Область техники
Настоящее изобретение относится к способу получения промежуточных соединений класса гербицидов, в частности к способу получения интермедиата урацилового соединения, содержащего изоксазолин.
Уровень техники
В патенте WO2016095768 сообщается, что соединение общей формулы I может эффективно контролировать ежовник обыкновенный, щетинник зеленый, осоку, водную осоку, Digitaria sanguinalis (L.) Scop., hispid arthraxon, piemarker, циннию, щирицу запрокинутую, портулак, дурнишник обыкновенный, Solanum nigrum L., Cassia tora Linn., Hibiscus trionum L., Glycine soja и другие сорняки, может достигать хорошего эффекта борьбы с сорняками в низких дозах и может использоваться в качестве гербицида в сельском хозяйстве.
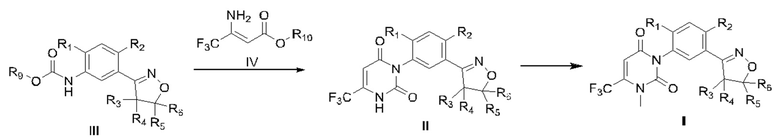
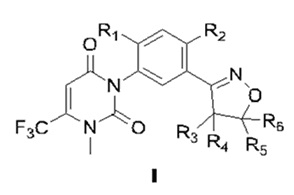
Соединение общей формулы II представляет собой интермедиат соединения общей формулы I.
Хотя соединение общей формулы I обладает превосходной гербицидной активностью, часть соединения общей формулы I является маслянистой при комнатной температуре, и ее трудно очистить перекристаллизацией, дистилляцией и другими промышленными способами, что приводит к большему количеству примесей, низкому содержанию и плохому внешнему виду продукта и влияет на использование продукта. Поэтому, необходим способ получения соединений с высоким содержанием общей формулы II. Соединение общей формулы I синтезируют из соединений с высоким содержанием общей формулы II, и соединение общей формулы I может удовлетворять требованиям без дополнительной очистки. Хотя соединение общей формулы II может удовлетворить требования по содержанию путем многократной перекристаллизации, это, несомненно, значительно снизит выход продукта и повысит стоимость и количество отходов. Поэтому необходим способ получения интермедиата урацилового соединения, содержащего изоксазолин, с общей формулой II с высоким выходом и высоким содержанием.
В патенте WO2016095768 описан способ синтеза соединения общей формулы II, но используемые циклоидальные реагенты, такие как дихлорметандиметиламмонийхлорид, коммерчески недоступны и дороги, поэтому промышленное производство невозможно.
В ссылочном патентном документе DE19543676 раскрыт способ получения аналогов соединения общей формулы II. В способе используется полярный апротонный растворитель с высокой температурой кипения, N,N-диметилформамид или N-метилпирролидон, в качестве растворителя и карбонат калия в качестве щелочи для реакции и дистилляция для удаления образовавшегося этанола. После реакции растворитель удаляют путем декомпрессии, смесь подкисляют и перекристаллизовывают с получением целевого продукта. Первый недостаток способа заключается в том, что следовые количества воды в реакционной системе не могут быть эффективно удалены из системы, а следовые количества воды могут вызвать серьезный гидролиз исходного вещества соединения общей формулы III, что приведет к снижению коэффициента использования исходного вещества и потере выхода. Второй недостаток заключается в том, что N,N-диметилформамид или N-метилпирролидон в качестве растворителя вызывают низкую селективность реакции и большее количество примесей, и продукт необходимо перекристаллизовывать несколько раз, что приводит к значительному увеличению в три раза отходов.
Ссылочный патент JP2002193914A улучшает вышеуказанный способ. Смешанная система растворителей N,N-диметилформамида и толуола с соответствующей массовой долей 50% используется для кипячения с обратным холодильником и удаления воды, а карбонат калия используется в качестве щелочи. После реакции растворитель удаляют путем декомпрессии, смесь подкисляют и промывают с получением целевого продукта. Хотя воду в определенной степени удаляют из системы раскрытым способом и гидролизованные продукты ингибируют, вся реакция также занимает слишком много времени из-за добавления толуола, неполярного агента для переноса воды, что приводит к разложению промежуточного соединения общей формулы III в разной степени. Таким образом, продукты имеют больше примесей и содержания смол. Последующая обработка требует многократной перекристаллизации для получения содержания выше 95%, и эффективность производства также значительно снижается.
Таким образом, в данной области надеются получить способ, который может хорошо ингибировать гидролиз субстрата, ускорить реакцию, уменьшить образование примесей и смолы и облегчить получение интермедиата урацилового соединения, содержащего изоксазолин, с высоким выходом и высоким содержанием.
Сущность изобретения
Для преодоления недостатков предшествующего уровня техники целью настоящего изобретения является создание способа получения интермедиата урацилового соединения, содержащего изоксазолин, с высоким выходом и высоким содержанием.
Для реализации вышеуказанной цели техническое решение настоящего изобретения состоит в следующем:
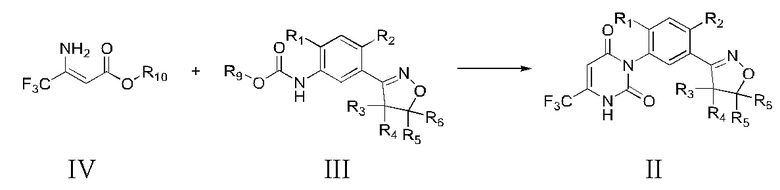
Способ получения интермедиата урацилового соединения, содержащего изоксазолин, включает: взаимодействие метилового эфира 3-амино-4,4,4-трифторкротоновой кислоты с замещенным арилкарбаматом; в процессе реакции непрерывное испарение воды и побочного продукта спирта в системе; и проведение обработки для получения интермедиата урацилового соединения, содержащего изоксазолин;
или, взаимодействие метилового эфира 3-амино-4,4,4-трифторкротоновой кислоты с замещенным арилкарбаматом в присутствии катализатора; в процессе реакции непрерывное испарение воды и побочного продукта спирта в системе; и проведение обработки для получения интермедиата урацилового соединения, содержащего изоксазолин.
Схема реакции представляет собой:
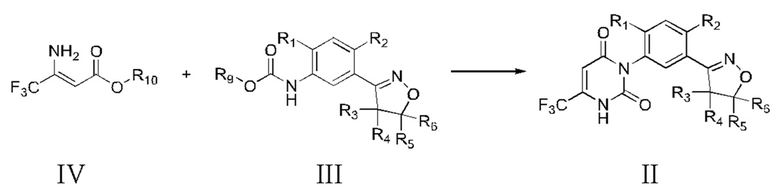
R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора;
R3 выбран из водорода или C1-C4 алкила;
R4 выбран из водорода, CO2R7 или CH2OR8;
R5 выбран из водорода, CO2R7 или CH2OR8;
R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R7 выбран из C1-C4 алкила, C1-C4 галогеналкила, аллила или пропаргила;
R8 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила или C1-C4 алкилкарбонила;
R9 выбран из C1-C4 алкила;
R10 выбран из C1-C4 алкила.
Предпочтительно, R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора;
R3 выбран из водорода или C1-C4 алкила;
R4 выбран из водорода;
R5 выбран из CO2R7;
R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R7 выбран из метила, этила, н-пропила, н-бутила, изопропила, изобутила, трет-бутила, трифторэтила, аллила и пропаргила;
R9 выбран из метила или этила;
R10 выбран из метила или этила.
Соединение формулы III и соединение формулы IV взаимодействуют в присутствии агента для переноса воды или смешанного растворителя. В процессе реакции вода и побочный продукт спирт в системе непрерывно испаряются, и после обработки получают интермедиат урацилового соединения, содержащего изоксазолина; или процесс реакции осуществляют под действием катализатора.
Щелочь добавляют в процессе реакции, где молярное отношение щелочи к соединению формулы III составляет 0,5:1-3:1.
Катализатор представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен, соль 1,8-диазабицикло[5.4.0]ундец-7-ена или раствор 1,8-диазабицикло[5.4.0]ундец-7-ена, предпочтительно 1,8-диазабицикло[5.4.0]ундец-7-ен, где используемое количество катализатора составляет 0,001%-10% от массы соединения, представленного формулой III.
Используемое количество катализатора составляет 0,1%-5% от массы соединения формулы III.
Щелочь представляет собой одну или две из карбоната калия, карбоната натрия, карбоната цезия, бикарбоната калия, бикарбоната натрия или бикарбоната цезия.
Щелочь представляет собой одну или две из карбоната калия, карбоната натрия, бикарбоната калия и бикарбоната натрия, где молярное отношение щелочи к соединению формулы III составляет 0,5:1-2:1.
Количество агента для переноса воды или смешанного растворителя в 2-20 раз превышает массу соединения формулы III;
Смешанный растворитель включает агент для переноса воды и полярный апротонный растворитель, где масса полярного апротонного растворителя в смешанном растворителе составляет 20%-70%.
Агент для переноса воды представляет собой один из н-пропилацетата, изопропилацетата, н-бутилацетата, метилизопропилкетона, метилизобутилкетона, диметилового эфира этиленгликоля, диэтилового эфира этиленгликоля, 2-метилтетрагидрофурана и ацетонитрила.
Агент для переноса воды в смешанном растворителе представляет собой один из толуола, хлорбензола, н-пропилацетата, изопропилацетата, метилизопропилкетона, метилизобутилкетона, диметилового эфира этиленгликоля, диэтилового эфира этиленгликоля, 2-метилтетрагидрофурана и ацетонитрила; и полярный апротонный растворитель в смешанном растворителе представляет собой N,N-диметилформамид, диметилсульфоксид или N-метилпирролидон.
Предпочтительно количество агента для переноса воды или смешанного растворителя в 3-8 раз превышает массу соединения формулы III;
Смешанный растворитель включает агент для переноса воды и полярный апротонный растворитель, где масса полярного апротонного растворителя в смешанном растворителе составляет 30%-60%.
Агент для переноса воды или смешанный растворитель могут быть дополнительно предпочтительно следующими: агент для переноса воды предпочтительно представляет собой один из изопропилацетата, метилизобутилкетона и ацетонитрила, и смешанный растворитель предпочтительно представляет собой смешанный растворитель из одного из агентов для переноса воды из толуола, н-пропилацетата, изопропилацетата, метилизобутилкетона, 2-метилтетрагидрофурана и ацетонитрила и одного из полярных апротонных растворителей из N, N-диметилформамида, диметилсульфоксида или N-метилпирролидона.
Инициатор общей формулы IV известен или может быть получен известными способами (см. J. Hetercycl. Chem. 9 (1972), 513-522).
Соединение общей формулы III можно получить по способу, описанному в патенте DE19543676.
В соединениях общих формул (I, II, III, IV), приведенных выше, используемые термины обычно определяются следующим образом: алкил относится к форме с прямой или разветвленной цепью, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил и другие группы. Галогеналкил: алкилы с прямой или разветвленной цепью, в которых атомы водорода могут быть частично или полностью заменены галогенами, такие как хлорметил, дихлорметил, трихлорметил, фторметил, дифторметил и трифторметил. Алкилкарбонил: Алкилы связаны со структурами через карбонилы, такие как CH3CO- или CH3CH2CO-.
Настоящее изобретение имеет преимущества:
В настоящем изобретении путем выбора агента для переноса воды или смешанного растворителя вода и спирт из системы может непрерывно удаляться, гидролиз исходных веществ и продуктов уменьшен и селективность реакции улучшена. Более того, добавление катализатора значительно сокращает время реакции. При совместном действии этих двух факторов повышается селективность реакции и коэффициент использования исходных веществ, уменьшается количество продуктов гидролиза, примесей и смол, значительно сокращается время реакции и значительно повышается производительность. Более того, после одной перекристаллизации неочищенных продуктов можно получить промежуточный продукт с чистотой более 97%, и количественный выход может составить более 85%, что соответствует требованиям синтетических продуктов и подходит для промышленного производства.
Подробное описание
Способ получения соединения формулы II, более подробно описан ниже путем перечисления вариантов осуществления, но настоящее изобретение не ограничивается этими вариантами осуществления. В настоящее изобретение могут быть внесены различные изменения и вариации для специалистов в данной области техники. Любая модификация, эквивалентная замена, улучшение и тому подобное, выполненные в соответствии с сущностью и принципом настоящего изобретения, должны быть включены в объем охраны настоящего изобретения.
В способе по настоящему изобретению улучшаются селективность реакции и коэффициент использования исходных веществ; уменьшается количество продуктов гидролиза, примесей и смол; время реакции значительно сокращается; и повышается производительность. При этом после одной перекристаллизации неочищенных продуктов можно получить промежуточный продукт с чистотой более 97%, и количественный выход может составлять более 85%, что подходит для промышленного производства.
Вариант осуществления 1 Синтез соединения II-1
39,3 г (100 ммоль) 3-(2-хлор-5-((этоксикарбонил)амино)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-карбоксилат этила, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 10,4 г (75 ммоль) карбоната калия, 80 г изопропилацетата, 80 г N,N-диметилформамида и 1,0 г 1,8-диазабицикло[5.4.0]ундец-7-ена добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и кипятили с обратным холодильником в течение 5 часов; в этот период из верхней части колонны отделяли небольшое количество низкокипящих веществ с температурой ниже 78°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; изопропилацетат добавляли для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя изопропилацетат выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 96,1%; смесь перекристаллизовывали из этанола с водой, и фильтровали при 0-5°С; фильтрпрессную лепешку промывали по каплям холодной водой с этанолом и сушили с получением 41,3 г; ВЭЖХ количественное содержание составило 98,3%; и количественный выход составил 87,5%.
Вариант осуществления 2 Синтез соединения II-1
39,3 г (100 ммоль) этил 3-(2-хлор-5-((этоксикарбонил) амино)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-карбоксилата, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 10,4 г (75 ммоль) карбоната калия, 120 г метилизобутилкетона и 1,0 г 1,8-диазабицикло[5.4.0]ундец-7-ена добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и кипятили с обратным холодильником в течение 10 часов; в этот период из верхней части колонны отделяли небольшое количество низкокипящих веществ с температурой ниже 78°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; метилизобутилкетон добавляли для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя метилизобутилкетон выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 95,0%; смесь перекристаллизовывали из этанола с водой, и фильтровали при 0-5°С; фильтрпрессную лепешку промывали по каплям холодной водой с этанолом и сушили с получением 41,7 г; ВЭЖХ количественное содержание составило 96,0%; и количественный выход составил 86,3%.
Вариант осуществления 3 Синтез соединения II-2
39,3 г (100 ммоль) (3-(2-хлор-4-фтор-5-((метоксикарбонил)амино)фенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 9,1 г (65 ммоль) карбоната калия, 80 г изопропилацетата, 80 г N-метилпирролидона и 1,0 г 1,8-диазабицикло[5.4.0]ундец-7-ена добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и кипятили с обратным холодильником в течение 5 часов; в этот период из верхней части колонны отделяли небольшое количество низкокипящих веществ с температурой ниже 78°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; изопропилацетат добавляли для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя изопропилацетат выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 95,1%; смесь перекристаллизовывали из этанола с водой, и фильтровали при 0-5°С; фильтрпрессную лепешку промывали по каплям холодной водой с этанолом и сушили с получением 42,0 г; ВЭЖХ количественное содержание составило 97,1%; и количественный выход составил 87,9%.
Вариант осуществления 4 Синтез соединения II-2
39,0 г (100 ммоль) (3-(2-хлор-5-((метоксикарбонил)амино)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 3,0 г тетрабутиламмоний бромида, 9,1 г (65 ммоль) карбоната калия, 80 г толуола и 80 г N, N-диметилформамида добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и кипятили с обратным холодильником в течение 25 часов; в этот период из верхней части колонны отделяли небольшое количество низкокипящих веществ с температурой ниже 80°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; толуол добавляли для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя толуол выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 86%; смесь перекристаллизовывали из этанола с водой три раза, и фильтровали при 0-5°С; фильтрпрессную лепешку промывали по каплям холодной водой с этанолом и сушили с получением 24,2 г; ВЭЖХ количественное содержание составило 96,1%; и количественный выход составил 50,1%.
Вариант осуществления 5 Синтез соединения II-2
3,1 кг (8 моль) (3-(2-хлор-4-фтор-5-((метоксикарбонил)амино)фенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 1,5 кг (8,1 моль) 3-амино-4,4,4-трифторкротоната, 0,75 кг (5,5 моль) карбоната калия, 70 кг изопропилацетата, 60 кг N,N-диметилформамида и 0,06 кг 1,8-диазабицикло[5.4.0]ундец-7-ена добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и кипятили с обратным холодильником в течение 5 часов; в этот период из верхней части колонны отделяли небольшое количество низкокипящих веществ с температурой ниже 78°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; изопропилацетат добавляли для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя изопропилацетат выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 94,5%; смесь перекристаллизовывали из этанола с водой, и фильтровали при 0-5°С; фильтрпрессную лепешку промывали по каплям холодной водой с этанолом и сушили с получением 3,3 кг; ВЭЖХ количественное содержание составило 97,0%; и количественный выход составил 86,3%.
Вариант осуществления 6 Синтез соединения I-3
33,4 г (0,07 моль, количественное содержание 97,2%) вышеуказанного соединения II-2 (3-(2-хлор-5-(2,6-диокси-4-трифторметил-3,6-дигидропиримидин-1(2H)-ил)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 7,10 г (0,084 моль) бикарбоната натрия, 150 г дихлорметана и 3,0 г 1, 4-диазабицикло[2.2.2]октан добавляли в автоклав; 5,0 г метилхлорида отмеряли и вводили; смесь перемешивали и нагревали до 70-80°С; манометр показывал 0,4 МПа; и реакция длилась 11 часов. Температуру снижали до 20°С, давление в автоклаве сбрасывали, избыток метилхлорида удаляли. В автоклав добавляли 50 г воды и перемешивали в течение 10 минут; после этого смесь разделялась на слои и слой воды удаляли; органический слой однократно промывали 50 г воды; органический слой фильтровали для удаления небольшого количества нерастворившихся веществ; десольвент подвергали декомпрессии с получением 33,9 г маслянистого вещества, с количественным содержанием 93,5% и выходом 94,5%.
Конкретные структуры соединений, полученных в вариантах осуществления 1, 2, 3 и 4:
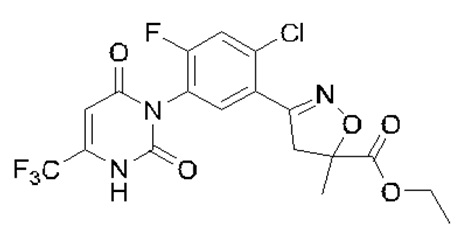
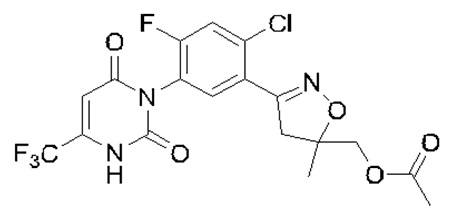
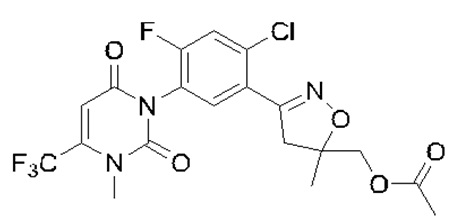
Ссылочный вариант осуществления 1 Синтез соединения II-2
39,0 г (100 ммоль) (3-(2-хлор-5-(метоксикарбонил)амино)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 9,1 г (65 ммоль) карбоната калия и 100 г N,N-диметилформамида добавляли в реакционную колбу с ректификационным устройством, перемешивали, нагревали и подвергали взаимодействию в течение 4 часов при 130°С; ВЭЖХ использовали для отслеживания окончания реакции; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; метилизобутилкетон использовали для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя метилизобутилкетон выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 42,2%;
Ссылочный вариант осуществления 2 Синтез соединения II-2
39,0 г (100 ммоль) (3-(2-хлор-5-(метоксикарбонил)амино)-4-фторфенил)-5-метил-4,5-дигидроизоксазол-5-ил)метилацетата, 19,2 г (103 ммоль) 3-амино-4,4,4-трифторкротоната, 9,1 г (65 ммоль) карбоната калия и 100 г N,N-диметилформамида добавляли в реакционную колбу с ректификационным устройством, перемешивали и нагревали; микроотрицательное давление прикладывали к реакционной системе через верхнюю часть колонны; воду и побочный продукт этанол удаляли из реакционной системы, и реакцию проводили при 105°С в течение 18 часов; большинство растворителей выпаривали при пониженном давлении; остатки подкисляли хлористоводородной кислотой; значение рН доводили до 2-4; метилизобутилкетон использовали для экстракции; после перемешивания в течение 20 минут нижний водный слой удаляли; органический слой промывали водой один раз; после удаления водного слоя метилизобутилкетон выпаривали при пониженном давлении; остатки нормализовали с помощью ВЭЖХ и содержание составило 64,3%;
Из приведенных выше вариантов осуществления и ссылочных вариантов осуществления видно, что способ получения интермедиата урацилового соединения, содержащего изоксазолин, является доступным по исходным веществам и мягким по условиям; с помощью агента для переноса воды или смешанного растворителя вода и спирт в системе могут непрерывно удаляться, гидролиз исходных веществ и продуктов уменьшен, а селективность реакции улучшена. Добавление катализатора значительно сокращает время реакции. При совместном действии этих двух факторов повышается коэффициент использования исходных веществ; уменьшается количество продуктов гидролиза, примесей и смол; время реакции значительно сокращается; и производительность значительно повышается, что подходит для промышленного производства.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛИЗОКСАЗОЛИНОВОГО СОЕДИНЕНИЯ | 2021 |
|
RU2818767C1 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
| ПОЛУЧЕНИЕ ЗАМЕЩЕННОГО АКРИЛАТНОГО СОЕДИНЕНИЯ | 2021 |
|
RU2830161C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ФАКТОРА СВЕРТЫВАНИЯ КРОВИ XIA И ЕГО ИНТЕРМЕДИАТА | 2019 |
|
RU2779013C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН- ИЛИ НАФТИРИДИНКАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2120940C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛ-2-ТЕТРАЗОЛ-2-ИЛКЕТОНА C УЛУЧШЕННОЙ СЕЛЕКТИВНОСТЬЮ | 2020 |
|
RU2808087C1 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛО-ГЕТЕРОАРИЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2748833C2 |
| ФТОРАЛКИЛИРУЮЩИЙ АГЕНТ | 2015 |
|
RU2716008C2 |
Изобретение относится к способу получения соединения формулы (II), являющегося интермедиатом в синтезе гербицидов. Предлагаемый способ включает взаимодействие эфира 3-амино-4,4,4-трифторкротоновой кислоты формулы (IV) с арилкарбаматом формулы (III) в присутствии агента для переноса воды и катализатора. В процессе реакции происходит непрерывное испарение воды и побочного продукта спирта в системе. Затем проводят обработку для получения соединения формулы (II). В указанных формулах R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора; R3 выбран из водорода или C1-C4 алкила; R4 выбран из водорода, CO2R7 или CH2OR8; R5 выбран из водорода, CO2R7 или CH2OR8; R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила; R7 выбран из C1-C4 алкила, C1-C4 галогеналкила, аллила или пропаргила; R8 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила или C1-C4 алкилкарбонила; R9 выбран из C1-C4 алкила; R10 выбран из C1-C4 алкила. Предлагаемый способ позволяет получать соединение формулы (II) с высоким выходом и высоким содержанием. Изобретение относится также к способу получения соединения формулы I из соединения формулы II. 2 н. и 9 з.п. ф-лы, 1 табл., 6 пр.
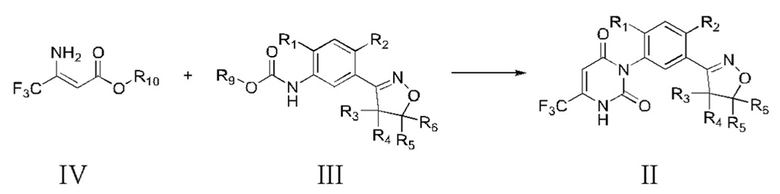
1. Способ получения соединения формулы (II), отличающийся тем, что включает:
взаимодействие эфира 3-амино-4,4,4-трифторкротоновой кислоты формулы (IV) с арилкарбаматом формулы (III) в присутствии агента для переноса воды и катализатора; в процессе реакции непрерывное испарение воды и побочного продукта спирта в системе; и проведение обработки для получения соединения формулы (II):
 ;
;
R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора;
R3 выбран из водорода или C1-C4 алкила;
R4 выбран из водорода, CO2R7 или CH2OR8;
R5 выбран из водорода, CO2R7 или CH2OR8;
R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R7 выбран из C1-C4 алкила, C1-C4 галогеналкила, аллила или пропаргила;
R8 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила или C1-C4 алкилкарбонила;
R9 выбран из C1-C4 алкила;
R10 выбран из C1-C4 алкила.
2. Способ по п. 1, отличающийся тем, что
R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора;
R3 выбран из водорода или C1-C4 алкила;
R4 выбран из водорода;
R5 выбран из CO2R7;
R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R7 выбран из метила, этила, н-пропила, н-бутила, изопропила, изобутила, трет-бутила, трифторэтила, аллила и пропаргила;
R9 выбран из метила или этила;
R10 выбран из метила или этила.
3. Способ по п. 1 или 2, отличающийся тем, что соединение формулы II получают после обработки.
4. Способ по п. 3, отличающийся тем, что в процессе реакции добавляют основание, где молярное отношение основания к соединению формулы III составляет 0,5:1-3:1.
5. Способ по п. 3, отличающийся тем, что катализатор представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен, соль 1,8-диазабицикло[5.4.0]ундец-7-ена или раствор 1,8-диазабицикло[5.4.0]ундец-7-ена, где используемое количество катализатора составляет 0,001-10% от массы соединения, представленного формулой III.
6. Способ по п. 5, отличающийся тем, что используемое количество катализатора составляет 0,1-5% от массы соединения, представленного формулой III.
7. Способ по п. 4, отличающийся тем, что основание представляет собой одно или два из карбоната калия, карбоната натрия, карбоната цезия, бикарбоната калия, бикарбоната натрия или бикарбоната цезия.
8. Способ по п. 7, отличающийся тем, что основание представляет собой одно или два из карбоната калия, карбоната натрия, бикарбоната калия и бикарбоната натрия, где молярное отношение основания к соединению, представленному формулой III, составляет 0,5:1-2:1.
9. Способ по п. 3, отличающийся тем, что количество агента для переноса воды или смешанного растворителя в 2-20 раз превышает массу соединения, представленного формулой III;
смешанный растворитель включает агент для переноса воды и полярный апротонный растворитель, где масса полярного апротонного растворителя в смешанном растворителе составляет 20-70%.
10. Способ по п. 9, отличающийся тем, что агент для переноса воды представляет собой один из н-пропилацетата, изопропилацетата, н-бутилацетата, метилизопропилкетона, метилизобутилкетона, диметилового эфира этиленгликоля, диэтилового эфира этиленгликоля, 2-метилтетрагидрофурана и ацетонитрила;
агент для переноса воды в смешанном растворителе представляет собой один из толуола, хлорбензола, н-пропилацетата, изопропилацетата, метилизопропилкетона, метилизобутилкетона, диметилового эфира этиленгликоля, диэтилового эфира этиленгликоля, 2-метилтетрагидрофурана и ацетонитрила; и полярный апротонный растворитель в смешанном растворителе представляет собой N,N-диметилформамид, диметилсульфоксид или N-метилпирролидон.
11. Способ получения соединения формулы I, включающий:
- получение соединения формулы II с использованием способа по любому из пп. 1-10, и
- метилирование соединения формулы I из соединения формулы II, полученного на предыдущей стадии
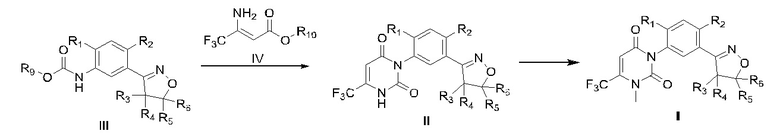 ,
,
где
R1 и R2 могут быть одинаковыми или различными и соответственно выбраны из водорода, фтора или хлора;
R3 выбран из водорода или C1-C4 алкила;
R4 выбран из водорода, CO2R7 или CH2OR8;
R5 выбран из водорода, CO2R7 или CH2OR8;
R6 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R7 выбран из C1-C4 алкила, C1-C4 галогеналкила, аллила или пропаргила;
R8 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила или C1-C4 алкилкарбонила;
R9 выбран из C1-C4 алкила;
R10 выбран из C1-C4 алкила.
| CN 105753853 A, 13.07.2016 | |||
| 0 |
|
SU190058A1 | |
| Устройство для предохранения от продольного пореза ленты конвейера | 1985 |
|
SU1283190A1 |
| CN 110818644, 21.02.2020 | |||
| RU 2003114858 A, 10.11.2004 | |||
| ПРОИЗВОДНЫЕ УРАЦИЛА, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ БОРЬБЫ С СОРНЯКАМИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ (ВАРИАНТЫ) | 2001 |
|
RU2264395C2 |

