ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям пирроло[2,3-d]пиримидина и изолированным формам указанных соединений. Изобретение также относится к получению указанных соединений и промежуточных продуктов, используемых в указанном получении, композициям, содержащим указанные соединения, применениям указанных соединений для ингибирования Янус-киназы (Janus Kinase - JAK) и способам лечения и профилактики состояний, опосредуемых JAK1.
УРОВЕНЬ ТЕХНИКИ
Протеинкиназы представляют собой семейства ферментов, которые катализируют фосфорилирование специфических остатков в белках и в широком смысле разделяются на тирозин-киназы и серии/треониновые киназы. Аномальная активность киназы, возникающая в результате мутации, избыточной экспрессии либо неадекватной регуляции, нарушения регуляции или отсутствия регуляции, а также избыточного или недостаточного продуцирования факторов роста или цитокинов, является причиной многих заболеваний, включая, но без ограничения указанным перечнем, рак, сердечно-сосудистые заболевания, аллергии, астму и другие респираторные заболевания, аутоиммунные заболевания, воспалительные заболевания, заболевания костей, нарушения обмена веществ, а также неврологические и нейродегенеративные расстройства, такие как болезнь Альцгеймера. Неадекватная активность киназы запускает различные биологические клеточные реакции, связанные с ростом клеток, дифференцировкой клеток, выживанием, апоптозом, митогенезом, контролем клеточного цикла и подвижностью клеток, что приводит к перечисленным выше и родственным заболеваниям.
Таким образом, протеинкиназы стали важным классом ферментов, рассматриваемых в качестве мишеней для терапевтического воздействия. В частности, семейство тирозинкиназ клеточных белков JAK (JAK1, JAK2, JAK3 и Tyk2) играют ключевую роль в передаче цитокиновых сигналов (Kisseleva et al., Gene, 2002, 285, 1; Yamaoka et al. Genome Biology 2004, 5, 253). При связывании со своими рецепторами цитокины активируют JAK, которые затем фосфорилируют рецепторы к цитокинам, создавая таким образом докинг-сайты для сигнальных молекул, в частности представителей семейства преобразователей сигналов и активаторов транскрипции (signal transducer and activator of transcription - STAT), что в конечном итоге приводит к экспрессии генов. Известно, семейство JAK активируют различные цитокины. К таким цитокинам относятся цитокины семейства IFN (IFN-альфа, IFN-бета, IFN-омега, Limitin, IFN-гамма, IL-10, IL-19, IL-20, IL-22), семейства gp130 (IL-6, IL-11, OSM, LIF, CNTF, NNT-1/BSF-3, G-CSF, СТ-1, лептин, IL-12, IL-23), семейства гамма С (IL-2, IL-7, TSLP, IL-9, IL-15, IL-21, IL-4, IL-13), семейства IL-3 (IL-3, IL-5, GM-CSF), семейства одноцепочечных (ЕРО, GH, PRL, ТРО), рецепторные тирозинкиназы (EGF, PDGF, CSF-1, HGF) и рецепторы, связанные с G-белком (ATI).
Сохраняется потребность в новых соединениях, эффективно и избирательно ингибирующих специфические ферменты JAK, в частности JAK1 в отличие от JAK2. JAK1 является представителем семейства протеинкиназ Янус (Janus), состоящего из JAK1, JAK2, JAK3 и TYK2. JAK1 экспрессируется на различных уровнях во всех тканях. Многие рецепторы цитокинов передают сигнал через пары JAK-киназ в следующих комбинациях: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/TYK2 или JAK2/JAK2. В этом контексте JAK1 является наиболее широко распространенной парной JAK-киназой и необходима для передачи сигналов общими γ-цепями рецепторов (IL-2Rγ) цитокинов, семейством рецепторов IL-6, семействами рецепторов I, II и III типов и семейством рецепторов IL-10. Исследования на животных показали, что JAK1 необходима для развития, функционирования и гомеостаза иммунной системы. Модуляция иммунной активности посредством ингибирования активности киназы JAK1 может быть полезной при лечении различных иммунных расстройств (Murray, P.J. J. 1 ммипо1., 178, 2623-2629 (2007); Kisseleva, Т. et al., Gene, 285, 1-24 (2002); O'Shea, J. J. et al., Cell, 109, (suppl.) S121-S131 (2002)), не допуская при этом передачи сигналов JAK2-зависимого эритропоэтина (ЕРО) и тромбопоэтина (ТРО) (Neubauer Н., et al., Cell, 93(3), 397-409 (1998); Parganas E., et al., Cell, 93(3), 385-95 (1998)).
Необходимы безопасные и эффективные средства для борьбы с расстройствами, связанными с JAK, такими как атопический дерматит, у людей и животных. На рынке средств для лечения атопического дерматита в настоящее время доминируют кортикостероиды, которые вызывают неприятные и нежелательные побочные эффекты. Используются также антигистаминные лекарственные средства, но они малоэффективны. Необходимы новые соединения, обладающие селективной ингибиторной активностью в отношении JAK1, обеспечивающие альтернативу применению стероидов и способствующие решению проблемы хронического зуда и воспаления, которые либо длительно сохраняются при атопическом дерматите, либо медленно регрессируют после удаления аллергена или возбудителя.
Недавно идентифицированный N-((1S,3S)-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид, широко известный как аброцитиниб, описан в Патенте США №9035074 правообладателя настоящего изобретения, содержание которого полностью включено в настоящее описании в качестве ссылки; химическая формула аброцитиниба - C14H21N5O2S, структурная формула представлена ниже:
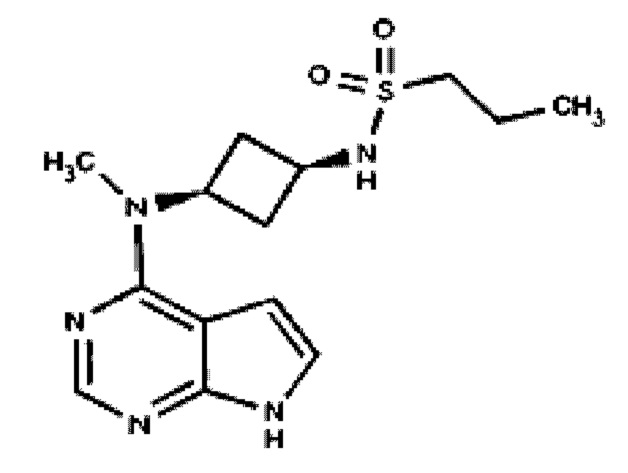
Известно, что аброцитиниб является перспективным ингибитором JRK и может применяться в качестве терапевтического средства для лечения различных заболеваний, включая атопический дерматит. Авторами настоящего изобретения были обнаружены новые метаболиты аброцитиниба, терапевтически активные в качестве ингибиторов JAK, которые обладают неожиданно высоким терапевтическим индексом и, соответственно, потенциально могут быть превосходными фармацевтическими средствами для лечения JAK-опосредуемых расстройств.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к пирроло[2,3-d]пиримидиновым соединениям и изолированным формам таких соединений. Было выявлено, что изолированные соединения по настоящему изобретению обладают неожиданно полезной активностью, включая более высокую эффективность и пониженную частоту проявления побочных эффектов. Изолированные соединения могут использоваться в фармацевтических композициях в комбинации с фармацевтически приемлемыми эксципиентами и в способах лечения состояний, опосредуемых JAK1.
Целью настоящего изобретения являются соединения формулы I представленной ниже структуры:

или их фармацевтически приемлемые соли, где R1 и R2 независимо представляют собой водород или гидроксильную группу; где если R1 представляет собой водород, то R2 является гидроксильной группой; и если R2 представляет собой водород, то R1 является гидроксильной группой.
В других аспектах целью настоящего изобретения также являются соединения формулы I или их фармацевтически приемлемые соли в изолированной форме.
В других аспектах целью настоящего изобретения также являются фармацевтические композиции, которые включают фармацевтически приемлемый эксципиент и соединение формулы I или его фармацевтически приемлемую соль.
В еще одном аспекте целью настоящего изобретения также являются способы лечения состояний или расстройств, включая миозит, васкулит, пузырчатку, болезнь Крона, волчанку, нефрит, псориаз, рассеянный склероз, большое депрессивное расстройство, аллергию, астму, болезнь Шегрена, синдром сухого глаза, отторжение трансплантата, рак, воспалительное заболевание кишечника, септический шок, сердечно-легочную дисфункцию, острое респираторное заболевание или кахексию, введением субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
В еще одном аспекте целью настоящего изобретения также являются способы лечения состояний или расстройств, включая атопический дерматит, экзему, псориаз, склеродермию, волчанку, зуд, другие зудящие состояния, аллергические реакции, включая аллергический дерматит у млекопитающих, воспалительное заболевание дыхательных путей, рецидивирующую обструкцию дыхательных путей, гиперчувствительность дыхательных путей и хроническую обструктивную болезнь легких, включающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
В еще одном аспекте целью настоящего изобретения также являются способы получения соединений по настоящему изобретению.
Настоящее изобретение станет более понятным из описания, приведенного далее только в качестве примера. Хотя настоящее изобретение не ограничивается указанным описанием, с помощью приведенного обсуждения и примеров будут оценены различные аспекты настоящего изобретения.
Термин «субъект» относится к человеку, домашнему скоту или животным-компаньонам.
Термин «лечение» или «излечение» означает облегчение симптомов, связанных с заболеванием, расстройством или состоянием, или остановку дальнейшего прогрессирования или развития этих симптомов. В зависимости от заболевания и состояния субъекта термин «лечение», используемый в настоящем описании, может включать один или несколько лечебных, паллиативных и профилактических методов. Лечение может также включать введение фармацевтической композиции по настоящему изобретению в комбинации с другими терапевтическими средствами.
Термин «терапевтически эффективное» указывает на способность средства предотвращать расстройство или облегчать его тяжесть, не вызывая при этом неблагоприятных побочных эффектов, обычно связанных с другими методами лечения. Фразу «терапевтически эффективный» следует понимать как эквивалентную фразе «эффективный для лечения, предотвращения или облегчения состояния», и обе эти фразы предназначены для определения количества каждого средства для применения в комбинированной терапии, которое позволит достичь цели улучшения тяжести рака, сердечно-сосудистых заболеваний, боли и воспаления, а также частоту их встречаемости по сравнению с лечением каждым средством отдельно, не вызывая при этом неблагоприятных побочных эффектов, обычно связанных с другими методами лечения.
Термин «фармацевтически приемлемый» означает подходящий для применения субъектом.
Если заместители описаны как «независимо выбранные» из группы, каждый заместитель выбран независимо от другого. Таким образом, каждый заместитель может быть идентичен другому заместителю(ям) или отличаться от него(них). В некоторых вариантах осуществления метаболиты соединений или их соли являются по существу изолированными.
Термины «изолированный», «очищенный», «в очищенной форме», «в изолированной форме» или «очищенное вещество», которые относятся к соединению, означают физическое состояние соединения после его выделения из синтетического процесса, например из реакционной смеси. Таким образом, термины «выделенное», «очищенное», «в очищенной форме», «в изолированной форме» или «очищенное вещество», относящиеся к соединению, относятся к физическому состоянию указанного соединения после получения в результате очистки или способами очистки, описанными в настоящем документе или хорошо известными специалисту (например, хроматографией, перекристаллизацией и т.п.), с чистотой, достаточной для получения его характеристик стандартными аналитическими методами, описанными в настоящем документе или хорошо известными квалифицированному специалисту. Например, способы очистки, раскрытые в настоящем документе (такие как методы ЖХ-МС и ЖК-МС/МС), приводят к получению изолированных форм рассматриваемых соединений. Ожидается, что такие методы выделения и очистки приведут к получения продукта, содержащего по меньшей мере примерно 70 %, по меньшей мере примере 80 %, по меньшей мере примерно 90 %, по меньшей мере примерно 95 %, по меньшей мере примерно 97 % или по меньшей мере примерно 99 % по массе указанного соединения или его соли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые являются селективными модуляторами JAK1, применимыми для лечения заболеваний и состояний, связанных с нарушением регуляции JAK1. Настоящее изобретение также относится к выделению соединений, которые являются селективными модуляторами JAK1, применимым для лечения заболеваний и состояний, связанных с нарушением регуляции JAK1. Настоящее изобретение также относится к пролекарствам и фармацевтическим композициям, содержащим указанные модуляторы JAK1, а также способам лечения и/или предотвращения таких заболеваний и состояний.
В первом аспекте изобретение относится к соединению формулы I, обладающему структурой:

или его фармацевтически приемлемой соли, где R1 и R2 независимо представляют собой водород или гидроксильную группу; где если R1 представляет собой водородом, то R2 является гидроксильной группой; и где если R2 представляет собой водород, то R1 является гидроксильной группой.
Ниже описаны варианты осуществления (Е) этого первого аспекта изобретения, где для удобства Е1 идентичен этому аспекту.
Е1. Соединение формулы I, обладающее структурой:

или его фармацевтически приемлемая соль, где R1 и R2 независимо представляют собой водород или гидроксильную группу; где если R1 представляет собой водород, тогда R2 является гидроксильной группой; и если R2 представляет собой водород, тогда R1 является гидроксильной группой.
Е2. Соединение согласно Е1 или его фармацевтически приемлемая соль, где R1 представляет собой гидроксильную группу и R2 представляет собой водород.
Е3. Соединение согласно Е1 или его фармацевтически приемлемая соль, где R1 представляет собой водород и R2 представляет собой гидроксильную группу.
Е4. Соединение формулы IA, обладающее структурой:

или его фармацевтически приемлемая соль.
Е5. Соединение формулы IB, обладающее структурой:

или его фармацевтически приемлемая соль.
Е6. Соединение, выбранное из группы, состоящей из следующих соединений:
(S) -2-гидрокси-N-(3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид;
3-гидрокси-N-(3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид;
или их фармацевтически приемлемые соли.
Е7. (S)-2-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид или его фармацевтически приемлемая соль.
Е8. 3-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид или его фармацевтически приемлемая соль.
Е9. (S)-2-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид.
Е10. 3-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид.
Е11. Соединение по любому из вариантов осуществления с Е1 по Е10 в изолированной форме.
Е12. Соединение по любому из вариантов осуществления с Е1 по E11 в кристаллической форме.
Е13. Способ лечения или предотвращения заболевания или состояния, для которого показан ингибитор JAK1, у субъекта, нуждающегося в таком лечении, который включает введение субъекту терапевтически эффективного количества соединения по любому из вариантов осуществления с E1 по Е12 или его фармацевтически приемлемой соли.
Е14. Способ лечения или предотвращения воспалительного или аутоиммунного состояния, включающий введение субъекту, страдающему таким состоянием, терапевтически эффективного количества соединения согласно любому из вариантов осуществления с E1 по Е12 или его фармацевтически приемлемой соли.
Е15. Фармацевтическая композиция, включающая соединение согласно любому из вариантов осуществления с E1 по Е12 и фармацевтически приемлемый эксципиент.
Е16. Способ лечения или предотвращения заболевания или состояния, выбранного из воспаления, аутоиммунного заболевания, нейровоспаления, артрита, ревматоидного артрита, спондилоартропатий, системной красной волчанки, волчаночного нефрита, остеоартрита, подагрического артрита, боли, лихорадки, саркоидоза легких, силикоза, сердечно-сосудистого заболевания, атеросклероза, инфаркта миокарда, тромбоза, застойной сердечной недостаточности и реперфузионного повреждения сердца, кардиомиопатии, инсульта, ишемии, реперфузионного повреждения, отека головного мозга, травмы головного мозга, нейродегенерации, заболевания печени, воспалительного заболевания кишечника, болезни Крона, язвенного колита, нефрита, ретинита, ретинопатии, дегенерации желтого пятна, глаукомы, диабета (1 и 2 типа), диабетической невропатии, вирусной и бактериальной инфекции, миалгии, эндотоксического шока, синдрома токсического шока, остеопороза, рассеянного склероза, эндометриоза, менструальных спазм, вагинита, кандидоза, рака, фиброза, ожирения, мышечной дистрофии, полимиозита, дерматомиозита, аутоиммунного гепатита, первичного билиарного цирроза, первичного склерозирующего холангита, витилиго, болезни Альцгеймера, гиперемии кожи, экземы, псориаза, атопического дерматита, солнечных ожогов, келоидных гипертрофических рубцов, ревматических заболеваний, крапивницы, дискоидной волчанки, кожной волчанки, волчанки центральной нервной системы, псориатического артрита, астмы, аллергической астмы, интерферонопатии I типа, включая синдром Айкарди-Гутьереса и другие менделевские заболевания, связанные с избыточной экспрессией интерферона I типа, первичного прогрессирующего рассеянного склероза, рецидивирующего ремиттирующего рассеянного склероза, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, склеродермии, очаговой алопеции, рубцовой алопеции, почесухи, узловатой почесухи, CPUO, болезней, вызываемых лишаями, красного плоского лишая, синдрома Стивена-Джонсона, спондилопатии, миозита, васкулита, пузырчатки, волчанки, большого депрессивного расстройства, аллергии, синдрома сухого глаза, отторжения трансплантата, рака, септического шока, сердечно-легочной дисфункции, острого респираторного заболевания, анкилозирующего спондилита, кахексии, хронического заболевания «трансплантат против хозяина», острого заболевания «трансплантат против хозяина», целиакии-спру, идиопатической тромбоцитопенической тромботической пурпуры, тромботической тромбоцитопенической пурпуры, миастении гравис, синдрома Шегрена, эпидермальной гиперплазии, воспаления хряща, дегенерации костной ткани, ювенильного артрита, ювенильного ревматоидного артрита, олигоартикулярного ювенильного ревматоидного артрита, полиартикулярного ювенильного ревматоидного артрита, ювенильного ревматоидного артрита с системным началом, ювенильного анкилозирующего спондилита, ювенильного энтеропатического артрита, ювенильного синдрома Ретера, синдрома SEA, ювенильного дерматомиозита, ювенильного псориатического артрита, ювенильной склеродермии, ювенильной системной красной волчанки, ювенильного васкулита, олигоартикулярного ревматоидного артрита, полиартикулярного ревматоидного артрита, ревматоидного артрита с системным началом, энтеропатического артрита, реактивного артрита, синдрома Ретера, myolitis, polymyolitis, dermatomyolitis, узелкового полиартериита, гранулематоза Вегенера, артериита, ревматической полимиалгии, саркоидоза, склероза, первичного билиарного склероза, склерозирующего холангита, дерматита, болезни Стилла, хронической обструктивной болезни легких, болезни Гийена-Барре, болезни Грейвса, болезни Аддисона, феномена Рейно, псориатической эпидермальной гиперплазии, бляшечного псориаза, каплевидного псориаза, инверсного псориаза, пустулезного псориаза, эритродермического псориаза, иммунного расстройства, связанного с активностью патогенных лимфоцитов или возникающего в результате такой активности, неинфекционного увеита, болезни Бехчета и синдрома Фогта-Коянаги-Харады, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из вариантов осуществления с E1 по Е12.
Е17. Способ лечения или предотвращения псориаза, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества композиции, включающей соединение или его фармацевтически приемлемую соль по любому из вариантов осуществления с E1 по Е12.
Е18. Способ лечения или предотвращения атопического дерматита, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества композиции, включающей соединение или его фармацевтически приемлемую соль по любому из вариантов осуществления с E1 по Е12.
Е19. Способ лечения или предотвращения экземы рук, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из вариантов осуществления с E1 по Е12.
Е20. Способ лечения или предотвращения зуда, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из вариантов с E1 по Е12.
Е21. Способ лечения или предотвращения кожной волчанки, включающий стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из вариантов осуществления с E1 по Е12.
Е22. Способ лечения расстройства или состояния, связанного с нарушением регуляции JAK, в частности JAK1, у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения или его фармацевтически приемлемой соли в по любому из вариантов осуществления с Е1 по Е12.
Е23. Способ согласно Е22, в котором расстройство или состояние, которое лечат или предотвращают, выбрано из группы, состоящей из воспаления, аутоиммунного заболевания, нейровоспаления, артрита, ревматоидного артрита, спондилоартропатий, системной красной волчанки, волчаночного нефрита, остеоартрита, подагрического артрита, боли, лихорадки, саркоидоза легких, силикоза, сердечно-сосудистого заболевания, атеросклероза, инфаркта миокарда, тромбоза, застойной сердечной недостаточности и реперфузионного повреждения сердца, кардиомиопатии, инсульта, ишемии, реперфузионного повреждения, отека головного мозга, травмы головного мозга, нейродегенерации, заболевания печени, воспалительного заболевания кишечника, болезни Крона, язвенного колита, нефрита, ретинита, ретинопатии, дегенерации желтого пятна, глаукомы, диабета (1 и 2 типа), диабетической невропатии, вирусной и бактериальной инфекции, миалгии, эндотоксического шока, синдрома токсического шока, остеопороза, рассеянного склероза, эндометриоза, менструальных спазм, вагинита, кандидоза, рака, фиброза, ожирения, мышечной дистрофии, полимиозита, дерматомиозита, аутоиммунного гепатита, первичного билиарного цирроза, первичного склерозирующего холангита, витилиго, болезни Альцгеймера, гиперемии кожи, экземы, псориаза, атопического дерматита, солнечных ожогов, келоидных гипертрофических рубцов, ревматических заболеваний, крапивницы, дискоидной волчанки, кожной волчанки, волчанки центральной нервной системы, псориатического артрита, астмы, аллергической астмы, интерферонопатий I типа, включая синдром Айкарди-Гутьереса и другие менделевские заболевания, связанные с избыточной экспрессией интерферона I типа, первичного прогрессирующего рассеянного склероза, рецидивирующего ремиттирующего рассеянного склероза, неалогольной жировой болезни печени, неалкогольного стеатогепатита, склеродермии, очаговой алопеции, спондилопатии, миозита, васкулита, пузырчатки, волчанки, большого депрессивного расстройства, аллергии, синдрома сухого глаза, отторжения трансплантата, рака, септического шока, сердечно-легочной дисфункции, острого респираторного заболевания, анкилозирующего спондилита, кахексии, хронического заболевания «трансплантат против хозяина», острого заболевания «трансплантат против хозяина», целиакии-спру, идиопатической тромбоцитопенической тромботической пурпуры, миастении гравис, синдрома Шегрена, эпидермальной гиперплазии, воспаления хряща, дегенерации костной ткани, ювенильного артрита, ювенильного ревматоидного артрита, олигоартикулярого ювенильного ревматоидного артрита, полиартикулярного ювенильного ревматоидного артрита, ювенильного ревматоидного артрита с системным началом, ювенильного анкилозирующего спондилита, ювенильного энтеропатического артрита, ювенильного синдрома Ретера, синдрома SEA, ювенильного дерматомиозита, ювенильного псориатического артрита, ювенильной склеродермии, ювенильной системной красной волчанки, ювенильного васкулита, олигоартикулярного ревматоидного артрита, полиартикулярного ревматоидного артрита, ревматоидного артрита с системным началом, энтеропатического артрита, реактивного артрита, синдрома Ретера, myolitis, polymyolitis, dermatomyolitis, узелкового полиартериита, гранулематоза Вегенера, артериита, ревматической полимиалгии, саркоидоза, склероза, первичного билиарного склероза, склерозирующего холангита, дерматита, болезни Стилла, хронической обструктивной болезни легких, болезни Гийена-Барре, болезни Грейвса, болезни Аддисона, феномена Рейно, псориатической эпидермальной гиперплазии, бляшечного псориаза, каплевидного псориаза, инверсного псориаза, пустулезного псориаза, эритродермического псориаза, иммунного расстройства, связанного с активностью патогенных лимфоцитов или возникающее в результате такой активности, неинфекционного увеита, болезни Бехчета и синдрома Вогта-Коянаги-Харады.
Е24. Способ согласно вариантам осуществления с Е13 по Е23, в котором терапевтически эффективное количество составляет от 0,01 мг/кг массы тела в сутки до 100 мг/кг массы тела в сутки.
Е25. Способ согласно Е24, в котором терапевтически эффективное количество составляет от 0,1 мг/кг массы тела в сутки до 10 мг/кг массы тела в сутки.
Е26. Применение соединения по любому из вариантов осуществления с E1 по Е12 для производства лекарственного средства для лечения расстройства, при котором показан ингибитор JAK1.
Е29. Соединение по любому из вариантов осуществления с Е1 по Е12 для применения в лечении расстройства, при котором показан ингибитор JAK1.
Е30. Применение соединения в изолированной форме по любому из вариантов осуществления с E1 по Е12 для производства лекарственного средства для лечения расстройства, при котором показан ингибитор JAK1.
Е31. Соединение в изолированной форме по любому из вариантов осуществления с E1 по Е12 для применения в лечении расстройства, при котором показан ингибитор JAK1
Е32. Фармацевтическая комбинация, включающая соединение по любому из вариантов осуществления с E1 по Е12 или его фармацевтически приемлемую соль и одно или несколько дополнительных фармакологически активных соединений.
Соединения или соединения в изолированной форме, у которых молекулярные формулы одинаковые, но разная природа или последовательность соединения атомов или разное расположение атомов в пространстве, называются «изомерами». Изомеры, которые отличаются расположением своих атомов в пространстве, называются «стереоизомерами». Специалистам в данной области будет понятно, что соединение или соединение в изолированной форме формулы I, IA или IB могут существовать в виде цис- и транс- ахиральных диастереомеров. Примером является соединение или соединение в изолированной форме формулы 1С.
В объем описанных соединений и соединений в изолированной форме включены все изомеры (например, цис-, транс- или диастереомеры) только описанных здесь соединений, а также любые их смеси. Все эти формы, в том числе энантиомеры, диастереомеры, цис-, транс-, син-, анти-, сольваты (включая гидраты), таутомеры и их смеси, включены в описанные соединения или соединения в изолированной форме. Стереоизомерные смеси, например смеси диастереомеров, могут быть разделены на соответствующие изомеры известными подходящими способами разделения. Диастереомерные смеси, например, могут быть разделены на индивидуальные диастереомеры фракционированной кристаллизацией, хроматографией, распределением в растворителях и аналогичными методами. Это разделение может проводиться либо на уровне одного из исходных соединений, либо в самом соединении формулы I, IA или IB. Энантиомеры могут быть разделены через образование диастереомерных солей, например посредством образования соли с энантиомерно чистой хиральной кислотой, или хроматографией, например ВЭЖХ, используя хроматографические субстраты с хиральными лигандами.
При терапевтическом применении для лечения расстройств у субъекта соединения или соединения в изолированной форме по настоящему изобретению или их фармацевтические композиции могут вводиться перорально, парентерально, местно, ректально, трансмукозально или интерстинально. Парентеральное введение включает непрямые инъекции для получения системного эффекта или прямые инъекции в пораженную область. Местное применение включает обработку кожи или органов, доступных для местного применения, например глаз или ушей. Оно также включает трансдермальную доставку для получения системного эффекта. Ректальное введение включает введение в форме суппозиториев. Предпочтительными способами введения являются пероральный и парентеральный.
Фармацевтически приемлемые соли соединений или соединений в изолированной форме формулы I, IA или IB включают их кислотно-аддитивные соли и соли оснований. Подходящие кислотно-аддитивные соли получают из кислот, которые образуют нетоксичные соли. Примеры кислотно-аддитивных солей включают ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат (naphthylate), 2-напсилат (2-napsylate), никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат.
Подходящие соли оснований получают из оснований, которые образуют нетоксичные соли. Примеры таких солей включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглюмина, оламина, калия, натрия, трометамина и цинка.
Могут быть получены также полусоли кислот и оснований, например гемисульфатные и гемикальциевые соли. Обзор подходящих солей см. в справочнике Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Фармацевтически приемлемые соли соединений и соединений в изолированной форме формулы I, IA или IB могут быть получены, соответственно, одним или более из трех способов: (i) взаимодействием соединения или соединения в изолированной форме формулы I, IA или IB с желаемой кислотой или основанием; (ii) удалением защитной группы, которое может проходить под действием кислоты или основания, из подходящего предшественника соединения или соединения в изолированной форме формулы I, IA или IB или раскрытием кольца подходящего циклического предшественника, например лактона или лактама, с использованием желаемой кислоты или основания; или (iii) превращением одной соли соединения или соединения в изолированной форме формулы I, IA или IB в другую взаимодействием с соответствующей кислотой или основанием или с помощью подходящей ионообменной колонки. Все три реакции обычно проводят в растворе. Образующаяся соль может выпадать в осадок и собираться фильтрованием или может быть выделена выпариванием растворителя. Степень ионизации в полученной соли может варьироваться от полностью ионизированной до почти неионизированной.
Фармацевтические композиции по настоящему изобретению могут быть получены способами, хорошо известными в данной области, например обычными методами смешивания, растворения, гранулирования, дражирования, измельчения, эмульгирования, инкапсулирования, улавливания, лиофилизации или распылительной сушки.
Фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть получены обычным способом с использованием одного или нескольких фармацевтически приемлемых носителей, включая эксципиенты и вспомогательные вещества, которые облегчают технологическую обработку активного соединения или соединения в изолированные формы для получения препаратов, которые могут быть использованы фармацевтически. Выбор подходящего препарата определяет выбор способа введению. Фармацевтически приемлемые эксципиенты и носители, как правило, известны специалистам в данной области и, таким образом, включены в настоящее изобретение. Такие вспомогательные вещества и носители описаны, например, в публикации "Remington's Pharmaceutical Sciences" Mack Pub. Co., New Jersey (1991). Препараты по настоящему изобретению могут разрабатываться в форме препаратов короткого действия, быстро высвобождающихся препаратов, препаратов длительного действия и препаратов с замедленным высвобождением действующего вещества. Таким образом, фармацевтические композиции также могут быть разработаны для контролируемого высвобождения или для медленного высвобождения действующего вещества.
Фармацевтические композиции, подходящие для применения в настоящем изобретении, включают композиции, в которых активные ингредиенты содержатся в количестве, достаточном для достижения заданной цели, т.е. для контроля или лечения расстройств или заболеваний. Точнее, термин «терапевтически эффективное количество» означает количество соединения или соединения в изолированной форме, эффективное для предотвращения, облегчения или ослабления симптомов/признаков заболевания или повышения выживаемости субъекта, который получает лечение.
Количество активного компонента, которым является соединение или соединение в изолированной форме по настоящему изобретению, в фармацевтической композиции и ее стандартной лекарственной форме может изменяться или регулироваться в широких пределах в зависимости от способа введения, эффективности конкретного соединения или соединения в изолированной форме и желаемой концентрации. Специалист в данной области техники сможет определить терапевтически эффективное количество. Как правило, количество активного компонента будет находиться в интервале от 0,01 % до 99 % по массе композиции.
Обычно, терапевтически эффективное количество дозы активного компонента будет находиться в интервале от примерно 0,01 до примерно 100 мг/кг массы тела в сутки, предпочтительно от примерно 0,1 до примерно 10 мг/кг массы тела в сутки, более предпочтительно от примерно 0,3 до 3 мг/кг массы тела в сутки, еще более предпочтительно примерно от 0,3 до 1,5 мг/кг массы тела в сутки. Следует иметь в виду, что дозы могут изменяться в зависимости от потребностей каждого субъекта и тяжести расстройств или заболеваний, подлежащих лечению.
Желаемая доза может быть удобно представлена в виде разовой дозы или разделяться на несколько доз, вводимых с соответствующими интервалами, например на две, три, четыре или более частей суточной дозы. Сама часть суточной дозы может быть дополнительно поделена, например, на ряд отдельных введений с небольшим интервалом, таких как многократные ингаляции из инсуффлятора или введение множества капель в глаз.
Следует также иметь в виду, что вводимая начальная доза может быть повышена относительно вышеуказанного верхнего уровня для быстрого достижения желаемой концентрации в плазме. С другой стороны, начальная доза может быть меньше оптимальной, и суточная доза может постепенно увеличиваться в течение курса лечения в зависимости от конкретной ситуации. При желании суточная доза также может разделяться на несколько доз для введения, например от двух до четырех раз в сутки.
Соединения и соединения в изолированной форме по настоящему изобретению могут вводиться в фармацевтически приемлемой форме либо отдельно, либо в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающих, или противовоспалительными средствами. Эти средства могут включать, но без ограничения представленным перечнем, антагонист белка, активирующего 5-липоксигеназу (FLAP); антагонист лейкотриена (LTRA), такой как антагонист LTB4, LTC4, LTD4, LTE4, CysLT1 или CysLT2, например монтелукаст или зафирлукаст; антагонист рецептора гистамина, такой как антагонист рецептора гистамина 1 типа или антагонист рецептора гистамина 2 типа, например лоратидин, фексофенадин, дезлоратидин, левоцетиризин, метапирилен или цетиризин; агонист al-адренорецепторов или агонист α2-адренорецепторов, например фенилэфрин, метоксамин, оксиметазолин или метилнорефрин; мускариновый М3 антагонист рецептора, например тиотропий или ипратропий; двойной антагонист мускаринового М3-рецептора/β2-агонист; ингибитор фосфодиэстеразы (ФДЭ), такой как ингибитор ФДЭЗ, ингибитор ФДЭ4 или ингибитор ФДЭ5, например теофиллин, силденафил, варденафил, тадалафил, ибудиласт, циломиласт или рофлумиласт; кромогликат натрия или недокромил натрия; ингибитор циклооксигеназы (ЦОГ), такой как неселективный ингибитор (например, аспирин или ибупрофен) или селективный ингибитор (например, целекоксиб или вальдекоксиб); глюкокортикостероид, например флутиказон, мометазон, дексаметазон, преднизолон, будесонид, циклесонид или бекламетазон; противовоспалительное моноклональное антитело, например инфликсимаб, адалимумаб, танезумаб, ранибизумаб, бевацизумаб или меполизумаб; β2-агонист, например сальметерол, альбутерол, сальбутамол, фенотерол или формотерол, в частности β2-агонист длительного действия; антагонист интегрина, например натализумаб; ингибитор адгезии молекул, такой как антагонист VLA-4; антагонист рецептора кинина В1 или В2; иммуносупрессивное средство, такое как ингибитор пути IgE (например, омализумаб) или циклоспорин; ингибитор матриксной металлопротеазы (ММП), такой как ингибитор ММП-9 или ММП-12; антагонист рецептора тахикинина NK1, NK2 или NK3; ингибитор протеазы, такой как ингибитор эластазы, химазы или катеопсина G; агонист рецептора аденозина А2а; антагонист рецептора аденозина A2b; ингибитор урокиназы; агонист рецептора дофамина (например, ропинирол), в частности агонист рецептора дофамина D2 (например, бромокриптин); модулятор пути NFkB, такой как ингибитор IKK; дополнительный модулятор пути передачи сигналов цитокинов, такой как ингибитор JAK-киназы, syk-киназы, р38-киназы, SPHK-1-киназы, Rho-киназы, EGF-R или MK-2; муколитическое, мукокинетическое или противокашлевое средство; антибиотик; противовирусное средство; вакцину; хемокин; блокатор эпителиальных натриевых каналов (ENaC) или ингибитор эпителиальных натриевых каналов (ENaC); агонист нуклеотидного рецептора, такой как агонист P2Y2; ингибитор тромбоксана; ниацин; ингибитор 5-липоксигеназы (5-LO), например зилеутон; фактор адгезии, такой как VLAM, ICAM или ELAM; антагонист рецептора CRTH2 (DP2); антагонист рецептора простагландина D2 (DPI); ингибитор гемопоэтической простагландин-D2-синтазы (HPGDS); интерферон-β; растворимый TNF рецептор человека, например этанерцепт; ингибитор HDAC; ингибитор фосфоинозитотид-3-киназы-гамма (PI3Kγ); ингибитор фосфоинозитотид 3-киназы дельта (PI3Kδ); антагонист рецептора CXCR-1 или CXCR-2; ингибитор IRAK-4; и ингибитор TLR-4 или TLR-9, включая фармацевтически приемлемый соли конкретно названных соединений. Указанные средства могут вводиться с другим активным агентом, причем второй активный агент может вводиться перорально или местно.
Подходящими конкретными лекарственными средствами для применения в комбинированной терапии с соединением или соединением в изолированной форме формулы I, IA или IB или их фармацевтически приемлемыми солями являются сульфасалазин, месалазин, преднизон, азатиоприн, инфликсимаб, адалимумаб, белимумаб, бецертолизумаб (becertolizumab), натализумаб, ведолизумаб, гидрокортизон, будесонид, циклоспорин, такролимус, фексофенадин, 6-меркаптопурин, метотрексат, урсодезоксихолевая кислота, обетихолевая кислота, антигистаминные препараты, рифампицин, преднизолон, метотрексат, азатиоприн, циклофосфамид, гидроксихлорохин, мофетил, микофенолат натрия, такролимус, лефлуномид, хлорохин и хинакрин, талидомид, ритуксан, НПВП, солумедрол, депомедрол и дексаметазон.
Химический синтез
На приведенных далее схемах и в их описании представлены общие сведения, касающиеся получения соединений по настоящему изобретению.
Схема 1

Способ синтеза, описанный в примере 1 и примере 2, представлен на схеме 1. Первичный амин (J. Med. Chem. 2018, 61(3), 1130-1152) подвергается обработке 2-оксо-1-пропансульфонилхлоридом (полученным в две стадии из хлорацетона) в дихлорметане, содержащем триэтиламин. Полученный сульфонамид после этого подвергается обработке борогидридом натрия в метаноле для восстановления кетона с получением изомерной смеси вторичных спиртов. После этого из пирролопиримидинового кольца удаляют защитную тозильную группу. Далее вторичные спирты разделяют с помощью сверхкритической флюидной хроматографии (SFC с получением соединения примера 1 (пик 1), соответствующего соединению IA, и соединения примера 2 (пик 2) в описанных условиях.
Альтернативный способ получения соединения примера 1 представлен на схеме 2.
Схема 2

В (S)-1-хлор-2-пропанол с использованием трет-бутилдиметилсилилхлорида в диметилформамиде в качестве растворителя и пиридина в качестве основания вводится защитная группа с получением простого силилового эфира. Первичный хлорид подвергается реакции замещения с использованием тиоацетата калия с получением сложного тиоэфира. Тиоэфир подвергается окислению газообразным хлором с получением сульфонилхлорида. В условиях реакции может одновременно удаляться силильная защитная группа. цис-N-Метил-N-{7-[(4-метилфенил)сульфонил]-7Н-пирроло[2,3-d]пиримидин-4-ил}циклобутан-1,3-диамин подвергается взаимодействию с сульфонилхлоридом с получением сульфонамида. После этого из пирролопиримидина удаляется защитная тозильная группа с использованием гидроксида лития в смеси воды и тетрагидрофурана с получением после очистки с помощью SFC соединения IA (пример 1).
Схема 3
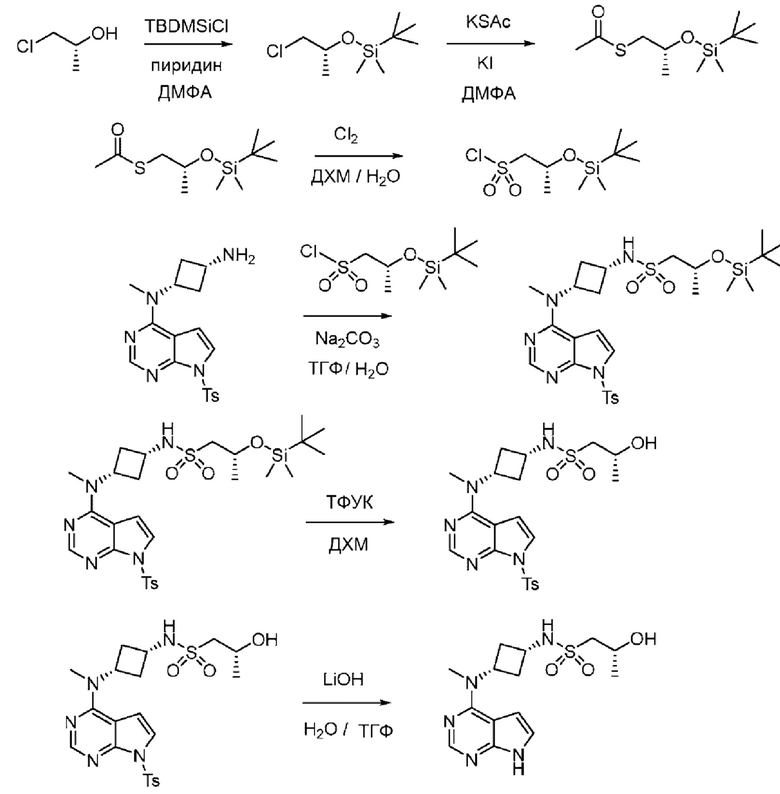
В (R)-1-Хлор-2-пропанол с использованием трет-бутилдиметилсилилхлорида с добавлением диметилформамида в качестве растворителя и пиридина в качестве основания вводится защитная группа с получением простого силилового эфира. Первичный хлорид подвергается реакции замещения с тиоацетатом калия с получением сложного тиоэфира. Тиоэфир подвергается окислению газообразным хлором с получением сульфонилхлорида; наблюдается некоторая потеря силильной защитной группы. цис-N-Метил-N-{7-[(4-метилфенил)сульфонил]-7Н-пирроло[2,3-d]пиримидин-4-ил}циклобутан-1,3-диамин подвергается взаимодействию с сульфонилхлоридом с получением сульфонамида. Силильная защитная трупа удаляется с помощью трифторуксусной кислоты с получением вторичного спирта. Далее из пирролопиримидина с помощью гидроксида лития в смеси воды и тетрагидрофурана удаляется защитная тозильная группа с получением после очистки SFC соединения примера 2.
Получение соединения IB представлено на схеме 4 и описано в примере 5. цис-N-Метил-N-{7-[(4-метилфенил) сульфонил]-7Н-пирроло[2,3-d]пиримидин-4-ил}циклобутан-1,3-диамин подвергается взаимодействию с метил-3-(хлорсульфонил)пропаноатом с получением сульфонамида. Сульфонамид подвергается восстановлению алюмогидридом лития с получением 3-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (IB).

Альтернативное получение соединения IB представлено на схеме 5 и описано в примере 6. Дигидробромид цис-N-метил-N-{7-[(4-метилфенил)сульфонил]-7Н-пирроло[2,3-d]-пиримидин-4-ил}циклобутан-1,3-диамина подвергается обработке основанием, таким как триэтиламин, и этил-3-(хлорсульфонил)пропаноатом с получением этил-3-(N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)сульфамоил)пропаноата. Полученный сульфамоилпропаноат подвергается восстановлению алюмогидридом лития и последующей обработке с получением сырого 3-гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, который используют далее без дополнительной очистки. Полученный сульфонамид подвергается обработке основанием, таким как LiOH, с получением 3-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (IB).

Специалисту в данной области техники понятно, что при осуществлении синтеза соединений по изобретению необходим отбор проб и анализ реакционных смесей перед обработкой, чтобы отслеживать ход реакций и решать, следует ли продолжать реакцию или реакционная смесь готова для обработки с получением желаемого продукта. Стандартные методы анализа реакционных смесей включают тонкослойную хроматографию (ТСХ), жидкостную хроматографию/масс-спектроскопию (ЖХМС) и ядерный магнитный резонанс (ЯМР).
Специалисту в данной области также понятно, что соединения и соединения в изолированной форме по изобретению могут быть получены в виде смесей диастереомеров или геометрических изомеров (например, в результате цис- и транс-замещения на циклоалкановом кольце). Эти изомеры могут быть разделены стандартными хроматографическими методами, такими как нормально-фазовая хроматография на силикагеле, препаративная жидкостная хроматография высокого давления с обращенной фазой или сверхкритическая жидкостная хроматография (supercritical fluid chromatography - SFC). Специалисту в данной области также понятно, что некоторые соединения по изобретению являются хиральными и, таким образом, могут быть получены в виде рацемических или скалемических смесей энантиомеров. Существует несколько способов разделения энантиомеров, которые хорошо известны специалистам в данной области. Предпочтительным методом обычного разделения энантиомеров является сверхкритическая жидкостная хроматография с использованием хиральной стационарной фазы.
ЭКСЕРИМЕНТАЛЬНАЯ ЧАСТЬ
За исключением особо оговоренных случаев, реакции проводят в атмосфере азота. Хроматографию на силикагеле проводят с использованием силикагеля 250-4 00 меш и азота под давлением (~10-15 фунтов на кв. дюйм (68,95-103,43 кПа)) для пропускания растворителя через колонку («флэш-хроматография»). Когда это указано, растворы и реакционные смеси концентрируют с помощью роторного испарения в вакууме.
Получение и примеры
Получение 1. Натриевая соль 2-оксопропан-1-сульфоновой кислоты

Смесь хлорацетона (9,25 г, 100 ммоль) и сульфита натрия (14,5 г, 115 ммоль) в воде (100 мл) перемешивают в течение 24 часов. Полученную смесь упаривают досуха и суспендируют твердый остаток в метаноле (300 мл). Смесь обрабатывают ультразвуком в течение 3 минут, затем фильтруют. Осадок на фильтре промывают метанолом и фильтраты концентрируют с получением указанного в заголовке соединения (15,5 г), которое используют далее без дополнительной очистки.
Получение 2. 2-Оксопропан-1-сульфонилхлорид
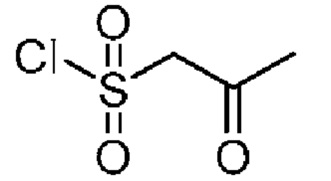
К перемешиваемой суспензии натриевой соли 2-оксопропан-1-сульфоновой кислоты (15,5 г, 97 ммоль) в сухом толуоле (40 мл) добавляют оксихлорид фосфора (40 мл). Смесь кипятят с обратным холодильником в течение 3 часов, затем нагрев удаляют и охлаждают смесь до 20°С. Смесь концентрируют при пониженном давлении и остаток обрабатывают дихлорметаном (100 мл). Смесь фильтруют и осадок на фильтре промывают дихлорметаном (20 мл). Фильтрат концентрируют с получением указанного в заголовке соединения (11,8 г). 1Н ЯМР (400 МГц, CDCl3-d) δ 4, 76-4, 53 (м, 2Н), 2,49 (с, 3Н).
Получение 3. цис-N-(3-(Метил-(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)-2-оксопропан-1-сульфонамид
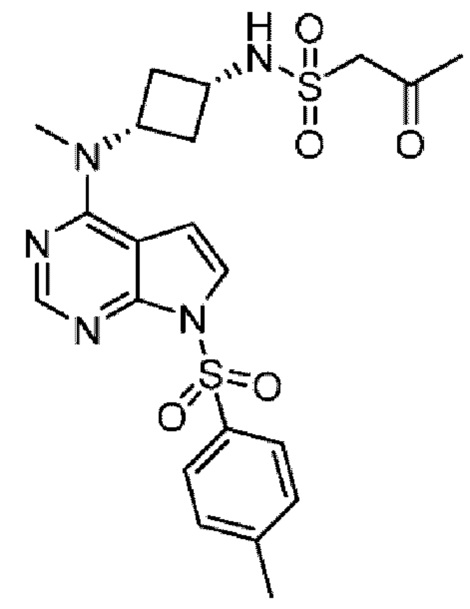
К раствору цис-N1-метил-N1-(7-тозил-7Н-пирроло [2,3-d]пиримидин-4-ил)циклобутан-1,3-диамина (2,4 г, 5,3 ммоль) в дихлорметане (45 мл) добавляют триэтиламин (2,68 г, 26,5 ммоль). К смеси при 0°С добавляют раствор 2-оксопропан-1-сульфонилхлорида (1,24 г, 6,27 ммоль) в дихлорметане (5 мл), затем смеси дают возможность нагреться до 20°С и перемешивают смесь в течение 3 часов. Полученную реакционную смесь гасят насыщенным раствором хлорида аммония (30 мл). Слои разделяют, водный слой дважды экстрагируют дихлорметаном (2×10 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (петролейный эфир : этилацетат, 1:2) с получением указанного в заголовке соединения (801 мг). ЖХ/МС [М+Н]=491,9.
Получение 4. 2-Гидрокси-N-{цис-3-(метил-(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
К раствору N-(3-{метил[7-(4-метилбензол-1-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]амино}циклобутил)-2-оксопропан-1-сульфонамида (0,801 г, 1,62 ммоль) в метаноле (10 мл) при 0°С добавляют борогидрид натрия (123 мг, 2,26 ммоль) в 10 % растворе гидроксида натрия (1,6 мл) в метаноле (1,6 мл). Смесь перемешивают в течение 30 минут, затем охлаждающую баню удаляют и перемешивают смесь при 20°С в течение 30 минут. Метанол удаляют при пониженном давлении. Остаток растворяют в этилацетате (15 мл), промывают насыщенным раствором соли (10 мл), сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (0-80 % этилацетата в петролейном эфире) с получением указанного в заголовке соединения (0,76 г). ЖХ/МС [М+Н]=494,2.
Получение 5. 2-Гидрокси-N-(цис-3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
К смеси 2-гидрокси-]-N-(цис-3-{метил-[7-(4-метилбензол-1-сульфонил)-7Н-пирроло[2,3-d]пиримидин-4-ил]амино}циклобутил)пропан-1-сульфонамида (1,13 г, 2,3 ммоль), тетрагидрофурана (10 мл) и воды (10 мл) при 20°С в течение 24 часов добавляют моногидрат гидроксида лития (273 мг, 11,4 ммоль). Смесь концентрируют и очищают флэш-хроматографией (0-10 % метанола в дихлорметане). Реакционную смесь растворяют в смеси вода/ацетонитрил, затем лиофилизируют. Остаток очищают препаративной тонкослойной хроматографией (дихлорметан : метанол, 10:1). Остаток растворяют в смеси вода/ацетонитрил и лиофилизируют. После этого остаток очищают суперкритической жидкостной хроматографией (Chiralpak AD-H™ 250×30 мм I.D., 5 мкм, подвижная фаза: 45 % метанол (0,1 % NH4OH) в CO2, скорость потока 50 мл/мин., темп. 35°С, RT 3,53 и 4,07 мин.) с получением указанного в заголовке соединения (552 мг, 71 %). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,64 (уш. с, 1Н), 8,11 (с, 1Н), 7,44 (д, J=9,0 Гц, 1Н), 7,16-7,15 (м, 1Н), 6, 65-6, 64 (м, 1Н), 5,02-4,81 (м, 2Н), 4.15- 3,94 (м, 1Н), 3, 67-3,54 (м, 1Н), 3,26 (с, 3Н), 3,14-2,95 (м, 2Н), 2,71-2,56 (м, 2Н), 2,31-2,19 (м, 2Н), 1,22 (д, J=6,0 Гц, 3Н), ЖХ/МС [М+Н]=339,9.
Пример 1. (S)-2-Гидрокси-N-(цис-3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид (IA)

2-Гидрокси-N-{3-[метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино]циклобутил}пропан-1-сульфонамид (532 мг, 1,57 ммоль) очищают с помощью SFC ((Chiralpak AD-H™ 250×30 мм I.D., 5 мкл подвижная фаза: 45 % этанол (0,1 %) NH4OH) в СО2, скорость потока 50 мл/мин., темп. 35°С, RT 3,44 мин.) с получением указанного в заголовке соединения (223, 7 мг, 42 %). 1Н ЯМР (400 МГц, ДМСО-d6) δ=11, 69-11,57 (м, 1Н), 8,10 (с, 1Н), 7,43 (д, J=9,0 Гц, 1Н), 7.16-7,12 (м, 1Н), 6,67-6,61 (м, 1Н), 4,92 (д, J=5,0 Гц, 1Н), 4, 90-4,84 (м, 1Н), 4,11-4,01 (м, 1Н), 3,63-3,53 (м, 1Н), 3,27-3,23 (м, 3Н), 3,11-2,96 (м, 2Н), 2, 63-2,53 (м, 2Н), 2,29-2,20 (м, 2Н), 1,21 (д, J=6,0 Гц, 3Н), ЖХ/МС [М+Н]=340,2.
Кристаллический продукт может быть получен из этилацетата.
Рентгеновская структура монокристалла
Сбор данных проводят на дифрактометре Bruker D8 Quest при комнатной температуре. Сбор данных включает омега- и фисканирования. Структуру определяют внутренним фазированием в триклинной пространственной группе Р1 с использованием пакета программ SHELX. Затем структуру уточняют методом наименьших квадратов в полноматричном приближении. Все атомы, не являющиеся атомами водорода, выявляют и их местоположение уточняют с использованием параметров анизотропного замещения.
Положения атомов водорода на атомах азота и кислорода определяют из разностной карты Фурье и уточняют с ограничением расстояний. Положения остальных атомов водорода рассчитывают и делают допущение их связи с атомами, которые являются их носителями. Конечное уточнение включает параметры изотропного замещения для всех атомов водорода. Анализ абсолютной структуры с использованием метода правдоподобия (Hooft 2008) проводят с помощью PLATON (Spek 2 010). При условии, что представленный образец является инантиомерно чистым, результаты показывают, что абсолютная структура определена правильно. Расчет с помощью указанного метода показывает, что вероятность правильного определения структуры составляет 100 %. Параметр Хоофта составляет 0,104 при Esd (15), параметр Парсона составляет 0,097 при Esd (16). Абсолютную конфигурацию при С13 и С2 7 для двух идентичных молекул на асимметрическую ячейку подтверждают как (-S) (-S). Конечный R-индекс составляет 5,9 %. Конечная разность Фурье не показывает отсутствующей или неуместной электронной плотности. Основные сведения о кристалле, совокупность полученных данных и уточнение представлены в таблице 1. Координаты атомов представлены в таблице 2.
Программное обеспечение и ссылки
SHELXTL, Version 5.1, Bruker AXS, 1997. PLATON, A.L. Spek, J. Appl. Cryst. 2003, 36, 7-13. MERCURY, C.F. Macrae, P.R. Edington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst. 39, 453-457, 2006. OLEX2, Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H., (2009). J. Appl. Cryst., 42, 339-341. R.W.W. Hooft et al. J. Appl. Cryst. (2008). 41. 96-103. H.D. Flack, Acta Cryst. 1983, A39, 867-881.




Пример 2. (R)-2-Гидрокси-N-(цис-3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид (IC)

2-Гидрокси-N-{3-[метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино]циклобутил}пропан-1-сульфонамид (532 мг, 1,57 ммоль) очищают с помощью SFC ((Chiralpak AD-H™ 250×30 мм I.D., 5 мкм, подвижная фаза: 45 % этанол (0,1 %) NH4OH) в CO2, скорость потока 50 мг/мин., темп. 35°С, RT, 3,88 мин.) с получением указанного в заголовке соединения (222, 7 мг, 42 %). 1Н ЯМР (400 МГц, ДМСО-d6) δ=11, 69-11,56 (м, 1Н), 8,10 (с, 1Н), 7,43 (д, J=9,0 Гц, 1Н), 7,18-7,11 (м, 1Н), 6, 66-6, 60 (м, 1Н), 4, 96-4, 84 (м, 2Н), 4,12-3,99 (м, 1Н), 3, 66-3,54 (м, 1Н), 3,24 (с, 3Н), 3,12-2,94 (м, 2Н), 2, 64-2, 56 (м, 2Н), 2,30-2,18 (м, 2Н), 1,20 (д, J=6,5 Гц, 3Н), ЖХ/МС [М+Н]=340,2. Кристаллический продукт может быть получен из этилацетата.
Рентгеновская структура монокристалла
Сбор данных проводят на дифрактометре Bruker D8 Quest при комнатной температуре. Сбор данных состоит из омега- и фи-сканирований. Этот лот дает пластинчатые кристаллы, сложенные вместе, кристалл отделяют и монтируют для сбора данных. Структуру определяют внутренним фазированием в триклинной пространственной группе Р1 с использованием пакета программ SHELX. Затем структуру уточняют методом наименьших квадратов в полноматричном приближении. Все атомы, не являющиеся атомами водорода, выявляют и уточняют их местоположение с использованием параметров анизотропного замещения. Положения атомов водорода на атомах азота и кислорода определяют из карты разностей Фурье и уточняют ограничением расстояний. Положения остальных атомов водорода рассчитывают и делают допущение об их связи с атомами, которые являются их носителями. Конечное уточнение включает параметры изотропного замещения для всех атомов водорода. Анализ абсолютной структуры с использованием методов правдоподобия (Hooft 2008) проводят с помощью PLATON (Spek 2010). При условии, что представленный образец является инантиомерно чистым, результаты показывают, что абсолютная структура определена правильно. Расчет с помощью указанного метода показывает, что вероятность правильного определения структуры составляет 100 %. Параметр Хоофта составляет 0,044 при Esd (18), параметр Парсона составляет 0,050 при Esd (19). Абсолютную конфигурацию при С13 и С27 для двух идентичных молекул на асимметричную ячейку подтверждают как (-R)_(-R). Псевдо-симметрия к Р-1. Конечный R-индекс составляет 4,5 %. Конечная разность Фурье не показывает отсутствующей или неуместной электронной плотности. Основные сведения о кристалле, совокупность полученных данных и уточнение представлены в таблице 3. Координаты атомов представлены в таблице 4.
Программное обеспечение и ссылки
SHELXTL, Version 5.1, Bruker AXS, 1997. PLATON, A.L. Spek, J. Appl. Cryst. 2003, 36, 7-13. MERCURY, C.F. Macrae, P.R. Edington, P. McCabe, E. Pidcock, G.P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Cryst.39, 453-457, 2006.OLEX2, Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.А.К.; Puschmann, H., (2009). J. Appl. Cryst., 42, 339-341. R.W.W. Hooft et al. J. Appl. Cryst. (2008). 41. 96-103. H.D. Flack, Acta Cryst. 1983, A39, 867-881.




Пример 3 - Альтернативное получение (S)-2-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (IA)
Стадия 1. (S)-трет-Бутил-((1-хлорпропан-2-ил)окси)диметилсилан
К раствору (S)-(+)-1-хлорпропан-2-ола (1,0 г, 10,6 ммоль) в N, N-диметилформамиде (20 мл) при 20°С добавляют пиридин (1,0 г, 12,7 ммоль) и трет-бутилдиметилсилилхлорид (1,91 г, 12,7 ммоль). После перемешивания при 20°С реакционную смесь разбавляют водой (40 мл). Полученную смесь экстрагируют метил-грет-бутиловым эфиром (2×40 мл). Органические экстракты объединяют, промывают насыщенным раствором соли (50 мл) и сушат (Na2SO4). Растворитель удаляют с получением указанного в заголовке соединения (2,2 г), которое используют далее без дополнительной очистки. 1Н ЯМР (4 00 МГц, CDCl3) δ 3,96 (м, 1Н), 3,42 (дд, J=10,8, 5,9 Гц, 1Н), 3,34 (дд, J=10,7, 5,9 Гц, 1Н), 1,23 (д, J=6,1 Гц, 3Н), 0,89 (с, 9Н), 0,08 (д, J=4,1 Гц, 6Н).
Стадия 2. (S)-S-(2-((трет-Бутилдиметилсилил)окси)пропил) этантиоат
К раствору (S)-трет-бутил((1-хлорпропан-2-ил)окси)диметилсилан (2,2 г, 10,5 ммоль) в N,N-диметилформамид (20 мл) при 20°С добавляют тиоацетат калия (2,41 г, 21,1 ммоль) и йодид калия (17,5 мг, 0,10 ммоль). Реакционную смесь нагревают до 80°С и перемешивают в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой (50 мл) и экстрагируют полученную смесь метил-грег-бутиловым эфиром (40 мл × 2). Органические экстракты охлаждают, промывают насыщенным раствором соли (60 мл), сушат (Na2SO4) и растворитель удаляют с получением указанного в заголовке соединения (2,40 г), которое используют далее без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 3,91 (м, 1Н), 3,00-2,91 (м, 2Н), 2,33 (с, 3Н), 1,18 (д, J=6,1 Гц, 3Н), 0,89 (с, 9Н), 0,07 (д, J=6,8 Гц, 6Н).
Стадия 3. (S)-2-Гидроксипропан-1-сульфонилхлорид
Cl2 (газ.) пропускают через раствор (S)-S-(2-((трет-бутилдиметилсилил)окси)пропил)этантиоата (0,5 г, 2,01 ммоль) в дихлорметане (20 мл) и воде (10 мл) в течение 5 минут при 0°С, в результате раствор приобретает желтый цвет. Поток Cl2 (газ.) останавливают и перемешивают реакционную смесь при 0°С в течение 5 часов. После этого N2 (газ.) барботируют через реакционную смесь с получением бесцветного раствора. ТСХ показывает израсходование исходного материала и появление нового пятна. К реакционной смеси добавляют воду (20 мл) и экстрагируют полученную смесь дихлорметаном (40 мл). Органический экстракт промывают 10 % NaHCO3 (50 мл), насыщенным раствором соли (50 мл) и сушат (Na2SO4). Растворитель удаляют с получением сырого указанного в заголовке соединения (0,6 г) в виде бесцветного масла, которое используют в следующей стадии без дополнительной очистки.
Стадия 4. (S)-2-Гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
В емкость объемом 40 мл добавляют цис-N1-метил-N1-(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)циклобутан-1,3-диамин (200 мг, 0,37 5 ммоль), Na2CO3 (199 мг, 1,8 8 ммоль), тетрагидрофуран (7 мл) и воду (2,5 мл). Спустя 2 минуты добавляют раствор, полученный на стадии 3 примера 1 (292 мг), в тетрагидрофуране (2 мл). Полученную суспензию нагревают до 50°С и перемешивают в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры и к смеси добавляют воду (20 мл). Полученную смесь экстрагируют этилацетатом (20 мл × 2). Органические экстракты охлаждают, сушат (Na2SO4), растворитель удаляют, полученный сырой продукт очищают хроматографией (диоксид кремния, EtOAc/PetEther, 20-100 %) с получением указанного в заголовке соединения (30 мг). ЖХ/МС m/z (М+Н)+=4 94, 3.
Стадия 5. (S)-2-Гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
К раствору (S)-2-гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (45 мг, 0,0 91 ммоль) в смеси тетрагидрофуран:вода (5 мл : 1 мл) при 15°С добавляют моногидрат гидроксида лития (23,0 мг, 0,547 ммоль). После этого реакционную смесь нагревают до 60°С и перемешивают при указанной температуре в течение 16 часов. К реакционной смеси добавляют воду (10 мл) и полученную смесь экстрагируют этилацетатом (15 мл × 4). Органические экстракты объединяют, сушат (Na2SO4) и растворитель удаляют, полученный сырой продукт (30 мг) очищают ОФ-ЖХВД (Phenomenex Gemini С-18™, 250 × 50 мм, 10 мкм, H2O/CH3CN + 0, 05 % NH4OH, 15-35 % в течение 10 min) с получением указанного в заголовке соединения (16 мг). 1Н ЯМР (400 МГц, ДМСО) δ 11,63 (с, 1Н), 8,09 (с, 1Н), 7,41 (с, 1Н), 7,14 (д, J=3,6 Гц, 1Н), 6,62 (д, J=3,6 Гц, 1Н), 4,89 (ддд, J=17,2, 7,9, 5,9 Гц, 2Н), 4,04 (м, 1Н), 3,58 (м, 1Н), 3,24 (с, 3Н), 3,14-2,86 (м, 2Н), 2,62-2,51 (м, 2Н), 2,23 (м, 2Н), 1,20 (д, J=6,2 Гц, 3Н); ЖХ/МС m/z (М+Н)+=340,1; хиральная SFC™ (Chiralpak AD-3™, 150 × 4,6 мм, 3μ, 40 % МеОН (0,05 % DEA) в CO2 (изократ.) 10 мин., 2,5 мг/мин., Т=35°С), Rt=5,54 мин., 99 % ээ.
Пример 4 - Альтернативное получение (R)-2-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (IC)

Стадия 1. (R)-трет-Бутил((1-хлорпропан-2-ил)окси)диметилсилан
К раствору (R)-(-)-1-хлорпропан-2-ола (2,0 г, 21,2 ммоль, CAS: 19141-39-0) в N,N-диметилформамиде (40 мл) при 20°С добавляют пиридин (2,0 г, 25,4 ммоль) и трет-бутилдиметилсилилхлорид (3,83 г, 25,4 ммоль). После перемешивания в течение 20 часов реакционную смесь разбавляют водой (60 мл) и полученную смесь экстрагируют метил-трет-бутиловым эфиром (2×80 мл). Органические экстракты охлаждают, промывают насыщенным раствором соли (100 мл) и сушат (Na2SO4). Растворитель удаляют с получением указанного в заголовке соединения (4,60 г), которое используют далее без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ 3, 96 (м, 1Н), 3,42 (дд, J=10,8, 5,9 Гц, 1Н), 3,34 (дд, J=10,7, 5,9 Гц, 1Н), 1,23 (д, J=6,l Гц, 3Н), 0,89 (с, 9Н), 0,08 (д, J=4,l Гц, 6Н).
Стадия 2. (R)-S-(2-((трет-Бутилдиметилсилил)окси)пропил)этантиоат
К раствору (R)-трет-бутил-((1-хлорпропан-2-ил)окси)диметилсилана (4,60 г, 22,03 ммоль) в N,N-диметилформамиде (40 мл) добавляют тиоацетат калия (5,03 г, 44,1 ммоль) и йодид калия (36,6 мг, 0,22 ммоль). Смесь выдерживают в течение 20 часов при 80°С, затем реакционную смесь охлаждают до комнатной температуры, разбавляют водой (100 мл) и полученную смесь экстрагируют метил-трет-бутиловым эфиром (100 мл × 2). Органические экстракты объединяют, сушат (Na2SO4) и растворитель удаляют с получением указанного в заголовке соединения (5,0 г). 1Н ЯМР (400 МГц, CDCl3) δ 3, 90 (м, 1Н), 3,00-2,91 (м, 2Н), 2,33 (с, 3Н), 1,18 (д, J=6,l Гц, 3Н), 0,88 (с, 9Н), 0,06 (д, J=6,7 Гц, 6Н).
Стадия 3. (R)-2-((трет-Бутилдиметилсилил)окси)пропан-1-сульфонилхлорид
Cl2 (газ.) пропускают в течение 5 минут при 0°С через раствор (R)-S-(2-((трет-бутилдиметилсилил)окси)пропил)этантиоата (0,5 г, 2,01 ммоль) в дихлорметане (20 мл) и воде (10,0 мл), в результате чего раствор приобретает желтую окраску. Поток С12 (газ.) останавливают и перемешивают реакционную смесь при 0°С в течение 0,5 часа. Через реакционную смесь пропускают газообразный N2 с получением бесцветного раствора. ТСХ показывает расходование исходного материала с образованием нового пятна. К реакционной смеси добавляют воду (20 мл) и экстрагируют реакционную смесь дихлорметаном (40 мл). Органический экстракт промывают 10 % NaHCO3 (50 мл), насыщенным раствором соли (50 мл) и сушат (Na2SO4). Растворитель удаляют с получением указанного в заголовке соединения (0,4 г) в виде бесцветного масла, которое используют в следующей стадии без дополнительной очистки.
Стадия 4. (R)-2-((трет-Бутилдиметилсилил)окси)-N-((1S,3S)-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4 -ил)амино)циклобутил)пропан-1-сульфонамид
В емкость объемом 40 мл добавляют цис-N1-метил-N1-(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)циклобутан-1,3-диамин (150 мг, 0,2 81 ммоль), Na2CO3 (149 мг, 1,41 ммоль), тетрагидрофуран (5 мл) и воду (2,0 мл). Спустя 2 минуты добавляют раствор соединения, полученного на стадии 3 примера 2 (292 мг, 0,7 5 ммоль) в ТГФ (2 мл). Полученную суспензию нагревают до 50°С и перемешивают в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры и к смеси добавляют воду (20 мл). Полученную смесь экстрагируют EtOAc (20 мл × 2). Органические экстракты охлаждают, сушат (Na2SO4) и растворитель удаляют, полученный сырой продукт очищают хроматографией (диоксид кремния, EtOAc/петролейный эфир, 20-100 %) с получением указанного в заголовке соединения (60 мг) и продукта без TBS-группы (20 мг). ЖХ/МС m/z (М+Н)+=608,3 (пик 1); ЖХ/МС m/z (М+Н)+=494,3 (пик 2).
Стадия 5. (R)-2-Гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
К раствору (R)-2-((трет-бутилдиметилсилил)окси)-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (60 мг, 0,099 ммоль) в дихлорметане (5 мл) при 0°С добавляют трифторуксусную кислоту (1,5 мл). Смесь выдерживают в течение 16 часов при 15°С, затем растворитель удаляют и остаток растворяют в этилацетате (15 мл). Смесь промывают насыщенным раствором NaHCO3(водн.), сушат (Na2SO4) и удаляют растворитель с получением указанного в заголовке соединения (50 мг), которое используют в следующей стадии без дополнительной очистки. ЖХ/МС m/z (М+Н)+=494,3.
Стадия 6. (R)-2-Гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
К раствору (R)-2-гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (50 мг, 0,10 ммоль) в смеси тетрагидрофуран : вода (5 мл: 1 мл) при 15°С добавляют моногидрат гидроксида лития (42,5 мг, 1,01 ммоль). Смесь выдерживают в течение 16 часов при 65°С, добавляют воду (10 мл) и экстрагируют полученную смесь этилацетатом (4×10 мл). Органические экстракты объединяют, сушат (Na2SO4) и растворитель удаляют, полученный сырой продукт очищают преп-ВЭЖХ (Phenomenex Gemini С-18™, 250 × 50 мм, 10 мкм, H2O/CH3CN+0,05 % NH4OH, 15-35 % в течение 10 минут) с получением указанного в заголовке соединения (18 мг). 1Н ЯМР (4 00 МГц, ДМСО) δ 11,63 (с, 1Н), 8,10 (с, 1Н), 7,43 (д, J=9,1 Гц, 1Н), 7,15 (дд, J=3,6, 2,4 Гц, 1Н), 6,64 (дд, J=3,6, 1,9 Гц, 1Н), 4,97-4,83 (м, 2Н), 4,12-3,98 (м, 1Н), 3,59 (м, 1Н), 3,25 (с, 3Н), 3,13-2,92 (м, 2Н), 2,58 (м, 2Н), 2,24 (м, 2Н), 1,21 (д, J=6,2 Гц, 3Н); ЖХ/МС m/z (М+Н)+=340,0; хиральная SFC (Chiralpak AD-3™, 150 × 4,6 мм, 3 мкм, 40 % МеОН (0,05 % DEA) в CO2 изократ. 10 минут, 2,5 мг/мин., Т=35°С), Rt=6,75 мин., 99 % ээ.
Пример 5-3-Гидрокси-N-(цис-3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид (IB)
К перемешиваемой смеси цис-N-Метил-N-7Н-пирроло[2,3-d]пиримидин-4-илциклобутан-1,3-диамина (190 мг) и карбоната калия (240 мг) в смеси 1:1 тетрагидрофуран: вода (по 5 мл), используемой в качестве растворителя, при 10°С добавляют метил-3-(хлорсульфонил)пропаноат (250 мг). Смесь нагревают до комнатной температуры и перемешивают в течение 10 минут. Смесь концентрируют в вакууме. Остаток очищают колоночной хроматографией (МеОН/ДХМ 1:20) с получением метил-3-(N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)сульфамоил)пропаноата (140 мг) в виде твердого белого вещества. 1Н ЯМР (400 МГц, метанол-d4): 8,13 (с, 1Н), 7,13 (д, 1Н), 6,70 (д, 1Н), 4,89 (м, 1Н), 3,73 (с, 3Н), 3,69 (м, 1Н), 3,4 (м, 5Н), 2,82 (м, 4Н), 2,34 (м, 2Н), МС m/z for C15H21N5O4S: 390,2 (M+Na)+. ВЭЖХ: колонка Ultimate ХВ-С18 3pm 3,0*50 мм, время удерживания: 2,13 мин., подвижная фаза: от 0 % ацетонитрила (0,1 % ТФУК) в воде до 60 % ацетонитрила (0,1 % ТФУК) в воде, длина волны: 220 нм.
К перемешиваемому раствору метил-3-(N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)сульфамоил)пропаноата (120 мг) в сухом тетрагидрофуране (5 мл) при 0°С в атмосфере азота добавляют алюмогидрид лития (50 мг). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 0,5 часов. Смесь гасят осторожным добавлением метанола и очищают колоночной хроматографией (МеОН/ДХМ 1:10) с получением 3-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (50 мг) в виде твердого белого вещества. 1Н ЯМР (400 МГц, метанол-d4): 8,12 (с, 1Н), 7,13 (д, 1Н), 6,70 (д, 1Н), 4,89 (м, 1Н), 3,73 (м, 3Н), 3,36 (с, 3Н), 3,15 (м, 2Н), 2,81 (м, 2Н), 2,35 (м, 2Н), 2,03 (м, 2Н); МС m/z для C14H21N5O3S: 340,2 (М+Н)+; ВЭЖХ: колонка Ultimate ХВ-С18 3 pm 3,0 × 50 мм, время удерживания: 1,85 мин., подвижная фаза: от 0 % ацетонитрила (0,1 % ТФУК) в воде до 60 % аацетонитрила (0,1 % ТФУК) в воде, длина волны: 22 0 нм.
Пример 6 - Альтернативное получение 3-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида
К смеси дигидробромида цис-N-метил-N-{7-[(4-метилфенил)сульфонил]-7Н-пирроло[2,3-d]-пиримидин-4-ил}циклобутан-1,3-диамина (988 мг) в 50 мл дихлорметана при 0°С добавляют триэтиламин (562 мг) и этил-3-(хлорсульфонил)пропаноат (743 мг). Реакционную смесь нагревают до 25°С и перемешивают при указанной температуре в течение 2 часов. Реакционную смесь упаривают в вакууме и остаток очищают хроматографией (силикагель, колонка Biotage, 20 г оксида кремния, петролейный эфир : этилацетат - от 1:0 до 0:1, этилацетат : метанол 20:1) с получением этил-3-(N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)сульфамоил)пропаноата. ЖХМС m/z для C23H29N5O6S2: 535, 9 (М+Н)+.
К раствору этил-3-(N-(цис-3-(метил-(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)сульфамоил)пропаноата (7 00 мг) в 50 мл тетрагидрофурана при 0°С добавляют алюмогидрид лития (74,4 мг). Реакционной смеси дают возможность нагреться до температуры 25°С в течение 15 часов. Реакционную смесь осторожно гасят добавлением 1 мл воды, затем фильтруют. Фильтрат упаривают в вакууме с получением в виде сырого продукта 3-гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, который используют далее без дополнительной очистки. ЖХМС m/z для C21H27N5O5S2: 494,0 (М+Н)+.
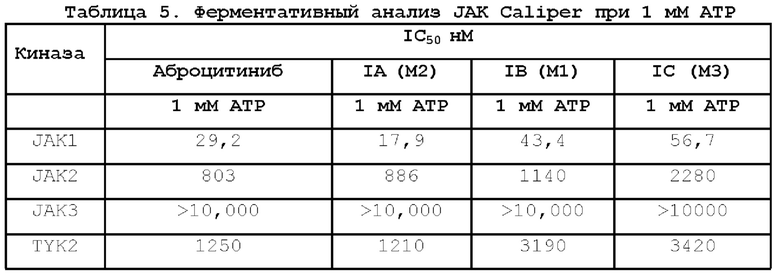
К раствору 3-гидрокси-N-(цис-3-(метил(7-тозил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида (400 мг) в 20 мл этанола и 10 мл воды добавляют LiOH (97 мг). Реакционную смесь нагревают до 90°С и перемешивают в течение 2 часов. Реакционную смесь охлаждают и упаривают в вакууме, полученный сырой продукт очищают преп-ВЭЖХ с получением 181 мг 3-гидрокси-N-(цис-3-(метил(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида в виде твердого белого вещества. Условия ВЭЖХ: колонка DuraShell™ 150 × 25 мм × 5 мкм, вода (0,05 % гидроксида аммония об./об.)-ацетонитрил, 1-41 %В, градиент 10 мин., время удерживания 1 мин. 100 %В, скорость потока 25 мг/мин.; 1Н ЯМР (400 МГц, ДМСО-d6): 11,6 (с, 1Н), 8,11 (с, 1Н), 7,53 (с, 1Н), 7,15 (д, 1Н), 6,65 (д, 1Н), 4,89 (м, 1Н), 4,68 (с, 1Н), 3,58 (м, 1Н), 3,49 (м, 2Н), 3,26 (с, 3Н), 3,0 (м, 2Н), 2,58 (м, 2Н), 2,24 (м, 2Н), 1,81 (м, 2Н). Оценка биологической активности Ферментативный анализ JAK Caliper при 1 мМ АТР Опытный образец растворяют в диметилсульфоксиде (ДМСО) до исходной концентрации 30 мМ. Приготавливают 11-точечную серию полулогарифмического разбавления в ДСМО с верхней концентрацией 500 мкМ. Планшет опытного соединения также содержит лунки положительного контроля, содержащие известный ингибитор для определения 100 % ингибирования, и лунки отрицательного контроля, содержащие ДМСО, для определения отсутствия ингибирования. Планшеты с соединением разбавляют 1 к 60, получая конечную концентрация опытного соединения 10 мкМ и концентрацию ДМСО 2 %. Опытное соединение и лунки с контролем помещают в 384-луночный планшет. Реакционные смеси содержат 20 мМ HEPES, рН 7,4, 10 мМ хлорида магния, 0,01 % альбумина бычьей сыворотки (BSA), 0,0005 % Tween 20, 1 мМ АТР и 1 мкМ пептидного субстрата. Тесты JAK1 и TYK2 содержат 1 мкМ пептида IRStide (5FAM-KKSRGDYMTMQID), тесты JAK2 и JAK3 содержат 1 мкМ пептида JAKtide (FITC-KGGEEEEYFELVKK). Анализы инициируют добавление 2 0 нМ JAK1, 1 нМ JAK2, 1 нМ JAK3 или 1 нМ фермента TYK2 и инкубируют при комнатной температуре в течение трех часов для JAK1, 60 минут для JAK2, 75 минут для JAK3 или 135 минут для TYK2. Концентрации ферментов и время инкубирования оптимизируют для каждого нового ферментного препарата и незначительно изменяют во времени для обеспечения 20 % - 30 % фосфорилирования. Тестирование останавливают с конечной концентрацией 10 мМ ЭДТА, 0,1 % реагента покрытия и 100 мМ HEPES, рН=7,4. Тестовые планшеты помещают на аппарат Caliper Life Science Lab Chip 3000 (LC3000) и из каждой лунки отбирают образец с использованием подходящих условий разделения для количественного определения нефосфорилированного и фосфорилированного пептида. Полученные данные представлены в таблице 5.
Оценка клеточной активности: индуцированное цитокинами фосфорилирование STAT в цельной крови человека, кератиноцитах человека и IFNγ-праймированнцх клетках ТНР-1
Цельную кровь человека собирают от здоровых доноров венозной пункцией в вакуумные пробирки для сбора, содержащие гепарин натрия, в соответствии с протоколами Pfizer (Protocol No. GOHW RDP-01), одобренными Институциональным наблюдательным советом Шульмана (Shulman Institutional Review Board). Кровь нагревают до 37°С перед использованием. Цельную кровь человека аликвотируют (90 мкл/лунка) в 96-луночные планшеты с глубокими лунками и V-образным дном и обрабатывают соединениями (5 мкл/лунка) в различных концентрациях до 60 мкМ (конечная концентрация 0,2 % ДМСО) при 37°С в течение 60 минут. Затем последовательно вводят IFNcx (5000 U/мл), IFNγ (100 нг/мл), IL-6 (50 нг/мл), IL-10 (30 нг/мл), IL-12 (30 нг/мл), IL-15 (30 нг/мл), IL-21 (50 нг/мл), IL-23 (25 нг/мл), IL-27 (1000 нг/мл), ЕРО (2U/мл), TSLP (50 нг/мл) или PBS и инкубируют в течение 15 минут. За 15 минут до стимуляции цитокинами добавляют антитела против клеточной поверхности; анти-СР3-ВУ421 (0,5 мкл/лунка) к образцам, обработанным IL-6, и образцам, обработанным TSLP, и анти-CD14-BV421 (0,5 мкл/лунка) к образцам, обработанным IFNγ. Образцы обрабатывают теплым буфером 1× Lyse/Fix (700 мкл/лунка) для прекращения активации и дополнительно инкубируют при 37°С в течение 2 0 минут для лизиса эритроцитов. Планшеты центрифугируют при 300 × g в течение 5 минут, супенратант аспирируют и клетки промывают 800 мкл/лунка окрашивающим буфером (D-PBS, содержащий 0,1 % FBS и 0,01 % азид натрия). Промытые клеточные пеллеты снова суспендируют в предварительно охлажденном 90 % метаноле (350 мкл/лунку) и инкубируют при 4°С в течение 30 минут. После удаления 90 % метанола клетки промывают однократно окрашивающим буфером. Затем все образцы суспендируют в растворах желаемых антител против фосфо-STAT (150 мкл/лунку) при разбавлении 1:150: анти-pSTAT1-AF647 в образцах, обработанных IFNγ; анти-pSTAT1-AF488 и анти-pSTAT3-AF647 в образцах, обработанных IL-6, анти-pSTAT3-AF647 в образцах, обработанных IFNα, IL-10, IL-21, IL-23 и IL-27, анти-pSTAT4-AF647 в образцах, обработанных IL-12, анти-pSTAT5-AF-647 в образцах, обработанных ЕРО, IL-15 и TSLP.
Первичные кератоциты человека культивируют в среде DermaLife™ с набором добавок для увеличения популяции клеток. В опытах используют клетки 2-5 пассажей. Клетки собирают при -80 % конфлюэнтности, суспендируют в теплой среде DermaLife™, алюквотируют (90 мкл/лунка) в 96-луночные планшеты с глубокими лунками и V-образным дном и инкубируют при 37°С в течение 30 минут. После этого клетки обрабатывают соединениями (от 0,0003 до 20 мкМ) в течение 60 минут с последующим стимулированием IL-4 (2 нг/мл) или IL-13 (20 нг/мл) при 37°С в течение 15 минут. Для анализа IL-22 индуцированного pSTAT3 кератоциты высевают в 24-луночные планшеты и культивируют в течение 18 часов. Среду клеток меняют на базальную среду DermaLife™, клетки обрабатывают соединениями (от 0, 0003 до 20 мкМ) в течение 60 минут и затем стимулируют IL-22 (100 нг/мл) в течение 30 минут. Клетки отделяют обработкой смесью 0,25 % трипсин/ЭДТА. Стимулированные кератиноциты фиклируют 2 % параформальдегидом и пермамбилизируют 90 % метанолом. Фиксированные и пермеамбилизированные клетки окрашивают AlexaFluor647™, меченым антителом против pSTAT6 (разведение 1 к 150), для образцов, обработанных IL-4 и IL-13, и AlexaFluor647, меченым антителом против pSTAT3 (разведение 1 к 150), для образцов, обработанных IL-22.
ТНР-1 клетки выдерживают в среде RPMI 1640, содержащей 10 % FBS, 50 μМ 2 меркаптоэтанола, 50 U/мл пенициллина, 50 мкг/мл стрептомицина и 2 мМ L-глютамина. ТНР 1 клетки обрабатывают IFNγ (20 нг/мл) в течение 18 часов. IFNγ-праймированные ТНР-1 клетки повторно суспендируют в свежую среду RPMI 1640, обрабатывают соединениями (от 0,0003 до 20 мкМ) в течение 60 минут с последующей стимуляцией IL-31 (1 мкг/мл) в течение дополнительных 10 минут. После этого клетки фиксируют 2 % параформальдегидом и пермеабилизируют 90 % метанолом.
Фиксированные и пермеабилизированные клетки окрашивают AlexaFluor647™, меченым антителом против pSTAT3 (разведение 1 к 150).
После инкубирования в течение ночи при 4°С образцы, окрашенные анти-pSTAT, переносят в 96-луночные полипропиленовые U-образные планшеты и проводят проточный цитометрический анализ на LSR Fortessa™, снабженном устройством для загрузки формных пластин HTS. В образцах цельной крови человека популяцию лимфоцитов выбирают для анализа pSTAT гистограммы для образцов, обработанных IFNα, IL-10, IL-12, IL-15, IL-21, IL-23 и IL-27; CD14+ клетки для образцов, обработанных IFNγ; CD3+ клетки для образцов, обработанных IL-6 и TSLP; все события (полные популяции) для образцов, обработанных ЭПО. Полные популяции кератиноцитов и ТНР-1 клетки отбирают для анализа IL-4, IL-13, IL-22 и IL-31. Фоновую флуоресценцию определяют с использованием нестимулированных клеток, и гейт помещают у подножия пика для включения -0,5 % популяции клеток, дающих сигнал выше порогового значения. Статистический анализ гистограммы проводят с использованием FACSDiva version 8.0. Относительную единицу флуоресценции, которая изменяет уровень pSTAT, вычисляют умножением процента положительной популяции на ее среднюю флуоресценцию. Кривые ингибирования и значения концентрации, при которой половина опытных клеток (IC50), определяют с использованием программного обеспечения Prism™ software (Version 8) или программного обеспечения для анализа данных Activity Base (ID Business Solutions).
Клеточная активность в отношении фосфорилирования STAT, вызванного цитокинами
Соединения формул IA и IB, полученные в соответствии с примерами 1 и 3 и примерами 5 и 6, соответственно, образуются in vivo в процессе метаболизма человека при приеме аброцитиниба и демонстрируют активность, сходную с аброцитинибом, мощным ингибитором JAK1, против фосфорилирования STAT, индуцированного IL-4, IL-13, IL-22, IL 31 и TSLP, в кератоцитах человека, интерферон-γ праймированных ТНР-1 клетках или человеческой цельной крови. Gooderham M.J. et al. JAMA Dermatology, 155(12), 1371-1379 (октябрь 2019). Примечательно, что, как и аброцитиниб, все эти соединения более эффективны в отношении цитокинов, которые передают свои сигналы через JAK1-зависимые пути, по сравнению с теми, которые передают свои сигналы через JAK1-зависимые пути, и поэтому, вероятно, способствуют фармакологической активности в основном через воздействие на JAK1. Сравнение клеточной активности аброцитиниба и соединений формул IA и IB по аналогичным сигнальным путям представлено в таблице 6.
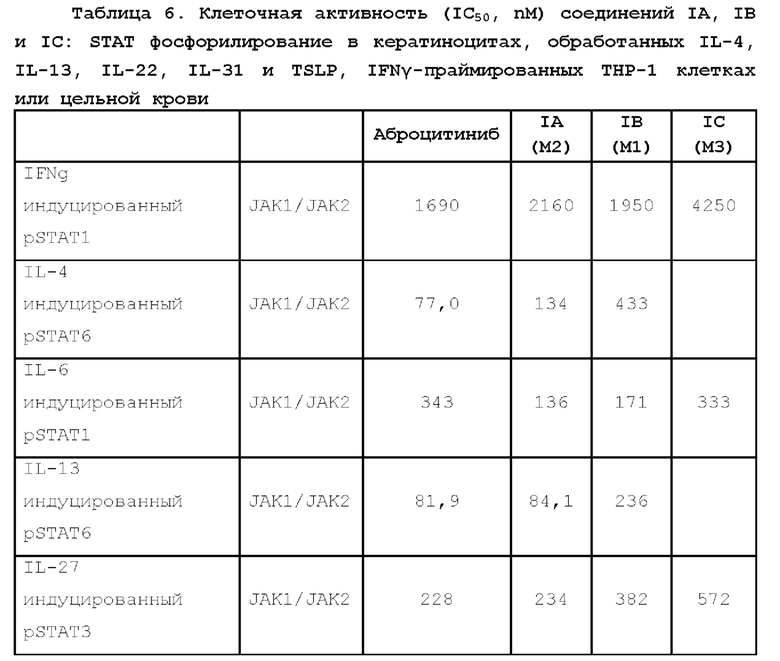

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 1, ИХ КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2824349C2 |
| ЛЕЧЕНИЕ ГИДРАДЕНИТА ИНГИБИТОРАМИ JAK | 2020 |
|
RU2805595C1 |
| БИСУЛЬФАТ ИНГИБИТОРА ЯНУС-КИНАЗЫ (JAK) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2665680C2 |
| ПИРРОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ | 2012 |
|
RU2631655C2 |
| ПРОИЗВОДНЫЕ 4-АМИНО-5-ФЕНИЛ-7- ЦИКЛОБУТИЛПИРРОЛО[2,3-D]ПИРИМИДИНА | 2002 |
|
RU2305682C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНИЛА | 2021 |
|
RU2817349C1 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| Соединение, ингибирующее активности киназ ВТК и/или JAK3 | 2014 |
|
RU2650512C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
Настоящее изобретение относится к производным пирроло{2,3-d}пиримидина


или их фармацевтически приемлемым солям, а также к фармацевтическим композициям, содержащим указанные соединения. Технический результат: получены новые производные пирроло{2,3-d}пиримидина, которые могут быть применимы для ингибирования Янус-киназы (Janus Kinase - JAK) и применимы в способах лечения состояний, опосредуемых JAK1. 5 н. и 1 з.п. ф-лы, 6 табл., 6 пр.
1. Соединение формулы IA, имеющее структуру

или его фармацевтически приемлемая соль.
2. Соединение формулы IB, имеющее структуру
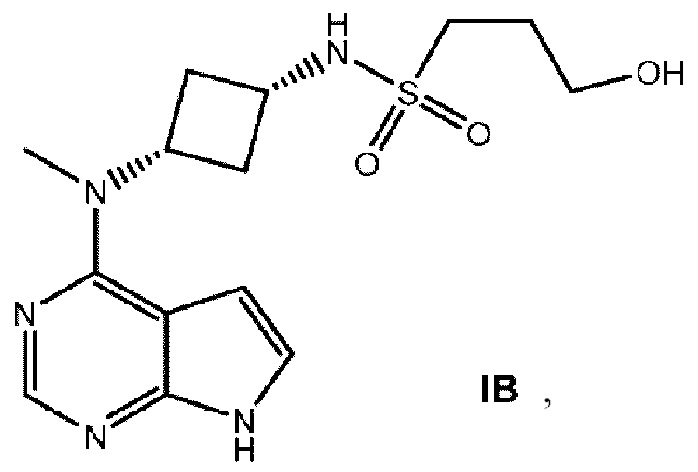
или его фармацевтически приемлемая соль.
3. (S)-2-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид или его фармацевтически приемлемая соль.
4. 3-Гидрокси-N-(3-(метил-(7Н-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид или его фармацевтически приемлемая соль.
5. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-4 в изолированной форме.
6. Фармацевтическая композиция для ингибирования Янус-киназы (JAK), содержащая эффективное количество соединения по пп. 1-5 или его фармацевтически приемлемой соли и фармацевтически приемлемый эксципиент.
| Ковшевой элеватор | 1930 |
|
SU27879A1 |
| US 20160045508 A1, 18.02.2016 | |||
| ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-d]ПИРИМИДИНА | 2009 |
|
RU2493157C2 |
| Способ изготовления стекол с незапотевающим слоем к противогазам | 1931 |
|
SU26201A1 |
| МОЙКА ДЛЯ СВЕКЛЫ | 1926 |
|
SU5852A1 |

