ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящая заявка содержит перечень последовательностей, который был подан в формате ASCII посредством EFS-Web и включен в данный документ посредством ссылки во всей своей полноте. Указанная копия ASCII, созданная 31 октября 2018 г. названа AVEX-003001WO_ST25.txt и имеет размер 14639 байтов.
РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет по предварительной заявке на патент США № 62/583035, поданной 8 ноября 2017 г., содержание которой включено в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам получения и очистки вирусных частиц и композициям и вариантам применения, предусматривающим таковые.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Аденоассоциированный вирус (AAV) относится к семейству Parvoviridae. Геном AAV состоит из линейной однонитевой молекулы ДНК, которая содержит приблизительно 4,7 тысячи пар нуклеотидов (т. п. н.) и состоит из двух основных открытых рамок считывания, кодирующих неструктурный белок Rep (репликация) и структурный белок Cap (капсид). Кодирующие участки AAV фланкированы двумя последовательностями инвертированного концевого повтора (ITR), действующими при размещении в цис-положении, длиной приблизительно 145 нуклеотидов, с разделенными спейсером палиндромными последовательностями, которые могут складываться в шпилечные структуры, которые функционируют в качестве праймеров во время инициации репликации ДНК. В дополнение к их роли в репликации ДНК, последовательности ITR, как было показано, необходимы для вирусной интеграции, освобождения из генома хозяина и инкапсулирования вирусной нуклеиновой кислоты в зрелые вирионы (Muzyczka, (1992) Curr. Top. Micro. Immunol. 158:97-129).
Существует множество серотипов AAV, которые обладают различным тканевым тропизмом. Известные серотипы включают, например, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, AAV10 и AAV11. AAV9 описан в пат. США № 7198951 и в Gao et al., J. Virol., 78: 6381-6388 (2004), которые включены в данный документ посредством ссылки во всей своей полноте. Достижения в области доставки AAV6 и AAV8 сделали возможным трансдукцию этими серотипами скелетной и сердечной мышцы вследствие простых системных внутривенных или внутрибрюшинных инъекций. См. Pacak et al., Circ. Res., 99(4): 3-9 (2006) и Wang et al., Nature Biotech. 23(3): 321-8 (2005). Однако применение AAV для нацеливания на типы клеток в центральной нервной системе потребовало хирургического интрапаренхиматозного введения. См. Kaplitt et al., "Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial." Lancet, 369:2097-2105; Marks et al., "Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomized, controlled trial." Lancet Neurol 9:1164-1172; и Worgall et al., "Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA." Hum Gene Ther, 19(5):463-74.
Нуклеотидная последовательность генома AAV серотипа 2 (AAV2) представлена в Srivastava et al., J. Virol, 45: 555-564 (1983), как исправлено Ruffing et al., J Gen Virol, 75: 3385-3392 (1994). Последовательности, действующие в цис-положении, управляющие репликацией (rep) вирусной ДНК, инкапсулированием/упаковкой и интеграцией в хромосому клетки-хозяина, содержатся в ITR. Три промотора AAV (названные p5, p19 и p40 по их относительным местоположениям на карте) инициируют экспрессию двух внутренних открытых рамок считывания AAV, кодирующих гены rep и cap. Два промотора rep (p5 и p19) в сочетании с дифференциальным сплайсингом единственного интрона AAV (по нуклеотидам 2107 и 2227) приводят к образованию четырех белков rep (rep 78, rep 68, rep 52 и rep 40) из гена rep. Белки Rep обладают несколькими ферментативными свойствами, которые по сути отвечают за репликацию вирусного генома. Ген cap экспрессируется с промотора p40, и он кодирует три капсидных белка: VP1, VP2 и VP3. Альтернативный сплайсинг и неконсенсусные стартовые сайты трансляции отвечают за продукцию трех родственных капсидных белков. Единственный консенсусный сайт полиаденилирования расположен в положении 95 на карте генома AAV. Жизненный цикл и генетика AAV рассматриваются в Muzyczka, Microbiology and Immunology, 158: 97-129 (1992).
Векторы, полученные из AAV, особенно привлекательны для доставки генетического материала, потому что (i) они способны инфицировать (трансдуцировать) широкий спектр типов неделящихся и делящихся клеток, включая мышечные волокна и нейроны; (ii) они лишены структурных генов вируса, предотвращая таким образом естественные ответы клетки-хозяина на вирусную инфекцию, например опосредованные интерфероном ответы; (iii) вирусы дикого типа никогда не были связаны с какой-либо патологией у человека; (iv) в отличие от представителей AAV дикого типа, которые способны интегрироваться в геном клетки-хозяина, дефектные по репликации векторы AAV обычно сохраняются в виде эписом, таким образом ограничивая риск инсерционного мутагенеза или активации онкогенов; и (v) в отличие от других векторных систем, векторы AAV не вызывают значительной иммунной реакции (см. ii), таким образом обеспечивая долгосрочную экспрессию терапевтических трансгенов (при условии, что их генные продукты не отторгаются).
Самокомплементарные аденоассоциированные векторы (scAAV) представляют собой вирусные векторы, сконструированные из встречающегося в природе аденоассоциированного вируса (AAV) для применения в генной терапии. scAAV называется "самокомплементарным", потому что кодирующий участок сконструирован таким образом, что он образует матрицу, представляющую собой внутримолекулярную двухнитевую ДНК. Лимитирующий скорость этап в случае жизненного цикла генома стандартного AAV включает синтез второй нити, поскольку геном типичного AAV представляет собой матрицу, являющуюся однонитевой ДНК. Однако это не относится к геномам scAAV. После инфицирования, не ожидая опосредованного клеткой синтеза второй нити, две комплементарные половины scAAV ассоциируют с образованием одной единицы двухнитевой ДНК (dsDNA), которая готова к немедленной репликации и транскрипции.
Сохраняется необходимость в разработке масштабируемого способа получения и очистки фармацевтического продукта на основе AAV, например, с низким содержанием пустых капсидов, низким содержанием белка клетки-хозяина и/или с низким содержанием загрязняющей ДНК, при сохранении высокой активности.
КРАТКОЕ ОПИСАНИЕ
Настоящее изобретение предусматривает способ получения очищенных препаратов вирусных частиц, включая препараты частиц AAV.
В некоторых вариантах осуществления настоящее изобретение предусматривает фармацевтическую композицию, содержащую (а) 1-8×1013 геномов вирусного вектора на основе AAV9/мл (г. в./мл), (b) менее чем приблизительно 7% пустых вирусных капсидов, (с) менее чем приблизительно 100 нг/мл белка клетки-хозяина на 1×1013 г. в./мл, и (d) менее чем приблизительно 5×106 пг/мл остаточной ДНК клетки-хозяина на 1×1013 г. в./мл, и при этом по меньшей мере приблизительно 80% от 1-8×1013 геномов вирусного вектора на основе AAV9/мл являются функциональными.
В одном варианте осуществления вирусный вектор на основе AAV9 содержит полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN). В одном варианте осуществления вирусный вектор на основе AAV9 содержит полинуклеотид, кодирующий белок, представляющий собой метил-СрG-связывающий белок 2 (MECP2). В одном варианте осуществления вирусный вектор на основе AAV9 содержит полинуклеотид, кодирующий короткую шпилечную РНК (shRNA), нацеливающуюся на супероксиддисмутазу 1 (SOD1). В одном варианте осуществления вирусный вектор на основе AAV9 содержит модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2.
Настоящее изобретение предусматривает фармацевтический состав. В некоторых вариантах осуществления водный фармацевтический состав содержит (а) вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), (b) Tris-буфер, (с) хлорид магния, (d) хлорид натрия и (е) полоксамер (например, полоксамер 188), где фармацевтическая композиция не содержит консерванта. В одном варианте осуществления состава вирусный вектор на основе AAV9 дополнительно содержит модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2. В одном варианте осуществления состава концентрация Tris-буфера составляет приблизительно 10-30 нМ, например приблизительно 20 мМ. В одном варианте осуществления pH состава составляет от приблизительно 7,7 до приблизительно 8,3, например, pH составляет приблизительно 8,0 (например, при измерении согласно <791> USP (включено посредством ссылки во всей своей полноте)). В одном варианте осуществления состава концентрация хлорида магния составляет приблизительно 0,5-1,5 мМ, например приблизительно 1 мМ. В одном варианте осуществления состава концентрация хлорида натрия составляет приблизительно 100-300 мМ, например, приблизительно 200 мМ. В одном варианте осуществления состав содержит приблизительно 0,005% вес./об. полоксамера 188.
Другой аспект настоящего изобретения направлен на способ лечения спинальной мышечной атрофии (SMA) типа I у пациента, нуждающегося в этом, включающий введение вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок SMN (например, композиции или состава, описанных в данном документе) пациенту интратекальным или внутривенным путем, где (а) возраст пациента составляет девять месяцев или менее, (б) вес тела пациента составляет по меньшей мере приблизительно 2,6 кг, (в) пациент имеет биаллельные нулевые мутации или делеции в SMN1 и (d) пациент имеет по меньшей мере одну функциональную копию SMN2. В одном варианте осуществления вирусный вектор на основе AAV9 содержит модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2. В одном варианте осуществления вес тела пациента составляет не более приблизительно 8,5 кг. В одном варианте осуществления у пациента нет замены c.859G>C в экзоне 7 по меньшей мере одной копии гена SMN2. В одном варианте осуществления средство для лечения вводят пациенту в возрасте до 6 месяцев. В одном варианте осуществления средство для лечения вводят пациенту до появления одного или нескольких симптомов SMA, выбранных из гипотонии, задержки развития двигательных навыков, слабого удержания головы, сутулой осанки и гипермобильности суставов. В одном варианте осуществления до введения у пациента имеются титры антител к AAV9 1:100 или ниже, при определении с помощью иммуноферментного анализа связывания ELISA.
В данном документе также раскрыт способ лечения пациента детского возраста со спинальной мышечной атрофией (SMA) типа I при наличии или отсутствии манифестации заболевания, включающий введение пациенту композиции или состава, содержащих вектор на основе аденоассоциированного вируса (AAV), описанный в данном документе.
Настоящее изобретение также направлено на способ лечения синдрома Ретта у пациента, нуждающегося в этом, включающий введение вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок, представляющий собой метил-CpG-связывающий белок 2 (MECP2), пациенту интратекальным или внутривенным путем.
Настоящее изобретение также направлено на способ лечения бокового амиотрофического склероза (ALS) у пациента, нуждающегося в этом, включающий введение вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий короткую шпилечную РНК (shRNA), нацеливающуюся на супероксиддисмутазу 1 (SOD1), пациенту интратекальным или внутривенным путем.
Другой аспект настоящего изобретения направлен на способ лечения пациента, страдающего от SMA типа I, включающий стадии (a) определения веса пациента, (b) получения набора, содержащего флаконы с фармацевтической композицией на основе вирусного вектора на основе AAV9, и (c) введение вирусного вектора на основе AAV9 из флаконов пациенту, при этом концентрация вирусного вектора в каждом флаконе составляет приблизительно 2,0×1013 г. в./мл, при этом вирусный вектор на основе AAV9 содержит полинуклеотид, кодирующий белок SMN; и при этом набор содержит следующее количество флаконов.
В одном варианте осуществления набор содержит вирусный вектор на основе AAV9, содержащий подвергнутый мутации ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и ITR AAV2. В одном варианте осуществления вирусный вектор на основе AAV вводят посредством инфузии в дозе, составляющей приблизительно 1,0×1014-2,5×1014 г. в./кг.
Другой аспект настоящего изобретения направлен на набор для лечения пациента, страдающего от спинальной мышечной атрофии (SMA) типа I, содержащий флаконы, содержащие композицию на основе вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), или состав, содержащий (a) вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), (b) Tris-буфер, (с) хлорид магния, (d) хлорид натрия и (е) полоксамер (например, полоксамер 188).
Другой аспект настоящего изобретения направлен на набор, содержащий флаконы, содержащие приблизительно 5,5 мл или приблизительно 8,3 мл вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), и составленного с концентрацией приблизительно 2,0×1013 г. в./мл в 20 мМ Tris, 1 мМ MgCl2, 200 мМ NaCl, 0,005% вес./об. полоксамера 188 при pH 7,7-8,3, например приблизительно 8,0.
Другой аспект настоящего изобретения направлен на способ лечения SMA типа I, включающий введение определенного объема композиции, причем композиция на основе вирусного вектора на основе AAV9 содержит полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), или состава, содержащего (a) вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), (b) Tris-буфер, (с) хлорид магния, (d) хлорид натрия и (е) полоксамер (например, полоксамер 188), посредством внутривенной инфузии пациенту, нуждающемуся в этом.
Другой аспект настоящего изобретения направлен на способы получения вирусного вектора на основе AAV. В одном варианте осуществления способ получения вирусного вектора на основе AAV включает стадии (a) культивирования адгезивных клеток, (b) трансфекции адгезивных клеток плазмидой(-ами) с обеспечением продуцирования вирусного вектора на основе AAV, (c) лизиса адгезивных клеток с выделением вирусного вектора на основе AAV, (d) подкисления и осветления клеточного лизата из (c), (e) очистки продукта из (d) с применением катионообменной хроматографии (CEX), (f) фильтрования продукта из (e) с применением фильтрации в тангенциальном потоке (TFF), (g) ультрацентрифугирования продукта из (f) в буфере с хлоридом цезия (CsCl); и (h) сбора вирусных векторов на основе AAV из продукта из (g).
Другой аспект настоящего изобретения направлен на способ очистки вирусного вектора на основе AAV от лизата культуры клеток, включающий стадии (a) подкисления и осветления клеточного лизата, (b) очистки продукта из (a) с применением катионообменной хроматографии (CEX), (c) фильтрования продукта из (b) посредством фильтрации в тангенциальном потоке, (d) ультрацентрифугирования продукта из (c) с применением 2-4 М буфера с хлоридом цезия (CsCl), (e) сбора вирусных векторов на основе AAV из продукта из (d), (f) фильтрования продукта из (e) посредством фильтрации в тангенциальном потоке. В одном варианте осуществления способ осуществляют в промышленном масштабе. В одном варианте осуществления способ дает выход, составляющий более 5×1015 г. в., или более 8×1015 г. в., или более 1×1016 г. в. на одну производственную партию.
Другой аспект настоящего изобретения направлен на способ лечения пациента, у которого имеется SMA типа 1, путем введения вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок SMN, полученного в соответствии с любым из способов, раскрытых в данном документе.
Другой аспект настоящего изобретения направлен на способ лечения пациента, у которого имеется синдром Ретта, путем введения вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий белок MECP2, полученного в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на способ лечения пациента, у которого имеется ALS, путем введения вирусного вектора на основе AAV9, содержащего полинуклеотид, кодирующий shRNA, нацеливающуюся на SOD1, полученного в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок SMN, полученный в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на фармацевтическую композицию, содержащую вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок SMN, полученную в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на водную фармацевтическую композицию, содержащую вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок SMN, Tris-буфер, раствор хлорида магния и раствор хлорида натрия, где фармацевтическая композиция не содержит консерванта, и где композиция получена в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок MECP2, полученный в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на фармацевтическую композицию, содержащую вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий белок MECP2, полученную в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на вирусный вектор на основе AAV9, содержащий полинуклеотид, кодирующий shRNA, нацеливающуюся на SOD1, полученный в соответствии с любым из способов, описанных в данном документе.
Другой аспект настоящего изобретения направлен на способ лечения SMA типа I у пациента, нуждающегося в этом, путем внутривенного введения фармацевтической композиции, содержащей (a) самокомплементарный вирусный вектор на основе AAV9, содержащий модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2, (b) 20 мM Tris при pH 8,0, (c) 1 мM MgCl2, (d) 200 мM NaCl и (e) 0,005% полоксамера 188, при этом вес тела пациента составляет от 2,6 кг до 8,5 кг. В одном варианте осуществления композиция не содержит консерванта.
В одном варианте осуществления пациент (а) в возрасте девять месяцев или менее, (b) имеет массу тела по меньшей мере приблизительно 2,6 кг, (c) имеет биаллельные нулевые мутации или делеции в SMN1 и (d) имеет по меньшей мере одну функциональную копию SMN2.
Другой аспект настоящего изобретения направлен на композицию, подходящую или полученную для внутривенного введения, при этом фармацевтическая композиция содержит (a) самокомплементарный вирусный вектор на основе AAV9, содержащий модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2, (b) 20 мМ Tris при pH 8,0, (c) 1 мМ MgCl2, (d) 200 мМ NaCl и (e) 0,005% полоксамера 188.
В некоторых вариантах осуществления в данном документе раскрыта композиция или состав, где композиция или состав содержат по меньшей мере одно из следующего: (a) менее приблизительно 0,09 нг бензоназы на 1,0×1013 г. в., (b) менее приблизительно 30 мкг/г (ppm) цезия, (c) приблизительно 20-80 ppm полоксамера 188, (d) менее приблизительно 0,22 нг BSA на 1,0×1013 г. в., (e) менее приблизительно 6,8×105 пг остаточной плазмидной ДНК на 1,0×1013 г. в., (f) менее приблизительно 1,1×105 пг остаточной hcDNA на 1,0×1013 г. в. и (g) менее приблизительно 4 нг rHCP на 1,0×1013 г. в.
В некоторых вариантах осуществления настоящее изобретение обеспечивает процесс накопления для получения промежуточного соединения (например, замороженного промежуточного продукта), полученного из рабочего банка клеток, причем процесс накопления включает стадии (a) культивирования клеток, (b) трансфицирования культивируемых клеток плазмидами (например, тремя плазмидами), (c) сбора увеличенного количества вирусных частиц из клеток после периода культивирования, (d) очистки вирусных частиц с помощью фильтрации для удаления любых интактных клеток или клеточного дебриса, (е) подвергания элюента из стадии (d) фильтрации в тангенциальном потоке, и (g) необязательно замораживания полученного промежуточного препарата очищенных вирусных частиц. В некоторых вариантах осуществления процесс накопления объединяют с дополнительными этапами обработки, в частности, например, дополнительными этапами очистки и составления для получения конечного фармацевтического продукта.
В одном варианте осуществления рабочий банк клеток содержит клетки HEK293. В других вариантах осуществления применяют другой тип клеток или производное, которое доступно в данной области техники и подходит для применения в способах, раскрытых в данном документе.
В одном варианте осуществления на стадии трансфекции применяют полиэтиленимин. В одном варианте осуществления стадия трансфекции включает трансфекцию тремя векторами с применением трансгенной плазмиды, например pSMN, плазмиды pAAV и плазмиды pHELP.
В одном варианте осуществления способ дополнительно включает лизис трансфицированных клеток буфером для лизиса и поверхностно-активным веществом.
В одном варианте осуществления стадия сбора включает обработку продукта эндонуклеазой, такой как бензоназа, например, в концентрации от 50 до 200 МЕ/мл, например, в концентрации от 75 до 150 МЕ/мл, для уменьшения содержания остаточной ДНК клетки хозяина.
В одном варианте осуществления стадия очистки включает глубинную фильтрацию и последующую фильтрацию через фильтр, который удаляет макромолекулярные загрязняющие вещества и клеточный дебрис, например фильтр с размером пор 0,45 микрона, но который позволяет геномам векторов проходить через него. Можно применять любой подходящий глубинный фильтр.
В одном варианте осуществления фильтрация в тангенциальном потоке ("TFF") позволяет достичь от 5 до 15 X, например, от 6 до 10 X концентрации элюента со стадии (d) и по меньшей мере 4 диаобъемов, например 6 диаобъемов диафильтрации, или 10 диаобъемов, или 12 диаобъемов, или 15 диаобъемов диафильтрации. Можно применять любой подходящий фильтр для TFF. В одном варианте осуществления мембрана для TFF представляет собой целлюлозную мембрану. В одном варианте осуществления мембрана для TFF характеризуется отсечением по 300 кДа.
Дополнительно в настоящем изобретении предусмотрен процесс выделения и очистки для обработки промежуточного продукта (например, замороженного промежуточного продукта) с получением отфильтрованного лекарственного вещества. Стадии процесса выделения и очистки включают стадию подкисления и осветления (с применением фильтрации), за которой следует катионообменная хроматография, фильтрация в тангенциальном потоке, ультрацентрифугирование с помощью CsCl и дополнительная стадия фильтрации в тангенциальном потоке для получения отфильтрованного лекарственного вещества, где очищенные частицы AAV суспендируют в фармацевтически приемлемом носителе.
В одном варианте осуществления стадия подкисления и осветления включает глубинную фильтрацию и последующую фильтрацию через фильтр, который удаляет макромолекулярные загрязняющие вещества и клеточный дебрис, например фильтр с размером пор 0,45 микрона. В некоторых вариантах осуществления контролируют регулирование рН. В качестве части этой стадии в одном варианте осуществления добавляют детергент, например Tween. В некоторых вариантах осуществления контролируют скорость добавления Tween и диапазон концентраций Tween.
В одном варианте осуществления катионообменная хроматография предусматривает смолу для мембранной хроматографии с применением композитной смолы с сульфонильными группами с размером пор 0,2 микрона.
В одном варианте осуществления на стадии ультрацентрифугирования с помощью хлорида цезия (CsCl) используют 2-4 М CsCl, например приблизительно 3 М CsCl.
В другом варианте осуществления стадию ультрацентрифугирования с помощью CsCl проводят при приблизительно 40-50 тыс. об/мин в течение приблизительно 20-25 часов. В другом варианте осуществления стадию ультрацентрифугирования с помощью CsCl проводят при приблизительно 45 тыс. об/мин в течение приблизительно 22 часов.
В одном варианте осуществления фильтрация в тангенциальном потоке ("TFF") позволяет достичь от 5 до 15 X, например, от 6 до 10 X концентрации элюента со стадии (d) и по меньшей мере 4 диаобъемов, например 6 диаобъемов диафильтрации, или 10 диаобъемов, или 12 диаобъемов, или 15 диаобъемов диафильтрации. В одном варианте осуществления мембрана для TFF характеризуется отсечением по 300 кДа.
В одном варианте осуществления элюент находится ниже уровня обнаружения цезия (Cs). В другом варианте осуществления уровень обнаружения Cs ниже 50 на миллион (ppm). В другом варианте осуществления уровень обнаружения Cs составляет приблизительно 50-70 ppm. В другом варианте осуществления уровень обнаружения Cs составляет приблизительно 70-90 частей на миллион (ppm). В другом варианте осуществления уровень обнаружения Cs составляет приблизительно 90-110 частей на миллион (ppm). В другом варианте осуществления уровень обнаружения Cs составляет приблизительно 110-130 частей на миллион (ppm). В другом варианте осуществления уровень обнаружения Cs составляет приблизительно 130-150 частей на миллион (ppm). В другом варианте осуществления уровень обнаружения Cs составляет менее 150 частей на миллион (ppm).
Такие способы очистки можно применять для получения вирусных препаратов с высоким выходом, включая препараты AAV (например, AAV9-SMN), которые содержат менее 5×106 пг/мл остаточной ДНК клетки хозяина (hcDNA) на 1×1013 геномов вектора ("г. в.")/мл, например, менее 1,2×106 пг/мл hcDNA на 1×1013 г. в./мл. Таким образом, пациент весом 5 кг, получающий 7,5×1015 г. в., получит не более чем вплоть до 1,2×106 пг/мл*7,5×1015 г. в. /(1×1013г. в./мл) = 8,4×107 пг hcDNA=84000 нг hcDNA на дозу 5 кг. В одном варианте осуществления препарат содержит менее 5,0×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., менее 2,0×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., менее 1,1×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., менее 1,0×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., менее 0,9×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., менее 0,8×105 пг остаточной ДНК клетки-хозяина на 1,0×1013 г. в., или любую концентрацию в диапазоне, ограниченном этими значениями.
В одном варианте осуществления AAV представляет собой дефектный по репликации AAV9, например scAAV9 с ITR, полученными из AAV2. В другом варианте осуществления вектор на основе AAV несет трансген SMN. В одном варианте осуществления ДНК, кодирующая SMN, представлена в GenBank под № доступа NM_000344.2. Также предусмотрены консервативные нуклеотидные замены в ДНК SMN (например, замена гуанина на аденин в положении 625, как указано в GenBank под № доступа NM_000344.2).
Другой аспект настоящего изобретения направлен на фармацевтическую композицию, содержащую частицы AAV в составе, подходящем либо для (a) внутривенной ("IV") инъекции, либо (b) интратекального ("IT") введения.
В другом варианте осуществления фармацевтическая композиция содержит менее 10% пустых капсидов, менее 8% пустых капсидов, менее 7% пустых капсидов, менее 5% пустых капсидов, менее 3% пустых капсидов или менее 1% пустых капсидов. В некоторых вариантах осуществления фармацевтическая композиция содержит менее приблизительно 5% пустых капсидов. В одном варианте осуществления количество пустых капсидов ниже предела обнаружения. В некоторых вариантах осуществления предпочтительно, чтобы фармацевтическая композиция характеризовалась низкими показателями количества пустых капсидов, потому что эти пустые капсиды могут вызывать неблагоприятную реакцию (например, иммунную реакцию, воспалительную реакцию, реакцию со стороны печени и/или реакцию со стороны сердца) без обеспечения терапевтического эффекта.
В другом варианте осуществления содержание остаточного белка клетки-хозяина ("rHCP") в указанной фармацевтической композиции составляет 100 нг/мл rHCP на 1×1013 г. в./мл или меньше, например, составляет 40 нг/мл rHCP на 1×1013 г. в./мл или меньше или 1-50 нг/мл rHCP на 1×1013 г. в./мл. В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, содержит менее 10 нг rHCP на 1,0×1013 г. в., или менее 5 нг rHCP на 1,0×1013 г. в., менее 4 нг rHCP на 1,0×1013 г. в., или менее 3 нг rHCP на 1,0×1013 г. в., или любую концентрацию в диапазоне, ограниченном этими значениями.
В другом варианте осуществления содержание остаточной ДНК клетки-хозяина ("hcDNA") в указанной фармацевтической композиции составляет 5×106 пг/мл hcDNA на 1×1013 г. в./мл или меньше, составляет 1,2×106 пг/мл rHDNA на 1×1013 г. в./мл или меньше или от 1×105 пг/мл rHDNA на 1×1013 г. в./мл до 1,2×106 пг/мл на 1×1013 г. в./мл. В одном варианте осуществления содержание остаточной ДНК клетки-хозяина в указанной фармацевтической композиции составляет менее 5,0×105 пг на 1,0×1013 г. в., менее 2,0×105 пг на 1,0×1013 г. в., менее 1,1×105 пг на 1,0×1013 г. в., менее 1,0×105 пг hcDNA на 1,0×1013 г. в., менее 0,9×105 пг hcDNA на 1,0×1013 г. в., менее 0,8×105 пг hcDNA на 1,0×1013 г. в., или любую концентрацию в диапазоне, ограниченном этими значениями.
В одном варианте осуществления содержание остаточной плазмидной ДНК в указанной фармацевтической композиции составляет 1,7×106 пг/мл на 1×1013 г. в./мл или меньше или от 1×105 пг/мл на 1×1013 г. в./мл до 1,7×106 пг/мл на 1×1013 г. в./мл. В одном варианте осуществления содержание остаточной плазмидной ДНК в указанной фармацевтической композиции составляет менее 10,0×105 пг на 1,0×1013 г. в., менее 8,0×105 пг на 1,0×1013 г. в. или менее 6,8×105 пг на 1,0×1013 г. в.
В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, содержит менее 0,5 нг на 1,0×1013 г. в., менее 0,3 нг на 1,0×1013 г. в., менее 0,22 нг на 1,0×1013 г. в. или менее 0,2 нг на 1,0×1013 г. в. или любую концентрацию в диапазоне, ограниченном этими значениями, бычьего сывороточного альбумина (BSA). В одном варианте осуществления содержание бензоназы в указанной фармацевтической композиции составляет менее 0,2 нг на 1,0×1013 г. в., менее 0,1 нг на 1,0×1013 г. в., менее 0,09 нг на 1,0×1013 г. в., менее 0,08 нг на 1,0×1013 г. в. или любую концентрацию в диапазоне, ограниченном этими значениями. В одном варианте осуществления содержание полоксамера 188 в указанной фармацевтической композиции составляет приблизительно 10-150 ppm, приблизительно 15-100 ppm или приблизительно 20-80 ppm. В одном варианте осуществления содержание цезия в указанной фармацевтической композиции составляет менее 50 мкг/г (ppm), менее 30 мкг/г (ppm) или менее 20 мкг/г (ppm) или любую концентрацию в диапазоне, ограниченном этими значениями.
В одном варианте осуществления общее содержание содержание примесей в фармацевтической композиции, раскрытой в данном документе, составляет, например, при определении с помощью SDS-PAGE, менее 10%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2% или любое процентное содержание в диапазоне, ограниченном этими значениями. В одном варианте осуществления общая чистота, например, при определении с помощью SDS-PAGE, составляет более 90%, более 92%, более 93%, более 94%, более 95%, более 96%, более 97%, более 98% или любое процентное значение в диапазоне, ограниченном этими значениями. В одном варианте осуществления фармацевтической композиции содержание ни одной из отдельных неименованных родственных примесей, например, при определении с помощью SDS-PAGE, не является больше 5%, больше 4%, больше 3% или больше 2% или любого процентного содержание в диапазоне, ограниченном этими значениями. В одном варианте осуществления фармацевтическая композиция предусматривает процентное содержание заполненных капсидов относительно общего количества капсидов (например, пик 1+пик 2 при измерении с помощью аналитического ультрацентрифугирования), составляющее более 85%, более 86%, более 87%, более 88%, более 89%, более 90%, более 91%, более 91,9%, более 92%, более 93% или любое процентное содержание в диапазоне, ограниченном этими значениями. В одном варианте осуществления фармацевтической композиции процентное содержание заполненных капсидов, измеренное в пике 1 с помощью аналитического ультрацентрифугирования, составляет 20-80%, 25-75%, 30-75%, 35-75% или 37,4-70,3%. В одном варианте осуществления фармацевтической композиции процентное содержание заполненных капсидов, измеренное в пике 2 с помощью аналитического ультрацентрифугирования, составляет 20-80%, 20-70%, 22-65%, 24-62% или 24,9-60,1%.
В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, содержит геномный титр, составляющий 1,0-5,0×1013 г. в./мл, 1,2-3,0×1013 г. в./мл или 1,7-2,3×1013 г. в./мл.
В одном варианте осуществления для фармацевтической композиции, раскрытой в данном документе, продемонстрирована бионагрузка, составляющая менее 5 КОЕ/мл, менее 4 КОЕ/мл, менее 3 КОЕ/мл, менее 2 КОЕ/мл или менее 1 КОЕ/мл или любую концентрацию в диапазоне, ограниченном этими значениями. В одном варианте осуществления количество эндотоксина в соответствии с USP, например статьей <85> USP (включенной посредством ссылки во всей своей полноте), составляет менее 1,0 ЕЭ/мл, менее 0,8 ЕЭ/мл или менее 0,75 ЕЭ/мл.
В одном варианте осуществления осмоляльность фармацевтической композиции раскрытой в данном документе, в соответствии с USP, например со статьей <785> USP (включенной посредством ссылки во всей своей полноте), составляет 350-450 мОсм/кг, 370-440 мОсм/кг или 390-430 мОсм/кг. В одном варианте осуществления фармацевтическая композиция содержит менее 1200 частиц, размер которых превышает 25 мкм, на контейнер, менее 1000 частиц, размер которых превышает 25 мкм, на контейнер, менее 600 частиц, размер которых превышает 25 мкм, на контейнер, менее 500 частиц, размер которых превышает 25 мкм, на контейнер или любое значение в диапазоне, ограниченном этими значениями. В одном варианте осуществления фармацевтическая композиция содержит менее 10000 частиц, размер которых превышает 10 мкм, на контейнер, менее 8000 частиц, размер которых превышает 10 мкм, на контейнер или менее 6000 частиц, размер которых превышает 10 мкм, на контейнер.
В одном варианте осуществления фармацевтическая композиция характеризуется геномным титром, составляющим 0,5-5,0×1013 г. в./мл, 1,0-4,0×1013 г. в./мл, 1,5-3,0×1013 г. в./мл или 1,7-2,3×1013 г. в./мл.
В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, предусматривает одно или несколько из следующего: менее приблизительно 0,09 нг бензоназы на 1,0×1013 г. в., менее приблизительно 30 мкг/г (ppm) цезия, приблизительно 20-80 ppm полоксамера 188, менее приблизительно 0,22 нг BSA на 1,0×1013 г. в., менее приблизительно 6,8×105 пг остаточной плазмидной ДНК на 1,0×1013 г. в., менее приблизительно 1,1×105 пг остаточной hcDNA на 1,0×1013 г. в., менее приблизительно 4 нг rHCP на 1,0×1013 г. в., pH 7,7-8,3, приблизительно 390-430 мОсм/кг, менее приблизительно 600 частиц размером ≥ 25 мкм, на контейнер, менее приблизительно 6000 частиц размером ≥ 10 мкм, на контейнер, геномный титр, составляющий приблизительно 1,7×1013-2,3×1013 г. в./мл, инфекционный титр, составляющий приблизительно 3,9×108-8,4×1010 МЕ на 1,0×1013 г. в., содержание общего белка, составляющее приблизительно 100-300 мкг на 1,0×1013 г. в., медианное значение выживаемости, составляющее ≥ 24 дней у мышей Δ7SMA при дозе вирусного вектора приблизительно 7,5×1013 г. в./кг, относительная активность, определяемая с помощью анализа на основе клеток in vitro, составляющая 70-130%, и/или менее приблизительно 5% пустых капсидов.
В различных вариантах осуществления фармацевтические композиции, раскрытые в данном документе, содержащие любые вирусные частицы, обсуждаемые в данном документе (например, вирусные частицы AAV SMN, AAV MECP2 или AAV SOD1), сохраняют активность в диапазоне+20%, в диапазоне+15%, в диапазоне+10%, или в диапазоне+5% от таковой эталонного стандарта. В некоторых вариантах осуществления активность измеряют с применением подходящего клеточного анализа in vitro или животной модели in vivo. Например, активность или % функциональных вирусных частиц AAV SMN могут быть определены с применением животной модели SMA, например мыши SMAΔ7, или количественного анализа на основе клеток с применением подходящей линии клеток, например первичных нейральных клеток-предшественниц (NPC), выделенных из коры головного мозга мышей SMAΔ7. В одном варианте осуществления активность оценивают по сравнению с эталонным стандартом с применением способов, описанных в Foust et al., Nat. Biotechnol., 28(3), pp. 271-274 (2010). Можно применять любой подходящий эталонный стандарт. Активность или % функциональной AAV MeCP2 можно анализировать с применением подходящего клеточного анализа in vitro или животной модели in vivo, например мыши с нокаутом гена Mecp2, как описано в Guy et al., "Reversal of neurological defects in a mouse model of Rett syndrome." Science, 315(5815):1143-7. Активность или % функциональной AAV SOD1 можно анализировать с применением подходящего клеточного анализа in vitro или животной модели in vivo, например мыши, мутантной по SOD1, как описано в Gurney et al., "Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation." Science, 264(5166):1772-5. В одном варианте осуществления фармацевтическая композиция обладает активностью in vivo, определяемой по медианному значению выживаемости у мыши SMAΔ7, получавшей дозу 7,5×1013 г. в./кг более 15 дней, более 20 дней, более 22 дней или более 24 дней. В одном варианте осуществления фармацевтическая композиция характеризуется относительной активностью in vivo, при исследовании с помощью анализа на основе клеток, составляющей 50-150%, 60-140% или 70-130% относительно эталонного стандарта и/или соответствующего контроля.
В одном варианте осуществления состав для внутривенного введения ("IV") характеризуется pH от 7,5 до 8,5, геномным титром от приблизительно 1 до 8×1013 геномов вирусного вектора/мл (г. в./мл) или от 2×1013 г. в./мл до 6×1013 г. в./мл и необязательно осмоляльностью 384-448 мОсм/кг. В одном варианте осуществления IV-состав содержит MgCl2, NaCl, плюроник F68 в Tris-буфере при pH 8,0.
В одном варианте осуществления для внутривенного введения вектор на основе AAV-9, несущий трансген SMN, вводят в стерильных условиях в подходящей обстановке (например, в интервенционном кабинете, операционной, выделенной комнате для процедур) однократно через венозный катетер, вставленный в периферическую вену конечности (руки или ноги) в указанной дозе и осуществляют медленную инфузию в течение приблизительно 30-60 минут,
В другом варианте осуществления в настоящем изобретении, представленном в данном документе, предусмотрены композиции и способы доставки полинуклеотида в центральную нервную систему пациента, нуждающегося в этом, включающие интратекальную ("IT") доставку rAAV9 и неионогенного низкоосмолярного контрастного средства пациенту, где rAAV9 содержит самокомплементарный геном, содержащий полинуклеотид. Полинуклеотид доставляется в, например, головной мозг, спинной мозг, глиальные клетки, астроцит и/или нижний мотонейрон. Неионогенное, низкоосмолярное контрастное средство представляет собой, например, йобитридол, йогексол, йомепрол, йопамидол, йопентол, йопромид, йоверсол или йоксилан. В некоторых вариантах осуществления полинуклеотид представляет собой белок выживаемости мотонейронов (SMN). В одном варианте осуществления контрастным средством является йогексол, например йогексол 180 (продается в виде омнипак 180, содержащего 388 мг йогексола, что эквивалентно 180 мг органического йода на мл).
В одном варианте осуществления для IT-введения вектор на основе scAAV9, несущий трансген SMN, разбавляют физиологическим раствором и предварительно смешивают с подходящей гипербарической контрастной средой, одобренной и маркированной для педиатрического применения при радиографическом мониторинге инъекции путем интратекальной инъекции в поясничный отдел (такой как омнипак 180). Общий объем водной композиции, содержащей вектор на основе AAV-9, несущий трансген SMN, плюс контрастную среду и/или физиологический раствор, не будет превышать 5 мл. Контрастное средство и вектор на основе scAAV-9, несущий трансген SMN, могут быть совместно составлены, совместно упакованы или упакованы и доставлены в центр работы с пациентами отдельно.
Пациенты получают вектор на основе scAAV-9, несущий трансген SMN, посредством интратекальной инъекции в стерильных условиях в палате PICU или в другой подходящей обстановке (например, в интервенционном кабинете, операционной, выделенной комнате для процедур) с непосредственным доступом к неотложной помощи при интенсивной терапии. На месте можно использовать атравматическую иглу, вставляемую скосом параллельно волокнам твердой мозговой оболочки, так как было показано, что это значительно уменьшает повреждение твердой мозговой оболочки и, следовательно, снижает риск вытекания спинномозговой жидкости после люмбальной пункции (Ebinger et al., "Headache and Backache After Lumbar Puncture in Children and Adolescents: A Prospective Study." Pediatrics, 113(6):1588-1592; Kiechl-Kohlendorfer et al., "Cerebrospinal Fluid Leakage After Lumbar Puncture in Neonates: Incidence and Sonographic Appearance." American Journal of Roentgenology, 181(1):231-234), в том числе у детей.
Седация/анестезия рекомендуется для всех пациентов, получающих IT-инъекции. Способ и медицинские препараты будут отводиться на усмотрение местного анестезиолога, но должны включать достаточную степень седации или анксиолизиса, чтобы обеспечить обезболивание и обездвиживание для расположения в положении Тренделенбурга во время процедуры и после процедуры. Пациентов будут располагать в положении Тренделенбурга с наклоном вниз под углом 30° на 15 минут после введения IT-терапевтического средства для улучшения распределения в области шейного отдела и головного мозга.
Пациентов помещают в положение лежа на боку и вводят катетер со стилетом путем люмбальной пункции в межостистое пространство L3-L4 или L4-L5 в субарахноидальное пространство. Субарахноидальная катетеризация подтверждается оттоком чистой спинномозговой жидкости (CSF) из катетера. CSF будет удалена и утилизирована в соответствии с правилами, принятыми в учреждении. Вектор на основе scAAV-9, несущий трансген SMN, в предварительно смешанном контрастном растворе, вводят непосредственно в субарахноидальное пространство.
В одном варианте осуществления настоящее изобретение предусматривает способ лечения неврологического заболевания у пациента, нуждающегося в этом, включающий внутривенную или интратекальную доставку фармацевтической композиции, раскрытой в данном документе, где парвовирус содержит самокомплементарный геном rAAV9, где сконструированный трансген содержит полинуклеотид SMN, и где заболевание представляет собой SMA.
В другом варианте осуществления настоящее изобретение предусматривает способ лечения неврологического заболевания у пациента, нуждающегося в этом, включающий интратекальную доставку фармацевтической композиции, раскрытой в данном документе, с контрастным средством, где парвовирус содержит самокомплементарный геном rAAV9, где сконструированный трансген содержит полинуклеотид SMN, где заболевание представляет собой SMA, и где контрастное средство представляет собой омнипак 180.
В другом варианте осуществления настоящее изобретение предусматривает способ лечения SMA типа II, III или IV у пациента, нуждающегося в этом, включающий интратекальную доставку фармацевтической композиции, раскрытой в данном документе, с контрастным средством, где парвовирус содержит самокомплементарный геном rAAV9, где сконструированный трансген содержит полинуклеотид SMN, где заболевание представляет собой SMA, и где контрастное средство представляет собой омнипак 180.
В другом варианте осуществления настоящее изобретение предусматривает способ лечения SMA типа I у пациента, нуждающегося в этом, включающий внутривенную доставку фармацевтической композиции, раскрытой в данном документе, где парвовирус содержит самокомплементарный геном rAAV9, и где сконструированный трансген содержит полинуклеотид SMN. В некоторых вариантах осуществления возраст пациента составляет 0-9 месяцев. В некоторых вариантах осуществления возраст пациента составляет 0-6 месяцев. В других вариантах осуществления вес пациента детского возраста составляет не более приблизительно 8 кг. В некоторых вариантах осуществления вес пациента детского возраста составляет не более приблизительно 8,5 кг или меньше. В некоторых вариантах осуществления вес пациента детского возраста составляет приблизительно 2,6 кг или больше.
В другом варианте осуществления настоящее изобретение предусматривает набор для лечения SMA типа I у пациента, нуждающегося в этом, предусматривающий внутривенное введение фармацевтической композиции, раскрытой в данном документе, содержащейся во флаконах. В некоторых вариантах осуществления измеряют вес пациента и дозу рассчитывают исходя из веса пациента.
Если не указано иное, все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понимают специалисты в области техники, к которой относится это изобретение.
Используемые в данном документе формы единственного числа слова также включают множественную форму слова, если контекст явно не предписывает иное; в качестве примеров, формы единственного и множественного числа следует понимать как формы единственного или множественного числа, а термин "или" следует понимать как включающий. В качестве примера, "элемент" означает один элемент или несколько элементов.
Во всем описании такое слово как "содержащий" или вариации, такие как "содержит", будут пониматься как предполагающие включение указанных элемента, целого числа, стадии или группы элементов, целых чисел или стадий без исключения любого другого элемента, целого числа, стадии или группы элементов, целых чисел или стадий. Во всем описании такие слова как "состоящий из" или вариации, такие как "состоит из", будут пониматься как предполагающие включение указанных элемента, целого числа, стадии или группы элементов, целых чисел или стадий, и исключение любого другого элемента, целого числа, стадии или группы элементов, целых чисел или стадий. Во всем описании такие слова как "по существу состоящий из" или варианты, такие как "по существу состоит из", будут пониматься как предполагающие включение указанных элемента, целого числа или стадии, или группы элементов, целых чисел или стадий и любого другого элемента, целого числа или стадии, или группы элементов, целых чисел или стадий, которые не оказывают существенного влияния на основные и новые характеристики настоящего изобретения и/или формулы изобретения.
"Приблизительно" можно понимать как в пределах 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,1%, 0,05% или 0,01% от заявленного значения. При использовании в отношении процентного значения "приблизительно" можно понимать как в пределах ± 1% (например, "приблизительно 5%" можно понимать как в пределах 4-6%) или ± 0,5% (например, "приблизительно 5%" можно понимать как в пределах 4,5% - 5,5%). Если иное не следует из контекста, все числовые значения, приведенные в данном документе, модифицируются термином "приблизительно".
Хотя при практическом осуществлении или тестировании настоящего изобретения можно применять способы и материалы, подобные или эквивалентные описанным в данном документе, подходящие способы и материалы описаны ниже. Все публикации, заявки на патенты, патенты и другие ссылки, упомянутые в данном документе, включены в данный документ посредством ссылки во всей своей полноте. Ссылки, цитируемые в данном документе, не признаются предшествующим уровнем техники в отношении заявленного изобретения. В случае конфликта настоящее описание, включая определения, будет иметь преимущественную силу. Кроме того, материалы, способы и примеры являются только иллюстративными и не предполагают ограничения. Другие особенности и преимущества настоящего изобретения будут очевидны из следующего подробного описания и формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Различные цели и преимущества, а также более полное понимание настоящего изобретения очевидны и более понятны со ссылкой на следующее подробное описание и прилагаемую формулу изобретения, взятые вместе с прилагаемым графическим материалом, где представлено следующее.
Фиг. 1. Карты плазмид pSMN, pHELP и pAAV.
На фиг. 1A показана карта плазмиды pSMN. pSMN представляет собой плазмиду, кодирующую информацию для рекомбинантного самокомплементарного ДНК-генома AAV, который обеспечивает экспрессию cDNA для белка выживаемости мотонейронов человека (SMN) под контролем гибридного промотора бета-актина курицы с элементом, представляющим собой непосредственно-ранний энхансер цитомегаловируса (CMV). cDNA SMN кодирует полноразмерный функциональный белок. Кассета экспрессии содержит модифицированную последовательность интрона, полученную из вируса обезьян 40 (SV40) и сигнал полиаденилирования гормона роста крупного рогатого скота (BGH). Кассета экспрессии (CMV-CB-SV40-SMN-BGHpA) фланкирована инвертированными концевыми повторами (ITR), полученными из AAV2. Левый ITR модифицирован для предпочтительной упаковки самокомплементарных геномов AAV. Участки между ITR, включая сами ITR, совместно упаковываются в рекомбинантные капсиды AAV9 во время получения лекарственного препарата по настоящему изобретению. Ключевые компоненты pSMN, которые не предназначены для упаковки в рекомбинантные геномы AAV, включают открытую рамку считывания, кодирующую устойчивость к канамицину (KanR) и точку начала репликации (ori), полученные из pUC. Участки ori и KanR полезны для получения плазмид.
На фиг. 1B показана плазмидная карта плазмиды pHELP. Плазмида pHELP содержит действующие в транс-положении компоненты аденовируса, необходимые для продукции рекомбинантных аденоассоциированных вирусов. Плазмида pHELP содержит участки генома аденовируса, которые обеспечивают факторы, которые являются важными для репликации AAV, а именно E2A, E4 и VA РНК. Функции E1 аденовируса, участвующие в репликации rAVV, обеспечивают путем трансфекции клеток-хозяев линии 293. Однако плазмида pHELP не содержит другие гены, ответственные за репликацию, или структурные гены аденовируса. Последовательности аденовируса, присутствующие в данной плазмиде, составляют только ~28% (9280/35938) генома аденовируса и не содержат цис-элементы, критические для репликации, такие как инвертированные концевые повторы. Следовательно, ожидается, что в такой системе продуцирования инфекционный аденовирус не образуется.
На фиг. 1C показана плазмидная карта плазмиды AAV. Геном AAV дикого типа содержит два некодирующих структурных элемента, называемых инвертированными концевыми повторами, которые фланкируют открытые рамки считывания rep и cap. Rep и cap кодируют белок репликации и белок капсида вируса соответственно. При производстве рекомбинантных аденоассоциированных вирусных векторов вирусные ITR представляют собой единственные элементы, применяемые в цис-положении, в то время как открытые рамки считывания вируса обеспечивают в транс-положении. При применении способа транзиентной трансфекции адгезивных клеток HEK293 для создания AAV цис/транс-роли для различных генетических элементов обеспечивают путем их распределения в отдельные плазмиды. Плазмида pAAV2/9 содержит открытые рамки считывания для гена rep AAV2 и гена cap AAV9.
На фиг. 2 показана схема технологического процесса отбора клеток HEK293 по исключительной адгезии и их хранения в качестве предварительного главного банка клеток (MCB).
На фиг. 3 показан перечень компонентов процесса обработки клеток для отбора клеток HEK293 по исключительной адгезии и хранения в качестве предварительного главного банка клеток (MCB).
На фиг. 4 описана технологическая схема процесса накопления для лекарственного вещества.
На фиг. 5 описана технологическая схема процесса выделения и очистки для лекарственного вещества.
На фиг. 6 показана инактивация XMuLV с помощью Tween 20, добавленного на не более чем 120 минут.
На фиг. 7 показана инактивация PRV с помощью Tween 20, добавленного на не более чем 120 минут.
На фиг. 8 описана схема технологического процесса обеспечения размножения клеток HEK 293 в ходе экспериментов по определению плотности посева клеток.
На фиг. 9A-E показаны профили роста и образования метаболитов. Клетки HEK 293 высевали в двух повторностях при 12000 и 8000 клеток/см2 в биореакторы (при pH 7,23, 37,0°C, 55% растворенного кислорода (DO)). Клетки трансфицировали ДНК-плазмидами/PEI через четыре дня (12000 клеток/см2) и пять дней (8000 клеток/см2) после посева. Сбор биомассы с биореакторов осуществляли через восемь дней (12000 клеток/см2) и девять дней (8000 клеток/см2) после посева. Данные pH и образования метаболитов считывали ежедневно с помощью Nova BioFlex.
На фиг. 10 показано продуцирование вирусного генома в виде зависимости от плотности посева клеток (8000 или 12000 клеток/см2) и четырех различных продолжительностей времени трансфекции (20 мин, 1 час или 2 часа).
На фиг. 11 показаны титры вируса из образцов промежуточных продуктов, отобранных на различных стадиях фильтрации в ходе всего процесса производства.
На фиг. 12 A-B показаны уровни извлечения вирусного вектора и удаления белка клетки-хозяина (HCP) на стадии TFF1.
На фиг. 13 описана схема технологического процесса обеспечения размножения клеток HEK 293 в ходе экспериментов по определению плотности посева клеток.
На фиг. 14 A-E показано, что клетки HEK 293 высевали в двух повторностях при 8000 клеток/см2, 9350 клеток/см2, 10700 клеток/см2, 12050 клеток/см2 в биореакторы (при pH 7,23, 37,0°C, 55% DO). Клетки трансфицировали ДНК-плазмидами/PEI (1:1 масса/масса) через пять дней после посева. Анализ pH и образования метаболитов проводили с применением NOVA BioProfile 400.
На фиг. 15 A-B показан уровень продуцирования лекарственного вещества при четырех начальных значениях плотности посева в биореакторах. Сравнение вирусного титра и количества векторного генома, собранного с единицы площади поверхности.
На фиг. 16 показаны способы получения фазы 1 (способ A) и фазы 3 испытания (способа B).
На фиг. 17 A-B представлена таблица, которая иллюстрирует результаты определения сопоставимости и однородности процесса производства - продукты способа A (фаза 1) и способа B (фаза 3). Показано, что продукты способа B имеют дополнительные преимущества по сравнению со способом A.
На фиг. 18 показана сопоставимость способа A и способа B с применением попарного сравнения способа A (фаза 1, серия NCHAAV9SMN0613) и способа B (фаза 3, серия 600156). Показано, что продукты способа B имеют дополнительные преимущества по сравнению со способом A.
На фиг. 19 показана оценка однородности процесса производства при попарном сравнении серий 600156 и 600307 способа B (фаза 3).
На фиг. 20 показан профиль стабильности серии NCHAAV9SMN0613 NCH, хранящейся в условиях хранения в реальном времени при ≤ -60°C в течение 12 месяцев.
На фиг. 21 показаны коэффициенты седиментации (с×10-13) для материала фазы 1 (NCHAAV9SMN0613) с указанием пустых капсидов (7%) с коэффициентом седиментации, составляющим приблизительно 60×10-13 с, и полных капсидов с диапазоном коэффициентов седиментации приблизительно 80-150×10-13 сек.
На фиг. 22 показаны коэффициенты седиментации (с×10-13) для материала фазы 3 (600156) с указанием пустых капсидов (2%) с коэффициентом седиментации, составляющим приблизительно 60×10-13 с, и полных капсидов с диапазоном коэффициентов седиментации приблизительно 80-150×10-13 сек.
На фиг. 23 показаны коэффициенты седиментации (с×10-13) для материала фазы 3 (600307) с указанием пустых капсидов (4%) с коэффициентом седиментации, составляющим приблизительно 60×10-13 с, и полных капсидов с диапазоном коэффициентов седиментации приблизительно 80-150×10-13 сек.
ПОДРОБНОЕ ОПИСАНИЕ
С целью продвижения развития генной терапии на основе AAV за пределы животных моделей и в клинические исследования и/или для терапевтических вариантов применения разработали масштабируемый процесс, обеспечивающий возможность получения вирусного материала, подходящего для применения у человека.
В некоторых вариантах осуществления под "вектором" подразумевают любой генетический элемент, такой как плазмида, фаг, транспозон, космида, хромосома, вирус, вирион и т.д., который способен к репликации при ассоциации с соответствующими элементами, осуществляющими контроль, и который может переносить последовательности генов между клетками. Таким образом, термин включает носители для клонирования и экспрессии, а также вирусные векторы.
В некоторых вариантах осуществления под "вектором на основе AAV" подразумевают вектор, полученный из серотипа аденоассоциированного вируса, включая без ограничения AAV-1, AAV-2, AAV-3, AAV-4, AAV-5, AAV-6, AAV-7, AAV-8 и AAV-9. Векторы на основе AAV могут иметь полную или частичную делецию одного или нескольких из генов AAV дикого типа, например, генов rep и/или cap, но сохранять функциональные фланкирующие последовательности ITR. Функциональные последовательности ITR необходимы для освобождения, репликации и упаковки вириона AAV. Таким образом, вектор на основе AAV определен в данном документе как включающий по меньшей мере те последовательности, которые в цис-положении обеспечивают репликацию и упаковку (например, функциональные ITR) вируса. ITR не обязательно должен представлять собой нуклеотидные последовательности дикого типа, а может быть измененным, например, посредством вставки, делеции или замены нуклеотидов, при условии, что последовательности обеспечивают функциональное освобождение, репликацию и упаковку. В одном варианте осуществления вектор представляет собой вектор на основе AAV-9 с ITR, полученными из AAV-2. Также под "вектором на основе AAV" подразумевают, белковую оболочку или капсид, которые обеспечивают эффективный носитель для доставки нуклеиновой кислоты, представляющей собой вектор, в ядро клеток-мишеней.
В некоторых вариантах осуществления под "scAAV" подразумевают самокомплементарный аденоассоциированный вирус (scAAV), который представляет собой вирусный вектор, сконструированный из встречающегося в природе аденоассоциированного вируса (AAV), для применения в генной терапии. scAAV называется "самокомплементарным", потому что кодирующий участок сконструирован таким образом, что он образует матрицу, представляющую собой внутримолекулярную двухнитевую ДНК.
В некоторых вариантах осуществления термин "связанные с вектором примеси" относится ко всем типам частиц AAV, отличных от полноценных рекомбинантных частиц AAV. Связанные с вектором примеси включают пустые капсиды AAV (также называемые "пустыми единицами" или "пустыми частицами") и частицы AAV, содержащие полинуклеотидные последовательности, отличные от желаемого генома вектора (также называемые "заключенными в капсид AAV примесями нуклеиновой кислоты" или "заключенными в капсид AAV примесями ДНК").
В некоторых вариантах осуществления под "рекомбинантным вирусом" подразумевают вирус, который был генетически изменен, например, посредством добавления или вставки конструкции на основе гетерологичной нуклеиновой кислоты в частицу. "Рекомбинантный" можно сокращать как "r", например, rAAV может относиться к рекомбинантному AAV. Используемый в данном документе термин "AAV" предназначен охватывать "рекомбинантный AAV" или "rAAV."
В некоторых вариантах осуществления под "вирионом AAV" подразумевают полную вирусную частицу, такую как вирусная частица AAV дикого типа (wt) (содержащая геном AAV в виде линейной однонитевой нуклеиновой кислоты, ассоциированный с капсидной белковой оболочкой AAV). При этом молекулы однонитевой нуклеиновой кислоты AAV любой комплементарной нити, например, "смысловой" или "антисмысловой" нитей, могут быть упакованы в любой вирион AAV, и обе нити являются одинаково инфекционными.
В некоторых вариантах осуществления термины "вирион рекомбинантного AAV", "вирион rAAV", "частица вектора на основе AAV", "полные капсиды" и "полные частицы" определены в данном документе как инфекционный дефектный по репликации вирус, содержащий белковую оболочку AAV, заключающую гетерологичную нуклеотидную последовательность, представляющую интерес, которая фланкирована с обеих сторон ITR AAV. Вирион rAAV продуцируется в подходящей клетке-хозяине, в которую введены последовательности, определяющие вектор на основе AAV, хелперные функциональные элементы AAV и добавочные функциональные элементы. Таким образом, клетка-хозяин становится способной к кодированию полипептидов AAV, которые обеспечивают упаковку вектора на основе AAV (содержащего рекомбинантную нуклеотидную последовательность, представляющую интерес) в частицы инфекционного рекомбинантного вириона для последующей доставки гена.
В некоторых вариантах осуществления термины "пустой капсид" и "пустая частица" относятся к вириону AAV, который содержит белковую оболочку AAV, но в котором отсутствует полностью или частично полинуклеотидная конструкция, содержащая гетерологичную нуклеотидную последовательность, представляющую интерес, фланкированную с обеих сторон ITR AAV.
Термин "клетка-хозяин" обозначает, например, микроорганизмы, клетки дрожжей, клетки насекомых и клетки млекопитающих, которые могут быть или были использованы в качестве реципиентов хелперной конструкции AAV, векторной плазмиды на основе AAV, вектора с добавочными функциональными элементами или другой переносимой ДНК. Термин включает потомство исходной клетки, которая была трансфицирована. Таким образом, используемая в данном документе "клетка-хозяин" в общем относится к клетке, которая была трансфицирована последовательностью экзогенной ДНК. Известно, что потомство одной родительской клетки необязательно может быть полностью идентичным по морфологии или по геномному или полному комплементу ДНК исходному родителю вследствие естественной, случайной или преднамеренной мутации.
В другом варианте осуществления термин "хелперные функциональные элементы AAV" относится к полученным из AAV кодирующим последовательностям, которые могут экспрессироваться с получением генных продуктов AAV, которые, в свою очередь, функционируют в транс-положении с обеспечением продуктивной репликации AAV. Таким образом, хелперные функциональные элементы AAV включают обе из основных открытых рамок считывания (ORF) AAV, rep и cap. Было показано, что продукты экспрессии Rep обладают множеством функций, включающих, среди прочих, распознавание, связывание и никирование точки начала репликации ДНК AAV; ДНК-геликазную активность и модуляцию транскрипции из промоторов AAV (или других гетерологичных). Продукты экспрессии Cap обеспечивают необходимые упаковывающие функциональные элементы. Хелперные функциональные элементы AAV используют в данном документе для дополнения функциональных элементов AAV в транс-положении, которые отсутствуют в векторах на основе AAV.
В одном варианте осуществления термин "хелперная конструкция AAV" относится в общем к молекуле нуклеиновой кислоты, которая содержит нуклеотидные последовательности, обеспечивающие функциональные элементы AAV, удаленные из вектора на основе AAV, который подлежит применению для получения трансдуцирующего вектора для доставки нуклеотидной последовательности, представляющей интерес. Хелперные конструкции AAV обычно используются для обеспечения транзиентной экспрессии генов rep и/или cap AAV для дополнения отсутствующих функциональных элементов AAV, которые необходимы для репликации AAV; однако в хелперных конструкциях отсутствуют ITR AAV, и они не могут ни реплицировать, ни упаковать себя. Хелперные конструкция AAV могут быть в форме плазмиды, фага, транспозона, космиды, вируса или вириона. Был описан ряд хелперных конструкций AAV, таких как широко используемые плазмиды pAAV/Ad и plM29+45, которые кодируют продукты экспрессии Rep и Cap. См., например, Samulski et al. (1989) J. Virol. 63:3822-3828 и McCarty et al. (1991) J. Virol. 65:2936-2945. Был описан ряд других векторов, которые кодируют продукты экспрессии Rep и/или Cap. См., например, патенты США №№ 5139941 и 6376237.
В другом варианте осуществления термин "трансфекция" используют для описания поглощения чужеродной ДНК клеткой, и клетка была "трансфицирована", если экзогенная ДНК была введена внутрь клетки через мембрану. Из уровня техники в общем известен ряд методик трансфекции. См., например, Graham et al. (1973) Virology, 52:456, Sambrook et al. (1989) Molecular Cloning, a laboratory manual, Cold Spring Harbor Laboratories, New York, Davis et al. (1986) Basic Methods in Molecular Biology, Elsevier и Chu et al. (1981) Gene 13:197. Такие методики можно применять для введения одного или нескольких фрагментов экзогенной ДНК в подходящие клетки-хозяева.
Используемый в данном документе термин "линия клеток" относится к популяции клеток, способной к постоянному или продолжительному росту и делению in vitro. Из уровня техники дополнительно известно, что спонтанные или индуцированные изменения могут возникнуть в кариотипе во время хранения или переноса таких клональных популяций. Следовательно, клетки, полученные из упомянутой линии клеток, могут не быть совершенно идентичными предковым клеткам или культурам, и упомянутая линия клеток включает такие варианты. В некоторых вариантах осуществления термины "клетки HEK293", "клетки линии 293" или их грамматические эквиваленты используются взаимозаменяемо в данном документе и относятся к линии клеток-хозяев/пакующих клеток, применяемой в способах, раскрытых в данном документе.
В некоторых вариантах осуществления термин "элюент" можно понимать в контексте для обозначения буфера, используемого для элюирования вещества. В некоторых вариантах осуществления термин "элюент" можно понимать в контексте для обозначения элюированного вещества, например, желаемого продукта или вещества, из предыдущей стадии очистки, например, для анализа или дополнительной очистки.
В некоторых вариантах осуществления способы, описанные в данном документе, осуществляют с применением надлежащей практики организации производства (GMP) и в промышленном масштабе. GMP представляет собой регуляторную практику, например, применяемую Федеральным управлением по качеству лекарственных средств (FDA), для обеспечения качества фармацевтических средств. Нормативные требования GMP устанавливают контроль над производственными процессами. Примеры текущих нормативных требований GMP опубликованы FDA. В некоторых вариантах осуществления в способах, описанных в данном документе, применяют процедуры GMP для получения вирусных векторов на основе AAV в промышленном масштабе. На сегодняшний день получение в промышленном масштабе вирусных векторов на основе AAV для генной терапии является сложной задачей из-за проблем с масштабируемостью. Таким образом, в некоторых вариантах осуществления способы, описанные в данном документе, обеспечивают преимущество посредством получения вирусных векторов на основе AAV, например, в адгезивных клетках, в промышленном масштабе и при значениях степени чистоты, достаточных для введения человеку. Термин "промышленный масштаб" относится к способам получения вирусных векторов в клетках в масштабе, превышающем лабораторный масштаб, например, коммерческом масштабе, например, где выход составляет более 5×1015 г. в., или более 8×1015 г. в., или более 1×1016 г. в. на одну производственную партию.
ПРОЦЕСС НАКОПЛЕНИЯ
В некоторых вариантах осуществления процесс накопления используется для получения промежуточного соединения, полученного из рабочего банка клеток, где процесс накопления включает стадии (a) культивирования клеток, например адгезивных клеток, (b) трансфицирования культивируемых клеток, например адгезивных клеток, тремя плазмидами, (c) осуществления сбора увеличенного количества вирусных частиц из клеток после периода культивирования, например, посредством полного лизиса клеток, (d) очистки вирусных частиц с помощью фильтрации для удаления любых интактных клеток или клеточного дебриса, (e) подвергания элюента из стадии (d) фильтрации в тангенциальном потоке и (f) необязательно замораживания полученного промежуточного препарата очищенных вирусных частиц. В некоторых вариантах осуществления промежуточный препарат можно замораживать. В других вариантах осуществления промежуточный препарат не нужно замораживать перед осуществлением процесса выделения и очистки. В некоторых вариантах осуществления AAV, полученный посредством процесса накопления, раскрытого в данном документе, представляет собой AAV, кодирующий shRNA, нацеливающуюся на SOD1, AAV, содержащий полинуклеотид, кодирующий MECP2, или AAV, содержащий полинуклеотид, кодирующий SMN, как описано в данном документе. В некоторых вариантах осуществления процесс накопления проводится согласно GMP и в промышленном масштабе.
1. Трансфекция и культивирование линии клеток
В одном аспекте в данном документе раскрыты геномы rAAV. Геномы rAAV содержат один или несколько ITR AAV, фланкирующих полинуклеотид, кодирующий полипептид (включающий без ограничения полипептид SMN) или кодирующий siRNA, shRNA, антисмысловую и/или miRNA, направленные на подвергнутые мутации белки или регуляторные последовательности их генов. Полинуклеотид функционально связан с последовательностями ДНК, обеспечивающими контроль транскрипции, в частности промоторной ДНК, энхансерной ДНК и сигнальной последовательностью полиаденилирования ДНК, которые функционируют в клетках-мишенях, с образованием генной кассеты. Генная кассета может также включать последовательности интронов для облегчения обработки РНК-транскрипта при экспрессии в клетках млекопитающих.
В некоторых вариантах осуществления геном rAAV (например, rAAV9) кодирует трофический или защитный фактор для лечения нейродегенеративных расстройств, включающих без ограничения болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, наряду с повреждением нервной системы, включающим травму/повреждение спинного мозга и головного мозга, инсульт и виды рака головного мозга. Неограничивающие примеры известных факторов роста нервной системы включают фактор роста нервов (NGF), нейротрофический фактор головного мозга (BDNF), нейротропин-3 (NT-3), нейротропин-4/5 (NT-4/5), нейротропин-6 (NT-6), цилиарный нейротрофический фактор (CNTF), нейротрофический фактор глиальной линии клеток (GDNF), фактор роста семейства фибробластов (например, FGF 1-15), фактор, ингибирующий лейкемию (LIF), некоторых членов семейства инсулин-подобного фактора роста (например, IGF-1), нейротурины, персефин, костные морфогенетические белки (BMP), иммунофилины, семейство трансформирующего фактора роста (TGF) факторов роста, нейрегулины, эпидермальный фактор роста (EGF), тромбоцитарный фактор роста (PDGF), семейство фактора роста сосудистого эндотелия (например, VEGF 165), фоллистатин, Hif1 и другие. Также обычно рассматриваются факторы транскрипции с "цинковыми пальцами", которые контролируют каждый из трофических или защитных факторов, рассматриваемых в данном документе. В дополнительных вариантах осуществления рассматриваются способы модуляции нейроиммунной функции, включающие без ограничения ингибирование микроглиальной и астроглиальной активации посредством, например, ингибирования NFkB или NFkB для нейропротекции (двойное действие NFkB и связанных путей в различных типах клеток) посредством siRNA, shRNA, антисмысловой или miRNA. В еще одних дополнительных вариантах осуществления геном rAAV (например, rAAV9) кодирует ингибитор апоптоза (например, bcl2, bclxL). Применение rAAV (например, rAAV9), кодирующего трофический фактор или белок, модулирующий повреждение спинного мозга, или супрессор ингибитора аксонального роста (например, супрессор Nogo [Oertle et al., The Journal of Neuroscience, 23(13):5393-5406 (2003)], также рассматривается для лечения повреждения спинного мозга.
Для лечения нейродегенеративных расстройств, таких как болезнь Паркинсона, геном rAAV (например, rAAV9) кодирует в различных вариантах осуществления дофа-декарбоксилазу ароматических кислот (AADC), тирозингидроксилазу, GTP-циклогидролазу 1 (gtpch1), ингибиторы апоптоза (например, bcl2, bclxL), нейротрофический фактор глиальной линии клеток (GDNF), аминомасляную кислоту, представляющую собой тормозной нейромедиатор (GABA), или ферменты, вовлеченные в биосинтез дофамина. В дополнительных вариантах осуществления геном rAAV (например, rAAV9) может кодировать, например, модификаторы паркина и/или синуклеина.
Для лечения нейродегенеративных расстройств, таких как болезнь Альцгеймера, в некоторых вариантах осуществления рассматриваются способы повышения продуцирования ацетилхолина. В некоторых вариантах осуществления рассматриваются способы повышения уровня ацетилхолинтрасферазы (ChAT) или подавления активности ацетилхолинэстеразы (AchE).
Геном rAAV (например, rAAV9) кодирует в некоторых вариантах осуществления siРНК, shRNA, антисмысловую и/или miRNA для применения в способах снижения экспрессии мутантного белка Хантингтона (htt) для лечения нейродегенеративного расстройства, такого как болезнь Хантингтона.
Геном rAAV (например, rAAV9) кодирует в различных вариантах осуществления siРНК, shRNA, антисмысловую и/или miRNA для применения в лечении нейродегенеративных расстройств, таких как ALS. Лечение приводит в результате к снижению экспрессии молекулярных маркеров заболевания, таких как TNF-α, оксид азота, пероксинитрит и/или синтаза оксида азота (NOS).
В некоторых вариантах осуществления векторы кодируют короткие шпилечные РНК (shRNA), направленные на подвергнутые мутации белки, такие как супероксиддисмутаза (SOD, например, SOD-1) в случае ALS, или нейротрофические факторы, такие как GDNF или IGF1, в случае ALS или болезни Паркинсона.
В одном варианте осуществления способы и материалы, описанные в данном документе, можно применять для лечения ALS. ALS представляет собой нейродегенеративное заболевание, приводящее к прогрессирующей потере мотонейронов в головном мозге и спинном мозге, при этом симптомы включают потерю способности говорить, питаться, двигаться и в конечном итоге дышать. Заболевание обычно приводит к смерти в течение 3-5 лет после постановки диагноза. Хотя причина 90-95% случаев ALS неизвестна, подгруппа ALS обусловлена генетическими мутациями в гене супероксиддисмутазы 1 (SOD1), где мутация вызывает токсическое доминантное приобретение функции. Исследования на мышах показывают, что нокаут SOD1 не приводит к заболеванию, и, следовательно, считается, что средства терапии, обеспечивающие снижение уровней мутантного SOD1, обеспечивают облегчение симптомов заболевания.
В некоторых вариантах осуществления вектор на основе AAV кодирует shRNA, нацеливающуюся на SOD1, для ALS. Иллюстративная конструкция на основе AAV, например scAAV9, кодирующая shRNA для SOD1, предусматривается в WO2015031392 и US2016272976, содержание которых включено в данный документ во всей их полноте. В некоторых вариантах осуществления конструкция на основе AAV, кодирующая shRNA для SOD1, может быть получена с применением способов, раскрытых в данном документе. В некоторых вариантах осуществления такие конструкции на основе AAV можно применять для лечения ALS. В некоторых вариантах осуществления для SOD1 AAV продемонстрировано менее 10%, например, менее 7%, 5%, 4%, 3%, 2% или 1% пустых капсидов. В некоторых вариантах осуществления для SOD1 AAV продемонстрированы низкие показатели количества остаточного белка клетки-хозяина, ДНК клетки-хозяина, плазмидной ДНК и/или эндотоксина, например, уровни, рассматриваемые в данном документе для получения и очистки векторов на основе AAV. Используемый в данном документе "AVXS-301" представляет собой неограничивающий пример вектора на основе scAAV9, т.е. содержащего полинуклеотид (например, pSOD1sh), кодирующий shRNA, нацеливающуюся на SOD1 человека, модифицированный ITR AAV2, промотор H1 человека и немодифицированный ITR AAV2. Модифицированные и немодифицированные ITR могут находиться в любой ориентации (т.е. 5' или 3') относительно кассеты экспрессии shRNA, нацеливающейся на SOD1 человека.
Используемая в данном документе векторная плазмида "pSOD1sh" содержит полинуклеотид, кодирующий короткую шпилечную РНК (shRNA), нацеливающуюся на экспрессию гена супероксиддисмутазы 1 (SOD1), т.е. кассету shRNA, нацеливающейся на SOD1, где кассета фланкирована последовательностями инвертированного концевого повтора (ITR) аденоассоциированного вируса, например, "слева" и "справа" от полинуклеотида, кодирующего pSOD1sh. В некоторых вариантах осуществления полинуклеотид, кодирующий pSOD1sh, транскрибируется в короткую шпилечную РНК, которая специфически нацеливается на mRNA SOD1 человека. В некоторых вариантах осуществления последовательности ITR, окружающие полинуклеотид, кодирующий pSOD1sh, являются нативными, вариантными или модифицированными последовательностями ITR AAV. В некоторых вариантах осуществления по меньшей мере одна последовательность ITR является нативной, вариантной или модифицированной последовательностью ITR AAV2. В некоторых вариантах осуществления ITR фланкируют полинуклеотид, кодирующий pSOD1sh. В некоторых вариантах осуществления обе из двух последовательностей ITR являются нативными, вариантными или модифицированными последовательностями ITR AAV2. В некоторых вариантах осуществления "левый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "правый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления "правый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "левый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления вектор pSOD1sh дополнительно содержит сегмент промотора РНК H1 человека, например, описанного Myslinksi. Myslinkski et al. "An unusually compact external promoter for RNA polymerase III transcription of the human H1RNA gene." Nucleic Acids Research, 29(12):2502-2509. В некоторых вариантах осуществления вектор pSOD1sh дополнительно содержит уникальную спейсерную последовательность, образованную из сегментов случайных плазмидных остовов, для увеличения размера кассеты экспрессии. В некоторых вариантах осуществления вектор pSOD1sh содержит полинуклеотид, кодирующий shRNA, нацеливающуюся на SOD1 человека, модифицированный ITR AAV2, промотор H1 человека и немодифицированный ITR AAV2.
В одном варианте осуществления способы и материалы, описанные в данном документе, можно применять для лечения нарушений нервно-психического развития, таких как синдром Ретта. Синдром Ретта представляет собой редкое неврологическое расстройство, впервые выявляемое в младенчестве, являющееся результатом мутаций в гене MECP2 на X-хромосоме в 90-95% случаев. Ruthie et al., "Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2". Nature Genetics, 23:185-188. Мальчики, у которых есть только одна копия X-хромосомы, как правило, умирают вскоре после рождения, тогда как девочки, у которых есть две копии X-хромосомы, обычно имеют одну функциональную копию гена. У них симптомы начинают развиваться в возрасте 6-18 месяцев, при этом характерными симптомами являются выкручивающие или сжимающие, хлопающие, трущие, моющие движения рук или движение руки в рот. Заболевание является прогрессирующим со значительной инвалидностью, что может включать поведение, подобное таковому при аутизме, нарушения дыхания, трудности с питанием и глотанием, задержку роста и судороги. Существует 200 известных мутаций гена MECP2, и в зависимости от уровня инактивации X и компенсации дозы, тяжесть заболевания широко варьируется от пациента к пациенту. Исследования на мышах показывают, что мутация MECP2 не обуславливает гибель нейронов, что позволяет предположить, что это не нейродегенеративное расстройство. Guy et al, "Reversal of Neurological Defects in a Mouse Model of Rett Syndrome". Science, 315(5815)"1143-1147.
Для вариантов осуществления, относящихся к синдрому Ретта, геном rAAV (например, rAAV9) может кодировать, например, связывающий метилцитозин белок 2 (MeCP2). Иллюстративная конструкция на основе AAV, например scAAV9, содержащая полинуклеотид, кодирующий MeCP2, предусматривается в патенте США № 9415121, содержание которого включено в данный документ во всей своей полноте. В некоторых вариантах осуществления конструкция на основе AAV, содержащая полинуклеотид, кодирующий MeCP2, может быть получена с применением способов, раскрытых в данном документе. В некоторых вариантах осуществления такие конструкции на основе AAV можно применять для лечения синдрома Ретта. В некоторых вариантах осуществления для MeCP2 AAV продемонстрировано менее 10%, например, менее 7%, 5%, 4%, 3%, 2% или 1% пустых капсидов. В некоторых вариантах осуществления для MeCP2 AAV продемонстрированы низкие показатели количества остаточного белка клетки-хозяина, ДНК клетки-хозяина, плазмидной ДНК и/или эндотоксина, например, уровни, рассматриваемые в данном документе для получения и очистки векторов на основе AAV.
Используемый в данном документе "AVXS-201" представляет собой неограничивающий пример вектора на основе scAAV9, т.е. содержащего полинуклеотид (например, pMECP2), содержащий кассету экспрессии MECP2 cDNA, модифицированный ITR AAV2, промотор Mecp2 мыши, модифицированный интрон SV40, минимальный сигнал полиаденилирования и немодифицированный ITR AAV2. Модифицированные и немодифицированные ITR могут находиться в любой ориентации (т.е. 5' или 3') относительно кассеты экспрессии MECP2 cDNA.
Используемая в данном документе векторная плазмида "pMECP2" содержит полинуклеотид, кодирующий белок MECP2, модифицированный ITR AAV2, промотор Mecp2 мыши, модифицированный интрон SV40, минимальный сигнал полиаденилирования и немодифицированный ITR AAV2. В некоторых вариантах осуществления pMECP2 представляет собой векторную конструкцию, содержащую полинуклеотид, кодирующий белок MECP2, т.е. кассету экспрессии MECP2 cDNA, где кассета фланкирована последовательностями инвертированного концевого повтора (ITR) аденоассоциированного вируса, например, "слева" и "справа" от полинуклеотида, кодирующего ген MECP2. В некоторых вариантах осуществления полинуклеотидом, кодирующим MECP2, является последовательность MECP2 человека, например, встречающаяся в природе последовательность MECP2 человека или ее изоформы, варианты или мутанты. В некоторых вариантах осуществления последовательности ITR являются нативными, вариантными или модифицированными последовательностями ITR AAV. В некоторых вариантах осуществления по меньшей мере одна последовательность ITR является нативной, вариантной или модифицированной последовательностью ITR AAV2. В некоторых вариантах осуществления обе из двух последовательностей ITR являются нативными, вариантными или модифицированными последовательностями ITR AAV2. В некоторых вариантах осуществления "левый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "правый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления "правый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "левый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления вектор pMECP2 дополнительно содержит сегмент, но не весь промотор Mecp2 мыши. В некоторых вариантах осуществления вектор pMECP2 дополнительно содержит интрон вируса обезьян 40 (SV40). В некоторых вариантах осуществления вектор pMECP2 дополнительно содержит минимальный сигнал полиаденилирования, например, определенный Levitt et al., "Definition of an efficient synthetic poly(A) site". Genes & Development, 3:1019-1025.
В некоторых вариантах осуществления в геномах rAAV, раскрытых в данном документе, отсутствует ДНК rep и cap AAV. ДНК AAV в геномах rAAV (например, ITR) может быть из любого серотипа AAV, для которого может быть получен рекомбинантный вирус, включая без ограничения серотипы AAV: AAV-1, AAV-2, AAV-3, AAV-4, AAV-5, AAV-6, AAV-7, AAV-8, AAV-9, AAV-10 и AAV-11. Нуклеотидные последовательности геномов серотипов AAV известны из уровня техники. Например, полный геном AAV-1 представлен в GenBank под № доступа NC_002077; полный геном AAV-2 представлен в GenBank под № доступа NC 001401 и в Srivastava et al., Virol., 45: 555-564 {1983); полный геном AAV-3 представлен в GenBank под № доступа NC_1829; полный геном AAV-4 представлен в GenBank под № доступа NC_001829; геном AAV-5 представлен в GenBank под № доступа AF085716; полный геном AAV-6 представлен в GenBank под № доступа NC_00 1862; по меньшей мере части геномов AAV-7 и AAV-8 представлены в GenBank под №№ доступа. AX753246 и AX753249 соответственно; геном AAV-9 представлен в Gao et al., J. Virol., 78: 6381-6388 (2004); геном AAV-10 представлен в Mol. Ther., 13(1): 67-76 (2006); геном AAV-11 представлен в Virology, 330(2): 375-383 (2004).
Используемая в данном документе векторная плазмида "pSMN" содержит полинуклеотид, кодирующий белок SMN, т.е. кассету экспрессии SMN cDNA, где эта кассета фланкирована последовательностями инвертированного концевого повтора (ITR) аденоассоциированного вируса, например, "слева" и "справа" от полинуклеотида, кодирующего ген SMN. В некоторых вариантах осуществления полинуклеотидом, кодирующим SMN, является последовательность SMN человека, например, встречающаяся в природе последовательность SMN человека или ее изоформы, варианты или мутанты. В некоторых вариантах осуществления последовательности ITR являются нативными, вариантными или модифицированными последовательностями ITR AAV. В некоторых вариантах осуществления по меньшей мере одна последовательность ITR является нативной, вариантной или модифицированной последовательностью ITR AAV2. В некоторых вариантах осуществления обе из двух последовательностей ITR являются нативными, вариантными или модифицированными последовательностями ITR AAV2. В некоторых вариантах осуществления "левый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "правый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления "правый" ITR представляет собой модифицированную последовательность ITR AAV2, которая обеспечивает продуцирование самокомплементарных геномов, и "левый" ITR представляет собой нативную последовательность ITR AAV2. В некоторых вариантах осуществления плазмида pSMN дополнительно содержит энхансер CMV/промотор бета-актина курицы ("CB"). В некоторых вариантах осуществления плазмида pSMN дополнительно содержит интрон вируса обезьян 40 (SV40). В некоторых вариантах осуществления плазмида pSMN дополнительно содержит сигнал терминации полиаденилирования (polyA) гормона роста крупного рогатого скота (BGH). Иллюстративные последовательности, которые можно применять в отношении одного или нескольких компонентов, рассмотренных выше, показаны в таблице 1 ниже. В некоторых вариантах осуществления применяют все из последовательностей, показанных в таблице 1 ниже. В некоторых вариантах осуществления "AVXS-101" представляет собой неограничивающий пример векторной конструкции, в которой применяются все последовательности из таблицы 1, и которая находится в пределах объема термина pSMN.
В некоторых вариантах осуществления вектор pSMN может содержать кассету экспрессии SMN cDNA, модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2. Модифицированные и немодифицированные ITR могут находиться в любой ориентации (т.е. 5' или 3') относительно кассеты экспрессии SMN cDNA.
В некоторых вариантах осуществления, например, во время процессов получения, описанных в данном документе, последовательность векторной конструкции заключается в капсид, например, в вирионы AAV9. В таких вариантах осуществления происходит заключение в капсид нереплицирующегося рекомбинантного AAV9, способного к доставке устойчивого функционального трансгена, например, полностью функционального трансгена SMN человека, трансгена MECP2 или shRNA, нацеливающейся на SOD1. В некоторых вариантах осуществления капсид состоит из 60 вирусных белков (VP1, VP2, VP3), например, в соотношении 1:1:10, полученных в результате альтернативного сплайсинга, таким образом, что VP2 и VP3 являются двумя усеченными формами VP1, все с общими С-концевыми последовательностями. В некоторых вариантах осуществления продукт процесса получения, например лекарственный продукт, может содержать капсид нереплицирующегося рекомбинантного AAV9 для доставки устойчивого полностью функционального трансгена SMN человека, трансгена MECP2 или shRNA, нацеливающейся на SOD1. В некоторых вариантах осуществления капсид состоит из 60 вирусных белков (VP1, VP2, VP3) в соотношении 1:1:10, полученных в результате альтернативного сплайсинга, таким образом, что VP2 и VP3 являются двумя усеченными формами VP1, все с общими С-концевыми последовательностями.
В некоторых вариантах осуществления количество функциональных вирусных векторов определяют по % функционального г. в./мл, измеренному с применением подходящего клеточного анализа in vitro или животной модели in vivo. Например, % функционального AAV SMN можно анализировать посредством относительной активности с применением животной модели SMA, например мыши SMAΔ7, или количественного анализа на основе клеток с применением подходящей линии клеток, например первичных нейральных клеток-предшественниц (NPC), выделенных из коры головного мозга мышей SMAΔ7. % функционального MeCP2 AAV можно анализировать с применением подходящего клеточного анализа in vitro или животной модели in vivo, например мыши с нокаутом Mecp2. % функционального AAV SOD1 можно анализировать с применением подходящего клеточного анализа in vitro или животной модели in vivo, например мыши, мутантной по SOD1.
Последовательность ДНК иллюстративной векторной конструкции, например AVXS-101, описана в таблице 1.
Таблица 1. Перечень компонентов последовательности ДНК векторной конструкции AVXS-101 (все нуклеотидные положения начала и конца указаны по отношению к SEQ ID NO: 1).
В другом аспекте последовательность ДНК векторной конструкции AVXS-101 предусмотрена под SEQ ID NO: 1:
ctgcgcgctc gctcgctcac tgaggccgcc cgggcaaagc ccgggcgtcg 50
ggcgaccttt ggtcgcccgg cctcagtgag cgagcgagcg cgcagagagg 100 gagtggaatt cacgcgtgga tctgaattca attcacgcgt ggtacctctg 150 gtcgttacat aacttacggt aaatggcccg cctggctgac cgcccaacga 200 cccccgccca ttgacgtcaa taatgacgta tgttcccata gtaacgccaa 250 tagggacttt ccattgacgt caatgggtgg agtatttacg gtaaactgcc 300 cacttggcag tacatcaagt gtatcatatg ccaagtacgc cccctattga 350 cgtcaatgac ggtaaatggc ccgcctggca ttatgcccag tacatgacct 400 tatgggactt tcctacttgg cagtacatct actcgaggcc acgttctgct 450 tcactctccc catctccccc ccctccccac ccccaatttt gtatttattt 500 attttttaat tattttgtgc agcgatgggg gcgggggggg ggggggggcg 550 cgcgccaggc ggggcggggc ggggcgaggg gcggggcggg gcgaggcgga 600 gaggtgcggc ggcagccaat cagagcggcg cgctccgaaa gtttcctttt 650 atggcgaggc ggcggcggcg gcggccctat aaaaagcgaa gcgcgcggcg 700 ggcgggagcg ggatcagcca ccgcggtggc ggcctagagt cgacgaggaa 750 ctgaaaaacc agaaagttaa ctggtaagtt tagtcttttt gtcttttatt 800 tcaggtcccg gatccggtgg tggtgcaaat caaagaactg ctcctcagtg 850 gatgttgcct ttacttctag gcctgtacgg aagtgttact tctgctctaa 900 aagctgcgga attgtacccg cggccgatcc accggtccgg aattcccggg 950 atatcgtcga cccacgcgtc cgggccccac gctgcgcacc cgcgggtttg 1000 ctatggcgat gagcagcggc ggcagtggtg gcggcgtccc ggagcaggag 1050 gattccgtgc tgttccggcg cggcacaggc cagagcgatg attctgacat 1100 ttgggatgat acagcactga taaaagcata tgataaagct gtggcttcat 1150 ttaagcatgc tctaaagaat ggtgacattt gtgaaacttc gggtaaacca 1200 aaaaccacac ctaaaagaaa acctgctaag aagaataaaa gccaaaagaa 1250 gaatactgca gcttccttac aacagtggaa agttggggac aaatgttctg 1300 ccatttggtc agaagacggt tgcatttacc cagctaccat tgcttcaatt 1350 gattttaaga gagaaacctg tgttgtggtt tacactggat atggaaatag 1400 agaggagcaa aatctgtccg atctactttc cccaatctgt gaagtagcta 1450 ataatataga acagaatgct caagagaatg aaaatgaaag ccaagtttca 1500 acagatgaaa gtgagaactc caggtctcct ggaaataaat cagataacat 1550 caagcccaaa tctgctccat ggaactcttt tctccctcca ccacccccca 1600 tgccagggcc aagactggga ccaggaaagc caggtctaaa attcaatggc 1650 ccaccaccgc caccgccacc accaccaccc cacttactat catgctggct 1700 gcctccattt ccttctggac caccaataat tcccccacca cctcccatat 1750 gtccagattc tcttgatgat gctgatgctt tgggaagtat gttaatttca 1800 tggtacatga gtggctatca tactggctat tatatgggtt ttagacaaaa 1850 tcaaaaagaa ggaaggtgct cacattcctt aaattaagga gaaatgctgg 1900 catagagcag cactaaatga caccactaaa gaaacgatca gacagatcta 1950 gaaagcttat cgataccgtc gactagagct cgctgatcag cctcgactgt 2000 gccttctagt tgccagccat ctgttgtttg cccctccccc gtgccttcct 2050 tgaccctgga aggtgccact cccactgtcc tttcctaata aaatgaggaa 2100 attgcatcgc attgtctgag taggtgtcat tctattctgg ggggtggggt 2150 ggggcaggac agcaaggggg aggattggga agacaatagc aggcatgctg 2200 gggagagatc gatctgagga acccctagtg atggagttgg ccactccctc 2250 tctgcgcgct cgctcgctca ctgaggccgg gcgaccaaag gtcgcccgac 2300 gcccgggctt tgcccgggcg gcctcagtga gcgagcgagc gcgcagagag 2350 ggagtggcc 2359 (SEQ ID NO: 1).
В некоторых вариантах осуществления аминокислотная последовательность белка SMN, кодируемого плазмидой pSMN, содержит
MAMSSGGSGGGVPEQEDSVLFRRGTGQSDDSDIWDDTALIKAYDKAVASFKHALKNGDICETSGKPKTTPKRKPAKKNKSQKKNTAASLQQWKVGDKCSAIWSEDGCIYPATIASIDFKRETCVVVYTGYGNREEQNLSDLLSPICEVANNIEQNAQENENESQVSTDESENSRSPGNKSDNIKPKSAPWNSFLPPPPPMPGPRLGPGKPGLKFNGPPPPPPPPPPHLLSCWLPPFPSGPPIIPPPPPICPDSLDDADALGSMLISWYMSGYHTGYYMGFRQNQKEGRCSHSLN (SEQ ID NO: 2).
В некоторых вариантах осуществления капсидные белки AAV: VP1, VP2, VP3, получают из одного и того же транскрипта. Они имеют альтернативные сайты начала, но общий карбоксиконец. Ниже специфические аминокислотные последовательности VP1 показаны черным и выделены жирным шрифтом. Аминокислотные последовательности, общие для VP1 и VP2, подчеркнуты и выделены курсивом. Аминокислоты, общие для всех трех капсидных белков выделены жирным шрифтом и курсивом.
1 MAADGYLPDW LEDNLSEGIR EWWALKPGAP QPKANQQHQD NARGLVLPGY KYLGPGNGLD
61 KGEPVNAADA AALEHDKAYD QQLKAGDNPY LKYNHADAEF QERLKEDTSF GGNLGRAVFQ
121 AKKRLLEPLG LVEEAAKTAP GKKRPVEQSP QEPDSSAGIG KSGAQPAKKR LNFGQTGDTE
181 SVPDPQPIGE PPAAPSGVGS LT MASGGGAP VADNNEGADG VGSSSGNWHC DSQWLGDRVI
241 TTSTRTWALP TYNNHLYKQI SNSTSGGSSN DNAYFGYSTP WGYFDFNRFH CHFSPRDWQR
301 LINNNWGFRP KRLNFKLFNI QVKEVTDNNG VKTIANNLTS TVQVFTDSDY QLPYVLGSAH
361 EGCLPPFPAD VFMIPQYGYL TLNDGSQAVG RSSFYCLEYF PSQMLRTGNN FQFSYEFENV
421 PFHSSYAHSQ SLDRLMNPLI DQYLYYLSKT INGSGQNQQT LKFSVAGPSN MAVQGRNYIP
481 GPSYRQQRVS TTVTQNNNSE FAWPGASSWA LNGRNSLMNP GPAMASHKEG EDRFFPLSGS
541 LIFGKQGTGR DNVDADKVMI TNEEEIKTTN PVATESYGQV ATNHQSAQAQ AQTGWVQNQG
601 ILPGMVWQDR DVYLQGPIWA KIPHTDGNFH PSPLMGGFGM KHPPPQILIK NTPVPADPPT
661 AFNKDKLNSF ITQYSTGQVS VEIEWELQKE NSKRWNPEIQ YTSNYYKSNN VEFAVNTEGV
721 YSEPRPIGTR YLTRNL (SEQ ID NO: 3).
В одном варианте осуществления капсидные белки AAV получают из транскрипта, кодирующего аминокислотную последовательность, представленную под SEQ ID NO: 3.
В другом аспекте в данном документе раскрыты ДНК-плазмиды, содержащие геномы rAAV. ДНК-плазмиды переносят в клетки, которые можно инфицировать вирусом-хелпером AAV (например, аденовирусом, аденовирусом с делецией E1 или герпесвирусом), для сборки генома rAAV с капсидными белками AAV9 с получением инфекционных вирусных частиц. Методики получения частиц rAAV, в которых геном AAV, подлежащий упаковке, гены rep и cap и функциональные элементы вируса-хелпера, доставляют в клетку, являются стандартными в данной области техники. В некоторых вариантах осуществления продуцирование rAAV предусматривает следующие компоненты, присутствующие в одной клетке (обозначенной в данном документе как упаковывающая клетка): геном rAAV, гены rep и cap AAV отдельно от генома rAAV (т.е. не в нем) и функциональные элементы вируса-хелпера. Получение псевдотипированного rAAV раскрыто, например, в WO 01/83692, который включен в данный документ посредством ссылки во всей своей полноте. В различных вариантах осуществления капсидные белки AAV можно модифицировать для облегчения доставки рекомбинантного вектора. Модификации капсидных белков общеизвестны из уровня техники. См., например, US 2005/0053922 и US 2009/0202490, раскрытие которых включено в данный документ посредством ссылки во всей своей полноте.
Общие принципы получения rAAV рассматриваются, например, в Carter, 1992, Current Opinions in Biotechnology, 1533-539; и Muzyczka, 1992, CUM Topics in Microbial. and Immunol., 158:97-129). Различные подходы описаны в Ratschin et al., Mol. Cell. Biol. 4:2072 (1984); Hennonat et al., Proc. Natl. Acad. Sci. USA, 81:6466 (1984); Tratschin et al., Mol. Cell. Biol. 5:3251 (1985); McLaughlin et al., J. Virol., 62:1963 (1988); и Lebkowski et al., 1988 Mol. Cell. Biol., 7:349 (1988). Samulski et al. (1989, J. Virol., 63:3822-3828); патенте США № 5173414; WO 95/13365 и соответствующем патенте США № 5658776; WO 95/13392; WO 96/17947; PCT/US98/18600; WO 97/09441 (PCT/US96/14423); WO 97/08298 (PCT/US96/13872); WO 97/21825 (PCT/US96/20777); WO 97/06243 (PCT/FR96/01064); WO 99/11764; Perrin et al. (1995) Vaccine 13:1244-1250; Paul et al. (1993) Human Gene Therapy 4:609-615; Clark et al. (1996) Gene Therapy 3:1124-1132; патенте США № 5786211; патенте США № 5871982 и патенте США № 6258595. Вышеуказанные документы тем самым включены в данный документ посредством ссылки во всей своей полноте, с особым выделением тех разделов документов, которые относятся к получению rAAV.
Иллюстративным способом получения упаковывающей клетки является создание линии клеток, которая стабильно экспрессирует необходимые компоненты для продуцирования частиц AAV. Например, плазмиду (или несколько плазмид), содержащую геном rAAV без генов rep и cap AAV, гены rep и cap AAV отдельно от генома rAAV и селектируемый маркер, такой как ген устойчивости к неомицину, интегрируют в геном клетки. Геномы AAV были введены в бактериальные плазмиды посредством процедур, таких как наращивание GC-хвоста (Samulski et al., 1982, Proc. Natl. Acad. S6. USA, 79:2077-2081), добавление синтетических линкеров, содержащих сайты расщепления рестрикционной эндонуклеазой (Laughlin et al., 1983, Gene, 23:65-73), или посредством прямого лигирования тупых концов (Senapathy & Carter, 1984, J. Biol. Chem., 259:4661-4666). Линию упаковывающих клеток затем инфицируют вирусом-хелпером, таким как аденовирус. Преимуществами данного способа является то, что клетки поддаются отбору и подходят для крупномасштабного получения rAAV. В других примерах подходящих способов для введения геномов rAAV и/или генов rep и cap в упаковывающие клетки используют аденовирус или бакуловирус, а не плазмиды.
Настоящее изобретение, представленное в данном документе, таким образом предусматривает в различных вариантах осуществления упаковывающие клетки, которые продуцируют инфекционный rAAV. Упаковывающие клетки могут являться неадгезивными клетками, культивируемыми в суспензии или адгезивными клетками. В одном варианте осуществления может применяться любая подходящая линия упаковывающих клеток, такая как клетки HeLa, клетки HEK 293 и клетки PerC.6 (родственная 293 линия). В одном варианте осуществления линия клеток представляет собой клетки HEK 293.
Чтобы увеличить выход продукции вирусного вектора, адгезивные клетки можно культивировать и отбирать на основании улучшенной адгезивности в отношении культуральных матрасов. В некоторых вариантах осуществления во время последующих стадий посева в биореакторе улучшается эффективность трансфекции и увеличивается количество клеток. Во время пересева клетки можно отделять от поверхности культуры клеток посредством способов, известных из уровня техники. Например, клетки можно поднимать посредством соскабливания или путем инкубирования в растворе, содержащем протеазы. В иллюстративном варианте осуществления клетки HEK293 можно промывать с помощью PBS и отделять с помощью трипсина в течение ~2 минут при комнатной температуре. Отделение можно останавливать посредством добавления питательных сред, содержащих сыворотку, и скопления клеток можно разделять путем повторного пипетирования суспензии. Суспензию клеток можно затем осаждать и выделенный осадок можно ресуспендировать в подходящих полных питательных средах. Клетки можно затем высевать в новые камеры для культивирования клеток и обеспечивать их прилипание. Клетки, которые не прилипают к поверхности после некоторого периода времени, можно удалять посредством осторожной аспирации со средами для культивирования клеток перед полной заменой сред для культивирования клеток питательными средами. В некоторых вариантах осуществления период времени, в течение которого обеспечивается прилипание клеток, может составлять приблизительно 2 часа, приблизительно 3 часа, приблизительно 4 часа, приблизительно 5 часов, приблизительно 6 часов или приблизительно 7 часов. Когда обеспечено размножение клеток, процесс может быть повторен для увеличения фракции клеток, которые сильно прилипают к культуральным матрасам. В некоторых вариантах осуществления процесс повторяют по меньшей мере 2 раза, по меньшей мере 3 раза, по меньшей мере 4 раза, по меньшей мере 5 раз или любое подходящее число раз. В иллюстративном варианте осуществления клетки HEK293 высевают в матрасы с площадью поверхности 75 см2, обеспечивают прилипание в течение 4 часов в инкубаторе при 37°C перед удалением низкоадгезивных клеток посредством аспирации и заменой сред для культивирования клеток. В иллюстративном варианте осуществления процесс отбора высокоадгезивных клеток повторяют в течение трех пересевов культуры клеток.
В других вариантах осуществления rAAV9 (т.е. инфекционные заключенные в капсид частицы rAAV9) содержит геном rAAV, раскрытый в данном документе. В одном аспекте, геном rAAV представляет собой самокомплементарный геном.
В другом аспекте предусматривается rAAV, такой как rAAV9 под названием "SMN rAAV". В некоторых вариантах осуществления геном SMN rAAV содержит в последовательности первый ITR AAV2, промотор β-актина курицы с энхансером цитомегаловируса, интрон SV40, полинуклеотид, кодирующий SMN, последовательность сигнала полиаденилирования из гормона роста крупного рогатого скота и второй ITR AAV2. В некоторых вариантах осуществления полинуклеотидом, кодирующим SMN, является ген SMN человека, например, приведенный в GenBank под номером доступа MN_000344.2, приведенный в GenBank под номером доступа NM_017411 или полученный из них, или любая другая подходящая изоформа SMN человека. Иллюстративная последовательность SMN содержит последовательность:
1 CCACAAATGT GGGAGGGCGA TAACCACTCG TAGAAAGCGT GAGAAGTTAC TACAAGCGGT
61 CCTCCCGGCC ACCGTACTGT TCCGCTCCCA GAAGCCCCGG GCGGCGGAAG TCGTCACTCT
121 TAAGAAGGGA CGGGGCCCCA CGCTGCGCAC CCGCGGGTTT GCTATGGCGA TGAGCAGCGG
181 CGGCAGTGGT GGCGGCGTCC CGGAGCAGGA GGATTCCGTG CTGTTCCGGC GCGGCACAGG
241 CCAGAGCGAT GATTCTGACA TTTGGGATGA TACAGCACTG ATAAAAGCAT ATGATAAAGC
301 TGTGGCTTCA TTTAAGCATG CTCTAAAGAA TGGTGACATT TGTGAAACTT CGGGTAAACC
361 AAAAACCACA CCTAAAAGAA AACCTGCTAA GAAGAATAAA AGCCAAAAGA AGAATACTGC
421 AGCTTCCTTA CAACAGTGGA AAGTTGGGGA CAAATGTTCT GCCATTTGGT CAGAAGACGG
481 TTGCATTTAC CCAGCTACCA TTGCTTCAAT TGATTTTAAG AGAGAAACCT GTGTTGTGGT
541 TTACACTGGA TATGGAAATA GAGAGGAGCA AAATCTGTCC GATCTACTTT CCCCAATCTG
601 TGAAGTAGCT AATAATATAG AACAGAATGC TCAAGAGAAT GAAAATGAAA GCCAAGTTTC
661 AACAGATGAA AGTGAGAACT CCAGGTCTCC TGGAAATAAA TCAGATAACA TCAAGCCCAA
721 ATCTGCTCCA TGGAACTCTT TTCTCCCTCC ACCACCCCCC ATGCCAGGGC CAAGACTGGG
781 ACCAGGAAAG CCAGGTCTAA AATTCAATGG CCCACCACCG CCACCGCCAC CACCACCACC
841 CCACTTACTA TCATGCTGGC TGCCTCCATT TCCTTCTGGA CCACCAATAA TTCCCCCACC
901 ACCTCCCATA TGTCCAGATT CTCTTGATGA TGCTGATGCT TTGGGAAGTA TGTTAATTTC
961 ATGGTACATG AGTGGCTATC ATACTGGCTA TTATATGGGT TTCAGACAAA ATCAAAAAGA
1021 AGGAAGGTGC TCACATTCCT TAAATTAAGG AGAAATGCTG GCATAGAGCA GCACTAAATG
1081 ACACCACTAA AGAAACGATC AGACAGATCT GGAATGTGAA GCGTTATAGA AGATAACTGG
1141 CCTCATTTCT TCAAAATATC AAGTGTTGGG AAAGAAAAAA GGAAGTGGAA TGGGTAACTC
1201 TTCTTGATTA AAAGTTATGT AATAACCAAA TGCAATGTGA AATATTTTAC TGGACTCTTT
1261 TGAAAAACCA TCTGTAAAAG ACTGGGGTGG GGGTGGGAGG CCAGCACGGT GGTGAGGCAG
1321 TTGAGAAAAT TTGAATGTGG ATTAGATTTT GAATGATATT GGATAATTAT TGGTAATTTT
1381 ATGGCCTGTG AGAAGGGTGT TGTAGTTTAT AAAAGACTGT CTTAATTTGC ATACTTAAGC
1441 ATTTAGGAAT GAAGTGTTAG AGTGTCTTAA AATGTTTCAA ATGGTTTAAC AAAATGTATG
1501 TGAGGCGTAT GTGGCAAAAT GTTACAGAAT CTAACTGGTG GACATGGCTG TTCATTGTAC
1561 TGTTTTTTTC TATCTTCTAT ATGTTTAAAA GTATATAATA AAAATATTTA ATTTTTTTTT
1621 A (SEQ ID NO: 4).
Также рассматриваются консервативные нуклеотидные замены в ДНК SMN (например, изменение гуанина на аденин в положении 625 указанного в GenBank под номером доступа NM_000344.2). В некоторых вариантах осуществления в геноме отсутствует ДНК rep и cap AAV, таким образом, нет ДНК rep или cap AAV между ITR генома. Рассматриваемые полипептиды SMN включают без ограничения полипептид SMN1 человека, приведенный в базе данных белков NCBI под номером NP_000335.1. В вариантах осуществления ДНК SMN содержит полинуклеотид, который кодирует полипептид SMN человека (например, белок SMN человека, идентифицированный в Uniprot под номером доступа Q16637, изоформа 1 (Q16637-1)). Также рассматривается полипептид пластин-3, модифицирующий SMN1 (PLS3) [Oprea et al., Science 320(5875): 524-527 (2008)]. Последовательности, кодирующие другие полипептиды, могут быть заменены на ДНК SMN.
Перед трансфекцией клетки размножают в подходящих культуральных средах, в матрасах или в подходящем биореакторе, или как в первом, так и во втором. В некоторых вариантах осуществления клетки можно размножать в биореакторах, которые обеспечивают непрерывную циркуляцию сред для культивирования клеток. В одном варианте осуществления клетки размножают в биореакторах iCELLis с площадью поверхности 200 м2, 333 м2 или 500 м2. Одна культуральная среда представляет собой DMEM с 5-10% FBS, 4,5 г/л глюкозы, 4 мМ L-глутамина. В некоторых вариантах осуществления адгезивные клетки добавляют к среде в пакете с рециркулирующей средой и подвергают циркуляции через биореактор. В некоторых вариантах осуществления осуществляют непрерывную рециркуляцию среды для культивирования клеток или любой другой среды через биореактор с применением перистальтического насоса. Клетки можно высевать при подходящей плотности в матрасы или биореакторы для культивирования и трансфекции. Плотность посева может зависеть от типа клеток и количества времени до трансфекции. В некоторых вариантах осуществления клетки высевают при приблизительно 8000-16000 клеток/см2. В одном варианте осуществления клетки HEK293 высевают при 8000-12000 клеток/см2.
Подходящие способы трансдукции и повторного введения трансдуцированных клеток субъекту известны из уровня техники. В одном варианте осуществления клетки можно трансдуцировать in vitro посредством объединения rAAV с клетками, например, в подходящих средах и осуществления скрининга в отношении тех клеток, которые несут ДНК, представляющую интерес, с применением традиционных методик, таких как варианты Саузерн-блота и/или ПЦР, или с применением селектируемых маркеров.
В некоторых вариантах осуществления линия упаковывающих клеток трансфицирована тремя плазмидами: плазмидой, кодирующей или содержащей векторную последовательность, подлежащую упаковке в вектор на основе AAV (например, pSMN, трансген pMECP2 или pSOD1sh), pHELP и pAAV2/9. Трансфекцию можно осуществлять с применением любой из методик, известных из уровня техники, включающих без ограничения электропорацию, липофекцию, например, липофектамином, катионными полимерами и катионными липидами. Можно применять любые подходящие среды для трансфекции. В одном варианте осуществления способа трансфекции адгезивные клетки почки эмбриона человека (HEK293) трансфицируют с помощью совместного осаждения трех ДНК-плазмид и полиэтиленимина (PEI). В одном варианте осуществления вектор scAAV9.CB.SMN (самокомплементарный вирусный вектор на основе AAV9, содержащий промотор CB и полинуклеотид, кодирующий SMN) получают с применением трансфекции тремя ДНК-плазмидами адгезивных клеток HEK293 посредством совместного осаждения с PEI в крупномасштабном биореакторе с адгезивными клетками. В одном варианте осуществления питательную среду DMEM, применяемую для размножения клеток, заменяют модифицированной средой DMEM для трансфекции. Данная среда составлена без кальция и L-глутамина. В одном варианте осуществления среда для трансфекции представляет собой DMEM без FBS, без кальция, без L-глутамина и 4,5 г/л глюкозы. В некоторых вариантах осуществления среда для трансфекции без сыворотка (например, без FBS) улучшает эффективность трансфекции. В одном варианте осуществления среда для трансфекции представляет собой OptiMEM (Invitrogen/Thermo Fisher). В одном варианте осуществления три плазмиды (pSMN, pHELP и pAAV2/9) смешивают вместе с PEI в среде для трансфекции и обеспечивают реакцию. В некоторых вариантах осуществления три плазмиды смешивают вместе в молярном соотношении приблизительно 1:1:1. В некоторых вариантах осуществления плазмиды и PEI смешивают в соотношении 1:1 по весу ДНК:PEI. В некоторых вариантах осуществления плазмиды и PEI смешивают в соотношении менее 1:1 по весу ДНК:PEI. В одном варианте осуществления pSMN, pHELP и pAAV2/9 смешивают в молярном соотношении 1:1:1 в среде OptiMEM. В таком варианте осуществления PEI добавляют так, чтобы соотношение ДНК:PEI составляло 1:1 по весу. В некоторых вариантах осуществления реакцию осуществляют в течение 0-60 минут, или 10-45 минут, или 20-30 минут. В одном варианте осуществления реакцию осуществляют в течение 15-30 минут.
В одном варианте осуществления настоящее изобретение предусматривает способ получения вирусного вектора на основе AAV, включающий стадии (i) культивирования адгезивных клеток HEK293 в биореакторе промышленного масштаба, (2) трансфекции адгезивных клеток плазмидами в течение менее 60 минут с обеспечением продуцирования вектора на основе AAV, и необязательно осуществления стадий дополнительной обработки, очистки, составления и заполнения для получения фармацевтического продукта. В одном варианте осуществления данного способа вектор scAAV9.CB.SMN получают с применением трансфекции тремя ДНК-плазмидами с применением совместного осаждения с полиэтилeнимином ("PEI"). В одном варианте осуществления 3 плазмиды, применяемые для этой трансфекции, представляют собой pSMN, pAAV2/9 и pHELP.
Трансфекцию можно осуществлять посредством приведения в контакт линия упаковывающих клеток с совместным осадком ДНК-PEI. В некоторых вариантах осуществления совместным осадком ДНК-PEI в среде для трансфекции наполняют пакет с рециркулирующей средой. В некоторых вариантах осуществления совместный осадок ДНК-PEI в среде для трансфекции циркулирует в биореакторе и полностью замещает питательную среду. В некоторых вариантах осуществления обеспечивают приведение совместного осадка ДНК-PEI в среде для трансфекции в контакт с адгезивными клетками в биореакторе. В некоторых вариантах осуществления обеспечивают приведение совместного осадка ДНК-PEI в среде для трансфекции в контакт с адгезивными клетками в биореакторе в течение не более двух часов. В некоторых вариантах осуществления трансфекция происходит в течение от одного до двух часов. В некоторых вариантах осуществления трансфекция происходит в течение менее одного часа, например, 10 минут, 20 минут, 30 минут, 40 минут или 50 минут. В некоторых вариантах осуществления трансфекция происходит в течение от одного до двух часов. В некоторых вариантах осуществления трансфекцию останавливают посредством рециркуляции полной питательной среды через биореактор и полного замещения среды для трансфекции.
2. Сбор увеличенного количества вирусных частиц
После подходящего периода размножения клеток после трансфекции в некоторых вариантах осуществления клетки подвергают лизису и собирают вирусные частицы. В некоторых вариантах осуществления клетки отделяют от реактора перед инициацией процесса лизиса клеток. В некоторых вариантах осуществления клетки лизируют in situ. Необязательно вирусные частицы собирают без лизиса. В некоторых вариантах осуществления добавляют эндонуклеазу, например, циркулируемую в биореактор до конечной целевой концентрации. Эндонуклеазой может являться та, которая разрушает как ДНК, так и РНК. В одном варианте осуществления эндонуклеаза представляет собой генетически сконструированную эндонуклеазу из Serratia marcescens (Eaves, G. N. et al. J. Bact. 1963, 85, 273-278; Nestle, M. et al. J. Biol. Chem. 1969, 244, 5219-5225), которую реализуют под названием Benzonase® (EMD Millipore). Фермент получают и очищают из штамма E. coli W3110, мутанта штамма K12, содержащего продуцирующую плазмиду pNUC1 (патент США № 5173418, который включен в данный документ посредством ссылки во всей своей полноте). Структурно белок является димером идентичных субъединиц, длиной 245 аминокислот, массой приблизительно 30 кДа, с двумя важными дисульфидными связями. Benzonase® разрушает все формы ДНК и РНК (однонитевые, двухнитевые, линейные и кольцевые) и является эффективным в широком диапазоне рабочих условий, разрушая нуклеиновые кислоты до олигонуклеотидов с 5'-монофосфатом на конце длиной 2-5 оснований. Benzonase® получают согласно текущей надлежащей практике организации производства (cGMP) и, таким образом, она может применяться в процессах промышленного масштаба для очистки белков и/или вирусных частиц. Другие эндонуклеазы, которые получают в условиях согласно cGMP можно подобным образом применять в способах очистки, раскрытых в настоящей заявке. В одном варианте осуществления бензоназу добавляют в биореактор до конечной концентрации от 50 до 200 МЕ/мл, например, 75-150 МЕ/мл, например, приблизительно 100 МЕ/мл. В некоторых вариантах осуществления добавление бензоназы в значительной степени снижает уровень ДНК клетки-хозяина, при этом обеспечивая высокое продуцирование г. в. в биореакторе.
В некоторых вариантах осуществления обеспечивают перемешивание эндонуклеазы перед добавлением буфера для лизиса в реактор. В некоторых вариантах осуществления обеспечивают смешивание раствора для лизиса клеток с адгезивными клетками в течение не более 1 часа, не более 2 часов, не более 3 часов, не более 4 часов или не более 5 часов. В некоторых вариантах осуществления буфер для лизиса может содержать хлорид магния и/или Tween-20 в подходящем буфере. В иллюстративном варианте осуществления буфер для лизиса представляет собой 500 мМ HEPES, 10% Tween 20, 20 мМ MgCl2, pH 8,0. Солевой раствор сахарозы (SSS), который гасит реакцию с бензоназой, можно добавлять для остановки реакции лизиса. В некоторых вариантах осуществления SSS добавляют в пакет для сбора биомассы, содержащий буфер для промывки, и смешивают в течение 15 минут. В некоторых вариантах осуществления биореактор ополаскивают с помощью буфера для промывки биореактора и смывы затем собирают в пакет для накопления собранной биомассы вместе с погашенным раствором для лизиса клеток и лизированным содержимым клеток, что все вместе составляет общую собранную биомассу. В некоторых вариантах осуществления буфер для промывки биореактора может содержать Tris, MgCl2, NaCl, Tween-20 и сахарозу. В иллюстративном варианте осуществления буфер для промывки биореактора содержит 20 мМ Tris, 1 мМ MgCl2, 500 мМ NaCl, 1% Tween-20 вес./об. и 1% сахарозы вес./об. при pH 8,1.
3. Очистка вирусных частиц
После сбора общую собранную биомассу вирусных частиц можно концентрировать и очищать, как правило посредством фильтрации. В одном варианте осуществления вирусные частицы фильтруют посредством глубинной фильтрации с последующей фильтрацией через фильтр, который удаляет макромолекулярные загрязняющие вещества и клеточный дебрис, например, фильтр 0,45 мкм, но который позволяет векторным геномам проходить через него. Можно применять любой подходящий глубинный фильтр.
Как известно в данной области техники, глубинная фильтрация относится к применению пористой фильтрационной среды для осветления растворов, содержащих значительные количества крупных частиц (например, интактных клеток или клеточного дебриса), по сравнению с мембранной фильтрацией, которая будет быстро забиваться в таких условиях. Разнообразные среды для глубинной фильтрации с различными размерами пор являются коммерчески доступными от различных производителей, таких как Millipore, Pall, General Electric и Sartorious.
Целевую скорость потока для глубинной фильтрации можно снизить для поддержания давления на входе фильтра в пределах технической характеристики. После того, как всю общую собранную биомассу отфильтровали, через глубинный фильтр можно в некоторых вариантах осуществления прогонять буфер для диафильтрации, применяемый для последующей первой стадии фильтрации в тангенциальном потоке ("TFF1"). Полученный с помощью глубинного фильтра пул смешивают. Полученный с помощью глубинного фильтра пул можно затем фильтровать через фильтр с размером пор 0,45 мкм для дополнительной очистки материала общей собранной биомассы. Затем через фильтр с размером пор 0,45 мкм прогоняют буфер для TFF1.
4. Фильтрация в тангенциальном потоке
В различных вариантах осуществления фильтрацию в тангенциальном потоке применяют для концентрации общей собранной биомассы и удаления солей и белков, например, с применением фильтрации в тангенциальном потоке. Фильтрация в тангенциальном потоке (TFF) (также называемая фильтрацией в перекрестном потоке, CFF) хорошо известна специалистам в данной области техники, и оборудование и протоколы для ее осуществления в широком диапазоне ситуаций коммерчески доступны от различных производителей, включая без ограничения Pall Corporation, Порт Вашингтон, штат Нью-Йорк, США, и Spectrum Labs, Ранчо Домингез, штат Калифорния, США. В общем, TFF может включать рециркуляцию ретентата через поверхность мембраны. Эта плавная подача поперечного потока в определенных вариантах осуществления может свести к минимуму загрязнение мембраны, поддерживать высокую скорость фильтрации и обеспечивать высокое извлечение продукта. В одном варианте осуществления стадию TFF можно осуществлять посредством системы плоских листов, приведенной в качестве примера в данном документе. Системы плоских листов можно применять в крупномасштабном производстве, где такие системы снабжены средствами (например, каналом для свободного потока) для предотвращения влияния избыточных усилий сдвига на вирусные частицы. В качестве альтернативы стадию TFF можно осуществлять посредством системы полых волокон, приведенной в качестве примера в данном документе. В одном варианте осуществления порог отсечения по молекулярной массе (MWCO) системы TFF составляет 200-400 кДа, например, приблизительно 300 кДа.
В одном варианте осуществления стадию TFF1 осуществляют с применением кассеты с мембранами из регенерированной целлюлозы с порогом отсечения, представляющим собой MW 300 кДа. Кассету промывают и дезинфицируют раствором NaOH и уравновешивают буфером для TFF1. В одном варианте осуществления буфер для TFF1 содержит 20 мМ Tris, 1 мМ MgCl2, 500 мМ NaCl, 1% сахарозы, pH 8,1.
В некоторых вариантах осуществления фаза концентрации стадии TFF1 выбрана для уменьшения объема осветленной собранной биомассы примерно в 10 раз. После достижения целевого объема ретентата можно начинать процессы диафильтрации. Ретентат в некоторых вариантах осуществления можно подвергать диафильтрации с помощью приблизительно 6 диаобъемов буфера для TFF1. В некоторых вариантах осуществления ретентат подвергают диафильтрации с помощью приблизительно 5-20, или 10-15, или 12 диаобъемов буфера для TFF1. После достижения 6 диаобъемов общего потока пермеата ретентат можно снова концентрировать и собирать. Полоскания, например, два последовательных полоскания мембраны можно проводить для повышения извлечения продукта из промежуточного лекарственного вещества.
5. Промежуточный продукт
В некоторых вариантах осуществления промежуточное лекарственное вещество можно затем замораживать на сухом льду или в морозильной камере и затем переносить на хранение при <-60°C. В других вариантах осуществления промежуточный продукт не нужно замораживать перед процессом выделения и очистки.
В некоторых вариантах осуществления несколько серий вещества промежуточного продукта объединяют вместе для дополнительной обработки (например, для очистки посредством процесса выделения и очистки, например, описанного в данном документе). Несколько серий вещества промежуточного продукта можно объединять до замораживания и хранения. В других вариантах осуществления несколько серий вещества промежуточного продукта можно объединять после размораживания замороженных и хранящихся серий.
ПРОЦЕСС ВЫДЕЛЕНИЯ И ОЧИСТКИ
В некоторых вариантах осуществления процесс выделения и очистки применяют для обработки промежуточного продукта (например, объединенного промежуточного продукта) с получением отфильтрованного лекарственного вещества. В некоторых вариантах осуществления стадии процесса выделения и очистки включают (a) подкисление и осветление (например, с применением фильтрации), (b) осуществление катионообменной хроматографии, (c) фильтрацию в тангенциальном потоке ("TFF2"), (d) ультрацентрифугирование с помощью CsCl, (e) сбор вирусного вектора и (f) дополнительную фильтрацию в тангенциальном потоке ("TFF3") для получения отфильтрованного лекарственного вещества, при этом очищенные частицы AAV суспендированы в фармацевтически приемлемом носителе. В некоторых вариантах осуществления процесс выделения и очистки включает следующие стадии получения, следующие после получения промежуточного продукта, полученного в результате TFF1: размораживание и объединения промежуточного продукта, полученного в результате TFF1, подкисление и осветление, осуществление катионообменной хроматографии (CEX), фильтрацию в тангенциальном потоке (TFF2), ультрацентрифугирование с помощью CsCl для разделения полных/пустых капсидов, фильтрацию в тангенциальном потоке (TFF3) для концентрирования/замены буфера, фильтрацию объединенного материала, полученного в результате TFF3, для образования лекарственного вещества, разбавление и фильтрацию лекарственного вещества для получения лекарственного продукта, хранение лекарственного продукта и заполнение лекарственным продуктом флаконов.
В некоторых вариантах осуществления процесс выделения и очистки, раскрытый в данном документе, можно применять для обработки промежуточного продукта, содержащего AAV SMN, AAV MECP2 или AAV, кодирующий shRNA, нацеливающуюся на SOD1, описанные в данном документе.
1. Подкисление и осветление промежуточного продукта
В вариантах осуществления, где промежуточное соединение заморожено, процесс выделения и очистки начинают путем размораживания промежуточного материала, полученного в результате TFF1. Детергент, например, Tween 20, может применять для стимуляции флоккуляции общей массы белков и ДНК клеток-хозяев в кислых условиях рН. pH промежуточного продукта, полученного в результате TFF1, содержащего детергент, затем можно снижать. Флокулянт и осадок, образованный при снижении pH, можно затем удалять посредством фильтрования раствора через глубинный фильтр и фильтр, который удаляет макромолекулярные загрязняющие вещества и клеточный дебрис, например, фильтр с размером пор 0,45 мкм, но который позволяет векторным геномам проходить через него. Можно применять любой подходящий глубинный фильтр.
В одном варианте осуществления Tween 20 медленно добавляют к раствору промежуточного продукта, полученного в результате TFF1, для достижения конечной концентрации 10-20% Tween 20. В некоторых вариантах осуществления целевая композиция после добавления Tween 20 представляет собой раствор 36% Tween 20 в 20 мМ Tris, 1 мМ MgCl2, 500 мМ NaCl, 1% сахарозы вес./об., pH 8,1. В некоторых вариантах осуществления Tween 20 добавляют медленно в течение периода приблизительно 1-6 часов. В некоторых вариантах осуществления Tween 20 добавляют медленно в течение 3-6 часов. В некоторых вариантах осуществления Tween 20 добавляют медленно в течение 4 часов. В некоторых вариантах осуществления обеспечивают инкубацию раствора Tween 20/промежуточного продукта, полученного в результате TFF1, в течение ночи при комнатной температуре. В некотором варианте осуществления обеспечивают инкубацию раствора Tween 20/промежуточного продукта, полученного в результате TFF1, в течение 8-20 часов при комнатной температуре. В иллюстративном вариант осуществления обеспечивают инкубацию раствора Tween 20/промежуточного продукта, полученного в результате TFF1, в течение 12-20 часов при комнатной температуре.
После инкубации pH содержащего Tween 20 раствора промежуточного продукта, полученного в результате TFF1, можно снизить посредством добавления любой подходящей кислоты. В некоторых вариантах осуществления 1 M глицина с pH 2,5 добавляют для достижения целевого значения pH 3,5 ± 0,1. В некоторых вариантах осуществления целевое значение pH составляет pH 3,0-4,0, приблизительно pH 3,3-3,7, приблизительно pH 3,4-3,6 или приблизительно pH 3,5. Как только рН находится в допустимом диапазоне, раствор можно пропустить через фильтр с любым размером пор. В иллюстративном варианте осуществления используют глубинный фильтр (например, Clarisolve POD), наряду с фильтром с размером пор 0,45 мкм (например, фильтр Opticap XL10 Durapore) или PES-фильтром с размером пор 0,8/0,45 мкм.
2. Катионообменная хроматография
В различных вариантах осуществления стадию осуществления катионообменной (CEX) захватной хроматографии применяют, например, для отделения вирусных капсидов от белков клетки-хозяина, ДНК клетки-хозяина, липидов клетки-хозяина, Tween 20 и других примесей, связанных с процессом. Принципы катионообменной хроматографии хорошо известны из уровня техники, но, вкратце, данный способ основан на взаимодействиях заряд-заряд между положительно заряженными частицами, которые подлежат выделению, и используемой отрицательно заряженной смолой. В целом, колонку сначала уравновешивают, пропуская несколько диаобъемов буфера до тех пор, пока pH и проводимость не стабилизируются. Затем загружают образец и колонку промывают с помощью загрузочного буфера. Наконец, элюирующий буфер применяют для элюирования представляющего интерес образца из колонки, и фракции, содержащие образец, собирают. Наличие образца, представляющего интерес, можно обнаружить посредством измерений оптической плотности элюента.
В одном варианте осуществления на стадии CEX применяли хроматографическую колонку CIMmultus S03-8000 Advanced Composite Column (с сульфонильными группами) (с размером пор 2 мкм). В одном варианте осуществления пик элюирования собирают, начиная с резкого повышения OD280. OD280 начинает расти, когда проводимость составляет 80-85 мСм/см. Элюат, полученный в результате CEX, можно собирать в соответствии со стандартной процедурой и его можно собирать в виде двух фракций. В одном варианте осуществления первая фракция начинается с резкого повышения OD280, и ее собирают до 1,5 объемов сбора (CV). В другом варианте осуществления вторая фракция начинается непосредственно после первой фракции, и ее собирают до 1,0 CV. Фракции объединяют и затем нейтрализуют до pH 8,0 ± 0,30. В одном варианте осуществления нейтрализующий буфер содержит 1,0 M Tris с pH 9,1 ± 0,1 при 20°C.
3. Фильтрация в тангенциальном потоке 2
В некоторых вариантах осуществления стадию фильтрации в тангенциальном потоке (TFF2) применяют для концентрирования, удаления примесей белка и замены буфера на подходящий буфер для последующей стадии ультрацентрифугирования с помощью CsCl. Можно применять любую подходящую мембрану для TFF. В одном варианте осуществления на стадии TFF2 используют мембраны из регенерированной целлюлозы с MWCO 300 кДа.
В некоторых вариантах осуществления фаза концентрирования на данной стадии разработана для уменьшения объема элюата, полученного в результате CEX. В одном варианте осуществления ретентат разбавляют в 2 раза буфером для диафильтрации, и ретентат концентрируют до его первоначального объема. В одном варианте осуществления буфер для диафильтрации представляет собой буфер на основе NaCl для диафильтрации при TFF2, который содержит 20 мM Tris, 2 мM MgCl2, 150 мM NaCl, 0,2% полоксамера 188, 1% сахарозы, pH 8,1 ± 0,1 при 20°C. В таких вариантах осуществления этот процесс может повторяться до тех пор, пока не будет завершена диафильтрация новым буфером. В одном варианте осуществления ретентат разбавляют в 2 раза буфером для диафильтрации, содержащим CsCl, и ретентат концентрируют до его первоначального объема. В одном варианте осуществления буфер для диафильтрации, содержащий CsCl, представляет собой буфер с CsCl для диафильтрации при TFF2, который содержит 20 мМ Tris, 2 мМ MgCl2, 3 M CsCl, 0,2% полоксамера 188, pH 8,1± 0,1 при 20°C. В таких вариантах осуществления этот процесс может повторяться до тех пор, пока не будет завершена диафильтрация новым буфером. Как только диафильтрация с CsCl завершается, ретентат затем концентрируют до заданного объема, который зависит от объема задержки в системе. В некоторых вариантах осуществления проводят ополаскивание, например, два последовательных ополаскивания мембраны для максимального извлечения продукта из системы TFF2.
4. Ультрацентрифугирование с помощью CsCl
В некоторых вариантах осуществления, где AAV применяют для трансдукции гена in vivo, конечный продукт rAAV может содержать минимальное количество примесей и пустых частиц. Два способа очистки вектора на основе AAV представляют собой ультрацентрифугирование с применением либо градиента йодиксанола, либо градиента CsCl. В одном исследовании, в котором сравнивали два способа, было продемонстрировано, что йодиксанол обеспечивает выход вирусных векторов на основе AAV с более высокой чистотой вектора, но дает большее количество пустых вирусных капсидов по сравнению с таковым в случае CsCl. Strobel et al. "Comparative Analysis of Cesium Chloride- and Iodixanol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applications." Human Gene Therapy Methods, 26(4):147-157. Несмотря на то, что применение CsCl приводит к меньшим количествам пустых вирусных капсидов, CsCl может быть токсичным для клеток и может потребоваться несколько стадий очистки для удаления остаточного CsCl, что приведет к процессу более длительному по времени (~3,5 дня) по сравнению с более быстрыми способами, такими как с йодиксанолом (~1 день). В другом исследовании было показано, что применение большого количества стадий очистки для удаления остаточного CsCl часто приводит к существенной потере rAAV, приводя к низким показателям выхода и скорости извлечения, что часто сводит на нет другие преимущества способа. Hermens et al. "Purification of Recombinant Adeno-Associated Virus by Iodixanol Gradient Ultracentrifugation Allows Rapid and Reproducible Preparation of Vector Stocks for Gene Transfer in the Nervous System." Human Gene Therapy, 10:1885-1891. Кроме того, в то время как эти два способа хорошо работают в лаборатории для получения образцов для доклинических испытаний, они не являются масштабируемыми и, таким образом, не подходят для крупномасштабного получения коммерческих продуктов. См., например, Tomono et al., "Ultracentrifugation-free chromatography-mediated large-scale purification of recombinant adeno-associated virus serotype 1 (rAAV1)." Molecular Therapy - Methods & Clinical Development, 3:15058 ("способы очистки с применением ультрацентрифугирования в градиенте плотности хлорида цезия (CsCl) или йодиксанола не подходят для крупномасштабного производства").
В некоторых вариантах осуществления стадию ультрацентрифугирования применяют, например, для отделения пустых капсидов от полных капсидов. Неожиданно оказалось, что способ ультрацентрифугирования с помощью CsCl, раскрытый в данном документе, является масштабируемым и подходит для крупномасштабного получения очищенных векторов на основе AAV. Ультрацентрифугирование может проводиться посредством аналитического ультрацентрифугирования и может включать применение буферов с градиентами. Примеры буферов с градиентами включают без ограничения таковые с CsCl, сахарозой, йодиксанолом и другие известные из уровня техники. Центрифугирование можно проводить в любой центрифуге, способной достигать требуемой динамической нагрузки, например, автоматическую систему ультрацентрифугирования Optima XPN 100 или эквивалентную систему, оснащенную ротором типа 50,2 Ti или эквивалентным ротором. После ультрацентрифугирования пустые капсиды и полные капсиды разделяются на отдельные полосы внутри пробирки, и их можно извлечь путем забора материала из конкретной полосы. В некоторых вариантах осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют при 241600-302000 g (~40000-50000 об/мин в роторе типа 50,2 Ti). В некоторых вариантах осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют в течение ночи. В некоторых вариантах осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют в течение 16-24 часов. В некоторых вариантах осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют в течение 20-24 часов. В некоторых вариантах осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют при 15-25°C. В одном варианте осуществления очищенный с применением TFF2 отфильтрованный материал центрифугируют при 302000 g (50000 об/мин в роторе типа 50,2 Ti) в течение 17 часов при 20°C. В некоторых вариантах осуществления буфер для центрифугирования с помощью CsCl может иметь в составе один или несколько из следующих ингредиентов, включающих (a) CsCl, дополнительно включающих один или несколько из (b) MgCl2, (c) полоксамера 188 и (d) Tris. В некоторых вариантах осуществления буфер для CsCl может включать все из (a), (b), (c) и (d). В некоторых вариантах осуществления буфер для CsCl характеризуется pH 7,5-8,5 или pH 7,9-8,2. В одном варианте осуществления подходящий буфер для центрифугирования с помощью CsCl представляет собой 20 мM Tris, 2 мM MgCl2, 3 M CsCl, 0,2% полоксамер 188, pH 8,1 ± 0,10. После завершения стадии центрифугирования, пробирки можно извлекать из ультрацентрифуги. В некоторых вариант осуществления полоса, занимающая самое верхнее положение, полоса A, содержит пустые капсиды. В некоторых вариантах осуществления следующие по высоте полосы, полосы B, C и D, содержат парные полосы с полными капсидами. В некоторых вариантах осуществления вирусные векторы на основе AAV собирают с применением шприца. В одном варианте осуществления полосы B, C и D удаляют иглой размером 18G, прикрепленной к шприцу объемом 30 мл, вставленной непосредственно под полосой D до середины пробирки. В других вариантах осуществления полосы можно анализировать на наличие полных или пустых капсидов с применением методик, известных из уровня техники, и/или так, как описано в данном документе, и собирать полосы, содержащие полные капсиды.
Соотношение пустых и не пустых вирусных капсидов можно измерять с помощью стандартных лабораторных методик. В некоторых вариантах осуществления измерение выполняют посредством измерений оптической плотности. В некоторых вариантах осуществления измерение выполняют посредством измерений оптической плотности в УФ-области. В некоторых вариантах осуществления общее количество капсидных белков и общее количество ДНК можно определить из измерений оптической плотности в УФ-области. В некоторых вариантах осуществления измерение выполняют посредством измерения оптического коэффициента преломления. В некоторых других вариантах осуществление измерение выполняют с помощью аналитического ультрацентрифугирования.
В одном варианте осуществления вирусный вектор на основе AAV, собранный после ультрацентрифугирования, содержит менее 8% пустых капсидов, менее 7% пустых капсидов, менее 5%, менее 3% или менее 1%. В одном варианте осуществления вирусный вектор на основе AAV, собранный после ультрацентрифугирования, содержит 1-10% пустых капсидов. В одном варианте осуществления вирусный вектор на основе AAV собранный после ультрацентрифугирования, содержит 2-8% пустых капсидов. В одном варианте осуществления количество пустых капсидов ниже предела обнаружения. В другом варианте осуществления процентное содержание пустых капсидов определяют в виде процентного содержания от общего количества капсидов.
5. Фильтрация в тангенциальном потоке 3 для получения отфильтрованного лекарственного вещества
В некоторых вариантах осуществления стадию фильтрации в тангенциальном потоке (TFF3) применяют для удаления CsCl и концентрирования полных капсидов с вектором. Фильтрацию в тангенциальном потоке можно проводить с применением подходящей мембраны. В одном варианте осуществления применяют мембраны из регенерированной целлюлозы с MWCO 300 кДа. Капсиды с вектором могут удерживаться мембранами. Фаза концентрирования процесса TFF3 может быть разработана для уменьшения концентрации остаточного CsCl и объема пула, получаемого в результате ультрацентрифугирования. В некоторых вариантах осуществления после достижения целевого объема ретентата начинают диафильтрацию. Ретентат подвергают диафильтрации с помощью не более 10 диаобъемов подходящего буфера для TFF3. В одном варианте осуществления подходящий буфер для TFF3 может содержать один или несколько из следующих компонентов, включающих (a) Tris, (b) MgCl2, (c) NaCl или (d) полоксамер 188. В одном варианте осуществления подходящий буфер для TFF3 может содержать все из перечисленного: (a), (b), (c) и (d). В одном варианте осуществления подходящий буфер для TFF3 характеризуется pH 7,5-8,5, pH 7,7-8,3 или pH 8,0. В одном варианте осуществления подходящий буфер для TFF3 содержит 20 мM Tris, 1 мМ MgCl2, 200 мМ NaCl, 0,001% полоксамера 188, имеет pH 8,0 ± 0,1 при 20°C. В другом варианте осуществления подходящий буфер для TFF3 содержит 20 мM Tris, 1 мM MgCl2, 200 мM NaCl, 0,005% полоксамера 188, имеет pH 8,0 ± 0,1 при 20°C. В одном варианте осуществления концентрированный ретентат подвергают фильтрации с применением фильтра, представляющего собой cтерилизующий фильтр Pall Supor® EKV (Mini Kleenpak) с размером пор 0,2 мкм, для получения отфильтрованного лекарственного вещества. В некоторых вариантах осуществления способы, описанные в данном документе, дают выход более 5×1015 г. в., или более 8×1015 г. в., или более 1×1016 г. в. rAAV на производственную партию.
Фармацевтические композиции
Вирусные (например, AAV) частицы, очищенные в соответствии со способами, раскрытыми в данном документе, можно производить с высоким выходом с чистотой, достаточной для того, чтобы вводить их субъекту-человеку. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 1-8×1013 геномов вирусного вектора/мл (г. в./мл) или приблизительно 1,7-2,3×1013 г. в./мл. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 1,9-2,1×1013 г. в./мл. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 2,0×1013 г. в./мл.
В некоторых вариантах осуществления в ходе процесса получения вирусного вектора могут образовываться пустые вирусные капсиды, которые не содержат материала нуклеиновой кислоты. Фармацевтические композиции, содержащие малые количества пустых вирусных капсидов, могут являться преимущественными, поскольку они позволяют избежать воздействия на пациентов, например младенцев с незрелой иммунной системой, антигенного материала (пустые капсиды, белок клетки-хозяина, ДНК клетки-хозяина) необязательно без обеспечения терапевтического эффекта. В некоторых вариантах осуществления такие фармацевтические композиции могут уменьшать возможные инфузионные реакции или полиспецифические иммунные реакции и могут улучшать терапевтическую эффективность. По сравнению с полными вирусными капсидами, содержащими материал генома, пустые капсиды имеют другие показатели плотности, что позволяет разделять два вида с помощью центрифугирования в градиенте или других способов, известных из уровня техники. В некоторых вариантах осуществления пустые капсиды разделяют с помощью ультрацентрифугирования. В некоторых вариантах осуществления пустые капсиды разделяют с помощью ультрацентрифугирования в градиенте CsCl. В других вариантах осуществления пустые капсиды разделяют с помощью ультрацентрифугирования в градиенте йодиксанола. В некоторых вариантах осуществления пустые капсиды разделяют с помощью ультрацентрифугирования в градиенте сахарозы.
Соотношение пустых и не пустых вирусных капсидов можно измерять с помощью стандартных лабораторных методик. В некоторых вариантах осуществления соотношение измеряют посредством измерений оптической плотности. В некоторых вариантах осуществления соотношение измеряют посредством измерений оптической плотности в УФ-области. В некоторых вариантах осуществления общее количество капсидных белков и общее количество ДНК можно определять с помощью измерений оптической плотности в УФ-области. В некоторых вариантах осуществления значение определяют посредством измерения оптического коэффициента преломления. В некоторых вариантах осуществления значение определяют с помощью аналитического ультрацентрифугирования.
Высокие уровни пустых капсидов могут представлять угрозу для эффективности средств для лечения на основе вирусного вектора. В одном варианте осуществления фармацевтическая композиция содержит менее 10% пустых капсидов, менее 8% пустых капсидов, менее 7%, менее приблизительно 5%, менее 3%, менее 1% пустых капсидов. В другом варианте осуществления фармацевтическая композиция содержит 1-10% пустых капсидов. В другом варианте осуществления фармацевтический композиция содержит 2-8% пустых капсидов. В другом варианте осуществления фармацевтическая композиция содержит 6% пустых капсидов или меньше, 5% пустых капсидов, 4% пустых капсидов, 3% пустых капсидов, 2% пустых капсидов или меньше. В одном варианте осуществления количество пустых капсидов ниже предела обнаружения. В другом варианте осуществления процентное содержание пустых капсидов определяют в виде процентного содержания от общего количества капсидов, например, с применением AUC. В некоторых вариантах осуществления такой низкий показатель процентного содержания пустых капсидов повышает эффективность лечения и/или уменьшает побочные явления (например, воспалительные реакции, поражение печени) после введения пациенту, например, по сравнению с композициями, у которых более высокие показатели процентного содержания пустых капсидов. В некоторых вариантах осуществления способы получения вирусных векторов, раскрытые в данном документе, обеспечивают такие улучшенные показатели процентного содержания пустых капсидов по сравнению с уровнями в предшествующих способах, например, в тех, где не используются адгезивные клетки и/или способы очистки, описанные в данном документе.
В ходе процесса получения вирусного вектора остаточный белок из адгезивных клеток (например, клеток HEK293), применяемых для получения вирусных векторов, может отделяться не полностью. Остаточные белки клетки-хозяина обладают потенциалом вызывать иммунную реакцию. Количество остатков клетки-хозяина можно измерять с помощью любых стандартных лабораторных методик, с помощью которых можно различить вирусные капсидные белки и остаточные белки клетки-хозяина. В некоторых вариантах осуществления количество остаточных белков клетки-хозяина можно измерять посредством эксклюзионной или ионообменной хроматографии. В некоторых вариантах осуществления измерение можно проводить с помощью вестерн-блоттинга с использованием антител, специфичных в отношении родительской клетки. В одном варианте осуществления количество остаточного белка клетки-хозяина можно измерять с помощью иммуноферментного анализа (ELISA). В некоторых вариантах осуществления количество остаточного белка клетки-хозяина можно измерять с помощью коммерческого набора для ELISA. В некоторых вариантах осуществления количество остаточного белка клетки-хозяина можно измерять с помощью набора для ELISA HEK293 HCP от Cygnus Technologies.
В другом варианте осуществления количество остаточного белка клетки-хозяина в указанной фармацевтической композиции составляет 5×106 пг/мл на 1×1013 г. в./мл или меньше, составляет 1,2×106 пг/мл на 1×1013 г. в./мл или меньше, или от 1×105 пг/мл на 1×1013 г. в./мл до 1,2×106 пг/мл на 1×1013 г. в./мл, или составляет 40 нг/мл на 1×1013 г. в./мл или меньше. В одном варианте осуществления фармацевтическая композиция содержит 5 нг или меньше, 4, 3, 2, 1 нг или меньше остаточного белка клетки-хозяина на 1,0×1013 г. в. В одном варианте осуществления фармацевтическая композиция содержит 4 нг или меньше остаточного белка клетки-хозяина на 1,0×1013 г. в.
В ходе процесса получения вирусного вектора остаточная ДНК клетки-хозяина из адгезивных клеток (например, клеток HEK293) или остаточная плазмидная ДНК, трансфекцию которой осуществляли для получения вирусных векторов, может удаляться не полностью. Способ очистки (например, подкисление, осветление, фильтрация в тангенциальном потоке и т. д.) удаляет общую массу остаточной ДНК клетки-хозяина или плазмиды. В одном варианте осуществления измерение количества остаточной ДНК клетки-хозяина или плазмиды проводят с помощью ПЦР. В другом варианте осуществления измерение количества остаточной ДНК клетки-хозяина или плазмиды проводят с помощью количественной ПЦР (qPCR) с праймерами, специфичными в отношении последовательностей клетки-хозяина или плазмиды. В другом варианте осуществления измерение количества остаточной ДНК клетки-хозяина или плазмиды проводят с помощью цифровой капельной ПЦР (ddPCR). В одном варианте осуществления количество ДНК плазмиды определяют с применением анализа qPCR с праймерами, специфичными к участку плазмиды, представляющему собой ген устойчивости к канамицину. В другом варианте осуществления количества остаточной ДНК клетки-хозяина определяют с помощью коммерческих наборов для анализа qPCR, например, resDNASEQ© Human Residual DNA Quantitation Kit от ThermoFisher, Residual DNA Quantification Supermix от Biorad или любого эквивалентного продукта. Уменьшение количества остаточной ДНК клетки-хозяина или плазмиды может улучшить терапевтический эффект, и такую композицию можно очищать и/или отбирать для применения в средствах для лечения, раскрытых в данном документе.
В одном варианте осуществления количество остаточной ДНК клетки-хозяина в указанной фармацевтический композиция составляет 1,7×106 пг/мл на 1×1013 г. в./мл или меньше, от 1×105 пг/мл на 1×1013 г. в./мл до 1,2×106 пг/мл на 1×1013 г. в./мл. В одном варианте осуществления количество остаточной ДНК клетки-хозяина в указанной фармацевтической композиции составляет 3×105 или меньше, 2×105, 1,1×105, 1×105 пг или меньше на 1,0×1013 г. в. В вариантах осуществления количество остаточной ДНК клетки-хозяина в указанной фармацевтической композиции составляет 1,1×105 пг на 1,0×1013 г. в. или меньше.
В другом варианте осуществления количество остаточной плазмидной ДНК в указанной фармацевтической композиции составляет 1,7×106 пг/мл на 1×1013 г. в./мл или меньше, от 1×105 пг/мл на 1×1013 г. в./мл до 1,7×106 пг/мл на 1×1013 г. в./мл. В другом варианте осуществления количество остаточной плазмидной ДНК в указанной фармацевтической композиции составляет 6,8×105 пг на 1,0×1013 г. в. или меньше.
В одном варианте осуществления количество остаточной ДНК клетки-хозяина в фармацевтической композиции составляет 1,1×105 пг на 1,0×1013 г. в. или меньше, и количество остаточной плазмидной ДНК в указанной фармацевтической композиции составляет 6,8×105 пг на 1,0×1013 г. в. или меньше.
В одном варианте осуществления количество остаточной ДНК клетки-хозяина в фармацевтической композиции составляет 1,1×105 пг на 1,0×1013 г. в. или меньше, и количество остаточной плазмидной ДНК в указанной фармацевтической композиции составляет 6,8×105 пг на 1,0×1013 г. в. или меньше, и количество остаточного белка клетки-хозяина в указанной фармацевтической композиции составляет 4 нг на 1,0×1013 г. в. или меньше.
В некоторых вариантах осуществления количество эндотоксина в фармацевтической композиции составляет менее приблизительно 1 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,75 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,5 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,4 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,35 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,3 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,25 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,2 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,15 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,1 МЕ/мл на 1,0×1013 г. в./мл, менее приблизительно 0,05 МЕ/мл на 1,0×1013 г. в./мл или менее приблизительно 0,02 МЕ/мл на 1,0×1013 г. в./мл. Способы определения количества эндотоксина известны из уровня техники, например, реакция с лизатом амебоцитов мечехвоста (LAL). В вариантах осуществления эндотоксин анализируют согласно <85> Фармакопеи США ("USP") (включено в данный документ посредством ссылки во всей своей полноте).
В одном варианте осуществления содержание бычьего сывороточного альбумина (BSA) в фармацевтической композиции составляет менее 0,5 нг на 1,0×1013 г. в., менее 0,3 нг на 1,0×1013 г. в. или менее 0,22 нг на 1,0×1013 г. в. В одном варианте осуществления содержание бензоназы в указанной фармацевтической композиции составляет менее 0,2 нг на 1,0×1013 г. в., менее 0,1 нг на 1,0×1013 г. в. или менее 0,09 нг на 1,0×1013 г. в.
В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, предусматривает одно или несколько из следующего: менее приблизительно 0,09 нг бензоназы на 1,0×1013 г. в., менее приблизительно 30 мкг/г (ppm) цезия, приблизительно 20-80 ppm полоксамера 188, менее приблизительно 0,22 нг BSA на 1,0×1013 г. в., менее приблизительно 6,8×105 пг остаточной плазмидной ДНК на 1,0×1013 г. в., менее приблизительно 1,1×105 пг остаточной hcDNA на 1,0×1013 г. в., менее приблизительно 4 нг rHCP на 1,0×1013 г. в., pH 7,7-8,3, приблизительно 390-430 мОсм/кг, менее приблизительно 600 частиц размером ≥ 25 мкм на контейнер, менее приблизительно 6000 частиц размером ≥ 10 мкм на контейнер, геномный титр, составляющий приблизительно 1,7×1013-2,3×1013 г. в./мл, инфекционный титр, составляющий приблизительно 3,9×108-8,4×1010 МЕ на 1,0×1013 г. в., содержание общего белка, составляющее приблизительно 100-300 мкг на 1,0×1013 г. в., медианное значение выживаемости, составляющее ≥ 24 дней у мышей Δ7SMA при дозе вирусного вектора приблизительно 7,5×1013 г. в./кг, относительная активность, определяемая с помощью анализа на основе клеток in vitro, составляющая 70-30%, и/или менее приблизительно 5% пустых капсидов.
В одном варианте осуществления фармацевтическая композиция, раскрытая в данном документе, предусматривает одно или несколько, например все, из следующего: pH 7,7-8,3 (например, при измерении согласно <791> USP), приблизительно 390-430 мОсм/кг (например, при измерении согласно <785> USP), менее приблизительно 600 частиц размером ≥ 25 мкм на контейнер (например, при измерении согласно <787>USP), менее приблизительно 6000 частиц размером ≥ 10 мкм на контейнер (например, при измерении согласно <787> USP), геномный титр, составляющий приблизительно 1,7×1013-2,3×1013 г. в./мл, инфекционный титр, составляющий приблизительно 3,9×108-8,4×1010 МЕ на 1,0×1013 г. в., общий белок, составляющий приблизительно 100-300 мкг на 1,0×1013 г. в., медианное значение выживаемости ≥ 24 дней у мышей Δ7SMA при дозе вирусного вектора приблизительно 7,5×1013, например, в тесте функциональности in vivo, например, описанном в данном документе, относительная активность, определенная с помощью анализа на основе клеток in vitro, составляющая приблизительно 70-130%, и/или менее приблизительно 5% пустых капсидов. В вариантах осуществления фармацевтическая композиция, раскрытая в данном документе, характеризуется общей чистотой, составляющей 95% или больше (например, при определении с помощью SDS-PAGE). В вариантах осуществления фармацевтическая композиция, раскрытая в данном документе, характеризуется тем, что содержание ни одной из отдельных неименованных родственных примесей не составляет больше 2% (например, при определении с помощью SDS-PAGE). В вариантах осуществления фармацевтическая композиция, раскрытая в данном документе, предусматривает уровни эндотоксина, равные 0,75 МЕ/мл или меньше (например, при измерении согласно <85>USP). В вариантах осуществления фармацевтическая композиция, раскрытая в данном документе, показывает отсутствие роста в тесте на стерильность, например, при измерении согласно <71> USP.
Высокие уровни остаточного белка клетки-хозяина, ДНК клетки-хозяина, плазмидной ДНК и/или эндотоксина могут представлять угрозу для эффективности средств для лечения на основе вирусного вектора. В некоторых вариантах осуществления такие низкие количества остаточного белка клетки-хозяина, ДНК клетки-хозяина, плазмидной ДНК и/или эндотоксина повышают эффективность лечения и/или уменьшают побочные явления (например, воспалительные реакции, поражение печени) после введения пациенту, например, по сравнению с композициями, у которых более высокие показатели количества. В некоторых вариантах осуществления способы получения вирусных векторов, раскрытые в данном документе, обеспечивают такие улучшенные уровни по сравнению с уровнями в предшествующих способах, например, в тех, где не используются адгезивные клетки и/или способы очистки, описанные в данном документе. В некоторых вариантах осуществления способы, описанные в данном документе, также позволяют получать вирусные векторы с уменьшенными показателями процентного содержания пустых капсидов в дополнение к малым количествам остаточного белка клетки-хозяина, ДНК клетки-хозяина, плазмидной ДНК и/или эндотоксина.
В некоторых вариантах осуществления количество остаточного цезия после TFF, например второй TFF, составляет меньше приблизительно 50 мкг/г. В некоторых вариантах осуществления количество остаточного цезия после TFF, например второй TFF, составляет меньше приблизительно 30 мкг/г. В некоторых вариантах осуществления количество остаточного цезия после TFF, например второй TFF, составляет меньше приблизительно 20 мкг/г. В некоторых вариантах осуществления количество остаточного цезия в фармацевтической композиции составляет 30 мкг/г (ppm) или меньше. В некоторых вариантах осуществления количество остаточного CsCl можно измерять с помощью масс-спектрометрии, масс-спектрометрии с индуктивно связанной плазмой (ICP-MS) и/или другого подходящего способа. В некоторых вариантах осуществления количество остаточного цезия после второй TFF меньше предела количественного определения, например, при применении ICP-MS.
В некоторых вариантах осуществления концентрация вирусных векторов на основе AAV, собранных после второй TFF, составляет приблизительно 5×1012 г. в./мл или больше, составляет 1×1013 г. в./мл или больше или составляет приблизительно 3×1013 г. в./мл или больше.
В одном варианте осуществления фармацевтическая композиция характеризуется одним или несколькими из следующего: менее 0,09 нг бензоназы на 1,0×1013 г. в., менее 30 мкг/г (ppm) цезия, приблизительно 20-80 ppm полоксамера 188, менее 0,22 нг BSA на 1,0×1013 г. в., менее 6,8×105 пг остаточной плазмидной ДНК на 1,0×1013 г. в., менее 1,1×105 пг остаточной hcDNA на 1,0×1013 г. в. и менее 4 нг rHCP на 1,0×1013 г. в.
В другом варианте осуществления фармацевтическая композиция сохраняет активность в диапазоне+20%, в диапазоне+15%, в диапазоне+10% или в диапазоне+5% от активности эталонного стандарта. В одном варианте осуществления активность оценивают по сравнению с эталонным стандартом с применением способов, описанных в Foust et al., Nat. Biotechnol., 28(3), pp. 271-274 (2010). Можно применять любой подходящий эталонный стандарт. В одном варианте осуществления фармацевтическая композиция обладает активностью in vivo при испытании на мышах SMAΔ7. В одном варианте осуществления подопытная мышь, которой давали дозу 7,5×1013 г. в./кг, имеет медианное значение выживаемости более 15 дней, более 20 дни, более 22 дней или более 24 дней. В одном варианте осуществления фармацевтическая композиция обладает относительной активностью in vitro при исследовании с помощью анализа на основе клеток, составляющей 50-150%, 60-140% или 70-130% относительно эталонного стандарта и/или соответствующего контроля.
Вирусные частицы, очищенные в соответствии с настоящим изобретением (например, вирусные частицы), можно составлять в соответствии с известными способами с получением фармацевтически применимой композиции. Композиции по настоящему изобретению можно составлять для введения субъекту-млекопитающему, например человеку, с применением методик, известных из уровня техники. В частности, системы доставки можно составить для внутримышечного, интрадермального введения, нанесения на слизистые оболочки, подкожного, внутривенного, интратекального введения, для введения с помощью типа устройств для инъекционных депо или местного применения.
Если система доставки составлена в виде раствора или суспензии, система доставки находится в приемлемом носителе, например водном носителе. Можно применять различные водные носители, например воду, забуференную воду, 0,8% солевой раствор, 0,3% глицин, гиалуроновую кислоту и т.п. Эти композиции можно стерилизовать с помощью традиционных, широко известных методик стерилизации или можно подвергать стерильной фильтрации. Полученные водные растворы могут быть упакованы для использования, как есть или лиофилизированы, причем лиофилизированный препарат объединяют со стерильным раствором перед введением.
Композиции, например фармацевтические композиции, могут содержать фармацевтически приемлемые вспомогательные вещества для обеспечения условий, приближенных к физиологическим, такие как средства для регулирования pH и буферные средства, средства для регулирования тоничности, смачивающие средства и т.п., например, ацетат натрия, лактат натрия, хлорид натрия, хлорид калия, хлорид кальция, сорбитанмонолаурат, триэтаноламинолеат и т.д. В некоторых вариантах осуществления фармацевтическая композиция содержит консервант. В некоторых других вариантах осуществления фармацевтическая композиция не содержит консервант.
Геномный титр вирусных векторов, например, в композициях и составах, раскрытых в данном документе, может быть определен множеством стандартных путей. ПЦР с праймерами, специфичными в отношении вирусного вектора, может обеспечить относительные измерения, но количественную ПЦР (qPCR) можно применять для небольших образцов и абсолютных измерений. Капельная цифровая ПЦР (ddPCR) представляет собой способ для проведения цифровой PCR, которая основана на технологии капель водно-масляной эмульсии. Образец разделяют на десятки тысяч капель, и ПЦР-амплификация молекул матрицы происходит в каждой отдельной капле. Нет необходимости в построении стандартной кривой или в праймерах с высокой эффективностью амплификации, поэтому в случае ddPCR обычно не используют так много вещества образца, как в случае традиционных методик на основе ПЦР. В одном варианте осуществления геномный титр вирусного вектора определяют с применением ПЦР. В другом варианте осуществления геномный титр вирусного вектора определяют с применением qPCR. В другом варианте осуществления геномный титр вирусного вектора определяют с применением ddPC. Способ определения титра вирусного генома с применением ddPCR описан, например, в Lock et al., "Absolute Determination of Single-Stranded and Self-Complementary Adeno-Associated Viral Vector Genome Titers by Droplet Digital PCR," Human Gene Therapy Methods, 25(2):115-125.
В некоторых вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на ген SMN. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на промотор бета-актина курицы. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на энхансер CMV. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на последовательности ITR. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на сигнал полиаденилирования гормона роста крупного рогатого скота.
В некоторых вариантах осуществления фармацевтическая композиция характеризуется pH приблизительно 7,7-8,3 и характеризуется осмоляльностью 390-430 мОсм/кг. В некоторых вариантах осуществления рН измеряют с применением рН-метра. В некоторых вариантах осуществления pH измеряют потенциометрически с применением микроэлектрода с температурной компенсацией в соответствии со стандартами, установленными Фармакопеей США (USP), например согласно <791> (включена посредством ссылки во всей своей полноте). В некоторых вариантах осуществления осмоляльность измеряют с применением понижения точки замерзания в соответствии с USP, например согласно <785> USP (включена посредством ссылки во всей своей полноте). В некоторых вариантах осуществления осмоляльность измеряют с применением осмометра снижения давления пара. В других вариантах осуществления осмоляльность измеряют с применением мембранного осмометра.
В одном варианте осуществления состав для внутривенного введения характеризуется pH от 7,5 до 8,5, геномным титром 2×1013 г. в./мл - 6×1013 г. в./мл и осмоляльностью 384-448 мОсм/кг. В другом варианте осуществления состав для внутривенного введения характеризуется pH от 7,5 до 8,5, геномным титром 1,5×1013 г. в./мл - 3,5×1013г. в./мл и осмоляльностью 384-448 мОсм/кг. В другом варианте осуществления состав для внутривенного введения характеризуется pH от 7,5 до 8,5, геномным титром 1,8×1013 г. в./мл - 2,2×1013г. в./мл и осмоляльностью 384-448 мОсм/кг. В одном варианте осуществления IV-состав содержит приблизительно 0,1-2,0 мМ MgCl2. В одном варианте осуществления IV-состав содержит приблизительно 100-300 мМ NaCl. В одном варианте осуществления IV-состав содержит приблизительно 0,001%-0,01% вес./об. полоксамера 188. В одном варианте осуществления IV-состав представляет собой водный состав в 10-30 мМ Tris-буфера, например, при pH 7,5-8,5.
В одном варианте осуществления IV-состав содержит 1 мМ MgCl2, 200 мМ NaCl, 0,005% вес./об. полоксамера 188 в 20 мМ Tris-буфера при pH 8,0. В вариантах осуществления IV-состав предусматривает геномный титр от приблизительно 1×1013 до 3×1013г. в./мл или от 1,7×1013 до 2,3×1013г. в./мл.
Варианты применения фармацевтических композиций
В других вариантах осуществления в данном документе раскрыты способы доставки полинуклеотида в центральную нервную систему пациента, включающие введение rAAV9 с геномом, содержащим полинуклеотид. В некоторых вариантах осуществления доставка представляет собой интратекальную доставку полинуклеотида в центральную нервную систему пациента, предусматривающую введение rAAV9 с геномом, содержащим полинуклеотид. В некоторых вариантах осуществления пациенту также вводят неионогенное низкоосмолярное контрастное средство. Неионогенное, низкоосмолярное контрастное средство увеличивает уровень трансдукции клеток-мишеней в центральной нервной системе пациента. В некоторых вариантах осуществления геном rAAV9 представляет собой самокомплементарный геном. В других вариантах осуществления геном rAAV9 представляет собой однонитевой геном.
В некоторых вариантах осуществления полинуклеотид доставляется в область головного мозга. Области головного мозга, предполагаемые для доставки, включают без ограничения двигательную область коры головного мозга и ствол головного мозга. В некоторых вариантах осуществления полинуклеотид доставляется в спинной мозг. В некоторых вариантах осуществления полинуклеотид доставляется в нижний мотонейрон. В вариантах осуществления настоящего изобретения rAAV9 применяют для доставки полинуклеотидов в нервные и глиальные клетки. В некоторых вариантах осуществления глиальная клетка представляет собой микроглиоцит, олигодендроглиоцит или астроцит. В некоторых вариантах осуществления rAAV9 применяют для доставки полинуклеотида в шванновскую клетку.
Варианты применения включают, например, лечение заболеваний с поражением нижних мотонейронов, таких как SMA и ALS, а также болезни Помпе, лизосомных болезней накопления, мультиформной глиобластомы и болезни Паркинсона. Лизосомные болезни накопления включают без ограничения дефицит активатора/Gm2-ганглиозидоз, альфа-маннизидоз, аспартилглюкозаминурию, болезнь накопления эфиров холестерина, хроническую недостаточность гексозаминидазы А, цистиноз, болезнь Данона, болезнь Фабри, болезнь Фарбера, фукозидоз, галактосиалидоз, болезнь Гоше (типа I, типа II, типа III), GM1-ганглиозидоз (ранний, поздний детский/ювенильный, взрослых/хронический), I-клеточную болезнь/муколипидоз II, болезнь накопления свободной сиаловой кислоты у детей/ISSD, детскую недостаточность гексозаминидазы А, болезнь Краббе (с ранней манифестацией, с поздним началом), метахроматическую лейкодистрофию, нарушения, представляющие собой мукополисахаридозы (псевдополидистрофия Гурлер/муколипидоз IIIA, MPSI, представляющий собой синдром Гурлер, MPSI представляющий собой синдром Шейе, MPS I, представляющий собой Пфаундлера-Гурлер-Эллиса, MPS II, представляющий собой болезнь Хантера, синдром Санфилиппо типа A/MPS III A, синдром Санфилиппо типа B/MPS III B, Синдром Санфилиппо C/MPS III C, синдром Санфилиппо тип D/MPS III D, синдром Моркио типа A/MPS WA, синдром Моркио типа B/MPS IVB, MPS IX, представляющий собой недостаточность гиалуронидазы, MPS VI, представляющий собой синдром Марото-Лами, MPS VII, представляющий собой синдром Слая, муколипидоз I/сиалидоз, муколипидоз IIIC, муколипидоз тип IV), множественную сульфатазную недостаточность, болезнь Ниманна-Пика (тип A, тип B, тип C), нейрональный цероид-липофусциноз (заболевание CLN6 (атипичное позднее детское, вариант с поздним началом, ранний детский), болезнь Шпильмайера-Фогта-Баттена/детское NCL/CLN3-заболевание, финский вариант поздней инфантильной CLN5, болезнь Бильшовского Янского/поздний детский CLN2/TPP1-заболевание, болезнь Куфса/приобретенный NCL/CLN4-заболевание, северную эпилепсию/вариант CLN8 позднего детского возраста, болезнь Сантавуори-Халтиа/детское CLN1/PPT-заболевание, бета-маннозидоз, болезнь Помпе/ гликогеноз типа II, пикнодизостоз, болезнь Сандхоффа/приобретенный/GM2-ганглиозидоз, болезнь Сандхоффа/GM2-ганглиозидоз-ранний, болезнь Сандхоффа/GM2-ганглиозидоз-детский, болезнь Шиндлера, болезнь Салла/болезнь накопления сиаловой кислоты, болезнь Тея-Сакса/GM2-ганглиозидоз, болезнь Вольмана.
В дополнительных вариантах осуществления применение способов и материалов показано для лечения заболевания нервной системы, такого как синдром Ретта, болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, или для лечения повреждения нервной системы, включая травму/повреждение спинного мозга и головного мозга, инсульт и виды рака мозга. В одном варианте осуществления применение способов и материалов показано для лечения спинальной мышечной атрофии (SMA).
Существует четыре типа SMA, которые обычно классифицируются по возрасту начала и самому высокому уровню двигательной функции, который достигается. Все формы SMA характеризуются аутосомно-рецессивным наследованием и вызваны
мутациями гена выживаемости мотонейронов 1 (SMN1). Люди также несут вторую почти идентичную копию гена SMN, называемую SMN2. Lefebvre et al. "Identification and characterization of a spinal muscular atrophy-determining gene." Cell, 80(1):155-65. Monani et al. "Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor-neuron specific disease." Neuron, 48(6):885-896. Оба гена, SMN1 и SMN2, экспрессируют белок SMN, однако SMN2 содержит трансляционно молчащую мутацию в экзоне 7, что приводит к неэффективному включению экзона 7 в транскрипты SMN2. Таким образом, SMN2 продуцирует как полноразмерный белок SMN, так и усеченную версию SMN, в которой отсутствует экзон 7, причем усеченная версия является преобладающей формой. В результате количество функционального полноразмерного белка, продуцируемого SMN2, значительно меньше (на 70-90%), чем продуцируемого SMN1. Lorson et al. "A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy". PNAS, 96(11) 6307-6311. Monani et al, "A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2". Hum Mol Genet 8(7):1177-1183. Хотя SMN2 не может полностью компенсировать потерю гена SMN1, пациенты с более легкими формами SMA обычно имеют более высокое число копий SMN2. Lefebvre et al., "Correlation between severity and SMN protein level in spinal muscular atrophy". Nat Genet 16(3):265-269. Park et al., "Spinal muscular atrophy: new and emerging insights from model mice". Curr Neurol Neurosci Rep 10(2):108-117. Предостережение заключается в том, что количество копий SMN2 не является единственным фенотипическим модификатором. В частности, вариант c.859G>C в экзоне 7 гена SMN2 указан в качестве положительного модификатора заболевания. Пациенты с данной конкретной мутацией имеют менее тяжелые фенотипы заболевания. Prior et al., "A positive modified of spinal muscular atrophy in the SMN2 gene". Am J Hum Genet 85(3):408-413.
SMA типа I (также называемый младенческой формой заболевания или заболеванием Верднига-Гоффмана) характеризуется тем, что симптомы SMA присутствуют при рождении или появляются до возраста 6 месяцев. При данном типе дети обычно имеют низкий мышечный тонус (гипотония), слабый крик и затрудненное дыхание. Им часто трудно глотать и сосать, и они не достигают этапа развития, когда они могут сидеть без посторонней помощи. Они часто демонстрируют один или несколько симптомов SMA, выбранных из гипотонии, задержки развития двигательных навыков, слабого удержания головы, сутулой осанки и гипермобильности суставов. Как правило, эти дети имеют две копии гена SMN2, по одной на каждую хромосому 5. Больше половины всех новых случаев SMA относятся к SMA типа I.
SMA типа II или промежуточная SMA характеризуется тем, что начало SMA приходится на возраст 7-18 месяцев и до того, как ребенок может стоять или ходить самостоятельно. Дети с SMA типа 2 обычно имеют по крайней мере три гена SMN2. SMA с поздним началом (также известная как SMA типа III и типа IV, легкая форма SMA, SMA с началом во взрослом возрасте и заболевание Кугельберга-Веландера) приводит к различным уровням слабости. Начало SMA типа III приходится на возраст после 18 месяцев, и дети могут стоять и ходить самостоятельно, хотя им может требоваться помощь. SMA типа IV возникает в зрелом возрасте, и люди могут ходить в течение взрослых лет. Люди со SMA типа III или IV обычно имеют от четырех до восьми генов SMN2, из которых можно получить достаточное количество полноразмерного белка SMN.
В одном варианте осуществления термин "лечение" включает стадию введения внутривенно или интратекальным путем эффективной дозы или эффективных многократных доз композиции, содержащей rAAV, как раскрыто в данном документе, животному (включая человека), нуждающемуся в этом. Если дозу вводят до развития расстройства/заболевания, введение является профилактическим. Если дозу вводят после развития расстройства/заболевания, введение является терапевтическим. В вариантах осуществления эффективная доза представляет собой дозу, которая облегчает (либо устраняет, либо уменьшает) по меньшей мере один симптом, ассоциированный с расстройством/болезненным состоянием, которое подвергают лечению, которая замедляет или предотвращает прогрессирование расстройства/болезненного состояния, которая замедляет или предотвращает прогрессирование расстройства/болезненного состояния, которая уменьшает степень заболевания, которая приводит к ремиссии (частичной или полной) заболевания и/или продлевает выживаемость. Примеры болезненных состояний, предполагаемых для лечения, приведены в данном документе.
В одном варианте осуществления композиции, содержащие rAAV по настоящему изобретению, вводят внутривенно пациенту, нуждающемуся в этом, имеющему SMA типа I. В другом варианте осуществления композиции, содержащие rAAV по настоящему изобретению, вводят интратекально пациенту, нуждающемуся в этом, имеющему SMA типов II, III или IV.
В данном документе раскрыт способ лечения SMA типа I у пациента, нуждающегося в этом, путем введения вирусного вектора на основе AAV9 интратекальным или внутривенным путем. В некоторых вариантах осуществления возраст пациента составляет 0-9 месяцев. В некоторых других вариантах осуществления возраст пациента составляет 0-6 месяцев. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, определяют вес пациента. В некоторых вариантах осуществления вес тела пациента составляет менее 8,5 кг. В некоторых вариантах осуществления вес тела пациента составляет более 2,6 кг. В некоторых вариантах осуществления вес тела пациента составляет 2,6-8,5 кг.
В некоторых вариантах осуществления пациент имеет мутации, например нулевую мутацию, в одной копии гена SMN1 (включая любую мутацию, которая приводит кодируемый SMN1 в нефункциональное состояние). В некоторых вариантах осуществления пациент имеет мутации, например нулевую мутацию, в двух копиях гена SMN1. В некоторых вариантах осуществления пациент имеет мутации, например нулевую мутацию, во всех копиях гена SMN1. В некоторых вариантах осуществления пациент имеет делецию в одной копии гена SMN1. В некоторых вариантах осуществления пациент имеет делецию в двух копиях гена SMN1. В некоторых вариантах осуществления пациент имеет биаллельные мутации SMN1, то есть либо делецию, либо замену SMN1 в обоих аллелях хромосомы. В некоторых вариантах осуществления пациент имеет по меньшей мере одну функциональную копию гена SMN2. В некоторых вариантах осуществления пациент имеет по меньшей мере две функциональные копии гена SMN2. В некоторых вариантах осуществления пациент имеет по меньшей мере две функциональные копии гена SMN2. В некоторых вариантах осуществления пациент имеет по меньшей мере три функциональные копии гена SMN2. В некоторых вариантах осуществления пациент имеет по меньшей четыре функциональные копии гена SMN2. В некоторых вариантах осуществления пациент имеет по меньшей пять функциональных копий гена SMN2. В некоторых вариантах осуществления у пациента нет замены c.859G> C в экзоне 7 по меньшей мере одной копии гена SMN2. В некоторых вариантах осуществления генетическая последовательность гена SMN1 или SMN2 может быть определена путем полного секвенирования генома. В других вариантах осуществления генетическая последовательность и количество копий гена SMN1 или SMN2 могут быть определены путем высокопроизводительного секвенирования. В некоторых вариантах осуществления генетическая последовательность и количество копий гена SMN1 или SMN2 могут быть определены с помощью микроматричного анализа. В некоторых вариантах осуществления генетическая последовательность и количество копий гена SMN1 или SMN2 могут быть определены с помощью секвенирования по Сенгеру. В некоторых вариантах осуществления количество копий гена SMN1 или SMN2 может быть определено флуоресцентной гибридизацией in situ (FISH).
В некоторых вариантах осуществления у пациента проявляется один или несколько симптомов SMA. Симптомы SMA могут включать гипотонию, задержку развития двигательных навыков, слабое удержание головы, сутулую осанку и гипермобильность суставов. В некоторых вариантах осуществления слабое удержание головы определяется путем помещения пациента в положение сидя с расположением ног в форме кольца с поддержкой в области плеч (спереди и сзади). Удержание головы оценивали по способности пациента держать голову вертикально. В некоторых вариантах осуществления спонтанное движение наблюдают, когда пациент находится в положении лежа на спине, а моторные навыки оцениваются по способности пациента поднимать свои локти, колени, руки и ноги с поверхности. В некоторых вариантах осуществления силу захвата у пациента измеряют путем помещения пальца в ладонь пациента и подъема пациента до тех пор, пока его плечо не оторвется от поверхности. Гипотонию и силу захвата измеряют тем, насколько быстро/долго пациент сохраняет хватку. В некоторых вариантах осуществления удержание головы оценивают путем помещения головы пациента в максимально возможное повернутое положение и измерения способности пациента поворачивать голову назад к средней линии. В некоторых вариантах осуществления сутулую осанку можно оценивать, помещая пациента в сидячее положение с поддержкой головы и туловища и наблюдая, сгибает ли пациент локти или плечо, чтобы достичь стимула, который находится на уровне плеч на расстоянии вытянутой руки. В некоторых вариантах осуществления сутулую осанку также можно оценивать путем помещения пациента в положение лежа на боку и наблюдая, сгибает ли пациент локти или плечо, чтобы достичь стимула, который находится на уровне плеч на расстоянии вытянутой руки. В некоторых вариантах осуществления двигательные навыки оценивают, наблюдая, сгибают ли пациенты свои бедра или колени, когда их ногу поглаживают, щекочут или сжимают. В некоторых вариантах осуществления сгибание плеча, сгибание локтя, отведение бедра, сгибание шеи, отведение головы, разгибание шеи и/или изгиб позвоночника можно оценивать с помощью известных клинических систем мер, например CHOP INTEND. Другие симптомы SMA можно оценивать в соответствии с известными клиническими системами мер, например CHOP INTEND.
В некоторых вариантах осуществления пациентов подвергают лечению после того, как у них проявляются симптомы SMA типа I (например, один или несколько симптомов), определенные с применением одного из тестов, описанных в данном документе. В некоторых вариантах осуществления пациенты проходят лечение до того, как у них появляются симптомы SMA типа I. В некоторых вариантах осуществления у пациентов диагностируют SMA типа I на основе генетического тестирования, прежде чем она проявится симптоматически.
Комбинированные средства терапии также рассматриваются в данном документе. Комбинация, используемая в данном документе, предусматривает либо одновременное лечение, либо последовательное применение средств для лечения. Комбинации способов могут предусматривать добавление определенных стандартных медицинских средств для лечения (например, рилузол при ALS), а также представляют собой комбинации с новыми средствами терапии. Например, другие средства терапии SMA включают антисмысловые олигонуклеотиды (ASO), которые изменяют связывание с pre-mRNA и изменяют их паттерны сплайсинга. Singh. et al., "A multi-exon-skipping detection assay reveals surprising diversity of splice isoforms of spinal muscular atrophy genes". Plos One, 7(11):e49595. В одном варианте осуществления может применяться нусинерсен (патенты США 8361977 и US 8980853, включенные в данный документ посредством ссылки). Нусинерсен представляет собой одобренный ASO, который нацеливается на интрон 6, экзон 7 или интрон 7 pre-mRNA SMN2, модулируя сплайсинг SMN2 для более эффективного продуцирования полноразмерного белка SMN. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации с мышечным энхансером. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации с нейропротектором. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации с лекарственным средством на основе антисмысловых олигонуклеотидов, нацеливающихся на SMN. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации с нусинерсеном. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации с лекарственным средством, ингибирующим миостатин. В некоторых вариантах осуществления способ лечения включает вирусный вектор на основе AAV9, который вводят в комбинации со стамулумабом.
Хотя доставка индивидууму, нуждающемуся в этом, предусматривается после рождения, также предусматривается внутриутробная доставка плоду.
Рассматриваются способы лечения пациентов со SMA типа I с применением фармацевтических композиций, содержащих вирусный вектор. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 1-8×1013 AAV9 геномов вирусного вектора/мл (г. в./мл). В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 1,7-2,3×1013 г. в./мл. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 1,9-2,1×1013 г. в./мл. В некоторых вариантах осуществления вирусный вектор составлен с концентрацией приблизительно 2,0×1013 г. в./мл.
В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV (например, AAV SMN) вводят пациенту в дозе, составляющей приблизительно 1,0-2,5×1014 г. в./кг. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV вводят пациенту в дозе, составляющей приблизительно 1,1×1014 г. в./кг. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, инфузию вирусного вектора на основе AAV пациенту осуществляют в течение приблизительно 45-70 мин. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, инфузию вирусного вектора на основе AAV пациенту осуществляют в течение приблизительно 60 мин. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, инфузию вирусного вектора на основе AAV пациенту осуществляют с применением инфузионного насоса, перистальтического насоса или любого другого оборудования, известного в данной области техники. В некоторых вариантах осуществления, где вирусный вектор применяют для лечения SMA типа I у пациента, инфузию вирусного вектора на основе AAV пациенту осуществляют с применением шприцевого насоса.
Титры вирусного вектора на основе rAAV, подлежащие введению, будут варьировать в зависимости, например, от конкретного rAAV, способа введения, цели лечения, индивидуума и целевого типа(-ов) клеток, и могут быть определены стандартными способами из уровня техники. Титры rAAV могут находиться в диапазоне от приблизительно 1×106, приблизительно 1×107, приблизительно 1×108, приблизительно 1×109, приблизительно 1×1010, приблизительно 1×1011, приблизительно 1×1012, приблизительно 1×1013, приблизительно 1×1014 или больше устойчивых к ДНКазе частиц (DRP) на мл. Дозы также могут быть выражены в единицах геномов вектора (г. в.). Геномный титр можно определять с применением ddPCR, как описано в настоящей заявке, у Lock et al., или любыми другими способами, известными из уровня техники.
Дозы также могут варьировать в зависимости от времени введения человеку. Такие дозы rAAV могут находиться в диапазоне от приблизительно 1×1011 г. в./кг, приблизительно 1×1012 г. в./кг, приблизительно 1×1013 г. в./кг, приблизительно 1×1014 г. в./кг, приблизительно 1×1015 г. в./кг, приблизительно 1×1016 г. в./кг или больше геномов вектора на килограмм веса тела у взрослых. Для новорожденного дозы rAAV могут находиться в диапазоне от приблизительно 1×1011 г. в./кг, приблизительно 1×1012 г. в./кг, приблизительно 3×1012 г. в./кг, приблизительно 1×1013 г. в./кг, приблизительно 3×1013 г. в./кг, приблизительно 1×1014 г. в./кг, приблизительно 3×1014 г. в./кг, приблизительно 1×1015 г. в./кг, приблизительно 3×1015 г. в./кг, приблизительно 1×1016 г. в./кг, приблизительно 3×1016 г. в./кг или больше геномов вектора на килограмм веса тела.
Дозы также могут варьировать в зависимости от времени введения человеку. Такие дозы rAAV могут находиться в диапазоне от приблизительно 1×1011 г. в./кг/неделя, приблизительно 1×1012 г. в./кг/неделя, приблизительно 1×1013 г. в./кг/неделя, приблизительно 1×1014 г. в./кг/неделя, приблизительно 1×1015 г. в./кг/неделя, приблизительно 1×1016 г. в./кг/неделя или больше геномов вектора на килограмм веса тела у взрослых. Для новорожденного дозы rAAV могут находиться в диапазоне от приблизительно 1×1011 г. в./кг/неделя, приблизительно 1×1012 г. в./кг/неделя, приблизительно 3×1012 г. в./кг/неделя, приблизительно 1×1013 г. в./кг/неделя, приблизительно 3×1013 г. в./кг/неделя, приблизительно 1×1014 г. в./кг/неделя, приблизительно 3×1014 г. в./кг/неделя, приблизительно 1×1015 г. в./кг/неделя, приблизительно 3×1015 г. в./кг/неделя, приблизительно 1×1016 г. в./кг/неделя, приблизительно 3×1016 г. в./кг/неделя или больше геномов вектора на килограмм веса тела в неделю. Дозы rAAV 1×1011 г. в./1,5 кг/неделя, приблизительно 1×1012 г. в./1,5 кг/неделя, приблизительно 1×1013 г. в./1,5 кг/неделя, приблизительно 1×1014 г. в./1,5 кг/неделя, приблизительно 1×1015 г. в./1,5 кг/неделя, приблизительно 1×1016 г. в./1,5 кг/неделя или больше геномов вектора на килограмм веса тела у взрослых. Для новорожденного дозы rAAV могут находиться в диапазоне от приблизительно 1×1011 г. в./1,5 кг/неделя, приблизительно 1×1012 г. в./1,5 кг/неделя, приблизительно 3×1012 г. в./кг/неделя, приблизительно 1×1013 г. в./1,5 кг/неделя, приблизительно 3×1013 г. в./1,5 кг/неделя, приблизительно 1×1014 г. в./1,5 кг/неделя, приблизительно 3×1014 г. в./1,5 кг/неделя, приблизительно 1×1015 г. в./1,5 кг/неделя, приблизительно 3×1015 г. в./1,5 кг/неделя, приблизительно 1×1016 г. в./1,5 кг/неделя, приблизительно 3×1016 г. в./1,5 кг/неделя или больше геномов вектора на 1,5 килограмма веса тела на неделю.
В одном варианте осуществления доза составляет приблизительно 1,1×1014 геномов вектора на кг ( г. в./кг) веса тела пациента. В одном варианте осуществления пациент весом 5 кг получал бы общую дозу от 0,5×1014 до 5,0×1014 геномов вектора. В одном варианте осуществления вирусный вектор вводят в солевом растворе, забуференном с помощью Tris. В одном варианте осуществления вирусный вектор вводят в приблизительно 5-20 мл/кг, приблизительно 10-20 мл/кг или приблизительно 5,5-6,5 мл/кг солевого раствора, забуференного с помощью Tris.
Доза может быть определена множеством стандартных путей. ПЦР с праймерами, специфичными в отношении вирусного вектора, может обеспечить относительные измерения, но qPCR можно применять для меньших образцов и абсолютных измерений. ddPCR представляет собой способ проведения цифровой PCR, которая основана на технологии капель водно-масляной эмульсии. Baker et al., "Digital PCR hits its stride." Nature Methods, 9(6):541-544. Sykes et al., "Quantitation of targets for PCR by use of limiting dilution". Biotechniques, 13(3)444-449. Образец разделяют на десятки тысяч капель, и ПЦР-амплификация молекул матрицы происходит в каждой отдельной капле. Нет необходимости в построении стандартной кривой или в праймерах с высокой эффективностью амплификации, поэтому в случае ddPCR обычно не используют так много вещества образца, как в случае традиционных методик на основе ПЦР. Примеры коммерчески доступных устройств для ddPCR включают без ограничения BioRad QX100 ddPCR и RainDance Raindrop Digital PCR. В одном варианте осуществления дозу определяют с применением ПЦР. В другом варианте осуществления дозу определяют с применением qPCR. В другом варианте осуществления дозу определяют с применением цифровой капельной ПЦР (ddPCR). В некоторых вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на ген SMN. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на промотор бета-актина курицы. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на энхансер CMV. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на последовательности ITR. В других вариантах осуществления с помощью способов на основе ПЦР обнаруживают и определяют количество заключенного в капсид вирусного генома AAV9 с применением специально разработанных праймеров и зондов, нацеливающихся на сигнал полиаденилирования гормона роста крупного рогатого скота.
В одном аспекте дозу вводят в соответствии со следующей таблицей с применением 2,0×1013 г. в/мл в качестве целевой концентрации лекарственного продукта.
В некоторых вариантах осуществления фармацевтическую композицию, содержащую вирусный вектор на основе AAV, вводят с помощью инфузии пациенту в течение приблизительно 20-70 минут, например в течение приблизительно 45-70 минут. В некоторых вариантах осуществления фармацевтическую композицию, содержащую вирусный вектор на основе AAV, вводят с помощью инфузии пациенту в течение приблизительно 60 мин. В некоторых вариантах осуществления фармацевтическую композицию, содержащую вирусный вектор на основе AAV, вводят с помощью инфузии пациенту с применением инфузионного насоса, перистальтического насоса или любого другого оборудования, известного из уровня техники. В некоторых вариантах осуществления фармацевтическую композицию, содержащую вирусный вектор на основе AAV, вводят с помощью инфузии пациенту с применением шприцевого насоса.
Также предусмотрен предварительный скрининг поддающихся лечению пациентов, а также введение средства для лечения пациентам, идентифицированным в соответствии с раскрытыми в данном документе критериями. AAV может вызывать как клеточный, так и гуморальный иммунный ответ. Вследствие этого доля предполагаемых пациентов для основанной на AAV генной терапии несет уже существующие антитела к AAV. Jeune et al., "Pre-existing anti-Adeno-Associated Virus antibodies as a challenge in AAV gene therapy". Hum Gene Ther Methods, 24(2):59-67. Boutin et al., "Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors". Hum Gene Ther, 21:704-712. Поскольку даже очень низкие уровни антител могут препятствовать успешной трансдукции, уже существующие антитела к AAV представляют серьезное препятствие повсеместному применению генной терапии на основе AAV. В некоторых вариантах осуществления уровни титров антител к AAV9 у пациента определяют до введения вирусного вектора на основе AAV. В некоторых вариантах осуществления уровни титров антител к AAV9 у пациента определяют с помощью иммуноферментного анализа связывания ELISA. В некоторых вариантах осуществления до введения средства для лечения у пациента имеются титры антител к AAV9 1:100 или ниже, при определении с помощью иммуноферментного анализа связывания ELISA. В некоторых вариантах осуществления до введения средства для лечения у пациента имеются титры антител к AAV9 1:50 или ниже, при определении с помощью иммуноферментного анализа связывания ELISA. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:100, при определении с помощью иммуноферментного анализа связывания ELISA после лечения, и при этом осуществляют мониторинг в течение 1-8 недель или до тех пор, пока титры не снизятся до значения ниже 1:100. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:100 при определении с помощью иммуноферментного анализа связывания ELISA после лечения, и при этом осуществляют мониторинг в течение 1-8 недель или до тех пор, пока титры не снизятся до значения ниже 1:50.
Одним подходом для преодоления высокого титра антител к AAV является применение иммунодепрессантов. Было показано, что моноклональное антитело к CD20, ритуксимаб, в комбинации с циклоспорином A является эффективным в снижении титров антител к AAV. Mingozzi et al., "Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B". Mol Ther, 20:1410-1416. Другим подходом является применение плазмафереза для истощения нейтрализующих антител к вводимому вектору. Monteilhet et al., "A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8". Mol Ther, 19(11):2084-2091. Во время плазмафереза кровь отбирают у пациента и плазму и клетки крови разделяют с помощью либо центрифугирования, либо фильтрации через полые волокна. Клетки крови затем возвращают пациенту вместе либо с обработанной плазмой, либо с замещающими жидкостями, такими как 4,5% человеческого альбумина в солевом растворе. Широкое применение терапевтического афареза представляет собой удаление нежелательных иммуноглобулинов, но в данном случае, плазмаферез представляет собой привлекательный подход для истощения антител к AAV. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:100, при определении с помощью иммуноферментного анализа связывания ELISA до или после лечения, и его лечат с помощью плазмафереза. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:50, при определении с помощью иммуноферментного анализа связывания ELISA до или после лечения, и его лечат с помощью плазмафереза.
Уже существующие материнские антитела к AAV9 могут быть перенесены пациенту-младенцу посредством грудного молока или плацентарного переноса в утробе. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:100, при определении с помощью иммуноферментного анализа связывания ELISA до или после лечения, и его переводят на искусственное вскармливание. В некоторых вариантах осуществления у пациента имеются титры антител к AAV9 выше 1:50, при определении с помощью иммуноферментного анализа связывания ELISA до или после лечения, и его переводят на искусственное вскармливание.
Мониторинг состояния пациента можно осуществлять до и после введения средства для лечения. У некоторых пациентов, которые получают средства для лечения на основе AAV, может проявляться тромбоцитопения, которая является состоянием, характеризующимся низким значением количества тромбоцитов. Тромбоцитопения может быть выявлена с помощью полного анализа крови с использованием разбавленного образца крови на гемоцитометре. Также тромбоцитопения может быть выявлена путем визуального осмотра микроскопического препарата, полученного из крови пациента (мазок крови или мазок периферической крови), под микроскопом. Нормальные значения количества тромбоцитов у человека находятся в диапазоне от 150000 клеток/мл до приблизительно 450000 клеток/мл.
В некоторых вариантах осуществления до введения у пациента имеются значения количества тромбоцитов выше приблизительно 67000 клеток/мл, или выше приблизительно 100000 клеток/мл, или выше приблизительно 150000 клеток/мл. В некоторых вариантах осуществления до введения у пациента имеются значения количества тромбоцитов ниже приблизительно 150000 клеток/мл, или ниже приблизительно 100000 клеток/мл, или ниже приблизительно 67000 клеток/мл, и при этом осуществляют мониторинг в течение 1-8 недель или до тех пор, пока значения количества тромбоцитов не увеличатся до значения выше приблизительно 67000 клеток/мл, или до значения выше приблизительно 100000 клеток/мл, или до значения выше приблизительно 150000 клеток/мл. В некоторых вариантах осуществления, где значения количества тромбоцитов ниже приблизительно 67000 клеток/мл после введения вирусного вектора, пациента могут лечить с помощью переливания тромбоцитарной массы. В некоторых вариантах осуществления у пациента отсутствует тромбоцитопения до введения вирусного вектора. В некоторых вариантах осуществления пациент имеет тромбоцитопению после введения вирусного вектора, и при этом осуществляют мониторинг в течение приблизительно 1-8 недель или до тех пор, пока у пациента не исчезнет тромбоцитопения. В некоторых вариантах осуществления пациент имеет тромбоцитопению после введения вирусного вектора, и его лечат с помощью переливания тромбоцитарной массы.
Мониторинг состояния пациентов также может включать стандартные анализы крови, с помощью которых измеряют уровни тромбоцитов, электрофореза сывороточных белков, сывороточной гамма-глутамилтрансферазы (GGT), аспартаттрансаминазы (AST) и аланинаминотрансферазы (ALT), общего билирубина, глюкозы, креатинкиназы (СК), креатинина, азота мочевины крови (BUN), электролитов, щелочной фосфатазы и амилазы. Уровни тропонина I являются общим показателем здоровья сердца, а повышенные уровни отражают повреждение сердца или связанные с сердцем состояния. В некоторых вариантах осуществления после введения вирусного вектора осуществляют мониторинг уровней тропонина I. В некоторых вариантах осуществления у пациентов могут быть уровни тропонина I менее приблизительно 0,3, 0,2, 0,15 или 0,1 мкг/мл до введения вирусного вектора. В некоторых вариантах осуществления у пациентов могут быть уровни тропонина I менее приблизительно 0,176 мкг/мл до введения вирусного вектора. В некоторых вариантах осуществления у пациентов могут быть уровни тропонина I выше приблизительно 0,176 мкг/мл после введения вирусного вектора. В некоторых вариантах осуществления пациенты проходят кардиомониторинг после введения вирусного вектора до тех пор, пока уровни тропонина I не будут менее приблизительно 0,176 мкг/мл.
Уровни аспартаттрансаминазы (AST), и аланинаминотрансферазы (ALT), и общего билирубина являются общим показателем функции печени, в то время как по уровню креатинина отслеживают функцию почек. Повышенные уровни AST, ALT или общего билирубина могут указывать на нарушение функции печени. В некоторых вариантах осуществления до введения вирусного вектора у пациента нормальная функция печени. В некоторых вариантах осуществления до введения вирусного вектора у пациента уровни трансаминазы печени составляют менее приблизительно 8-40 МЕ/л. В некоторых вариантах осуществления до введения вирусного вектора у пациента уровни AST или ALT составляют менее приблизительно 8-40 МЕ/л. В некоторых вариантах осуществления до введения вирусного вектора у пациента уровни билирубина составляют 3,0 мг/дл. В некоторых вариантах осуществления до введения вирусного вектора у пациентов уровни креатинина составляют менее 1,8 мг/дл. В некоторых вариантах осуществления до введения вирусного вектора у пациентов уровни гемоглобина (Hgb) составляют от 8 до 18 г/дл. В некоторых вариантах осуществления до введения вирусного вектора у пациента количества лейкоцитов в крови (WBC) составляют менее 20000 на мм3.
Эффективность способа лечения может быть определена с применением разнообразных тестов двигательных навыков до и после лечения. В частности, для оценки двигательных навыков пациентов с SMA типа I был разработан тест детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорожденных (CHOP INTEND). Glanzman et al., "The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability". Neuromuscular Disorders, 20(3):155-161. Тест CHOP INTEND был разработан после оценки 26 младенцев с SMA типа I, средний возраст 11,5 месяца (1,4-37,9 месяца) с помощью теста двигательной активности младенцев (TIMP) и теста силы при SMA детской больницы Филадельфии (CHOP TOSS), недавно разработанной оценки двигательной функции для SMA. Тестирование эффективности лечения не ограничено тестом CHOP INTEND, но также может включать другие тесты двигательных навыков, известные из уровня техники, в том числе без ограничения TIMP, CHOP TOSS, шкалы связанной с развитием двигательной активности Пибоди, оценочный тест неонатального поведения Бразелтона, анализ двигательного развития с использованием указателя расстояния, оценку способности улавливания с помощью интерактивного видео (ACTIVE), шкалу Бейли для оценки развития младенца и измерения потенциалов составных двигательной активности (CMAP).
В некоторых вариантах осуществления исходное тестирование до лечения проводят с применением шкалы CHOP INTEND. В одном варианте осуществления эффективность лечения определяют с применением шкалы CHOP INTEND во время контрольных визитов. В некоторых вариантах осуществления CHOP INTEND включает такие показатели, как удержание головы, реакции выпрямления, движения туловища при сидении с поддержкой, положения лежа на спине и на животе. В некоторых вариантах осуществления CHOP INTEND включает такие показатели, как преодолевающие гравитацию движения при переворачивании с помощью, положение на весу на животе и стояние с поддержкой.
Во многих исследованиях генной терапии, включающих векторы на основе AAV, наблюдают антиген-специфический ответ T-клеток на вектор на основе AAV, и он может быть ожидаем в пределах 2-4 недель после переноса генов. Одним возможным следствием такого антиген-специфического ответа T-клеток является клиренс трансдуцированных клеток и утрата экспрессии трансгена. В попытке ослабить иммунный ответ хозяина на терапию на основе AAV пациентам могут предоставляться иммуносупрессоры. В некоторых вариантах осуществления пациентам могут предоставляться глюкокортикоиды перед введением вирусного вектора. В некоторых вариантах осуществления пациентам может предоставляться кортикостероид перед введением вирусного вектора. В некоторых вариантах осуществления пациентам может предоставляться пероральный стероид перед введением вирусного вектора. Примеры пероральных стероидов включают без ограничения преднизон, преднизолон, метилпреднизолон, триамцинолон, бетаметазон, дексаметазон и гидрокортизон. В некоторых вариантах осуществления пероральный стероид представляет собой преднизолон или содержит его. В некоторых вариантах осуществления пациент начинает прием профилактического стероида за по меньшей мере 24 часа до введения вирусного вектора. В некоторых вариантах осуществления пациенту предоставляется пероральный стероид в течение по меньшей мере 30 дней после введения вирусного вектора. В некоторых вариантах осуществления пероральный стероид вводят один раз в день. В некоторых вариантах осуществления пероральный стероид вводят два раза в день. В некоторых вариантах осуществления пероральный стероид предоставляется в дозе приблизительно 0,1-10 мг/кг, например приблизительно 1 мг/кг. В некоторых вариантах осуществления пероральный стероид предоставляется в дозе приблизительно 0,1-10 мг/кг/день, например приблизительно 1 мг/кг/день. В некоторых вариантах осуществления после введения вирусного вектора осуществляют мониторинг уровней AST и ALT. В таких вариантах осуществления средство для лечения на основе перорального стероида вводят, когда уровни AST и ALT в два раза превышают верхний предел нормы, например которая определена с помощью клинических стандартов и способов, известных из уровня техники, или составляют приблизительно 120 МЕ/л. В некоторых вариантах осуществления средство для лечения на основе перорального стероида вводят в течение более 30 дней, при условии что уровни AST и ALT в два раза превышают верхний предел нормы, например которая определена с помощью клинических стандартов и способов, известных из уровня техники, или превышают приблизительно 120 МЕ/л. Во время длительного лечения кортикостероидами надпочечные железы естественным образом снижают уровень продуцирования кортизола. В тех случаях, когда лечение кортикостероидами внезапно прекращается, организм может испытывать дефицит кортизола. В некоторых вариантах осуществления, где пероральный стероид предоставляется пациенту в течение по меньшей мере 30 дней, дозу стероида медленно снижают согласно графику. В некоторых вариантах осуществления дозу перорального стероида снижают, когда уровни AST и ALT падают ниже уровня, в два раза превышающего верхний предел нормы, например которая определена с помощью клинических стандартов и способов, известных из уровня техники, или приблизительно 120 МЕ/л. В некоторых вариантах осуществления снижение дозы предусматривает пошаговое уменьшение до 0,5 мг/кг/день в течение 2 недель и далее до 0,25 мг/кг/день в течение еще 2 недель. В некоторых других вариантах осуществления снижение дозы перорального стероида происходит по усмотрению врача.
Набор
Настоящее изобретение также предусматривает набор для лечения SMA у пациента, нуждающегося в этом, где набор содержит одну или несколько доз фармацевтической композиции, содержащей эффективное количество или дозу вирусного вектора, содержащего полинуклеотид SMN, раскрытый в данном документе, и в зависимости от типа SMA (как дополнительно раскрыто в данном документе) набор дополнительно содержит контрастное средство (например, омнипак 180) и инструкции по применению фармацевтического препарата или композиции и контрастного средства.
В некоторых вариантах осуществления набор содержит флаконы с фармацевтической композицией на основе вирусного вектора. В некоторых вариантах осуществления фармацевтическую композицию на основе вирусного вектора предоставляют в концентрации приблизительно 1,7-2,3×1013 г. в./мл. В некоторых вариантах осуществления фармацевтическую композицию на основе вирусного вектора предоставляют в концентрации приблизительно 1,9-2,1×1013 г. в./мл. В некоторых вариантах осуществления фармацевтическую композицию на основе вирусного вектора предоставляют в концентрации приблизительно 2,0×1013 г. в./мл. В некоторых вариантах осуществления флаконы содержат приблизительно 5,9 мл фармацевтической композиции на основе вирусного вектора. В некоторых вариантах осуществления флаконы содержат приблизительно 8,7 мл фармацевтической композиции на основе вирусного вектора. В некоторых вариантах осуществления набор не содержит флакона с 5,9 мл, содержит по меньшей мере один флакон с 5,9 мл, по меньшей мере два флакона с 5,9 мл или по меньшей мере три флакона с 5,9 мл. В некоторых вариантах осуществления набор не содержит флакона с 8,7 мл, содержит по меньшей мере один флакон с 8,7 мл, по меньшей мере два флакона с 8,7 мл, по меньшей мере три флакона с 8,7 мл, по меньшей мере четыре флакона с 8,7 мл, по меньшей мере пять флаконов с 8,7 мл, по меньшей мере шесть флаконов с 8,7 мл.
В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, определяют вес пациента. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вес пациента составляет по меньшей мере приблизительно 2,6 кг. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вес пациента составляет не более приблизительно 8,5 кг. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вес пациента составляет приблизительно 2,6-8,5 кг. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят пациенту. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят пациенту в дозе, составляющей приблизительно 1,0-2,5×1014 г. в./кг. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят пациенту в дозе, составляющей приблизительно 1,1×1014 г. в./кг. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят с помощью инфузии пациенту в течение приблизительно 45-70 мин. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят с помощью инфузии пациенту в течение приблизительно 60 мин. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят с помощью инфузии пациенту с применением инфузионного насоса, перистальтического насоса или любого другого оборудования, известного из уровня техники. В некоторых вариантах осуществления, где набор применяют для лечения SMA типа I у пациента, вирусный вектор на основе AAV из флаконов в наборе вводят с помощью инфузии пациенту с применением шприцевого насоса.
В одном варианте осуществления вектор вводят внутривенно или интратекально. В одном варианте осуществления вектор вводят внутривенно. В одном варианте осуществления вектор вводят внутривенно совместно с омнипак 180. В другом варианте осуществления вектор вводят интратекально совместно с омнипак 180.
В другом аспекте в данном документе предусмотрены способы трансдуцирования клеток-мишеней пациента (в том числе без ограничения нервных или глиальных клеток) с помощью rAAV.
Трансдукция клеток пациента с помощью rAAV, раскрытого в данном документе, может привести к длительной экспрессии полипептида или РНК, кодируемой rAAV. Таким образом, в настоящем изобретении предусмотрены способы введения/доставки rAAV (например, кодирующего белок SMN) пациенту-животному или -человеку. Такие способы включают трансдукцию нервных и/или глиальных клеток одним или несколькими rAAV. Трансдукцию можно проводить с помощью кассет генов, содержащих специфичные в отношении ткани контрольные элементы. Например, промоторы, которые способствуют экспрессии конкретно в нейронах или конкретно в астроцитах. Примеры включают промоторы специфической в отношении нейрона энолазы и глиофибриллярного кислого белка. Также можно разработать индуцибельные промоторы под контролем поглощенного лекарственного средства.
В некоторых аспектах предполагается, что трансдукция клеток повышается, когда вектор по настоящему изобретению применяют в комбинации с контрастным средством, которое описано в данном документе, относительно трансдукции вектора по настоящему изобретению, когда его применяют не в комбинации с контрастным средством. В различных вариантах осуществления трансдукция клеток повышается на по меньшей мере приблизительно 1% или по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%, по меньшей мере приблизительно 120%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 180%, по меньшей мере приблизительно 200%, по меньшей мере приблизительно 250%, по меньшей мере приблизительно 300%, по меньшей мере приблизительно 350%, по меньшей мере приблизительно 400%, по меньшей мере приблизительно 450%, по меньшей мере приблизительно 500% или больше, когда вектор по настоящему изобретению применяют в комбинации с контрастным средством, которое описано в данном документе, относительно трансдукции вектора по настоящему изобретению, когда его применяют не в комбинации с контрастным средством. В дополнительных вариантах осуществления трансдукция клеток повышается на от приблизительно 10% до приблизительно 50%, или на от приблизительно 10% до приблизительно 100%, или на от приблизительно 5% до приблизительно 10%, или на от приблизительно 5% до приблизительно 50%, или на от приблизительно 1% до приблизительно 500%, или на от приблизительно 10% до приблизительно 200%, или на от приблизительно 10% до приблизительно 300%, или на от приблизительно 10% до приблизительно 400%, или на от приблизительно 100% до приблизительно 500%, или на от приблизительно 150% до приблизительно 300%, или на от приблизительно 200% до приблизительно 500%, когда вектор по настоящему изобретению применяют в комбинации с контрастным средством, которое описано в данном документе, относительно трансдукции вектора по настоящему изобретению, когда его применяют не в комбинации с контрастным средством.
Настоящее изобретение также предусматривает аспекты, в которых интратекальное введение вектора по настоящему изобретению и контрастного средства в центральную нервную систему пациента, нуждающегося в этом, таким образом, приводит к увеличению выживаемости пациента по сравнению с выживаемостью пациента, когда вектор по настоящему изобретению вводят в условиях отсутствия контрастного средства. В различных вариантах осуществления вектор по настоящему изобретению и контрастное средство вводят интратекально по отдельности в центральную нервную систему пациента, нуждающегося в этом. В других вариантах осуществления вектор и контрастное средство совместно составляют и вводят интратекально в центральную нервную систему или пациенту, нуждающемуся в этом. В других вариантах осуществления вектор и контрастное средство предусмотрены в одной и той же упаковке для введения интратекально в центральную нервную систему или пациенту, нуждающемуся в этом. В различных вариантах осуществления введение вектора по настоящему изобретению и контрастного средства в центральную нервную систему пациента, нуждающегося в этом, таким образом, приводит к увеличению выживаемости пациента на по меньшей мере приблизительно 1%, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 200% или больше относительно выживаемости пациента, когда вектор по настоящему изобретению вводят при условии отсутствия контрастного средства.
В некоторых аспектах предполагается, что трансдукция клеток дополнительно повышается, когда вектор по настоящему изобретению применяют в комбинациях с контрастным средством и когда пациент принимает положение Тренделенберга (положение головой вниз). В некоторых вариантах осуществления, например, пациенты помещены в положение головой вниз под углом от приблизительно 1 градуса до приблизительно 30 градусов, от приблизительно 15 до приблизительно 30 градусов, от приблизительно 30 до приблизительно 60 градусов, от приблизительно 60 до приблизительно 90 градусов или от приблизительно 90 до приблизительно 180 градусов) во время или после интратекальной инфузии вектора. В различных вариантах осуществления трансдукция клеток повышается на по меньшей мере приблизительно 1% или по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%, по меньшей мере приблизительно 120%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 180%, по меньшей мере приблизительно 200%, по меньшей мере приблизительно 250%, по меньшей мере приблизительно 300%, по меньшей мере приблизительно 350%, по меньшей мере приблизительно 400%, по меньшей мере приблизительно 450%, по меньшей мере приблизительно 500% или больше, когда вектор по настоящему изобретению применяют в комбинации с контрастным средством и положением Тренделенберга, которые описаны в данном документе, относительно трансдукции вектора по настоящему изобретению, когда его применяют не в комбинации с контрастным средством и положением Тренделенберга. В дополнительных вариантах осуществления трансдукция клеток увеличивается на от приблизительно 10% до приблизительно 50%, или на от приблизительно 10% до приблизительно 100%, или на от приблизительно 5% до приблизительно 10%, или на от приблизительно 5% до приблизительно 50%, или на от приблизительно 1% до приблизительно 500%, или на от приблизительно 10% до приблизительно 200%, или на от приблизительно 10% до приблизительно 300%, или на от приблизительно 10% до приблизительно 400%, или на от приблизительно 100% до приблизительно 500%, или на от приблизительно 150% до приблизительно 300%, или на от приблизительно 200% до приблизительно 500%, когда вектор по настоящему изобретению применяют в комбинации с контрастным средством и положением Тренделенберга, которые описаны в данном документе, относительно трансдукции вектора по настоящему изобретению, когда его применяют не в комбинации с контрастным средством и положением Тренделенберга.
Настоящее изобретение также предусматривает аспекты, в которых интратекальное введение вектора по настоящему изобретению и контрастного средства в центральную нервную систему пациента, нуждающегося в этом, помещенного в положение Тренделенберга, приводит к дальнейшему увеличению выживаемости пациента по сравнению с выживаемостью пациента, когда вектор по настоящему изобретению вводят в условиях отсутствия контрастного средства и положения Тренделенберга. В различных вариантах осуществления введение вектора по настоящему изобретению и контрастного средства в центральную нервную систему пациента, нуждающегося в этом, помещенного в положение Тренделенберга, приводит к увеличению выживаемости пациента на по меньшей мере приблизительно 1%, по меньшей мере приблизительно 5%, по меньшей мере приблизительно 10%, по меньшей мере приблизительно 20%, по меньшей мере приблизительно 30%, по меньшей мере приблизительно 40%, по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 100%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 200% или больше относительно выживаемости пациента, когда вектор по настоящему изобретению вводят при условии отсутствия контрастного средства и положения Тренделенберга.
Используемая в настоящем изобретении и прилагаемой формуле изобретения форма единственного числа включает ссылки на множественное число, если контекст явно не предписывает иное. "Необязательный" или "необязательно" означает, что описанное впоследствии событие или обстоятельство может произойти или может не происходить, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, когда этого не происходит. Например, фраза "композиция необязательно может содержать комбинацию" означает, что композиция может содержать комбинацию различных молекул или может не включать комбинацию, так что описание включает как комбинацию, так и отсутствие комбинации (т.е. отдельные элементы комбинации). Диапазоны могут быть выражены в данном документе в виде от приблизительно одного конкретного значения и/или до приблизительно другого конкретного значения. Когда такой диапазон выражен, другой аспект включает от одного конкретного значения и/или до другого конкретного значения. Подобным образом, когда значения выражены в виде аппроксимаций с применением предшествующего значения, будет понятно, что конкретное значение образует другой аспект. Далее будет понятно, что конечные точки каждого из диапазонов являются значимыми как по отношению к другой конечной точке, так и независимо от другой конечной точки.
Настоящее изобретение дополнительно проиллюстрировано следующими примерами, которые не должны рассматриваться как ограничивающие. Содержание всех ссылок, патентов и опубликованных заявок на патент, процитированных в настоящей заявке, а также графических материалов, включено в данный документ посредством ссылки в их полном объеме для всех целей.
ПРИМЕРЫ
Следующие примеры следует рассматривать как иллюстративные и не ограничивающие объем описанного выше изобретения.
Пример 1. Получение предшественника главного банка клеток стандарта GMP
Способы
Размораживание. Флакон с отдельными клетками (1×106 клеток) размораживали на водяной бане при 37°C в течение приблизительно 1 минуты и содержимое разбавляли в 5 мл предварительно нагретой полной питательной среды. Клетки переносили в Т-матрас с площадью поверхности 25 см2 и выращивали в инкубаторе при 37°C в течение 4 дней с ежедневной заменой культуральных сред предварительно нагретой полной питательной средой.
Отбор на основании повышенной адгезивности. Для отбора высокоадгезивных клеток, клетки культивировали с применением следующей методики. По достижении клетками 95% конфлюэнтности в матрасе с площадью поверхности 25 см2 клетки пересевали. Клетки промывали с помощью 5 мл PBS, затем диссоциировали с помощью 0,5-1 мл HyQTase в течение ~2 минут при комнатной температуре. Диссоциацию останавливали добавлением 5 мл полной питательной среды и многократно пипетировали для диссоциации клеточных агломератов. Затем клеточную суспензию центрифугировали в течение 4 минут при 200 x g. Надосадочную жидкость отбрасывали, а клеточный осадок ресуспендировали в 10 мл полной питательной среды. Клетки переносили в матрас с площадью поверхности 75 см2 после 4 часов инкубации в инкубаторе при 37°C, низкоадгезивные клетки вымывали путем аспирации среды для культивирования клеток с удалением низкоадгезивных и неадгезивных клеток. Культуральную среду заменяли с помощью 10 мл предварительно нагретой полной питательной среды. По результатам визуального осмотра этот процесс уменьшал клеточную массу на величину до 35% от количества клеток. Перед повторным пересевом клетки инкубировали в течение дополнительных 2 дней. Этот процесс отбора, состоящий из замены среды через 4 часа после посева, проводили три раза перед обеспечением размножения отобранной клеточной популяции (фигура 2 и фигура 3). На последней стадии отбора клетки высевали в 2 матраса с площадью поверхности 175 см2 с конечным объемом 25 мл. Отмечали, что после последней замены среды через 4 часа после посева происходило снижение уровня потери клеток.
Обеспечение размножения клеток. Впоследствии, после достижения клетками в 2 матрасах с площадью поверхности 175 см2 состояния конфлюэнтности, обеспечивали размножение клеток. Клетки промывали с помощью 15 мл PBS, затем диссоциировали с помощью 3 мл HyQTase и инкубировали в течение ~2 минут при комнатной температуре. Диссоциацию останавливали путем добавления 10 мл полной питательной среды. Затем клеточную суспензию центрифугировали с получением 2 клеточных осадков после аспирации надосадочной жидкости. Каждый осадок ресуспендировали в 8 мл полной питательной среды и 2 мл этой концентрированной клеточной суспензии добавляли в 8 матрасов с площадью поверхности 175 см2. Матрасы подготавливали путем добавления 20 мл полной питательной среды, в результате чего получали 22 мл клеточной суспензии и коэффициент разделения 1:4. На следующей стадии обеспечения размножения применяли ту же процедуру со следующими изменениями: в 4 матрасах с площадью поверхности 175 см2 обеспечивали размножение с коэффициентом разделения 1:2, и в 4 матрасах с площадью поверхности 175 см2 обеспечивали размножение с коэффициентом разделения 1:3. Это давало в общей сложности 20 матрасов с площадью поверхности 175 см2.
Сбор биомассы. Собирали клетки из 20 матрасов с площадью поверхности 175 см2. Клетки промывали с помощью 15 мл PBS, затем диссоциировали с помощью 3 мл HyQTase, как было описано ранее. Диссоциацию клеток останавливали путем добавления 10 мл полной питательной среды и собирали в пробирки объемом 50 мл, при этом клеточную суспензию из 4 матрасов с площадью поверхности 175 см2 добавляли в 1 пробирку объемом 50 мл. Это давало в результате 5 пробирок по 50 мл с 40 мл клеточной суспензии в каждой. Пробирки центрифугировали для получения клеточных осадков, надосадочные жидкости аспирировали и клетки ресуспендировали с помощью 10 мл полной питательной среды, в результате чего получали 50 мл клеточной суспензии.
Объем разделяли на 2 пробирки по 50 мл, при этом общий объем клеточной суспензии в каждой пробирке составлял по 25 мл. Для подсчета количества жизнеспособных клеток на пробирку с помощью гемоцитометра и толудинового (трипанового) синего использовали образцы, разбавленные 1:2. Образец из пробирки 1 имел количество жизнеспособных клеток, составляющее 1,99×106 клеток/мл, что давало концентрацию клеток 3,98×106 клеток/мл, а образец из пробирки 2 имел количество жизнеспособных клеток, составляющее 2,4×106 клеток/мл (всего 1×108 клеток), что давало концентрацию клеток 4,8×106 клеток/мл (всего 1,2×108 клеток). Таким образом, всего собрали 2,2×108 клеток. Обе пробирки снова центрифугировали (6 минут, 200 x g), и осадки ресуспендировали в 10 мл (пробирка 1) и 12 мл (пробирка 2) среды для замораживания соответственно с регулированием концентрации клеток до 1×107 клеток/мл. Две клеточные суспензии объединяли и аликвотами по 1 мл (каждая из которых содержала 1×107 клеток) заполняли 22 стерильные криофлакона (таблица 3).
Таблица 3. Расчет общего количества собранных клеток.
в образце 1:2
при сборе
сборе
Заполненные флаконы затем переносили на ночь в морозильную камеру со свежим изопропанолом в морозильной камере при -80°C для заморозки с контролируемой скоростью. Затем замороженные флаконы переносили в газообразный жидкий азот в резервуар с жидким азотом. Десять флаконов переносили на сухой лед для хранения в помещении стандарта GMP.
Клетки HEK293 от ATCC размораживали и успешно адаптировали для повышения адгезивности за 3 пересева перед обеспечением размножения и успешным созданием банка посевного материала. Банк посевного материала тестировали в отношении роста и наличия занесенных возбудителей инфекционных заболеваний (микоплазм, грибов и бактерий). В результате исследования было показано, что банк посевного материала был пригодным для хранения в качестве главного банка клеток в помещении стандарта GMP.
Пример 2. Процесс накопления
Процесс накопления (см., например, фигуру 4) применяли для получения промежуточного продукта, полученного из рабочего банка клеток, при этом процесс накопления включал стадии (а) культивирования адгезивных клеток, (b) трансфицирования культивируемых клеток тремя плазмидами, как показано на фигуре 1 (например, включая описанный в данном документе AAV SMN), для обеспечения продуцирования вирусного вектора на основе AAV, (c) обеспечения лизиса адгезивных клеток с выделением вирусного вектора на основе AAV, (d) очистки вирусных частиц с помощью фильтрации для удаления любых интактных клеток или клеточного дебриса, и (е) подвергания очищенного продукта из стадии (d) фильтрации в тангенциальном потоке и (f) замораживания полученного промежуточного препарата очищенных вирусных частиц. В альтернативных вариантах осуществления AAV, полученный с помощью процесса накопления, раскрытого в данном документе, кодирует shRNA, нацеливающуюся на SOD1 или MECP2, раскрытую в данном документе.
(а) Культивирование адгезивных клеток
Клетки HEK293 размораживали и обеспечивали их размножение в ходе семи пересевов в одноразовых матрасах с применением CO2-инкубаторов. Размороженные клетки промывали питательной средой для размножения клеток, центрифугировали и ресуспендировали свежей питательной средой для размножения клеток. Ресуспендированные клетки высевали в матрас, содержащий питательную среду для размножения клеток, и инкубировали.
После достижения клетками состояния конфлюэнтности их промывали с помощью DPBS и извлекали из матрасов с помощью раствора фермента TrypLE Select. Добавляли питательную среду для размножения клеток для нейтрализации раствора фермента и суспендированные клетки разделяли и пересевали в новые матрасы, содержащие питательную среду для размножения клеток. Этот процесс обеспечения размножения повторяли 7 раз. При последнем повторении суспендированные клетки не пересевали в матрасы, а вместо этого клеточную взвесь инокулировали в биореактор для дальнейшего размножения.
Перед инокуляцией биореактор для адгезивных клеток iCELLis 500/200 м2 или iCELLis 500/333 м2 подготавливали к инокуляции. Подготовительные работы включали распаковку одноразового биореактора, физический осмотр, тестирование на протечку, присоединение системы трубок и уравновешивание датчиков. Для уравновешивания биореактора загружали питательную среду для размножения клеток. После того, как было подтверждено, что значения рН (рН 6,9-7,5), температуры (от 35°С до 39°С) и растворенного кислорода (40-125%) находятся в заданных диапазонах, биореактор засевали при целевой плотностью посева 4800-7000 клеток/см2 (для реактора размером 200 м2) или 5000-12000 клеток/см2 (для реактора размером 333 м2). Клеточную взвесь из предыдущей стадии добавляли к среде в пакете с рециркулирующей средой и подвергали циркуляции через биореактор.
(b) Трансфекция адгезивных клеток
В дни 4, 5 или 6 от момента инокуляции в биореактор адгезивные клетки HEK293 трансфицировали совместным осаждением с тремя ДНК-плазмидами и PEI. 3 плазмиды, применяемые для этой трансфекции, представляли собой pSMN, pAAV2/9 и pHELP. Применяемую для размножения клеток питательную среду DMEM удаляли из биореактора и заменяли средой для трансфекции. Вектор scAAV9.CB.SMN получали с применением трансфекции трех ДНК-плазмид в адгезивные клетки почки эмбриона человека (HEK293) путем совместного осаждения с полиэтиленимином ("PEI") в крупномасштабном биореакторе для адгезивных клеток. Векторная плазмида pSMN содержала cDNA человеческого белка выживаемости мотонейронов (SMN). 3 плазмиды, применяемые для этой трансфекции, представляли собой pSMN (222 мг), pAAV2/9 (333 мг) и pHELP (444 мг). Плазмидами можно было трансфицировать при их соотношении 1:1:1 моль. Перед добавлением PEI и плазмид для совместного осаждения среду для трансфекции оставляли для уравновешивания в биореакторе до тех пор, пока температура в биореакторе не составляла >30°C. Процесс совместного осаждения PEI и плазмид предусматривал добавление плазмид в среду для трансфекции и фильтрацию через фильтр с размером пор 0,2 мкм в реакционный пакет. PEI добавляли в среду для трансфекции, а затем в реакционный пакет. Соотношение PEI и плазмиды составляло приблизительно 1:1 по весу. Реакционную среду из PEI и плазмид вручную перемешивали до получения гомогенной суспензии, и реакция проходила за период продолжительностью 15-30 минут. В конце времени реакции PEI и плазмиды для совместного осаждения переносили из реакционного мешка в биореактор. PEI и плазмиды для совместного осаждения оставляли смешиваться в биореакторе на 1-2 часа (альтернативные значения продолжительности описаны в примере 7) до возобновления перемешивания. Cреду для трансфекции подвергали рециркуляции в биореакторе в течение 18-24 часов перед заменой следующей средой.
В день 6 в биореакторе, через 18-24 часа после трансфекции, биореактор опустошали, а среду для трансфекции в пакете с рециркулирующей средой заменяли средой для применения после трансфекции. Биореактор повторно заполняли средой для применения после трансфекции путем рециркуляции в биореакторе. В день 7, через 18-24 часа после замены среды в день 6, среду для применения после трансфекции в рециркуляционном пакете заменяли свежим пакетом среды для применения после трансфекции. В ходе этой стадии биореактор не опустошали. Рециркуляцию среды продолжали до сбора биомассы, как правило, в день 9.
(c) Обеспечение лизиса трансфицированных адгезивных клеток
Спустя 9 дней в биореакторе из реактора отбирали последние образцы перед сбором биомассы и начинали процесс полного лизиса клеток. В биореактор добавляли бензоназу до конечной концентрации 100 МЕ/мл. Бензоназу оставляли перемешиваться в реакторе и в реактор добавляли буфер для лизиса. Буфер для лизиса перемешивали в реакторе при 15-25°С в течение 2 часов до переноса содержимого биореактора в пакет для сбора биомассы. В пакет для сбора биомассы добавляли солевой раствор сахарозы (SSS), который гасит реакцию с бензоназой, и перемешивали в течение 15 минут. Затем биореактор ополаскивали буфером для промывки биореактора в течение 15 минут, а затем смыв собирали в пакет для накопления собранной биомассы вместе с погашенным раствором для лизиса клеток. После добавления смыва в пакет для накопления его содержимое перемешивали в течение 15 минут и отбирали образцы общей собранной биомассы.
(d) Получение вирусных частиц путем фильтрации и фильтрации в тангенциальном потоке
Смешанную общую собранную биомассу фильтровали через глубинный фильтр POD в пакет для накопления. После того, как всю общую собранную биомассу отфильтровали, через глубинный фильтр прогоняли буфер для TFF1. Пул, полученный с помощью глубинного фильтра, перемешивали и отбирали из него образцы. Полученный с помощью глубинного фильтра пул затем фильтровали через фильтр с размером пор 0,45 мкм для дополнительного осветления материала общей собранной биомассы. Затем через фильтр с размером пор 0,45 мкм прогоняли буфер для TFF1.
В ходе стадии TFF1 5,0 м2 кассеты с мембранами из регенерированной целлюлозы с порогом отсечения, представляющим собой MW 300 кДа, промывали, дезинфицировали раствором NaOH и уравновешивали буфером для TFF1. Фаза концентрирования в данном процессе предназначалась для уменьшения объема осветленной собранной биомассы примерно в 10 раз. После достижения целевого объема ретентата начинали процесс диафильтрации. Ретентат подвергали диафильтрации с помощью 6 диаобъемов буфера для TFF1. Альтернативно, ретентат можно было подвергнуть диафильтрации с помощью более чем 6 диаобъемов буфера для TFF1, например, 10 диаобъемов, 12 диаобъемов или 15 диаобъемов. После достижения 6 диаобъемов общего потока пермеата ретентат снова концентрировали и собирали в пакет для накопления. Проводили два последовательных ополаскивания мембраны для максимального извлечения продукта из системы TFF с получением промежуточного лекарственного вещества.
(e) Замораживание промежуточного продукта
Отбирали аликвоту промежуточного продукта, полученного в результате TFF1, в стерильные бутылки из PETG объемом 1 или 2 литра в вытяжном шкафу LFH, а затем замораживали их на сухом льду или в морозильной камере и переносили на хранение при -60°C.
Таблица 4. Буферы, применяемые в процессе накопления
без L-глутамина и 4,5 г/л глюкозы
4 мM L-глутамина и без FBS
Пример 3. Процесс выделения и очистки
Процесс выделения и очистки (см., например, фигуру 5) применяли для обработки промежуточного продукта, полученного в результате TFF1, с получением отфильтрованного лекарственного вещества. В некоторых вариантах осуществления процесс выделения и очистки, раскрытый в данном документе, можно применять для обработки промежуточного продукта, содержащего AAV SMN, AAV MECP2 или AAV, кодирующий shRNA, нацеливающуюся на SOD1, описанные в данном документе. Стадии процесса выделения и очистки включают: (а) подкисление и осветление промежуточного продукта (с применением фильтрации), (b) очистку с применением катионообменной хроматографии, (с) фильтрование с применением фильтрации в тангенциальном потоке ("TFF2"), (d) ультрацентрифугирование с применением буфера с CsCl для разделения заполненных и пустых вирусных капсидов (e) сбор вирусных векторов на основе AAV и (d) фильтрование собранных вирусных векторов на основе AAV с помощью второй стадии фильтрации в тангенциальном потоке ("TFF3").
(a) Подкисление и осветление
Промежуточный материал, полученный в результате TFF1, из процесса накопления (размороженный до комнатной температуры, если предварительно он был заморожен) закачивали в пакет с мешалкой. Перемешивали объединенный промежуточный продукт, полученный в результате TFF1, и отбирали из него образцы для определения титра. Сразу после этого обрабатывали объединенный промежуточный продукт, полученный в результате TFF1, путем добавления 11-14% Tween 20. Tween 20 использовали для стимуляции флоккуляции общей массы белков и ДНК клеток-хозяев в кислых условиях рН. Смесь оставляли для инкубации в течение 12-20 часов. Затем рН снижали путем добавления подкисляющего буфера (1 М глицина) до рН 3,3-3,7. Образовавшийся после снижения рН осадок затем удаляли фильтрованием раствора через глубинные фильтры Clarisolve с размером 1,1 м2 и Millistak+C0HC с размером 2,2 м2 и фильтры тонкой очистки с размером пор 0,45 мкм. Этот процесс приводил к получению подкисленного и осветленного промежуточного продукта, полученного в результате TFF.
(b) Очистка с помощью катионообменной хроматографии
Стадию катионообменной хроматографии (CEX) применяли для отделения вирусных капсидов от белков, ДНК и других технологических примесей, например, липидов клеток-хозяев, TWEEN-20. На этой стадии применяли хроматографическую колонку CIMmultus S03-8000 Advanced Composite Column (с сульфонильными группами) (с размером пор 0,2 мкм) (8,0 л), которая управлялась с применением хроматографической системы с автоматизированным процессом. Буферы и растворы описаны в следующей таблице.
Таблица 5. Буферы и растворы для одного цикла CEX
pH 3,5 ± 0,1 при 20°C
pH 3,5 ± 0,1 при 20°C
pH 9,1 ± 0,1 при 20°C
Подкисленный и осветленный промежуточный продукт, полученный в результате TFF (т.е. загрузку CEX), загружали в очищенную и уравновешенную колонку для CEX. Условия были такими, что вирусные векторы связывались с монолитной колонкой. Несвязанный материал вымывали из колонки с помощью буфера A для CEX. Продукт элюировали из смолы с помощью градиента буфера B для CEX в буфере A для CEX. Накопление фракции 1 начинали в начале градиента элюирования в течение 10 объемов колонки (CV) до заданного объема 2,3-2,7 CV. Хроматографическую колонку утилизировали после каждой партии (т.е. хроматографическую колонку повторно не использовали). Элюат продукта CEX (фракцию 2) затем нейтрализовали с применением нейтрализующего буфера до рН 7,7-8,3.
(c) Фильтрование путем фильтрации в тангенциальном потоке (TFF2)
На стадии TFF2 концентрировали вирусный вектор, удаляли белковые примеси и заменяли буфер на подходящий буфер для стадии ультрацентрифугирования с помощью CsCl. Нейтрализованный элюат, полученный в результате CEX, обрабатывали с применением системы TFF, снабженной мембраной из регенерированной целлюлозы с MWCO 300 кДа размером 0,3 м2.
Объем нейтрализованного элюата, полученного в результате CEX, уменьшали до целевого объема ретентата. После достижения целевого объема ретентата начинали диафильтрацию в периодическом режиме TFF (порционном режиме). Ретентат разбавляли в 2 раза с помощью буфера на основе NaCl для диафильтрации при TFF2 и концентрировали ретентат до его первоначального объема. Это повторяли до завершения диафильтрации с помощью буфера на основе NaCl для диафильтрации при TFF2. Затем ретентат разбавляли в 2 раза буфером с CsCl для диафильтрации при TFF2 и концентрировали ретентат до его первоначального объема. Это повторяли до завершения диафильтрации с помощью буфера на основе NaCl для диафильтрации при TFF2.
Ретентат дополнительно концентрировали до конечной массы исходя из физического титра нейтрализованного элюата, полученного в результате CEX, объема задержки в системе, объема промывки системы и плотности ретентата для достижения требуемой целевой концентрации вектора и извлекали в пакет для накопления. После одного цикла промывки системы с помощью буфера с CsCl для диафильтрации при TFF2 следовало выдувание продукта для максимизации извлечения продукта из системы TFF. Для измерения физического титра брали образец ретентата, полученного в результате TFF2, который содержал ретентат и смыв. Кассеты с мембранами для TFF2 утилизировали после каждой партии (т.е. мембраны для TFF повторно не использовали).
Таблица 6. Буферы для TFF2
(d) Ультрацентрифугирование с помощью CsCl
Целью стадии ультрацентрифугирования было удаление пустых капсидов от полных капсидов путем применения ультрацентрифугирования в градиенте хлорида цезия. Ретентат, полученный в результате TFF2, добавляли в пробирки для ультрацентрифугирования и пробирки герметично закрывали. Пробирки помещали в ультрацентрифугу, например, в автоматическую систему ультрацентрифугирования Optima XPN 100 или эквивалентную систему, оснащенную ротором типа 50,2 Ti или эквивалентным ротором. Заполненные пробирки центрифугировали при скорости 45000 об/мин в течение 22 часов при 20°С.
(e) Сбор вирусных векторов на основе AAV
После завершения стадии центрифугирования пробирки извлекали из ультрацентрифуги и помещали в бокс микробиологической безопасности. Содержащие продукт пробирки ставили на кольцевых стойках над контейнером для отходов. Непосредственно под пробиркой размещали лампу с целью визуализации полосы пустых капсидов (полосы А, полосы, занимающей самое верхнее положение), парных полос полных капсидов (полосы В и полосы С, верхней и нижней полос пары) и полосы, занимающей самое нижнее положение под парой (полосы D). Пробирки прокалывали иглой, прикрепленной к шприцу, для выпуска воздуха из пробирок и полосы B, C и D извлекали с помощью иглы. Собранный материал переносили в пакет для накопления. Собранный подвергнутый ультрацентрифугированию пул (UC-пул), разбавляли буфером для TFF2 до достижения соответствующей начальной концентрации CsCl в материале загрузки для TFF2. Разбавленный UC-пул обрабатывали на стадии TFF3. Буфер для стадии ультрацентрифугирования с помощью CsCl указан в таблице ниже.
Таблица 7. Буфер для ультрацентрифугирования с помощью CsCl
(f) Фильтрование путем фильтрации в тангенциальном потоке (TFF3)
На стадии TFF3 удаляли CsCl и концентрировали полный вектор с применением буфера для получения конечного состава. Систему фильтрации в тангенциальном потоке применяли в сочетании с мембранами из регенерированной целлюлозы с MWCO 300 кДа размером 50 см2. Вирусный вектор удерживался мембранами.
Объем разбавленного UC-пула уменьшали до целевого объема ретентата. После достижения целевого объема начинали непрерывную диафильтрацию при постоянном объеме ретентата. Ретентат подвергали диафильтрации с помощью буфера для TFF3. Образец подвергнутого диафильтрации ретентата отбирали для
измерения физического титра. Ретентат дополнительно концентрировали, ориентируясь на массу пермеата, которую рассчитывали по 1) объему ретентата в системе TFF в конце диафильтрации, 2) физическому титру разбавленного UC-пула, 3) целевой концентрации лекарственного вещества (DS), 4) объединенному объему промывок системы и промывок фильтров и 5) плотности буфера для TFF3. Кассеты с мембранами для TFF3 утилизировали после каждой партии (т.е. кассеты повторно не использовали).
Таблица 8. Буферы для TFF3
Для выделения вектора из системы TFF производили две последовательных ополаскивания мембран для TFF с помощью 20 мл буфера для TFF3. Смывы извлекали через стерилизующий фильтр Pall Supor® EKV с размером пор 0,2 мм (Mini Kleenpak). Ополаскивание фильтра выполняли буфером для TFF3 для извлечения вектора, оставшегося в фильтре, при наличии такового, и для регулирования конечного объема отфильтрованного пула, полученного в результате TFF3 (т.е. лекарственного вещества, DS). Отбирали аликвоту DS в бутылки из PETG объемом 125 или 250 мл и замораживали при < -60°C.
Пример 4. Составление и заполнение
Лекарственный продукт (DP) представлял собой однократную дозу не содержащего консервантов, стерильного, от прозрачного до такового с небольшой степенью непрозрачности и от бесцветного до бледно-белого раствора для внутривенной инфузии нереплицирующегося, самокоплементарного вектора на основе AAV9 в целевой концентрации 2,0×1013 г. в./мл. DP содержал 20 мM Tris, 1 мM MgCl2, 200 мM NaCl, 0,005% вес./об. полоксамера 188. Диапазон pH раствора находился в пределах от 7,7 до 8,3.
Таблица 9. Типовой процесс получения лекарственного продукта - состав буфера
DP разливали в стерильные, готовые к использованию флаконы объемом 10 мл Crystal Zenith (CZ), закупоривали стерильными, готовыми к использованию хлорбутилэластомерными пробками и герметично закрывали стерильными алюминиевыми колпачками с отрывной накладкой диаметром 20 мм. Флаконы заполняли при номинальном объеме наполнения либо 5,5 мл, либо 8,3 мл. Целевое значение переполнения составляло 0,4 мл, и флаконы заполняли до 5,9 ± 0,1 мл или 8,7 ± 0,1 мл.
Пример 5. Анализ активности
Относительную активность лекарственного продукта измеряли с применением количественного анализа in vivo. В анализе применяли общепринятую модель заболевания SMA на мышах. Пары для разведения линии мышей SMAΔ7 (Jackson Laboratories, № 005025) имеют нормальный фенотип, но ~25% их потомков являются гомозиготными по целевой мутации гена SMN и проявляют SMA-подобный фенотип. Ко дню 5 у них появляются признаки мышечной слабости, а на следующей неделе развивается аномальная походка и тенденция к падению. По сообщениям Jackson Laboratories, среднее значение выживаемости животных с SMA-подобным фенотипом составляет ~15 ± 2 дня. В поисковых исследованиях было продемонстрировано, что медианное время выживания для животных с SMA-подобным фенотипом без обработки составляло 16,3 дня (среднее геометрическое; n=3 исследования; 10 мышей на исследование).
Биологически активный лекарственный продукт, вводимый путем внутривенной (IV) инфузии, приводит к увеличению времени выживания, которое зависит от дозы (г. в./кг). Активность лекарственного продукта измеряли относительно эталонного материала (предыдущей партии вектора). Титр лекарственного продукта и эталонного материала (геномов вектора/мл; г. в./мл) определяли с помощью капельной цифровой полимеразной цепной реакции (ddPCR). Вектор разбавляли солевым раствором для достижения каждого из трех указанных уровней доз, которые будут введены мышам с SMA-подобным фенотипом.
Результаты анализа считали приемлемыми, если анализ удовлетворял критериям пригодности. Критерии пригодности анализа заключались в следующем.
1. Предел приемлемости для образца отрицательного контроля (15 ± 2 дня, медианное значение выживаемости);
2. Предел приемлемости для образца положительного контроля (>40 дней, медианное значение выживаемости);
3. Пределы приемлемости на кривой зависимости ответа от дозы для медианного значения выживаемости при применении эталонного стандарта.
В данном исследовании применяли предыдущую партию вектора (далее по тексту "предыдущая партия") для определения линейной корреляции между медианным значением выживаемости (в днях) мыши SMAΔ7 при введении дозы лекарственного продукта (далее по тексту "исследуемая партия") при пяти различных уровнях дозы, включая 0 (нулевую) дозу с применением 0,9% солевого раствора (группа без обработки). Относительную активность лекарственного продукта из исследуемой партии определяли путем сравнения кривой линейной регрессии эталонного стандарта из предыдущей партии с кривой линейной регрессии лекарственного продукта из исследуемой партии. Это осуществляли путем применения соотношения точки пересечения с осью Y и углового коэффициента каждой линии линейной регрессии (т.е. эталонного стандарта и исследуемого препарата). Расчет процента относительной активности описан в уравнении (1) ниже.
%RP = [(точка пересечения с осью Y/угловой коэффициент исследуемого препарата) ÷ (точка пересечения с осью Y/угловой коэффициент эталонного стандарта)] X 100 (1)
Предыдущую партию, которую применяли в клиническом испытании фазы 1, применяли в качестве партии эталонного стандарта и ее активность принимали за 100%.
Для демонстрации эффективности терапевтических средств против SMA, в том числе лекарственного продукта, применяли модель на мышах Δ7. Контрольные животные, обработанные буферным раствором для TFF3 (средой-носителем), обеспечивали надежный контроль исходного уровня, относительно которого можно было измерять активность продукта в виде увеличения медианного значения выживаемости. При разработке лекарственного продукта были выявлены три (3) дозы (исключая дозу при обработке средой-носителем), определенные по геномному титру с применением капельной цифровой ПЦР (ddPCR), которые влияют на выживаемость в модели на мышах с линейной корреляцией, если значение вводимой дозы (г. в./кг) подвергнуть логарифмическому преобразованию и нанести на график в зависимости от медианного значения выживаемости (в днях) у подвергнутой обработке новорожденной мыши SMAΔ7. См. стандартные титры (г. в./мл) в таблице 11 для стандартов низкого, среднего и высокого титра. Кроме того, буферный раствор для TFF (среду-носитель) применяли как в качестве нулевой точки (0) калибровочной кривой, так и в качестве отрицательного контроля. В качестве положительного контроля также включали дозу, для которой наблюдали выживаемость ≥40 дней (больше, чем в случае дозы, для которой наблюдали удвоение медианного значения выживаемости).
Таблица 11. Целевые дозы
(без обработки)
Препараты растворов доз (см. пример схемы разбавления в таблице 12).
Отрицательный контроль - в качестве отрицательного контроля применяли буферный раствор для TFF3 (буфер для получения конечного состава лекарственного продукта)
Положительный контроль - серию исследуемого препарата получали с концентрацией 1,10 х 1014 г. в./кг с применением буферного раствора для TFF3. Пример схемы разбавления см. в таблице 12.
Растворы эталонного стандарта - серию эталонного стандарта готовили в трех концентрациях, указанных в таблице 11, с применением буферного раствора для TFF3.
Таблица 12. Схема разбавления эталонного стандарта и исследуемого препарата (пример)
зование в г. в./мкл
исследуемого препарата для применения (мкл)
(мкл)
(мкл)
Подготовка исследуемого препарата - исследуемый препарат разбавляли буферным раствором для TFF3. Разбавления рассчитывали для получения тестовых доз (г. в./кг), указанных в таблице 11, на мышь в общем конечном объеме 50 мкл. Разбавления делали для 12 мышей одновременно с одним дополнительным объемом в качестве положительного контроля, нацеленного на увеличение минимальной продолжительности жизни подвергнутых обработке мышей до медианного значения выживаемости (дни), составляющего ≥40 дней.
Пределы приемлемости на контрольных образцах
Отрицательный контроль (мыши без обработки) - предел приемлемости анализа для группы отрицательного контроля состоял в том, что мыши SMAΔ7 соответствовали критерию медианного значения выживаемости, составляющего 15±2 дня. Кроме того, любую мышь, погибшую через ≤10 дней, исключали из анализа.
Положительный контроль (группа, которую обрабатывали целевой клинической дозой) - предел приемлемости анализа для группы положительного контроля заключался в том, что минимальная продолжительность жизни подвергнутых обработке мышей должна была составлять ≥40 дней, что представляло собой медианное значение выживаемости. Кроме того, любую мышь, погибшую через ≤10 дней, исключали из анализа.
Пределы приемлемости на кривой зависимости ответа от дозы для эталонного стандарта
Критерии пригодности анализа для линейной кривой зависимости ответа от дозы для эталонного стандарта, построенной в виде зависимости медианного значения выживаемости (дни) от вводимой дозы (г. в./мл), будут определены. Соотношение точка пересечения с осью Y/угловой коэффициент - определяли кривую линейной регрессии зависимости медианного значения выживаемости (в днях) от вводимой дозы (г. в./кг) для эталонного стандарта и исследуемого препарата. Для каждого случая линейной регрессии рассчитывали соотношение точки пересечения с осью Y и углового коэффициента.
Представление результатов
Качественное представление результатов определения относительной активности - оценивали критерии пригодности анализа для каждого анализа перед определением медианного значения выживаемости в одной точке (в днях), определяемого через ≥40 дней по материалу положительного контроля. Если медианное значение выживаемости у группы положительного контроля составляло ≥40 дней, исследуемый препарат можно использовать, если соблюдены критерии, представленные ниже.
Количественное представление результатов определения относительной активности - перед количественным определением относительной активности исследуемого препарата для каждого анализа оценивали критерии пригодности анализа. Относительную активность для исследуемого препарата можно было регистрировать, когда положительный контроль достигал ≥40 дней, и медианное значение выживаемости для группы мышей, представляющей верхнюю стандартную дозу 7,5×1013 г. в./кг, составляло 31±3 дня. Относительную активность в процентах (% RP) для исследуемого препарата рассчитывали с применением точки пересечения с осью Y и углового коэффициента линейной регрессии кривой зависимости ответа от дозы для медианного значения выживаемости (в днях) следующим образом:
%RP=100% * [(точка пересечения с осью Y/угловой коэффициент исследуемого препарата) ÷ (точка пересечения с осью Y/угловой коэффициент эталонного стандарта)]
Пример 6. Исследование инактивации поверхностно-активным веществом
Для того, чтобы разделить влияние низкого рН и Tween, для исследования инактивации поверхностно-активным веществом применяли образец промежуточного продукта лекарственного вещества, полученного в результате TFF1, который характеризовался рН 7,6, и содержал 4-8% Tween 20. Концентрация Tween 20 в промежуточном продукте, полученном в результате TFF1, была приблизительно в 2,5 раза ниже, чем при полном процессе, когда Tween 20 добавляли к промежуточному продукту, полученному в результате TFF1 до его подкисления. Такую более низкую концентрацию Tween 20 считали наихудшим условием для инактивации, активируемой поверхностно активным веществом, в процессе получения лекарственного вещества. Исследуемый препарат для стадии инактивации вируса путем обработки поверхностно-активным веществом представлял собой промежуточный продукт, полученный в результате TFF-1, содержащий 4-8% Tween 20. Для того, чтобы оценить способность к инактивации вируса посредством стадии обработки поверхностно-активным веществом, применяли как XMuLV, так и PRV для внесения вируса в исследуемый препарат в дублирующих экспериментах. Количественное определение вируса осуществляли с применением анализа на инфекционность по бляшкообразованию.
На стадии получения TFF1 получали диапазон концентрации поверхностно-активного вещества 4-8% Tween 20. Промежуточный продукт, полученный в результате TFF1, объединяли и к промежуточному продукту, полученному в результате TFF1, добавляли еще 12% Tween 20 при рабочей температуре 16-20°C при перемешивании в течение 12-20 часов. Процесс удаление вирусов осуществляли при концентрации Tween 20 4-8% и при контролируемой температуре 16,0 ± 0,1°С. Продолжительность процесса инактивации составляла 120 минут в сравнении со стандартным временем процесса, составляющим 16 часов.
В ходе процесса инактивации инактивационную нагрузку получали путем измерения объема исследуемого препарата, уравновешивания до целевой температуры, а затем внесения вируса. В шесть (6) моментов времени отбирали образцы с внесенной инактивационной нагрузкой для наблюдения за кинетикой инактивации с течением времени: <1 минуты, 15 минут, 30 минут, 60 минут, 90 минут и 120 минут. После отбора каждый образец разбавляли в питательной среде для прекращения инактивации вируса и анализировали на наличие вируса. Для увеличения чувствительности анализа через 90 минут и 120 минут на вирус анализировали большие объемы образцов, собранных в эти моменты времени. В связи с наличием Tween 20 в исследуемом препарате на этой стадии титр инактивационной нагрузки рассчитывали по титру вносимого вируса и объему внесенного вируса.
Было показано, что эффективность инактивации вируса путем обработки поверхностно-активным веществом (Tween 20) являлась эффективной, о чем свидетельствовали значения LRV, превышающие 4 log10, для обоих вирусов в момент времени 90 минут. Кинетика инактивации путем обработки поверхностно-активным веществом проиллюстрирована на фигуре 6 и фигуре 7 в виде графиков скорости инактивации вируса в течение 120 минут во время обработки поверхностно-активным веществом Tween 20.
Пример 7. Влияние более высокой плотности посева, более ранней трансфекции и более раннего сбора и времени смешивания ДНК/PEI на продуцирование лекарственного вещества
Оценивали влияние более высокой плотности посева, трансфекции и сбора на один день раньше и времени смешивания ДНК/PEI на продуцирование DS. Каждое условие оценивали в двух повторностях в биореакторе размером 1,6 м2.
Материалы и способы
Увеличение клеточной массы
Клетки HEK 293 размораживали и ресуспендировали в DMEM с добавлением 10% FBS. Клетки центрифугировали при 209 x g, 5 мин, при комнатной температуре, затем удаляли надосадочную жидкость и добавляли свежую DMEM+10% FBS. Клетки подсчитывали в отношении плотности жизнеспособных клеток и их жизнеспособности, и высевали в 2 Т-матраса с площадью поверхности 175 см2, и инкубировали при 37,0°С, 5% СО2 в течение трех дней, пока культуры не достигали ~90% конфлюэнтности. В случае каждого пересева клеток использованную среду удаляли, матрасы промывали с помощью PBS (-CaCl2, -MgCl2) в количестве 0,08 мл/см2, а затем матрасы обрабатывали TrypLE Select (0,04 мл/см2) и инкубировали при 37,0°С, 5% СО2 в течение 2-3 мин. Трипсин гасили с помощью DMEM+10% FBS (0,04 мл/см2). Клетки размножали и высеивали в соответствии с диаграммой, представленной на фигуре 8.
Инокуляция и мониторинг клеток
Биореакторы инокулировали в двух повторностях клетками НЕК 293 с целевой плотностью 8000 клеток/см2 и 12000 клеток/см2 в питательной среде (DMEM с высоким содержанием глюкозы+10% FBS из Австралии+смесь 1:100 пенициллина и стрептомицина (Pen Strep) с перемешиванием. Задавали следующие параметры обработки: pH 7,23, 37,0°C, 55% растворенного кислорода (DO) и линейная скорость 2 см/с. Через 24 часа после посева запускали рециркуляцию питательной среды DMEM (0,188 мл/см2) на скорости рециркуляции (12,5 мл/мин.). Ежедневные образцы, отбираемые для определения pH, метаболитов и питательных веществ с помощью автономного прибора, считывали с применением Nova BioFlex. На четвертый день (12000 клеток/см2), на пятый день (8000 клеток/см2) и на девятый день после посева отбирали три волокна и лизировали с помощью PBS, растворов A100 и B100 (ChemoMetec) в соотношении 1:1:1 об./об. и подсчитывали (NucleoCounter NC-200) общее количество ядер для мониторинга роста культуры.
Трансфекция
На четвертый день (12000 клеток/см2) и на пятый день (8000 клеток/см2) после инокуляции клеток рециркуляцию прекращали и клетки в каждом биореакторе трансфицировали с помощью плазмидной ДНК и полиэтиленимина (PEI). ДНК и PEI смешивали в соотношении 1:1 мг/мг. Трансфекцию плазмидными ДНК осуществляли в массовом соотношении 1:1,5:2, при этом плазмиду pSMN, плазмиду pAAV2/9 и плазмиду pHELP добавляли в среду DMEM -/- с высоким содержанием глюкозы, -CaCl2, -L-глутамином, и фильтровали через фильтр с размером пор 0,2 мкм, и перемешивали путем переворачивания. В среду DMEM -/- добавляли PEI и перемешивали путем переворачивания. Затем PEI добавляли к ДНК, перемешивали путем переворачивания и инкубировали при комнатной температуре в течение 20 минут в случае 8000 клеток/см2 (контроля) и 12000 клеток/см2. ДНК/PEI инкубировали в течение 1 часа и 2 часов для двух остальных условий с 8000 клеток/см2. Эту смесь комплекса ДНК/PEI использовали для трансфекции двух биореакторов для каждого из четырех условий. Комплекс ДНК/PEI вносили в каждый биореактор и инкубировали при параметрах обработки в течение двух часов. Через два часа после трансфекции снова включали рециркуляционную петлю.
Замена среды после трансфекции
Через 24 часа после трансфекции все среды из биореакторов и рециркуляционных петель удаляли и заменяли на OptiMEM+1:100 Pen Strep (0,132 мл/см2) и продолжали рециркуляцию в течение 24 часов при параметрах обработки. Через 48 часов после трансфекции всю среду удаляли только из системы рециркуляции и заменяли на OptiMEM+1:100 Pen Strep (12 мл/мин) и продолжали рециркуляцию при параметрах обработки.
Сбор
На восьмой день (12000 клеток/см2) и на девятый день (8000 клеток/см2) после инокуляции клеток добавляли бензоназу (100 МЕ/мл), прогоняли буфером для лизиса (50 мМ HEPES, 1% Tween 20) и инкубировали в течение двух часов при параметрах обработки. Биореакторы опустошали и добавляли солевой раствор сахарозы (500 мМ NaCl, 1% вес./об. сахарозы), и перемешивали их путем переворачивания. Биореакторы промывали буфером для промывки биореактора (500 мМ NaCl, 1% вес./об. сахарозы, 20 мМ основания Tris, 1% об./об. Tween 20, 1 мМ MgCl2.6H2O) в течение приблизительно 15 мин при параметрах обработки. Биореакторы опустошали, а буфер для промывки биореактора объединяли с неочищенной общей собранной биомассой, перемешивали путем переворачивания и отбирали образцы для ddPCR-анализа.
Глубинная фильтрация и фильтрация в тангенциальном потоке
На восьмой день (12000 клеток/см2) и на девятый день (контроль 8000 клеток/см2) после инокуляции клеток из биореакторов собирали биомассу, отбирали образцы и неочищенный лизат каждого условия объединяли (n=2 биореактора). Объединенный лизат затем осветляли посредством фильтра Millistak C0HC Pod, 270 см2, и фильтра тонкой очистки Millipak 40 с размером пор 0,45 мкм, Durapore, 200 см2 (EMD Millipore). После C0HC+0,45 образцы отбирали и замораживали при -80,0°С. Осветленный лизат затем концентрировали с помощью тангенциального потока на фильтре Pellicon® 2 Ultrafiltration Module PLCMK C 0,1 м2 (EMD Millipore). Для диафильтрации конечного продукта использовали по меньшей мере 6 диаобъемов. После фильтрации TFF1 образцы получали и замораживали при -80,0°С. Все образцы подвергали титрованию в отношении AAV2/9 и белка клетки-хозяина.
Для получения DS в биореакторах использовали плазмиды. Данными представлено каждое условие в двух повторностях, соответствующих номеру биореактора и условию, указанному в таблице 14.
Таблица 14. Номера биореакторов и соответствующие условия
клеток/см2
(мин)
Рост клеток. Клетки подсчитывали в день посева (день 0), а ядра подсчитывали в день 4 (12000 клеток/см2), день 5 (8000 клеток/см2) и день 9 после посева. Данные свидетельствовали, что клетки во всех реакторах росли экспоненциально с дня 0 по день 5. После трансфекции результаты подсчета ядер в день 9 свидетельствовали о том, что общее количество ядер в биореакторе 221 выросло в 2,0 раза с дня 5 по день 9. Все остальные реакторы (с 222 по 228) не характеризовались значительным ростом с дня 5 по день 9. Увеличение роста в биореакторе 221 могло быть артефактом, основанным на неравномерном распределении клеток по отдельным волокнам, используемым для подсчета общего количества ядер. Возможно, что клетки во всех биореакторах росли схожим образом, исходя из данных о метаболитах, показанных на фигурах 9A-E.
pH, питательные вещества и метаболиты. Потребление глюкозы одинаково изменялось во всех биореакторных культурах, что позволяло предположить, что, несмотря на увеличение кривой роста для биореактора 221, клетки потребляли глюкозу со схожими значениями скорости. Значение рН для биореакторов, засеянных при 12000 клеток/см2, составляло в среднем 7,06, а при 8000 клеток/см2 составляло в среднем 7,18 в течение первых трех дней. Значение pH немного снижалось с повышением метаболизма питательных веществ и увеличивалось в день 9 одновременно с повышением уровней ионов аммония. Уровень лактата увеличивался до дня 5 (биореакторы 221, 223, 224, 225, 227) и дня 6 (биореакторы 222, 226, 228), а затем выравнивался к концу продуцирования, что свидетельствовало о использовании лактата в качестве источника энергии на этой стадии.
Титры продукции
Вирусные геномы из собранного материала измеряли с помощью цифровой капельной ПЦР (ddPCR). Титры были приблизительно в 1,5 раза выше в биореакторах, засеянных при 12000 клеток/см2, со средним показателем титра 6,37E+10 г. в./мл (n=2), по сравнению с контрольными биореакторами, засеянными при 8000 клеток/см2, со средним показателем титра 4,33Е+10 г. в./мл (n=2). Данные по титру свидетельствуют о том, что посев при более высокой плотности, трансфекция и сбор биомассы на один день раньше обеспечивают более высокие выходы продукции DS. Титр на выходе для ДНК/PEI, инкубированных в течение одного часа, характеризовался снижением в 1,4 раза измеренного среднего титра (3,17E+10 г. в./мл, n=2), а для двухчасовой инкубации измеренный средний титр уменьшался в 1,6 раза (2,67E+10 г. в./мл, n=2) по сравнению с контролем, в котором ДНК/PEI инкубировали в течение 20 минут (4,33E+10 г. в./мл, n=2). Данные свидетельствовали о том, что более длительное время инкубации приводит к снижению титра. Это могло быть связано с тем, что ДНК и PEI образуют большие комплексы, которые не были способны эффективно трансфицировать клетки HEK293. Продуцирование вируса на мл и значения площади поверхности приведены на фигуре 10 в сравнении с продуцированием в известном процессе в качестве положительного контроля.
Титр вируса, измеряемый на каждой стадии стадий осветления и концентрирования, показан на фигуре 11. Остаточный белок клетки-хозяина на каждой стадии в течение стадии TFF1 показан на фигурах 12A-B.
Пример 8. Эффект плотности посева в отношении продуцирования лекарственного вещества
Оценивали эффект плотности посева в отношении продуцирования DS. Оценивали четыре значения плотности посева, и каждое значение плотности посева было представлено в биореакторе в двух повторностях.
Увеличение клеточной массы
Клетки HEK 293 размораживали и ресуспендировали в DMEM, дополненной 10% FBS. Клетки центрифугировали при 209 x g, 5 мин, при комнатной температуре, затем удаляли надосадочную жидкость и добавляли свежую DMEM+10% FBS. Клетки подсчитывали в отношении плотности жизнеспособных клеток и жизнеспособности, и высевали в 2 Т-матраса с площадью поверхности 175 см2, и инкубировали при 37,0°С, 5% СО2 в течение четырех дней, пока культуры не достигали ~90% конфлюэнтности. В случае каждого пересева клеток использованную среду удаляли, матрасы промывали с помощью PBS (-CaCl2, -MgCl2) в количестве 0,08 мл/см2, а затем матрасы обрабатывали TrypLE Select (0,04 мл/см2) и инкубировали при 37,0°С, 5% СО2 в течение 2-3 мин. Трипсин гасили DMEM+10% FBS (0,04 мл/см2). Клетки размножали и высеивали в соответствии с диаграммой, представленной на фигуре 13.
Инокуляция и мониторинг клеток
Биореакторы инокулировали клетками НЕК 293 при четырех целевых значениях плотности, каждое в двух повторностях: 8000 клеток/см2, 9350 клеток/см2, 10700 клеток/см2 и 12050 клеток/см2 в 700 мл питательной среды (DMEM с высоким содержанием глюкозы+10% FBS из Австралии+1:100 Pen Strep) при перемешивании. Задавали следующие параметры обработки: pH 7,23, 37,0°C, 55% растворенного кислорода (DO) и линейная скорость 2 см/с. Через 24 часа после посева запускали рециркуляцию питательной среды DMEM (общий объем теперь 0,188 мл/см2) на скорости рециркуляции (12,5 мл/мин). Отбирали ежедневные образцы для определения pH, метаболитов и питательных веществ с помощью автономного прибора и считывали их с применением Nova BioProfile 400. На пятый день и на девятый день после посева отбирали три волокна и лизировали с помощью PBS, растворов A100 и B100 (ChemoMetec) в соотношении 1:1:1 об./об. и подсчитывали (NucleoCounter NC-200) общее количество ядер для мониторинга роста культуры.
Трансфекция
На пятый день после инокуляции клеток рециркуляцию прекращали и среду внутри каждой камеры биореактора (бутылки без рециркуляции) заменяли на 600 мл среды DMEM -/- (с высоким содержанием глюкозы, - CaCl2, - L-глутамин). Содержимое каждого реактора подвергали трансфекции плазмидной ДНК и полиэтиленимином (PEIpro) в весовом соотношении 1:1. Плазмидные ДНК смешивали в весовом соотношении 1:1,5:2 (pSMN - 3,56 мг, pAAV2/9-5,34 мг и pHELP - 7,1 мг), добавляли к 300 мл среды DMEM -/-, фильтровали через фильтр с размером пор 0,2 мкм и перемешивали путем переворачивания. К 300 мл среды DMEM -/- добавляли PEI (16 мл) и перемешивали путем переворачивания. Смеси PEI и ДНК объединяли, перемешивали путем переворачивания и инкубировали при комнатной температуре в течение 20 минут. Каждые 600 мл смеси комплекса PEI/ДНК использовали для трансфекции содержимого двух биореакторов, повторяли для каждой соответствующей плотности посева. Комплекс PEI/ДНК вносили в каждый биореактор и инкубировали при параметрах обработки в течение двух часов. Рециркуляционную петлю снова включали через два часа после трансфекции (12,5 мл/мин.).
Замена среды после трансфекции
Через 24 часа после трансфекции всю среду в биореакторах и рециркуляционных петлях удаляли и заменяли на OptiMEM (0,132 мл/см2) и продолжали рециркуляцию (12,5 мл/мин) в течение 24 часов при параметрах обработки. Через 48 часов после трансфекции среду в рециркуляционной бутылке заменяли свежей OptiMEM и продолжали рециркуляцию при параметрах обработки (12 мл/мин).
Сбор
На девятый день после инокуляции клеток добавляли бензоназу (100 МЕ/мл), прогоняли буфером для лизиса (50 мМ HEPES, 1% Tween 20) и инкубировали в течение двух часов при параметрах обработки. Биореакторы опустошали и добавляли солевой раствор сахарозы (500 мМ NaCl, 1% вес./об. сахарозы), перемешивали путем переворачивания. Биореактор промывали буфером для промывки биореактора (500 мМ NaCl, 1% вес./об. сахарозы, 20 мМ основания Tris, 1% об./об. Tween 20, 1 мМ MgCl2.6H2O) в течение 15 мин при параметрах обработки. Биореактор опустошали, а буфер для промывки биореактора объединяли с неочищенной общей собранной биомассой, перемешивали путем переворачивания и отбирали образцы для ddPCR-анализа.
Для получения лекарственного вещества в биореакторах использовали плазмиды. Реакторы засевали при различных значениях плотности, трансфицировали и собирали биомассу по одному и тому же графику (день 5, день 9 после посева соответственно). Данными представлено две повторности каждой плотности посева, которые показаны в приведенной ниже таблице 15.
Таблица 15. Номер биореактора с начальной плотностью посева
Рост клеток. Клетки подсчитывали в день посева (день 0), а ядра подсчитывали в день 5 и день 9 после посева. Данные свидетельствовали, что клетки во всех реакторах росли экспоненциально с дня 0 по день 5. Несмотря на различия в начальных значениях плотности посева, количество ядер не указывало на существенные различия в количествах клеток среди групп в день 5. После трансфекции результаты подсчета ядер в день 9 свидетельствовали о том, что в пяти реакторах (221, 224, 225, 226, 228) удваивалось общее количество ядер с дня 5 по день 9. В двух реакторах (222, 223) общее количество ядер увеличивалось в 1,4 раза, как видно на фиг. 14А. В реакторе 227 общее количество ядер увеличивалось в 3,8 раза с дня 5 по день 9. Такое различие могло быть артефактом, основанным на неравномерном распределении клеток по отдельным волокнам, используемым для подсчета. Возможно, что клетки во всех реакторах росли схожим образом, исходя из данных о метаболитах, показанных на фигурах 14B-E.
pH, питательные вещества и метаболиты. Потребление глюкозы (фиг. 14В) одинаково изменялось во всех биореакторных культурах, что позволяло предположить, что, несмотря на различия в начальных значениях плотности посева, культуры потребляли глюкозу со схожими значениями скорости. Значение pH, измеренное с помощью автономного прибора (фиг. 14С), оставалось постоянным (рН 7,25) в течение первых трех дней во всех культурах, снижалось при увеличении метаболизма питательных веществ и увеличивалось после дня 8 одновременно с повышением уровней ионов аммония (фиг. 14Е). Уровень лактата (фиг. 14D) увеличивался до дня 6, а затем выравнивался к концу продуцирования, что позволяло предположить об использовании лактата в качестве источника энергии на этой стадии. Значимого различия в профилях метаболитов не наблюдали между реакторами, засеянными при различных начальных значениях плотности. Это могло быть связано с тем, что различие начальных значений плотности посева было минимальным, т.е. различие в <1,2 раза.
Титры продукции
Вирусные геномы из собранного материала измеряли с помощью цифровой капельной ПЦР (ddPCR). Титры были сопоставимыми между начальными значениями плотности посева 8000 клеток/см2 и 10070 клеток/см2, в среднем составляя 3,99E+10 ± 2,1E+09 г. в./мл (n=2) и 3,70E+10 ± 7,4E+09 г. в./мл (n=2) соответственно. По неидентифицируемой причине реакторы, засеянные при 9350 клеток/см2, характеризовались средним показателем титра 5,02E+08 г. в./мл (n=2), примерно на 2 log ниже, чем средние титры у реакторов, засеянных при соседних значениях плотности. Это различие, вероятно, является следствием неидентифицированной операционной ошибки во время трансфекции или сбора биомассы, а не недостаточной продуктивности при такой плотности посева. У продублированных реакторов, засеянных при 12050 клеток/см2, наблюдали двукратное различие между друг другом, причем один реактор наблюдали в диапазоне для более низких значений плотности посева, 3,2E+10 г. в./мл, в то время как второй давал титр лишь 1,4E+10 г. в./мл. Продуцирование вируса на мл и значения площади поверхности приведены на фигуре 15А.
Плотность посева и продуцирование DS оценивали, как показано на фиг. 15B. Клетки HEK 293, высеянные в диапазоне от 8E+03 до 10E+03 клеток/см2, демонстрировали постоянные профили роста, pH, потребление глюкозы, образование лактата и аммония. Кроме того, были получены сопоставимые титры, свидетельствующие о том, что немного более высокая плотность посева не оказывает отрицательного влияния на продуцирование. Напротив, реакторы, засеянные при более высокой плотности, составляющей 12×103 клеток/см2, характеризовались большей вариабельностью между повторностями, включая более низкий средний титр по сравнению с другими условиями, что свидетельствовало о том, что подход, использованный в этом эксперименте, может не быть оптимальным для продуцирования. Эти результаты обосновывали посев клеток при плотности в диапазоне от 8×103 и 1×104 клеток/см2 для экспериментов с биореактором.
Пример 9. Оценка сопоставимости
Сопоставимость между лекарственным продуктом AVXS-101, использованным в клинических исследованиях фазы 1 (способ A), и лекарственным продуктом, использованным в базовых клинических исследованиях (способ B), оценивали как основную цель со вторичной целью оценки однородности процесса производства с применением способа B путем сравнения серий 600156 и 600307 лекарственного продукта. На фигуре 16 представлены последовательности производственных процессов фазы 1 (способ А) и фазы 3 (способ В) и различия между ними.
Оценку сопоставимости проводили с использованием клинического лекарственного продукта фазы 1, серии NCHAAV9SMN0613, изготовленного в Nationwide Children's Hospital (NCH), и лекарственного продукта серии 600156, изготовленного в AveXis.
Продукт
Оценивали следующие серии материала, которые обобщены в таблице 16. Оценка включала прямое сравнение полученных качественных признаков у клинического лекарственного продукта фазы 1 серии NCHAAV9SMN0613, полученного с применением способа А, и лекарственного продукта AVXS-101 серии 600156, полученного с применением способа В. Кроме того, результаты испытания при выпуске продукции серии 600156 и серии 600307 оценивали в целом в отношении воспроизводимости и однородности процессов в крупном масштабе.
Таблица 16. Лекарственный продукт AVXS-101, подлежащий оценке в отношении сопоставимости между способом A (фазы 1) и способом B (фазы 3) и в отношении однородности процесса производства в случае способа B
Обзор производственного процесса
На фигуре 17A-B представлено обобщение результатов по однородности.
Оценка сопоставимости и однородности процесса производства
Материалы из способа A и способа B были оценены как сопоставимые, а материалы из способа B были оценены как совместимые. Также было определено, что материалы из способа B обладают дополнительными преимуществами, например, для производства в промышленных масштабах.
Способы тестирования
pH
Анализ pH проводили на серии NCHAAV9SMN0613 (способ A), серии 600156 (способ B) и серии 600307 (способ B). Результаты обоих процессов варьировали от 7,9 до 8,0. Это продемонстрировало, что значения pH материалов из способа A и способа B были сопоставимы. и что материалы из способа B были совместимы.
Внешний вид
Визуальную оценку внешнего вида проводили на серии NCHAAV9SMN0613 (способ A), серии 600156 (способ B) и серии 600307 (способ B). Очевидные различия в результатах оценки внешнего вида между способом A и способом В были обусловлены различными концентрациями вектора (геномный титр). Серия NCH AAV9SMN0613 характеризовалась более низкой концентрацией вектора, чем серии способа B. В результате, серия NCH AAV9SMN0613 была более разбавленной, что приводило к получению раствора, являющегося в большей степени прозрачным и бесцветным, тогда как в случае серий способа В наблюдения от бесцветного до белого и с небольшой степенью непрозрачности являлись результатом примерно 4-кратного увеличения концентрации вирусных частиц в растворе на мл.
Принимая во внимание различие концентрации, внешний вид материалов из способа A и способа B были оценены как сопоставимые, а материалы из способа B были оценены как совместимые.
Осмоляльность
Оценку осмоляльности по снижению температуры замерзания проводили на серии NCHAAV9SMN0613 (способ A), серии 600156 (способ B) и серии 600307 (способ B). Результаты обоих процессов варьировали от 410 до 415 мОсм/кг. Это продемонстрировало, что значения осмоляльности материалов из способа A и способа B были сопоставимы и что материалы из способа B были совместимы.
Частицы, невидимые невооруженным глазом
Оценку частиц, невидимых невооруженным глазом, методом светотени проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Результаты обоих процессов были значительно ниже рекомендуемых пределов в монографии USP для инъекционных лекарственных продуктов. Это продемонстрировало, что количества частиц, невидимых невооруженным глазом, для материалов из способа А и способа В были сопоставимы, и что материалы из способа В были совместимы.
Геномный титр
Оценку геномного титра с помощью ddPCR проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Согласно ожиданиям, геномный титр для серий AVXS-101 должен был колебаться в зависимости от целевых концентраций при производстве. Геномный титр, полученный с помощью способа В (3,7×1013 г. в./мл и 4,0×1013 г. в./мл), был как минимум в 3 раза выше, чем в случае способа А (1,1×1013 г. в./мл), поэтому способ В был лучшим способом крупномасштабного производства AVXS-101.
Инфекционный титр
Оценку инфекционного титра с помощью TCID50 осуществляли на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Способ B (1,3×1010 МЕ/мл и 6,7×109 МЕ/мл) обеспечивал в среднем на 66% более высокий инфекционный титр, чем способ А (5,9×1010 МЕ/мл), что может быть преимущественным, например, в случае крупномасштабного производства rAAV, например, AVXS-101.
Общий белок
Оценку общего белка с помощью микропланшетного анализа BCA проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Нормализовали к 1,0×1013 г. в./мл, при этом результаты, полученные для обоих процессов, варьировали от 167 до 179 мкг/мл. По нормализованным значениям общего белка было видно, что способ А и способ В были сопоставимы, и что материалы из способа В были совместимы.
Идентичность по результатам вестерн-блоттинга
Оценку идентичности с помощью вестерн-блоттинга проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Профиль блоттинга и значения кажущейся молекулярной массы для основных полос (VP1, VP2 и VP3) были оценены как сопоставимые для материалов из способа А и способа В, а также было оценено, что материалы из способа В были совместимыми.
% Пустых капсидов по AUC
Оценку % пустых капсидов с помощью AUC проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). Результат для серии NCHAAV9SMN0613 (способа А) составлял 7%. Результаты для серий 600156 и 600307 (способа В) составляли 2% и 4% соответственно. Способ B (2% и 4%) обеспечивал в приблизительно два раза меньше пустых капсидов, по результатам измерения AUC, чем способ А (7%). Следовательно, способ В позволял получить улучшенную композицию, содержащую более низкую концентрацию пустых капсидов.
Идентичность и чистота по SDS-PAGE
Оценку идентичности и чистоты с помощью SDS-PAGE проводили на серии NCHAAV9SMN0613 (способ А),
серии 600156 (способ В) и серии 600307 (способ В). % Общей чистоты в обоих процессах составлял ≥ 98%, а картина разделения на полосы, а также кажущаяся молекулярная масса для каждого из трех капсидных белков были в высокой степени совместимыми. По этим результатам было видно, что способ А и способ В были сопоставимы, и что материалы из способа В были совместимы.
Остаточный белок клетки-хозяина
Оценку остаточного белка клетки-хозяина с помощью ELISA проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). В случае данного анализа результаты для всех протестированных серий были < LOQ (8 нг/мл). По этим результатам было видно, что способ А и способ В были сопоставимы, и что материалы из способа В были совместимы.
Остаточный бычий сывороточный альбумин (BSA)
Оценку остаточного бычьего сывороточного альбумина (BSA) проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). В случае данного анализа результаты для всех протестированных серий были < LOQ (0,50 нг/мл). По этим результатам было видно, что способ А и способ В были сопоставимы, и что материалы из способа В были совместимы.
Остаточная бензоназа
Оценку остаточной бензоназы с помощью ELISA проводили на серии NCHAAV9SMN0613 (способ А), серии 600156 (способ В) и серии 600307 (способ В). В случае данного анализа результаты для всех протестированных серий были < LOQ (0,20 нг/мл). По этим результатам было видно, что способ А и способ В были сопоставимы, и что материалы из способа В были совместимы.
Остаточная ДНК клетки-хозяина
Оценку остаточной ДНК клетки-хозяина с помощью qPCR проводили на серии NCHAAV9SMN0613 (способ А),
серии 600156 (способ В) и серии 600307 (способ В). Нормализовали к 1,0×1013 г. в./мл, результат для способа А составлял 3,7×105 пг/мл, тогда как результаты для способа В составляли 0,76×105 пг/мл и 0,68×105 пг/мл соответственно. Следовательно, способ В обеспечивал вирусные векторы со значительно меньшим содержанием остаточной hcDNA, что может быть преимущественным, например, в случае крупномасштабного производства rAAV, например, AVXS-101.
Статистический анализ
Статистический анализ проводили по количественным и качественным признакам. Сравнения проводились попарно между серией способа А (NCHAAV9SMN0613) и каждой серией способа В (600156 и 600307), как указано ниже. Эти результаты показаны на фигурах 18 и 19.
По результатам этих исследований видно, что способ В является превосходным способом получения вирусных векторов. Способ B согласованно обеспечивал большее количество вирусных векторов (как измерено по геномному титру и инфекционному титру) с небольшим количеством примесей (более низкое содержание остаточной hcDNA) с меньшим количеством пустых капсидов.
Секвенирование следующего поколения
Также проводили секвенирование следующего поколения (NGS) для установления идентичности (определения и/или подтверждения геномной последовательности) и для оценки, существуют ли варианты последовательностей (субпопуляции) в случае материала фазы 3 лекарственного продукта AVXS-101 из способа В. В результате выравнивания набора данных последовательностей относительно предоставленной спонсором эталонной последовательности (pscSMN) выявляли полную (100%) ширину и достаточную глубину охвата по всей длине генома, что обеспечивает возможность обнаружения вариантов. Всего было отмечено четыре минорных варианта положений, однако они, судя по всему, представляли собой ошибки секвенирования в труднодоступных для секвенирования областях (например, инвертированных концевых повторах (ITR) у AAV, которые, как известно, трудно секвенировать из-за высокого содержания в них GC и палиндромных последовательностей), а не истинные варианты. Результаты секвенирования см. в таблице 17.
Таблица 17. Результаты секвенирования ДНК серии 600156 AVXS-101 фазы 3, полученной посредством способа B
считываний,
использованное для
картирования
последовательность
Профиль стабильности серии NCHAAV9SMN0613 фазы 1
Серию NCHAAV9SMN0613 хранили в течение 12 месяцев при ≤ -60°C. Данную серию анализировали в каждый момент времени. Никаких неблагоприятных тенденций отмечено не было. На фигуре 20 показаны результаты по стабильности на сегодняшний день.
Исследование сопоставимости проводили для AVXS-101, использованного в клинических исследованиях фазы 1. Оценку проводили с использованием клинического лекарственного продукта фазы 1, серии NCHAAV9SMN0613, изготовленного в Nationwide Children's Hospital, и лекарственного продукта AVXS-101 серии 600156, изготовленного в AveXis. Дополнительно оценивали однородность процесса производства, используя серии 600156 и 600307 из способа В. Как для оценки сопоставимости (способ А по сравнению со способом В), так и для оценки однородности процесса производства (серии 600156 по сравнению с серией 600307 способа В) в исследовании оценивали идентичность, качество, чистоту материала клинического испытания AVXS-101 с использованием недавно улучшенного процесса и аналитических способов, обеспечивающих получение надежной оценки сопоставимости и однородности процесса производства.
Статистический анализ проводили по количественным и качественным признакам. Сравнения проводились попарно между серией способа А NCHAAV9SMN0613 и серией способа В 600156. Способ B был лучшим способом, который давал большее количество вирусных векторов с более высокой чистотой, чем способ А. Например, по сравнению со способом A вирусные векторы, полученные с помощью способа В, имели инфекционный титр на 48% выше, геномный титр на 8% выше, на 92% меньше частиц, невидимых невооруженным глазом, размером более 10 мкм, на 50% меньше частиц, невидимых невооруженным глазом, размером более 25 мкм, на 100% меньше пустых капсидов и на 11% меньше остаточной hcDNA. Все результаты были совместимы относительно предела испытания для каждого качественного признака.
Более того, для установления однородности процесса производства с использованием способа В проводили попарное сравнение с применением серий 600156 и 600307. Результат, полученный в результате такого начального попарного сравнения между серией 600156 и 600307 из способа В, демонстрирует однородность в процессе производства. Все результаты были также совместимы относительно предела испытания для каждого качественного признака.
Исходя из результатов оценки, полученные качественные признаки серии клинического лекарственного продукта NCHAAV9SMN0613 фазы 1, полученной с применением способа А, и серии 600156 лекарственного продукта AVXS-101, полученной
с применением способа В, демонстрировали, что способ В обеспечивал более высокие количества вирусного вектора и улучшенную чистоту, что может быть преимущественным, например, в случае крупномасштабного производства rAAV. Кроме того, было обнаружено, что две серии материала, полученные с помощью способа В (серии 600156 и 600307), были воспроизводимы, что дополнительно характеризует однородность процесса производства.
Пример 10. Анализ с использованием ультрацентрифугирования для аналитического исследования (AUC)
С применением способа AUC анализировали материал из фазы 1 (способ А, серия NCHAAV9SMN0613) и фазы 3 (способ В, серии 600156 и 600307). Профили AUC (проанализированные в двух повторностях) для NCHAAV9SMN0613, 600156 и 600307, показаны на фигуре 21, фигуре 22 и фигуре 23 соответственно.
AUC-анализ каждого материала обеспечивал схожие коэффициенты седиментации для пустых и полных капсидов с материалом фазы 1 (способ А, серия NCHAAV9SMN0613), при этом демонстрируя повышенное содержание пустых капсидов (7%) по сравнению с материалом фазы 3 (способ В, серии 600156 и 600307) с содержанием пустых капсидов 2% и 4% соответственно. Это связано с возможностью посредством ультрацентрифугирования в градиенте CsCl на стадии получения способа B более эффективно отделять пустые капсиды от полных капсидов по сравнению с ультрацентрифугированием в градиенте йодиксанола на стадии получения, которая используется в способе A.
Серии продукции AVXS-101, при использовании клинического и коммерческого представления, сопоставимо характеризовались тремя видимыми полосами капсидов, если их подвергнуть процессу очистки в градиенте CsCl с использованием ультрацентрифугирования, как в материале клинического испытания фазы 1, произведенном в Nationwide Children's Hospital (NCHAAV9SMN0613), так и в каждой из последующих серий продукции, произведенных AveXis. Исходя из профилей AUC для клинического лекарственного продукта фазы 1 серии NCHAAV9SMN0613, полученного с применением способа А, и лекарственного продукта AVXS-101, серии 600156, полученного с применением способа В, эти материалы были расценены как сопоставимые. Кроме того, профили AUC для двух серий материала, полученных с помощью способа В (серии 600156 и 600307), были оценены как совместимые.
Пример 11. Процесс накопления
Процесс накопления применяли для получения промежуточного продукта, полученного из рабочего банка клеток, где процесс накопления включает стадии (а) культивирования клеток, (b) трансфекции культивированных клеток тремя плазмидами, которые показаны на фигуре 1, (c) осуществления сбора увеличенного количества вирусных частиц из клеток после периода культивирования, (d) очистки вирусных частиц с помощью фильтрации для удаления любых интактных клеток или клеточного дебриса, (е) подвергания элюента из стадии (d) фильтрации в тангенциальном потоке и (f) замораживания полученного промежуточного препарата очищенных вирусных частиц.
Перед трансфекцией клетки размножали в подходящих культуральных средах, в матрасах или в подходящем биореакторе, или как в первом, так и во втором. Одна культуральная среда представляла собой DMEM с 10% FBS, 4,5 г/л глюкозы, 4 мM L-глутамина. В одном варианте осуществления адгезивные клетки сначала выращивали в матрасах, а затем переносили в биореактор iCELLis для дополнительного размножения адгезивных клеток в биореакторе.
После размножения клеток адгезивные клетки HEK293 трансфицировали совместным осаждением с тремя ДНК-плазмидами и PEI. 3 плазмиды, применяемые для этой трансфекции, представляли собой pSMN, pAAV2/9 и pHELP. Питательную среду DMEM, применяемую для размножения клеток, заменяли модифицированной средой DMEM для трансфекции. Среда для трансфекции DMEM не содержала FBS, кальция, L-глутамина и содержала 4,5 г/л глюкозы. Вектор scAAV9.CB.SMN получали с применением трансфекции тремя ДНК-плазмидами в адгезивные клетки HEK293 путем совместного осаждения с PEI в крупномасштабном биореакторе для адгезивных клеток. Векторная плазмида pSMN содержала cDNA для SMN человека. 3 плазмиды, применяемые для этой трансфекции, представляли собой pSMN (222 мг), pAAV2/9 (333 мг) и pHELP (444 мг). Перед добавлением PEI и плазмид для совместного осаждения среду для трансфекции оставляли для уравновешивания в биореакторе до тех пор, пока температура в биореакторе не составляла >30°C. Процесс совместного осаждения PEI и плазмид предусматривал добавление плазмид в среду для трансфекции и фильтрацию через фильтр с размером пор 0,2 мкм в реакционный пакет. PEI добавляли в среду для трансфекции, а затем в реакционный пакет. Реакционную среду из PEI и плазмид вручную перемешивали до получения гомогенной суспензии, и реакция проходила за период продолжительностью 15-30 минут. В конце времени реакции PEI и плазмиды для совместного осаждения переносили из реакционного пакета в биореактор. Обеспечивали перемешивание PEI и плазмид для совместного осаждения в биореакторе в течение 1-2 часов перед возобновлением рециркуляции. Питательную среду DMEM подвергали рециркуляции в биореакторе в течение 18-24 часов перед заменой следующей средой.
Геном SMN rAAV (нуклеотиды 980-3336 из SEQ ID NO: 1) имеет в последовательности ITR AAV2, промотор β-актина курицы с цитомегаловирусным энхансером, интрон SV40, ДНК, кодирующую SMN, представленную под (номером доступа NM_000344.2 в GenBank), сигнальную последовательность полиаденилирования из бычьего гормона роста и другой ITR AAV2. Также рассматриваются консервативные нуклеотидные замены в ДНК SMN (например, замена гуанина на аденин в положении 625 указанного в GenBank под номером доступа NM_000344.2). В геноме отсутствует ДНК rep и cap AAV, т.е. нет ДНК rep или cap AAV между ITR генома. Рассматриваемые полипептиды SMN включают без ограничения полипептид SMN1 человек, приведенный в базе данных белков NCBI под номером NP_000335.1. Вектор rAAV9 SMN описан в Foust et al., Nature Biotechnology 28(3): 271-274 (2010), причем последовательность вставки векторного генома показана в виде нуклеотидов 980-3336 из SEQ ID NO: 1).
В день 6 в биореакторе, через 18-24 часа после трансфекции, биореактор опустошали, а DMEM в пакете с рециркулирующей средой заменяли на 200 литров свежей OptiMEM для применения после трансфекции. Биореактор повторно заполняли 64 литрами и возобновляли в биореакторе рециркуляцию. В день 7, через 18-24 часа после замены в день 6, OptiMEM для применения после трансфекции в рециркуляционном пакете (~135 литров) заменяли свежим пакетом среды OptiMEM. В ходе этой стадии биореактор не опустошали. Рециркуляцию среды продолжали до сбора биомассы в день 9.
Спустя 9 дней в биореакторе из реактора отбирали последние образцы перед сбором биомассы и начинали процесс лизиса клеток. В биореактор добавляли бензоназу до конечной концентрации 100 МЕ/мл. После этого обеспечивали перемешивание бензоназы в реакторе и в реактор добавляли 7,1 литра раствора для лизиса. Раствор для лизиса перемешивали в реакторе в течение 2 часов перед первой стадией сбора биомассы. В конце 2-часового лизиса содержимое биореактора переносили в пакет для сбора биомассы. В пакет для сбора биомассы добавляли 8,9 литра солевого раствора сахарозы (SSS) и перемешивали в течение 15 минут. Раствор SSS гасил бензоназу в собранной среде. Затем биореактор ополаскивали буфером для промывки биореактора. Для ополаскивания биореактора в биореактор добавляли 64 литра буфера для промывки биореактора и перемешивали в течение 15 минут. Затем смыв переносили в общий пакет для накопления собранной биомассы. После добавления смыва в пакет для накопления его содержимое перемешивали в течение 15 минут и отбирали образцы общей собранной биомассы.
Смешанную общую собранную биомассу фильтровали через глубинный фильтр в пакет для накопления. После того, как всю общую собранную биомассу отфильтровали, через глубинный фильтр прогоняли 50 литров буфера для диафильтрации при TFF1. Пул, полученный с помощью глубинного фильтра, перемешивали и отбирали из него образцы. Полученный с помощью глубинного фильтра пул затем фильтровали через фильтр с размером пор 0,45 мкм для дополнительного осветления материала общей собранной биомассы. Затем через фильтр с размером пор 0,45 мкм прогоняли 6 литров буфера для TFF1.
В ходе стадии TFF1 5,0 м2 кассеты с мембранами из регенерированной целлюлозы с порогом отсечения, представляющим собой MW 300 кДа, промывали, дезинфицировали раствором NaOH и уравновешивали буфером для TFF1. Фаза концентрирования в данном процессе предназначалась для уменьшения объема осветленной собранной биомассы примерно в 10 раз. После достижения целевого объема ретентата начинали процесс диафильтрации. Ретентат подвергали диафильтрации с помощью 6 диаобъемов буфера для TFF1. После достижения 6 диаобъемов общего потока пермеата ретентат снова концентрировали и собирали в пакет для накопления. Проводили два последовательных ополаскивания мембраны для максимального извлечения продукта из системы TFF с получением промежуточного лекарственного вещества. Отбирали аликвоту промежуточного продукта, полученного в результате TFF1, в стерильные бутылки из PETG объемом 1 или 2 литра в вытяжном шкафу LFH, а затем замораживали их на сухом льду или в морозильной камере и переносили на хранение при -60°C.
Таблица 18. Буферы, применяемые в процессе накопления
без L-глутамина и 4,5 г/л глюкозы
4 мM L-глутамина и без FBS
Пример 12. Процесс выделения и очистки
Процесс выделения и очистки использовали для обработки промежуточного продукта с получением отфильтрованного лекарственного вещества. Стадии процесса выделения и очистки включали стадию подкисления и осветления (с применением фильтрации) с последующей катионообменной хроматографией, фильтрацией в тангенциальном потоке ("TFF2"), ультрацентрифугированием с помощью CsCl и дополнительной стадией фильтрации в тангенциальном потоке ("TFF3") с получением отфильтрованного лекарственного вещества, при этом очищенные частицы AAV суспендированы в фармацевтически приемлемом носителе. В частности, процесс выделения и очистки включал следующие стадии получения, следующие после получения промежуточного продукта в результате TFF1: размораживание и объединение промежуточного продукта, полученного в результате TFF1, подкисление и осветление, осуществление катионообменной хроматографии (CEX), фильтрацию в тангенциальном потоке (TFF2), ультрацентрифугирование с помощью CsCl для разделения полных/пустых капсидов, фильтрацию в тангенциальном потоке (TFF3) для концентрирования/замены буфера, фильтрацию полученного в результате TFF3 объединенного материала с образованием лекарственного вещества, разбавление и фильтрацию лекарственного вещества с получением лекарственного продукта, хранение лекарственного продукта и заполнение лекарственным продуктом флаконов.
Промежуточный материал, полученный в результате TFF1, размораживали и осторожно перемешивали. Tween 20 использовали для стимуляции флоккуляции основной массы белков и ДНК клеток-хозяев в кислом рН. Значение pH объединенного промежуточного продукта, полученного в результате TFF1, содержащего 15% Tween 20, снижали для CEX-хроматографии (pH 3,5). Образовавшийся после снижения рН осадок затем удаляли фильтрованием раствора через глубинные фильтры и фильтры с размером пор 0,45 мкм.
Tween 20 (36% раствор Tween 20 в 20 мМ Tris, 1 мМ MgCl2, 500 мМ NaCl, 1% сахарозы вес./об., pH 8,1) медленно добавляли к раствору промежуточного продукта, полученного в результате TFF1, в течение 4 часов до достижения конечной концентрации 20% Tween 20. После инкубации в течение ночи при комнатной температуре (к.т.) значение рН промежуточного продукта, полученного в результате TFF1, содержащего Tween 20, снижали путем добавления примерно 4 г 1 М глицина, рН 2,5, на кг объединенного промежуточного продукта, полученного в результате TFF1/внесенного Tween до достижения целевого значения рН, составляющего 3,5±0,1. После достижения значением pH допустимого диапазона раствор пропускали через глубинный фильтр Clarisolve POD наряду с фильтром с размером пор 0,45 мкм Opticap XL10 Durapore или PES-фильтром с размером пор 0,8/0,45 мкм с последующей промывкой фильтров двойным объемом задержки фильтра POD плюс одним объемом задержки фильтра тонкой очистки буфером A для CEX.
Стадию захватной катионообменной хроматографии (CEX) применяли для отделения вирусных капсидов от белка, ДНК и других технологических примесей. На этой стадии использовали хроматографическую (с сульфонильными группами) колонку CIMmultus S03-8000 Advanced Composite Column (с размером пор 2 мкм) (8,0 л), которая управлялась с применением хроматографической системы с автоматизированным процессом. Буферы и растворы описаны в следующей таблице.
Таблица 19. Буферы и растворы для одного цикла CEX
pH 3,5 ± 0,1 при 20°C
pH 3,5 ± 0,1 при 20°C
pH 9,1 ± 0,1 при 20°C
Загрузку в колонке для CEX определяли по содержанию белка в осветленном подкисленном промежуточном продукте, полученном в результате TFF1. Загрузку белка для колонки для CEX устанавливали на уровне 70% максимальной емкости колонки.
Пик элюирования собирали вручную, начиная с резкого повышения OD280. OD280 повышается при коэффициенте проводимости 80-85 мСм/см. Примерный объем элюата, полученного в результате CEX (продукта), составлял ~20 литров или 2,5 CV (объема колонки). Элюат, полученный в результате CEX, собирали в виде двух фракций. Первая фракция начиналась с резкого повышения OD280, и ее собирали за 1,5 CV. Вторая фракция начиналась сразу после первой фракции, и ее собирали за 1,0 CV. Две фракции нейтрализовали до рН 8,0 ± 0,30 с применением нейтрализующего буфера с рН 9,0.
Продукт стадии TFF2 концентрировали, удаляли белковые примеси и заменяли буфер на подходящий буфер для стадии ультрацентрифугирования с помощью CsCl. Систему фильтрации в тангенциальном потоке применяли в сочетании с мембранами из регенерированной целлюлозы с MWCO 300 кДа размером 0,4 м2 (два цикла CEX) или 0,2 м2 (один цикл CEX).
В ходе фазы концентрирования в данном процессе уменьшали объем элюатов, полученных в результате CEX. После достижения целевого объема ретентата начинали диафильтрацию в периодическом режиме TFF (порционном режиме). Ретентат разбавляли в 2 раза и подвергали диафильтрации 8 диаобъемов буфера на основе NaCl для диафильтрации при TFF2 и после этого 8 диаобъемов буфера с CsCl для диафильтрации TFF2 в периодическом режиме TFF. После завершения диафильтрации с CsCl ретентат концентрировали до заданного объема, который зависел от объема задержки в системе. Проводили два последовательных ополаскивания мембраны для максимального извлечения продукта из системы TFF2.
Скорость подачи ретентата устанавливали на 5 л/м2/мин (500 мл/мин на 0,1 м2 кассеты) с 20% коэффициентом преобразования в пермеат (скорость потока пермеата 100 мл на 500 мл скорости подачи ретентата). Скорость потока пермеата контролировали с помощью зажима на трубке для пермеата, поддерживая скорость потока пермеата на уровне 20% от скорости потока подачи ретентата.
Таблица 20. Буферы для TFF2
Ультрацентрифугирование можно было использовать для удаления пустых капсидов от полых капсидов путем применения ультрацентрифугирования в градиенте хлорида цезия. Для стадии ультрацентрифугирования с помощью CsCl использовали автоматизированную систему ультрацентрифугирования Optima XPN 100 или эквивалентную систему, оснащенную ротором типа 50.2 Ti или эквивалентным ротором. Очищенный отфильтрованный материал TFF2 медленно добавляли в пробирки для ультрацентрифуги по внутренней части стенки пробирки без введения пузырьков в раствор. Заполненные пробирки герметично закрывали ручным устройством для термогерметизации и центрифугировали при 302000 g (50000 об/мин в роторе 50.2 Ti) в течение 17 часов при 20°C. После завершения стадии центрифугирования пробирки извлекали из ультрацентрифуги и помещали в бокс микробиологической безопасности. Содержащие продукт пробирки ставили на кольцевых стойках над контейнером для отходов. Непосредственно под пробиркой размещали лампу и на пробирках отмечали полосы пустых капсидов (полоса А является полосой, занимающей самое верхнее положение), парные полосы полных капсидов (полосы В и полосы С, верхней и нижней полосы пары) и полосу, занимающую самое нижнее положение под парой. Полосы B, C и D удаляли иглой размером 18G, прикрепленной к шприцу объемом 30 мл, вставленной непосредственно под полосу D в середине пробирки. Собранный материал переносили в стерильную бутылку из PETG объемом 1 л. Материал из всех центрифужных пробирок собирали в стерильную бутылку из PETG объемом 1 л с получением ультрацентрифужного (UC) пула. Буфер для стадии ультрацентрифугирования с помощью CsCl указан в таблице ниже.
Таблица 21. Буфер для ультрацентрифугирования с помощью CsCl
На стадии TFF3 удаляли CsCl и концентрировали полный вектор с применением буфера для получения конечного состава. Систему фильтрации в тангенциальном потоке использовали в сочетании с двумя мембранами из регенерированной целлюлозы с MWCO 300 кДа размером 50 см2. Фаза концентрирования процесса TFF3 предназначалась для уменьшения концентрации остаточного CsCl и объема UC-пула. После достижения целевого объема ретентата начинали диафильтрацию. Ретентат подвергали диафильтрации с помощью 10 диаобъемов буфера для TFF3. По завершении диафильтрации концентрированный ретентат переносили во вторичную коническую пробирку через стерилизующий фильтр с размером пор 0,2 мкм Pall Supor® EKV (Mini Kleenpak).
Проводили последовательное ополаскивание мембраны для извлечения вектора из системы TFF3. Вносили буфер для TFF3 в первичную коническую пробирку, в которой ранее содержался ретентат TFF3. Этот материал подвергали рециркуляции через целлюлозные мембраны. После рециркуляции смыв переносили во вторичную коническую пробирку через стерилизующий фильтр с размером пор 0,2 мкм Pall Supor® EKV (Mini Kleenpak). Концентрат TFF3 и частичный пул смешивали для достижения конечной концентрации вектора ≥ 4,5 X 1013 г. в./мл лекарственного вещества (объединенный ретентат TFF3+два ополаскивания).
Проводили последовательное ополаскивание мембраны для максимального извлечения продукта из системы TFF3. Вносили буфер для TFF3 в первичную коническую пробирку, в которой ранее содержался ретентат TFF3 и материал первоначального смыва. Этот материал подвергали рециркуляции через целлюлозные мембраны. Вторичный смыв переносили во вторичную коническую трубку через стерилизующий фильтр с размером пор 0,2 мкм Pall Supor® EKV (Mini Kleenpak) до тех пор, пока во вторичной конической пробирке не достигался определенный вес. Конечный концентрированный раствор назывался лекарственным веществом (DS).
Таблица 22. Буферы для TFF3
DS фильтровали с помощью стерилизующего фильтра Pall Supor® EKV (Mini Kleenpak) в стерильную стеклянную бутылку объемом 1 л, используя стерилизованную одноразовую сборную конструкцию. Перед фильтрацией пула TFF3 фильтр промывали, пропуская буфер для TFF3 через фильтр с использованием перистальтического насоса и отбрасывая его в пакет для смыва. Затем лекарственное вещество (DS) фильтровали через промывной фильтр с использованием перистальтического насоса и собирали в стерильную стеклянную бутылку объемом 1 л. Исходя из целевой концентрации DS в 5×1013 г. в./мл, добавляли буфер для TFF3 во вторичную коническую пробирку, которая содержала DS, и пропускали через фильтр для получения разбавленного лекарственного продукта ("DP") с целевой концентрацией 3,5×1013 г. в./мл.
Буфер для TFF3, использованный при промывке фильтра и разбавлении DS для получения DP, имел следующий состав.
Таблица 23. Типовой процесс получения лекарственного продукта - состав буфера
DP разливали в стерильные, готовые к использованию флаконы объемом 5 мл Crystal Zenith (CZ), закупоривали стерильными, готовыми к использованию пробками и герметично закрывали стерильными, готовыми к использованию колпачками.
Пример 13. Анализ активности
Относительную активность лекарственного продукта измеряли с применением количественного анализа in vivo. В анализе применяли общепринятую модель заболевания SMA на мышах. Пары для разведения линии мышей SMAΔ7 (Jackson Laboratories, № 005025) имеют нормальный фенотип, но ~25% их потомков являются гомозиготными по целевой мутации гена SMN и проявляют SMA-подобный фенотип. Ко дню 5 у них появляются признаки мышечной слабости, а на следующей неделе развивается аномальная походка и тенденция к падению. По сообщениям Jackson Laboratories, среднее значение выживаемости животных с SMA-подобным фенотипом составляет ~15 ± 2 дня. В поисковых исследованиях было продемонстрировано, что медианное время выживания для животных с SMA-подобным фенотипом без обработки составляло 16,3 дня (среднее геометрическое; n=3 исследования; 10 мышей на исследование).
Биологически активный лекарственный продукт, вводимый путем внутривенной (IV) инфузии, приводит к увеличению времени выживания, которое зависит от дозы (г. в./кг). Активность лекарственного продукта измеряли относительно эталонного материала (предыдущей партии вектора). Титр лекарственного продукта и эталонного материала (геномов вектора/мл; г. в./мл) определяли с помощью капельной цифровой полимеразной цепной реакции (ddPCR). Вектор разбавляли солевым раствором для достижения каждого из трех указанных уровней доз, которые будут введены мышам с SMA-подобным фенотипом.
Результаты анализа считали приемлемыми, если анализ удовлетворял критериям пригодности. Критерии пригодности анализа заключались в следующем.
4. Предел приемлемости для образца отрицательного контроля (15 ± 2 дня, медианное значение выживаемости);
5. Предел приемлемости для образца положительного контроля (> 40 дней, медианное значение выживаемости);
6. Пределы приемлемости на кривой зависимости ответа от дозы для медианного значения выживаемости при применении эталонного стандарта.
В данном исследовании применяли предыдущую партию вектора (далее по тексту "предыдущая партия") для определения линейной корреляции между медианным значением выживаемости (в днях) мыши SMAΔ7 при введении дозы лекарственного продукта при пяти различных уровнях дозы, включая 0 (нулевую) дозу с применением 0,9% солевого раствора (группа без обработки). Это осуществляли путем применения соотношения точки пересечения с осью Y и углового коэффициента каждой линии линейной регрессии (т.е. эталонного стандарта и исследуемого препарата). Расчет процента относительной активности описан в уравнении (1) ниже.
%RP = [(точка пересечения с осью Y/угловой коэффициент исследуемого препарата) ÷ (точка пересечения с осью Y/угловой коэффициент эталонного стандарта)] X 100 (1)
Для демонстрации эффективности терапевтических средств против SMA, в том числе лекарственного продукта, применяли модель на мышах Δ7. Контрольные животные, обработанные солевым раствором или не подвергнутые обработке, обеспечивали надежный контроль исходного уровня, относительно которого можно было измерять активность продукта в виде увеличения медианного значения выживаемости . При разработке лекарственного продукта были выявлены три (3) дозы (исключая дозу при обработке средой-носителем), определенные по геномному титру с применением капельной цифровой ПЦР (ddPCR), которые влияют на выживаемость в модели на мышах с линейной корреляцией, если значение вводимой дозы (г. в./кг) подвергнуть логарифмическому преобразованию и нанести на график в зависимости от медианного значения выживаемости (в днях) у подвергнутой обработке новорожденной мыши SMAΔ7. См. стандартные титры (г. в./мл) в таблице 26 для стандартов низкого, среднего и высокого титра. Кроме того, буферный раствор для TFF (среду-носитель) применяли как в качестве нулевой точки (0) калибровочной кривой, так и в качестве отрицательного контроля. В качестве положительного контроля также включали дозу, для которой наблюдали выживаемость ≥40 дней (больше, чем в случае дозы, для которой наблюдали удвоение медианного значения выживаемости).
Таблица 25. Целевые дозы
(без обработки)
Препараты растворов доз (см. пример схемы разбавления в таблице 26).
Отрицательный контроль - в качестве отрицательного контроля использовали 0,9% солевой раствор.
Положительный контроль - серию исследуемого препарата готовили в концентрации 1,5×1014 г. в./кг с применением солевого раствора.
Растворы эталонного стандарта - серию эталонного стандарта готовили в трех концентрациях, указанных в таблице 25, с применением солевого раствора.
Таблица 26. Схема разбавления эталонного стандарта и исследуемого препарата (пример)
исследуемого препарата по ddPCR (г. в./мл)
исследуемого препарата для применения (мкл)
(мкл)
(мкл)
Препарат исследуемого препарата - исследуемый препарат разбавляли солевым раствором. Разбавления рассчитывали для получения тестовых доз (г. в./кг), указанных в таблице 26, на мышь в общем конечном объеме 50 мкл. Разбавления делали для 10 мышей одновременно с одним дополнительным объемом в качестве положительного контроля, нацеленного на увеличение минимальной продолжительности жизни подвергнутых обработке мышей до медианного значения выживаемости (дни), составляющего ≥40 дней.
Пределы приемлемости на контрольных образцах
Отрицательный контроль (мыши без обработки) - предел приемлемости анализа для группы отрицательного контроля состоял в том, что мыши SMAΔ7 соответствовали критерию медианного значения выживаемости 15 ± 2 дня. Кроме того, любую мышь, погибшую через ≤ 10 дней, исключали из анализа. Если в группе использовали более 7 мышей, в течение <= 10 дней по причине гибели можно было исключить максимум 2 мыши.
Положительный контроль (группа, которую обрабатывали целевой клинической дозой) - предел приемлемости анализа для группы положительного контроля заключался в том, что минимальная продолжительность жизни подвергнутых обработке мышей должна была составлять ≥ 40 дней, что представляло собой медианное значение выживаемости. Кроме того, любую мышь, погибшую через ≤ 10 дней, исключали из анализа. Если в группе использовали более 7 мышей, в течение <= 10 дней по причине гибели можно было исключить максимум 2 мыши.
Пределы приемлемости на кривой зависимости ответа от дозы для эталонного стандарта
Критерии пригодности анализа для линейной кривой зависимости ответа от дозы для эталонного стандарта, построенной в виде зависимости медианного значения выживаемости (дни) от вводимой дозы (г. в./мл), будут определены. Соотношение точка пересечения с осью Y/угловой коэффициент - определяли кривую линейной регрессии зависимости медианного значения выживаемости (в днях) от вводимой дозы (г. в./кг) для эталонного стандарта и исследуемого препарата. Для каждого случая линейной регрессии рассчитывали соотношение точки пересечения с осью Y и углового коэффициента.
Представление результатов
Качественное представление результатов определения относительной активности - оценивали критерии пригодности анализа для каждого анализа перед определением медианного значения выживаемости в одной точке (в днях), определяемого через ≥40 дней по материалу положительного контроля. Если медианное значение выживаемости у группы положительного контроля составляло ≥ 40 дней, исследуемый препарат можно использовать, если соблюдены критерии, представленные ниже.
Количественное представление результатов определения относительной активности - перед количественным определением относительной активности исследуемого препарата для каждого анализа оценивали критерии пригодности анализа. Относительную активность для исследуемого препарата можно было регистрировать, когда положительный контроль достигал ≥ 40 дней, и медианное значение выживаемости для группы мышей, представляющей верхнюю стандартную дозу 7,5×1013 г. в./кг, составляло 31 ± 3 дня. Относительную активность в процентах (% RP) для исследуемого препарата рассчитывали с применением точки пересечения с осью Y и углового коэффициента линейной регрессии кривой зависимости ответа от дозы для медианного значения выживаемости (в днях) следующим образом:
%RP=100% * [(точка пересечения с осью Y/угловой коэффициент исследуемого препарата) ÷ (точка пересечения с осью Y/угловой коэффициент эталонного стандарта)]
Пример 14. Генная заместительная терапия с однократной дозой при спинальной мышечной атрофии: исследование дозы
Спинальная мышечная атрофия (SMA) является тяжелым детским моногенным заболеванием, возникающим в результате утраты или дисфункции гена, кодирующего фактор выживаемости мотонейронов 1 (SMN1). Частота возникновения этого заболевания составляет примерно 1 на 10000 живорожденных, а частота встречаемости носителей составляет 1 на 54. SMA характеризуется дегенерацией и потерей нижних моторных нейронов, что приводит к мышечной атрофии. Заболевание делится на четыре подтипа (1-4), исходя из возраста и достижения ключевых точек. SMA типа 1 (SMA1) является наиболее тяжелой формой и наиболее распространенной генетической причиной смерти среди младенцев. Существует две формы SMN; SMN1 является основным геном, ответственным за функциональное продуцирование белка SMN. SMN2 преимущественно исключает экзон 7 во время сплайсинга и, как результат, продуцирует лишь небольшую долю функционального белка SMN по сравнению с SMN1. Следовательно, количество копий SMN2 модифицирует фенотип заболевания, а наличие двух копий SMN2 связано с SMA1.
Младенцы с биаллельными делециями SMN1 и двумя копиями SMN2 характеризуются 97% риском развития SMA1.
Недавние исследования по естественному течению SMA1 (группа течения заболевания) показали, что медианное значение возраста
при появлении симптомов среди младенцев с заболеванием составляло 1,2 месяца (диапазон от 0 до 4 месяцев), и
заболевание характеризовалось гипотонией, сильной слабостью с грудного возраста и неспособностью сидеть без поддержки. У младенцев с SMA1, у которых есть две копии SMN2, медианный возраст на момент смерти или необходимости неинвазивной вентиляции в течение по меньшей мере 16 часов в день в течение по меньшей 14 последовательных дней (считается эквивалентным постоянной вентиляции) составлял 10,5 месяца. В одной группе пораженных детей через 13,6 месяца лишь 25% выжило без постоянной искусственной вентиляции легких и 8% выжило без этой поддержки к 20 месяцам. Другое проспективное многоцентровое исследование течения заболевания, спонсируемое Национальным институтом здоровья (NeuroNEXT) с участием пациентов с двумя копиями SMN2, показало медианное значение выживаемости без трахеостомии, составляющее 8 месяцев (95% доверительный интервал, 6-17). Все пациенты с SMA1 характеризуются резким снижением респираторных и глотательных функций после рождения и, в конечном итоге, нуждаются в механическом парентеральном питании (через назогастральную или гастростомическую трубку) для поддержания надлежащего питания и уменьшения респираторных рисков, связанных с аспирацией. В случае пациентов с SMA1, у которых появление симптомов происходит в возрасте 3 месяца, большинство пациентов нуждается в кормлении до 12 месяцев.
Пациенты с SMA1 также не достигают ключевых точек в двигательной функции и имеют снижение функции, по результатам измерения по шкале CHOP INTEND (тест детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорожденных), которая варьирует от 0 до 64, при этом более высокие показатели указывают на более хорошую двигательную функцию, инструмент, который чувствителен к незначительным изменениям двигательной функции, таким как антигравитационные движения конечностей. В ретроспективном анализе 34 пациентов с SMA1 все, кроме 1 пациента, не достигали показателя по меньшей мере 40 баллов после возраста 6 месяцев. В группе NeuroNEXT показатели CHOP INTEND снижались в среднем на 10,7 балла в возрасте от 6 до 12 месяцев.
Терапевтические стратегии увеличения уровня белка SMN в двигательных нейронах фокусируются на повышении эффективности SMN2. Один из подходов заключался в доставке в центральную нервную систему нусинерсена (Ionis Pharmaceuticals/Biogen), антисмыслового олигонуклеотида, который был разработан для ингибирования сплайсинга экзона 7 в SMN2. Было показано, что это лекарственное средство уменьшает слабость в мышиной модели тяжелой SMA и увеличивает медианную продолжительность жизни пораженных мышей с 16 до 25 дней. В декабре 2016 года Управление по контролю за пищевыми продуктами и лекарственными средствами одобрило нусинерсен для лечения SMA. Это лекарственное средство вводят путем многократных интратекальных инъекций после четырех нагрузочных доз в течение первых 2 месяцев жизни.
Потенциальным альтернативным лечением SMA1 является генная терапия, проводимая в форме однократного внутривенного введения, с помощью которого доставляют копию SMN в самокомплементарном аденоассоциированном вирусе серотипа 9 (scAAV9). (Кодирующая область этого рекомбинантного вируса образует внутримолекулярную двухнитевую ДНК [или самокомплементарную] матрицу.) С помощью такого подхода индуцировали экспрессию SMN в двигательных нейронах и периферических тканях, что противодействовало эффектам SMA в мышиной модели и увеличивало среднюю выживаемость в этой модели с 15 дней до 28,5 дня при низкой дозе (6,7×1013 г. в. на килограмм веса тела) и более 250 дней при более высоких дозах вектора (2,0×1014 и 3,3×1014 г. в. на килограмм).
Помимо прохождения гематоэнцефалического барьера и целенаправленного воздействия на нейроны центральной нервной системы во всех областях спинного мозга, системное введение средства AAV9-опосредованной генной терапии может быть преимущественным с учетом того факта, что белок SMN экспрессируется повсеместно, а SMA1 поражает множество систем (например, вегетативную и энтерическую нервные системы, сердечно-сосудистую систему и поджелудочную железу), наряду со многими типами клеток (например, сердца, поджелудочной железы и скелетной мышцы). Признак самокомплементарности вектора в сочетании с гибридным цитомегаловирусным энхансером-промотором гена бета-актина курицы обеспечивает быструю и устойчивую экспрессию SMN. В апреле 2014 года авторы настоящего изобретения начали исследование генной заместительной терапии с участием младенцев с SMA1, которые получали однократную дозу scAAV9 с доставкой гена выживаемости мотонейронов человека (hSMN) под контролем промотора гена бета-актина курицы (scAAV9.CB.hSMN) (AVXS-101).
Способы
Процедуры исследования и пациент. Для данного исследования все пациенты имели генетически подтвержденный диагноз SMA1, гомозиготные делеции экзона 7 SMN1 и две копии SMN2. Исключали пациентов с модификатором c.859G→C в экзоне 7 SMN2. У пациентов, которые были отобраны, было выявлено начало заболевания с рождения до возраста 6 месяцев, характеризующееся гипотонией, которую определяли по клинической оценке, сопровождающейся задержкой двигательных навыков, плохим удержанием головы, сутулой осанкой и разболтанностью суставов. Из исследования исключали пациентов с активными вирусными инфекциями (в том числе HIV или положительной на гепатит B или C серологией) или сопутствующим заболеванием, которое создавало ненужные риски передачи генов. Также исключали пациентов, нуждающихся в инвазивной вспомогательной искусственной вентиляции легких (трахеотомия с положительным давлением) или пульсовой оксиметрии <95% насыщения при скрининговом визите.
Пациентов включали в две группы в соответствии с дозой средства генной терапии, которое вводили. Пациенты из группы 1 получали низкую дозу (6,7×1013 г. в. на килограмм) и участвовали в исследовании в течение пяти месяцев; пациенты из группы 2 получали высокую дозу (2,0×1014 г. в. на килограмм) и участвовали в исследовании в течение одного года. В день 30 после введения дозы посредством ELISpot-анализа на IFN-γ пациента 1 из группы 1 выявили Т-клеточный ответ и показывали внезапный всплеск количества образующих "ореол" клеток (SFC) на 106 мононуклеарных клеток периферической крови (РВМС), которые были >50 направлены против капсида AAV9 (в норме <50 SFC на 106 PBMC). Начинали введение преднизолона в количестве 2 мг/кг и продолжали в течение 35 дней, пока не снизились Т-клеточный ответ и уровень трансаминазы в сыворотке крови. В результате протокол эксперимента был изменен, и пациенты 2-15 получали преднизолон для перорального введения в дозе 1 мг на килограмм в день в течение приблизительно 30 дней, начиная с 24 часов до введения генного вектора. Лечение продолжали с сохранением преднизолона до тех пор, пока уровень ферментов AST и ALT не опускался ниже уровня 120 МЕ/л, а Т-клеточный ответ не опускался ниже 100 SFC на 106 РВМС, после чего дозу преднизолона уменьшали, исходя из решения лечащего врача.
Вектор доставляли в нормальном солевом растворе (примерно 10-20 мл на килограмм), который вводили внутривенной инфузией в течение примерно 60 минут. Во время участия некоторым пациентам требовалось энтеральное питание с помощью гастростомической трубки или назогастрального зонда, выбор которого основывался на предпочтениях родителей или основного врача. После включения в исследование все пациенты, которым требовалось парентеральное питание, проходили установку гастростомической трубки, и трубки не были удалены во время исследования.
Результаты. Первичным результатом было определение безопасности на основе любых связанных с лечением неблагоприятных явлений 3-й степени или выше. Вторичным результатом было время до смерти или необходимость постоянной вентиляционной поддержки. Последнюю определяли как по меньшей мере 16 часов непрерывной респираторной поддержки в день в течение по меньшей мере 14 дней при отсутствии острого обратимого заболевания или периоперационного состояния. Исследовательские результаты включали достижения ключевых точек, связанных с двигательной активностью (в частности, сидение без посторонней помощи), и показатели CHOP INTEND.
Сохранение показателей, превышающих 40 баллов, считали клинически значимым в SMA при применении шкалы CHOP INTEND. Сидение без посторонней помощи оценивали и классифицировали в соответствии со следующими критериями: сидение без посторонней помощи в течение по меньшей мере 5 секунд согласно разделу 22 подтеста крупной моторики с использованием шкал Бэйли для оценки развития младенцев и детей возрастом 1-3 года ("сидение без посторонней помощи"); сидение без посторонней помощи в течение по меньшей мере 10 секунд в соответствии с критериями Всемирной организации здравоохранения (WHO) ("сидение без посторонней помощи в соответствии с критериями ВОЗ") и сидение без посторонней помощи в течение по меньшей мере 30 секунд в соответствии с разделом 26 упомянутых выше шкал Бейли ("независимое функциональное сидение"). Основные ключевые точки, связанные с двигательной активностью, подтверждали путем изучения видеозаписей пациентов независимым экспертом с помощью Ability Captured Through Interactive Video Evaluation-mini (ACTIVE-mini). Электрические вызванные ответы мышцы (CMAP) регистрировали с поверхностных электродов на исходном уровне и каждые 6 месяцев после инфузии. Патологический статус мышц количественно оценивали с помощью электроимпедансной миографии (EIM).
Статистический анализ. Анализы безопасности проводили для всех пациентов, которые также были включены в первичный анализ выживания (как определено выше и в протоколе) и в анализы изменений по шкале CHOP INTEND от исходного уровня до 1 месяца и 3 месяцев. Такие изменения от исходного уровня до каждого визита исследования анализировали с применением модели смешанных эффектов для повторных измерений. Смешанная модель включала фиксированные эффекты группы и визита, а также ковариату показателя на исходном уровне. В группе 2 анализировали достижения ключевых точек, а также парентеральное питание и вспомогательную искусственную вентиляцию легких. Статистический анализ проводили с использованием программного обеспечения SAS, версии 9.4. Все сравнения с группами течения заболевания были исключительно описательными.
Результаты
Пациенты. Из 16 пациентов, которые были подвергнуты скринингу, 1 был исключен из-за постоянно повышенных титров антитела к AAV9 (> 1:50). Из 15 пациентов, которые были включены в исследование, 3 были включены в группу 1 с низкой дозой, а 12 были включены в группу 2 с высокой дозой. Средний возраст пациентов на момент лечения составлял 6,3 месяца (диапазон от 5,9 до 7,2) в группе 1 и 3,4 месяца (диапазон от 0,9 до 7,9) в группе 2 (таблица 28).
Таблица 28. Демографические и клинические характеристики 15 пациентов
Выживаемость и постоянная вентиляция. На конец исследования всеми пациентами был достигнут возраст по меньшей мере 20 месяцев, и они не нуждались в постоянной искусственной вентиляции легких; медианный возраст при последней легочной оценке составлял 30,8 месяца в группе 1 и 25,7 месяца в группе 2. В отличие от этого, лишь 8% пациентов в группе течения заболевания не нуждались в постоянной механической вентиляции. В возрасте 29 месяцев одному пациенту из группы 1 потребовалась постоянная вентиляция из-за гиперсаливации. После перевязки слюнных желез потребность в применении неинвазивной вентиляции была снижена на 25% до 15 часов в день.
Оценки двигательных функций. Все пациенты в группах 1 и 2 по сравнению с исходным уровнем имели повышенные показатели по шкале CHOP INTEND и сохраняли эти изменения в ходе исследования. У пациентов в группе 2 среднее увеличение составляло 9,8 балла на 1 месяце и 15,4 балла на 3 месяце (P < 0,001 для обоих сравнений); 11 пациентов достигали и сохраняли показатели более 40 баллов. В заключительный день исследования 7 августа 2017 года пациенты в группе 1 характеризовались средним увеличением на 7,7 балла относительно среднего значения на исходном уровне 16,3 балла, а пациенты в группе 2 характеризовались средним увеличением на 24,6 балла относительно среднего значения на исходном уровне 28,2 балла.
Связанные с двигательной активностью контрольные точки в группе 2. В общей сложности 11 из 12 пациентов в группе 2 могли сидеть без посторонней помощи в течение по меньшей мере 5 секунд, 10 - в течение по меньшей мере 10 секунд, и 9 - в течение по меньшей мере 30 секунд (таблица 31). В общей сложности 11 достигали удержания головы, 9 могли перевернуться, и 2 смогли ползти, тянуться в попытке встать, стоять независимо и самостоятельно ходить. Одиннадцать пациентов достигали способности говорить. Ни один из пациентов в группах течения заболевания не достигал ни одной из этих контрольных точек, связанных с двигательной активностью, и редко достигал способности говорить.
Таблица 29. Выживаемость без неблагоприятных явлений и связанные с двигательной активностью и другие контрольные точки у 12 пациентов из группы 2.
Статус функции легких и питания в группе 2. Среди 12 пациентов в группе 2 10 не нуждались в неинвазивной вентиляции на исходном уровне в сравнении с 7, которые не зависели от искусственной вентиляции легких на последнем контрольном визите (таблица 29). На исходном уровне 7 пациентов не нуждались в энтеральном питании, в том числе 1 пациент, которому позже потребовалось размещение гастростомической трубки после генной заместительной терапии, возможно, в связи с хирургическим вмешательством по причине сколиоза. Из 5 пациентов, которые получали энтеральное питание до генной заместительной терапии, на последнем контрольном посещении 11 из 12 пациентов достигали или сохраняли способность самостоятельно глотать, а 4 были в состоянии питаться перорально.
Безопасность. По состоянию на конец исследования у 13 пациентов в двух группах было зарегистрировано в общей сложности 56 серьезных неблагоприятных явлений. Из этих явлений исследователями было определено, что 2 явления были связанными с лечением явлениями 4-й степени на основе лабораторных значений в соответствии с Общими терминологическими критериями для неблагоприятных явлений (таблица 30). Пациент 1 в группе 1 характеризовался повышениями уровней аминотрансферазы в сыворотке крови (в 31 раз выше верхнего предела нормального диапазона для аланинаминотрансферазы (ALT) и в 14 раз выше верхнего предела для аспартатаминотрансферазы (AST)) без других нарушений функции печени (т.е. общего и свободного билирубина и щелочной фосфатазы) и без клинических проявлений. Как описано выше, эти повышения смягчали лечением преднизолоном, которое впоследствии назначали оставшимся пациентам. Один пациент из группы 2 нуждался в дополнительном преднизолоне для снижения повышенных уровней ALT и AST в сыворотке крови (в 35 раз выше верхнего предела нормального диапазона для ALT и в 37 раз для AST). Из 241 несерьезного неблагоприятного явления 3 посчитали связанными с лечением, и они заключались в бессимптомных повышениях уровней аминотрансферазы в сыворотке крови у 2 пациентов (ALT и AST, обе менее чем в 10 раз превышали верхний предел нормального диапазона), которые разрешались без дополнительного лечения преднизолоном (таблица 0. Других нарушений при тестировании функции печени выявлено не было. Из 15 пациентов у 14 были респираторные заболевания, которые у детей с SMA1 зачастую приводят к смерти или необходимости трахеостомии.
Таблица 30. Неблагоприятные явления.
Однократная внутривенная инфузия вектора на основе аденоассоциированного вируса, содержащего ДНК, кодирующую SMN, у пациентов с SMA1 приводила к более длительному выживанию, чем в группах течения заболевания с этим заболеванием. Все 15 пациентов преодолевали ранее сообщенный медианный возраст выживания без постоянной вентиляции, составлявший 10,5 месяца для пациентов с SMA1 с двумя копиями SMN2. Все пациенты также преодолевали контрольную точку в 20 месяцев, при этом лишь 8% пациентов с этим заболеванием обычно доживают без постоянной вентиляции. Из 12 пациентов в группе 2 все, кроме 1, достигали ключевых точек, связанных с двигательной активностью, чего не было отмечено в группах течения заболевания. Достигнутая двигательная функция была клинически значимой, что отражалось в питании (поднесение руки ко рту), сидении и говорении. Большинство пациентов, которые не нуждались в поддерживающем уходе на момент включения в исследование, не имели парентерального питания (6 из 7 пациентов) и вспомогательной искусственной вентиляции легких (7 из 10 пациентов) на последнем контрольном визите. В двух группах пациенты имели увеличения показателя по шкале CHOP INTEND относительно исходного уровня. За первый месяц в группе 2 среднее увеличение составляло 9,8 балла, в отличие от снижения среднего значения более чем на 10 баллов в возрасте от 6 до 12 месяцев в группе течения заболевания в исследовании NeuroNEXT.
Доклинические исследования заместительной по гену SMN терапии в модели на мышах SMNΔ7 демонстрировали улучшение выживаемости и двигательной функции при раннем лечении, возможно в тот момент, когда двигательные нейроны все еще были интактными. Клинические результаты, полученные в исследовании авторов настоящего изобретения раннего лечения, отражали направленность результатов, полученных в доклинических исследованиях. После раннего лечения два пациента были способны ползать, стоять и ходить без поддержки. Оба этих пациента имели семейный анамнез SMA, что, вероятно, способствовало ранней диагностике. Хотя у всех пациентов из двух групп в исследовании авторов настоящего изобретения продолжала улучшаться двигательная функция, доклинические и клинические данные свидетельствовали о преимуществах раннего лечения и скрининга новорожденного в отношении SMA.
Примерно через 3 недели после лечения у двух пациентов серьезные неблагоприятные явления, вызванные генной заместительной терапией на основе AAV, были ограничены повышенными уровнями аминотрансферазы в сыворотке крови без нарушений других ферментов печени; у двух других пациентов были повышения, которые не достигали предела для того, чтобы подпадать под определение серьезных неблагоприятных явлений (т.е. в >10 раз выше нормального диапазона). Повышения уровней ферментов печени снижали лечением преднизолоном. Один пациент не прошел скрининг из-за наличия антитела к AAV9, что согласовывалось с результатами популяционных исследований, которые свидетельствуют о низкой частоте встречаемости сероположительной реакции к AAV9 у детей и молодых людей и повышении частоты встречаемости сероположительной реакции к AAV9 среди лиц, которые старше 40 лет. Тем не менее наличие антител к вирусу может быть ограничением генной заместительной терапии на основе AAV.
В этом исследовании использовали схему с одной группой с группой течения заболевания в качестве контроля, которая является одним из ограниченного числа вариантов, когда естественное течение заболевания хорошо охарактеризовано и приводит к летальному исходу. Для включения в исследование однородной выборки, которая была аналогична выборкам из опубликованных исследований течения заболевания, авторы настоящего изобретения ограничивали включение в исследование включением только симптоматических пациентов с SMA1, у которых были биаллельные мутации SMN1 и две копии SMN2, и не включали в исследование пациентов с генетическим модификатором c.859G→C в экзоне 7 SMN2, поскольку этот генетический модификатор определяет более мягкий фенотип заболевания. Однако эта генная заместительная терапия не должна ограничиваться симптоматическими пациентами или пациентами с конкретным подтипом генома.
В заключение необходимо отметить, что однократная внутривенная инфузия высокой дозы вектора на основе аденоассоциированного вируса, содержащего ДНК, кодирующую SMN, у пациентов с SMA1 приводила к увеличенному выживанию, улучшенной двигательной функции и увеличенным показателям по шкале CHOP INTEND до уровней, которые ранее не наблюдались при данном заболевании. Такие улучшения приводили к более низкому проценту пациентов, которые нуждались в поддерживающей терапии, чем в исследованиях течения заболевания. При последующем наблюдении в течение до 2-х лет не было замечено ослабления эффекта или клинической регрессии двигательной функции. Несколько пациентов имели временные и бессимптомные повышения уровней аминотрансферазы. Необходимы дополнительные исследования для оценки долгосрочной безопасности и долговечности результатов генной заместительной терапии у пациентов с SMA1.
Пример 15. Фармакокинетика scAAV9.CB.hSMN
Традиционные клинические фармакокинетические исследования не применимы к продуктам генной заместительной терапии. Тем не менее, результаты исследований по снижению активности вектора scAAV9.CB.hSMN, в которых оценивали количество вектора, выделяемого из организма посредством жидкостей и выделений, являются показателем, который можно использовать вместо традиционных фармакокинетических исследований в случае генной заместительной терапии.
Снижение активности вектора после инфузии посредством scAAV9.CB.hSMN исследовали в нескольких моментах времени во время клинического исследования. Образцы слюны, мочи и кала собирали еженедельно в течение 30 дней, а затем ежемесячно в течение 12 месяцев и каждые 3 месяца после этого. Для анализа снижения активности вектора scAAV9.CB.hSMN с помощью капельной цифровой полимеразной цепной реакции использовали образцы от 5 пациентов, полученные во время визита на 18-ом месяце. Всем 5 пациентам, которых анализировали в отношении снижения активности вектора SMN scAAV9.CB.h, вводили терапевтическую дозу 1,1 × 1014 г. в./кг.
scAAV9.CB.hSMN поддавался обнаружению в образцах по изучению снижения активности после инфузии. Концентрации scAAV9.CB.hSMN в моче и слюне составляли от 0,1 до 0,01% от начальной концентрации в организме в день 1 после инфузии, после чего концентрации падали ниже предела количественного определения. В кале в день 1 после инфузии были обнаружены уровни от 10% до 30% от начальной концентрации в организме. У одного пациента наблюдали пиковую концентрацию в кале в день 14 после инфузии, составляющую 280% от начальной концентрации в организме. Напротив, у 3 пациентов, от которых были доступны данные, в день 14 после инфузии наблюдали концентрацию < 1% от начальной концентрации в организме, при этом концентрация снижалась примерно на 4 log (в 10000 раз) на протяжении 30 дней после инфузии. В целом, scAAV9.CB.hSMN в основном выводился из организма в кале, а к дню 60 после инфузии был ниже предела количественного определения в кале.
Пример 16. Неклинические токсикологические тесты
Фармакология на животных. После инфузии вектора scAAV9.CB.hSMN на модели заболевания на мышах дельта 7 SMA (мыши SMN Δ7) вес тела увеличивался, выпрямительные рефлексы улучшались, значительно дозозависимым образом увеличивалось выживаемость, а связанная с SMA сердечная недостаточность возвращалась к нормальному состоянию по сравнению с мышами SMN Δ7 без обработки.
Токсикология на животных. После внутривенной инфузии мыши, вектор и трансген были широко распределены с наиболее высокой экспрессией, обычно наблюдаемой в сердце и печени, и существенной экспрессией в головном мозге и спинном мозге. В 3-месячных базовых исследованиях токсикологии на мышах, соответствующих надлежащей лабораторной практике (GLP), основными целевыми органами оценки токсичности были сердце и печень. Связанные с вектором scAAV9.CB.hSMN результаты в желудочках сердца охватывали связанные с дозой воспаление, отек и фиброз, а в предсердии - воспаление и тромбоз. Результаты исследования печени охватывали гепатоклеточную гипертрофию, активацию купферовых клеток и рассеянный гепатоклеточный некроз. Не вызывающий вредного воздействия уровень (NoAEL) не был выявлен для связанных с вектором scAAV9.CB.hSMN результатов для печени и сердца мыши, а максимальная переносимая доза была определена как составляющая 1,5 × 1014 г. в./кг, что обеспечивало запас безопасности в примерно 1,4 раза относительно рекомендованной терапевтической дозы 1,1 × 1014 г. в./кг. Переносимость наблюдаемых результатов у мышей на приматов в настоящее время неизвестна.
Пример 17. Спинальная мышечная атрофия у пациентов детского возраста
Данное испытание представляло собой исследование фазы 1, в котором оценивали безопасность и эффективность вектора scAAV9.CB.hSMN у пациентов с SMA типа 1, генетически протестированных для подтверждения биаллельных делеций SMN1, 2 копий гена выживаемости мотонейронов 2 (SMN2), отрицательных результатов по модификации c.859G>C в экзоне 7 и с появлением клинических симптомов в возрасте до 6 месяцев. Вектор scAAV9.CB.hSMN доставлялся внутривенно во время инфузии однократной дозы пациентам в возрасте от 0,9 до 7,9 месяца. Дозы вводили двум группам: группа 1 (n=3) получала низкую дозу, используемую в этом исследовании, а группа 2 (n=12) получала высокую дозу (терапевтическая доза: 1,1×1014 г. в./кг), используемую в этом исследовании. Опубликованные результаты исследования отражают группу 2 и включают последующее наблюдение за всеми пациентами в течение периода до 24 месяцев после инфузии вектора scAAV9.CB.hSMN.
Смертность и выживаемость без явлений
Результаты анализов выживаемости и времени до явления подтверждали эффективность вектора scAAV9.CB.hSMN. В группе 2 все 12 пациентов (100%) были старше 24 месяцев и не имели явлений, в отличие от только 8% пациентов в исследовании естественного течения заболевания. Это указывало на значительное и клинически значимое увеличение общей выживаемости у пациентов, которым инфузией вводили вектор scAAV9.CB.hSMN, по сравнению с пациентами без лечения. Через 2 года после инфузии не было зарегистрировано ни одного случая смерти пациента.
Связанные с двигательной активностью контрольные точки в процессе развития
Изучали связанные с двигательной активностью контрольные точки в процессе развития; оценки в случае всех 15 пациентов записывали на видео для обеспечения подтверждения достижения связанных с двигательной активностью контрольных точек в процессе развития. Пациенты в группе 2 неизменно достигали ключевых связанных с двигательной активностью контрольных точек в процессе развития и сохраняли достигнутый результат. Через 24 месяца последующего наблюдения после введения дозы 11 пациентов (91,7%) смогли держать свою голову в вертикальном положении в течение ≥ 3 секунд и сидеть без поддержки в течение ≥ 5 секунд, 10 пациентов (83,3%) могли сидеть без поддержки в течение ≥ 10 секунд, 9 пациентов (75,0%) могли сидеть без поддержки в течение ≥ 30 секунд, а каждый из 2 пациентов (16,7%) смог самостоятельно стоять, ходить с помощью и ходить самостоятельно. Пациенты из группы 2, которые в настоящее время участвуют в длительном эмпирическом наблюдении в этом исследовании, сохраняли свои связанные с двигательной активностью контрольные точки в процессе развития, а некоторые достигали дополнительных связанных с двигательной активностью контрольных точек.
Группа 2
(N=12)
n (%)
Легкие
Из 10 пациентов в группе 2, которые не использовали неинвазивную вентиляцию легких (NIV) на исходном уровне, 7 не нуждались в ежедневном использовании NIV через 24 месяца последующего наблюдения. Почти всеми пациентами были перенесены общие детские респираторные заболевания, которые у детей с SMA типа 1 обычно приводят к трахеостомии или смерти. Всеми пациентами были пережиты респираторные госпитализации без трахеостомии или необходимости постоянной вентиляции.
Парентеральное питание
Также наблюдали за необходимостью парентерального питания. В группе 2 семь пациентов не получали энтеральное питание до генной заместительной терапии. У одного (1) из этих 7 пациентов было парентеральное питание для облегчения заживления ран после сложного восстановления от хирургического вмешательства в связи со сколиозом, но также у него было и пероральное кормление. Четыре (4) из 5 пациентов в группе 2, которые получали энтеральное питание до генной заместительной терапии, в конце исследования могли питаться перорально; таким образом, в общей сложности 11 из 12 пациентов в группе 2 были в состоянии питаться перорально, за исключением 6-го пациента.
Двигательная функция (CHOP-INTEND)
Пациенты, получавшие терапевтическую дозу, достигали статистически значимого улучшения двигательной функции к месяцу 1 и месяцу 3; среднее увеличение значений теста детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорожденных (CHOP-INTEND) составляло 9,8 балла (n=12, P < 0,001) и 15,4 балла (n=12, P < 0,001) соответственно.
У пациентов, которым была введена инфузия вектора scAAV9.CB.hSMN, улучшения двигательных функций сохранялись с течением времени. Одиннадцать из двенадцати (91,7%) пациентов из группы 2 через 24 месяца достигали показателя CHOP-INTEND ≥ 50 баллов. Раннее вмешательство и доза, судя по всему, положительно влияли на ответ. В общей клинической практике дети без лечения и с SMA типа 1 в возрасте 6 месяцев и старше не превышают 40 баллов по CHOP-INTEND. Более того, среди младенцев без лечения было зарегистрировано среднее снижение на 10,7 балла в возрасте от 6 до 12 месяцев, что частично согласовывалось с предполагаемым естественным течением заболевания.
Пример 18. Измерение с помощью qPCR остаточной ДНК клетки-хозяина в векторах на основе вируса AAV9
Способ
Данный способ применяли для количественного определения с помощью qPCR остаточной ДНК клетки-хозяина в лекарственном веществе на основе AAV, например, AVXS-101, и в образцах, взятых в процессе. На одном планшете тестировали до шести образцов. Анализ qPCR проводили с применением зонда TaqMan. Зонд TaqMan имеет флуорогенный репортерный краситель, связанный с 5'-концом, и нефлуоресцентный гаситель, связанный с 3'-концом. Пока зонд находится в интактном состоянии, близость гасителя к репортерному красителю значительно снижает флуоресценцию, испускаемую репортерным красителем. Расщепление зонда разделяет репортерный краситель и гаситель, увеличивая флуоресценцию репортера.
В реакционную смесь добавляли фланкирующие прямой и обратный праймеры, предназначенные для связывания с повторяющейся последовательностью в геноме человека, и гибридизировали с целевой последовательностью, присутствующей в образце и стандартах. Флюорогенный зонд TaqMan гибридизировался между праймерными участками. Посредством последовательных циклов денатурации матрицы, гибридизации праймеров и элонгации продукта амплифицировали целевую последовательность. Во время стадии элонгации цикла амплификации посредством экзонуклеазной активности ДНК-полимеразы Taq высвобождали репортерный краситель из зонда, освобождая краситель от гасителя, что приводило к флуоресцентному излучению, пропорциональному количеству матрицы.
Флуоресценцию каждой лунки 96-луночного планшета измеряли с помощью прибора для qPCR. С помощью дополнительных циклов ПЦР получали увеличивающиеся количества целевой последовательности, и в результате высвобождалось больше репортерного красителя из зонда и имела место более высокая флуоресценция в каждом последующем цикле ПЦР. Измеряли количество циклов амплификации, необходимых для достижения флуоресцентным сигналом предопределенного порогового значения. Этот цикл называют пороговым циклом или значением CT. Чем выше начальная концентрация ДНК в лунке с образцом или со стандартом, тем меньше число циклов ПЦР, необходимых для достижения порогового уровня флуоресценции, и тем ниже значение CT. Калибровочную кривую создавали путем построения графика log10 (концентрации ДНК) в зависимости от значения CT, измеренного для каждой стандартной точки. Значение CT использовали для определения количества ДНК, присутствующего в образце, путем использования индивидуального значения CT для образца и определения значения ДНК.
Сначала проводили подготовку образцов с использованием набора Wako DNA Extractor (Wako, 295-50201). Вкратце, образцы для тестирования хорошо перемешивали и разбавляли в 1000 раз. Разбавленные образцы разделяли на четыре пробирки (по 500 мкл каждая) и в две пробирки (копии без добавок) добавляли 50 мкл воды, в то время как в две другие пробирки (контроль с добавкой) добавляли 50 мкл стандарта ДНК с концентрацией 30000 пг/мл. Солюбилизацию белка проводили путем добавления в каждую пробирку 20 мкл раствора N-лауроилсаркозината натрия, встряхивая ее на вортексе в течение 5 секунд, затем кратковременно центрифугируя. Раствор NaI, содержащий гликоген и Pellet Paint (Novagen, 70748), получали таким образом, чтобы они были представлены в соотношении 2000:5:4 NaI:гликоген:Pellet Paint. В каждую пробирку добавляли 500 мкл смеси NaI и инкубировали при 53°С ± 1°С в течение 15 минут. Пробирки снимали с нагрева, содержимое пробирок смешивали с 900 мкл изопропанола и инкубировали при комнатной температуре в течение 15 минут. Затем пробирки центрифугировали при 10000 g в течение 15 минут при 18°C и сливали надосадочную жидкость. Оставшийся осадок промывали 800 мкл промывочного раствора A, центрифугировали и еще дважды повторяли процедуру осаждения. Наконец, осадок промывали 1500 мкл охлажденного промывочного раствора изопропанола, содержащего гликоген, центрифугировали и повторяли процедуру осаждения. Конечную осажденную центрифугированием массу ресуспендировали в 500 мкл не содержащей нуклеаз воды.
Реакцию qPCR проводили с применением набора resDNA SEQ Human Quantitative (Applied Biosystems, A26366). Реакционную смесь получали путем объединения 2x мастер-микса Environmental Master Mix, 10x смесь для анализа ДНК человека и отрицательного контроля согласно тому, как указано в инструкциях набора. 20 мкл реакционной смеси смешивали с 10 мкл подготовленного образца и добавляли в каждую лунку планшета для ПЦР. Каждый образец вносили в планшет в трех повторностях. Планшет герметично закрывали оптической клейкой пленкой. Во время термоциклирования сначала осуществляли плавление при 95°С в течение 10 минут, а затем образцы запускали в температурный цикл от 95°С в течение 15 с до 60°С в течение 1 минуты в течение 40 циклов.
Калибровочную кривую получали путем построения графика зависимости значения CT от количества ДНК в log([пг/мл]). Данные аппроксимировали до прямой линии, задаваемой следующим уравнением:
значение CT=m x log10 (x) + b,
где x=концентрация стандарта в пг/мл, m обозначает угловой коэффициент, а b обозначает точку пересечения с осью Y. Концентрацию ДНК клетки-хозяина рассчитывали обратно из значения CT в лунке с помощью приведенного выше уравнения, затем корректировали с помощью коэффициента разбавления.
Результаты
Остаточная ДНК клетки-хозяина, измеренная с помощью qPCR, составляла 3,7×105 пг/мл для предыдущей партии вектора, 0,76×105 пг/мл для AVXS-101 серии 600156, 0,68×105 пг/мл для AVXS1-101 серии 600307 и 1,3×105 пг/мл для AVXS-201 DS.
Пример 19. Измерение с помощью ELISA остаточного белка клетки-хозяина (HCP) в векторах на основе вируса AAV9
Способ
Концентрацию белка клетки-хозяина (HCP) в образцах AVXS-101 измеряли с использованием коммерческого набора для иммуноферментного анализа (ELISA). Набор для ELISA Cygnus Technologies Human Embryonic Kidney 293 HCP представляет собой набор для твердофазного двухстадийного иммуноферментного анализа. Он основан на прямом сэндвич-методе, в котором два поликлональных антитела направлены против раздельных антигенных детерминант у HCP. Во время инкубации HCP в образце связывали с антителами к HCP, связанными с лункой микропланшета, и с конъюгированными с пероксидазой антителами к HCP в растворе.
После инкубационного периода лунки промывали для удаления любого несвязанного антитела, конъюгированного с ферментом. Затем в лунки добавляли раствор субстрата 3,3',5,5'-тетраметилбензидин (TMB). Связанный с пероксидазой конъюгат катализировал реакцию с изменением цвета субстрата. Реакцию останавливали путем добавления кислоты, что давало колориметрическую конечную точку, которую можно было считать на спектрофотометре при 450 нм. Количество гидролизованного субстрата было прямо пропорционально концентрации присутствующего HCP.
Подлежащие тестированию образцы разбавляли до приведения в соответствие диапазону данного способа, т.е. от 4 нг/мл до 200 нг/мл. Затем каждый образец разбавляли в 2 раза в SDB (110 мкл образца и 110 мкл SDB) и перемешивали. Для проверки согласованности также получали контроли с добавками. В контролях с добавками 110 мкл каждого образца смешивали с 27,5 мкл стандарта HCP в концентрации 200 нг/мл и 82,5 мкл SDB. Наконец, 50 мкл каждого стандарта, контрольного или тестируемого образца добавляли в лунку 96-луночного планшета и смешивали с 100 мкл конъюгата антитела к HEK 293 с HRP. Все условия вносили в планшет в трех повторностях. Планшет герметично закрывали герметизирующей лентой и встряхивали при 400-600 об/мин в течение 2 часов при комнатной температуре. После инкубации удаляли растворы в лунках, стряхивая планшет вверх дном и промакивая его впитывающим полотенцем. Лунки промывали раствором из промывочной бутылки, быстро промакивали, не давая промывному раствору впитаться в лунки. Промывку повторяли 4 раза и оставляли в покое верх дном приблизительно на 20 сек. для стекания раствора после последней промывки. Наконец, в каждую лунку планшета вносили 100 мкл субстрата TMB и инкубировали в течение 20-30 минут при комнатной температуре без перемешивания. Реакцию останавливали путем добавления в каждую лунку 100 мкл стоп-раствора. Планшет загружали в планшет-ридер в течение 45 мин. после добавления стоп-раствора и планшет считывали при 450 нм и 650 нм.
Средние значения оптической плотности стандартов наносили на график относительно теоретической концентрации HCP стандартов на полулогарифмическом графике для получения четырехпараметрической логистической аппроксимированной кривой (4PL), исходя из следующего уравнения:
Y = [(A - D) / (1+(X/C)^B)] + D,
где A обозначает нижнюю асимптоту, B обозначает угловой коэффициент кривой, C обозначает концентрацию, соответствующую значениям оптической плотности в средней точке между двумя асимптотами (нг/мл), D обозначает верхнюю асимптоту, X обозначает концентрацию образца (нг/мл), а Y обозначает оптическую плотность. Затем использовали калибровочную кривую для определения концентрации HCP в контрольном образце с добавкой и в тестовых образцах без добавок с использованием программного обеспечения SoftMax Pro. Тест считали признанным только в том случае, если r2 стандартной кривой составлял ≥ 0,98, средняя скорректированная оптическая плотность стандарта в концентрации 200 нг/мл составляла ≥1,0 OD, средняя скорректированная оптическая плотность стандарта в концентрации 0 нг/мл составляла ≤0,2 OD, а коэффициент вариации скорректированной оптической плотности для лунок в 3 повторностях составлял ≤15%. Конечную концентрацию HCP для каждого образца рассчитывали с применением уравнения:
концентрация HCPобразца (нг/мл) = коэффициент разбавления x средняя измеренная концентрация HCP (нг/мл).
Результаты
Остаточный белок клетки-хозяина, измеренный с помощью ELISA, был ниже предела количественного определения (8 нг/мл) для предыдущей партии вектора, AVXS-101 серии 600156, AVXS1-101 серии 600307 и AVXS-201 DS.
Пример 20. Измерение с помощью ELISA остаточной бензоназы в векторах на основе вируса AAV9
Способ
Концентрацию остаточной бензоназы в продукте на основе AAV, например AVXS-101, измеряли с использованием коммерческого набора для иммуноферментного анализа (ELISA). Набор Merck Benzonase Endonuclease ELISA Kit II представляет собой набор для твердофазного двухстадийного иммуноферментного анализа. Он основан на прямом сэндвич-методе, в котором два поликлональных антитела направлены против раздельных антигенных детерминант у бензоназы. Во время инкубации бензоназу в образце связывали с антителами к бензоназе, связанными с лункой микропланшета, и с конъюгированными с пероксидазой антителами к бензоназе в растворе.
После инкубационного периода лунки промывали для удаления любого несвязанного антитела, конъюгированного с ферментом. Затем в лунки добавляли раствор субстрата 3,3',5,5'-тетраметилбензидин (TMB).
Связанный с пероксидазой конъюгат катализировал реакцию с изменением цвета субстрата. Реакцию останавливали путем добавления кислоты, что давало колориметрическую конечную точку, которую можно было считать на спектрофотометре при 450 нм. Количество гидролизованного субстрата было прямо пропорционально концентрации присутствующей бензоназы.
Вкратце, образцы разбавляли в 2 раза путем объединения 175 мкл образца с 175 мкл PBST. Параллельно также готовили контрольный образец с добавлением бензоназы путем объединения 175 мкл образца с 35 мкл стандарта бензоназы в концентрации 10 нг/мл и 140 мкл PBST. Стрипы из набора для ELISA с предварительно нанесенным покрытием устанавливали в держатель стрипов, и в каждую лунку загружали 100 мкл каждой тестируемой смеси. Для холостых проб загружали 100 мкл PBST вместо образца. Образец каждого условия загружали в планшет в трех повторностях. Планшет герметично закрывали и инкубировали при комнатной температуре в течение 2 часов ± 5 минут при перемешивании на планшетном шейкере (450 об/мин). После инкубации содержимое отбрасывали, а планшет промывали путем внесения ~350 мкл PBST, используя промыватель для микропланшетов, и инкубировали в течение 1 минуты, затем переворачивали и простукивали по впитывающему полотенцу. В общей сложности производили 3 промывки, прежде чем в каждую лунку добавляли 100 мкл разбавленного антитела, конъюгированного с HRP. Планшет герметично закрывали и инкубировали при комнатной температуре в течение 1 часа ± 5 минут при перемешивании на планшетном шейкере (450 об/мин). После инкубации содержимое отбрасывали, а планшет промывали путем внесения ~350 мкл PBST, используя промыватель для микропланшетов, и инкубировали в течение 1 минуты, затем переворачивали и простукивали по впитывающему полотенцу. В общей сложности производили 3 промывки, прежде чем в каждую лунку добавляли 100 мкл субстрата TMB. Планшет герметично закрывали и содержимое инкубировали в течение 15-40 минут при комнатной температуре без перемешивания в темноте. Реакцию останавливали путем добавления в каждую лунку 100 мкл стоп-раствора 0,2 н. H2SO4. Оптическую плотность планшета измеряли с использованием спектрофотометра на 450 нм в пределах 45 минут после добавления стоп-раствора.
Средние значения оптической плотности стандартов наносили на график относительно теоретической концентрации бензоназы в стандартах на полулогарифмическом графике для получения четырехпараметрической логистической аппроксимированной кривой (4PL), исходя из следующего уравнения:
Y = [(A - D) / (1+(X/C)^B)] + D,
где A обозначает нижнюю асимптоту, B обозначает угловой коэффициент кривой, C обозначает концентрацию, соответствующую значениям оптической плотности в средней точке между двумя асимптотами (нг/мл), D обозначает верхнюю асимптоту, X обозначает концентрацию образца (нг/мл), а Y обозначает оптическую плотность. Затем использовали калибровочную кривую для определения концентрации HCP в контрольном образце с добавкой и в тестовых образцах без добавок с использованием программного обеспечения SoftMax Pro. Тест считали признанным только в том случае, если r2 стандартной кривой составлял ≥ 0,98, средняя скорректированная оптическая плотность стандарта в концентрации 2,5 нг/мл составляла ≥ 1,0 OD, средняя скорректированная оптическая плотность стандарта в концентрации 0,10 нг/мл превышала среднюю OD у холостой пробы с PBST, а коэффициент вариации скорректированной оптической плотности для лунок в 3 повторностях составлял ≤ 15%. Конечную концентрацию HCP для каждого образца рассчитывали с применением уравнения:
концентрация бензоназыобразца (нг/мл) = коэффициент разбавления x средняя измеренная концентрация бензоназы (нг/мл).
Результаты
Концентрация остаточной бензоназы, измеренная с помощью ELISA, была ниже предела количественного определения (0,2 нг/мл) для предыдущей партии вектора, AVXS-101 серии 600156, AVXS1-101 серии 600307 и AVXS-201 DS.
Пример 21. Измерение с помощью микропланшетного анализа BCA концентрации белка в векторах на основе вируса AAV9
Способ
Количество белков во взятых в процессе образцах лекарственного вещества и лекарственного продукта, например AVXS-101, измеряли с помощью микропланшетного анализа BCA, используя в качестве эталонного белка бычий сывороточный альбумин в концентрации 2 мг/мл (Thermo Fisher Scientific, 23209) и набор Micro BCA Protein Assay (Thermo Fisher Scientific, 23235). Анализ основан на совместимом с детергентом составе с бицинхониновой кислотой (BCA) для колориметрического выявления и количественного определения общего белка. BCA выявляет Cu1+, который образуется при восстановлении Cu2+ белком в щелочной среде. Пурпурный продукт реакции образуется при хелатировании двух молекул BCA одним ионом меди (Cu1+), который характеризуется сильным поглощением при 562 нм, которое является линейным с увеличением концентраций белка.
Вкратце, стандарты получали путем проведения серийных разведений 2 мг/мл BSA в разбавителе (разбавления в 20 раз буфера для составления, 200 мМ NaCl, 20 мМ Tris, 1 мМ MgCl2, 0,001% вес./об. плюроника F-68, pH 8,0). Тестируемые образцы AVXS-101 также разбавляли в 20 раз водой и делали серийные разведения разбавителем. Конечная концентрация составляла приблизительно 7,5 мкг/мл. Рабочий реагент (WR) получали путем смешивания 25 частей реагента A для микропланшетного анализа BCA, 24 частей реагента B и 1 части реагента C из набора. 150 мкл каждого из стандарта и тестируемого образца загружали в трех повторностях в 96-луночный планшет и смешивали с 150 мкл WR. Планшет герметично закрывали и встряхивали при 300 об/мин на планшетном шейкере в течение 30 секунд. Затем планшет инкубировали без встряхивания при 37°С ± 2°С в течение 2 часов. После инкубации планшет центрифугировали при 1000 об/мин в течение 2 минут для сбора конденсата и планшет охлаждали в течение 15-60 мин. после инкубации. Планшет считывали в планшет-ридере при 562 нм и данные анализировали с помощью SoftMax Pro.
Среднюю оптическую плотность стандартов относительно теоретической концентрации белка в стандартах наносили на полулогарифмический график и получали квадратичную аппроксимацию. Квадратичная аппроксимация основана на уравнении:
Y=A+Bx+Cx2,
где A, B, C обозначают параметры аппроксимации кривой, x обозначает концентрацию образца в мкг/мл, а Y обозначает оптическую плотность в OD. Тест считали признанным только в том случае, если r2 стандартной кривой составлял ≥0,98, средняя оптическая плотность холостой пробы была меньше, чем у самого низкого стандарта (1 мкг/мл), а коэффициент вариации оптической плотности для каждого стандарта в 3 повторностях составлял ≤10%. Затем для определения концентрации белка в тестируемых образцах использовали калибровочную кривую. Конечную концентрацию белка рассчитывали с применением уравнения:
концентрация общего белка (мкг/мл) = коэффициент разбавления x средняя измеренная концентрация белка (мкг/мл).
Результаты
Концентрация общего белка, измеренная с помощью микропланшетного анализа BCA, составляла 167 мкг/мл на 1,0×1013 г. в./мл для предыдущей партии вектора, 179 мкг/мл на 1,0×1013 г. в./мл для AVXS-101 серии 600156, 176 мкг/мл на 1,0×1013 г. в./мл для AVXS1-101 серии 600307, 182 мкг/мл на 1,0×1013 г. в./мл для AVXS-201 DP и 418 мкг/мл на 1,0×1013 г. в./мл для AVXS-301 DP.
Пример 22. Чистота и спецификация при выпуске AVXS-101, AVXS-201 и AVXS-301
Лекарственное вещество AVXS-101 и лекарственный продукт AVXS-101 из примеров 1-4 тестировали в отношении чистоты. В таблицах 32 и 33 приведены спецификация и критерии выпуска для этих продуктов.
Таблица 32. Спецификация при выпуске для лекарственного вещества AVXS-101
Таблица 33. Спецификации при выпуске для лекарственного продукта AVXS-101
≤ 6000 частиц ≥ 10 мкм на контейнер
- Примесь 1A (~71-73 кДа)
- Примесь 1 (~61-67 кДа)
- Примесь 2 (~56-64 кДа)
- Примесь 3 (~48-58 кДа)
- Примесь 4 (~33-38 кДа)
- Примесь 5 (~30-34 кДа)
AVXS-201 и AVXS-301 можно получить и очистить с применением того же процесса, который описан для AVXS-101, например, который описан в примерах 1-4. Спецификация и критерии выпуска для партий каждого продукта, полученного в биореакторах, показаны в таблицах 34-36.
Таблица 34. Лекарственное вещество AVXS-201 серии 283-0218-005
совместно мигрируют с контролем AVXS-201
% Примесей с MW:
53,2 кДа, < 1,0%
36,9 кДа, < 1,0%
29,9 кДа, < 1,0%
Таблица 35. Лекарственный продукт AVXS-201 серии 283-0218-006
• 0 частиц размером, составляющим 10 мкм на контейнер или больше
MW основной полосы
VP1: 79,0 кДа
VP2: 65,0 кДа
VP3: 57,7 кДа
совместно мигрируют с контролем AVXS-201
% Примесей с MW:
52,5 кДа, < 1,0%
36,4 кДа, < 1,0%
29,1 кДа, < 1,0%
Таблица 36. Чистота исследуемой партии лекарственного продукта AVXS-301
• 0 частиц размером, составляющим 10 мкм на контейнер или больше
микропланшетного анализа BCA
SDS-PAGE
совместно мигрируют с контролем AVXS-301
SDS-PAGE
Примесей с MW:
- Примесь 1 (~61-67 кДа) 2%
Высокая чистота, достигнутая для AVXS-201 и AVXS-301, демонстрировала, что способы получения и очистки вирусных векторов на основе AAV, описанных в настоящей заявке, характеризуются широкой применимостью к вирусным векторам на основе AAV с различной полезной нагрузкой.
При прочтении описанных вариантов осуществления со ссылкой на прилагаемые графические материалы следует понимать, что настоящее изобретение не ограничено точными вариантами осуществления, и что специалистами в данной области техники в него могут быть внесены различные изменения и модификации без отклонения от объема или сущности настоящего изобретения и вариантов осуществления, определенных в прилагаемой формуле изобретения.
Перечень иллюстративных вариантов осуществления
1. Фармацевтическая композиция, содержащая:
(a) 1-8 X 1013 геномов вектора/мл парвовируса, сконструированного с включением трансгена;
(b) менее 5,0% пустых капсидов;
(c) менее 40 нг/мл остаточного белка клетки-хозяина на 1 X 1013 г. в./мл;
(d) менее 1,2 X 106 пг/мл остаточной ДНК клетки-хозяина на 1 X 1013 г. в./мл и при этом
(e) по меньшей мере 80% из 1-8 X 1013 геномов вектора/мл являются функциональными.
2. Композиция согласно варианту осуществления 1, где количество остаточной плазмидной ДНК составляет менее 1,7 X 106 пг на 1 X 1013 г. в.
3. Композиция согласно варианту осуществления 1, где количество геномов вектора/мл парвовируса составляет 1,8-2,2 X 1013 геномов вектора/мл.
4. Способ очистки частиц AAV из культуры клеток-хозяев млекопитающих с получением замороженного промежуточного лекарственного вещества, включающий стадии:
(a) культивирования клеток, которые были трансфицированы рекомбинантным вирионом AAV;
(b) осуществления сбора увеличенного количества вирусных частиц из клеток после периода культивирования;
(c) очистки вирусных частиц с помощью фильтрации для удаления каких-либо интактных клеток или клеточного дебриса;
(d) подвергания элюента из стадии (с) фильтрации в тангенциальном потоке и
(e) замораживания полученного промежуточного препарата очищенных вирусных частиц.
5. Способ согласно варианту осуществления 4, где в ходе стадии осуществления сбора применяют эндонуклеазу.
6. Способ согласно варианту осуществления 5, где эндонуклеаза представляет собой бензоназу.
7. Способ согласно варианту осуществления 4, где на стадии очистки применяют глубинную фильтрацию с последующей фильтрацией через фильтр, который удаляет макромолекулярные загрязняющие вещества и клеточный дебрис.
8. Способ согласно варианту осуществления 4, где на стадии фильтрации в тангенциальном потоке применяют целлюлозные мембраны.
9. Способ очистки образца частиц AAV из культуры клеток-хозяев млекопитающих с получением лекарственного продукта, включающий стадии:
(a) стадию подкисления и осветления;
(b) стадию осуществления катионообменной хроматографии;
(c) стадию фильтрации в тангенциальном потоке;
(d) стадию ультрацентрифугирования с помощью CsCl для удаления пустых капсидов и
(e) стадию фильтрации в тангенциальном потоке.
10. Способ согласно варианту осуществления 9, где стадия подкисления и осветления предусматривает регулирование образца до рН 3,5, а белки и ДНК клетки-хозяина удаляют посредством флокуляции с детергентом.
11. Способ по варианту осуществления 9, где катионообменная колонка содержит смолу с сульфонильными группами.
12. Способ согласно варианту осуществления 9, где на стадии (с) фильтрации в тангенциальном потоке применяют целлюлозные мембраны с отсечением по молекулярной массе, представляющим собой MW 300 кДа, и уменьшают объем элюата из катионообменной стадии в по меньшей мере шесть раз.
13. Способ согласно варианту осуществления 9, где на стадии ультрацентрифугирования с помощью CsCl удаляют по меньшей мере 80% пустых капсидов в образце, применяют глубинную фильтрацию с последующей фильтрацией через фильтр с размером пор 0,45 микрона.
14. Способ согласно варианту осуществления 9, где на стадии (e) фильтрации в тангенциальном потоке применяют целлюлозные мембраны с порогом отсечения по молекулярной массе, представляющим собой MW 300 кДа.
15. Способ лечения неврологического заболевания у пациента, нуждающегося в этом, включающий внутривенную или интратекальную доставку фармацевтической композиции согласно любому из вариантов осуществления 1-3, где парвовирус содержит самокомплементарный геном AAV9, и где сконструированный трансген содержит полинуклеотид SMN, и где заболевание представляет собой SMA.
16. Способ лечения неврологического заболевания у пациента, нуждающегося в этом, включающий интратекальную доставку фармацевтической композиции согласно любому из вариантов осуществления 1-3 с контрастным средством, где парвовирус содержит самокомплементарный геном AAV9, и где сконструированный трансген содержит полинуклеотид SMN, где заболевание представляет собой SMA, и где контрастное средство представляет собой омнипак 180.
17. Способ согласно любому из вариантов осуществления 15-16, где SMA представляет собой SMA II типа, III типа или IV типа.
18. Способ лечения SMA II, III или IV типа у пациента, нуждающегося в этом, включающий интратекальную доставку фармацевтической композиции по любому из вариантов осуществления 1-3 с контрастным средством, где парвовирус содержит самокомплементарный геном AAV9, где сконструированный трансген содержит полинуклеотид SMN, и где контрастное средство представляет собой омнипак 180.
19. Способ лечения SMA I типа у пациента, нуждающегося в этом, включающий внутривенную доставку фармацевтической композиции согласно любому из вариантов осуществления 1-3, где парвовирус содержит самокомплементарный геном AAV9, и где сконструированный трансген содержит полинуклеотид SMN.
20. Способ согласно варианту осуществления 19, где возраст пациента составляет 0-9 месяцев.
21. Способ согласно варианту осуществления 20, где возраст пациента составляет 0-6 месяцев.
22. Способ согласно варианту осуществления 19, где вес пациента детского возраста составляет не более приблизительно 8 кг.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> AveXis Inc.
<120> СРЕДСТВА И СПОСОБ ПОЛУЧЕНИЯ ВИРУСНЫХ ВЕКТОРОВ И ВАРИАНТЫ
ИХ ПРИМЕНЕНИЯ
<130> AVEX-003/001WO
<150> 62/583,035
<151> 2017-11-08
<160> 4
<170> PatentIn версия 3.5
<210> 1
<211> 2359
<212> ДНК
<213> Искусственная
<220>
<223> векторная конструкция
<400> 1
ctgcgcgctc gctcgctcac tgaggccgcc cgggcaaagc ccgggcgtcg ggcgaccttt 60
ggtcgcccgg cctcagtgag cgagcgagcg cgcagagagg gagtggaatt cacgcgtgga 120
tctgaattca attcacgcgt ggtacctctg gtcgttacat aacttacggt aaatggcccg 180
cctggctgac cgcccaacga cccccgccca ttgacgtcaa taatgacgta tgttcccata 240
gtaacgccaa tagggacttt ccattgacgt caatgggtgg agtatttacg gtaaactgcc 300
cacttggcag tacatcaagt gtatcatatg ccaagtacgc cccctattga cgtcaatgac 360
ggtaaatggc ccgcctggca ttatgcccag tacatgacct tatgggactt tcctacttgg 420
cagtacatct actcgaggcc acgttctgct tcactctccc catctccccc ccctccccac 480
ccccaatttt gtatttattt attttttaat tattttgtgc agcgatgggg gcgggggggg 540
ggggggggcg cgcgccaggc ggggcggggc ggggcgaggg gcggggcggg gcgaggcgga 600
gaggtgcggc ggcagccaat cagagcggcg cgctccgaaa gtttcctttt atggcgaggc 660
ggcggcggcg gcggccctat aaaaagcgaa gcgcgcggcg ggcgggagcg ggatcagcca 720
ccgcggtggc ggcctagagt cgacgaggaa ctgaaaaacc agaaagttaa ctggtaagtt 780
tagtcttttt gtcttttatt tcaggtcccg gatccggtgg tggtgcaaat caaagaactg 840
ctcctcagtg gatgttgcct ttacttctag gcctgtacgg aagtgttact tctgctctaa 900
aagctgcgga attgtacccg cggccgatcc accggtccgg aattcccggg atatcgtcga 960
cccacgcgtc cgggccccac gctgcgcacc cgcgggtttg ctatggcgat gagcagcggc 1020
ggcagtggtg gcggcgtccc ggagcaggag gattccgtgc tgttccggcg cggcacaggc 1080
cagagcgatg attctgacat ttgggatgat acagcactga taaaagcata tgataaagct 1140
gtggcttcat ttaagcatgc tctaaagaat ggtgacattt gtgaaacttc gggtaaacca 1200
aaaaccacac ctaaaagaaa acctgctaag aagaataaaa gccaaaagaa gaatactgca 1260
gcttccttac aacagtggaa agttggggac aaatgttctg ccatttggtc agaagacggt 1320
tgcatttacc cagctaccat tgcttcaatt gattttaaga gagaaacctg tgttgtggtt 1380
tacactggat atggaaatag agaggagcaa aatctgtccg atctactttc cccaatctgt 1440
gaagtagcta ataatataga acagaatgct caagagaatg aaaatgaaag ccaagtttca 1500
acagatgaaa gtgagaactc caggtctcct ggaaataaat cagataacat caagcccaaa 1560
tctgctccat ggaactcttt tctccctcca ccacccccca tgccagggcc aagactggga 1620
ccaggaaagc caggtctaaa attcaatggc ccaccaccgc caccgccacc accaccaccc 1680
cacttactat catgctggct gcctccattt ccttctggac caccaataat tcccccacca 1740
cctcccatat gtccagattc tcttgatgat gctgatgctt tgggaagtat gttaatttca 1800
tggtacatga gtggctatca tactggctat tatatgggtt ttagacaaaa tcaaaaagaa 1860
ggaaggtgct cacattcctt aaattaagga gaaatgctgg catagagcag cactaaatga 1920
caccactaaa gaaacgatca gacagatcta gaaagcttat cgataccgtc gactagagct 1980
cgctgatcag cctcgactgt gccttctagt tgccagccat ctgttgtttg cccctccccc 2040
gtgccttcct tgaccctgga aggtgccact cccactgtcc tttcctaata aaatgaggaa 2100
attgcatcgc attgtctgag taggtgtcat tctattctgg ggggtggggt ggggcaggac 2160
agcaaggggg aggattggga agacaatagc aggcatgctg gggagagatc gatctgagga 2220
acccctagtg atggagttgg ccactccctc tctgcgcgct cgctcgctca ctgaggccgg 2280
gcgaccaaag gtcgcccgac gcccgggctt tgcccgggcg gcctcagtga gcgagcgagc 2340
gcgcagagag ggagtggcc 2359
<210> 2
<211> 294
<212> БЕЛОК
<213> Homo sapiens
<400> 2
Met Ala Met Ser Ser Gly Gly Ser Gly Gly Gly Val Pro Glu Gln Glu
1. 5 10 15
Asp Ser Val Leu Phe Arg Arg Gly Thr Gly Gln Ser Asp Asp Ser Asp
20 25 30
Ile Trp Asp Asp Thr Ala Leu Ile Lys Ala Tyr Asp Lys Ala Val Ala
35 40 45
Ser Phe Lys His Ala Leu Lys Asn Gly Asp Ile Cys Glu Thr Ser Gly
50 55 60
Lys Pro Lys Thr Thr Pro Lys Arg Lys Pro Ala Lys Lys Asn Lys Ser
65 70 75 80
Gln Lys Lys Asn Thr Ala Ala Ser Leu Gln Gln Trp Lys Val Gly Asp
85 90 95
Lys Cys Ser Ala Ile Trp Ser Glu Asp Gly Cys Ile Tyr Pro Ala Thr
100 105 110
Ile Ala Ser Ile Asp Phe Lys Arg Glu Thr Cys Val Val Val Tyr Thr
115 120 125
Gly Tyr Gly Asn Arg Glu Glu Gln Asn Leu Ser Asp Leu Leu Ser Pro
130 135 140
Ile Cys Glu Val Ala Asn Asn Ile Glu Gln Asn Ala Gln Glu Asn Glu
145 150 155 160
Asn Glu Ser Gln Val Ser Thr Asp Glu Ser Glu Asn Ser Arg Ser Pro
165 170 175
Gly Asn Lys Ser Asp Asn Ile Lys Pro Lys Ser Ala Pro Trp Asn Ser
180 185 190
Phe Leu Pro Pro Pro Pro Pro Met Pro Gly Pro Arg Leu Gly Pro Gly
195 200 205
Lys Pro Gly Leu Lys Phe Asn Gly Pro Pro Pro Pro Pro Pro Pro Pro
210 215 220
Pro Pro His Leu Leu Ser Cys Trp Leu Pro Pro Phe Pro Ser Gly Pro
225 230 235 240
Pro Ile Ile Pro Pro Pro Pro Pro Ile Cys Pro Asp Ser Leu Asp Asp
245 250 255
Ala Asp Ala Leu Gly Ser Met Leu Ile Ser Trp Tyr Met Ser Gly Tyr
260 265 270
His Thr Gly Tyr Tyr Met Gly Phe Arg Gln Asn Gln Lys Glu Gly Arg
275 280 285
Cys Ser His Ser Leu Asn
290
<210> 3
<211> 736
<212> БЕЛОК
<213> Аденоассоциированный вирус 9
<400> 3
Met Ala Ala Asp Gly Tyr Leu Pro Asp Trp Leu Glu Asp Asn Leu Ser
1. 5 10 15
Glu Gly Ile Arg Glu Trp Trp Ala Leu Lys Pro Gly Ala Pro Gln Pro
20 25 30
Lys Ala Asn Gln Gln His Gln Asp Asn Ala Arg Gly Leu Val Leu Pro
35 40 45
Gly Tyr Lys Tyr Leu Gly Pro Gly Asn Gly Leu Asp Lys Gly Glu Pro
50 55 60
Val Asn Ala Ala Asp Ala Ala Ala Leu Glu His Asp Lys Ala Tyr Asp
65 70 75 80
Gln Gln Leu Lys Ala Gly Asp Asn Pro Tyr Leu Lys Tyr Asn His Ala
85 90 95
Asp Ala Glu Phe Gln Glu Arg Leu Lys Glu Asp Thr Ser Phe Gly Gly
100 105 110
Asn Leu Gly Arg Ala Val Phe Gln Ala Lys Lys Arg Leu Leu Glu Pro
115 120 125
Leu Gly Leu Val Glu Glu Ala Ala Lys Thr Ala Pro Gly Lys Lys Arg
130 135 140
Pro Val Glu Gln Ser Pro Gln Glu Pro Asp Ser Ser Ala Gly Ile Gly
145 150 155 160
Lys Ser Gly Ala Gln Pro Ala Lys Lys Arg Leu Asn Phe Gly Gln Thr
165 170 175
Gly Asp Thr Glu Ser Val Pro Asp Pro Gln Pro Ile Gly Glu Pro Pro
180 185 190
Ala Ala Pro Ser Gly Val Gly Ser Leu Thr Met Ala Ser Gly Gly Gly
195 200 205
Ala Pro Val Ala Asp Asn Asn Glu Gly Ala Asp Gly Val Gly Ser Ser
210 215 220
Ser Gly Asn Trp His Cys Asp Ser Gln Trp Leu Gly Asp Arg Val Ile
225 230 235 240
Thr Thr Ser Thr Arg Thr Trp Ala Leu Pro Thr Tyr Asn Asn His Leu
245 250 255
Tyr Lys Gln Ile Ser Asn Ser Thr Ser Gly Gly Ser Ser Asn Asp Asn
260 265 270
Ala Tyr Phe Gly Tyr Ser Thr Pro Trp Gly Tyr Phe Asp Phe Asn Arg
275 280 285
Phe His Cys His Phe Ser Pro Arg Asp Trp Gln Arg Leu Ile Asn Asn
290 295 300
Asn Trp Gly Phe Arg Pro Lys Arg Leu Asn Phe Lys Leu Phe Asn Ile
305 310 315 320
Gln Val Lys Glu Val Thr Asp Asn Asn Gly Val Lys Thr Ile Ala Asn
325 330 335
Asn Leu Thr Ser Thr Val Gln Val Phe Thr Asp Ser Asp Tyr Gln Leu
340 345 350
Pro Tyr Val Leu Gly Ser Ala His Glu Gly Cys Leu Pro Pro Phe Pro
355 360 365
Ala Asp Val Phe Met Ile Pro Gln Tyr Gly Tyr Leu Thr Leu Asn Asp
370 375 380
Gly Ser Gln Ala Val Gly Arg Ser Ser Phe Tyr Cys Leu Glu Tyr Phe
385 390 395 400
Pro Ser Gln Met Leu Arg Thr Gly Asn Asn Phe Gln Phe Ser Tyr Glu
405 410 415
Phe Glu Asn Val Pro Phe His Ser Ser Tyr Ala His Ser Gln Ser Leu
420 425 430
Asp Arg Leu Met Asn Pro Leu Ile Asp Gln Tyr Leu Tyr Tyr Leu Ser
435 440 445
Lys Thr Ile Asn Gly Ser Gly Gln Asn Gln Gln Thr Leu Lys Phe Ser
450 455 460
Val Ala Gly Pro Ser Asn Met Ala Val Gln Gly Arg Asn Tyr Ile Pro
465 470 475 480
Gly Pro Ser Tyr Arg Gln Gln Arg Val Ser Thr Thr Val Thr Gln Asn
485 490 495
Asn Asn Ser Glu Phe Ala Trp Pro Gly Ala Ser Ser Trp Ala Leu Asn
500 505 510
Gly Arg Asn Ser Leu Met Asn Pro Gly Pro Ala Met Ala Ser His Lys
515 520 525
Glu Gly Glu Asp Arg Phe Phe Pro Leu Ser Gly Ser Leu Ile Phe Gly
530 535 540
Lys Gln Gly Thr Gly Arg Asp Asn Val Asp Ala Asp Lys Val Met Ile
545 550 555 560
Thr Asn Glu Glu Glu Ile Lys Thr Thr Asn Pro Val Ala Thr Glu Ser
565 570 575
Tyr Gly Gln Val Ala Thr Asn His Gln Ser Ala Gln Ala Gln Ala Gln
580 585 590
Thr Gly Trp Val Gln Asn Gln Gly Ile Leu Pro Gly Met Val Trp Gln
595 600 605
Asp Arg Asp Val Tyr Leu Gln Gly Pro Ile Trp Ala Lys Ile Pro His
610 615 620
Thr Asp Gly Asn Phe His Pro Ser Pro Leu Met Gly Gly Phe Gly Met
625 630 635 640
Lys His Pro Pro Pro Gln Ile Leu Ile Lys Asn Thr Pro Val Pro Ala
645 650 655
Asp Pro Pro Thr Ala Phe Asn Lys Asp Lys Leu Asn Ser Phe Ile Thr
660 665 670
Gln Tyr Ser Thr Gly Gln Val Ser Val Glu Ile Glu Trp Glu Leu Gln
675 680 685
Lys Glu Asn Ser Lys Arg Trp Asn Pro Glu Ile Gln Tyr Thr Ser Asn
690 695 700
Tyr Tyr Lys Ser Asn Asn Val Glu Phe Ala Val Asn Thr Glu Gly Val
705 710 715 720
Tyr Ser Glu Pro Arg Pro Ile Gly Thr Arg Tyr Leu Thr Arg Asn Leu
725 730 735
<210> 4
<211> 1621
<212> ДНК
<213> Homo sapiens
<400> 4
ccacaaatgt gggagggcga taaccactcg tagaaagcgt gagaagttac tacaagcggt 60
cctcccggcc accgtactgt tccgctccca gaagccccgg gcggcggaag tcgtcactct 120
taagaaggga cggggcccca cgctgcgcac ccgcgggttt gctatggcga tgagcagcgg 180
cggcagtggt ggcggcgtcc cggagcagga ggattccgtg ctgttccggc gcggcacagg 240
ccagagcgat gattctgaca tttgggatga tacagcactg ataaaagcat atgataaagc 300
tgtggcttca tttaagcatg ctctaaagaa tggtgacatt tgtgaaactt cgggtaaacc 360
aaaaaccaca cctaaaagaa aacctgctaa gaagaataaa agccaaaaga agaatactgc 420
agcttcctta caacagtgga aagttgggga caaatgttct gccatttggt cagaagacgg 480
ttgcatttac ccagctacca ttgcttcaat tgattttaag agagaaacct gtgttgtggt 540
ttacactgga tatggaaata gagaggagca aaatctgtcc gatctacttt ccccaatctg 600
tgaagtagct aataatatag aacagaatgc tcaagagaat gaaaatgaaa gccaagtttc 660
aacagatgaa agtgagaact ccaggtctcc tggaaataaa tcagataaca tcaagcccaa 720
atctgctcca tggaactctt ttctccctcc accacccccc atgccagggc caagactggg 780
accaggaaag ccaggtctaa aattcaatgg cccaccaccg ccaccgccac caccaccacc 840
ccacttacta tcatgctggc tgcctccatt tccttctgga ccaccaataa ttcccccacc 900
acctcccata tgtccagatt ctcttgatga tgctgatgct ttgggaagta tgttaatttc 960
atggtacatg agtggctatc atactggcta ttatatgggt ttcagacaaa atcaaaaaga 1020
aggaaggtgc tcacattcct taaattaagg agaaatgctg gcatagagca gcactaaatg 1080
acaccactaa agaaacgatc agacagatct ggaatgtgaa gcgttataga agataactgg 1140
cctcatttct tcaaaatatc aagtgttggg aaagaaaaaa ggaagtggaa tgggtaactc 1200
ttcttgatta aaagttatgt aataaccaaa tgcaatgtga aatattttac tggactcttt 1260
tgaaaaacca tctgtaaaag actggggtgg gggtgggagg ccagcacggt ggtgaggcag 1320
ttgagaaaat ttgaatgtgg attagatttt gaatgatatt ggataattat tggtaatttt 1380
atggcctgtg agaagggtgt tgtagtttat aaaagactgt cttaatttgc atacttaagc 1440
atttaggaat gaagtgttag agtgtcttaa aatgtttcaa atggtttaac aaaatgtatg 1500
tgaggcgtat gtggcaaaat gttacagaat ctaactggtg gacatggctg ttcattgtac 1560
tgtttttttc tatcttctat atgtttaaaa gtatataata aaaatattta attttttttt 1620
a 1621
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ВИРУСНЫЕ ВЕКТОРЫ НА ОСНОВЕ AAV И ПУТИ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2796274C2 |
| ИНТРАТЕКАЛЬНАЯ ДОСТАВКА РЕКОМБИНАНТНОГО АДЕНОАССОЦИИРОВАННОГО ВИРУСА, КОДИРУЮЩЕГО МЕТИЛ-CPG-СВЯЗЫВАЮЩИЙ БЕЛОК 2 | 2017 |
|
RU2788084C2 |
| АДЕНОАССОЦИИРОВАННЫЙ ВИРУСНЫЙ ВЕКТОР, СОСТОЯЩИЙ ИЗ БЕЛКОВ КАПСИДА РНР.В, НУКЛЕИНОВОЙ КИСЛОТЫ, КОДИРУЮЩЕЙ БЕЛОК SMN, И ЕГО ПРИМЕНЕНИЕ | 2022 |
|
RU2833225C2 |
| ГЕННАЯ ТЕРАПИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ НАРУШЕНИЙ | 2021 |
|
RU2836835C2 |
| ГЕННАЯ ТЕРАПИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ НАРУШЕНИЙ | 2010 |
|
RU2743398C2 |
| ГЕННАЯ ТЕРАПИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ НАРУШЕНИЙ | 2010 |
|
RU2603740C2 |
| Синергетическое действие SMN1 и miR-23a при лечении спинальной мышечной атрофии | 2021 |
|
RU2839898C2 |
| Кодон-оптимизированная нуклеиновая кислота, которая кодирует белок SMN1, и ее применение | 2020 |
|
RU2742837C1 |
| МАСШТАБИРУЕМЫЕ СПОСОБЫ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ВЕКТОРА НА ОСНОВЕ АДЕНОАССОЦИИРОВАННОГО ВИРУСА (AAV) В СИСТЕМЕ БЕССЫВОРОТОЧНОЙ СУСПЕНЗИОННОЙ КУЛЬТУРЫ КЛЕТОК, ПОДХОДЯЩЕГО ДЛЯ КЛИНИЧЕСКОГО ПРИМЕНЕНИЯ | 2016 |
|
RU2766583C2 |
| ПОЛУЧЕНИЕ УЛУЧШЕННОЙ ЧЕЛОВЕЧЕСКОЙ PAH ДЛЯ ЛЕЧЕНИЯ ТЯЖЕЛОЙ PKU С ПОМОЩЬЮ ГЕННОЙ ЗАМЕСТИТЕЛЬНОЙ ТЕРАПИИ, НАПРАВЛЕННОЙ НА ПЕЧЕНЬ | 2019 |
|
RU2839587C2 |
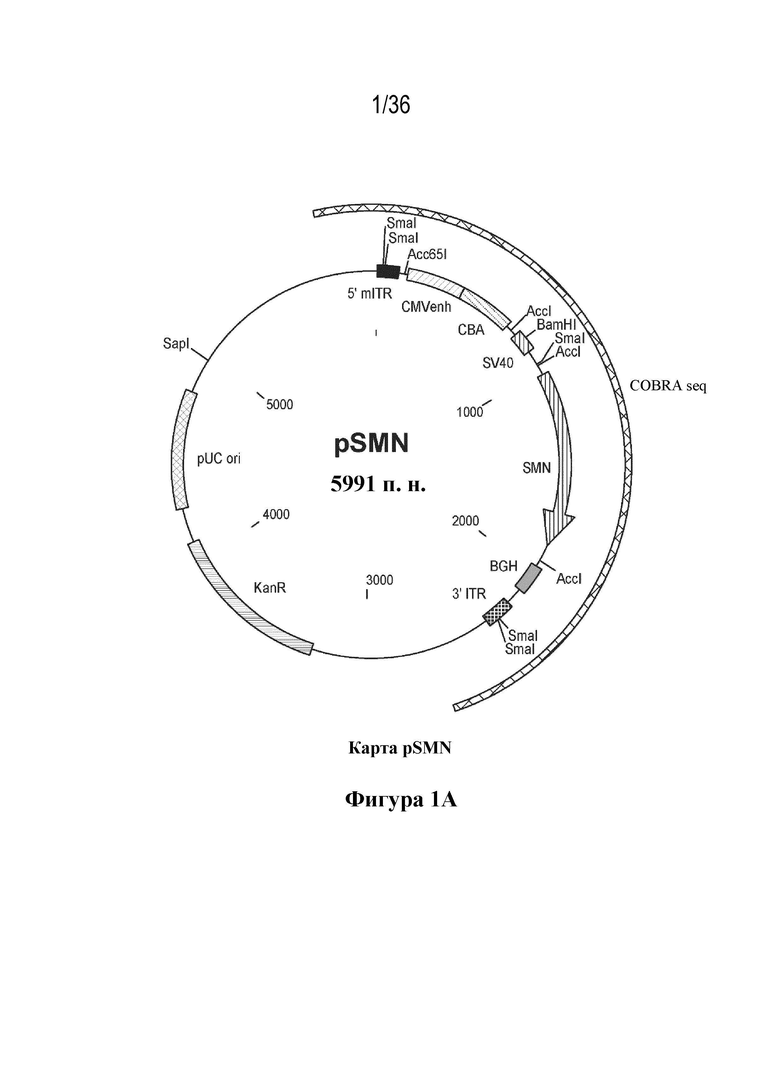
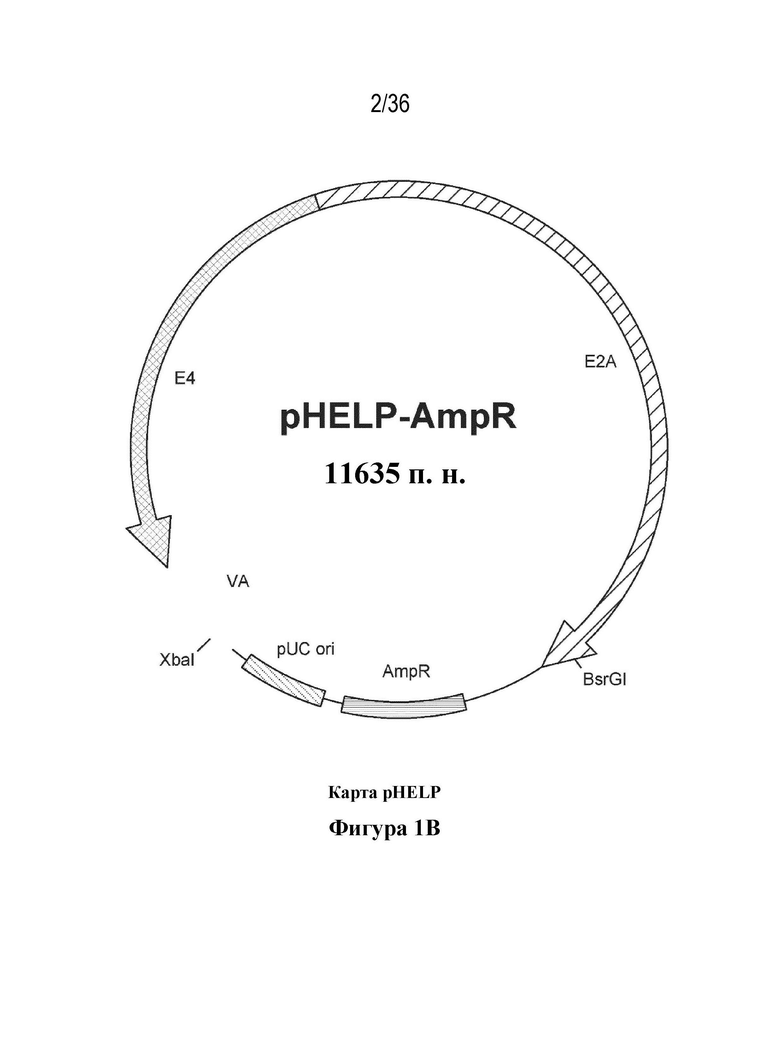
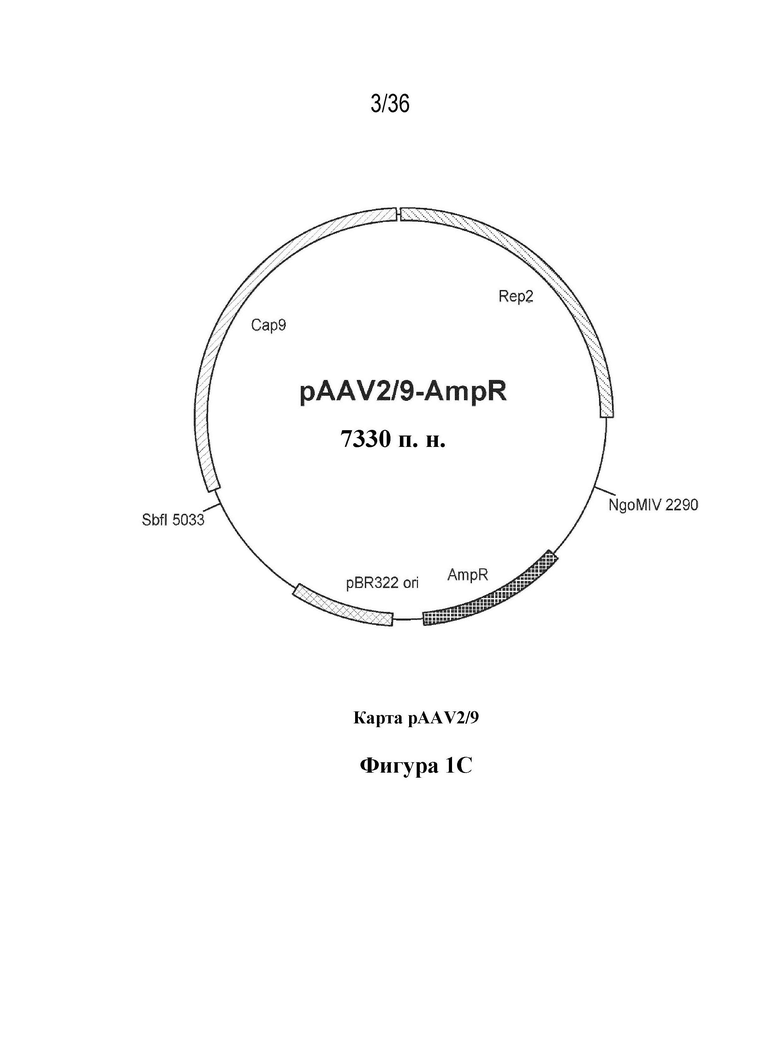
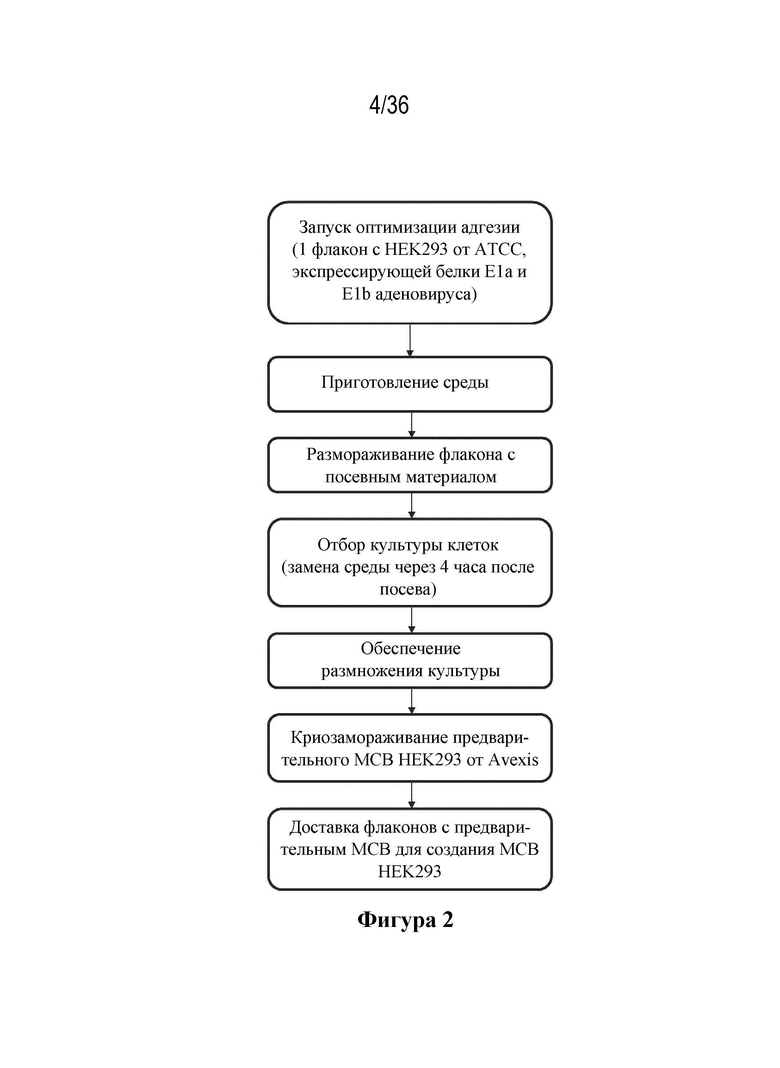
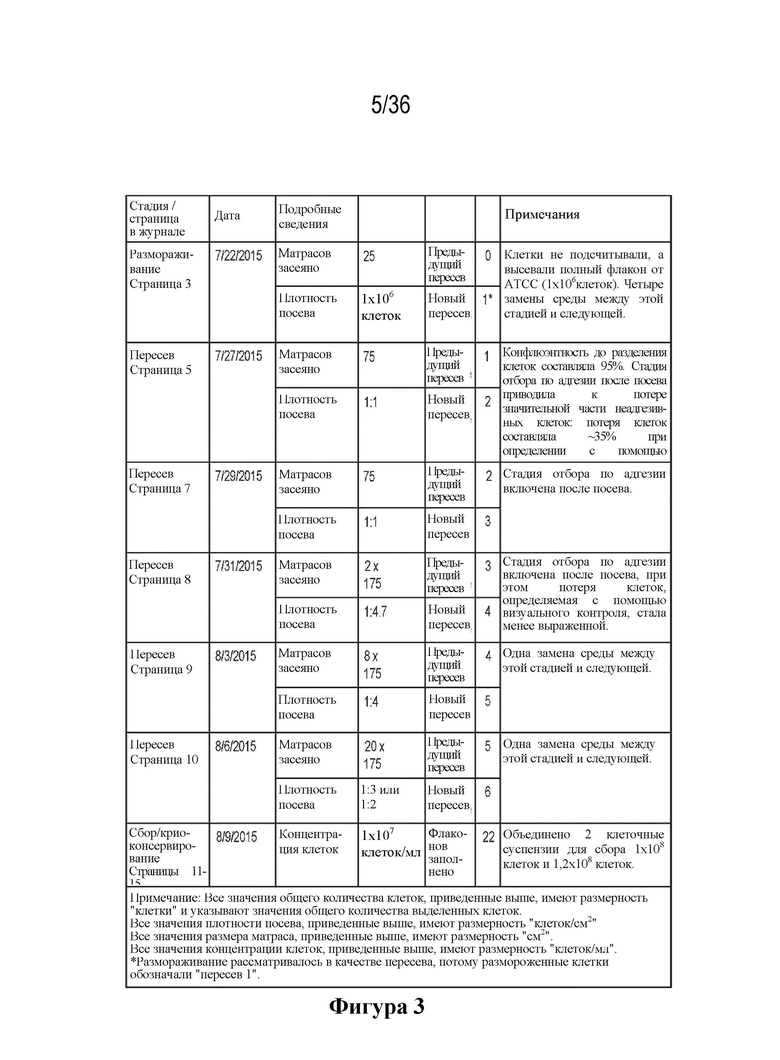
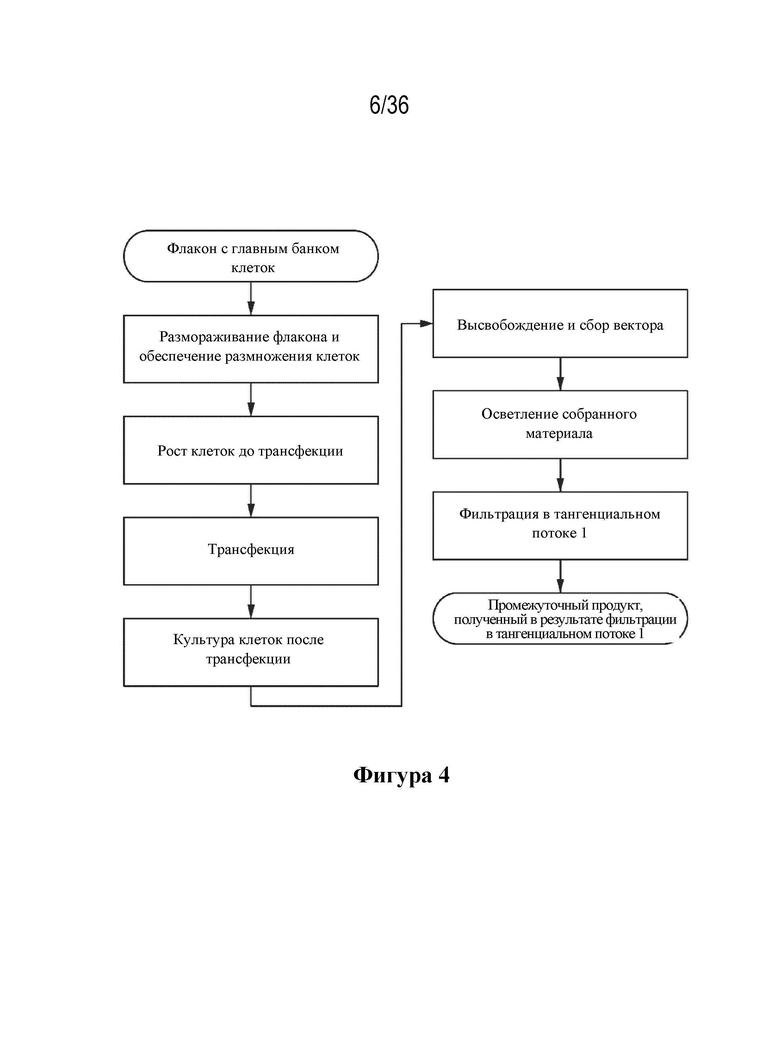
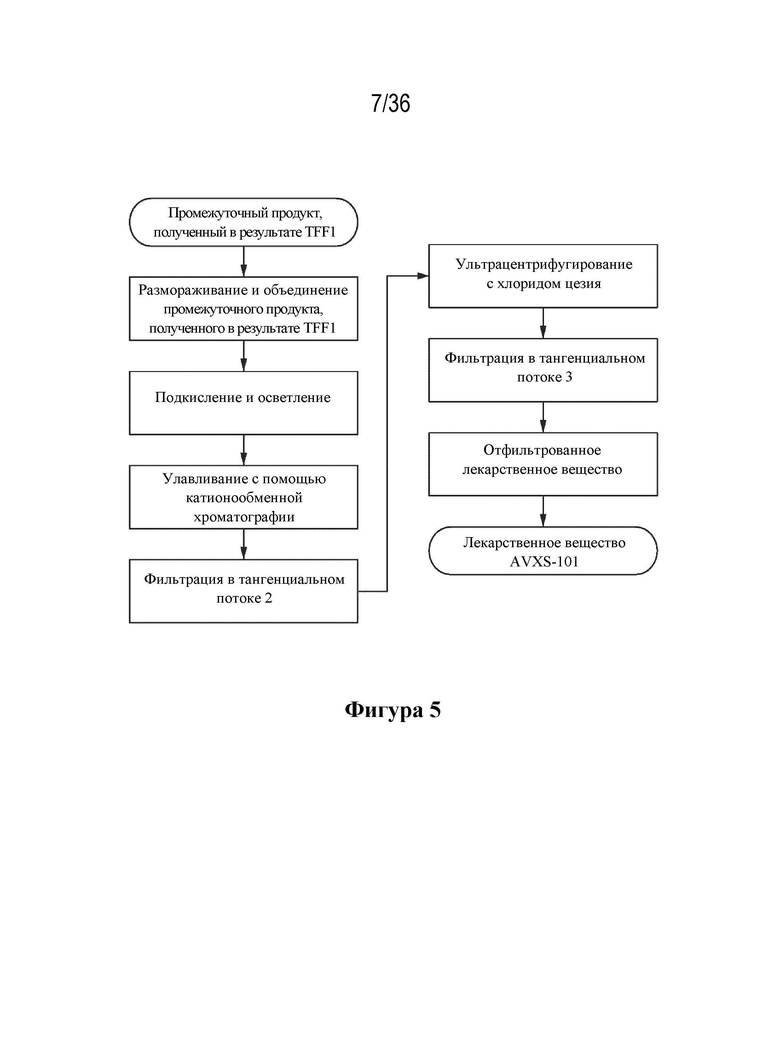
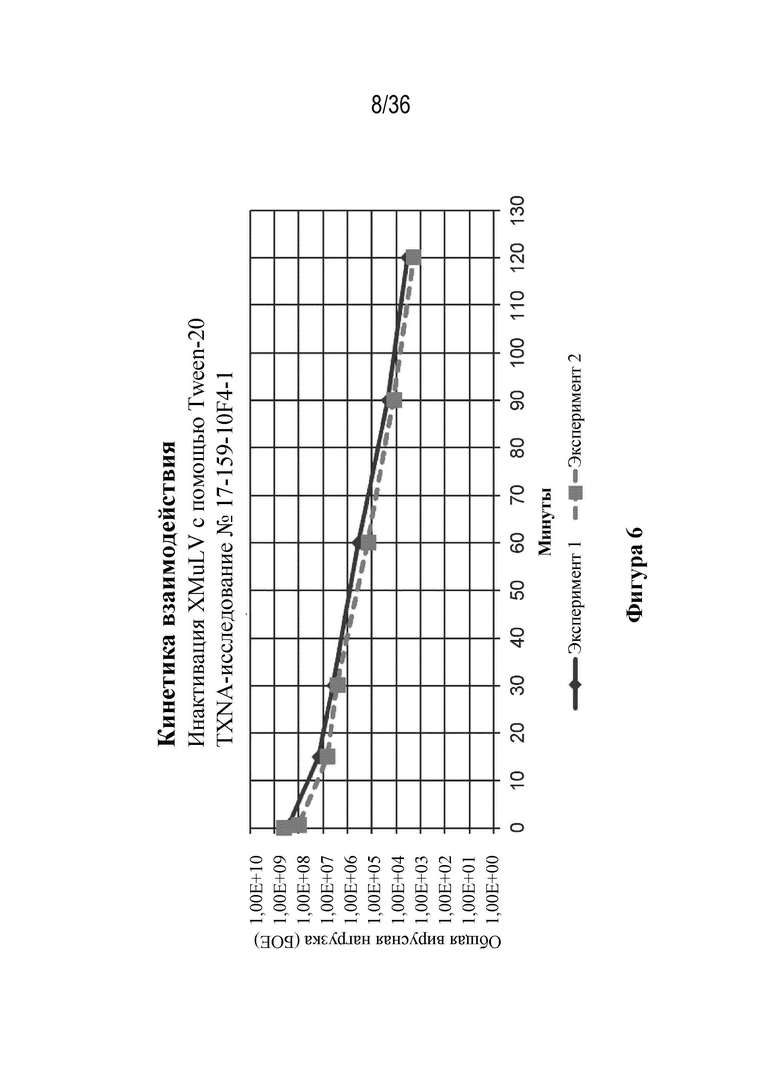
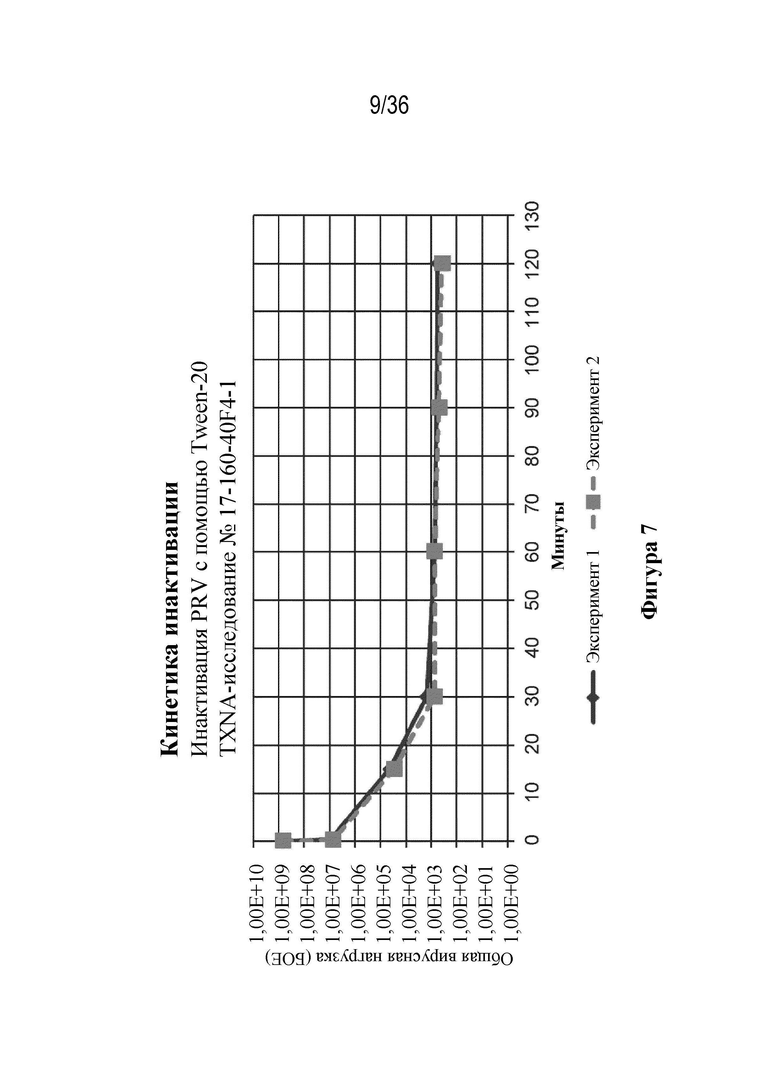
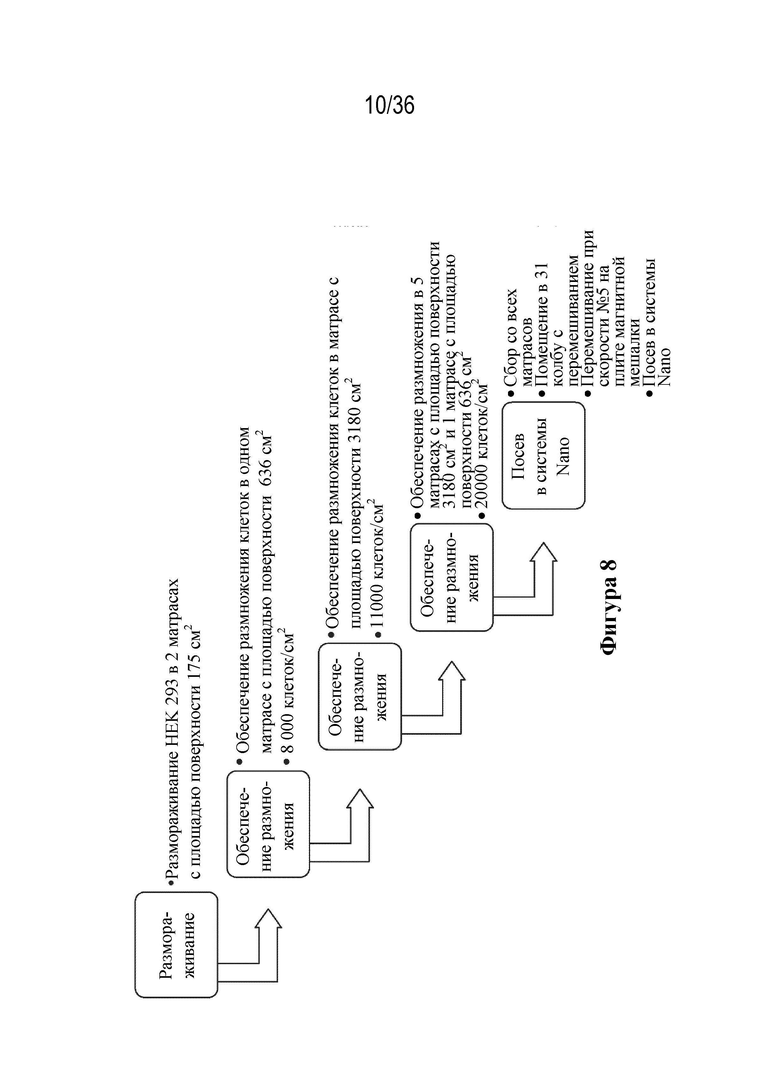
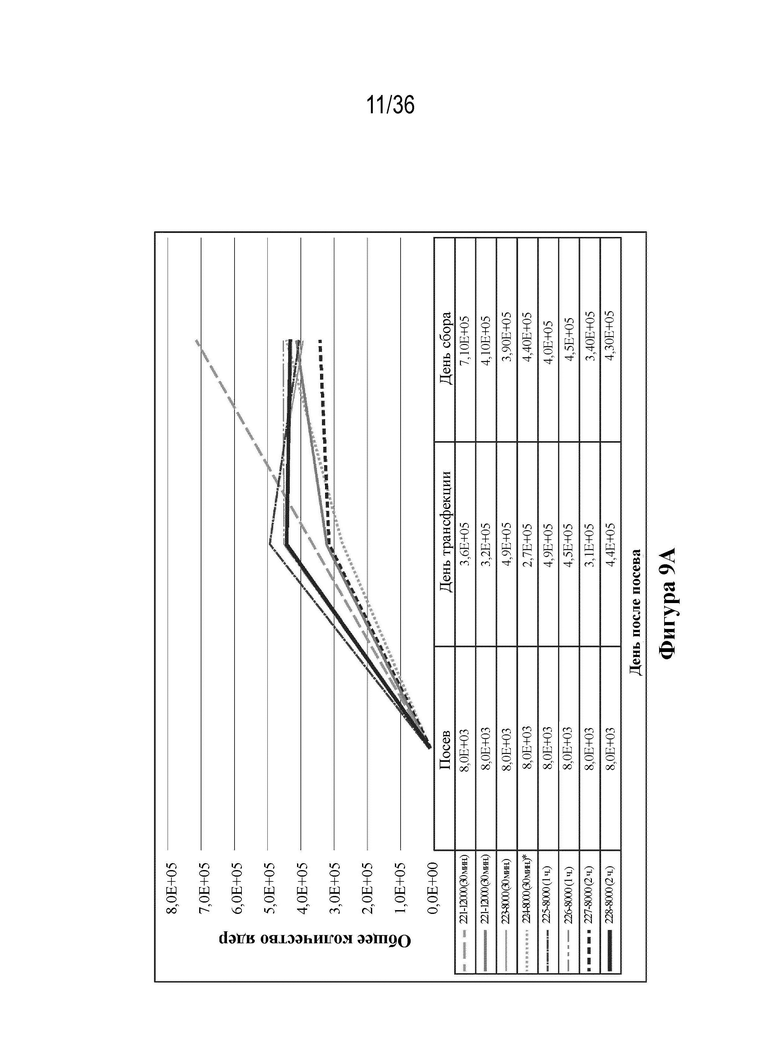
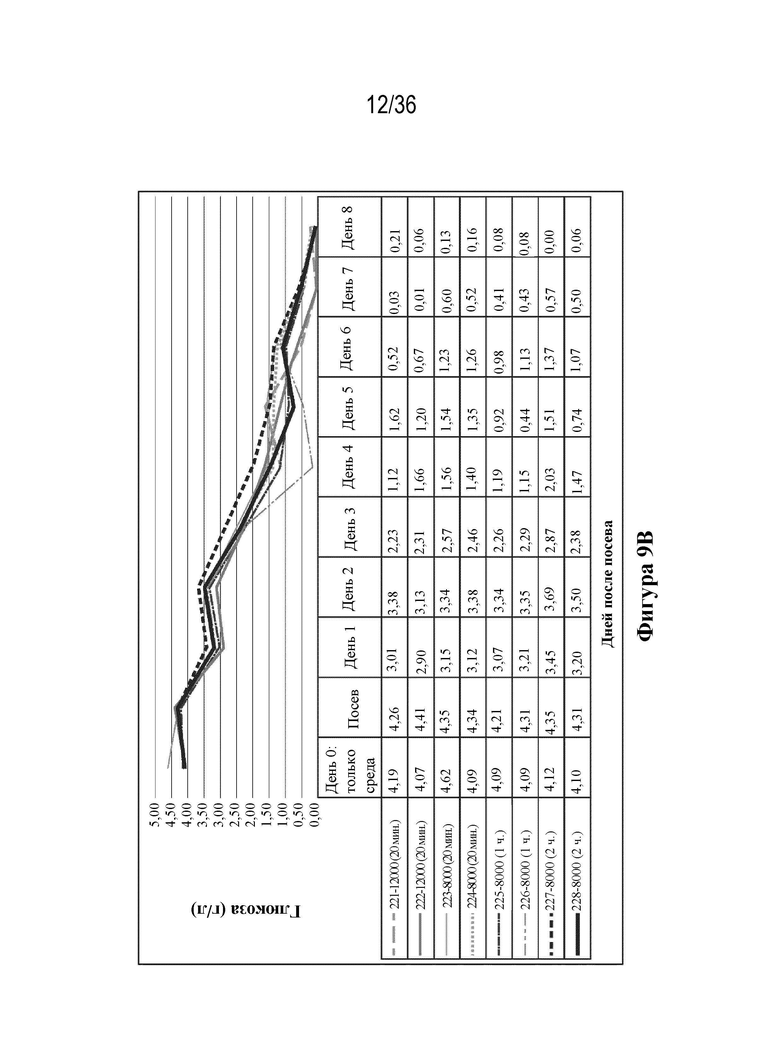
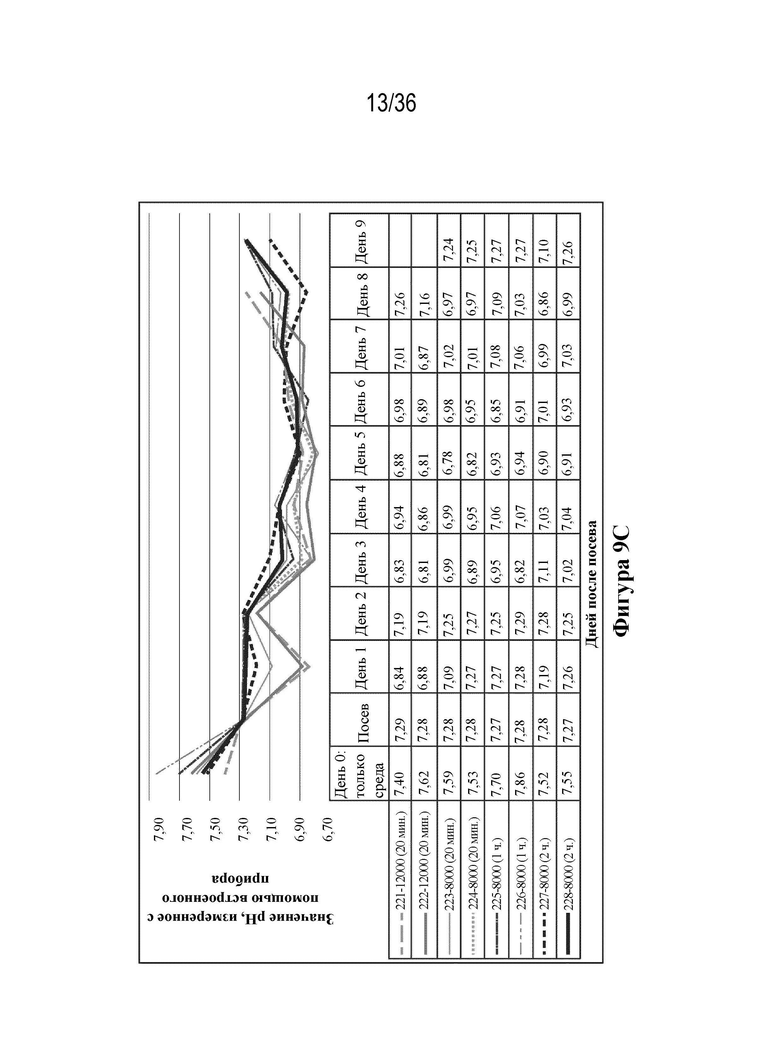
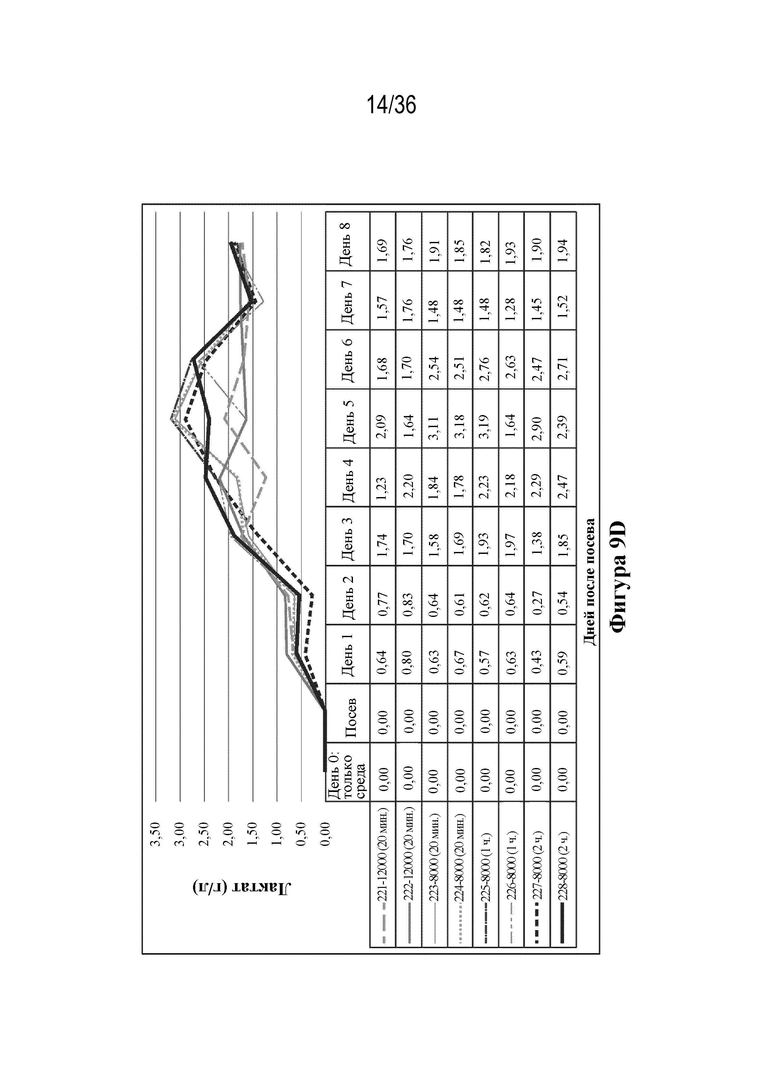

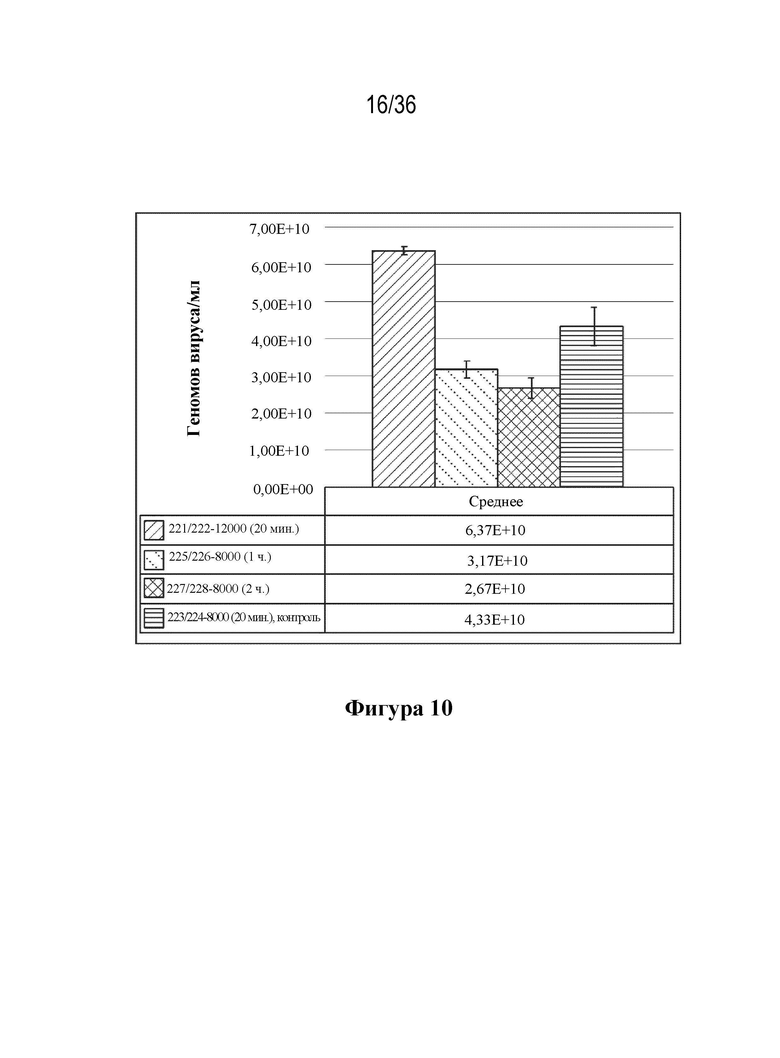
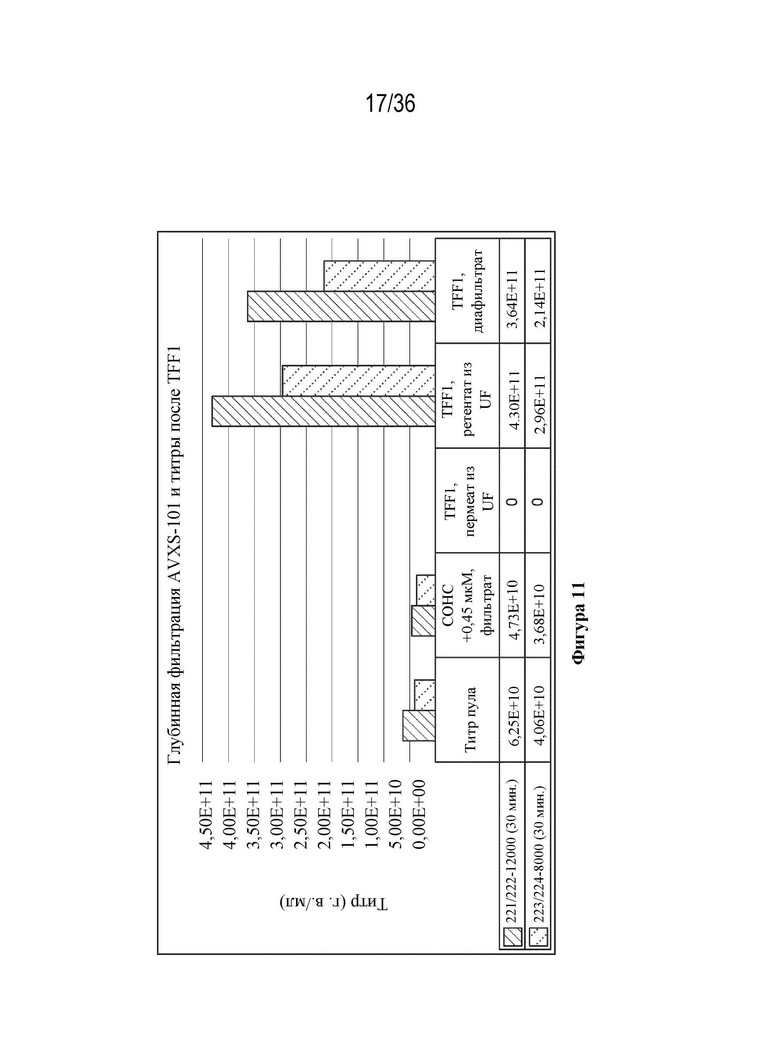
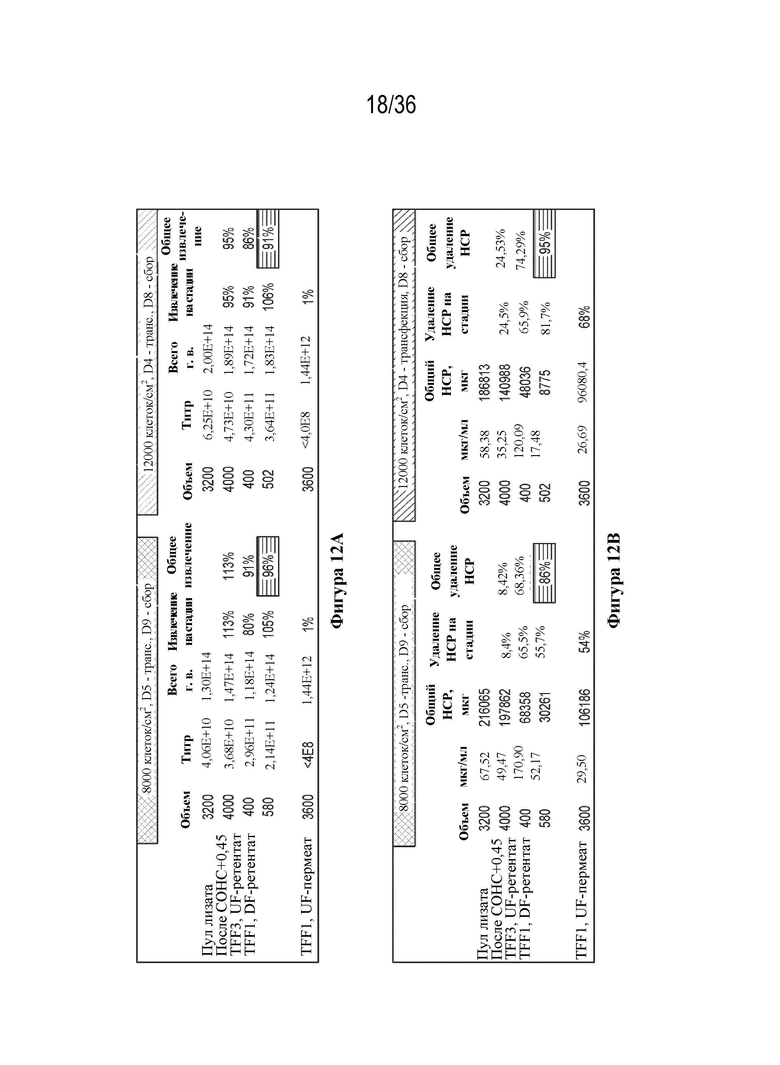
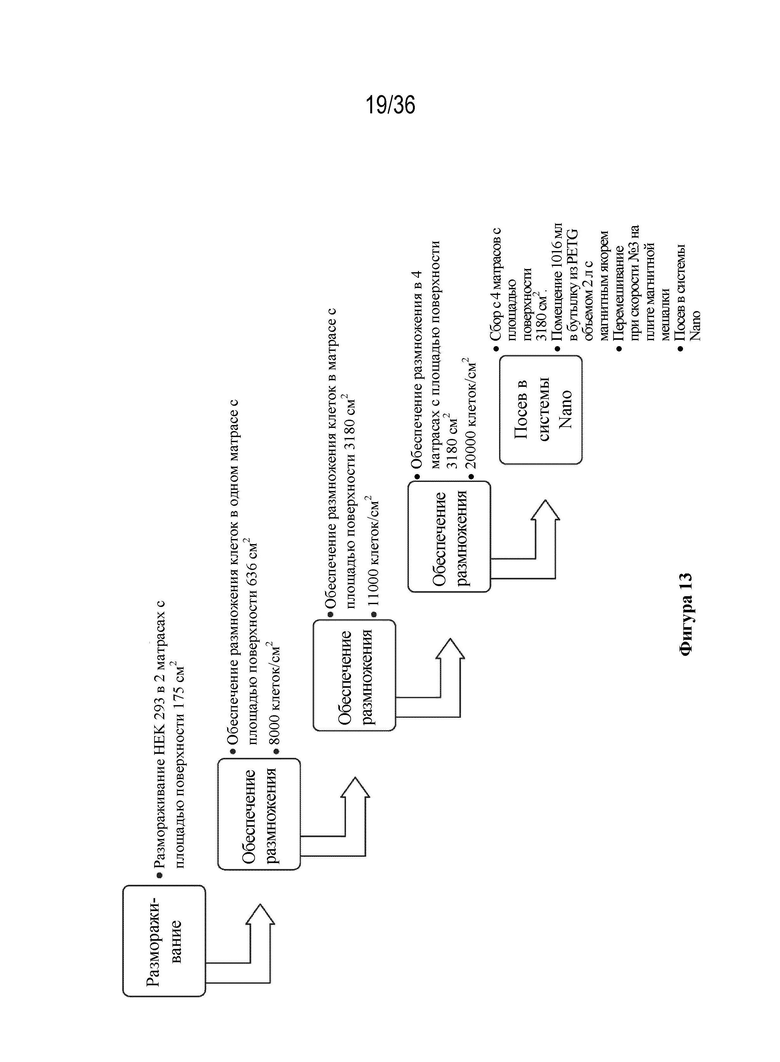
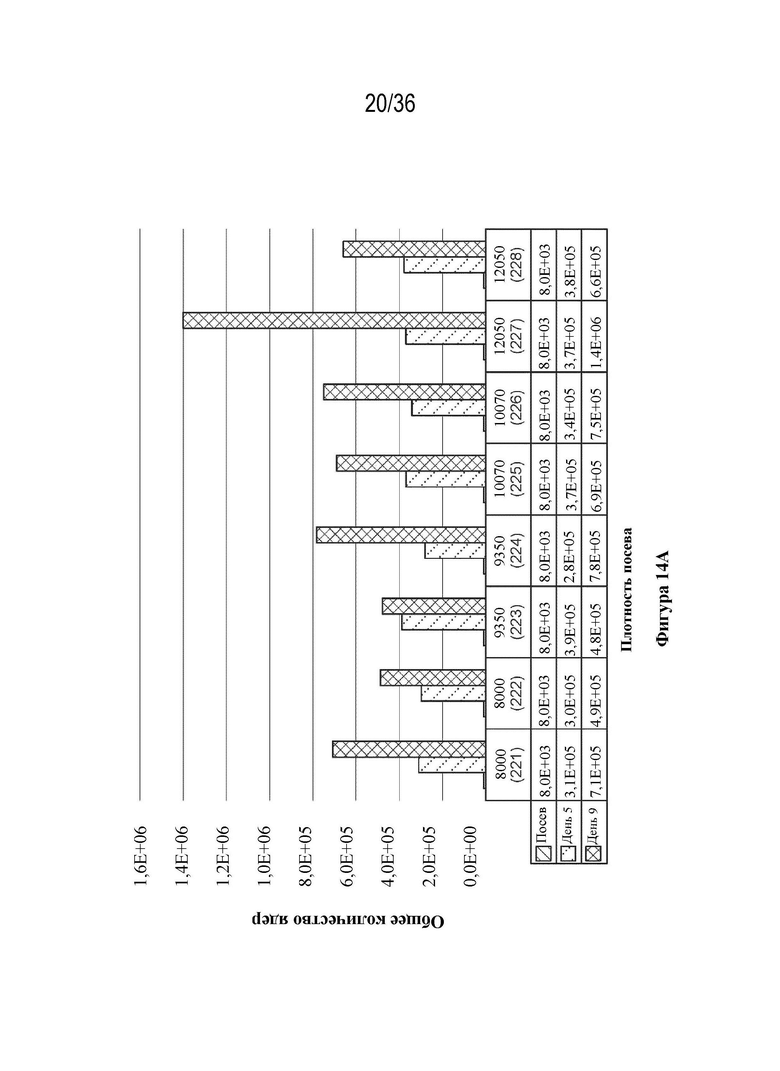
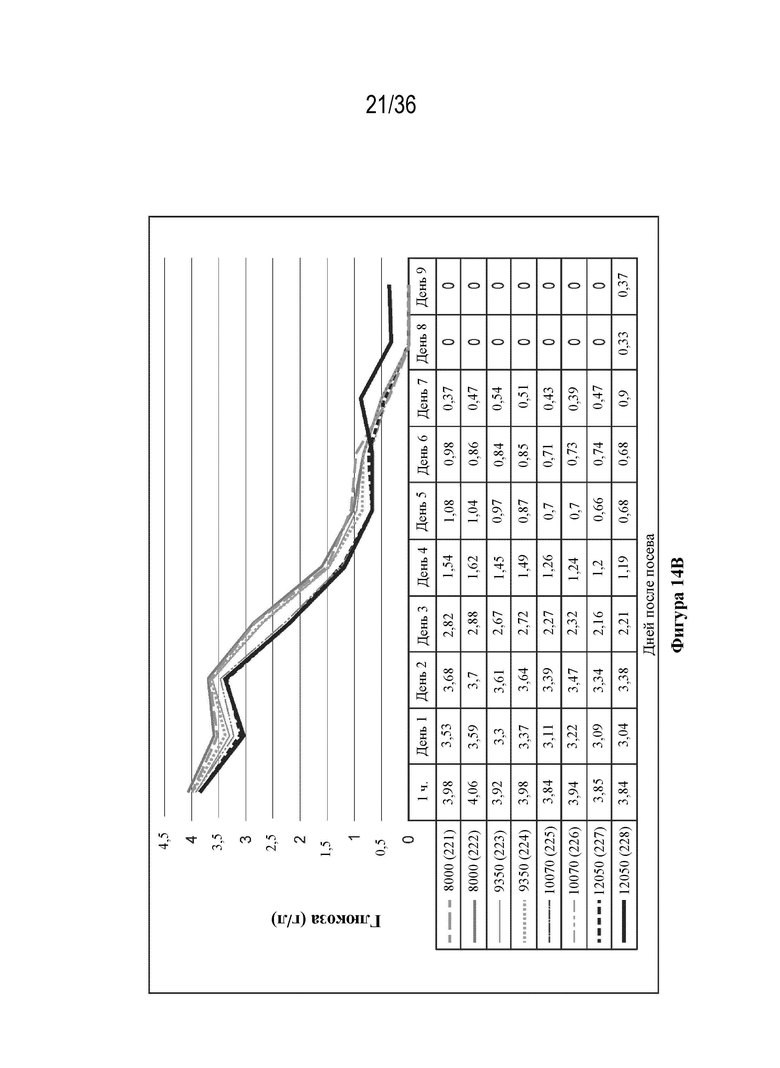
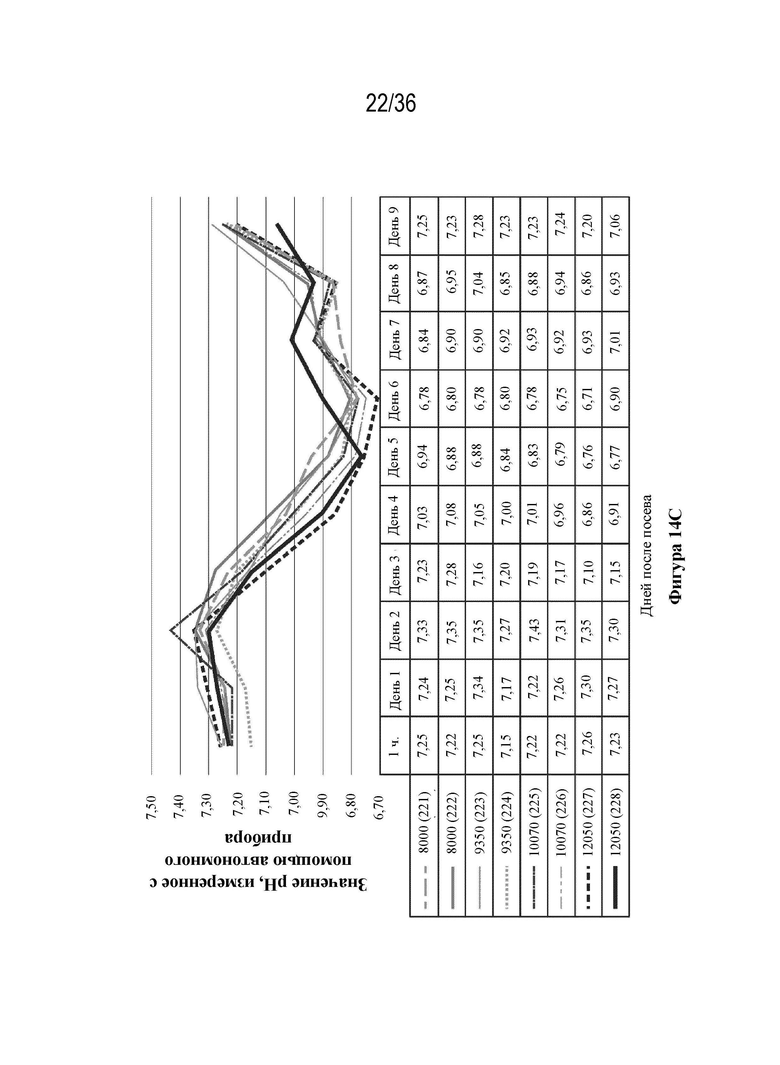
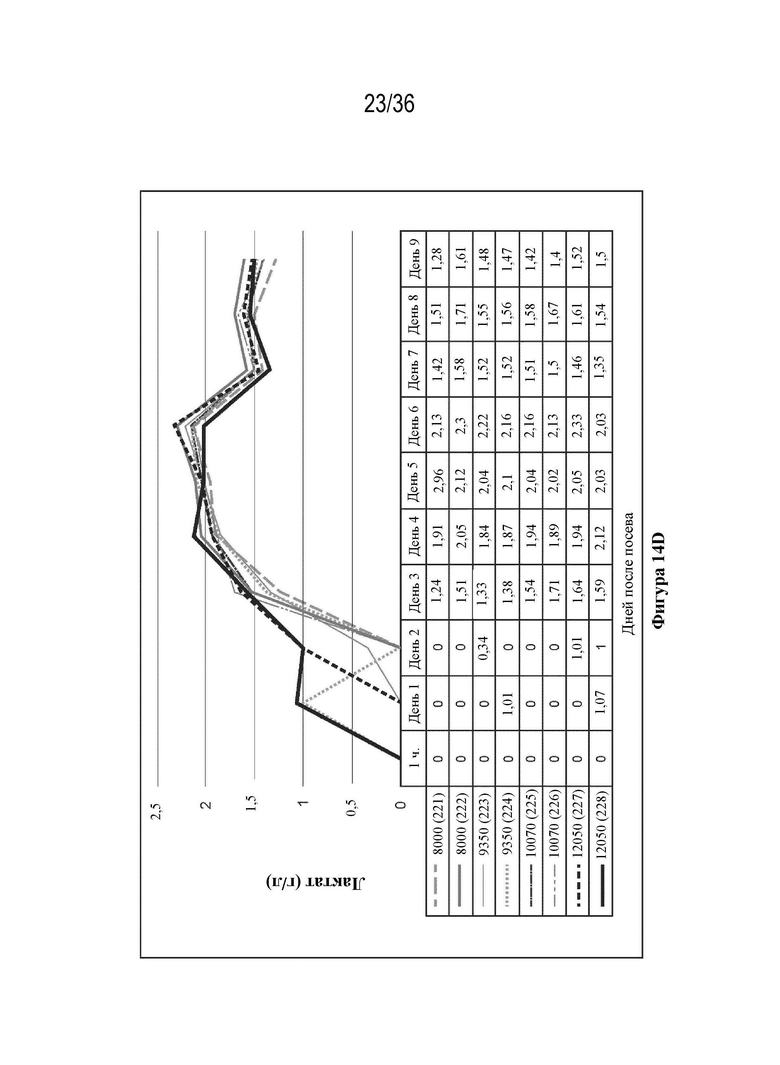

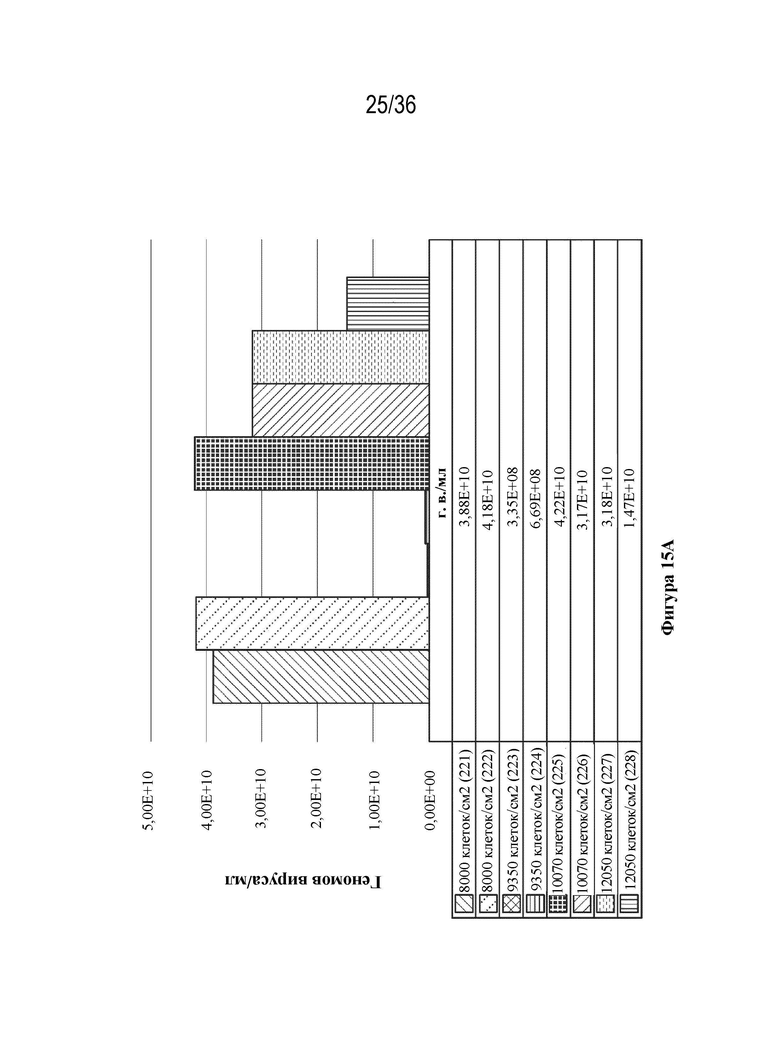


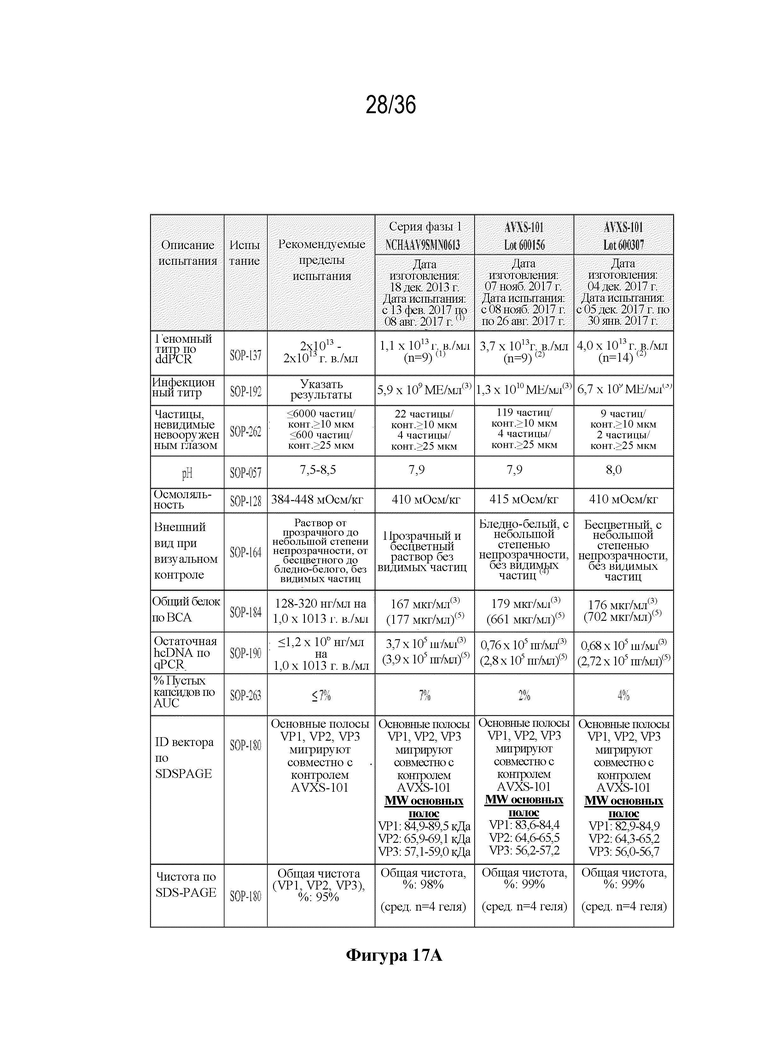
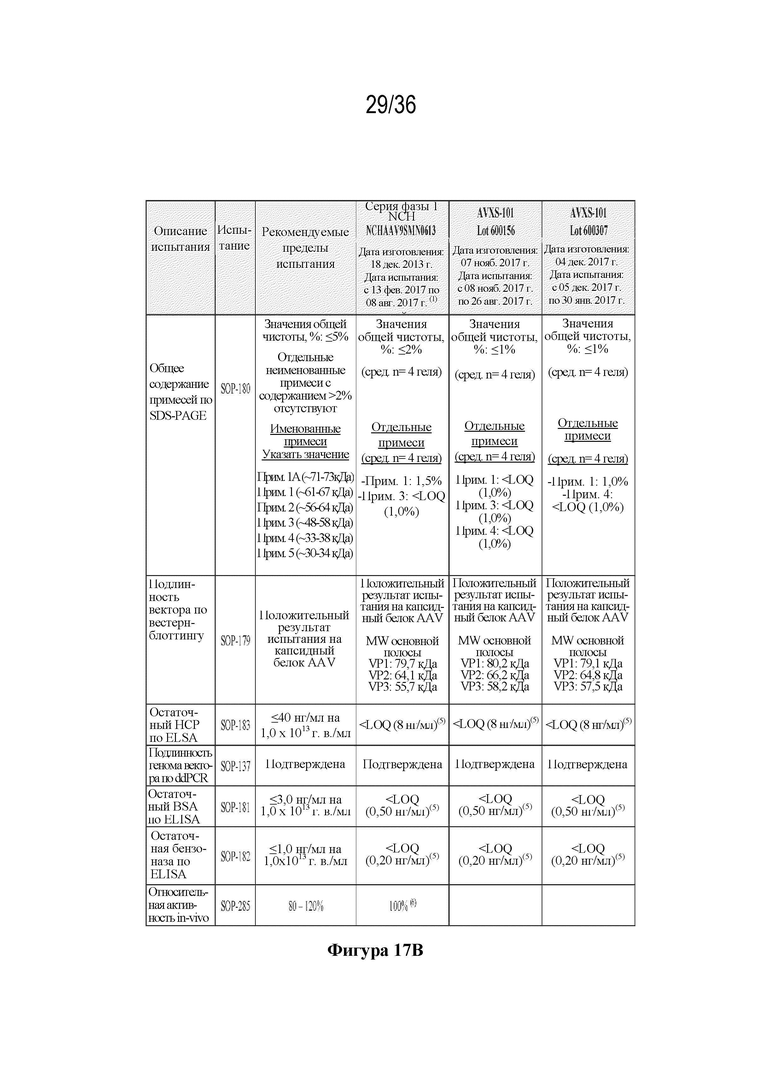

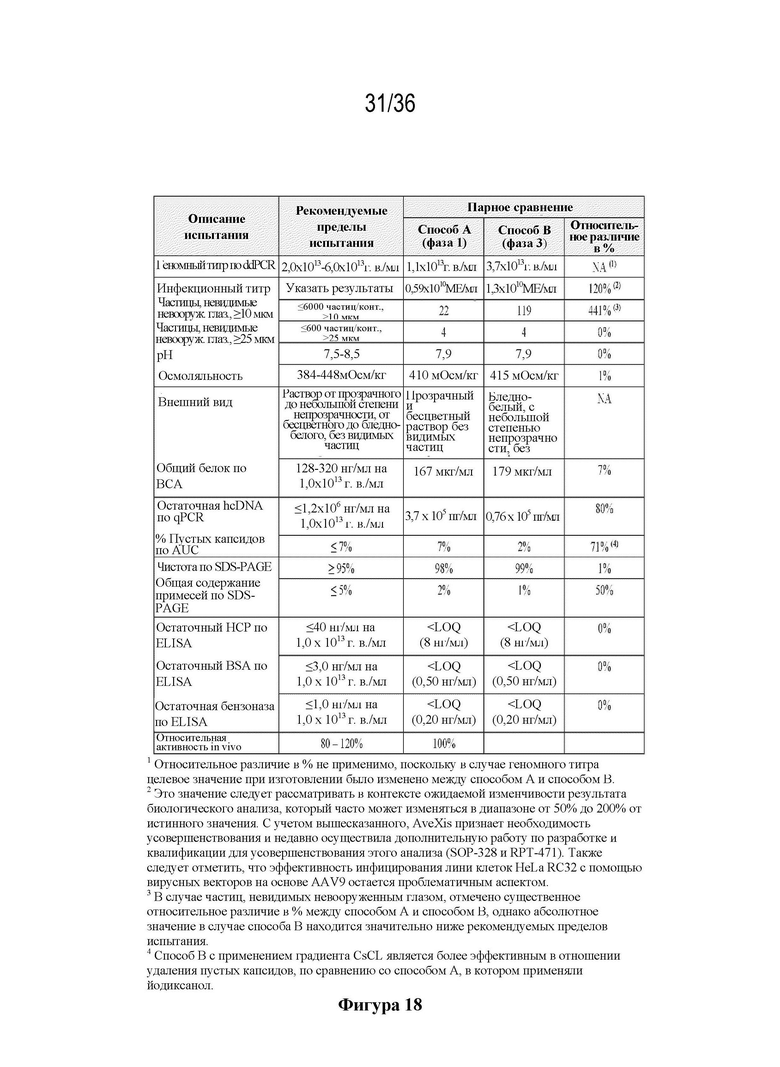
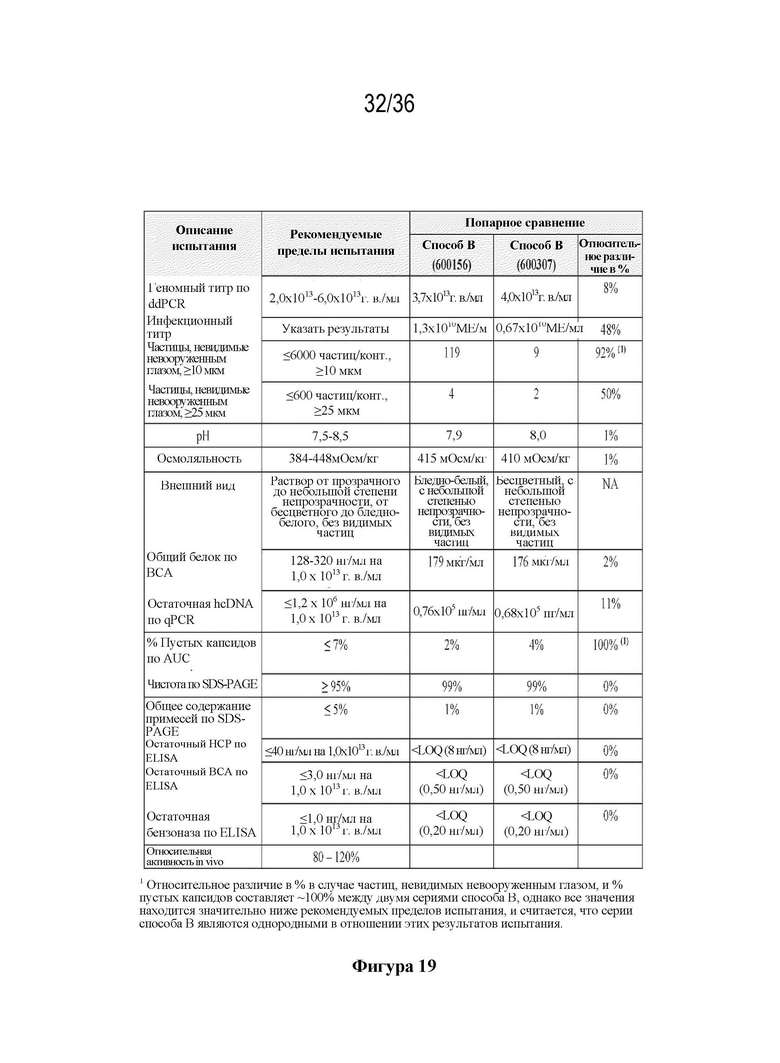
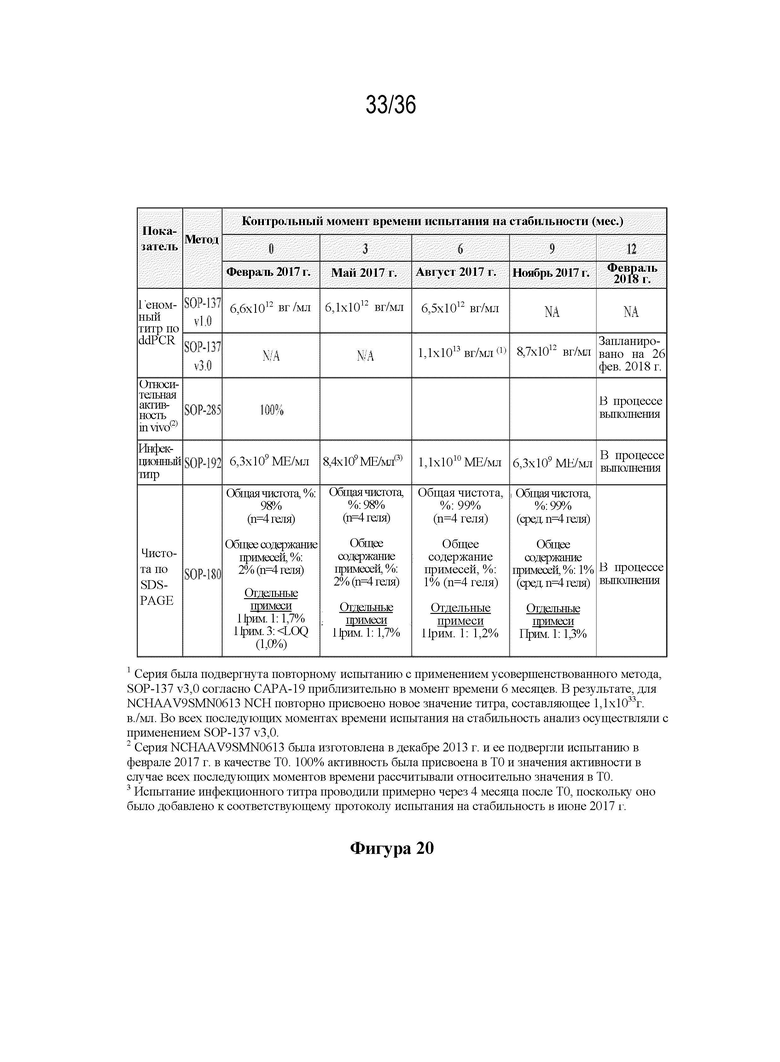
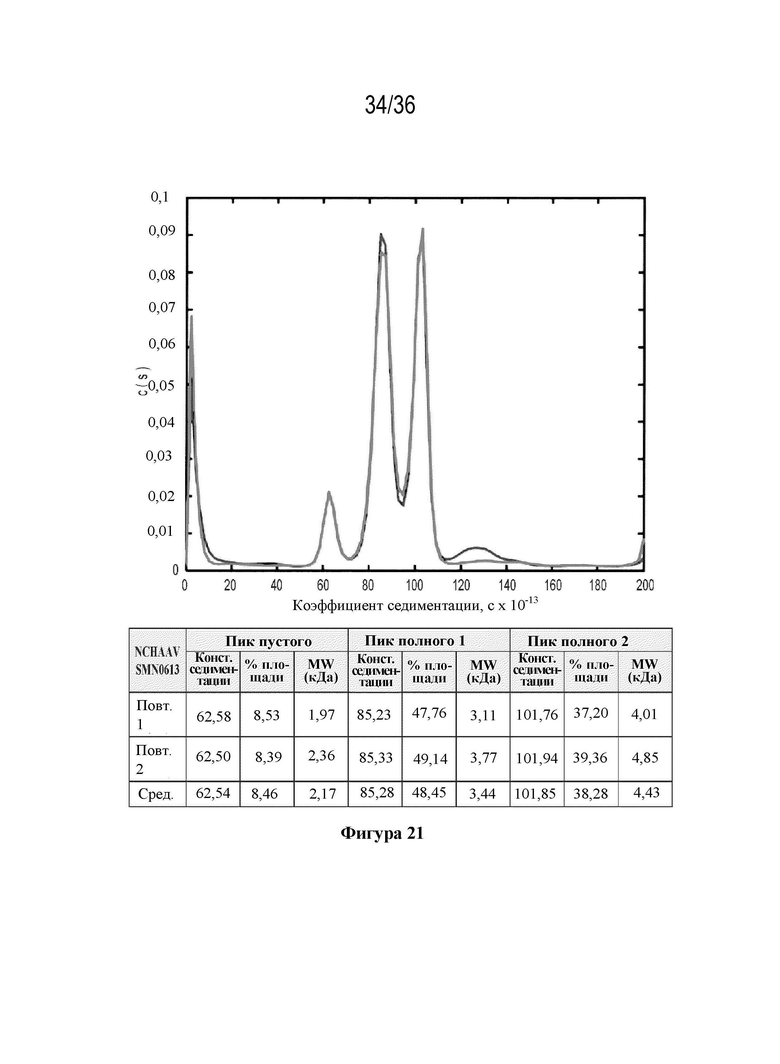
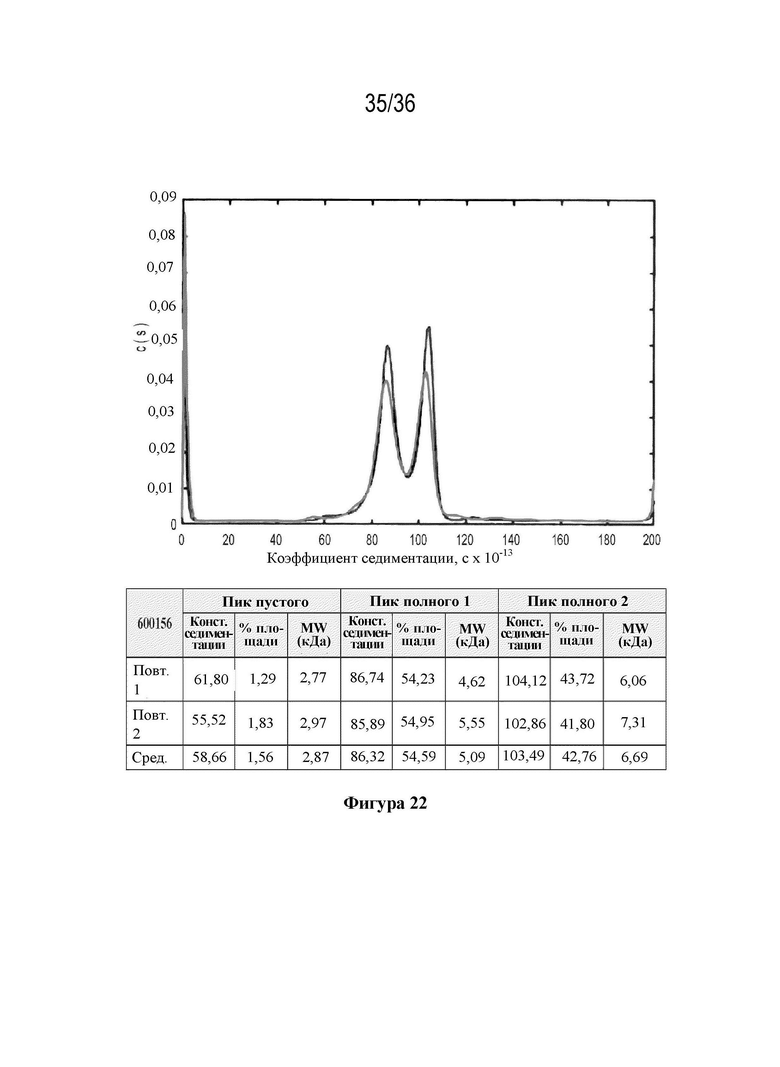
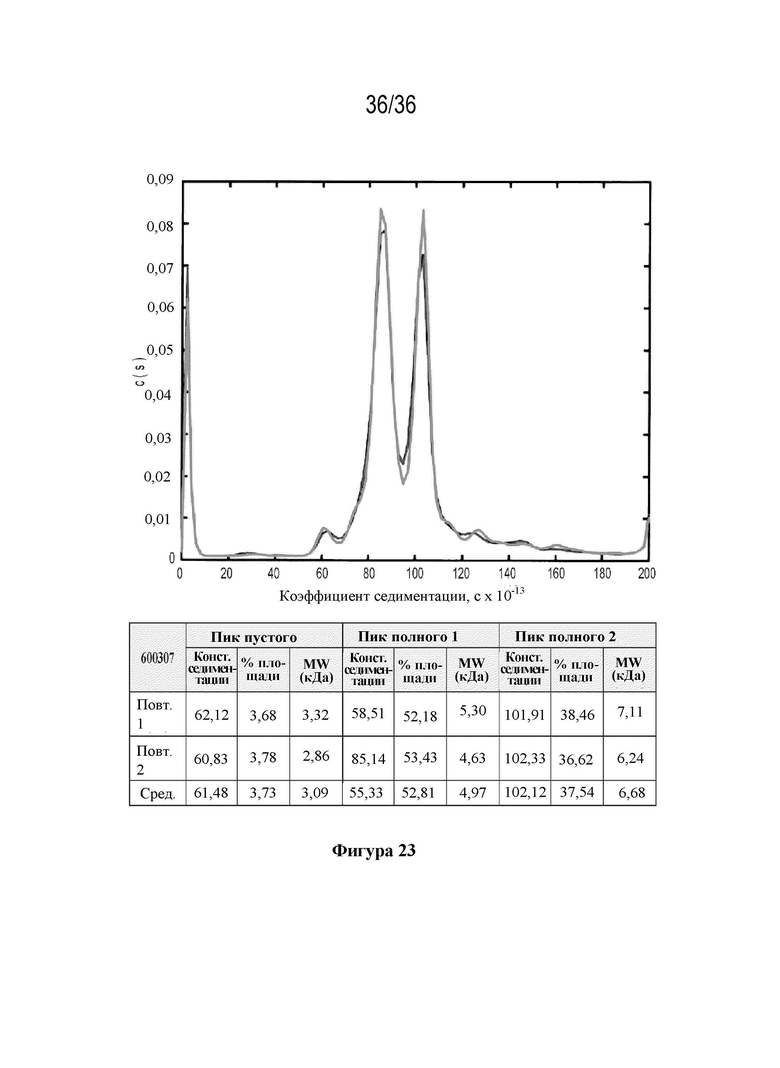
Группа изобретений относится к способам получения и очистки вирусных частиц AAV и к их применению. Способ получения вирусного вектора на основе AAV включает культивирование адгезивных клеток; их трансфекцию плазмидой(-ами) с обеспечением продуцирования вирусного вектора на основе AAV; лизис адгезивных клеток с выделением вирусного вектора на основе AAV; подкисление и осветление полученного клеточного лизата; очистку полученного продукта с применением катионообменной хроматографии (CEX); фильтрование с применением фильтрации в тангенциальном потоке (TFF); ультрацентрифугирование в буфере с хлоридом цезия CsCl и сбор вирусных векторов на основе AAV. Способ лечения спинальной мышечной атрофии SMA типа I включает введение композиции, содержащей вектор на основе AAV, пациенту, где вектор на основе AAV содержит полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN). Группа изобретений обеспечивает получение фармацевтического продукта на основе AAV, с низким содержанием пустых капсидов, низким содержанием белка клетки-хозяина и/или с низким содержанием загрязняющей ДНК, при сохранении высокой активности. 3 н. и 37 з.п. ф-лы, 36 ил., 36 табл., 22 пр.
1. Способ получения вирусного вектора на основе AAV, включающий:
a. культивирование адгезивных клеток;
b. трансфекцию адгезивных клеток плазмидой(-ами) с обеспечением продуцирования вирусного вектора на основе AAV;
c. лизис адгезивных клеток с выделением вирусного вектора на основе AAV;
d. подкисление и осветление клеточного лизата из (c);
e. очистку продукта из (d) с применением катионообменной хроматографии (CEX);
f. фильтрование продукта из (e) с применением фильтрации в тангенциальном потоке (TFF);
g. ультрацентрифугирование продукта из (f) в буфере с хлоридом цезия (CsCl); и
h. сбор вирусных векторов на основе AAV из продукта из (g).
2. Способ по п. 1, где AAV представляет собой AAV9.
3. Способ по п. 1 или 2, где AAV является самокомплементарным (scAAV).
4. Способ по любому из пп. 1-3, где адгезивные клетки представляют собой клетки HEK293.
5. Способ по п. 4, где стадия (а) включает посев клеток в крупномасштабный биореактор, который может обеспечивать непрерывную циркуляцию среды для культивирования клеток.
6. Способ по п. 5, где плотность посева составляет 8000-12000 клеток/см2.
7. Способ по любому из пп. 1-6, где стадия трансфекции (b) включает приведение в контакт адгезивных клеток с хелперной плазмидой (pHELP) для аденовируса, и с плазмидой, кодирующей гены rep AAV и cap AAV.
8. Способ по п. 7, где ген rep AAV представляет собой rep2, и ген cap AAV представляет собой cap9.
9. Способ по любому из пп. 1-8, где стадия трансфекции (b) включает приведение в контакт адгезивных клеток с плазмидой, содержащей полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), полинуклеотид, кодирующий MeCP2, или полинуклеотид, кодирующий SOD1 shRNA.
10. Способ по п. 9, где плазмида, содержащая полинуклеотид, кодирующий белок SMN, содержит модифицированный ITR AAV2, промотор бета-актина курицы (CB), немедленно-ранний энхансер цитомегаловируса (CMV), модифицированный интрон поздней 16s SV40, сигнал полиаденилирования гормона роста крупного рогатого скота (BGH) и немодифицированный ITR AAV2.
11. Способ по п. 9 или 10, где полинуклеотид кодирует белок SMN под SEQ ID NO: 2.
12. Способ по любому из пп. 1-11, где вирусный вектор на основе AAV9 содержит SEQ ID NO: 1.
13. Способ по любому из пп. 1-12, где стадия трансфекции включает приведение в контакт адгезивной клетки со средством для трансфекции, представляющим собой полиэтиленимин (PEI).
14. Способ по любому из пп. 1-13, где стадия трансфекции включает приведение в контакт адгезивной клетки со средой для трансфекции, которая не содержит сыворотку, кальций или глутамин.
15. Способ по любому из пп. 1-14, где стадия лизиса включает применение буфера для лизиса с добавлением бензоназы и TWEEN.
16. Способ по любому из пп. 1-15, дополнительно включающий замораживание клеточного лизата из стадии (c) до стадии подкисления (d).
17. Способ получения вирусного вектора на основе AAV из лизата культуры клеток, включающий стадии:
a. подкисления и осветления клеточного лизата;
b. очистки продукта из (a) с использованием катионообменной хроматографии (CEX);
c. фильтрования продукта из (b) посредством фильтрации в тангенциальном потоке (TFF);
d. ультрацентрифугирования продукта из (c) с применением 2 - 4 М буфера с хлоридом цезия (CsCl);
e. сбора вирусных векторов на основе AAV из продукта из (d);
f. фильтрования продукта из (e) посредством второй фильтрации в тангенциальном потоке.
18. Способ по любому из пп. 1-17, где стадия подкисления включает подкисление клеточного лизата до значения pH, составляющего 3,0 - 4,0.
19. Способ по любому из пп. 1-18, где ультрацентрифугирование проводят при 40000 - 50000 об/мин.
20. Способ по любому из пп. 1-19, где буфер с CsCl содержит 3 M CsCl, Tris, MgCl2 и полоксамер 188, и pH составляет рН от 7,5 до 8,5.
21. Способ по любому из пп. 1-20, где клеточный лизат инкубируют с Tween до стадии подкисления.
22. Способ по любому из пп. 1-21, где стадия осветления включает фильтрование клеточного лизата посредством глубинного фильтра.
23. Способ по любому из пп. 1-22, где CEX включает смолу с сульфонильными группами.
24. Способ по любому из пп. 1-23, где по меньшей мере одна стадия фильтрации в тангенциальном потоке (TFF) включает применение целлюлозных мембран с порогом отсечения по молекулярной массе, составляющим MW 300 кДа.
25. Способ по любому из пп. 1-24, где количество пустых вирусных капсидов составляет менее 7%, менее 5%, менее 3% или менее 1% от общего количества вирусных капсидов после сбора вирусных векторов на основе AAV из клеточного лизата, подвергнутого ультрацентрифугированию.
26. Способ по любому из пп. 17-25, где вирусные векторы на основе AAV, собранные после стадии второй TFF, хранят в растворе, содержащем Tris, MgCl2, NaCl и полоксамер 188, и pH составляет рН от 7,5 до 8,5.
27. Способ по любому из пп. 17-26, где вирусные векторы на основе AAV, собранные после второй TFF, содержат менее 30 мкг/г или менее 20 мкг/г CsCl.
28. Способ по любому из пп. 17-27, где концентрация вирусных векторов на основе AAV, собранных после второй TFF, составляет 3×1013 г. в./мл или больше.
29. Способ по любому из пп. 1-28, где получаемый выход AAV составляет более 5×1015 г. в., или более 8×1015 г. в., или более 1×1016 г. в. на одну производственную партию.
30. Способ по любому из пп. 1-29, где вирусный вектор на основе AAV содержит полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), полинуклеотид, кодирующий белок MeCP2, или полинуклеотид, кодирующий SOD1 shRNA.
31. Способ лечения спинальной мышечной атрофии (SMA) типа I у пациента, нуждающегося в этом, включающий введение композиции, содержащей вектор на основе AAV, полученный в соответствии со способом по любому из пп. 1-30 , пациенту, где вектор на основе AAV содержит полинуклеотид, кодирующий белок выживаемости мотонейронов (SMN), и где пациент:
a. характеризуется возрастом, составляющим девять месяцев или меньше;
b. имеет массу тела по меньшей мере приблизительно 2,6 кг;
c. имеет биаллельные нулевые мутации или делеции в SMN1 и
d. имеет по меньшей мере одну функциональную копию SMN2.
32. Способ по п. 31, где вирусный вектор вводят в дозе, составляющей 1-2,5×1014 г. в./кг.
33. Способ по п. 31 или 32, где масса тела пациента составляет не более 8,5 кг.
34. Способ по любому из пп. 31-33, где у пациента нет замены c.859G> C в экзоне 7 по меньшей мере одной копии гена SMN2.
35. Способ по любому из пп. 31-34, где средство для лечения вводят пациенту до возраста 6 месяцев или до появления одного или более симптомов SMA, выбранных из гипотонии, задержки развития двигательных навыков, слабого удержания головы, сутулой осанки и гипермобильности суставов.
36. Способ по любому из пп. 31-35, где вирусный вектор вводят в 5-20 мл/кг, 10-20 мл/кг или 5,5-6,5 мл/кг солевого раствора, забуференного с помощью Tris.
37. Способ по любому из пп. 31-36, где эффективность определяют с использованием шкалы CHOP-INTEND.
38. Способ по пп. 31-37, включающий введение объема дозы, выбранного из группы, состоящей из дозы 16,5 мл для пациента с массой тела 2,6-3,0 кг, дозы 19,3 мл для пациента с массой тела 3,1-3,5 кг, дозы 22,0 мл для пациента с массой тела 3,6-4,0 кг, дозы 24,8 мл для пациента с массой тела 4,1-4,5 кг, дозы 27,5 мл для пациента с массой тела 4,6-5,0 кг, дозы 30,3 мл для пациента с массой тела 5,1-5,5 кг, дозы 33,0 мл для пациента с массой тела 5,6-6,0 кг, дозы 35,8 мл для пациента с массой тела 6,1-6,5 кг, дозы 38,5 мл для пациента с массой тела 6,6-7,0 кг, дозы 41,3 мл для пациента с массой тела 7,1-7,5 кг, дозы 44,0 мл для пациента с массой тела 7,6-8,0 кг и дозы 46,8 мл для пациента с массой тела 8,1-8,5 кг.
39. Способ по любому из пп. 31-38, где композицию вводят внутривенной или интратекальной инфузией пациенту, нуждающемуся в этом.
40. Способ по п. 39, где инфузию вирусного вектора осуществляют в течение 45-75 минут.
| WO 2011094198 A1 04.08.2011 | |||
| J FRASER WRIGHT et al., "Manufacturing and Regulatory Strategies for Clinical AAV2-hRPE65", CURRENT GENE THERAPY,Vol | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кардочесальная машина | 1923 |
|
SU341A1 |
| RASHNONEJAD AFROOZ et al., "Large-Scale Production of Adeno-Associated Viral Vector Serotype-9 Carrying the Human Survival Motor Neuron Gene", 25.11.2015, Vol | |||
| Способ окисления боковых цепей ароматических углеводородов и их производных в кислоты и альдегиды | 1921 |
|
SU58A1 |

