ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет согласно предварительной заявке на выдачу патента США № 62/869389, которая была подана 1 июля 2019 г. Данная заявка включена посредством ссылки, как если бы она была полностью переписана в данном документе.
УРОВЕНЬ ТЕХНИКИ
Понимание механизма и кинетики клиренса новых химических веществ при разработке лекарственных средств важно для обеспечения безопасного и эффективного воздействия на пациента новыми химическими веществами и устранения клинически неблагоприятных взаимодействий, которые могут быть вызваны совместным введением с другими лекарственными средствами. Сообщалось о применении соединения 1, которое представляет собой диаммониевую соль E7766, показанного ниже, при лечении рака. См., например, патент США № 10246480, который включен в данный документ посредством ссылки.
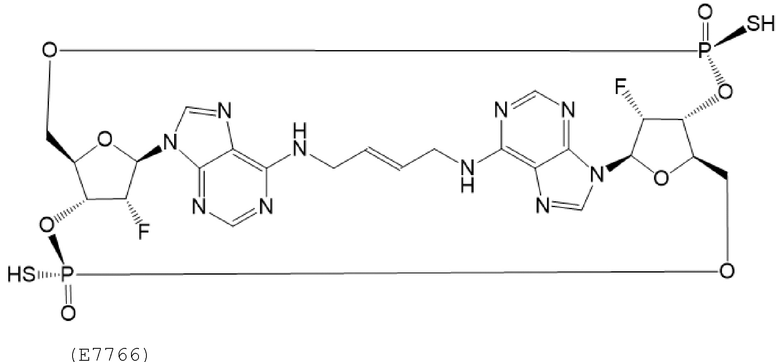
E7766 также называют (1R,3R,15E,28R,29R,30R,31R,34R,36R,39S,41R)-29,41-дифтор-34,39-бис(сульфанил)-2,33,35,38,40,42-гексаокса-4,6,9,11,13,18,20,22,25,27-декааза-34λ5,39λ5-дифосфаоктацикло[28.6.4.13,36.128,31.04,8.07,12.019,24.023,27]дотетраконта-5,7,9,11,15,19,21,23,25-нонаен-34,39-дионом. Если существуют какие-либо расхождения между данным химическим названием и представленной выше структурой, то указанная выше структура будет иметь преимущественную силу.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Для облегчения в подготовке к клиническим испытаниям авторами настоящего изобретения были использованы доклинические модели для измерения клиренса соединения 1 как отдельно, так и при потенциальном воздействии межлекарственных взаимодействий. Не желая привязываться к теории, исходя из обзора соединения, авторы настоящего изобретения пришли к выводу, что соединение будет обладать низким LogP (<1), низкой способностью к проникновению (<1×10-6 см/с), pKa в диапазоне 3-4 и MW >600. Это приведет к тому, что соединение 1 будет отнесено к классу 3B в соответствии с Системой классификации соединений с продленным клиренсом. Соединения этого класса преимущественно выводятся путем активного поглощения с последующим выведением с желчью или мочой.
Ниже представлено E7766. Соединение 1, которое представляет собой диаммониевую соль E7766, имеет молекулярную массу 780,7, измеренное значение pKa 3,41, измеренное значение LogD 1,31, PSA 200, растворимость 150 мкмоль/л, значение Papp 0,36×106 см/с и является соединением класса 3B по ECCS при использовании шкалы, описанной в Varma et al., Pharm Res (2015) 32:3785-3802, которая включена в данный документ посредством ссылки. В представленных в данном документе примерах применяли соединение 1, диаммониевую соль. Как правило, для разных исследований применяли разные партии.
В одном варианте осуществления предусмотрена система для уменьшения количества фармакологических ошибок и повышения степени соблюдения терапевтической схемы индивидуумом, страдающим от рака, содержащая по меньшей мере один контейнер, при этом указанный контейнер включает некоторое количество фармацевтической композиции, содержащей E7766 или его фармацевтически приемлемую соль
и по меньшей мере информационное сообщение, прилагаемое к указанному контейнеру, при этом указанное информационное сообщение содержит инструкцию по применению, содержащую информацию о потенциальном межлекарственном взаимодействии, где информация о потенциальном межлекарственном взаимодействии содержит информацию, указывающую, что введение фармацевтической композиции приводит к потенциально иному эффекту у индивидуума, получающего лечение ингибитором транспортного полипептида органических анионов, чем можно было бы ожидать у индивидуума, не получающего ингибитор транспортного полипептида органических анионов. В дополнительном варианте осуществления ингибитор транспортного полипептида органических анионов ингибирует транспортный полипептид органических анионов, выбранный из OATP1B1, OATP1B3 и комбинации OATP1B1 и OATP1B3.
В дополнительных вариантах осуществления ссылка на "OATP" ограничена до ингибиторов OATP1B1 и/или OATP1B3.
В дополнительном варианте осуществления в информации о потенциальном межлекарственном взаимодействии указано, что не следует вводить совместно ингибитор транспортного полипептида органических анионов и данную фармацевтическую композицию. В дополнительном варианте осуществления в информации о потенциальном межлекарственном взаимодействии указано, что один или оба из ингибитора транспортного полипептида органических анионов и фармацевтической композиции следует вводить в более низкой дозе и/или реже, чем если бы любой из них вводили без другого.
В некоторых вариантах осуществления дозу фармацевтической композиции снижают на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%. В дополнительных вариантах осуществления дозу фармацевтической композиции снижают на 5%-75%; снижают на 10%-50% или снижают на 20%-40%. В некоторых вариантах осуществления дозу ингибитора OATP снижают на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%. В дополнительных вариантах осуществления дозу ингибитора OATP снижают на 5%-75%; снижают на 10%-50% или снижают на 20%-40%.
В дополнительном варианте осуществления вышеупомянутой системы ингибитор транспортного полипептида органических анионов выбран из группы, состоящей из фимасартана, кларитромицина, рифампина, клопидогреля, эсликарбазепина, CP-778875, изавуконазола, итраконазола, омбитасвира, асунапревира, боцепревира, даклатасвира, дазабувира, элбасвира, фалдапревира, глекапревира, гразопревира, летермовира, омбитасвира, паритапревира, пибрентасвира, триметоприма, ритонавира, симепревира, софосбувира, телапревира, велпатасвира, воксилапревира, лопинавира, пефицитиниба, кверцетина, типранавира, метформина, дилтиазема, сакубитрила, валсартана, фуросемида, гемфиброзила, элюксадолина, циклоспорина, такролимуса, элтромбопага, грейпфрутового сока, урсодезоксихолевой кислоты, расторопши (Silybum marianum), эмтрицитабина, тенофовира, верцирнона (GSK1605786), телмисартана, эпигаллокатехина галлата, эзетимиба, амлодипина, обетихолевой кислоты, омега-3-карбоновых кислот, иделалисиба, байкалина, эмпаглифлозина, элвитегравира и кобицистата.
В дополнительном варианте осуществления предусмотрен способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой солью, предусматривающий введение E7766 или его фармацевтически приемлемой соли указанному пациенту, если указанному пациенту при этом не вводят лекарственное средство, которое является ингибитором OATP1B1 или OATP1B3.
В дополнительном варианте осуществления предусмотрен способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой солью, предусматривающий введение E7766 или его фармацевтически приемлемой соли указанному пациенту, если указанному пациенту вводят некоторое количество лекарственного средства, которое представляет собой ингибитор OATP1B1 или OATP1B3, которое меньше количества ингибитора OATP1B1 или OATP1B3, которое указанному пациенту вводили бы в отсутствие введения E7766 или его фармацевтически приемлемой соли.
В дополнительном варианте осуществления предусмотрен способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий введение E7766 или его фармацевтически приемлемой соли пациенту, которому вводят некоторое количество лекарственного средства, которое представляет собой ингибитор OATP1B1 или OATP1B3, предусматривающий введение указанному пациенту количества E7766 или его фармацевтически приемлемой соли, которое меньше количества E7766 или его фармацевтически приемлемой соли, которое указанному пациенту вводили бы в отсутствие введение ингибитора OATP1B1 или ингибитора OATP1B3.
В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 1 день после введения указанного лекарственного средства; через по меньшей мере 2 дня после введения указанного лекарственного средства; через по меньшей мере 3 дня после введения указанного лекарственного средства; через по меньшей мере 4 дня после введения указанного лекарственного средства; через по меньшей мере 5 дней после введения указанного лекарственного средства; через по меньшей мере 6 дней после введения указанного лекарственного средства; через по меньшей мере 7 дней после введения указанного лекарственного средства; через по меньшей мере 2 недели после введения указанного лекарственного средства; через по меньшей мере 3 недели после введения указанного лекарственного средства или через по меньшей мере 1 месяц после введения указанного лекарственного средства.
В вариантах осуществления способа, описанного в данном документе, указанное лекарственное средство может быть выбрано из группы, состоящей из фимасартана, кларитромицина, рифампина, клопидогреля, эсликарбазепина, CP-778875, изавуконазола, итраконазола, омбитасвира, асунапревира, боцепревира, даклатасвира, дазабувира, элбасвира, фалдапревира, глекапревира, гразопревира, летермовира, омбитасвира, паритапревира, пибрентасвира, триметоприма, ритонавира, симепревира, софосбувира, телапревира, велпатасвира, воксилапревира, лопинавира, пефицитиниба, кверцетина, типранавира, метформина, дилтиазема, сакубитрила, валсартана, фуросемида, гемфиброзила, элюксадолина, циклоспорина, такролимуса, элтромбопага, грейпфрутового сока, урсодезоксихолевой кислоты, расторопши (Silybum marianum), эмтрицитабина, тенофовира, верцирнона (GSK1605786), телмисартана, эпигаллокатехина галлата, эзетимиба, амлодипина, обетихолевой кислоты, омега-3-карбоновых кислот, иделалисиба, байкалина, эмпаглифлозина, элвитегравира и кобицистата.
Дополнительные варианты осуществления предусматривают способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий отслеживание экспозиции E7766 или его фармацевтически приемлемой соли в отношении указанного пациента и поддержание указанной экспозиции на уровне значения, которое ниже 12800 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 9600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 6400 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 3200 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 2400 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 2000 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 1750 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 1600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 1200 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 800 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 300 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 150 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 75 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; поддержание указанной экспозиции на уровне значения, которое ниже 50 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; или поддержание указанной экспозиции на уровне значения, которое ниже 25 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента. В некоторых вариантах осуществления экспозицию E7766 или его фармацевтически приемлемой соли, которые указаны в данном абзаце, устанавливали у пациента, которому вводят ингибитор OATP1B1 и/или ингибитор OATP1B3.
В некоторых способах стадия поддержания предусматривает снижение величины дозы E7766 или его фармацевтически приемлемой соли. Ее можно снизить, например, на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% относительно предыдущего значения.
В некоторых способах указанную экспозицию отслеживают путем количественной оценки наличия E7766 или его фармацевтически приемлемой соли в крови указанного пациента. В некоторых способах указанную экспозицию оценивают с использованием плазмы крови указанного пациента.
Дополнительный вариант осуществления предусматривает систему или способ, которые изложены в данном документе, где указанная фармацевтически приемлемая соль E7766 представляет собой диаммониевую соль E7766.
В дополнительном варианте осуществления предусмотрен способ лечения рака у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий введение E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент еще не получал лечения ингибитором транспортного полипептида органических анионов (OATP).
В дополнительном варианте осуществления предусмотрен способ лечения рака у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий введение E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент ранее получал лечения ингибитором OATP или все еще получает ингибитор OATP, и одновременное прекращение или уменьшение введения указанного ингибитора OATP для устранения или уменьшения частоты связанных неблагоприятных явлений.
В еще дополнительном варианте осуществления предусмотрен способ лечения рака у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий введение E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент ранее получал лечение ингибитором OATP или все еще получает ингибитор OATP, и одновременное прекращение или уменьшение введения указанного E7766 или его фармацевтически приемлемой соли для устранения или уменьшения частоты связанных неблагоприятных явлений.
В некоторых вариантах осуществления ингибитор OATP выбран из группы, состоящей из фимасартана, кларитромицина, рифампина, клопидогреля, эсликарбазепина, CP-778875, изавуконазола, итраконазола, омбитасвира, асунапревира, боцепревира, даклатасвира, дазабувира, элбасвира, фалдапревира, глекапревира, гразопревира, летермовира, омбитасвира, паритапревира, пибрентасвира, триметоприма, ритонавира, симепревира, софосбувира, телапревира, велпатасвира, воксилапревира, лопинавира, пефицитиниба, кверцетина, типранавира, метформина, дилтиазема, сакубитрила, валсартана, фуросемида, гемфиброзила, элюксадолина, циклоспорина, такролимуса, элтромбопага, грейпфрутового сока, урсодезоксихолевой кислоты, расторопши (Silybum marianum), эмтрицитабина, тенофовира, верцирнона (GSK1605786), телмисартана, эпигаллокатехина галлата, эзетимиба, амлодипина, обетихолевой кислоты, омега-3-карбоновых кислот, иделалисиба, байкалина, эмпаглифлозина, элвитегравира и кобицистата.
В некотором варианте осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 1 день после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 2 дня после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 3 дня после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 4 дня после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 5 дней после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 6 дней после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 7 дней после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 2 недели после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 3 недели после введения ингибитора OATP. В некоторых вариантах осуществления E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 1 месяц после введения ингибитора OATP. В некоторых вариантах осуществления пациент все еще принимает ингибитор OATP. В изложенных выше вариантах осуществления ингибитор OATP обычно представляет собой OATP1B1 и/или OATP1B3.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1A - фиг. 1D показаны результаты in vitro фенотипирования гепатобилиарного транспортера и кинетических исследований соединения 1.
На фиг. 2A - фиг. 2C показаны in vivo PK-профили и элиминация соединения 1 с одновременным приемом рифампина ((7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,27,29-пентагидрокси-11-метокси-3,7,12,14,16,18,22-гептаметил-26-{(E)-[(4-метилпиперазин-1-ил)имино]метил}-6,23-диоксо-8,30-диокса-24-азатетрацикло[23.3.1.14,7.05,28]триаконта-1(28),2,4,9,19,21,25(29),26-октаен-13-ил-ацетата) или без него.
На фиг. 3A показана доля, транспортируемая посредством OATP1B1/1B3; на фиг. 3B и фиг. 3C показаны смоделированные значения концентрации соединения 1 в печени и плазме при одновременном введении с рифампином или без него. На фиг. 3D показаны средние значения синусоидального клиренса при поглощении соединения 1 с взаимодействием и без него с течением времени с применением рафампицина. Моделирования производили с применением модели PBPK, разработанной с помощью моделирования Simcyp™.
На фиг. 4A показано поглощение печенью соединения 1 в гепатоцитах человека, культивируемых сэндвич-способом (SCHH), с течением времени. На фиг. 4B показано выведение соединения 1 с желчью в SCHH спустя 20 минут воздействия.
На фиг. 5 показана AUC соединения 1 в печени мышей дикого типа после внутривенного введения соединения 1 и совместного введения соединения 1 и рифампина.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Авторами настоящего изобретения была изучена фармакокинетика и распределение соединения 1 в системах in vitro, а также в доклинических испытаниях in vivo. По фармакокинетике у крыс и собак с канюлированными желчными протоками было видно, что соединение 1 преимущественно выводилось в неизменном виде с желчью (>80%) и, в меньшей степени, с мочой (<20%). По результатам исследования поглощения гепатоцитами человека было видно зависящее от температуры активное поглощение, которое можно ингибировать посредством рифампина, но не тетраэтиламмония, что указывает на участие транспортного полипептида органических анионов (OATP) в элиминации соединения 1. Дополнительные исследования на клетках HEK293, сверхэкспрессирующих OATP1B1 и OATP1B3 человека, подтвердили, что соединение 1 является субстратом для OATP1B1 и OATP1B3.
Исследования in vitro везикул, экспрессирующих белок 2, ассоциированный с множественной лекарственной устойчивостью (MRP2), продемонстрировали, что соединение 1 является субстратом для выводящего с желчью транспортера MRP2. Фармакокинетику соединения 1 также оценивали на гуманизированных мышах с нокаутом OATP1B1/1B3, Oatp1a/1b или мышах дикого типа. У мышей дикого типа увеличение (в 5,4 раза) экспозиции соединения 1 в плазме крови наблюдали в присутствии рифампина, тогда как у мышей дикого типа экспозиция соединения 1 в печени была сравнимой в присутствии или отсутствии рифампина. У гуманизированных мышей OATP1B1/1B3 в присутствии рифампина концентрация соединения 1 в плазме крови увеличивалась в 4,5 раза. Данные доклинические результаты позволяют спрогнозировать, что OATP-опосредованное поглощение печенью является ограничивающей скорость стадией при клиренсе соединения 1, и данные доклинические результаты также позволяют спрогнозировать, что ингибирование OATP в клинических условиях приведет к значительному увеличению системного воздействия соединения 1.
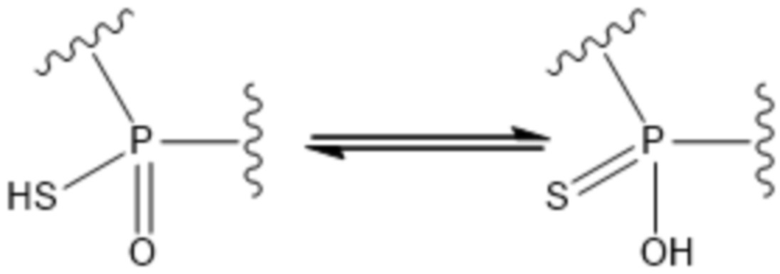
Специалисты в данной области поймут, что если заместители, связанные с атомами фосфора (P1,P2), имеют как одинарные, так и двойные связи, то они могут подвергаться таутомеризации. Например, соединения могут таутомеризоваться при равновесии. Ниже представлен один пример:
 .
.
Такие таутомеры следует рассматривать как входящие в объем формулы изобретения. Структурное представление любого таутомера для данного соединения будет представлять собой одно и то же соединение.
Способы лечения
В некоторых вариантах осуществления E7766 или фармацевтически приемлемую соль вводят нуждающемуся в лечении пациенту. В некоторых вариантах осуществления вводимое соединение представлено в виде NH4-соли, свободной кислоты или ее фармацевтически приемлемой соли. В некоторых вариантах осуществления соединение представлено в виде NH4-соли. При введении E7766 в виде NH4-соли соединение называют соединением 1.
Дозы
Оптимальную дозу для лечения рака можно определить эмпирически для каждого индивидуума с помощью известных способов, и она будет зависеть от ряда факторов, в том числе активности средств, возраста, массы тела, общего состояния здоровья, пола и рациона индивидуума, времени и пути введения, а также других лекарственных препаратов, которые принимает индивидуум. Оптимальные дозы можно установить с помощью стандартного тестирования и процедур, которые хорошо известны в уровне техники. Введение вышеупомянутых соединений можно осуществлять любым подходящим путем.
"Межлекарственное взаимодействие" в контексте данного документа относится к фармакокинетическим или фармакодинамическим эффектам, которые могут возникать при совместном введении двух или более лекарственных средств. Такие эффекты, как правило, не возникают при введении отдельных лекарственных средств (т.е. не в присутствии других лекарственных средств). Неограничивающие примеры фармакокинетических эффектов межлекарственного взаимодействия могут включать, например, изменения абсорбции, распределения, метаболизма или выведения одного или обоих совместно вводимых лекарственных средств. Неограничивающие примеры фармакодинамических эффектов межлекарственного взаимодействия могут включать, например, интерференцию одного лекарственного средства (например, конкурентная или аллостерическая) в отношении другого лекарственного средства в участке связывания белка (или рецептора) или косвенную интерференцию посредством связывания белка (или рецептора) в родственном биологическом пути. Неограничивающие примеры межлекарственных взаимодействий могут включать ожидаемые побочные эффекты, неожиданные побочные эффекты, клинические нежелательные явления и противопоказания, все из которых можно контролировать во время введения лекарственных средств, потенциально участвующих в межлекарственном взаимодействии.
"Фармацевтически приемлемая соль" в контексте данного документа относится к солям присоединения кислот или солям присоединения оснований соединений по настоящему изобретению. Фармацевтически приемлемая соль представляет собой любую соль, которая сохраняет активность исходного соединения и не оказывает какого-либо чрезмерно вредного или нежелательного эффекта на субъект, которому ее вводят, и в окружении, в котором ее вводят. Фармацевтически приемлемые соли включают без ограничения комплексы металлов и соли как неорганических, так и карбоновых кислот. Фармацевтически приемлемые соли также включают соли металлов, таких как алюминий, кальций, железо, магний, марганец, и комплексные соли. Кроме того, фармацевтически приемлемые соли включают без ограничения кислые соли, такие как соль уксусной, аспарагиновой, алкилсульфоновой, арилсульфоновой, аксетиловой, бензолсульфоновой, бензойной, биугольной, бисерной, битартратной, масляной кислоты, эдетат кальция, соль камзиловой, угольной, хлорбензойной, лимонной, этилендиаминтетрауксусной, эдисиловой, эстоловой, эзиловой, муравьиной, фумаровой, глюцептовой, глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексамовой, гексилрезорциновой, гидрабаминовой, бромистоводородный, хлористоводородной, гидрохлоридной, йодистоводородной, гидроксинафтойной, изэтиновой, молочной, лактобионовой, малеиновой, яблочной, малоновой, миндальной, метансульфоновой, метилазотной, метилсерной, муциновой, муконовой, напсиловой, азотной, щавелевой, п-нитрометансульфоновой, памовой, пантотеновой, фосфорной, моногидрофосфорной, дигидрофосфорной, фталевой, полигалактоуроновой, пропионовой, салициловой, стеариновой, янтарной, сульфаминовой, сульфаниловой, сульфоновой, серной, дубильной, винной, теокловой, толуолсульфоновой кислоты и др. Также можно получить натриевые соли и калиевые соли.
Вариантами осуществления могут быть диаммониевые соли E7766. Фармацевтически приемлемые соли можно получить из аминокислот, в том числе без ограничения цистеина. Способы получения соединений в виде солей известны специалистам в данной области (см., например, Stahl et al., Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH; Verlag Helvetica Chimica Acta, Zurich, 2002; Berge et al., J. Pharm. Sci. 66: 1, 1977).
“Эффективное количество” или “терапевтически эффективное количество” терапевтического средства представляет собой количество, достаточное для получения наблюдаемого терапевтического эффекта в сравнении с раковым образованием у субъекта или пациента, который не получал лечения.
Активные средства, описываемые в данном документе, можно объединить с фармацевтически приемлемым носителем для получения фармацевтических составов на их основе. Конкретный выбор носителя и состава будет зависеть от конкретного пути введения, для которого предназначена композиция.
"Фармацевтически приемлемый носитель" в контексте данного документа относится к нетоксичным носителю, вспомогательному веществу или среде-носителю, которые не нарушают фармакологическую активность соединения, с которым они составлены. Фармацевтически приемлемые носители, вспомогательные вещества или среды-носители, которые могут использоваться в композициях по настоящему изобретению, включают без ограничения сорбиновую кислоту, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, полиэтиленгликоль и ланолин.
Композиции по настоящему изобретению могут быть пригодны для парентерального, перорального введения, ингаляционного спрея, местного, ректального, назального, трансбуккального, вагинального, внутрипузырного, интравезикального, внутриопухолевого введения или введения в виде имплантируемого резервуара и т.д. В некоторых вариантах осуществления состав содержит ингредиенты, которые получены из природных или неприродных источников. В некоторых вариантах осуществления состав или носитель могут быть представлены в стерильной форме. Неограничивающие примеры стерильного носителя включают не содержащую эндотоксинов воду или апирогенную воду. Композиции можно вводить путем внутрипузырного, интравезикального или внутриопухолевого введения.
Термин “парентеральный” в контексте данного документа включает методики подкожных, внутривенных, внутримышечных, внутрисуставных, внутрисиновиальных, внутригрудинных, интратекальных, внутрипеченочных, внутриочаговых и внутричерепных инъекций или инфузий. В конкретных вариантах осуществления соединения вводят внутривенно, перорально, подкожно или посредством внутримышечного введения. Стерильные инъекционные формы композиций по настоящему изобретению могут представлять собой водную или маслянистую суспензию. Данные суспензии могут быть составлены в соответствии с методиками, известными из уровня техники, с применением подходящих диспергирующих или смачивающих средств и суспендирующих средств. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе. К приемлемым средам-носителям и растворителям, которые могут использоваться, относятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или среды для суспендирования традиционно используются стерильные нелетучие масла.
Для данной цели можно использовать любое легкое нелетучее масло, в том числе синтетические моно- или диглицериды. При получении инъекционных препаратов применимы жирные кислоты и их глицеридные производные, также как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно их полиоксиэтилированные варианты. Данные масляные растворы или суспензии могут также содержать длинноцепочечный спиртовой разбавитель или диспергатор, такой как карбоксиметилцеллюлоза или подобные диспергирующие средства, которые обычно используются при составлении фармацевтически приемлемых лекарственных форм, в том числе эмульсий и суспензий. Другие широко используемые поверхностно-активные вещества, такие как разновидности Tween, Span, и другие эмульгирующие средства, которые широко используются при изготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут применяться для целей получения состава.
В случае перорального введения соединение или соль может быть представлена в виде лекарственной формы, приемлемой для перорального введения, включая без ограничения капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения широко используемые носители включают лактозу и кукурузный крахмал. Также можно добавлять смазывающие средства, такие как стеарат магния. В случае перорального введения в форме капсулы применимые разбавители включают лактозу и высушенный кукурузный крахмал. Если водные суспензии предназначены для перорального введения, то активный ингредиент может быть объединен с эмульгирующими и суспендирующими средствами. При необходимости также могут быть добавлены определенные подсластители, ароматизаторы или красители. Кроме того, также можно добавить консерванты. Подходящие примеры фармацевтически приемлемых консервантов включают без ограничения различные противобактериальные и противогрибковые средства, такие как растворители, например этанол, пропиленгликоль, бензиловый спирт, хлорбутанол, соли четвертичного аммония и парабены (такие как метилпарабен, этилпарабен, пропилпарабен и т.д.).
“Немедленное высвобождение” понимают как включающее традиционное высвобождение, при котором высвобождение лекарственного средства начинается сразу после введения. В контексте данного документа термин “немедленное высвобождение” включает лекарственные формы, которые позволяют лекарственному средству растворяться в содержимом желудочно-кишечного тракта без стремления замедлить или продлить растворение или всасывание лекарственного средства. Цель заключается в быстром высвобождении лекарственного средства после введения, например, чтобы было возможно высвобождение по меньшей мере 80% лекарственного средства в течение примерно 30 минут после начала растворения в тесте на растворимость.
"С замедленным высвобождением" или "с продленным высвобождением" охватывает лекарственные формы, у которых характеристики высвобождения лекарственного средства в зависимости от времени и/или местоположения выбраны так, чтобы достигались терапевтические цели или цели удобства, которые не обеспечиваются традиционными лекарственными формами, такими как раствор или лекарственная форма с немедленным высвобождением.
Термин "равновесное состояние" означает, что для данного активного средства был достигнут некоторый уровень в плазме крови и его поддерживают последующими дозами активного средства на уровне, который равен минимальному эффективному терапевтическому уровню или превышает его и ниже минимального токсичного уровня в плазме крови для данного активного средства.
Термин “диапазон доз” в контексте данного документа относится к верхнему и нижнему пределу приемлемого варьирования количества указанного средства. Как правило, пациентам, проходящим лечение, можно вводить дозу средства в любом количестве в пределах указанного диапазона.
Термин "лечить" в данном документе применяют для обозначения облегчения, уменьшения или ослабления по меньшей мере одного симптома заболевания у субъекта. Например, в отношении рака термин "лечить" может означать остановку, отсрочку начала (т. е. период до клинического проявления заболевания или симптома заболевания) и/или уменьшение риска развития или ухудшения симптома рака. Термин "защищать" в данном документе применяют для обозначения предупреждения отсрочки или лечения, или всего сразу, в зависимости от ситуации, развития, или продолжения, или ухудшения симптомов рака у субъекта.
Термин "субъект" или "пациент" подразумевают как включающий животных, которые способны страдать от рака или подвержены раку. Примеры субъектов или пациентов включают млекопитающих, например людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных животных, отличных от человека. В определенных вариантах осуществления субъектом является человек, например человек, страдающий от рака, подверженный риску пострадать от рака или потенциально способный пострадать от рака.
Термин "информационное сообщение", используемый в данном документе, может включать документацию как часть одобренного регуляторными органами фармацевтического продукта, в том числе без ограничения этикетку продукта или вкладыш продукта. Такая документация может включать инструкции, меры предосторожности или предупреждения для пациента или лечащего врача, например, в разделах "Взаимодействие с другими лекарственными средствами", "Клиническая фармакология", "Доза и схема приема", "Предупреждения и меры предосторожности", "Противопоказания" или "Особые предупреждения".
Термин "приблизительно" или "примерно" обычно означает в пределах 20%, более предпочтительно в пределах 10% и наиболее предпочтительно в пределах 5% от заданного значения или диапазона. В качестве альтернативы, особенно в биологических системах, термин “приблизительно” означает примерно в пределах log (т. е. порядок величины), предпочтительно в пределах двукратной величины от заданного значения.
Применение термина в форме единственного числа и его синонимов в контексте описания настоящего изобретения (особенно в контексте последующей формулы изобретения) следует истолковывать как охватывающее как форму единственного, так и форму множественного числа, если только в данном документе не указано иное или это явно противоречит контексту. Термины "охватывающий", "имеющий", "включающий" и "содержащий" следует истолковывать как термины с открытой формулировкой (т. е. означающие "включающий без ограничения"), если не указано иное. Указание диапазонов значений в данном документе подразумевают лишь как служащее в качестве сокращенного способа индивидуальной отсылки к каждому отдельному значению, подпадающему в этот диапазон, если в данном документе не указано иное, и каждое отдельное значение включено в настоящее описание, как если бы оно было отдельно указано в данном документе.
Иллюстративные нарушения пролиферации клеток, которые можно лечить с применением одного или нескольких описанных в данном документе соединений, включают без ограничения рак, предрак или предраковое состояние и метастатические поражения в ткани и органах организма. Нарушения пролиферации клеток могут включать гиперплазию, метаплазию и дисплазию.
Раскрываемое в данном документе соединение или его фармацевтически приемлемую соль можно применять для лечения или предупреждения нарушения пролиферации клеток или для лечения или предупреждения рака у субъекта, имеющего повышенный риск развития рака по сравнению со всем населением, или применять для идентификации подходящих кандидатов для таких целей.
Фармацевтические составы и пути введения
В данном документе представлены фармацевтические составы, содержащие E7766 или его фармацевтически приемлемую соль, для лечения рака. Фармацевтические составы могут дополнительно содержать носитель или вспомогательное вещество, стабилизатор, ароматизатор и/или краситель.
E7766 или его фармацевтически приемлемую соль можно вводить с помощью ряда путей введения, известных специалистам в данной области. Пути введения включают пероральное введение, внутриопухолевое введение, внутрипузырное введение и интравезикальное введение. В определенных вариантах осуществления фармацевтический состав, содержащий соединение или его фармацевтически приемлемую соль, можно принимать перорально в форме жидкости, сиропа, таблетки, капсулы, порошка, обсыпки в капсулах, жевательной таблетки или растворимого диска. В качестве альтернативы, фармацевтические составы по настоящему изобретению можно вводить внутривенно или трансдермально. Дополнительные пути введения известны специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, Gennaro A. R., Ed., 20th Edition, Mack Publishing Co., Easton, Pa.).
В некоторых вариантах осуществления соединение или фармацевтически приемлемая соль составлены в виде пасты, желе или суспензии. Например, лекарственное средство растворено, захвачено или суспендировано в форме частиц лекарственного средства, микроинкапсулированных частиц или частиц лекарственного средства-полимера в гелеобразном растворе или полутвердой форме. Преимуществом перорального желеобразного состава является то, что пациентам, которым трудно глотать таблетки, капсулы или пилюли, легче вводить такое лекарственное средство. В определенных вариантах осуществления соединение тщательно перемешано и суспендировано в подходящей среде с образованием пасты или геля. Для придания вкусоароматических свойств при пероральном введении необязательно можно примешивать дополнительные средства. Примерами многих подходящих средств, маскирующих вкус, являются арахисовое масло или альгинат с ароматизатором "малина" и подсластителем. В различных вариантах осуществления паста или желе также могут быть составлены с подходящими связующими веществами или вспомогательными веществами, известными в уровне техники для местного применения.
Способы получения составов с замедленным высвобождением в форме таблеток, капсул или пилюль известны из уровня техники. В некоторых вариантах осуществления состав с замедленным высвобождением получают путем нанесения на активный ингредиент лекарственного средства покрытия из полимера, предпочтительно водонерастворимого полимера. Например, водонерастворимого полимера, применяемого в фармацевтической области в качестве покрывающего средства для замедленного высвобождения, энтеросолюбильного покрывающего средства или растворяющегося в желудке покрывающего средства. Водонерастворимый полимер может включать, например, этилцеллюлозу, очищенный шеллак, белый шеллак, сополимер аминоалкилметакрилата RS, фталат гидроксипропилметилцеллюлозы, ацетосукцинат гидроксипропилметилцеллюлозы, карбоксиметилэтилцеллюлозу, ацетофталат целлюлозы, сополимер метакриловой кислоты L, сополимер метакриловой кислоты LD, сополимер метакриловой кислоты S, сополимер аминоалкилметакрилата E или поливинилацетальдиэтиламиноацетат.
Тип, степень замещения и молекулярная масса водонерастворимых полимеров могут зависеть от растворимости активного ингредиента в воде или спирте, требуемого уровня замедленного высвобождения и др. Водонерастворимые полимеры можно применять либо по отдельности, либо в комбинации. Дополнительно в качестве вспомогательного средства для нанесения покрытия можно включить гидрогенизированное масло, стеариновую кислоту или цетанол и в качестве пластификатора триглицерид со средней длиной цепи, триацетин, триэтилцитрат или цетанол.
В некоторых вариантах осуществления состав с замедленным высвобождением представляет собой таблетку или гранулу матричного типа. Активный ингредиент можно покрыть вплоть до 3-мя различными типами полимеров. Данные три различных типа полимеров могут включать: 1) водонерастворимый полимер, такой как этилцеллюлоза; 2) рН-независимый гелеобразующий полимер, такой как гидроксипропилметилцеллюлоза; и 3) рН-зависимый гелеобразующий полимер, такой как альгинат натрия. Эти три различных типа полимеров можно применять совместно для уменьшения скорости высвобождения лекарственных средств.
Внутриопухолевые дозы и схемы введения
В одном варианте осуществления внутриопухолевого введения E7766 или его фармацевтически приемлемую соль вводят пациенту за несколько циклов, каждый продолжительностью 3 недели. E7766 или его фармацевтически приемлемую соль вводят в индукционном цикле (цикл 1) в дни 1, 8 и 15; и вводят в день 1 каждого последующего поддерживающего цикла (цикл 2 и далее). Общая доза, вводимая каждый раз, может составлять 25 мкг, 50 мкг, 75 мкг, 150 мкг, 300 мкг, 600 мкг, 1200 мкг или 1750 мкг. Вводимая доза может быть в одном из следующих диапазонов доз: 75 мкг - 1750 мкг, 75 мкг - 1200 мкг, 75 мкг - 600 мкг, 75 мкг - 300 мкг, 75 мкг - 150 мкг, 150 мкг - 1750 мкг, 150 мкг - 1200 мкг, 150 мкг - 600 мкг, 150 мкг - 300 мкг, 300 мкг - 1750 мкг, 300 мкг - 1200 мкг, 300 мкг - 600 мкг, 1200 мкг - 1750 мкг, 75 мкг - 200 мкг, 75 мкг - 150 мкг или 100 мкг - 150 мкг.
В одном варианте осуществления E7766 или его фармацевтически приемлемая соль представлены в виде твердого или концентрированного раствора, и для внутриопухолевого введения его разводят нормальным солевым раствором до конечного объема 1 мл.
В одном варианте осуществления E7766 или его фармацевтически приемлемая соль представлены в форме для внутриопухолевого введения для лечения рака молочной железы (в том числе трижды негативного рака молочной железы (TNBC), рака толстой кишки, колоректального рака, глиомы, плоскоклеточной карциномы головы и шеи, рака печени, лимфомы, меланомы, рака предстательной железы, рака поджелудочной железы, рака почек или других солидных опухолей.
Внутрипузырные или интравезикальные дозы и схемы введения
В одном варианте осуществления внутрипузырного введения или варианте осуществления интравезикального введения E7766 или его фармацевтически приемлемую соль вводят пациенту сначала в индукционном цикле (цикл 1), продолжительностью 6 недель. В индукционном цикле E7766 или его фармацевтически приемлемую соль вводят в дни 1, 8, 15, 22, 29 и 36. Последующие поддерживающие циклы (цикл 2 и далее) инициируют так, как указано в представленной ниже таблице 1.
Таблица 1
Общая доза, вводимая каждый раз, может составлять 600 мкг, 800 мкг, 1600 мкг, 2000 мкг, 2400 мкг, 3200 мкг, 6400 мкг, 9600 мкг или 12800 мкг. Вводимая доза может быть в одном из следующих диапазонов доз: 800 мкг - 12800 мкг; 800 мкг - 9600 мкг; 800 мкг - 6400 мкг; 800 мкг - 3200 мкг; 800 мкг - 2400 мкг; 800 мкг - 2000 мкг; 800 мкг - 1600 мкг; 1600 мкг - 12800 мкг; 1600 мкг - 9600 мкг; 1600 мкг - 6400 мкг; 1600 мкг - 3200 мкг; 1600 мкг - 2400 мкг; 1600 мкг - 2000 мкг; 2000 мкг - 12800 мкг; 2000 мкг - 9600 мкг; 2000 мкг - 6400 мкг; 2000 мкг - 3200 мкг; 2000 мкг - 2400 мкг; 2400 мкг - 12800 мкг; 2400 мкг - 9600 мкг; 2400 мкг - 6400 мкг; 2400 мкг - 3200 мкг; 3200 мкг - 12800 мкг; 3200 мкг - 9600 мкг; 3200 мкг - 6400 мкг; 6400 мкг - 12800 мкг; 6400 мкг - 9600 мкг или 9600 мкг - 12800 мкг. В другом варианте осуществления E7766 или его фармацевтически приемлемая соль представлена в виде твердого или концентрированного раствора, и для внутрипузырного введения его разводят нормальным солевым раствором до конечного объема 25 мл. В другом варианте осуществления E7766 или его фармацевтически приемлемая соль представлена в виде твердого или концентрированного раствора, и для интравезикального введения его разводят нормальным солевым раствором до конечного объема 25 мл.
В одном варианте осуществления E7766 или его фармацевтически приемлемая соль представлены в форме для внутрипузырного введения для лечения рака (в том числе рака мочевого пузыря с инвазией в мышечный слой и без инвазии в мышечный слой (NMIBC, в том числе NMIBC, не отвечающий на терапию с применением бациллы Кальмета-Герена (BGC)), переходно-клеточной карциномы мочевого пузыря, папиллярной болезни Та или Т1 с карцимой in situ (CIS) или без нее и новообразований мочевого пузыря). В одном варианте осуществления E7766 или его фармацевтически приемлемая соль представлены в форме для интравезикального введения для лечения рака мочевого пузыря (в том числе рака мочевого пузыря с инвазией в мышечный слой и без инвазии в мышечный слой (NMIBC, в том числе NMIBC, не отвечающий на терапию с применением бациллы Кальмета-Герена (BGC)), переходно-клеточной карциномы мочевого пузыря, папиллярной болезни Та или Т1 с карцимой in situ (CIS) или без нее и новообразований мочевого пузыря).
Лекарственные формы: свойства высвобождения
Составы с замедленным высвобождением могут достигать некоторой степени долговременного эффекта. Тем не менее, экспозиция и/или биодоступность активного ингредиента может варьироваться в зависимости от ряда факторов, таких как, например, интервал всасывания, носители или вспомогательные вещества, применяемые в составе, способ доставки состава и/или время прохождения активного ингредиента через желудочно-кишечный тракт пациента.
Средство терапии может содержать по меньшей мере одну часть с замедленным высвобождением для осуществления функции замедленного высвобождения и одну часть с немедленным высвобождением для осуществления функции немедленного высвобождения. В определенных вариантах осуществления, если средство терапии представлено в виде однократной лекарственной формы, то оно может иметь форму таблеток, образованных из смеси гранул с замедленным высвобождением, составляющих часть с замедленным высвобождением, и гранул с немедленным высвобождением, составляющих часть с немедленным высвобождением, препарата в виде капсулы, полученного при заполнении капсулы гранулами с замедленным высвобождением и гранулами с немедленным высвобождением, или спрессованных таблеток с покрытием, в которых внешний слой, составляющий часть с немедленным высвобождением, образован на внутреннем ядре, составляющем часть с замедленным высвобождением. Тем не менее, для вышеупомянутых вариантов осуществления нет никаких ограничений.
Более того, нет никаких конкретных ограничений в отношении состояния содержания лекарственного средства в композиции, или в части с немедленным высвобождением, или в части с замедленным высвобождением; соединение может быть равномерно диспергировано в композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может содержаться только в одной части композиции, части с немедленным высвобождением или части с замедленным высвобождением, или может содержаться так, чтобы получался градиент концентрации.
Часть с замедленным высвобождением в композиции по настоящему изобретению может содержать по меньшей мере одно pH-независимое полимерное вещество или pH-зависимое полимерное вещество для контроля высвобождения лекарственного средства.
pH-независимое полимерное вещество, применяемое в данном документе, может включать полимерное вещество, состояние заряда которого практически не изменяется в условиях pH, обычно встречающихся в желудочно-кишечном тракте, в частности от pH 1 до pH 8. Это означает, например, что состояние заряда полимерного вещества, которое не имеет функциональных групп, изменяется в зависимости от pH, как, например, основных функциональных групп, таких как аминогруппы, или кислотных функциональных групп, таких как группы карбоновых кислот. Следует отметить, что pH-независимое полимерное вещество может быть включено для придания композиции по настоящему изобретению функции замедленного высвобождения, но также может быть включено и для другой цели. Более того, pH-независимое полимерное вещество, применяемое в настоящем изобретении, может быть водонерастворимым, или может набухать в воде, или растворяться в воде с образованием геля.
Примеры водонерастворимых pH-независимых полимерных веществ включают без ограничения простые эфиры целлюлозы, сложные эфиры целлюлозы и сополимеры метакриловой кислоты и акриловой кислоты (торговое название Eudragit, производства компании Rohm GmbH & Co. KG, Дармштадт, Германия). Примеры включают без ограничения простые алкиловые эфиры целлюлозы, такие как этилцеллюлоза (торговое название Ethocel, производства компании Dow Chemical Company, США), этилметилцеллюлоза, этилпропилцеллюлоза или изопропилцеллюлоза и бутилцеллюлоза, простые аралкиловые эфиры целлюлозы, такие как бензилцеллюлоза, простые цианоалкиловые эфиры целлюлозы, такие как цианоэтилцеллюлоза, сложные эфиры целлюлозы и органических кислот, такие как ацетобутират целлюлозы, ацетат целлюлозы, пропионат целлюлозы или бутират целлюлозы и ацетопропионат целлюлозы, сополимеры этилакрилата и метилметакрилата (торговое название Eudragit NE, производства компании Rohm GmbH & Co. KG, Дармштадт, Германия) и сополимер аминоалкилметакрилата RS (торговые названия Eudragit RL, Eudragit RS).
Нет никаких конкретных ограничений в отношении среднего диаметра частиц водонерастворимого полимера, применяемого в настоящем изобретении, но обычно чем ниже данный средний диаметр частиц, тем лучше характеристики, при этом средний диаметр частиц предпочтительно составляет от 0,1 до 100 мкм, более предпочтительно от 1 до 50 мкм, особенно предпочтительно от 3 до 15 мкм, наиболее предпочтительно от 5 до 15 мкм. Более того, примеры водорастворимых или набухающих в воде pH-независимых полимерных веществ включают без ограничения полиэтиленоксид (торговое название Polyox, производства компании Dow Chemical Company, молекулярная масса от 100000 до 7000000), гидроксипропилцеллюлозу с низкой степенью замещения (торговое название L-HPC, производства компании Shin-Etsu Chemical, Япония), гидроксипропилцеллюлозу (торговое название HPC, производства компании Nippon Soda, Co., Ltd, Япония), гидроксипропилметилцеллюлозу (торговые названия Metolose 60SH, 65SH, 90SH, производства компании Shin-Etsu Chemical, Япония) и метилцеллюлозу (торговое название Metolose SM, производства компании Shin-Etsu Chemical, Япония).
В некоторых вариантах осуществления в композиции может содержаться одно pH-независимое полимерное вещество или может содержаться множество pH-независимых полимерных веществ. PH-независимое полимерное вещество, при его применении в вариантах осуществления, описанных в данном документе, может быть водонерастворимым полимерным веществом, более предпочтительно этилцеллюлозой, сополимером этилакрилата и метилметакрилата (торговое название Eudragit NE) или сополимером аминоалкилметакрилата RS (торговое название Eudragit RL, Eudragit RS). Особенно предпочтительным является по меньшей мере одно из этилцеллюлозы и сополимера аминоалкилметакрилата RS. Наиболее предпочтительной является этилцеллюлоза. Нет никаких конкретных ограничений в отношении количества pH-независимого полимерного вещества, содержащегося в композиции; данное количество можно при необходимости корректировать в соответствии с такой целью, как контроль замедленного высвобождения лекарственного средства.
pH-зависимое полимерное вещество, которое можно применять в вариантах осуществления, описанных в данном документе, может представлять собой полимерное вещество, состояние заряда которого изменяется в условиях pH, которые обычно встречаются в желудочно-кишечном тракте, в частности от pH 1 до pH 8. Это означает, например, что состояние заряда полимерного вещества, имеющего функциональные группы, изменяется в зависимости от pH, как, например, основных функциональных групп, таких как аминогруппы, или кислотных функциональных групп, таких как группы карбоновых кислот. pH-зависимые функциональные группы pH-зависимого полимерного вещества предпочтительно представляют собой кислотные функциональные группы, причем pH-зависимое полимерное вещество наиболее предпочтительно имеет группы карбоновых кислот.
pH-зависимое полимерное вещество, применяемое в настоящем изобретении, может быть водонерастворимым, или может набухать в воде, или растворяться в воде с образованием геля. Примеры pH-зависимых полимерных веществ, применяемых в настоящем изобретении, включают без ограничения энтеросолюбильные полимерные вещества. Примеры энтеросолюбильных полимерных веществ включают без ограничения сополимеры метакриловой кислоты и метилметакрилата (Eudragit L100, Eudragit S100, производства компании Rohm GmbH & Co. KG, Дармштадт, Германия), сополимеры метакриловой кислоты и этилакрилата (Eudragit L100-55, Eudragit L30D-55, производства компании Rohm GmbH & Co. KG, Дармштадт, Германия), фталат гидроксипропилметилцеллюлозы (HP-55, HP-50, производства компании Shin-Etsu Chemical, Япония), ацетосукцинат гидроксипропилметилцеллюлозы (AQOAT, производства компании Shin-Etsu Chemical, Япония), карбоксиметилэтилцеллюлозу (CMEC, производства компании Freund Corporation, Япония) и ацетофталат целлюлозы.
Примеры pH-зависимых полимерных веществ, которые набухают в воде или растворяются в воде с образованием геля, включают без ограничения альгиновую кислоту, пектин, карбоксивиниловый полимер и карбоксиметилцеллюлозу. В настоящем изобретении в композиции может содержаться одно pH-зависимое полимерное вещество или может содержаться множество pH-зависимых полимерных веществ. pH-зависимое полимерное вещество, применяемое в настоящем изобретении, предпочтительно представляет собой энтеросолюбильное полимерное вещество, более предпочтительно сополимер метакриловой кислоты и этилакрилата, сополимер метакриловой кислоты и метилметакрилата, фталат гидроксипропилметилцеллюлозы или ацетосукцинат гидроксипропилметилцеллюлозы, особенно предпочтительно сополимер метакриловой кислоты и этилакрилата.
При применении pH-зависимого полимерного вещества в способе изготовления композиции по настоящему изобретению коммерчески доступный продукт порошкового типа, или гранулированного типа, или суспензионного типа, в котором pH-зависимое полимерное вещество было заранее диспергировано в растворителе, можно применять как есть или такой коммерчески доступный продукт можно применять диспергированным в воде или органическом растворителе. Чем меньше диаметр частиц pH-зависимого полимерного вещества, тем лучше характеристики, при этом pH-зависимое полимерное вещество предпочтительно относится к порошковому типу. В случае сополимера метакриловой кислоты и этилакрилата примером является Eudragit L100-55. Нет никаких конкретных ограничений в отношении среднего диаметра частиц pH-зависимого полимерного вещества, применяемого в настоящем изобретении, но средний диаметр частиц предпочтительно составляет от 0,05 до 100 мкм, более предпочтительно от 0,05 до 70 мкм, наиболее предпочтительно от 0,05 до 50 мкм. Более того, нет никаких конкретных ограничений в отношении количества pH-зависимого полимерного вещества, например, в случае энтеросолюбильного полимерного вещества, количество обычно составляет от 0,1 до 90 частей по массе, предпочтительно от 1 до 70 частей по массе, более предпочтительно от 5 до 60 частей по массе, особенно предпочтительно от 10 до 50 частей по массе на 100 частей по массе композиции.
Средство терапии согласно вариантам осуществления, описанным в данном документе, при необходимости может дополнительно содержать любую из различных добавок, такую как любой из различных фармакологически приемлемых носителей, таких как разбавители, скользящие вещества, связующие вещества и разрыхлители, а также консерванты, красители, подсластители, пластификаторы, пленкообразователи и так далее. Примеры разбавителей включают без ограничения лактозу, маннит, двухосновный фосфат кальция, крахмал, прежелатинизированный крахмал, кристаллическую целлюлозу, легкий кремниевый ангидрид, синтетический силикат алюминия, метасиликат алюмината магния или т.п. Примеры скользящих веществ включают без ограничения стеарат магния, стеарат кальция, тальк, стеарилфумарат натрия или т.п. Примеры связующих веществ включают без ограничения гидроксипропилцеллюлозу, метилцеллюлозу, карбоксиметилцеллюлозу натрия, гидроксипропилметилцеллюлозу, поливинилпирролидон или т.п. Примеры разрыхлителей включают без ограничения карбоксиметилцеллюлозу, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, карбоксиметилкрахмал натрия, гидроксипропилцеллюлозу с низкой степенью замещения или тому подобное.
Примеры консервантов включают без ограничения сложные эфиры параоксибензойной кислоты, хлорбутанол, бензиловый спирт, фенэтиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту или т.п. Предпочтительные примеры красителей включают без ограничения водонерастворимые красочные пигменты, природные пигменты (например, бета-каротин, хлорофилл, красный оксид железа), желтый оксид железа, красный оксид железа, черный оксид железа или т.п. Предпочтительные примеры подсластителей включают без ограничения сахарин натрия, глицирризат калия, аспартам, стевию или т.п. Примеры пластификаторов включают без ограничения сложные эфиры глицерина и жирных кислот, триэтилцитрат, пропиленгликоль, полиэтиленгликоль или т.п. Примеры пленкообразователей включают без ограничения гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу или т.п.
Способы изготовления
Что касается вариантов осуществления, относящихся к изготовлению, которые описаны в данном документе, то можно применять единственный традиционный способ или комбинацию традиционных способов. Например, при изготовлении гранул, содержащих лекарственное средство, в качестве части с замедленным высвобождением или части с немедленным высвобождением, гранулирование является основной операцией, но ее можно сочетать с другими операциями, такими как смешивание, сушка, просеивание и классификация. В качестве способа гранулирования можно применять, например, способ влажного гранулирования, при котором к порошку добавляют связующее вещество и растворитель и производят гранулирование, способ сухого гранулирования, при котором порошок спрессовывают и производят гранулирование, способ гранулирования в расплаве, при котором добавляют связующее вещество, которое плавится при нагревании, и производят нагревание и гранулирование, или т.п.
Более того, в соответствии со способом гранулирования, можно применять оперативный способ, такой как способ гранулирования с перемешиванием с применением планетарного смесителя, шнекового смесителя или т.п., способ гранулирования с перемешиванием на высокой скорости с применением смесителя Henschel, смесителя Super или т.п., способ гранулирования с экструзией с применением цилиндрического гранулятора, роторного гранулятора, шнекового экструдера-гранулятора, гранулятора по типу окомкователя или т.п., способ влажного гранулирования с большим усилием сдвига, способ гранулирования в псевдоожиженном слое, способ гранулирования с прессованием, способ гранулирования с дроблением или способ гранулирования с распылением. После гранулирования можно произвести сушку с применением сушилки, псевдоожиженного слоя или т.п., разламывание и просеивание с получением гранул или мелких гранул для применения. Более того, при получении композиции согласно настоящему изобретению можно применять растворитель для гранулирования. Нет никаких особых ограничений в отношении такого растворителя для гранулирования, которым может быть вода или любой из различных органических растворителей, например, вода, низший спирт, такой как метанол или этанол, кетон, такой как ацетон или метилэтилкетон, метиленхлорид или их смесь.
В случае гранул с замедленным высвобождением, содержащихся в вариантах осуществления, по меньшей мере одно лекарственное средство и по меньшей мере одно, выбранное из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, смешивают вместе, при необходимости добавляют разбавитель и связующее вещество и проводят грануляцию с получением гранулированного материала. Полученный гранулированный материал сушат с применением лотковой сушилки, сушилки с псевдоожиженным слоем или т.п. и проводят просеивание с применением мельницы или осциллятора, в результате чего можно получить гранулы с замедленным высвобождением. В качестве альтернативы, в качестве способа изготовления гранул с замедленным высвобождением в настоящем изобретении можно добавить по меньшей мере одно лекарственное средство, по меньшей мере одно, выбранное из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, и, при необходимости, разбавитель и связующее вещество с применением пресса для сухого прессования, такого как роликовый пресс или машина для таблетирования штампованием, и произвести формование прессованием при перемешивании, а затем произвести гранулирование путем разламывания до подходящего размера. Гранулированный материал, полученный с применением такого гранулятора, можно применять как есть в виде гранул или мелких гранул в соответствии с настоящим изобретением или можно дополнительно подвергнуть разламыванию с применением силовой мельницы, валковой дробилки, роторной скоростной мельницы или тому подобного и просеять с получением гранул с замедленным высвобождением. Следует отметить, что гранулы с немедленным высвобождением можно изготовить так же, как и гранулы с замедленным высвобождением.
Сформованный прессованием продукт можно изготовить в качестве содержащей лекарственное средство части с замедленным высвобождением или части с немедленным высвобождением или в виде композиции, описанной в данном документе, с применением единственного традиционного способа или комбинации традиционных способов. Например, применяют по меньшей мере одно лекарственное средство, по меньшей мере одно, выбранное из pH-независимых полимерных веществ и pH-зависимых полимерных веществ, разбавитель, такой как маннит или лактоза, связующее вещество, такое как поливинилпирролидон или кристаллическая целлюлоза, разрыхлитель, такой как кармеллоза натрия или кросповидон, и скользящее вещество, такое как стеарат магния или тальк, и производят таблетирование с помощью обычного способа, в результате чего можно получить сформованный прессованием продукт. В данном случае таблетирование является основной операцией в способе изготовления сформованного прессованием продукта, но его можно комбинировать с другими операциями, такими как смешивание, сушка, формирование сахарного покрытия и нанесение покрытия.
Примеры способа таблетирования включают без ограничения прямое формование прессованием, при котором по меньшей мере одно лекарственное средство и фармакологически приемлемые добавки смешивают вместе, а затем смесь непосредственно формуют прессованием в таблетки с применением таблетировочной машины, и сухое прессование гранул или влажное прессование гранул, при котором гранулы с замедленным высвобождением или гранулы с немедленным высвобождением по настоящему изобретению подвергают формованию прессованием после добавления при необходимости скользящего вещества или разрыхлителя. Нет никаких особых ограничений в отношении таблетировочной машины, применяемой при формовании прессованием; например, можно применять таблетировочную машину с одним пуансоном, роторную таблетировочную машину или таблетировочную машину с нанесением покрытия прессованием.
Содержащие лекарственное средство гранулы с замедленным высвобождением, или гранулы с немедленным высвобождением, или формованный прессованием продукт в соответствии с вариантами осуществления, описанными в данном документе, можно применять как есть в форме гранул или таблетки в качестве композиции, но также можно подвергнуть дополнительной обработке для изготовления композиции. Например, на сформованный прессованием продукт или гранулы можно нанести пленочное покрытие с применением материала основы пленки, такого как этилцеллюлоза, казеин, метилцеллюлоза, гидроксипропилметилцеллюлоза, сополимер метакриловой кислоты L, ацетофталат целлюлозы, шеллак или т.п., или нанести сахарное покрытие с применением жидкости для нанесения сахарного покрытия, содержащей сахарозу, сахарный спирт, порошок гуммиарабика, тальк или т.п., с получением, таким образом, таблеток с пленочным покрытием или таблеток с сахарным покрытием. Одним растворителем в данной методике нанесения покрытия может быть очищенная вода, но также можно применять органический растворитель, такой как спирт, кетон, простой эфир или хлорированный углеводород или их смесь. Например, в качестве органического растворителя можно применять этанол, ацетон, метиленхлорид или т.п. Более того, в качестве устройства для нанесения покрытия можно применять устройство, обычно применяемое в методиках нанесения покрытия в процессе изготовления лекарственных препаратов, при этом примеры включают устройство для нанесения покрытия распылением, в котором нанесение покрытия выполняют путем распыления жидкости для нанесения покрытия или т.п., и роторный гранулятор с псевдоожиженным слоем для нанесения слоев.
В случае изготовления препаратов в форме капсул препараты в форме капсул можно изготавливать путем заполнения твердых желатиновых капсул или капсул из HPMC гранулами с замедленным высвобождением или гранулами с немедленным высвобождением, которые описаны выше, или мини-таблетками с применением автоматической машины для заполнения капсул. В качестве альтернативы, в случае препаратов для введения через систему трубок или в форме сухого сиропа, который применяют в смеси с водой или тому подобного при приеме, гранулы с замедленным высвобождением или гранулы с немедленным высвобождением, которые описаны выше, можно смешивать с загустителем или диспергатором, с тем чтобы диспергировать данные гранулы, при этом смесь затем формируют в гранулы или таблетки. Кроме того, жидкость или желе можно получить с применением воды и веществ, выбранных из диспергаторов, эмульгаторов, загустителей, консервантов, регуляторов pH, подсластителей, ароматизаторов, отдушек и т.д. Тем не менее, что касается других способов изготовления, нет никаких ограничений для вышеупомянутого.
Как сообщается более подробно ниже, в механистических экспериментах в условиях in vivo и in vitro было определено следующее: 1) стадия определения скорости системного клиренса соединения 1; 2) относительные вклады транспортеров, задействованных в захвате и клиренсе с желчью соединения 1; и 3) потенциальные межлекарственные взаимодействия, связанные с OATP1B1/1B3, у людей.
Фармакокинетику и распределение соединения 1 изучали в условиях in vivo на крысах и собаках с канюлированными желчными протоками, а также на гуманизированных мышах OATP1B1/1B3. Фенотипирование транспортера соединения 1 проводили с применением трансфицированных клеточных линий и везикул. Выведение с желчью и клиренс с поглощением соединения 1 также определяли на гепатоцитах человека, культивируемых сэндвич-способом. Также использовали моделирование в условиях in silico, в частности с помощью simCYP™, для физиологически обоснованного фармакокинетического (PBPK) моделирования и прогнозирования клинического межлекарственного взаимодействия (DDI). PBPK-моделирование применяли для оценки потенциальных межлекарственных взаимодействий с ингибиторами OATP в клинических условиях.
Исходя из результатов исследований, представленных в данном документе, E7766 или его фармацевтически приемлемая соль и, в частности, соединение 1 могут вызывать потенциальное межлекарственное взаимодействие с ингибиторами OATP. Данные ингибиторы OATP могут быть ингибиторами OATP1B1 и/или ингибиторами OATP1B3. Совместное введение ингибиторов OATP с E7766 или его фармацевтически приемлемой солью может привести к изменению величин дозы и режимов введения E7766 или его фармацевтически приемлемой соли и/или введения ингибитора OATP, или данное совместное введение может быть совершенно неприемлемо.
Ингибиторы OATP, которые могут характеризоваться потенциальным межлекарственным взаимодействием с E7766 или его фармацевтически приемлемой солью, могут включать, например, без ограничения фимасартан, кларитромицин, рифампин, клопидогрель, эсликарбазепин, CP-778875, изавуконазол, итраконазол, омбитасвир, асунапревир, боцепревир, даклатасвир, дазабувир, элбасвир, фалдапревир, глекапревир, гразопревир, летермовир, омбитасвир, паритапревир, пибрентасвир, триметоприм, ритонавир, симепревир, софосбувир, телапревир, велпатасвир, воксилапревир, лопинавир, пефицитиниб, кверцетин, типранавир, метформин, дилтиазем, сакубитрил, валсартан, фуросемид, гемфиброзил, элюксадолин, циклоспорин, такролимус, элтромбопаг, грейпфрутовый сок, урсодезоксихолевую кислоту, расторопшу (Silybum marianum), эмтрицитабин, тенофовир, верцирнон (GSK1605786), телмисартан, эпигаллокатехин галлата, эзетимиб, амлодипин, обетахолиевую кислоту, омега-3-карбоновые кислоты, иделалисиб, байкалин, эмпаглифлозин, элвитегравир и кобицистат. Ингибиторы OATP описаны, в целом, в Karlgren et al., “Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (OATPs): Influence of Protein Expression on Drug-Drug Interactions,” J Med Chem., 2012 May 24; 55(10): 4740-4763, которая включена в данный документ посредством ссылки.
Раскрыта система, которая дает информацию о потенциальных межлекарственных взаимодействиях E7766 или его фармацевтически приемлемой соли и по меньшей мере одного ингибитора OATP. Делая данную информацию доступной одновременно с E7766 или его фармацевтически приемлемой солью или фармацевтической композицией, содержащей E7766 или его фармацевтически приемлемую соль, можно контролировать потенциальные межлекарственные взаимодействия (в том числе возможные клинические неблагоприятные явления). Контроль можно осуществлять, например, путем изменения величины дозы, типа или режима приема ингибитора OATP и/или Е7766 или его фармацевтически приемлемой соли или путем прекращения введения либо Е7766 или его фармацевтически приемлемой соли, либо ингибитора OATP.
В некоторых вариантах осуществления контроль потенциального межлекарственного взаимодействия осуществляют путем уменьшения количества вводимого ингибитора OATP и/или частоты введения ингибитора OATP. В некоторых вариантах осуществления контроль потенциального межлекарственного взаимодействия осуществляют путем уменьшения количества вводимых E7766 или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей E7766 или его фармацевтически приемлемую соль, и/или путем уменьшения частоты введения E7766 или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей E7766 или его фармацевтически приемлемую соль.
Для того чтобы описанные в данном документе варианты осуществления можно было понять полнее, ниже представлены следующие примеры. Следует понимать, что данные примеры предназначены лишь для иллюстративных целей и не должны истолковываться как ограничивающие.
ПРИМЕРЫ
Способы и материалы
Получали состав с соединением 1 в стерильном фосфатно-буферном солевом растворе (PBS) для исследований на крысах и собаках и в 0,5% 0,1 н. HCl, 5% DMSO, 10% EtOH, 84,5% солевого раствора для исследований на мышах WT (дикого типа) и гуманизированных мышах. Получали состав с рифампином в 0,5% 0,1 н. HCl, 5% DMSO, 10% EtOH, 84,5% солевого раствора. Не содержащие примесей мочу и желчь, а также плазму крови, содержащую гепарин натрия в качестве антикоагулянта, приобретали у BioreclamationIVT (Вестбери, штат Нью-Йорк, США). Не содержащие примеси фекалии получали от крыс, приобретенных у компании Charles River Laboratories (Вилмингтон, штат Массачусетс, США).
Ниже представлены условия масс-спектрометрии и HPLC для анализа соединения 1 в биологических матрицах.
B: 2 ммоля/л бикарбоната аммония в 95:5 MeOH:H2O (об.:об.)
Пример 1. Фармакокинетика у крыс и собак
Самцам собак породы бигль или крыс линии Sprague Dawley (n=3) с канюлированными желчными протоками вводили однократную внутривенную (IV) дозу соединения 1. У собак собирали образцы плазмы крови, мочи и желчи, а у крыс собирали образцы крови, мочи, желчи и кала в течение 48 часов включительно после введения дозы. После сбора образцы крови хранили на влажном льду до центрифугирования для отделения плазмы крови. Образцы мочи собирали в пробирки для сбора образцов на влажном льду с интервалами 0-4, 4-8, 8-24 и 24-48 часов после введения дозы. Образцы желчи собирали в пробирки для сбора образцов, охлаждаемые с помощью пакета со льдом, с интервалами 0-4, 4-8, 8-24 и 24-48 часов после введения дозы. Образцы фекалий крыс собирали в течение интервала 0-24 часа. Все образцы переносили в надлежащим образом помеченные пробирки и хранили при -70ºC до отправки в биоаналитический центр. Все образцы анализировали с помощью LC-MS/MS и фармакокинетические параметры соединения 1 определяли с помощью Phoenix WinNonlin с применением некомпартментного анализа.
В таблице 2 показан процент введенной дозы, экскретируемый (% Ae) с желчью, мочой или фекалиями крысы или собаки с канюлированными желчными протоками после внутривенного введения 1 мг/кг или 0,075 мг/кг соответственно. Данные представлены в виде среднего значения ± SD. NC обозначает "не собирали".
Таблица 2
Пример 2. Фармакокинетика у нокаутных мышей, гуманизированных мышей и мышей дикого типа
Соответствующих по возрасту мышей с нокаутом по кластеру Oatp1a/1b, мышей с нокином по OATP1B1 или OATP1B3, гуманизированных на фоне нокаута по Oatp1a/1b, и самцов мышей FVB дикого типа приобретали у Taconic Biosciences (Гудзон, штат Нью-Йорк, США). На момент исследования возраст мышей составлял от 8 до 10 недель (22-34 г). Соединение 1 вводили в дозе 0,5 мг/кг совместно либо со средой-носителем, либо с рифампином (30 мг/кг). Образцы крови и печени собирали через 0,083, 0,25, 0,5, 1, 1,5, 3, 6 часов. Плазму крови из образцов крови выделяли с помощью центрифугирования. Все образцы до биоанализа хранили при -80°C.
Соединение 1 вводили мышам WT, гуманизированным мышам и KO мышам посредством внутривенного введения. В течение 6 часов у животных собирали кровь, мочу и фекалии. Кровяные капли собирали и переносили на карточку FTA™ DMPK-B DBS (GE Healthcare, Life Sciences, Whatman™) в пределах соответствующего круга образца. Также оценивали фармакокинетику соединения 1 у животных WT после совместного введения с рифампином. Также у животных WT собирали образцы печени и замораживали их до анализа. Все образцы анализировали с помощью LC-MS/MS и фармакокинетические параметры соединения 1 определяли с помощью Phoenix WinNonlin с применением некомпартментного анализа.
PK соединения 1 в условиях in vivo у нокаутных по Oatp1b2 и гуманизированных на фоне OATP1B мышей
На фиг. 2A-2C показан PK-профиль в условиях in vivo и устранение соединения 1 с совместной дозой ингибитора Oatp/OATP рифампина и без нее на различных мышиных моделях. На фиг. 2A показан PK-профиль соединения 1 и совместной дозы с рифампином у мышей дикого типа. На фиг. 2В показан PK-профиль соединения 1 и совместной дозы с рифампином у мышей, гуманизированных на фоне различных OATP1B. На фиг. 2C показано выведение соединения 1 с мочой и фекалиями у мышей с фиг. 2A и фиг. 2B.
Исходя из данных результатов, авторами настоящего изобретения было отмечено, что совместный прием дозы ингибитора Oatp/OATP рифампина с соединением 1 увеличивал системное воздействие соединения 1 в 4,5 раза, при этом уменьшая выведение с желчью на 66-79%. Это указывает на то, что опосредованное транспортером поглощение определяется скоростью для клиренса и распределения соединения 1.
Дополнительный обзор, представленный на фиг. 5, свидетельствовал, что AUC (площадь под кривой или биодоступность) соединения 1 в печени после IV введения мышам дикого типа не претерпевала значимых изменений после совместного введения с рифампином. В присутствии рифампина значение Kp (печень/плазма крови) у соединения 1 снижалось в 5 раз. В присутствии рифампина Cmax соединения 1 возрастала приблизительно в 2 раза.
Пример 3. Оценка поглощения и выведения с желчью с помощью анализа гепатоцитов человека, культивируемых сэндвич-способом
Поглощение печенью и гепатобилиарное распределение соединения 1 оценивали в культивируемых сэндвич-способом гепатоцитах человека (SCHH), полученных от одного донора, JEL. Для оценки поглощения печенью соединение 1 инкубировали в течение 1, 5 и 10 минут и собирали и замораживали растворы при -80°C до обработки для биоанализа. Затем лунки три раза промывали ледяным плюсовым (+) буфером и замораживали при -80°C до обработки для биоанализа. Выведение соединения 1 с желчью оценивали в SCHH с использованием технологии B-CLEAR®. Вкратце: удаляли среду для культивирования клеток и гепатоциты дважды промывали теплым плюсовым (+) или минусовым (-) буфером для поддержания или разрушения плотных контактов, соответственно. Удаляли промывочные растворы и заменяли их свежим плюсовым (+) буфером или минусовым (-) буфером. Гепатоциты кондиционировали в течение 10 минут при 37°C. Удаляли растворы для кондиционирования и заменяли их растворами для введения дозы соединения 1. После 20-минутной инкубации собирали растворы и замораживали их при -80°C до обработки для биоанализа. Затем лунки три раза промывали ледяным плюсовым (+) буфером. Планшеты замораживали при -80°C до обработки для биоанализа. Индекс выведения с желчью (BEI) рассчитывали по следующему уравнению:
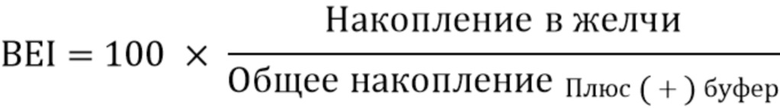
После инкубаций при 4°C поглощение соединения 1 печенью заметно снижалось до <7,7% при 37°C. Эти результаты позволяли предположить, что поглощение соединения 1 печенью в первую очередь опосредовано активными механизмами поглощения. Поглощение соединения 1 печенью было примерно дозопропорциональным в диапазоне уровней доз 0,3-1 мкМ после 1-5 минут воздействия. Тем не менее данная дозопропорциональность утрачивалась при концентрации 10 мкМ, что позволяло предположить о насыщении поглощения печенью при концентрациях >1 мкМ. Индекс выведения с желчью (BEI) у соединения 1 находился в диапазоне 70,9-86,2% во всем диапазоне оцениваемых концентраций, что указывало на высокий отток с желчью для данного соединения. Результаты представлены на фиг. 4A и фиг. 4B.
Пример 4. Фенотипирование гепатобилиарного транспортера in vitro - оценка транспорта в характеризующихся сверхэкспрессией клетках или везикулах
Клетки TransportoCells™ (Корнинг, штат Нью-Йорк, США), экспрессирующие OATP2B1, NTCP и HEK293-FT, стабильно трансфицированные вектором, содержащим cDNA OATP1B1 или cDNA OATP1B3, либо пустым вектором, выращивали в среде Игла, модифицированной по Дульбекко, с добавлением 10% фетальной телячьей сыворотки и 2 ммоль/л бутирата натрия (только в случае NTCP) в увлажняемом термостате при 37°C и с 5% CO2. Клетки собирали при 90% конфлюэнтности, а затем за 24 ч. до анализа транспортера высевали в покрытые поли-D-лизином 24-луночные планшеты. Клетки дважды промывали и предварительно инкубировали с 200 мкл предварительно нагретого буфера Кребса-Хенселейта. После предварительной инкубации клетки инкубировали с 3 или 10 мкмоль⋅л-1 соединения 1 в присутствии или в отсутствие 100 мкмоль⋅л-1 ингибиторов (рифамицина SV в случае OATP2B1, троглитазона в случае NTCP, рифамицина в случае OATP1B1 и OATP1B3). Транспортную реакцию прекращали путем аспирации буфера из лунок в назначенное время. После трехкратной промывки посредством 200 мкл ледяного буфера Кребса-Хенселейта клетки лизировали и полученные клеточные лизаты анализировали с помощью LC-MS/MS.
Зависимое от концентрации поглощение соединения 1 посредством OATP1B1 и OATP1B3 проводили с диапазоном концентраций 0,25-100 мкмоль⋅л-1 при линейном поглощении. Все эксперименты проводили в трех повторностях.
Для изучения взаимодействия соединения 1 с транспортерами выведения печенью (ABC) мембранные везикулы TransportoCells™, экспрессирующие BCRP, BSEP, MRP2, и везикулы с контрольным вектором предварительно инкубировали с буфером для поглощения везикул (47 ммоль⋅л-1 MOPs-Tris, 65 ммоль·л-1 KCl, 7 ммоль·л-1 MgCl2, pH 7,4 в случае BCRP; 47 ммоль·л-1 MOPs-Tris, 2,5 ммоль·л-1 GSH, 65 ммоль·л-1 KCl, 7 ммоль·л-1 MgCl2, pH 7,4 в случае MRP2; и 10 ммоль·л-1 HEPES-Tris, 100 ммоль·л-1 KNO3, 12,5 ммоль·л-1 Mg(NO3)2 и 50 ммоль·л-1 сахарозы, pH 7,4 в случае BSEP) при 37°C в течение 10 минут. Транспорт инициировали добавлением предварительно нагретого 25 ммоль/л MgATP, 3 мкмоль·л-1 соединения 1 в присутствии или в отсутствие ингибиторов (3 мкмоль·л-1 новобионцина в случае BCRP, 100 мкмоль·л-1 MK-571 в случае MRP2 и 20 мкмоль·л-1 кетоконазола в случае BSEP). Транспорт прекращали в назначенное время добавлением 200 мкл ледяного буфера для поглощения везикул. Затем все содержимое быстро подвергали фильтрации с помощью вакуумного коллектора со множеством фильтров для высокоэффективной фильтрации с последующими 5 промывками и фильтрациями. Планшету давали полностью высохнуть, а затем помещали на 96-луночный планшет-приемник. В каждую аналитическую лунку вносили 50 мкл элюирующего раствора (75% метанол, содержащий внутренний стандарт) с последующим центрифугированием на 2000 об/мин в течение 5 минут. Данную процедуру лизиса и центрифугирования повторяли еще один раз для максимизации экстракции соединения. Образцы продукта на выходе после двух центрифугирований объединяли и анализировали с помощью LC-MS/MS. Все эксперименты проводили в трех повторностях.
На фиг. 1A - фиг. 1D показаны результаты фенотипирования гепатобилиарного транспортера in vitro исследований соединения 1. На фиг. 1(A) показано поглощение соединения 1, оцениваемое на экспрессирующих транспортер SLC клетках HEK293; на фиг. 1(B) показан транспорт соединения 1, оцениваемый на мембранных везикулах, экспрессирующих транспортер ABC; на фиг. 1(C) показан график кинетики и параметры Михаэлиса-Ментена OATP1B1-опосредованного поглощения соединения 1; а на фиг. (D) показан график кинетики и параметры Михаэлиса-Ментена OATP1B3-опосредованного поглощения соединения 1.
Результаты, показанные на фиг. 1A - фиг. 1D, позволяют предположить, что транспортеры SLC OATP1B1 и OATP1B3, а также транспортер ABC MRP2 ответственны за поглощение печенью и последующее выведение соединения 1 с желчью.
Пример 5. PBPK-моделирование для прогнозирования OATP1B-опосредованного межлекарственного взаимодействия соединения 1 в клинических условиях
PBPK-моделирование проводили с помощью восходящего PBPK-моделирования в условиях in silico с применением Simcyp™. Результаты PBPK-моделирования позволяли предположить, что могут иметь место изменения в системном и печеночном воздействии, если соединение 1 вводить совместно с ингибиторами различных OATP1B. Более того, из-за преобладающего вклада OATP1B3, но не OATP1B1, в гепатобилиарный клиренс соединения 1 гепатобилиарный клиренс соединения 1 вряд ли будет подвергаться PK-изменению из-за полиморфизма PATP1B1.
На фиг. 4А показана средняя доля (ft%) соединения 1, транспортируемая в печень. На фиг. 3В показаны средние значения внутрипеченочной концентрации соединения 1 с взаимодействием с рифампином и без него с течением времени. На фиг. 3C показаны средние значения системной концентрации в плазме соединения 1 с взаимодействием с рифампином и без него с течением времени. На фиг. 4D показаны средние значения синусоидального клиренса с поглощением соединения 1 с взаимодействием с рифампином и без него с течением времени. Применяли программное обеспечение Simcyp™ версии 17.0.0. Ниже в таблице 3 представлены параметры Simcyp™.
Таблица 3
*Для DDI-моделирования использованы встроенная рабочая среда simCYP для рифампицина "SV-рифампицин-SD" и встроенные параметры.
Краткие выводы
По фармакокинетике у крыс с канюлированными желчными протоками было видно, что соединение 1 преимущественно выводилось в неизменном виде с желчью (>80%) и, в меньшей степени, с мочой (<20%). По результатам исследования с применением гепатоцитов человека, культивируемых сэндвич-способом, было видно зависящее от температуры активное поглощение с последующим выведением с порциями желчи. Дополнительные исследования in vitro подтверждали, что соединение 1 было субстратом для OATP1B1, OATP1B3 человека и выводящего транспортера MRP2. У гуманизированных мышей OATP1B1/1B3 в присутствии рифампина экспозиция соединения 1 в плазме крови увеличивалась в 4,5 раза. Исследования по фенотипированию демонстрировали преобладающий вклад OATP1B3 по сравнению с OATP1B1 в гепатобилиарный клиренс соединения 1. Таким образом, изменения в системном воздействии из-за полиморфизма в OATP1B1 являются маловероятными. PBPK-моделирование и имитация продемонстрировали потенциально двукратное увеличение системного воздействия соединения 1 при одновременном приеме с ингибиторами OATP1B1/1B3.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
| ТВЕРДЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ СПОСОБСТВУЮЩЕЕ АПОПТОЗУ | 2010 |
|
RU2550134C2 |
| СПОСОБ ЛЕЧЕНИЯ ВИЧ КАБОТЕГРАВИРОМ И РИЛПИВИРИНОМ | 2020 |
|
RU2840867C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
| УЛУЧШЕННЫЙ ТЕРАПЕВТИЧЕСКИЙ ИНДЕКС ИНГИБИТОРОВ ПРОТИВ ИММУННОЙ КОНТРОЛЬНОЙ ТОЧКИ С ИСПОЛЬЗОВАНИЕМ КОМБИНИРОВАННОЙ ТЕРАПИИ, ВКЛЮЧАЮЩЕЙ ЭКСТРАКТ PHY906, ЭКСТРАКТ Scutellaria baicalensis Georgi (S) ИЛИ СОЕДИНЕНИЕ ИЗ ТАКИХ ЭКСТРАКТОВ | 2017 |
|
RU2753523C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩАЯ ФЕБУКСОСТАТ | 2011 |
|
RU2602188C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2018 |
|
RU2764724C2 |
| КОМБИНАЦИИ ИАДАДЕМСТАТА ДЛЯ ЛЕЧЕНИЯ РАКА | 2020 |
|
RU2833564C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ ГЕПАТОЦЕЛЛЮЛЯРНОЙ КАРЦИНОМЫ | 2017 |
|
RU2769251C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ИНГИБИТОРОМ EZH2 | 2018 |
|
RU2754131C1 |
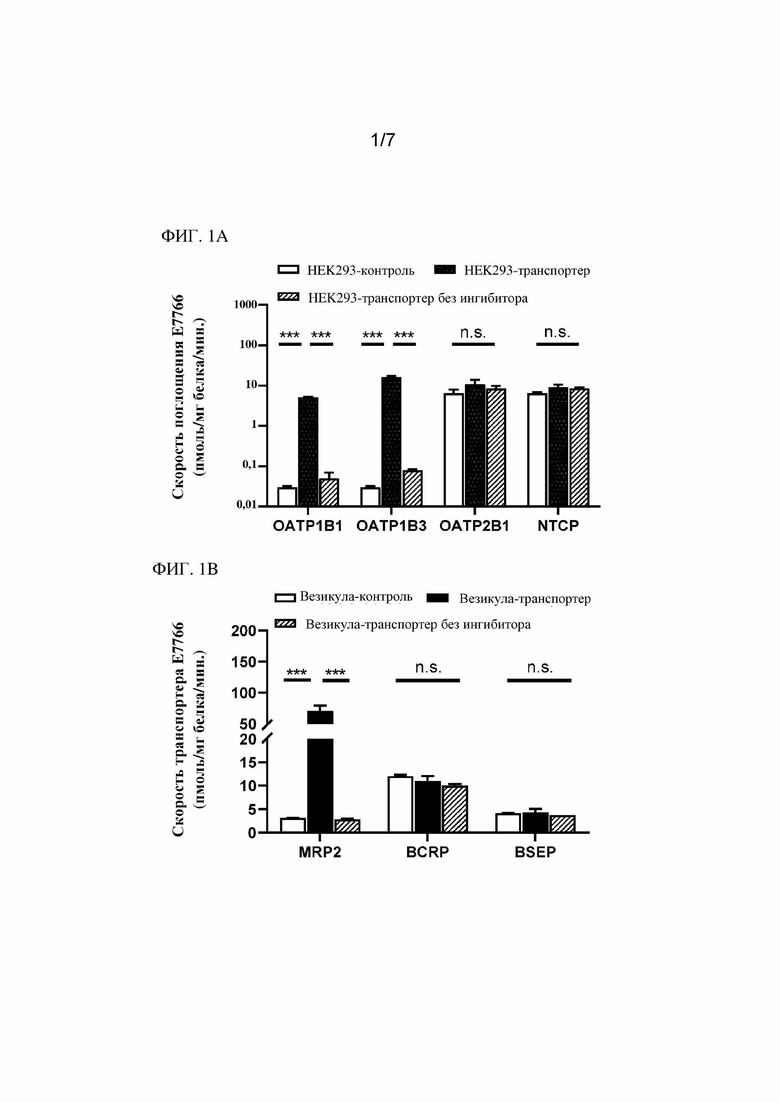
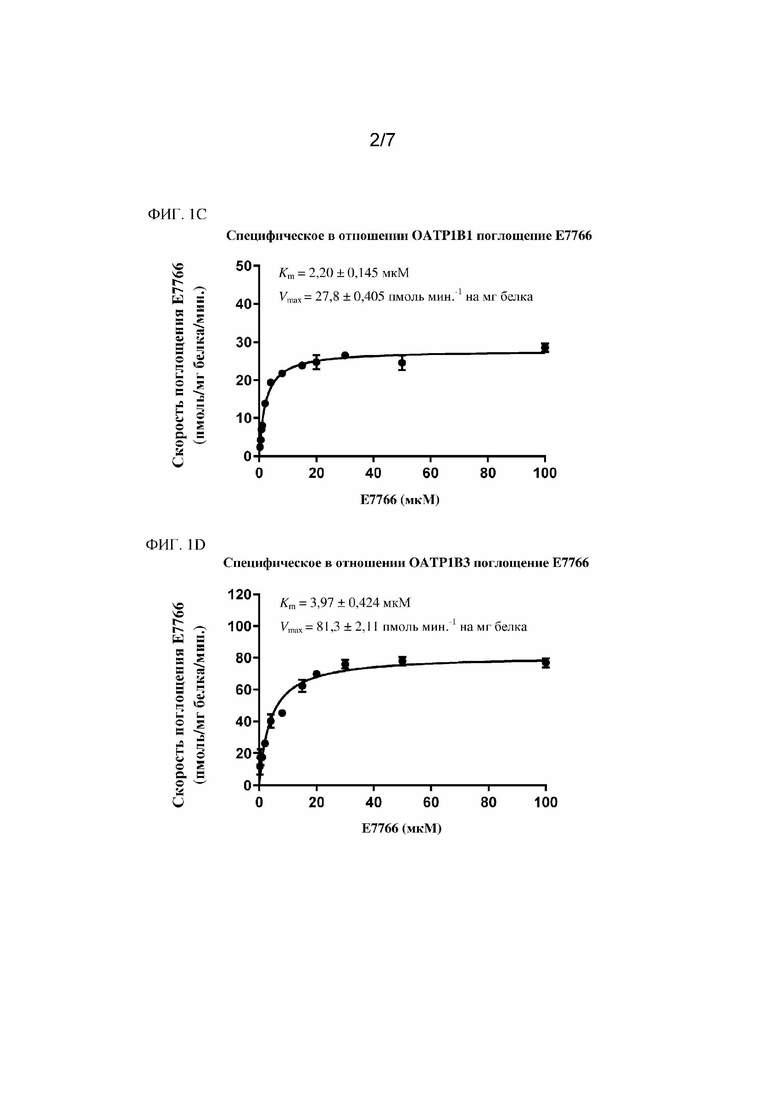
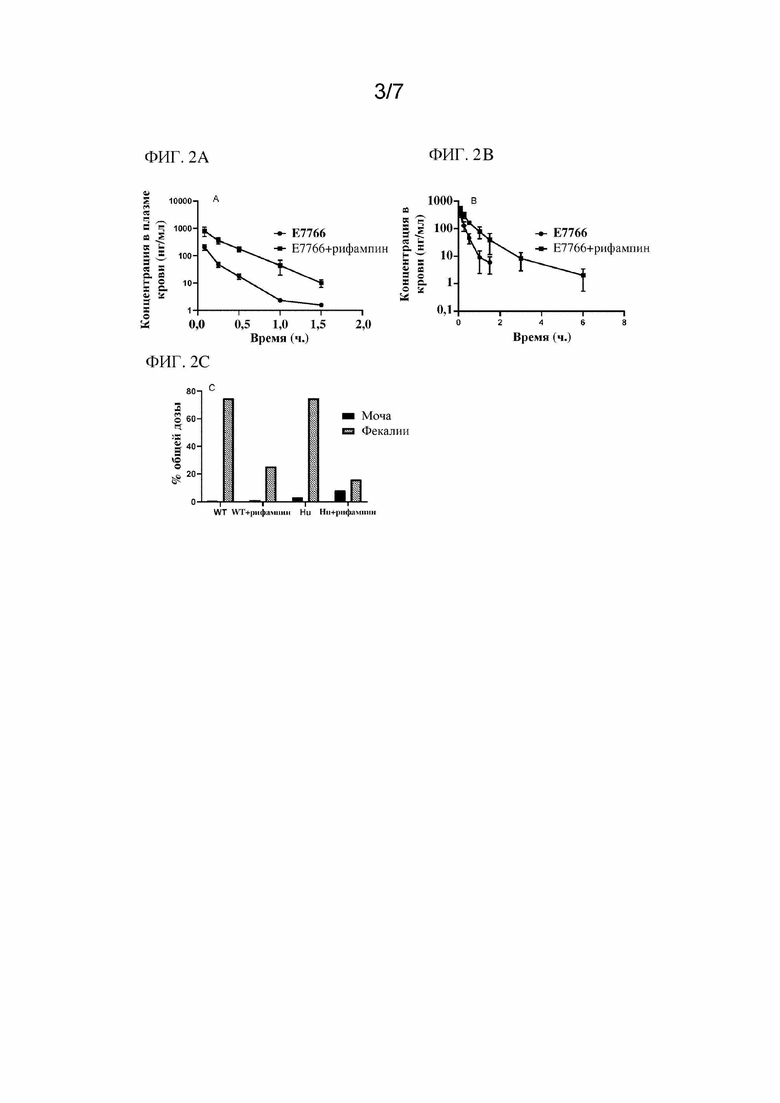
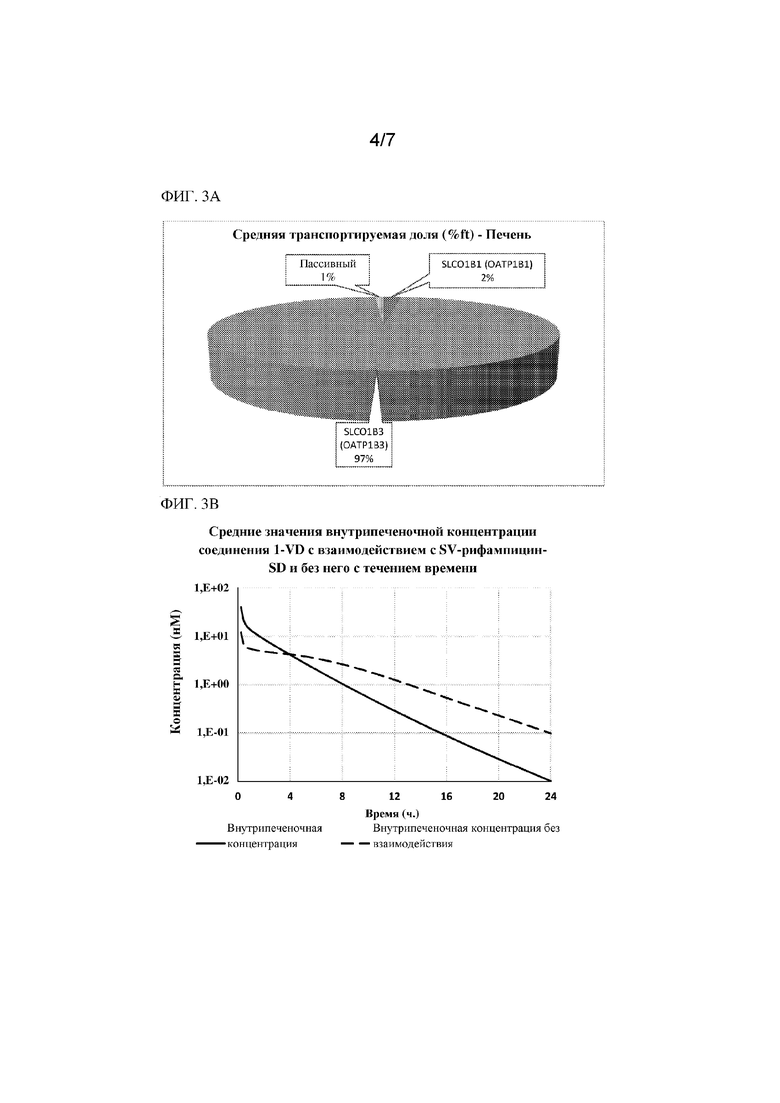
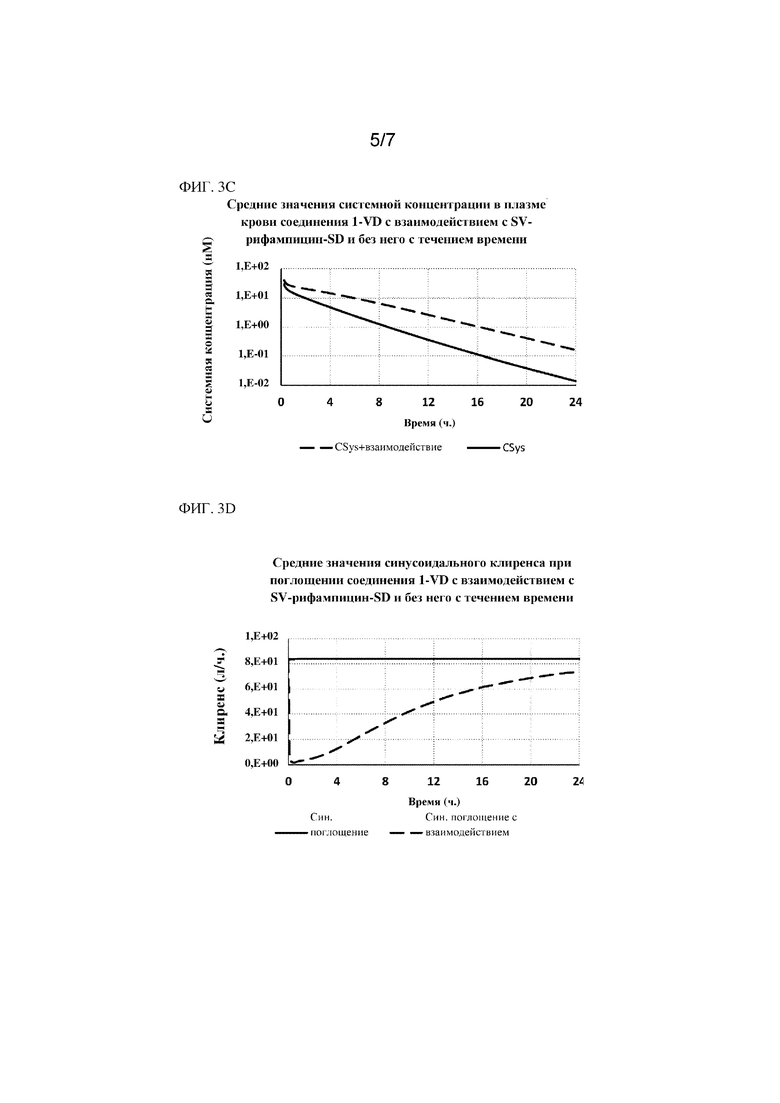
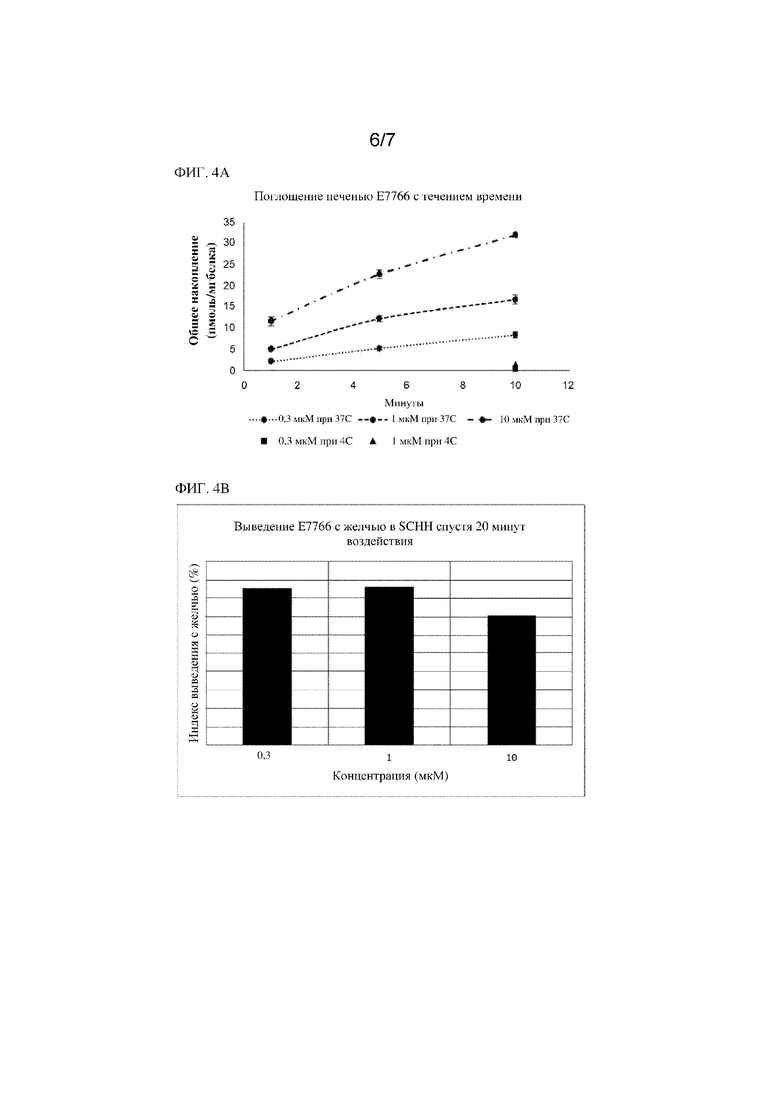
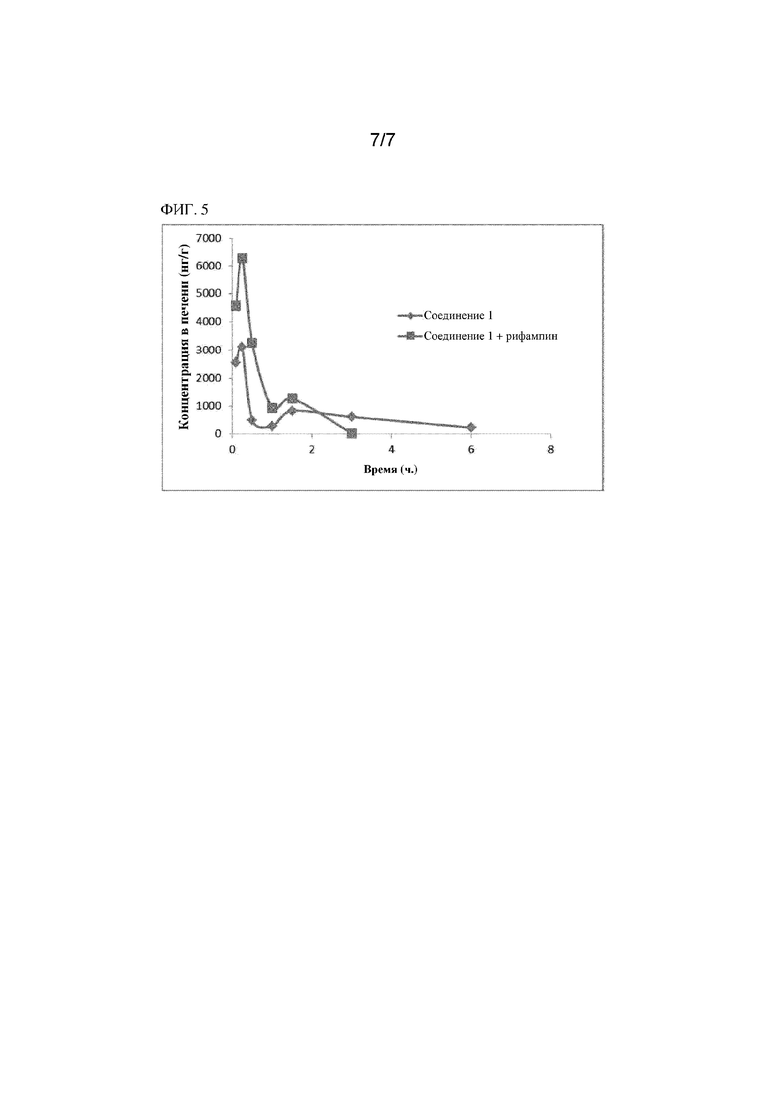
Изобретение относится к двум вариантам способа предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли. Способ по одному из вариантов предусматривает введение начального количества E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент получает ингибитор OATP1B1 или OATP1B3. Также изобретение относится к способу лечения рака. Совместное использование отмеченных соединений позволяет значительно увеличить системное воздействие соединения Е7766. 3 н. и 41 з.п. ф-лы, 5 ил., 3 табл., 5 пр.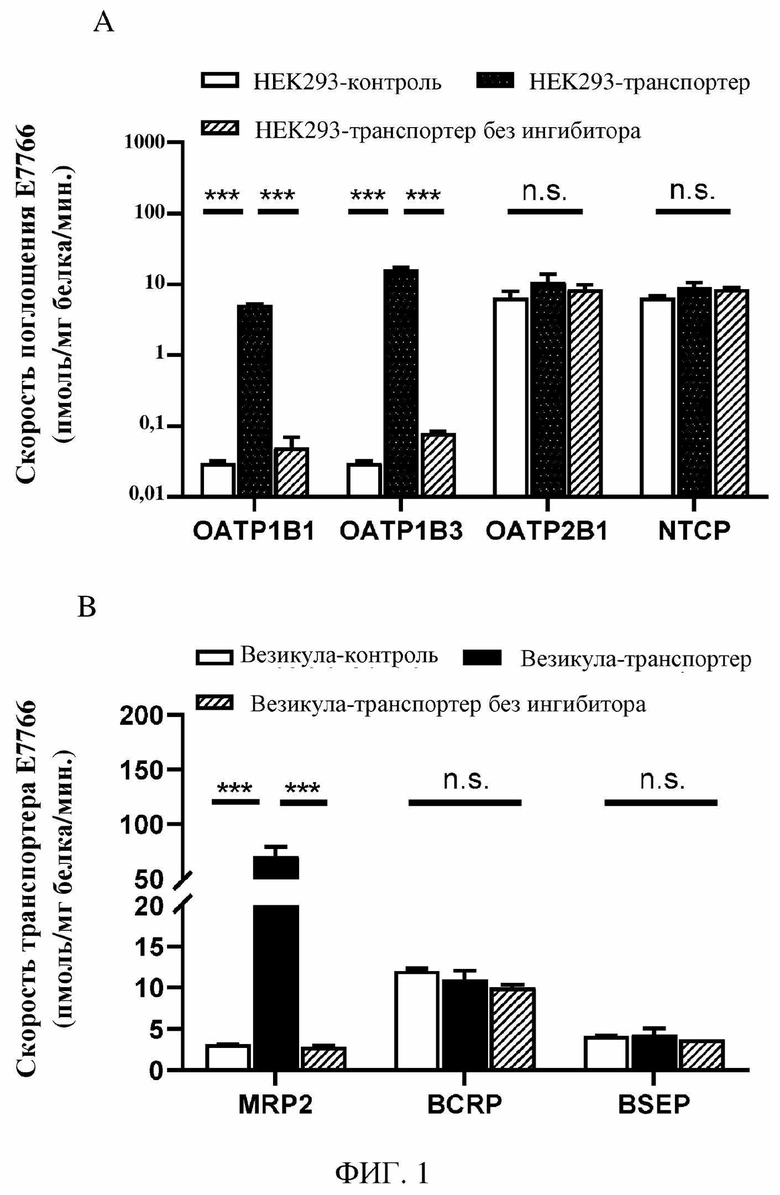
1. Способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий:
введение начального количества E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент получает ингибитор OATP1B1 или OATP1B3;
одновременное прекращение или уменьшение введения E7766 или его фармацевтически приемлемой соли в случае возникновения связанного с ним неблагоприятного явления в результате введения E7766 или его фармацевтически приемлемой соли.
2. Способ по п. 1, где указанное уменьшение предусматривает введение количества E7766 или его фармацевтически приемлемой соли, которое на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95% меньше начального количества, введенного до возникновения связанного с ним неблагоприятного явления.
3. Способ по п. 1, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 1 день после введения ингибитора OATP1B1 или OATP1B3.
4. Способ по п. 3, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 2 дня после введения ингибитора OATP1B1 или OATP1B3.
5. Способ по п. 4, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 3 дня после введения ингибитора OATP1B1 или OATP1B3.
6. Способ по п. 5, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 4 дня после введения ингибитора OATP1B1 или OATP1B3.
7. Способ по п. 6, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 5 дней или через по меньшей мере 6 дней после введения ингибитора OATP1B1 или OATP1B3.
8. Способ по п. 7, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 7 дней после введения ингибитора OATP1B1 или OATP1B3.
9. Способ по п. 8, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 2 недели после введения ингибитора OATP1B1 или OATP1B3.
10. Способ по п. 9, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 3 недели после введения ингибитора OATP1B1 или OATP1B3.
11. Способ по п. 10, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 1 месяц после введения ингибитора OATP1B1 или OATP1B3.
12. Способ по любому из пп. 1-11, где ингибитор OATP1B1 или OATP1B3 выбран из группы, состоящей из фимасартана, кларитромицина, рифампина, клопидогреля, эсликарбазепина, CP-778875, изавуконазола, итраконазола, омбитасвира, асунапревира, боцепревира, даклатасвира, дазабувира, элбасвира, фалдапревира, глекапревира, гразопревира, летермовира, омбитасвира, паритапревира, пибрентасвира, триметоприма, ритонавира, симепревира, софосбувира, телапревира, велпатасвира, воксилапревира, лопинавира, пефицитиниба, кверцетина, типранавира, метформина, дилтиазема, сакубитрила, валсартана, фуросемида, гемфиброзила, элюксадолина, циклоспорина, такролимуса, элтромбопага, грейпфрутового сока, урсодезоксихолевой кислоты, расторопши (Silybum marianum), эмтрицитабина, тенофовира, верцирнона (GSK1605786), телмисартана, эпигаллокатехина галлата, эзетимиба, амлодипина, обетихолевой кислоты, омега-3-карбоновых кислот, иделалисиба, байкалина, эмпаглифлозина, элвитегравира и кобицистата.
13. Способ предупреждения передозировки E7766 или его фармацевтически приемлемой соли у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий:
отслеживание экспозиции E7766 или его фармацевтически приемлемой соли, когда пациенту также вводят ингибитор OATP1B1 или OATP1B3;
поддержание указанной экспозиции на уровне значения, которое ниже 12800 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента; и
одновременное прекращение или уменьшение введения E7766 или его фармацевтически приемлемой соли в случае возникновения связанного с ним неблагоприятного явления в результате введения E7766 или его фармацевтически приемлемой соли.
14. Способ по п. 13, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 9600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
15. Способ по п. 14, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 6400 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
16. Способ по п. 15, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 3200 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
17. Способ по п. 16, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 2400 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
18. Способ по п. 17, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 2000 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
19. Способ по п. 18, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 1750 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
20. Способ по п. 19, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 1600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
21. Способ по п. 20, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 1200 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
22. Способ по п. 21, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 800 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
23. Способ по п. 22, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 600 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
24. Способ по п. 23, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 300 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
25. Способ по п. 24, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 150 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
26. Способ по п. 25, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 75 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
27. Способ по п. 26, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 50 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
28. Способ по п. 27, предусматривающий поддержание указанной экспозиции на уровне значения, которое ниже 25 мкг E7766 или его фармацевтически приемлемой соли на 100 кг массы тела пациента.
29. Способ по любому из пп. 13-28, где указанную экспозицию отслеживают путем количественной оценки наличия E7766 или его фармацевтически приемлемой соли в крови указанного пациента.
30. Способ по п. 29, где снижение экспозиции E7766 или его фармацевтически приемлемой соли предусматривает уменьшение дозы E7766 или его фармацевтически приемлемой соли на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%.
31. Способ по п. 29, где указанную экспозицию оценивают с использованием плазмы крови указанного пациента.
32. Способ по любому из пп. 1-31, где указанная фармацевтически приемлемая соль E7766 представляет собой диаммониевую соль E7766.
33. Способ лечения рака у пациента, выбранного для лечения посредством E7766 или его фармацевтически приемлемой соли, предусматривающий:
введение начального количества E7766 или его фармацевтически приемлемой соли указанному пациенту, где указанный пациент получает ингибитор OATP1B1 или OATP1B3; и
одновременное прекращение или уменьшение введения E7766 или его фармацевтически приемлемой соли в случае возникновения связанного с ним неблагоприятного явления в результате введения E7766 или его фармацевтически приемлемой соли.
34. Способ по п. 33, где ингибитор OATP1B1 или OATP1B3 выбран из группы, состоящей из фимасартана, кларитромицина, рифампина, клопидогреля, эсликарбазепина, CP-778875, изавуконазола, итраконазола, омбитасвира, асунапревира, боцепревира, даклатасвира, дазабувира, элбасвира, фалдапревира, глекапревира, гразопревира, летермовира, омбитасвира, паритапревира, пибрентасвира, триметоприма, ритонавира, симепревира, софосбувира, телапревира, велпатасвира, воксилапревира, лопинавира, пефицитиниба, кверцетина, типранавира, метформина, дилтиазема, сакубитрила, валсартана, фуросемида, гемфиброзила, элюксадолина, циклоспорина, такролимуса, элтромбопага, грейпфрутового сока, урсодезоксихолевой кислоты, расторопши (Silybum marianum), эмтрицитабина, тенофовира, верцирнона (GSK1605786), телмисартана, эпигаллокатехина галлата, эзетимиба, амлодипина, обетихолевой кислоты, омега-3-карбоновых кислот, иделалисиба, байкалина, эмпаглифлозина, элвитегравира и кобицистата.
35. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 1 день после введения ингибитора OATP1B1 или OATP1B3.
36. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 2 дня после введения ингибитора OATP1B1 или OATP1B3.
37. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 3 дня после введения ингибитора OATP1B1 или OATP1B3.
38. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 4 дня после введения ингибитора OATP1B1 или OATP1B3.
39. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 5 дней после введения ингибитора OATP1B1 или OATP1B3.
40. Способ по п. 33 или 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 6 дней после введения ингибитора OATP1B1 или OATP1B3.
41. Способ по п. 33 или п. 34, где начальное количество E7766 или его фармацевтически приемлемой соли вводят через по меньшей мере 7 дней после введения ингибитора OATP1B1 или OATP1B3.
42. Способ по п. 33 или п. 34, где E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 2 недели после введения ингибитора OATP1B1 или OATP1B3.
43. Способ по п. 33 или п. 34, где E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 3 недели после введения ингибитора OATP1B1 или OATP1B3.
44. Способ по п. 33 или п. 34, где E7766 или его фармацевтически приемлемую соль вводят через по меньшей мере 1 месяц после введения ингибитора OATP1B1 или OATP1B3.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| MARIA KARLGREN et al., "CLASSIFICATION OF INHIBITORS OF HEPATIC ORGANIC ANION TRANSPORTING POLYPEPTIDES (OATPS): INFLUENCE OF PROTEIN EXPRESSION ON DRUG-DRUG INTERACTIONS", JOURNAL OF MEDICINAL CHEMISRTY, vol | |||
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| АВТОМАТИЧЕСКИЙ СПРИНКЛЕР | 1926 |
|
SU4740A1 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА, ИМЕЮЩЕГО ГЕМИЗИГОТНУЮ ПОТЕРЮ ТР53 | 2016 |
|
RU2721953C2 |

