитиниб, эрлотиниб и афатиниб являются одобренными терапевтическими агентами для лечения немелкоклеточного рака легкого с активирующими мутациями киназы EGFR. Однако существует проблема в том, что резистентность к этим лекарственным средствам развивается быстро, и часто возникает мутация Т790М в АТФ сайте рецептора. Соответственно, недавно разработанный необратимый, селективный к мутациям ингибитор демонстрирует высокую активность против мутации T790M, но его эффективность может быть нейтрализована приобретенной мутацией C797, цистеинового остатка, который образует ключевую ковалентную связь.
Внутриклеточный EGFR2 человека, известный как HER2/neu или ErbB2, представляет собой рецептор тирозинкиназы, принадлежащий к семейству рецепторов эпидермального фактора роста человека (HER/EGFR/ERBB). Обычно он участвует в путях передачи сигнала и индуцирует рост и дифференциацию клеток. HER2 демонстрирует очень высокое структурное сходство с другими тремя членами семейства EGFR (EGFR, HER3 и HER4).
Однако, в отличие от других противораковых мишеней, в случае HER2 (эпидермального фактора роста человека 2) лиганд, который с ним связывается, еще неизвестен, и он сотрудничает с другими рецепторами HER, которые связываются с лигандом, образует гетеродимер и участвует в увеличении клеточного цикла, регуляции клеточной пролиферации, дифференциации и выживания посредством различных сигнальных путей. В случае антител, таргетирующих Her2, были разработаны и коммерчески доступны терапевтические агенты на основе антител типа IgG2. Основное терапевтическое действие заключается не в антителозависимой клеточной цитотоксичности (ADCC) или комплементзависимой цитотоксичности (CDC), а в нейтрализующей активности, обусловленной антителом.
Соответственно, хотя авторы настоящего изобретения пытались разработать противораковый терапевтический агент, который ингибирует множественные мутации EGFR, было обнаружено, что производное пиримидин-2,4-диамина по настоящему изобретению демонстрирует относительно низкое ингибирование EGFR дикого типа и высокую ингибирующую способность против мутации EGFR и мутации HER2, и, таким образом, его можно с успехом использовать для профилактики или лечения рака. На основании вышеизложенного, авторы настоящего изобретения завершили настоящее изобретение.
Подробное описание изобретения
Техническая проблема
Объектом настоящего изобретения является производное пиримидин-2,4-диамина.
Другим объектом настоящего изобретения является способ получения производного пиримидин-2,4-диамина.
Другим объектом настоящего изобретения является фармацевтическая композиция для профилактики или лечения рака, содержащая производное пиримидин-2,4-диамина в качестве активного ингредиента.
Другим объектом настоящего изобретения является создание функционального оздоровительного питания для профилактики или облегчения рака, содержащего производное пиримидин-2,4-диамина в качестве активного ингредиента.
Решение проблемы
Для достижения вышеуказанного объекта в соответствии с одним аспектом настоящего изобретения предложено соединение, представленное Формулой 1 ниже, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемая соль:
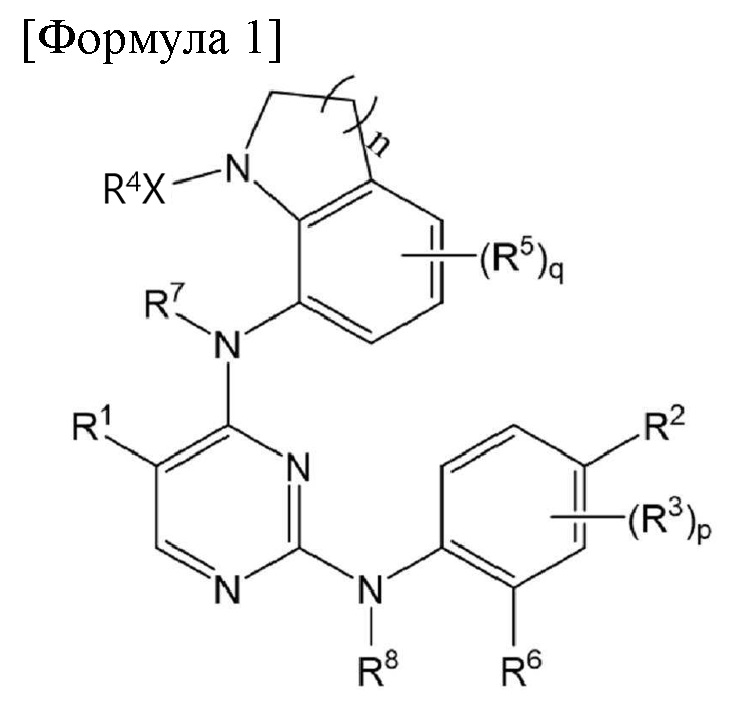
в Формуле 1 ,
n представляет собой целое число от 0 до 3;
X представляет собой сульфонил или карбонил;
R1 представляет собой водород, галоген, нитрил, C1-10 алкил с прямой или разветвленной цепью, незамещенный или замещенный одним или несколькими галогенами, карбоксилом или C1-10 алкоксикарбонилом с прямой или разветвленной цепью;
R2 представляет собой -ORa или -NRb1Rb2,
Ra представляет собой -(CH2)m-NRb1Rb2, где m представляет собой целое число от 1 до 3,
Rb1 и Rb2 каждый независимо представляет собой водород, незамещенный или замещенный C1-10 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, где заместитель замещенного C1-10 алкила с прямой или разветвленной цепью может представлять собой -NRc1Rc2, и заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-10 алкил с прямой или разветвленной цепью, и
Rc1 и Rc2 каждый независимо представляет собой водород, C1-10 алкил с прямой или разветвленной цепью, или Rc1 и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, где заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой C1-10 алкил с прямой или разветвленной цепью; и
R3, R5 и R6 каждый независимо представляет собой водород, галоген, C1-10алкокси с прямой или разветвленной цепью или C1-10 алкил с прямой или разветвленной цепью, незамещенный или замещенный одним или несколькими галогенами;
R4 представляет собой -NH2, C1-10 алкил с прямой или разветвленной цепью или C3-7 циклоалкил;
R7 и R8 каждый независимо представляет собой водород или C1-10 алкил с прямой или разветвленной цепью;
n представляет собой целое число от 0 до 3; и
p и q каждый независимо представляет собой целое число от 1 до 3.
Кроме того, настоящее изобретение предлагает способ получения соединения, представленного Формулой 1 по пункту 1, включающий: взаимодействие соединения, представленного Формулой 2, с соединением, представленным Формулой 3, с получением соединения, представленного Формулой 1, как показано на Схеме реакции 1 ниже:
[Схема реакции 1]
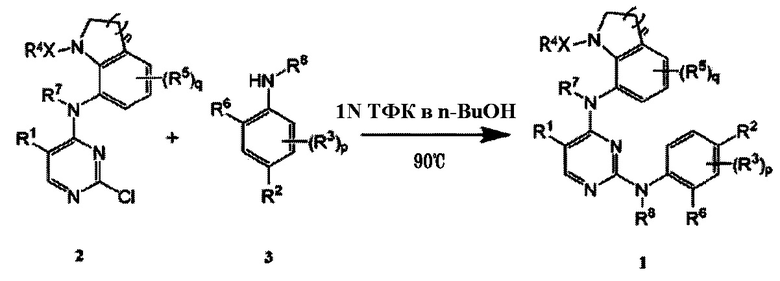
на Схеме реакции 1 выше, X, R1, R2, R3, R4, R5, R6, R7, R8, n, p и q такие, как определены в Формуле 1 по пункту 1.
Согласно другому аспекту настоящего изобретения, предложена фармацевтическая композиция для профилактики или лечения рака, содержащая соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль в качестве активного ингредиента.
Согласно другому аспекту настоящего изобретения, предложена функциональная оздоровительная продукция для профилактики или улучшения состояния рака, содержащая соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль в качестве активного ингредиента.
Согласно другому аспекту настоящего изобретения, предложен способ профилактики или лечения рака, включающий: введение фармацевтической композиции или функциональной оздоровительной продукции, содержащей соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль в качестве активного ингредиента, субъекту, нуждающегося в этом.
Согласно другому аспекту настоящего изобретения, предложено применение фармацевтической композиции или функциональной оздоровительной продукции, содержащей соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль, для профилактики или лечение рака.
Эффекты изобретения
Производное пиримидин-2,4-диамина по настоящему изобретению демонстрирует высокую ингибирующую способность против мутаций EGFR и HER2 и, таким образом, может быть успешно использовано при лечении рака, при котором возникли мутации EGFR и HER2. Кроме того, производное пиримидин-2,4-диамина демонстрирует значительный синергический эффект при комбинированном введении и, таким образом, может с успехом использоваться в комбинированной терапии.
Лучший способ осуществления изобретения
Далее настоящее изобретение будет описано подробно.
В одном аспекте настоящего изобретения, предложено соединение, представленное Формулой 1 ниже, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемая соль:
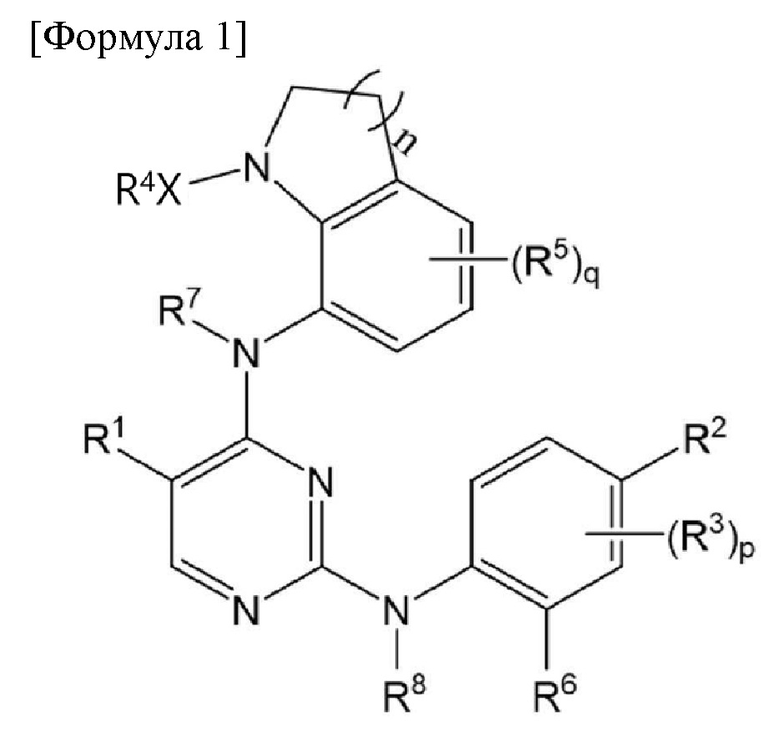
в Формуле 1 выше,
X представляет собой сульфонил или карбонил;
R1 представляет собой водород, галоген, нитрил, C1-10 алкил с прямой или разветвленной цепью незамещенный или замещенный одним или несколькими галогенами, карбоксил или C1-10алкоксикарбонил с прямой или разветвленной цепью;
R2 представляет собой -ORa или -NRb1Rb2,
Ra представляет собой -(CH2)m-NRb1Rb2, где m представляет собой целое число от 1 до 3,
Rb1 и Rb2 каждый независимо представляет собой водород, незамещенный или замещенный C1-10 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, где заместитель замещенного C1-10 алкила с прямой или разветвленной цепью может представлять собой -NRc1Rc2, и заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-10 алкил с прямой или разветвленной цепью, и
Rc1 и Rc2 каждый независимо представляет собой водород, C1-10 алкил с прямой или разветвленной цепью, или Rc1 и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, где заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой C1-10 алкил с прямой или разветвленной цепью; и
R3, R5 и R6 каждый независимо представляет собой водород, галоген, C1-10 алкокси с прямой или разветвленной цепью или C1-10 алкил с прямой или разветвленной цепью незамещенный или замещенный одним или несколькими галогенами;
R4 представляет собой -NH2, C1-10 алкил с прямой или разветвленной цепью или C3-7 циклоалкил;
R7 и R8 каждый независимо представляет собой водород или C1-10 алкил с прямой или разветвленной цепью;
n представляет собой целое число от 0 до 3; и
p и q каждый независимо представляет собой целое число от 1 до 3.
Кроме того, R1 может представлять собой водород, галоген, C1-3 алкил с прямой или разветвленной цепью незамещенный или замещенный одним или несколькими галогенами или C1-6алкоксикарбонил с прямой или разветвленной цепью;
R2 может представлять собой -NRb1Rb2,
Rb1 и Rb2 каждый независимо может представлять собой водород, незамещенный или замещенный C1-3 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 могут быть взяты вместе с азотом, к которому они присоединены, с образованием незамещенного или замещенного 5-6-членного гетероциклоалкила, где заместитель замещенного C1-3 алкила с прямой или разветвленной цепью может представлять собой -NRc1Rc2, и заместитель замещенного 5-6-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-3 алкил с прямой или разветвленной цепью, и
Rc1 и Rc2 каждый независимо может представлять собой водород, C1-3 алкил с прямой или разветвленной цепью, или Rc1 и Rc2 могут быть взяты вместе с азотом, к которому они присоединены, с образованием незамещенного или замещенного 5-6-членного гетероциклоалкила, где заместитель замещенного 5-6-членного гетероциклоалкил может представлять собой C1-3 алкил с прямой или разветвленной цепью; и
R3 и R5 каждый независимо может представлять собой водород или C1-3 алкил с прямой или разветвленной цепью незамещенный или замещенный одним или несколькими галогенами;
R4 может представлять собой -NH2, C1-3 алкил с прямой или разветвленной цепью или C4-6 циклоалкил;
R6 может представлять собой C1-3 алкокси с прямой или разветвленной цепью; и
n может представлять собой целое число от 1 до 3.
Кроме того, R1 может представлять собой водород, галоген, C1-2 алкил с прямой или разветвленной цепью незамещенный или замещенный одним или несколькими галогенами или C1-3 алкоксикарбонил с прямой или разветвленной цепью;
R2 может представлять собой -NRb1Rb2,
Rb1 и Rb2 каждый независимо может представлять собой водород, незамещенный или замещенный C1-2 алкил, или Rb1 и Rb2 могут быть взяты вместе с азотом, к которому они присоединены, с образованием незамещенного или замещенного 6-членного гетероциклоалкила, где заместитель замещенного C1-2 алкила может представлять собой -NRc1Rc2, и заместитель замещенного 6-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-2 алкил, и
Rc1 и Rc2 каждый независимо может представлять собой водород, C1-3 алкил с прямой или разветвленной цепью, или Rc1 и Rc2 могут быть взяты вместе с азотом, к которому они присоединены, с образованием незамещенного или замещенного 6-членного гетероциклоалкила, где заместитель замещенного 6-членного гетероциклоалкила может представлять собой C1-2 алкил; и
R3 и R5 каждый независимо может представлять собой водород или C1-2 алкил;
R4 может представлять собой C1-2 алкил;
R6 может представлять собой метокси;
R7 и R8 каждый независимо может представлять собой водород или C1-2 алкил;
n может представлять собой 1 или 2; и
p и q каждый независимо может представлять собой целое число от 1 до 3.
Кроме того, R1 может представлять собой метил, F, Cl, CF3, 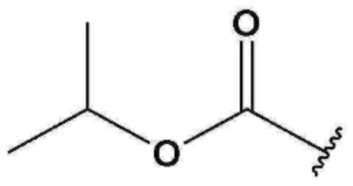 или
или 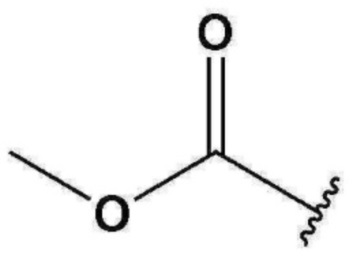 ;
;
R2 может представлять собой 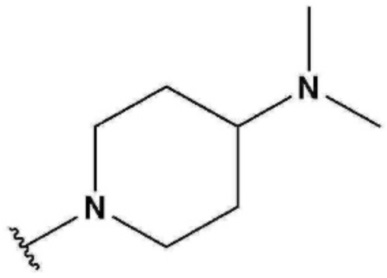 ,
, 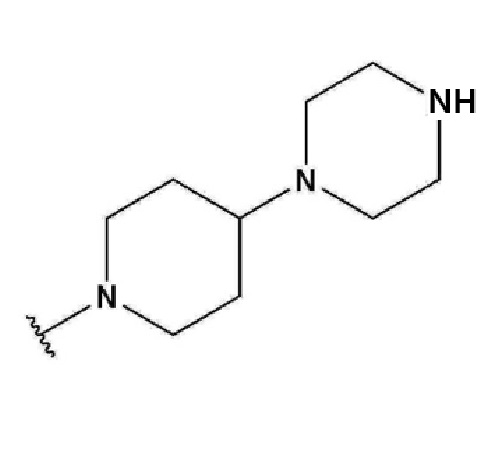 ,
, 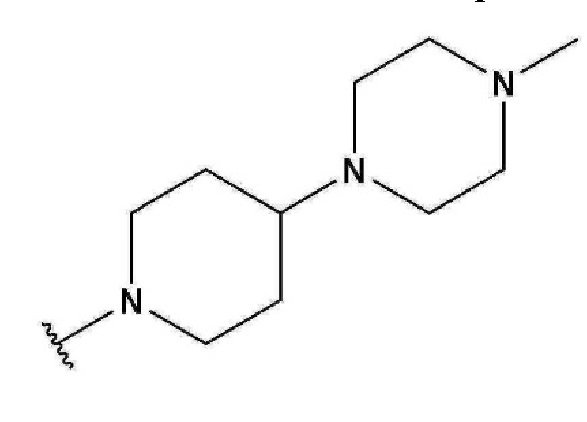 или
или  ;
;
R3 может представлять собой водород;
R4 может представлять собой метил или этил;
R5 может представлять собой водород;
R6 может представлять собой метокси;
R7 может представлять собой водород;
R8 может представлять собой водород;
n может представлять собой 1 или 2; и
p и q каждый независимо может представлять собой целое число от 1 до 3.
Термин «гетероциклоалкил», если не указано иное, включает одновалентную насыщенную группу, состоящую из 1-3 колец, содержащих 1, 2, 3 или 4 гетероатома, выбранных из N, O или S. Два или три кольца могут содержать мостиковый, конденсированный или спиро гетероциклоалкил, и могут представлять собой, например, пиридин, пиразин, пиримидин, пиридазин, пиперазин, пиперидин, пиразол, оксазол, тиазол или морфолин.
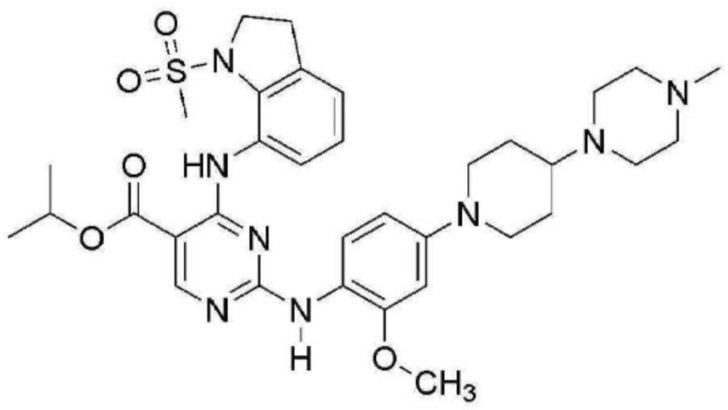
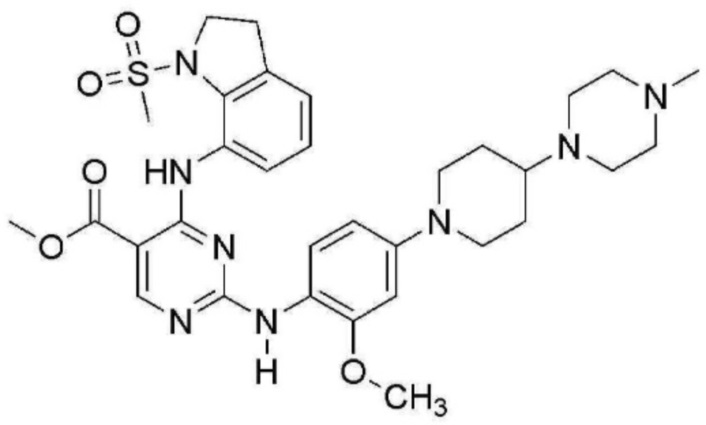
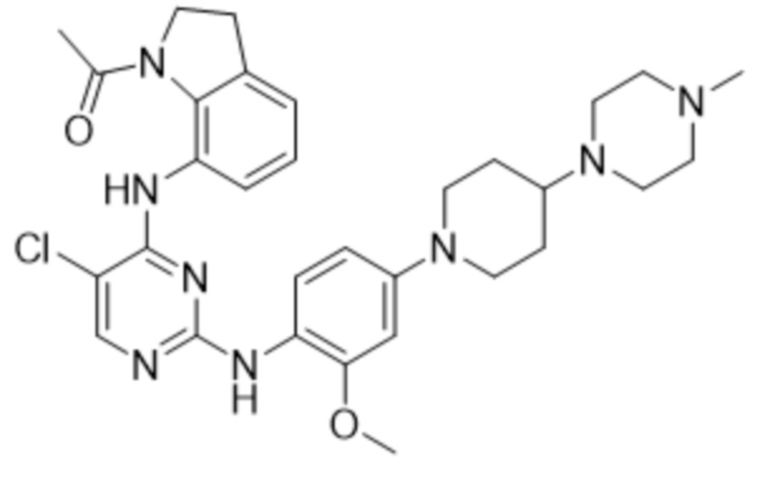
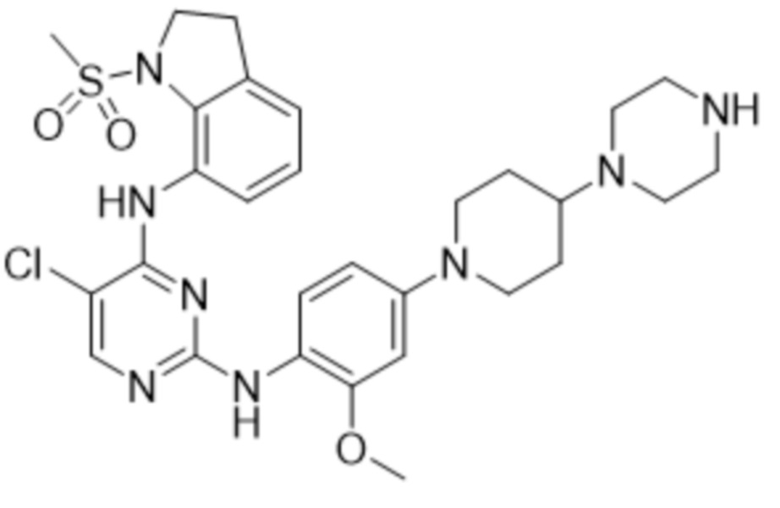
Примеры соединения, представленного Формулой 1, по настоящему изобретению, могут включать следующие соединения:
<1> 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<2> 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<3> 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<4> 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<5> 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<6> 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<7> N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-5-метил-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<8> 5-фтор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<9> N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)-5-(трифторметил)пиримидин-2,4-диамин;
<10> изопропил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилат;
<11> метил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилат;
<12> 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)этан-1-он;
<13> 5-хлор-N4-(1-(этилсульфонил)индолин-7-ил)-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пиримидин-2,4-диамин;
<14> 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)пропан-1-он;
<15> 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)этан-1-он;
<16> 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)пропан-1-он; и
<17> 5-хлор-N2-(2-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин.
Соединение, представленное Формулой 1 по настоящему изобретению, можно использовать в форме фармацевтически приемлемой соли, и в качестве соли полезна кислотно-аддитивная соль, образованная фармацевтически приемлемой свободной кислотой. Кислотно-аддитивные соли получают из неорганических кислот, таких как хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, иодистоводородная кислота, азотистая кислота, фосфористая кислота и подобные, нетоксичных органических кислот, таких как алифатические моно- и дикарбоксилаты, фенилзамещенный алканоат, гидроксиалканоат и алкандиоат, ароматические кислоты, алифатические и ароматические сульфоновые кислоты, и органических кислот, таких как трифторуксусная кислота, ацетат, бензойная кислот, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, метансульфоновая кислота, 4-толуолсульфоновая кислота, винная кислота, фумаровая кислота и подобные. Эти фармацевтически нетоксичные соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфатхлорид, бромид, йодид, фторид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себацинат, фумарат, малеат, бутин-1,4-диоат, гексан-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, бензолсульфонат, толуолсульфонат, хлорбензолсульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, β-гидроксибутират, гликолат, малат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и тому подобные.
Кислотно-аддитивную соль по настоящему изобретению можно получить обычными способами. Кислотно-аддитивную соль по настоящему изобретению можно получить, например, путем растворения производного Формулы 1 в органическом растворителе, таком как метанол, этанол, ацетон, метиленхлорид, ацетонитрил и подобные, добавления органической кислоты или неорганической кислоты, фильтрования и сушки полученного осадка, или ее можно получить путем дистилляции растворителя и избытка кислоты при пониженном давлении, затем сушки и кристаллизации в органическом растворителе.
Кроме того, фармацевтически приемлемая соль металла может быть получена с использованием основания. Соль щелочного металла или щелочноземельного металла получают, например, растворением соединения в избытке раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, фильтрованием нерастворенной соли соединения, выпариванием и сушкой фильтрата. В этом случае, фармацевтически целесообразно приготовить соль натрия, калия или кальция в виде соли металла. Кроме того, соответствующую соль получают путем взаимодействия соли щелочного или щелочноземельного металла с подходящей отрицательной солью (например, нитратом серебра).
Кроме того, настоящее изобретение включает не только соединение, представленное Формулой 1, и его фармацевтически приемлемую соль, но также сольват, стереоизомер, гидрат и подобные, которые могут быть получены из него.
Термин «гидрат» относится к соединению по настоящему изобретению или его соли, содержащей стехиометрическое или не стехиометрическое количество воды, связанной нековалентными межмолекулярными силами. Гидрат соединения, представленного Формулой 1 по настоящему изобретению, может содержать стехиометрическое или не стехиометрическое количество воды, связанной нековалентными межмолекулярными силами. Гидрат может содержать более 1 эквивалента воды, предпочтительно, от 1 до 5 эквивалентов воды. Гидрат можно получить кристаллизацией соединения, представленного Формулой 1 по настоящему изобретению, его стереоизомера или его фармацевтически приемлемой соли из воды или водосодержащего растворителя.
Термин «сольват» относится к соединению по настоящему изобретению или его соли, содержащей стехиометрическое или не стехиометрическое количество растворителя, связанного нековалентными межмолекулярными силами. Предпочтительные растворители для этого включают растворители, которые являются летучими, нетоксичными и/или пригодными для введения людям.
Термин «изомер» относится к соединению по настоящему изобретению или его соли, которое имеет ту же химическую формулу или молекулярную формулу, но отличается структурно или пространственно. Эти изомеры включают структурные изомеры, такие как таутомеры, стереоизомеры, такие как R или S изомеры с асимметричными углеродными центрами, геометрические изомеры (транс, цис) и оптические изомеры (энантиомеры). Все эти изомеры и их смеси также включены в объем настоящего изобретения.
В другом аспекте настоящего изобретения, предложена фармацевтическая композиция для профилактики или лечения рака, содержащая соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль в качестве активного ингредиента.
В этом случае, рак представляет собой по меньшей мере один рак, выбранный из группы, состоящей из псевдомиксомы, рака внутрипеченочных желчных путей, гепатобластомы, рака печени, рака щитовидной железы, рака толстой кишки, рака яичек, миелодиспластического синдрома, глиобластомы, рака полости рта, рака губы, грибовидного микоза, острого миелоидного лейкоза, острого лимфоцитарного лейкоза, базальноклеточного рака, рака эпителия яичников, герминогенного рака яичников, рака молочной железы, рака головного мозга, аденомы гипофиза, множественной миеломы, рака желчного пузыря, рака желчевыводящих путей, рака толстой кишки, хронического миелоидного лейкоза, хронического лимфоцитарного лейкоза, ретинобластомы, меланомы хориоидеи, рака фатерового соска, рака мочевого пузыря, рака брюшины, рака паращитовидной железы, рака надпочечников, рака придаточных пазух носа, немелкоклеточного рака легкого, рака языка, астроцитомы, мелкоклеточного рака легкого, детского рака головного мозга, детской лимфомы, детского лейкоза, рака тонкой кишки, менингиомы, рака пищевода, глиомы, рака почечной лоханки, рака почки, рака сердца, рака двенадцатиперстной кишки, злокачественного рака мягких тканей, злокачественного рака кости, злокачественной лимфомы, злокачественной мезотелиомы, злокачественной меланомы, рака глаза, рака вульвы, рака мочеточника, рака уретры, рака неизвестной первичной локализации, лимфомы желудка, рака желудка, карциноида желудка, стромального рака желудочно-кишечного тракта, рака Вильмса, рака молочной железы, саркомы, рака полового члена, рака глотки, гестационной трофобластической болезни, рака шейки матки, рака эндометрия рак, саркомы матки, рака предстательной железы, метастатического рака кости, метастатического рака головного мозга, рака средостения, рака прямой кишки, карциноида прямой кишки, рака влагалища, рака спинного мозга, акустической невромы, рака поджелудочной железы, рака слюнной железы, саркомы Капоши, болезни Педжета, рака небных миндалин, плоскоклеточного рака, аденокарциномы легких, рака легких, плоскоклеточного рака легких, рака кожи, анального рака, рабдомиосаркомы, рака гортани, рака плевры, рака крови и рака тимуса, и рак может представлять собой рак, экспрессирующий мутацию, по меньшей мере одну, выбранную из группы, состоящей из EGFR, HER2, ALK, FAK, FLT3, JAK3, KIT и PLK4.
Кроме того, соединение, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемая соль могут ингибировать мутацию EGFR (рецептора эпидермального фактора роста), где мутация EGFR может представлять собой по меньшей мере одну мутацию, выбранную из группы, состоящей из инсерционной мутации EGFR del19, EGFR T790M, EGFR C797S, EGFR L858R и EGFR Ex20, и предпочтительно, она может представлять собой по меньшей мере одну, выбранную из группы, состоящей из EGFR del19, EGFR del19/T790M, EGFR del19/T790M/C797S, EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S, EGFR A763_Y764insFHEA, EGFR V769_D770insASV и EGFR D770_N771insSVD и т. д.
Кроме того, соединение, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемая соль могут ингибировать мутацию HER2, где мутация HER2 может представлять собой по меньшей мере одну, выбранную из группы, состоящей из HER2 A775_G776insYVMA, HER2 G776_delinsVC и т. д.
Кроме того, фармацевтическая композиция для профилактики или лечения рака, содержащая соединение, представленное Формулой 1, его стереоизомер или его фармацевтически приемлемую соль в качестве активного ингредиента, может вводиться в качестве индивидуального терапевтического агента или в комбинации с другими применяемыми противораковыми агентами.
Кроме того, фармацевтическая композиция для профилактики или лечения рака, содержащая соединение, представленное Формулой 1, его стереоизомер или его фармацевтически приемлемую соль в качестве активного ингредиента, может усиливать противораковый эффект путем введения ее в комбинации с противораковым агентом.
Соединение, представленное Формулой 1 по настоящему изобретению, демонстрирует относительно слабое ингибирующее действие на активность EGFR против EGFR дикого типа, одновременно демонстрируя высоко селективное ингибирующее действие против мутаций EGFR и HER2 и особенно высокую ингибирующую способность против тройной мутации EGFR del19/T790M/C797S или EGFR L858R/T790M/C797S, и инсерционной мутации Ex20 EGFR и/или мутации HER2.
Можно видеть, что соединение, представленное Формулой 1 по настоящему изобретению, при отдельном введении, имеет более низкую степень выживаемости клеток в отношении клеточной линии Ba/F3 Del19/T790M/C797S с тройной мутацией EGFR, чем обычное лекарственное средство. Кроме того, соединение, представленное Формулой 1 по настоящему изобретению, имеет значительно сниженную степень выживаемости клеток при введении в комбинации с традиционным лекарственным средством по сравнению с отдельным введением. Таким образом, можно видеть, что соединение, представленное Формулой 1 по настоящему изобретению, не только обладает превосходной способностью уничтожать раковые клетки в отношении клеточных линий с тройной мутацией EGFR, инсерционной мутацией Ex20 EGFR и мутацией HER2 при введении отдельно, но и также оказывает значительно усиленный противораковый эффект при введении в комбинации с обычным лекарственным средством.
Таким образом, соединение, представленное Формулой 1 по настоящему изобретению, демонстрирует более высокую ингибирующую способность против мутаций EGFR и HER2, чем EGFR дикого типа, и, в частности, демонстрирует значительно более высокий ингибирующий эффект против мутации EGFR в EGFR L858R/T790M/C797S, и может быть успешно использован при лечении рака, экспрессирующего мутации EGFR del19, EGFR del19/T790M, EGFR del19/T790M/C797S, EGFR L858R, EGFR L858R/T790MS, EGFR L858R/T790M/C797S, EGFR A763_Y764insFHEA, EGFR V769_D770insASV и EGFR D770_N771insSVD и т.д. В частности, соединение, представленное Формулой 1 по настоящему изобретению, обладает значительной превосходной ингибирующей способностью против тройной мутации EGFR del19/T790M/C797S или EGFR L858R/T790M/C797S и, таким образом, также может быть успешно использовано при лечении рака, экспрессирующего EGFR del19/T790M/C797S или EGFR L858R/T790M/C797S, EGFR A763_Y764insFHEA, EGFR V769_D770insASV и EGFR D770_N771insSVD.
Кроме того, при введении в сочетании с традиционным лекарственным средством, соединение, представленное Формулой 1 по настоящему изобретению, демонстрирует синергический эффект и, таким образом, может быть успешно использовано в комбинации с традиционным лекарственным средством.
Соединение, представленное Формулой 1, или его фармацевтически приемлемую соль можно вводить в различных пероральных и парентеральных дозированных формах во время клинического введения. При составлении, его готовят с использованием разбавителей или эксципиентов, таких как обычно используемые наполнители, экстендеры, связующие агенты, смачивающие агенты, разрыхлители и поверхностно-активные вещества. Твердые препараты для перорального введения включают таблетки, пилюли, порошки, гранулы, капсулы и подобные. Эти твердые препараты готовят путем смешивания одного или нескольких соединений по меньшей мере с одним или несколькими эксципиентами, такими как крахмал, карбонат кальция, сахароза или лактоза, желатин и т. д. Помимо простых эксципиентов также используются смазывающие агенты, такие как стеарат магния и тальк. Жидкие препараты для перорального применения включают суспензии, растворы для внутреннего применения, эмульсии и сиропы. В дополнение к обычно используемым простым разбавителям, таким как вода и жидкий парафин, могут быть включены различные эксципиенты, такие как смачивающие агенты, подсластители, ароматизаторы и консерванты. Препараты для парентерального введения включают стерилизованные водные растворы, неводные растворители, суспензии и эмульсии. Неводные растворители и суспензии могут включать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, и сложные эфиры для инъекций, такие как этилолеат.
Фармацевтическую композицию, содержащую соединение, представленное Формулой 1, или его фармацевтически приемлемую соль в качестве активного ингредиента, можно вводить парентерально, и парентеральное введение осуществляют посредством подкожной инъекции, внутривенной инъекции, внутримышечной инъекции или внутригрудной инъекции.
В этом случае, чтобы составить состав для парентерального введения, соединение, представленное Формулой 1, или его фармацевтически приемлемую соль смешивают с водой вместе со стабилизатором или буфером для приготовления раствора или суспензии, которую можно приготовить в стандартной дозированной форме в ампуле или флаконе. Композиция может быть стерилизована и/или может содержать вспомогательные вещества, такие как консерванты, стабилизаторы, смачивающие агенты или ускорители эмульгирования, соли и/или буферы для регулирования осмотического давления и другие терапевтически полезные вещества, и может быть составлена в соответствии с обычными методами, такими как смешивание, гранулирование или покрытие.
Составы для перорального введения включают, например, таблетки, пилюли, твердые/мягкие капсулы, растворы, суспензии, эмульсии, сиропы, гранулы, эликсиры, пастилки и подобные. В дополнение к активному ингредиенту, эти составы содержат разбавители (например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин), смазывающие агенты (например, диоксид кремния, тальк, стеариновую кислоту и ее магниевые или кальциевые соли и/или полиэтиленгликоль). Таблетки могут содержать связующие агенты, такие как алюмосиликат магния, крахмальную пасту, желатин, метилцеллюлозу, карбоксиметилцеллюлозу натрия и/или поливинилпирролидин, и в некоторых случаях, могут содержать разрыхлители или шипучие смеси, такие как крахмал, агар, альгиновая кислота или ее натриевая соль, и/или абсорбенты, красители, ароматизаторы и подсластители.
В другом аспекте настоящего изобретения, предложена функциональная оздоровительная продукция для профилактики или улучшения состояния рака, содержащая соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль в качестве активного ингредиента.
Рак может представлять собой по меньшей мере один, выбранный из группы, состоящей из псевдомиксомы, рака внутрипеченочных желчных путей, гепатобластомы, рака печени, рака щитовидной железы, рака толстой кишки, рака яичек, миелодиспластического синдрома, глиобластомы, рака полости рта, рака губы, грибовидного микоза, острого миелоидного лейкоза, острого лимфоцитарного лейкоза, базальноклеточного рака, рака эпителия яичников, герминогенного рака яичников, рака молочной железы, рака головного мозга, аденомы гипофиза, множественной миеломы, рака желчного пузыря, рака желчевыводящих путей, рака толстой кишки, хронического миелоидного лейкоза, хронического лимфоцитарного лейкоза, ретинобластомы, меланомы хориоидеи, рака фатерового соска, рака мочевого пузыря, рака брюшины, рака паращитовидной железы, рака надпочечников, рака придаточных пазух носа, немелкоклеточного рака легкого, рака языка, астроцитомы, мелкоклеточного рака легкого, детского рака головного мозга, детской лимфомы, детского лейкоза, рака тонкой кишки, менингиомы, рака пищевода, глиомы, рака почечной лоханки, рака почки, рака сердца, рака двенадцатиперстной кишки, злокачественного рака мягких тканей, злокачественного рака кости, злокачественной лимфомы, злокачественной мезотелиомы, злокачественной меланомы, рака глаза, рака вульвы, рака мочеточника, рака уретры, рака неизвестной первичной локализации, лимфомы желудка, рака желудка, карциноида желудка, стромального рака желудочно-кишечного тракта, рака Вильмса, рака молочной железы, саркомы, рака полового члена, рака глотки, гестационной трофобластической болезни, рака шейки матки, рака эндометрия рак, саркомы матки, рака предстательной железы, метастатического рака кости, метастатического рака головного мозга, рака средостения, рака прямой кишки, карциноида прямой кишки, рака влагалища, рака спинного мозга, акустической невромы, рака поджелудочной железы, рака слюнной железы, саркомы Капоши, болезни Педжета, рака небных миндалин, плоскоклеточного рака, аденокарциномы легких, рака легких, плоскоклеточного рака легких, рака кожи, анального рака, рабдомиосаркомы, рака гортани, рака плевры, рака крови и рака тимуса, и рак может представлять собой рак, экспрессирующий мутации в EGFR и HER2.
Соединение, представленное Формулой 1 по настоящему изобретению, демонстрирует высокую ингибирующую способность против мутаций EGFR и HER2 и, таким образом, может быть добавлено в функциональные продукты питания, такие как продукты питания и напитки, в качестве функциональной пищевой композиции для профилактики или улучшения рака.
Соединение, представленное Формулой 1 по настоящему изобретению, может быть добавлено в пищу как есть или использовано вместе с другими продуктами питания или пищевыми ингредиентами, и может быть использовано соответствующим образом в соответствии с обычными способами. Количество смешиваемого активного ингредиента может быть соответствующим образом определено в зависимости от цели применения (профилактика или улучшение). В общем, вышеуказанное соединение в оздоровительном пищевом продукте может быть добавлено в количестве от 0,1 до 90 массовых частей от общей массы пищевого продукта. Однако в случае длительного приема с целью поддержания здоровья и гигиены или контроля за здоровьем, указанное количество может быть ниже указанного выше диапазона, и, поскольку проблем с точки зрения безопасности нет, активный ингредиент можно использовать в количестве выше вышеуказанного диапазона.
Кроме того, композиция функционального оздоровительного напитка по настоящему изобретению не имеет особых ограничений в отношении других ингредиентов, кроме содержания вышеуказанного соединения в качестве активного ингредиента в указанной пропорции, и может содержать различные ароматизаторы или натуральные углеводы в качестве дополнительных ингредиентов, как и обычные напитки. Примеры вышеупомянутых природных углеводов включают моносахариды, такие как глюкоза, фруктоза и т.д.; дисахариды, такие как мальтоза, сахароза и т.д.; и полисахариды, такие как обычные сахара, такие как декстрин и циклодекстрин, и сахарные спирты, такие как ксилит, сорбит и эритрит. В дополнение к упомянутым выше, в качестве вкусовых добавок можно успешно использовать натуральные вкусовые добавки (тауматин, экстракт стевии (например, ребаудиозид А, глицирризин и т.д.)) и синтетические вкусовые добавки (сахарин, аспартам и т.д.). Доля природных углеводов обычно составляет примерно от 1 до 20 г, предпочтительно, примерно от 5 до 12 г на 100 г композиции по настоящему изобретению.
Кроме того, в дополнение к вышесказанному, соединение, представленное Формулой 1 по настоящему изобретению, может содержать различные питательные вещества, витамины, минералы (электролиты), ароматизаторы, такие как синтетические и натуральные ароматизаторы, красители и усилители (сыр, шоколад, и т.д.), пектиновую кислоту и ее соли, альгиновую кислоту и ее соли, органические кислоты, защитные коллоидные загустители, регуляторы pH, стабилизаторы, консерванты, глицерин, спирт, карбонизаторы, используемые в газированных напитках, и подобные. Кроме того, соединение, представленное Формулой 1 по настоящему изобретению, может содержать мякоть для производства натуральных фруктовых соков, напитков из фруктовых соков и овощных напитков.
В другом аспекте настоящего изобретения, предложен способ профилактики или лечения рака, включающий: введение фармацевтической композиции или функциональной оздоровительной продукции, содержащей соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат, или его фармацевтически приемлемую соль в качестве активного ингредиента, субъекту, нуждающемуся в этом.
В другом аспекте настоящего изобретения, предложено применение фармацевтической композиции или функциональной оздоровительной продукции, содержащей соединение, представленное Формулой 1, его стереоизомер, его сольват, его гидрат или его фармацевтически приемлемую соль, для профилактики или лечения рака.
Фармацевтическую композицию по настоящему изобретению вводят в «фармацевтически эффективном количестве». В настоящем документе, термин «фармацевтически эффективное количество» относится к количеству, достаточному для лечения заболевания с разумным соотношением польза/риск, применимому для медицинского лечения или улучшения, и уровень эффективной дозы может быть определен на основе факторов, включая тип и тяжесть заболевания субъекта, возраст, пол, активность лекарственного средства, чувствительность к лекарственному средству, время введения, путь введения и скорость выведения, продолжительность лечения, одновременно применяемые лекарственные средства и другие факторы, хорошо известные в области медицины. Например, включено эффективное количество от 0,001 мг/кг до 1000 мг/кг, от 0,01 мг/кг до 100 мг/кг или от 0,1 до 20 мг/кг или от 0,1 до 500 мг/кг. Верхний количественный предел фармацевтической композиции по настоящему изобретению может быть выбран и реализован специалистом в данной области техники в соответствующем диапазоне.
Способ осуществления изобретения
Далее настоящее изобретение будет подробно описано посредством примеров и экспериментальных примеров.
Однако следующие примеры и экспериментальные примеры являются только иллюстративными для настоящего изобретения, и содержание настоящего изобретения не ограничено следующими примерами и экспериментальными примерами.
[Схема реакции A]
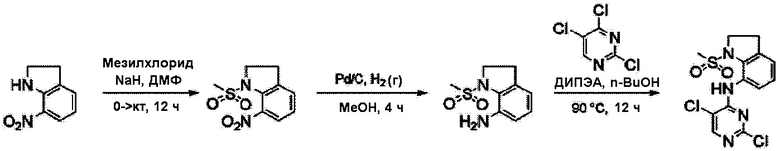
Согласно Схеме реакции A выше, получают соединения Примеров получения 1-3.
<Пример получения 1> Получение 1-(метилсульфонил)-7-нитроиндолина
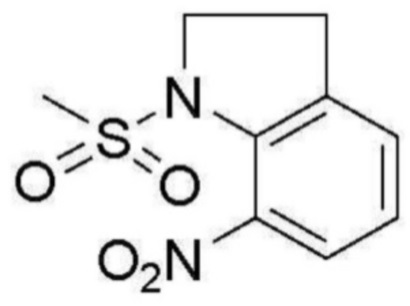
К перемешиваемому раствору 7-нитроиндолина (2,0 г, 1,21 ммоль) в ДМФ добавляют NaH (0,8 г, 3,6 ммоль) и мезилхлорид (4,7 мл, 6,0 ммоль) при 0°C. Полученную смесь нагревают до комнатной температуры и перемешивают в течение 12 часов. Реакционную смесь гасят холодной водой. Выпавшее в осадок твердое вещество фильтруют, промывают водой и сушат с получением 1-(метилсульфонил)-7-нитроиндолина (87%) в виде чистого желтого твердого вещества.
1H ЯМР (500 MГц, Хлороформ-d) δ 7,79 (д, J=8,3 Гц, 1 H), 7,57-7,48 (м, 1 H), 7,22 (т, J=7,8 Гц, 1 H), 4,29 (т, J=7,8 Гц, 2 H), 3,26 (с, 3 H), 3,22 (т, J=7,8 Гц, 2 H); ЖХМС: 243,0 [M+H+].
<Пример получения 2> Получение 1-(метилсульфонил)индолин-7-амина
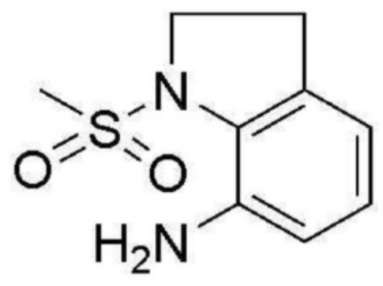
К перемешиваемому раствору 1-(метилсульфонил)-7-нитроиндолина (2,2 г, 9,0 ммоль) в метаноле добавляют 10% Pd/C (0,1 г, 0,9 ммоль). Полученную смесь перемешивают под 1 атм. газообразного водорода в течение 4 часов при комнатной температуре. Реакционную смесь фильтруют через Целит и концентрируют при пониженном давлении с получением 1-(метилсульфонил)индолин-7-амина (90%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 7,00 (тд, J=7,8, 2,3 Гц, 1 H), 6,70 (д, J=7,5 Гц, 1 H), 6,61 (дд, J=8,1, 2,3 Гц, 1 H), 4,60 (с, 2 H), 4,11 (тд, J=7,8, 2,3 Гц, 2 H), 3,03 (тд, J=7,6, 2,3 Гц, 2 H), 2,87 (с, 3H); ЖХМС: 213,0 [M+H+].
<Пример получения 3> Получение N-(2,5-дихлорпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина
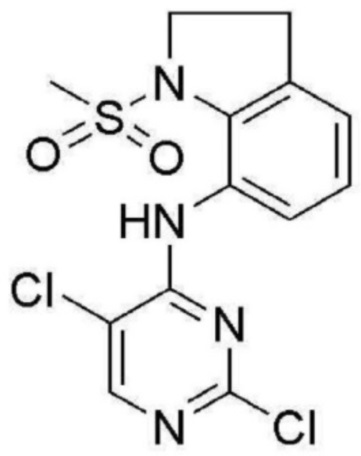
К перемешиваемому раствору 1-(метилсульфонил)индолин-7-амина (500 мг, 2,35 ммоль) в n-BuOH (5 мл) добавляют диизопропилэтиламин (1,3 мл, 7,05 ммоль) и 2,4,5-трихлорпиримидин (518 мг, 2,82 ммоль) при комнатной температуре. Реакционную смесь нагревают до 90°C в течение 14 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. Полученный продукт фильтруют и промывают n-BuOH с получением N-(2,5-дихлорпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина в виде не совсем белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 9,35 (с, 1 H), 8,21 (с, 1 H), 8,01 (д, J=8,4 Гц, 1 H), 7,42-7,26 (м, 2 H), 7,18 (д, J=7,5 Гц, 1 H), 4,16 (тд, J=7,5, 2,2 Гц, 2 H), 3,15 (т, J=7,7 Гц, 2 H), 2,92 (с, 3 H); ЖХМС: 359,8 [M+H+].
[Схема реакции B]
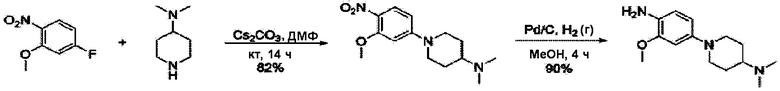
Согласно Схеме реакции B выше, получают соединения Примеров получения 4 и 5.
<Пример получения 4> Получение (3-метокси-4-нитрофенил)-N, N-диметилпиперидин-4-амина
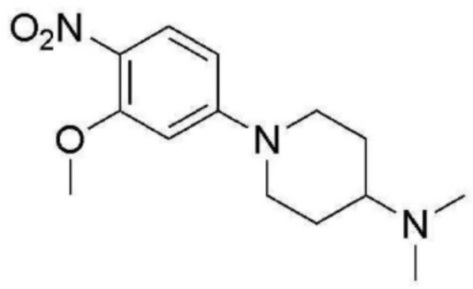
К перемешиваемому раствору 4-фтор-2-метокси-1-нитробензола (10 г, 58,4 ммоль) в ДМФ (150 мл) добавляют N, N-диметилпиперидин-4-амин (7,5 г, 58,4 ммоль) и карбонат цезия (3,8 г, 116,8 моль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь гасят холодной водой и перемешивают в течение 15 минут. Выпавшее в осадок твердое вещество фильтруют и концентрируют при пониженном давлении с получением 1-(3-метокси-4-нитрофенил)-N, N-диметилпиперидин-4-амина (14,0 г, 85%) в виде желтого твердого вещества.
1H ЯМР (500 MГц, Хлороформ-d) δ 8,01 (д, J=9,4 Гц, 1 H), 6,44 (дд, J=9,4, 2,6 Гц, 1 H), 6,33 (д, J=2,5 Гц, 1 H), 3,97 (с, 3 H), 3,97-3,86 (м, 2 H), 3,03-2,94 (м, 2 H), 2,50-2,36 (м, 1 H), 2,33 (с, 6 H), 2,04-1,89 (м, 2 H), 1,68-1,51 (м, 2 H); ЖХМС: 279,0 [M+H+].
<Пример получения 5> Получение 1-(4-амино-3-метоксифенил)-N, N-диметилпиперидин-4-амина
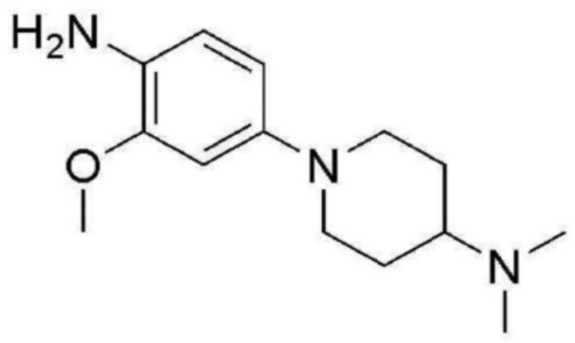
К перемешиваемому раствору 1-(3-метокси-4-нитрофенил)-N, N-диметилпиперидин-4-амина (14,0 г, 50,1 ммоль) в метаноле (150 мл) добавляют 10% Pd/C (1,0 г, 10,0 ммоль). Полученную смесь перемешивают под 1 атм. газообразного водорода при комнатной температуре в течение 4 часов. Реакционную смесь фильтруют через Целит и концентрируют при пониженном давлении с получением 1-(4-амино-3-метоксифенил)-N, N-диметилпиперидин-4-амина (11,2 г, 90%) в виде коричневого твердого вещества.
1H ЯМР (500 MГц, Хлороформ-d) δ 6,65 (д, J=8,3 Гц, 1 H), 6,55 (д, J=2,4 Гц, 1 H), 6,45 (дд, J=8,4, 2,5 Гц, 1 H), 3,86 (с, 3 H), 3,57-3,50 (м, 4 H), 2,67-2,60 (м, 2H), 2,34 (с, 6H), 2,89-2,19 (м, 1 H), 1,96-1,90 (м, 2 H); ЖХМС: 249,0 [M+H+].
[Схема реакции C]

Согласно Схеме реакции C выше, получают соединения Примеров получения 6 и 7.
<Пример получения 6> Получение N1-(3-метокси-4-нитрофенил)-N1,N2,N2-триметилэтан-1,2-диамина
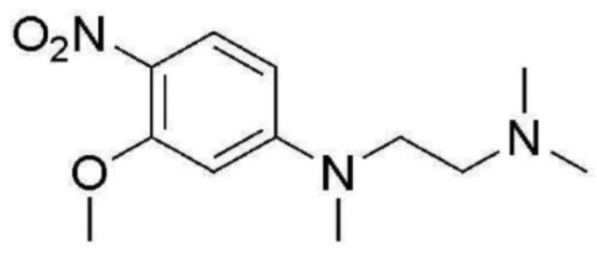
Применяют тот же способ, как в Примере получения 4, с получением N1-(3-метокси-4-нитрофенил)-N1,N2,N2-триметилэтан-1,2-диамина (84%).
1H ЯМР (500 MГц, Хлороформ-d) δ 8,03 (д, J=9,3 Гц, 1 H), 6,25 (дд, J=9,4, 2,6 Гц, 1 H), 6,13 (д, J=2,6 Гц, 1 H), 3,96 (с, 3 H), 3,59-3,53 (м, 2 H), 3,11 (с, 3 H), 2,53 (т, J=7,3 Гц, 2 H), 2,32 (с, 6 H); ЖХМС: 253,0 [M+H+].
<Пример получения 7> Получение N1-(2-(диметиламино)этил)-3-метокси-N1-метилбензол-1,4-диамина
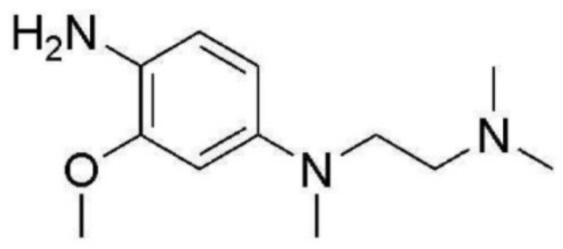
Применяют тот же способ, как в Примере получения 5, с получением N1-(2-(диметиламино)этил)-3-метокси-N1-метилбензол-1,4-диамина (96%).
1H ЯМР (400 MГц, Метанол-d4) δ 7,23 (д, J=8,7 Гц, 1 H), 6,61 (д, J=2,6 Гц, 1 H), 6,52 (дд, J=8,8, 2,7 Гц, 1 H), 4,02 (с, 3 H), 3,84 (т, J=7,3 Гц, 2 H), 3,43-3,35 (м, 5 H), 3,06 (с, 3 H), 2,98 (с, 6 H); ЖХМС: 223,0 [M+H+].
[Схема реакции D]
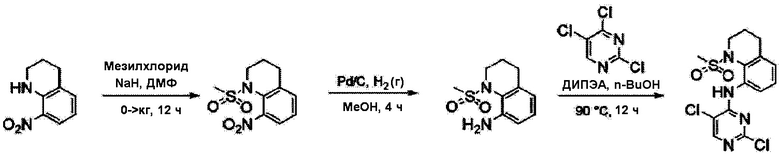
Согласно Схеме реакции D выше, получают соединения Примеров получения 8-10.
<Пример получения 8> Получение 1-(метилсульфонил)-8-нитро-1,2,3,4-тетрагидрохинолина
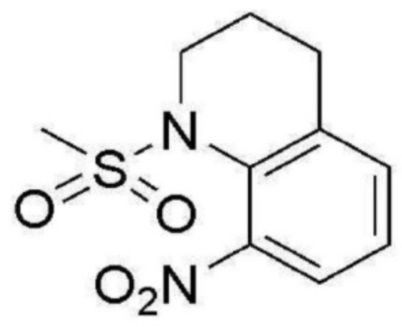
Применяют тот же способ, как в Примере получения 1, с получением 1-(метилсульфонил)-8-нитро-1,2,3,4-тетрагидрохинолина (69%).
1H ЯМР (400 MГц, Хлороформ-d) δ 7,80-7,73 (м, 1 H), 7,40 (д, J=7,8 Гц, 1 H), 7,25 (дкв, J=7,4, 3,5, 2,5 Гц, 1 H), 3,72 (с, 2 H), 3,14 (с, 3 H), 2,95 (тд, J=7,3, 2,2 Гц, 2 H), 2,23 (с, 2 H), 1,43-1,34 (м, 2 H); ЖХМС: 257,0 [M+H+].
<Пример получения 9> Получение 1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-амина
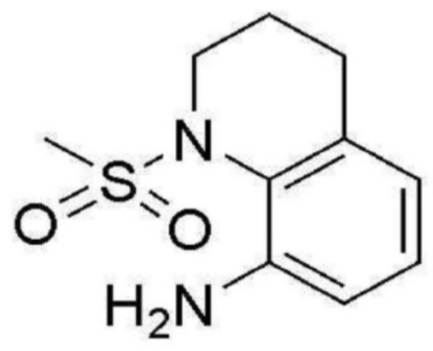
Применяют тот же способ, как в Примере получения 2, с получением 1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-амина (92%).
1H ЯМР (400 MГц, Хлороформ-d) δ 7,03 (тд, J=7,8, 2,2 Гц, 1 H), 6,66 (д, J=8,1 Гц, 1 H), 6,59 (д, J=7,5 Гц, 1 H), 4,44 (с, 2 H), 3,73 (с, 2 H), 2,94 (с, 3 H), 2,83-2,71 (м, 2 H), 2,17-2,00 (м, 2 H); ЖХМС: 227,0 [M+H+].
<Пример получения 10> Получение N-(2,5-дихлорпиримидин-4-ил)-1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-амина
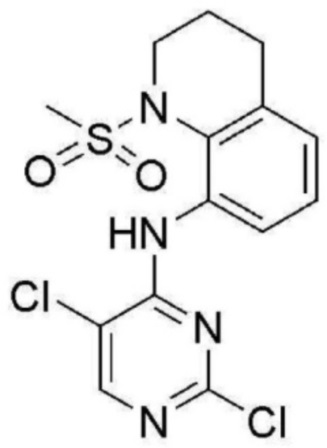
Применяют тот же способ, как в Примере получения 3, с получением N-(2,5-дихлорпиримидин-4-ил)-1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-амина (77%).
1H ЯМР (400 MГц, Хлороформ-d) δ 9,05 (с, 1 H), 8,21 (с, 1 H), 7,93 (д, J=8,3 Гц, 1 H), 7,41-7,29 (м, 1 H), 7,09 (д, J=7,7 Гц, 1 H), 3,89-3,59 (м, 2 H), 2,97 (с, 3 H), 2,89 (т, J=7,5 Гц, 2 H), 2,18 (д, J=9,8 Гц, 2 H); ЖХМС: 374,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера получения 11.
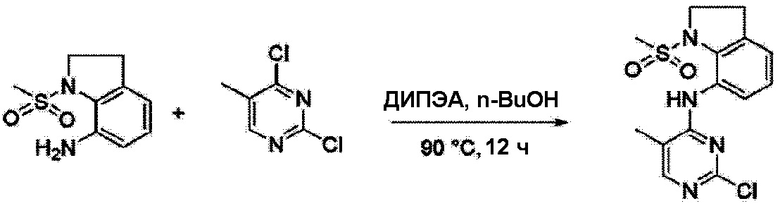
<Пример получения 11> Получение N-(2-хлор-5-метилпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина
Применяют тот же способ, как в Примере получения 3, с получением N-(2-хлор-5-метилпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина (29%).
1H ЯМР (500 MГц, Хлороформ-d) δ 8,86 (с, 1 H), 8,16 (дд, J=8,4, 1,1 Гц, 1 H), 8,02 (с, 1 H), 7,35-7,27 (м, 1 H), 7,13 (дкв, J=7,4, 1,1 Гц, 1 H), 4,17 (т, J=7,5 Гц, 2 H), 3,13 (т, J=7,5 Гц, 2 H), 2,89 (с, 3 H), 2,22 (д, J=0,9 Гц, 3 H); ЖХМС: 338,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера получения 12.
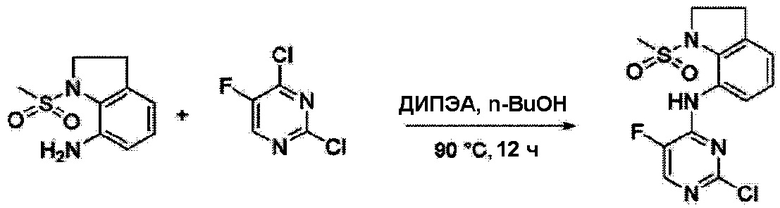
<Пример получения 12> Получение N-(2-хлор-5-фторпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина
Применяют тот же способ, как в Примере получения 3, с получением N-(2-хлор-5-фторпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина (82%).
1H ЯМР (400 MГц, Хлороформ-d) δ 9,28 (с, 1 H), 8,14 (шс, 2 H), 7,31 (д, J=12,3 Гц, 1 H), 4,18 (т, J=7,7 Гц, 2 H), 3,15 (т, J=7,8 Гц, 2 H), 2,91 (с, 3 H); ЖХМС: 342,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера получения 13.
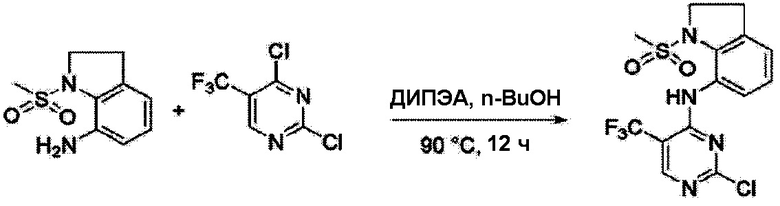
<Пример получения 13> Получение N-(2-хлор-5-(трифторметил)пиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина
Применяют тот же способ, как в Примере получения 3, с получением N-(2-хлор-5-(трифторметил)пиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина (24%), который отделяют с применением ВЭЖХ.
1H ЯМР (400 MГц, Хлороформ-d) δ 9,20 (с, 1 H), 8,44 (кс, 1 H), 7,80 (дд, J=8,2, 1,1 Гц, 1 H), 7,35-7,29 (м, 1 H), 7,22 (дкв, J=7,4, 1,1 Гц, 1 H), 4,13 (т, J=7,5 Гц, 2 H), 3,17 (т, J=7,4 Гц, 2 H), 2,94 (с, 3 H); ЖХМС: 394,0 [M+H2+].
Согласно следующей схеме реакции, получают соединение Примера получения 14.
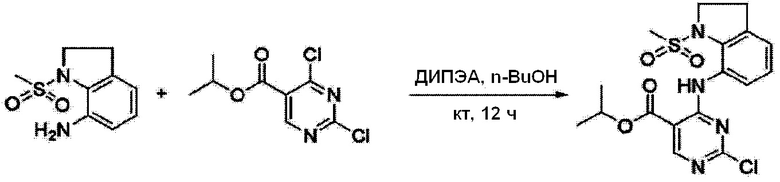
<Пример получения 14> Получение изопропил 2-хлор-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата
Применяют тот же способ, как в Примере получения 3, с получением изопропил 2-хлор-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата (84%).
1H ЯМР (500 MГц, Хлороформ-d) δ 10,71 (с, 1 H), 8,83 (с, 1 H), 7,94 (дд, J=8,3, 1,0 Гц, 1 H), 7,31-7,27 (м, 2 H), 7,18 (дкв, J=7,4, 1,1 Гц, 1 H), 5,41-5,29 (м, 1 H), 4,13 (т, J=7,4 Гц, 2 H), 3,20-3,15 (м, 2 H), 3,00 (с, 3 H), 1,41 (д, J=6,3 Гц, 6 H); ЖХМС: 411,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера получения 15.
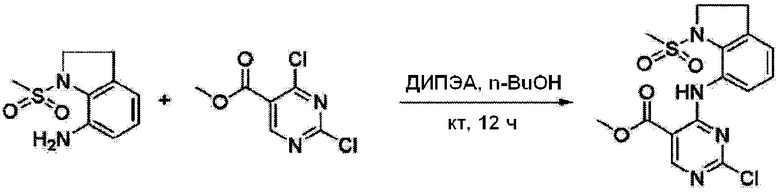
<Пример получения 15> Получение метил 2-хлор-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата
Применяют тот же способ, как в Примере получения 3, с получением метил 2-хлор-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата (88%).
1H ЯМР (500 MГц, ДМСО-d6) δ 10,44 (с, 1H), 8,80 (с, 1H), 7,72 (дд, J=7,3, 2,1 Гц, 1H), 7,31-7,24 (м, 2H), 4,05 (т, J=7,4 Гц, 2H), 3,90 (с, 3H), 3,12 (т, J=7,4 Гц, 2H), 3,07 (с, 3H); ЖХМС: 383,1 [M+H+].
[Схема реакции E]
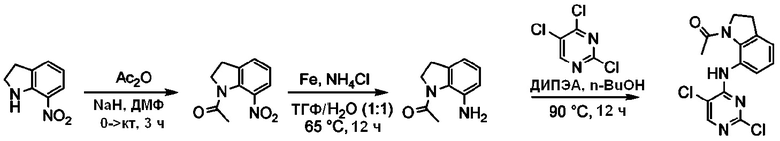
Согласно Схеме реакции E выше, получают соединения Примеров получения 16-18.
<Пример получения 16> Получение 1-(7-нитроиндолин-1-ил)этан-1-она
Применяют тот же способ, как в Примере получения 1, с получением 1-(7-нитроиндолин-1-ил)этан-1-она.
1H ЯМР (300 MГц, Хлороформ-d) δ 7,64 (дд, J=8,2, 1,1 Гц, 1 H), 7,46-7,39 (м, 1 H), 7,15 (т, J=7,8 Гц, 1 H), 4,25 (т, J=8,1 Гц, 2 H), 3,23 (т, J=8,0 Гц, 2 H), 2,27 (с, 3 H). ЖХМС: 207,2 [M+H+].
<Пример получения 17> Получение 1-(7-аминоиндолин-1-ил)-этан-1-она
К перемешиваемому раствору 1-(метилсульфонил)-7-нитроиндолина (1,0 г, 4,84 ммоль) в 20 мл ТГФ/H2O (1:1) добавляют железный порошок (1,3 г, 24,24 ммоль) и NH4Cl (1,3 г, 24,24 ммоль) при комнатной температуре с получением 1-(7-аминоиндолин-1-ил)-этан-1-она (90%) в виде коричневого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 6,97 (т, J=7,6 Гц, 1 H), 6,66 (дт, J=7,3, 1,1 Гц, 1 H), 6,60 (дд, J=8,1, 1,0 Гц, 1 H), 4,82 (с, 2 H), 4,07 (т, J=7,8 Гц, 2 H), 3,07 (т, J=7,7 Гц, 2 H), 2,33 (с, 3 H). ЖХМС: 177,8 [M+H+].
<Пример получения 18> Получение 1-(7-((2,5-дихлорпиримидин-4-ил)амино)индолин-1-ил)этан-1-она
Применяют тот же способ, как в Примере получения 3, для 1-(7-аминоиндолин-1-ил)-этан-1-она (1,0 г, 5,67 ммоль), с получением 1-(7-((2,5-дихлорпиримидин-4-ил)амино)индолин-1-ил)этан-1-она (80%) в виде темно-зеленого твердого вещества.
1H ЯМР (400 MГц, ДМСО-d6) δ 10,47 (с, 1 H), 8,38 (с, 1 H), 7,64-7,57 (м, 1 H), 7,22 (т, J=7,7 Гц, 1 H), 7,15 (дд, J=7,3, 1,3 Гц, 1 H), 4,16 (т, J=7,8 Гц, 2 H), 3,11 (т, J=7,7 Гц, 2 H), 2,32 (с, 3 H).. ЖХМС: 324,8 [M+H+].
[Схема реакции F]
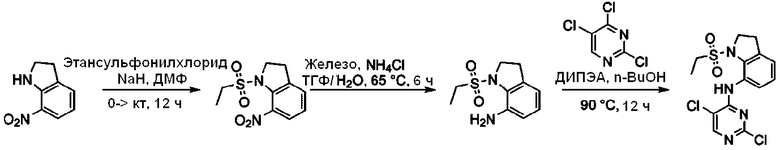
Согласно Схеме реакции F выше, получают соединения Примеров получения 19-21.
<Пример получения 19> Получение 1-(этилсульфонил)-7-нитроиндолина
Применяют тот же способ, как в Примере получения 1, с получением 1-(этилсульфонил)-7-нитроиндолина (59%).
1H ЯМР (400 MГц, Хлороформ-d) δ 7,78 (дд, J=8,3, 1,1 Гц, 1 H), 7,49 (дд, J=7,4, 1,2 Гц, 1 H), 7,21 (дд, J=8,2, 7,4 Гц, 1 H), 4,28 (т, J=7,7 Гц, 2 H), 3,42 (кв, J=7,4 Гц, 2 H), 3,21 (тт, J=7,7, 1,0 Гц, 2 H), 1,52 (т, J=7,4 Гц, 3 H). ЖХМС: 257,0 [M+H+].
<Пример получения 20> Получение 1-(этилсульфонил)индолин-7-амина
Применяют тот же способ, как в Примере получения 17, с получением 1-(этилсульфонил)индолин-7-амина (92%).
1H ЯМР (400 MГц, Хлороформ-d) δ 7,01-6,89 (м, 1 H), 6,69 (т, J=8,2 Гц, 1 H), 6,60 (т, J=8,7 Гц, 1 H), 4,61 (с, 2 H), 4,20-3,93 (м, 2 H), 3,16-2,76 (м, 4 H), 1,50-1,20 (м, 3 H). ЖХМС: 227,0 [M+H+].
<Пример получения 21> Получение N-(2,5-дихлорпиримидин-4-ил)-1-(этилсульфонил)индолин-7-амина
Применяют тот же способ, как в Примере получения 3, с получением N-(2,5-дихлорпиримидин-4-ил)-1-(этилсульфонил)индолин-7-амина (71%).
1H ЯМР (500 MГц, Хлороформ-d) δ 9,33 (с, 1 H), 8,21 (с, 1 H), 7,92 (д, J=8,2 Гц, 1 H), 7,33-7,24 (м, 2 H), 7,16 (д, J=7,4 Гц, 1 H), 4,13 (т, J=7,4 Гц, 2 H), 3,16 (т, J=7,5 Гц, 2 H), 3,11 (кв, J=7,4 Гц, 2 H), 1,50-1,40 (м, 3 H). ЖХМС: 374,0 [M+2H+].
[Схема реакции G]
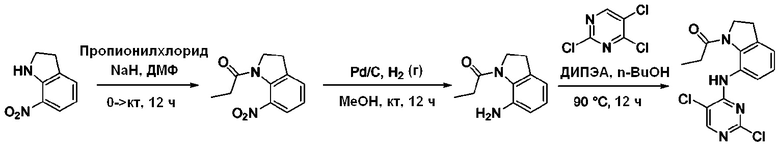
Согласно Схеме реакции G выше, получают соединения Примеров получения 22-24.
<Пример получения 22> Получение 1-(7-нитроиндолин-1-ил)пропан-1-она
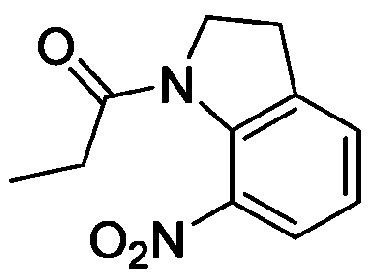
Применяют тот же способ, как в Примере получения 1, с получением 1-(7-нитроиндолин-1-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 7,66-7,56 (м, 2 H), 7,23 (т, J=7,8 Гц, 1 H), 4,24 (т, J=8,1 Гц, 2 H), 3,21 (т, J=8,2 Гц, 2 H), 2,54 (дд, J=8,7, 6,2 Гц, 2 H), 1,05 (тд, J=7,4, 2,5 Гц, 3 H). ЖХМС: 220,0 [M+H+].
<Пример получения 23> Получение 1-(7-аминоиндолин-1-ил)пропан-1-она
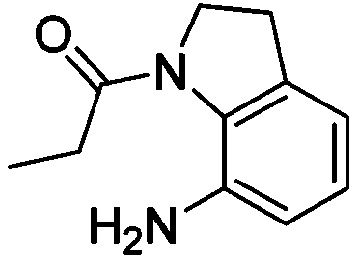
Применяют тот же способ, как в Примере получения 2, с получением 1-(7-аминоиндолин-1-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 6,86 (т, J=7,6 Гц, 1 H), 6,61-6,50 (м, 7,6 Гц, 2 H), 5,42 (с, 2 H), 4,03 (т, J=7,8 Гц, 2 H), 2,96 (т, J=7,9 Гц, 2 H), 2,56 (кв, J=7,5 Гц, 2 H), 1,10 (т, J=7,4 Гц, 3 H). ЖХМС: 190,2 [M+H+].
<Пример получения 24> Получение 1-(7-((2,5-дихлорпиримидин-4-ил)амино)индолин-1-ил)пропан-1-она
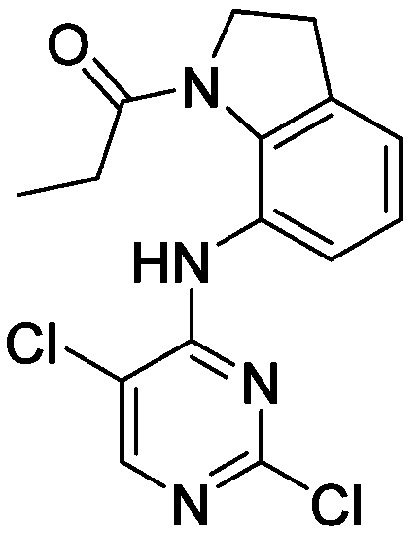
Применяют тот же способ, как в Примере получения 3, с получением 1-(7-((2,5-дихлорпиримидин-4-ил)амино)индолин-1-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 10,35 (с, 1 H), 8,38 (д, J=2,4 Гц, 1 H), 7,62 (д, J=8,2 Гц, 1 H), 7,22 (д, J=7,8 Гц, 1 H), 7,17 (д, J=7,5 Гц, 1 H), 4,16 (с, 2 H), 3,12 (с, 2 H), 2,64 (д, J=7,4 Гц, 2 H), 1,11 (т, J=7,4 Гц, 3 H). ЖХМС: 337,8 [M+H+].
[Схема реакции H]
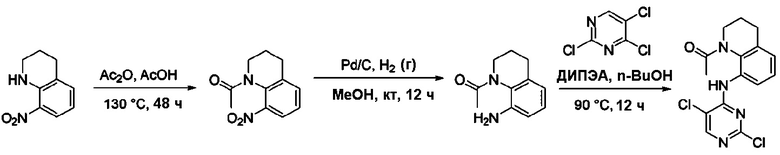
Согласно Схеме реакции H выше, получают соединения Примеров получения 25-27.
<Пример получения 25> Получение 1-(8-нитро-3,4-дигидрохинолин-1(2H)-ил)этан-1-она
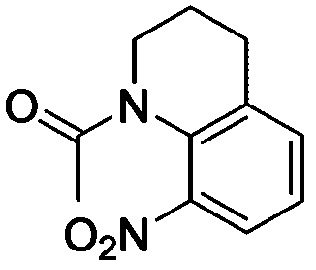
Применяют тот же способ, как в Примере получения 16, с применением 8-нитро-3,4-дигидрохинолина, с получением 1-(8-нитро-3,4-дигидрохинолин-1(2H)-ил)этан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 7,76-7,65 (м, 1 H), 7,53 (д, J=7,6 Гц, 1 H), 7,30 (т, J=7,9 Гц, 1 H), 3,82 (с, 2 H), 2,83 (с, 2 H), 2,19 (д, J=2,2 Гц, 3 H), 1,98 (с, 2 H). ЖХМС: 220,4 [M+H+].
<Пример получения 26> Получение 1-(8-амино-3,4-дигидрохинолин-1(2H)-ил)этан-1-она
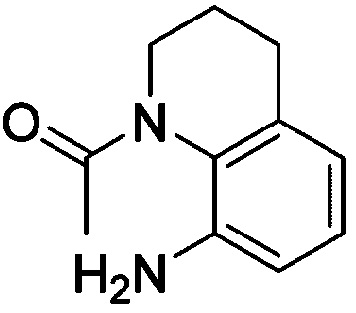
Применяют тот же способ, как в Примере получения 2, с получением 1-(8-амино-3,4-дигидрохинолин-1(2H)-ил)этан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 6,89 (т, J=7,6 Гц, 1 H), 6,61 (д, J=7,9 Гц, 1 H), 6,44 (д, J=7,3 Гц, 1 H), 5,12 (с, 2 H), 4,63-4,48 (м, 2 H), 2,68-2,59 (м, 1H), 2,58-2,53 (м, 1 H), 2,33-2,26 (м, 1 H), 2,15-2,04 (м, 1H), 1,90 (с, 3 H), 1,61-1,49 (м, 1 H). ЖХМС: 190,2 [M+H+].
<Пример получения 27> Получение 1-(8-((2,5-дихлорпиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)этан-1-она
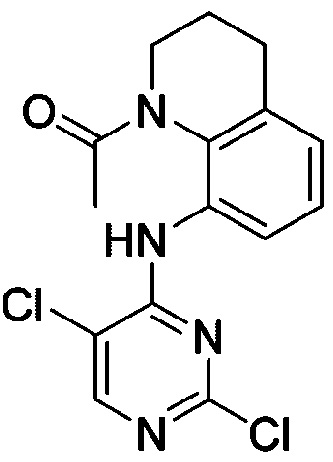
Применяют тот же способ, как в Примере получения 3, с получением 1-(8-((2,5-дихлорпиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)этан-1-она.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,72 (с, 1 H), 8,37 (с, 1 H), 7,52-7,08 (м, 3 H), 4,31-3,98 (м, 2 H), 3,12-2,69 (м, 2 H), 2,20 (д, J=49,1 Гц, 3 H), 1,91-1,53 (м, 2 H). ЖХМС: 337,8 [M+H+].
[Схема реакции I]
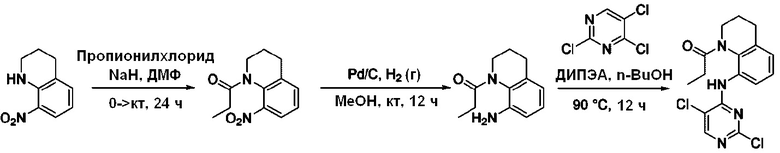
Согласно Схеме реакции I выше, получают соединения Примеров получения 28-30.
<Пример получения 28> Получение 1-(8-нитро-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она
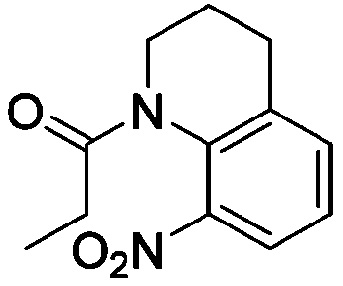
Применяют тот же способ, как в Примере получения 1, с получением 1-(8-нитро-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 7,70 (д, J=8,1 Гц, 1 H), 7,53 (д, J=7,6 Гц, 1H), 7,29 (т, J=8,0 Гц, 1 H), 3,83 (шс, 2 H), 2,82 (с, 2 H), 2,54 (с, 2 H), 2,12-1,79 (м, 2 H), 1,01 (т, J=7,5 Гц, 3 H). ЖХМС: 234,4 [M+H+].
<Пример получения 29> Получение 1-(8-амино-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она
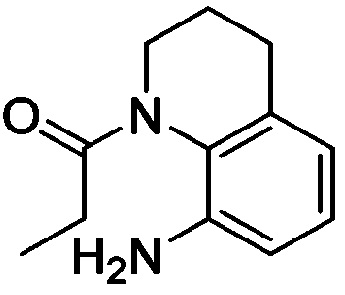
Применяют тот же способ, как в Примере получения 2, с получением 1-(8-амино-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 6,89 (т, J=7,7 Гц, 1 H), 6,60 (д, J=8,1 Гц, 1 H), 6,44 (д, J=7,5 Гц, 1 H), 5,10 (с, 2 H), 4,68-4,55 (м, 2 H), 2,70-2,59 (м, 1 H), 2,54 (д, J=4,9 Гц, 1 H), 2,41 (дкв, J=15,4, 7,6 Гц, 1 H), 2,24 (кв, J=12,2, 11,0 Гц, 1 H), 2,10 (с, 1 H), 2,02 (дт, J=15,9, 7,6 Гц, 1 H), 1,53 (с, 1 H), 1,09 (т, J=7,5 Гц, 1 H), 0,90 (т, J=7,5 Гц, 3 H). ЖХМС: 204,4 [M+H+].
<Пример получения 30> Получение 1-(8-((2,5-дихлорпиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она
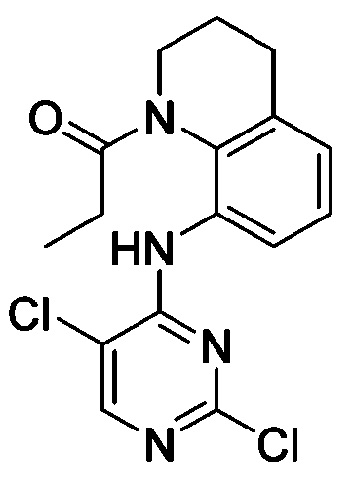
Применяют тот же способ, как в Примере получения 3, с получением 1-(8-((2,5-дихлорпиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,69 (с, 1 H), 8,36 (с, 1 H), 7,44 (с, 1 H), 7,33-7,09 (м, 2 H), 4,17 (д, J=78,3 Гц, 2 H), 3,28 (с, 1 H), 2,83 (с, 1 H), 2,74 (с, 1 H), 2,58 (с, 1 H), 2,33-1,52 (м, 2 H ), 1,05 (с, 2 H), 0,83 (с, 1 H). ЖХМС: 351,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 1.
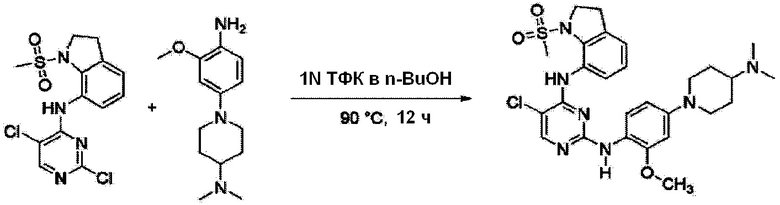
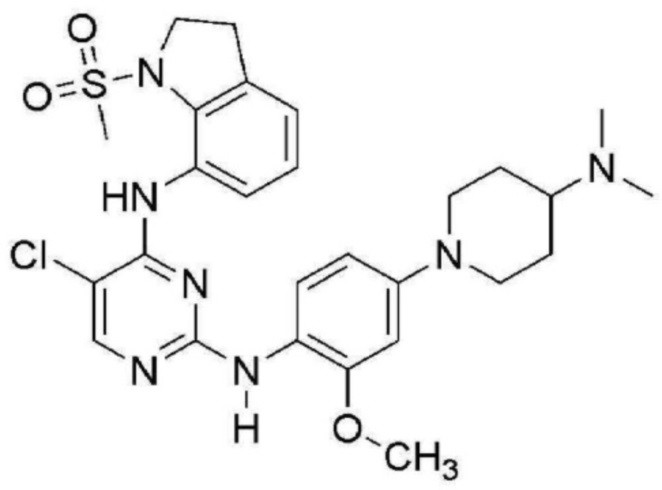
<Пример 1> Получение 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
К перемешиваемому раствору N-(2,5-дихлорпиримидин-4-ил)-1-(метилсульфонил)индолин-7-амина (100 мг, 0,27 ммоль) в 1 N ТФК в n-BuOH (3 мл) добавляют 1-(4-амино-3-метоксифенил)-N, N-диметилпиперидин-4-амин (81,3 мг, 0,27 ммоль) при комнатной температуре. Полученную смесь нагревают до 90°C в течение 14 часов. Реакционную смесь разбавляют ДХМ. Органический слой промывают насыщенным водным раствором NaHCO3, затем водой и насыщенным раствором соли, сушат над MgSO4, и затем выпаривают при пониженном давлении. Полученный продукт очищают с использованием колоночной хроматографии (5-10% метанол в ДХМ в качестве элюента) с получением 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина (49%) в виде белого твердого вещества.
1H ЯМР (400 MГц, Хлороформ-d) δ 8,95 (с, 1 H), 8,14-8,03 (м, 2 H), 8,04-7,98 (м, 1 H), 7,36-7,24 (м, 3 H), 7,15 (дд, J=7,4, 1,2 Гц, 1 H), 6,54 (д, J=2,6 Гц, 1 H), 6,43 (дд, J=8,8, 2,6 Гц, 1 H), 4,17 (т, J=7,5 Гц, 2 H), 3,87 (с, 3 H), 3,66 (д, J=11,9 Гц, 2 H), 3,16 (т, J=7,5 Гц, 2 H), 2,94 (с, 3 H), 2,72 (тд, J=12,3, 2,4 Гц, 2 H), 2,64-2,40 (м, 6 H), 2,08 (д, J=12,3 Гц, 2 H), 1,79 (квд, J=12,1, 4,1 Гц, 2 H); ЖХМС: 572,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 2.
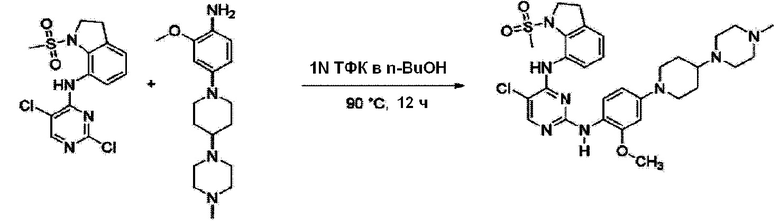
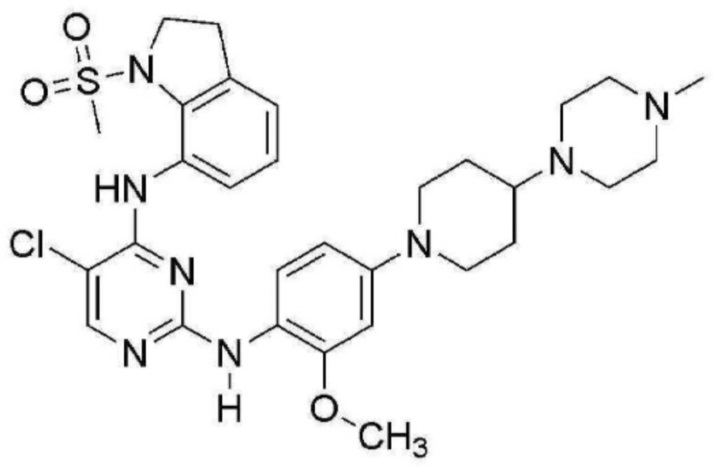
<Пример 2> Получение 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина (54%).
1H ЯМР (400 MГц, Хлороформ-d) δ 8,94 (с, 1 H), 8,10-7,99 (м, 3 H), 7,33-7,23 (м, 3 H), 7,17-7,12 (м, 1 H), 6,54 (д, J=2,5 Гц, 1 H), 6,42 (дд, J=8,8, 2,5 Гц, 1 H), 4,17 (т, J=7,5 Гц, 2 H), 3,87 (с, 3 H), 3,65 (д, J=12,1 Гц, 2 H), 3,15 (т, J=7,5 Гц, 2 H), 2,94 (с, 3 H), 2,87-2,57 (м, 9 H), 2,47 (м, 1 H), 2,41 (с, 3 H), 1,99 (д, J=12,1 Гц, 2 H), 1,82-1,67(м, 2 H); ЖХМС: 627,9 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 3.
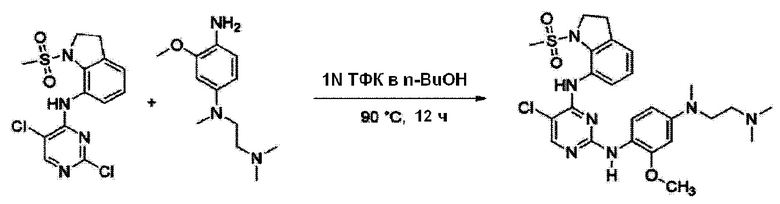
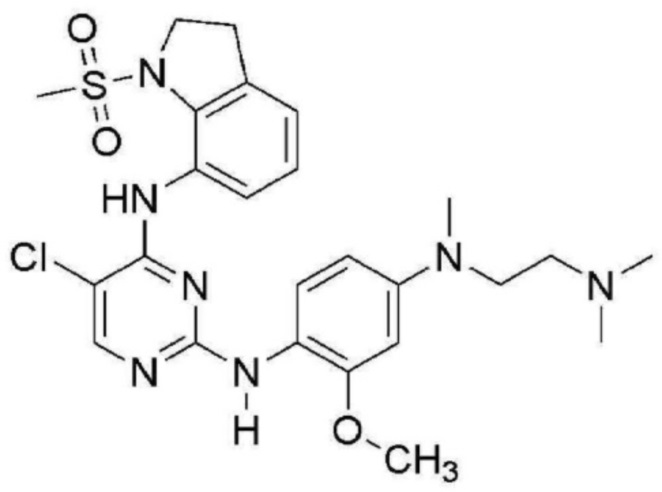
<Пример 3> Получение 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина (43%).
1H ЯМР (400 MГц, Хлороформ-d) δ 8,93 (с, 1 H), 8,08-8,02 (м, 2 H), 7,97 (д, J=8,9 Гц, 1 H ), 7,28-7,24 (м, 1 H), 7,18 (с, 1 H), 7,15-7,10 (м, 1 H), 6,39 (д, J=2,6 Гц, 1 H), 6,26 (дд, J=8,9, 2,7 Гц, 1 H), 4,17 (т, J=7,5 Гц, 2 H), 3,88 (с, 3 H), 3,53 (т, J=7,5 Гц, 2 H), 3,15 (т, J=7,4 Гц, 2 H), 2,96 (с, 3 H), 2,94 (с, 3 H), 2,68-2,60 (м, 2 H), 2,44 (с, 6 H); ЖХМС: 546,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 4.
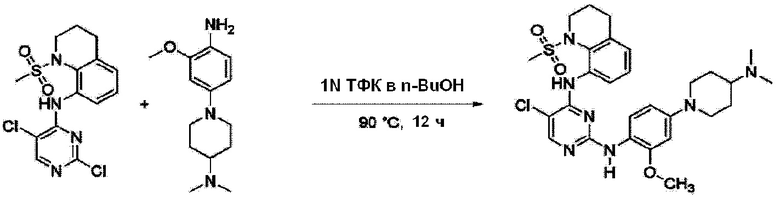
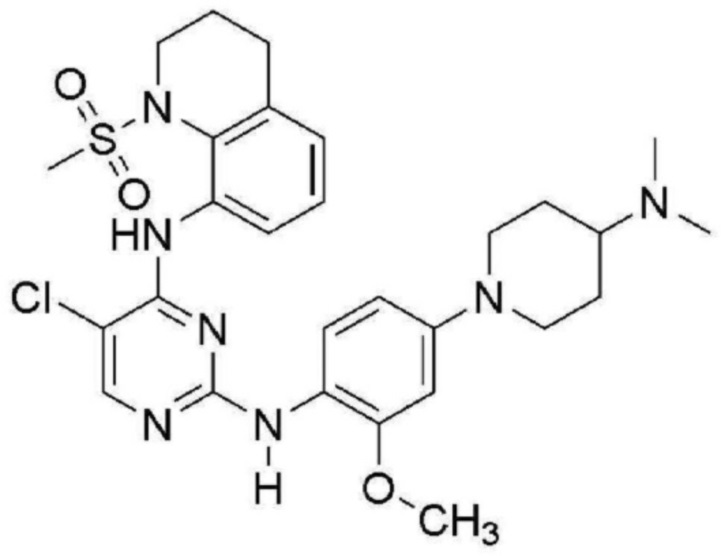
<Пример 4> Получение 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина (55%).
1H ЯМР (400 MГц, Хлороформ-d) δ 8,47 (с, 1 H), 8,06 (с, 1 H), 7,99 (д, J=8,9 Гц, 1 H), 7,93 (дд, J=8,2, 1,4 Гц, 1 H), 7,33-7,28 (м, 3 H), 7,10-7,05 (м, 1 H), 6,54 (д, J=2,6 Гц, 1 H), 6,39 (дд, J=8,9, 2,6 Гц, 1 H), 3,86 (с, 3 H), 3,76-3,60 (м, 4 H), 3,02 (с, 3 H), 2,89 (т, J=7,2 Гц, 2 H), 2,71 (тд, J=12,2, 2,4 Гц, 2 H), 2,50-2,37 (м, 6 H), 2,24-2,11 (м, 2 H), 2,03 (д, J=12,5 Гц, 2 H), 1,75 (квд, J=12,1, 3,9 Гц, 2 H); ЖХМС: 586,9 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 5.
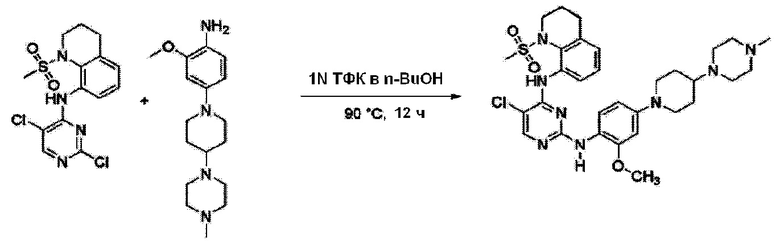
<Пример 5> Получение 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)фенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)фенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина (49%).
1H ЯМР (400 MГц, Хлороформ-d) δ 8,46 (с, 1H), 8,05 (с, 1 H), 7,97 (д, J=8,8 Гц, 1 H), 7,93 (дд, J=8,2, 1,4 Гц, 1 H), 7,34-7,29 (м, 2 H), 7,08 (дд, J=7,4, 1,4 Гц, 1 H), 6,53 (д, J=2,6 Гц, 1 H), 6,38 (дд, J=8,9, 2,5 Гц, 1 H), 3,86 (с, 3 H), 3,75-3,60 (м, 4 H), 3,02 (с, 3 H), 2,89 (т, J=7,2 Гц, 2 H), 2,84-2,52 (м, 11 H), 2,39 (с, 3 H), 2,23-2,11 (м, 2 H), 1,99 (д, J=12,4 Гц, 2 H), 1,74 (дд, J=12,0, 3,9 Гц, 2 H); ЖХМС: 641,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 6.
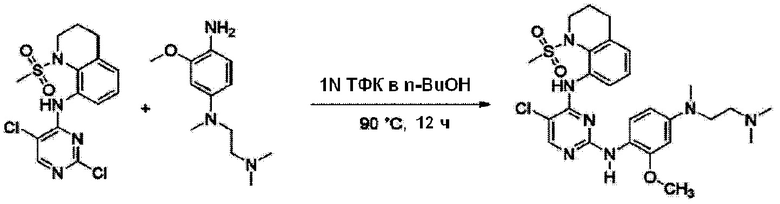
<Пример 6> Получение 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамина (34%).
1H ЯМР (400 MГц, Хлороформ-d) δ 8,45 (с, 1H), 8,05 (с, 1 H), 7,97 (дд, J=8,2, 1,4 Гц, 1 H), 7,92 (д, J=8,9 Гц, 1 H), 7,31 (д, J=7,8 Гц, 1 H), 7,19 (с, 1 H), 7,06 (дд, J=7,5, 1,4 Гц, 1 H), 6,40 (д, J=2,6 Гц, 1 H), 6,23 (дд, J=8,9, 2,6 Гц, 1 H), 3,88 (с, 3 H), 3,77-3,64 (шс, 2 H), 3,62-3,51 (шс, 2 H), 3,02 (с, 3 H), 2,96 (с, 3 H), 2,89 (т, J=7,1 Гц, 2 H), 2,69 (с, 2 H), 2,48 (с, 6 H), 2,23-2,12 (м, 2 H); ЖХМС: 561,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 7.
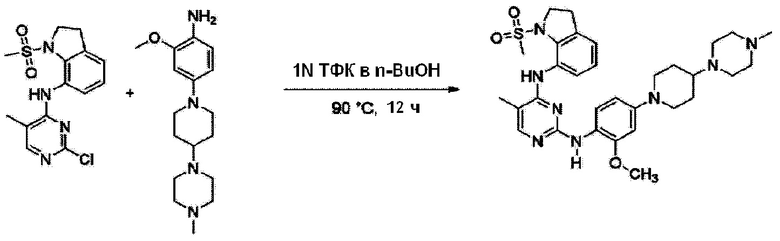
<Пример 7> Получение N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-5-метил-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-5-метил-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина (54%).
1H ЯМР (400 MГц, Метанол-d4) δ 7,76 (д, J=8,0 Гц, 1 H), 7,67 (с, 1 H), 7,40 (д, J=8,8 Гц, 1 H), 7,37-7,32 (м, 1 H), 7,29 (т, J=7,7 Гц, 1 H), 6,83 (д, J=2,5 Гц, 1H ), 6,62 (дд, J=8,8, 2,5 Гц, 1 H), 4,12 (т, J=7,6 Гц, 2 H), 3,88 (с, 3 H), 3,83 (д, J=2,5 Гц, 2 H ) 3,27-3,06 (м, 6 H), 2,96 (м, 6 H), 2,88 (с, 3 H), 2,57-2,52 (м, 5 H) 2,21 (с, 3 H), 2,14 (д, J=12,6 Гц, 2 H), 1,91-1,71 (м, 2 H); ЖХМС: 606,9 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 8.
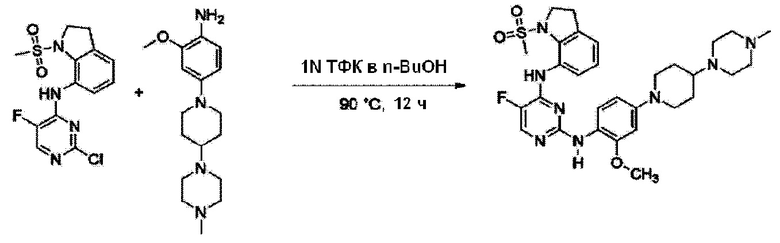
<Пример 8> Получение 5-фтор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-фтор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина (45%).
1H ЯМР (500 MГц, ДМСО-d6) δ 8,92 (д, J=2,5 Гц, 1 H), 8,06 (д, J=3,3 Гц, 1 H), 7,98 (д, J=8,0 Гц, 1 H), 7,69 (с, 1 H), 7,56 (д, J=8,7 Гц, 1 H), 7,22-7,12 (м, 2 H), 6,61 (д, J=2,5 Гц, 1 H), 6,42 (дд, J=8,8, 2,5 Гц, 1 H), 4,07 (т, J=7,5 Гц, 2 H), 3,78 (с, 3 H), 3,68 (д, J=12,1 Гц, 2 H), 3,12 (т, J=7,5 Гц, 2 H), 3,05 (с, 3 H), 2,64 (тд, J=12,2, 2,4 Гц, 2 H), 2,57-2,52 (м, 4 H), 2,46-2,27 (м, 4 H) 2,20 (с, 3 H), 1,86 (д, J=12,5 Гц, 2 H), 1,52 (дт, J=13,3, 9,6 Гц, 2 H); ЖХМС: 610,8 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 9.
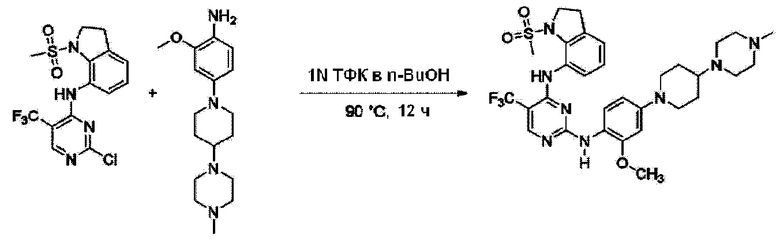
<Пример 9> Получение N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)-5-(трифторметил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)-5-(трифторметил)пиримидин-2,4-диамина (48%).
1H ЯМР (400 MГц, Метанол-d4) δ 8,35 (с, 1 H), 7,71 (д, J=8,9 Гц, 1 H), 7,59 (д, J=7,9 Гц, 1 H), 7,37 (дд, J=7,4, 1,4 Гц, 1 H), 7,32 (д, J=7,8 Гц, 1 H), 6,99 (д, J=2,5 Гц, 1 H), 6,72 (дд, J=8,8, 2,5 Гц, 1 H), 4,10 (т, J=7,6 Гц, 2 H), 3,93 (с, 3 H), 3,82 (д, J=12,5 Гц, 2 H), 3,45-3,33 (м, 3 H), 3,26 (д, J=12,0 Гц, 3 H), 3,24-3,08 (м, 6 H), 2,98 (с, 3 H), 2,92 (с, 3 H), 2,57-2,52 (м, 4 H), 2,20 (д, J=13,0 Гц, 2 H), 2,06-1,89 (м, 2 H); ЖХМС: 661,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 10.
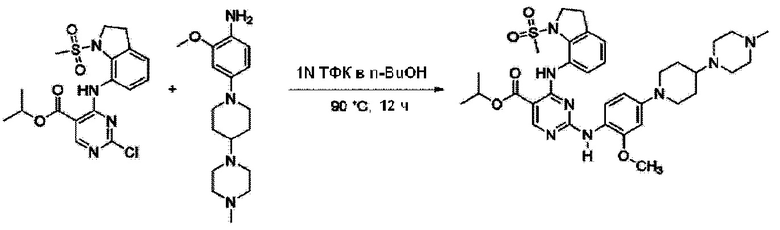
<Пример 10> Получение изопропил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата
Используют тот же способ, как в Примере 1, с получением изопропил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата (52%).
1H ЯМР (400 MГц, ДМСО-d6) δ 10,35 (с, 1 H), 8,60 (д, J=9,5 Гц, 2 H), 7,32 (д, J=8,6 Гц, 1 H), 7,16-6,93 (м, 2 H), 6,62 (д, J=2,5 Гц, 1 H), 6,43 (дд, J=8,9, 2,5 Гц, 1 H), 5,18-5,06 (м, 1 H), 4,00 (т, J=7,1 Гц, 2 H), 3,75 (с, 5 H), 3,12-3,01 (м, 5 H), 2,76-2,62 (м, 2 H), 2,57-2,52 (м, 4 H), 2,42-2,23 (м, 5 H), 2,15 (с, 3 H), 1,91-1,80 (м, 2 H), 1,58-1,45 (м, 2 H), 1,32 (д, J=6,2 Гц, 6 H); ЖХМС: 679,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 11.
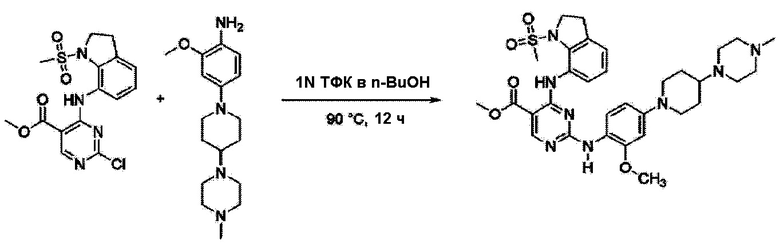
<Пример 11> Получение метил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата
Используют тот же способ, как в Примере 1, с получением метил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилата (44%).
1H ЯМР (400 MГц, ДМСО-d6) δ 10,32 (с, 1 H), 8,62 (д, J=10,4 Гц, 2 H), 7,87 (с, 1 H), 7,31 (д, J=8,6 Гц, 1 H), 7,12-6,93 (м, 2 H), 6,61 (д, J=2,5 Гц, 1 H), 6,50-6,38 (м, 1 H), 4,00 (т, J=7,2 Гц, 2 H), 3,82 (с, 3 H), 3,78-3,68 (м, 5 H), 3,13-3,02 (м, 5 H), 2,69 (т, J=11,7 Гц, 2 H), 2,57-2,52 (м, 4 H), 2,41-2,24 (м, 5 H), 2,15 (с, 3 H), 1,86 (д, J=12,2 Гц, 2 H), 1,64-1,42 (м, 2 H); ЖХМС: 651,0 [M+H+].
Согласно следующей схеме реакции, получают соединение Примера 12.
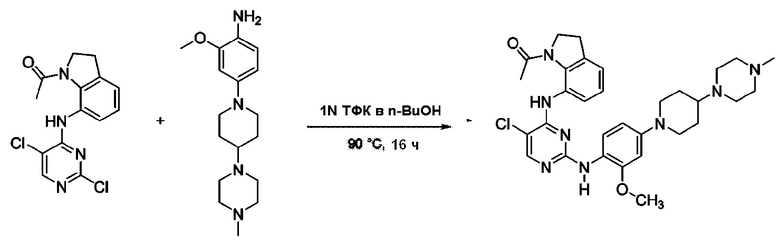
<Пример 12> Получение 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)этан-1-она
Используют тот же способ, как в Примере 1, с получением 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)этан-1-она.
1H ЯМР (400 MГц, ДМСО-d6) δ 9,76 (с, 1 H), 8,03 (с, 1 H), 7,74-7,63 (м, 2 H), 7,54 (д, J=8,7 Гц, 1 H), 7,13 (т, J=7,7 Гц, 1 H), 7,06 (д, J=7,2 Гц, 1 H), 6,58 (д, J=2,5 Гц, 1 H), 6,37 (дд, J=8,8, 2,5 Гц, 1 H), 4,13 (т, J=7,7 Гц, 2 H), 3,77 (с, 3 H), 3,67 (д, J=12,2 Гц, 2 H), 3,07 (т, J=7,7 Гц, 2 H), 2,63 (т, J=11,9 Гц, 2 H), 2,31 (с, 8 H), 2,14 (с, 3 H), 1,84 (д, J=12,3 Гц, 2 H), 1,51 (тт, J=12,7, 6,4 Гц, 2 H).
Согласно следующей схеме реакции, получают соединение Примера 13.
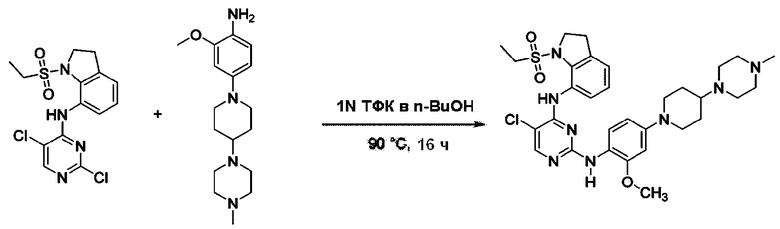
<Пример 13> Получение 5-хлор-N4-(1-(этилсульфонил)индолин-7-ил)-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением 5-хлор-N4-(1-(этилсульфонил)индолин-7-ил)-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пиримидин-2,4-диамина.
1H ЯМР (400 MГц, ДМСО-d6) δ 8,88 (с, 1 H), 8,08 (с, 1 H), 7,86 (с, 1 H), 7,82 (т, J=4,7 Гц, 1 H), 7,44 (д, J=8,7 Гц, 1 H), 7,19-7,10 (м, 2 H), 6,59 (д, J=2,4 Гц, 1 H), 6,38 (дд, J=8,7, 2,4 Гц, 1 H), 4,03 (т, J=7,4 Гц, 2 H), 3,76 (с, 3 H), 3,68 (д, J=12,3 Гц, 2 H), 3,27 (т, J=7,3 Гц, 2 H), 3,09 (т, J=7,4 Гц, 2 H), 2,64 (т, J=11,7 Гц, 2 H), 2,49 (с, 5 H) 2,30 (т, J=11,6 Гц, 4 H), 2,15 (с, 3 H), 1,84 (д, J=11,8 Гц, 2 H), 1,51 (тт, J=13,6, 6,9 Гц, 2 H), 1,20 (т, J=7,3 Гц, 3 H).
Согласно следующей схеме реакции, получают соединение Примера 14.
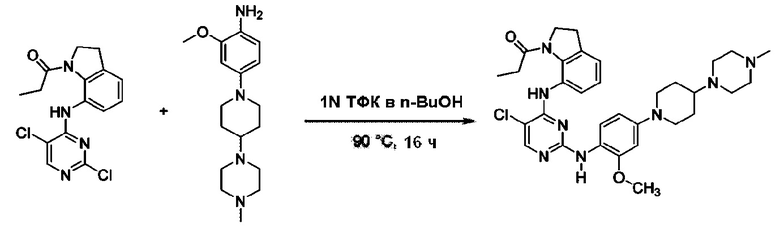
<Пример 14> Получение 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)пропан-1-она
Используют тот же способ, как в Примере 1, с получением 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 9,88 (с, 1 H), 8,31 (с, 1 H), 8,06 (д, J=8,1 Гц, 1 H), 7,93 (д, J=8,6 Гц, 1 H), 7,72 (с, 1 H), 7,43 (д, J=7,3 Гц, 1 H), 7,38 (д, J=7,5 Гц, 1 H), 6,89 (д, J=2,7 Гц, 1 H), 6,70 (дд, J=8,7, 2,8 Гц, 1 H), 4,45 (т, J=7,9 Гц, 2 H), 4,10 (с, 3 H), 3,95 (д, J=12,3 Гц, 2 H), 3,41 (т, J=7,8 Гц, 2 H), 3,05-2,97 (м, 2 H), 2,93 (кв, J=7,3 Гц, 2H), 2,85 (д, J=4,7 Гц, 4 H), 2,68 (т, J=4,8 Гц, 4 H), 2,55 (с, 3 H) 2,17 (д, J=11,8 Гц, 2 H), 1,93-1,80 (м, 2 H), 1,74 (кв, J=6,9 Гц, 1 H), 1,65 (кв, J=7,4 Гц, 1 H), 1,47 (т, J=7,3 Гц, 3 H).
Согласно следующей схеме реакции, получают соединение Примера 15.
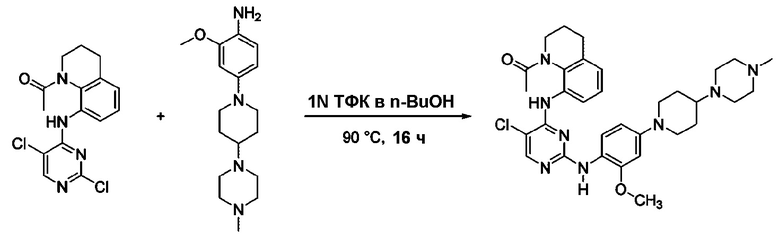
<Пример 15> Получение 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)этан-1-она
Используют тот же способ, как в Примере 1, с получением 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)этан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 8,02 (с, 1 H), 7,61 (д, J=8,2 Гц, 1 H), 7,54 (д, J=8,8 Гц, 1 H), 7,43 (с, 1 H), 7,21 (т, J=7,8 Гц, 1 H), 7,08 (д, J=7,6 Гц, 1 H), 6,57 (д, J=2,6 Гц, 1 H), 6,34 (д, J=8,4 Гц, 1 H), 3,79 (с, 3H ), 3,64 (д, J=12,1 Гц, 2 H), 2,81-2,62 (м, 4 H), 2,57-2,52 (м, 4 H), 2,51-2,42 (м, 4 H), 2,39-2,26 (м, 5 H), 2,18 (с, 5 H), 1,95 (с, 2 H), 1,86 (д, J=12,9 Гц, 2 H), 1,63-1,46 (м, 2H).
Согласно следующей схеме реакции, получают соединение Примера 16.
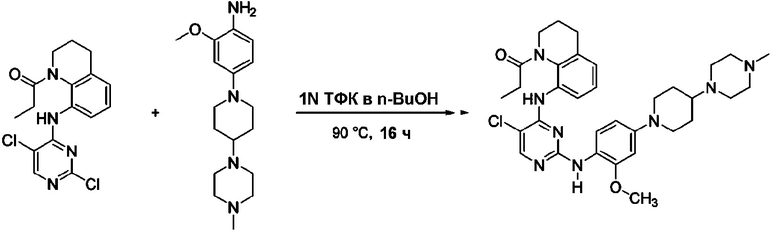
<Пример 16> Получение 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она
Используют тот же способ, как в Примере 1, с получением 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2H)-ил)пропан-1-она.
1H ЯМР (500 MГц, ДМСО-d6) δ 7,99 (д, J=7,7 Гц, 1 H), 7,92 (с, 1 H), 7,64-7,57 (м, 1 H), 7,55-7,48 (м, 1 H), 7,40 (д, J=9,1 Гц, 1 H), 7,24-7,13 (м, 1 H), 7,07 (д, J=8,2 Гц, 1 H), 6,55 (д, J=8,6 Гц, 1 H), 6,32 (д, J=8,9 Гц, 1 H), 3,78 (с, 3 H), 3,62 (с, 2 H), 2,68 (т, J=11,1 Гц, 4 H), 2,37-2,26 (м, 6 H), 2,17 (с, 3 H), 1,92 (с, 2 H), 1,85 (д, J=11,7 Гц, 2 H), 1,54 (т, J=11,6 Гц, 2 H), 1,13-0,98 (м, 3 H).
Согласно следующей схеме реакции, получают соединение Примера 17.
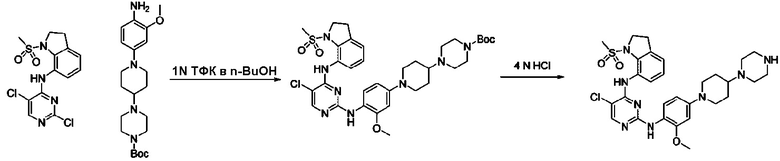
<Пример 17> Получение 5-хлор-N2-(2-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина
Используют тот же способ, как в Примере 1, с получением промежуточного соединения трет-бутил 4-(1-(4-((5-хлор-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-2-ил)амино)-3-метоксифенил)пиперидин-4-ил)пиперазин-1-карбоксилата. Затем проводят реакцию снятия защиты с применением 4N HCl с получением целевого соединения 5-хлор-N2-(2-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамина.
1H ЯМР (500 MГц, Метанол-d4) δ 8,10 (с, 1 H), 7,71 (д, J=7,9 Гц, 1 H), 7,48 (д, J=8,6 Гц, 1 H), 7,36 (д, J=7,6 Гц, 1 H), 7,30 (т, J=7,8 Гц, 1 H), 6,88 (с, 1H), 6,67 (д, J=8,7 Гц, 1 H), 4,13 (т, J=7,6 Гц, 2 H), 3,94-3,84 (м, 5H), 3,56 (д, J=5,1 Гц, 4 H), 3,50 (с, 4 H), 3,37 (м, 1 H), 3,20 (т, J=7,5 Гц, 2 H), 3,09 (т, J=12,5 Гц, 2 H), 2,98 (с, 3 H), 2,28 (д, J=12,3 Гц, 2 H), 2,05-1,95 (м, 2 H).
В таблице 1 ниже суммированы структурные формулы соединений, полученных в Примерах 1-17.
[Таблица 1]
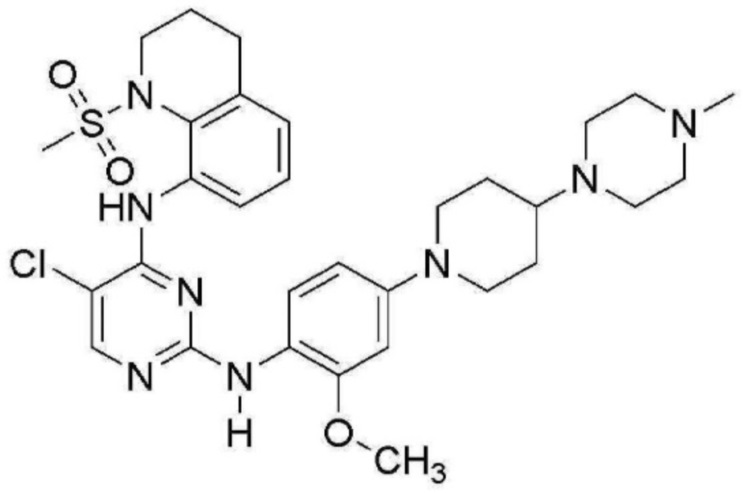
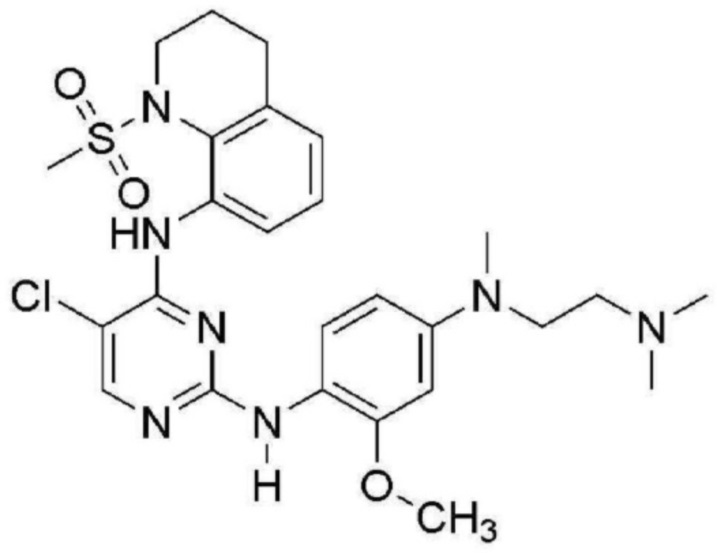
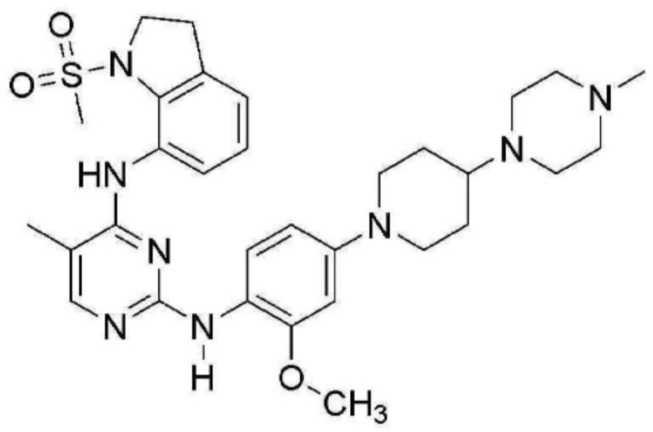
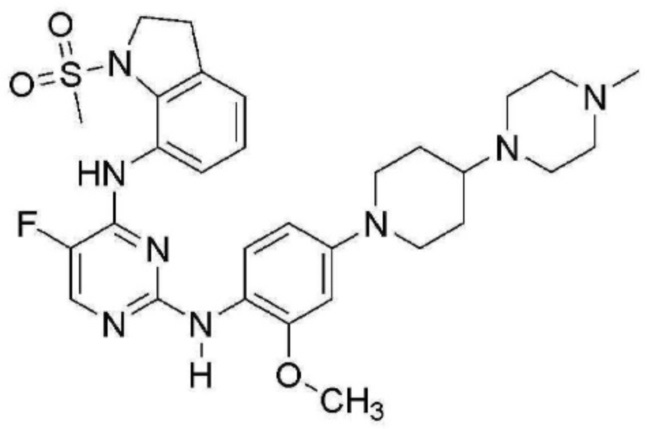
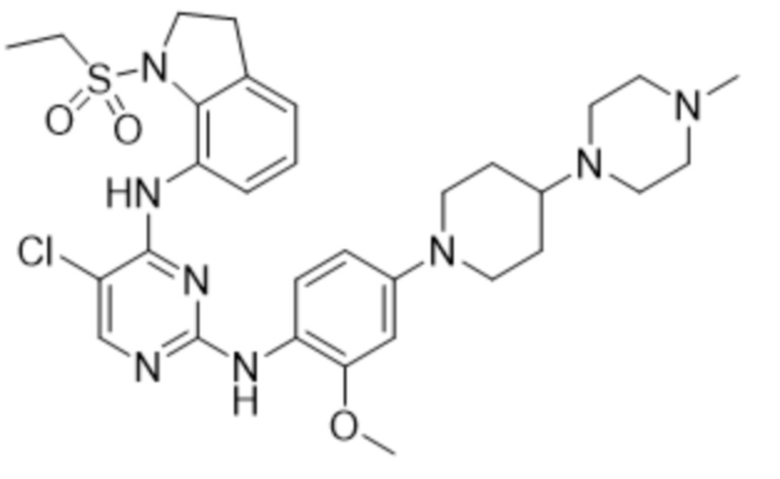
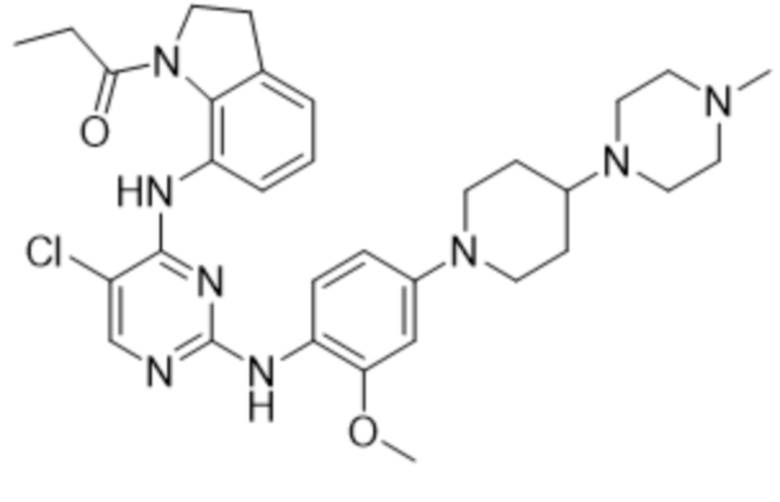
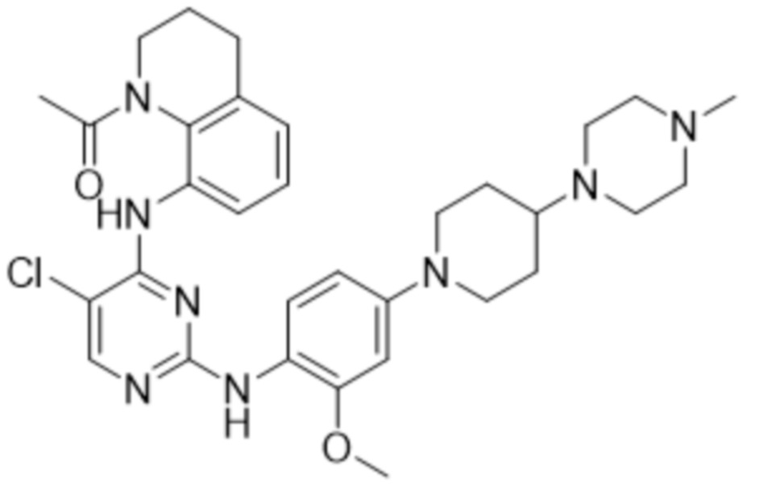
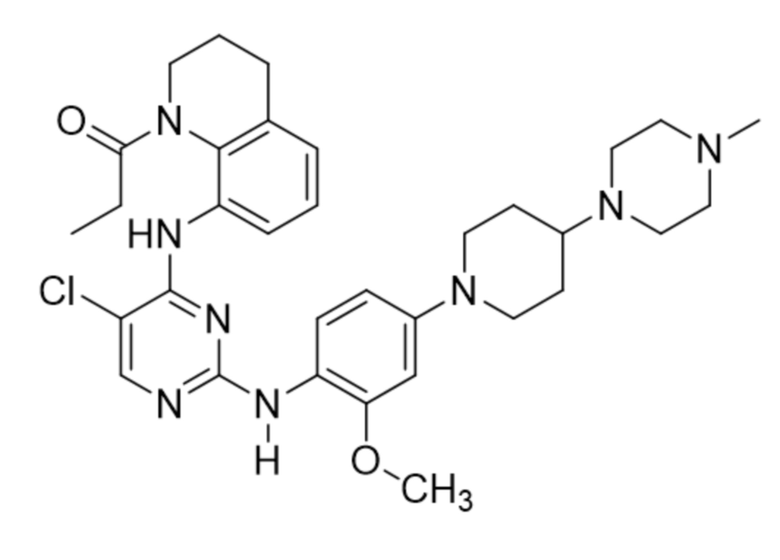
<Экспериментальный пример 1> Измерение ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению, против EGFR дикого типа и мутантов EGFR
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению, против различных мутаций EGFR, проводят следующий эксперимент. Результаты показаны в Таблице 2 ниже.
Эксперимент по измерению активности соединений настоящего изобретения против мутантных ферментов EGFR проводят следующим образом с использованием системы HTRF, продаваемой Cisbio. Для мутантного фермента EGFR del19/T790M приобретают и используют рекомбинантный белок, предоставленный Carna Biosciences, и для мутантного фермента EGFR del19/T790M/C797S приобретают и используют белок, предоставленный SignalChem, в качестве источника фермента.
Состав аналитического буфера, используемого для измерения активности, включает 50 мМ Трис-HCl, pH 7,5, 100 мМ NaCl, 7,5 мМ MgCl2, 3 мМ KCl, 0,01% Tween 20, 0,1% BSA и 1 мМ DTT. В настоящем документе, ферментативную реакцию проводят с использованием АТФ в концентрации 50 мМ и пептидного субстрата, меченного биотином в концентрации 0,5 мМ. Анализ ингибирующего действия соединений на активность EGFR проводят согласно следующему протоколу реакции анализа.
Компонент 1: 4 мкл мутантного фермента EGFR
Компонент 2: 2 мкл раствора соединения
Компонент 3: 4 мкл АТФ и пептида, меченного биотином
Ферментативную реакцию начинают сначала смешиванием Компонента 1 и Компонента 2, и затем добавлением Компонента 3. После реакции при 37°C в течение 2 часов, 10 мл измерительного раствора, состоящего из стрептавидина-XL665 и меченных европием антител против фосфотирозина, предоставленных Cisbio, добавляют к ферментному реакционному раствору и подвергают реакции при комнатной температуре в течение 1 часа. Наконец, соотношение значений флуоресценции при 615 нм и 665 нм получают с использованием оборудования Envision от Perkin-Elmer для количественного измерения активности фермента и подтверждения ингибирующей способности соединений. Значения измерений, измеренные при семи концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем ингибирующей способности соединения.
[Таблица 2]
Как показано в Таблице 2 выше, можно видеть, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность против двойных и тройных мутаций EGFR, EGFR del19/T790M и EGFR del19/T790M/C797S.
Таким образом, соединения, представленные Формулой 1 по настоящему изобретению, обладают превосходным ингибирующим действием против двойных и тройных мутаций EGFR, EGFR del19/T790M и EGFR del19/T790M/C797S, и, таким образом, могут быть с успехом использованы при лечении рака, заболевания, связанного с мутациями EGFR, и, в частности, его можно с успехом использовать при лечении рака, экспрессирующего EGFR del19/T790M/C797S.
<Экспериментальный пример 2> Измерение ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению, против мутации EGFR в клеточной линии Ba/F3
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению, против мутации EGFR в клеточной линии Ba/F3, проводят следующий эксперимент. Результаты показаны в Таблице 3 ниже.
Эксперимент по измерению активности соединений по настоящему изобретению против мутантной клеточной линии Ba/F3 EGFR проводят следующим образом с использованием системы CellTiter-Glo, продаваемой Promega. Анализ CellTiter-Glo представляет собой способ подтверждения жизнеспособности клеток путем измерения АТФ, присутствующего в клетках в клеточной культуре. Для мутантной клеточной линии Ba/F3 EGFR del19, Ba/F3 EGFR del19/T790M, Ba/F3 del19/T790M/C797S приобретают и используют клеточную линию, предоставленную Crown Bioscience. Мутантную клеточную линию Ba/F3 EGFR del19, Ba/F3 EGFR del19/T790M, Ba/F3 del19/T790M/C797S культивируют в RPMI, содержащем 10% FBS и 1% пенициллин-стрептомицин с 1 мкг пуромицина в инкубаторе при 37°С и 5% CO2.
Анализ действия ингибирующей способности соединений против EGFR проводят согласно следующему протоколу реакции анализа.
2500 клеток/90 мкл пассируют и культивируют в 96-луночном культуральном планшете и через 24 часа обрабатывают соединением, представленным Формулой 1, в концентрации 0, 0,01, 0,03, 0,1, 0,3, 1, 3 и 10 (мкМ) . После реакции в течение 72 часов, пластину, обработанную соединением, оставляют при комнатной температуре на 30 минут, затем дополнительно обрабатывают 100 мкл реагента и встряхивают при комнатной температуре в течение 10 минут. Наконец, соотношение значений флуоресценции при 570 нм получают с использованием оборудования для количественного измерения и подтверждения ингибирующей способности соединений. Значения измерений, измеренные при восьми концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем ингибирующей способности соединения.
[Таблица 3]
IC50(мкМ)
IC50(мкМ)
IC50(мкМ)
Как показано в Таблице 3, подтверждено, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность против различных мутаций EGFR, включая тройную мутацию EGFR del19/T790M/C797S.
<Экспериментальный пример 3> Измерение 2 ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению, против мутации EGFR в клеточной линии Ba/F3
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению, против мутации EGFR в клеточной линии Ba/F3, дополнительно проводят следующий эксперимент. Результаты показаны в Таблице 4 ниже.
Эксперимент по измерению активности соединений настоящего изобретения против мутантной клеточной линии Ba/F3 EGFR проводят следующим образом с использованием системы CellTiter-Glo, продаваемой Promega. Анализ CellTiter-Glo представляет собой способ подтверждения жизнеспособности клеток путем измерения АТФ, присутствующего в клетках в клеточной культуре. Используют мутантную клеточную линию Ba/F3 EGFR L858R, Ba/F3 EGFR L858R/T790M, Ba/F3 EGFR L858R/T790M/C797S. Мутантные клеточные линии Ba/F3 EGFR L858R, Ba/F3 EGFR L858R/T790M и Ba/F3 EGFR L858R/T790M/C797S культивируют в инкубаторе в среде RPMI, содержащей 10% FBS и 1% пенициллин-стрептомицин с 1 мкг пуромицина при 37°C и 5% CO2.
Анализ действия ингибирующей способности соединений против EGFR проводят согласно следующему протоколу реакции анализа.
2500 клеток/90 мкл пассируют и культивируют в 96-луночном культуральном планшете и через 24 часа обрабатывают соединением, представленным Формулой 1, в концентрациях 0, 0,01, 0,03, 0,1, 0,3, 1, 3 и 10 (мкМ). После реакции в течение 72 часов, планшет, обработанный соединением, оставляют при комнатной температуре на 30 минут, затем дополнительно обрабатывают 100 мкл реагента и встряхивают при комнатной температуре в течение 10 минут. Наконец, соотношение значений флуоресценции при 570 нм получают с использованием оборудования для количественного измерения и подтверждения ингибирующей способности соединений. Значения измерений, измеренные при восьми концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем ингибирующей способности соединения.
[Таблица 4]
IC50(мкМ)
IC50(мкМ)
IC50(мкМ)
Как показано в Таблице 4, подтверждено, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность против различных мутаций EGFR , включая тройную мутацию Ba/F3 EGFR L858R/T790M/C797S.
Следовательно, соединения, представленные Формулой 1 по настоящему изобретению, демонстрируют высокую ингибирующую способность против мутаций EGFR и, таким образом, могут быть успешно использованы при лечении рака, экспрессирующего мутации EGFR, такие как EGFR del19, EGFR del19/T790M, EGFR del19/T790M/C797S, EGFR L858R, EGFR L858R/T790MS и EGFR L858R/T790M/C797S. В частности, эти соединения демонстрируют превосходную ингибирующую способность против тройной мутации. EGFR del19/T790M/C797S или EGFR L858R/T790M/C797S. Соответственно, соединение, представленное Формулой 1 по настоящему изобретению, также можно успешно использовать при лечении рака, экспрессирующего EGFR del19/T790M/C797S или EGFR L858R/T790M/C797S.
Кроме того, при введении в комбинации с обычным лекарственным средством соединение, представленное Формулой 1 по настоящему изобретению, демонстрирует синергетический эффект и, таким образом, также может быть успешно использовано в комбинации с традиционным лекарственным средством.
<Экспериментальный пример 4> Измерение ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению, против инсерционного мутантного фермента Ex20 EGFR
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению, против инсерционного мутантного фермента Ex20 EGFR, проводят следующий эксперимент. Результаты показаны в Таблице 5 ниже.
Эксперимент по измерению активности соединений настоящего изобретения против мутантных ферментов EGFR проводят следующим образом с использованием системы HTRF, продаваемой Cisbio. Для мутантного фермента EGFR, фермента EGFR A763_Y764insFHEA, приобретают и используют рекомбинантный белок, предоставленный SignalChem.
Состав аналитического буфера, используемого для измерения активности, включает 50 мМ Трис-HCl, pH 7,5, 100 мМ NaCl, 7,5 мМ MgCl2, 3 мМ KCl, 0,01% Tween 20, 0,1% BSA и 1 мМ DTT. В настоящем документе, ферментативную реакцию проводят с использованием АТФ в концентрации 50 мМ и пептидного субстрата, меченного биотином в концентрации 0,5 мМ. Анализ ингибирующего действия соединений на активность EGFR проводят согласно следующему протоколу реакции анализа.
Компонент 1: 4 мкл мутантного фермента EGFR
Компонент 2: 2 мкл раствора соединения
Компонент 3: 4 мкл АТФ и пептида, меченного биотином
Ферментативную реакцию начинают сначала смешиванием Компонента 1 и Компонента 2, и затем добавлением Компонента 3. После реакции при 37°C в течение 2 часов, 10 мл измерительного раствора, состоящего из стрептавидина-XL665 и меченных европием антител против фосфотирозина, предоставленных Cisbio, добавляют к ферментному реакционному раствору и подвергают реакции при комнатной температуре в течение 1 часа. Наконец, соотношение значений флуоресценции при 615 нм и 665 нм получают с использованием оборудования Envision от Perkin-Elmer для количественного измерения активности фермента и подтверждения ингибирующей способности соединений. Значения измерений, измеренные при семи концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем ингибирующей способности соединения.
[Таблица 5]
Как показано в Таблице 5, подтверждено, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность в отношении инсерционной мутации EGFR Ex20 , мутации EGFR A763_Y764insFHEA.
<Экспериментальный пример 5> Измерение ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению против роста клеток инсерционного мутанта EGFR Ex20 Ba/F3
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению против роста клеток инсерционного мутанта EGFR Ex20 Ba/F3, проводят следующий эксперимент. Результаты показаны в Таблице 6 ниже.
Эксперимент по измерению активности соединений по настоящему изобретению против мутантной клеточной линии Ba/F3 EGFR V769_D770insASV проводят следующим образом с использованием системы CellTiter-Glo, продаваемой Promega. Анализ CellTiter-Glo представляет собой способ подтверждения жизнеспособности клеток путем измерения АТФ, присутствующего в клетках в клеточной культуре. Для линии мутантных клеток Ba/F3 EGFR V769_D770insASV приобретают и используют клеточную линию, предоставленную Crown Bioscience. Мутантную клеточную линию Ba/F3 EGFR V769_D770insASV культивируют в RPMI, содержащем 10% FBS и 1% пенициллин-стрептомицин с 1 мкг/мл пуромицина в инкубаторе при 37°С и 5% CO2.
Анализ действия ингибирующей способности соединений на рост мутантных клеток EGFR проводят в соответствии со следующим протоколом реакции анализа.
1000 клеток/100 мкл пассируют и культивируют в 96-луночном культуральном планшете и через 24 часа обрабатывают соединением, представленным Формулой 1, в концентрации 0, 0,01, 0,03, 0,1, 0,3, 1, 3 и 10 (мкМ). После реакции в течение 72 часов, планшет, обработанный соединением, оставляют при комнатной температуре на 30 минут, затем дополнительно обрабатывают 100 мкл реагента и встряхивают при комнатной температуре в течение 10 минут. Наконец, соотношение значений флуоресценции при 570 нм получают с использованием оборудования для количественного измерения и подтверждения способности соединений ингибировать рост клеток. Значения измерений, измеренные при восьми концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем способности соединения ингибировать рост клеток.
[Таблица 6]
Как показано в Таблице 6, подтверждено, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность против роста мутантных клеток Ba/F3 EGFR V769_D770insASV.
<Экспериментальный пример 6> Измерение ингибирующей способности соединений, представленных Формулой 1 по настоящему изобретению, против мутации HER2.
Чтобы подтвердить ингибирующую способность соединений, представленных Формулой 1 по настоящему изобретению, против мутации HER2, проводят следующий эксперимент. Результаты показаны в Таблице 7 ниже.
Эксперимент по измерению активности соединений настоящего изобретения против мутантных ферментов HER проводят следующим образом с использованием системы HTRF, продаваемой Cisbio. Для мутантного фермента HER2, мутантного фермента HER2 A775_G776insYVMA, приобретают и используют рекомбинантный белок, предоставленный Carna Biosciences.
Состав аналитического буфера, используемого для измерения активности, включает 50 мМ Трис-HCl, pH 7,5, 100 мМ NaCl, 7,5 мМ MgCl2, 3 мМ KCl, 0,01% Tween 20, 0,1% BSA и 1 мМ DTT. В настоящем документе, ферментативную реакцию проводят с использованием АТФ в концентрации 50 мМ и пептидного субстрата, меченного биотином, в концентрации 0,5 мМ. Анализ ингибирующего действия соединений против активности мутанта HER2 A775_G776insYVMA проводят согласно следующему протоколу реакции анализа.
Компонент 1: 4 мкл мутантного фермента HER2 A775_G776insYVMA
Компонент 2: 2 мкл раствора соединения
Компонент 3: 4 мкл АТФ и пептида, меченного биотином
Ферментативную реакцию начинают сначала смешиванием Компонента 1 и Компонента 2, и затем добавлением Компонента 3. После реакции при 37°C в течение 2 часов, 10 мл измерительного раствора, состоящего из стрептавидина-XL665 и меченных европием антител против фосфотирозина, предоставленных Cisbio, добавляют к ферментному реакционному раствору и подвергают реакции при комнатной температуре в течение 1 часа. Наконец, соотношение значений флуоресценции при 615 нм и 665 нм получают с использованием оборудования Envision от Perkin-Elmer для количественного измерения активности фермента и подтверждения ингибирующей способности соединений. Значения измерений, измеренные при семи концентрациях соединения, анализируют с использованием программы Prism (версия 5,01, Graphpad Software, Inc.) и рассчитывают значение IC50, которое является показателем ингибирующей способности соединения.
[Таблица 7]
Как показано в Таблице 7, подтверждено, что примеры соединений по настоящему изобретению демонстрируют высокую ингибирующую способность в отношении мутации HER2 A775_G776insYVMA. Следовательно, соединение, представленное Формулой 1 по настоящему изобретению, демонстрируют высокую ингибирующую способность против мутации EGFR и, таким образом, может быть успешно использовано при лечении рака, экспрессирующего мутации EGFR, такие как EGFR del19/T790M, EGFR del19/T790M/C797S и EGFR A763_Y764insFHEA, Ba/F3 EGFR del19, Ba/F3 EGFR del19/T790M, Ba/F3 EGFR del19/T790M/C797S, Ba/F3 EGFR L858R, Ba/F3 EGFR L858R/T790M, Ba/F3 EGFR L858R/T790M/C797S, Ba/F3 EGFR A763_Y764insFHEA, Ba/F3 EGFR V769_D770insASV и Ba/F3 EGFR D770_N771insSVD.
Кроме того, соединения, представленные Формулой 1 по настоящему изобретению, демонстрируют высокую ингибирующую способность против мутации HER2 и, таким образом, также могут быть успешно использованы при лечении рака, экспрессирующего мутации HER2, такие как HER2 A775_G776insYVMA, Ba/F3 HER2 A775_G776insYVMA и Ba/F3 HER2 G776_delinsVC.
Из приведенных выше результатов, видно, что производное пиримидин-2,4-диамина по настоящему изобретению может эффективно ингибировать мутации EGFR и HER2 и, таким образом, может быть успешно использовано в качестве фармацевтической композиции для профилактики или лечения рака.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
| НОВОЕ ЗАМЕЩЕННОЕ ДЕЙТЕРИЕМ ПРОИЗВОДНОЕ ПИРИМИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2811770C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА С ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ПРОТЕИНКИНАЗ И СОДЕРЖАЩАЯ ЕГО ТЕРАПЕВТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2833427C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2015 |
|
RU2727700C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| МОДУЛЯТОРЫ ПРОТЕИН-ТИРОЗИНКИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2656591C2 |
| ПРОИЗВОДНЫЕ 2,4-ДИЗАМЕЩЕННОГО ФЕНИЛЕН-1,5-ДИАМИНА И ИХ ПРИМЕНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КОМПОЗИЦИИ, ПОЛУЧЕННЫЕ ИЗ НИХ | 2015 |
|
RU2649001C1 |
Изобретение относится к производному пиримидин-2,4-диамина, представленному формулой 1, способу его получения и фармацевтической композиции для профилактики или лечения рака, содержащей его в качестве активного ингредиента. В формуле 1 X представляет собой сульфонил или карбонил; R1 представляет собой галоген, C1-10 алкил с прямой или разветвленной цепью, незамещенный или замещенный одним или несколькими галогенами, карбоксил или С1-10 алкоксикарбонил с прямой или разветвленной цепью; R2 представляет собой -NRb1Rb2, Rb1 и Rb2 каждый независимо представляет собой, незамещенный или замещенный С1-10 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, где заместитель замещенного С1-10 алкила с прямой или разветвленной цепью может представлять собой -NRc1Rc2, и заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-10 алкил с прямой или разветвленной цепью, и Rc1 и Rc2 каждый независимо представляет собой C1-10 алкил с прямой или разветвленной цепью, или Rc1 и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, содержащего 1 или 2 гетероатома, представляющих собой N, где заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой C1-10 алкил с прямой или разветвленной цепью; и R3, R5 и R6 каждый независимо представляет собой водород, или С1-10 алкокси с прямой или разветвленной цепью; R4 представляет собой С1-10 алкил с прямой или разветвленной цепью; R7 и R8 каждый независимо представляет собой водород; n представляет собой целое число от 0 до 3; и р и q каждый независимо представляет собой целое число от 1 до 3. Производное пиримидин-2,4-диамина демонстрирует высокую ингибирующую способность против мутаций EGFR и HER2 и может быть успешно использовано при лечении рака, при котором возникли мутации EGFR и HER2. 3 н. и 9 з.п. ф-лы, 7 табл., 53 пр.
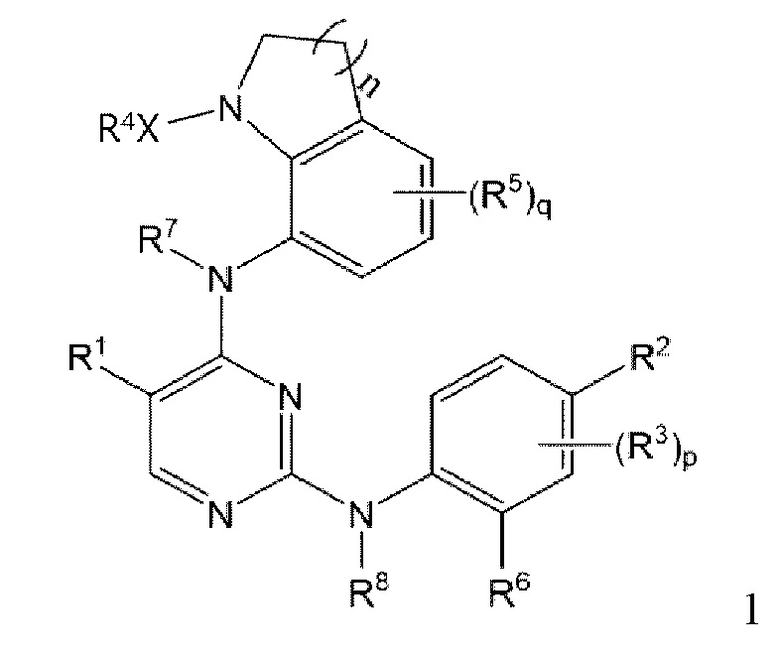
1. Соединение, представленное Формулой 1, или его фармацевтически приемлемая соль:
[Формула 1]
в Формуле 1,
X представляет собой сульфонил или карбонил;
R1 представляет собой галоген, C1-10 алкил с прямой или разветвленной цепью, незамещенный или замещенный одним или несколькими галогенами, карбоксил или С1-10 алкоксикарбонил с прямой или разветвленной цепью;
R2 представляет собой -NRblRb2,
Rb1 и Rb2 каждый независимо представляет собой незамещенный или замещенный С1-10 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, где заместитель замещенного С1-10 алкила с прямой или разветвленной цепью может представлять собой -NRclRc2, и заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-10 алкил с прямой или разветвленной цепью, и
Rc1 и Rc2 каждый независимо представляет собой C1-10 алкил с прямой или разветвленной цепью, или Rcl и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 3-7-членного гетероциклоалкила, содержащего 1 или 2 гетероатома, представляющих собой N, где заместитель замещенного 3-7-членного гетероциклоалкила может представлять собой C1-10 алкил с прямой или разветвленной цепью; и
R3, R5 и R6 каждый независимо представляет собой водород, или С1-10 алкокси с прямой или разветвленной цепью;
R4 представляет собой С1-10 алкил с прямой или разветвленной цепью;
R7 и R8 каждый независимо представляет собой водород;
n представляет собой целое число от 0 до 3; и
р и q каждый независимо представляет собой целое число от 1 до 3.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где
R1 представляет собой галоген, С1-3 алкил с прямой или разветвленной цепью, незамещенный или замещенный одним или несколькими галогенами, или C1-3 алкоксикарбонил с прямой или разветвленной цепью;
R2 представляет собой -NRb1Rb2,
Rb1 и Rb2 каждый независимо представляет собой незамещенный или замещенный C1-3 алкил с прямой или разветвленной цепью, или Rb1 и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 5-6-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, где заместитель замещенного C1-3 алкила с прямой или разветвленной цепью может представлять собой -NRc1Rc2, и заместитель замещенного 5-6-членного гетероциклоалкила может представлять собой -NRc1Rc2 или C1-3 алкил с прямой или разветвленной цепью, и
Rc1 и Rc2 каждый независимо представляет собой C1-3 алкил с прямой или разветвленной цепью, или Rcl и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 5-6-членного гетероциклоалкила, содержащего 1 или 2 гетероатома, представляющего собой N, где заместитель замещенного 5-6-членного гетероциклоалкила может представлять собой С1-3 алкил с прямой или разветвленной цепью; и
R3 и R5 каждый независимо представляет собой водород;
R4 представляет собой С1-3 алкил с прямой или разветвленной цепью;
R6 представляет собой С1-3 алкокси с прямой или разветвленной цепью; и
n представляет собой целое число от 1 до 3.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой галоген, С1-2 алкил,
незамещенный или замещенный одним или несколькими галогенами или С1-3 алкоксикарбонил;
R2 представляет собой -NRblRb2,
Rb1 и Rb2 каждый независимо представляет собой незамещенный или замещенный С1-2 алкил, или Rbl и Rb2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 6-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, где заместитель замещенного C1-2 алкила может представлять собой -NRclRc2, и заместитель замещенного 6-членного гетероциклоалкила может представлять собой -NRclRc2 или С1-2 алкил, и
Rcl и Rc2 каждый независимо представляет собой С1-2 алкил, или Rcl и Rc2 взяты вместе с атомом азота, к которому они присоединены, с образованием незамещенного или замещенного 6-членного гетероциклоалкила, содержащего 2 гетероатома, представляющих собой N, где заместитель замещенного 6-членного гетероциклоалкила может представлять собой С1-2 алкил; и
R3 и R5 каждый независимо представляет собой водород;
R4 представляет собой С1-2 алкил;
R6 представляет собой метокси;
R7 и R8 каждый независимо представляет собой водород; и
n равен 1 или 2.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где
R1 представляет собой метил, F, Cl, CF3,  или
или 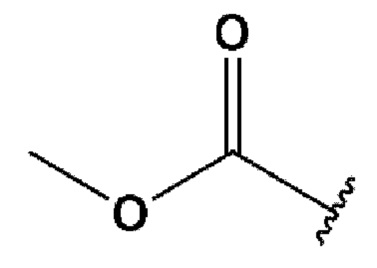 ;
;
R2 представляет собой 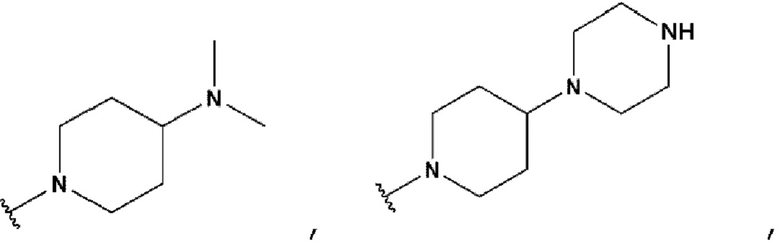
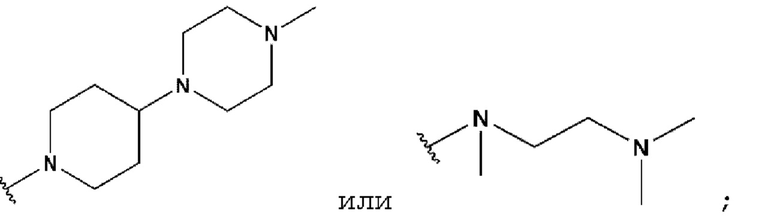
R3 представляет собой водород;
R4 представляет собой метил или этил;
R5 представляет собой водород;
R6 представляет собой метокси;
R7 представляет собой водород;
R8 представляет собой водород; и
n равен 1 или 2.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение, представленное Формулой 1, представляет собой любое из соединений, выбранных из группы, состоящей из следующих соединений:
<1> 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<2> 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<3> 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<4> 5-хлор-N2-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<5> 5-хлор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<6> 5-хлор-N2-(4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)-N4-(1-(метилсульфонил)-1,2,3,4-тетрагидрохинолин-8-ил)пиримидин-2,4-диамин;
<7> N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-5-метил-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<8> 5-фтор-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин;
<9> N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)-5-(трифторметил)пиримидин-2,4-диамин;
<10> изопропил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилат;
<11> метил 2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)-4-((1-(метилсульфонил)индолин-7-ил)амино)пиримидин-5-карбоксилат;
<12> 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино) пиримидин-4-ил)амино)индолин-1-ил)этан-1-он;
<13> 5-хлор-N4-(1-(этилсульфонил)индолин-7-ил)-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пиримидин-2,4-диамин;
<14> 1-(7-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)индолин-1-ил)пропан-1-он;
<15> 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2Н)-ил)этан-1-он;
<16> 1-(8-((5-хлор-2-((2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)амино)пиримидин-4-ил)амино)-3,4-дигидрохинолин-1(2Н)-ил)пропан-1-он; и
<17> 5-хлор-N2-(2-метокси-4-(4-(пиперазин-1-ил)пиперидин-1-ил)фенил)-N4-(1-(метилсульфонил)индолин-7-ил)пиримидин-2,4-диамин.
6. Способ получения соединения, представленного Формулой 1 по п. 1, включающий:
взаимодействие соединения, представленного Формулой 2, с соединением, представленным Формулой 3, с получением соединения, представленного Формулой 1, как показано на Схеме реакции 1 ниже:
[Схема реакции 1]
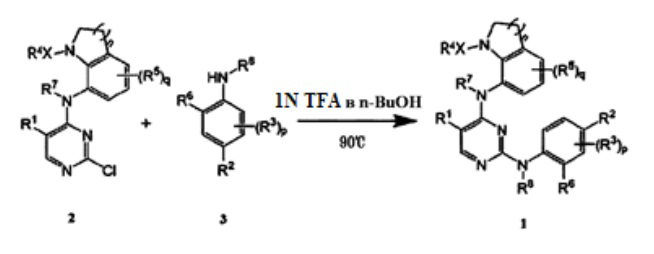
в Схеме реакции 1 X, R1, R2, R3, R4, R5, R6, R7, R8, n, р и q такие, как определены в Формуле 1 по п. 1.
7. Фармацевтическая композиция для профилактики или лечения рака, содержащая соединение, представленное Формулой 1, или его фармацевтически приемлемую соль по п. 1 в качестве активного ингредиента, где рак представляет собой рак, экспрессирующий мутацию EGFR или HER2.
8. Фармацевтическая композиция по п. 7, где соединение ингибирует мутации EGFR (рецептора эпидермального фактора роста) или HER2.
9. Фармацевтическая композиция по п. 8, отличающаяся тем, что
мутация EGFR представляет собой по меньшей мере одну мутацию, выбранную из группы, состоящей из EGFR de119, EGFR Т790М, EGFR C797S, EGFR L858R, EGFR Ex20 инсерционных мутаций, EGFR A763_Y764insFHEA, EGFR V769_D770insASV и EGFR D770_N771insSVD; и
мутация HER2 представляет собой по меньшей мере одну мутацию, выбранную из группы, состоящей из HER2 A775_G776insYVMA, HER2 G776_delinsVC.
10. Фармацевтическая композиция по п. 7, где рак представляет собой по меньшей мере один рак, выбранный из группы, состоящей из псевдомиксомы, рака внутрипеченочных желчных путей, гепатобластомы, рака печени, рака щитовидной железы, рака толстой кишки, рака яичек, миелодиспластического синдрома, глиобластомы, рака полости рта, рака губы, грибовидного микоза, острого миелоидного лейкоза, острого лимфоцитарного лейкоза, базальноклеточного рака, рака эпителия яичников, герминогенного рака яичников, рака молочной железы, рака головного мозга, аденомы гипофиза, множественной миеломы, рака желчного пузыря, рака желчевыводящих путей, рака толстой кишки, хронического миелоидного лейкоза, хронического лимфоцитарного лейкоза, ретинобластомы, меланомы хориоидеи, рака фатерового соска, рака мочевого пузыря, рака брюшины, рака паращитовидной железы, рака надпочечников, рака придаточных пазух носа, немелкоклеточного рака легкого, рака языка, астроцитомы, мелкоклеточного рака легкого, детского рака головного мозга, детской лимфомы, детского лейкоза, рака тонкой кишки, менингиомы, рака пищевода, глиомы, рака почечной лоханки, рака почки, рака сердца, рака двенадцатиперстной кишки, злокачественного рака мягких тканей, злокачественного рака кости, злокачественной лимфомы, злокачественной мезотелиомы, злокачественной меланомы, рака глаза, рака вульвы, рака мочеточника, рака уретры, рака неизвестной первичной локализации, лимфомы желудка, рака желудка, карциноида желудка, стромального рака желудочно-кишечного тракта, рака Вильмса, рака молочной железы, саркомы, рака полового члена, рака глотки, гестационной трофобластической болезни, рака шейки матки, рака эндометрия, саркомы матки, рака предстательной железы, метастатического рака кости, метастатического рака головного мозга, рака средостения, рака прямой кишки, карциноида прямой кишки, рака влагалища, рака спинного мозга, акустической невромы, рака поджелудочной железы, рака слюнной железы, саркомы Капоши, болезни Педжета, рака небных миндалин, плоскоклеточного рака, аденокарциномы легких, рака легких, плоскоклеточного рака легких, рака кожи, анального рака, рабдомиосаркомы, рака гортани, рака плевры, рака крови и рака тимуса.
11. Фармацевтическая композиция по п. 7, где соединение ингибирует мутации EGFR (рецептора эпидермального фактора роста) и мутации HER2.
12. Фармацевтическая композиция по п. 7, где противораковый эффект усиливается введением фармацевтической композиции в комбинации с противораковым агентом.
| KR 1020190114910 A, 10.10.2019 | |||
| WO 2016022460 A1, 11.02.2016 | |||
| WO 2008118822 A1, 02.10.2008 | |||
| KR 1020180135781 A, 21.12.2018 | |||
| Комбинация ингибитора EGFR и ингибитора MEK для применения в лечении рака, вызванного мутировавшим NRAS | 2015 |
|
RU2683276C2 |

