Область техники, к которой относится изобретение
Настоящее изобретение касается нового способа синтеза билирубина.
Предшествующий уровень техники
Билирубин представляет собой компонент желчи и генерируется в организме главным образом из гемоглобина. Билирубин представляет собой желтоватый финальный метаболит, образующийся из гема, и, несмотря на множество гидроксильных групп, он чрезвычайно гидрофобный из-за внутримолекулярных водородных связей.
Билирубин считался нежелательным веществом, поскольку он вызывает желтуху, когда его концентрация в крови повышена. Однако в недавно опубликованном исследовании было обнаружено, что слегка повышенное содержание билирубина в крови значительно снижает риск развития сердечно-сосудистых заболеваний или рака. Кроме того, был подтвержден тканезащищающий эффект билирубина в экспериментах на животных, поскольку он удаляет активный кислород и контролирует иммуноциты, связанные с воспалением.
Несмотря на то, что билирубин представляет собой промышленно используемое вещество, его получали экстракцией из животного сырья, и он никогда не был успешно синтезирован. Когда билирубин экстрагируют из животного сырья, сложно получить билирубин в больших количествах из-за высоких производственных затрат. Кроме того, поскольку билирубин экстрагируется из животного сырья в виде смеси трех региоизомеров, необходимо проводить дополнительное разделение и очистку, чтобы иметь возможность применять его в медицине. Имеется острая потребность в разработке способа химического получения билирубина.
Краткое описание изобретения
Проблемы, на решение которых направлено изобретение
Целью настоящего изобретения является разработка способа синтеза билирубина.
Способы решения проблем
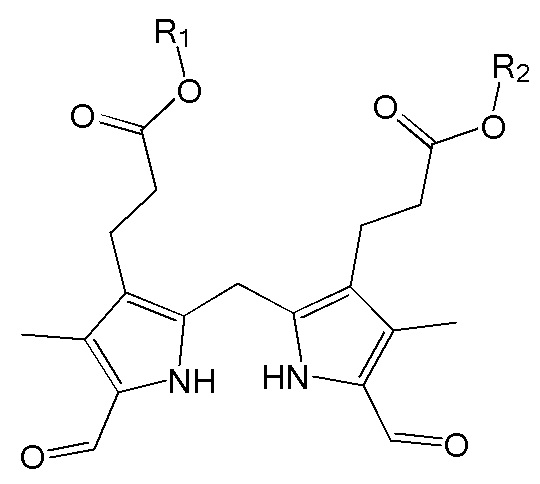
1. Способ синтеза билирубина, включающий получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2:
[Формула 1]
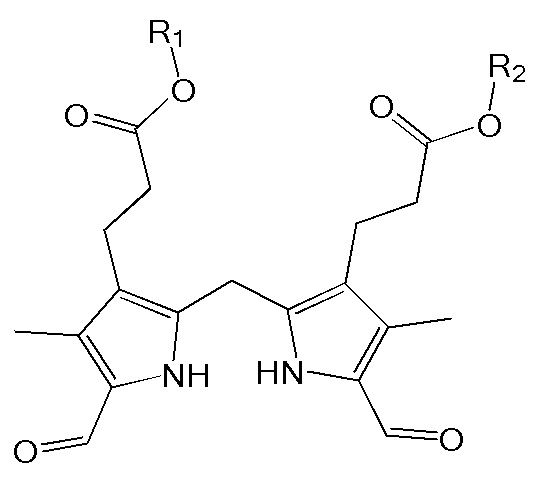
[Формула 2]
[Формула 3]
 ,
,
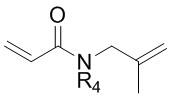
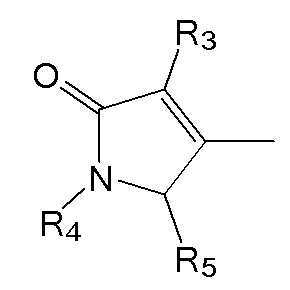
где в формулах 1, 2 и 3, R1 и R2 каждый независимо представляют собой атом водорода, алкильную группу, содержащую 1 - 12 атомов углерода, арильную группу, содержащую 6 - 20 атомов углерода, гетероарильную группу, содержащую 2 - 20 атомов углерода, арилалкильную группу, содержащую 7 - 20 атомов углерода, или гетероарилалкильную группу, содержащую 3 - 20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу.
2. Способ по п. 1, дополнительно включающий реакцию соединения формулы 3 с полиэтиленгликолем (ПЭГ).
3. Способ по п. 1, где соединение формулы 1 реагирует с полиэтиленгликолем (ПЭГ) и затем вступает в реакцию сочетания с соединением формулы 2.
4. Способ по п. 1, дополнительно включающий димеризацию изображенного ниже соединения формулы 6 с получением соединения формулы 1:
[Формула 6]
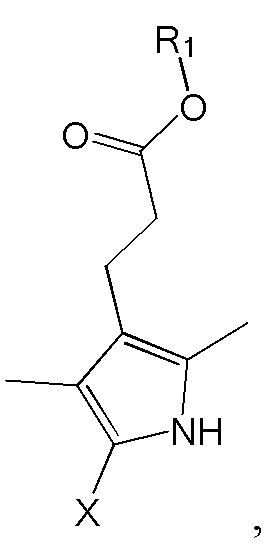
где R1 такой же как R1 в формуле 1, x представляет собой арилалкил сложноэфирную группу, содержащую 8 - 20 атомов углерода, -CH2OH, -COOH, атом галогена или атом водорода.
5. Способ по п. 1, дополнительно включающий проведение циклизации изображенного ниже соединения формулы 8 с получением соединения формулы 2:
[Формула 8]
 ,
,
где R4 такой же как R4 в формуле 2.
6. Способ по п. 1, дополнительно включающий восстановление ацетильной группы в изображенном ниже соединении формулы 11 с получением соединения формулы 2:
[Формула 11]
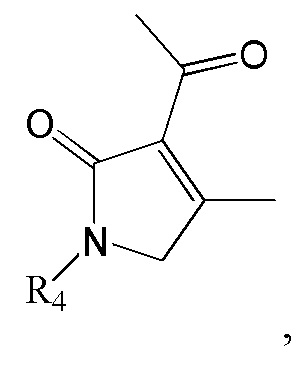
где R4 такой же как R4 в формуле 2.
7. Способ по п. 1, дополнительно включающий дегидратацию гидроксильной группы в изображенном ниже соединении формулы 12 с получением соединения формулы 2:
[Формула 12]
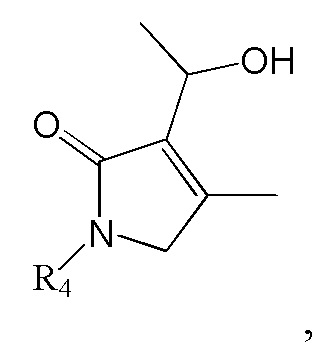 ,
,
где R4 такой же как R4 в формуле 2.
8. Способ по п. 1, дополнительно включающий проведение циклизации изображенного ниже соединения формулы 13 с получением соединения формулы 2:
[Формула 13]
 ,
,
где Y представляет собой селенид, и R4 такой же как R4 в формуле 2.
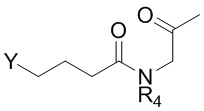
9. Способ по п. 1, дополнительно включающий окисление изображенного ниже соединения формулы 14 с получением соединения формулы 2:
[Формула 14]
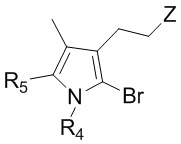 ,
,
где Z представляет собой сульфид, R4 и R5 такие же как R4 и R5 в формуле 2.
10. Способ по п. 1, дополнительно включающий получение билирубина из соединения формулы 3.
11. Способ по п. 1, где стадию получения соединения проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан.
12. Способ по п. 1, где стадию получения соединения проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды.
13. Способ по п. 1, где стадию получения соединения проводят при температуре от -20 до 200°C.
14. Способ по п. 1, где стадию получения соединения проводят в течение 0.5 - 120 часов.
15. Способ по п. 11, где основание добавляют в количестве от 2 до 20 моль относительно 1 моля соединения формулы 1.
Выгодные эффекты изобретения
Способ синтеза билирубина по настоящему изобретению можно экономически выгодно использовать в мягких условиях.
Способ синтеза билирубина по настоящему изобретению обеспечивает высокий выход и подходит для крупномасштабного производства.
Краткое описание чертежей
Фиг. 1 - 5 представляют собой 2D ЯМР-спектры соединения F-3a, полученного в Примере 29, где фиг. 2 представляет собой HSQC спектр F-3a; фиг. 3 представляет собой COSY спектр; фиг. 4 представляет собой HMBC спектр; и фиг. 5 представляет собой NOESY 2D ЯМР спектр.
Фиг. 6 представляет собой масс-спектр соединения F-3a, полученного в Примере 54.
Фиг. 7 представляет собой LCMS спектр соединения D-Ed, полученного в Примере 59.
Фиг. 8 представляет собой LCMS спектр соединения F-3a, полученного в Примере 60.
Фиг. 9 представляет собой LCMS спектр соединения F-3a, полученного в Примере 76.
Фиг. 10 представляет собой масс-спектр соединения FP-3a, полученного в Примере 77.
Фиг. 11 представляет собой ВЭЖХ хроматограмму соединения FdP-3a, полученного в Примере 90.
Фиг. 12 - 14 представляют собой масс-спектры соединения DF, полученного в Примере 93.
Фиг. 15 представляет собой масс-спектр соединения FP-3a, полученного в Примере 94.
Способ реализации изобретения
Настоящее изобретение касается нового способа синтеза билирубина.
При использовании в настоящем тексте, термин «алкил» означает линейный или разветвленный, замещенный или незамещенный углеводород, такой как метил, этил, н-пропил, изопропил, циклопропил, н-бутил, втор-бутил, трет-бутил, циклобутил, циклопропилметил, н-пентил, изопентил, неопентил, трет-пентил, циклопентил, циклобутилметил, н-гексил, изогексил, циклогексил, циклопентилметил и т.п.
Термин «циклоалкил» означает моноциклический или бициклический, замещенный или незамещенный циклический углеводород, такой как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.п.
Термин «гетероциклоалкил» означает моноциклический или бициклический, замещенный или незамещенный циклический углеводород, содержащий один или больше гетероатомов, выбранных из B, N, O, S, P(=O), Si и P, такой как тетрагидропиранил, азетидил, 1,4-диоксанил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, дигидрофуранил, дигидроимидазолил, дигидроиндолил, дигидроизоксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил, дигидропиразинил, дигидропиразолил, дигидропиридил, дигидропиримидинил, дигидропирролил, дигидрохинолил, дигидротетразолил, дигидротиадиазолил, дигидротиазолил, дигидротиенил, дигидротриазолил, дигидро-азетидил, метилендиоксибензоил, тетрагидрофуранил, тетрагидротиенил и т.п.
Термин «арил» означает моноциклическую или бициклическую, замещенную или незамещенную ароматическую группу, такую как фенил, бифенил, терфенил, нафтил, бинафтил, фенилнафтил, нафтилфенил, фенилтерфенил, флуоренил, фенилфлуоренил, дифенилфлуоренил, бензофлуоренил, дибензофлуоренил, фенантренил, фенилфенантренил, антраценил, инденил, трифениленил, пиренил, тетраценил, периленил, хризенил, нафтаценил, флуоратенил, спиробифлуоренил, азиуренил и т.п.
«Арил» включает, например, фенил, 1-нафтил, 2-нафтил, 1-антрил, 2-антрил, 9-антрил, бензантрил, 1-фенантрил, 2-фенантрил, 3-фенантрил, 4-фенантрил, 9-фенантрил, нафтацетил, пиренил, 1-хризенил, 2-хризенил, 3-хризенил, 4-хризенил, 5-хризенил, 6-хризенил, бензо[c]фенантрил, бензо[g]хризенил, 1-трифениленил, 2-трифениленил, 3-трифениленил, 4-трифениленил, 1-флуоренил, 2-флуоренил, 3-флуоренил, 4-флуоренил, 9-флуоренил, бензофлуоренил, дибензофлуоренил, 2-бифенилил, 3-бифенилил, 4-бифенилил, o-терфенил, м-терфенил-4-ил, м-терфенил-3-ил, м-терфенил-2-ил, п-терфенил-4-ил, п-терфенил-3-ил, п-терфенил-2-ил, м-кватерфенил, 3-фторантенил, 4-фторантенил, 8-фторантенил, 9-фторантенил, бензофторантенил, o-толил, м-толил, п-толил, 2,3-ксилил, 3,4-ксилил, 2,5-ксилил, мезитил, o-куменил, м-куменил, п-куменил, п-трет-бутилфенил, п-(2-фенилпропил)фенил, 4’-метилбифенилил, 4”-трет-бутил-п-терфенил-4-ил, 9,9-диметил-1-флуоренил, 9,9-диметил-2-флуоренил, 9,9-диметил-3-флуоренил, 9,9-диметил-4-флуоренил, 9,9-дифенил-1-флуоренил, 9,9-дифенил-2-флуоренил, 9,9-дифенил-3-флуоренил, 9,9-дифенил-4-флуоренил и т.п.
Термин «гетероарил» означает моноциклическую или бициклическую, замещенную или незамещенную ароматическую группу, содержащую один или больше гетероатомов, выбранных из B, N, O, S, P(=O), Si и P, такую как бензотиенил, бензоксазолил, бензофуранил, бензимидазолил, бензтиазолил, бензотриазолил, синнолинил, фурил, имидазолил, тетразолил, индазолил, индолил, изоксазолил, изохинолинил, изотиазолил, нафтиридинил, оксадиазолил, оксазолил, изоксазолил, пуринил, тиазолил, изотиазолил, тиенопиридинил, тиенил, тиадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, пиридо[2,3-d]пиримидинил, пирроло[2,3-b]пиридинил, хиназолинил, хинолинил, тиено[2,3-c]пиридинил, триазинил и т.п.
Термин «арилалкил» означает алкильную группу, в которой по меньшей мере один из заместителей замещен арилом, где «арил» и «алкил» имеют приведенные выше значения. Например, данный термин включает бензил, фенилэтил, фенилпропил, фенилбутил, фенилгексил, нафтилэтил, нафтилпропил, нафтилбутил, нафтилгексил, антраценилметил, антраценилэтил, антраценилпропил, антраценилбутил, фенантрилметил, фенантрилэтил, фенантрилпропил, трифенилметил, трифенилэтил, трифенилпропил, пиренилметил, пиренилэтил, пиренилпропил, фенилантраценметил, фенилантраценэтил, фенилантраценпропил, периленилметил, периленилэтил, периленилпропил, хризенилметил, хризенилэтил, хризенилпропил, флуоренилметил, флуоренилэтил, флуоренилпропил и т.п.
Термин «гетероарилалкил» означает алкильную группу, в которой по меньшей мере один из заместителей замещен гетероарилом, где гетероарил и алкил имеют приведенные выше значения. Например, данный термин включает пиридинилметил, пиридинилэтил, пиридинилпропил, пиридинилбутил, пиримидинилметил, пиримидинилэтил, пиримидинилпропил, пиразолилметил, пиразолилэтил, пиразолилметил, пиразолилэтил, пиразолилпропил, хинолинилметил, хинолинилэтил, хинолинилпропил и т.п.
Термин «замещенный» означает наличие по меньшей мере одного заместителя, например, одного или двух или больше заместителей, выбранных из атома галогена, нитрогруппы, гидроксила, циано-группы, аминогруппы, тиола, карбоксила, амида, нитрила, сульфида, дисульфида, сульфенила, формила, формилокси-группы, формиламино-группы, арила или замещенного арила.
В случае когда не описано заместителей в формуле по настоящему изобретению, при том что есть сайт, требующий заместителя, считается, что опущен водородный заместитель.
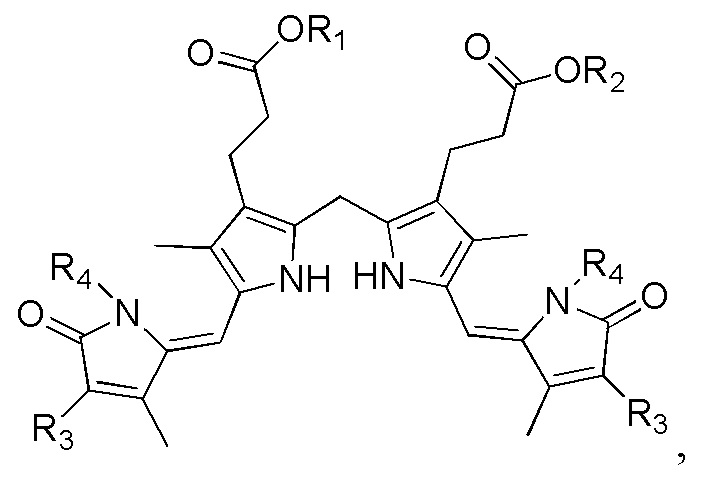
Настоящее изобретение касается способа синтеза билирубина, который включает получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2.
[Формула 1]
[Формула 2]
[Формула 3]
 ,
,
где в формулах 1, 2 и 3, R1 и R2 каждый независимо представляют собой атом водорода, алкильную группу, содержащую 1 - 12 атомов углерода, арильную группу, содержащую 6 - 20 атомов углерода, гетероарильную группу, содержащую 2 - 20 атомов углерода, арилалкильную группу, содержащую 7 - 20 атомов углерода, или гетероарилалкильную группу, содержащую 3 - 20 атомов углерода.
Число атомов углерода в R1 и R2 можно надлежащим образом выбирать из диапазона, который не влияет на реакцию сочетания между соединением формулы 1 и соединением формулы 2.
Например, R1 и R2 каждый независимо могут представлять собой алкильную группу, содержащую 1 - 10 атомов углерода, арильную группу, содержащую 6 - 10 атомов углерода, гетероарильную группу, содержащую 2 - 10 атомов углерода, арилалкильную группу, содержащую 7 - 10 атомов углерода, или гетероарилалкильную группу, содержащую 3 - 10 атомов углерода.
Кроме того, R1 и R2 каждый независимо могут быть выбраны из алкильной группы, содержащей 1 - 5 атомов углерода, арильной группы, содержащей 6 - 10 атомов углерода, гетероарильной группы, содержащей 4 - 10 атомов углерода, арилалкильной группы, содержащей 7 - 10 атомов углерода, или гетероарилалкильной группы, содержащей 5 - 10 атомов углерода.
R3 может представлять собой атом водорода, винильную или ацетильную группу; или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом.
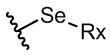
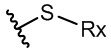
В настоящем тексте селенид представляет собой функциональную группу, содержащую структуру изображенной ниже формулы 4, а сульфид представляет собой функциональную группу, содержащую структуру изображенной ниже формулы 5.
[Формула 4]
[Формула 5]
 ,
,
где в формулах 4 и 5, RX может представлять собой атом водорода или замещенную или незамещенную, линейную или разветвленную алкильную, циклоалкильную, гетероциклоалкильную, арильную, гетероарильную, арилалкильную или гетероарилалкильную группу.
Например, RX представляет собой алкильную группу, содержащую 1 - 12 атомов углерода, циклоалкильную группу, содержащую 5 - 20 атомов углерода, гетероциклоалкильную группу, содержащую 2 - 20 атомов углерода, арильную группу, содержащую 5 - 20 атомов углерода, гетероарильную группу, содержащую 2 - 20 атомов углерода, арилалкильную группу, содержащую 6 - 20 атомов углерода, или гетероциклоалкильную группу, содержащую 3 - 20 атомов углерода.
Например, RX представляет собой фенильную или п-толильную группу.
R3 может представлять собой этильную группу, замещенную гидроксильной группой, например, может представлять собой функциональную группу, в которой гидроксильная группа является заместителем у углерода в положении 1 этильной группы.
R3 может представлять собой этильную группу, замещенную селенидом, например, может представлять собой функциональную группу, в которой селенид является заместителем у углерода в положении 2 этильной группы.
R3 может представлять собой этильную группу, замещенную сульфидом, например, может представлять собой функциональную группу, в которой сульфид является заместителем у углерода в положении 2 этильной группы.
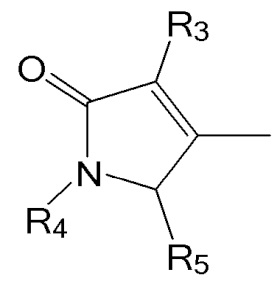
R4 представляет собой атом водорода или азот-защитную группу.
В настоящем тексте, азот-защитная группа не ограничивается какой-то определенной группой, при условии что она представляет собой заместитель, способный защитить атом азота, с которым связан R4, и например, она может быть выбрана из группы, состоящей из -COORx (Rx имеет указанное выше значение), трет-бутилоксикарбонила (Boc), тритила (-CPh3), тозила (SOOPhCH3), 9-флуоренилметилоксикарбонила (Fmoc), карбоксибензильной группы (Cbz), п-метоксибензилкарбонила (Moz), ацетила (Ac), бензоила (Bz), п-метоксибензила (PMB), 3,4-диметоксибензила (DMPM), п-метоксифенила (PMP), 2-нафтилметилового эфира (Nap) и трихлорэтил хлорформиата (Troc).
R5 представляет собой атом водорода, тозильную (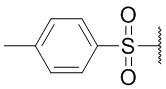 , Ts или Tos) или метильную группу (
, Ts или Tos) или метильную группу (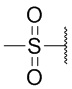 , Ms).
, Ms).
Когда азот-защитная группа R4 в формуле 2 сохраняется после реакции сочетания соединения формулы 1 и соединения формулы 2, может потребоваться дополнительная стадия снятия азот-защитной группы.
Соединение формулы 1 и соединение формулы 2 могут связываться в мольном соотношении 1:2. Соединение формулы 1 и соединение формулы 2 можно вводить в реакцию в мольном соотношении 1: 2 - 10, 1: 2 - 5, 1: 2 - 4 или 1: 2 - 3, в зависимости от конкретной реакции.
Реакцию сочетания проводят в присутствии растворителя и основания.
Растворитель представляет собой неорганический растворитель или органический растворитель. Органический растворитель может представлять собой, например, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы или амиды. Растворители, принадлежащие к этим классам, перечислены, например, в таблице 1. Неорганическим растворителем является, например, вода.
Таблица 1
Основание представляет собой органическое основание или неорганическое основание. Основание предпочтительно является более сильным основанием, чем соединение формулы 2.
В качестве органического основания предпочтительно применять органическое основание, представляющее собой амин. Например, это может быть линейный органический амин, такой как метиламин, этиламин, диметиламин, диэтиламин, этилметиламин, пропиламин, дипропиламин, метилпропиламин, этилпропиламин, диизопропиламин, N-метилциклогексиламин или триметиламин и т.д.; или циклический органический амин, такой как азиридин, азетидин, оксазиридин, азетидин, диазетидин, имидазолидин, пиразолидин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, пиперидин, 2-метилпиперидин, 2-этилпиперидин, 2,6-диметилпиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, N-пропилпиперазин, морфолин, тиоморфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, азокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метилазокан-2-карбоксилат, 1-метилазокан, 2-(2-метилфенил) азокан или пролин и т.д.
Органическое основание предпочтительно представляет собой пиперидин, пирролидин, морфолин, пиперазин, азепан, азокан, N-метилпиперидин, N-этилпиперидин или пролин.
Неорганическое основание может представлять собой, например, LiOH, KOH или NaOH.
Основание может применяться в количестве от 2 до 20 моль, от 2 до 15 моль, 2 до 10 моль, 4 до 20 моль, 4 до 15 моль, 4 до 10 моль или 5 до 20 моль, 5 до 15 моль, 5 до 10 моль, 6 до 20 моль, 6 до 15 моль или 6 до 10 моль относительно 1 моля соединения формулы 1.
Температура реакции сочетания по настоящему изобретению составляет от -20 до 200°C. Например, она может составлять от 30 до 180°C, от 30 до 150°C, от 30 до 120°C, от 30 до 100°C, от 40 до 150°C, от 40 до 140°C, от 40 до 120°C, от 40 до 100°C, от 50 до 150°C, от 50 до 120°C или от 50 до 100°C. Оптимальная температура реакции зависит от растворителя и основания, которые применяются в реакции.
Время прохождения реакции сочетания по настоящему изобретению может составлять от 10 минут до 120 часов, например, от 1 до 72 часов, от 1 до 48 часов, от 1 до 24 часов, от 3 до 72 часов, от 3 до 48 часов, от 3 до 24 часов, от 6 до 72 часов, от 6 до 48 часов или от 6 до 24 часов. Оптимальное время прохождения реакции зависит от растворителя и основания, которые применяются в реакции.
Способ синтеза билирубина по настоящему изобретению может дополнительно включать превращение R1 и/или R2 в соединении формулы 3 в атом водорода по реакции омыления. Например, когда R1 и R2 соединения формулы 3 представляют собой метильные группы, к соединению формулы 3 можно добавлять основание, такое как LiOH, KOH или NaOH, для замены метильной группы на атом водорода.
Растворитель, применяемый для реакции омыления, не ограничен каким-либо особым образом. В качестве растворителя для реакции омыления можно применять тот же растворитель, что и для реакции сочетания. Например, это может быть метанол, этанол, 2-пропанол, тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран (2-МеТГФ), 1,4-диоксан, ацетонитрил, N,N-диметилформамид (ДМФА), трет-бутанол, диметоксиэтан (ДМЭ), дихлорметан (ДХМ), изопропиловый спирт и т.п.
Реакцию омыления можно проводить в условиях, известных в данной области. Например, ее можно проводить при температуре от 10 до 150°C в течение 1 - 72 часов или при температуре от 10 до 60°C в течение 1 - 48 часов.
Способ синтеза билирубина по настоящему изобретению может дополнительно включать стадию ПЭГилирования, представляющую собой реакцию соединения формулы 3 с полиэтиленгликолем (ПЭГ).
Способ синтеза билирубина по настоящему изобретению может включать стадию ПЭГилирования соединения формулы 1 полиэтиленгликолем (ПЭГ) и последующее сочетание полученного продукта с соединением формулы 2.
ПЭГилированный билирубин имеет улучшенную растворимость в воде.
Полиэтиленгликоль представляет собой, например, мПЭГn-NH2 (метокси полиэтиленгликоль-амин, n = 5 - 60). В данном случае n - это число повторяющихся фрагментов -CH2-CH2-O- в метоксиполиэтиленгликоль-амине, которое может составлять от 5 до 60, от 10 до 50, от 10 до 40, от 20 до 40, от 10 до 30 или от 20 до 30.
ПЭГилирование включает моно-ПЭГилирование, при котором O-R1 или O-R2 ПЭГилированный, и би-ПЭГилирование, при котором они оба ПЭГилированные.
В реакции ПЭГилирования, полиэтиленгликоль добавляют в нужном количестве относительно числа молей соединения формулы 1 или формулы 3. Например, полиэтиленгликоль можно добавлять в количестве 0.1 - 10 моль, 0.1 - 8 моль, 0.1 - 5 моль, 0.3 - 8 моль, 0.3 - 5 моль, 0.3 - 4 моль или 0.3 - 3 моль относительно 1 моля соединения формулы 1 или формулы 3.
В качестве реагентов для реакции ПЭГилирования можно применять CDI (1,1-карбонилдиимидазол), CMPI (2-хлор-1-метилпиридиний иодид), BEP (2-бром-1-этил-пиридиний тетрафторборат), EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид), HATU (1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат; гексафторфосфат азабензотриазол триметилурония, DCC (N,N’-дициклогексилкарбодиимид) или HOBt (гидроксибензотриазол) и т.д., но не ограничиваясь только ими.
Реагент для реакции ПЭГилирования можно добавлять в количестве от 0.3 до 5 моль, от 0.3 до 3 моль, от 0.5 до 5 моль, от 0.5 до 3 моль, от 0.5 до 2.5 моль или от 0.5 до 2 моль относительно 1 моля соединения формулы 1 или формулы 3, но не ограничиваясь только этими соотношениями.
Растворитель для реакции ПЭГилирования не ограничен каким-либо особым образом. В качестве растворителя для реакции ПЭГилирования можно применять тот же растворитель, что и для реакции сочетания. Например, это может быть ДМСО (диметилсульфоксид), ДМФА (диметилформамид), ДМА (диметилацетамид) или пиридин.
Реакцию ПЭГилирования можно проводить в присутствии основания. Основание можно выбрать из ряда, приведенного выше в качестве примеров оснований для реакции сочетания, и предпочтительным является DIPEA (N,N-диизопропилэтиламин) или пиридин.
Реакцию ПЭГилирования можно проводить при температуре от 10 до 100°C, например от 10 до 80°C, от 20 до 60°C, от 20 до 50°C или от 20 до 30°C.
Реакцию ПЭГилирования можно проводить в течение 1 - 24 часов, 1 - 18 часов и 1 - 12 часов, но не ограничиваясь только этими значениями.
В одном варианте осуществления, реакцию ПЭГилирования можно проводить путем добавления 0.3 - 5 моль полиэтиленгликоля и 0.5 - 5 моль каплинг-агента (CDI, EDCI, CMPI и т.д.) относительно 1 моля соединения формулы 1 или формулы 3, и реакцию можно проводить при температуре от 20 до 40°C в течение 0.5 - 24 часов.
Соединение формулы 1, как и соединение формулы 2, в качестве реагентов в способе синтеза билирубина по настоящему изобретению можно получить как описано ниже.
Соединение формулы 1 можно получить димеризацией изображенного ниже соединения формулы 6 и замены x в продукте на -C(=O)H.
[Формула 6]
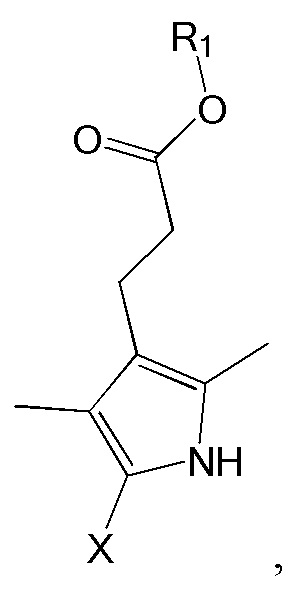
где R1 в формуле 6 такой же, как R1 в формуле 1, и x представляет собой арилалкил сложноэфирную группу, содержащую 8 - 20 атомов углерода, -CH2OH, -COOH, атом галогена или атом водорода.
Арилалкил сложноэфирная группа представляет собой функциональную группу, в которой RY в изображенной ниже формуле 7 представляет собой арилалкильную группу.
[Формула 7]
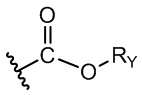
Арилалкильная группа формулы 7 такая же, как арилалкильная группа формулы 1.
Число атомов углерода в арилалкил сложноэфирной группе можно выбрать из диапазона, который не мешает димеризации соединения формулы 6. Например, она может содержать 8 - 20 атомов углерода, 8 - 18 атомов углерода, 8 - 15 атомов углерода или 8 - 12 атомов углерода.
Способ замены x в продукте на альдегидную группу может представлять собой, например, описанные ниже способы (1) - (5).
(1) Когда x представляет собой арилалкил сложноэфирную группу, то арилалкил сложноэфирную группу восстанавливают до -COOH путем гидрирования, затем -COOH восстанавливают или удаляют. Затем можно ввести -C(=O)H для замены x на альдегидную группу.
В одном варианте осуществления, гидрирование можно проводить с катализатором Pd/С. Например, арилалкил сложноэфирная группа представляет собой -C(=O)OBn (Bn=бензил), затем -C(=O)OBn восстанавливают до -COOH с помощью реакции гидрирования с катализатором Pd/C. После этого -COOH удаляют для замены арилалкил сложноэфирной группы на -C(=O)H.
(2) Когда x представляет собой -CH2OH, × окисляют известным методом и заменяют на -C(=O)H.
(3) Когда x представляет собой -COOH, -COOH восстанавливают или удаляют, затем вводят -C(=O)H для замены x на альдегидную группу.
(4) Когда x представляет собой атом галогена, то атом галогена заменяют на альдегидную группу по реакции карбонилирования. Для реакции карбонилирования можно применять известный метод. Например, реакцию карбонилирования можно проводить с использованием монооксида углерода и палладия.
(5) Когда x представляет собой атом водорода, то атом водорода заменяют на альдегидную группу по реакции введения альдегидной группы. Можно применять известный метод для проведения реакции введения альдегидной группы. Например, реакцию введения альдегидной группы можно проводить с использованием BuLi и ДМФА.
Димеризацию соединения формулы 6 можно проводить, например, в условиях присутствия брома (Br2). Растворитель для димеризации не ограничивается каким-либо специальным образом. Например, в качестве органического растворителя можно использовать растворители, приведенные в Таблице 1 в качестве примеров растворителей для реакции сочетания.
Реакцию димеризации можно проводить при температуре от 10 до 100°C, например, от 10 до 80°C, от 20 до 60°C, от 20 до 50°C или от 20 до 30°C.
Реакцию димеризации можно проводить в течение 1 - 24 часов, 1 - 18 часов или 1 - 12 часов, но не ограничиваясь только этими значениями.
Соединение формулы 2 (R3 = водород) можно получить циклизацией соединения формулы 8.
[Формула 8]
,
где R4 такой же, как R4 в формуле 2.
Реакцию циклизации соединения формулы 8 можно проводить с катализатором Граббса. Когда циклизуют соединение формулы 8, образуется соединение формулы 2.

Соединение формулы 2 (R3 = ацетил) можно получить реакцией соединения формулы 9 с изображенным ниже соединением формулы 10.
[Формула 9]
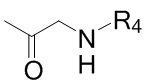 ,
,
где R4 такой же, как R4 в формуле 2
[Формула 10]
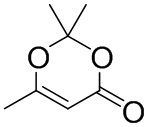
Соединение формулы 2 (R3 = этильная группа, замещенная гидроксильной группой) можно получить восстановлением ацетильной группы в изображенном ниже соединении формулы 11.
[Формула 11]
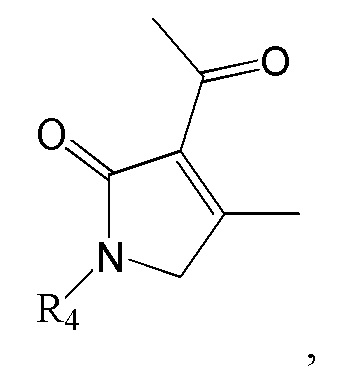
где R4 такой же, как R4 в формуле 2.
Реакцию восстановления ацетильной группы можно провести известным методом, используя тот же ряд растворителей, температуру, время реакции и т.п., как описано для реакции сочетания. Например, ацетильную группу можно восстановить, используя MeOH как растворитель и NaBH4 и CeCl3⋅7H2O как восстановитель, или ТГФ как растворитель и DIBAL как восстановитель.
Соединение формулы 2 (R3 = винильная группа) можно получить дегидратацией гидроксильной группы в изображенном ниже соединении формулы 12.
[Формула 12]
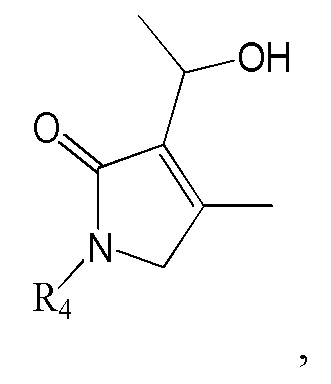
где R4 такой же, как R4 в формуле 2.
Реакцию дегидратации гидроксильной группы можно провести известным методом, используя тот же ряд растворителей, температуру, время реакции и т.п., как описано для реакции сочетания. Например, гидроксильную группу можно дегидратировать, используя ДХМ (дихлорметан) как растворитель и POCl3 и ТЭА как реагенты.
Соединение формулы 2 (R3 = этильная группа, замещенная селенидом) можно получить циклизацией соединения формулы 13.
[Формула 13]
,
где Y представляет собой селенид, и R4 такой же как R4 в формуле 2.
Реакцию циклизации соединения формулы 13 можно проводить, используя тот же ряд растворителей, температуру, время реакции и т.п., как описано для реакции сочетания. Когда циклизуют соединение формулы 13, образуется соединение формулы 2.
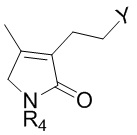
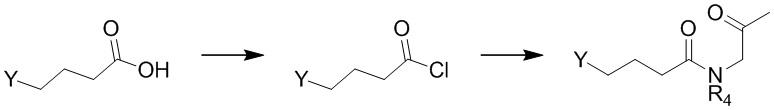
Соединение формулы 13 можно получить, например, следующим образом.
Соединение формулы 2 (R3 = этильная группа, замещенная сульфидом) можно получить окислением изображенного ниже соединения формулы 14.
[Формула 14]
,
где Z представляет собой сульфид, и R4 и R5 такие же как R4 и R5 в формуле 2.
Соединение формулы 2, полученное окислением соединения формулы 14 (R3 = этильная группа, замещенная сульфид), представляет собой следующее:
Реакцию окисления соединения формулы 14 можно проводить путем добавления NaI после взаимодействия с mCPBA. Реакцию окисления соединения формулы 14 можно проводить, используя тот же ряд растворителей, температуру, время реакции и т.п., как описано для реакции сочетания, но не ограничиваясь только ими.
Соединение формулы 14 можно получить следующим образом:
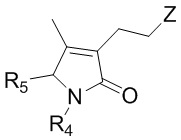
Когда R3 в соединении формулы 3 по настоящему изобретению представляет собой атом водорода или ацетильную группу; или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом, то необходимо дополнительно проводить стадию превращения этих заместителей в винильную группу.
(1) Когда R3 представляет собой атом водорода, превращение в винильную группу можно проводить путем бромирования и реакции углерод-углеродного сочетания. Бромирование можно проводить известным методом, и например, можно использовать Br2 как реагент для реакции бромирования.
Реакция углерод-углеродного сочетания может представлять собой, например, реакцию сочетания по Стилле, реакцию Сузуки-Мияуры, реакцию Хека или реакцию Гриньяра.
Реакцию углерод-углеродного сочетания можно проводить, например, следующим образом:
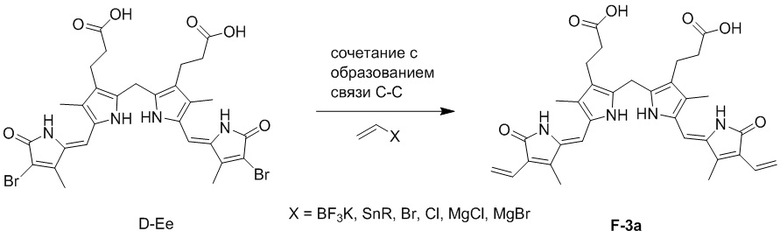
Реагент для реакции углерод-углеродного сочетания может представлять собой, например, BF3K, SnR (R представляет собой C1 - C20 алкил), Br, Cl, MgCl или MgBr, но не ограничивается только ими.
Реакция сочетания по Стилле может быть проведена известным методом и может представлять собой, например, реакцию с изображенным ниже соединением формулы 15, но не ограничивается только этим.
[Формула 15]
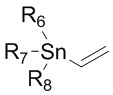 ,
,
где R6 - R8 каждый независимо представляют собой алкильную группу, содержащую 1 - 20 атомов углерода, 1 - 15 атомов углерода или 1 - 10 атомов углерода.
Реакция Сузуки может быть проведена известным методом, и может представлять собой, например, реакцию с изображенным ниже соединением формулы 16, но не ограничивается только этим.
[Формула 16]
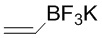
(2) Когда R3 представляет собой ацетильную группу, превращение в винильную группу можно проводить путем восстановления и дегидратации ацетильной группы. Восстановление и дегидратацию ацетильной группы можно проводить известными методами.
(3) Когда R3 представляет собой этильную группу, замещенную гидроксильной группой, превращение в винильную группу можно проводить по реакции дегидратации гидроксильной группы. Реакция дегидратации гидроксильной группы может быть проведена известным методом.
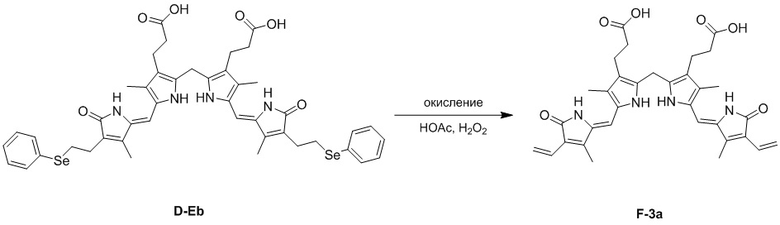
(4) Когда R3 представляет собой этильную группу, замещенную селенидом, превращение в винильную группу можно проводить реакцией окисления селенида. Например, соединение формулы 3 можно превратить в винильную группу путем окисления в присутствии HOAc и H2O2, как изображено ниже.
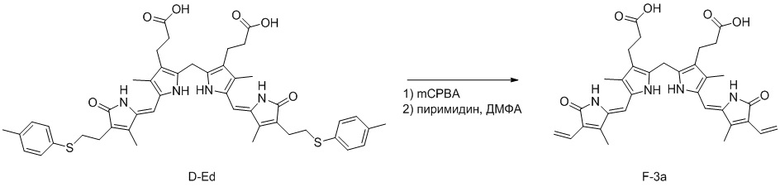
(5) Когда R3 представляет собой этильную группу, замещенную сульфидом, превращение в винильную группу можно проводить реакцией окисления сульфида. Например, соединение формулы 3 можно окислить с помощью mCPBA и затем снять защиту пиридином, как показано ниже.
Далее настоящее изобретение описано более подробно с помощью примеров.
Примеры
1. Получение соединения формулы 1
Соединение C, соединение D, соединение C1 и соединение C2, соответствующие соединению формулы 1 по настоящему изобретению, получали как описано ниже.
Пример 1. Получение соединения C
(1-1) Получение соединения A
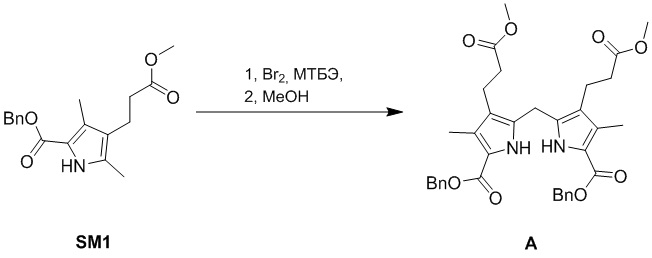
Смесь Br2 (53.2 г, 333 ммоль, 1.4 экв.) в МТБЭ (375 мл) добавляли по каплям в смесь соединения SM1 (75.0 г, 238 ммоль, 1.0 экв.) в МТБЭ (1125 мл) при 20°C в атмосфере азота. Полученную смесь перемешивали при 20°C в течение 1 часа, после чего подтверждали полноту прохождения реакции методом ТСХ. Растворитель удаляли при пониженном давлении. Затем добавляли в смесь метанол (546 мл). Полученную смесь перемешивали при 50°C в течение 12 часов, после чего подтверждали полное исчезновение реагентов методом ТСХ. Реакционную смесь охлаждали до 20°C и упаривали при пониженном давлении. Полученную смесь растирали в метаноле (100 мл) при 20°C, и отфильтрованный продукт промывали метанолом (50 мл × 2), получая соединение A в виде серого твердого вещества (59.0 г, 95.9 ммоль, выход: 81%).
1H ЯМР (400 МГц, CDCl3) δ 9.11 (с, 2H), 7.41 - 7.25 (м, 10H), 5.26 (с, 4H), 3.97 (с, 2H), 3.58 (с, 6H), 2.77 (т, J = 7.2 Гц, 4H), 2.52 (т, J = 6.8 Гц, 4H), 2.29 (с, 6H).
(1-2) Получение соединения B
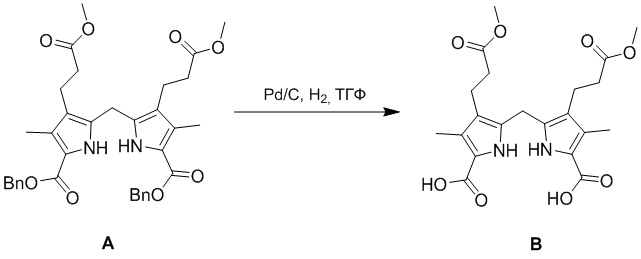
В смесь соединения A (50.0 г, 81.3 ммоль, 1.0 экв.), полученного как описано выше, в ТГФ (650 мл), добавляли Pd/C (5.00 г, 10 мол.%) в атмосфере азота. Полученную смесь несколько раз дегазировали в вакууме и заполняли водородом. Полученную смесь перемешивали при 20°C в течение 16 часов в атмосфере водорода (15 фунт/кв.дюйм). Смесь Na2CO3 (8.62g, 81.3 ммоль) и H2O (50 мл) добавляли в реакционную смесь и перемешивали 0.5 часа. Полученную смесь фильтровали и доводили фильтрат до pH 7 добавлением уксусной кислоты (около 10 мл). Осадок отфильтровывали и сушили, получая соединение B в виде розового твердого вещества (34.0 г, 78.3 ммоль, выход: 96%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.09 (с, 2H), 3.78 (с, 2H), 3.56 (с, 6H), 2.56 (т, J = 7.2 Гц, 4H), 2.16 - 2.10 (м, 10H).
(1-3) Получение соединения C
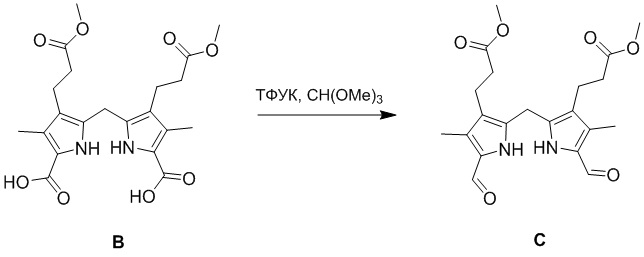
Соединение B (20.0 г, 46.1 ммоль, 1.0 экв.), полученное как описано выше, добавляли в ТФУК (190 мл) при 0°C. Полученную смесь перемешивали при 0°C в течение 1 часа в атмосфере азота и добавляли триметоксиэтан (55.2 г, 520 ммоль, 11.3 экв.) при 0°C. Полученную смесь затем перемешивали при 0°C в течение 1 часа и отслеживали методом LCMS. Реакционную смесь выливали в 1.7 л воды и перемешивали 10 минут. Выпавший осадок отфильтровывали и промывали 0.3 л воды. Отфильтрованное твердое вещество растирали в этаноле (0.2 л) и гидроксиде аммония (0.4 л) при 20°C в течение 30 минут. Осадок отфильтровывали, получая желтый порошок, который затем промывали водой (0.3 л). Метанол (0.4 л) добавляли в полученный продукт и кипятили 10 минут. После охлаждения смеси до комнатной температуры осадок отфильтровывали и промывали холодным метанолом (0.1 л), получая соединение C, соответствующее соединению формулы 1 по настоящему изобретению, в виде коричневого твердого вещества (12.0 г, 29.8 ммоль, выход: 65%).
1H ЯМР (400 МГц, CDCl3) δ 10.19 - 10.00 (м, 2H), 9.47 (с, 2H), 4.05 (с, 2H), 3.71 (с, 6H), 2.80 (т, J = 7.2 Гц, 4H), 2.61 - 2.45 (м, 4H), 2.29 (с, 6H).
Пример 2. Получение соединения D
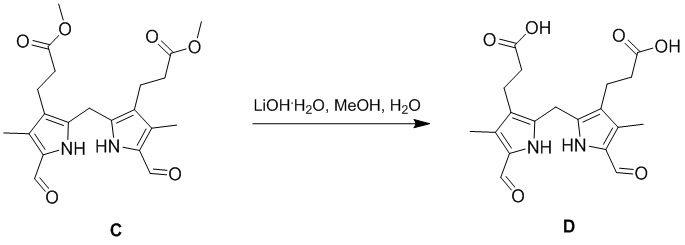
Гидроксид лития (LiOH⋅H2O) (2.75 г, 65.6 ммоль, 6.6 экв.) добавляли в смесь метанола (100 мл), воды (100 мл) и соединения C (4.00 г, 9.94 ммоль, 1.0 экв.) из Примера 1. Полученную смесь перемешивали при 25°C в течение 16 часов и разводили водой (100 мл). 1 М раствор соляной кислоты добавляли по каплям в смесь, доводя pH до 2-3. Затем осадок отфильтровывали и сушили, получая соединение D, соответствующее соединению формулы 1 по настоящему изобретению, в виде фиолетового твердого вещества (3.49 г, 9.32 ммоль, выход: 94%).
1H ЯМР (400 МГц, CDCl3) δ 12.03 (ушир.с, 2H), 11.51 (с, 2H), 9.48 (с, 2H), 3.91 (с, 2H), 2.54 (перекрывается с сигналом ДМСО-d6, 4H), 2.18 (с, 6H), 2.06 (т, J = 8.0 Гц, 4H).
C19H22N2OS m/z [M+H]+ = 375
Пример 3. Получение соединения C1
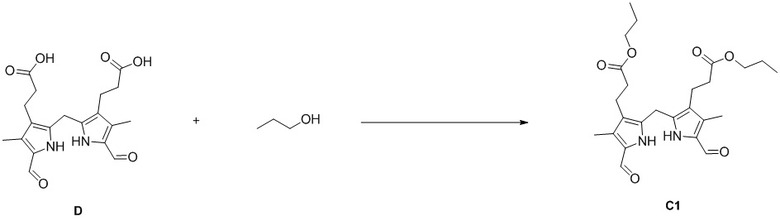
DIPEA (103.56 мг, 0.80 ммоль, 3.0 экв.), HOBt (108.28 мг, 0.80 ммоль, 3.0 экв.) и EDCI (153.61 мг, 0.80 ммоль, 3.0 экв.) добавляли в смесь соединения D (100 мг, 0.26 ммоль, 1.0 экв.) из Примера 2 в ДМСО (3 мл) и перемешивали при 25°C в течение 30 минут. Добавляли по каплям 1-пропанол (80.26 мг, 1.34 ммоль, 5.0 экв.) и перемешивали смесь в течение 12 часов. Добавляли в смесь EtOAc (140 мл) и промывали водой (120 мл × 5). Объединенные органические слои промывали насыщенным раствором хлорида натрия (120 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный осадок растирали в MeOH (1 мл) и фильтровали при пониженном давлении, получая соединение C1, соответствующее соединению формулы 1 по настоящему изобретению, в виде розового твердого вещества (27 мг, 0.046 ммоль, выход: 21%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.53 (ушир.с, 2H), 9.47 (с, 2H), 3.91 (т, J = 6.8 Гц, 6H), 2.54 (перекрывается с сигналом ДМСО-d6, 4H), 2.17 (с, 6H), 2.06 (т, J = 8.0 Гц, 4H), 1.58 - 1.49 (м, 4H), 0.84 (т, J = 7.6 Гц, 3H).
Пример 4. Получение соединения C2
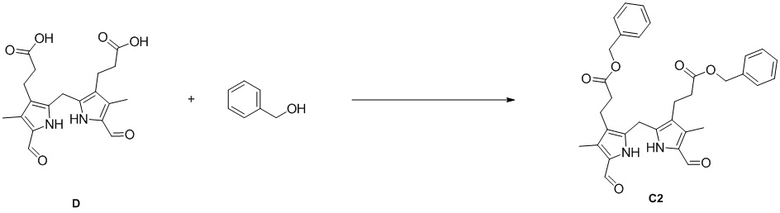
DIPEA (165.56 мг, 1.28 ммоль, 3.0 экв.), HOBt (173.24 мг, 1.28 ммоль, 3.0 экв.) и EDCI (254.78 мг, 1.28 ммоль, 3.0 экв.) добавляли в смесь соединения D (160 мг, 0.42 ммоль, 1.0 экв.) из Примера 2 в ДМСО (5 мл) и перемешивали при 25°C в течение 30 минут. Добавляли бензиловый спирт (231.07 мг, 2.14 ммоль, 5.0 экв.) по каплям, и реакционную смесь перемешивали в течение 12 часов. Добавляли EtOAc (180 мл) и промывали смесь водой (130 мл × 5). Объединенные органические слои промывали насыщенным раствором хлорида натрия (130 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный осадок растирали в MeOH/МТБЭ (об/об = 1/1, 10 мл) и затем фильтровали при пониженном давлении, получая соединение C2, соответствующее соединению формулы 1 по настоящему изобретению, в виде коричневого твердого вещества (110 мг, 0.198 ммоль, выход: 47%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.53 (с, 2H), 9.45 (с, 2H), 7.37 - 7.26 (м, 10H), 5.03 (с, 4H), 3.87 (с, 2H), 2.53 (перекрывается с сигналом ДМСО-d6, 4H), 2.17 - 2.14 (м, 10H).
2. Получение соединения формулы 2
Соединения (Примеры 5 - 28), соответствующие соединению формулы 2 по настоящему изобретению, получали как описано ниже.
2.1. Получение соединений Ea-2, Ea-3, Ea-4 и Ea
Пример 5. Получение соединения Ea-2
(5-1) Получение соединения Ea-1
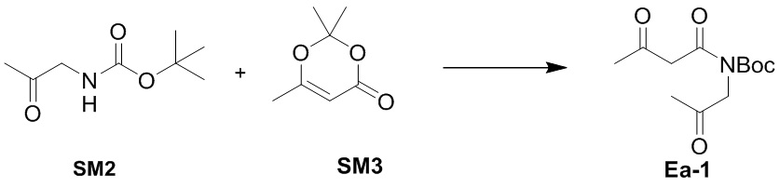
В смесь соединения SM2 (40.0 г, 231 ммоль, 1.0 экв.) в ксилоле (241 мл) добавляли соединение SM3 (32.8 г, 231 ммоль, 1.0 экв.). Полученную смесь перемешивали при 150°C в течение 10 минут в атмосфере азота. Реакционную смесь упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-1 в виде коричневого масла (51.0 г, 198 ммоль, выход: 85%).
1H ЯМР (400 МГц, CDCl3) δ 4.48 (с, 2H), 4.00 (с, 2H), 2.21 (с, 3H), 2.13 (с, 3H), 1.39 (с, 9H).
(5-2) Получение соединения Ea-2
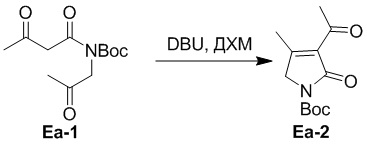
Смесь DBU (34.8 мл, 233 ммоль, 0.5 экв.) в ДХМ (34.8 мл) добавляли по каплям в смесь соединения Ea-1 (120 г, 466 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (600 мл) при 0°C и перемешивали 40 минут. Реакцию останавливали добавлением водного раствора KH2PO4 (480 мл), органический слой экстрагировали дихлорметаном (600 мл × 2) и промывали водой (480 мл) и насыщенным раствором хлорида натрия (1.2 л). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растворяли в МТБЭ (60 мл) и добавляли петролейный эфир (720 мл) при 0°C. Выпавший осадок отфильтровывали при пониженном давлении, получая соединение Ea-2, соответствующее соединению формулы 2 по настоящему изобретению, в виде коричневого твердого вещества (90 г, 372 ммоль, выход: 80%).
1H ЯМР (400 МГц, CDCl3) δ 4.27 (с, 2H), 2.56 (с, 3H), 2.39 (с, 3H), 1.57 (с, 9H).
Пример 6. Получение соединения Ea-3
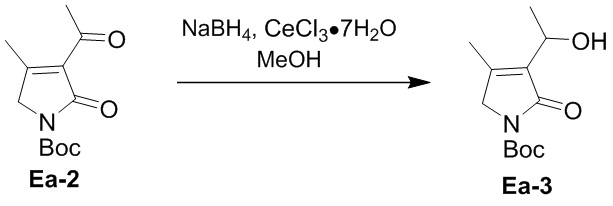
CeCl3⋅7H2O (126g, 338 ммоль, 2.0 экв.) растворяли в метаноле (600 мл) и перемешивали 5 минут. Добавляли соединение Ea-2 (53.9 г, 16 9 ммоль, 1.0 экв.), полученное как описано выше, и перемешивали 5 минут. Полученную смесь охлаждали до 0°C и медленно добавляли NaBH4 (12.8 г, 338 ммоль, 2.0 экв.) в течение примерно 1 часа. Полученную смесь перемешивали при 0°C в течение 3 часов в атмосфере азота (N2). Добавляли в смесь 1 M водный раствор HCl (300 мл), затем последовательно экстрагировали этилацетатом (500 мл × 5) и ДХМ (500 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (300 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ea-3, соответствующее соединению формулы 2 по настоящему изобретению, в виде коричневого масла (23.0 г, 95.3 ммоль, выход: 56%).
1H ЯМР (400 МГц, CDCl3) δ 4.72 - 4.64 (м, 1H), 4.13 (с, 2H), 3.49 (д, J = 9.6 Гц, 1H), 2.05 (с, 3H), 1.56 (с, 9H), 1.48 (д, J = 6.8 Гц, 3H).
Пример 7. Получение соединения Ea-4
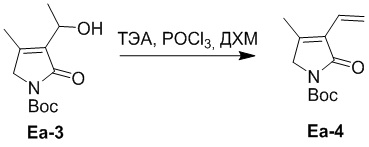
Триэтиламин (ТЭА) (116 г, 1.14 моль, 12.0 экв.) добавляли в смесь соединения Ea-3 (23.0 г, 95.3 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (700 мл), и затем добавляли по каплям смесь POCl3 (58.4 г, 381 ммоль, 4.0 экв.) в ДХМ (180 мл) при 0°C в течение 1 часа. Полученную смесь перемешивали при 20°C в течение 3 часов в атмосфере азота (N2) и контролировали методом ТСХ. Полученную смесь упаривали при пониженном давлении и разводили водой (300 мл), затем экстрагировали дихлорметаном (500 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (200 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая остаток от упаривания. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ea-4, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (8.0 г, 24.2 ммоль, выход: 38%).
1H ЯМР (400 МГц, CDCl3) δ 6.45 - 6.38 (м, 1H), 6.31 - 6.26 (м, 1H), 5.46 - 5.42 (м, 1H), 4.15 (с, 2H), 2.11 (с, 3H), 1.56 (с, 9H).
Пример 8. Получение соединения Ea
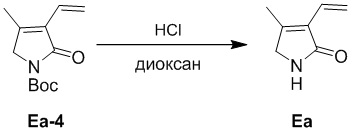
HCl/1,4-диоксан (4 M, 18 мл) добавляли в смесь соединения Ea-4 (4.0 г, 17.9 ммоль), полученного как описано выше, в EtOAc (18 мл) при 0°C, после этого реакционную смесь перемешивали при 20°C в течение 1 часа. Реакцию останавливали путем добавления водного раствора NaHCO3 (80 мл). Полученную смесь экстрагировали этилацетатом (80 мл × 2) и затем дихлорметаном (150 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали, получая соединение Ea, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (4.6 г, выход: >99 %).
1H ЯМР (400 МГц, CDCl3) δ 6.87 (ушир.с, 1H), 6.48 (дд, J = 17.6, 11.6 Гц, 1H), 6.24 (дд, J = 18.0, 2.0 Гц, 1H), 5.41 (дд, J = 11.6, 2.0 Гц, 1H), 3.86 (с, 2H), 2.09 (с, 3H).
2.2. Получение соединений Ea-3a и Ea-2a
Пример 9. Получение соединения Ea-3a
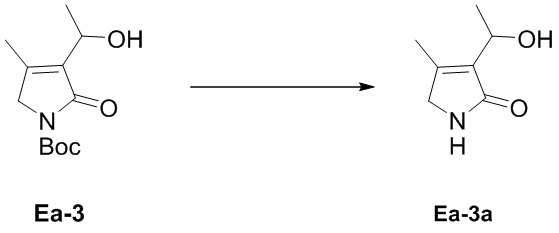
В смесь соединения Ea-3 (1.6 г, 6.63 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (10 мл), добавляли 4 M HCl/1,4-диоксан (6.63 мл, 26.52 ммоль, 4.0 экв.) при 0°C и затем перемешивали при 0°C в течение 2 часов. Доводили значение pH раствора до 7 водным раствором NaHCO3 (100 мл), органический слой экстрагировали дихлорметаном (125 мл × 2) и промывали насыщенным раствором хлорида натрия (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение Ea-3a, соответствующее соединению формулы 2 по настоящему изобретению, в виде коричневого твердого вещества (650 мг, 4.61 ммоль, выход: 70%).
1H ЯМР (400 МГц, CDCl3) δ 6.85 (ушир.с, 1H), 5.02 (кв, J = 6.8 Гц, 1H), 3.85 (с, 2H), 2.17 (с, 3H), 1.82 (д, J = 6.8 Гц, 3H).
Пример 10. Получение соединения Ea-2a
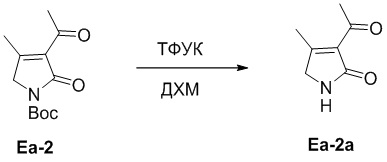
Добавляли ТФУК (0.32 мл, 4.18 ммоль, 5.0 экв.) в смесь соединения Ea-2 (200 мг, 0.83 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (16 мл), и смесь перемешивали при 25°C в течение 1 часа. Реакционную смесь доводили до pH 7 путем добавления водного раствора NaHCO3 (50 мл). После экстракции дихлорметаном (50 мл × 3), объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли ДХМ (3 мл) в упаренный фильтрат и перемешивали при 25°C в течение 5 минут, затем медленно добавляли гексан (20 мл) по каплям. Осадок отфильтровывали при пониженном давлении, получая соединение Ea-2a, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (70 мг, 0.50 ммоль, выход: 60%).
1H ЯМР (400 МГц, CDCl3) δ 7.15 (ушир.с, 1H), 3.97 (с, 2H), 2.55 (с, 3H), 2.36 (с, 3H).
2.3. Получение соединений Ea-2b, Ea-3b и Ea-4b
Пример 11. Получение соединения Ea-2b
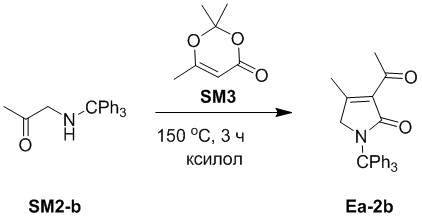
Смесь соединений SM2-b (1 г, 3.17 ммоль, 1.0 экв.) и SM3 (450.70 мг, 3.17 ммоль, 1.0 экв.) в ксилоле (15 мл) перемешивали при 150°C в течение 3 часов. Ксилол удаляли при пониженном давлении, и остаток очищали методом хроматографии на силикагеле, получая соединение Ea-2b, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (30 мг, 75.10 мкмоль, выход: 3%).
1H ЯМР (400 МГц, CDCl3) δ 7.31 - 7.22 (м, 15H), 3.91 (с, 2H), 2.47 (с, 3H), 2.29 (с, 3H).
Пример 12. Получение соединения Ea-3b
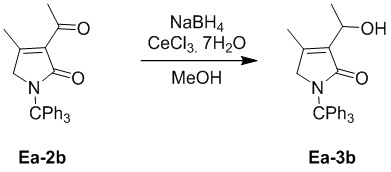
В смесь соединения Ea-2b (500 мг, 1.31 ммоль, 1.0 экв.), полученного как описано выше, и CeCl3 7H2O (976.71 мг, 2.62 ммоль, 2.0 экв.) в метаноле (5 мл) добавляли NaBH4 (99.18 мг, 2.62 ммоль, 2.0 экв.) при 0°C. Полученную смесь перемешивали при 0°C в течение 4 часов. Реакцию останавливали добавлением 1 M водного раствора HCl (10 мл), добавляли в смесь воду (20 мл), затем экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-3b, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (102 мг, 0.27 ммоль, выход: 20%).
1H ЯМР (400 МГц, CDCl3) δ 7.32 - 7.21 (м, 15H), 4.66 - 4.62 (м, 1H), 3.75 (с, 2H), 1.93 (с, 3H), 1.40 (д, J = 6.8 Гц, 3H).
Пример 13. Получение соединения Ea-4b
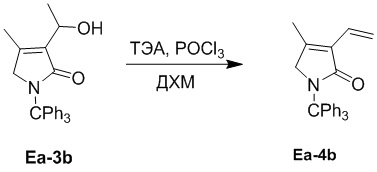
В смесь соединения Ea-3b (214 мг, 0.55 ммоль, 1.0 экв.), полученного как описано выше, и ТЭА (0.93 мл, 6.70 ммоль, 12.0 экв.) в ДХМ (5 мл), добавляли раствор POCl3 (0.27 мл, 2.23 ммоль, 4.0 экв.) в ДХМ (2 мл) при 0°C и перемешивали при 25°C в течение 2 часов. Полученную смесь упаривали при пониженном давлении и экстрагировали дихлорметаном (30 мл × 2) после добавления воды (10 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-4b, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (30 мг, 82.1 мкмоль, выход: 15%).
1H ЯМР (400 МГц, CDCl3) δ 7.32 - 7.21 (м, 15H), 6.42 (дд, J = 18.0, 11.6 Гц, 1H), 6.16 (дд, J = 18.0, 2.0 Гц, 1H), 5.34 (дд, J = 11.6, 2.0 Гц, 1H), 3.79 (с, 2H), 2.03 (с, 3H).
2.4. Получение соединений Ea-2c, Ea-3c и Ea-4c
Пример 14. Получение соединения Ea-2c
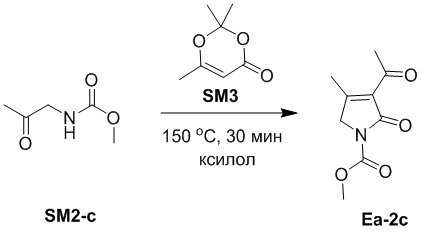
Смесь соединений SM2-c (598 мг, 4.56 ммоль, 1.0 экв.) и SM3 (648.27 мг, 4.56 ммоль, 1.0 экв.) в ксилоле (6 мл) перемешивали при 150°C в течение 30 минут. Ксилол удаляли при пониженном давлении, и остаток очищали методом хроматографии на силикагеле, получая соединение Ea-2c, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (484 мг, 2.45 ммоль, выход: 53%).
1H ЯМР (400 МГц, CDCl3) δ 4.33 (с, 2H), 3.92 (с, 3H), 2.55 (с, 3H), 2.43 (с, 3H), 2.03 (с, 3H).
Пример 15. Получение соединения Ea-3c
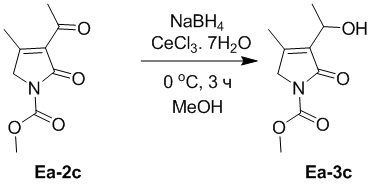
В смесь соединения Ea-2c (1.14 г, 5.78 ммоль, 1.0 экв.), полученного как описано выше, и CeCl3⋅7H2O (4.31 г, 11.56 ммоль, 2.0 экв.) в метаноле (10 мл) добавляли NaBH4 (437.44 мг, 11.56 ммоль, 2.0 экв.) при 0°C и перемешивали при 0°C в течение 3 часов. Реакцию останавливали добавлением 1 M водного раствора HCl (20 мл). Полученную смесь разводили водой (100 мл) и затем экстрагировали этилацетатом (150 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-3c, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого масла (121 мг, 0.61 ммоль, выход: 10%).
1H ЯМР (400 МГц, CDCl3) δ 4.73 - 4.66 (м, 1H), 4.19 (с, 2H), 3.90 (с, 3H), 3.36 (д, J = 9.2 Гц, 1H), 2.07 (с, 3H), 1.46 (т, J = 6.8 Гц, 3H).
Пример 16. Получение соединения Ea-4c
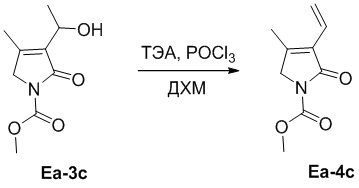
В смесь соединения Ea-3c (198 мг, 0.99 ммоль, 1.0 экв.), полученного как описано выше, и ТЭА (1.66 мл, 11.93 ммоль, 12.0 экв.) в ДХМ (5 мл) добавляли смесь POCl3 (0.37 мл, 3.98 ммоль, 4.0 экв.) в ДХМ (3 мл) при 0°C. Полученную смесь перемешивали при 25°C в течение 2 часов. Полученную смесь упаривали при пониженном давлении, разводили водой (20 мл) и экстрагировали дихлорметаном (30 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-4c, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (94 мг, 0.51 ммоль, выход: 52%).
1H ЯМР (400 МГц, CDCl3) δ 6.43 (дд, J = 17.6, 11.6 Гц, 1H), 6.30 (дд, J = 17.6, 2.0 Гц, 1H), 5.47 (дд, J = 11.6, 2.0 Гц, 1H), 4.22 (с, 2H), 3.91 (с, 3H), 2.13 (с, 3H).
2.5. Получение соединений Ea-2d, Ea-3d и Ea-4d
Пример 17. Получение соединения Ea-2d
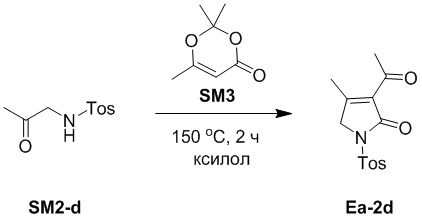
Смесь соединений SM2-d (5.0 г, 22.0 ммоль, 1.0 экв.) и SM3 (3.13 г, 22.0 ммоль, 1.0 экв.) в ксилоле (40 мл) перемешивали при 150°C в течение 2 часов в атмосфере азота. Полученную смесь упаривали при пониженном давлении, и остаток очищали методом хроматографии на силикагеле, получая соединение Ea-2d, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (1.41 г, 4.81 ммоль, выход: 22%).
1H ЯМР (400 МГц, CDCl3) δ 7.98 (д, J = 8.4 Гц, 2H), 7.38 (д, J = 8.0 Гц, 2H), 4.43 (с, 2H), 2.50 (с, 3H), 2.47 (с, 3H), 2.40 (с, 3H).
Пример 18. Получение соединения Ea-3d
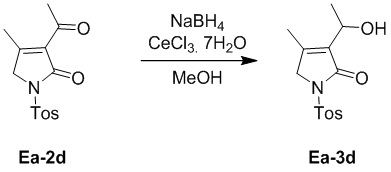
В смесь CeCl3·7H2O (2.82 г, 7.57 ммоль, 2.0 экв.) в метаноле (20 мл) добавляли соединение Ea-2d (1.11 г, 3.78 ммоль, 1.0 экв.), полученное как описано выше, и NaBH4 (99.18 мг, 2.62 ммоль, 2.0 экв.). Полученную смесь перемешивали при 0°C в течение 3 часов. Реакцию останавливали путем добавления 1 M водного раствора HCl (10 мл), затем экстрагировали этилацетатом (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-3d, соответствующее соединению формулы 2 по настоящему изобретению, в виде бесцветного твердого вещества (681 мг, 2.31 ммоль, выход: 61%).
1H ЯМР (400 МГц, CDCl3) δ 7.94 (д, J = 8.4 Гц, 2H), 7.35 (д, J = 8.0 Гц, 2H), 4.65 - 4.58 (м, 1H), 4.26 (кв, J = 16.0 Гц, 2H), 3.02 (д, J = 8.8 Гц, 2H), 2.44 (с, 3H), 2.06 (с, 3H), 1.42 (д, J = 6.8 Гц, 3H).
C14H17NO4S m/z [M+H]+ = 295.9.
Пример 19. Получение соединения Ea-4d
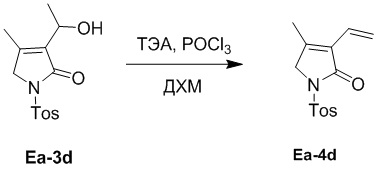
В смесь соединения Ea-3d (680 мг, 2.30 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (20 мл) добавляли ТЭА (3.85 мл, 27.63 ммоль, 12.0 экв.) и POCl3 (0.86 мл, 9.21 ммоль, 4.0 экв.), и перемешивали при 0°C в течение 2 часов в атмосфере азота. Полученную смесь упаривали при пониженном давлении и экстрагировали дихлорметаном (50 мл × 4) после добавления воды (50 мл). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-4d, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (276 мг, 0.99 ммоль, выход: 43%).
1H ЯМР (400 МГц, CDCl3) δ 7.97 (д, J = 8.4 Гц, 2H), 7.34 (д, J = 8.4 Гц, 2H), 6.33 (дд, J = 18.0, 11.6 Гц, 1H), 6.22 (дд, J = 18.0, 2.0 Гц, 1H), 5.43 (дд, J = 11.2, 2.0 Гц, 1H), 4.30 (с, 2H), 2.43 (с, 3H), 2.10 (с, 3H).
2.6. Получение соединений Ea-2e, Ea-3e и Ea-4e
Пример 20. Получение соединения Ea-2e
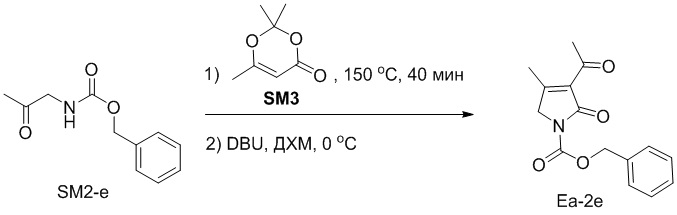
Смесь соединений SM2-e (360 мг, 1.74 ммоль, 1.0 экв.) и SM3 (321 мг, 2.26 ммоль, 1.0 экв.) перемешивали при 150°C в течение 40 минут. После окончания реакции очищали остаток методом хроматографии на силикагеле. В раствор очищенного остатка в ДХМ (5 мл) добавляли DBU (130 мг) при 0°C. Перемешивали реакционную смесь при 0°C в течение 40 минут и останавливали реакцию добавлением водного раствора K2HPO4 (100 мл). Полученную смесь экстрагировали дихлорметаном (100 мл × 2), промывали насыщенным раствором хлорида натрия и сушили над безводным Na2SO4. Высушенный органический слой фильтровали и упаривали. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-2e, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (280 мг, 1.02 ммоль, выход: 60%).
C15H15NO4 m/z [M+H]+ = 274.
Пример 21. Получение соединения Ea-3e
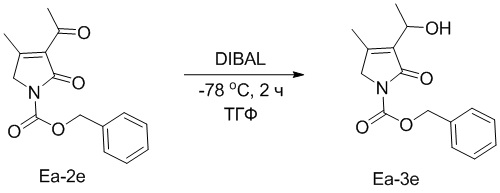
В смесь соединения Ea-2e (100 мг, 0.380 ммоль, 1.0 экв.), полученного как описано выше, в ТГФ (2 мл) добавляли DIBAL (660 мл, 0.680 ммоль, 2.0 экв.) при -78°C и перемешивали при -78°C в течение 2 часов. Реакцию останавливали путем добавления водного раствора NH4Cl (50 мл), затем смесь экстрагировали дихлорметаном (100 мл × 2). Затем объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл) и сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ea-3e, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого масла (22 мг, 0.08 ммоль, выход: 21%).
C15H17NO4 m/z [M+H]+ = 276.
Пример 22. Получение соединения Ea-4e
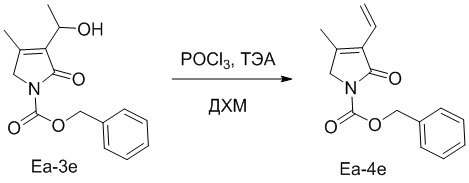
В смесь соединения Ea-3e (22.0 мг, 0.08 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (1.0 мл) последовательно добавляли ТЭА (111 мкл, 0.8 ммоль, 10.0 экв.) и POCl3 (11 мкл, 0.24 ммоль, 3.0 экв.) при 0°C. Реакционную смесь перемешивали при 20°C в течение 3 часов в атмосфере азота (N2). Полученную смесь упаривали при пониженном давлении, разводили водой (50 мл) и экстрагировали дихлорметаном (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая остаток от упаривания. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ea-4e, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (7.0 мг, 0.027 ммоль, выход: 34%).
1H ЯМР (400 МГц, CDCl3) δ 7.45 - 7.31 (м, 5H), 6.41 (дд, J = 17.7, 11.5 Гц, 1H), 6.30 (дд, J = 17.8, 2.1 Гц, 1H), 5.49 - 5.43 (м, 1H), 4.21 (с, 2H), 2.10 (с, 3H).
2.7. Получение соединений Eb-5 и Eb
Пример 23. Получение соединения Eb-5
(23-1) Получение соединения Eb-2
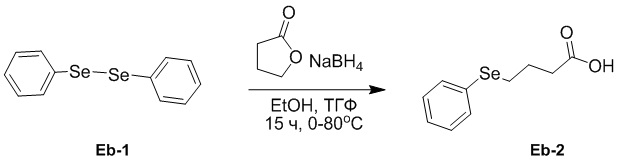
В раствор соединения Eb-1 (30.0 г, 96.1 ммоль, 1.0 экв.) в EtOH (300 мл) медленно добавляли NaBH4 (7.27 г, 192 ммоль, 2.0 экв.) при 0°C и перемешивали смесь при 0°C в течение 1 часа в атмосфере азота. Затем добавляли раствор тетрагидрофуран-2-она (16.6 г, 192 ммоль, 2.0 экв.) в ТГФ (20 мл). Полученную смесь перемешивали при 80°C в течение 15 часов в атмосфере азота. После охлаждения до 25°C, разводили смесь водой (200 мл), затем доводили до pH 2 добавлением 2 M водного раствора HCl, затем экстрагировали дихлорметаном (75 мл × 3). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Eb-2 в виде белого твердого вещества (15.0 г, 61.7 ммоль, выход: 64%).
1H ЯМР (400 МГц, CDCl3) δ 7.53 - 7.51 (м, 2H), 7.29 - 7.26 (м, перекрывается с сигналом CDCl3, 3H), 2.97 (т, J = 7.2 Гц, 2H), 2.53 (т, J = 7.2 Гц, 2H), 2.06 - 1.99 (м, 2H).
(23-2) Получение соединения Eb-3
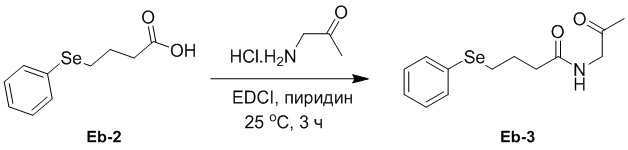
В смесь соединения Eb-2 (14.0 г, 57.6 ммоль, 1.0 экв.), полученного как описано выше, в пиридине (100 мл) добавляли EDCI (13.2 г, 69.1 ммоль, 1.2 экв.) и 1-аминопропан-2-он гидрохлорид (6.31 г, 57.6 ммоль, 1.0 экв.), и реакционную смесь перемешивали при 25°C в течение 3 часов в атмосфере азота. Добавляли воду (50 мл) и экстрагировали полученную смесь дихлорметаном (60 мл × 3). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Eb-3 в виде светло-желтого твердого вещества (6.30 г, 21.1 ммоль, выход: 37%).
1H ЯМР (400 МГц, CDCl3) δ 7.54 - 7.45 (м, 2H), 7.35 - 7.20 (м, перекрывается с сигналом CDCl3, 3H), 6.23 (ушир.с, 1H), 4.20 - 4.10 (м, 2H), 2.96 (т, J = 7.2 Гц, 2H), 2.39 (т, J = 7.2 Гц, 2H), 2.20 (с, 3H), 2.07 - 2.00 (м, 2H).
(23-3) Получение соединения Eb-4
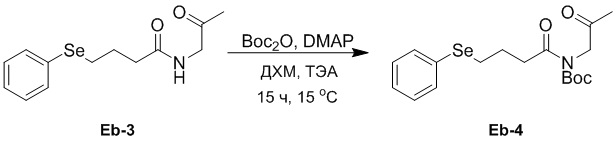
В смесь соединения Eb-3 (4.49 г, 15.1 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (30 мл) добавляли Boc2O (6.57 г, 30.1 ммоль, 2.0 экв.), ТЭА (1.52 г, 15.1 ммоль, 1.0 экв.) и DMAP (1.84 г, 15.1 ммоль, 1.0 экв.), и реакционную смесь перемешивали при 15°C в течение 15 часов в атмосфере азота. Удаляли растворитель при пониженном давлении и очищали остаток методом флэш-хроматографии на силикагеле, получая соединение Eb-4 в виде ярко-желтого твердого вещества (5.40 г, 13.6 ммоль, выход: 90%).
1H ЯМР (400 МГц, CDCl3) δ 7.53 - 7.49 (м, 2H), 7.29 - 7.22 (м, перекрывается с сигналом CDCl3, 3H), 4.51 (2H, с), 3.10 (т, J = 7.2 Гц, 2H), 2.97 (т, J = 7.2 Гц, 2H), 2.16 (с, 3H), 2.09 - 2.01 (м, 2H), 1.49 (с, 9H).
(23-4) Получение соединения Eb-5
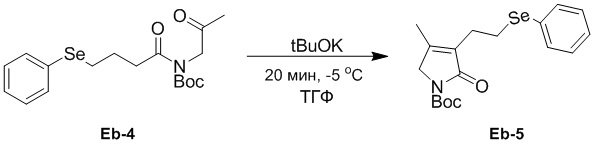
В смесь соединения Eb-4 (1.48 г, 3.72 ммоль, 1.0 экв.), полученного как описано выше, в ТГФ (40 мл) добавляли t-BuOK (7.43 мл, 2.0 экв., 1 M в ТГФ) при -5°C в атмосфере азота и перемешивали смесь при -5°C в течение 20 минут. Добавляли в реакционную смесь холодную воду (70 мл) и экстрагировали дихлорметаном (50 мл × 4). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ea-5, соответствующее соединению формулы 2 по настоящему изобретению, в виде бесцветного и прозрачного масла (413 мг, 1.09 ммоль, выход: 29%).
1H ЯМР (400 МГц, CDCl3) δ 7.51 - 7.46 (м, 2H), 7.29 - 7.20 (м, перекрывается с сигналом CDCl3, 3H), 3.98 (с, 2H), 3.15 (т, J = 7.2 Гц, 2H), 2.69 (т, J = 7.2 Гц, 2H), 1.94 (с, 3H), 1.56 (с, 9H).
Пример 24. Получение соединения Eb
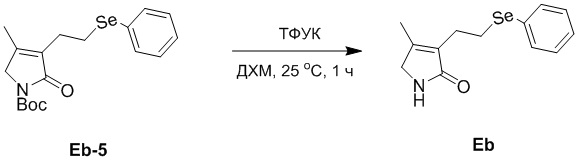
В смесь соединения Eb-5 (697 мг, 1.83 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (10 мл), добавляли ТФУК (627 мг, 5.50 ммоль, 3.0 экв.) и перемешивали при 25°C в течение 1 часа в атмосфере азота. Реакционную смесь доводили до pH 8 добавлением водного раствора NaHCO3 (20 мл) и затем экстрагировали дихлорметаном (20 мл × 3). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение Eb, соответствующее соединению формулы 2 по настоящему изобретению, в виде оранжевого масла (595 мг, выход: >99%).
1H ЯМР (400 МГц, CDCl3) δ 7.50 - 7.35 (м, 2H), 7.20 - 7.11 (м, перекрывается с сигналом CDCl3, 3H), 6.93 (ушир.с, 1H), 3.66 (с, 2H), 3.07 (т, J = 7.2 Гц, 2H), 2.62 (т, J = 7.2 Гц, 2H), 1.85 (с, 3H).
2.8. Получение соединений Ed-11 и Ed
Пример 25. Получение соединения Ed-11
(25-1) Получение соединения Ed-2
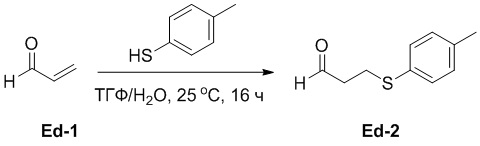
В смесь 4-метилбензолтиола (36.9 г, 297 ммоль, 0.83 экв.) в ТГФ (300 мл) и воде (150 мл) медленно добавляли Ed-1 (20.1 г, 358 ммоль, 1.0 экв.) при 0°C, и реакционную смесь перемешивали при 25°C в течение 16 часов в атмосфере азота. Добавляли водный раствор NaHCO3 (200 мл) и экстрагировали смесь этилацетатом (200 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл × 2), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение Ed-2 в виде желтого масла (59.4 г, 330 ммоль, выход: 92%).
1H ЯМР (400 МГц, CDCl3) δ 9.74 (с, 1H), 7.27 (д, J = 8.0 Гц, 2H), 7.11 (д, J = 7.6 Гц, 2H), 3.13 (т, J = 7.2 Гц, 2H), 2.75 - 2.71 (м, 2H), 2.32 (с, 3H).
(25-2) Получение соединения Ed-3
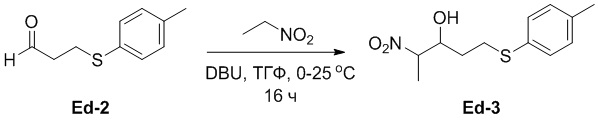
В смесь соединения Ed-2 (58 г, 322 ммоль, 1.0 экв.), полученного как описано выше, и DBU (4.90 г, 32.2 ммоль, 0.1 экв.) в ТГФ (400 мл) добавляли раствор 1-нитроэтана (24.0 г, 322 ммоль, 1.0 экв.) в ТГФ (50 мл) при 0°C, и реакционную смесь перемешивали при 25°C в течение 16 часов. Полученную смесь разводили водой (200 мл), затем экстрагировали этилацетатом (400 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (200 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ed-3 в виде желтого масла (51.7 г, 202 ммоль, выход: 63%).
1H ЯМР (400 МГц, CDCl3) δ 7.28 (д, J = 7.6 Гц, 2H), 7.13 (д, J = 8.0 Гц, 2H), 4.54 - 4.46 (м, 1H), 4.15 - 4.12 (м, 1H), 3.15 - 3.08 (м, 1H), 3.03 - 2.96 (м, 1H), 2.33 (с, 3H), 1.80 - 1.64 (м, 2H), 1.53 (т, J = 8.0 Гц, 3H).
(25-3) Получение соединения Ed-4
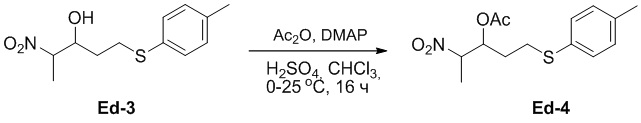
В смесь соединения Ed-3 (51.7 г, 202 ммоль, 1.0 экв.), полученного как описано выше, и H2SO4 (199 мг, 2.02 ммоль, 0.01 экв.) в хлороформе (500 мл) медленно добавляли уксусный ангидрид (31.0 г, 304 ммоль, 1.5 экв.) при 0°C и перемешивали при 25°C в течение 16 часов. Реакцию останавливали путем добавления водного раствора NaHCO3 (100 мл) и экстрагировали смесь дихлорметаном (50 мл × 4). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл × 2), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение Ed-4 в виде коричневого масла (65.5 г, выход: >99%).
1H ЯМР (400 МГц, CDCl3) δ 7.27 (д, J = 8.4 Гц, 2H), 7.12 (д, J = 8.0 Гц, 2H), 5.45 - 5.40 (м, 1H), 4.75 - 4.66 (м, 1H), 2.98 - 2.78 (м, 2H), 2.33 (с, 3H), 2.07 (д, J = 8.8 Гц, 3H), 1.99 - 1.80 (м, 2H), 1.49 (дд, J = 6.8, 2.0 Гц, 3H).
(25-4) Получение соединения Ed-5
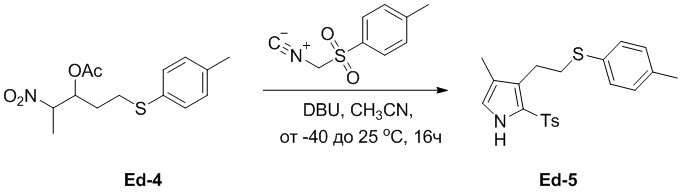
В смесь соединения Ed-4 (3.61 г, 18.5 ммоль, 1.0 экв.), полученного как описано выше, и DBU (5.63 г, 37.0 ммоль, 2.0 экв.) в ACN (50 мл) добавляли раствор TosMIC (5.00 г, 16.8 ммоль, 0.9 экв.) в ACN (10 мл) по каплям при -40°C в атмосфере азота и перемешивали при 25°C в течение 16 часов в атмосфере азота. Полученную смесь разводили водой (100 мл), затем экстрагировали этилацетатом (100 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ed-5 в виде красного масла (3.47 г, 9.00 ммоль, выход: 53%).
1H ЯМР (400 МГц, CDCl3) δ 8.99 (с, 1H), 7.65 (д, J = 8.0 Гц, 2H), 7.31 (д, J = 8.0 Гц, 2H), 7.21 (д, J = 8.0 Гц, 2H), 7.14 (д, J = 8.0 Гц, 2H), 6.69 (д, J = 2.0 Гц, 1H), 2.87 - 2.81 (м, 4H), 2.37 (с, 3H), 2.35 (с, 3H), 1.93 (с, 3H).
(25-5) Получение соединения Ed-6
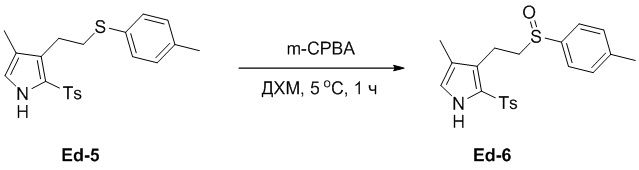
В смесь соединения Ed-5 (10.0 г, 25.9 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (40 мл) добавляли m-CPBA (5.27 г, 25.9 ммоль, 85% чистота, 1.0 экв.) по каплям при 5°C в атмосфере азота и перемешивали при 5°C в течение 1 часа. Реакцию останавливали путем добавления водного раствора Na2SO3 (100 мл), и смесь экстрагировали дихлорметаном (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая остаток от упаривания. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ed-6 в виде белого твердого вещества (5.0 г, 12.5 ммоль, выход: 48%).
1H ЯМР (400 МГц, CDCl3) δ 9.20 (с, 1H), 7.57 - 7.52 (м, 4H), 7.36 (д, J = 8.4 Гц, 2H), 7.21 (д, J = 8.0 Гц, 2H), 6.69 (д, J = 2.8 Гц, 1H), 2.97 - 2.93 (м, 2H), 2.89 - 2.69 (м, 2H), 2.45 (с, 3H), 2.39 (с, 3H), 1.94 (с, 3H).
(25-6) Получение соединения Ed-7
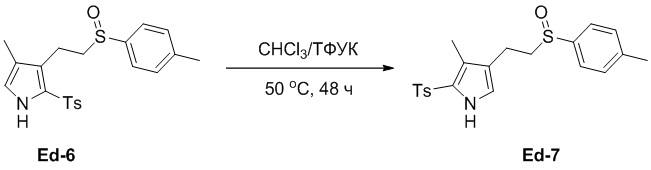
Смесь соединения Ed-6 (5.0 г, 12.5 ммоль, 1.0 экв.), полученного как описано выше, и ТФУК (5 мл) в хлороформе (45 мл) перемешивали при 50°C в течение 48 часов в атмосфере азота. Реакцию останавливали путем добавления воды (100 мл) и затем экстрагировали смесь дихлорметаном (100 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 × 2 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая остаток от упаривания. Остаток очищали методом флэш-хроматографии на силикагеле, получая соединение Ed-7 в виде белого твердого вещества (1.90 г, 4.68 ммоль, выход: 37%).
1H ЯМР (400 МГц, CDCl3) δ 9.02 (с, 1H), 7.75 (д, J = 8.4 Гц, 2H), 7.49 (д, J = 8.4 Гц, 2H), 7.33 - 7.27 (м, 4H), 6.74 (д, J = 2.8 Гц, 1H), 2.91 - 2.64 (м, 4H), 2.42 (с, 3H), 2.40 (с, 3H), 2.10 (с, 3H).
(25-7) Получение соединения Ed-8
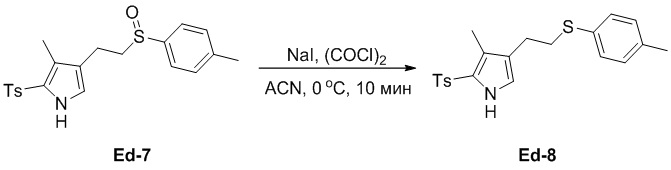
В смесь соединения Ed-7 (3 г, 7.47 ммоль, 1.0 экв.), полученного как описано выше, в ACN (48 мл) добавляли NaI (2.82 г, 18.67 ммоль, 2.5 экв.) при 0°C и перемешивали 10 минут. Добавляли по каплям (COCl)2 (0.77 мл, 9.38 ммоль, 1.2 экв.) при 0°C и перемешивали смесь 10 минут в тех же условиях. Реакцию останавливали путем добавления воды (30 мл), затем экстрагировали смесь этилацетатом (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ed-8 в виде коричневого твердого вещества (2.21 г, 5.71 ммоль, выход: 76%).
1H ЯМР (400 МГц, CDCl3) δ 8.83 (с, 1H), 7.76 (д, J = 8.0 Гц, 2H), 7.29 (д, J = 8.0 Гц, 2H), 7.23 (д, J = 8.4 Гц, 2H), 7.09 (д, J = 8.0 Гц, 2H), 6.75 (д, J = 2.8 Гц, 1H), 3.00 - 2.96 (м, 2H), 2.65 (т, J = 8.0 Гц, 2H), 2.41 (с, 3H), 2.32 (с, 3H), 2.12 (с, 3H).
(25-8) Получение соединения Ed-9
В смесь соединения Ed-8 (2.2 г, 5.71 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (62 мл) добавляли PhMe3NBr3 (2.36 г, 5.71 ммоль, 1.0 экв.) при 0°C и перемешивали в течение 1 часа. Добавляли водный раствор NaHSO3 (30 мл) и экстрагировали смесь дихлорметаном (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ed-9 в виде коричневого твердого вещества (2.65 г, 5.23 ммоль, выход: 99%).
1H ЯМР (400 МГц, CDCl3) δ 8.93 (с, 1H), 7.76 (д, J = 8.0 Гц, 2H), 7.30 (д, J = 8.4 Гц, 2H), 7.25 (д, J = 8.0 Гц, 2H), 7.09 (д, J = 8.0 Гц, 2H), 2.92 (т, J = 7.6 Гц, 2H), 2.62 (т, J = 7.6 Гц, 2H), 2.42 (с, 3H), 2.32 (с, 3H), 2.13 (с, 3H).
(25-9) Получение соединения Ed-10
В смесь соединения Ed-9 (2.59 г, 5.57 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (50 мл) добавляли m-CPBA (1.05 г, 6.13 ммоль, 1.1 экв.) при 0°C и перемешивали при 25°C в течение 1 часа. Добавляли водный раствор NaHSO3 (30 мл), затем экстрагировали смесь дихлорметаном (60 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. В остаток добавляли ДХМ/Гексан, и смесь фильтровали, получая соединение Ed-10 в виде белого твердого вещества (2.9 г, выход: >99%).
1H ЯМР (500 МГц, CDCl3) δ 9.00 (с, 1H), 7.68 (д, J = 8.0 Гц, 2H), 7.43 (д, J = 7.9 Гц, 2H), 7.24 (дд, J = 12.2, 8.0 Гц, 4H), 2.86 - 2.78 (м, 1H), 2.78 - 2.67 (м, 2H), 2.58 - 2.48 (м, 1H), 2.34 (с, 3H), 2.33 (с, 3H), 2.04 (с, 3H).
(25-10) Получение соединения Ed-11
Смесь соединения Ed-10 (1.0 г, 2.08 ммоль, 1.0 экв.), полученного как описано выше, и ТФУК (1.5 мл) перемешивали при 25°C в течение 30 мин в атмосфере азота. Добавляли NaI (1.56 г, 10.4 ммоль, 5.0 экв.) и перемешивали при 25°C в течение 10 мин, нейтрализовывали смесь добавлением водного раствора K2CO3 при 0°C и экстрагировали дихлорметаном (150 мл × 2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ed-11, соответствующее соединению формулы 2 по настоящему изобретению, в виде темно-зеленого твердого вещества (550 мг, 1.37 ммоль, выход: 66%).
1H ЯМР (500 МГц, CDCl3) δ 7.59 (д, J = 8.2 Гц, 2H), 7.23 (д, J = 8.0 Гц, 2H), 7.10 (д, J = 8.1 Гц, 2H), 7.04 (д, J = 7.9 Гц, 2H), 6.30 (с, 1H), 2.71 - 2.65 (м, 1H), 2.41 - 2.32 (м, 2H), 2.30 (с, 3H), 2.25 (с, 3H), 2.21 - 2.14 (м, 1H), 2.05 (с, 3H).
Пример 26. Получение соединения Ed
В смесь соединения Ed-11 (0.55 г, 1.37 ммоль, 1.0 экв.), полученного как описано выше, в EtOH (50 мл) добавляли NaBH4 (67 мг, 1.78 ммоль, 1.3 экв.) при 0°C и перемешивали при 25°C в течение 1 часа. Добавляли водный раствор NH4Cl (30 мл) и экстрагировали смесь дихлорметаном (60 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли в остаток ДХМ/Гексан, и выпавший белый осадок отфильтровывали, получая соединение Ed, соответствующее соединению формулы 2 по настоящему изобретению (305 мг, 1.23 ммоль, выход: 90%).
1H ЯМР (500 МГц, ДМСО-d6) δ 7.92 (с, 1H), 7.25 (д, J = 7.9 Гц, 2H), 7.13 (д, J = 7.9 Гц, 2H), 3.70 (с, 2H), 3.01 (т, J = 7.2 Гц, 2H), 2.40 (т, J = 6.8 Гц, 2H), 2.26 (с, 3H), 1.86 (с, 3H).
2.9. Получение соединений Ee-5 и Ee
Пример 27. Получение соединения Ee-5
(27-1) Получение соединения Ee-3
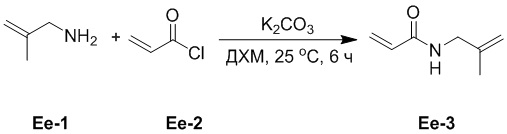
В смесь соединения Ee-1 (20.0 г, 281 ммоль, 1.0 экв.) в ДХМ (300 мл), добавляли K2CO3 (81.6 г, 591 ммоль, 2.1 экв.) при 0°C и перемешивали 15 минут. Затем добавляли соединение Ee-2 (25.5 г, 281 ммоль, 1.0 экв.) при 0°C и перемешивали при 25°C в течение 6 часов. Полученную смесь фильтровали при пониженном давлении, и остаток очищали методом колоночной хроматографии на силикагеле, получая соединение Ee-3 (35.0 г, 279 ммоль, выход: 99%).
1H ЯМР (400 МГц, CDCl3) δ 6.28 (дд, J = 16.8, 1.6 Гц, 1H), 6.16 (дд, J = 16.8, 10.0 Гц, 1H), 6.15 (ушир.с, 1H), 5.63 (дд, J = 10.4, 1.6 Гц, 1H), 4.83 (с, 2H), 3.86 (д, J = 6.0 Гц, 2H), 1.72 (с, 3H).
(27-2) Получение соединения Ee-4
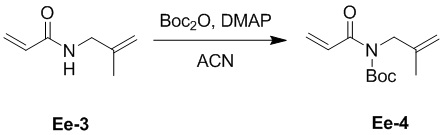
В смесь соединения Ee-3 (36.0 г, 288 ммоль, 1.0 экв.), полученного как описано выше, и Boc2O (75.3 г, 345 ммоль, 1.2 экв.) в ACN (300 мл), добавляли раствор DMAP (3.51 г, 28.8 ммоль, 0.1 экв.) в ACN (50 мл) при 25°C и перемешивали 16 часов. Упаривали реакционную смесь при пониженном давлении для удаления ACN, остаток разводили водой (100 мл), затем экстрагировали этилацетатом (150 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл) и сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая соединение Ee-4 в виде желтого твердого вещества (46.8 г, 207 ммоль, выход: 72%).
1H ЯМР (400 МГц, CDCl3) δ 7.05 (дд, J = 16.8, 10.4 Гц, 1H), 6.34 (дд, J = 16.8, 1.6 Гц, 1H), 5.71 (дд, J = 10.4, 1.6 Гц, 1H), 4.83 (с, 1H), 4.70 (с, 1H), 4.26 (с, 2H), 1.73 (с, 3H), 1.50 (с, 9H).
(27-3) Получение соединения Ee-5
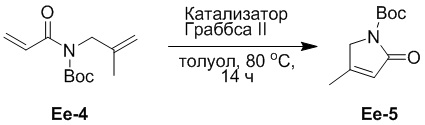
В смесь соединения Ee-4 (5.0 г, 22.2 ммоль, 1.0 экв.), полученного как описано выше, в толуоле (50 мл) добавляли катализатор Граббса второго поколения (1.32 г, 1.55 ммоль, 0.07 экв.) и перемешивали смесь при 80°C в течение 14 часов. Реакционную смесь упаривали при пониженном давлении для удаления толуола. Остаток очищали на обращенно-фазной колонке, получая соединение Ee-5, соответствующее соединению формулы 2 по настоящему изобретению, в виде желтого твердого вещества (2.14 г, 10.9 ммоль, выход: 49%).
1H ЯМР (400 МГц, CDCl3) δ 5.83 (с, 1H), 4.19 (с, 2H), 2.08 (с, 3H), 1.53 (с, 9H).
Пример 28. Получение соединения Ee
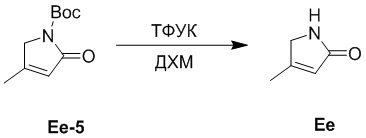
ТФУК (0.3 мл, 6.08 ммоль, 3.0 экв.) добавляли в смесь соединения Ee-5 (0.4 г, 2.02 ммоль, 1.0 экв.), полученного как описано выше, в ДХМ (4 мл) и перемешивали при 25°C в течение 3 часов. Значение pH реакционной смеси доводили до 7 добавлением водного раствора NaHCO3 (10 мл) и экстрагировали дихлорметаном (20 мл × 2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение Ee, соответствующее соединению формулы 2 по настоящему изобретению, в виде белого твердого вещества (0.12 г, 1.23 ммоль, выход: 61%).
1H ЯМР (400 МГц, CDCl3) δ 6.46 (с, 1H), 5.85 (с, 1H), 3.92 (с, 2H), 2.09 (с, 3H).
3. Получение соединения формулы 3
Соединения формулы 3 (Примеры 29 - 76), соответственно, получали путем сочетания соединения формулы 1 и соединения формулы 2 по настоящему изобретению.
3.1. Получение соединения F-3a путем сочетания соединения D и соединения Ea
Соединение D из Примера 2 и соединение Ea из Примера 8 вводили в реакцию сочетания в различных условиях, получая F-3a, соответствующее соединению формулы 3.
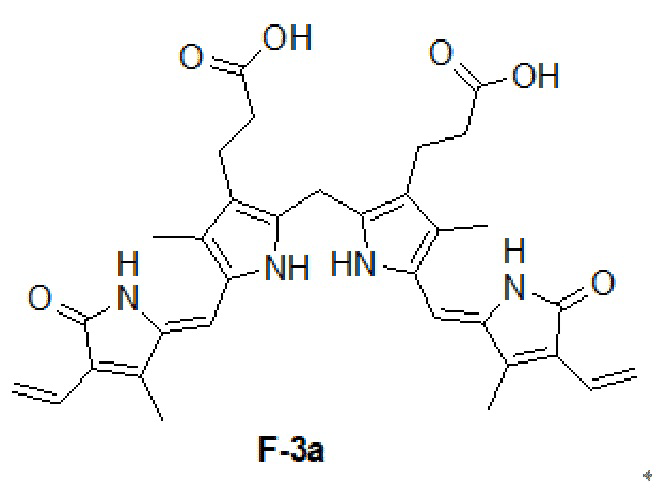
Пример 29. Получение F-3a
В смесь соединения D (1.29 г, 3.44 ммоль, 1.0 экв.) в 1,4-диоксане (20 мл) добавляли пиперидин (2.35 г, 27.5 ммоль, 8.0 экв.) и соединение Ea (1.06 г, 8.61 ммоль, 2.5 экв.) и перемешивали при 100°C в течение 14 часов в атмосфере азота. Удаляли растворитель при пониженном давлении, добавляли в остаток CHCl3 (600 мл), перемешивали 0.5 часа и промывали 0.2 M водным раствором соляной кислоты (150 мл × 2). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в метаноле (50 мл) при 20°C. Красный осадок отфильтровывали и промывали метанолом (10 мл × 2), получая соединение F-3a в виде красного твердого вещества (1.20 г, 2.05 ммоль, выход: 60%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.52 (ушир.с, 2H), 9.95 (ушир.с, 2H), 6.58 (дд, J = 17.6, 11.6 Гц, 2H), 6.20 (дд, J = 17.6, 2.8 Гц, 2H), 6.09 (с, 2H), 5.29 (дд, J = 11.6, 2.8 Гц, 2H), 3.99 (с, 2H), 2.50 - 2.43 (м, 4H), 2.16 (с, 6H), 2.03 (с, 6H), 2.00 - 1.85 (м, 4H).
Пример 30. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пирролидин (8.0 экв.) и соединение Ea (2.5 экв.) и перемешивали реакционную смесь при 100°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 9%.
Пример 31. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли пиперазин (8.0 экв.) и соединение Ea (2.5 экв.) и перемешивали 100°C в течение 16 часов в атмосфере азота. Удаляли растворитель при пониженном давлении, добавляли в остаток CHCl3 (600 мл), перемешивали 0.5 часа и промывали 0.2 М раствором соляной кислоты (150 мл × 2). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в метаноле (50 мл) при 20°C. Красный осадок отфильтровывали и промывали метанолом (10 мл × 2), получая соединение F-3a в виде красного твердого вещества (выход: 15%).
Пример 32. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли морфолин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 37%.
Пример 33. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 20%.
Пример 34. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 11%.
Пример 35. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали 8 часов в атмосфере азота при 100°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 38%.
Пример 36. Получение F-3a
В смесь соединения D (1.0 экв.) и MeOH добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 11%.
Пример 37. Получение F-3a
В смесь соединения D (1.0 экв.) и MeOH добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 9%.
Пример 38. Получение F-3a
В смесь соединения D (1.0 экв.) и MeOH добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 65°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 7%.
Пример 39. Получение F-3a
В смесь соединения D (1.0 экв.) и MeOH добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 66°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 4%.
Пример 40. Получение F-3a
В смесь соединения D (1.0 экв.) и ТГФ добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 22%.
Пример 41. Получение F-3a
В смесь соединения D (1.0 экв.) и ТГФ добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 25°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 17%.
Пример 42. Получение F-3a
В смесь соединения D (1.0 экв.) и ТГФ добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 66°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 51%.
Пример 43. Получение F-3a
В смесь соединения D (1.0 экв.) и ТГФ добавляли пиперидин (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 66°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 10%.
Пример 44. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли азепан (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 60°C в течение 22 часов в атмосфере азота. Удаляли растворитель при пониженном давлении, добавляли в остаток CHCl3 (600 мл), перемешивали 0.5 часа и промывали 0.2 М раствором соляной кислоты (150 мл × 2). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в метаноле (50 мл) при 20°C. Красный осадок отфильтровывали и промывали метанолом (10 мл × 2), получая соединение F-3a в виде красного твердого вещества (выход: 25%).
Пример 45. Получение F-3a
В смесь соединения D (1.0 экв.) и 1,4-диоксана добавляли азокан (8.0 экв.) и соединение Ea (2.5 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов в атмосфере азота. Удаляли растворитель при пониженном давлении, добавляли в остаток CHCl3 (600 мл), перемешивали 0.5 часа и затем промывали 0.2 М раствором соляной кислоты (150 мл × 2). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в метаноле (50 мл) при 20°C. Красный осадок отфильтровывали и промывали метанолом (10 мл × 2), получая соединение F-3a в виде красного твердого вещества (выход: 26%).
3.2. Получение соединения F-3a путем сочетания соединения D и соединения Ea-3a
F-3a, соответствующее соединению формулы 3, получали путем сочетания соединения D из Примера 2 с соединением Ea-3a из Примера 9 в различных условиях.
Пример 46. Получение F-3a
В смесь соединения D (50 мг, 0.13 ммоль, 1.0 экв.) в 1,4-диоксане (2 мл) добавляли пиперидин (90.97 мг, 1.07 ммоль, 8.0 экв.) и соединение Ea-3a (56.56 мг, 0.40 ммоль, 3.0 экв.) и перемешивали при 100°C в течение 16 часов в атмосфере азота. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали. Остаток растирали в MeOH (6 мл), получая соединение F-3a в виде коричневого твердого вещества (18.1 мг, 30.9 мкмоль, выход: 23%).
Пример 47. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пиперидин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 15%.
Пример 48. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пирролидин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 12%.
Пример 49. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пирролидин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 11%.
Пример 50. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли морфолин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов в атмосфере азота. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали. Остаток растирали в MeOH, получая соединение F-3a в виде коричневого твердого вещества (выход: 13%).
Пример 51. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли морфолин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов в атмосфере азота. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали. Остаток растирали в MeOH, получая соединение F-3a в виде коричневого твердого вещества (выход: 10%).
Пример 52. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пиперазин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов в атмосфере азота. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали. Остаток растирали в MeOH, получая соединение F-3a в виде коричневого твердого вещества (выход: 18%).
Пример 53. Получение F-3a
В смесь соединения D (1.0 экв.) в 1,4-диоксане добавляли пиперазин (8.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов в атмосфере азота. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали. Остаток растирали в MeOH, получая соединение F-3a в виде коричневого твердого вещества (выход: 17%).
3.3. Получение соединения F-3a путем сочетания соединения D и соединения Ea-3c
F-3a, соответствующее соединению формулы 3, получали путем сочетания соединения D из Примера 2 с соединением Ea-3c из Примера 15 в различных условиях.
Пример 54. Получение F-3a
В смесь соединения D (30 мг, 80.13 мкмоль, 1.0 экв.) и соединения Ea-3c (47.89 мг, 0.24 ммоль, 3.0 экв.) в 1,4-диоксане (3 мл) добавляли пиперидин (54.58 мг, 0.64 ммоль, 8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов. Упаривали смесь при пониженном давлении, добавляли CHCl3 (110 мл × 2) и промывали 0.3 M водным раствором HCl (20 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали при пониженном давлении и упаривали, затем подтверждали структуру F-3a методом LCMS.
C33H36N4O6 m/z [M+H]+ = 585.
3.4. Получение соединения D-Ea-2a путем сочетания соединения D и соединения Ea-2a
D-Ea-2a, соответствующее соединению формулы 3, получали путем сочетания соединения D из Примера 2 с соединением Ea-2a из Примера 10, и таким образом получали соединение F.
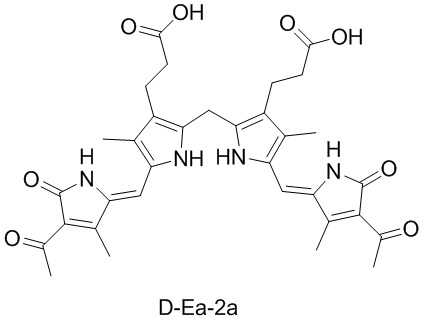
Пример 55. Получение соединения D-Fa-2a
Смесь соединения D (64 мг, 0.17 ммоль, 1.0 экв.) и азепана (0.11 мл, 1.02 ммоль, 6.0 экв.) в 1,4-диоксане (3.6 мл) перемешивали при 25°C в течение 10 минут. Добавляли соединение Ea-2a (60 мг, 0.43 ммоль, 2.5 экв.) и перемешивали смесь при 40°C в течение 16 часов. Добавляли CHCl3 (100 мл), и органический слой промывали 0.2 M водным раствором HCl (80 мл × 2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Растворяли остаток в EtOAc (5 мл) и перемешивали 5 минут, добавляли по каплям гексан (30 мл). Выпавший красный осадок отфильтровывали, получая соединение D-Ea-2a (100 мг, 0.16 ммоль, выход: 94%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.94 (ушир.с, 2H), 10.84 (с, 2H), 10.14 (с, 2H), 6.45 (с, 2H), 4.08 (с, 2H), 2.46 (с, 6H), 2.45 (с, 6H), 2.45 - 2.40 (м, 4H), 2.09 (с, 6H), 1.95 - 1.85 (м, 4H).
Пример 56. Получение соединения F-3a из соединения D-Ea-2a
(56-1) Получение соединения D-Ea-3a
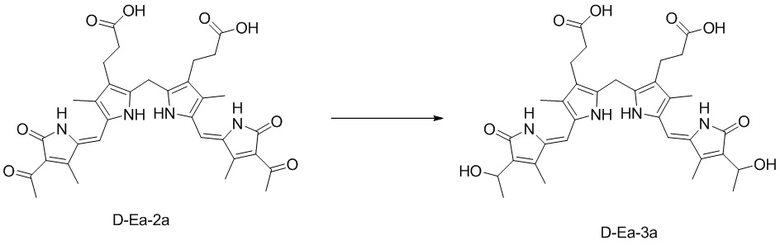
В смесь соединения D-Ea-2a (100 мг, 0.16 ммоль, 1.0 экв.), полученного как описано выше, в ДМСО (6 мл) добавляли NaBH4 (24.2 мг, 0.64 ммоль, 4.0 экв.), и реакционную смесь перемешивали при 25°C в течение 16 часов. Реакцию останавливали добавлением водного раствора NH4Cl (50 мл), экстрагировали полученную смесь хлороформом (100 мл × 3) и промывали насыщенным раствором хлорида натрия (50 мл × 5). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение D-Ea-3a (20 мг, 0.03 ммоль, выход: 20%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.87 (ушир.с, 2H), 10.35 (ушир.с, 2H), 9.77 (с, 2H), 5.98 (с, 2H), 4.67 - 4.65 (м, 2H), 3.96 (с, 2H), 2.43 - 2.41 (м, 4H), 2.29 (с, 6H), 2.00 (с, 6H), 1.97 - 1.94 (м, 4H), 1.31 (д, J = 6.8 Гц, 6H).
(56-2) Получение соединения F-3a

В смесь соединения D-Ea-3a (10 мг, неочищенное), полученного как описано выше, и ДХМ (1 мл) добавляли ТЭА (27 мкл, 0.19 ммоль, 12.0 экв.), затем добавляли раствор POCl3 (6.02 мкл. 0.064 ммоль, 4.0 экв.) в ДХМ (30 мкл) при 0°C. Полученную смесь перемешивали при 25°C в течение 3 часов в атмосфере азота. Реакцию останавливали путем добавления воды (30 мл), затем экстрагировали хлороформом (50 мл × 2). Экстракт сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в смеси ДХМ:MeOH (1:1) и фильтровали, получая соединение F-3a в виде оранжевого твердого вещества.
3.5. Получение соединения D-Eb путем сочетания соединения D и соединения Eb
D-Eb, соответствующее соединению формулы 3, получали путем сочетания соединения D из Примера 2 с соединением Eb из Примера 24, и из него получали соединение F-3a.
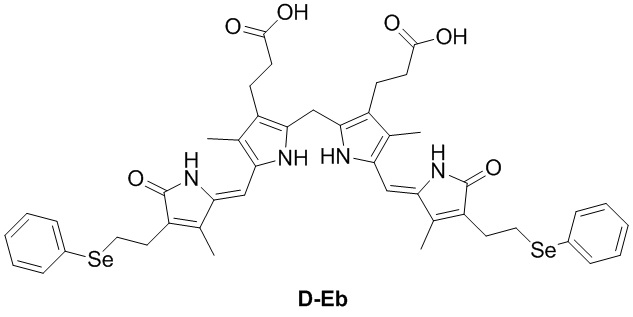
Пример 57. Получение соединения D-Eb
В смесь соединения D (268 мг, 0.71 ммоль, 1.0 экв.) и пиперидина (10.0 экв.) в 1,4-диоксане (15 мл) добавляли соединение Eb (501 мг, 1.80 ммоль, 2.5 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов. Добавляли 0.1 M водный раствор HCl (71.6 мл), затем экстрагировали смесь этилацетатом (50 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растирали в MeOH (20 мл) при 20°C. Выпавший осадок отделяли фильтрованием и промывали метанолом (10 мл × 2), получая соединение D-Eb в виде желтого твердого вещества (471 мг, 0.52 ммоль, выход: 73%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.91 (ушир.с, 2H), 10.36 (с, 2H), 9.82 (с, 2H), 7.50 - 7.48 (м, 4H), 7.31 - 7.23 (м, 6H), 5.97 (с, 2H), 3.96 (с, 2H), 3.09 (т, J = 7.6 Гц, 4H), 2.63 (т, J = 7.6 Гц, 4H), 2.41 (т, J = 7.2 Гц, 4H), 2.01 (с, 6H), 2.00 (с, 6H), 1.96 (т, J = 8.0 Гц, 4H).
Пример 58. Получение соединения F-3a из соединения D-Eb
В раствор соединения D-Eb (100 мг, 0.11 ммоль, 1.0 экв.) в ТГФ (8 мл) добавляли HOAc (20 мг, 0.33 ммоль, 3.0 экв.) и H2O2 (51 мг, 0.45 ммоль, 4.0 экв.), реакционную смесь перемешивали при 25°C в течение 1 часа. Упаривали смесь при пониженном давлении, остаток растирали в MeOH (10 мл) при 20°C. Осадок отделяли фильтрованием и промывали метанолом (5 мл × 2), получая F-3a, соответствующее соединению формулы 3 по настоящему изобретению в виде красного твердого вещества (10 мг, 0.017 ммоль, выход: 15%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.74 (ушир.с, 2H), 10.51 (с, 2H), 9.95 (с, 2H), 6.58 (дд, J = 17.6, 11.6 Гц, 2H), 6.20 (дд, J = 17.6, 2.8 Гц, 2H), 6.08 (с, 2H), 5.29 (дд, J = 11.6, 2.8 Гц, 2H), 3.98 (с, 2H), 2.43 (т, J = 8.4 Гц, 4H), 2.16 (с, 6H), 2.03 (с, 6H), 1.94 (т, J = 8.4 Гц, 4H).
3.6. Получение соединения D-Ed путем сочетания соединения D и соединения Ed
D-Ed, соответствующее соединению формулы 3 получали путем сочетания соединения D из Примера 2 с соединением Ed из Примера 26, и из него получали соединение F-3a.
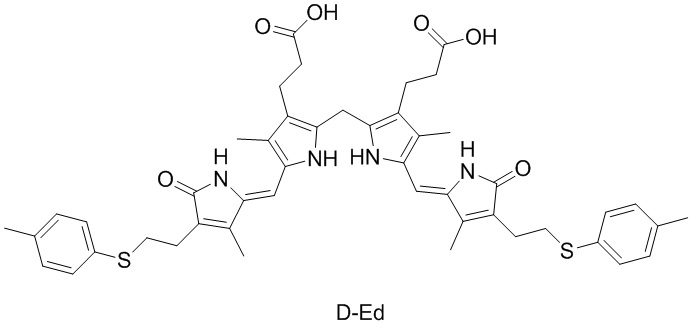
Пример 59. Получение соединения D-Ed
Смесь соединения D (16.3 мг, 0.04 ммоль, 1.0 экв.) и азепана (29.8 мкл, 0.26 ммоль, 6.0 экв.) в 1,4-диоксане (5 мл) перемешивали при 25°C в течение 10 минут. Добавляли соединение Ed (21.6 мг, 0.09 ммоль, 2 экв.), полученное как описано выше, и реакционную смесь перемешивали при 80°C в течение 16 часов. Добавляли 0.1 M водный раствор HCl (10 мл) и экстрагировали смесь хлороформом (10 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, получая соединение D-Ed.
C47H52N4O6S2 m/z [M+2H]+ = 834.
Пример 60. Получение соединения F-3a из соединения D-Ed
Смесь D-Ed (30 мг), полученного как описано выше, в 1,4-диоксане (8 мл) охлаждали до 0°C и добавляли m-CPBA (9 мг, 0.04 ммоль). Затем реакционную смесь перемешивали при 0°C в течение 1 часа. Добавляли водный раствор NaHSO3, затем экстрагировали смесь хлороформом, и объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Растворяли остаток в ДМФА (8 мл), добавляли пиридин (3 мл), кипятили в течение 2 часов. Полученную смесь разделяли на слои между 0.1 M HCl и CHCl3, органический слой сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении, затем подтверждали структуру соединения F-3a методом LCMS.
C33H36N4O6 m/z [M+2H]+ = 586.
3.7. Получение соединения D-Ee путем сочетания соединения D и соединения Ee
D-Ee, соответствующее соединению формулы 3, получали путем сочетания соединения D из Примера 2 с соединением Ee из Примера 28.
Пример 61. Получение соединения D-Ee путем сочетания соединения D и соединения Ee
В смесь соединения D (100 мг, 0.26 ммоль, 1.0 экв.) в 1,4-диоксане (9 мл) добавляли азепан (0.18 мл, 1.60 ммоль, 6.0 экв.), и реакционную смесь перемешивали при 25°C в течение 10 минут. Добавляли соединение Ee (64.8 мг, 0.67 ммоль, 2.5 экв.) и перемешивали реакционную смесь при 40°C в течение 15 часов. Добавляли в смесь CHCl3 (100 мл) и промывали 0.1 М раствором соляной кислоты (30 мл × 2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный осадок очищали методом хроматографии на силикагеле, получая соединение D-Ee в виде зеленого твердого вещества (32 мг, 0.057 ммоль, выход: 22%).
1H ЯМР (400 МГц, ДМСО-d6) δ 11.90 (ушир.с, 2H), 10.40 (с, 2H), 9.77 (с, 2H), 5.99 (с, 2H), 5.73 (с, 2H), 3.97 (с, 2H), 2.41 (т, J = 6.0 Гц, 4H), 2.14 (с, 6H), 2.01 (с, 6H), 1.95 (т, J = 6.1 Гц, 4H).
3.8. Получение соединения C-Ea путем сочетания соединения C и соединения Ea
Соединение C-Ea, соответствующее соединению формулы 3 (Примеры 62 - 67) получали путем сочетания соединения C из Примера 1 с соединением Ea из Примера 8 в различных условиях, и из него получали соединение F-3a (Пример 68).
Пример 62. Получение соединения C-Ea
В смесь соединения C (1.09 г, 2.71 ммоль, 1.0 экв.) и соединения Ea (1.00 г, 8.12 ммоль, 3.0 экв.) в 1,4-диоксане (10 мл) добавляли пиперидин (1.84 г, 21.7 ммоль, 8.0 экв.), и реакционную смесь перемешивали при 80°C в течение 12 часов. Реакционную смесь упаривали при пониженном давлении и очищали методом преп-ВЭЖХ. Остатки растворителя удаляли лиофилизациоей, получая соединение C-Ea в виде черного твердого вещества (10 мг, 16.3 мкмоль, выход: 0.6%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.62 (с, 2H), 9.96 (с, 2H), 6.65 (дд, J = 17.2, 11.2 Гц, 2H), 6.28 (дд, J = 17.2, 2.4 Гц, 2H), 6.15 (с, 2H), 5.37 (д, J = 12.0 Гц, 2H), 4.05 (с, 2H), 3.49 (с, 6H), 2.51 - 2.41 (м, 4H), 2.23 (с, 6H), 2.08 (с, 6H), 1.98 - 1.94 (м, 4H).
Пример 63. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) и соединения Ea (3.0 экв.) в 1,4-диоксане добавляли пиперазин (8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 27%.
Пример 64. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) и соединения Ea (3.0 экв.) в 1,4-диоксане добавляли пиперазин (8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 21%.
Пример 65. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) и соединения Ea (3.0 экв.) в 1,4-диоксане добавляли морфолин (8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 8 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 35%.
Пример 66. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) и соединения Ea (3.0 экв.) в 1,4-диоксане добавляли морфолин (8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 38%.
Пример 67. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) и соединения Ea (3.0 экв.) в 1,4-диоксане добавляли пролин (8.0 экв.), и реакционную смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь упаривали при пониженном давлении и очищали методом преп-ВЭЖХ. Остатки растворителя удаляли лиофилизацией, получая соединение C-Ea в виде черного твердого вещества (выход: 0.9%).
Пример 68. Получение соединения F-3a из соединения C-Ea
В раствор соединения C-Ea (50 мг, 81.60 мкмоль, 1.0 экв.), полученного как описано выше, в метаноле (5 мл) добавляли раствор LiOH⋅H2O (20.55 мг, 0.49 ммоль, 6.0 экв.) в воде (1 мл), и реакционную смесь перемешивали при 60°C в течение 2 часов в атмосфере азота. Добавляли CHCl3 (20 мл) и pH смеси доводили до кислого добавлением 0.1 M водного раствора HCl (10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Метанол (10 мл) добавляли в остаток, и выпавший осадок отфильтровывали при пониженном давлении, получая соединение F-3a (билирубин) в виде оранжевого твердого вещества (10 мг, 17.10 мкмоль, выход: 21%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.50 (с, 2H), 9.93 (с, 2H), 6.58 (дд, J = 17.2, 11.2 Гц, 2H), 6.21 (дд, J = 17.6, 2.4 Гц, 2H), 6.07 (с, 2H), 5.30 (дд, J = 11.6, 2.8 Гц, 2H), 3.99 (с, 2H), 2.42 - 2.30 (м, 4H), 2.16 (с, 6H), 2.03 (с, 6H), 1.95 - 1.91 (м, 4H).
3.9. Получение соединения C-Ea путем сочетания соединения C и соединения Ea-3a
Соединение C-Ea, соответствующее соединению формулы 3 (Примеры 69 - 72), получали путем сочетания соединения C из Примера 1 с соединением Ea-3a из Примера 9 в различных условиях.
Пример 69. Получение соединения C-Ea
В смесь соединения C (1.33 г, 3.31 ммоль, 1.0 экв.) в 1,4-диоксане (20 мл) добавляли пиперидин (2.81 г, 33.1 ммоль, 3.26 мл, 10.0 экв.) и соединение Ea-3a (1.4 г, 9.92 ммоль, 3.0 экв.), и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Добавляли CHCl3 (350 мл) и промывали полученную смесь 0.1 M водным раствором HCl (150 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом преп-ВЭЖХ. Растворитель удаляли лиофилизацией, получая соединение C-Ea в виде черного твердого вещества (415 мг, 0.68 ммоль, выход: 20%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.55 (с, 2H), 9.88 (с, 2H), 6.58 (дд, J = 17.2, 11.6 Гц, 2H), 6.21 (дд, J = 17.2, 2.8 Гц, 2H), 6.07 (с, 2H), 5.30 (дд, J = 11.6, 2.4 Гц, 2H), 3.98 (с, 2H), 3.28 (с, перекрывается с пиком H2O, 6H), 2.50 (т, перекрывается с сигналом ДМСО-d6, 4H), 2.16 (с, 6H), 2.01 (с, 6H), 1.88 (т, J = 8.4 Гц, 4H).
Пример 70. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) в 1,4-диоксане добавляли пирролидин (10.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 23%.
Пример 71. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) в 1,4-диоксане добавляли пиперазин (10.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 37%.
Пример 72. Получение соединения C-Ea
В смесь соединения C (1.0 экв.) в 1,4-диоксане добавляли морфолин (10.0 экв.) и соединение Ea-3a (3.0 экв.), и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 32%.
3.10. Получение соединения C1-Ea путем сочетания соединения C1 и соединения Ea
C1-Ea, соответствующее соединению формулы 3, получали путем сочетания соединения C1 из Примера 3 с соединением Ea из Примера 8, и из него получали соединение F-3a.
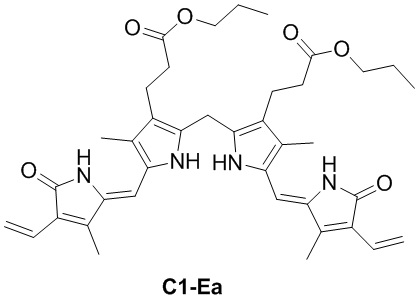
Пример 73. Получение соединения C1-Ea
В смесь соединения C1 (80 мг, 0.17 ммоль, 1.0 экв.) в 1,4-диоксане (1 мл) добавляли пиперидин (0.14 г, 1.7 ммоль, 10.0 экв.) и соединение Ea (62.8 мг, 0.51 ммоль, 3.0 экв.), и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Добавляли CHCl3 (30 мл) и промывали полученную смесь 0.1 M водным раствором HCl (20 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли в остаток по каплям ДХМ/Гексан и отфильтровывали выпавший осадок, получая соединение C1-Ea в виде красного твердого вещества (40 мг, 0.059 ммоль, выход: 35%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.58 (ушир.с, 2H), 9.89 (ушир.с, 2H), 6.58 (дд, J = 17.2, 11.6 Гц, 2H), 6.21 (2H, д, J = 17.2 Гц), 6.06 (2H, с), 5.30 (д, J = 11.6 Гц, 2H), 3.98 (с, 2H), 3.77 (т, J = 6.0 Гц, 4H), 2.50 - 2.40 (м, перекрывается с сигналом ДМСО-d6, 4H), 2.15 (с, 6H), 2.01 (с, 6H), 1.89 - 1.77 (м, 4H), 1.45 - 1.40 (м, 4H), 0.76 (т, J = 7.2 Гц, 6H).
Пример 74. Получение соединения F-3a из соединения C1-Ea
В смесь соединения C1-Ea (50 мг, 0.074 ммоль, 1.0 экв.), полученного как описано выше, в метаноле (1 мл) добавляли смесь LiOH⋅H2O (18.45 мг, 0.44 ммоль, 6.0 экв.) в воде (1 мл), и реакционную смесь перемешивали при 60°C в течение 3 часов в атмосфере азота. Добавляли CHCl3 (20 мл) и pH смеси доводили до кислого добавлением 0.1 M водного раствора HCl (10 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Добавляли в остаток ДХМ/Гексан и отфильтровывали выпавший осадок при пониженном давлении, получая соединение F-3a (билирубин) в виде оранжевого твердого вещества (15 мг, 0.025 ммоль, выход: 34%).
3.11. Получение соединения C2-Ea путем сочетания соединения C2 и соединения Ea
Соединение C2-Ea, соответствующее соединению формулы 3, получали путем сочетания соединения C2 из Примера 4 с соединением Ea из Примера 8, и из него получали соединение F-3a.
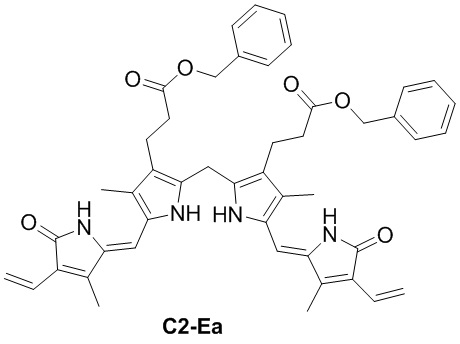
Пример 75. Получение соединения C2-Ea
В смесь соединения C2 (90 мг, 0.16 ммоль, 1.0 экв.) в 1,4-диоксане (1 мл) добавляли пиперидин (0.13 г, 1.6 ммоль, 10.0 экв.) и соединение Ea (59.1 мг, 0.48 ммоль, 3.0 экв.), полученное как описано выше, и реакционную смесь перемешивали при 101°C в течение 16 часов в атмосфере азота. Добавляли CHCl3 (30 мл) и промывали смесь 0.1 M водным раствором HCl (20 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток затвердевал при обработке смесью EtOAc/Гексан, при этом получали соединение C2-Ea в виде темно-оранжевого твердого вещества (45 мг, 0.058 ммоль, выход: 36%).
1H ЯМР (400 МГц, ДМСО-d6) δ 10.59 (ушир.с, 2H), 9.92 (ушир.с, 2H), 7.32-7.02 (м, 10H), 6.58 (дд, J = 17.6 Гц, J = 11.6 Гц, 2H), 6.22 (дд, J = 17.2, 2.4 Гц, 2H), 6.06 (с, 2H), 5.31 (дд, J = 12.0, 2.0 Гц, 2H), 4.92 (с, 4H), 4.00 (с, 2H), 2.50 - 2.40 (м, перекрывается с сигналом ДМСО-d6, 4H), 2.10 (с, 6H), 2.02-1.97 (м, 10H).
Пример 76. Получение соединения F-3a из соединения C2-Ea
В смесь соединения C2-Ea (60 мг, 0.078 ммоль, 1.0 экв.), полученного как описано выше, в ТГФ (2 мл) добавляли Pd/C (5.0 мг, 10 мол.%) в атмосфере азота. Полученную смесь несколько раз дегазировали в вакууме и заполняли водородом. Реакционную смесь перемешивали при 25°C в течение 6 часов в атмосфере H2 (15 фунт/кв.дюйм), затем подтверждали структуру полученного соединения F-3a методом LCMS.
C33H36N4O6 m/z [M+H]+ = 585.
4. ПЭГилирование соединения формулы 3
F-3a, соответствующее соединению формулы 3, ПЭГилировали в различных условиях.
Пример 77ю Получение FP-3a (моно-ПЭГилирование)
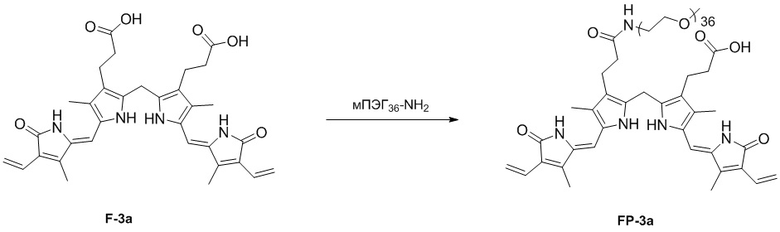
Смесь соединения F-3a (440 мг, 0.75 ммоль, 1.0 экв.) в ДМСО (22 мл) перемешивали при 25°C в течение 15 минут. Добавляли раствор CDI (183 мг, 1.13 ммоль, 1.5 экв.) в ДМСО (8.8 мл) по каплям. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (487 мг, 0.30 ммоль, 0.4 экв.) в ДМСО (4.4 мл), и реакционную смесь перемешивали при 25°C в течение 4 часов. Добавляли водный раствор Na2CO3 (220 мл) и перемешивали реакционную смесь до получения прозрачного желтого раствора. Полученный раствор экстрагировали хлороформом (200 мл × 3), объединенные органические слои сушили над безводным MgSO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии, получая соединение FP-3a (220 мг, 0.25 ммоль, выход: 33%), которое представляло собой продукт моно-ПЭГилирования соединения, соответствующего соединению формулы 3 по настоящему изобретению, в виде оранжевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 11.91 (с, 1H), 10.50-10.40 (м, 2H), 9.92 (с, 2H), 7.62 - 7.60 (м, 1H), 6.58 (дд, J = 14.0, 10.0 Гц, 2H), 6.21 (д, J = 14.0 Гц, 2H), 6.09 (с, 2H), 5.29 (д, J = 9.2 Гц, 2H), 3.98 (с, 2H), 3.65 - 3.41 (м, 142H), 3.24 (с, 3H), 3.13 - 3.12 (м, 2H), 2.47 - 2.40 (м, 4H), 2.16 (с, 6H), 2.04 (с, 6H), 1.96 - 1.93 (м, 4H).
Пример 78. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО перемешивали при 25°C в течение 15 минут. Добавляли по каплям CDI (1.5 экв.) в ДМСО. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в ДМСО и перемешивали реакционную смесь при 25°C в течение 16 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 30%.
Пример 79. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО/ДМФА перемешивали при 25°C в течение 15 минут. Затем добавляли по каплям раствор EDCI (1.5 экв.) в ДМСО/ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (0.4 экв.) в ДМСО/ДМФА и пиридин (3.0 экв.), и реакционную смесь перемешивали при 25°C в течение 4.5 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 62%.
Пример 80. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор HATU (1.2 экв.) в ДМСО. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в ДМСО и DIPEA (3.0 экв.), и реакционную смесь перемешивали при 20°C в течение 12 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 10%.
Пример 81. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор CMPI (0.9 экв.) в ДМСО. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в ДМСО и DIPEA (3.0 экв.), и реакционную смесь перемешивали при 25°C в течение 1 часа. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 45%.
Пример 82. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО/ДМФА перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор EDCI (1.5 экв.) в ДМСО/ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в ДМСО/ДМФА и пиридин (3.0 экв.), и реакционную смесь перемешивали при 20°C в течение 12 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 56%.
Пример 83. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМФА перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор CDI (1.6 экв.) в ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (0.4 экв.) в ДМФА, и реакционную смесь перемешивали при 25°C в течение 6.5 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 12%.
Пример 84. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в пиридине перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор EDCI (0.8 экв.) в пиридине. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в пиридине, и реакционную смесь перемешивали при 0°C в течение 2 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 9%.
Пример 85. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в пиридине перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор EDCI (1.1 экв.) в пиридине. Перемешивали при 25°C в течение 2 часов, добавляли раствор мПЭГ36-NH2 (0.4 экв.) в пиридине, и реакционную смесь перемешивали при 25°C в течение 6.5 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 18%.
Пример 86. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в пиридине перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор EDCI (2.0 экв.) и HOBt (2.2 экв.) в ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (0.4 экв.) в ДМФА и DIPEA (3.0 экв.), и реакционную смесь перемешивали при 25°C в течение 65 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 20%.
Пример 87. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМФА перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор DCC (1.1 экв.) и HOBt (1.1 экв.) в ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (0.4 экв.) в ДМФА, и реакционную смесь перемешивали при 25°C в течение 24 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 26%.
Пример 88. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМФА перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор DCC (1.0 экв.) и HOBt (1.0 экв.) в ДМФА. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36 -NH2 (0.4 экв.) в ДМФА и DIPEA (3.0 экв.), и реакционную смесь перемешивали в светозащитном чехле при 25°C в течение 16 часов. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 17%.
Пример 89. Получение FP-3a (моноПЭГилирование)
Смесь соединения F-3a (1.0 экв.) в ДМСО перемешивали при 25°C в течение 15 минут. Добавляли по каплям раствор BEP (0.9 экв.) в ДМСО. Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (0.4 экв.) в ДМСО и DIPEA (3.0 экв.), и реакционную смесь перемешивали при 25°C в течение 1 часа. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 43%.
Пример 90. Получение соединения FdP-3a (биПЭГилирование)
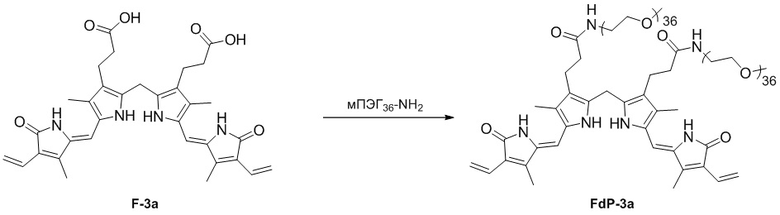
Смесь полученного как описано выше соединения F-3a (200 мг, 0.34 ммоль, 1.0 экв.), которое соответствует соединению формулы 3 по настоящему изобретению, HOBt (138.67 мг, 1.03 ммоль, 3.0 экв.), мПЭГ36-NH2 (1.38 г, 0.86 г, 2.5 экв.) и EDCI (196.73 мг, 1.03 ммоль, 3.0 экв.) в ДМСО (10 мл) перемешивали при 25°C в течение 30 минут. Добавляли раствор DIPEA (0.18 мл, 1.03 ммоль, 3.0 экв.) в ДМСО (10 мл), и реакционную смесь перемешивали при 25°C в течение 12 часов в атмосфере азота. Добавляли в смесь хлороформ (400 мл) и воду (300 мл), затем экстрагировали и промывали. Объединенные органические слои промывали насыщенным раствором хлорида натрия (100 мл × 2), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом преп-ВЭЖХ, получая соединение FdP-3a (358.31 мг, 0.94 ммоль, выход: 27%), которое представляло собой би-ПЭГилированное соединение, соответствующее соединению формулы 3 в настоящем тексте, в виде красного твердого вещества.
Пример 91. Получение соединения FdP-3a (биПЭГилирование)
Смесь соединения F-3a (1.0 экв.), которое соответствует описанному выше соединению формулы 3 по настоящему изобретению, HOBt (3.0 экв.), мПЭГ36-NH2 (2.5 экв.) и EDCI (3.0 экв.) в ДМСО перемешивали при 25°C в течение 30 минут. Смесь DIPEA (3.0 экв.) в ДМСО перемешивали при 25°C в течение 42 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия в реакции после стандартизации была определена равной 36%.
Пример 92. Получение соединения FdP-3a (биПЭГилирование)
Смесь соединения F-3a (1.0 экв.), которое соответствует описанному выше соединению формулы 3 по настоящему изобретению, мПЭГ36-NH2 (2.5 экв.) и CDI (2.5 экв.) в ДМСО перемешивали при 25°C в течение 30.5 часов в атмосфере азота. Оценивали конверсию в реакции методом жидкостной хроматографии/масс-спектрометрии (LCMS), и конверсия после стандартизации была определена равной 34%.
5. Сочетание ПЭГилированного соединения формулы 1 и соединения формулы 2
После ПЭГилирования соединения D из Примера 2, проводили сочетание с Ea из Примера 8, получая FP-3a, которое представляет собой ПЭГилированное соединение формулы 3.
Пример 93. ПЭГилирование соединения D
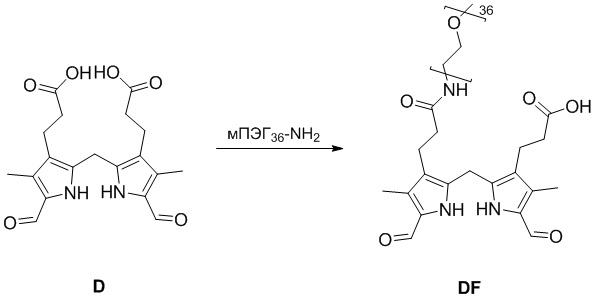
Соединение D (500 мг, 1.34 ммоль, 1.0 экв.) растворяли в ДМСО (20 мл) и перемешивали реакционную смесь при 25°C в течение 15 минут. Добавляли по каплям смесь CDI (325 мг, 2.00 ммоль, 1.5 экв.) в ДМСО (10 мл). Перемешивали при 25°C в течение 2 часов, добавляли смесь мПЭГ36-NH2 (2.16 г, 1.34 ммоль, 1.0 экв.) в ДМСО (10 мл) и перемешивали реакционную смесь при 25°C в течение 4 часов. Органический слой экстрагировали хлороформом (250 мл) и водой (50 мл × 2). Объединенные органические слои промывали насыщенным раствором хлорида натрия (50 мл × 2), сушили над безводным Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали на обращенно-фазной колонке, получая соединение DF (270 мг, 0.137 ммоль, выход: 10%), которое представляло собой моноПЭГилированное соединение, соответствующее соединению формулы 1 по настоящему изобретению, в виде ярко-желтого твердого вещества.
C92H169N3O41 m/z [M]+ = 1972
Пример 94. Получение соединения FP-3a из соединения DF
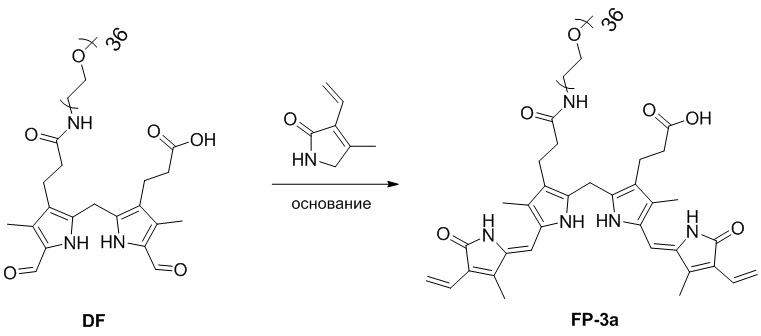
Пиперидин (116 мг, 1.37 ммоль, 10.0 экв.) добавляли в смесь соединения Ea (51 мг, 0.41 ммоль, 3.0 экв.) и соединения DF (270 мг, 0.14 ммоль, 1.0 экв.), полученного как описано выше, в 1,4-диоксане (10 мл) и перемешивали при 100°C в течение 16 часов в атмосфере азота. Полученную смесь упаривали при пониженном давлении. Остаток очищали методом препаративной ВЭЖХ, получая соединение FP-3a (16 мг, выход за 2 стадии: 0.5%), которое представляло собой моноПЭГилированное соединение, соответствующее соединению формулы 3 по настоящему изобретению, в виде коричневого твердого вещества.
C106H183N5O41 m/z [M]+ = 2182.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2792163C2 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С PI3 КИНАЗАМИ, СОДЕРЖАЩАЯ ДАННОЕ ДЕЙСТВУЮЩЕЕ ВЕЩЕСТВО | 2016 |
|
RU2719367C2 |
| ИНГИБИТОРЫ МАТРИКСНОЙ МЕТАЛЛОПРОТЕИНАЗЫ (MMP) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2797558C2 |
| ТЕТРАЗОЛ-СОДЕРЖАЩИЕ ИНГИБИТОРЫ РЕГУЛИРУЮЩЕЙ АПОПТОТИЧЕСКИЕ СИГНАЛЫ КИНАЗЫ 1 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2807545C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| ПРОИЗВОДНЫЕ НОР-СЕКО ХИМБАЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293735C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛОТРИАЗИНА И/ИЛИ ПИРАЗОЛОПИРИМИДИНА | 2019 |
|
RU2818563C2 |
| ИНГИБИТОРЫ МАТРИКСНОЙ МЕТАЛЛОПРОТЕИНАЗЫ (MMP) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2820540C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
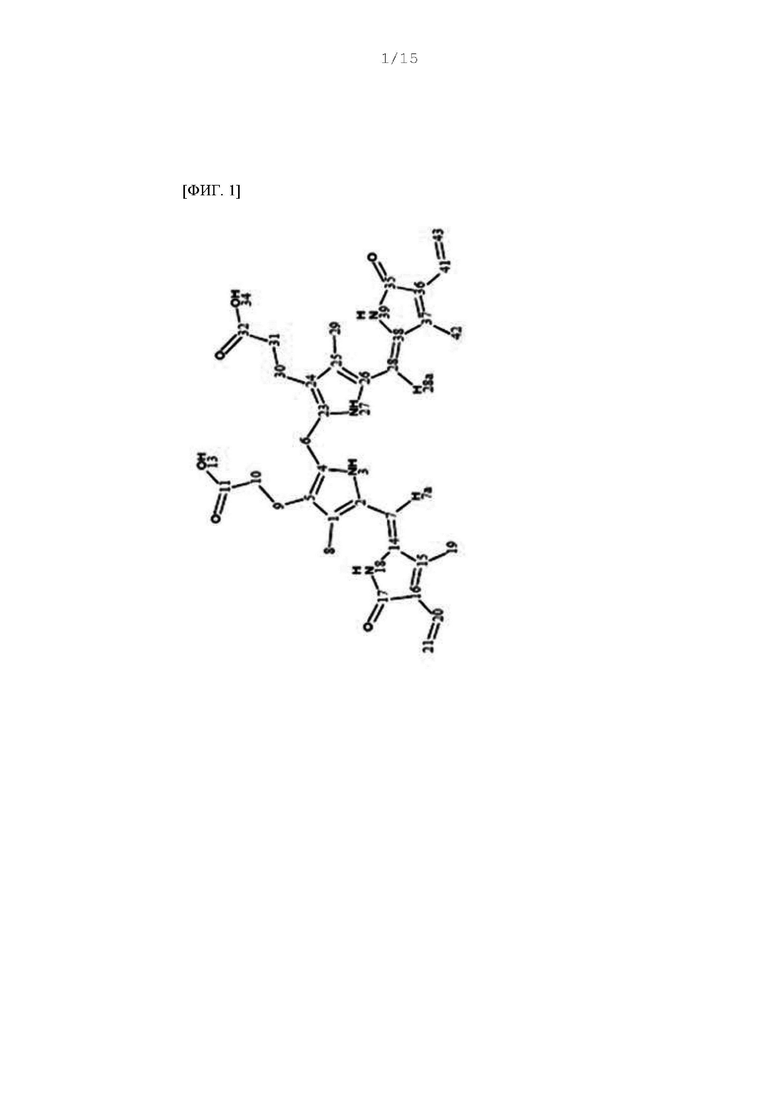
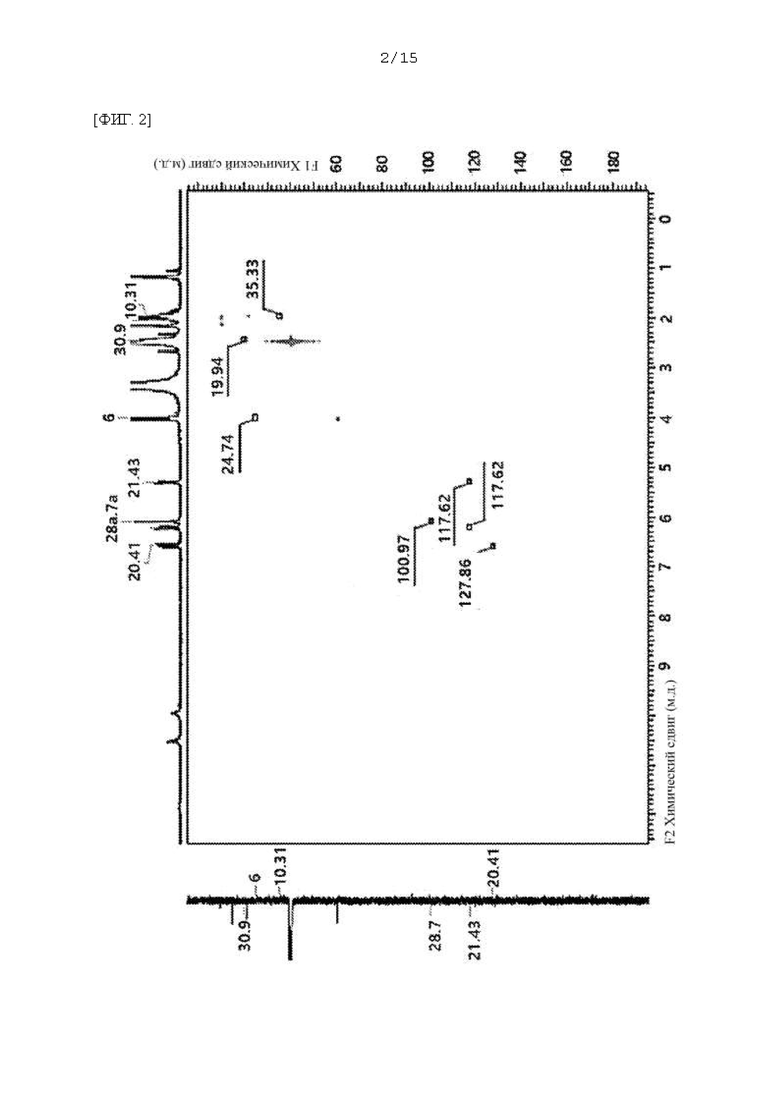
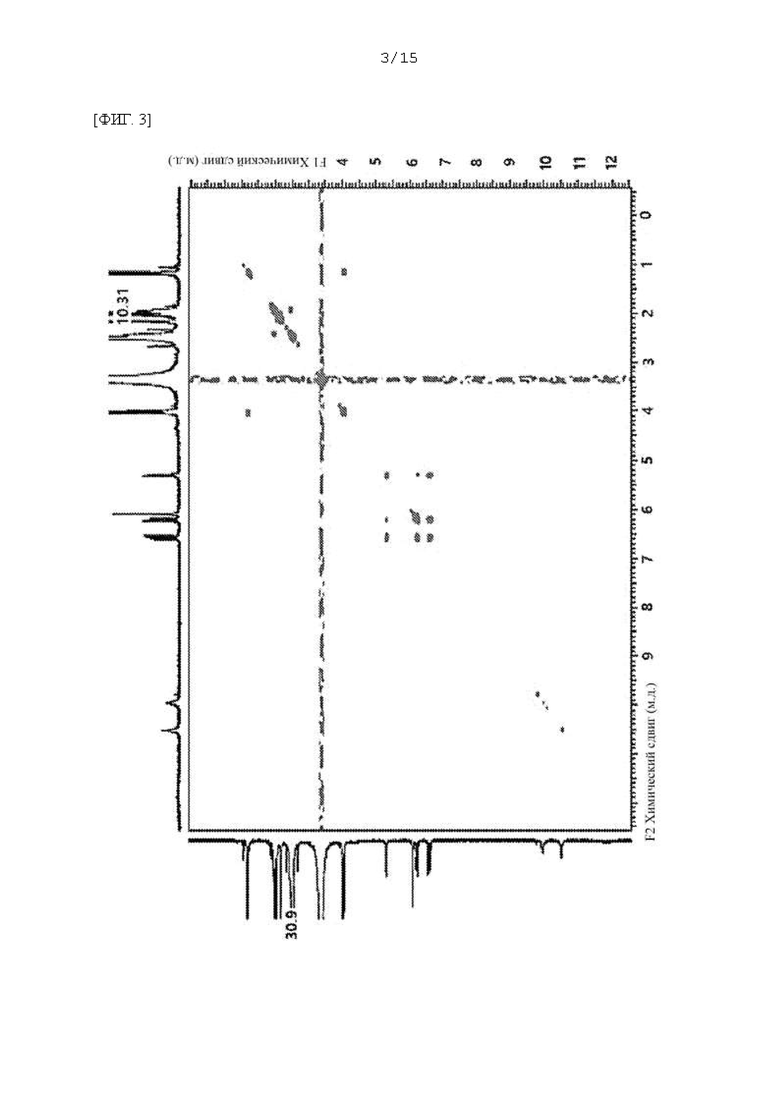
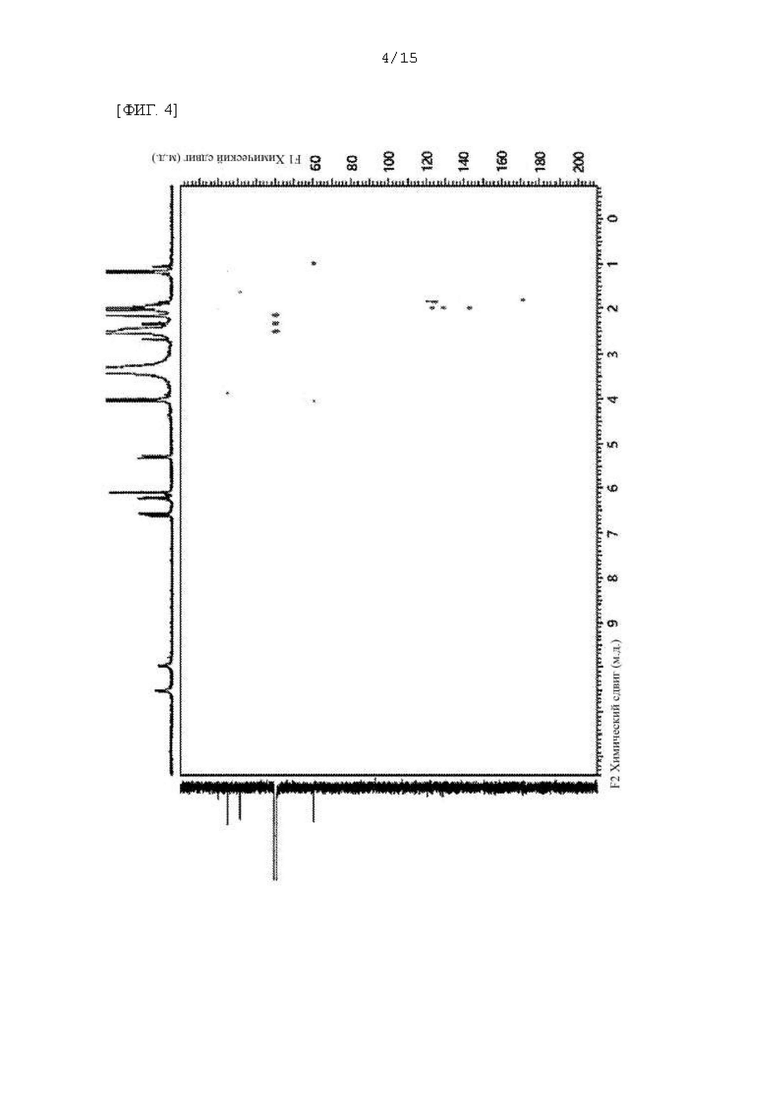
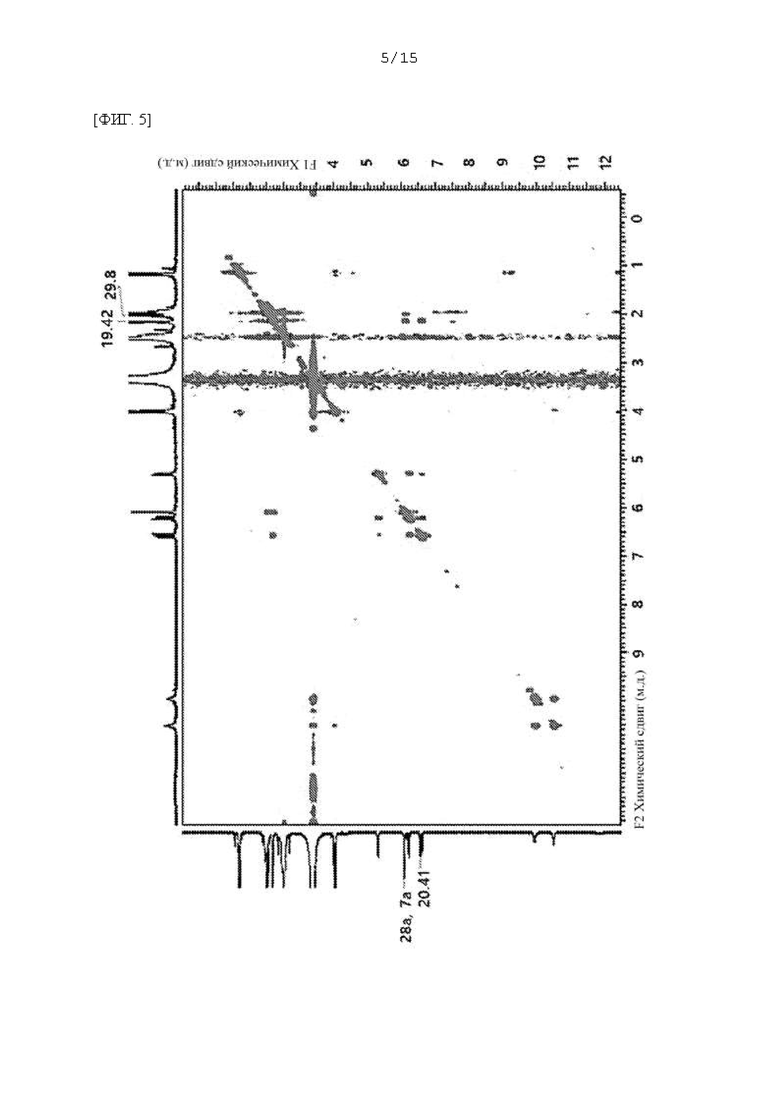
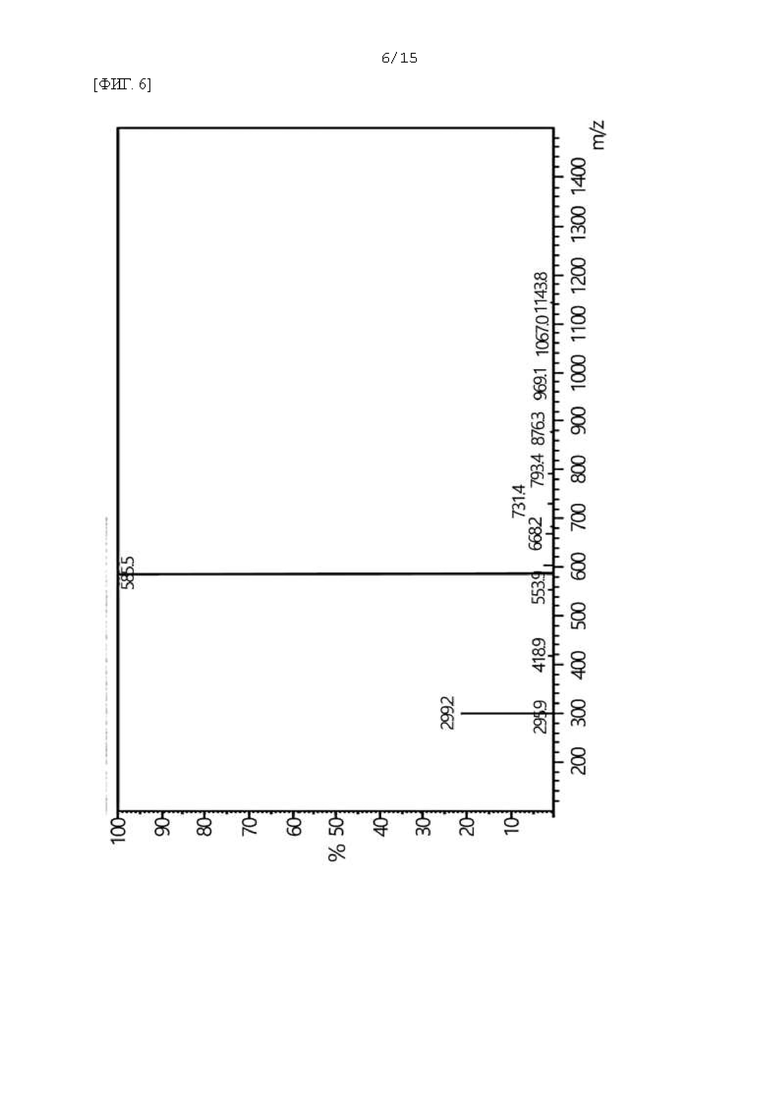
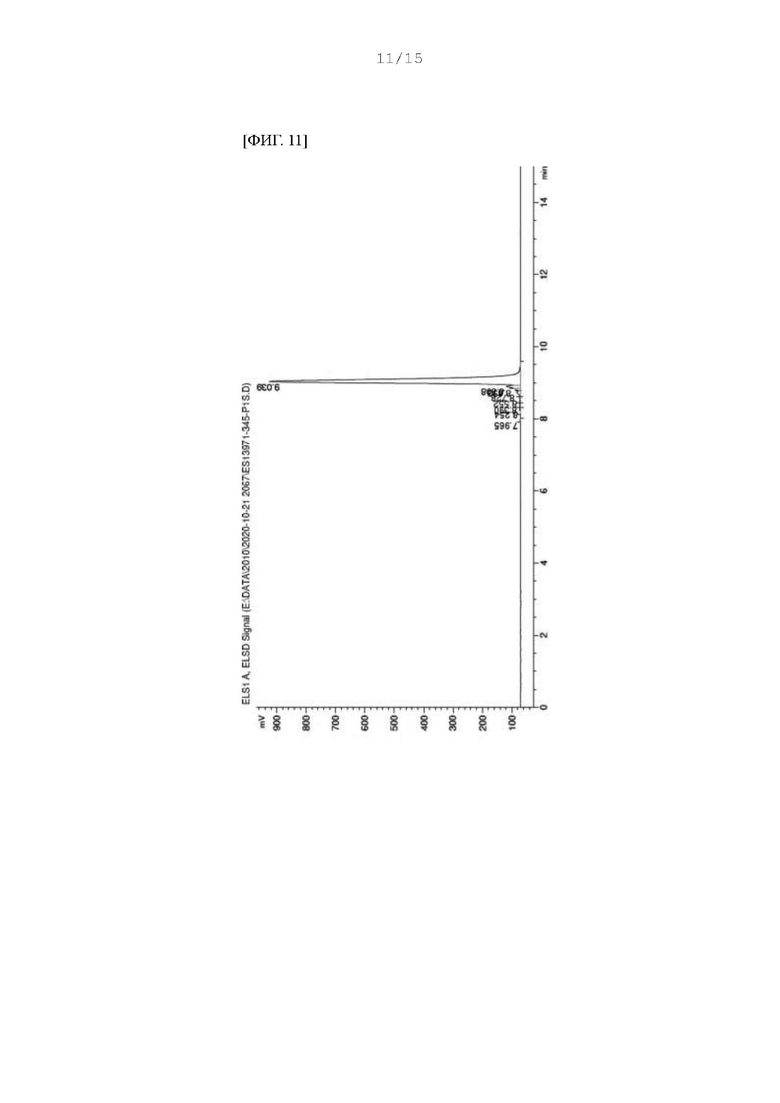
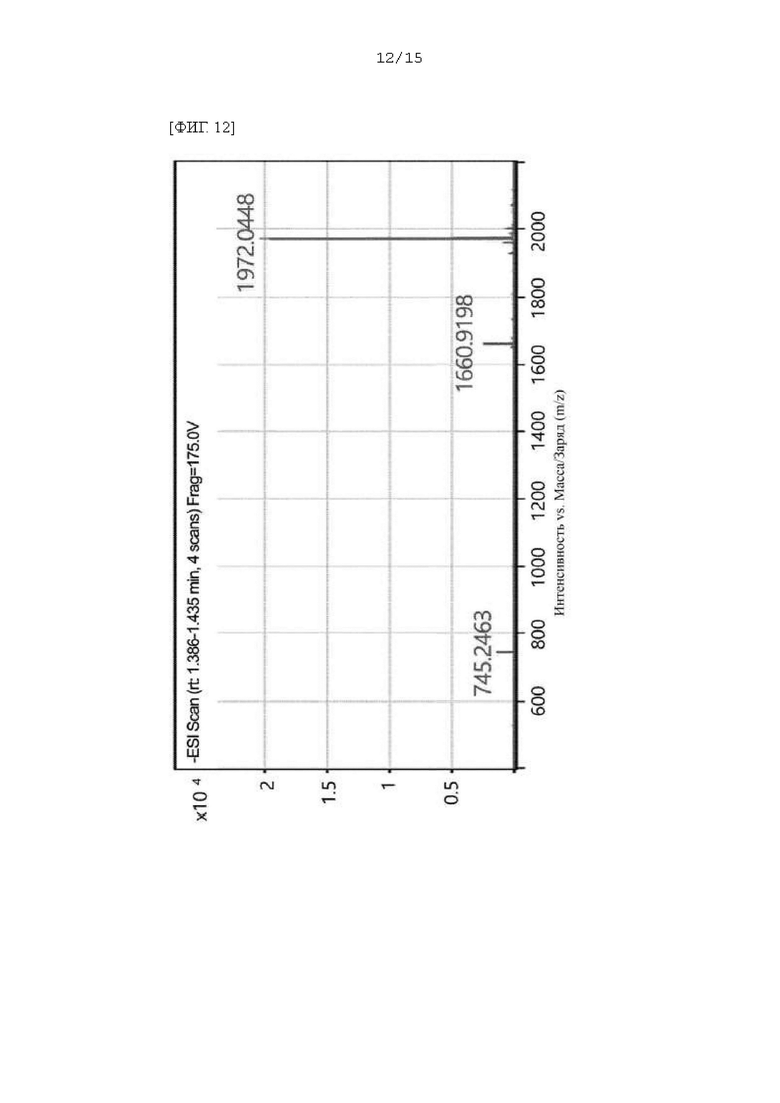
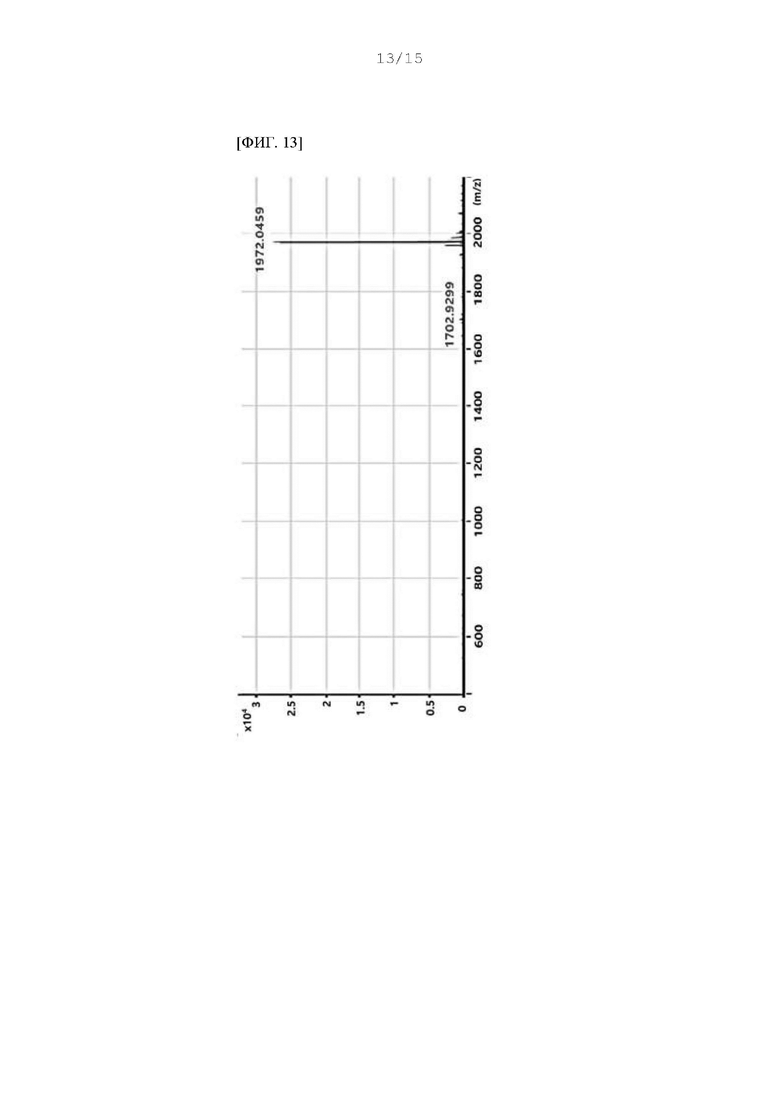
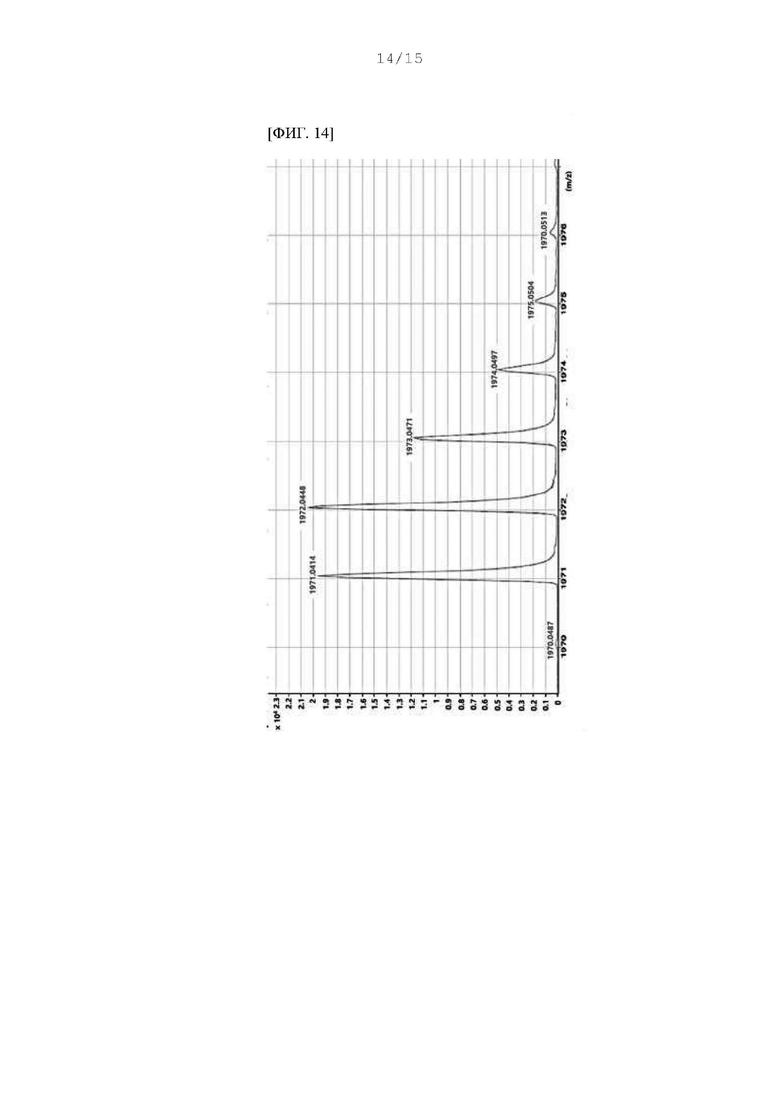
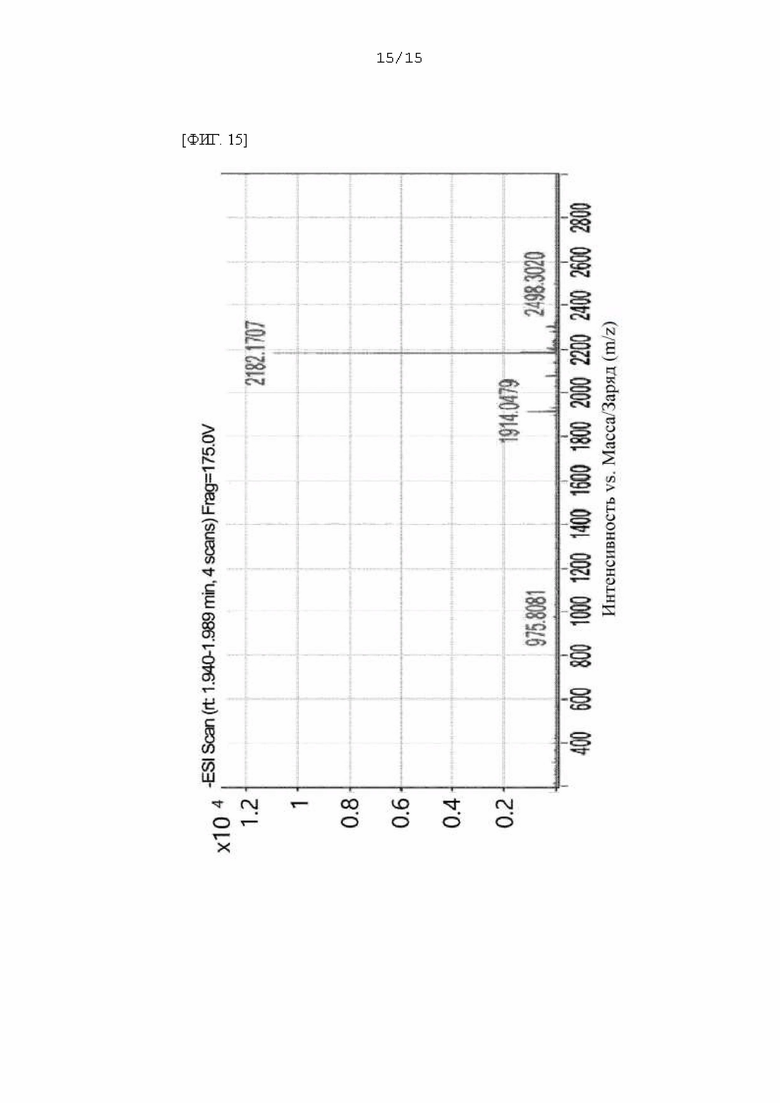
Предложена группа изобретений, касающаяся способов получения синтетического билирубина. Способ синтеза билирубина включает димеризацию соединения формулы 6 и замены Х в продукте на -C(=O)H с получением соединения формулы 1, получение соединения формулы 3 путем сочетания соединения формулы 1 с соединением формулы 2 в присутствии основания и растворителя и получение билирубина из соединения формулы 3. Заместители R1-R5 и X описаны в формуле изобретения. Технический результат – обеспечение способом синтеза целевого соединения. 6 н. и 5 з.п. ф-лы, 15 ил., 1 табл., 94 пр.
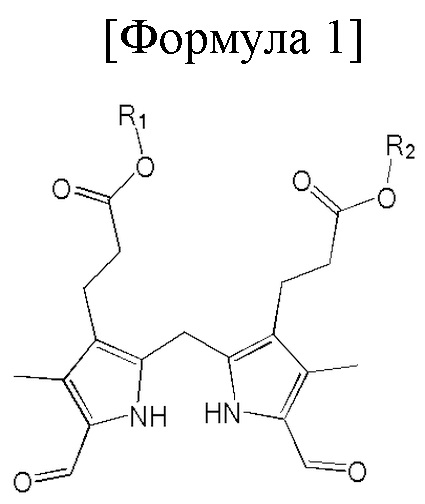
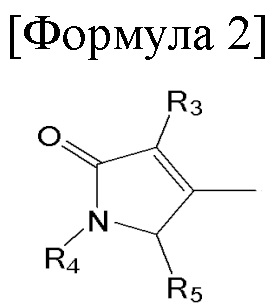
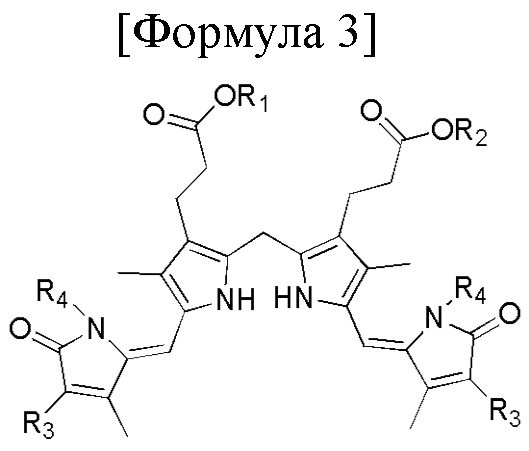
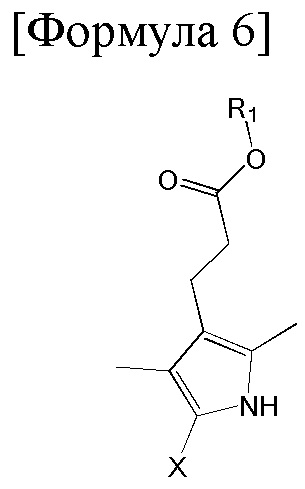
1. Способ синтеза билирубина, включающий димеризацию изображенного ниже соединения формулы 6 с получением соединения формулы 1, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
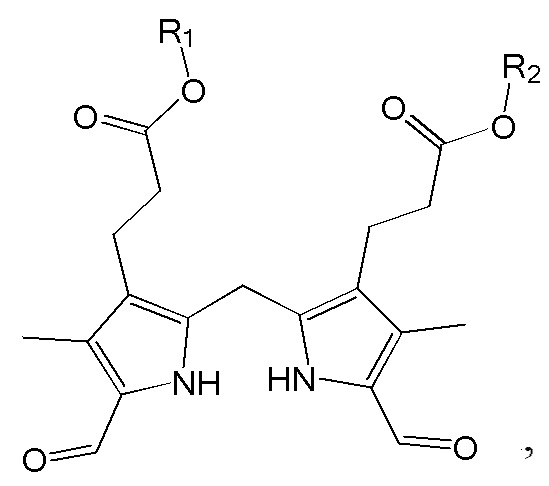
[Формула 2]
[Формула 3]
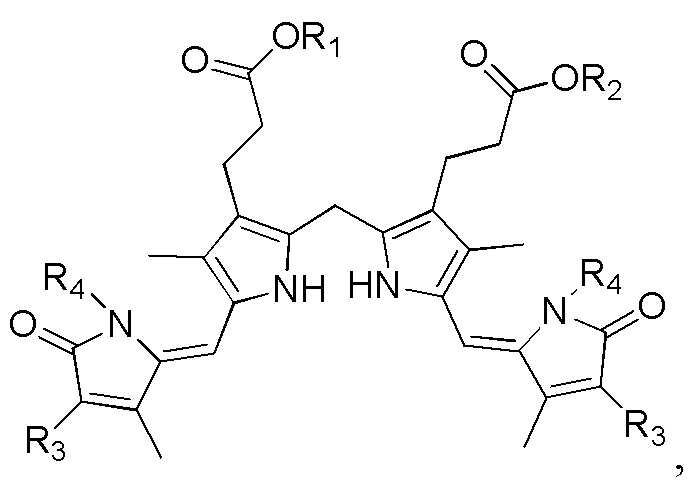
[Формула 6]
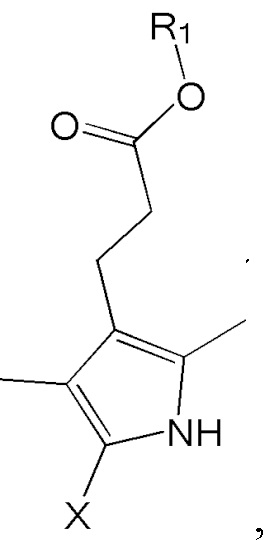
где в формулах 1, 2, 3 и 6 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу и
X представляет собой арилалкил сложноэфирную группу, содержащую 8-20 атомов углерода, -CH2OH, -COOH, атом галогена или атом водорода.
2. Способ по п. 1, дополнительно включающий реакцию соединения формулы 3 с полиэтиленгликолем (ПЭГ).
3. Способ по п. 1, где соединение формулы 1 реагирует с полиэтиленгликолем (ПЭГ) и затем вступает в реакцию сочетания с соединением формулы 2.
4. Способ синтеза билирубина, включающий проведение циклизации изображенного ниже соединения формулы 8 с получением соединения формулы 2, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
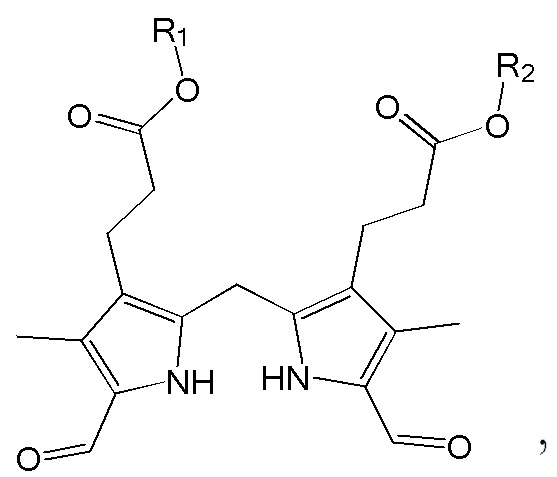
[Формула 2]
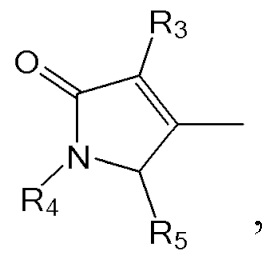
[Формула 3]
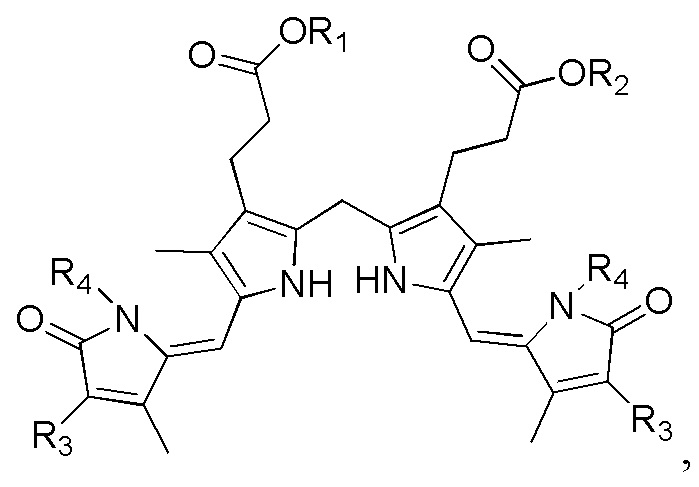
[Формула 8]
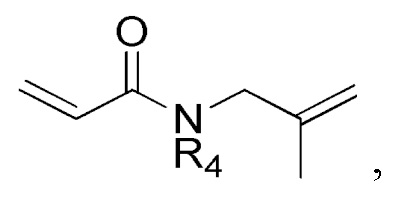
где в формулах 1, 2, 3 и 8 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу.
5. Способ синтеза билирубина, включающий восстановление ацетильной группы в изображенном ниже соединении формулы 11 с получением соединения формулы 2, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
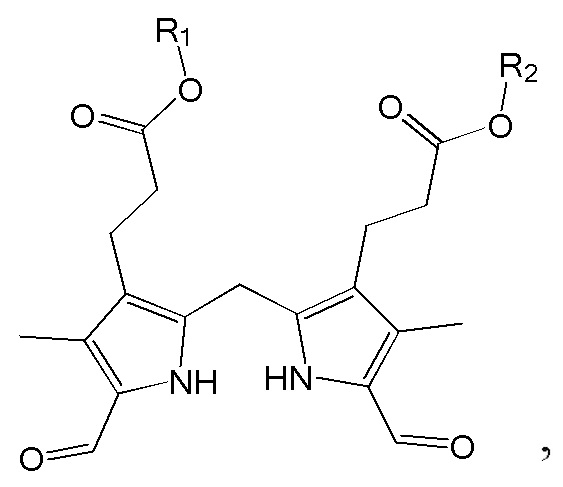
[Формула 2]
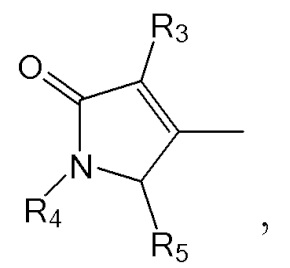
[Формула 3]
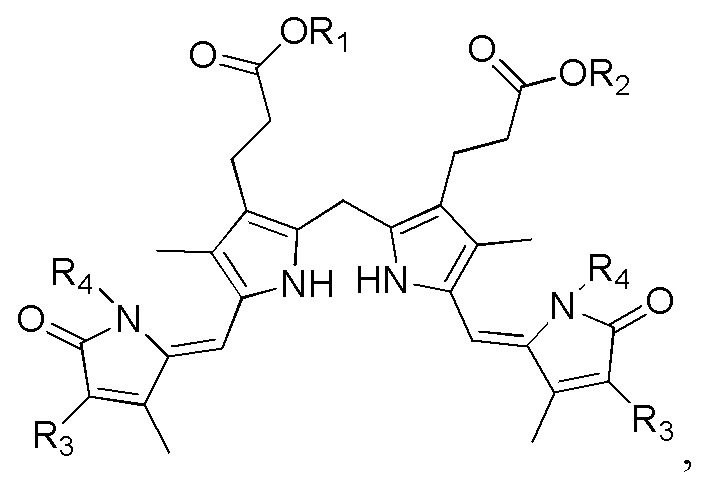
[Формула 11]
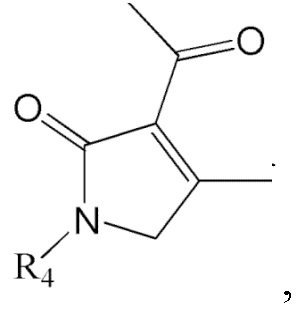
где в формулах 1, 2, 3 и 11 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу.
6. Способ синтеза билирубина, включающий дегидратацию гидроксильной группы в изображенном ниже соединении формулы 12 с получением соединения формулы 2, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
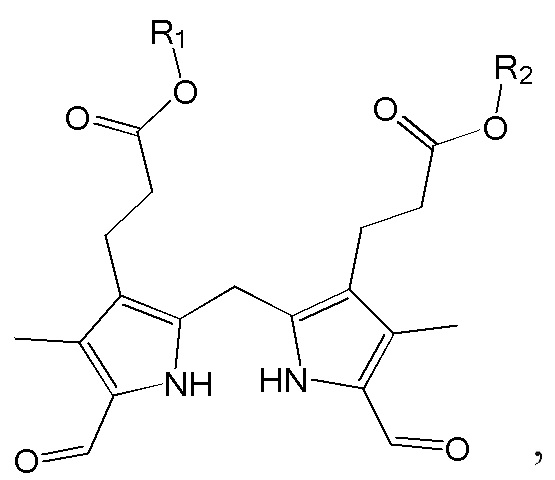
[Формула 2]
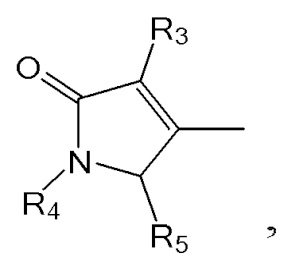
[Формула 3]
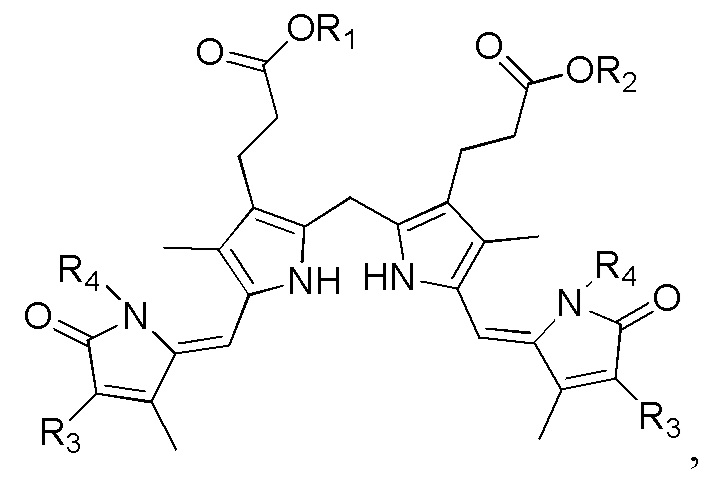
[Формула 12]
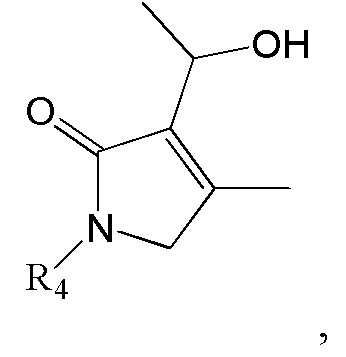
где в формулах 1, 2, 3 и 12 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу.
7. Способ синтеза билирубина, включающий проведение циклизации изображенного ниже соединения формулы 13 с получением соединения формулы 2, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
[Формула 2]
[Формула 3]
[Формула 13]
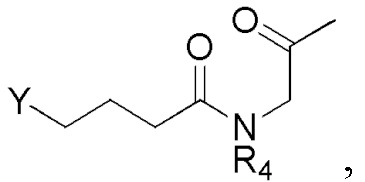
где в формулах 1, 2, 3 и 13 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу,
Y представляет собой селенид.
8. Способ синтеза билирубина, включающий окисление изображенного ниже соединения формулы 14 с получением соединения формулы 2, получение изображенного ниже соединения формулы 3 путем сочетания изображенного ниже соединения формулы 1 с изображенным ниже соединением формулы 2, где стадию получения соединения формулы 3 проводят в присутствии основания, выбранного из группы, состоящей из следующих: пиперидин, N-метилпиперидин, N-этилпиперидин, 2,6-диметилпиперидин, 2,2,6,6-тетраметилпиперидин, 3-метилпиперидин, 3-этилпиперидин, 1-метил-4-(метиламино)пиперидин, 4-аминопиперидин, пирролидин, 2-пирролидин карбоксамид, пирролидин-3-ол, пиперазин, 2,6-диметилпиперазин, 1-бензилпиперазин, 1-изопропилпиперазин, 2-этилпиперазин, морфолин, 4-метилморфолин, 2,6-диметилморфолин, этилморфолин, азепан, 2-метилазепан, 4-метилазепан, 2,2,7,7-тетраметилазепан, 1,2,2-триметилазепан, 1,2-диметилазепан, 2,7-диметилазепан, метил азепан-4-карбоксилат, азокан, 2-метилазокан, 1,2-диметилазокан, 1,2,2-триметилазокан, метил азокан-2-карбоксилат, 1-метилазокан и 2(2-метилфенил)азокан, и где стадию получения соединения формулы 3 проводят в присутствии растворителя, выбранного из группы, состоящей из следующих: вода, спирты, простые эфиры, кетоны, алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, алкоксилированные соединения, нитрилы и амиды, и получение билирубина из изображенного ниже соединения формулы 3:
[Формула 1]
[Формула 2]
[Формула 3]
[Формула 14]
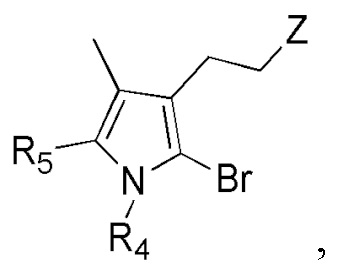
где в формулах 1, 2, 3 и 14 R1 и R2, каждый независимо, представляют собой атом водорода, алкильную группу, содержащую 1-12 атомов углерода, арильную группу, содержащую 6-20 атомов углерода, гетероарильную группу, содержащую 2-20 атомов углерода, арилалкильную группу, содержащую 7-20 атомов углерода, или гетероарилалкильную группу, содержащую 3-20 атомов углерода; R3 представляет собой атом водорода, винильную группу, ацетильную группу или этильную группу, замещенную гидроксильной группой, селенидом или сульфидом; R4 представляет собой атом водорода или азот-защитную группу; и R5 представляет собой атом водорода, тозильную или мезильную группу,
Z представляет собой сульфид.
9. Способ по любому из пп. 1 и 4-8, где стадию получения соединения формулы 3 проводят при температуре от -20 до 200°C.
10. Способ по любому из пп. 1 и 4-8, где стадию получения соединения формулы 3 проводят в течение 0,5-120 ч.
11. Способ по любому из пп. 1 и 4-8, где основание добавляют в количестве 2-20 моль относительно 1 моля соединения формулы 1.
| Brower J | |||
| O | |||
| et al.: Synthesis of a New Lipophilic Bilirubin | |||
| Conformation, Transhepatic Transport and Glucuronidation | |||
| Tetrahedron, 56, 2000, p | |||
| Способ получения хлористого водорода и окиси магния из шестиводного хлористого магния | 1925 |
|
SU7869A1 |
| Brower J | |||
| O | |||
| et al.: Aromatic Congeners of Bilirubin: Synthesis, Stereochemistry, Glucuronidation and Hepatic Transport | |||
| Tetrahedron, 57, 2001, 7p | |||
| Плитки для тротуаров, мостовых и облицовок и на приспособление для их изготовления | 1923 |
|
SU813A1 |
| Dey S | |||
| K | |||
| et al.: Lipid-and | |||

