Описание уровня техники
Ранняя диагностика РЛ призвана увеличить эффективность фармакотерапевтического и хирургического лечения. Для неинвазивного скрининга РЛ используют рентгенографию и компьютерную томографию. Они могут давать как ложноположительные результаты, влекущие за собой не всегда обоснованную травматичную процедуру тканевой биопсии, так и давать ложноотрицательные результаты из-за погрешностей детектирования небольших опухолевых очагов. Надежных методов лабораторной диагностики РЛ на данный момент не существует, несмотря на открытие большого числа кандидатных биомаркеров, изучаемых в доклинических и клинических исследованиях. Существующие разработки в области неинвазивной лабораторной диагностики с использованием концепции «жидкостной биопсии» пока недостаточно совершенны для внедрения в клиническую практику, поскольку не отличаются высокими параметрами чувствительности и/или специфичности или не прошли клиническую валидацию. Под термином «жидкостная биопсия» следует понимать тесты, ориентированные на анализ молекул в биологических жидкостях организма, таких как, например, кровь или моча. Использование диагностического подхода, основанного на исследовании «жидкостной биопсии», в перспективе позволит дополнить лучевые методы диагностики РЛ и для ряда случаев заменить тяжело переносимую процедуру биопсии ткани легкого. С другой стороны, исследование образцов жидкостной биопсии дает возможность реализации малоинвазивного тестирования на этапе долучевой диагностики РЛ.
На молекулярном уровне, злокачественные изменения в ткани легкого сопряжены с нестабильностью геномной ДНК, а именно, связаны с появлением патогенных вариантов генов и аномальных эпигенетических изменений ДНК [Malhotra J. et al, Risk factors for lung cancer worldwide // Eur Respir J. - 2016. - V. 48. - No. 3. - P. 889-902; Daniel P.T., et al. Detection of genomic DNA fragmentation during apoptosis (DNA ladder) and the simultaneous isolation of RNA from low cell numbers // Anal Biochem. - 1999. - V. 266. - No. 1. P. 110-115], а также связаны с нарушением регуляции апоптоза, что приводит к накоплению фрагментированной ДНК [Cristiano S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer // Nature. - 2019. - Vol 570. - No. 7761. - P. 385-389; Underhill HR et al. Fragment length of circulating tumor DNA // PLoS Genet. - 2016. - Vol.12. - No. 7. - P. е1006162]. Поскольку фрагменты ДНК трансформированных клеток попадают в системный кровоток или в другие биологические жидкости организма, то такая фракция ДНК получила название свободно циркулирующей ДНК (сцДНК). Под термином фрагментом сцДНК следует понимать совокупность фрагментов сцДНК, находящихся вне источника происхождения, например, фрагментом сцДНК плазмы крови.
Анализ фрагментома сцДНК позволяет косвенно оценить экспрессию генов и предсказать локализацию опухоли, поскольку фрагменты сцДНК разного тканевого происхождения характеризуются разными длинами и нуклеотидными последовательностями [Cristiano S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer // Nature. - 2019. - V. 570. - No. 7761. - P. 385-389; Underhill H.R. et al. Fragment length of circulating tumor DNA // PLoS Genet. - 2016. - Vol.12. - No. 7. - P. е1006162]. Содержание фрагментированной сцДНК в системном кровотоке по мере прогрессирования злокачественного новообразования (ЗНО) существенно возрастает благодаря увеличению поступления фрагментированной сцДНК из опухолевого очага. Это делает возможным выделение из образцов крови сцДНК в количестве, достаточном для выполнения секвенирования. Таким образом, количество сцДНК и ее профиль фрагментации в образцах жидкостной биопсии может коррелировать со стадиями, агрессивностью и локализацией ЗНО. Дополнительно, по профилю фрагментации сцДНК плазмы крови можно судить об особенностях нуклеосомной укладки, которая, в свою очередь, является отражением эпигенетического и транскриптомного профилей. Поскольку наиболее многообещающим методом изучения фрагментома сцДНК в образцах жидкостной биопсии является секвенирование нового поколения (Next Generation Sequencing, NGS), то создание алгоритмов для анализа и интерпретации данных NGS-анализа позволит создать классификационную модель для точного различия вариантов нормы и наличия ЗНО. С методологической точки зрения, лабораторный этап анализа сцДНК, выделенной из образцов биоматериала, хорошо воспроизводится при использовании коммерчески доступных NGS-платформ, что делает данный подход крайне перспективным благодаря возможности стандартизации многих преаналитических и собственно аналитических этапов. Что касается анализа NGS-данных, то описано достаточно много уникальных способов их биоинформатической и статистической обработки. Одна часть таких способов нацелена на идентификацию источника происхождения фрагментов сцДНК через анализ мутационных биомаркеров, что снижает диагностическую ценность способа, поскольку, на ранних стадиях злокачественной трансформации клеток представленность циркулирующей опухолевой ДНК в образцах жидкостной биопсии относительно низкая. Другая часть способов направлена на раскрытие особенностей анализа NGS-данных для извлечения максимальной диагностической ценности. Однако, не описаны классификационные модели, позволяющие с высокой точностью разделять группы субъектов только по профилю фрагментации сцДНК в образцах жидкостной биопсии, следовательно, разработка способа анализа NGS-данных на основе комбинации методов машинного обучения, ориентированного на получение четких различий между нормой и патологией, поможет преодолеть многие технические и биологические затруднения в области диагностики РЛ.
Сведения об аналогичных изобретениях.
Информационный поиск в базах данных GooglePatent, EspaceNet, USPTO и базы данных ФИПС позволил выявить, по крайней мере, ряд аналогов, которые имеют некоторые формальные сходства с настоящим изобретением: US 20210238668 A1, US 20210189494 A1, WO 2021231455 A1, WO 2021262770 A1, WO 2020094775 A1, WO 2021071638 A1, СА 3134519 А1, CN 109112191, CN 112599197, CN 111575347 А, JP 2017522908 A, WO 2013060762 A1, ЕР 3728582 А4, US 20190256924, US 20190390253, WO 2020176659 A1, WO 2018009723, ЕР 3004383 В1, US 20210239685, WO 2021007462 A1, US 20200232010, WO 2015048535 A1, US 10741270 B2, US 9892230 B2, US 20200294624 A1, ЕР 3899956 А2, CN 106156543, CN 111243673, СА 2824387 С, US 20180307796 A1, US 20170220735 A1, US 10364467 B2, US 20190352695 A1, JP 2019531700 A5, СА 3119980 А1, WO 2022140386 A1, WO 2019222657 A1, US 20220136062 A1, US 20190287654 A1. Найденные аналоги можно условно разделить на три группы, которые включают описание: 1) способов секвенирования нуклеотидной последовательности сцДНК; 2) способов секвенирования сцДНК и обработки NGS-данных; 3) способов обработки NGS-данных. В группах 2 и 3 были выявлены восемь близких аналогов.
В патенте WO 2021262770 A1 раскрыт способ обработки данных секвенирования сцДНК. Поскольку «горячие точки» (от англ. hotspots) аномальной фрагментации сцДНК чаще встречаются в участках открытого хроматина, вблизи микросателлитных повторов, гена CTCF и генов, участвующих в иммунных процессах, то акцент был сделан на анализе фрагментов сцДНК, соответствующих этим участкам. Выбор «горячих точек» фрагментации сцДНК был сделан на основе баллов, учитывающих длину фрагмента и отношение средней длины фрагмента в скользящем окне к длине фрагмента во всей хромосоме.
В патенте WO 2020094775 A1 раскрыт способ обработки NGS-данных с созданием классификационной модели на основе данных о числе копий генов и генетических вариантах. При включении в модель данных по фрагментому сцДНК значение AUC (площадь под кривой, от англ. Area Under Curve,) возросло с 0,8 до 0,99. Алгоритм классификации фрагментов сцДНК заключался в расчете соотношения фрагментов сцДНК в интервалах длин 20-150 пар нуклеотидов (п.н.); 100-150 п.н.; 160-180 п.н.; 180-220 п.н.; 250-320 п.н. и соотношений Р (20-150) / Р (160-180); Р (100-150) / 163-169; Р (20-150) / (180-220).
В патенте US 20210238668 A1 раскрыт способ обработки данных секвенирования фрагментов сцДНК из плазмы крови для определения источника происхождения фрагментов сцДНК на основе наблюдений о том, что последовательности концевых мотивов фрагментов сцДНК демонстрируют тканеспецифичные профили и разные частоты встречаемости.
В патенте US 20210189494 A1 описан способ анализа нуклеотидной последовательности концевых мотивов фрагментов сцДНК определенного размера, которые являются интегральным отражением функционирования клеточных нуклеаз и/или нуклеаз крови в ответ на фармакологическое воздействие. Таким образом, реализация способа обеспечивает оценку потенциального генотоксического действия экспериментальных лекарств, но способ лишен диагностической ценности.
В патенте СА 3134519 А1 раскрыт способ мониторинга вероятности рецидива опухоли и эффективности терапии на основе данных секвенирования ДНК, выделенной из крови. В некоторых вариантах воплощения способ предусматривает анализ проб, обогащенных фракциями три-нуклеосомной, ди-нуклеосомной, моно-нуклеосомной ДНК. Аннотацию данных секвенирования фрагментов сцДНК предполагается проводить по соматическим генетическим вариантам. Способ больше ориентирован на раскрытие общих особенностей пробоподготовки и лабораторного анализа сцДНК на основе полимеразной цепной реакции, чем на раскрытие особенностей обработки данных о фрагментоме сцДНК.
В патенте WO 2021007462 А1 раскрыт способ детекции сцДНК, выделенной из образцов крови или мочи, взятых от здоровых людей (отсутствие заболевания) и субъектов с диагностированным заболеванием. Он состоит в секвенировании фрагментированной сцДНК, обработке данных о фрагментоме сцДНК, анализе распределения последовательностей концевых мотивов фрагментов сцДНК, сопоставительном анализе нуклеотидной последовательности сайтов инициации транскрипции генов и уровней тканеспецифичной экспрессии генов для создания дифференциальной карты позиций фрагментов сцДНК в составе как связанной так и не связанной с нуклеосомами ДНК.
В патенте US 20180307796 A1 описан способ обработки данных секвенирования фрагментов сцДНК с последующей разбивкой всего множества фрагментов сцДНК в бины (диапазоны). Способ учитывает частоты встречаемости аллельных вариантов генов в целевых бинах для разделения усредненных вариантов нормы и наличия патологии по профилю патогенных соматических вариантов и профилю распределения фрагментов сцДНК по длинам.
В патенте WO 2021071638 А1 раскрыт способ обработки данных секвенирования для различения терминальных и соматических вариантов сцДНК, происходящих из гемопоэтических клеток, на основе анализа распределения фрагментов сцДНК по длинам. В одном из воплощений способ позволяет идентифицировать источник происхождения фрагментов сцДНК.
В научной литературе также были найдены аналоги. В статье Вао и соавторов было выполнено секвенирование фрагментома сцДНК плазмы крови, полученной от пациентов с раком печени, раком легких и колоректальным раком [Вао Н., et al. Letter to the Editor: An ultra-sensitive assay using cell-free DNA fragmentomics for multi-cancer early detection // Mol Cancer. - 2022. - V. 21. - No. 1. - P. 129]. Для анализа профиля фрагментации сцДНК использовали четыре группы длин фрагментов (65-150, 151-260, 261-400, 65-400 п.н.). После сопоставления результатов секвенирования с референсным геномом человека (сборка hgl9) были введены показатели EDM (EnD Motif) и ВРМ (Break Point Motif). Затем были рассчитаны частоты встречаемости концевых мотивов фрагментов сцДНК. Сумма всех частот EDM была равна 1. ВРМ учитывал последовательности геномной ДНК длиной 3 п.н. выше и ниже 5'-концевых точек разрыва сцДНК, а сумма всех частот ВРМ также была равна 1. EDM и ВРМ позволяют классифицировать субъектов обследования на основе данных о последовательностях концевых мотивов фрагментов сцДНК.
В работе Cirmena и соавторов [Cirmena G, et al. Plasma Cell-Free DNA Integrity Assessed by Automated Electrophoresis Predicts the Achievement of Pathologic Complete Response to Neoadjuvant Chemotherapy in Patients With Breast Cancer // JCO Precis Oncol. - 2022. - V. 6. - P. e2100198] описан способ обработки данных о фрагментоме сцДНК из плазмы крови. Отличия аналога заключаются в прогнозе патоморфологического ответа опухоли на неоадъювантную химиотерапию у пациентов с РЛ, а количественную оценку фрагментома проводили методом капиллярного электрофореза на рабочей станции TapeStation 2200 (Agilent Technologies, Santa Clara, CA). В настоящем изобретении также используют высокопроизводительное секвенирование, что значительно повышает пределы детекции сцДНК методом капиллярного электрофореза. Профиль фрагментации включал пять диапазонов длин фрагментов сцДНК: 90-150, 150-220, 100-300, 221-320 и 321-1000 п.н. Показатель cfDI был использован для прогноза патоморфологического ответа опухоли на терапию и представлял собой отношение концентрации фрагментов размером 321-1000 п.н. к концентрации фрагментов размером 150-220 п.н. в точке завершения терапии.
Итак, следует однозначно считать существование различий формулы настоящего изобретения от приведенных выше близких аналогов. В настоящем изобретении используют уникальную комбинацию методов биоинформатической и статистической и обработки данных о фрагментоме сцДНК плазмы крови: методов снижения размерности массивов данных и методов машинного обучения. Работа диагностического алгоритма основана на комбинации модифицированного метода главных компонент, логистической регрессии с L1- и L2-регуляризацией, а также вероятностной классификационной модели, основанной на гауссовских процессах и ядерной логистической регрессии. Ближайший аналог
В публикации Mathios и соавторов [Mathios D., et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes // Nat Commun. - 2021. - V. 12. - No. 1. - P. 5060] раскрыт способ диагностики онкологического заболевания по профилю фрагментации сцДНК. Способ описывает следующие стадии: получение образцов цельной крови, выделение фракции плазмы с последующим выделением сцДНК, подготовку ДНК-библиотек для секвенирования и проведение процедуры секвенирования. Обработка данных секвенирования включает расчет соотношения количества коротких (100-150 п.н.) и длинных (151-220 п.н.) фрагментов с введением поправки на содержание гуанина и цитозина и размер ДНК-библиотек, анализ данных с использованием референсных данных TCGA (The Cancer Genome Atlas) и алгоритмов машинного обучения (см. раздел «Methods» на стр. 10-11) для обнаружения опухолевой сцДНК. Классификационная модель была валидирована на независимой когорте из 385 здоровых субъектах и 46 субъектах с РЛ. Комплексный анализ данных о профиле фрагментации сцДНК, клинико-анамнестических характеристик, уровней ракового эмбрионального антигена в плазме крови, данных лучевой диагностики позволил выявить 94% пациентов с РЛ, в том числе 91% пациентов - стадии I и II и 96% пациентов - стадии III и IV со специфичностью 80%. Таким образом, способ позволяет реализовать малоинвазивную диагностику рака легкого. Более детально ближайший аналог раскрывает сбор крови у субъекта исследования (см. подраздел «Sample collection and preservation)) на стр. 10); выделение фракции плазмы крови (см. подраздел «Sample collection and preservation)) на стр. 10); выделение сцДНК из плазмы крови; количественную оценку содержания сцДНК в пробах (см. подраздел «Sequencing library preparation)) на стр. 10-11); анализ качества выделенной сцДНК (см. подраздел «Sequencing library preparation)) на стр. 10-11); подготовку ДНК-библиотек для секвенирования (см. подраздел ((Sequencing library preparation)) на стр. 10-11); выполнение секвенирования (см. подраздел «Low coverage whole-genome sequencing and alignment)) на стр. 11); биоинформатическую обработку данных секвенирования, включающую преобразование последовательностей фрагментома (прочтения) в профиль фрагментации как численное выражение степени фрагментации ДНК в зависимости от положения в геноме и картирование прочтений на референсную последовательность человеческого генома, определение количества длинных и коротких фрагментов (см. подраздел «Whole-genome fragment features)) на стр. 11); определение статуса пациента по профилю фрагментации сцДНК с помощью классификационной модели, которая различает вариант нормы и наличие РЛ (см. раздел «Methods» на стр. 11-12).
Итак, с одной стороны, настоящее изобретение имеет сходство с теми аналогами, в которых описаны лабораторные способы выделения сцДНК, подготовки ДНК-библиотек и процедуры проведения NGS. С другой стороны, настоящее изобретение принципиально отличается от ближайшего аналога особенностями алгоритма биоинформатической и статистической обработки NGS-данных и созданием классификационной модели в части использования позиционно-весовых матриц, заключающих сведения о профиле последовательностей концевых мотивов фрагментов сцДНК, и комбинации статистических методов и моделирования. Также следует отметить, что настоящее изобретение имеет диагностическую ценность в отношении рака легкого I, II и III стадий без учета информации об иных опухоль-ассоциированных биомаркерах плазмы крови, таких как раковый эмбриональный антиген, что имеет важное преимущество перед ближайшим аналогом.
Список иллюстраций:
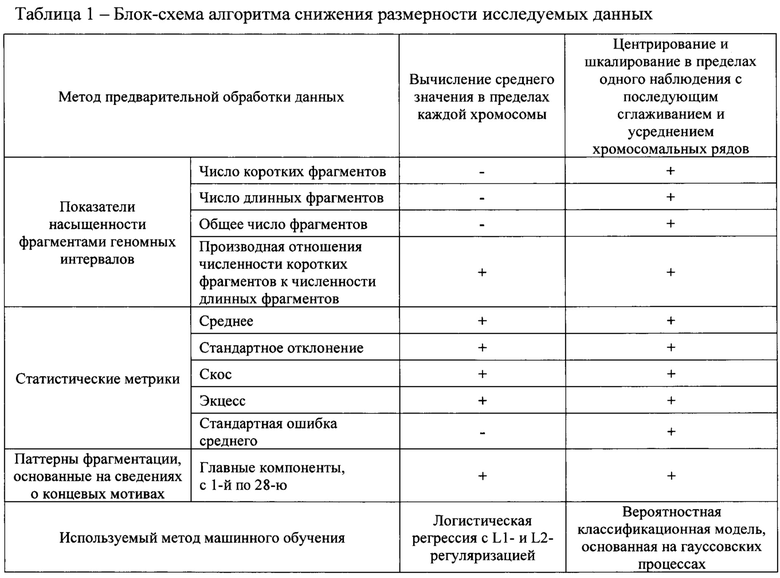
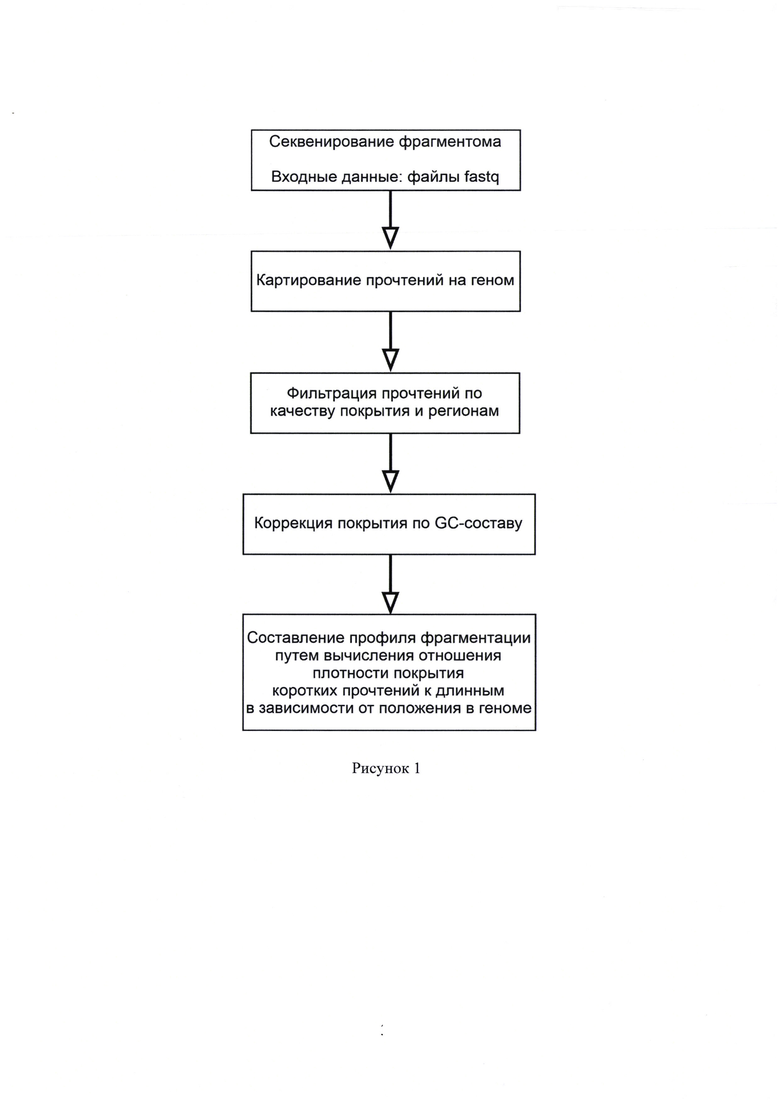
1. Рисунок 1 - Блок-схема алгоритма биоинформатической обработки NGS-данных о фрагментоме сцДНК.
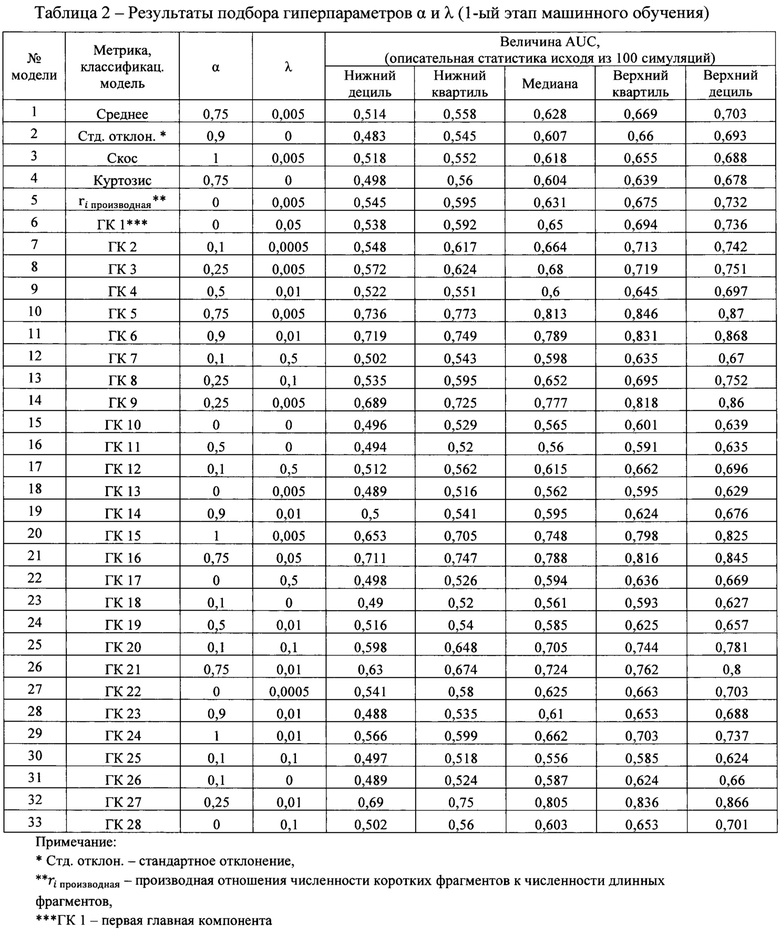
2. Рисунок 2 - Схематичное представление позиционно-весовой матрицы. По вертикали расположены нуклеотиды, а по горизонтали представлены позиции в выравнивании последовательности концевых мотивов фрагментов сцДНК.
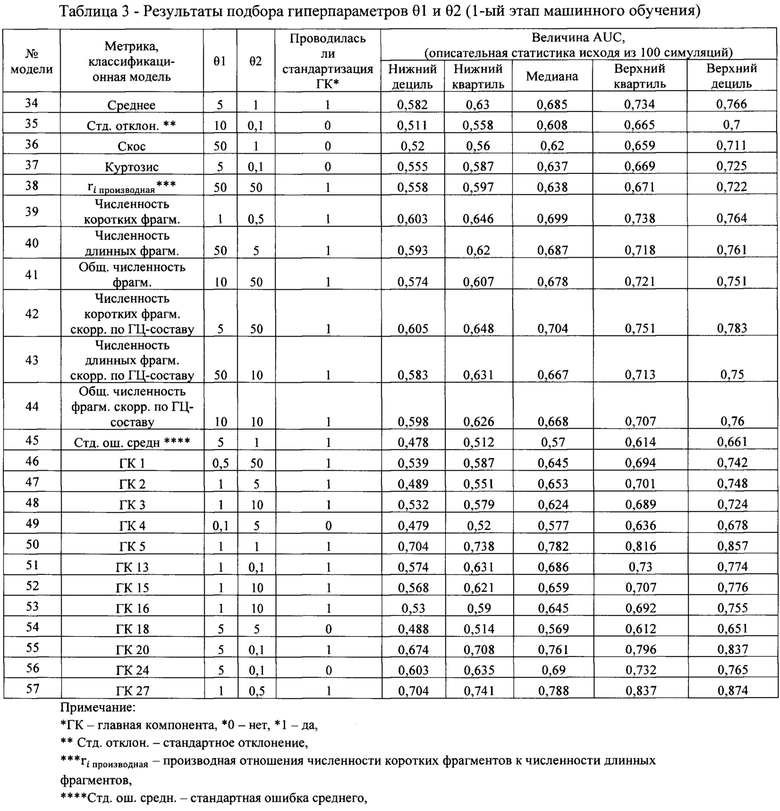
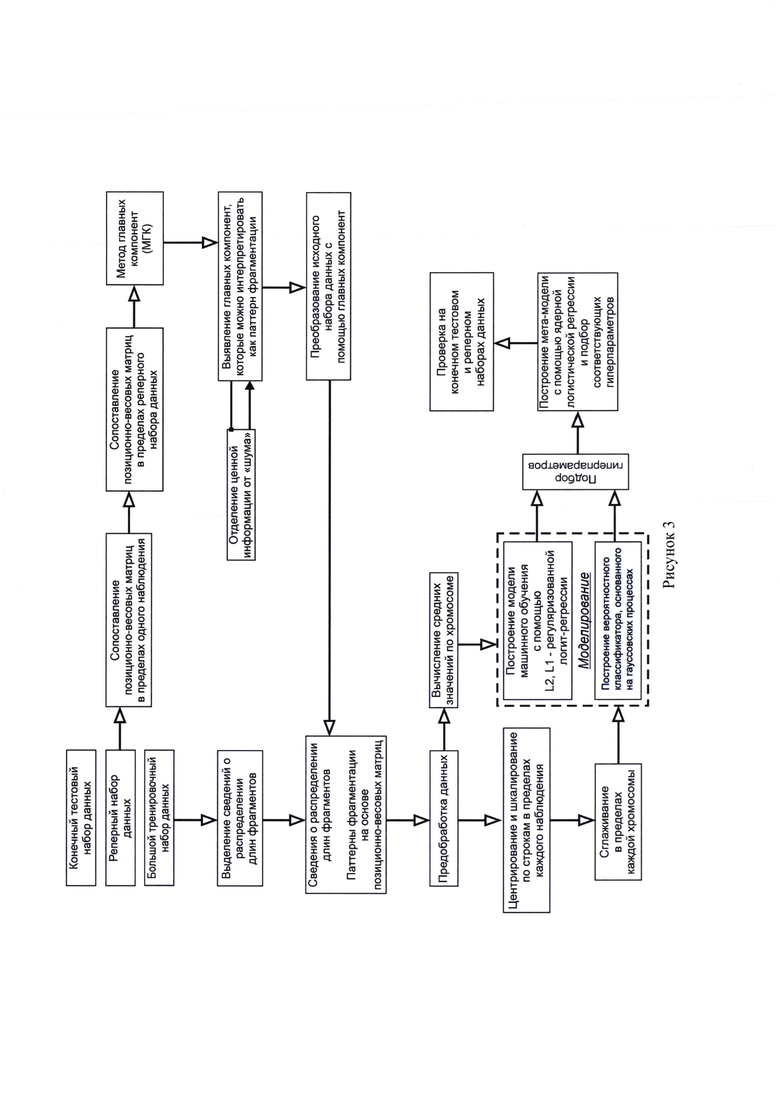
3. Рисунок 3 - Блок-схема алгоритма статистической обработки данных о фрагментоме сцДНК.
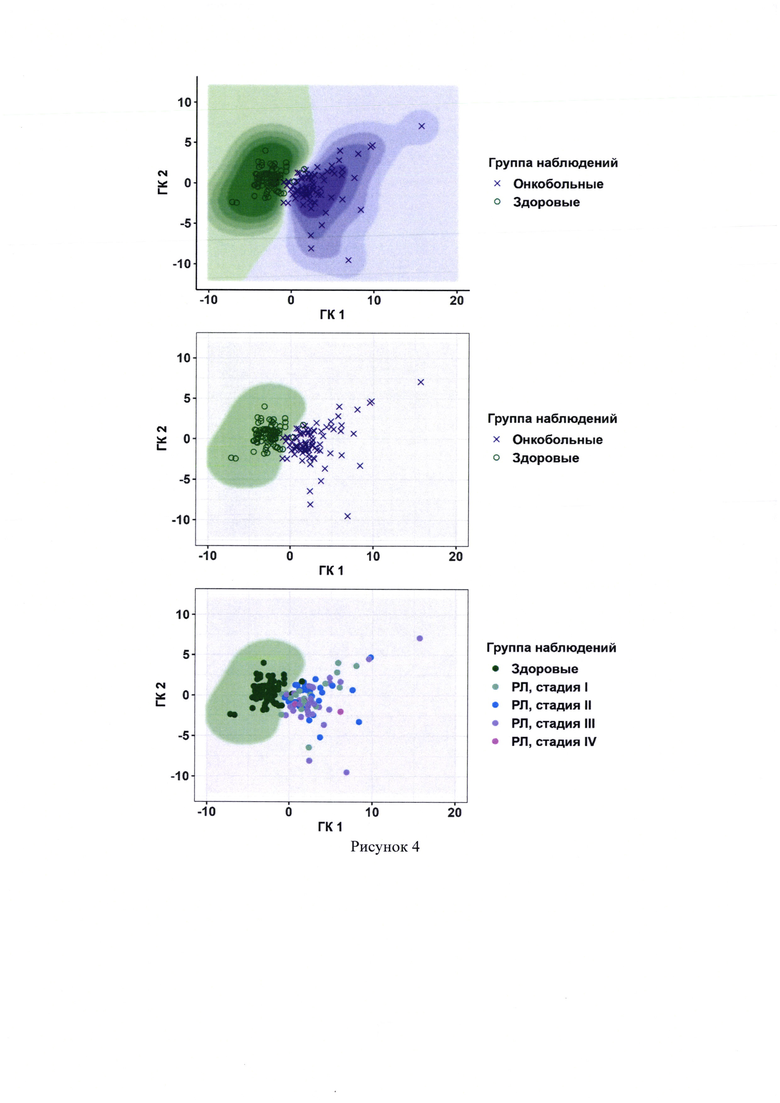
4. Рисунок 4 - Расположение наблюдений тренировочного набора данных в координатах главных компонент.Показаны результаты работы модели II этапа, использующей математический аппарат ядерной логистической регрессии. Оттенками фиолетового и зеленого показаны участки, расположение в которых указывает на высокую и низкую вероятность наличия рака легкого (РЛ) у пациента, соответственно. Зеленые точки соответствуют здоровым, фиолетовые точки - онкобольным. На нижнем и среднем графиках серая область соответствует значению метрики р, большему 0,35. Пациенты со значением метрики р выше либо равным 0,35 считаются онкобольными, пациенты со значением метрики р ниже 0,35 считаются здоровыми.
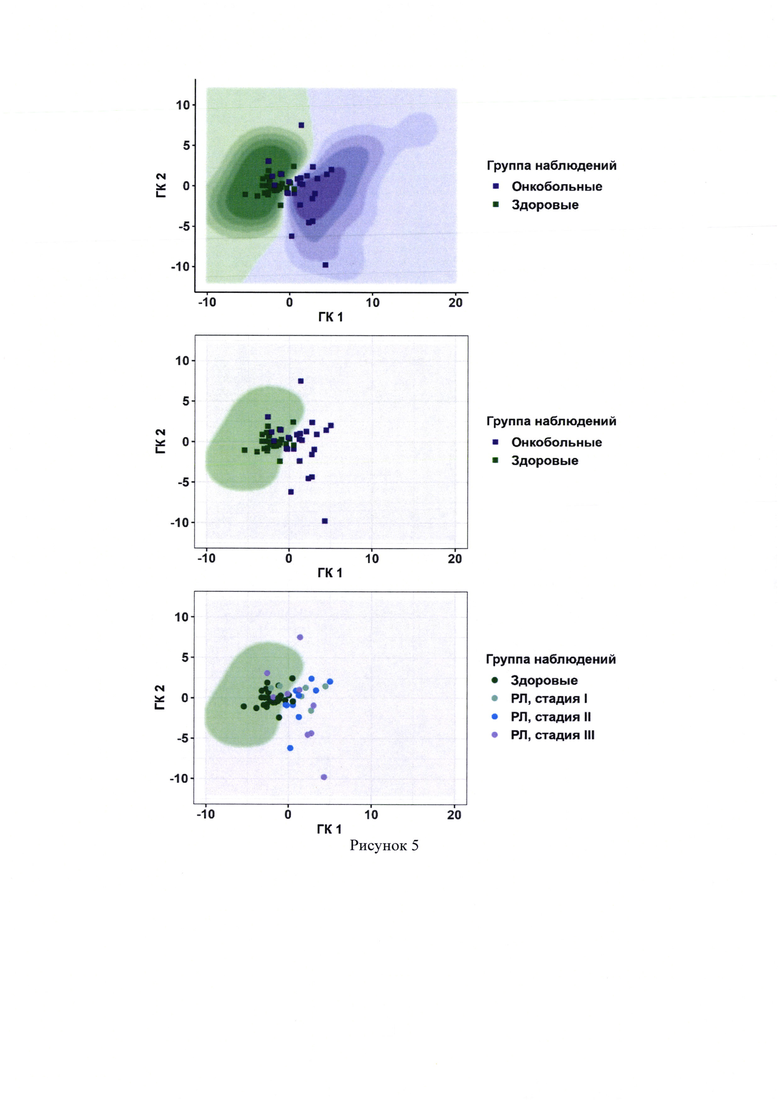
5. Рисунок 5 - Расположение наблюдений тестового набора данных в координатах главных компонент.Показаны результаты работы модели II этапа, использующей математический аппарат ядерной логистической регрессии. Оттенками фиолетового и зеленого показаны участки, расположение в которых указывает на высокую и низкую вероятность наличия рака легкого (РЛ) у пациента соответственно. Точки соответствуют наблюдениям тестового набора данных. Зеленые точки соответствуют здоровым, фиолетовые точки - онкобольным (пациенты с РЛ). На нижнем и среднем графиках серая область соответствует значению метрики р, большему 0,35. Пациенты со значением метрики р выше либо равным 0,35 считаются онкобольными, пациенты со значением метрики р ниже 0,35 считаются здоровыми.
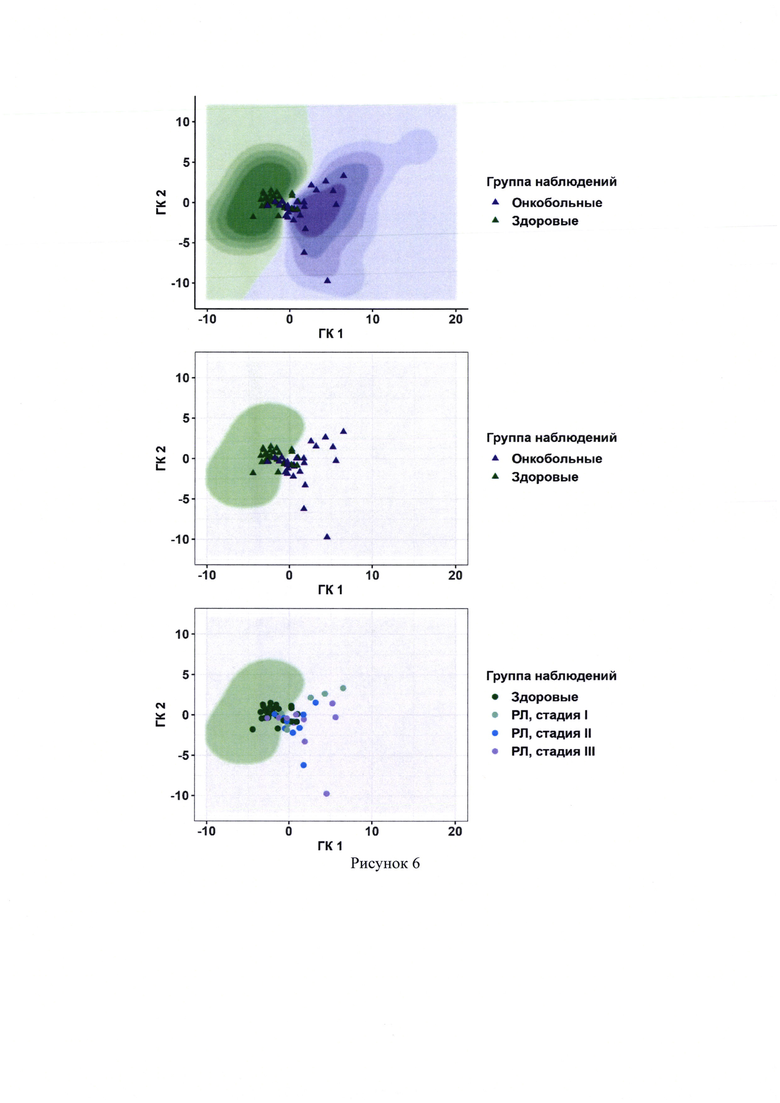
6. Рисунок 6 - Расположение наблюдений реперного набора данных в координатах главных компонент. Показаны результаты работы модели II этапа, использующей математический аппарат ядерной логистической регрессии. Оттенками фиолетового и зеленого показаны участки, расположение в которых указывает на высокую и низкую вероятность наличия рака легкого (РЛ) у пациента соответственно. Точки соответствуют наблюдениям реперного набора данных. Зеленые точки соответствуют здоровым, фиолетовые точки - онкобольным (пациенты с РЛ). На нижнем графике серая область соответствует значению метрики р, большему 0,35. Пациенты со значением метрики р выше либо равным 0,35 считаются онкобольными, пациенты со значением метрики р ниже 0,35 считаются здоровыми.
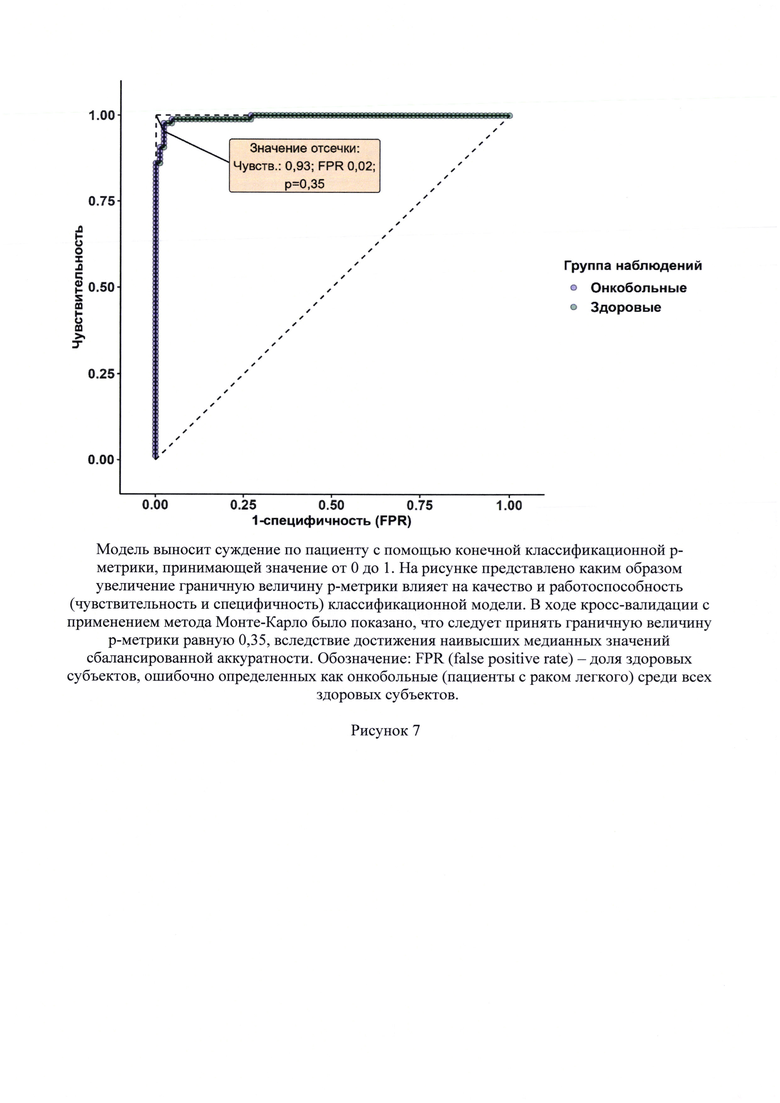
7. Рисунок 7 - ROC-кривая, демонстрирующая применение модели на тренировочной выборке.
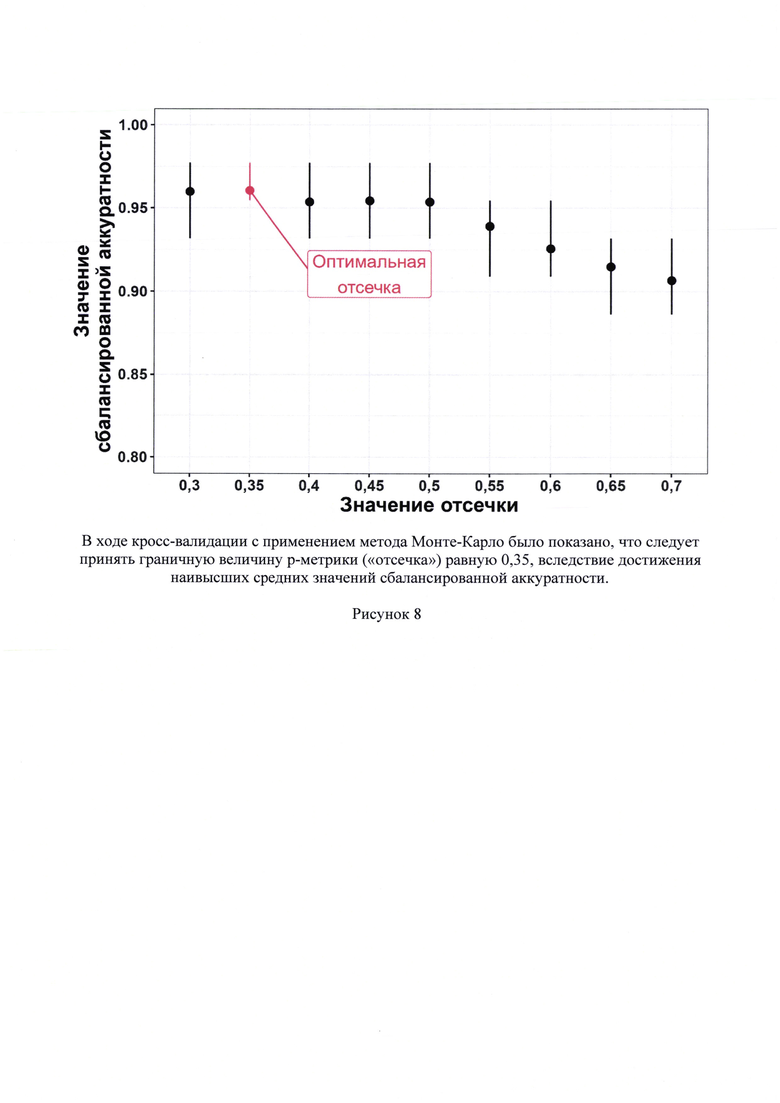
8. Рисунок 8 - Графическое представление результата поиска оптимальной граничной величины р-метрики на тренировочной выборке. Точками обозначены средние значения сбалансированной аккуратности, нижней и верхней вертикальной чертой обозначается соответственно нижний и верхний квартиль сбалансированной аккуратности.
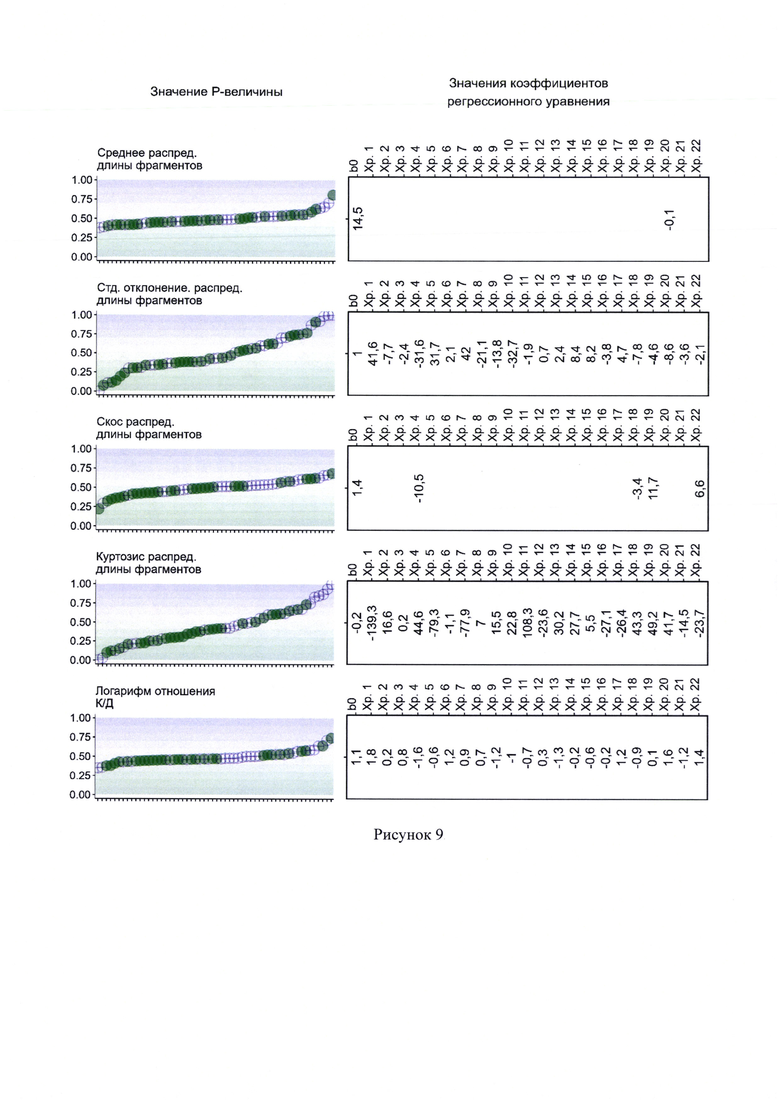
9. Рисунок 9 - Результаты работы классификационных моделей I этапа, основанных на логистической регрессии с L1 и L2-регуляризации. По правой стороне расположены группы переменных и соответствующие им коэффициенты логистических регрессионных моделей. Пробел соответствует отсутствию влияния переменной на результат предсказания модели. По левой стороне представлены результаты проверки классификационных моделей I этапа на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые перекрестья - онкобольным (пациенты с раком легкого). Наблюдения тестовой выборки выстроены по горизонтальной оси в порядке возрастания вероятности наличия РЛ, рассчитанной с помощью соответствующей классификационной модели I этапа. Структура рисунка 9 и обозначения, принятые на нем, аналогичны также для рисунков 10-13.
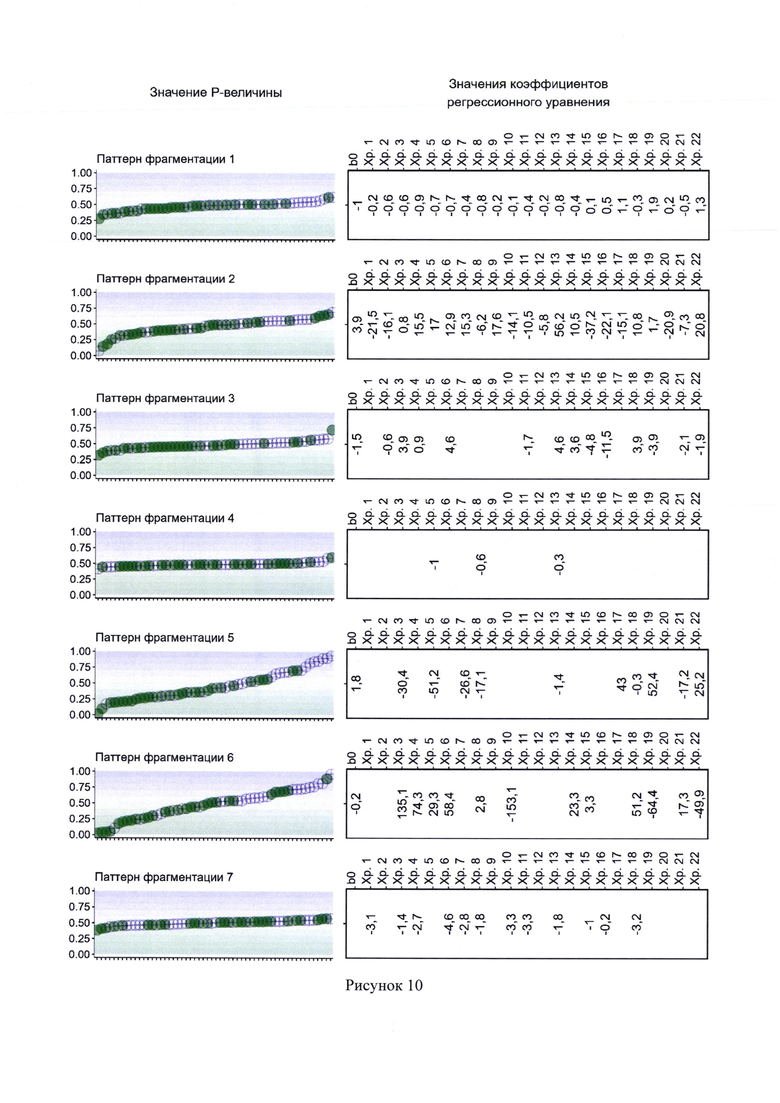
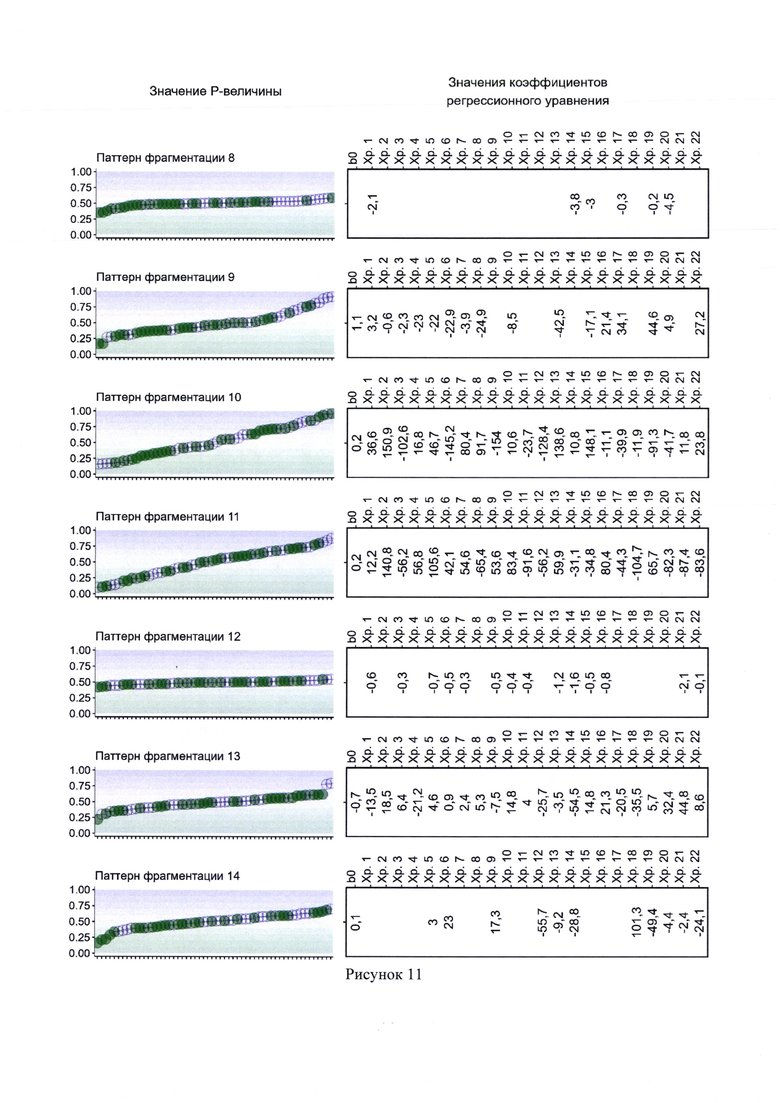
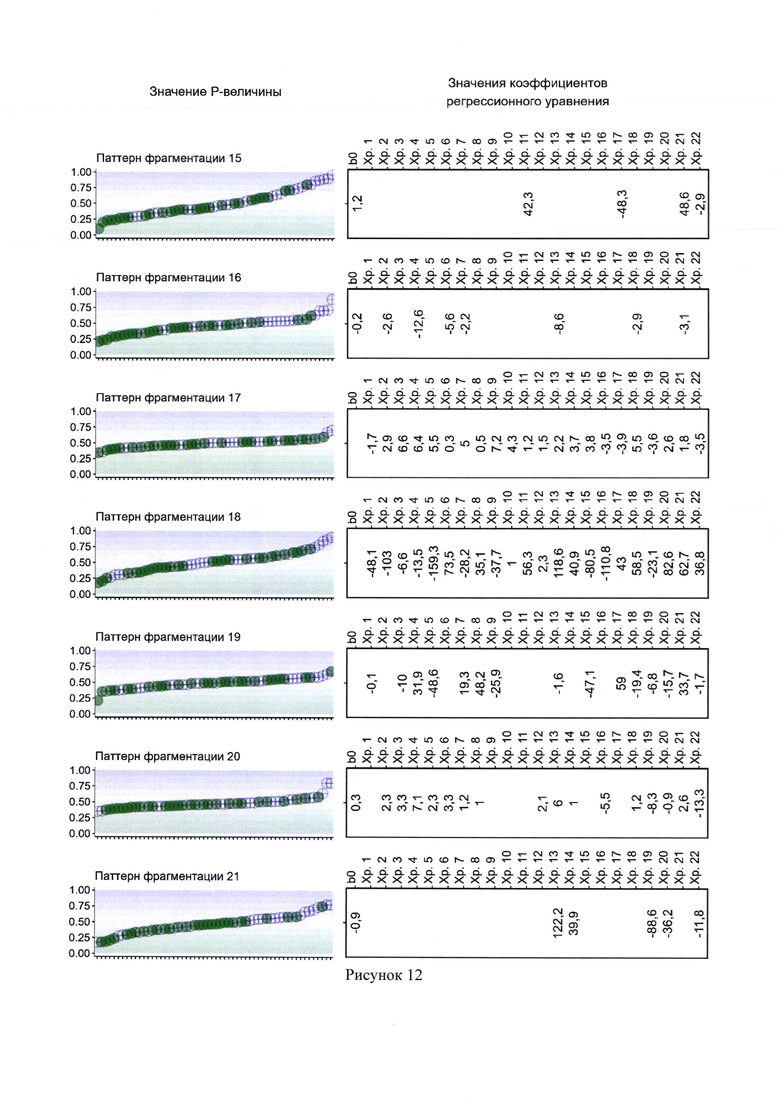
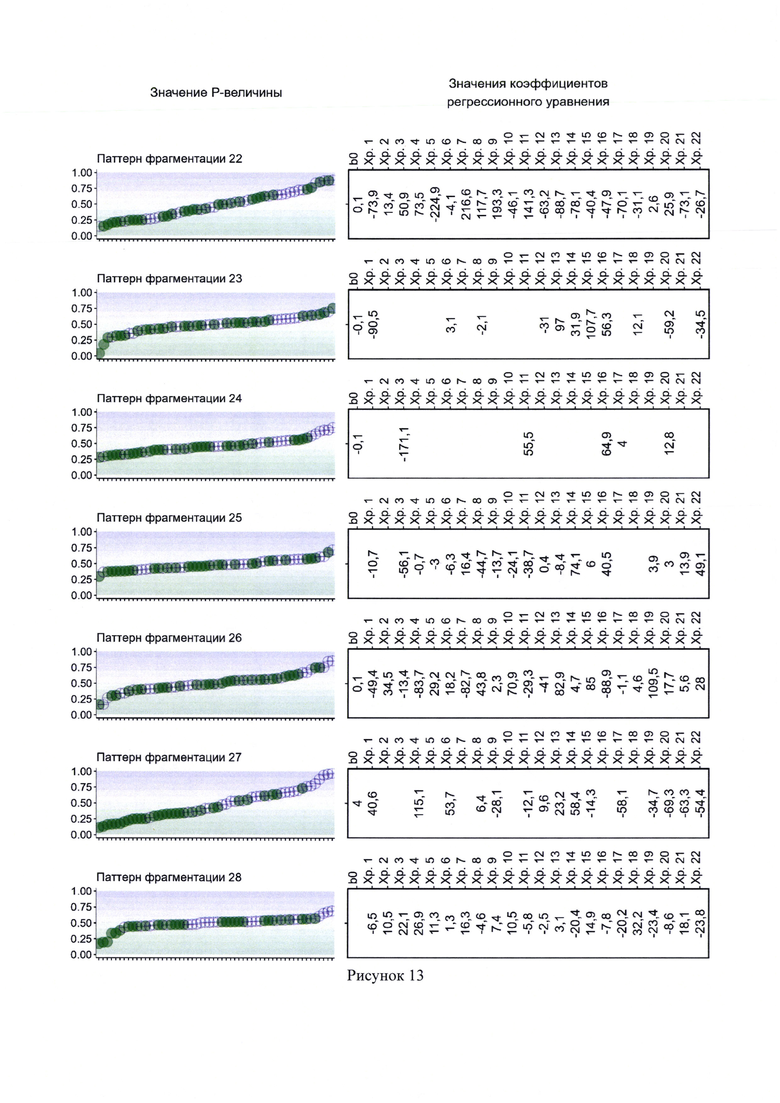
10. Рисунок 10 - Результаты работы классификационных моделей I этапа, основанных на логистической регрессии с L1 и L2-регуляризации. Для каждой модели представлены коэффициенты при переменных. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые перекрестья - онкобольным (пациенты с раком легкого).
11. Рисунок 11 - Результаты работы классификационных моделей I этапа, основанных на логистической регрессии с L1 и L2-регуляризации. Для каждой модели представлены коэффициенты при переменных. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые перекрестья - онкобольным (пациенты с раком легкого).
12. Рисунок 12 - Результаты работы классификационных моделей I этапа, основанных на логистической регрессии с L1 и L2-регуляризации. Для каждой модели представлены коэффициенты при переменных. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые перекрестья - онкобольным (пациенты с раком легкого).
13. Рисунок 13 - Результаты работы классификационных моделей I этапа, основанных на логистической регрессии с L1 и L2-регуляризации. Для каждой модели представлены коэффициенты при переменных. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые перекрестья - онкобольным (пациенты с раком легкого).
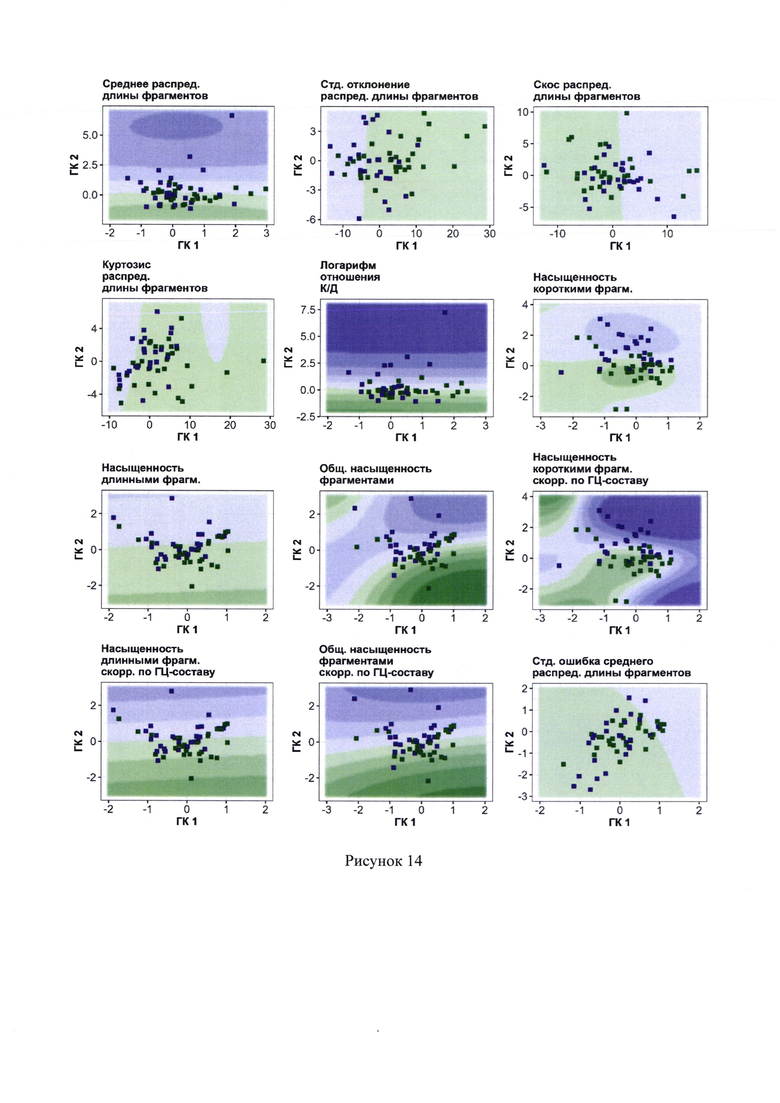
14. Рисунок 14 - Результаты работы моделей I этапа, представляющих собой вероятностные классификаторы, основанные на гауссовских процессах. Оттенками фиолетового и зеленого показаны участки, расположение в которых, указывает на высокую и низкую вероятность наличия рака легкого (РЛ) у пациента соответственно. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые точки - онкобольным (пациенты с РЛ). Структура рисунка 14 и обозначения, принятые на нем, аналогичны также для рисунка 15.
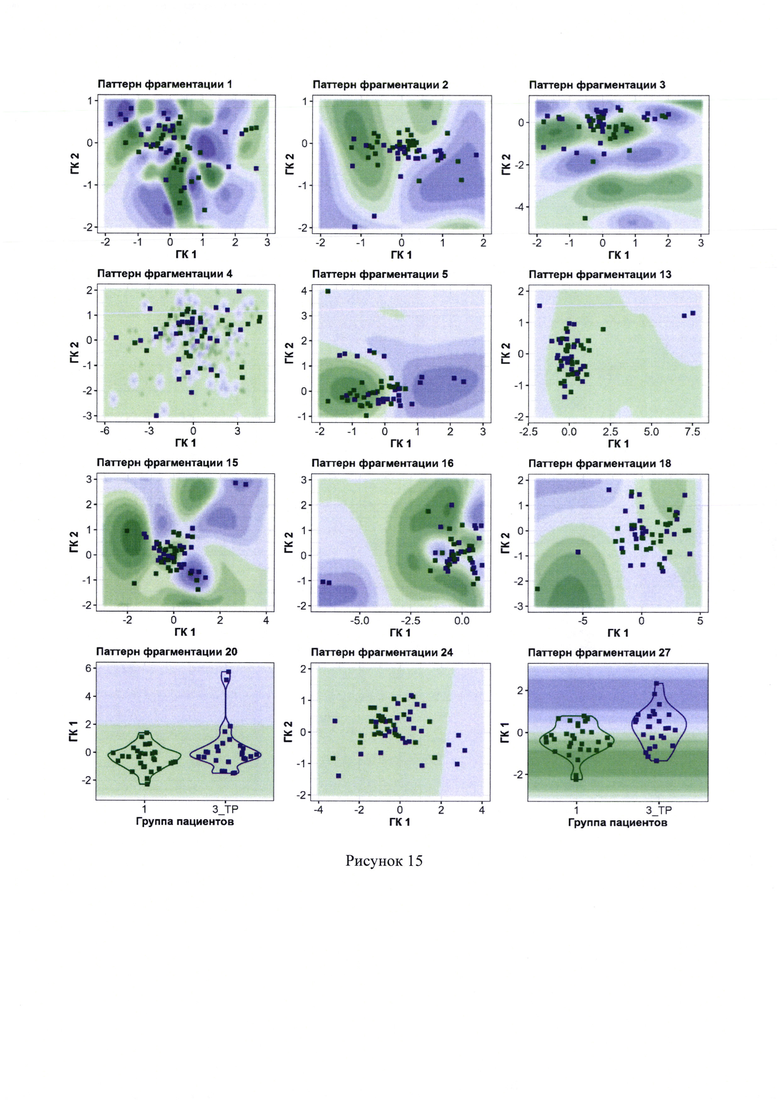
15. Рисунок 15 - Результаты работы моделей I этапа, представляющих собой вероятностные классификаторы, основанные на гауссовских процессах. Оттенками фиолетового и зеленого показаны участки, расположение в которых, указывает на высокую и низкую вероятность наличия рака легкого (РЛ) у пациента соответственно. Точки обозначают результаты проверки модели на тестовой выборке. Зеленые точки соответствуют здоровым, фиолетовые точки - онкобольным (пациенты с РЛ).
ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Задача изобретения состояла в разработке способа малоинвазивной диагностики РЛ, осуществляемого через обработки массива NGS-данных о всей совокупности фрагментов сцДНК, выделенных из плазмы крови субъекта обследования. Создаваемая классификационная модель должна хорошо различать вариант нормы и случаи с установленным РЛ на разных стадиях, в том числе и на ранних стадиях (I-II стадии), только по профилю фрагментации сцДНК и профилю последовательностей концевых мотивов фрагментов сцДНК со следующими основными операционными характеристиками: площадь под ROC-кривой >0,7 [Metz С.Е. Basic principles of ROC analysis // Semin Nucl Med. - 1978. - V. 8. - No. 4. - P. 283-298] и сбалансированной аккуратностью (от англ. balanced accuracy)>0,7 [Rai S.N., et al. Multi-group diagnostic classification of high-dimensional data using differential scanning calorimetry plasma thermograms // PLoS One. - 2019. - V. 14. - No. 8. - P. e0220765; Yildiz P.B., et al. Diagnostic accuracy of MALDI mass spectrometric analysis of unfractionated serum in lung cancer // J Thorac Oncol. - 2007. - V. 2. - No. 10. - P. 893-901].
Поставленная задача решается тем, что предлагаемый способ осуществляется через уникальный алгоритм биоинформатической и статистической обработки массива NGS-данных о фрагментоме сцДНК из плазмы крови. Алгоритм включает следующие основные этапы: картирование чтений совокупности фрагментов ДНК на референсный геном человека (hg38); разделение генома на участки длиной в 100 тыс.п.н., внутри которых проводится подсчет количества коротких и длинных фрагментов сцДНК; коррекция фрагментов на содержание гуанина и цитозина; составление набора позиционно-весовых матриц для участка генома, содержащих информацию о пятинуклеотидных мотивах с обоих концов фрагмента сцДНК, по которым чаще всего происходит фрагментация ДНК внутри этого участка; описание профиля фрагментации сцДНК с помощью линейных комбинаций значений позиционного-весовых матриц; описание профиля фрагментации ДНК с помощью характеристик распределения длин фрагментов ДНК в расчете на геномный интервал; построение классификационных моделей с помощью логистической регрессии с L1- и L2-регуляризацией; создание вероятностных классификационных моделей, основанных на гауссовских процессах; построение конечной классификационной модели с использованием ядерной логистической регрессии.
Достигаемый технический результат заключается в создании способа малоинвазивной диагностики РЛ, в том числе и на ранних стадиях, по профилю фрагментации сцДНК и профилю последовательностей концевых мотивов фрагментов сцДНК, выделенных из плазмы крови, на основе: 1) алгоритма с уникальной комбинацией методов машинного обучения; 2) учета позиционно-весовых матриц; 3) учета данных о длинах фрагментов сцДНК и их соотношении. Важно подчеркнуть, что по перечисленным пунктам реализуется основное преимущество технического результата перед аналогами и обеспечивается его новизна.
Первое преимущество технического результата реализуется в определении абсолютной длины всех фрагментов сцДНК, что позволяет повысить диагностическую ценность классификационной модели по сравнению с аналогами, которые определяют мутационный и/или метиломный профиль. Следует заметить, что в выборках пациентов частоты отдельных патогенных генетических вариантов и сайтов аномального метилирования обычно варьируют ввиду гетерогенности молекулярных подтипов ЗНО и лабильности эпигенетических биомаркеров, соответственно.
Второе преимущество технического результата реализуется в относительно более быстром и дешевом варианте классификации субъекта обследования вне зависимости от пола и возраста на здорового или онкобольного, что достигается путем обработки NGS-данных только одного типа детектируемых биомолекул - фрагментов сцДНК, без сбора информации о клинико-анамнестических характеристиках субъекта, без использования вспомогательных данных о генетических вариантах, о метилировании промоторных областей генов, об изменении копийности отдельных генов, о количественной оценке протеомных или метаболомных биомаркеров и иных известных биомаркеров в плазме крови.
Третье преимущество технического результата заключается в малоинвазивном способе забора образцов крови от субъекта и низкой вероятности возникновения осложнений при такого рода манипуляциях.
Ввиду вышесказанного, есть целесообразность внедрения рассматриваемого технического решения в существующий набор анализов для диагностики РЛ после клинической валидации предлагаемого способа. Так принадлежность субъекта обследования к классу «онкобольные» по результатам NGS-анализа фрагментов сцДНК плазмы крови может привести к дальнейшей верификации диагноза посредством компьютерной томографии, что выгодно с точки зрения раннего выявления ЗНО; и наоборот, визуализация неоднозначно интерпретируемых очаговых образований в ткани легкого, сопряженная с получением информации о принадлежности субъекта обследования к классу «здоровые» по результатам NGS-анализа, может способствовать принятию врачебного решения в пользу выполнения мониторинга пациента, вместо проведения травматичной процедуры биопсии легкого. Таким образом, промышленная применимость настоящего способа связана с расширением арсенала способов диагностики РЛ.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Алгоритм обработки данных о фрагментоме сцДНК состоит из биоинформатической обработки NGS-данных (часть I) и статистической обработки данных (часть II). Конечной целью алгоритма является создание классификационной модели (классификатора), которая позволяет различить вариант нормы и наличие РЛ по профилю фрагментации сцДНК и профилю последовательностей концевых мотивов фрагментов сцДНК.
Алгоритм обработки данных секвенирования фрагментов сцДНК 1. Биоинформатическая обработка NGS-данных (Часть I)
Первой характеристикой фрагментации генома является профиль распределения фрагментов сцДНК по длинам и их соотношение. Для трансформации NGS-данных о фрагментоме сцДНК плазмы крови в характеристики фрагментации генома необходимо выполнить несколько этапов обработки выходных NGS-данных. Во-первых, чтения фрагментома сцДНК картируют на референсный геном человека (hg38). Картированные чтения фильтруют по качеству картирования (чтения с показателем качества картирования MAPQ<=30 подлежат удалению), также удаляют ПЦР-дубликаты и чтения, картированные на участки неоднозначного картирования, такие как теломеры, центромеры и участки, попадающие в ENCODE Blacklist [Amemiya, Н.М., et al. The ENCODE Blacklist: Identification of Problematic Regions of the Genome // Sci Rep.- 2019. - V. 9. - P. 9354]. Портал ENCODE Blacklist содержит полный набор областей генома человека, которые имеют аномальный, неструктурированный или высокий сигнал в NGS-анализе, независимо от клеточной линии или особенностей эксперимента. Во-вторых, геном разделяют на участки длиной в 100 тыс.п.н., внутри которых подсчитывают количества коротких (100-150 п.н.) и длинных (150-220 п.н.) фрагментов. В-третьих, полученные количества фрагментов корректируют на содержание гуанина и цитозина согласно протоколу, описанному в статье Benjamini и соавторов [Benjamini Y, et al. Summarizing and correcting the GC content bias in high-throughput sequencing // Nucl Acids Res. - 2012. - V. 40. - No. 10. - P. е72]. Исследуют локальную полиномиальную регрессию [Cleveland W.S. Robust Locally Weighted Regression and Smoothing Scatterplots // JASA. - 1979. - V. 74. - No. 368. - P. 829-836] зависимости количества фрагментов в участке генома от содержания гуанина и цитозина. Затем из реального числа фрагментов вычитают предсказанное регрессией количества фрагментов и к полученному числу прибавляют медианное число фрагментов по всем участкам генома. Данную процедуру проводят по отдельности для коротких и длинных фрагментов. Полученные скорректированные количества коротких и длинных фрагментов образуют профиль фрагментации вместе с нескорректированным распределением длин фрагментов в диапазоне от 100 п.н. до 220 п.н. внутри тех же участков генома. Данный способ для построения профиля фрагментации отличается от метода DELFI (DNA evaluation of fragments for early interception), описанного в статье Cristiano и соавторов [Cristiano S., et al. Genome-wide cell-free DNA fragmentation in patients with cancer // Nature. - 2019. - V. 570. - P. 385-389] тем, что конечным результатом DELFI является набор отношений числа коротких к длинным фрагментов, которые проходят двойную коррекцию на содержание гуанина и цитозина (ГЦ-состав или GC-состав): вначале коррекцию применяют к числу коротких и длинных фрагментов, затем коррекцию применяют к полученному отношению скорректированного числа фрагментов. В настоящем изобретении коррекцию применяют единственный раз, и ее результатом являются количества фрагментов в двух диапазонах длин. Схематическое представление алгоритма в части биоинформатической обработки NGS-данных показано на рисунке 1.
Второй характеристикой фрагментации генома является набор позиционно-весовых матриц (ПВМ), которые определяют по способу, описанному в статье Claverie и соавторов [Claverie J.M. & Audic S. The statistical significance of nucleotide position-weight matrix matches // Comput Appl Biosci. - 1996. - V. 12. - No. 5. - P. 431-439]. Создание ПВМ - биоинформатический метод, который применяется для поиска мотивов в биологических последовательностях. ПВМ представляет собой матрицу, количество строк которой соответствует размеру алфавита (4 нуклеотида для нуклеиновых кислот), а количество столбцов - длине мотива. Итак, ПВМ характеризует последовательности концевых мотивов фрагментов внутри участков генома длиной 100 тыс. п.н. ДНК. Из каждого участка генома длиной 100 тыс.п.н. извлекают концевые мотивы фрагментов сцДНК длиной 5 п.н. с 5` - и 3`-концов. Из этих концевых мотивов для каждого участка генома составляют выравнивание, которое затем преобразуют в ПВМ следующим образом: в каждой позиции выравнивания подсчитывают частоты встречаемости каждого нуклеотида. Полученные частоты делят на частоты соответствующих нуклеотидов во всем выравнивании, после чего результат логарифмируют по основанию 2. Пример итоговой ПВМ представлен на рисунке 2. Таким образом, ПВМ участка генома содержит информацию о мотивах, по которым чаще происходит фрагментация ДНК внутри этого участка. Для одного анализируемого образца составляют набор таких ПВМ по всем участкам генома.
2. Статистическая обработка данных (часть II)
Блок-схема алгоритма в части статистической обработки преобразованных NGS-данных, полученных по окончании биоинформатической обработки данных (Часть I) представлена на рисунке 3. Конечной точкой статистической обработки данных является бинарная классификационная модель, которая позволяет различить классы здоровых субъектов и онкобольных по профилю фрагментации сцДНК и профилю последовательностей концевых мотивов фрагментов сцДНК. Итак, классификационная модель рассматривает каждый исследуемый образец как отдельно взятое наблюдение, описываемое множеством переменных. Данные, предоставленные для построения классификационной модели, первоначально были организованы в виде отдельных таблиц в формате «csv». Исследуемому образцу, взятому от определенного субъекта, соответствует одна таблица со сведениями о распределении длин фрагментов ДНК на протяжении всего генома и одна ПВМ.
Целевой фрагментом сцДНК представляет собой совокупность фрагментов ДНК длиной от 100 до 220 п.н., картированных на геном. В свою очередь, геном разделен на 26460 неперекрывающихся интервалов. По результатам секвенирования, внутри каждого интервала обнаруживается определенное количество фрагментов ДНК. Множество фрагментов ДНК внутри одного геномного интервала можно охарактеризовать сразу с двух сторон. Во-первых, описать распределение длин этих фрагментов с помощью таких статистических метрик как среднее, стандартное отклонение, скос (асимметрия), куртозис (коэффициент эксцесса, числовая характеристика степени остроты пика распределения случайной величины), стандартная ошибка среднего. Во-вторых, с помощью ПВМ дается характеристика концевых мотивов всех фрагментов, присутствующих в том или ином геномном интервале.
Таким образом, описание отдельно взятого геномного интервала для отдельно взятого образца представляет собой таблицу, состоящую из одной строки и 52 столбцов, из которых: 40 принадлежат ПВМ; 3 столбца - это общее количество фрагментов, число прочитанных коротких фрагментов (100-150 п.н.) и число прочитанных длинных фрагментов (151-220 п. н); 3 столбца - это общее количество фрагментов, число прочитанных коротких фрагментов, число прочитанных длинных фрагментов (все эти показатели скорректированы с учетом изменения содержания гуанина и цитозина на протяжении всего генома); 5 столбцов - это среднее, стандартное отклонение, скос (асимметрия), куртозис и стандартная ошибка среднего выборочного распределения длин фрагментов; 1 столбец - это производная величина от логарифма отношения числа коротких фрагментов к числу длинных фрагментов.
Теоретически возможна такая ситуация, что для определенного наблюдения на отдельно взятом участке генома могут быть отмечены в весьма малом количестве или вовсе отсутствовать либо длинные, либо короткие фрагменты. В таком случае, натуральный логарифм отношения количества прочитанных коротких и длинных фрагментов может принимать сколь угодно большие положительные или отрицательные значения. Следовательно, необходимо ограничение значений производной величины как сверху, так и снизу, и, поэтому, если значение логарифма оказывалось менее -10, то оно заменялось на -10, если же оно оказывалось больше 10, то оно также заменялось на 10.
Если умножить число столбцов, характеризующих отдельно взятый геномный интервал на число геномных интервалов, то становится ясно, что одному наблюдению сопоставляется более чем миллионное количество переменных. Возникает задача снижения размерности, которая решается отдельно для метрик выборочного распределения длин фрагментов и отдельно для переменных, порождаемых ПВМ.
2.1 Снижение размерности метрик выборочного распределения длин фрагментов
Снижение размерности метрик выборочного распределения длин фрагментов сцДНК предоставляет в итоге 110 переменных. Для каждой из 22 хромосом (аутосом) рассматривается 5 метрик (среднее, стандартное отклонение, скос и куртозис выборочного распределения длин фрагментов, а также производная величина от логарифма отношения числа коротких фрагментов к числу длинных фрагментов). Далее для более полного описания профиля фрагментации сцДНК ряды метрик были преобразованы в хромосомальные ряды. Для отдельно взятой метрики в пределах каждого наблюдения рассчитывали медианное значение и межквартильный размах метрики по всему геному. Описываемое преобразование, называемое центрированием и шкалированием, затрагивает каждую метрику и каждое наблюдение по отдельности и основывается только на тех данных, что охватываются отдельно взятой метрикой и наблюдением. Затем, набор статистических метрик Zi,metric,chr подразделяют на хромосомальные ряды. Единая таблица, состоящая из одного ряда и множества столбцов, подразделяется на 22 отдельные таблицы, по числу хромосом. Внутри каждой таблицы последовательно проводят две операции:
1) по хромосомальному ряду несколько раз проходит скользящее среднее с шириной окна в два наблюдения. Последовательно, несколько раз осуществляется скользящее сглаживание. Каждый раз, после прохождения скользящего среднего, длина хромосомального ряда уменьшается на одно наблюдение. Когда длина хромосомального ряда станет кратной десяти, прохождение скользящего среднего прекращается.
2) хромосомальный ряд делят на неперекрывающиеся участки шириной в 10 наблюдений. Поскольку длина хромосомального ряда кратна 10, он оказывается полностью покрыт этими неперекрывающимися участками. Внутри каждого участка вычисляют среднее. Полученные средние образуют новый хромосомальный ряд, являющийся своего рода «сверткой» своего предшественника. Преобразованные хромосомальные ряды соединяют в новый набор переменных Ui,metric. В результате, для i-го наблюдения формируется новый набор переменных:
Ui,metric={Xi,metric,среднее1, …, Xi,metric,среднее22, …, Ui,metric,chr1,1, …, Ui,metric,chr22,h},
где {Xi,metric,среднее1, …, Xi,metric,среднее22} - среднее значение метрики с 1-й по 22-ю хромосому, {Ui,metric,chr1,1, …, Ui,metric,chr22,h} - результат преобразования хромосомальных рядов, h - конечная длина набора переменных, который был получен в ходе преобразования хромосомальных рядов.
2.2 Снижение размерности позиционно-весовых матриц (ПВМ)
NGS-данные разбиты на 26460 неперекрывающихся интервалов и каждый интервал характеризуется схожей структурой данных. Каждый интервал описывается ПВМ, сформированной из одних и тех же 40 переменных. Возникает вопрос, можно ли в каждом интервале заменить эту формирующую матрицу на ее аналог меньшей размерности, сохранив при этом как можно больше представленной в ней информации. Подобное преобразование возможно выполнить с помощью модификации метода главных компонент (МГК), то есть для каждого наблюдения и для каждого геномного интервала вычисляют одинаковые линейные комбинации исходных переменных, составлявших ПВМ. МГК является технологией многомерного статистического анализа, используемой для сокращения размерности пространства признаков с минимальной потерей полезной информации. Таким образом, исходный набор данных для отдельно взятого наблюдения состоял из 26460 позиционно-весовых матриц и обладал размерностью в одну строку и 26460*40 столбцов. Преобразованный набор данных для отдельно взятого наблюдения стал матрицей размерностью 26460 строк и 40 столбцов. Размерность такой матрицы может быть еще снижена при помощи МГК согласно блок-схеме, представленной в таблице 1.
2.3 Создание классификационных моделей
При создании классификационной модели использовали метамоделирование (модель, используемая для создания прототипа моделирования классификационной модели). Метамоделирование заключается в том, что на первом этапе создается несколько десятков относительно простых в интерпретации классификационных моделей, каждая из которых по отдельности вполне может демонстрировать посредственные результаты классификации субъектов. Кроме того, модель, создаваемая на первом этапе, использует далеко не весь набор признаков, а охватывает какую-либо его часть, например, определенную статистическую метрику или главную компоненту, отражающую профиль фрагментации. На втором этапе результаты моделей первого этапа используют в качестве переменных для построения конечной модели.
2.3.1 Создание классификационных моделей первого этапа машинного обучения
Классификационные модели первого этапа машинного обучения были основаны либо на логистической регрессии с L1- и L2-регуляризацией либо представляли собой вероятностные классификационные модели, основанные на гауссовских процессах.
Назначение логистической регрессии состоит в анализе связи между несколькими независимыми переменными (называемыми также регрессорами или предикторами) и зависимой переменной. L1-регуляризация способствует разреженности функции, когда лишь немногие факторы не равны нулю. L2-регуляризация способствует появлению малых весовых коэффициентов модели, но не способствует их точному равенству нулю. Классификационные модели, реализующие алгоритм логистической регрессии с регуляризацией, использовали в качестве независимых переменных средние значения метрик в пределах каждой хромосомы. Все классификационные модели этой разновидности на входе обрабатывали массив из 22 переменных. Каждая классификационная модель специализируется на отдельно взятой метрике и переменные, которые она обрабатывает, были ограничены исключительно этой метрикой. Результатом логистической регрессии является вероятность принадлежности к классу онкобольных. Для того, чтобы не допустить переобучения L1- и L2-регуляризованной логистической регрессии, было необходимо провести подбор гиперпараметров α и λ. Для решения этой задачи был проведен поиск гиперпараметров по сетке, сочетанный с кросс-валидацией по методу Монте-Карло. При рассмотрении одной комбинации гиперпараметров выполнялось 100 симуляций. Критерием качества классификации являлась площадь под ROC-кривой. ROC-кривая - это график, позволяющий оценить качество бинарной классификации и отображает соотношение между долей объектов от общего количества носителей признака, верно классифицированных как несущие признак, и долей объектов от общего количества объектов, не несущих признака, ошибочно классифицированных как несущие признак при варьировании порога решающего правила. Метод Монте-Карло, также известный как перекрестная проверка с разделением в случайном порядке и перекрестная проверка с повторной случайной подвыборкой, включает в себя разделение всех данных на обучающие данные и тестовые данные. Поскольку разделение выполняется независимо для каждого запуска, одна и та же точка может появляться в наборе тестов несколько раз, что является основным отличием Монте-Карло от перекрестной проверки. Следует также отметить, что разделение на обучающую и тестовую выборку являлось стратифицированным по стадии рака легкого, то есть в обучающей и тестовой выборке присутствовало одинаковое распределение по стадиям рака легкого.
Вероятностные классификационные модели, основанные на гауссовских процессах, использовали в качестве входящей информации массивы данных, полученные при сглаживании и усреднении хромосомальных рядов. Поскольку подобные массивы могут состоять из нескольких сотен переменных, то перед началом обучения классификационной модели проводили снижение размерности с помощью МГК. Размерность снижали до одной либо двух переменных. Структура данных была такова, что только первые две компоненты охватывали сколько-нибудь значимую долю разброса наблюдений в многомерном пространстве, остальные же компоненты, фактически, отражали шум. Для того, чтобы не допустить переобучения или, наоборот, недообучения, вероятностной классификационной модели было необходимо провести подбор гиперпараметров θ1, θ2, а также параметра стандартизации. Для решения этой задачи был проведен поиск по сетке, сочетанный с Монте-Карло кросс-валидацией. При рассмотрении одной комбинации гиперпараметров выполняли 100 симуляций. Разделение на обучающую и тестовую выборку при каждой симуляции являлось стратифицированным по стадии рака легкого. Критерием качества классификации являлась площадь под ROC-кривой.
2.3.2 Создание классификационных моделей второго этапа машинного обучения Предсказания моделей первого этапа были использованы в качестве переменных на втором этапе машинного обучения. Результатом логистической регрессии и вероятностной классификационной модели является вероятность. Далее эту метрику преобразовывали в логарифм отношения шансов (ОШ). ОШ представляет собой метрику, которая количественно определяет силу связи между двумя событиями из одной статистической совокупности. Поскольку ОШ не ограничена какими-либо пределами сверху и снизу, то во избежание появления бесконечных значений, ограничивали значение логарифма ОШ: по модулю его значение не превышало 10.
Результаты работы классификационной модели были сведены в таблицу, которая состояла из 57 столбцов (по числу классификаторов первого этапа моделирования), а в каждом столбце был представлен соответствующий логарифм ОШ. Эта таблица была, опять же, преобразована с помощью МГК, и первые две компоненты были использованы в качестве входящих переменных для финальной классификационной модели на основе ядерной логистической регрессии (позволяет проводить поиск нелинейного отношения между парой случайных величин). Для того, чтобы не допустить переобучения регрессионной модели, проводился подбор гиперпараметров λ и σ2. Для решения этой задачи был проведен поиск гиперпараметров по сетке сочетанный с кросс-валидацией по методу Монте-Карло. При рассмотрении одной комбинации гиперпараметров выполняли 100 симуляций. Разделение на обучающую и тестовую выборку при каждой симуляции являлось стратифицированным по стадии рака легкого. Критерием качества классификационной модели являлась площадь под ROC-кривой. Результатом работы финальной классификационной модели является конечная классификационная р-метрика, принимающая значение от 0 до 1. Физический смысл р-метрики заключается в том, что чем ближе значение этой метрики к 1, тем больше уверенности в том, что исследуемый субъект является онкобольным, и, напротив, чем ближе значение к нулю, тем больше уверенности, что субъект является здоровым.
Итак, прогнозирование статуса пациента осуществляется в два этапа.
На первом этапе статус пациента определяется с помощью 24 вероятностных классификационных моделей на основе гауссовских процессов и 33 классификаторов на основе логистической регрессии с L1 и L2-регуляризацией. Все эти классификаторы действуют независимо друг от друга. Результатом работы каждого из классификаторов является число, принимающее значение от -10 до +10. Чем выше значение этого числа, тем выше вероятность того, что у пациента РЛ. Таким образом, на первом этапе для отдельно взятого наблюдения создается 57 переменных, каждая из которых является индикатором статуса пациента.
На втором этапе, с помощью метода главных компонент, из этих 57 переменных рассчитываются две линейные комбинации, то есть две новые переменные, которые являются линейными комбинациями результатов работы классификаторов первого этапа.
В свою очередь, на основе этих двух линейных комбинаций, с помощью метода ядерной логистической регрессии рассчитывается конечная классификационная р-метрика, принимающая значения от 0 до 1. Конечная классификационная р-метрика является оценкой вероятности наличия у пациента РЛ.
Как следует из представленного ниже Примера 2, если р-метрика принимает значение ниже 0,35, то это означает, что пациент здоров. Значение р-метрики выше 0,35 включительно указывает на наличие у пациента РЛ. Граничное значение р-метрики, равное 0,35, было определено с помощью симуляционного моделирования посредством метода Монте-Карло. Было показано, что при таком граничном значении конечной р-метрики созданный нами классификатор демонстрирует наивысшие медианные значения сбалансированной аккуратности.
ПРИМЕРЫ ОСУЩЕСТВЛЕНИЯ СПОСОБА
Пример 1.
Сбор периферической венозной крови (30 мл) от субъектов осуществляли в пробирки типа PAXgene Blood ccfDNA (QIAGEN, Германия) объемом 10 мл. В пробирках PAXgene Blood ccfDNA содержатся добавки, которые препятствуют свертыванию крови и стабилизируют клетки крови, что помогает предотвратить контаминацию образца внутриклеточной геномной ДНК в процессе хранения. После сбора крови содержимое пробирок сразу перемешивали вручную плавными движениями, не встряхивая пробирки в соответствии с инструкцией изготовителя пробирок. Хранение пробирок с кровью до момента выделения фракции плазмы крови осуществляли при температуре +4°С до 10°С не более 7 суток с момента забора крови.
Фракцию плазмы крови получали по протоколу QIAsymphony PAXgene Blood ccfDNA Kit Handbook (QIAGEN, Германия). Пробирки PAXgene Blood ccfDNA центрифугировали при комнатной температуре (15-25°С) в течение 15 минут при 1900g. Далее супернатант (фракция плазмы) переносили в пробирки объемом 15 мл с коническим дном центрифугировали при комнатной температуре в течение 10 минут при 1900g. Супернатант переносили в полистироловые криопробирки, которые хранили при температуре -80°С
Для выделения сцДНК из образцов плазмы крови был использован набор реагентов QIAamp MinElute ccfDNA Circulating Nucleic Acid Kit (QIAGEN, Германия), принцип которого основан на селективной обратимой твердофазной иммобилизации фрагментов сцДНК на кремнийсодержащей мембране специальной колонки. Все стандартные операционные процедуры выполняли в соответствие с протоколом производителя. Выделенную сцДНК помещали в криопробирки и хранили при температуре -80°С.
Измерение концентрации сцДНК в пробах осуществляли при помощи флуориметра Quantus (Promega, США) и флуоресцентного красителя QuantiFluor ONE dsDNA Dye (Promega, США), который связывается с двуцепочечной ДНК (λ=504 нм возбуждение / λ=531 нм, эмиссия). Калибровку флуориметра Quantus выполняли следующим образом: готовили две пробы, одна из которых содержала 200 мкл красителя QuantiFluor ONE dsDNA Dye (Promega Corporation), а вторая - 200 мкл QuantiFluor ONE dsDNA Dye и 1 мкл ДНК-стандарта QuantiFluor ONE Lambda DNA в концентрации 400 нг/мкл. В качестве «Blank» и «Standard» устанавливали выходные сигналы при измерении флуоресценции проб, содержащих краситель и краситель с ДНК-стандартом, соответственно. К 200 мкл раствора красителя QuantiFluor ONE dsDNA Dye добавляли 2 мкл образца с неизвестной концентрацией сцДНК и далее измеряли сигнал флуоресценции пробы. Измерения повторяли трижды с вычислением среднего значения.
Контроль чистоты выделенной сцДНК осуществляется при помощи спектрофотометра NanoDrop 8000 путем измерения поглощение пробы по нескольким длинам волн (А230, А260, А280) и затем рассчитывали соотношения А260/А280 и А260/А230. При использовании NanoDrop 8000 измерительную ячейку прибора промывали водой, не содержащей нуклеаз, а затем устанавливали «Blank» для измерения фонового сигнала растворителя. Далее в измерительную ячейку наносили 1 мкл пробы, после чего измеряли поглощение. Измерения повторяют трижды с вычислением среднего значения.
Оценку распределения выделенных фрагментов сцДНК по длинам выполняли методом капиллярного электрофореза на рабочей станции Agilent 2200 TapeStation (Agilent Technologies, Inc., Германия) по протоколу «Agilent High Sensitivity D1000 ScreenTape System Quick Guide» (протокол адаптирован для анализа фрагментов ДНК в диапазоне от 35 до 1000 п.н.).
Приготовление ДНК-библиотек для геномного секвенирования выполняли по протоколу NEBNext Ultra II DNA Library Prep Kit for Illumina (Illumina, США).
Для подготовки концов ДНК (выполнение протокола инкубации) использовали термоциклер CFX96 Touch (Bio-Rad Laboratories, Inc). Реакционная смесь (объемом 60 мкл в расчете на 1 образец) содержала 3 мкл NEBNext Ultra II End Prep Enzyme Mix, 7 мкл NEBNext Ultra II End Prep Reaction Buffer и 50 мкл фрагментированной ДНК.
Лигирование адаптеров к фрагментам ДНК выполняли следующим образом. Была приготовлена реакционная смесь (End Prep Reaction Mixture) 60 мкл образца ДНК с подготовленными концами, 2,5 мкл NEBNext Adaptor for Illumina, 30 мкл NEBNext Ultra II Ligation Master Mix, 1 мкл NEBNext Ligation Enhancer. Образцы инкубировали в термоциклере CFX96 Touch (Bio-Rad Laboratories, Inc., США) при 37°C в течение 15 минут с включенным нагревом крышки при температуре ≥47°С. Очистка адаптер-лигированной ДНК была выполнена на магнитных частицах SPRIselect (Beckman Coulter, Inc., США) согласно протоколу производителя. Финальный объем пробы после очистки составлял 15 мкл.
Амплификация адаптер-лигированной ДНК выполняли в объеме 50 мкл в расчете на один образец: адаптер-лигированные фрагменты ДНК (15 мкл), NEBNext Ultra II Q5 Master Mix (25 мкл), праймер (10 мкмоль, 10 мкл). Количество циклов амплификации при стандартной подготовке ДНК-библиотек определяли в зависимости от входного количества ДНК в реакции приготовления концов ДНК. Например, при содержании ДНК 1 нг и 100 нг количество циклов составляет 10 и 3, соответственно.
Пост-амплификационная очистка была выполнена на магнитных частицах SPRIselect (Beckman Coulter, Inc., США) согласно протоколу производителя.
Процедура пулирования была выполнена по следующему протоколу. Аликвоту красителя QuDye в составе набора Lumiprobe QuDye ssDNA разводили в 200 раз 1х буфером ТЕ. В каждую пробирку для стандартов вносили 190 мкл рабочего раствора красителя QuDye и 10 мкл Quantitative standard, 0 нг/мкл (Стандарт #1) и ssDNA quantitative standard, 20 нг/мкл (Стандарт #2), соответственно. В каждую пробирку для образцов вносили 180-199 мкл рабочего раствора для красителя QuDye ssDNA и от 20 до 1 мкл образца, соответственно. Пробирки инкубировали в течение 2 минут при комнатной температуре. Для пулирования в пробирку объемом на 1,5 мл вносили 16 образцов и доводили 0,1х буфером ТЕ до молярной концентрации пула 1,5-3 нМ. Далее, отбирали 100 мкл в новую пробирку на 1,5 мл.
Денатурацию ДНК-библиотек проводили по следующему протоколу. В качестве контроля была использована адаптер-лигированная библиотека PhiX, полученная из хорошо охарактеризованного бактериофага PhiX с одноцепочечной ДНК. В пробирку с объединенными неденатурированными ДНК-библиотеками был добавлен 0,6 мкл 2,5 нМ неденатурированный PhiX в буфере 10 мМ Трис-HCl (рН 8,5). Далее, 25 мкл свежего раствора NaOH (0,2 Н) добавляли к 100 мкл образцу, содержащего ДНК-библиотеки и PhiX, после чего инкубировали при комнатной температуре в течение 8 минут. К реакционной смеси добавляли 25 мкл 400 мМ раствора Трис-HCl (рН 8,0) для нейтрализации щелочи. Далее полный объем денатурированного пула ДНК-библиотек или денатурированного пула ДНК-библиотек и PhiX был перенесен в пробирку Library Tube, поставляемую в составе комплекта реактивов NovaSeq 6000.
Для проведения запуска секвенатора NovaSeq 6000 (Illumina, США) использовали набор реагентов NovaSeq 6000 S1 Reagent Kit (200 cycles) v1.5 (Illumina, США). Высокопроизводительное секвенирование было выполнено для 286 подготовленных проб, содержащих фрагменты сцДНК. Образцы плазмы были получены от 148 субъектов без диагностированного РЛ (класс «здоровые») и 138 субъектов с установленным РЛ (класс «онкобольные»). Секвенирование проводили с покрытием не менее 100 млн прочтений в расчете на образец.
По окончании осуществления биоинформатической части алгоритма обработки NGS-данных одному наблюдению соответствовало 106074 переменных. Исследуемый массив данных охватывал 286 наблюдений, из них:
175 наблюдений были отнесены к тренировочной выборке;
55 наблюдений были отнесены к реперной выборке;
56 наблюдений были отнесены к тестовой выборке.
В тренировочную выборку был включен набор данных по 89 здоровым субъектам и 86 онкобольным.
В тестовую выборку включен набор данных по 30 здоровым субъектам и 26 онкобольным.
В реперную выборку был включен набор данных по 29 здоровым субъектам и 26 онкобольным.
Следует отметить, что формирование наборов данных было случайным и стратифицированным по полу, возрасту и стадии рака легких. Стратифицированный отбор позволяет сохранять распределение исследуемых градаций во всех создаваемых выборках таким же, каким оно было в исходной выборке, из которой происходило извлечение наблюдений.
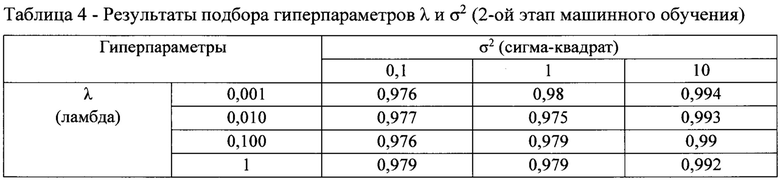
На тренировочном наборе данных был выполнен подбор гиперпараметров для классификационной модели. Наилучшие значения подобранных гиперпараметров α и λ, приведены в таблице 2. Наилучшие значения подобранных гиперпараметров θ1 и θ2 приведены в таблице 3. Результат подбора гиперпараметров λ, и σ2 приведен в таблице 4. Наилучшим вариантом оказалось применение классификационной модели с параметрами λ=0,001 и σ2=10, при этом медианное значение площади под ROC-кривой (AUC) при проверке на тестовой выборке составило 0,994 (таблица 4).
Результаты проверки работы классификационной модели для разделения классов здоровых субъектов и онкобольных на тестовом наборе данных (n=56) представлены в таблице 5. Было установлено, что значение AUC было равным 0,87; значение сбалансированной аккуратности (от англ. balanced accuracy)=0,85 (с учетом дисбаланса численности классов); чувствительность=А/(А+С)=0,81; специфичность=D/(B+D)=0,9.
На основе реперного набора данных была создана матрица размерностью 1455300 строк и 40 столбцов. Наблюдения, которые были использованы для формирования реперного набора данных не использовались для последующего обучения классификационных моделей и рассматривались в дальнейшем как дополнительная проверочная выборка. Созданная реперная матрица размерностью была преобразована с помощью МГК. Было отобрано 28 главных компонент (ГК), охватывающих в сумме более 95% дисперсии исходных реперных данных в многомерном пространстве. Каждая из ГК являлась линейной комбинацией 40 переменных, составляющих ПВМ. Все ПВМ, составлявшие структуру каждого из 286 наблюдений, были преобразованы в соответствии с результатами МГК. Вместо 40 исходных переменных было получено 28 метрик, каждая из которых отражает паттерн фрагментации, встречающийся на протяжении генома как здоровых субъектов, так и у онкобольных. В первую очередь, этот паттерн имеет отношение не к длине фрагмента, а к характеристике его пятинуклеотидных концевых мотивов. Эти метрики были преобразованы по тем же правилам, что и статистические метрики (см. раздел Раскрытие изобретения).
Результаты проверки работы классификационной модели на реперном наборе данных (n=55) представлены в таблице 6. Показано, что AUC=0,875; сбалансированная аккуратность=0,8 (с учетом дисбаланса численности классов); чувствительность=А/(А+С)=0,81; специфичность=D/(B+D)=0,79.
Результаты работы классификационной модели (расположение наблюдений по трем использованным выборкам) можно представить графически в координатах первой (ГК1) и второй (ГК2) главных компонент (рисунки 4-6). На рисунках оттенки зеленого и фиолетового отражают результаты ядерной логистической регрессии, а чем темнее оттенок, тем выше вероятность принадлежности субъекта к классу (группе) здоровых субъектов и классу онкобольных. Результат применения созданной классификационной модели на тренировочной выборке можно продемонстрировать на ROC-кривой (рисунок 7).
Результатом работы финальной классификационной модели является конечная классификационная р-метрика (от англ. probability), принимающая значение от 0 до 1. Физический смысл метрики р заключается в том, что чем ближе значение этой метрики к 1, тем больше оценка вероятности того, что исследуемый субъект является онкобольным, и, напротив, чем ближе значение р-метрики к нулю, тем больше оценка вероятности того, что субъект является здоровым. Далее, на тренировочной выборке была выбрана оптимальная граничная величина р-метрики, с помощью которой, после завершения выполнения алгоритма биоинформатической и статистической обработки NGS-данных о фрагментоме сцДНК из плазмы крови неизвестного образца, выносится суждение о том, является ли субъект обследования здоровым или онкобольным. Расчет значения граничной величины р-метрики проводили посредством поиска по сетке гиперпараметров λ и σ2 (см. подробно таблицу 4), который выполнялся путем кросс-валидации по методу Монте-Карло. При рассмотрении каждого варианта граничной величины р-метрики выполняли 100 симуляций. В качестве критерия качества классификационной модели было медианное значение сбалансированной аккуратности. При определенном значении граничной величины р-метрики в ходе кросс-валидации было достигнуто наивысшее значение сбалансированной аккуратности. Итак, оптимальная граничная величина р-метрики составила 0,35 (рисунок 8).
Пример 2.
В настоящем примере показано функционирование классификационной модели - каким образом на основе паттерна фрагментации происходит постановка диагноза и принимается решение о принадлежности пациента к группе больных раком легких или группе здоровых.
Каждому пациенту соответствует 106074 переменных. Все эти переменные подразделяются на несколько групп. В свою очередь, для каждой группы переменных реализован собственный алгоритм, на основе которого определяется статус пациента. Здесь стоит еще раз особо отметить, что классификационная модель состоит из нескольких относительно простых моделей, каждая из которых предсказывает статус пациента, исходя из определенной части от общего числа используемых переменных. Эти относительно простые модели разумно обозначить как классификационные модели первого (I) этапа. Результаты, полученные на классификационных моделях I этапа, сводятся в таблицу и используются в качестве переменных для финальной оценки статуса пациента с помощью классификационной модели второго (II) этапа.
Классификационные модели I этапа являются либо логистическими регрессионными моделями с L1- и L2-регуляризацией, либо реализованы в виде вероятностных классификаторов, основанных на гауссовских процессах.
Используются следующие группы переменных:
1) Средние значения показателей распределения длин фрагментов в пределах каждой хромосомы. 5 групп переменных и, соответственно 5 классификаторов I этапа, реализованные в виде логистической регрессионной модели.
- Средние длин фрагментов в пределах каждой хромосомы;
- Стандартные отклонения, рассчитанные по выборкам длин фрагментов в пределах каждой хромосомы;
- Скос распределения длин фрагментов в пределах каждой хромосомы;
- Куртозис распределения длин фрагментов в пределах каждой хромосомы;
- Производная натурального логарифма отношения численности коротких фрагментов к численности длинных в пределах каждой хромосомы;
2) Средние значения показателей, полученных на основе анализа ПВМ, в пределах каждой хромосомы. 28 групп переменных, каждая группа описывает определенный паттерн фрагментации, который был ранее получен в ходе преобразования ПВМ. Каждой группе соответствует определенный классификатор I этапа, реализованный в виде логистической регрессионной модели. Каждая группа охватывает по 22 переменных, по числу хромосом (аутосом);
3) Массивы переменных, полученные при сглаживании и усреднении хромосомальных рядов. Каждый массив охватывает определенную характеристику фрагментома, т.е. ее изменение на протяжении всей длины генома. Всего рассматривается 24 массива, каждый размером в несколько сотен переменных. Каждому массиву соответствует определенный вероятностный классификатор I этапа, основанный на гауссовских процессах.
Пример работы всех классификаторов I этапа представлен на рисунках 9-15.
На рисунках 9-13 по правой стороне расположены группы переменных и соответствующие им коэффициенты логистических регрессионных моделей. В направлении слева направо указывается свободный коэффициент, затем коэффициенты при переменных для отдельно взятых хромосом, с 1 по 22. Пробел соответствует отсутствию влияния переменной на результат предсказания модели, то есть значение коэффициента при такой переменной равно нулю.
По левой стороне рисунков 9-13 представлены результаты проверки классификационных моделей I этапа на тестовой выборке. Зеленые точки соответствуют здоровым пациентам, фиолетовые перекрестья - онкобольным. Наблюдения тестовой выборки выстроены по горизонтальной оси в порядке возрастания вероятности наличия РЛ, рассчитанной с помощью соответствующей классификационной модели I этапа.
Как можно видеть из определения, тестовая выборка не участвовала в построении модели, и в случае каждого классификатора I этапа заметно явное несовершенство в разделении двух классов пациентов. Простая модель вполне может указать высокую вероятность наличия онкологического заболевания у пациента, который в действительности здоров или наоборот, указать низкую вероятность наличия РЛ у пациента, который в действительности им болен.
На рисунках 14 и 15 показаны результаты работы классификаторов I этапа, основанных на гауссовских процессах. Каждая панель соответствует отдельно взятому классификатору. Классификатор преобразует массив из нескольких сотен переменных в маломерный массив данных размерностью в одну либо две переменные. Эти переменные можно отобразить на плоскости в виде декартовых координат. Наблюдения, соответствующие здоровым или онкобольным, проецируются в определенные регионы этих одномерных или двухмерных пространств. Оттенками фиолетового показаны участки, расположение в которых указывает на высокую вероятность наличия РЛ у пациента. Оттенками зеленого показаны участки, расположение в которых указывает на низкую вероятность наличия РЛ у пациента и, следовательно, высокую вероятность того, что пациент здоров. Точки обозначают результаты проверки модели на тестовой выборке, каждая точка соответствует определенному пациенту. Заметно, что модели несовершенны, в «фиолетовой» области могут находиться здоровые пациенты, в «зеленой» области - онкобольные (пациенты с РЛ). Обозначения: "1" - группа здоровых пациентов, "3_ТР" - группа онкобольных пациентов.
На рисунке 5 показаны результаты работы классификатора II этапа, основанного на ядерной логистической регрессии и использующего в качестве исходных данных результаты, полученные при работе классификаторов I этапа. Результатом работы финальной классификационной модели является конечная классификационная метрика р, принимающая значение от 0 до 1. Физический смысл метрики р заключается в том, что чем ближе значение этой метрики к 1, тем больше уверенности в том, что исследуемый субъект является онкобольным (т.е. в данном случае пациентом с РЛ), и, напротив, чем ближе значение р к нулю, тем больше уверенности, что субъект является здоровым. Метрику р разумно интерпретировать как вероятность наличия РЛ у исследуемого пациента. Оттенками фиолетового показаны участки, соответствующие высоким значениям метрики р, расположение в которых указывает на высокую вероятность наличия РЛ у пациента. Оттенками зеленого показаны участки, соответствующие низким значениям метрики р расположение в которых указывает на низкую вероятность наличия РЛ у пациента и, следовательно, высокую вероятность того, что пациент здоров. Точки обозначают результаты проверки модели на тестовой выборке. Серая область соответствует значению метрики р выше 0,35. Пациенты со значением метрики р выше 0,35 считаются онкобольными, пациенты со значением метрики р ниже 0,35 считаются здоровыми.
ВЫВОДЫ
При технической реализации алгоритма биоинформатической и статистической обработки NGS-данных в рамках осуществления способа малоинвазивной диагностики РЛ по фрагментированной сцДНК из плазмы крови была создана бинарная классификационная модель на основе уникальной комбинации методов машинного обучения (L1, L2 - регуляризованной логистической регрессии, вероятностных классификаторов, основанных на гауссовских процессах, метода главных компонент и ядерной логистической регрессии).
Классификационная модель характеризуется значениями AUC в пределах от 0,87 до 0,875 (качество - «выдающееся», от англ. excellent) и позволяет различать здоровых субъектов и субъектов с раком легкого I-III стадий по профилю фрагментации сцДНК и профилю последовательностей концевых мотивов фрагментов сцДНК, которые установлены при помощи высокопроизводительного секвенирования. По данным проверки классификационной модели на тестовом и реперном наборах данных, можно заключить, что значения параметров чувствительность и специфичность равны 0,81 и 0,79-0,9, соответственно.
Классификационная модель функционирует без использования сведений о дополнительных геномных, транскриптомных, протеомных и метаболомных биомаркерах, клинико-анамнестических характеристиках субъектов и результатов иных лабораторных или инструментальных исследований, в том числе посредством методик визуализации на основе рентгенографии и компьютерной томографии.
Следует отметить, что в мета-анализе Не X. и соавторов [Не X. et al. A systematic review and meta-analysis of circulating cell-free DNA as a diagnostic biomarker for non-small cell lung cancer // J Thorac Dis. - 2022. - V. 14. - No. 6. - P. 2103-2111], посвященному оценке аналитической точности биомаркеров на основе сцДНК для диагностики РЛ, было установлено усредненное значение AUC равное 0,89, что позволяет позиционировать созданную классификационную модель как соответствующую мировому уровню техники и имеющую хорошую диагностическую ценность.


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ создания таргетной панели для исследования геномных регионов для выявления терапевтических биомаркеров ингибиторов иммунных контрольных точек (ИКТ) | 2023 |
|
RU2818360C1 |
| Способ обработки данных полногеномного секвенирования | 2023 |
|
RU2806429C1 |
| Система обработки данных полногеномного секвенирования | 2023 |
|
RU2804535C1 |
| Способ выделения целевых фрагментов ДНК из многокомпонентной смеси | 2024 |
|
RU2832884C1 |
| Способ неинвазивного пренатального скрининга анеуплоидий плода | 2019 |
|
RU2712175C1 |
| Способ прогнозирования риска когнитивных нарушений у долгожителей | 2023 |
|
RU2822168C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АНЕУПЛОИДИИ ПЛОДА В ОБРАЗЦЕ КРОВИ БЕРЕМЕННОЙ ЖЕНЩИНЫ | 2021 |
|
RU2777072C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИСТОЧНИКА АНЕУПЛОИДНЫХ КЛЕТОК ПО КРОВИ БЕРЕМЕННОЙ ЖЕНЩИНЫ | 2016 |
|
RU2674700C2 |
| Способ прогнозирования риска тяжелого течения COVID-19 у пациента | 2022 |
|
RU2791487C1 |
| СПОСОБ И СИСТЕМА ГЕНЕРАЦИИ ИНДИВИДУАЛЬНЫХ РЕКОМЕНДАЦИЙ ПО ДИЕТЕ НА ОСНОВАНИИ АНАЛИЗА СОСТАВА МИКРОБИОТЫ | 2019 |
|
RU2724498C1 |

Изобретение относится к области онкологии, биомедицинской статистики и диагностики злокачественных новообразований (ЗНО). Изобретение включает в себя способ малоинвазивной диагностики рака легкого по профилю фрагментации свободно циркулирующей ДНК (сцДНК) и профилю последовательностей концевых мотивов фрагментов сцДНК. Способ осуществляется посредством выделения сцДНК из плазмы крови, подготовки ДНК-библиотек и их секвенированием, а также путем применения уникального алгоритма биоинформатической и статистической обработки данных секвенирования для создания бинарной классификационной модели. Модель позволяет различать здоровых субъектов (без диагностированного рака легкого, РЛ) и онкобольных (субъекты с установленным РЛ) со значением AUC от 0,87 до 0,875 при значениях чувствительности 0,81 и специфичности - от 0,79 до 0,9. Профиль фрагментации сцДНК содержит записи о длинах фрагментов сцДНК и соотношении их длин. Профиль последовательностей концевых мотивов фрагментов сцДНК содержит записи о линейных комбинациях значений позиционно-весовых матриц для участка генома, по которым чаще всего происходит фрагментация ДНК внутри этого участка. Классификационная модель была создана на основе методов машинного обучения с помощью L1, L2-регуляризованной логистической регрессии, вероятностных классификаторов, основанных на гауссовских процессах, метода главных компонент и ядерной логистической регрессии. Таким образом, модель имеет хорошую диагностическую значимость и не требует наличия клинических данных и данных об иных опухоль-ассоциированных биомаркерах. Настоящий способ может быть положен в основу разработки диагностического теста, который может быть использован для помощи в принятии врачебных решений. 1 з.п. ф-лы, 15 ил., 6 табл.
1. Способ малоинвазивной диагностики рака легкого по пулу фрагментированной свободно циркулирующей ДНК (сцДНК), выделенной, прежде всего, из плазмы крови, а также других биологических жидкостей организма человека, включающий выделение сцДНК из плазмы крови методами селективной обратимой твердофазной иммобилизации фрагментов сцДНК на кремнийсодержащих инертных носителях и иных носителях, далее проводят высокопроизводительное секвенирование (NGS) сцДНК с получением выходных NGS-данных, включающих сведения о паттерне фрагментации сцДНК и паттерне последовательности концевых мотивов сцДНК, затем проводят биоинформатическую и статистическую части обработки выходных NGS-данных, при этом биоинформатическая часть алгоритма реализуется в обработке NGS-данных о распределении коротких 100-150 п.н. и длинных 151-220 п.н. фрагментов сцДНК, с картированием чтений на геном, с коррекцией или без коррекции покрытия по GC-составу, коррекцией на глубину прочтения, коррекцией с фильтрацией прочтений по качеству покрытия и регионам генома; и обработке NGS-данных по последовательности концевых мотивов каждого фрагмента сцДНК, которые выражаются в сопоставлении позиционно-весовых матриц в пределах как одного наблюдения, так и набора данных, статистическая часть алгоритма реализуется в обработке преобразованных NGS-данных и включает в себя применение метода главных компонент для преобразования исходного набора данных для выявления таких главных компонент, которые можно интерпретировать как паттерн фрагментации сцДНК; вычисление средних значений по хромосоме; центрирование и шкалирование в пределах каждого наблюдения; сглаживание в пределах каждой хромосомы; статистическая часть алгоритма реализуется в виде комплексной классификационной модели, элементами которой являются классификаторы, построенные с помощью L2, L1-регуляризованной логистической регрессии, вероятностные классификаторы, основанные на гауссовских процессах, и ядерная логистическая регрессия с подбором оптимального соответствия гиперпараметров лямбда и сигма-квадрат путем кросс-валидации по методу Монте-Карло.
2. Способ по п. 1, где статистическую часть алгоритма проводят в два этапа, на первом этапе статус пациента определяется с помощью 24 вероятностных классификаторов, основанных на гауссовских процессах и 33 классификаторов на основе логистической регрессии с L1 и L2-регуляризацией, при этом результатом работы каждого из классификаторов является число, принимающее значение от -10 до +10, для отдельно взятого наблюдения создается 57 переменных, каждая из которых является индикатором статуса пациента, на втором этапе, с помощью метода главных компонент, из этих 57 переменных рассчитываются две линейные комбинации, то есть две новые переменные, которые являются линейными комбинациями результатов работы классификаторов первого этапа, далее на основе этих двух линейных комбинаций с помощью метода ядерной логистической регрессии рассчитывается конечная классификационная р-метрика для оценки вероятности наличия у пациента рака легких, принимающая значения от 0 до 1.
| Малоинвазивный способ диагностики рака легкого на основании изменения копийности локуса мтДНК HV2 | 2018 |
|
RU2678227C1 |
| СПОСОБ ДИАГНОСТИКИ НЕМЕЛКОКЛЕТОЧНОГО РАКА ЛЕГКОГО И НАБОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2008 |
|
RU2390780C1 |
| WO 2013174948 A1, 28.11.2013 | |||
| RU 2015109526 A, 10.10.2016. | |||

