Область техники
Настоящее изобретение относится к области органического синтеза, в частности, к способу получения фенилизоксазолинового соединения.
Уровень техники
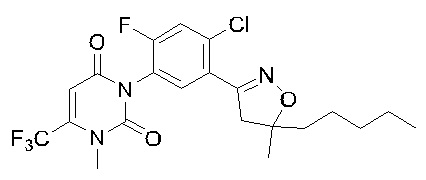
В патенте WO 2016095768 описано фенилизоксазолиновое соединении, представленное общей формулой I:
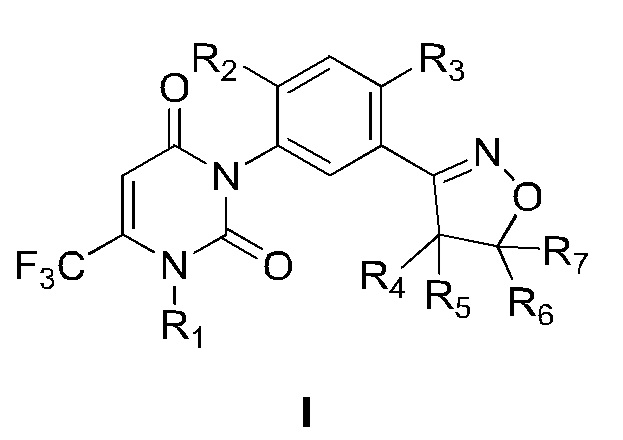
Соединение общей формулы I обладает хорошей гербицидной активностью, может эффективно контролировать Echinochloa crusgalli, Setaria viridis, Cyperus difformis, Juncellus serotinus, Digitaria sanguinalis (L.) Scop., Arthraxon hispidus, Abutilon theophrasti, Zinnia elegans, Amaranthus retroflexus, Portulaca oleracea, Xanthium sibiricum, Solanum nigrum L., Cassia tora Linn., Hibiscus trionum L., Glycine soja и другие сорняки, может достигать хорошего эффекта борьбы с сорняками в низких дозах и может использоваться в качестве гербицида в сельском хозяйстве. Патенты WO 2016095768 и CN 108570041 также включают получение таких соединений, но соединения синтезируют, сначала синтезируя изоксазолиновое кольцо, а затем синтезируя урациловое кольцо. Недостатками вышеуказанных способов синтеза являются: стабильность изоксазолинового кольца, синтезированного первым, не очень хорошая; и требования к температуре и щелочи для циклизации урацила относительно жесткие, что легко приводит к образованию побочных продуктов, что приводит к длительному времени реакции и низкому выходу.
Сущность изобретения
Целью настоящего изобретения является обеспечение способа получения фенилизоксазолинового соединения с дешевым и доступным сырьем и простым процессом синтеза.
Для реализации вышеуказанной цели техническое решение настоящего изобретения состоит в следующем:
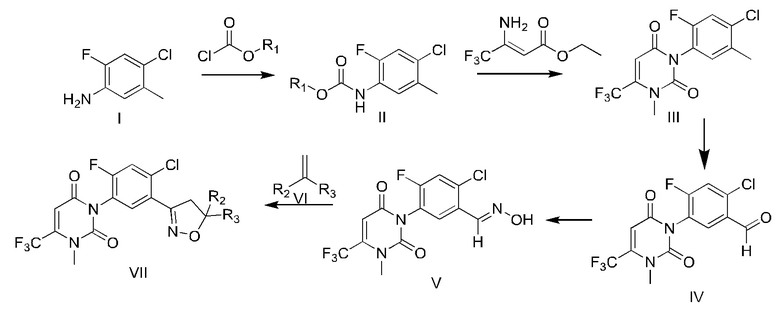
Способ синтеза фенилизоксазолинового соединения включает:
1) применение 2-фтор-4-хлор-5-метиланилина и соединения хлорформиата в качестве исходных веществ для реакции с получением соединения карбамата;
2) взаимодействие карбамата, полученного на стадии 1), с 3-амино-4,4,4-трифторкротонатом, трифтораминокротонатом с последующим метилированием метилирующим реагентом с получением урацила;
3) обработка урацила, полученного на стадии 2), путем окисления или дигалогенирования гидролиза с получением урацилбензальдегида;
4) взаимодействие урацилбензальдегида (IV), полученного на стадии 3), с гидроксиламин гидрохлоридом с получением урацилбензальдоксима;
5) проведение хлорирования NCS урацилбензальдоксима, полученного на стадии 4), с последующей циклизацией с алкеновым соединением с получением фенилизоксазолинового соединения.
Путь синтеза является следующим:
В формуле
R1 выбран из метила, этила, фенила, 4-нитрофенила или бензила;
R2 выбран из водорода, C1-C4 алкила, CO2R4 или CH2OR5;
R3 выбран из водорода, циано, C1-C4 алкила, C1-C4 галогеналкила, CO2R4 или CH2OR5;
R4 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила, C3-C4 алкенила, C3-C4 алкила, C1-C4 алкокси C1-C4 алкила, C1-C4 алкилкарбонилокси C2-C3 алкила, и незамещенного бензила, незамещенного фуранметилена, незамещенного тетрагидрофуранметилена, и замещенного бензила, фуранметилена или тетрагидрофуранметилена следующими 1-4 группами независимых заместителей: галоген, CN, NO2, C1-C4 алкил или C1-C4 галогеналкил;
R5 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкоксикарбонила, C1-C4 алкилкарбонила, C1-C4 галогеналкилкарбонила, C3-C6 циклоалкилкарбонила, C3-C6 галогенциклоалкилкарбонила, C1-C4 алкилсульфонила, C1-C4 галогеналкилсульфонила, C1-C3 алкиламиносульфонила, ди(C1-C3) алкиламиносульфонила, C1-C3 алкиламинокарбонила, ди(C1-C3) алкиламинокарбонила, ди(C1-C3)алкиламинотиокарбонила, C1-C2 алкилсульфурил C2-C4 алкилкарбонила и фенил C1-C2 алкила, фенилкарбонила, фенил C1-C2 алкилкарбонила, фенил C2-C4 алкилкарбонила, фенокси C1-C2 алкилкарбонила, тиофенилкарбонила, пиразолкарбонила и хинолинкарбонила, которые являются незамещенными или замещенными 1-4 группами, независимо замещенными из следующих групп; следующие группы представляют собой галоген, CN, NO2, C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкокси, C1-C4 галогеналкокси, C1-C4 алкоксикарбонил, C1-C4 алкил, C1-C4 алкилсульфонил, или фенокси, который независимо замещен 1-4 галогенами, CN, NO2, C1-C4 алкилом, C1-C4 галогеналкилом, C1-C4 алкокси или C1-C4 галогеналкокси.
Также, в общих формулах II и VII,
R1 выбран из метила, этила, фенила, 4-нитрофенила или бензила;
R2 выбран из водорода, C1-C4 алкила, CO2R4 или CH2OR5;
R3 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R4 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила, C3-C4 алкенила, C3-C4 алкила, C1-C4 алкокси C1-C4 алкила, C1-C4 алкилкарбонилокси C2-C3 алкила, бензила, фуранметилена или тетрагидрофуранметилена;
R5 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила, C1-C4 алкоксикарбонила, C1-C4 алкилкарбонила, C1-C4 галогеналкилкарбонила, C3-C6 циклоалкилкарбонила, C3-C6 галогенциклоалкилкарбонила, C1-C4 алкилсульфонила и C1-C4 галогеналкилсульфонила.
Кроме того, в общих формулах II и VII,
R1 выбран из метила или этила;
R2 выбран из водорода, C1-C4 алкила, CO2R4 или CH2OR5;
R3 выбран из водорода, C1-C4 алкила или C1-C4 галогеналкила;
R4 выбран из водорода, C1-C4 алкила, C1-C4 галогеналкила, C3-C4 алкенила, C3-C4 алкила, C1-C4 алкокси C1-C4 алкила, C1-C4 алкилкарбонилокси C2-C3 алкила, бензила, фуранметилена или тетрагидрофуранметилена;
R5 выбран из водорода, C1-C4 алкилкарбонила или C3-C6 циклоалкилкарбонила.
Предпочтительно в общих формулах II и VII,
R1 выбран из этила;
R2 выбран из водорода, цианида, метила, этила, н-пропила, н-бутила, изопропила, изобутила, трет-бутила, трифторэтила, трифторметил или CO2R4;
R3 выбран из водорода, метила, этила, пропила, изопропила, трет-бутила или трифторметила;
R4 выбран из водорода, метила, этила, н-пропила, н-бутила, изопропила, изобутила, трет-бутила, трифторэтила, аллила, пропаргила, метоксиэтила, этоксиэтила, метилкарбонилоксиэтила, 2-тетрагидрофуранметилена или 3-тетрагидрофуранметилена.
Кроме того, на стадии 1) 2-фтор-4-хлор-5-метиланилин (I) нагревают до 60-100°С в растворителе и в щелочных условиях и по каплям добавляют соединение хлорформиата для реакции в течение 1-4 ч с получением соединения карбамата (II), где молярное соотношение 2-фтор-4-хлор-5-метиланилина (I), щелочи и соединения хлорформиата составляет 1:(1-4):(1-2).
На стадии 2) соединение карбамата (II), полученное на стадии 1), подвергают взаимодействию с 3-амино-4,4,4-трифторкротонатом в растворителе и в щелочных условиях с катализатором при 100-140°С в течение 3-8 часов; затем температуру снижают до комнатной температуры, добавляют метилирующий реагент и добавляют щелочь для реакции при 20-80°С в течение 2-8 часов с получением урацила (III).
При этом молярное соотношение карбамата (II), 3-амино-4,4,4-трифторкротоната, щелочи, катализатора и метилирующего реагента составляет 1: (1-1,2): (1,5-3): (0,01-0,1): (1-2).
На стадии 3) урацил (III), полученный на стадии 2), галогенированный реагент, растворитель и катализатор смешивают для взаимодействия при 50-150°С в течение 2-10 часов; затем температуру снижают до комнатной температуры и проводят экстракцию для сбора органической фазы; вакуумную дистилляцию проводят с получением дигалогенида; добавляют кислоту для гидролиза; реакцию проводят при 50-100°С в течение 4-12 часов; затем проводят вакуумную дистилляцию; рН системы нейтрализуют до нейтрального значения; и продукт фильтруют с получением урацилбензальдегида (IV).
При этом молярное соотношение урацила (III), галогенированного реагента, катализатора и кислоты составляет 1: (2,5-3,5): (0,01-0,1): (10-30).
На стадии 4), урацилбензальдегид (IV) взаимодействует с гидроксиламин гидрохлоридом в спирте при комнатной температуре в течение 1-6 часов, и продукт фильтруют с получением урацилбензальдоксима (V), где молярное отношение урацилбензальдегида (IV) к гидроксиламин гидрохлориду составляет 1:(1-1,5).
На стадии 5), урацилбензальдоксим (V), полученный на стадии 4), добавляют к растворителю; галогенированный реагент добавляют при 20-40°С для реакции при этой температуре в течение 1-2 часов; температуру снижают до 0-15°С и при этой температуре добавляют алкеновое соединение (VI) и щелочь, выдерживают в течение 1-4 часов; реактанты экстрагируют и разделяют на слои; и органическую фазу промывают и затем перегоняют под вакуумом с получением продукта фенилизоксазолинового соединения (VII);
где молярное соотношение урацилбензальдоксима (V), галогенированного реагента, алкенового соединения (VI) и щелочи составляет 1: (1-1,5): 1: (1-2).
Растворитель на стадии 1) выбран из ацетонитрила, тетрагидрофурана, 1,4-диоксана, диметилового эфира этиленгликоля, этилацетата, 2-бутанона, N, N-диметилформамида или диметилсульфоксида; щелочь выбрана из карбоната калия, карбоната натрия, бикарбоната калия, бикарбоната натрия, гидроксида натрия, гидроксида калия, гидроксида лития, трет-бутоксида натрия, трет-бутоксида калия, этоксида натрия, метоксида натрия, триэтиламина, пиридина или 4-диметиламинопиридина.
Предпочтительно, на стадии 1) молярное соотношение 2-фтор-4-хлор-5-метиланилина (I), щелочи и соединения хлорформиата 1: (1,5-3): (1-1,5); растворитель выбран из ацетонитрила, этилацетата или 2-бутанона; и щелочь выбрана из карбоната калия, карбоната натрия, бикарбоната калия или бикарбоната натрия;
Растворитель на стадии 2) выбран из одного или двух из ацетонитрила, тетрагидрофурана, 1,4-диоксана, диметилового эфира этиленгликоля, этилацетата, 2-бутанона, N, N-диметилформамида или диметилсульфоксида; щелочная среда и щелочь с добавками выбрана из карбоната калия, карбоната натрия, бикарбоната калия, бикарбоната натрия, гидроксида натрия, гидроксида калия, гидроксида лития, трет-бутоксида натрия, трет-бутоксида калия, этоксида натрия, метоксида натрия, триэтиламина, пиридина или 4-диметиламинопиридина; катализатор выбран из одного или двух из катализатора фазового переноса полиэфира, катализатора фазового переноса циклического краун-эфира, катализатора фазового переноса соли четвертичного аммония, катализатора фазового переноса третичного амина, катализатора фазового переноса четвертичного аммониевого основания и катализатора фазового переноса четвертичного фосфина; и метилирующий реагент выбран из йодметана, диметилсульфата или хлорметана.
Предпочтительно, на стадии 2), ректификационное устройство может быть использовано для разделения воды и низкокипящего растворителя в реакционной смеси; молярное соотношение карбамата (II), трифтораминокротоната, щелочи составляет 1: (1-1,1): (1-2,5); растворитель выбран из одного или двух из ацетонитрила, 2-бутанона, N, N-диметилформамида или диметилсульфоксида; щелочная среда и щелочь с добавками выбрана из карбоната калия, карбоната натрия, бикарбоната калия или бикарбоната натрия; и катализатор выбран из одного или двух из PEG-200, PEG-400, PEG-600, 18-краун-6, 15-краун-5, циклодекстрина, бензилтриэтиламмоний хлорида, тетрабутиламмоний бромида, тетрабутиламмоний хлорида, тетрабутиламмоний бисульфата, тетраметиламмоний бромида, трибутилметиламмоний хлорида, триоктиламмоний хлорида, додецилтриметиламмоний хлорида, миристилтриметиламмоний хлорида, пиридина, трибутиламин, 1,8-диазодициклодекундекан-7-ена (DBU) или триэтилендиамина.
Более предпочтительно катализатор на стадии 2) выбран из одного или двух из тетрабутиламмоний бромида, трибутилметиламмоний хлорида или DBU.
На стадии 3), галогенированный реагент выбран из NBS, NCS, Cl2 или Br2; растворитель выбран из четыреххлористого углерода, трихлорметана, ацетонитрила, этилацетата, изопропилацетата, тетрагидрофурана, 1,4-диоксана, диметилового эфира этиленгликоля или бензола; катализатор выбран из азодиизобутиронитрила или бензоилпероксида; кислота выбрана из хлористоводородной кислоты, серной кислоты и муравьиной кислоты; и щелочь выбрана из гидроксида натрия, карбоната натрия, бикарбоната натрия, гидроксида калия, карбоната калия или бикарбоната калия.
Предпочтительно, на стадии 3), галогенированный реагент выбран из NBS; растворитель выбран из четыреххлористого углерода или 1,4-диоксана; и щелочь выбрана из гидроксида натрия или гидроксида калия.
На стадии 4) спирт выбран из метанола, этанола или изопропилового спирта.
Предпочтительно, на стадии 4) время реакции составляет 1-3 часа.
На стадии 5), реактанты экстрагируют и разделяют на слои, и органическую фазу последовательно промывали 1н. хлористоводородной кислотой и насыщенным раствором соли; вакуумную дистилляцию проводят с получением продукта фенилизоксазолинового соединения (VII); галогенированный реагент выбиран из NBS, NCS, хлора или бромида; щелочь выбрана из бикарбоната натрия, бикарбоната калия, карбоната натрия, карбоната калия, триэтиламина или пиридина; и растворитель выбран из одного или двух из дихлорметан, трихлорметан, диметилового эфира этиленгликоля, этилацетата или N, N-диметилформамида;
Предпочтительно, на стадии 5), галогенированный реагент выбран из NCS или бромата; и щелочь выбрана из бикарбоната натрия, бикарбоната калия или триэтиламина.
В процессе получения содержание продукта определяют методом высокоэффективной жидкостной хроматографии по методу внешнего стандарта.
Кроме того, исходные вещества хлорформиата, 2-фтор-4-хлор-5-метиланилина, 3-амино-4,4,4-трифторкротоната и алкенового соединения (VI), используемые в настоящем изобретении, могут быть коммерчески доступными.
Настоящее изобретение относится к промежуточному соединению для синтеза фенилизоксазолинового соединения. Структурная формула промежуточного соединения представлена в формуле V схемы реакции. Заместители выбирают, как описано выше.
Настоящее изобретение кроме того относится к применению соединения в синтезе изоксазолиновых соединений, содержащих урацил.
Настоящее изобретение имеет следующие преимущества:
В способе получения по настоящему изобретению сначала синтезируют урациловое кольцо и, наконец, синтезируют изооксазолиновое кольцо без использования дорогостоящего дихлорметилендиметиламмонийхлорида. Используемые исходные вещества являются легко доступными, а стоимость низкая, что может эффективно снизить производственные затраты. Кроме того, реакции, используемые в способе по настоящему изобретению, представляют собой обычные операционные установки, которые просты в эксплуатации и легко адаптируются к промышленным масштабам. В процессе реакции родственные промежуточные соединения стабильны, и из них нелегко получить побочные продукты. В процессе реакции некоторые промежуточные соединения не нуждаются в специальной очистке и могут быть непосредственно использованы в следующей реакции, что способствует непрерывной работе промышленности. Выход значительно выше, чем в предшествующем уровне техники, а общий выход увеличивается в 3 раза.
Подробное описание
Следующие конкретные варианты осуществления используются для дополнительной иллюстрации настоящего изобретения, но настоящее изобретение не ограничивается этими примерами. Проценты, используемые в следующих вариантах осуществления, представляют собой массовые проценты, такие как содержание и чистота.
Пример 1 Синтез промежуточного соединения V
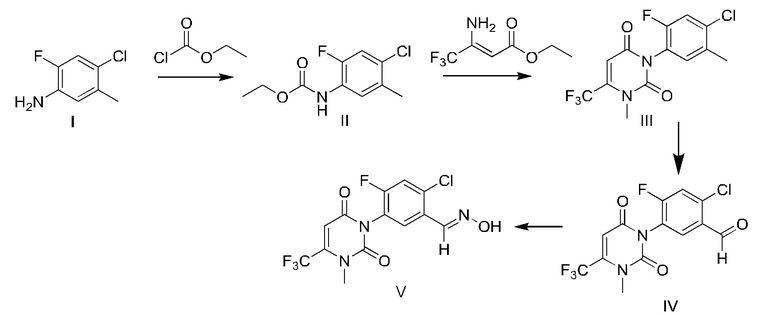
63,8 г (0,4 моль) 2-фтор-4-хлор-5-метиланилина и 67,2 г (0,8 моль) бикарбоната натрия последовательно добавляли к 300 мл этилацетата, и нагревали до микрокипячения с обратным холодильником; добавляли по каплям 48,8 г (0,45 моль) этилхлорформиата, и кипятили с обратным холодильником в течение 4 часов; ВЭЖХ показала, что реакция завершилась, и температуру снижали до комнатной температуры добавлением воды; продукт экстрагировали и разделяли на слои; органическую фазу промывали насыщенным раствором соли; органическую фазу сушили безводным сульфатом магния; 94,2 г промежуточного соединения II получали десольватацией при пониженном давлении, которое представляло собой масло с содержанием 98% (нормализовано с помощью ВЭЖХ, то же самое ниже).
Вышеупомянутое масло и 200 мл ацетонитрила добавляли в реакционную колбу, содержащую 200 мл DMF, 56,6 г (0,41 моль) карбоната калия, 75 г (0,41 моль) этил 3-амино-4,4,4-трифторкротоната и 4,98 г (15,46 ммоль) тетрабутиламмоний бромида и имеющую дистилляционную колонну и конденсатор. Температуру повышали до температуры кипения с обратным холодильником и отделяли низкокипящий растворитель; через 4 часа ВЭЖХ показала, что реакция завершилась; после охлаждения до комнатной температуры добавляли 56,6 г (0,41 моль) карбоната калия, и добавляли по каплям 85,2 г (0,6 моль) метан йодида, и перемешивали при комнатной температуре в течение 6 часов; после того, как ВЭЖХ показала, что реакция завершилась, реакционный раствор медленно выливали в воду, перемешивали в течение 30 минут, фильтровали и сушили с получением 128 г промежуточного соединения III, которое представляло собой светло-желтое твердое вещество с содержанием 97,8%, выходом 93% (измеренный по 2-фтор-4-хлор-5-метиланилину) и температурой плавления 117-119°С.
В реакционную колбу последовательно добавляли 68,8 г (0,2 моль) II, 78,5 г (0,44 моль) NBS, 3,5 г (21,3 ммоль) азодиизобутиронитрила и 300 мл четыреххлористого углерода и нагревали до температуры кипения с обратным холодильником. Через 2 часа добавляли 11 г (0,06 моль) NBS и 0,5 г (3,05 ммоль) азодиизобутиронитрила, и реакцию продолжали в течение 2 часов; ВЭЖХ показала, что реакция завершилась; содержание дибромида составляло 91,8% и содержание монобромида составляло 3,85%. Температуру снижали до комнатной температуры; 100 мл 1 н HCl добавляли для отделения органической фазы; 200 мл дихлорметана добавляли к водной фазе для экстрагирования органической фазы; органические фазы объединяли и концентрировали при пониженном давлении; затем добавляли 150 мл 88% муравьиной кислоты и смешанный раствор нагревали до температуры кипения с обратным холодильником; температуру поддерживали в течение 8 часов; растворитель концентрировали при пониженном давлении и осторожно добавляли к воде; значение pH доводили до 9 с помощью гидроксида натрия; продукт перемешивали в течение 15 минут, фильтровали и сушили с получением 66,7 г IV, которое представляло собой светло-желтое твердое вещество, с содержанием 94,5%, выходом 89,9% и температурой плавления 176-177°С.
66,4 г (0,18 моль) IV добавляли к 200 мл этанола и перемешивали при комнатной температуре в течение 10 минут; затем добавляли смешанный раствор 14,4 г (0,207 моль) гидрохлорида гидроксиламина и 50 мл воды, и перемешивали при комнатной температуре до постепенного образования бледно-желтой мутной жидкости. После реакции в течение 1 часа, ВЭЖХ показала, что реакция завершена, и реакция останавливали. Продукт выстаивали, фильтровали, промывали 50 мл воды и сушили с получением 64,9 г V, которое представляло собой светло-желтое твердое вещество, с содержанием 96,1%, выходом 94,8%, и температурой плавления 182-185°С.
В пересчете на 2-фтор-4-хлор-5-метиланилина выход составил 79,3%.
Пример 2 Синтез соединения VII-1
0,76 г (2 ммоль) урацилбензальдоксима (V) растворяли в 20 мл дихлорметана и 5 мл N, N-диметилформамида. Температуру повышали до 35°С, и при этой температуре осторожно добавляли 0,28 г (2,1 ммоль) NCS, и реакционную смесь поддерживали при этой температуре в течение 1 часа. Температуру снижали до 0-5°С; по каплям добавляли смешанный раствор 0,23 г (2 ммоль) этилметакрилата, 0,22 г (2,2 ммоль) триэтиламинаа и 5 мл метиленхлорида; реакцию проводили при температуре в течение 1,5 ч; после того, как ВЭЖХ показала, что реакция завершилась, продукт последовательно промывали 1н. хлористоводородной кислотой, водой и насыщенным раствором соли, и органическую фазу сушили безводным сульфатом магния и десольвентизировали с получением 0,81 г светло-желтого маслянистого вещества, с содержанием 96,7% и выходом 81,7%. 1H-ЯМР(300 МГц, внутренний стандарт TMS, растворитель CDCl3) δ(ppm):1,35 (т, 3H), 1,68 (с, 3H), 3,38 (д, 1H), 3,60(с, 3H), 3,90(д, 1H), 4,30(м, 2H), 6,25 (с, 1H), 7,38(д, 1H), 7,79(д, 1H).
В пересчете на 2-фтор-4-хлор-5-метиланилин, общий выход составил 64,8%.
Ссылочный вариант осуществления 1 Получение соединения VII-1 (WO2016095768)
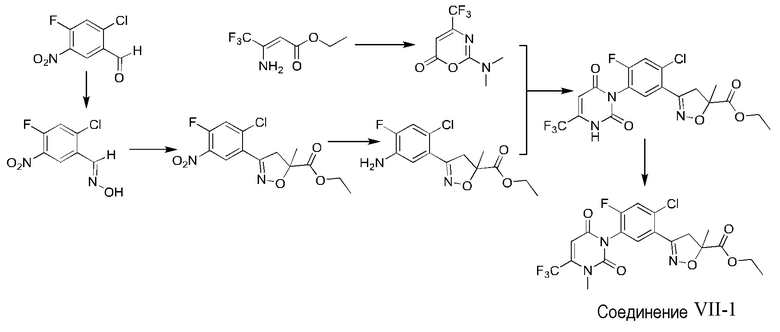
1) Получение 2-хлор-4-фтор-5-нитробензальдоксима
42 г (0,206 моль) 2-хлор-4-фтор-5-нитробензальдегида растворяли в 200 мл этанола, доводили до 0°С, и и при перемешивании добавляли 17,4 г (0,25 моль) водного раствора гидрохлорида гидроксиламина; и затем температуру повышали до комнатной температуры для перемешивания реакционной смеси. Через 2 часа TLC показала, что реакция завершилась. Раствор выливали в воду, и фильтровали с получением 38,3 г (98%) белого твердого вещества с выходом 83,4%.
2) Получение 3-(2-хлор-4-фтор-5-нитрофенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата
43,7 г (0,2 моль) 2-хлор-4-фтор-5-нитробензальдоксима растворяли в 150 мл N, N-диметилформамида и нагревали до 30°С; и 32 г (0,24 моль) NCS добавляли порциями при этой температуре с образованием светло-желтого раствора, и реакцию поддерживали при 35°С в течение 1 часа. Температуру снижали до комнатной температуры; добавляли 300 мл дихлорметана; затем раствор дважды промывали 1н хлористоводородной кислотой, промывали дважды насыщенным раствором соли, сушили безводным сульфатом магния и фильтровали с отсасыванием; раствор дихлорметана доводили до 0-5°С, и по каплям добавляли смешанный раствор 34,2 г (0,3 моль) этилметакрилата и 31 г (0,3 моль) триэтиламина, и реакционную смесь поддерживали при температуре в течение 1 часа. Раствор последовательно промывали 1н хлористоводородной кислотой и насыщенным раствором соли; органическую фазу сушили безводным сульфатом магния; и после десольватации проводили колоночную хроматографию (этилацетат: петролейный эфир =1:3) с получением 57 г (97%) светло-желтого твердого вещества, с выходом 83,6%.
3) Получение (2-хлор-4-фтор-5-аминофенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата
57 г (0,18 моль) 3-(2-хлор-4-фтор-5-нитрофенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата растворяли в 300 мл этилацетата, и 163 г (0,72 моль) дигидрата двухлористого олова добавляли порциями при нагревании, и затем реакционную смесь кипятили с обратным холодильником в течение 8 часов. TLC показала, что реакция завершилась. Раствор охлаждали до комнатной температуры, добавляли к воде со льдом, доводили гидроксидом натрия до значения pH 8, экстрагировали этилацетатом, промывали насыщенным раствором соли, сушили безводным сульфатом магния и затем перегоняли при пониженном давлении с получением 31 г маслянистого вещества, которое непосредственно использовали для следующей реакции без очистки, с содержанием 90% и выходом 51,6%.
4) Получение 2-диметиламино-4-трифторметил-6H-1,3-оксазин-6-она
25 г (0,15 моль) дихлорметилендиметиламмонийхлорида добавляли к 100 мл хлороформа, и нагревали до 60°С; добавляли по каплям смешанный раствор 25 г (0,14 моль) 3-амино-4,4,4-трифторкротоната и 15 мл хлороформа; реакцию нагревания с обратным холодильником продолжали; раствор постепенно менялся от бледно-желтого мутного до прозрачного; и через 4 часа TLC показала, что реакция завершилась. Раствор охлаждали до комнатной температуры; добавляли насыщенный водный раствор бикарбоната натрия; органическую фазу отделяли; раствор промывали насыщенным раствором соли; органическую фазу сушили безводным сульфатом магния, и перегоняли при пониженном давлении с получением 30,8 г светло-желтого твердого вещества.
5) Получение 3-(2-хлор-5-(2,6-диокси-4-трифторметил-3,6-дигидропиримидин-1(2H)-ил)-4-фторфенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата
13,2 г (0,046 моль) 3-(2-хлор-4-фтор-5-аминофенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата и 9,8 г (0,047 моль) 2-диметиламино-4-трифторметил-6H-1,3-оксазин-6-она последовательно добавляли в реакционную колбу, содержащую 100 мл уксусной кислоты; температуру повышали до реакции кипения с обратным холодильником с образованием темного раствора; и реакцию проводили при этой температуре в течение 6 ч, и растворитель выпаривали при пониженном давлении; добавляли водный раствор бикарбоната натрия для доведения значения рН до 7; раствор экстрагировали этилацетатом и сушили безводным сульфатом магния; и растворитель выпаривали при пониженном давлении с получением неочищенного продукта; и неочищенный продукт перекристаллизовывали с этанолом с получением 14,5 г (95%) белого твердого вещества, с выходом 64,6%.
6) Получение соединения VII-1
14 г (0,031 моль) 3-(2-хлор-5-(2,6-диокси-4-трифторметил-3,6-дигидропиримидин-1(2H)-ил)-4-фторфенил)-5-метил-4,5-дигидроизооксазол-5-этилкарбоксилата и 12,9 г (0,094 моль) карбоната калия последовательно добавляли в реакционную колбу со 150 мл N, N-диметилформамида, и охлаждали до 0°С; добавляли по каплям 8,9 г (0,062 моль) йодметана, и затем температуру повышали до комнатной температуры для перемешивания реакционной смеси в течение 6 ч. TLC показала, что реакция завершилась. Раствор выливали в воду, экстрагировали этилацетатом и промывали насыщенным раствором соли; органическую фазу сушили безводным сульфатом магния, перегоняли при пониженном давлении, и подвергали колоночной хроматографии (этилацетат: петролейный эфир =1:5) с получением 13,2 г маслянистого вещества (94%), с выходом 83,8%.
В пересчете на 2-хлор-4-фтор-5-нитробензальдегид, общий выход составил 19,5%.
В соответствии со способами, описанными выше в вариантах осуществления 1 и 2, этилметакрилат в варианте осуществления синтеза 2 заменяли метилметакрилатом, изопропилметакрилатом, н-бутилметакрилатом, н-пропилметакрилатом, трет-бутилметакрилатом, 2-этоксиэтилметакрилатом, 2-(трифторметил)метилакрилатом, этилакрилатом, 2-метил-1-гептеном и 2,4-диметил-1-пентеном для получения соединений, относящихся к формуле VII, отличных от соединения VII-1. Конкретные данные ядерно-магнитного резонанса и выходы соединений формулы VII представлены в таблице 1:
Таблица 1
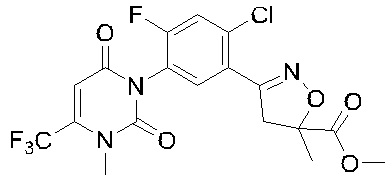
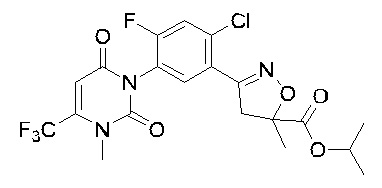
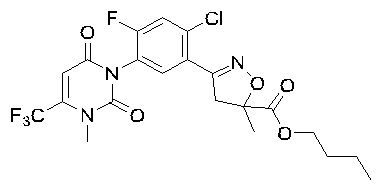
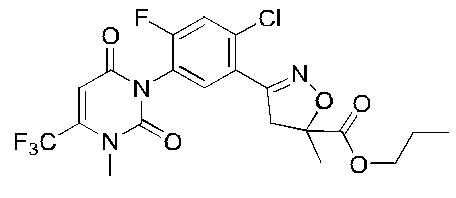
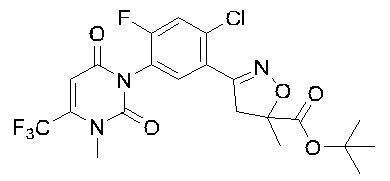
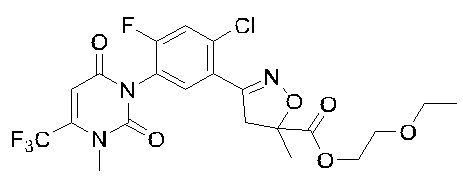
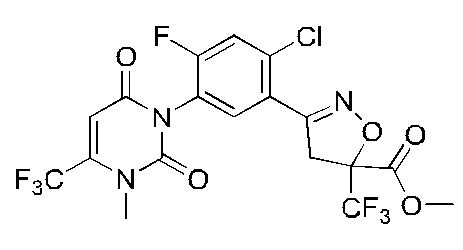
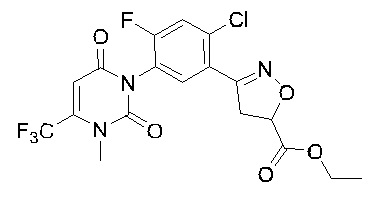
Кроме того, различные заместители исходных веществ изменяются в формуле и в соответствии с записями процесса получения могут быть также получены соединения формулы I, представленные различными заместителями, что также показывает универсальность применения способа по настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИНТЕРМЕДИАТА УРАЦИЛОВОГО СОЕДИНЕНИЯ, СОДЕРЖАЩЕГО ИЗОКСАЗОЛИН | 2021 |
|
RU2818999C1 |
| КОНДЕНСИРОВАННОЕ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2729636C2 |
| ПИРАЗОЛ ИЛИ ЕГО СОЛЬ И СПОСОБ ПОЛУЧЕНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2707086C1 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| ИНГИБИТОР EGFR | 2021 |
|
RU2817044C1 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ПРОИЗВОДНОЕ ТИОФЕНКАРБОКСАМИДА И СРЕДСТВО ДЛЯ КОНТРОЛЯ ЗАБОЛЕВАНИЯ РАСТЕНИЙ, СОДЕРЖАЩЕЕ ЕГО | 2019 |
|
RU2776177C1 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДОКСИМА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО ДЛЯ ЗАЩИТЫ СЕЛЬСКОХОЗЯЙСТВЕННЫХ РАСТЕНИЙ | 1995 |
|
RU2140908C1 |
| НОВОЕ КОНДЕНСИРОВАННОЕ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2017 |
|
RU2770727C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545074C1 |
Изобретение относится к способу синтеза фенилизоксазолинового соединения, отличающемуся тем, что включает: 1) применение 2-фтор-4-хлор-5-метиланилина и соединения хлорформиата в качестве исходных веществ для реакции с получением соединения карбамата, причем 2-фтор-4-хлор-5-метиланилин нагревают до 60-100°С в растворителе и в щелочных условиях и по каплям добавляют соединение хлорформиата, где молярное соотношение 2-фтор-4-хлор-5-метиланилина (I), щелочи и соединения хлорформиата составляет 1:(1-4):(1-2); 2) взаимодействие карбамата, полученного на стадии 1), с 3-амино-4,4,4-трифторкротонатом в растворителе и в щелочных условиях с катализатором при 100-140°С, с последующим метилированием метилирующим реагентом с получением урацила, причем молярное соотношение карбамата (II), 3-амино-4,4,4-трифторкротоната, щелочи, катализатора и метилирующего реагента составляет 1:(1-1,2):(1,5-3):(0,01-0,1):(1-2); 3) обработка урацила, полученного на стадии 2), путем окисления или дигалогенирования гидролиза с получением урацилбензальдегида, причем галогенированный реагент, растворитель и катализатор смешивают для взаимодействия при 50-150°С; затем температуру снижают до комнатной температуры и проводят экстракцию для сбора органической фазы; добавляют кислоту для гидролиза; где молярное соотношение урацила (III), галогенированного реагента, катализатора и кислоты составляет 1:(2,5-3,5):(0,01-0,1):(10-30); 4) взаимодействие урацилбензальдегида (IV), полученного на стадии 3), с гидроксиламин гидрохлоридом с получением урацилбензальдоксима, причем урацилбензальдегид (IV) взаимодействует с гидроксиламин гидрохлоридом в спирте при комнатной температуре с получением урацилбензальдоксима (V), где молярное отношение урацилбензальдегида (IV) к гидроксиламин гидрохлориду составляет 1:(1-1,5); 5) проведение хлорирования NCS урацилбензальдоксима, полученного на стадии 4), с последующей циклизацией с алкеновым соединением с получением фенилизоксазолинового соединения, причем урацилбензальдоксим (V), полученный на стадии 4), добавляют к растворителю; галогенированный реагент добавляют при 20-40°С и при этой температуре добавляют алкеновое соединение (VI) и щелочь; где молярное соотношение урацилбензальдоксима (V), галогенированного реагента, алкенового соединения (VI) и щелочи составляет 1:(1-1,5):1:(1-2), причем путь синтеза является следующим: 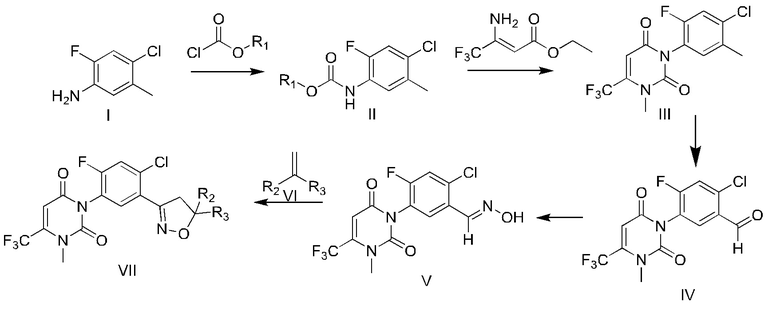 ; R1 выбран из метила или этила; R2 выбран из водорода, С1-С4 алкила или CO2R4; R3 выбран из водорода, С1-С4 алкила или С1-С4 галогеналкила; R4 выбран из С1-С4 алкила или С1-С4 галогеналкила. Технический результат: способ обеспечивает получение фенилизоксазолинового соединения с дешевым и доступным сырьем и простым процессом синтеза. 3 н. и 6 з.п. ф-лы, 1 табл., 2 пр.
; R1 выбран из метила или этила; R2 выбран из водорода, С1-С4 алкила или CO2R4; R3 выбран из водорода, С1-С4 алкила или С1-С4 галогеналкила; R4 выбран из С1-С4 алкила или С1-С4 галогеналкила. Технический результат: способ обеспечивает получение фенилизоксазолинового соединения с дешевым и доступным сырьем и простым процессом синтеза. 3 н. и 6 з.п. ф-лы, 1 табл., 2 пр.
1. Способ синтеза фенилизоксазолинового соединения, отличающийся тем, что включает:
1) применение 2-фтор-4-хлор-5-метиланилина и соединения хлорформиата в качестве исходных веществ для реакции с получением соединения карбамата, причем 2-фтор-4-хлор-5-метиланилин нагревают до 60-100°С в растворителе и в щелочных условиях и по каплям добавляют соединение хлорформиата, где молярное соотношение 2-фтор-4-хлор-5-метиланилина (I), щелочи и соединения хлорформиата составляет 1:(1-4):(1-2);
2) взаимодействие карбамата, полученного на стадии 1), с 3-амино-4,4,4-трифторкротонатом в растворителе и в щелочных условиях с катализатором при 100-140°С, с последующим метилированием метилирующим реагентом с получением урацила, причем молярное соотношение карбамата (II), 3-амино-4,4,4-трифторкротоната, щелочи, катализатора и метилирующего реагента составляет 1:(1-1,2):(1,5-3):(0,01-0,1):(1-2);
3) обработка урацила, полученного на стадии 2), путем окисления или дигалогенирования гидролиза с получением урацилбензальдегида, причем галогенированный реагент, растворитель и катализатор смешивают для взаимодействия при 50-150°С; затем температуру снижают до комнатной температуры, и проводят экстракцию для сбора органической фазы; добавляют кислоту для гидролиза;
где молярное соотношение урацила (III), галогенированного реагента, катализатора и кислоты составляет 1:(2,5-3,5):(0,01-0,1):(10-30);
4) взаимодействие урацилбензальдегида (IV), полученного на стадии 3), с гидроксиламин гидрохлоридом с получением урацилбензальдоксима, причем урацилбензальдегид (IV) взаимодействует с гидроксиламин гидрохлоридом в спирте при комнатной температуре с получением урацилбензальдоксима (V), где молярное отношение урацилбензальдегида (IV) к гидроксиламин гидрохлориду составляет 1:(1-1,5);
5) проведение хлорирования NCS урацилбензальдоксима, полученного на стадии 4), с последующей циклизацией с алкеновым соединением с получением фенилизоксазолинового соединения, причем урацилбензальдоксим (V), полученный на стадии 4), добавляют к растворителю; галогенированный реагент добавляют при 20-40°С и при этой температуре добавляют алкеновое соединение (VI) и щелочь;
где молярное соотношение урацилбензальдоксима (V), галогенированного реагента, алкенового соединения (VI) и щелочи составляет 1:(1-1,5):1:(1-2), причем путь синтеза является следующим:
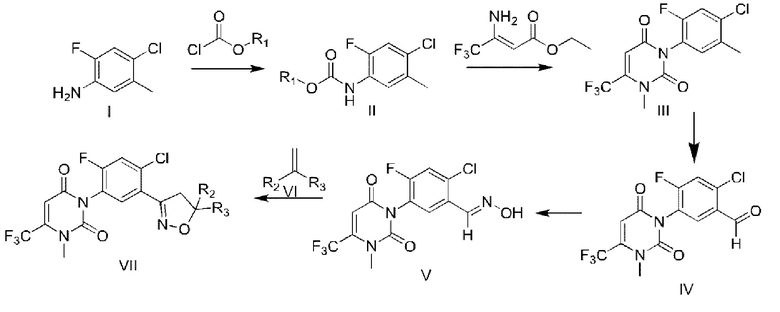
R1 выбран из метила или этила;
R2 выбран из водорода, С1-С4 алкила или CO2R4;
R3 выбран из водорода, С1-С4 алкила или С1-С4 галогеналкила;
R4 выбран из С1-С4 алкила или С1-С4 галогеналкила.
2. Способ синтеза фенилизоксазолинового соединения по п. 1, отличающийся тем, что путь синтеза является следующим:
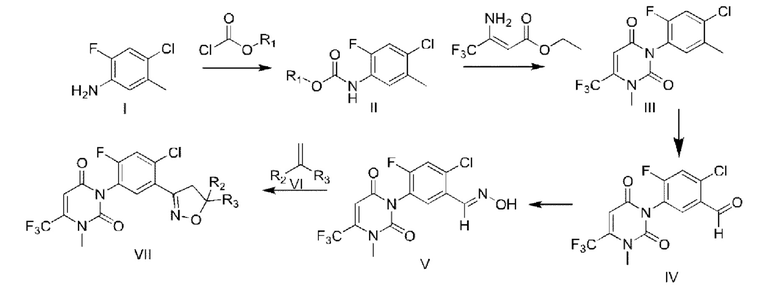
В формуле
R1 выбран из этила;
R2 выбран из водорода, С1-С4 алкила или CO2R4;
R3 выбран из водорода, С1-С4 алкила или С1-С4 галогеналкила;
R4 выбран из С1-С4 алкила или С1-С4 галогеналкила.
3. Способ синтеза фенилизоксазолинового соединения по п. 1 или 2, отличающийся тем, что на стадии 1) 2-фтор-4-хлор-5-метиланилин (I) нагревают до 60-100°С в растворителе и в щелочных условиях и по каплям добавляют соединение хлорформиата для реакции в течение 1-4 ч с получением соединения карбамата (II), где молярное соотношение 2-фтор-4-хлор-5-метиланилина (I), щелочи и соединения хлорформиата составляет 1:(1-4):(1-2).
4. Способ синтеза фенилизоксазолинового соединения по п. 1 или 2, отличающийся тем, что на стадии 2) соединение карбамата (II), полученное на стадии 1), подвергают взаимодействию с 3-амино-4,4,4-трифторкротонатом в растворителе и в щелочных условиях с катализатором при 100-140°С в течение 3-8 часов; затем температуру снижают до комнатной температуры, добавляют метилирующий реагент и добавляют щелочь для реакции при 20-80°С в течение 2-8 часов с получением урацила (III);
где молярное соотношение карбамата (II), 3-амино-4,4,4-трифторкротоната, щелочи, катализатора и метилирующего реагента составляет 1:(1-1,2):(1,5-3):(0,01-0,1):(1-2).
5. Способ синтеза фенилизоксазолинового соединения по п. 1 или 2, отличающийся тем, что на стадии 3) урацил (III), полученный на стадии 2), галогенированный реагент, растворитель и катализатор смешивают для взаимодействия при 50-150°С в течение 2-10 часов; затем температуру снижают до комнатной температуры и проводят экстракцию для сбора органической фазы; вакуумную дистилляцию проводят с получением дигалогенида; добавляют кислоту для гидролиза; реакцию проводят при 50-100°С в течение 4-12 часов; затем проводят вакуумную дистилляцию; значение рН системы нейтрализуют до нейтрального значения; и продукт фильтруют с получением урацилбензальдегида (IV);
где молярное соотношение урацила (III), галогенированного реагента, катализатора и кислоты составляет 1:(2,5-3,5):(0,01-0,1):(10-30).
6. Способ синтеза фенилизоксазолинового соединения по п. 1 или 2, отличающийся тем, что на стадии 4) урацилбензальдегид (IV) взаимодействует с гидроксиламин гидрохлоридом в спирте при комнатной температуре в течение 1-6 часов, и продукт фильтруют с получением урацилбензальдоксима (V), где молярное отношение урацилбензальдегида (IV) к гидроксиламин гидрохлориду составляет 1:(1-1,5).
7. Способ синтеза фенилизоксазолинового соединения по п. 1 или 2, отличающийся тем, что на стадии 5) урацилбензальдоксим (V), полученный на стадии 4), добавляют к растворителю; галогенированный реагент добавляют при 20-40°С для реакции при этой температуре в течение 1-2 часов; температуру снижают до 0-15°С, и при этой температуре добавляют алкеновое соединение (VI) и щелочь, выдерживают в течение 1-4 часов; реактанты экстрагируют и разделяют на слои; и органическую фазу промывают и затем перегоняют под вакуумом с получением продукта фенилизоксазолинового соединения (VII);
где молярное соотношение урацилбензальдоксима (V), галогенированного реагента, алкенового соединения (VI) и щелочи составляет 1:(1-1,5):1:(1-2).
8. Промежуточное соединение для синтеза фенилизоксазолинового соединения, отличающееся тем, что структурная формула промежуточного соединения представлена формулой V по п. 2:
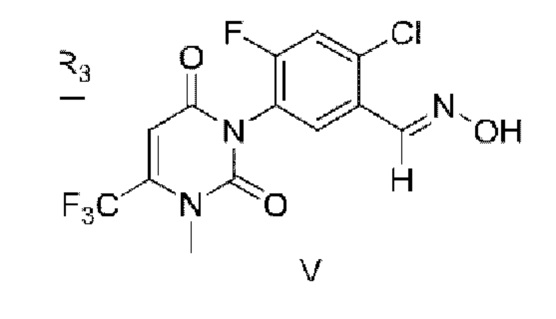
9. Применение соединения по п. 8 в синтезе изоксазолиновых соединений, содержащих урацил.
| CN05753853 A, 13.07.2016 | |||
| WO2019240082 A1, 19.12.2019 | |||
| WO2020063613 A1, 02.04.2020 | |||
| N-(3-КАРБАМОИЛФЕНИЛ)-1Н-ПИРАЗОЛ-5-КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ ДЛЯ БОРЬБЫ С ЖИВОТНЫМИ-ВРЕДИТЕЛЯМИ | 2012 |
|
RU2600739C2 |

