Область техники
Настоящее изобретение относится к новому гетероциклическому соединению, фармацевтической композиции, содержащей его, способу его получения и его применению для предотвращения или лечения заболевания или состояния, связанного с активностью RET (англ.: Rearranged during transfection, перестройка во время трансфекции).
Уровень техники
Протеинкиназы представляют собой класс ферментов, катализирующих реакции фосфорилирования белков. Опосредуя процесс передачи клеточного сигнала, фосфорилирование белка регулирует физиологические активности клеток, такие как выживаемость, пролиферация, дифференцировка, апоптоз и метаболизм. Дисфункция протеинкиназ тесно связана со многими заболеваниями, включая опухоли, аутоиммунные заболевания, воспалительные реакции, заболевания центральной нервной системы, сердечно-сосудистые заболевания, диабет и тому подобное.
В качестве протоонкогена RET кодирует белок RET, который является трансмембранной рецепторной тирозиновой протеинкиназой и состоит из богатого цистеином кадгерин-подобного внеклеточного домена (связывание с лигандами), трансмембранного домена и внутриклеточного домена с тирозинкиназной активностью. Активированный белок RET может активировать несколько нижестоящих сигнальных путей, включая путь RAS/RAF/ERK, путь PI3K/Akt и путь JNK, тем самым приводя к пролиферации, миграции и дифференцировке клеток. Изменение (мутация или слияние) гена RET и аномальная экспрессия гена RET дикого типа приводят к аномальной активации белков RET, так что сигнальные пути становятся сверхактивными, что является одним из основных механизмов канцерогенеза. Аномально активированные белки RET участвуют в пролиферации и инвазии различных опухолевых клеток через множество сигнальных путей, тем самым влияя на возникновение и развитие опухолей. Изменение гена RET оказывает более значительное влияние на последующие каскадные реакции, где мутация гена RET в основном связана с медуллярным раком щитовидной железы и папиллярным раком щитовидной железы, а слияние гена RET в основном связано с немелкоклеточным раком легких и хроническим миелоидным лейкозом. Следовательно, ингибирование активности RET имеет большое медицинское значение (Nature Reviews Cancer, 2014, 14 (3): 173-86).
Ингибиторы RET обладают большим потенциалом для лечения и предотвращения различных заболеваний (таких как опухоли и синдром раздраженного кишечника). В настоящее время пять соединений находятся на стадии клинических испытаний, а соединения многих компаний - на стадии доклинических исследований. Однако в настоящее время на рынке нет ингибиторов, нацеленных в основном для RET. Следовательно, для удовлетворения клинических потребностей необходимо разработать новые ингибиторы RET с высокой эффективностью и низкой токсичностью.
Сущность изобретения
В настоящем изобретении предложено новое гетероциклическое соединение, которое оказывает желаемое ингибирующее действие на RET и имеет желаемые фармакокинетические свойства, безопасность и т. п.
В одном аспекте настоящее изобретение предусматривает соединение формулы I, стереоизомер, таутомер или их смесь, его N-оксид, фармацевтически приемлемую соль, эвтектику, полиморф или его сольват, или производное со стабильным изотопом, метаболит, или его пролекарство:
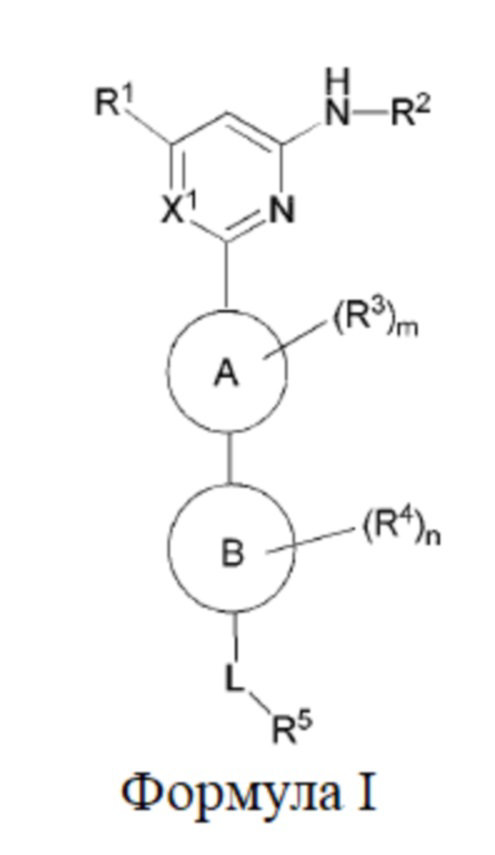
где:
кольцо A выбрано из C6-10 ароматического кольца и 5-6-членного гетероароматического кольца;
кольцо B выбрано из C3-8 циклоалкила и 4-11-членного гетероциклила;
X1 выбран из CH и N;
R1 выбран из группы, состоящей из H, галогена, гидрокси, циано, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси), C3-8 циклоалкила, 4-10-членного гетероциклила и -NR20aR20b, и каждый из алкила, гетероалкила (например, алкокси), циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси и C1-4 гетероалкила (например, C1-4 алкокси);
R2 выбран из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, C3-8 циклоалкила, 4-10-членного гетероциклила, 5-10-членного гетероарила и -C(=O)R21, и каждый из алкила, гетероалкила, циклоалкила, гетероциклила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила и C3-6 циклоалкила;
R3 и R4 отсутствуют или в каждом случае независимо выбраны из группы, состоящей из гидрокси, галогена, CN, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси) и C3-6 циклоалкила, каждый из алкила, гетероалкила (например, алкокси) и циклоалкила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; если m больше 1, два R3 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил; и/или если n больше 1, два R4 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил;
L выбран из группы, состоящей из -O-, -S-, -S(O)-, -S(O)2-, -N=CR21-, -N(R23a)-C(O)-, C1-6 алкилена, C1-6 гетероалкилена, C2-6 алкенилена, C2-6 алкинилена,
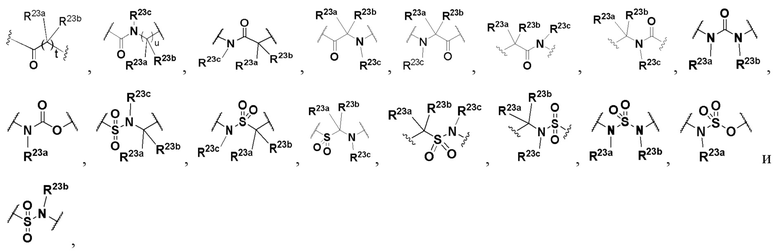
каждый из алкилена, гетероалкилена, алкенилена и алкинилена необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-6 алкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 галогеналкокси, C1-6 гетероалкила (например, C1-6 алкокси) и C3-8 циклоалкила; или L представляет собой -N(R23a)-;
R5 выбран из группы, состоящей из гидрокси, галогена, CN, NO2, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси), C2-6 алкенила, C2-6 алкинила, C3-8 циклоалкила, C3-8 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR20aR20b, -OR21, -SR21, -S(=O)R22, -S(=O)2R22, -S(=O)NR20aR20b, -S(=O)2NR20aR20b, -NR20aS(=O)R20b, -NR20aS(=O)2R20b, -C(=O)R21, -C(=O)NR23aR23b, -NR23aC(=O)R23b, -OC(=O)NR23aR23b и -NR24aC(=O)NR25aR25b, и каждый из алкила, гетероалкила (например, алкокси), алкенила, алкинила, циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4-10-членного гетероциклила;
каждый из R20a, R20b, R23a, R23b, R23c, R24a, R25a и R25bнезависимо выбран из группы, состоящей из H, OH, C1-6 алкила, C1-6 алкокси, и C3-8 циклоалкила; или R20a и R20b, R23a и R23b, или R25a и R25b образуют вместе с атомом, к которому они присоединены, 3-8-членный циклоалкил или гетероциклил, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, NO2, C1-4 алкила, C1-4 алкокси, C1-4 гидроксиалкила, C1-4 галогеналкила и C1-4 галогеналкокси;
каждый из R30a, R30b, R33a, R33b, R34a, R35a и R35b независимо выбран из группы, состоящей из H, C1-6 алкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 алкокси и C1-6 галогеналкокси;
каждый из R21, R22, R31 и R32 независимо выбран из группы, состоящей из C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 4-10-членного гетероциклила, C6-12 арила и 5-10-членного гетероарила, и каждый из алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C3-6 циклоалкила, 4-10-членного гетероциклила;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4;
t равно 0, 1, 2, 3 или 4; и
u равно 0, 1, 2, 3 или 4;
при условии, что если кольцо B представляет собой пиперазиновое кольцо и X1 представляет собой CH, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил.
В другом аспекте настоящее изобретение предусматривает фармацевтическую композицию,содержащую профилактически или терапевтически эффективное количество соединения по настоящему изобретению, стереоизомера, таутомера или их смеси, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства. Необязательно фармацевтическая композиция дополнительно содержит один или более фармацевтически приемлемых носителей.
В другом аспекте настоящее изобретение предусматривает применение соединения по настоящему изобретению, стереоизомера, таутомера или их смесь, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства, или фармацевтической композиции, как описано выше, в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET.
В другом аспекте настоящее изобретение предусматривает соединение по настоящему изобретению, стереоизомер, таутомер или их смесь, его N-оксид, фармацевтически приемлемая соль, эвтектика, полиморф или его сольват, или производное со стабильным изотопом, метаболит, или его пролекарство, или фармацевтическая композиция, как описано выше, которые применяются в предотвращении или лечении заболевания или состояния, связанного с активностью RET.
В другом аспекте настоящее изобретение предусматривает способ предотвращения или лечения заболевания или состояния, связанного с активностью RET, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по настоящему изобретению, стереоизомера, таутомера или их смеси, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства, или фармацевтическую композицию, как описано выше.
В другом аспекте настоящее изобретение предусматривает способ получения соединения по настоящему изобретению.
Краткое описание графических материалов
На фиг. 1 показаны результаты испытаний in vivo эффективности соединения 17 и контрольного соединения BLU-667 на модели подкожного ксенотрансплантата ТТ-клеток медуллярной карциномы щитовидной железы.
Подробное описание изобретения
Определения
Если иное не определено в контексте, все используемые в данном документе технические термины и научные термины имеют то же значение, которое обычно понимается специалистами в данной области. Ссылка на технологию, используемую в данном документе, предназначена для ссылки на технологию, обычно понимаемую в данной области техники, включая те технологические изменения или эквивалентные технологические замены, которые очевидны для специалистов в данной области техники. Хотя считается, что следующие термины хорошо понятны специалистам в данной области техники, следующие определения все же приводятся для лучшего объяснения настоящего изобретения.
Термин «включающий», «содержащий», «имеющий», «содержащий» или «относящийся к» и его дополнительные вариации в данном документе являются включающими или неограничивающими и не исключают дополнительных элементов, не включенных в список, или стадий способа, хотя дополнительные элементы, не указанные в списке, или стадии способа не обязательно существуют (т. е. эти термины также включают термины «по существу состоящий из» и «состоящий из»).
Как используется в данном документе, термин «алкил» определен как линейный или разветвленный насыщенный алифатический углеводород. В некоторых вариантах осуществления арильная группа имеет от 1 до 12, например, от 1 до 6 атомов углерода. Например, как используется в данном документе, термины «C1-6 алкил» и «C1-4 алкил» относятся к линейной или разветвленной радикальной группы, имеющей от 1 до 6 атомов углерода, и линейной или разветвленной радикальной группе, имеющей от 1 до 4 атомов углерода соответственно (например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил или н-гексил), которые необязательно замещены одним или более (например, от 1 до 3) подходящими заместителями, например, галогеном (в этом случае радикальная группа называется «галогеналкилом») (например, CH2F, CHF2, CF3, CCl3, C2F5, C2Cl5, CH2CF3, CH2Cl или -CH2CH2CF3). Термин «C1-4 алкил» относится к линейной или разветвленной алифатической углеводородной цепи, имеющей от 1 до 4 атомов углерода (т. е. метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил). Термин «алкилен» представляет соответствующую двухвалентную радикальную группу, включая, например, «C1-8 алкилен», «C1-6 алкилен», «C1-4 алкилен» и т. п., и его конкретные примеры включают, но не ограничиваются ими: метилен (-CH2-), этилиден (-CH2CH2- или -CH(CH3)-), пропилиден (-CH2CH2CH2-), изопропилиден (-CH(CH3)CH2-), бутилиден, пентилиден, гексилиден и т. п. Алкилен необязательно замещен одним или более (например, от 1 до 3) одинаковыми или разными заместителями.
Как используется в данном документе, термин «гетероалкил» относится к необязательно замещенному алкильному радикалу, который имеет один или несколько атомов основной цепи, выбранных из атомов, отличных от углерода, таких как кислород, азот, сера, фосфор или их комбинации. Числовой диапазон (например, C1-6 гетероалкил), который может быть указан, относится к количеству атомов углерода в цепи, включая от 1 до 6 атомов углерода в этом примере. Например, группа -CH2OCH2CH3 называется C3 гетероалкилом. Точки присоединения к остальной части молекулы могут быть через гетероатом или атом углерода в гетероалкильной цепи. Термин «гетероалкилен» относится к соответствующей двухвалентной радикальной группе, включая, например, «C1-6 гетероалкилен», «C1-4 гетероалкилен» и т. п.
Как используется в данном документе, термин «галогеналкил» относится к алкильному радикалу, замещенному одним или несколькими (например, от 1 до 3) одинаковыми или разными атомами галогена, и термин «C1-8 галогеналкил», «C1-6 галогеналкил» и «C1-4 галогеналкил» относится к галогеналкильному радикалу, имеющему от 1 до 8 атомов углерода, галогеналкильному радикалу, имеющему от 1 до 6 атомов углерода, и галогеналкильному радикалу, имеющему от 1 до 4 атомов углерода соответственно, например, -CF3, -C2F5, -CHF2, -CH2F, -CH2CF3, -CH2Cl или -CH2CH2CF3.
Как используется в данном документе, термин «гидроксиалкил» относится к радикальной группе, образованной путем замещения атома(-ов) водорода в алкильном радикале одним или несколькими из гидрокси, например, C1-4 гидроксиалкила или C1-3 гидроксиалкила, и его примеры включают, но не ограничиваются ими, гидроксиметил, гидроксиэтил, гидроксипропил, гидроксибутил, -CH(OH)CH3, -C(CH3)2OH и т. п.
Как используется в данном документе, термин «алкокси» относится к радикальной группе, образованной путем вставки атома кислорода в любое подходящее положение алкильного радикала (как определено выше), и предпочтительно представляет собой C1-8 алкокси, C1-6 алкокси, C1-4 алкокси или C1-3алкокси. Иллюстративные примеры C1-6 алкокси включают, но не ограничиваются ими, метокси, этокси, пропокси, изопропокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, пентилокси, гексилокси, -CH2-OCH3 и т. п., и алкокси необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями.
Как используется в данном документе, термин «алкоксилен» относится к двухвалентной алкоксигруппе, например, -OCH2-, -OCH(CH3)CH2-, -OCH2CH2O- и -CH2CH2O-, и алкоксилен необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями.
Как используется в данном документе, термин «алкенил» относится к линейному или разветвленному одновалентному гидрокарбилу, содержащему одну или несколько двойных связей и имеющему от 2 до 6 атомов углерода («C2-6 алкенил»). Алкенил представляет собой, например, -CH=CH2, -CH2CH=CH2, -C(CH3)=CH2, -CH2-CH=CH-CH3, 2-пентенил, 3-пентенил, 4-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 2-метил-2-пропенил и 4-метил-3-пентенил. Когда соединение по настоящему изобретению содержит алкенильный радикал, соединение может существовать в чистой E (entgegen) форме, чистой Z (zusammen) форме или в форме любой их смеси. Термин «алкенилен» представляет собой соответствующую двухвалентную радикальную группы, включая, например, «C2-6алкенилен» и «C2-4 алкенилен», и его конкретные, но не ограничиваются ими: -CH=CH-, -CH2CH=CH-, -C(CH3)=CH-, бутенилен, пентенилен, гексенилен, циклопентенилен, циклогексенилен и т. п.
Как используется в данном документе, термин «алкинил» относится к одновалентному гидрокарбилу, содержащему одну или несколько тройных связей, и предпочтительно имеющему 2, 3, 4, 5 или 6 атомов углерода, например, этинил, 2-пропинил, 2-бутинил, 1,3-бутадиинил. Алкинил необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями. Термин «алкинилен» представляет собой соответствующую двухвалентную радикальную группу, включающую, например, «C2-8 алкинилен», «C2-6 алкинилен» и «C2-4 алкинилен». Его примеры включают, но не ограничиваются ими,  ,
,  ,
, 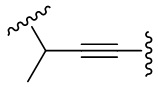 ,
, 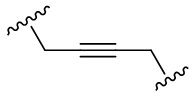 , и т. п. Алкинилен необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями.
, и т. п. Алкинилен необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями.
Как используется в данном документе, термин «конденсированное кольцо» или «слитое кольцо» относится к кольцевой системе, образованной двумя или более чем двумя кольцевыми структурами, имеющими общие два смежных атома.
Как используется в данном документе, термин «спирокольцо» относится к кольцевой системе, образованной двумя или более чем двумя кольцевыми структурами, имеющими один общий атом в кольце друг с другом.
Как используется в данном документе, термин «мостиковое кольцо» относится к кольцевой системе, образованной двумя или более чем двумя кольцевыми структурами, имеющими два общих атома (которые не соединены непосредственно) друг с другом.
Как используется в данном документе, термин «циклоалкил» относится к насыщенному или ненасыщенному неароматическому моноциклическому или полициклическому (например, бициклического) углеводородного кольцевого радикала, включая, но не ограничиваясь ими, моноциклоалкил (например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и циклононил) и бициклоалкил, включая спирокольцо, слитое кольцо или мостиковую кольцевую систему (т.е. спироциклоалкил, конденсированный (слитый) циклоалкил и мостиковый циклоалкил, например, бицикло[1.1.1]пентил и бицикло[2.2.1]гептил). В настоящему изобретении циклоалкильный радикал необязательно замещен одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями. Атом углерода на циклоалкильном радикале необязательно замещен оксо-группой (т. е. образуя C=O). Термин «C3-8 циклоалкил» относится к циклоалкильному радикалу, имеющему от 3 до 8 кольцо-образующих атомов углерода, таких как C3-6 циклоалкил, который может быть моноциклоалкильным радикалом, таким как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил, и может также быть бициклоалкильным радикалом, таким как C5-8 спироциклоалкил, C5-8 мостиковый циклоалкил, C5-8 слитый циклоалкил, C5-6 спироциклоалкил, C5-6 мостиковый циклоалкил или C5-6 конденсированный циклоалкил.
Как используется в данном документе, термин «циклоалкокси» означает -O-циклоалкил, где циклоалкил определен выше. Иллюстративные примеры циклоалкокси-групп включают, но не ограничиваются ими, циклопропокси, циклобутокси, циклопентокси, циклогексокси и т. п.
Как используется в данном документе, термин «гетероциклил» или «гетероциклическое кольцо» относится к моноциклической или полициклической (например, конденсированной, спиро- или мостиковой циклической) радикальной группе, имеющей 2 или более 2 (например, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14) атомов углерода, и один или несколько (например, 1, 2, 3 или 4) гетероатомов, где гетероатомы включают, но не ограничиваются ими, атом кислорода, атом азота и атом серы, и атом углерода и гетероатом на гетероциклиле необязательно замещены оксо-группой (например, образуя C=O, S(=O) или S(=O)2).
Как используется в данном документе, термин «4-11-членный гетероциклил» означает гетероциклил, содержащий от 4 до 11 атомов в кольце, включая, но без ограничения, 4-10-членный гетероциклил, 4-9-членный гетероциклил, 4-8-членный гетероциклил, 4-7-членный гетероциклил, 5-6-членный гетероциклил, 3-8-членный гетероциклил, 3-7-членный гетероциклил, 4-7-членный азот-содержащий гетероциклил, 4-7-членный кислород-содержащий гетероциклил, 4-7-членный серо-содержащий гетероциклил, 5-6-членный азот-содержащий гетероциклил, 5-6-членный кислород-содержащий гетероциклил, 5-6-членный серо-содержащий гетероциклил и т. п. «Азот-содержащий гетероциклил», «кислород-содержащий гетероциклил» и «серо-содержащий гетероциклил» каждый необязательно дополнительно содержит один или несколько дополнительных гетероатомов, выбранных их кислорода, азота и серы. Примеры 4-11-членного гетероциклила включают, но не ограничиваются ими, оксиранил, азиридинил, азациклобутил, оксациклобутил, тетрагидрофурил, пирролидинил, пирролидонил (например, 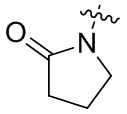 ), имидазолидинил, пиразолидинил, тетрагидропиранил, пиперидинил, морфолинил, дитианил, тиоморфолинил, пиперазинил и тритианил.
), имидазолидинил, пиразолидинил, тетрагидропиранил, пиперидинил, морфолинил, дитианил, тиоморфолинил, пиперазинил и тритианил.
Как используется в данном документе, термин «гетероциклил» охватывает конденсированную кольцевую структуру, и точки присоединения конденсированной кольцевой структуры к дополнительным радикальным группам могут находиться на любом кольце в конденсированной кольцевой структуре. Соответственно, гетероциклил по настоящему изобретению дополнительно включает, но не ограничивается ими, гетероциклил-гетероциклил, гетероциклил-циклоалкил, моногетероциклил-моногетероциклил, моногетероциклил-моноциклоалкил, например, 3-7-членный (моно)гетероциклил-3-7-членный (моно)гетероциклил, 3-7-членный (моно)гетероциклил-(моно)циклоалкил и 3-7-членный (моно)гетероциклил-C4-6 (моно)циклоалкил. Его примеры включают, но не ограничиваются ими, пирролидинил-циклопропил, циклопентил-азациклопропил, пирролидинил-циклобутил, пирролидинил-пирролидинил, пирролидинил-пиперидинил, пирролидинил-пиперазинил, пиперидинил-морфолинил, 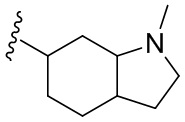 ,
,  или
или  .
.
Как используется в данном документе, термин «гетероциклил» охватывает мостиковый гетероциклил и спирогетероциклил.
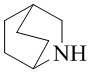
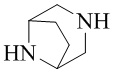
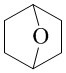
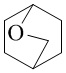
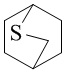
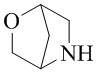
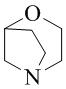
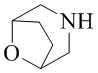
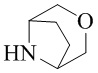
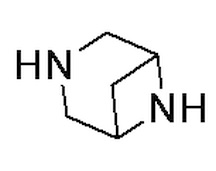
Как используется в данном документе, термин «мостиковое гетероциклическое кольцо» относится к циклической структуре, которая образована двумя насыщенными кольцами, имеющими два общих атома в кольце, которые не связаны напрямую, и которая включает один или несколько (например, 1, 2, 3 или 4) гетероатомов (таких, как атом кислорода, атом азота и/или атом серы), включая, но не ограничиваясь ими, 7-10-членное мостиковое гетероциклическое кольцо, 8-10-членное мостиковое гетероциклическое кольцо, 7-10-членное азот-содержащее мостиковое гетероциклическое кольцо, 7-10-членное кислород-содержащее мостиковое гетероциклическое кольцо, 7-10-членное серо-содержащее мостиковое гетероциклическое кольцо и т. п., например,  ,
,  ,
, 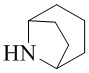 ,
, 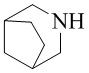 ,
, 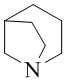 ,
, 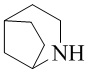 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 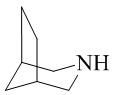 и
и 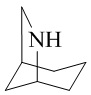 . «Азот-содержащее мостиковое гетероциклическое кольцо», «кислород-содержащее мостиковое гетероциклическое кольцо» и «серо-содержащее мостиковое гетероциклическое кольцо» необязательно дополнительно включают один или несколько дополнительных гетероатомов, выбранных из кислорода, азота и серы.
. «Азот-содержащее мостиковое гетероциклическое кольцо», «кислород-содержащее мостиковое гетероциклическое кольцо» и «серо-содержащее мостиковое гетероциклическое кольцо» необязательно дополнительно включают один или несколько дополнительных гетероатомов, выбранных из кислорода, азота и серы.
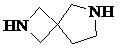
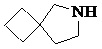
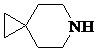
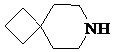
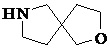
Как используется в данном документе, термин «спирогетероциклическое кольцо» относится к циклической структуре, которая образована двумя или более чем двумя насыщенными кольцами, имеющими один общий атом в кольце, и которая включает один или несколько (например, 1, 2, 3 или 4) гетероатомов (таких, как атом кислорода, атом азота и/или атом серы), включая, но не ограничиваясь ими, 5-10-членное спирогетероциклическое кольцо, 6-10-членное спирогетероциклическое кольцо, 6-10-членное азот-содержащее спирогетероциклическое кольцо, 6-10-членное кислород-содержащее спирогетероциклическое кольцо, 6-10-членное серо-содержащее спирогетероциклическое кольцо и т. п., например, 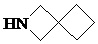 ,
, 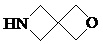 ,
, 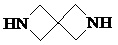 ,
, 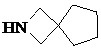 ,
, 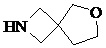 ,
, 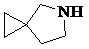 ,
, 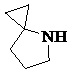 ,
, 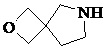 ,
,  ,
,  ,
,  ,
,  ,
, 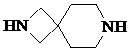 ,
, 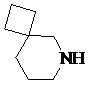 ,
, 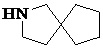 ,
, 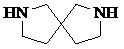 ,
,  ,
, 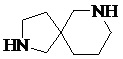 и
и 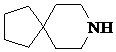 . «Азот-содержащее спирогетероциклическое кольцо», «кислород-содержащее спирогетероциклическое кольцо» и «серо-содержащее спирогетероциклическое кольцо» необязательно дополнительно содержат один или несколько дополнительных гетероатомов, выбранных из кислорода, азота и серы. Термин «6-10-членный азот-содержащий спирогетероциклил» относится к спирогетероциклилу, который полностью содержит от 6-10 атомов в кольце, и где по меньшей мере один из атомов в кольце представляет собой атом азота.
. «Азот-содержащее спирогетероциклическое кольцо», «кислород-содержащее спирогетероциклическое кольцо» и «серо-содержащее спирогетероциклическое кольцо» необязательно дополнительно содержат один или несколько дополнительных гетероатомов, выбранных из кислорода, азота и серы. Термин «6-10-членный азот-содержащий спирогетероциклил» относится к спирогетероциклилу, который полностью содержит от 6-10 атомов в кольце, и где по меньшей мере один из атомов в кольце представляет собой атом азота.
В настоящем изобретении гетероциклил может быть слит с арильной группой с образованием слитой кольцевой структуры. Примеры слитых кольцевых структур включают, но не ограничиваются ими: 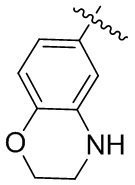 ,
,  ,
,  , и
, и  .
.
Как используется в данном документе, термин «арил» или «ароматическое кольцо» относится к полностью углеродной моноциклической или слитой полициклической ароматической группе, имеющей сопряженную π-электронную систему. Как используется в данном документе, термин «C6-12 арил (ароматическое кольцо)» означает арильную группу (ароматическое кольцо), содержащую от 6 до 12 атомов углерода, и предпочтительно представляет собой C6-10 арильную группу (ароматическое кольцо), предпочтительно фенил или нафтил. Арильная группа необязательно замещена одним или несколькими (например, от 1 до 3) одинаковыми или разными заместителями (например, галоген, OH, CN, NO2 или C1-C6 алкил).
Как используется в данном документе, термин «гетероарил» или «гетероароматическое кольцо» относится к моноциклической или полициклической ароматической группе, содержащей один или несколько одинаковых или разных гетероатомов, включая моноциклическую гетероарильную группу и бициклическую или полициклическую кольцевую систему, содержащую по меньшей мере одно гетероароматическое кольцо (ароматическая кольцевая система, содержащая по меньшей мере один гетероатом), которая может иметь 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 атомов в кольце, например, 5, 6, 7, 8, 9 или 10 атомов в кольце. Гетероатом может представлять собой кислород, азот или серу. Атом углерода и гетероатом на гетероариле необязательно замещены оксо-группой (например, образуя C=O, S(=O) или S(=O)2).
Как используется в данном документе, термин «5- 10-членный гетероарил» или «5- 10-членное гетероароматическое кольцо» означает гетероарильную группу (гетероароматическое кольцо), содержащую от 5 до 10 (например, от 5 до 6) атомов в кольце, включая 5- 10-членную азот-содержащую гетероарильную группу, 5-10-членную кислород-содержащую гетероарильную группу, 5-10-членную серо-содержащую гетероарильную группу, 5-6-членную азот-содержащую гетероарильную группу, 5-6-членную кислород-содержащую гетероарильную группу, 5-6-членную серо-содержащую гетероарильную группу, и т. п. «Азот-содержащий гетероарил», «кислород-содержащий гетероарил» и «серо-содержащий гетероарил» каждый необязательно содержит один или несколько дополнительных гетероатомов, выбранных из кислорода, азота и серы. Его примеры включают, но не ограничиваются ими, тиенил, фурил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, триазолил, тетразолил, оксадиазолил, тиадиазолил и т. д., или пиридил, пиридазинил, пиримидинил, пиразинил, триазинил и т. д., и 5-10-членные конденсированные кольцевые группы, содержащие эти группы.
Как используется в данном документе, термин «гетероарил» охватывает конденсированную кольцевую структуру, и точки присоединения конденсированной кольцевой структуры к дополнительным радикальным группам могут находиться на любом кольце в конденсированной кольцевой структуре. Таким образом, гетероарильные группы по настоящему изобретению дополнительно включают, но не ограничиваются ими, (моно)гетероарил-(моно)гетероарил, (моно)гетероарил-(моноцикло)арил, (моно)гетероарил-(моно)гетероциклил и (моно)гетероарил-(моно)циклоалкил, например, 5-6-членный (моно)гетероарил-5-6-членный (моно)гетероарил, 5-6 членный (моно)гетероарил-фенил, 5-6-членный (моно)гетероарил-5-6-членный (моно)гетероциклил или 5-6-членный (моно)гетероарил-C4-6(моно)циклоалкил (например, 5-6-членный гетероарил-циклобутил, 5-6-членный гетероарил-циклопентил или 5-6-членный гетероарил-циклогексил). Примеры гетероарильных групп включают, но не ограничиваются ими, индолил, изоиндолил, индазолил, бензимидазолил, хинолинил, изохинолинил,
 и т. п.
и т. п.
Как используется в данном документе, термин «галогенидная» или «галогеновая» группа определена как охватывающая F, Cl, Br или I.
Термин «замещение» означает, что один или несколько (например, один, два, три или четыре) атомов водорода на указанном атоме заменяются выбором из указанной группы при условии, что нормальная валентность указанного атома в текущем случае не превышена, и замещение образует стабильное соединение. Комбинация заместителей и/или переменных допустима только тогда, когда такая комбинация образует стабильное соединение.
Если заместитель описан как «необязательно ... замещенный», заместитель может быть (1) незамещенным или (2) замещенным. Если углерод заместителя описан как необязательно замещенный одним или несколькими заместителями в списке заместителей, один или несколько атомов водорода на углероде (в пределах любых присутствующих атомов водорода) могут быть независимо и/или вместе заменены независимо выбранными необязательными заместителями. Если азот заместителя описан как необязательно замещенный одним или несколькими заместителями в списке заместителей, один или несколько атомов водорода на азоте (в пределах любых присутствующих атомов водорода) каждый может быть заменен независимо выбранными необязательными заместителями.
Если заместитель описан как «независимо выбранный из» группы, каждый заместитель выбирается независимо от другого. Следовательно, каждый заместитель может быть таким же или отличаться от другого заместителя(-ей).
Как используется в данном документе, термин «один или несколько» означает 1 или более 1, например 2, 3, 4, 5 или 10, при подходящих условиях.
Если не указано иное, как используется в данном документе, точки присоединения заместителя могут находиться в любых подходящих положениях заместителя.
Когда связь заместителя показана как связь, соединяющая два атома через кольцо, такой заместитель может связываться с любым образующим кольцо атомом в замещаемом кольце.
Настоящее изобретение дополнительно включает все фармацевтически приемлемые меченные изотопом соединения, которые аналогичны соединениям по настоящему изобретению, за исключением того, что один или несколько атомов заменены атомами, которые имеют тот же атомный номер, но атомная масса или массовое число отличаются от атомной массы или массового числа, преимущественно встречающегося в природе. Примеры изотопов, подходящих для включения в соединения по настоящему изобретению, включают, но не ограничиваются ими, изотопы водорода (например, дейтерий (2H), тритий (3H)); изотопы углерода (например, 11C, 13C и 14C); изотопы хлора (например, 36Cl); изотопы фтора (например, 18F); изотопы йода (например, 123I и 125I); изотопы азота (например, 13N и 15N); изотопы кислорода (например, 15O, 17O и 18O); изотопы фосфора (например, 32P); и изотопы серы (например, 35S). Некоторые меченые изотопами соединения (например, те, которые включены в радиоизотоп) по настоящему изобретению пригодны в исследовании распределения лекарственных средств и/или субстратов в тканях (например, в анализе). Изотопы трития (т. е. 3H) и углерода-14 (т. е. 14C) особенно предпочтительны из-за простоты их включения и обнаружения. Замены изотопами позитронной эмиссии (такими как 11C, 18F, 15O и 13N) могут быть использованы для проверки степени занятости рецептора субстратом в исследованиях позитронно-эмиссионной томографии (ПЭТ). Меченные изотопами соединения по настоящему изобретению могут быть получены способами, аналогичными тем, которые описаны в сопровождающих путях синтеза и/или примерах и получениях, путем замены реагента, не меченного изотопами, подходящим реагентом, меченным изотопами. Фармацевтически приемлемые сольваты по настоящему изобретению включают те, в которых растворитель кристаллизации может быть заменен изотопом, например, D2O, ацетон-d6 или ДМСО-d6,
Термин «стереоизомер» означает изомер, образованный по меньшей мере одним асимметричным центром. В соединении с одним или несколькими (например, одним, двумя, тремя или четырьмя) асимметричными центрами могут быть получены его экзо/мезо-смеси, отдельные энантиомеры и смеси диастереомеров и отдельные диастереомеры. Конкретные отдельные молекулы также могут существовать в виде геометрических изомеров (цис/транс). Аналогично, соединение по настоящему изобретению может существовать в смеси двух или более структурно различных форм (обычно называемых таутомерами) в быстром равновесии. Типичные примеры таутомеров включают кето-енольные таутомеры, фенол-кето-таутомеры, нитрозо-оксимные таутомеры, имин-енаминные таутомеры и т. п. Например, нитрозооксимы могут существовать в равновесии в следующих таутомерных формах в растворе:
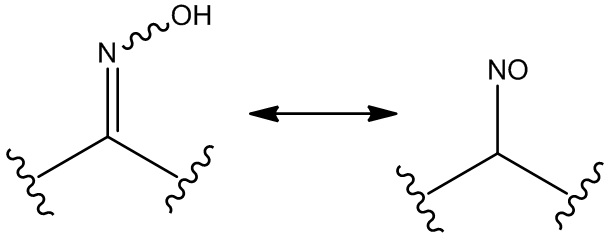 .
.
Следует понимать, что объем настоящего изобретения охватывает все такие изомеры или их смеси в любых пропорциях (например, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96 %, 97%, 98%, 99%).
Сплошная линия ( ), сплошной клин (
), сплошной клин ( ) или пунктирный клин (
) или пунктирный клин ( ) могут использоваться в данном документе для изображения химических связей соединений по настоящему изобретению. Использование сплошных линий для изображения связей с асимметричными атомами углерода предназначено для обозначения того, что включены все возможные стереоизомеры у этого атома углерода (например, определенные энантиомеры или рацемические смеси). Использование сплошных или пунктирных клиньев для изображения связей с асимметричными атомами углерода предназначено для обозначения того, что показанные стереоизомеры существуют. Когда они присутствуют в рацемической смеси, сплошные и пунктирные клинья используются для определения относительной стереохимии, а не абсолютной стереохимии. Если не указано иное, соединение по настоящему изобретению предназначено для существования в форме стереоизомеров (которые включают цис- и транс-изомеры, оптические изомеры (такие как R- и S-энантиомеры), диастереомеры, геометрические изомеры, ротамеры, конформационные изомеры, атропоизомеры, и их смеси). Соединение по настоящему изобретению может проявлять более одного типа изомерии и состоит из их смесей (например, рацемических смесей и диастереомерных пар).
) могут использоваться в данном документе для изображения химических связей соединений по настоящему изобретению. Использование сплошных линий для изображения связей с асимметричными атомами углерода предназначено для обозначения того, что включены все возможные стереоизомеры у этого атома углерода (например, определенные энантиомеры или рацемические смеси). Использование сплошных или пунктирных клиньев для изображения связей с асимметричными атомами углерода предназначено для обозначения того, что показанные стереоизомеры существуют. Когда они присутствуют в рацемической смеси, сплошные и пунктирные клинья используются для определения относительной стереохимии, а не абсолютной стереохимии. Если не указано иное, соединение по настоящему изобретению предназначено для существования в форме стереоизомеров (которые включают цис- и транс-изомеры, оптические изомеры (такие как R- и S-энантиомеры), диастереомеры, геометрические изомеры, ротамеры, конформационные изомеры, атропоизомеры, и их смеси). Соединение по настоящему изобретению может проявлять более одного типа изомерии и состоит из их смесей (например, рацемических смесей и диастереомерных пар).
Настоящее изобретение охватывает все возможные кристаллические формы или полиморфы соединения настоящего изобретения, которые могут быть одним полиморфом или смесью более чем одного полиморфа в любом соотношении.
Эвтектическая кристаллизация относится к тому факту, что активные молекулы лекарственного средства и дополнительные физиологически приемлемые молекулы кислот, оснований, солей и неионных соединений связаны водородными связями, π-π-стэкингом, силами Ван-дер-Ваальса и дополнительными ковалентными связями, объединяемыми в одну кристаллическую решетку.
Также следует понимать, что некоторые соединения по настоящему изобретению могут существовать в свободной форме для лечения или, где это уместно, в форме их фармацевтически приемлемых производных. В настоящем изобретении фармацевтически приемлемые производные включают, но не ограничиваются ими, фармацевтически приемлемые соли, сложные эфиры, сольваты, N-оксиды, метаболиты или пролекарства, которые после введения пациентам, нуждающимся в этом, могут прямо или косвенно обеспечивать соединение по настоящему изобретению или его метаболиты или остатки. Следовательно, когда в данном документе упоминается «соединение по настоящему изобретению», оно также подразумевает охват вышеуказанных различных производных форм соединения.
Фармацевтически приемлемые соли соединения по настоящему изобретению включают его соли присоединения кислот и соли присоединения оснований, например, гексафторфосфат и соль меглумина. Для обзора подходящих солей см. Stahl and Wermuth, «Handbook of Pharmaceutical Salts: Properties, Selection, and Use» (Wiley-VCH, 2002).
Как используется в данном документе, термин «сложный эфир» означает сложный эфир, полученный из соединения каждой общей формулы в настоящем изобретении, который включает физиологически гидролизуемые сложные эфиры (гидролизуемые в физиологических условиях для высвобождения соединения по настоящему изобретению в форме свободной кислоты или спирта). Соединение по настоящему изобретению само по себе может также представлять собой сложный эфир.
Соединение по настоящему изобретению может существовать в форме сольвата (предпочтительно гидрата), где соединение по настоящему изобретению включает полярный растворитель в качестве структурного элемента кристаллической решетки указанного соединения, особенно, например, воду, метанол или этанол. Количество полярного растворителя, особенно воды, может присутствовать в стехиометрическом или нестехиометрическом соотношении.
Специалисты в данной области поймут, что, поскольку для окисления азота с образованием оксидов требуются доступные неподеленные пары электронов, не все азот-содержащие гетероциклические кольца могут образовывать N-оксиды. Специалисты в данной области техники распознают азот-содержащие гетероциклические кольца, которые могут образовывать N-оксиды. Специалисты в данной области техники также поймут, что третичные амины могут образовывать N-оксиды. Способы синтеза для получения N-оксидов гетероциклических колец и третичных аминов хорошо известны специалистам в данной области техники, включая, но не ограничиваясь ими, использование пероксикислот, таких как пероксиуксусная кислота и м-хлорпероксибензойная кислота (MCPBA), пероксид водорода, алкилгидропероксидов, таких как трет-бутилгидропероксид и перборат натрия, и диоксирана, такого как диметилдиоксиран, для окисления гетероциклических колец и третичных аминов. Эти методы получения N-оксидов широко описаны и обобщены в литературе (см., например: T. L. Gilchrist, Comprehensive Organic Synthesis, vol. 7, pp 748-750; A. R. Katritzky and A. J. Boulton, Eds., Academic Press; and G. W. H. Cheeseman and E. S. G. Werstiuk, Advances in Heterocyclic Chemistry, vol. 22, pp 390-392, A. R. Katritzky and A. J. Boulton, Eds., Academic Press.)
Настоящее изобретение дополнительно включает в свой объем метаболиты соединения по настоящему изобретению, то есть вещества, образующиеся in vivo при введении соединения по настоящему изобретению. Такие продукты могут быть получены, например, путем окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, ферментолиза и т. п. вводимого соединения. Следовательно, настоящее изобретение включает метаболиты соединения по настоящему изобретению, включая соединения, полученные контактированием соединения по настоящему изобретению с млекопитающим в течение времени, достаточного для образования его метаболитов.
Настоящее изобретение дополнительно включает, в пределах своего объема, пролекарства соединения по настоящему изобретению, которые представляют собой определенные производные соединения по настоящему изобретению, которые сами могут иметь меньшую фармакологическую активность или не иметь фармакологической активности при введении в организм человека или на него, и это может быть преобразовано в соединение по настоящему изобретению, обладающее желаемой активностью, например, путем гидролитического расщепления. Обычно такие пролекарства представляют собой производные функциональной группы соединения, которые легко превращаются в необходимое терапевтически активное соединение in vivo. Дополнительную информацию об использовании пролекарств можно найти в «Pro-drugs as Novel Delivery Systems», Volume 14, ACS Symposium Series (T. Higuchi and V. Stella). Пролекарства по настоящему изобретению могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединении по настоящему изобретении, определенными фрагментами, известными специалистам в данной области техники как «про-фрагмент» (например, как описано в «Design of Prodrugs», H. Bundgaard (Elsevier, 1985))».
Настоящее изобретение дополнительно включает соединение по настоящему изобретению, содержащее защитные группы. В любом процессе получения соединения по настоящему изобретению защита чувствительных групп или реактивных групп на любых родственных молекулах может быть необходимой и/или желательной, таким образом образуя химически защищенную форму соединения по настоящему изобретению. Это может быть достигнуто с помощью обычных защитных групп, например, тех защитных групп, которые описаны в T. W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991. Эти ссылки включены в данный документ посредством ссылки. Защитные группы могут быть удалены на соответствующей последующей стадии с использованием способов, известных в данной области техники.
Термин «около» означает в пределах ± 10%, предпочтительно в пределах ± 5%, более предпочтительно в пределах ± 2% от указанного значения.
Соединения
В одном аспекте настоящее изобретение предусматривает соединение формулы I, стереоизомер, таутомер или их смесь, его N-оксид, фармацевтически приемлемую соль, эвтектику, полиморф или его сольват, или производное со стабильным изотопом, метаболит, или его пролекарство:
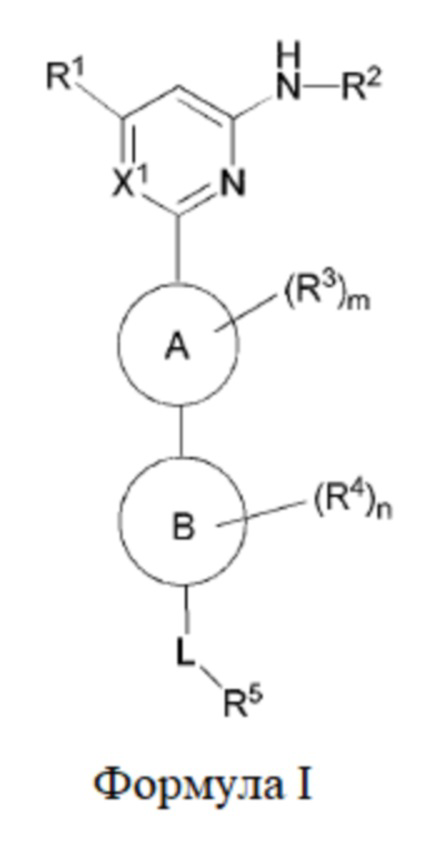
где:
кольцо A выбрано из C6-10 ароматического кольца и 5-6-членного гетероароматического кольца;
кольцо B выбрано из C3-8 циклоалкила и 4-11-членного гетероциклила;
X1 выбран из CH и N;
R1 выбран из группы, состоящей из H, галогена, гидрокси, циано, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси), C3-8 циклоалкила, 4-10-членного гетероциклила и -NR20aR20b, и каждый из алкила, гетероалкила (например, алкокси), циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси и C1-4 гетероалкила (например, C1-4 алкокси);
R2 выбран из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, C3-8 циклоалкила, 4-10-членного гетероциклила, 5-10-членного гетероарила и -C(=O)R21, и каждый из алкила, гетероалкила, циклоалкила, гетероциклила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила и C3-6 циклоалкила;
R3 и R4 отсутствуют или в каждом случае независимо выбраны из группы, состоящей из гидрокси, галогена, CN, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси) и C3-6 циклоалкила, каждый из алкила, гетероалкила (например, алкокси) и циклоалкила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; если m больше 1, два R3 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил; и/или если n больше 1, два R4 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил;
L выбран из группы, состоящей из -O-, -S-, -S(O)-, -S(O)2-, -N=CR21-, -N(R23a)-C(O)-, C1-6 алкилена, C1-6 гетероалкилена, C2-6 алкенилена, C2-6 алкинилена,
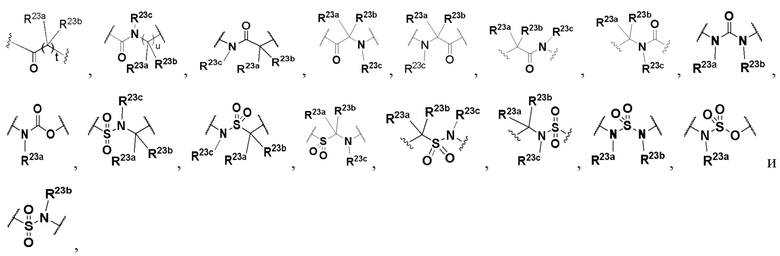
каждый из алкилена, гетероалкилена, алкенилена и алкинилена необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-6 алкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 галогеналкокси, C1-6 гетероалкила (например, C1-6 алкокси) и C3-8 циклоалкила; или L представляет собой -N(R23a)-;
R5 выбран из группы, состоящей из гидрокси, галогена, CN, NO2, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси), C2-6 алкенила, C2-6 алкинила, C3-8 циклоалкила, C3-8 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR20aR20b, -OR21, -SR21, -S(=O)R22, -S(=O)2R22, -S(=O)NR20aR20b, -S(=O)2NR20aR20b, -NR20aS(=O)R20b, -NR20aS(=O)2R20b, -C(=O)R21, -C(=O)NR23aR23b, -NR23aC(=O)R23b, -OC(=O)NR23aR23b и -NR24aC(=O)NR25aR25b, и каждый из алкила, гетероалкила (например, алкокси), алкенила, алкинила, циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4-10-членного гетероциклила;
каждый из R20a, R20b, R23a, R23b, R23c, R24a, R25a и R25bнезависимо выбран из группы, состоящей из H, OH, C1-6 алкила, C1-6 алкокси, и C3-8 циклоалкила; или R20a и R20b, R23a и R23b, или R25a и R25b образуют вместе с атомом, к которому они присоединены, 3-8-членный циклоалкил или гетероциклил, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, NO2, C1-4 алкила, C1-4 алкокси, C1-4 гидроксиалкила, C1-4 галогеналкила и C1-4 галогеналкокси;
каждый из R30a, R30b, R33a, R33b, R34a, R35a и R35b независимо выбран из группы, состоящей из H, C1-6 алкила, C1-6 галогеналкила, C1-6 гидроксиалкила, C1-6 алкокси и C1-6 галогеналкокси;
каждый из R21, R22, R31 и R32 независимо выбран из группы, состоящей из C1-6 алкила, C1-6 алкокси, C3-8 циклоалкила, 4-10-членного гетероциклила, C6-12 арила и 5-10-членного гетероарила, и каждый из алкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C3-6 циклоалкила, 4-10-членного гетероциклила;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4;
t равно 0, 1, 2, 3 или 4; и
u равно 0, 1, 2, 3 или 4;
при условии, что если кольцо B представляет собой пиперазиновое кольцо и X1 представляет собой CH, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил.
В некоторых вариантах осуществления кольцо A представляет собой бензольное кольцо или 5-6-членное гетероароматическое кольцо; предпочтительно кольцо A представляет собой бензольное кольцо, тиазольное кольцо, пиридиновое кольцо, пиразиновое кольцо или пиримидиновое кольцо; и более предпочтительно кольцо A представляет собой 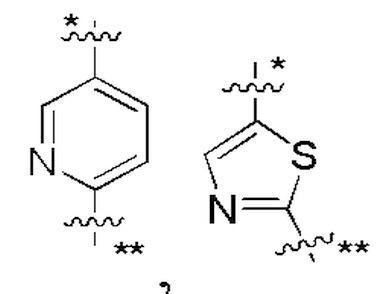 или
или 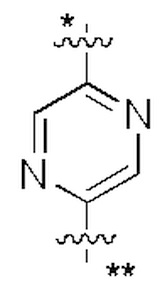 , соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом B в позиции, отмеченной **.
, соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом B в позиции, отмеченной **.
В некоторых вариантах осуществления кольцо B представляет собой C3-6 циклоалкил или 5-7-членный гетероциклил; предпочтительно кольцо B представляет собой пиперидиновое кольцо, пиперазиновое кольцо, азациклогептановое мостиковое кольцо или диазациклогептановое мостиковое кольцо; и более предпочтительно кольцо B представляет собой 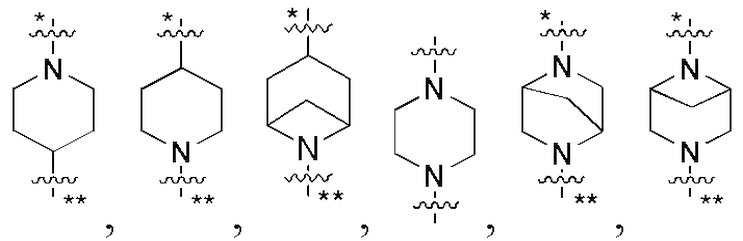 или
или 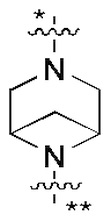 , которое соединено с кольцом A в позиции, отмеченной *, и соединено с L в позиции, отмеченной **.
, которое соединено с кольцом A в позиции, отмеченной *, и соединено с L в позиции, отмеченной **.
В некоторых вариантах осуществления X1 представляет собой CH или N, и предпочтительно X1 представляет собой N.
В некоторых вариантах осуществления R1 выбран из группы, состоящей из H, галогена, гидрокси, циано, C1-4 алкила, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила, и каждый из алкила, гетероалкила (например, алкокси), циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси и C1-4 гетероалкила (например, C1-4 алкокси).
В некоторых вариантах осуществления R1 выбран из группы, состоящей из C1-4 алкила, 5-членного азот-содержащего гетероциклила и C1-4 гетероалкила (например, C1-4 алкокси), и каждый из алкила, гетероциклила и гетероалкила (например, алкокси) необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси и C1-3 гетероалкила (например, C1-4 алкокси).
В некоторых вариантах осуществления R1 выбран из группы, состоящей из C1-3 алкила (например, метила), пирролидинила (например, пирролидин-1-ила) и C1-3 алкокси (например, этокси).
В некоторых вариантах осуществления R2 выбран из группы, состоящей из C1-4 алкила, C1-4 гетероалкила, C3-6 циклоалкила, 4-6-членного гетероциклила, 5-6-членного гетероарила и -C(=O)R21, и каждый из алкила, гетероалкила, циклоалкила, гетероциклила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила и C3-6 циклоалкила.
В некоторых вариантах осуществления R2 выбран из группы, состоящей из C1-3 алкила, 5-6-членного гетероарила и -C(=O)CH3, и каждый из алкила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 гетероалкила и C3-6 циклоалкила.
В некоторых вариантах осуществления R2 выбран из группы, состоящей из C1-3 алкила (например, метила), -C(=O)CH3, тиенила, пирролила, пиразолила, имидазолила, тиазолиила, тиадиазолил, изотиазолила, оксазолила, оксадиазолила, изоксазолила и пиридила, и каждый из алкила, тиенила, пирролила, пиразолила, имидазолила, тиазолила, тиадиазолила, изотиазолила, оксазолила, оксадиазолила, изоксазолила и пиридила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила (например, метила), C1-3 галогеналкила, C1-3 галогеналкокси, C1-3 гетероалкила (например, C1-3 алкокси) и C3-6 циклоалкила; и предпочтительно R2 представляет собой метил-замещенный пиразолил (например, 5-метил-1H-пиразол-3-ил или 1-метил-1H-пиразол-4-ил), циклопропил-замещенный пиразолил (например, 5-циклопропил-1H-пиразол-3-ил) или -C(O)CH3.
В некоторых вариантах осуществления R3 и R4 отсутствуют или в каждом случае независимо выбраны из группы, состоящей из гидрокси, галогена, CN, C1-4 алкила и C1-4 алкокси, каждый из алкила и алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 алкокси и C1-4 галогеналкокси; если m больше 1, два R3 необязательно образуют вместе с атомом, к которому они присоединены, a C3-6 циклоалкил или 4-10-членный гетероциклил; и/или если n больше 1, два R4 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил.
В некоторых вариантах осуществления R3 и R4 отсутствуют или в каждом случае независимо выбраны из группы, состоящей из гидрокси, галогена, CN, C1-3 алкила, C1-3 алкокси, где каждый из алкила и алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN и C1-3 алкила; если m больше 1, два R3 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил; и/или если n больше 1, два R4 необязательно образуют вместе с атомом, к которому они присоединены, C3-6 циклоалкил или 4-10-членный гетероциклил.
В некоторых вариантах осуществления R3 и R4 отсутствуют или в каждом случае независимо выбраны из группы, состоящей из: F, Cl, CN, OH, C1-3 алкила и C1-3 алкокси; и предпочтительно R3 и R4 отсутствуют.
В некоторых вариантах осуществления L выбран из группы, состоящей из -O-, -S-, -C(O)-, -N(R23a)-C(O)-, -C(O)-N(R23c)-, C1-4 алкилена, C1-4 гетероалкилена,
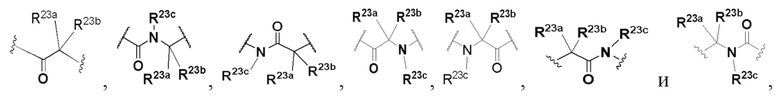
и каждый из алкилена и гетероалкилена необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси) и C3-6 циклоалкила.
В некоторых вариантах осуществления L выбран из группы, состоящей из -O-, -C(O)-, -NHC(O)-, -C(O)NH-, C1-3 алкилена, C1-3 гетероалкилена,
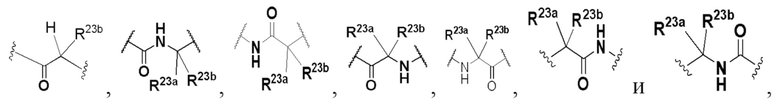
и каждый из алкилена и гетероалкилена необязательно замещен одним или несколькими заместителями, выбранными из группы: гидрокси, галогена, CN, NO2, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 гетероалкила (например, C1-3 алкокси) и C3-6 циклоалкила, где R23a и R23b предпочтительно представляют собой H или C1-3 алкил.
В некоторых вариантах осуществления L выбран из группы, состоящей из -O-, -C(O)-, -NHC(O)-, -C(O)NH-, C1-3 алкилена,
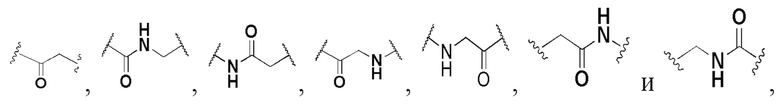
и алкилен необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила и C1-3 галогеналкила. Предпочтительно L представляет собой -CH2-, -CH(CH3)-, -O-, -C(O)-, 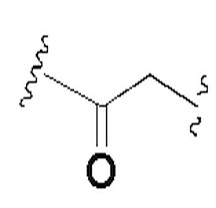 , -C(O)NH- или
, -C(O)NH- или 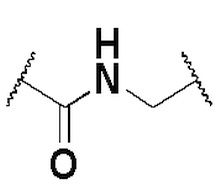 .
.
В некоторых вариантах осуществления R5 выбран из группы, состоящей из гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR20aR20b, -OR21, -SR21, -S(=O)R22, -S(=O)2R22, -S(=O)NR20aR20b, -S(=O)2NR20aR20b, -NR20aS(=O)R20b, -NR20aS(=O)2R20b, -C(=O)R21, -C(=O)NR23aR23b, -NR23aC(=O)R23b, -OC(=O)NR23aR23b и -NR24aC(=O)NR25aR25b, и каждый из алкила, гетероалкила (например, алкокси), алкенила, алкинила, циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4-10-членного гетероциклила.
В некоторых вариантах осуществления R5 выбран из группы, состоящей из C3-6 циклоалкила, 4-10-членного гетероциклила, C6-12 арила и 5-10-членного гетероарила, и каждый из циклоалкила, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4-10-членного гетероциклила.
В некоторых вариантах осуществления R5 выбран из группы, состоящей из C6-10 арила и 5-6-членного гетероарила, и каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 гетероалкила (например, C1-3 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -C(=O)R31, -C(=O)NR33aR33b и -NR33aC(=O)R33b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4- 6-членного гетероциклила.
В некоторых вариантах осуществления R5 выбран из фенила и 5-6-членного гетероарила (например, пиридила, пиримидила, пиразинила, пиридазинила, пиразолила, оксазолила, имидазолила или тиазолила), и каждый из фенила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 гетероалкила (например, C1-3алкокси), C3-6 циклоалкила, C3-6 циклоалкокси, 4- 6-членного гетероциклила, 5- 8-членного гетероарила (например, пиридила, пирролила, пиразолила, фурила, оксазолила, имидазолила, тиазолила или циклопентил-пиразолила), -NR30aR30b, -OR31, -C(=O)R31, -C(=O)NR33aR33b и -NR33aC(=O)R33b, где каждый из циклоалкила, циклоалкокси, гетероциклил и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 гетероалкила (например, C1-3алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4- 6-членного гетероциклила.
В некоторых вариантах осуществления R5 выбран из группы, состоящей из фенила, пиридила, пиразолила и тиазолила, и каждый из фенила, пиридила, пиразолила и тиазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 алкокси, C3-6 циклоалкила, C3-6 циклоалкокси, 4-6-членного гетероциклила, 5-8-членного гетероарила (например, пиридила, пирролила, пиразолила, фурила, оксазолила, имидазолила, тиазолила или циклопентил-пиразолила), -NR30aR30b и -OR31, где каждый из гетероциклила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 алкокси, C3-6 циклоалкила, C3-6 циклоалкокси и 4-6-членного гетероциклила. Предпочтительно R5 представляет собой фенил, пиридил, пиразолил или тиазолил, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), CN, C1-3 алкила (например, метил или этил), C1-3 галогеналкила (например, трифторметил), C1-3 алкокси (например, метокси или этокси), C3-6 циклоалкила (например, циклопропил), C3-6 циклоалкокси (например, циклопропокси) и 5-6-членного гетероарила (например, пиридил, пирролил, пиразолил, фурил, оксазолил, имидазолил или тиазолил), где 5-6-членный гетероарил необязательно дополнительно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), C1-3 алкила (например, метил, этил или изопропил), C1-3 галогеналкила (например, фторметил), C1-3 гидроксиалкила (например, гидроксиметил или гидроксипропил), C1-3 алкокси (например, метокси), C3-6 циклоалкила (например, циклопропил) и C3-6 циклоалкокси (например, циклопропокси или циклобутокси).
В некоторых вариантах осуществления R5 выбран из группы, состоящей из фенила, пиридила, пиразолила и тиазолила, и каждый из фенила, пиридила, пиразолила и тиазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 галогеналкокси, C1-3 алкокси, C3-6 циклоалкила, C3-6 циклоалкокси, 4-6-членного гетероциклила, 5-6-членного гетероарила (например, пиридила, пирролила, фурила, пиразолила, оксазолила, имидазолила или тиазолила), -NR30aR30b и -OR31, где каждый из гетероциклила и гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-3 алкила, C1-3 галогеналкила, C1-3 галогеналкокси, C1-3 алкокси, C3-6 циклоалкила и 4-6-членного гетероциклила. Предпочтительно R5 представляет собой фенил, пиридил, пиразолил или тиазолил, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), CN, C1-3 алкила (например, метил или этил), C1-3 галогеналкила (например, трифторметил), C1-3 алкокси (например, метокси или этокси), C3-6 циклоалкила (например, циклопропил), C3-6 циклоалкокси (например, циклопропокси) и пяти-членный гетероарил (например, пиразолил, имидазолил или тиазолил), где пяти-членный гетероарил необязательно дополнительно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), C1-3 алкила (например, метил) и C1-3 гидроксиалкила (например, гидроксиметил или гидроксипропил).
В некоторых вариантах осуществления каждый из R20a, R20b, R23a, R23b, R23c, R24a, R25a и R25bнезависимо выбран из группы, состоящей из H, C1-4 алкила, C1-4 алкокси, и C3-8 циклоалкила; или R20a и R20b, R23a и R23b, или R25a и R25b образуют вместе с атомом, к которому они присоединены, 3-8-членный циклоалкил или гетероциклил, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, NO2, C1-4 алкила, C1-4 алкокси, C1-4 гидроксиалкила, C1-4 галогеналкила и C1-4 галогеналкокси.
В некоторых вариантах осуществления каждый из R20a, R20b, R23a, R23b, R23c, R24a, R25a и R25b независимо представляет собой H, C1-4 алкил или C1-4 алкокси.
В некоторых вариантах осуществления каждый из R23a и R23b независимо выбран из группы, состоящей из H, C1-3 алкила, C1-3 алкокси и C3-6 циклоалкила; или R23a и R23b образуют вместе с атомом C, к которому они присоединены, C3-6 циклоалкил или гетероциклил, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, C1-3 алкила, C1-3 алкокси, C1-3 гидроксиалкила, C1-3 галогеналкила и C1-3 галогеналкокси.
В некоторых вариантах осуществления каждый из R21, R22, R31 и R32 независимо выбран из группы, состоящей из C1-4 алкила, C1-4 алкокси, C3-8 циклоалкила и 4-10-членного гетероциклила, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, галогена, CN, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, C3-6 циклоалкила, 4-10-членного гетероциклила.
В некоторых вариантах осуществления каждый из R21, R22, R31 и R32 независимо выбран из C1-4 алкила.
В некоторых вариантах осуществления каждый из R30a, R30b, R33a, R33b, R34a, R35a и R35b независимо выбран из группы, состоящей из H, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси и C1-4 галогеналкокси.
В некоторых вариантах осуществления каждый из R30a, R30b, R33a, R33b, R34a, R35a и R35b независимо выбран из H и C1-4 алкила.
В некоторых вариантах осуществления m равно 0.
В некоторых вариантах осуществления n равно 0, 1 или 2.
В некоторых вариантах осуществления t равно 0 или 1.
В некоторых вариантах осуществления u равно 0 или 1.
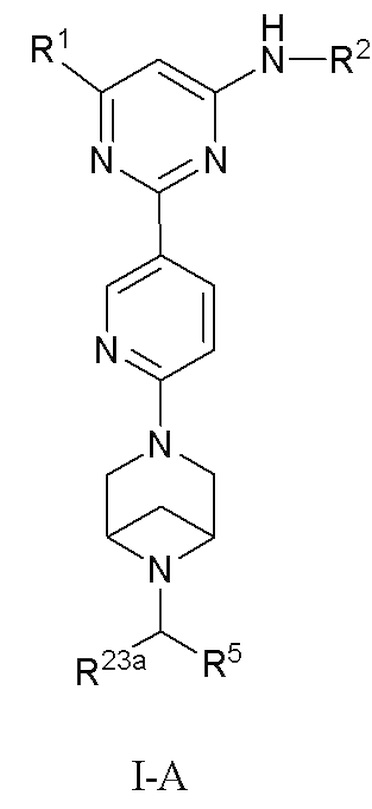
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-A:
 ;
;
где:
R1 и R2 определены выше в формуле I; и
R5 выбран из группы, состоящей из C6-12 арила и 5-10-членного гетероарила, где (1) C6-12 арил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: C3-6 циклоалкокси, C6-12 арила, 5-10-членного гетероарила, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкокси, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила; и (2) 5-10-членный гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила, C3-6 циклоалкокси и 4-10-членного гетероциклила; и
R23a, R30a, R30b, R31, R32, R33a, R33b, R34a, R35a, и R35b определены выше в формуле I, и R23a предпочтительно представляет собой H или C1-3 алкил.
В некоторых вариантах осуществления R5 выбран из C6-12 арила и 5-10-членного гетероарила, где (1) C6-12 арил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: C3-6 циклоалкокси, C6-12 арила, 5-10-членного гетероарила, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкокси, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила, и (2) 5-10-членный гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила; и
R23a, R30a, R30b, R31, R32, R33a, R33b, R34a, R35a, и R35b определены выше в формуле I, и R23a предпочтительно представляет собой H или C1-3 алкил.
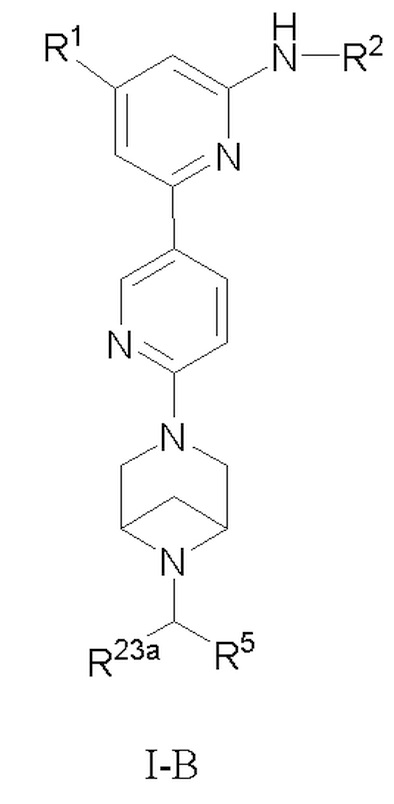
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-B:
где:
R1, R2, R5 и R23a определены выше в формуле I, и R23a предпочтительно представляет собой H или C1-3 алкил.
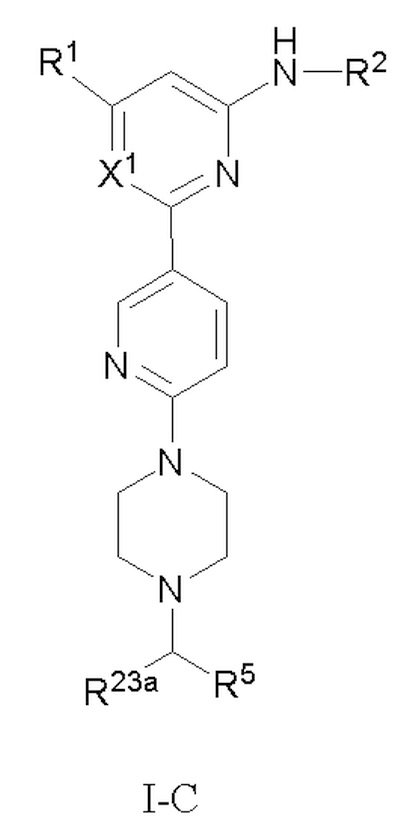
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-C:
где:
если X1 представляет собой CH, R1, R2, R5 и R23a определены выше в формуле I, R23a предпочтительно представляет собой H или C1-3 алкил; и если X1 представляет собой N, R1, R2, R5 и R23a определены выше в формуле I-A.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-D:
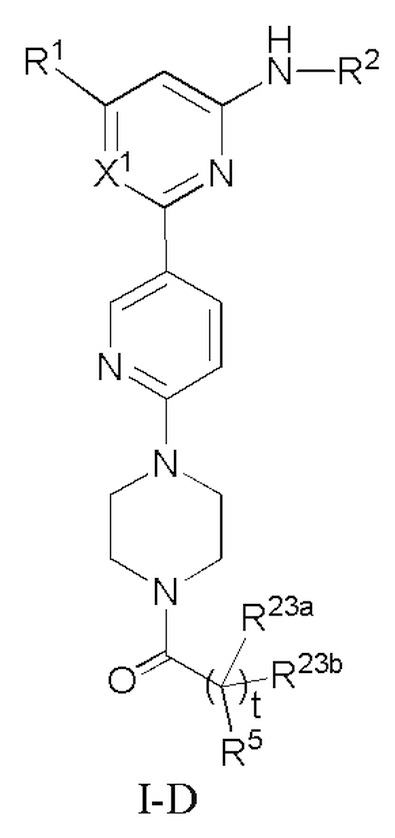
где:
R1, R2, R23a, R23b и t определены выше в формуле I;
если X1 представляет собой CH, R5 определен выше в формуле I; и
если X1 представляет собой N, R5 представляет собой C6-12 арил или 5-10-членный гетероарил, где
(i) если t равно 0, каждый из C6-12 арила и 5-10-членного гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6циклоалкила и 4-10-членного гетероциклила,
(ii) если t равно 1, (1) C6-12 арил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила, и (2) 5-10-членный гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: NO2, C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкокси, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила; и
R30a, R30b, R31, R32, R33a, R33b, R34a, R35a, и R35b определены выше в формуле I.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-E:
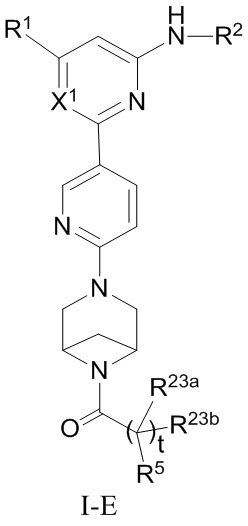
где:
R1, R2, R5, R23a, R23b, X1 и t определены выше в формуле I-D.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-F:
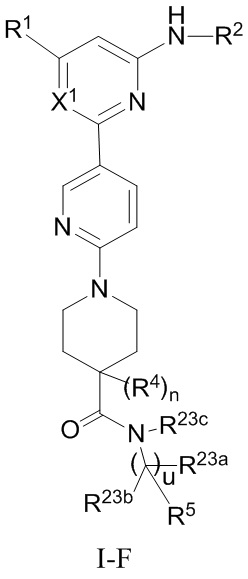
где:
R1, R2, R5, R23a, R23b и X1 определены выше в формуле I-D;
R4 определен выше в формуле I и предпочтительно представляет собой C1-3 алкил или C1-3 алкокси;
R23c представляет собой H, C1-3 алкил или C1-3 алкокси, и каждый из алкила и алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, C1-4 алкокси и C1-4 гидроксиалкила;
u равно 0 или 1; и
n равно 0 или 1.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I-G:
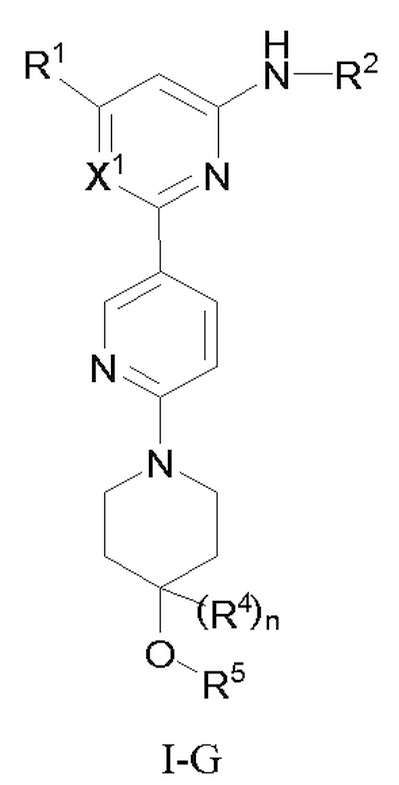
где:
X1 представляет собой CH или N;
R1, R2 и R4 определены выше в формуле I, и R4 предпочтительно представляет собой C1-3 алкил или C1-3 алкокси;
n равно 0 или 1;
R5 выбран из C6-12 арила и 5-10-членного гетероарила, где (1) C6-12 арил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: C3-6 циклоалкокси, C6-12 арила, 5-10-членного гетероарила, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкокси, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила; и (2) 5-10-членный гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C2-6 алкенила, C2-6 алкинила, C3-6 циклоалкила, C3-6 циклоалкокси, 4-10-членного гетероциклила, C6-12 арила, 5-10-членного гетероарила, -NR30aR30b, -OR31, -SR31, -S(=O)R32, -S(=O)2R32, -S(=O)NR30aR30b, -S(=O)2NR30aR30b, -NR30aS(=O)R30b, -NR30aS(=O)2R30b, -C(=O)R31, -C(=O)NR33aR33b, -NR33aC(=O)R33b, -OC(=O)NR33aR33b и -NR34aC(=O)NR35aR35b, где каждый из циклоалкила, циклоалкокси, гетероциклила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, NO2, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила (например, C1-4 алкокси), C3-6 циклоалкила и 4-10-членного гетероциклила; и
R30a, R30b, R31, R32, R33a, R33b, R34a, R35a, и R35b определены выше в формуле I.
В некоторых вариантах осуществления соединение по настоящему изобретению имеет структуру, показанную в формуле I, где:
кольцо A представляет собой 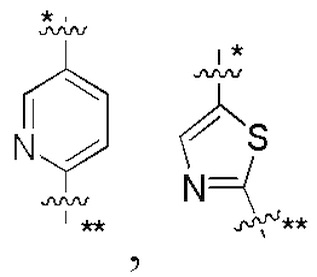 или
или 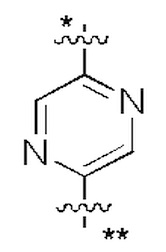 , соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом B в позиции, отмеченной **;
, соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом B в позиции, отмеченной **;
кольцо B представляет собой 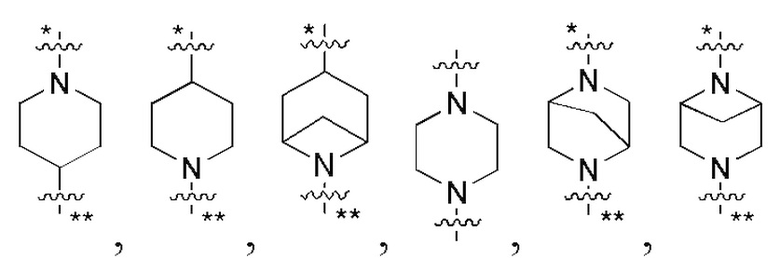 или
или 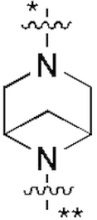 , соединено с кольцом A в позиции, отмеченной *, и соединено с L посредством в позиции, отмеченной **;
, соединено с кольцом A в позиции, отмеченной *, и соединено с L посредством в позиции, отмеченной **;
X1 представляет собой N;
R1 выбран из группы, состоящей из C1-3 алкила (например, метила), пирролидинила (например, пирролидин-1-ила) и C1-3 алкокси (например, этокси);
R2 представляет собой метил-замещенный пиразолил (например, 5-метил-1H-пиразол-3-ил или 1-метил-1H-пиразол-4-ил), циклопропил-замещенный пиразолил (например, 5-циклопропил-1H-пиразол-3-ил) или -C(O)CH3;
R3 и R4 отсутствуют;
L представляет собой -CH2-, -CH(CH3)-, -O-, -C(O)-,  , -C(O)NH- или
, -C(O)NH- или  ; и
; и
R5 представляет собой фенил, пиридил, пиразолил или тиазолли, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), CN, C1-3 алкила (например, метил или этил), C1-3 галогеналкила (например, трифторметил), C1-3 алкокси (например, метокси или этокси), C3-6 циклоалкила (например, циклопропил), C3-6 циклоалкокси (например, циклопропокси) и 5-6-членного гетероарила (например, пиридил, пирролил, пиразолил, фурил, оксазолил, имидазолил или тиазолил), где 5-6-членный гетероарил необязательно дополнительно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена (например, фтор или хлор), C1-3 алкила (например, метил, этил или изопропил), C1-3 галогеналкила (например, фторметил), C1-3 гидроксиалкила (например, гидроксиметил или гидроксипропил), C1-3 алкокси (например, метокси), C3-6 циклоалкила (например, циклопропил) и C3-6 циклоалкокси (например, циклопропокси или циклобутокси).
Настоящее изобретение охватывает любую комбинацию вышеупомянутых вариантов осуществления.
В некоторых вариантах осуществления соединение по настоящему изобретению содержит, но без ограничения:
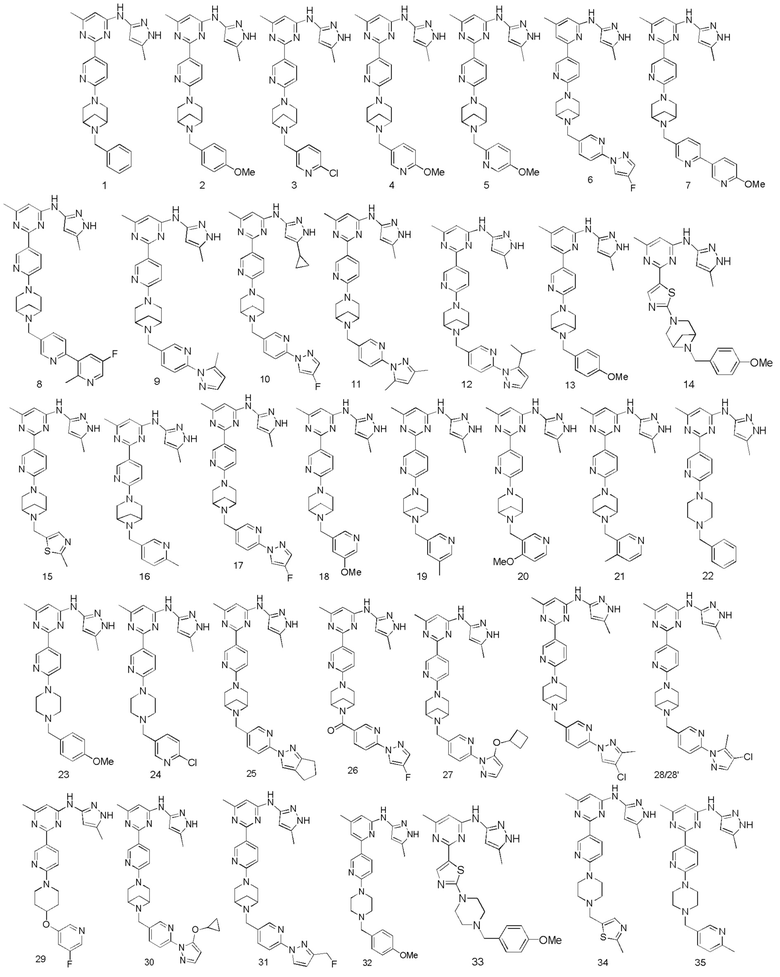

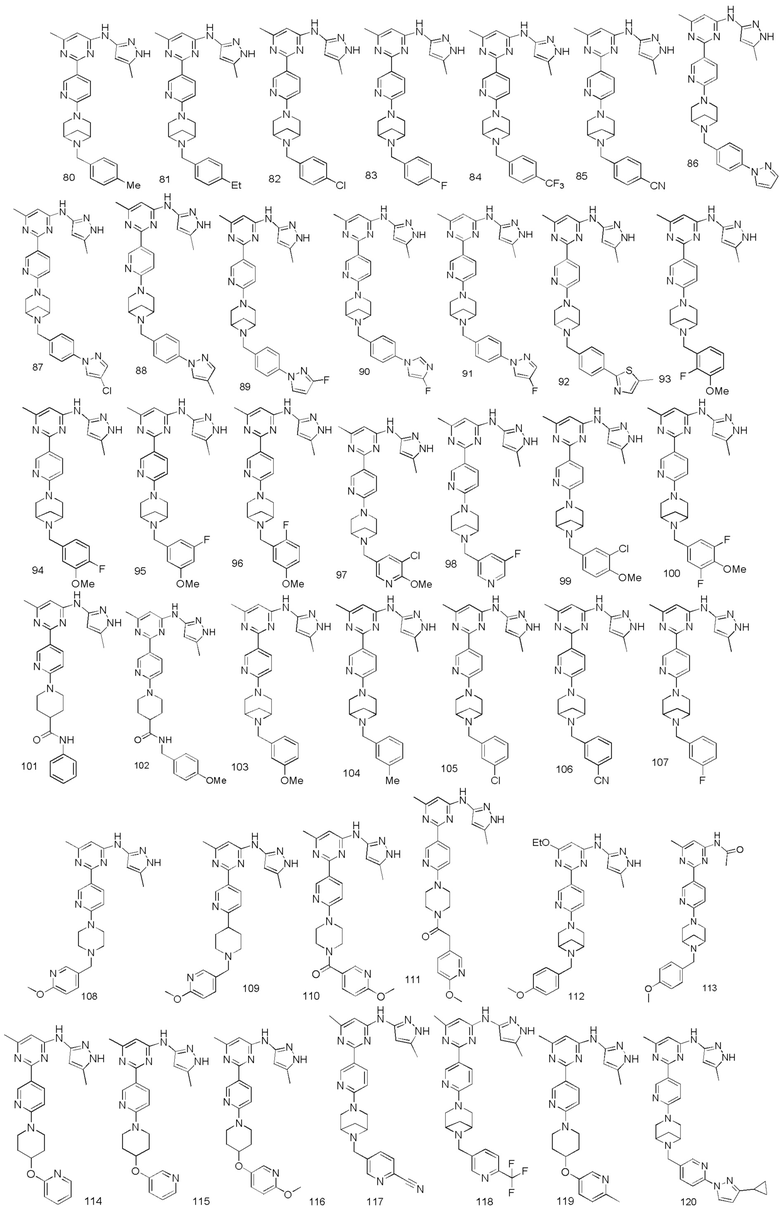
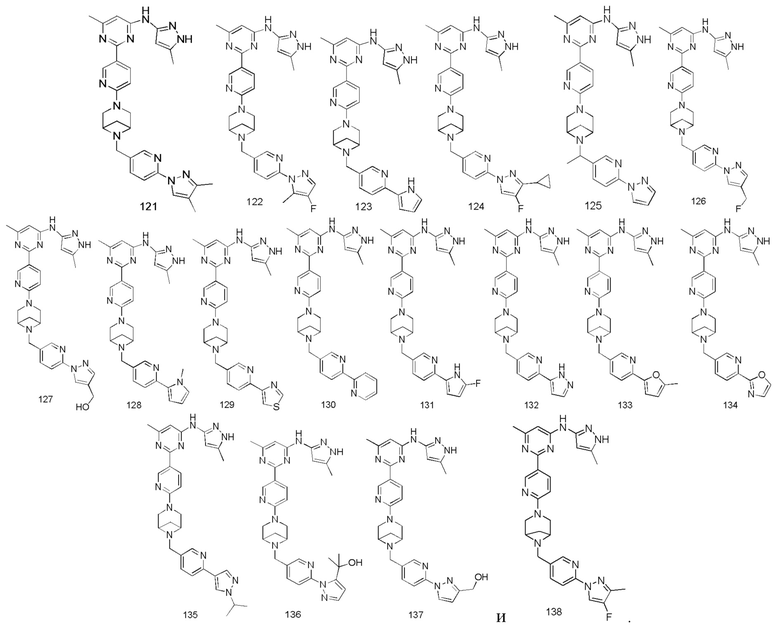
Способы получения
В некоторых вариантах осуществления соединение формулы I-A может быть синтезировано с использованием способа, показанного в пути синтеза A следующим образом:
Путь синтеза A
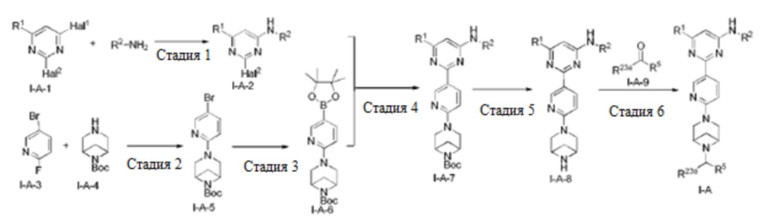
где:
каждый из Hal1 и Hal2 независимо представляют собой F, Cl, Br или I; и предпочтительно Hal1 представляет собой F, Cl, Br или I, и Hal2 представляет собой Cl, Br или I;
R1 выбран из группы, состоящей из H, циано, C1-6 алкила, C1-6 гетероалкила (например, C1-6 алкокси), C3-8 циклоалкила, 4-6-членного гетероциклила и -NR20aR20b, и каждый из алкила, гетероалкила (например, алкокси), циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 галогеналкокси, и C1-4 гетероалкила (например, C1-4 алкокси);
R2 выбран из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, C3-8 циклоалкила, 4- 6-членного гетероциклила и 5-6-членного гетероарила, и каждый из алкила, гетероалкила, циклоалкила, гетероциклила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: гидрокси, галогена, CN, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 галогеналкокси, C1-4 гетероалкила и C3-6 циклоалкила;
R23a выбран из группы, состоящей из H, C1-6 алкила, C1-6 алкокси и C3-8 циклоалкила, и каждый из алкила, алкокси и циклоалкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, C1-4 алкила, C1-4 алкокси, C1-4 гидроксиалкила, C1-4 галогеналкила и C1-4 галогеналкокси;
R20a и R20b определены выше в формуле I; и
R5 определен выше в формуле I-A.
Стадия 1. Введение в реакцию соединения I-A-1 с R2-NH2 посредством реакции замещения или сочетания (например, реакции Бухвальда, реакции Сузуки, или реакции Ульмана) в присутствии основания с образованием соединения I-A-2.
Для реакции замещения используемые основания представляют собой, например, tBuONa, tBuOK, tBuOLi, Cs2CO3, DIPEA, LiHMDS, LDA, NaHMDS, KHMDS, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3; используемые растворители представляют собой, например, третичный бутанол, толуол, ксилол, ТГФ, DME, 1,4-диоксан, DMF, ДМСО или NMP; и температура реакции составляет от 40°C до 140°C.
Для реакции Бухвальда используемые катализаторы представляют собой, например, Pd(OAc)2, Pd2(dba)3, Pd(dba)2, PdCl2, Pd(PPh3)4, Pd(dppf)Cl2, Pd(acac)2 или Pd(allyl)2; используемые лиганды представляют собой, например, PPh3, XPhos, SPhos, RuPhos, XantPhos, Dppf, BINOL, BINAP или Pcy3; используемые основания представляю собой, например, tBuONa, tBuOK, tBuOLi, Cs2CO3, LiHMDS, LDA, NaHMDS, KHMDS, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3; используемые растворители представляют собой, например, толуол, ксилол, ТГФ, DME, 1,4-диоксан, DMF, ДМСО или NMP; и температура реакции составляет от 40°C до 140°C.
Для реакции Сузуки используемые катализаторы представляют собой, например, Pd(PPh3)4 или Pd(dppf)Cl2; используемые основания представляют собой, например, Cs2CO3, K3PO4, Na2CO3, AcOK, NaHCO3 или K2CO3; используемые растворители представляют собой, например, 1,4-диоксан/H2O, DMF/H2O, ДМСО/H2O или CH3CN/H2O; и температура реакции составляет от 60°C до 120°C;
Для реакции Ульмана используемые катализаторы представляют собой, например, CuCl, CuBr, CuI или Cu2O; используемые лиганды представляют собой, например, салицилалдоксим, циклогександиамин, N,N’-диметилэтандиамин, TMEDA или этандиамин; используемые основания представляют собой, например, tBuONa, tBuOK, tBuOLi, Cs2CO3, LiHMDS, LDA, NaHMDS, KHMDS, K3PO4, Na2CO3, KOAc, NaHCO3, или K2CO3; используемые растворители представляют собой, например, толуол, ксилол, ТГФ, DME, 1,4-диоксан, DMF, ДМСО, или NMP; и температура реакции составляет от 40°С до 140 °C.
Стадия 2. Введение в реакцию соединения I-A-3 с соединением I-A-4 в присутствии основания с образованием соединения I-A-5.
Используемые основания представляют собой, например, tBuONa, tBuOK, tBuOLi, Cs2CO3, DIPEA, LiHMDS, LDA, NaHMDS, KHMDS, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3. Используемые растворители представляют собой, например, третичный бутанол, толуол, ксилол, ТГФ, DME, 1,4-диоксан, DMF, ДМСО или NMP. Температура реакции составляет от 40°C до 140°C.
Стадия 3. Введение в реакцию соединения I-A-5 с бор-содержащим реагентом с образованием соединения I-A-6.
Используемые бор-содержащие реагенты представляют собой, например, B2(pin)2. Используемые катализаторы представляют собой, например, Pd(PPh3)4, Pd(dppf)Cl2 или Pd(dppf)2Cl2·ДХМ. Используемые основания представляют собой, например, Cs2CO3, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3. Используемые растворители представляют собой, например, 1,4-диоксан, DMF, ДМСО или CH3CN. Температура реакции составляет от 50 °C до 120 °C.
Стадия 4. Введение в реакцию соединения I-A-2 с соединением I-A-6 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-A-7.
Используемые катализаторы представляют собой, например, Pd(PPh3)4, Pd(dppf)Cl2 или Pd(dppf)2Cl2·ДХМ. Используемые основания представляют собой, например, Cs2CO3, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3. Используемые растворители представляют собой, например, 1,4-диоксан, DMF, ДМСО или CH3CN, или смесь любых из вышеуказанных растворителей и H2O. Температура реакции составляет от 50 °C до 120 °C.
Стадия 5. Снятие защиты с соединения I-A-7 в кислых условиях с образованием соединения I-A-8.
Используемые кислоты представляют собой, например, раствор HCl в 1,4-диоксане, раствор HCl в EA или раствор TFA в ДХМ. Температура реакции составляет от 0 °C до 80 °C.
Стадия 6. Введение в реакцию соединения I-A-8 с соединением I-A-9 посредством реакции восстановительного аминирования с образованием соединения I-A.
Используемые растворители представляют собой, например, метанол, этанол, ТГФ, ДХМ, DCE, DMA или смесь их и уксусной кислоты в любом соотношении. Используемые восстанавливающие агенты представляют собой, например, NaBH4, NaBH3CN или NaBH(OAc)3. Температура реакции составляет от 0°C до 80°C. В некоторых вариантах осуществления реакция может быть проведена в присутствии основания или кислоты, основание представляет собой, например, TEA или DIPEA, и кислота представляет собой, например, AcOH, HCl или Ti(OiPr)4.
В некоторых вариантах осуществления соединение формулы I-A может быть синтезировано с использованием способа, показанного в пути синтеза B следующим образом:
Путь синтеза B
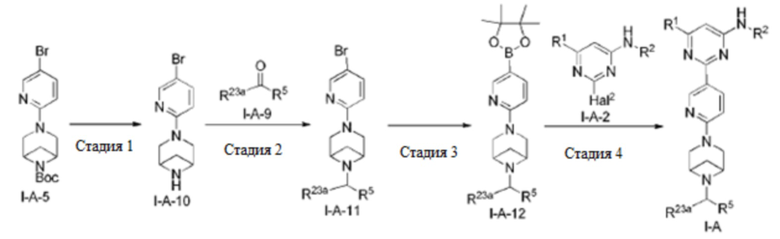
где:
Hal2, R1, R2, R5 и R23a определены выше в пути синтеза A.
Стадия 1. Снятие защиты с соединения I-A-5 в кислых условиях с образованием соединения I-A-10.
Используемые кислоты представляют собой, например, раствор HCl в 1,4-диоксане, раствор HCl в EA или раствор TFA в ДХМ. Температура реакции составляет от 0°C до 80°C.
Стадия 2. Введение в реакцию соединения I-A-10 с соединением I-A-9 посредством реакции восстановительного аминирования с образованием соединения I-A-11.
Используемые основания представляют собой, например, DIPEA или TEA. Используемые восстанавливающие агенты представляют собой, например, NaBH3CN или NaBH(OAc)3. Используемые растворители представляют собой, например, MeOH, EtOH или DCE. Температура реакции составляет от 0°C до 80°C.
Используемые растворители представляют собой, например, метанол, этанол, ТГФ, ДХМ, DCE, DMA или смесь их и уксусной кислоты в любом соотношении. Используемые восстанавливающие агенты представляют собой, например, NaBH4, NaBH3CN или NaBH(OAc)3. Температура реакции составляет от 0°C до 80°C. В некоторых вариантах осуществления реакция может быть проведена в присутствии основания или кислоты, основание представляет собой, например, TEA или DIPEA, и кислота представляет собой, например, AcOH, HCl или Ti(OiPr)4.
Стадия 3. Введение в реакцию соединения I-A-11 с бор-содержащим реагентом с образованием соединения I-A-12.
Используемые бор-содержащие реагенты представляют собой, например, B2(pin)2. Используемые катализаторы представляют собой, например, Pd(PPh3)4, Pd(dppf)Cl2 или Pd(dppf)2Cl2·ДХМ. Используемые основания представляют собой, например, Cs2CO3, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3. Используемые растворители представляют собой, например, 1,4-диоксан, DMF, ДМСО или CH3CN. Температура реакции составляет от 50°C до 120 °C.
Стадия 4. Введение в реакцию соединения I-A-12 с соединением I-A-2 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-A.
Используемые катализаторы представляют собой, например, Pd(PPh3)4, Pd(dppf)Cl2 или Pd(dppf)2Cl2·ДХМ. Используемые основания представляют собой, например, Cs2CO3, K3PO4, Na2CO3, KOAc, NaHCO3 или K2CO3. Используемые растворители представляют собой, например, 1,4-диоксан, DMF, ДМСО или CH3CN, или смесь любых из вышеуказанных растворителей и H2O. Температура реакции составляет от 50 °C до 120 °C.
В некоторых вариантах осуществления соединение формулы I-B может быть синтезировано с использованием способа, показанного в пути синтеза C следующим образом:
Путь синтеза C
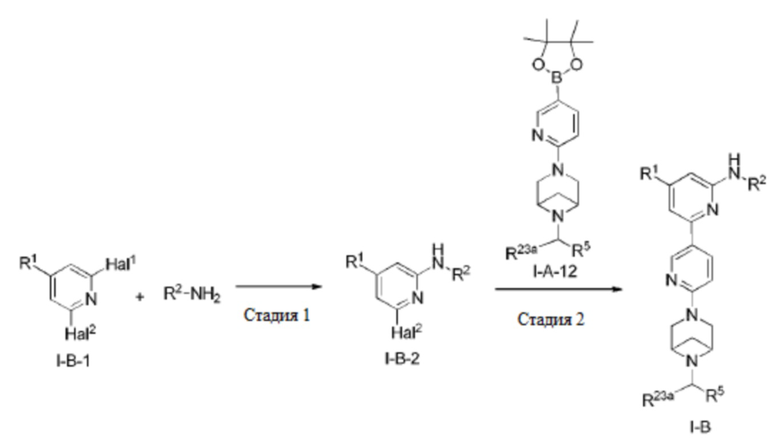
где:
Hal1, Hal2, R1, R2, R5, R23a определены выше в пути синтеза A.
Стадия 1. Введение в реакцию соединения I-B-1 с R2-NH2 посредством реакции замещения или сочетания (например, реакции Бухвальда, реакции Сузуки, или реакции Ульмана) в присутствии основания с образованием соединения I-B-2.
Условия реакции описаны на стадии 1 пути синтеза A для получения соединения формулы I-A.
Стадия 2. Введение в реакцию соединения I-B-2 с соединением I-A-12 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-B.
Условия реакции описаны на стадии 4 пути синтеза A для получения соединения формулы I-A.
В некоторых вариантах осуществления соединение формулы I-C может быть синтезировано с использованием способа, показанного в пути синтеза D следующим образом:
Путь синтеза D
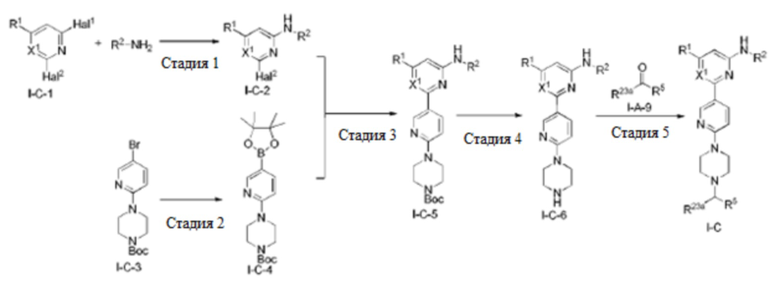
где:
Hal1, Hal2, R1, R2, R5 и R23a определены выше в пути синтеза A; и
X1 выбран из CH и N.
Стадия 1. Введение в реакцию соединения I-C-1 с R2-NH2 посредством реакции замещения или сочетания (например, реакции Бухвальда, реакции Сузуки, или реакции Ульмана) в присутствии основания с образованием соединения I-C-2.
Условия реакции описаны на стадии 1 пути синтеза A для получения соединения формулы I-A.
Стадия 2. Введение в реакцию соединения I-C-3 с бор-содержащим реагентом с образованием соединения I-C-4.
Условия реакции описаны на стадии 3 пути синтеза A для получения соединения формулы I-A.
Стадия 3. Введение в реакцию соединения I-C-2 с соединением I-C-4 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-C-5.
Условия реакции описаны на стадии 4 пути синтеза A для получения соединения формулы I-A.
Стадия 4. Снятие защиты с соединения I-C-5 в кислых условиях с образованием соединения I-C-6.
Условия реакции описаны на стадии 5 пути синтеза A для получения соединения формулы I-A.
Стадия 5. Введение в реакцию соединения I-C-6 с соединением I-A-9 посредством реакции восстановительного аминирования с образованием соединения I-C.
Условия реакции описаны на стадии 6 пути синтеза A для получения соединения формулы I-A.
В некоторых вариантах осуществления соединение формулы I-D может быть синтезировано с использованием способа, показанного в пути синтеза E следующим образом:
Путь синтеза E
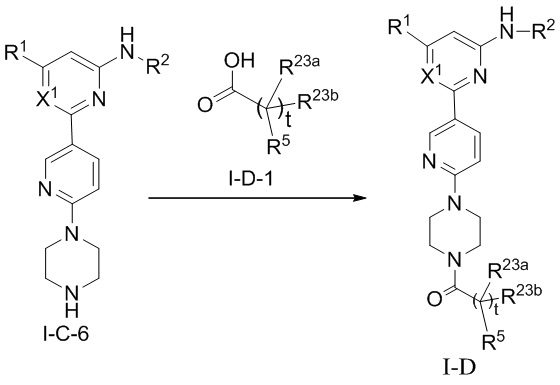
где:
R1 и R2 определены выше в пути синтеза A;
R5 определен выше в формуле I-D;
каждый из R23a и R23b независимо выбран из группы, состоящей из H, C1-6 алкила, C1-6 алкокси и C3-8 циклоалкила; или R23a и R23b образуют вместе с атомом C, к которому они присоединены, 3-8-членный циклоалкил или гетероциклил, и каждый из алкила, алкокси, циклоалкила и гетероциклила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: CN, галогена, C1-4 алкила, C1-4 алкокси, C1-4 гидроксиалкила, C1-4 галогеналкила и C1-4 галогеналкокси;
X1 выбран из CH и N; и
t равно 0 или 1.
Соединение I-C-6 вступает в реакцию с соединением I-D-1 посредством реакции конденсации с образованием соединения I-D.
Используемые конденсирующие агенты представляют собой, например, HATU, CDI, HOBt, DMAP, DCC, DIC, EDC, HBTU, HCTU или PyBOP. Используемые основания представляют собой, например, TEA, DIPEA, tBuOK, tBuONa, tBuOLi, NaH, NaOH, Cs2CO3, K3PO4 или Na2CO3. Используемые растворители представляют собой, например, ТГФ, ДХМ, DCE, MeOH, EtOH, DMF, ДМСО, пропанон, CH3CN, 1,4-диоксан или толуол. Температура реакции составляет от 0°C до 120°C, например, комнатная температура.
Альтернативно, соединение I-D-1 сначала вступает в реакцию с ацилирующим реагентом с образованием ацилгалогенида, который затем вступает в реакцию с соединением I-C-6, необязательно в присутствии основания, с образованием соединения формулы I-D. Используемые ацилирующие агенты представляют собой, например, тионилхлорид или оксалилхлорид. Реакцию также можно проводить при катализе небольшим количеством DMF. Используемые основания представляют собой, например, TEA или DIPEA. Используемые растворители представляют собой, например, ТГФ, ДХМ, DCE, CH3CN, 1,4-диоксан или толуол. Температура реакции составляет от 0°C до 100°C.
В некоторых вариантах осуществления соединение формулы I-E может быть синтезировано с использованием способа, показанного в пути синтеза F, следующим образом:
Путь синтеза F
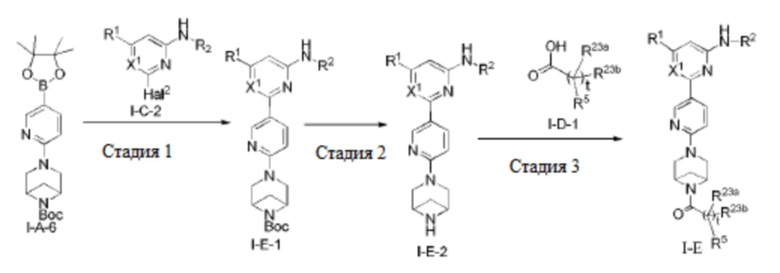
где:
R1, R2, R5, R23a, R23b и t определены выше в пути синтеза E;
X1 выбран из CH и N; и
Hal2 представляет собой F, Cl, Br или I; и предпочтительно Hal2 представляет собой Cl, Br или I.
Стадия 1. Введение в реакцию соединения I-C-2 с соединением I-A-6 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-E-1.
Условия реакции описаны на стадии 4 пути синтеза A для получения соединения формулы I-A.
Стадия 2. Снятие защиты с соединения I-E-1 в кислых условиях с образованием соединения I-E-2.
Условия реакции описаны на стадии 5 пути синтеза A для получения соединения формулы I-A.
Стадия 3. Введение в реакцию соединения I-E-2 с соединением I-D-1 посредством реакции конденсации с образованием соединения I-E.
Условия реакции описаны в пути синтеза E для получения соединения формулы I-D.
В некоторых вариантах осуществления соединение формулы I-F может быть синтезировано с использованием способа, показанного в пути синтеза G следующим образом:
Путь синтеза G
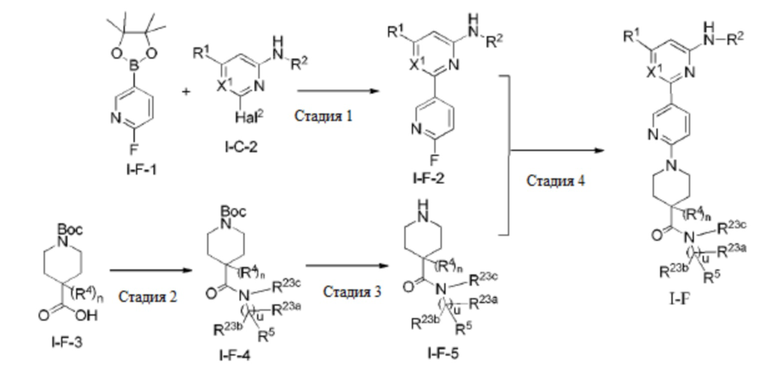
где:
R1, R2, R23a, R23b и R5 определены выше в пути синтеза E;
X1 выбран из CH и N;
R4 отсутствует или выбран из группы, состоящей из гидрокси, CN, C1-6 алкила, C1-6 галогеналкила и C1-6 гетероалкила (например, C1-6 алкокси);
R23c представляет собой H, C1-3 алкил или C1-3 алкокси, и каждый из алкила и алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: OH, CN, галогена, C1-4 алкокси и C1-4 гидроксиалкила;
Hal2 представляет собой F, Cl, Br или I; и предпочтительно Hal2 представляет собой Cl, Br или I;
u равно 0 или 1; и
n равно 0 или 1.
Стадия 1. Введение в реакцию соединения I-C-2 с соединением I-F-1 посредством реакции сочетания (например, реакции Сузуки) с образованием соединения I-F-2.
Условия реакции описаны на стадии 4 пути синтеза A для получения соединения формулы I-A.
Стадия 2. Введение в реакцию соединения I-F-3 с амином посредством реакции конденсации с образованием соединения I-F-4.
Условия реакции описаны в пути синтеза E для получения соединения формулы I-D.
Стадия 3. Снятие защиты с соединения I-F-4 в кислых условиях с образованием соединения I-F-5.
Условия реакции описаны на стадии 5 пути синтеза A для получения соединения формулы I-A.
Стадия 4. Введение в реакцию соединения I-F-2 с соединением I-F-5 посредством реакции нуклеофильного замещения в присутствии основания с образованием соединения I-F.
Условия реакции описаны на стадии 2 пути синтеза A для получения соединения формулы I-A.
В некоторых вариантах осуществления соединение формулы I-G может быть синтезировано с использованием способа, показанного в пути синтеза H следующим образом:
Путь синтеза H
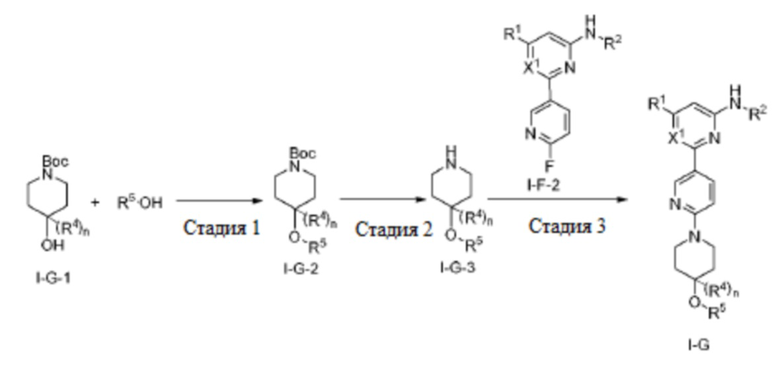
где:
R1 и R2 определены выше в пути синтеза A;
X1 выбран из CH и N;
R4 выбран из группы, состоящей из H, C1-6 алкила, C1-6 галогеналкила и C1-6 гетероалкила;
R5 определен выше в формуле I-G; и
n равно 0 или 1.
Стадия 1. Введение в реакцию соединения I-G-1 с R5-OH посредством реакции Мицунобу с образованием соединения I-G-2.
Используемые реакционные реагенты представляют собой, например, PPh3, PMe3, DIAD, DEAD или DBAD. Используемые растворители представляют собой апротонные растворители, такие как ТГФ, диэтиловый эфир, ДХМ, DMF или толуол. Температура реакции составляет от -20 °C до 100 °C, например, комнатная температура.
Стадия 2. Снятие защиты с соединения I-G-2 в кислых условиях с образованием соединения I-G-3.
Используемые кислоты представляют собой, например, раствор HCl в 1,4-диоксане, раствор HCl в EA или раствор TFA в ДХМ, или реакцию проводят в смеси вышеуказанного раствора кислоты и, например, любого одного растворителя, выбранного из ТГФ, MeOH и EtOH. Температура реакции составляет от 0°C до 80°C.
Стадия 3. Введение в реакцию соединения I-F-2 с соединением I-G-3 посредством реакции нуклеофильного замещения в присутствии основания с образованием соединения I-G.
Условия реакции описаны на стадии 2 пути синтеза A для получения соединения формулы I-A.
Фармацевтические композиции, составы и терапевтические способы
В некоторых вариантах осуществления настоящее изобретение предусматривает фармацевтическую композицию, содержащую профилактически или терапевтически эффективное количество соединения по настоящему изобретению, стереоизомера, таутомера или их смеси, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства. Необязательно фармацевтическая композиция дополнительно содержит один или более фармацевтически приемлемых носителей.
В некоторых вариантах осуществления настоящее изобретение предусматривает фармацевтический состав, который предпочтительно представляет собой твердый состав, полутвердый состав, жидкий состав или газообразный состав. В некоторых вариантах осуществления фармацевтическая композиция может дополнительно содержать одно или несколько дополнительных терапевтических средств.
В некоторых вариантах осуществления фармацевтическую композицию или фармацевтический состав предпочтительно вводят пероральным, внутривенным, внутриартериальным, подкожным, внутрибрюшинным, внутримышечным или трансдермальным путем.
В некоторых вариантах осуществления настоящее изобретение предусматривает применение соединения по настоящему изобретению, стереоизомера, таутомера или их смесь, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства, или фармацевтической композиции, как описано выше, или фармацевтического состава по настоящему изобретению в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET.
В некоторых вариантах осуществления настоящее изобретение предусматривает применение соединения по настоящему изобретению, стереоизомера, таутомера или их смесь, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита или его пролекарства, или фармацевтической композиции, как описано выше, или фармацевтического состава по настоящему изобретению в получении лекарственного средства для регулирования (например, уменьшения или ингибирования) активности RET.
В некоторых вариантах осуществления настоящее изобретение предусматривает соединение по настоящему изобретению, стереоизомер, таутомер или их смесь, его N-оксид, фармацевтически приемлемая соль, эвтектика, полиморф или его сольват, или производное со стабильным изотопом, метаболит, или его пролекарство, или фармацевтическая композиция, как описано выше, или фармацевтический состав по настоящему изобретению, которые применяются в предотвращении или лечении заболевания или состояния, связанного с активностью RET.
В некоторых вариантах осуществления настоящее изобретение предусматривает способ предотвращения или лечения заболевания или состояния, связанного с активностью RET, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по настоящему изобретению, стереоизомера, таутомера или их смеси, его N-оксида, фармацевтически приемлемой соли, эвтектики, полиморфа или его сольвата, или производного со стабильным изотопом, метаболита, или его пролекарства, или фармацевтическую композицию, как описано выше, или фармацевтический состав по настоящему изобретению.
В некоторых вариантах осуществления заболевание или состояние, связанное с активностью RET, предпочтительно представляет собой рак или опухоль, или синдром раздраженного кишечника.
В некоторых вариантах осуществления рак или опухоль, кроме того, предпочтительно представляют собой рак легких (например, немелкоклеточный рак легких), рак молочной железы, рак головы и шеи, рак прямой кишки, рак печени, лимфому, рак щитовидной железы (например, медуллярный рак щитовидной железы или папиллярный рак щитовидной железы), рак толстой кишки, множественную миелому, меланому, глиому, опухоль головного мозга или саркому.
«Фармацевтически приемлемые носители» в настоящем изобретении относятся к разбавителям, адъювантам, вспомогательным веществам или носителям, которые вводятся вместе с терапевтическим средством и, в рамках здравого медицинского заключения, подходят для контакта с тканями человека и/или других животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримых с разумным соотношением польза/риск.
Фармацевтически приемлемые носители, используемые в фармацевтической композиции по настоящему изобретению, включают, но не ограничиваются ими, стерильную жидкость. Примеры подходящих фармацевтически приемлемых носителей описаны в Remington's Pharmaceutical Sciences (1990).
Фармацевтические композиции по настоящему изобретению могут действовать системно и/или локально. С этой целью их можно вводить подходящим путем.
Для этих способов введения фармацевтическая композиция по настоящему изобретению может вводиться в подходящей лекарственной форме.
Термин «эффективное количество», как используется в данном документе, относится к количеству соединения, которое после введения в определенной степени облегчит один или несколько симптомов состояния, подлежащего лечению.
Эта схема дозирования может быть скорректирована для обеспечения оптимального желаемого ответа. Например, можно вводить один болюс, можно вводить несколько разделенных доз с течением времени, или доза может быть пропорционально уменьшена или увеличена в зависимости от необходимости терапевтической ситуации. Следует отметить, что величина дозы может варьироваться в зависимости от типа и тяжести состояния, которое необходимо облегчить, и может включать однократную или многократную дозу. Кроме того, следует понимать, что для любого конкретного человека конкретная схема дозирования должна корректироваться с течением времени в соответствии с потребностями субъекта и профессиональным суждением лица, вводящего композицию или контролирующего введение композиции.
Количество вводимого соединения по настоящему изобретению будет зависеть от субъекта, подлежащего лечению, тяжести расстройства или состояния, скорости введения, утилизации соединения и заключения лечащего врача. Обычно эффективная доза находится в диапазоне от около 0,0001 до около 50 мг на кг массы тела в день. В некоторых случаях уровни дозировки, не превышающие нижний предел вышеуказанного диапазона, могут быть соответствующими, в то время как в других случаях могут использоваться еще большие дозы, не вызывая каких-либо вредных побочных эффектов, при условии, что такие большие дозы сначала разделены на несколько небольших доз для введения в течение дня.
Содержание или количество соединения по настоящему изобретению в фармацевтической композиции может составлять от около 0,01 мг до около 1000 мг.
Если не указано иное, термин «лечение», как используется в данном документе, означает обращение, облегчение, ингибирование прогрессирования или предотвращение расстройства или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого расстройства или состояния.
«Субъект», как используется в данном документе, включает человека или животных, отличных от человека. Примеры людей включают людей-субъектов (называемых пациентами), страдающих заболеваниями (такими как заболевания, описанные в данном документе), или здоровых субъектов. В настоящем изобретении «животные, отличные от человека», включают всех позвоночных, например, не млекопитающих (таких как птицы, земноводные, рептилии) и млекопитающих, например, приматов, не относящихся к человеку, домашний скот и/или домашних животных (таких как овцы, собаки, кошки, коровы и свиньи).
В некоторых вариантах осуществления фармацевтическая композиция по настоящему изобретению может дополнительно содержать один или несколько дополнительных терапевтических средств или профилактических средств (например, дополнительные лекарственные средства для лечения рака или неопластического заболевания). В некоторых вариантах осуществления способ по настоящему изобретению может также включать введение одного или нескольких дополнительных терапевтических средств или профилактических средств (например, дополнительных лекарственных средств для лечения рака или неопластического заболевания).
Примеры
Настоящее изобретение будет дополнительно описано ниже в комбинации с примерами, но предоставление этих примеров не предназначено для ограничения объема настоящего изобретения.
Сокращения, как используется в данном документе, имеют следующие значения.
Соединение по настоящему изобретению разделяют и очищают препаративной ТСХ, колоночной хроматографией на силикагеле, препаративной ВЭЖХ и/или колоночной флеш-хроматографией, и его структуру подтверждают с помощью 1H ЯМР и/или МС. Реакцию отслеживали посредством ТСХ или ЖХ-МС.
Для спектроскопии 1H ЯМР используется сверхпроводящий спектрометр ядерного магнитного резонанса Bruker (модель AVACE III HD 400 МГц).
Квадруполь Agilent 1260 Infinity/Agilent 6120 используется для ЖХ/МС.
Силикагель GF 254 используется в качестве стационарной фазы для ТСХ.
Силикагель 200-300 меш (Qingdao Haiyang) обычно используется в качестве стационарной фазы для колоночной хроматографии.
Колоночный флеш-хроматограф Biotage используется для колоночной флеш-хроматографии.
Хроматографические инструменты Agilent 1260, Waters 2489 и GeLai 3500 используются для препаративной ВЭЖХ.
Микроволновой реактор Biotage Initiator используется для реакции в условиях микроволнового излучения.
В следующих примерах, если не указано иное, температура реакции составляет комнатную температуру (от 15 до 30°C).
Реагенты, используемые в настоящем изобретении, приобретены у таких компаний, как Acros Organics, Aldrich Chemical Company или Terbo Chemical.
Пример 1 2-(6-(6-Бензил-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 1)
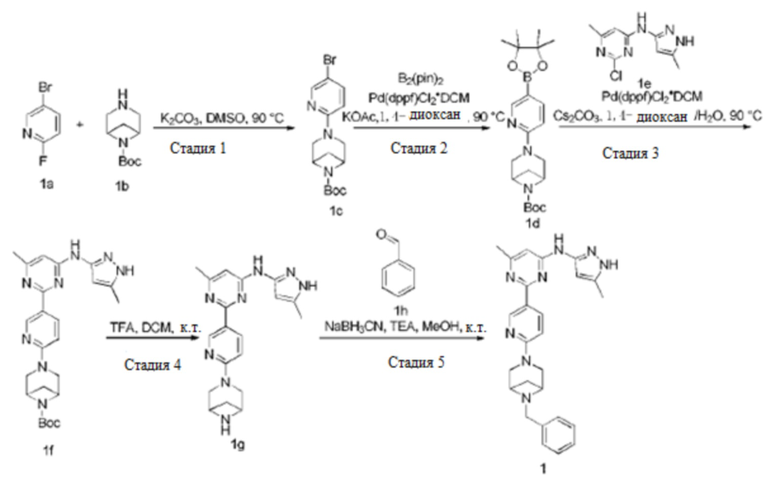
Стадия 1 Получение трет-бутил-3-(5-бромпиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилата (соединение 1c)
Соединение 1a (1,50 g) и соединение 1b (1,77 g) последовательно добавляли в одногорлую колбу объемом 100 мл, и затем ДМСО (20,0 мл) и K2CO3 (5,83 g) последовательно добавляли. Смесь нагревали до 90 °C и перемешивали в условиях защиты азотом при этой же температуре в течение 20 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью EA. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=5:1) с получением соединения 1c (2,03 г). МС m/z (ИЭР): 354,1 [M+H]+.
Стадия 2 Получение трет-бутил-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилата (соединение 1d)
Соединение 1c (2,03 g), B2(pin)2 (4,01 г), KOAc (1,55 г), 1,4-диоксан (15,0 мл) и Pd(dppf)Cl2·ДХМ (644,67 мг) последовательно добавляли в одногорлую колбу объемом 100 мл и нагревали до 90 °C для реакции в условиях защиты азотом. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (40 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=15:1) с получением соединения 1d (2,11 г). МС m/z (ИЭР): 402,3 [M+H]+.
Стадия 3 Получение трет-бутил-3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-карбоксилата (соединение 1f)
Соединение 1e (950 мг) растворяли в 1,4-диоксане (50,0 мл), соединение 1d (2,11 г), Cs2CO3 (3,15 г) и воду (5,0 мл) последовательно добавляли, и затем Pd(dppf)Cl2·ДХМ (477,83 мг) добавляли. Смесь нагревали до 90°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 14 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью EA (60 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, и затем концентрировали досуха при пониженном давлении с получением соединения 1f (587,0 мг). МС m/z (ИЭР): 463,3 [M+H]+.
Стадия 4 Получение 2-(6-(3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 1g)
Соединение 1f (1,36 г) растворяли в ДХМ (20,0 мл), TFA (20,0 мл) затем добавляли, и смесь удерживали для реакции в условиях защиты азотом при комнатной температуре. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением трифторацетата соединения 1g (587,0 мг). МС m/z (ИЭР): 363,3 [M+H]+.
Стадия 5 Получение 2-(6-(6-бензил-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 1)
Трифторацетат соединения 1g (31,58 мг) и соединения 1h (27,74 мг) растворяли в MeOH (0,5 мл), TEA (8,46 мг) и цианоборогидрид натрия (26,27 мг) последовательно добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 1 (11,0 мг). МС m/z (ИЭР): 453,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,15 (уш., 1H), 9,67 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,44 (дд, J = 8,8, 2,4 Гц, 1H),7,43-7,21 (м, 5H), 6,78 (д, J = 8,8 Гц, 2H), 6,30 (уш., 1H), 3,98-3,57 (м, 8H), 2,72-2,61 (м, 1H), 2,33 (с, 3H), 2,24 (с, 3H), 1,72-1,64 (м, 1H).
Пример 2 2-(6-(6-(4-Метоксибензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 2)
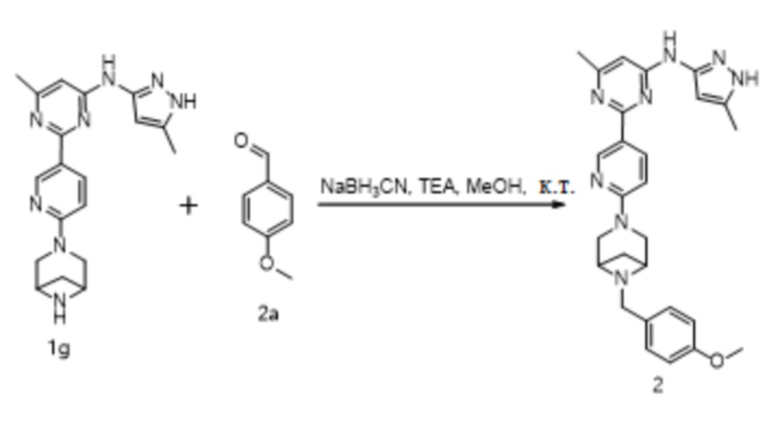
Трифторацетат соединения 1g (30,0 мг) и соединения 2a (33,81 мг) растворяли в MeOH (0,5 мл), TEA (8,12 мг) и цианоборогидрид натрия (25,21 мг) последовательно добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 2 (15,0 мг). МС m/z (ИЭР): 483,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,14 (уш., 1H), 9,66 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 7,26 (д, J = 8,8 Гц, 2H), 6,88-6,76 (м, 4H), 6,30 (уш., 1H), 3,79-3,72 (м, 7H), 3,58-3,53 (м, 4H), 2,59-2,55 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 3 2-(6-(6-((6-Хлорпиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 3)
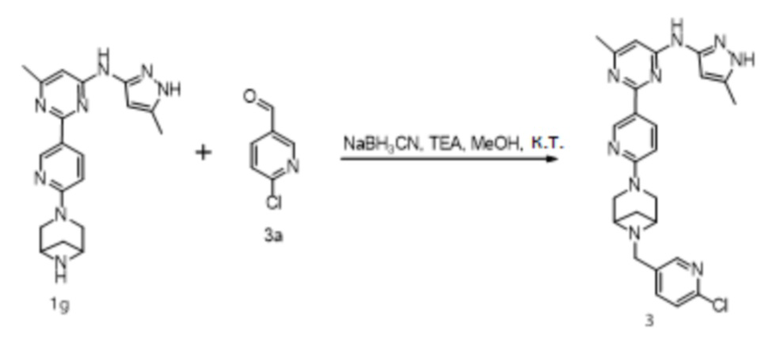
Трифторацетат соединения 1g (30,0 мг) и соединения 3a (35,15 мг) растворяли в MeOH (0,5 мл), TEA (8,12 мг) и цианоборогидрид натрия (25,21 мг) последовательно добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 3 (20,0 мг). МС m/z (ИЭР): 488,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,12 (уш., 1H), 9,67 (с, 1H), 9,11 (д, J= 2,0 Гц, 1H), 8,46-8,42 (м, 2H), 7,87-7,80 (м, 1H), 7,50-7,47 (м, 1H), 6,80-6,77 (м, 2H), 6,31 (уш., 1H), 4,01-3,52 (м, 8H), 2,66-2,57 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,69-1,62 (м, 1H).
Пример 4 2-(6-(6-((6-Метоксипиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 4)
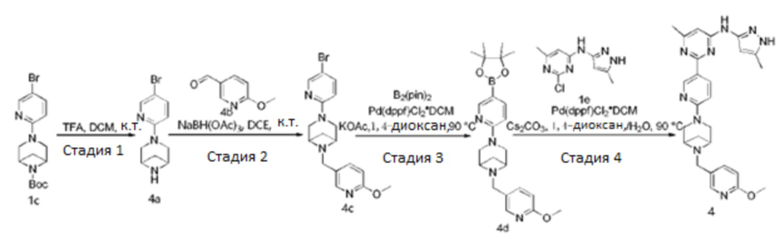
Стадия 1 Получение 3-(5-бромпиридин-2-ил)-3,6-диазабицикло[3.1.1]гептана (соединение 4a)
Соединение 1c (460,0 мг) растворяли в ДХМ (5,0 мл) в условиях защиты азотом, TFA (5,0 мл) добавляли и смесь выдерживали для реакции при комнатной температуре в течение 2 ч. до полного превращения исходных материалов. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, промывали насыщенным раствором карбоната натрия, экстрагировали с помощью ДХМ (30 мл x 3), сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 4a (250,0 мг). МС m/z (ИЭР): 254,0 [M+H]+.
Стадия 2 Получение 3-(5-бромпиридин-2-ил)-6-((6-метоксипиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептана (соединение 4c)
Соединение 4a (250,0 мг) и соединение 4b (275,0 мг) растворяли в DCE (5,0 мл), NaBH(OAc)3 (1,06 г) добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 10 ч. После завершения реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью EA (50 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=10:1-1:3) с получением соединения 4c (320,0 мг). МС m/z (ИЭР): 375,1 [M+H]+.
Стадия 3 Получение 6-((6-метоксипиридин-3-ил)метил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептана (соединение 4d)
Соединение 4c (332,0 мг) растворяли в 1,4-диоксана (5,0 мл), B2(pin)2 (619,76 мг) и KOAc (237,11 мг) последовательно добавляли, и Pd(dppf)Cl2·ДХМ (99,65 мг) добавляли в условиях защиты азотом. Смесь нагревали до 90 °C и удерживали для реакции при этой же температуре в течение 4 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью EA (60 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=10:1) с получением соединения 4d (320,0 мг). МС m/z (ИЭР): 423,3 [M+H]+.
Стадия 4 Получение 2-(6-(6-((6-метоксипиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 4)
Соединение 4d (129,8 мг) растворяли в 1,4-диоксане (5,0 мл), соединение 1e (55 мг), Cs2CO3 (149 мг) и воду (5,0 мл) последовательно добавляли, и Pd(dppf)Cl2·ДХМ (27,93 мг) добавляли в условиях защиты азотом. Смесь нагревали до 90°C и удерживали для реакции при этой же температуре в течение 5 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью EA (60 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 4 (18,0 мг). МС m/z (ИЭР): 484,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 8,07 (д, J = 2,0 Гц, 1H), 7,68 (дд, J = 8,4, 2,4 Гц, 1H), 6,78-6,75 (м, 3H), 6,30 (уш., 1H), 3,82 (с, 3H), 3,74 (д, J = 11,6 Гц, 2H), 3,66 (д, J = 6,0 Гц, 2H), 3,61-3,45 (м, 4H), 2,53-2,51 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,57 (д, J = 8,0 Гц, 1H).
Пример 5 2-(6-(6-((5-Метоксипиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 18)
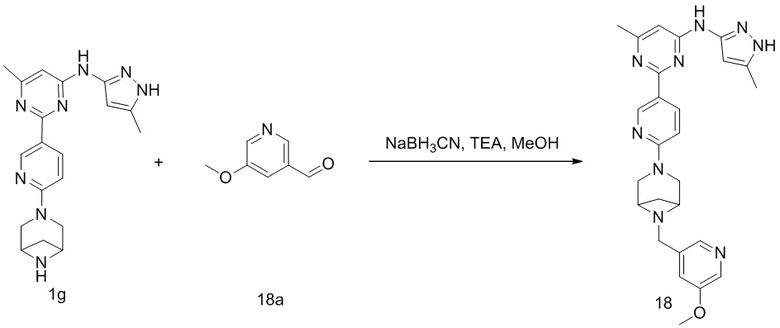
Трифторацетат соединения 1g (35,00 мг) и соединения 18a (27,75 мг) добавляли в метанол (1,0 мл), и затем триэтиламин (6,83 мг) и цианоборогидрид натрия (17,00 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 18 (6,0 мг). МС m/z (ИЭР): 484,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,13 (уш., 1H), 9,65 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,43 (дд, J = 8,92, 2,32 Гц, 1H), 8,21 (с, 1H), 8,16-8,12 (м, 2H), 7,35-7,31 (м, 1H), 6,77 (д, J = 9,0 Гц, 1H), 6,31 (уш., 1H), 3,81 (с, 3H), 3,78-3,69 (м, 4H), 3,59 (уш., 4H), 2,59-2,52 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,36 Гц, 1H).
Пример 6 2-(6-(6-((6-(4-Фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 17)
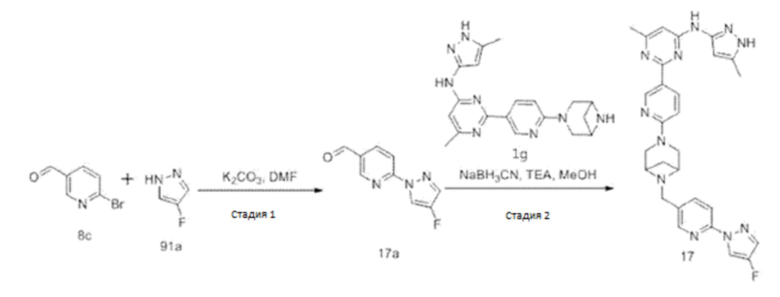
Стадия 1 Получение 6-(4-фтор-1H-пиразол-1-ил)никотинальдегида (соединение 17a)
Соединение 8c (2,0 г), гидрохлорид соединения 91a (1,58 г) и карбонат калия (4,45 г) последовательно добавляли в DMF (15 мл), нагревали до 80°C и перемешивали при этой же температуре в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью ДХМ (50 мл×2). Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=10:1) с получением соединения 17a (0,81 г). МС m/z (ИЭР): 192,1 [M+H]+.
Стадия 2 Получение 2-(6-(6-((6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 17)
Трифторацетат соединения 1g (22,82 мг) и соединение 17a (27,47 мг) добавляли в метанол (1,0 мл), и затем триэтиламин (4,45 мг) и цианоборогидрид натрия (13,86 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 14 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 17 (7,0 мг). МС m/z (ИЭР): 538,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,16 Гц, 1H), 8,67 (дд, J = 4,54, 0,64 Гц, 1H), 8,43 (дд, J = 8,94, 2,28 Гц, 1H), 8,41 (д, J = 1,68, 1H), 7,98 (дд, J = 8,48 Гц, 2,12 1H), 7,92 (д, J = 4,28, 1H), 7,87 (д, J = 8,4, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,31 (уш., 1H), 3,78-3,71 (м, 4H), 3,68-3,52 (м, 4H), 2,59-2,52 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,60 (д, J= 8,36 Гц, 1H).
Пример 7 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-метилпиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 16)
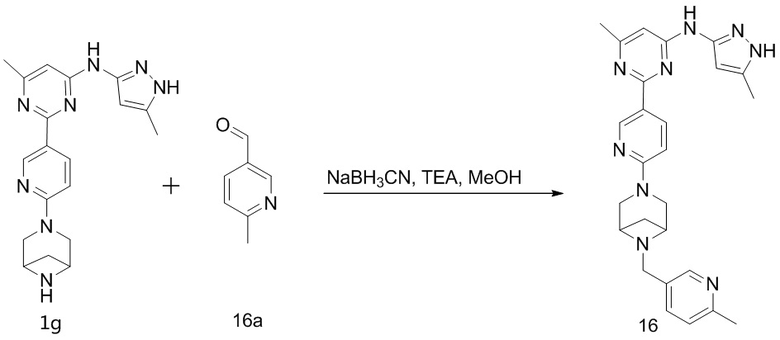
Трифторацетат соединения 1g (35,0 мг) и соединение 16a (35,1 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,5 мг) и цианоборогидрид натрия (29,4 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 16 (15,0 мг). МС m/z (ИЭР): 468,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 8,38 (с, 1H), 7,63 (дд, J = 7,6, 1,6 Гц, 1H), 7,18 (д, J = 8,0 Гц, 1H), 6,77 (д, J = 8,8 Гц, 2H), 6,29 (уш., 1H), 3,83-3,64 (м, 4H), 3,63-3,45 (м, 4H), 2,57-2,52 (м, 1H), 2,43 (с, 3H), 2,33 (с, 3H), 2,26 (с, 3H), 1,58 (д, J = 8,0 Гц, 1H).
Пример 8 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((2-метилтиазол-5-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 15)
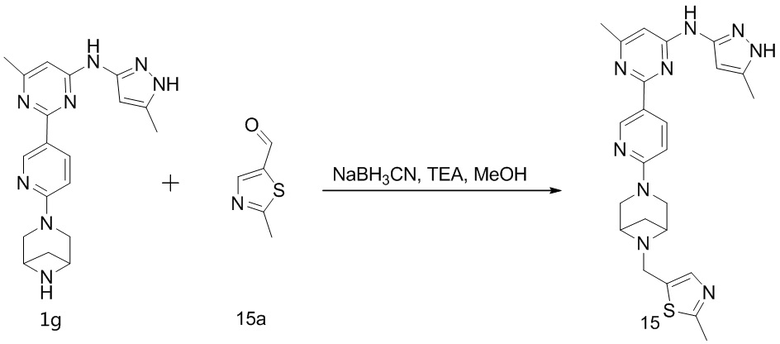
Трифторацетат соединения 1g (35,0 мг) и соединение 15a (36,8 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,5 мг) и цианоборогидрид натрия (29,4 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 15 (16,0 мг). МС m/z (ИЭР): 474,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,19 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,0 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 8,17 (с, 1H), 7,46 (с, 1H), 6,76 (д, J = 8,8 Гц, 1H), 6,29 (уш., 1H), 3,78-3,66 (м, 6H), 3,64-3,51 (м, 2H), 2,59 (с, 3H), 2,49-2,44 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,57 (д, J = 8,4 Гц, 1H).
Пример 9 2-(6-(4-((6-Метоксипиридин-3-ил)метил)пиперазин-1-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 49)
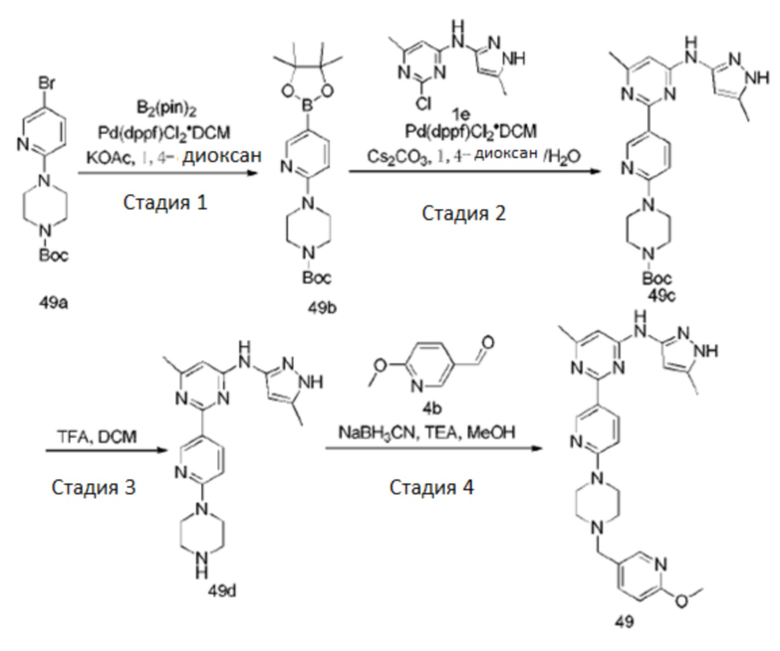
Стадия 1 Получение трет-бутил-4-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)-пиперазин-1-карбоксилата (соединение 49b)
Соединение 49a (2,00 г), B2(pin)2 (4,09 г), KOAc (1,58 г), 1,4-диоксан (15,0 мл) и Pd(dppf)Cl2·ДХМ (657,43 мг) последовательно добавляли в реакционную колбу. Смесь нагревали до 90°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 3 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (40 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=15:1) с получением соединения 49b (2,77 г). МС m/z (ИЭР): 390,3 [M+H]+.
Стадия 2 Получение трет-бутил-4-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)пиперазин-1-карбоксилата (соединение 49c)
Соединение 1e (1,30 г) растворяли в 1,4-диоксане (50,0 мл), соединение 49b (2,80 г), Cs2CO3 (3,44 г) и воду (2,0 мл) последовательно добавляли, и затем Pd(dppf)Cl2·ДХМ (647,3 мг) добавляли. Смесь нагревали до 90°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 4 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали с помощью EA (60 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, и затем концентрировали досуха при пониженном давлении с получением соединения 49c (587,0 мг). МС m/z (ИЭР): 451,3 [M+H]+.
Стадия 3 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(пиперазин-1-ил)пиридин-3-ил)пиримидин-4-амина (соединение 49d)
Соединение 49c (2,7 г) растворяли в ДХМ (20,0 мл), и затем TFA (20,0 мл) добавляли. Смесь выдерживали для реакции в условиях защиты азотом при комнатной температуре в течение 4 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением трифторацетата соединения 49d (761,0 мг). МС m/z (ИЭР): 351,2 [M+H]+.
Стадия 4 Получение 2-(6-(4-((6-метоксипиридин-3-ил)метил)пиперазин-1-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 49)
Трифторацетат соединение 49d (35,0 мг) и соединение 4b (38,9 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,3 мг) и цианоборогидрид натрия (28,8 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 49 (25,0 мг). МС m/z (ИЭР): 472,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,20 (уш., 1H), 9,65 (с, 1H), 9,03 (д, J = 2,4 Гц, 1H), 8,36 (дд, J = 8,8, 2,0 Гц, 1H), 8,09 (д, J = 2,0 Гц, 1H), 7,68 (дд, J = 8,4, 2,4 Гц, 1H), 6,94-6,78 (м, 3H), 6,29 (уш., 1H), 3,84 (с, 3H), 3,64-3,56 (м, 4H), 3,49 (с, 2H), 2,49-2,44 (м, 4H), 2,31 (с, 3H), 2,24 (с, 3H).
Пример 10 2-(6-(4-(4-Метоксибензил)пиперазин-1-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 23)
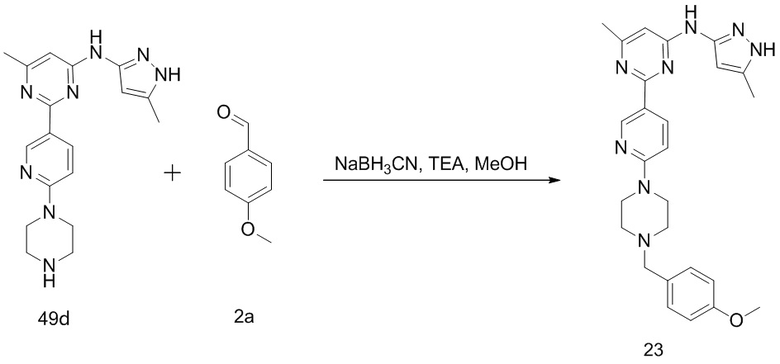
Трифторацетат соединение 49d (35,0 мг) и соединение 2a (38,9 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,3 мг) и цианоборогидрид натрия (28,8 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 23 (18,0 мг). МС m/z (ИЭР): 471,3 [M+H]+.
1H ЯМР (CD3OD, 400 МГц) δ 9,07 (д, J= 2,4 Гц, 1H), 8,45 (дд, J = 8,8, 2,4 Гц, 1H), 8,28 (с, 1H), 7,42-7,35 (м, 2H), 7,02-6,92 (м, 3H), 6,76 (с, 1H), 6,26 (с, 1H), 3,99 (с, 2H), 3,82 (с, 7H), 3,08-3,00 (м, 4H), 2,40 (с, 3H), 2,31 (с, 3H).
Пример 11 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((4-метилпиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 21)
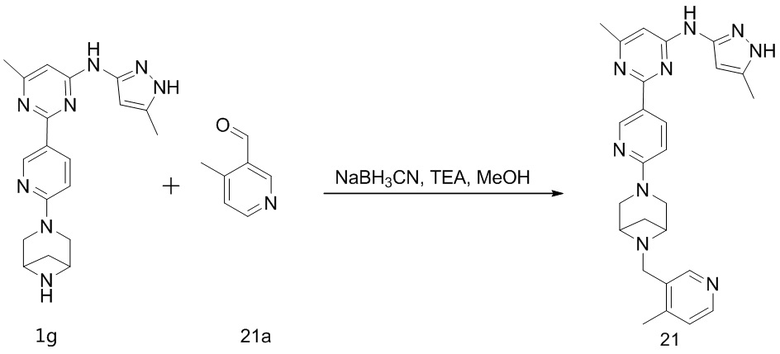
Трифторацетат соединения 1g (35,0 мг) и соединение 21a (27,52 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,08 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 21 (10,0 мг). МС m/z (ИЭР): 468,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,93 (с, 1H), 9,61 (с, 1H), 9,07 (д, J = 1,2 Гц, 1H), 8,39 (дд, J = 8,8, 2,2 Гц, 1H), 8,35 (с, 1H), 8,25 (д, J = 4,8 Гц, 1H), 7,11 (д, J = 4,8 Гц, 1H), 7,01-6,63 (м, 2H), 6,27 (уш., 1H), 3,82-3,69 (м, 2H), 3,63 (д, J = 6,4 Гц, 2H), 3,59-3,42 (м, 4H), 2,52-2,46 (м, 1H), 2,28 (с, 3H), 2,25 (с, 3H), 2,21 (с, 3H), 1,53 (д, J = 8,4 Гц, 1H).
Пример 12 2-(6-(6-(2-Фтор-5-метоксибензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 96)
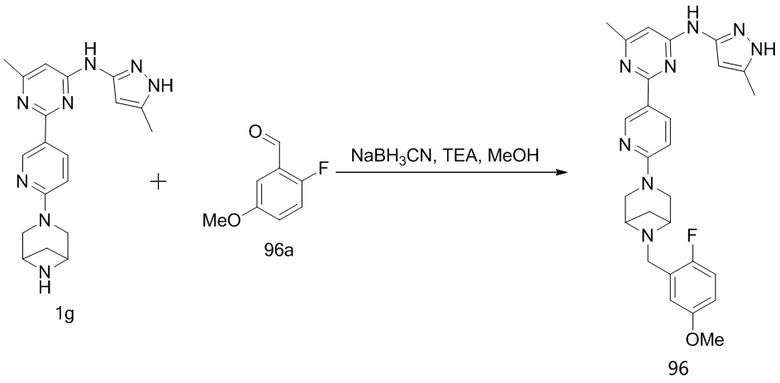
Трифторацетат соединения 1g (30,0 мг) и соединения 96a (19,41 мг) растворяли в MeOH (0,5 мл), TEA (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 96 (10,0 мг). МС m/z (ИЭР): 501,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,0 Гц, 1H), 8,43 (дд, J = 9,2, 2,4 Гц, 1H), 7,06 (т, J = 9,2 Гц, 1H), 7,03-7,00 (м, 1H), 6,83-6,78 (м, 1H), 7,03-6,64 (м, 2H), 6,32 (уш., 1H), 3,77-3,71 (м, 7H), 3,58-3,53 (м, 4H), 2,58-2,53 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,58 (д, J = 8,4 Гц, 1H).
Пример 13 (6-Метоксипиридин-3-ил)(4-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)амино)пиримидин-2-ил)пиридин-2-ил)пиперазин-1-ил)метанон (соединение 110)
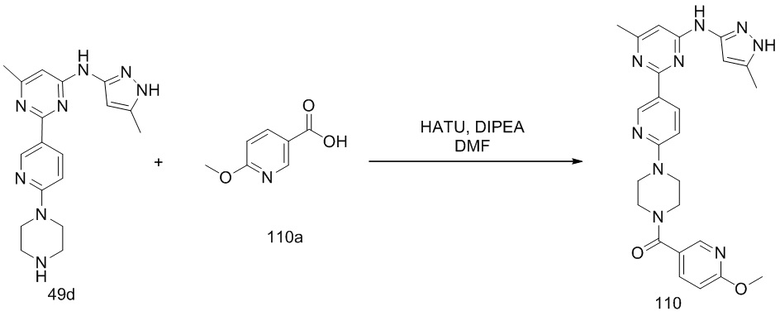
Соединение 110a (16,5 мг), HATU (53,2 мг) и DIPEA (41,7 мг) добавляли в DMF (3,0 мл), и смесь выдерживали для реакции при комнатной температуре в течение 5 мин. Затем трифторацетат соединения 49d (50,0 мг) добавляли, и смесь вводили в реакцию при комнатной температуре в течение 0,5 ч. После завершения реакции реакционную смесь разбавляли с помощью EA, промывали насыщенным раствором хлорида натрия 3 раза, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 110 (19,0 мг). МС m/z (ИЭР): 486,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,01 (уш., 1H), 9,69 (с, 1H), 9,08 (д, J = 2,2 Гц, 1H), 8,41 (дд, J = 9,0, 2,3 Гц, 1H), 8,34 (д, J = 2,1 Гц, 1H), 7,84 (дд, J = 8,5, 2,3 Гц, 1H), 7,12-6,63 (м, 3H), 6,30 (с, 1H), 3,92 (с, 3H), 3,72 (с, 8H), 2,34 (с, 3H), 2,26 (с, 3H).
Пример 14 2-(6-Метоксипиридин-3-ил)-1-(4-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)амино)пиримидин-2-ил)пиридин-2-ил)пиперазин-1-ил)этан-1-он (соединение 111)

Соединение 111a (18,0 мг), HATU (53,2 мг) и DIPEA (41,7 мг) добавляли в DMF (3,0 мл), и смесь выдерживали для реакции при комнатной температуре в течение 5 мин. Затем трифторацетат соединения 49d (50,0 мг) добавляли, и смесь вводили в реакцию при комнатной температуре в течение 0,5 ч. После завершения реакции реакционную смесь разбавляли с помощью EA, промывали насыщенным раствором хлорида натрия 3 раза, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 111 (6,0 мг). МС m/z (ИЭР): 500,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,02 (уш., 1H), 9,68 (с, 1H), 9,06 (д, J = 2,3 Гц, 1H), 8,39 (дд, J = 9,0, 2,3 Гц, 1H), 8,02 (д, J = 2,2 Гц, 1H), 7,56 (дд, J = 8,5, 2,4 Гц, 1H), 6,94 (д, J = 9,0 Гц, 1H), 6,90-6,69 (м, 2H), 6,27 (с, 1H), 3,83 (с, 3H), 3,74 (с, 2H), 3,70-3,59 (м, 8H), 2,32 (с, 3H), 2,25 (с, 3H).
Пример 15 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-(4-метилбензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 80)
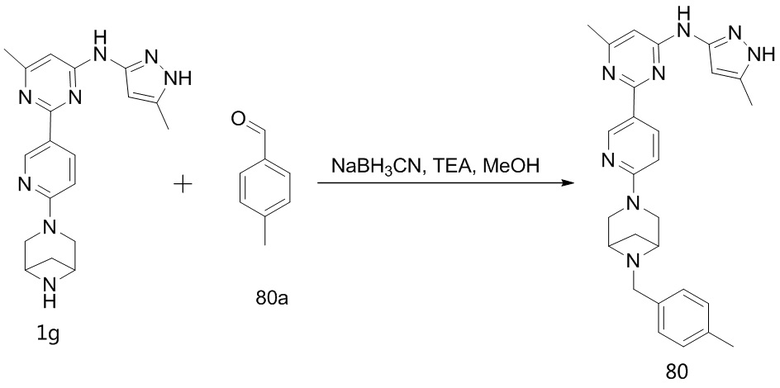
Трифторацетат соединения 1g (30 мг) и соединение 80a (22,70 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 80 (10,0 мг). МС m/z (ИЭР): 467,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,16 (с, 1H), 9,66 (с, 1H), 9,11 (д, J = 2,2 Гц, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 7,22 (д, J = 7,9 Гц, 2H), 7,11 (д, J = 7,9 Гц, 2H), 6,76 (д, J = 9,0 Гц, 2H), 6,31 (уш., 1H), 3,78-3,65 (м, 4H), 3,64-3,49 (м, 4H), 2,60-2,49 (м, 1H), 2,33 (с, 3H), 2,27 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 16 5-((3-(5-(4-Метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)-2-цианопиридин (соединение 117)
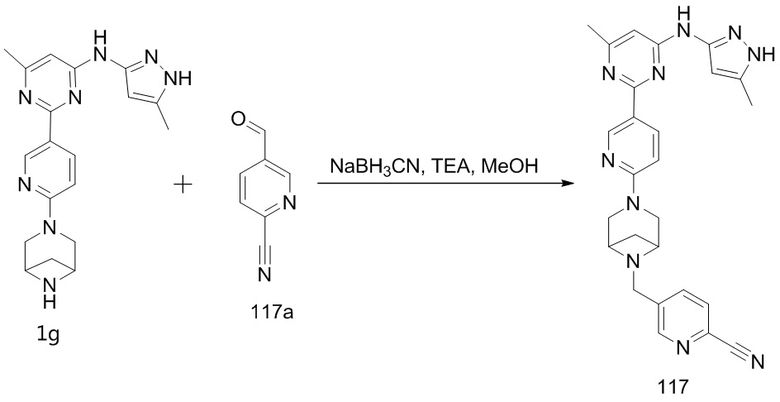
Трифторацетат соединения 1g (30 мг) и соединение 117a (24,96 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 117 (2,0 мг). МС m/z (ИЭР): 479,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,11 (д, J = 2,2 Гц, 1H), 8,74 (д, J = 1,2 Гц, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 8,01 (дт, J = 17,2, 5,0 Гц, 2H), 6,77 (д, J = 9,0 Гц, 2H), 6,32 (уш., 1H), 3,81-3,66 (м, 6H), 3,65-3,52 (м, 2H), 2,61-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 17 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(трифторметил) пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 118)
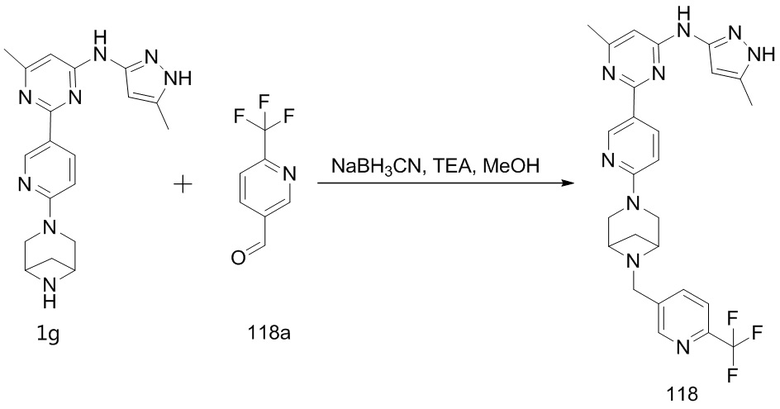
Трифторацетат соединения 1g (30 мг) и соединение 118a (43,48 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 118 (6,0 мг). МС m/z (ИЭР): 522,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,74 (с, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 8,06 (д, J = 7,1 Гц, 1H), 7,84 (д, J = 8,1 Гц, 1H), 6,77 (д, J = 9,0 Гц, 2H), 6,30 (уш., 1H), 3,84-3,65( m, 6H), 3,60-3,48 (м, 2H), 2,61-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,61 (д, J = 8,4 Гц, 1H).
Пример 18 6-Метил-2-(6-(6-((6-(4-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 62)
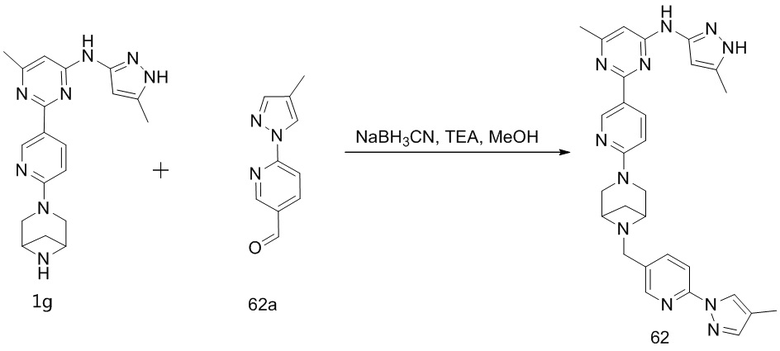
Трифторацетат соединения 1g (30 мг) и соединение 62a (46,49 мг, получено посредством ссылки на способ получения соединения 17a в примере 6, за исключением того, что 4-фторпиразол был заменен 4-метилпиразолом) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 62 (6,0 мг). МС m/z (ИЭР): 534,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,37 (м, 2H), 7,93 (дд, J = 8,5, 2,2 Гц, 1H), 7,82 (д, J = 8,4 Гц, 1H), 7,62 (с, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,30 (уш., 1H), 3,83-3,67 (м, 4H), 3,66-3,50 (м, 4H), 2,60-2,52 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 2,11 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 19 2-(6-(6-((6-(1H-Пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло [3.1.1] гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 60)
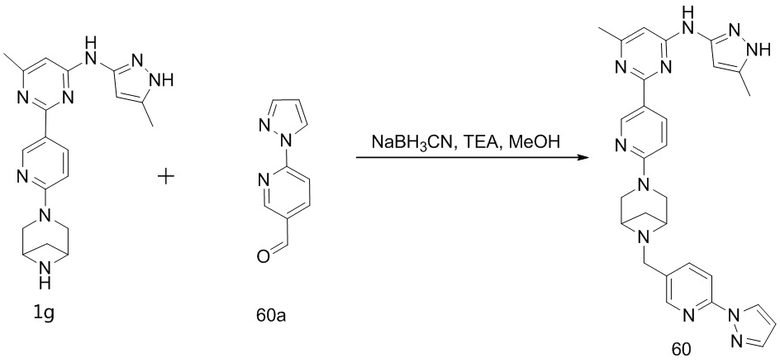
Трифторацетат соединения 1g (30 мг) и соединение 60a (43,00 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, и отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 60 (11,0 мг). МС m/z (ИЭР): 520,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,65 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,59 (дд, J = 2,6, 0,5 Гц, 1H), 8,46-8,42 (дд, J = 8,8, 2,4 Гц, 1H), 8,41-8,39 (д, J = 2,0 Гц, 1H), 7,97 (дд, J = 8,4, 2,2 Гц, 1H), 7,88 (д, J = 8,4 Гц, 1H), 7,81 (д, J = 1,0 Гц, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,56 (дд, J = 2,5, 1,7 Гц, 1H), 6,29 (уш., 1H), 3,82-3,68 (м, 4H), 3,67-3,52 (м, 4H), 2,59-2,52 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 20 2-(6-(6-((5-Фторпиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 98)
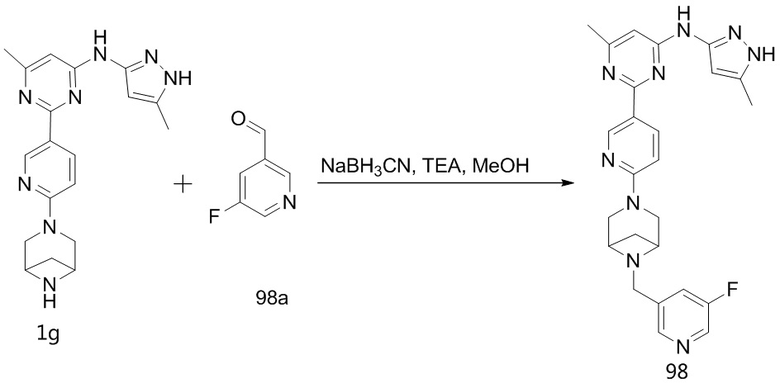
Трифторацетат соединения 1g (35,0 мг) и соединение 98a (36,24 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,77 мг) и цианоборогидрид натрия (30,34 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 98 (15,0 мг). МС m/z (ИЭР): 472,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (уш., 1H), 9,66 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,45-8,41 (м, 3H), 7,73-7,65 (м, 1H), 6,77 (д, J = 9,0 Гц, 2H), 6,29 (уш., 1H), 3,74 (т, J = 8,7 Гц, 4H), 3,64 (с, 2H), 3,58 (д, J = 6,5 Гц, 2H), 2,55-2,51 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J= 8,4 Гц, 1H).
Пример 21 2-(6-(6-((5-Хлорпиридин-2-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 69)
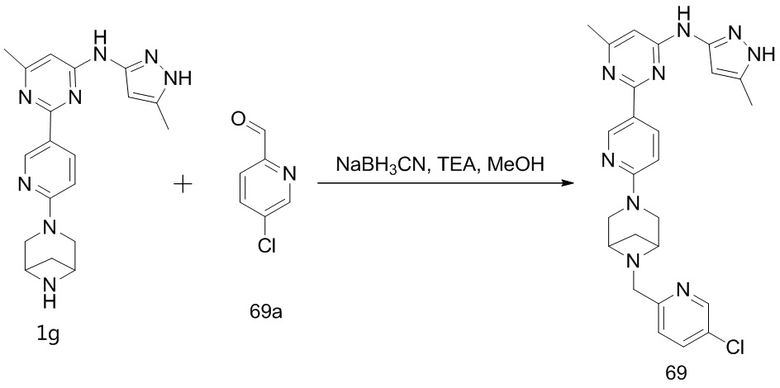
Трифторацетат соединения 1g (35,0 мг) и соединение 69a (41,0 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,77 мг) и цианоборогидрид натрия (30,34 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 69 (16,0 мг). МС m/z (ИЭР): 488,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,01 (уш., 1H), 9,66 (с, 1H), 9,11 (д, J = 2,2 Гц, 1H), 8,50 (д, J = 2,3 Гц, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 7,89 (дд, J = 8,4, 2,5 Гц, 1H), 7,52 (д, J = 8,4 Гц, 1H), 6,76 (д, J = 9,0 Гц, 2H), 6,30 (с, 1H), 3,79 (д, J = 11,7 Гц, 2H), 3,73 (д, J = 5,9 Гц, 2H), 3,65 (с, 2H), 3,57 (д, J= 10,0 Гц, 3H), 2,55-2,51 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 22 2-(6-(6-((5-Метоксипиридин-2-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 67)
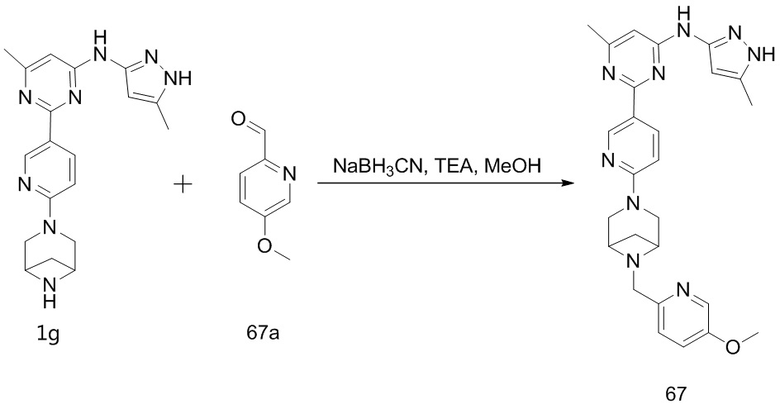
Трифторацетат соединения 1g (30 мг) и соединение 67a (25,90 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 67 (10,0 мг). МС m/z (ИЭР): 484,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6)) δ 11,97 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,1 Гц, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 8,16 (дд, J = 2,8, 0,6 Гц, 1H), 7,39 (д, J = 8,3 Гц, 1H), 7,35 (дд, J = 8,6, 2,9 Гц, 1H), 6,76 (д, J = 9,0 Гц, 2H), 6,30 (с, 1H), 3,85-3,77 (м, 5H), 3,69 (д, J = 5,9 Гц, 2H), 3,60-3,47 (м, 4H), 2,55-2,51 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,58 (д, J= 8,4 Гц, 1H).
Пример 23 2-(6-(6-(4-Этоксибензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 41)

Трифторацетат соединения 1g (30 мг) и соединение 41a (28,37 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 41 (15,0 мг). МС m/z (ИЭР): 497,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 7,22 (д, J = 8,8 Гц, 2H), 6,88-6,76 (м, 4H), 6,32 (уш., 1H), 3,98 (к, J = 6,9 Гц, 2H), 3,68 (дд, J = 30,7, 8,6 Гц, 4H), 3,51 (д, J = 30,4 Гц, 4H), 2,59-2,55 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,56 (д, J = 8,3 Гц, 1H), 1,31 (т, J = 7,0 Гц, 3H).
Пример 24 2-(6-(6-(1-(4-Метоксифенил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 50)
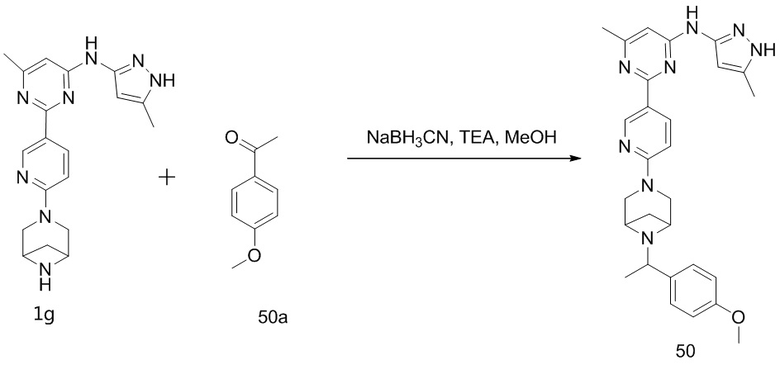
Трифторацетат соединения 1g (30 мг) и соединение 50a (28,37 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 50 (15,0 мг). МС m/z (ИЭР): 497,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,63 (с, 1H), 9,11 (д, J = 2,1 Гц, 1H), 8,42 (дд, J = 8,9, 2,2 Гц, 1H), 7,25 (д, J = 8,6 Гц, 2H), 7,02-6,57 (м, 4H), 6,30 (уш., 1H), 3,97-3,77 (м, 2H), 3,72 (с, 3H), 3,67-3,50 (м, 2H), 3,50-3,36 (м, 2H), 2,49-2,39 (м, 2H), 2,33 (с, 3H), 2,26 (с, 3H), 1,51 (д, J = 8,3 Гц, 1H), 1,12 (д, J = 6,2 Гц, 3H).
Пример 25 2-(6-(6-(4-Фторбензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 83)
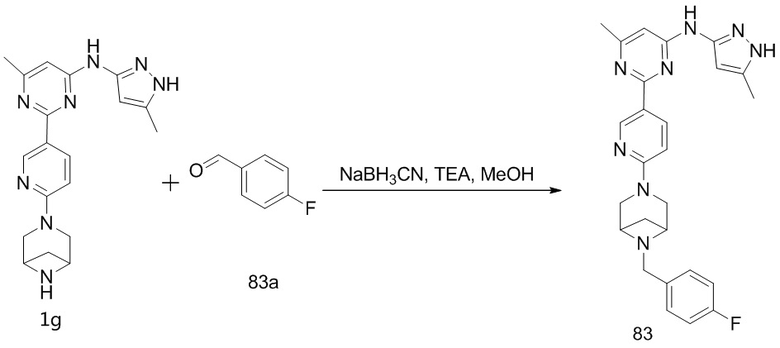
Трифторацетат соединения 1g (35,0 мг) и соединение 83a (18,2 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 83 (26,0 мг). МС m/z (ИЭР): 471,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,64 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,42 (дд, J = 9,2, 2,4 Гц, 1H), 7,93-7,35 (м, 2H), 7,13-7,09 (м, 2H), 6,83 (уш., 1H), 6,76 (д, J = 9,2 Гц, 1H), 6,30 (уш., 1H), 3,74-3,66 (м, 4H), 3,62-3,53 (м, 4H), 2,55-2,52 (м, 1H), 2,32 (с, 3H), 2,25 (с, 3H), 1,57 (д, J = 8,4 Гц, 1H).
Пример 26 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-(4-(трифторметил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 84)
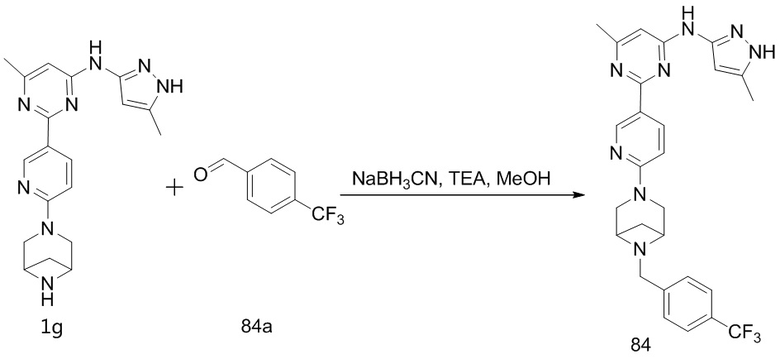
Трифторацетат соединения 1g (35,0 мг) и соединение 84a (25,6 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 84 (17,0 мг). МС m/z (ИЭР): 521,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,90 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,42 (дд, J = 8,8, 2,4 Гц, 1H), 7,66 (д, J = 8,0, 2H), 7,58 (д, J = 8,4, 2H), 6,94 (уш., 1H), 6,76 (д, J = 9,2 Гц, 1H), 6,30 (уш., 1H), 3,74-3,57 (м, 8H), 2,57-2,55 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 27 4-((3-(5-(4-Метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)бензонитрил (соединение 85)
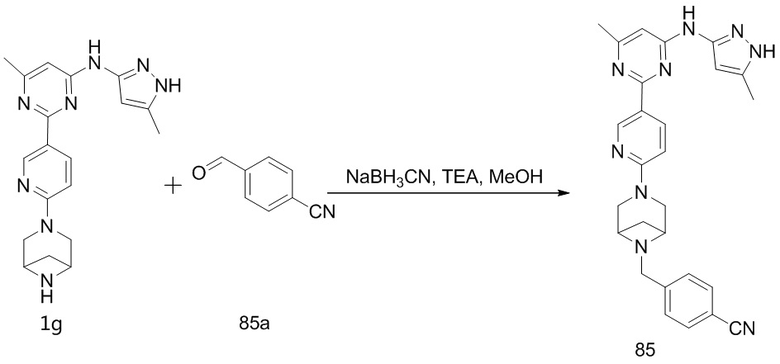
Трифторацетат соединения 1g (35,0 мг) и соединение 85a (19,3 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 85 (12,0 мг). МС m/z (ИЭР): 478,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,08 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,42 (дд, J = 9,2, 2,4 Гц, 1H), 7,77 (д, J = 8,4, 2H), 7,56 (д, J = 8,0, 2H), 6,89 (уш., 1H), 6,76 (д, J = 8,8 Гц, 1H), 6,30 (уш., 1H), 3,74-3,56 (м, 8H), 2,59-2,58 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,61 (д, J = 8,4 Гц, 1H).
Пример 28 2-(6-(6-(4-(1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил) пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 86)
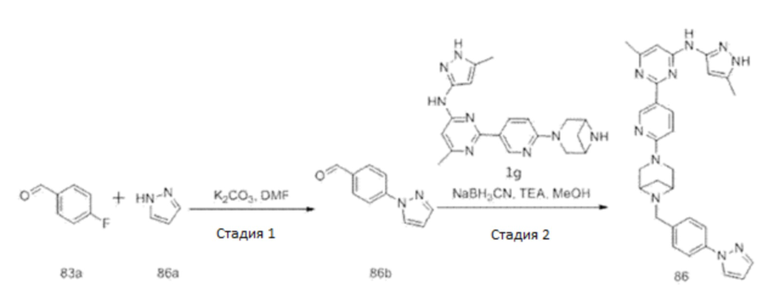
Стадия 1 Получение 4-(1H-пиразол-1-ил)бензальдегида (соединение 86b)
Соединение 83a (1,0 г) и соединение 86a (0,8 г) растворяли в DMF (10,0 мл) и безводный карбонат калия (2,2 г) добавляли. Смесь нагревали до 120°C и удерживали для реакции при этой же температуре в течение 16 ч. до полного превращения исходных материалов. После завершения реакции реакционную смесь промывали насыщенным раствором карбоната натрия, экстрагировали с помощью EA (30 мл x 3), сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 86b (1,1 г). МС m/z (ИЭР): 173,1 [M+H]+.
Стадия 2 Получение 2-(6-(6-(4-(1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 86)
Трифторацетат соединения 1g (35,0 мг) и соединение 86b (25,3 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 86 (12,0 мг). МС m/z (ИЭР): 519,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,46-8,42(м, 2H), 7,77-7,72 (м, 3H), 7,45 (д, J = 8,8 Гц, 2H), 6,89 (уш., 1H), 6,77 (д, J = 9,2 Гц, 1H), 6,52 (т, J = 2,0 Гц, 1H), 6,30 (уш., 1H), 3,77-3,59 (м, 8H), 2,56-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 29 2-(6-(6-(4-Хлорбензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 82)
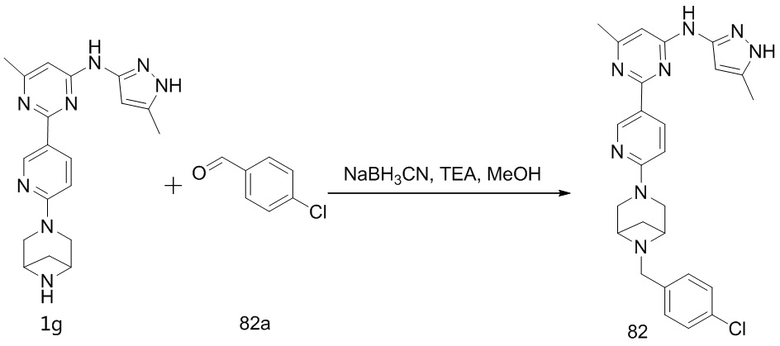
Трифторацетат соединения 1g (35,0 мг) и соединение 82a (20,6 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 82 (26,0 мг). МС m/z (ИЭР): 487,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,21 (уш., 1H), 9,65 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 7,41-7,36 (м, 4H), 6,86 (уш., 1H), 6,76 (д, J = 9,2 Гц, 1H), 6,30 (уш., 1H), 3,75-3,63 (м, 8H), 2,60-2,56 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,63 (д, J = 8,4 Гц, 1H).
Пример 30 2-(6-(6-(4-(4-Фтор-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 91)
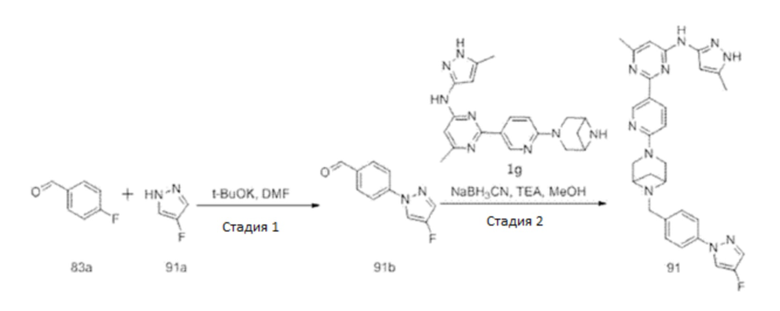
Стадия 1 Получение 4-(4-фтор-1H-пиразол-1-ил)бензальдегида (соединение 91b)
Соединение 83a (0,1 г) и соединение 91a (0,1 г) растворяли в DMF (5,0 мл) и трет-бутоксид калия (0,3 г) добавляли. Смесь нагревали до 120°C и удерживали для реакции при этой же температуре в течение 16 ч. до полного превращения исходных материалов. После завершения реакции реакционную смесь промывали насыщенным раствором карбоната натрия, экстрагировали с помощью EA (30 мл x 3), сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 91b (60 мг). МС m/z (ИЭР): 190,1 [M+H]+.
Стадия 2 Получение 2-(6-(6-(4-(4-фтор-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 91)
Трифторацетат соединения 1g (35,0 мг) и соединение 91b (27,9 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (7,4 мг) и цианоборогидрид натрия (23,1 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 91 (23,0 мг). МС m/z (ИЭР): 537,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) 11,97 (с, 1H), 9,63 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,61 (д, J = 4,8 Гц, 1H), 8,43 (дд, J = 8,8, 2,0 Гц, 1H), 7,80 (д, J = 4,0 Гц, 1H), 7,71 (д, J = 8,8 Гц, 2H), 7,46 (д, J = 6,8 Гц, 2H), 6,89 (уш., 1H), 6,77 (д, J = 8,8 Гц, 1H), 6,30 (уш., 1H), 3,76-3,59 (м, 8H), 2,56-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 31 6-Метил-2-(6-(6-(4-(4-метил-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 88)
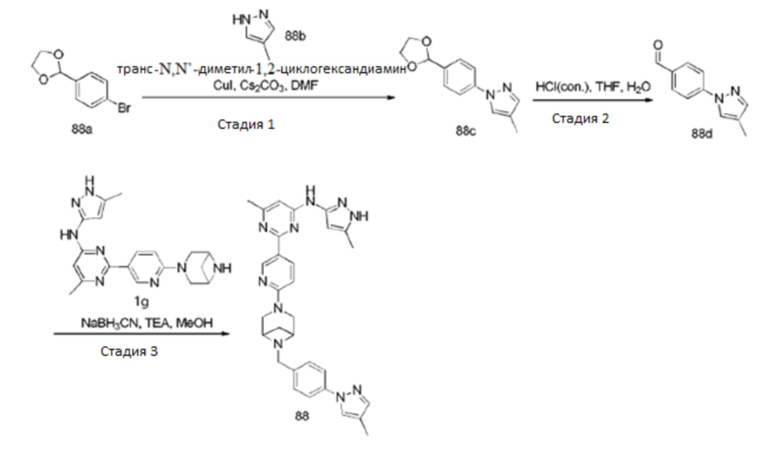
Стадия 1 Получение 1-(4-(1,3-диоксолан-2-ил)фенил)-4-метил-1H-пиразола (соединение 88c)
Соединение 88a (300 мг), соединение 88b (161,3 мг), транс-N,N'-диметил-1,2-циклогександиамин (74,5 мг), CuI (50,0 мг) и карбонат цезия (1,71 г) добавляли в DMF (5,0 мл). Смесь нагревали до 115 °C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 7 ч. H2O (10 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (10 мл×3). Органические фазы объединяли, промывали водой (10 мл ×3), сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 88c (120 мг). МС m/z (ИЭР): 231,2 [M+H]+.
Стадия 2 Получение 4-(4-метил-1H-пиразол-1-ил)бензальдегида (соединение 88d)
Концентрированную хлористоводородную кислоту (1,5 мл) добавляли по каплям в раствор соединения 88c (120 мг) в ТГФ (10 мл) и H2O (8 мл). Смесь нагревали до 65°C и удерживали для реакции при этой же температуре в течение 1,5 ч. После завершения реакции реакционную смесь охлаждали на ледяной бане, и затем насыщенный раствор бикарбоната натрия медленно добавляли по каплям для регулирования значения pH реакционной смеси до около 8. Растворитель ТГФ удаляли при пониженном давлении и затем смесь экстрагировали с помощью ДХМ (10 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=5:1) с получением соединения 88d (90 мг). МС m/z (ИЭР): 187,1 [M+H]+.
Стадия 3 Получение 6-метил-2-(6-(6-(4-(4-фтор-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 88)
Трифторацетат соединения 1g (35,0 мг) и соединение 88d (54,0 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (9,8 мг) и цианоборогидрид натрия (30,3 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 88 (10,0 мг). МС m/z (ИЭР): 533,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,14 (с, 1H), 8,45 (д, J = 8,4 Гц, 1H), 8,23 (с, 1H), 7,71 (д, J = 8,0 Гц, 2H), 7,54 (с, 1H), 7,44 (д, J = 8,0 Гц, 2H), 6,97-6,65 (м, 2H), 6,32 (уш., 1H), 3,82-3,67 (м, 4H), 3,65-3,50 (м, 4H), 2,62-2,55 (м, 1H), 2,35 (с, 3H), 2,27 (с, 3H), 2,11 (с, 3H), 1,60 (д, J = 8,0 Гц, 1H).
Пример 32 2-(6-(6-((6-(3,4-Диметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 121)
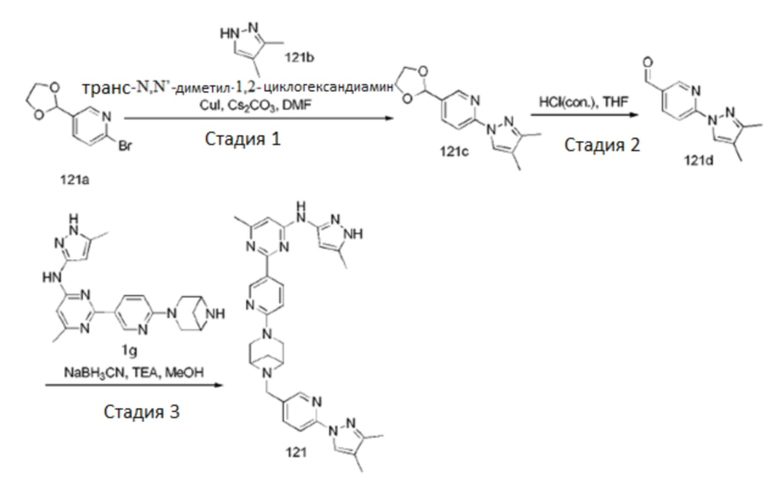
Стадия 1 Получение 2-(3,4-диметил-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 121c)
Соединение 121a (300 мг), соединение 121b (188,0 мг), транс-N,N'-диметил-1,2-циклогександиамин (74,2 мг), CuI (49,7 мг) и карбонат цезия (1,70 г) добавляли в DMF (5,0 мл). Смесь нагревали до 120°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 5 ч. H2O (10 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (10 мл×3). Органические фазы объединяли, промывали водой (10 мл ×3), сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=4:1) с получением соединения 121c (305 мг). МС m/z (ИЭР): 246,1 [M+H]+.
Стадия 2 Получение 6-(3,4-диметил-1H-пиразол-1-ил)никотинальдегида (соединение 121d)
Концентрированную хлористоводородную кислоту (2,0 мл) добавляли по каплям в раствор соединения 121c (305 мг) в ТГФ (10 мл). Смесь нагревали до 65°C и удерживали для реакции при этой же температуре в течение 1,5 ч. После завершения реакции реакционную смесь охлаждали на ледяной бане, затем регулировали значение pH до около 8 путем медленного добавления карбоната калия, и затем экстрагировали с помощью EA (10 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=4:1) с получением соединения 121d (200 мг). МС m/z (ИЭР): 202,2 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(3,4-диметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 121)
Трифторацетат соединения 1g (40,4 мг) и соединения 121d (53,4 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (8,7 мг) и цианоборогидрид натрия (26,9 мг) последовательно добавляли. Смесь выдерживали для реакции при 20°C в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 121 (3,0 мг). МС m/z (ИЭР): 548,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,96 (с, 1H), 9,63 (с, 1H), 9,12 (д, J = 2,4 Гц, 1H), 8,43 (дд, J = 8,8, 2,4 Гц, 1H), 8,32 (д, J = 1,6 Гц, 1H), 8,27 (с, 1H), 7,88 (дд, J = 8,4, 2,4 Гц, 1H), 7,79-7,70 (м, 1H), 7,03-6,60 (м, 2H), 6,29 (уш., 1H), 3,83-3,70 (м, 4H), 3,66-3,47 (м, 4H), 2,58-2,52 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 2,19 (с, 3H), 2,02 (с, 3H), 1,58 (д, J = 8,4 Гц, 1H).
Пример 33 2-(6-(6-((5-Фторпиридин-2-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 70)
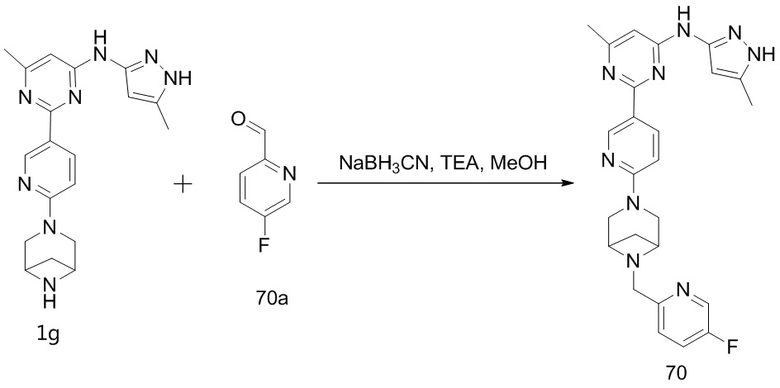
Трифторацетат соединения 1g (30 мг) и соединение 70a (23,63 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 70 (12,0 мг). МС m/z (ИЭР): 472,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6)) δ 11,97 (уш., 1H), 9,64 (с, 1H), 9,11 (д, J = 2,2 Гц, 1H), 8,43 (дд, J = 9,6, 2,7 Гц, 2H), 7,69 (тд, J = 8,8, 3,0 Гц, 1H), 7,54 (дд, J = 8,7, 4,7 Гц, 1H), 6,77 (д, J = 9,0 Гц, 2H), 6,31 (с, 1H), 3,80 (д, J = 11,9 Гц, 2H), 3,72 (д, J = 5,9 Гц, 2H), 3,64 (с, 2H), 3,56 (д, J = 10,2 Гц, 2H), 2,57-2,51 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 34 2-(6-(6-((6-(3-Фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 63)
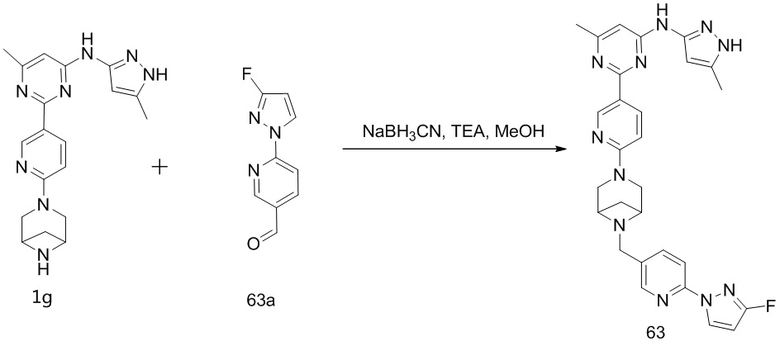
Трифторацетат соединения 1g (30 мг) и соединение 63a (47,47 мг, получено посредством ссылки на способ получения соединения 17a в примере 6, за исключением того, что 4-фторпиразол был заменен 3-фторпиразолом) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь выдерживали для реакции при 20°C в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 63 (10,0 мг). МС m/z (ИЭР): 538,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,55 (м, 1H), 8,47-8,38 (м, 2H), 7,98 (д, J = 8,6 Гц, 1H), 7,70 (д, J= 8,4 Гц, 1H), 6,80 (д, J = 8,0 Гц, 2H), 6,56 (дд, J =8, 4 Гц, 1H), 6,29 (уш., 1H), 3,84-3,68 (м, 4H), 3,67-3,47 (м, 4H), 2,60-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,60 (д, J = 8,2 Гц, 1H).
Пример 35 2-(6-(6-((6-(4-Фтор-1H-имидазол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 64)
Стадия 1 Получение 5-(1,3-диоксолан-2-ил)-2-(4-фтор-1H-имидазол-1-ил)пиридина (соединение 64b)
Соединение 64a (230 мг), соединение 121a (86 мг), N,N'-диметилэтандиамин (38 мг), CuI (83 мг) и карбонат цезия (425 мг) добавляли в DMF (1 мл). Смесь нагревали до 115°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 3 ч. Воду добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA. Органическую фазу промывали водой, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 64b (120 мг, неочищенное), которое непосредственно использовали на следующей стадии реакции без очистки. МС m/z (ИЭР): 236,1 [M+H]+.
Стадия 2 Получение 6-(4-фтор-1H-имидазол-1-ил)никотинальдегида (соединение 64c)
Концентрированную хлористоводородную кислоту (12 N, 3,0 мл) добавляли по каплям в раствор соединения 64b (120 мг) в ТГФ (10 мл) и воды (10 мл). Смесь выдерживали для реакции при комнатной температуре в течение 18 ч. После завершения реакции реакционную смесь регулировали с помощью насыщенного раствора бикарбоната натрия до значения pH около 8, экстрагировали с помощью EA и сушили над безводным сульфатом натрия. Высушенный продукт фильтровали и затем концентрировали при пониженном давлении с получением соединения 64c (60 мг, неочищенное), которое непосредственно использовали на следующей стадии реакции без очистки. МС m/z (ИЭР): 192,1 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(4-фтор-1H-имидазол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 64)
Трифторацетат соединения 1g (30 мг) и соединение 64c (47,47 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь выдерживали для реакции при 20°C в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 64 (10,0 мг). МС m/z (ИЭР): 538,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,64 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,44 (дд, J = 9,1, 2,4 Гц, 2H), 8,28 (м, 1H), 7,99 (д, J = 8,4 Гц, 1H), 7,76-7,74(д, J = 8,0 Гц, 1H), 7,69-7,67 (дд, J = 8,4, 1,6 Гц, 1H), 6,86 (уш., 1H), 6,77 (д, J = 9,2 Гц, 1H), 6,30 (уш., 1H), 3,78-3,71(м, 4H), 3,67-3,51(м, 4H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 36 2-(6-(6-(1-(6-(4-Фтор-1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 52)
Соединение 1g (320 мг), соединение 52a (181,17 мг, получено посредством ссылки на способ получения соединения 17a в примере 6, за исключением того, что 6-бромникотинальдегид заменяли 1-(6-бромпиридин-3-ил)этаноном), и тетраизопропилтитанат (752,81 мг) добавляли в сухой ТГФ (25 мл) и после замещения азотом три раза перемешивали при 75°C в течение 24 ч. Затем триацетоксиборогидрид натрия (935,64 мг) добавляли в систему по частям, сухой ТГФ (15 мл) дополняли и смесь перемешивали при 75°C в течение дополнительных 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии (MeOH:ДХМ=1:9) с получением неочищенного соединения 52, которое дополнительно отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 52 (90,0 мг). МС m/z (ИЭР): 552,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,96 (с, 1H), 9,63 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,66 (д, J = 4,4 Гц, 1H), 8,46-8,39 (м, 2H), 8,01 (дд, J= 8,5, 2,0 Гц, 1H), 7,90 (дд, J = 14,7, 6,2 Гц, 2H), 6,82(уш., 1H), 6,75(д, J = 12,0 Гц, 1H), 6,27(уш., 1H),3,96-3,82 (м, 2H), 3,77 (к, J = 6,1 Гц, 1H), 3,63 (м, 1H), 3,49-3,37 (м, 3H), 2,55-2,51 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,55 (д, J = 8,4 Гц, 1H), 1,23 (д, J = 6,2 Гц, 3H).
Пример 37 2-(6-(6-((6-(3-Циклопропил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 120)
Трифторацетат соединения 1g (30 мг) и соединения 120a (22,95 мг, получено посредством ссылки на способ получения соединения 64c в примере 35, за исключением того, что исходный материал 4-фторимидазол был заменен 3-циклопропилимидазолом) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли. Смесь перемешивали при 20°C в течение 16 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 120 (13,0 мг). МС m/z (ИЭР): 560,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,64 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,44 (дд, J = 8,9, 2,4 Гц, 2H), 8,35 (д, J = 1,3 Гц, 1H), 7,92 (дд, J = 8,5, 1,8 Гц, 1H), 7,78 (д, J = 8,4 Гц, 1H), 6,8(уш., 1H), 6,78 (д, J = 9,0 Гц, 1H), 6,28(уш., 1H), 6,26 (д, J = 2,5 Гц, 1H), 3,81-3,66 (м, 4H), 3,67-3,52 (м, 4H), 2,58-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 2,03-1,95 (м, 1H), 1,59 (д, J = 8,4 Гц, 1H), 0,98-0,91 (м, 2H), 0,79-0,73 (м, 2H).
Пример 38. 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(4-((6-метилпиридин-3-ил)окси)пиперидин-1-ил)пиридин-3-ил)пиримидин-4-амин (соединение 119)
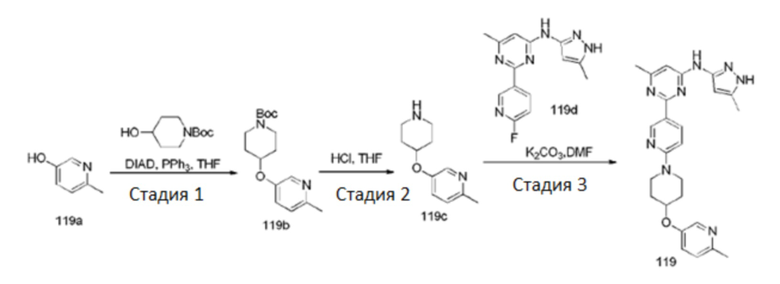
Стадия 1 Получение трет-бутил-4-((6-метилпиридин-3-ил)-окси)пиперидин-1-карбоксилата (соединение 119b)
Соединение 119a (100 мг), N-Boc-4-гидроксипиперидин (276,65 мг) и трифенилфосфин (480,18 мг) добавляли в сухой ТГФ (5 мл) и охлаждали до 0°C. DIAD (370,21 мг) добавляли по каплям в систему, и смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции систему концентрировали при пониженном давлении и очищали посредством колоночной флеш-хроматографии (PE:EA=1:1) с получением соединения 119b (65 мг). МС m/z (ИЭР): 293,2 [M+H]+.
Стадия 2 Получение 2-метил-5-(пиперидин-4-илокси)пиридина (соединение 119c)
Соединение 119b (65 мг) добавляли в перемешанный раствор хлорида водорода в 1,4-диоксане (4 н., 2 мл) и ТГФ (1 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. и концентрировали досуха при пониженном давлении с получением гидрохлорида соединения 119c (54 мг). МС m/z (ИЭР): 193,2 [M+H]+.
Стадия 3 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(4-((6-метилпиридин-3-ил)окси)пиперидин-1-ил)пиридин-3-ил)пиримидин-4-амина (соединение 119)
Гидрохлорид соединения 119c (50 мг), соединение 119d (50 мг) и карбонат калия (73 мг) добавляли в DMF (2 мл), и смесь нагревали до 100°C и перемешивали при этой же температуре в течение 16 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (30 мл×2). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 119 (13 мг). МС m/z (ИЭР): 457,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,96 (с, 1H), 9,63 (с, 1H), 9,04 (д, J = 2,4 Гц, 1H), 8,37 (дд, J = 9,0, 2,4 Гц, 1H), 8,19 (д, J = 2,9 Гц, 1H), 7,38 (дд, J = 8,5, 3,0 Гц, 1H), 7,18 (д, J = 8,5 Гц, 1H), 6,96 (д, J = 9,1 Гц, 1H), 6,82 (с, 1H), 6,28 (с, 1H), 4,72-4,65 (м, 1H), 4,13-4,01 (м, 2H), 3,49-3,40 (м, 2H), 2,40 (с, 3H), 2,32 (с, 3H), 2,25 (с, 3H), 2,06-1,98 (м, 2H), 1,69-1,58 (м, 2H).
Пример 39 2-(6-(6-(4-(3-Фтор-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 89)

Стадия 1 Получение 1-(4-(1,3-диоксолан-2-ил)фенил)-3-фтор-1H-пиразола (соединение 89b)
Соединение 89a (80 мг), соединение 88a (212,92 мг), N,N'-диметилэтандиамин (81,94 мг), карбонат цезия (908,55 мг) и CuI (177,02 мг) последовательно добавляли в DMF (6 мл). Смесь нагревали до 110°C и перемешивали при этой же температуре в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали с помощью ДХМ (50 мл×2). Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии (PE:EA=63:37) с получением соединения 89b (35 мг). МС m/z (ИЭР): 235,1 [M+H]+.
Стадия 2 Получение 4-(3-фтор-1H-пиразол-1-ил)бензальдегида (соединение 89c)
Соединение 89b (35 мг) добавляли в перемешанный раствор хлорида водорода в 1,4-диоксане (4 н., 2 мл) и ДХМ (1 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением соединения 89c (28 мг). МС m/z (ИЭР): 191,1 [M+H]+.
Стадия 3 Получение 2-(6-(6-(4-(3-фтор-1H-пиразол-1-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 89)
Соединение 89c (14,37 мг), трифторацетат соединения 1g (30 мг), триэтиламин (6,37 мг) и цианоборогидрид натрия (19,78 мг) последовательно добавляли в метанол (0,5 мл) и перемешивали при комнатной температуре в течение 36 ч. Насыщенный водный раствор хлорида аммония (0,1 мл) добавляли для гашения реакционной смеси. Реакционную смесь концентрировали и предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=10:1) с получением 5 мг неочищенного продукта (Rf=0,15-0,25), который дополнительно отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 89 (4 мг). МС m/z (ИЭР): 537,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,64 (с, 1H), 9,12 (д, J = 2,3 Гц, 1H), 8,49-8,39 (м, 2H), 7,68 (д, J = 8,6 Гц, 2H), 7,46 (д, J = 8,5 Гц, 2H), 6,78 (д, J = 9,0 Гц, 2H), 6,31 (дд, J = 5,8, 2,6 Гц, 2H), 3,80-3,67 (м, 4H), 3,64-3,48 (м, 4H), 2,59-2,54 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 40 6’-(6-((6-(4-Фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)-4-метил-N-(5-метил-1H-пиразол-3-ил)-[2,3’-дипиридил]-6-амин (соединение 6)
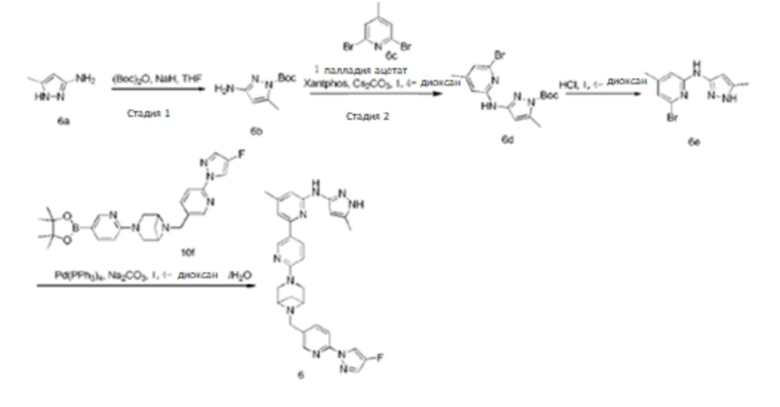
Стадия 1 Получение трет-бутил-3-амино-5-метил-1H-пиразол-1-карбоксилата (соединение 6b)
Соединение 6a (2,4 г) растворяли в сухом ТГФ (50 мл), NaH (988,39 мг, чистота 60%) добавляли порциями и затем смесь перемешивали в течение 10 мин. Ди-трет-бутил-дикарбонат (5,39 г) добавляли по каплям, и смесь перемешивали в условиях защиты азотом при 25°C в течение 2 ч. Воду добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=5:1) с получением соединения 6b (3,95 г).
Стадия 2 Получение трет-бутил-3-((6-бром-4-метилпиридин-2-ил)амино)-5-метил-1H-пиразол-1-карбоксилата (соединение 6d)
Соединение 6c (1,0 г), соединение 6b (864,65 мг), ацетат палладия (89,47 мг), Xantphos (461,20 мг) и карбонат цезия (2,60 г) последовательно добавляли в 1,4-диоксан (10 мл), и перемешивали в условиях защиты азотом при 95°C в течение 2 ч. ЖХ-МС продемонстрировала, что исходные материалы были полностью превращены в целевой продукт. Реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 6d (415 мг). МС m/z (ИЭР): 367 [M+H]+.
Стадия 3 Получение 6-бром-4-метил-N-(5-метил-1H-пиразол-3-ил)пиридин-2-амина (соединение 6e)
Соединение 6d (300 мг) добавляли в раствор хлорида водорода в 1,4-диоксане (4 н., 2 мл). Смесь перемешивали при 25°C в течение 1 ч. и непосредственно концентрировали досуха. Неочищенный продукт растворяли в метаноле (5 мл). Затем триэтиламин (1 мл) добавляли, и смесь перемешивали при комнатной температуре в течение 15 мин. Реакционную смесь концентрировали и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=97:3) с получением соединения 6e (145 мг). МС m/z (ИЭР): 267 [M+H]+.
Стадия 4 Получение 6’-(6-((6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)-4-метил-N-(5-метил-1H-пиразол-3-ил)-[2,3’-дипиридил]-6-амина (соединение 6)
Соединение 6e (30 мг), соединение 10f (85 мг), Na2CO3 (27,85 мг), Pd(PPh3)4 (12,98 мг), H2O (1 мл) и 1,4-диоксан (5 мл) последовательно добавляли в реакционную колбу, и после замещения азотом три раза перемешивали при 95°C в течение 5 ч. После завершения реакции реакционную смесь разбавляли водой и экстрагировали с помощью EA. Органический слой собирали, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали при всасывании, концентрировали, предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=9:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 6 (6 мг). МС m/z (ИЭР): 537,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,63 (с, 1H), 9,02 (с, 1H), 8,86 (д, J = 2,3 Гц, 1H), 8,67 (дд, J = 4,5, 0,6 Гц, 1H), 8,41 (д, J = 1,7 Гц, 1H), 8,21 (дд, J = 8,9, 2,4 Гц, 1H), 7,98 (дд, J = 8,5, 2,2 Гц, 1H), 7,92 (д, J = 4,3 Гц, 1H), 7,86 (д, J = 8,4 Гц, 1H), 7,03 (с, 1H), 6,88 (с, 1H), 6,77 (д, J = 9,0 Гц, 1H), 6,22 (с, 1H), 3,81-3,69 (м, 4H),3,66-3,53 (м, 4H), 2,58-2,53 (м, 1H), 2,26 (с, 3H), 2,22 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 41 2-(6-(6-((6’-Метокси-[2,3’-дипиридил]-5-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 7)
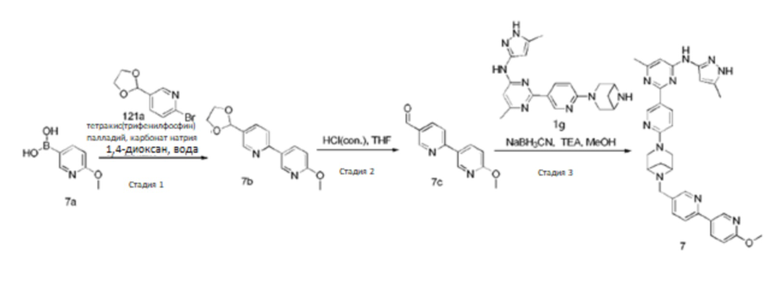
Стадия 1 Получение 5-(1,3-диоксолан-2-ил)-6’-метокси-2,3’-дипиридина (соединение 7b)
Соединение 7a (499 мг), соединение 121a (500 мг), Na2CO3 (691 мг), Pd(PPh3)4 (126 мг), H2O (3 мл) и 1,4-диоксан (17 мл) последовательно добавляли в реакционную колбу и после замещения азотом три раза перемешивали при 95°C в течение 5 ч. После завершения реакции реакционную смесь концентрировали досуха, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=1:1) с получением соединения 7b (350 мг).
Стадия 2 Получение 6’-метокси-[2,3’-дипиридил]-5-карбальдегида (соединение 7c)
Концентрированную хлористоводородную кислоту (12 N, 3,0 мл) добавляли по каплям в раствор соединения 7b (200 мг) в ТГФ (11 мл) и воды (9 мл). Смесь выдерживали для реакции при комнатной температуре в течение 18 ч. После завершения реакции значение pH реакционной смеси регулировали раствором карбоната калия до около 10 и затем экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=2:1) с получением соединения 7c (60 мг). МС m/z (ИЭР): 215,1 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6’-метокси-[2,3’-дипиридил]-5-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 7)
Трифторацетат соединения 1g (50 мг) и соединение 7c (67,44 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (10,62 мг) и цианоборогидрид натрия (32,97 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 7 (10,0 мг). МС m/z (ИЭР): 561,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (уш., 1H), 9,65 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,86 (д, J = 2,2 Гц, 1H), 8,60 (д, J = 1,5 Гц, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,36 (дд, J = 8,7, 2,5 Гц, 1H), 7,90 (д, J = 8,1 Гц, 1H), 7,83 (дд, J = 8,2, 2,1 Гц, 1H), 6,85 (дд, J = 56,9, 8,8 Гц, 3H), 6,33 (с, 1H), 3,91 (с, 3H), 3,75 (дд, J = 20,2, 8,9 Гц, 4H),3,65-3,49 (м, 4H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 42 2-(6-(6-((5’-Фтор-2’-метил-[2,3’-дипиридил]-5-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 8)
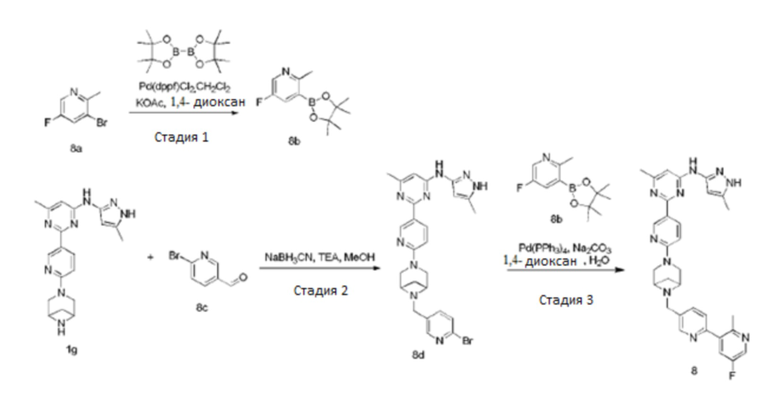
Стадия 1 Получение сложного пинаколового эфира 5-фтор-2-метилпиридин-3-бороновой кислоты (соединение 8b)
Бис(пинаколато)дибор (1,20 г), соединение 8a (300 мг), Pd(dppf)Cl2·ДХМ (128,93 мг), ацетат калия (464,85 мг) и сухой 1,4-диоксан (10 мл) последовательно добавляли в реакционную колбу. Смесь нагревали до 100°C в условиях защиты азотом и перемешивали при этой же температуре в течение 4 ч. После завершения реакции смесь фильтровали. Фильтрат разбавляли водой и экстрагировали с помощью EA. Органическую фазу промывали водой дважды, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=1:1) с получением соединения 8b (300 мг). МС m/z (ИЭР): 238,2 [M+H]+.
Стадия 2 Получение 2-(6-(6-((6-бромпиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 8d)
Трифторацетат соединения 1g (200 мг) и соединение 8c (195,20 мг) добавляли в метанол (10 мл), и триэтиламин (42,48 мг) добавляли при перемешивании при комнатной температуре. После перемешивания в течение 30 мин. цианоборогидрид натрия (131,89 мг) добавляли, и смесь перемешивали при 20°C в течение 16 ч. ЖХ-МС продемонстрировала отсутствие очевидного пика продукта. Смесь отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=10:1) с получением соединения 8d (220 мг). МС m/z (ИЭР): 532,1[M+H]+.
Стадия 3 Получение 2-(6-(6-((5’-фтор-2’-метил-[2,3’-дипиридил]-5-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 8)
Соединение 8b (222,63 мг), соединение 8d (100 мг), Na2CO3 (69,87 мг), Pd(PPh3)4 (32,55 мг), воду (1 мл) и 1,4-диоксан (5 мл) последовательно добавляли в реакционную колбу и перемешивали в условиях защиты азотом при 95°C в течение 5 ч. После завершения реакции реакционную смесь выпаривали ротационно, предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=9:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 8 (25 мг). МС m/z (ИЭР): 563,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,66 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,66 (д, J = 1,5 Гц, 1H), 8,51 (д, J = 2,9 Гц, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 7,90 (дд, J = 8,1, 2,1 Гц, 1H), 7,76 (дд, J = 9,5, 2,8 Гц, 1H), 7,62 (д, J = 8,0 Гц, 1H), 6,81 (уш., 1H), 6,79 (д, J = 9,0 Гц, 1H), 6,32 (уш., 1H), 3,85-3,71(м, 4H), 3,70-3,51 (м, 4H), 2,61-2,53 (м, 1H), 2,51 (с, 3H), 2,33 (с, 3H), 2,26 (с, 3H), 1,61 (д, J= 8,4 Гц, 1H).
Пример 43 6-Метил-2-(6-(6-((6-(5-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 9)
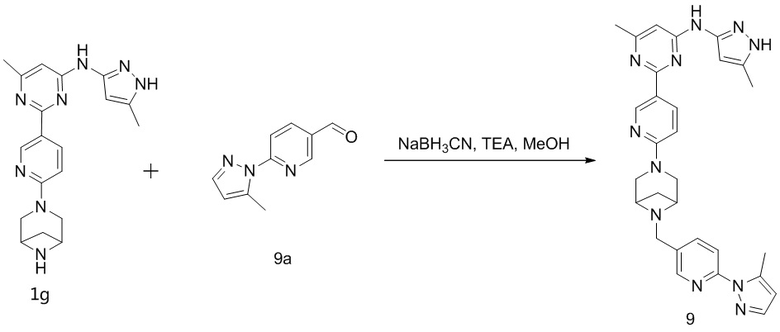
Трифторацетат соединения 1g (30 мг) и соединение 9a (17,68 мг, получено посредством ссылки на способ получения соединения 64c в примере 35, за исключением того, что исходный материал 4-фторимидазол заменяли 5-метилимидазолом) добавляли в метанол (0,5 мл), и затем триэтиламин (8,38 мг) добавляли. После перемешивания в течение 30 мин. цианоборогидрид натрия (19,78 мг) добавляли, и смесь перемешивали при 20°C в течение 16 ч. ЖХ-МС продемонстрировала, что исходные материалы были полностью превращены, и очевидный пик продукта отсутствовал. Смесь отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 9 (7 мг). МС m/z (ИЭР): 534,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,99 (с, 1H), 9,67 (с, 1H), 9,14 (д, J= 2,1 Гц, 1H), 8,49 (д, J = 2,4 Гц, 1H), 8,45 (дд, J = 9,2, 2,4 Гц, 1H), 8,38 (д, J = 1,6 Гц, 1H), 7,94 (дд, J = 8,5, 2,1 Гц, 1H), 7,81 (д, J = 8,4 Гц, 1H), 6,83(уш., 1H), 6,79 (д, J = 9,2 Гц, 1H), 6,38 (д, J = 2,4 Гц, 1H), 6,33 (уш., 1H), 3,85-3,71 (м, 4H), 3,69-3,51 (м, 4H), 2,61-2,55 (м, 1H), 2,35 (с, 3H), 2,30 (с, 3H), 2,27 (с, 3H), 1,61 (д, J = 8,4 Гц, 1H).
Пример 44 N-(5-Циклопропил-1H-пиразол-3-ил)-2-(6-(6-((6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метилпиримидин-4-амин (соединение 10)
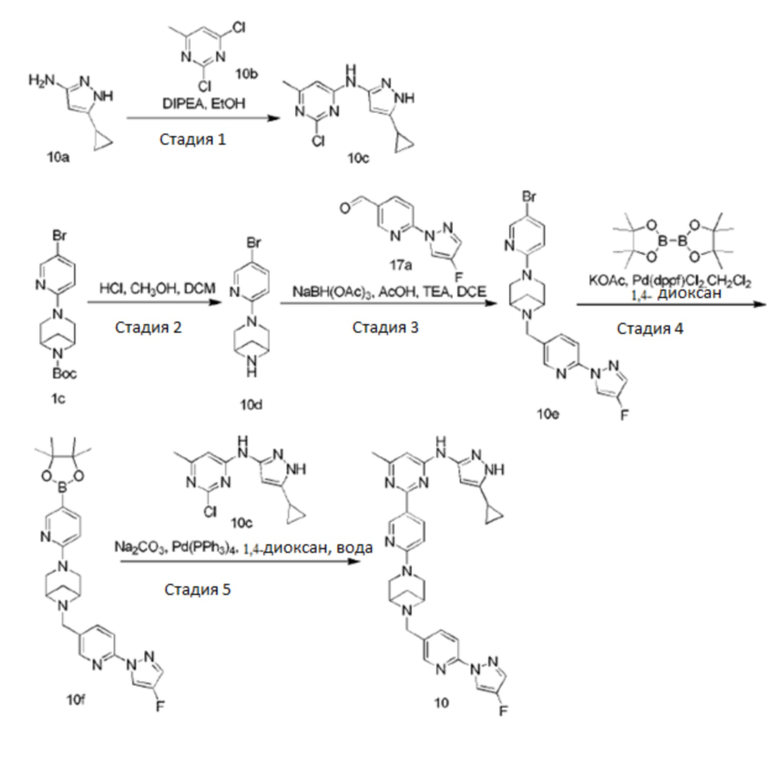
Стадия 1 Получение 2-хлор-N-(5-циклопропил-1H-пиразол-3-ил)-6-метилпиримидин-4-амина (соединение 10c)
Соединение 10b (1,0 г) и соединение 10a (755,53 мг) растворяли в этаноле (20,00 мл), и затем N,N-диизопропилэтиламин (1590,00 мг) добавляли. Смесь нагревали до 70°C и перемешивали в условиях защиты азотом при этой же температуре в течение 48 ч. Реакционную смесь концентрировали при пониженном давлении и разбавляли с помощью EA (300,00 мл). Органическую фазу промывали водой три раза, дополнительно промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=9:1) с получением соединения 10c (820,00 мг). МС m/z (ИЭР): 250,1 [M+H]+.
Стадия 2 Получение 3-(5-бромпиридин-2-ил)-3,6-диазабицикло[3.1.1]гептана (соединение 10d)
Соединение 1c (1180,00 мг) растворяли в перемешанном растворителе метанола (5,00 мл) и ДХМ (10,00 мл), и затем раствор хлорида водорода в растворе 1,4-диоксана (4 н., 6,00 мл) медленно добавляли по каплям на ледяной бане. Смесь перемешивали при 25°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида соединения 10d (1040,00 мг). МС m/z (ИЭР): 254,0 [M+H]+.
Стадия 3 Получение 3-(5-бромпиридин-2-ил)-6-((6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептана (соединение 10e)
Гидрохлорид соединения 10d (300,00 мг) и соединение 17a (297,04 мг) растворяли в 1,2-дихлорэтане (10,00 мл), и триэтиламин (104,82 мг) медленно добавляли по каплям. После перемешивания в течение 10 мин. уксусную кислоту (31,10 мг) добавляли по каплям, и смесь перемешивали в течение дополнительных 30 мин. Затем триацетоксиборогидрид натрия (878,21 мг) добавляли, и смесь перемешивали при 25°C в течение 20 ч. Насыщенный водный раствор хлорида аммония добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь разбавляли водой и экстрагировали с помощью ДХМ. Органический слой сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=1:1) с получением соединения 10e (363,00 мг). МС m/z (ИЭР): 429,1 [M+H]+.
Стадия 4 Получение 6-((-6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептана (соединение 10f)
Соединение 10e (187,00 мг) и бис(пинаколато)дибор (221,23 мг) растворяли в 1,4-диоксане (15,00 мл), и затем ацетат калия (106,88 мг) и Pd(dppf)Cl2·ДХМ (35,57 мг) добавляли. Смесь нагревали до 95°C в условиях защиты азотом и перемешивали при этой же температуре в течение 5 ч. Реакционную смесь разбавляли с помощью EA (100,00 мл) и промывали водой три раза. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 10f (100,00 мг). МС m/z (ИЭР): 477,3 [M+H]+.
Стадия 5 Получение N-(5-циклопропил-1H-пиразол-3-ил)-2-(6-(6-((6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метилпиримидин-4-амина (соединение 10)
Соединение 10c (26,21 мг) и соединение 10f (50,00 мг) растворяли в 1,4-диоксане (4,00 мл), и затем Pd(PPh3)4 (18,19 мг) и водный раствор карбоната натрия (33,38 мг растворяли в 1,00 мл воды) последовательно добавляли. Смесь нагревали до 95°C и перемешивали в условиях защиты азотом при этой же температуре в течение 12 ч. После завершения реакции реакционную смесь разбавляли с помощью EA (20,00 мл) и промывали водой три раза. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением неочищенного продукта. Неочищенный продукт отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 10 (9,00 мг). МС m/z (ИЭР): 564,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,02 (с, 1H), 9,64 (с, 1H), 9,11 (д, J = 2,4 Гц, 1H), 8,67 (дд, J= 4,8, 0,8 Гц, 1H), 8,42 (дд, J = 9,2, 2,0 Гц, 2H), 7,99 (дд, J = 8,4, 2,0 Гц, 1H), 7,92 (дд, J = 4,4, 0,8 Гц, 1H), 7,87 (д, J = 8,4 Гц, 1H), 6,78 (д, J = 9,2 Гц, 2H), 6,18 (уш., 1H), 3,79-3,71 (м, 4H), 3,64-3,53 (м, 4H), 2,56-2,51 (м, 1H), 2,33 (с, 3H), 1,95-1,89 (м, 1H), 1,60 (д, J = 8,4 Гц, 1H), 0,97-0,92 (м, 2H), 0,75-0,69 (м, 2H).
Пример 45 2-(6-(6-((6-(3,5-Диметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 11)

Стадия 1 Получение 2-(3,5-диметил-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 11b)
Соединение 11a (1,0 г), соединение 121a (1,74 г), N,N'-диметилэтандиамин (664,83 мг), CuI (1,44 г) и карбонат цезия (7,37 г) добавляли в DMF (30 мл). Смесь нагревали до 98 °C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 16 ч. Воду добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 11b (0,69 г). МС m/z (ИЭР): 246,2 [M+H]+.
Стадия 2 Получение 6-(3,5-диметил-1H-пиразол-1-ил)никотинальдегида (соединение 11c)
Хлористоводородную кислоту (2 н., 3,0 мл) добавляли по каплям в раствор соединения 11b (250 мг) в ТГФ (6 мл). Смесь выдерживали для реакции при комнатной температуре в течение 3 ч. После завершения реакции реакционную смесь охлаждали на ледяной бане, регулировали значение pH до около 8 путем медленного добавления карбоната калия, и затем экстрагировали с помощью EA (10 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 11c (150 мг).
Стадия 3 Получение 2-(6-(6-((6-(3,5-диметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 11)
Соединение 1g (30 мг) и соединение 11c (25 мг) добавляли в метанол (1 мл), и затем уксусную кислоту (5 мг) добавляли. Смесь перемешивали при комнатной температуре в течение 0,5 ч. Цианоборогидрид натрия (15,6 мг) добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 20 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении и предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=96:4) с получением неочищенного продукта, который дополнительно отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 11 (7 мг). МС m/z (ИЭР): 548,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6)) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,44 (дд, J = 8,9, 2,2 Гц, 1H), 8,37 (д, J = 1,8 Гц, 1H), 7,91 (дд, J = 8,5, 2,2 Гц, 1H), 7,74 (д, J = 8,4 Гц, 1H), 6,96-6,67 (м, 2H), 6,31 (с, 1H), 6,09 (с, 1H), 3,85-3,68 (м, 4H), 3,66-3,49 (м, 4H), 2,56 (с, 3H), 2,54 (с, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 2,19 (с, 3H), 1,60 (д, J = 8,5 Гц, 1H).
Пример 46 2-(6-(6-((6-(5-Изопропил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 12)
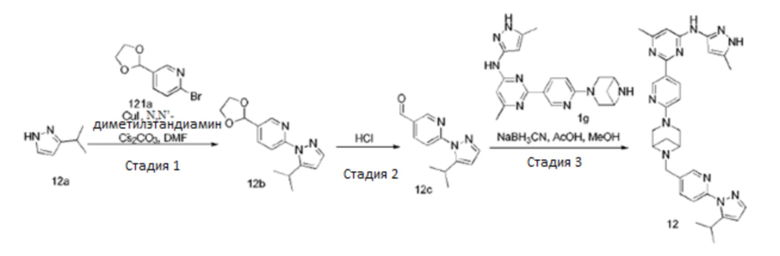
Стадия 1 Получение 5-(1,3-диоксолан-2-ил)-2-(5-изопропил-1H-пиразол-1-ил)пиридина (соединение 12b)
Соединение 12a (210,68 мг), соединение 121a (400 мг), N,N'-диметилэтандиамин (153,27 мг), карбонат цезия (1,13 г) и CuI (331,13 мг) последовательно добавляли в DMF (10 мл). Смесь нагревали до 110°C и перемешивали при этой же температуре в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью ДХМ (50 мл×2). Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=55:45) с получением соединения 12b (300 мг). МС m/z (ИЭР): 260,2 [M+H]+.
Стадия 2 Получение 6-(5-изопропил-1H-пиразол-1-ил)никотинальдегида (соединение 12c)
Соединение 12b (300,00 мг) добавляли в перемешанный раствор хлорида водорода (4 н., 6 мл) в 1,4-диоксане и ДХМ (4 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали посредством колоночной флеш-хроматографии (PE:EA=80:20) с получением соединения 12c (120 мг). МС m/z (ИЭР): 216,2 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(5-изопропил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 12)
Соединение 12c (26,73 мг), соединение 1g (30 мг) и цианоборогидрид натрия (26,01 мг) последовательно добавляли в перемешанный растворитель метанола (1 мл) и уксусной кислоты (0,1 мл). Смесь нагревали до 40°C и перемешивали при этой же температуре в течение 16 ч. Насыщенный водный раствор хлорида аммония (0,1 мл) добавляли для гашения реакционной смеси. Реакционную смесь концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением трифторацетата соединения 12 (15 мг), который дополнительно отделяли посредством ЖХСД с получением соединения 12 (8 мг) в свободном состоянии. МС m/z (ИЭР): 562,3 [M+H]+.
Условия ЖХСД:
Модель прибора: Biotage Isolera Prime 2,3,1, хроматографическая колонка: Agela Technologies C18, сферическая 20-35 мкм 100A, 12 g; температура хроматографической колонки: 25°C; расход: 15,0 мл/мин.; длина волны обнаружения: 254 нм; градиент для элюирования: (0 мин.: 0% A, 100% B; 3,0 мин.: 0% A, 100% B; 20 мин.: 80% A, 20% B); подвижная фаза A: 100% ацетонитрил; подвижная фаза B: 0,5% водный раствор бикарбоната аммония; время сбора соединения: 10,4 мин.-11,8 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,16-9,09 (м, 1H), 8,49-8,40 (м, 2H), 8,36 (д, J = 1,9 Гц, 1H), 7,93 (дд, J = 8,5, 2,3 Гц, 1H), 7,81 (дд, J = 8,4, 0,7 Гц, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,42 (д, J = 2,5 Гц, 1H), 6,33 (с, 1H), 3,83-3,68 (м, 4H), 3,66-3,50 (м, 4H), 2,99 (p, J = 6,9 Гц, 1H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H), 1,27 (с, 3H), 1,25 (с, 3H).
Пример 47 2-(6-(6-(4-(5,6-Дигидроциклопентено[c]пиразол-2(4H)-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 25)
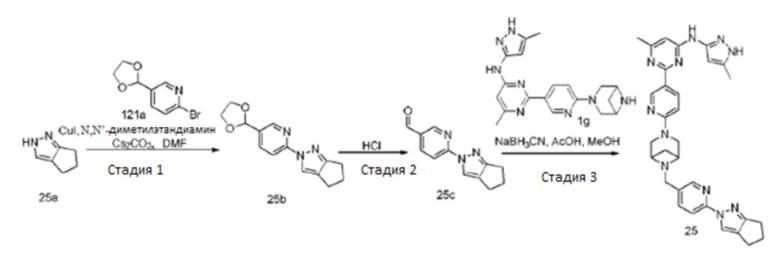
Стадия 1 Получение 2-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-2,4,5,6-тетрагидроциклопенено[c]пиразола (соединение 25b)
Соединение 25a (206,83 мг), соединение 121a (400 мг), N,N'-диметилэтандиамин (153,27 мг), карбонат цезия (1,13 г) и CuI (331,13 мг) последовательно добавляли в DMF (10 мл). Смесь нагревали до 110°C и перемешивали при этой же температуре в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали с помощью ДХМ (50 мл×2). Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 25b (525 мг). МС (ИЭР, m/z): 258,2 [M+H]+.
Стадия 2 Получение 6-(5,6-дигидроциклопенено[c]пиразол-2(4H)-ил)никотинальдегида (соединение 25c)
Соединение 25b (315 мг) добавляли в перемешанный раствор хлорида водорода (4 N, 10 мл), раствора 1,4-диоксана и ДХМ (10 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и очищали посредством колоночной флеш-хроматографии (PE:EA=80:20) с получением соединения 25c (200 мг). МС m/z (ИЭР): 214,2 [M+H]+.
Стадия 3 Получение 2-(6-(6-(4-(5,6-дигидроциклопенено[c]пиразол-2(4H)-ил)бензил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 25)
Соединение 25c (29,42 мг), соединение 1g (50 мг) и цианоборогидрид натрия (43,35 мг) последовательно добавляли в перемешанный растворитель метанола (0,5 мл) и уксусной кислоты (0,05 мл). Смесь нагревали до 40°C и перемешивали при этой же температуре в течение 16 ч. Насыщенный водный раствор хлорида аммония (0,1 мл) добавляли для гашения реакционной смеси. Реакционную смесь концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением трифторацетата соединения 25 (25 мг), который дополнительно отделяли посредством ЖХСД с получением соединения 25 (15 мг) в свободном состоянии. МС m/z (ИЭР): 560,3 [M+H]+.
Условия ЖХСД:
Модель прибора: Biotage Isolera Prime 2,3,1, хроматографическая колонка: Agela Technologies C18, сферическая 20-35 мкм 100A, 12 g; температура хроматографической колонки: 25°C; расход: 15,0 мл/мин.; длина волны обнаружения: 254 нм; градиент для элюирования: (0 мин.: 0% A, 100% B; 3,0 мин.: 0% A, 100% B; 20 мин.: 80% A, 20% B); подвижная фаза A: 100% ацетонитрил; подвижная фаза B: 0,5% водный раствор бикарбоната аммония; время сбора соединения: 11,4 мин.-12,8 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,15-9,10 (м, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,35-8,30 (м, 1H), 8,19 (д, J = 1,2 Гц, 1H), 7,89 (дд, J = 8,5, 2,3 Гц, 1H), 7,76 (дд, J = 8,4, 0,7 Гц, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,31 (с, 1H), 3,82-3,69 (м, 4H), 3,66-3,50 (м, 4H), 2,74-2,62 (м, 4H), 2,57-2,54 (м, 2H), 2,38 (к, J = 7,4 Гц, 2H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 48 (6-(4-Фтор-1H-пиразол-1-ил)пиридин-3-ил)(3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метанон (соединение 26)

Стадия 1 Получение метил 6-(4-фтор-1H-пиразол-1-ил)никотината (соединение 26b)
Соединение 26a (1 g) и соединение 91a (478,08 мг) добавляли в ацетонитриле (20 мл), и затем салицилалдоксим (129,96 мг), карбонат цезия (3,77 г) и оксид меди (132,47 мг) последовательно добавляли. Смесь нагревали до 85 °C и перемешивали в условиях защиты азотом при этой же температуре в течение 16 ч. После завершения реакции разбавленную хлористоводородную кислоту добавляли в реакционную смесь для регулирования pH до около 5. Силикагель добавляли для смешивания с образцами, которые отделяли и очищали посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=20:1) с получением соединения 26b (602 мг). МС m/z (ИЭР): 221,9 [M+H]+.
Стадия 2 Получение 6-(4-фтор-1H-пиразол-1-ил)никотиновой кислоты (соединение 26c)
Соединение 26b (580 мг) добавляли в ТГФ (10 мл) и H2O (5 мл), и затем NaOH (314,66 мг) добавляли. Смесь перемешивали при 25°C в течение 14 ч. После завершения реакции разбавленную хлористоводородную кислоту добавляли в реакционную смесь для регулирования pH до около 5. Реакционную смесь экстрагировали с помощью EA (80 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 26c (312 мг). МС m/z (ИЭР): 208,1 [M+H]+.
Стадия 3 (6-(4-Фтор-1H-пиразол-1-ил)пиридин-3-ил)(3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метанон (соединение 26)
Соединение 26c (20 мг) добавляли в DMF (5 мл), и затем HBTU (29,41 мг), DIPEA (37,43) и трифторацетат соединения 1g (50,60 мг) последовательно добавляли. Смесь перемешивали при 25°C в течение 1 ч. Воду (25 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (80 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 26 (25 мг). МС m/z (ИЭР): 551,8 [M+H]+.
1H ЯМР (400 МГц, ДМСО) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,04 (с, 1H), 8,77-8,73 (м, 2H), 8,38 (дд, J = 8,8, 2,0 Гц, 1H), 8,25 (дд, J = 8,4, 2,0 Гц, 1H), 8,03 (д, J = 4,4 Гц, 1H), 7,97 (д, J = 8,8 Гц, 1H), 7,05-6,47 (м, 2H), 6,27 (уш., 1H), 4,96 (с, 1H), 4,67 (с, 1H), 4,17 (д, J = 9,6 Гц, 1H), 3,80-3,65 (м, 2H), 3,63-3,50 (м, 1H), 2,92-2,82 (м,1H), 2,31 (с, 3H), 2,24 (с, 3H), 1,72 (д, J = 8,4 Гц, 1H).
Пример 49 2-(6-(6-((6-(5-Циклобутокси-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 27)

Стадия 1 Получение трет-бутил-3-гидроксил-1H-пиразол-1-карбоксилата (соединение 27b)
Соединение 27a (500 мг), Boc2O (1,30 г), TEA (1,81 г) и DMAP (145,31 мг) последовательно добавляли в реакционную колбу, и затем ТГФ (20 мл) добавляли. Смесь перемешивали при комнатной температуре в течение 16 ч., так что желтая мутная реакционная смесь постепенно становилась прозрачной. После завершения реакции смесь непосредственно концентрировали при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=20:1) с получением соединения 27b (425 мг).
Стадия 2 Получение трет-бутил-3-циклобутокси-1H-пиразол-1-карбоксилата (соединение 27d)
Соединение 27b (360 мг), соединение 27c (215,71 мг) и PPh3 (776,71 мг) растворяли в толуоле (10 мл) и охлаждали на ледяной бане. DIAD (628,74 мг) добавляли, и смесь нагревали в условиях защиты азотом при 110°C и проводили реакцию в течение 6 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=1:20) с получением соединения 27d (374 мг).
Стадия 3 Получение 3-циклобутокси-1H-пиразола (соединение 27e)
Соединение 27d (374 мг) растворяли в метаноле (3 мл), и затем раствор хлорида водорода в 1,4-диоксане (4 н., 3 мл) добавляли. Реакционную смесь удерживали для реакции в условиях защиты азотом при комнатной температуре. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении с получением гидрохлорида соединения 27e (280 мг). МС m/z (ИЭР): 139,1 [M+H]+.
Стадия 4 Получение 2-(5-циклобутокси-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 27f)
Соединение 121a (315 мг), гидрохлорид соединения 27e (239 мг), Cs2CO3 (675,94 мг) и DMF (10 мл) добавляли в реакционную колбу и полностью перемешивали, и затем N,N’-диметилэтандиамин (48,77 мг) и CuI (52,68 мг, 273,84 мкмоль) добавляли. Смесь нагревали до 100 °C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 14 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (40 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=1:1) с получением соединения 27f (300 мг). МС m/z (ИЭР): 287,9 [M+H]+.
Стадия 5 Получение 6-(5-циклобутокси-1H-пиразол-1-ил)никотинальдегида (соединение 27g)
Соединение 27f (300 мг) растворяли в ТГФ (5 мл) и затем хлористоводородную кислоту (2 н., 5 мл) добавляли в раствор. Смесь выдерживали для реакции при комнатной температуре в течение 8 ч. После завершения реакции реакционную смесь разбавляли водой (20 мл), регулировали pH насыщенным водным раствором NaHCO3 до 7-8 и экстрагировали с помощью EA (40 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=1:15) с получением соединения 27g (140 мг). МС m/z (ИЭР): 243,9 [M+H]+.
Стадия 6 Получение 2-(6-(6-((6-(5-циклобутокси-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 27)
Соединение 1g (65 мг), соединение 27g (50,66 мг) и тетраизопропилтитанат (195,65 мг) растворяли в сухом ТГФ (3 мл), и смесь нагревали до 75°C и удерживали для реакции при этой же температуре в течение 1 ч. Триацетоксиборогидрид натрия (182,37 мг) добавляли в реакционную колбу, и смесь выдерживали для реакции при 75°C в течение 18 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (20 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении и очищали посредством препаративной ВЭЖХ с получением соединения 27 (20 мг). МС m/z (ИЭР): 589,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,13 (д, J = 2,4 Гц, 1H), 8,44 (дд, J = 8,8, 2,4 Гц, 1H), 8,40 (д, J = 2,4 Гц, 1H), 8,33 (д, J = 2,0 Гц, 1H), 7,91 (дд, J = 8,4, 2,4 Гц, 1H), 7,66 (д, J = 8,4 Гц, 1H), 7,12-6,69 (м, 2H), 6,49-6,14 (м, 1H), 6,02 (д, J = 2,8 Гц, 1H), 4,90 (p, J = 7,6 Гц, 1H), 3,83-3,73 (м, 2H), 3,73-3,66 (м, 2H), 3,65-3,47 (м, 4H), 2,60-2,53 (м, 1H), 2,46-2,38 (м, 2H), 2,33 (с, 3H), 2,26 (с, 3H), 2,13-2,03 (м, 2H), 1,82-1,73 (м, 1H), 1,67-1,56 (м, 2H).
Пример 50 2-(6-(6-((6-(4-Хлор-3-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин и 2-(6-(6-((6-(4-хлор-5-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 28/соединение 28’)
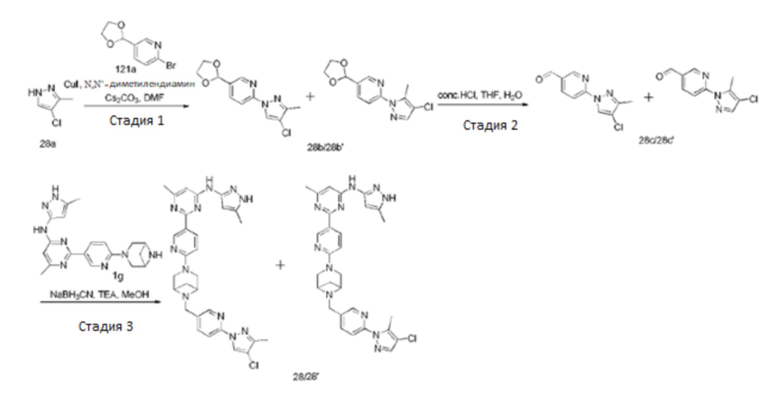
Стадия 1 Получение 2-(4-хлор-3-метил-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина и 2-(4-хлор-5-метил-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 28b/соединение 28b’)
Соединение 121a (202,0 мг), соединение 28a (102,3 мг), CuI (167,2 мг), N,N’-диметилэтандиамин (77,3 мг) и Cs2CO3 (856,1 мг) растворяли в DMF (5,0 мл) в условиях защиты азотом. Смесь нагревали до 120°C и удерживали для реакции при этой же температуре до полного превращения исходных материалов. После завершения реакции реакционную смесь промывали насыщенным водным раствором карбоната натрия и экстрагировали с помощью EA (30 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ТСХ с получением смеси соединения 28b и соединения 28b’ (230,0 мг). МС m/z (ИЭР): 266,0 [M+H]+.
Стадия 2 Получение 6-(4-хлор-3-метил-1H-пиразол-1-ил)никотинальдегида и 6-(4-хлор-5-метил-1H-пиразол-1-ил)никотинальдегида (соединение 28c/соединение 28c’)
Смесь (230,0 мг) соединения 28b и соединения 28b’ добавляли в перемешанный растворитель концентрированной хлористоводородной кислоты (3 мл), ТГФ (10 мл) и воды (10 мл), и смесь выдерживали для реакции при 25°C, до полного превращения исходных материалов. Реакционную смесь концентрировали досуха при пониженном давлении с получением смеси (39,0 мг) соединения 28c и соединения 28c’. МС m/z (ИЭР): 222,1 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(4-хлор-3-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина и 2-(6-(6-((6-(4-хлор-5-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 28/соединение 28’)
Смесь (29,0 мг) соединения 28c и соединения 28c’ и трифторацетат соединения 1g (30,0 мг) добавляли в метанол (0,5 мл), и затем триэтиламин (6,4 мг) и цианоборогидрид натрия (19,8 мг) последовательно добавляли. Смесь выдерживали для реакции при комнатной температуре до полного превращения исходных материалов. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 28 (2,0 мг, время сбора 5,6-6,0 мин.), МС m/z (ИЭР): 568,2 [M+H]+, и соединения 28' (1,0 мг, время сбора 5,0-5,4 мин.), МС m/z (ИЭР): 568,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,64 (с, 1H), 9,13 (д, J = 2,0 Гц, 1H), 8,69 (с, 1H), 8,45 (д, J = 8,0 Гц, 1H), 8,40 (с, 1H), 7,98 (д, J = 8,8 Гц, 1H), 7,82 (д, J = 8,4 Гц, 1H), 6,89 (д, J = 8,8 Гц, 1H), 6,76-6,67 (м, 1H), 6,38-6,23 (м, 1H), 3,80-3,55 (м, 8H), 2,62-2,58 (м, 1H), 2,34 (с, 3H), 2,28 (с, 3H), 2,27 (с, 3H), 1,61 (д, J = 8,0 Гц, 1H) (Соединение 28).
1H ЯМР (400 МГц, ДМСО-d6) δ 12,03 (с, 1H), 9,68 (с, 1H), 9,18 (д, J = 2,4 Гц, 1H), 8,51 (д, J = 2,4 Гц, 1H), 8,48 (д, J = 2,4 Гц, 1H), 8,05 (дд, J = 8,4, 2,4 Гц, 1H), 7,90 (с, 1H), 7,83 (д, J = 8,4 Гц, 1H), 6,85 (д, J = 9,2 Гц, 1H), 6,82-6,69 (м, 1H), 6,43-6,29 (м, 1H), 3,86-3,56 (м, 8H), 2,63 (с, 3H), 2,62-2,58 (м, 1H), 2,39 (с, 3H), 2,31 (с, 3H), 1,66 (д, J = 8,4 Гц, 1H) (Соединение 28').
Пример 51 2-(6-(4-((5-Фторпиридин-3-ил)окси)пиперидин-1-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 29)
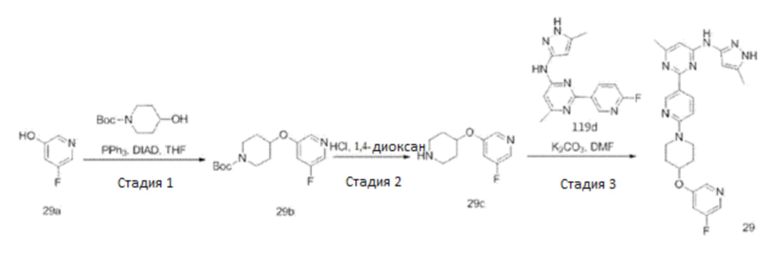
Стадия 1 Получение трет-бутил-4-((5-фторпиридин-3-ил)-окси)пиперидин-1-карбоксилата (соединение 29b)
В условиях защиты азотом соединение 29a (425,6 мг), N-Boc-4-гидроксипиперидин (505,0 мг) и PPh3 (1,3 г) растворяли в сухом ТГФ (5,0 мл) и охлаждали до 0°C. DIAD (1,0 г) добавляли по каплям. Затем смесь медленно нагревали до 25°C и выдерживали для реакции при этой же температуре до полного превращения исходных материалов. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной хроматографии с получением соединения 29b (736,2 мг). МС m/z (ИЭР): 297,1 [M+H]+.
Стадия 2 Получение гидрохлорида 3-фтор-5-(пиперидин-4-илокси)пиридина (соединение 29c)
Соединение 29b (555,5 мг) добавляли в раствор хлорида водорода в 1,4-диоксане (4 н., 20 мл). Смесь выдерживали для реакции при 25°C до полного превращения исходных материалов. Реакционную смесь концентрировали досуха при пониженном давлении с получением гидрохлорида соединения 29c (390,0 мг). МС m/z (ИЭР): 197,1 [M+H]+.
Стадия 3 Получение 2-(6-(4-((5-фторпиридин-3-ил)окси)пиперидин-1-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 29)
Гидрохлорид соединения 29c (86,0 мг), соединение 119d (55,5 мг) и K2CO3 (122,7 мг) добавляли в DMF (5 мл), нагревали до 125°C и выдерживали для реакции при этой же температуре до полного превращения исходных материалов. После завершения реакции реакционную смесь промывали насыщенным водным раствором карбоната натрия и экстрагировали с помощью EA (10 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 29 (17,0 мг). МС m/z (ИЭР): 460,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,41 (с, 1H), 11,12 (с, 1H), 8,97 (д, J = 2,4 Гц, 1H), 8,31 (дд, J = 9,2, 2,4 Гц, 1H), 8,25 (т, J = 1,6 Гц, 1H), 8,20 (д, J = 2,4 Гц, 1H), 7,59 (дт, J = 11,6, 2,4 Гц, 1H), 7,14 (д, J = 9,2 Гц, 1H), 6,90-6,80 (м, 2H), 4,87-4,81 (м, 1H), 4,19-4,13 (m , 2H), 3,85-3,73 (м, 2H), 2,47 (с, 3H), 2,28 (с, 3H), 2,11-2,05 (м, 2H), 1,72-1,63 (м, 2H).
Пример 52 2-(6-(6-((R)-1-(6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин и 2-(6-(6-((S)-1-(6-(4-фтор-1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 52-1/соединение 52-2)
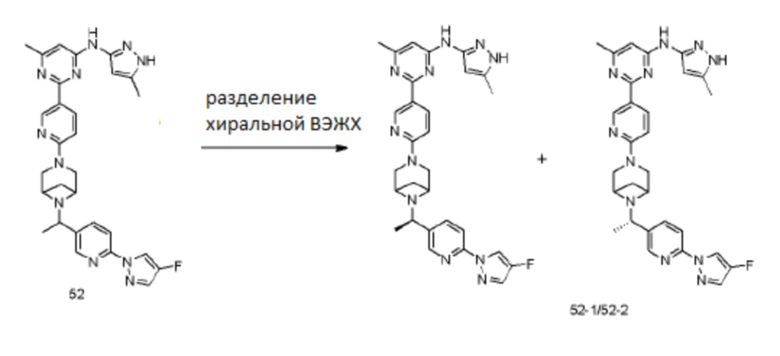
Соединение 52 (100 мг) разделяли посредством хиральной препаративной ВЭЖХ с получением соединения 52-1 (время удерживания 6,788 мин.) и соединения 52-2 (время удерживания 10,115 мин.). Два соединения выделяли и определяли на основе времени удерживания хирального повторного раствора. Таким образом, получали соединение 52-1 (14 мг, ee: 98,85%), MS m/z (ИЭР): 552,3 [M+H]+; и соединение 52-2 (20 мг, ee: 99,22%), MS m/z (ИЭР): 552,3 [M+H]+.
Условия расщепления хиральной ВЭЖХ:
Модель прибора: Shimadzu LC-20AD; хроматографическая колонка: CHIRALPAK IE (IE00CD-RH008), 0,46 см I.D. ×15 см L; температура хроматографической колонки: 35°C; расход: 1,0 мл/мин.; длина волны обнаружения: 254 нм; подвижная фаза: MeOH:CAN:DEA=80:20:0,1 (об./об./об.); время появления соединения 52-1: 6,788 мин., время появления соединения 52-2: 10,115 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,11 (д, J = 2,0 Гц, 1H), 8,66 (д, J = 4,6 Гц, 1H), 8,43 (д, J = 2 Гц, 1H), 8,41 (д, J = 2,4 Гц, 1H), 8,01 (дд, J = 8,5, 1,9 Гц, 1H), 7,90 (дд, J = 16,8, 6,4 Гц, 2H), 6,82(уш., 1H), 6,75(д, J = 8,8 Гц, 1H), 6,29(уш., 1H), 3,95-3,81 (м, 2H), 3,77 (к, J = 6,1 Гц, 1H), 3,63 (с, 1H), 3,53-3,38 (м, 3H), 2,55-2,53 (м, 1H), 2,32 (с, 3H), 2,25 (с, 3H), 1,55 (д, J = 8,4 Гц, 1H), 1,22 (д, J = 6,2 Гц, 3H). (соединение 52-1)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,11 (д, J = 2,0 Гц, 1H), 8,66 (д, J = 4,6 Гц, 1H), 8,43 (д, J = 2 Гц, 1H), 8,41 (д, J = 2,4 Гц, 1H), 8,00 (дд, J = 8,5, 1,9 Гц, 1H), 7,89 (дд, J = 16,8, 6,4 Гц, 2H), 6,82(уш., 1H), 6,75(д, J = 8,8 Гц, 1H), 6,27(уш., 1H), 3,95-3,81 (м, 2H), 3,76 (к, J = 6,1 Гц, 1H), 3,62 (с, 1H), 3,53-3,38 (м, 3H), 2,55-2,51 (м, 1H), 2,32 (с, 3H), 2,25 (с, 3H), 1,54 (д, J = 8,4 Гц, 1H), 1,21 (д, J = 6,2 Гц, 3H). (Соединение 52-2)
Пример 53 2-(6-(6-((6-(5-Циклопропокси-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 30)
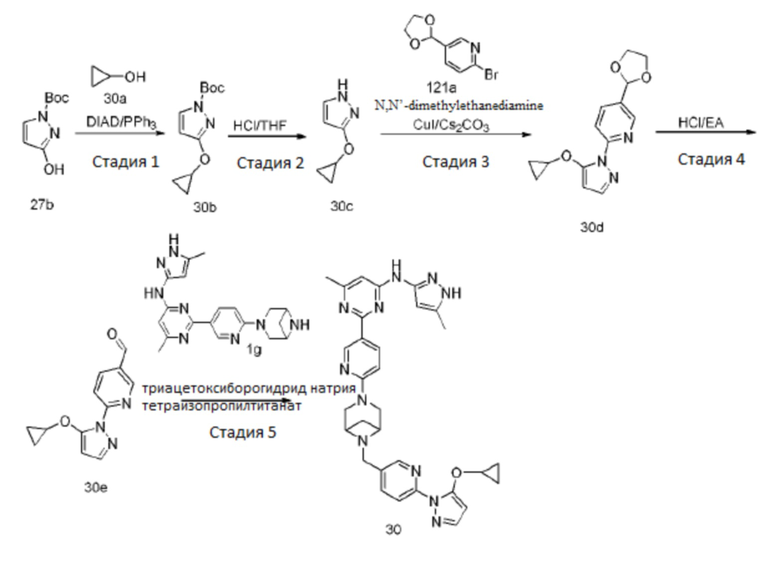
Стадия 1 Получение трет-бутил-3-циклопропокси-1H-пиразол-1-карбоксилата (соединение 30b)
Соединение 27b (300 мг), соединение 30a (141,89 мг) и трифенилфосфин (640,09 мг) добавляли в толуол (10 мл) и охлаждали до 0°C. DIAD (493,51 мг) добавляли по каплям. Смесь нагревали до 110°C и выдерживали для реакции при этой же температуре в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=23:77) с получением соединения 30b (60 мг).
Стадия 2 Получение 3-циклопропокси-1H-пиразола (соединение 30c)
Соединение 30b (102 мг) добавляли в перемешанный раствор хлорида водорода (2 н., 2 мл) в 1,4-диоксане и ТГФ (4 мл). Смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида соединения 30c (73 мг). МС m/z (ИЭР): 125,1 [M+H]+.
Стадия 3 Получение 2-(5-циклопропокси-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 30d)
Гидрохлорид соединения 30c (69,81 мг), соединение 121a (100 мг), N,N'-диметилэтандиамин (38,32 мг), карбонат цезия (424,87 мг) и CuI (82,78 мг) последовательно добавляли в DMF (10 мл). Смесь нагревали до 110°C и перемешивали при этой же температуре в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью ДХМ (50 мл×2). Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 30d (80 мг). МС m/z (ИЭР): 273,9 [M+H]+.
Стадия 4 Получение 6-(5-циклопропокси-1H-пиразол-1-ил)никотинальдегида (соединение 30e)
Соединение 30d (80 мг) добавляли в перемешанный раствор хлорида водорода (4 н., 3 мл) в 1,4-диоксане и EA (2 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли с помощью ДХМ (30 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 30e (55 мг). МС m/z (ИЭР): 229,9 [M+H]+.
Стадия 5 Получение 2-(6-(6-((6-(5-циклопропокси-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 30)
Сухой ТГФ (2 мл) добавляли в соединение 30e (40 мг), соединение 1g (40 мг) и тетраизопропилтитанат (26,01 мг), присутствующий в реакционной колбе объемом 5 мл. Смесь нагревали до 75°C и перемешивали при этой же температуре в течение 16 ч. Затем триацетоксиборогидрид натрия (26,01 мг) добавляли, и смесь перемешивали при 75 °C в течение 8 ч. Реакционную смесь охлаждали до комнатной температуры, и насыщенный водный раствор хлорида аммония (0,1 мл) добавляли для гашения реакционной смеси. Реакционную смесь концентрировали и предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=10:1) с получением 25 мг неочищенного продукта (Rf=0,35-0,45), который дополнительно отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 30 (10 мг). МС m/z (ИЭР): 575,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,95 (с, 1H), 9,62 (с, 1H), 9,12 (д, J = 2,3 Гц, 1H), 8,46-8,41 (м, 2H), 8,34 (д, J = 2,2 Гц, 1H), 7,92 (дд, J = 8,4, 2,2 Гц, 1H), 7,67 (д, J = 8,4 Гц, 1H), 6,78 (д, J = 9,0 Гц, 2H), 6,29 (с, 1H), 6,17 (д, J = 2,7 Гц, 1H), 4,10 (tt, J = 6,0, 3,2 Гц, 1H), 3,84-3,68 (м, 4H), 3,66-3,48 (м, 4H), 2,60-2,54 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H), 0,79-0,68 (м, 4H).
Пример 54 2-(6-(6-((6-(3-(Фторметил)-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 31)
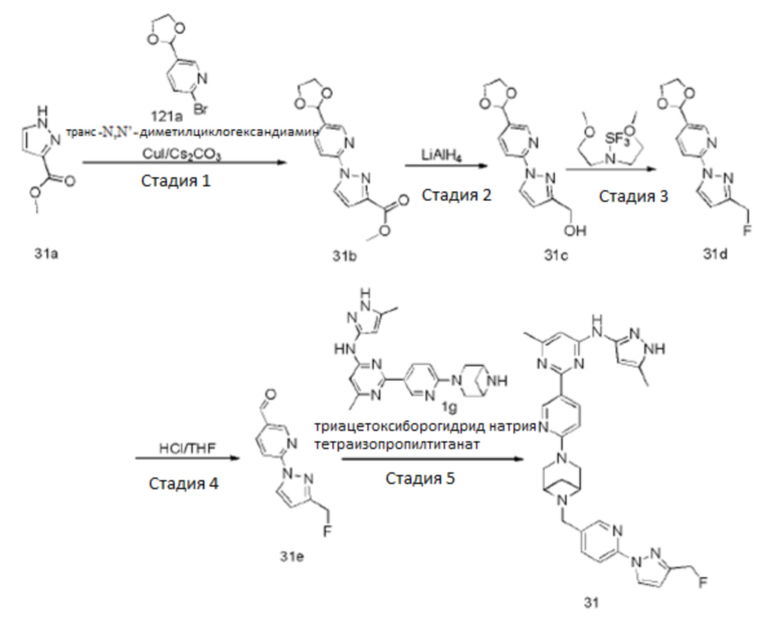
Стадия 1 Получение метил-1-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пиразол-3-карбоксилата (соединение 31b)
Соединение 121a (2,0 г), соединение 31a (1,12 г), Cs2CO3 (5,72 г), транс-N,N’-диметилциклогександиамин (504,72 мг), CuI (337,89 мг) и DMF (10 мл) добавляли в реакционную колбу. Смесь нагревали до 90°C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 2 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, промывали водой, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=20:80) с получением соединения 31b (2,2 г). МС m/z (ИЭР): 276,0 [M+H]+.
Стадия 2 Получение (1-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пиразол-3-ил)метанола (соединение 31c)
Соединение 31b (2,2 г) растворяли в ТГФ (20 мл) и охлаждали до -20°C. LiAlH4 (464,31 мг) медленно добавляли в реакционную смесь порциями, и смесь выдерживали для реакции при этой температуре в течение 15 мин. После завершения реакции EA (1 мл) медленно добавляли по каплям для поглощения избытка LiAlH4. Затем воду (1 мл) добавляли по каплям для гашения реакционной смеси. Смесь разбавляли водой (20 мл) и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=50:50) с получением соединения 31c (1,24 г). МС m/z (ИЭР): 248,0 [M+H]+.
Стадия 3 Получение 5-(1,3-диоксолан-2-ил)-2-(3-(фторметил)-1H-пиразол-1-ил)пиридина (соединение 31d)
В условиях защиты азотом сухой ДХМ (20 мл) охлаждали до -40°C, и затем бис(2-метоксиэтил)аминосеры трифторид (2,86 г) медленно добавляли по каплям. Раствор соединения 31c (0,8 г) в ДХМ (20 мл) добавляли по каплям в реакционную смесь. Затем реакционную смесь медленно нагревали до 25°C и удерживали для реакции при этой же температуре в течение 20 ч. После завершения реакции реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия и после завершения высвобождения пузырьков экстрагировали с помощью ДХМ (20 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=20:80) с получением соединения 31d (200 мг). МС m/z (ИЭР): 249,9 [M+H]+.
Стадия 4 Получение 6-(3-(фторметил)-1H-пиразол-1-ил)никотинальдегида (соединение 31e)
Соединение 31d (200 мг) растворяли в ТГФ (5 мл) и затем хлористоводородную кислоту (2 н., 2,3 мл) добавляли в раствор. Смесь выдерживали для реакции при комнатной температуре в течение 16 ч. После завершения реакции реакционную смесь разбавляли водой (20 мл), регулировали pH насыщенным водным раствором NaHCO3 до 7-8, непосредственно концентрировали, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=15:85) с получением соединения 31e (110 мг). МС m/z (ИЭР): 206,0 [M+H]+.
Стадия 5 Получение 2-(6-(6-((6-(3-(фторметил)-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 31)
Соединение 1g (87 мг), соединение 31e (110 мг) и тетраизопропилтитанат (278,46 мг) растворяли в сухом ТГФ (10 мл), нагревали до 75°C и выдерживали для реакции при этой же температуре в течение 8 ч. Реакционную смесь охлаждали до 25°C. Триацетоксиборогидрид натрия (259,57 мг) добавляли в реакционную колбу, и смесь выдерживали для реакции при 25°C в течение 12 ч. Воду (1 мл) добавляли по каплям для гашения реакционной смеси. Смесь концентрировали, предварительно очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=90:10), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 31 (25 мг). МС m/z (ИЭР): 551,8 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,63 (с, 1H), 9,13 (д, J = 2,0 Гц, 1H), 8,62 (д, J = 2,4 Гц, 1H), 8,49-8,39 (м, 2H), 8,15 (с, 1H), 7,99 (дд, J = 8,4, 1,6 Гц, 1H), 7,90-7,84 (м, 1H), 7,07-6,74 (м, 2H), 6,70 (с, 1H), 6,30 (уш., 1H), 5,53 (с, 1H), 5,41 (с, 1H), 3,81-3,73 (м, 4H), 3,67-3,58 (м, 4H), 2,63-2,53 (м, 1H), 2,34 (с, 3H), 2,26 (с, 3H), 1,61 (д, J = 8,4 Гц, 1H).
Пример 55 2-(6-(6-((6-(5-Этил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 46)
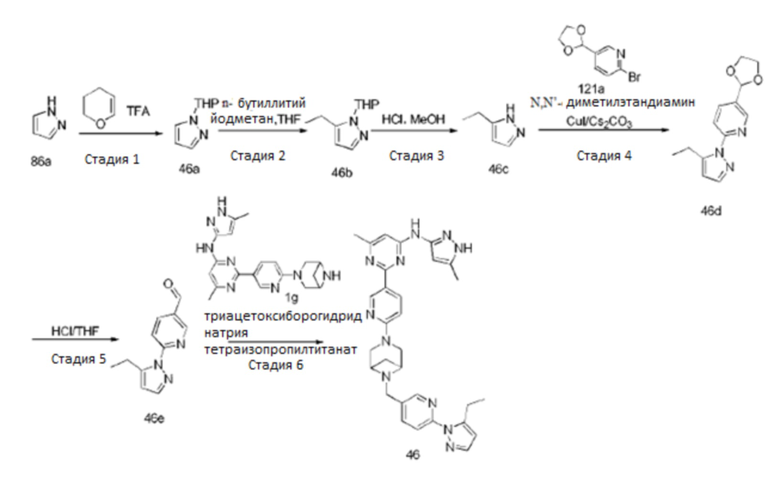
Стадия 1 Получение 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (соединение 46a)
Соединение 86a (2 г) добавляли в 3,4-дигидропиран (6 мл), и затем TFA (334,97 мг) добавляли. Смесь нагревали до 100°C и перемешивали при этой же температуре в течение 16 ч. Воду (100 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=60:1-10:1) с получением соединения 46a (2,4 г). МС m/z (ИЭР): 153,2 [M+H]+.
Стадия 2 Получение 5-этил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (соединение 46b)
Соединение 46a (2 г) добавляли в сухой ТГФ (30 мл), и затем n-бутиллитий (2,5 M, 6,31 мл) добавляли при -78 °C. Смесь перемешивали в течение 1 ч. Затем йодоэтан (3,07 г) медленно добавляли при -78°C. Смесь медленно нагревали до комнатной температуры и перемешивали при этой же температуре в течение 5 ч. Метанол (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Силикагель непосредственно смешивали с образцами, который отделяли и очищали посредством колоночной хроматографии (PE:EA=40:1-5:1) с получением соединения 46b (721 мг). МС m/z (ИЭР): 181,0 [M+H]+.
Стадия 3 Получение 5-этил-1H-пиразола (соединение 46c)
Соединение 46b (721 мг) добавляли в MeOH (7 мл), и затем раствор хлорида водорода в 1,4-диоксане (4 н., 1 мл) добавляли. Смесь перемешивали при 25°C в течение 1 ч. Насыщенный водный раствор бикарбоната натрия (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (80 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Затем органические фазы сушили в центрифуге с получением соединения 46c (185 мг). МС m/z (ИЭР): 97,1 [M+H]+.
Стадия 4 Получение 5-(1,3-диоксолан-2-ил)-2-(5-этил-1H-пиразол-1-ил)пиридина (соединение 46d)
Соединение 121a (401 мг) и соединение 46c (184,31 мг) добавляли в DMF (15 мл), и затем N,N-диметилэтандиамин (153,39 мг), карбонат цезия (1,7 г) и CuI (331,96 мг) последовательно добавляли. Смесь нагревали до 110°C и перемешивали в условиях защиты азотом при этой же температуре в течение 10 ч. Воду (100 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=40:1-3:1) с получением соединения 46d (199 мг). МС m/z (ИЭР): 245,9 [M+H]+.
Стадия 5 Получение 6-(5-этил-1H-пиразол-1-ил)никотинальдегида (соединение 46e)
Соединение 46d (200 мг) добавляли в ТГФ (4 мл) и H2O (2 мл), и затем раствор хлорида водорода в 1,4-диоксан (4 н., 1 мл) добавляли. Смесь перемешивали при 25°C в течение 5 ч. Насыщенный водный раствор бикарбоната натрия (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=40:1-3:1) с получением соединения 46c (91 мг). МС m/z (ИЭР): 202,1 [M+H]+.
Стадия 6 Получение 2-(6-(6-((6-(5-этил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 46)
Соединение 1g (40,93 мг), соединение 46e (25 мг) и тетраизопропилтитанат (128,41 мг) добавляли в сухой ТГФ (10 мл), и после замещения азотом три раза перемешивали при 75°C в течение 10 ч. Затем триацетоксиборогидрид натрия (119,69 мг) добавляли порциями, и смесь перемешивали при 75°C в течение дополнительных 6 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 46 (8 мг). МС m/z (ИЭР): 547,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,0 Гц, 1H), 8,47 (д, J = 2,4 Гц, 1H), 8,44 (дд, J = 9,20, 2,4 Гц, 1H), 8,36 (д, J = 1,6 Гц, 1H), 7,93 (дд, J = 8,4, 2,0 Гц, 1H), 7,81 (д, J = 8,4 Гц, 1H), 7,20-6,69 (м, 2H), 6,40 (д, J = 2,8 Гц, 1H), 6,32 (с, 1H), 3,83-3,68 (м, 4H), 3,66-3,53 (м, 4H), 2,66 (к, J = 7,6 Гц, 2H), 2,61-2,53 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H), 1,23 (т, J = 7,6 Гц, 3H).
Пример 56 2-(6-(6-((6-(4-Фтор-5-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 122)
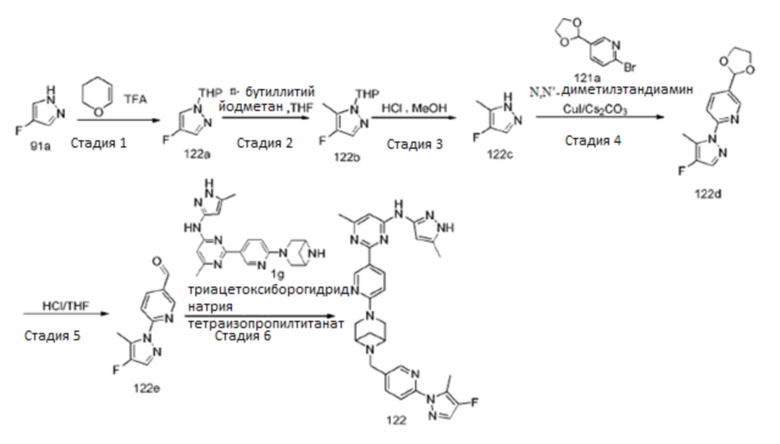
Стадия 1 Получение 4-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (соединение 122a)
Соединение 91a (500 мг) добавляли в 3,4-дигидропиран (2,5 мл), и затем TFA (129,96 мг) добавляли. Смесь нагревали до 100°C и перемешивали при этой же температуре в течение 16 ч. Воду (100 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=50:1-10:1) с получением соединения 122a (901 мг). МС m/z (ИЭР): 171,1 [M+H]+.
Стадия 2 Получение 4-фтор-5-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (соединение 122b)
Соединение 122a (900 мг) добавляли в сухой ТГФ (10 мл), и затем н-бутиллитий (2,5 M, 2,33 мл) добавляли при -78°C. Смесь перемешивали в течение 1 ч. Затем йодметан (1,13 г) медленно добавляли при -78°C. Смесь медленно нагревали до комнатной температуры и перемешивали при этой же температуре в течение 3 ч. Метанол добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (80 ×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=60:1-10:1) с получением соединения 122b (845 мг) в виде светло-желтой жидкости. МС m/z (ИЭР): 185,0 [M+H]+.
Стадия 3 Получение 4-фтор-5-метил-1H-пиразола (соединение 122c)
Соединение 122b (800 мг) добавляли в MeOH (7 мл), и затем раствор хлорида водорода (4 н., 7 мл) в 1,4-диоксане добавляли. Смесь перемешивали при 25°C в течение 1 ч. Насыщенный водный раствор бикарбоната натрия (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (80 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 122c (300 мг). МС m/z (ИЭР): 101,2 [M+H]+.
Стадия 4 Получение 5-(1,3-диоксолан-2-ил)-2-(4-фтор-5-метил-1H-пиразол-1-ил)пиридина (соединение 122d)
Соединение 121a (300 мг) и соединение 122c (143,58 мг) добавляли в DMF (15 мл), и затем N,N-диметилэтандиамин (114,75 мг), карбонат цезия (1,27 г) и CuI (248,35 мг) последовательно добавляли. Смесь нагревали до 110°C и перемешивали в условиях защиты азотом при этой же температуре в течение 10 ч. Воду (100 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=50:1-10:1) с получением соединения 122d (254 мг). МС m/z (ИЭР): 249,9 [M+H]+.
Стадия 5 Получение 6-(4-(фтор-5-метил)-1H-пиразол-1-ил)никотинальдегида (соединение 122e)
Соединение 122d (240 мг) добавляли в ТГФ (5 мл) и H2O (2 мл), и затем раствор хлорида водорода (4 н., 991,74 мкл) в 1,4-диоксане добавляли. Смесь перемешивали при 25°C в течение 5 ч. Насыщенный водный раствор бикарбоната натрия (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 122e (55 мг). МС m/z (ИЭР): 205,9 [M+H]+.
Стадия 6 Получение 2-(6-(6-((6-(4-фтор-5-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 122)
Соединение 1g (101 мг), соединение 122e (51,98 мг) и тетраизопропилтитанат (288,01 мг) добавляли в сухой ТГФ (25 мл), и после замещения азотом три раза перемешивали при 75°C в течение 10 ч. Затем триацетоксиборогидрид натрия (268,46 мг) добавляли порциями, и смесь перемешивали при 75°C в течение дополнительных 6 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 122 (22 мг). МС m/z (ИЭР): 551,8 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,99 (с, 1H), 9,65 (с, 1H), 9,13 (д, J = 4,0 Гц, 1H), 8,57 (д, J = 4,8 Гц, 1H), 8,44 (дд, J = 8,8, 2,0 Гц, 1H), 8,37 (с, 1H), 7,94 (дд, J = 8,4, 1,6 Гц, 1H), 7,80 (д, J = 8,4 Гц, 1H), 6,97-6,74 (м, 2H), 6,31 (с, 1H), 3,81-3,69 (м, 4H), 3,66-3,52 (м, 4H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,28 (с, 3H), 2,24 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 57 2-(6-(6-((6-(1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло [3.1.1] гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 123)
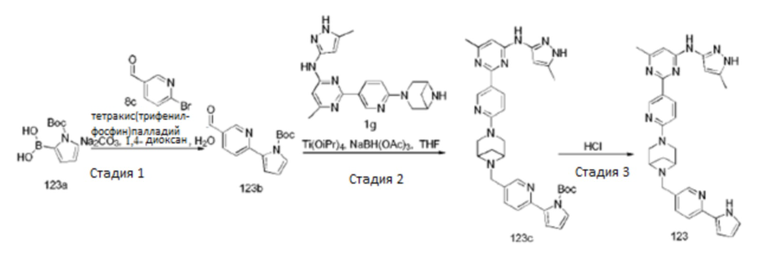
Стадия 1 Получение трет-бутил-2-(5-формилпиридин-2-ил)-1H-пиррол-1-карбоксилата (соединение 123b)
Соединение 123a (1,13 г), соединение 8c (1,0 g), тетракис(трифенилфосфин)палладий (310,62 мг) и карбонат натрия (1,71 г) добавляли в 1,4-диоксан (60 мл) и воду (15 мл). Смесь нагревали до 95°C и выдерживали для реакции в условиях защиты азотом при этой же температуре в течение 14 ч. После завершения реакции смесь концентрировали с удалением части органического растворителя и экстрагировали с помощью EA. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=5:1) с получением соединения 123b (1,25 г).
Стадия 2 Получение трет-бутил-2-(5-((3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиррол-1-карбоксилата (соединение 123c)
Соединение 1g (50 мг), соединение 123b (37,57 мг) и тетраизопропилтитанат (156,84 мг) добавляли в сухой ТГФ (5 мл), и смесь перемешивали при 72°C в течение 18 ч. Затем триацетоксиборогидрид натрия (146,19 мг) добавляли, и смесь выдерживали для реакции при 72°C в течение 4 ч. После завершения реакции смесь очищали непосредственно посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=93:7) с получением соединения 123c (40 мг). МС m/z (ИЭР): 618,9 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 123)
Раствор хлорида водорода в 1,4-диоксане (4 н., 2,0 мл) добавляли по каплям в раствор соединения 123c (40 мг) в метаноле (2 мл). Смесь выдерживали для реакции при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь концентрировали досуха. Неочищенный продукт растворяли в метаноле, избыток карбоната калия добавляли, и смесь перемешивали при комнатной температуре в течение 0,5 ч. до свободного гидрохлорида. Смесь фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 123 (22 мг). МС m/z (ИЭР): 518,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 11,43 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,49-8,35 (м, 2H), 7,70 (д, J = 7,9 Гц, 1H), 7,61 (д, J = 8,2 Гц, 1H), 6,93-6,59 (м, 4H), 6,31 (с, 1H), 6,12 (дд, J = 5,7, 2,5 Гц, 1H), 3,85-3,68 (м, 4H), 3,65-3,49 (м, 4H), 2,59-2,54 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,2 Гц, 1H).
Пример 58 2-(6-(6-((6-(3-Циклопропил-4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 124)
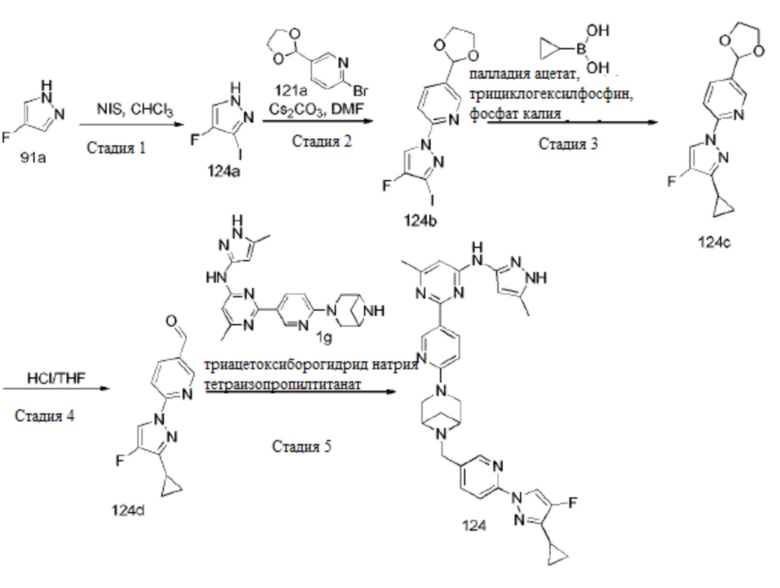
Стадия 1 Получение 4-фтор-3-йод-1H-пиразола (соединение 124a)
4-Фторпиразол (1,5 г), NIS (4,31 г) и хлороформ (30 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали в условиях защиты азотом при 80°C в течение 7 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры. Силикагель непосредственно смешивали с образцами, которые отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=4:1) с получением соединения 124a (1,2 г). МС m/z (ИЭР): 213 [M+H]+.
Стадия 2 Получение 5-(1,3-диоксолан-2-ил)-2-(4-фтор-3-йод-1H-пиразол-1-ил)пиридина (соединение 124b)
Соединение 121a (576,33 мг), соединение 124a (354 мг), карбонат цезия (1,63 г) и DMF (15 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали при 90°C в течение 12 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали с помощью EA. Органическую фазу промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колонки C18 (ацетонитрил:0,05% водный раствор бикарбоната аммония=68:32) с получением соединения 124b (300 мг). МС m/z (ИЭР): 362 [M+H]+.
Стадия 3 Получение 2-(3-циклопропил-4-фтор-1H-пиразол-1-ил)-5-(1,3-диоксолан-2-ил)пиридина (соединение 124c)
Циклопропилбороновую кислоту (89,20 мг), соединение 124b (125 мг), палладия ацетат (15,54 мг), фосфат калия (257,17 мг), трициклогексилфосфин (9,71 мг), толуол (5 мл) и воду (1 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали в условиях защиты азотом при 90°C в течение 12 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры. Силикагель непосредственно смешивали с образцами, которые отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 124c (70 мг). МС m/z (ИЭР): 276 [M+H]+.
Стадия 4 Получение 6-(3-циклопропил-4-фтор-1H-пиразол-1-ил)никотинальдегида (соединение 124d)
Соединение 124c (70 мг) растворяли в перемешанном растворе ТГФ (8 мл) и воде (8 мл), и затем концентрированную хлористоводородную кислоту (5 мл, 37%) добавляли по каплям. Смесь перемешивали при 25°C в течение 18 ч. После завершения реакции реакционную смесь сушили в центрифуге с удалением части растворителя, регулировали pH насыщенным водным раствором бикарбоната натрия до около 9 и затем экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=3:1) с получением соединения 124d (18 мг). МС m/z (ИЭР): 232 [M+H]+.
Стадия 5 Получение 2-(6-(6-((6-(3-циклопропил-4-фтор-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 124)
Соединение 124d (18 мг), соединение 1g (31,04 мг), изопропилтитанат (88,50 мг) и сухой ТГФ (5 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали в условиях защиты азотом при 75°C в течение 18 ч. Затем триацетоксиборогидрид натрия (82,49 мг) добавляли порциями, и смесь перемешивали при 75°C в течение дополнительных 6 ч. После завершения реакции реакционную смесь сушили в центрифуге с удалением части растворителя, регулировали pH насыщенным раствором бикарбоната натрия до около 9 и затем экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали, предварительно очищали посредством препаративной ТСХ (ДХМ:MeOH=9:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 124 (15 мг). МС m/z (ИЭР): 577,9 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,65 (с, 1H), 9,12 (д, J = 2,1 Гц, 1H), 8,55 (д, J = 4,6 Гц, 1H), 8,44 (дд, J = 8,9, 2,2 Гц, 1H), 8,35 (д, J = 1,5 Гц, 1H), 7,93 (дд, J = 8,5, 2,0 Гц, 1H), 7,76 (д, J = 8,5 Гц, 1H), 6,82 (уш., 1H), 6,79 (д, J = 12Гц, 1H), 6,30 (уш., 1H),3,84-3,75 (м, 4H), 3,73-3,49 (м, 4H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 2,02-1,93 (м, 1H), 1,59 (д, J = 8,4 Гц, 1H), 1,03-0,95 (м, 2H), 0,94-0,86 (м, 2H).
Пример 59 2-(6-(6-((R)-1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин и 2-(6-(6-((S)-1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 125-1/соединение 125-2)
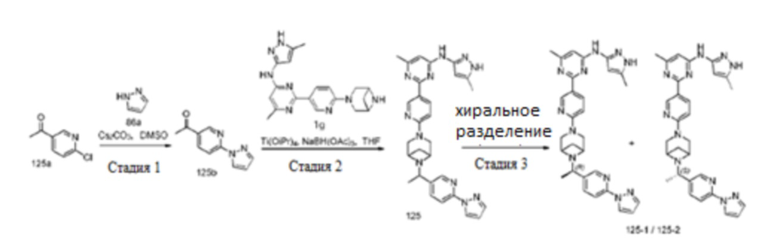
Стадия 1 Получение 1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этанона (соединение 125b)
Соединение 86a (328,18 мг), соединение 125a (500 мг) и карбонат цезия (1,57 г) последовательно добавляли в ДМСО (5 мл). Смесь нагревали до 100 °C и перемешивали при этой же температуре в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали с помощью EA. Органическую фазу промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали посредством колоночной флеш-хроматографии на силикагеле (PE:EA=50:50) с получением соединения 125b (364 мг). МС m/z (ИЭР): 188,1 [M+H]+.
Стадия 2 Получение 2-(6-(6-(1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 125)
Сухой ТГФ (20 мл) добавляли в соединение 125b (309,90 мг), соединение 1g (400 мг) и тетраизопропилтитанат (1,25 г), присутствующий в реакционной колбе объемом 250 мл. Смесь нагревали до 75°C и перемешивали при этой же температуре в течение 16 ч. Затем триацетоксиборогидрид натрия (1,17 мг) добавляли, и смесь перемешивали при 75 °C в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, и насыщенный водный раствор хлорида аммония (3 мл) добавляли для гашения реакционной смеси. Реакционную смесь концентрировали, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=9:1) с получением соединения 125 (270 мг). МС m/z (ИЭР): 533,9 [M+H]+.
Стадия 3 Получение 2-(6-(6-((R)-1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина и 2-(6-(6-((S)-1-(6-(1H-пиразол-1-ил)пиридин-3-ил)этил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 125-1/соединение 125-2)
Соединение 125 (250 мг) разделяли посредством хиральной препаративной ВЭЖХ с получением соединения 125-1 (время удерживания 7,647 мин.) и соединения 125-2 (время удерживания 11,638 мин.). Два соединения выделяли и определяли на основе времени удерживания хирального повторного раствора, и стереоструктуры не были определены. Таким образом, соединение 125-1 (101 мг, ee: 100 %), MS m/z (ИЭР): 533,9 [M+H]+; соединение 125-2 (100 мг, ee: 99,93 %), MS m/z (ИЭР): 533,9 [M+H]+.
Условия расщепления хиральной ВЭЖХ:
Модель прибора: Shimadzu LC-20AD; хроматографическая колонка: CHIRALPAK IE-3 (IE30CD-UL006), 0,46 см I.D. ×15 см L; температура хроматографической колонки: 25 °C; расход: 1,0 мл/мин.; длина волны обнаружения: 254 нм; подвижная фаза: MeOH:ACN:DEA=70:30:0,1 (об./об./об.); время появления соединения 125-1: 7,647 мин., время появления соединения 125-2: 11,638 мин.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,3 Гц, 1H), 8,59 (с, 1H), 8,47-8,39 (м, 2H), 7,99 (д, J = 6,4 Гц, 1H), 7,89 (д, J = 7,4 Гц, 1H), 7,81 (с, 1H), 6,75 (д, J = 9,0 Гц, 2H), 6,56 (с, 1H), 6,31 (с, 1H), 3,96-3,83 (м, 2H), 3,75 (д, J = 3,3 Гц, 1H), 3,63 (с, 1H), 3,54-3,36 (м, 3H), 2,57-2,53 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,54 (д, J = 6,9 Гц, 1H), 1,22 (д, J = 2,5 Гц, 3H). (Соединение 125-1)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,3 Гц, 1H), 8,59 (д, J = 2,6 Гц, 1H), 8,47-8,39 (м, 2H), 8,00 (дд, J = 8,5, 2,2 Гц, 1H), 7,90 (д, J = 8,4 Гц, 1H), 7,81 (д, J = 1,6 Гц, 1H), 6,75 (д, J = 9,0 Гц, 2H), 6,57 (т, J = 2,1 Гц, 1H), 6,31 (с, 1H), 3,96-3,83 (м, 2H), 3,76 (к, J = 6,2 Гц, 1H), 3,63 (с, 1H), 3,54-3,36 (м, 3H), 2,57-2,53 (м, 1H), 2,33 (с, 3H), 2,25 (с, 3H), 1,55 (д, J = 8,4 Гц, 1H), 1,23 (д, J = 6,1 Гц, 3H). (Соединение 125-2)
Пример 60 2-(6-(6-((6-(4-Фторметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 126) и (1-(5-((3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-4-ил)метанол (соединение 127)
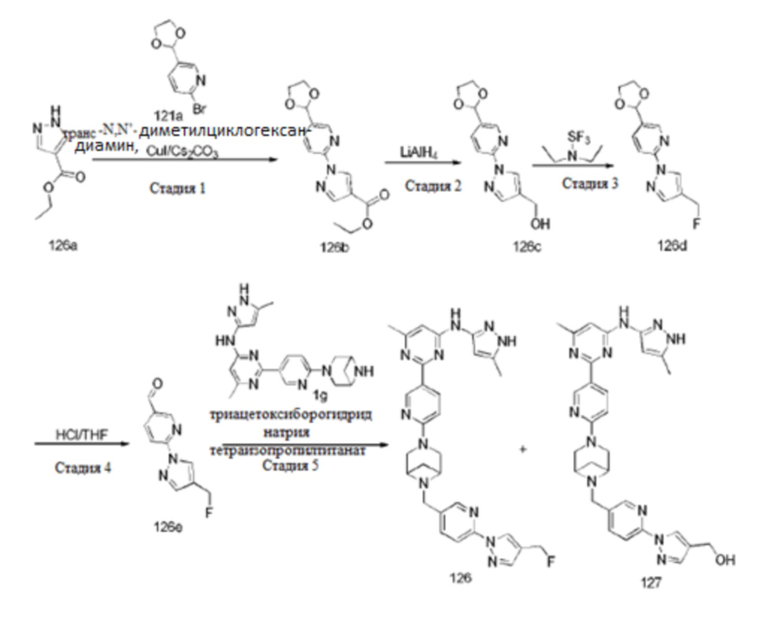
Стадия 1 Получение этил-1-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пиразол-4-карбоксилата (соединение 126b)
Соединение 121a (3,0 г), соединение 126a (1,86 г), Cs2CO3 (8,67 г), транс-N,N’-диметилциклогександиамин (757,08 мг), CuI (506,84 мг) и DMF (15 мл) добавляли в реакционную колбу. Смесь нагревали до 90 °C и удерживали для реакции в условиях защиты азотом при этой же температуре в течение 2 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, промывали водой, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=20:80) с получением соединения 126b (3,31 г). МС m/z (ИЭР): 290,0 [M+H]+.
Стадия 2 Получение (1-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пиразол-4-ил)метанола (соединение 126c)
Соединение 126b (3,31 г) растворяли в ТГФ (30 мл) и охлаждали до -20°C. LiAlH4 (651,40 мг, 16,99 ммоль) медленно добавляли в реакционную смесь порциями, и реакционную смесь выдерживали для реакции при этой температуре в течение 15 мин. После завершения реакции EA (2 мл) медленно добавляли по каплям для поглощения избытка LiAlH4. Затем воду (1 мл) добавляли по каплям для гашения реакционной смеси. Смесь разбавляли водой (20 мл) и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=50:50) с получением соединения 126c (2,30 г). МС m/z (ИЭР): 248,0 [M+H]+.
Стадия 3 Получение 5-(1,3-диоксолан-2-ил)-2-(4-(фторметил)-1H-пиразол-1-ил)пиридина (соединение 126d)
В условиях защиты азотом сухой ДХМ (30 мл) охлаждали до -40°C, и затем диэтиламиносеры трифторид (6,06 г) медленно добавляли по каплям. Раствор соединения 126c (2,30 г) в ДХМ (30 мл) добавляли по каплям в реакционную смесь. Затем реакционную смесь медленно нагревали до 25°C и удерживали для реакции при этой же температуре в течение 20 ч. После завершения реакции реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия и после завершения высвобождения пузырьков экстрагировали с помощью ДХМ (30 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, отделяли и очищали посредством колоночной флеш-хроматографии на силикагеле (EA:PE=20:80) с получением соединения 126d (684 г). МС m/z (ИЭР): 249,9 [M+H]+.
Стадия 4 Получение 6-(4-(фторметил)-1H-пиразол-1-ил)никотинальдегида (соединение 126e)
Соединение 126d (684 мг) растворяли в ТГФ (10 мл) и затем хлористоводородную кислоту (1 н., 5 мл) добавляли в раствор. Смесь выдерживали для реакции при 25 °C в течение 16 ч. После завершения реакции реакционную смесь разбавляли водой (20 мл), регулировали pH насыщенным водным раствором NaHCO3 до 7-8. Смесь непосредственно концентрировали с удалением ТГФ, и затем экстрагировали с помощью ДХМ (30 мл x 3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали при всасывании и концентрировали с получением соединения 126e (460 мг). МС m/z (ИЭР): 206,0 [M+H]+.
Стадия 5 Получение 2-(6-(6-((6-(4-фторметил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 126) и (1-(5-((3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-4-ил)метанола (соединение 127)
Соединение 1g (200 мг), соединение 126e (182 мг) и тетраизопропилтитанат (640 мг) растворяли в сухом ТГФ (40 мл), смесь нагревали до 75°C и выдерживали для реакции при этой же температуре в течение 8 ч. Реакционную смесь охлаждали до 25°C. Триацетоксиборогидрид натрия (597 мг) добавляли в реакционную колбу, и смесь выдерживали для реакции при 25°C в течение 12 ч. Воду (1 мл) добавляли по каплям для гашения реакционной смеси. Реакционную смесь концентрировали, отделяли и предварительно очищали посредством колоночной флеш-хроматографии на силикагеле (ДХМ:MeOH=90:10) с получением неочищенного продукта (смеси соединения 126 и соединения 127), который дополнительно отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 126 (21 мг), МС m/z (ИЭР): 552,3 [M+H]+; соединение 127 (19 мг), МС m/z (ИЭР): 550,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,00 (уш., 1H), 9,65 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,77 (д, J = 3,2 Гц, 1H), 8,44 (дд, J = 8,8, 2,2 Гц, 2H), 7,99 (дд, J = 8,4, 2,0 Гц, 1H), 7,94 (с, 1H), 7,91-7,96 (м, 1H), 6,95-6,69 (м, 2H), 6,30 (уш., 1H), 5,46 (с, 1H), 5,34 (с, 1H), 3,82-3,70 (м, 4H), 3,68-3,52 (м, 4H), 2,59-2,54 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H). (Соединение 126)
1H ЯМР (400 МГц, ДМСО-d6) δ 12,04 (уш., 1H), 9,66 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,52-8,35 (м, 3H), 7,95 (дд, J = 8,4, 1,6 Гц, 1H), 7,88-7,81 (м, 1H), 7,73 (с, 1H), 6,99-6,64 (м, 2H), 6,31 (уш., 1H), 5,05 (уш., 1H), 4,45 (с, 2H), 3,93-3,70 (м, 4H), 3,67-3,51 (м, 4H), 2,59-2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H). (Соединение 127)
Пример 61 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(1-метил-1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 128)
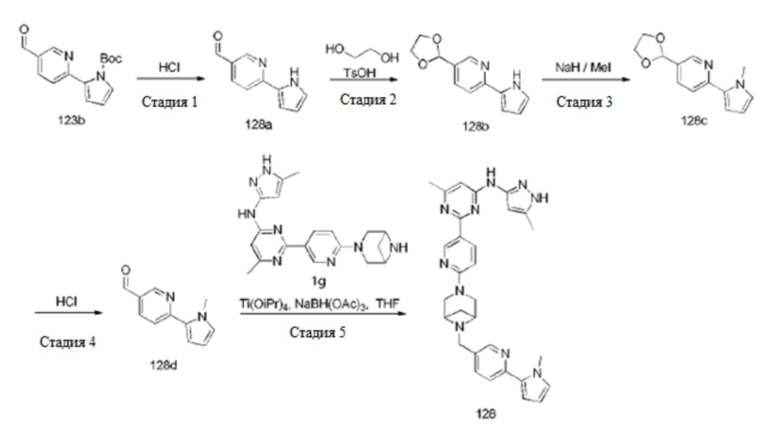
Стадия 1 Получение 6-(1H-пиррол-2-ил)никотинальдегида (соединение 128a)
Раствор хлорида водорода в 1,4-диоксане (4 н., 6,0 мл) добавляли по каплям в раствор соединения 123b (500 мг) в метаноле (10 мл). Смесь выдерживали для реакции при комнатной температуре в течение 2 ч. После завершения реакции реакционную смесь концентрировали досуха. Неочищенный продукт растворяли в метаноле, избыток карбоната калия добавляли, и смесь перемешивали при комнатной температуре в течение 0,5 ч. до свободного гидрохлорида. Смесь фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=4:1) с получением соединения 128a (250 мг). МС m/z (ИЭР): 173,0 [M+H]+.
Стадия 2 Получение 5-(1,3-диоксолан-2-ил)-2-(1H-пиррол-2-ил)пиридина (соединение 128b)
Соединение 128a (250 мг), этиленгликоль (180 мг) и п-метилфенилсульфоновую кислоту (27,6 мг) добавляли в толуол (15 мл), и смесь нагревали с обратным холодильником для отклонения воды в течение 18 ч. После завершения реакции реакционную смесь разбавляли с помощью EA и промывали насыщенным водным раствором бикарбоната натрия. Органические фазы сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением соединения 128b (290 мг). МС m/z (ИЭР): 217,0 [M+H]+.
Стадия 3 Получение 5-(1,3-диоксолан-2-ил)-2-(1-метил-1H-пиразол-2-ил)пиридина (соединение 128c)
Соединение 128b (250 мг) растворяли в сухом DMF (5 мл), NaH (138,7 мг, чистота 60%) добавляли в условиях защиты азотом при 0°C, и затем смесь перемешивали в течение 15 мин. Йодметан (820 мг) добавляли, и смесь выдерживали для реакции при комнатной температуре в течение 5 ч. После завершения реакции реакционную смесь выливали в насыщенный солевой раствор, экстрагировали с помощью EA, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=5:1) с получением соединения 128c (125 мг). МС m/z (ИЭР): 231,0 [M+H]+.
Стадия 4 Получение 6-(1-метил-1H-пиррол-2-ил)никотинальдегид (соединение 128d)
Разбавленную хлористоводородную кислоту (2 н., 2,0 мл) добавляли по каплям в раствор соединения 128c (110 мг) в ТГФ (4,0 мл). Смесь выдерживали для реакции при комнатной температуре в течение 3 ч. После завершения реакции реакционную смесь концентрировали досуха. Неочищенный продукт растворяли в метаноле, избыток карбоната калия добавляли, и смесь перемешивали при комнатной температуре в течение 0,5 ч. до свободного гидрохлорида. Смесь фильтровали, концентрировали при пониженном давлении, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=3:1) с получением соединения 128d (75 мг).
Стадия 5 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(1-метил-1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 128)
Соединение 1g (40 мг), соединение 128d (25 мг), и тетраизопропилтитанат (125 мг) добавляли в сухой ТГФ (5 мл), и смесь перемешивали при 72°C в течение 10 ч. Затем триацетоксиборогидрид натрия (117 мг) добавляли, и смесь выдерживали для реакции при 72°C в течение 6 ч. После завершения реакции смесь повторно очищали непосредственно посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=93:7), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 128 (34 мг). МС m/z (ИЭР): 532,9 [M+H]+.
1H ЯМР (400 МГц, CD3OD) δ 9,14 (д, J = 2,1 Гц, 1H), 8,59-8,43 (м, 2H), 7,78 (дд, J = 8,2, 2,2 Гц, 1H), 7,55 (д, J = 8,2 Гц, 1H), 6,85 (д, J = 9,0 Гц, 1H), 6,81-6,65 (м, 2H), 6,50 (дд, J = 3,7, 1,8 Гц, 1H), 6,35 (с, 1H), 6,10 (дд, J = 3,7, 2,7 Гц, 1H), 4,02-3,77 (м, 7H), 3,77-3,59 (м, 4H), 2,73 (д, J = 6,3 Гц, 1H), 2,41 (с, 3H), 2,32 (с, 3H), 1,71 (д, J = 8,8 Гц, 1H).
Пример 62 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(тиазол-4-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 129)
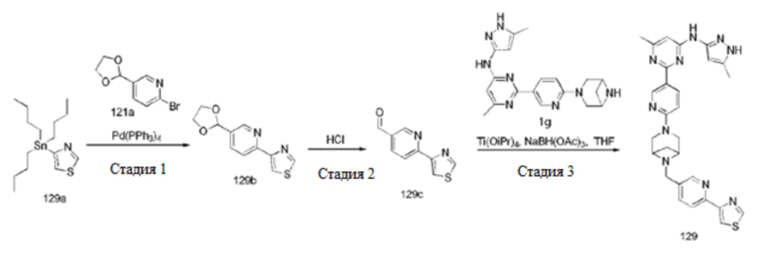
Стадия 1 Получение 4-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)тиазола (соединение 129b)
Соединение 129a (325 мг), соединение 121a (200 мг) и тетракис(трифенилфосфин)палладий (50 мг) добавляли в толуол (10 мл), и смесь выдерживали при 120°C в течение 6 ч. После завершения реакции реакционную смесь концентрировали досуха, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=2:1) с получением соединения 129b (115 мг). МС m/z (ИЭР): 235,1 [M+H]+.
Стадия 2 Получение 6-(тиазол-4-ил)никотинальдегида (соединение 129c)
Разбавленную хлористоводородную кислоту (2,0 мл, 3 N) добавляли по каплям в раствор соединения 129b (115 мг) в ТГФ (3,0 мл). Смесь выдерживали для реакции при комнатной температуре в течение 15 ч. После завершения реакции pH реакционной смеси регулировали до около 10 путем медленного добавления насыщенного раствора бикарбоната натрия по каплям и затем экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 129c (83 мг), которое непосредственно использовали на следующей стадии реакции без очистки. МС m/z (ИЭР): 191,1 [M+H]+.
Стадия 3 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(тиазол-4-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 129)
Соединение 1g (50 мг), соединение 129c (26,2 мг) и тетраизопропилтитанат (156,8 мг) добавляли в сухой ТГФ (5 мл), и смесь перемешивали при 72°C в течение 10 ч. Затем триацетоксиборогидрид натрия (146,2 мг) добавляли, и смесь выдерживали для реакции при 72°C в течение 6 ч. После завершения реакции смесь грубо очищали непосредственно посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=8:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 129 (51 мг). МС m/z (ИЭР): 537,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,22 (д, J = 2,1 Гц, 1H), 9,13 (д, J = 2,1 Гц, 1H), 8,58 (д, J = 1,6 Гц, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,29 (д, J = 2,0 Гц, 1H), 8,06 (д, J = 8,0 Гц, 1H), 7,88 (дд, J = 8,1, 2,2 Гц, 1H), 7,01 - 6,64 (м, 2H), 6,33 (с, 1H), 3,87 - 3,49 (м, 8H), 2,61-2,54 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 63 2-(6-(6-([[2,2’-Дипиридил]-5-илметил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 130)
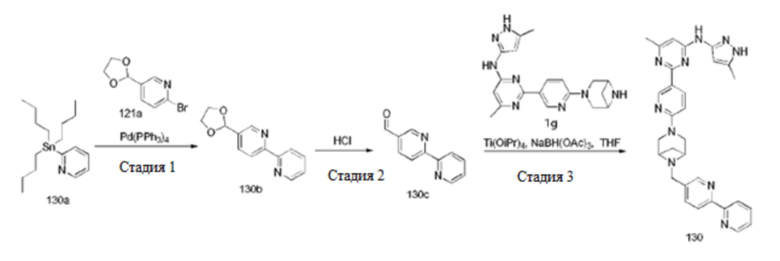
Стадия 1 Получение 5-(1,3-диоксолан-2-ил)-2,2’-дипиридина (соединение 130b)
Соединение 130a (320 мг), соединение 121a (200 мг), и тетракис(трифенилфосфин)палладий (50 мг) добавляли в толуол (10 мл), и смесь выдерживали при 120° для реакции в условиях микроволнового излучения в течение 4 ч. После завершения реакции реакционную смесь концентрировали досуха, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=2:1) с получением соединения 130b (100 мг). МС m/z (ИЭР): 229,1 [M+H]+.
Стадия 2 Получение [2,2’-дипиридил]-5-карбальдегида (соединение 130c)
Разбавленную хлористоводородную кислоту (5,0 мл, 3 N) добавляли по каплям в раствор соединения 130b (100 мг) в ТГФ (5,0 мл). Смесь выдерживали для реакции при комнатной температуре в течение 15 ч. После завершения реакции pH реакционной смеси регулировали до около 10 путем медленного добавления насыщенного раствора бикарбоната натрия по каплям и затем экстрагировали с помощью EA. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 130c (45 мг), которое непосредственно использовали на следующей стадии реакции без очистки. МС m/z (ИЭР): 185,2 [M+H]+.
Стадия 3 Получение 2-(6-(6-([[2,2’-дипиридил]-5-илметил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 130)
Соединение 1g (50 мг), соединение 130c (25,4 мг) и тетраизопропилтитанат (156,8 мг) добавляли в сухой ТГФ (5 мл), и смесь перемешивали при 72°C в течение 10 ч. Затем триацетоксиборогидрид натрия (146,2 мг) добавляли, и смесь выдерживали для реакции при 72°C в течение 6 ч. После завершения реакции смесь грубо очищали непосредственно посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=10:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 130 (40 мг). МС m/z (ИЭР): 531,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1H), 9,65 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,75 - 8,57 (м, 2H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,35 (дд, J = 9,7, 8,2 Гц, 2H), 7,93 (ддд, J = 8,3, 5,9, 1,9 Гц, 2H), 7,44 (ддд, J = 7,5, 4,8, 1,1 Гц, 1H), 7,08 - 6,57 (м, 2H), 6,31 (с, 1H), 3,82-3,57 (м, 8H), 2,63 - 2,56 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,61 (д, J = 8,4 Гц, 1H).
Пример 64 2-(6-(6-((6-(5-Фтор-1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 131)
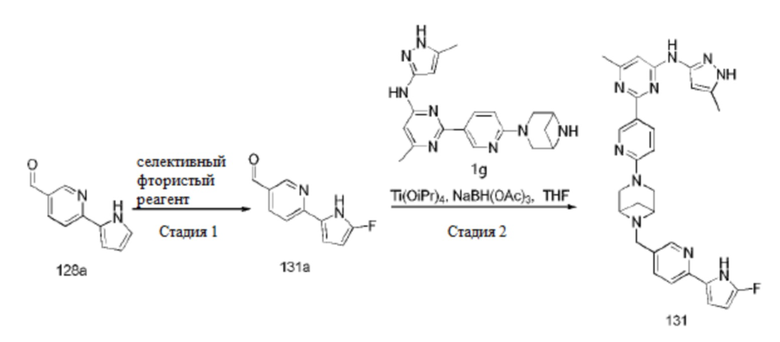
Стадия 1 Получение 6-(5-фтор-1H-пиррол-2-ил)никотинальдегида (соединение 131a)
Соединение 128a (200 мг) и 1-хлорметил-4-фтор-1,4-диазабицикло[2.2.2]октан бис(тетрафторборат) (432 мг, селективный фтористый реагент) добавляли в ацетонитрил (15 мл), и выдерживали при 70°C для реакции в условиях микроволнового излучения в течение 10 мин. После завершения реакции реакционную смесь концентрировали досуха, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=5:1) с получением соединения 131a (60 мг). МС m/z (ИЭР): 191,1 [M+H]+.
Стадия 2 Получение 2-(6-(6-((6-(5-фтор-1H-пиррол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 131)
Соединение 1g (50 мг), соединение 131a (26,2 мг) и тетраизопропилтитанат (156,8 мг) добавляли в сухой ТГФ (5 мл), и смесь перемешивали при 72°C в течение 10 ч. Затем триацетоксиборогидрид натрия (146,2 мг) добавляли, и смесь выдерживали для реакции при 72°C в течение 6 ч. После завершения реакции смесь грубо очищали непосредственно посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=8:1), и затем отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 131 (9 мг). МС m/z (ИЭР): 537,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,06 (с, 1H), 11,98 (с, 1H), 9,65 (с, 1H), 9,12 с J = 2,1 Гц, 1H), 8,43 (дд, J = 8,9, 2,3 Гц, 1H), 8,39 (д, J = 1,3 Гц, 1H), 7,74 - 7,65 (м, 1H), 7,57 (д, J = 8,2 Гц, 1H), 6,92 - 6,72 (м, 2H), 6,57 (т, J = 4,2 Гц, 1H), 6,31 (с, 1H), 5,57 (т, J = 3,8 Гц, 1H), 3,76 (д, J = 11,2 Гц, 2H), 3,70 (д, J = 5,7 Гц, 2H), 3,63 - 3,52 с 4H), 2,60-2,55 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,2 Гц, 1H).
Пример 65 2-(6-(6-((6-(1H-Пиразол-5-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1] гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 132)
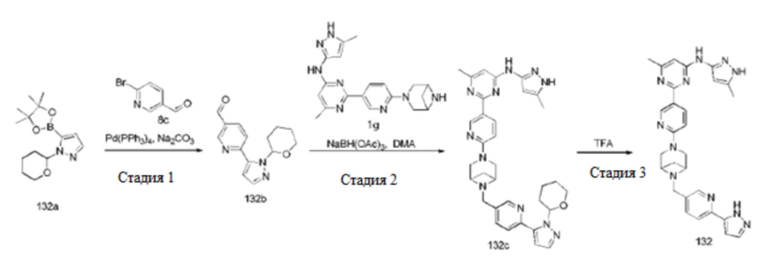
Стадия 1 Получение 6-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотинальдегида (соединение 132b)
Соединение 132a (2,24 г), соединение 8c (1 г), тетракис(трифенилфосфин)палладий (310,6 мг) и 1,4-диоксан (20 мл) последовательно добавляли в реакционную колбу, и затем раствор карбоната натрия (1,71 г) в воде (5 мл) добавляли. Смесь перемешивали в условиях защиты азотом при 95°C в течение 5 ч. После завершения реакции реакционную смесь разбавляли водой и экстрагировали с помощью EA (20 мл×3). Органические фазы объединяли, последовательно промывали водой и насыщенным солевым раствором один раз, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной флеш-хроматографии (ДХМ:MeOH=96:4) с получением соединения 132b (1,14 г).
Стадия 2 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-((6-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 132c)
Соединение 1g (500 мг) и соединение 132b (477,8 мг) растворяли в DMA (10 мл), и смесь перемешивали при 25 °C для реакции в течение 2 ч. Затем триацетоксиборогидрид натрия (1,17 г) добавляли, и смесь перемешивали при 25°C для реакции в течение дополнительных 16 ч. После завершения реакции воду добавляли для гашения реакционной смеси, и смесь экстрагировали с помощью EA. Органические фазы промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 132c (697 мг). МС m/z (ИЭР): 604,3 [M+H]+.
Стадия 3 Получение 2-(6-(6-((6-(1H-пиразол-5-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 132)
Соединение 132c (697 мг) растворяли в MeOH (15 мл), и затем TFA (0,86 мл) добавляли по каплям. Смесь выдерживали для реакции при 25°C в течение 16 ч. После завершения реакции насыщенный раствор бикарбоната натрия добавляли для гашения реакционной смеси, и pH раствора регулировали до около 9. Раствор концентрировали с удалением метанола, разбавляли водой и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 132 (279 мг). МС m/z (ИЭР): 521,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 13,48 (с, 0,3H, таутомер 1), 13,01 (с, 0,7H, таутомер 2), 11,98 (с, 1H), 9,66 (с, 1H), 9,13 (д, J = 2,2 Гц, 1H), 8,52 (с, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 8,01-7,64 (м, 3H), 7,01 - 6,59 (м, 3H),6,32 (уш., 1H), 3,85-3,68(м, 4H), 3,68 - 3,45 (м, 4H), 2,60 - 2,53 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H).
Пример 66 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(5-метилфуран-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 133)
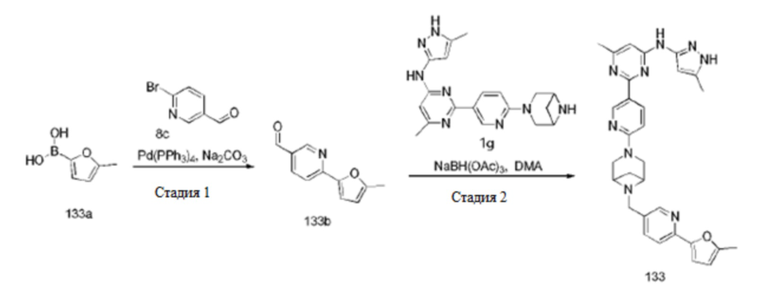
Стадия 1 Получение 6-(5-метилфуран-2-ил)никотинальдегида (соединение 133b)
Соединение 133a (203,1 мг), соединение 8c (200 мг), Na2CO3 (341,9 мг), тетракис(трифенилфосфин)палладий (62,1 мг), воду (2,5 мл) и 1,4-диоксан (10 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали при 95°C в течение 2 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, отделяли и очищали непосредственно посредством колоночной хроматографии на силикагеле (PE:EA=87:13) с получением соединения 133b (124 мг). МС m/z (ИЭР): 188,1 [M+H]+.
Стадия 2 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(5-метилфуран-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 133)
Соединение 133b (23,2 мг), соединение 1g (30 мг) и DMA (1 мл) последовательно добавляли в реакционную колбу, и смесь перемешивали при 25°C в течение 2 ч. Затем триацетоксиборогидрид натрия (70,2 мг) добавляли, и смесь перемешивали при 25°C для реакции в течение дополнительных 16 ч. После завершения реакции реакционную смесь отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 133 (9 мг). МС m/z (ИЭР): 534,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,66 (с, 1H), 9,12 (д, J = 2,2 Гц, 1H), 8,48 (д, J = 1,5 Гц, 1H), 8,44 (дд, J = 8,9, 2,3 Гц, 1H), 7,78 (дд, J = 8,2, 2,1 Гц, 1H), 7,60 (д, J = 8,1 Гц, 1H), 6,96 (д, J = 4,5 Гц, 1H), 6,84(уш., 1H), 6,78 (д, J = 9,0 Гц, 1H), 6,31(уш., 1H), 6,25 (дд, J = 3,2, 1,0 Гц, 1H), 3,82-3,68 (м, 4H), 3,68-3,45 (м, 4H), 2,60 - 2,53 (м, 1H), 2,36 (с, 3H), 2,33 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 67 6-Метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(оксазол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амин (соединение 134)
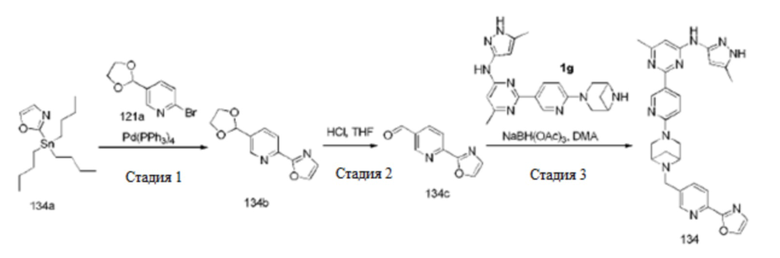
Стадия 1 Получение 2-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)оксазола (соединение 134b)
Соединение 121a (300,0 мг) и соединение 134a (467,0 мг) растворяли в толуоле (15,0 мл), и затем тетракис(трифенилфосфин)палладий (75,3 мг) добавляли. Смесь перемешивали в условиях защиты азотом при 120°C в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, и затем разбавляли с помощью EA (200,0 мл). Органическую фазу промывали водой 3 раза, дополнительно промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=7:3) с получением соединения 134b (78,0 мг). МС m/z (ИЭР): 219,1 [M+1]+.
Стадия 2 Получение 6-(оксазол-2-ил)никотинальдегида (соединение 134c)
Соединение 134b (78,0 мг) растворяли в смешанном растворителе ТГФ (2,0 мл) и воды (2,0 мл) и затем концентрированную хлористоводородную кислоту (1,0 мл, 12 н.) медленно добавляли по каплям. Смесь перемешивали при 25°C в течение 8 ч. Реакционную смесь регулировали водным раствором карбоната калия до щелочного pH и экстрагировали с помощью EA (100,0 мл). Органическую фазу промывали водой 3 раза, дополнительно промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (ДХМ:MeOH=9:1) с получением соединения 134c (51,0 мг). МС m/z (ИЭР): 175,1 [M+1]+.
Стадия 3 Получение 6-метил-N-(5-метил-1H-пиразол-3-ил)-2-(6-(6-((6-(оксазол-2-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)пиримидин-4-амина (соединение 134)
Соединение 134c (28,8 мг) и соединение 1g (30,0 мг) растворяли в N,N-диметилацетамиде (1,0 мл), и смесь перемешивали при 25°C в течение 1 ч. Затем триацетоксиборогидрид натрия (70,2 мг) добавляли в реакционную систему, и смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь разбавляли с помощью EA (20 мл), промывали водой 3 раза, дополнительно промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 134 (17,0 мг). МС m/z (ИЭР): 521,3 [M+1]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,00 (с, 1H), 9,68 (с, 1H), 9,14 (д, J = 2,4 Гц, 1H), 8,66 (с, 1H), 8,45 (дд, J = 9,2, 2,4 Гц, 1H), 8,29 (с, 1H), 8,06 (д, J = 8,4 Гц, 1H), 7,95 (д, J = 8,0 Гц, 1H), 7,45 (с, 1H), 6,79 (уш., 1H), 6,78 (д, J = 8,8 Гц, 1H), 6,32 (уш., 1H), 3,79-3,75 (м, 4H), 3,67-3,60 (м, 4H), 2,58-2,56 (м,1H), 2,34 (с, 3H), 2,26 (с, 3H), 1,61 (д, J = 8,0 Гц, 1H).
Пример 68 2-(6-(6-((6-(1-Изопропил-1H-пиразол-4-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 135)
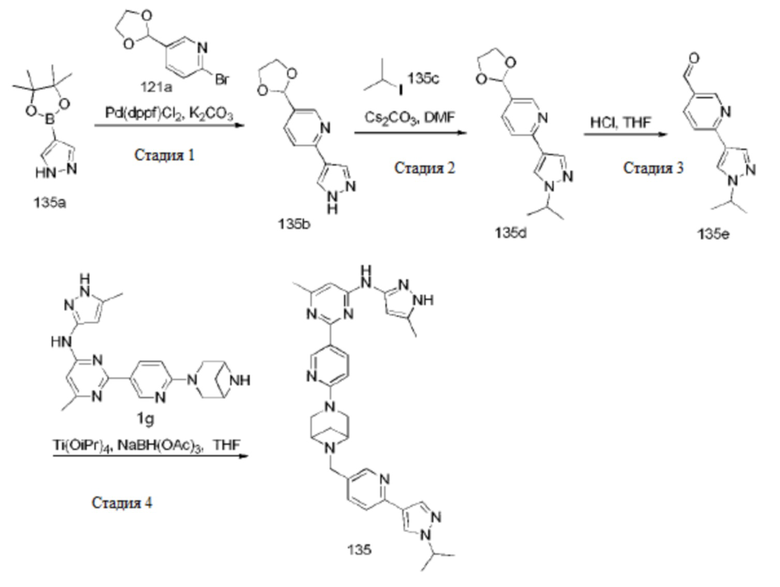
Стадия 1 Получение 5-(1,3-диоксолан-2-ил)-2-(1H-пиразол-4-ил)пиридина (соединение 135b)
Соединение 121a (1,0 г) и соединение 135a (1,0 г) растворяли в 1,4-диоксане (30 мл) и H2O (6 мл), и затем Pd(dppf)Cl2 (130 мг) и K2CO3 (1,5 г) последовательно добавляли. Смесь перемешивали в условиях защиты азотом при 95°C в течение 2 ч. После завершения реакции смесь охлаждали на ледяной бане и фильтровали через целит. Фильтрат концентрировали досуха, отделяли и очищали посредством ЖХСД с получением соединения 135b (452 мг). МС m/z (ИЭР): 218,2 [M+H]+.
Условия ЖХСД:
Модель прибора: Biotage Isolera Prime 2,3,1, хроматографическая колонка: Agela Technologies C18, сферическая 20-35 мкм 100A, 120 g; температура хроматографической колонки: 25°C; расход: 30,0 мл/мин.; длина волны обнаружения: 254 нм; градиент для элюирования: (0 мин.: 20% A, 80% B; 3,0 мин.: 20% A, 80% B; 25 мин.: 90% A, 10% B); подвижная фаза A: ацетонитрил, подвижная фаза B: 0,05% водный раствор TFA.
Стадия 2 Получение 5-(1,3-диоксолан-2-ил)-2-(1-изопропил-1H-пиразол-4-ил)пиридина (соединение 135d)
Соединение 135b (100 мг) и Cs2CO3 (374,9 мг) добавляли в сухой DMF (10 мл), и затем соединение 135c (195,6 мг) добавляли. Смесь перемешивали при 80 °C в течение 12 ч. После завершения реакции воду (100 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством колоночной хроматографии на силикагеле (PE:EA=1:1), с получением соединения 135d (52 мг). МС m/z (ИЭР): 260,0 [M+H]+.
Стадия 3 Получение 6-(1-изопропил-1H-пиразол-4-ил)никотинальдегида (соединение 135e)
Соединение 135d (240 мг) добавляли в ТГФ (4 мл) и H2O (2 мл), и затем раствор HCl в 1,4-диоксане (4 н., 1 мл) добавляли. Смесь перемешивали при 25°C в течение 5 ч. После завершения реакции насыщенный водный раствор бикарбоната натрия (50 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (50 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали досуха с получением соединения 135e (40 мг). МС m/z (ИЭР): 216,1 [M+H].
Стадия 4 Получение 2-(6-(6-((6-(1-изопропил-1H-пиразол-4-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амина (соединение 135)
Соединение 1g (40 мг), соединение 135e (26,1 мг) и тетраизопропилтитанат (31,4 мг) добавляли в сухой ТГФ (25 мл), и смесь перемешивали в условиях защиты азотом при 75°C в течение 10 ч. Затем триацетоксиборогидрид натрия (23,4 мг) добавляли в реакционную систему, и смесь перемешивали при 75°C в течение дополнительных 6 ч. После завершения реакции реакционную смесь концентрировали досуха при пониженном давлении, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 135 (2,1 мг). МС m/z (ИЭР): 562,1 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,98 (с, 1H), 9,67 (с, 1H), 9,12 (д, J = 2,0 Гц, 1H), 8,56-8,39 (м, 2H), 8,32 (с, 1H), 7,97 (с, 1H), 7,71 (дд, J = 8,0, 2,0 Гц, 1H), 7,59 (д, J = 8,0 Гц, 1H),7,05-6,65 (м, 2H), 6,33 (с, 1H), 4,64-4,36 (м, 1H), 3,94-3,63 (м, 4H), 3,64-3,45 (м, 4H), 2,62-2,50 (м, 1H), 2,31 (с, 3H), 2,29 (с, 3H), 1,56 (д, J = 8,0 Гц, 1H), 1,45 (д, J = 6,4 Гц, 6H).
Пример 69 2-(1-(5-((3-(5-(4-Метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-5-ил)пропан-2-ол (соединение 136)
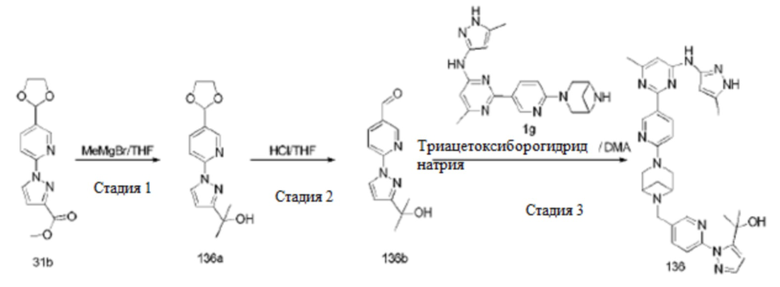
Стадия 1 Получение 2-(1-(5-(1,3-диоксолан-2-ил)пиридин-2-ил)-1H-пиразол-3-ил)пропан-2-ола (соединение 136a)
Соединение 31b (200 мг) добавляли в сухой ТГФ (10 мл) и охлаждали в бане сухой лед-этанол в течение 15 мин. Затем раствор метилмагния бромида в диэтиловом эфире (3 н., 0,65 мл) медленно добавляли по каплям, смесь выдерживали для реакции при температуре в течение 15 мин., затем нагревали до комнатной температуры и выдерживали для реакции при температуре в течение дополнительных 4 ч. Насыщенный водный раствор хлорида аммония (1 мл) добавляли в реакционную смесь для гашения реакционной смеси. Затем реакционную смесь разбавляли водой (30 мл) и экстрагировали с помощью EA (30 мл x 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 136a (200 мг). МС m/z (ИЭР): 276,1 [M+H]+.
Стадия 2 Получение 6-(3-(2-гидроксипропан-2-ил)-1H-пиразол-1-ил)никотинальдегида (соединение 136b)
Соединение 136a (200 мг) добавляли в ТГФ (5 мл), и затем хлористоводородную кислоту (2 н., 4,5 мл) добавляли. Смесь перемешивали при 25°C в течение 12 ч. Реакционную смесь регулировали насыщенным водным раствором бикарбоната натрия до pH от 7 до 8 и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 136b (165 мг). МС m/z (ИЭР): 232,1 [M+H]+.
Стадия 3 Получение 2-(1-(5-((3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-5-ил)пропан-2-ола (соединение 136)
Соединение 136b (50,5 мг) и соединение 1g (30 мг) добавляли в DMA (3 мл), и смесь перемешивали в условиях защиты азотом при комнатной температуре в течение 1 ч. Затем триацетоксиборогидрид натрия (101 мг) добавляли, и смесь выдерживали при комнатной температуре в течение ночи. После завершения реакции воду (60 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (30 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 136 (12 мг). МС m/z (ИЭР): 578,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,14 (уш., 1H), 9,67 (с, 1H), 9,12 (д, J = 2,0 Гц, 1H), 8,50-8,41 (м, 2H), 8,41-8,35 (м, 1H), 7,99-7,92 (м, 1H), 7,82 (д, J = 8,4 Гц, 1H), 6,79 (д, J = 9,2 Гц, 2H), 6,52 (д, J = 2,4 Гц, 1H), 6,31 (уш., 1H), 5,10 (с, 1H), 3,82-3,70 (м, 4H), 3,68-3,55 (м, 4H), 2,61-2,54 (м, 1H), 2,33 (с, 3H), 2,26 (с, 3H), 1,60 (д, J = 8,4 Гц, 1H), 1,49 (с, 6H).
Пример 70 (1-(5-((3-(5-(4-Метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-3-ил)метанол (соединение 137)
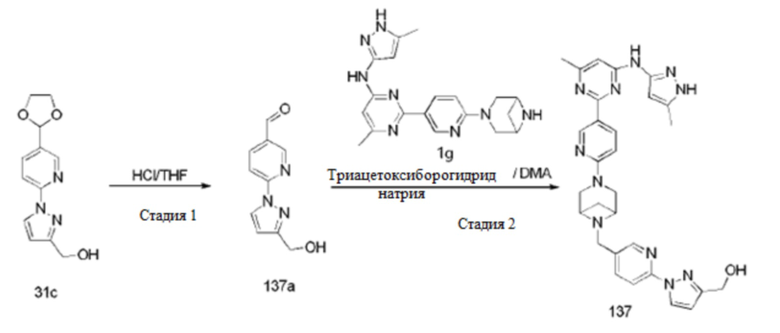
Стадия 1 Получение 6-(3-(гидроксиметил)-1H-пиразол-1-ил)никотинальдегида (соединение 137a)
Соединение 31c (189 мг) добавляли в ТГФ (5 мл), и затем разбавленную хлористоводородную кислоту (2 N, 5 мл) добавляли. Смесь перемешивали при 25 °C в течение 2 ч. Реакционную смесь регулировали насыщенным водным раствором бикарбоната натрия до pH от 7 до 8 и экстрагировали с помощью EA (30 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали с получением соединения 137a (157 мг). МС m/z (ИЭР): 204,1 [M+H]+.
Стадия 2 Получение (1-(5-((3-(5-(4-метил-6-((5-метил-1H-пиразол-3-ил)-амино)пиримидин-2-ил)пиридин-2-ил)-3,6-диазабицикло[3.1.1]гептан-6-ил)метил)пиридин-2-ил)-1H-пиразол-3-ил)метанола (соединение 137)
Соединение 137a (50,5 мг) и соединение 1g (50 мг) добавляли в DMA (2 мл), и смесь перемешивали в условиях защиты азотом при комнатной температуре в течение 1 ч. Затем триацетоксиборогидрид натрия (159 мг) добавляли, и смесь выдерживали при комнатной температуре в течение ночи. После завершения реакции воду (60 мл) добавляли в реакционную смесь для гашения реакционной смеси. Реакционную смесь экстрагировали с помощью EA (30 мл × 3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали, отделяли и очищали посредством препаративной ВЭЖХ с получением соединения 137 (10 мг). МС m/z (ИЭР): 550,3 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ 12,00 (с, 1H), 9,68 (с, 1H), 9,13 (д, J = 2,0 Гц, 1H), 8,52 (д, J = 2,4 Гц, 1H), 8,44 (дд, J = 8,8, 2,4 Гц, 1H), 8,38 (д, J = 2,4 Гц, 1H), 7,94 (дд, J = 8,4, 2,0 Гц, 1H), 7,82 (д, J = 8,4 Гц, 1H), 7,06-6,65 (м, 2H), 6,51 (д, J = 2,4 Гц, 1H), 6,29 (уш., 1H), 5,26 (т, J = 6,0 Гц, 1H), 4,53 (д, J = 5,6 Гц, 2H), 3,81-3,69 (м, 4H), 3,67-3,49 (м, 4H), 2,59-2,53 (м, 1H), 2,34 (с, 3H), 2,26 (с, 3H), 1,59 (д, J = 8,4 Гц, 1H).
Пример 71 2-(6-(6-((6-(4-Фтор-3-метил-1H-пиразол-1-ил)пиридин-3-ил)метил)-3,6-диазабицикло[3.1.1]гептан-3-ил)пиридин-3-ил)-6-метил-N-(5-метил-1H-пиразол-3-ил)пиримидин-4-амин (соединение 138)
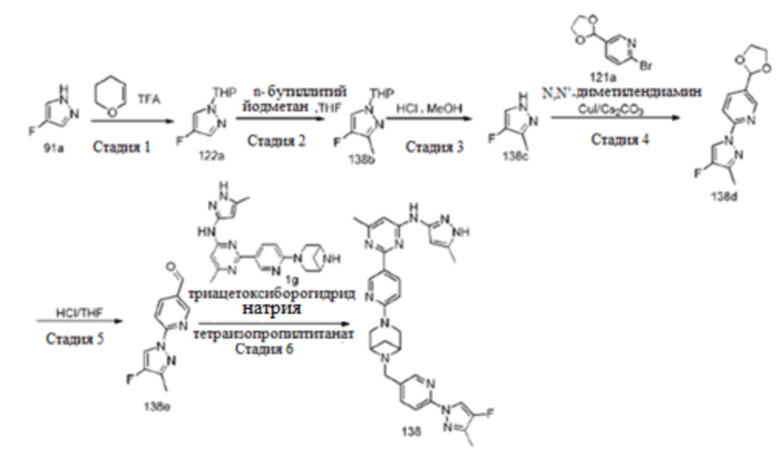
Соединение 138 получали путем ссылки на способ синтеза в примере 56. МС m/z (ИЭР): 551,8 [M+H]+.
Способ разделения
Очистку соединений в примерах с 1 по 51, с 53 по 58 и с 60 по 71 посредством препаративной ВЭЖХ проводили с использованием ВЭЖХ Aglient 1260, Waters 2489 или GeLai 3500 при температуре колонки 25°C и длине волны обнаружения 214 нм, 254 нм или 280 нм, и с дополнительными условиями разделения, как показано в таблице ниже.
Градиент: 0 мин. 10% A, 90% B
4 мин. 10% A, 90% B
6 мин. 23,4% A, 76,6% B
Градиент: 0 мин. 10% A, 90% B
2 мин. 10% A, 90% B
5 мин. 20,7% A, 79,3% B
Градиент: 0 мин. 10% A, 90% B
2 мин. 10% A, 90% B
16 мин. 60% A, 40% B
Градиент: 0 мин. 70% A, 30% B
4 мин. 70% A, 30% B
16 мин. 10% A, 90% B
Градиент: 0 мин.: 10% A, 90% B
7,0 мин.: 20% A, 80% B
Градиент: 0 мин.: 30% A, 70% B
4,0 мин.: 30%A, 70%B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
4 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
2 мин.: 10% A, 90% B
16 мин.: 70% A, 30% B
Градиент: 0 мин.: 10% A, 90% B
2 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 60% A, 40% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
10 мин.: 36% A, 64% B
Градиент: 0 мин.: 10% A, 90% B
10 мин.: 32% A, 68% B
Градиент: 0 мин.: 10% A, 90% B
2 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 20% A, 80% B
3 мин.: 20% A, 80% B
16 мин.: 47% A, 53% B
Градиент: 0 мин.: 20% A, 80% B
3 мин.: 20% A, 80% B
16 мин.: 47% A, 53% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
4 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 60% A, 40% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
4 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 70% A, 30% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 70% A, 30% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
2 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
18 мин.: 60% A, 40% B
Градиент: 0 мин.: 10% A, 90% B
15,0 мин.: 60% A, 40% B
Градиент: 0 мин.: 10% A, 90% B
8,0 мин.: 70% A, 30% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16,0 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16,0 мин.: 70% A, 30% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 70% A, 30% B
Градиент: 0 мин.: 10% A, 90% B
16,0 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16,0 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 70% A, 30% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 80% A, 20% B
Градиент: 0 мин.: 20% A, 80% B
16 мин.: 80% A, 20% B
Градиент: 0 мин.: 20% A, 80% B
16 мин.: 80% A, 20% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 90% A, 10% B
Градиент: 0 мин. 25% A, 75% B
5 мин. 25% A, 75% B
50 мин. 70% A, 30% B
Градиент: 0 мин.: 30% A, 70% B
16 мин.: 80% A, 20% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 60% A, 40% B
Градиент: 0 мин.: 10% A, 90% B
16 мин.: 90% A, 10% B
Градиент: 0 мин.: 10% A, 90% B
16,0 мин.: 90% A, 10% B
Следующие промежуточные соединения в примерах очищали с помощью ВЭЖХ GeLai 3500 при температуре колонки 25°C, при длине волны обнаружения 214 нм, 254 нм или 280 нм и в дополнительных условиях разделения, как показано в таблице ниже.
Градиент: 0 мин. 8% A, 92% B
10 мин. 8% A, 92% B
50 мин. 50% A, 50% B
Градиент: 0 мин. 8% A, 92% B
5 мин. 8% A, 92% B
50 мин. 50% A, 50% B
Биологическая оценка
Экспериментальный пример 1: Эксперимент по ингибированию RET
Экспериментальный метод: В соответствии с инструкциями к набору HTRF KinEASE-TK (Cisbio) соединения по настоящему изобретению были протестированы относительно их ингибирующего действия на активность фермента RET дикого типа, мутантного фермента RET (RET-V804M, RET-V804L и RET-M918T) и фермента RET слитого типа (RET-CCDC6). После предварительной инкубации различных ферментов RET и различных концентраций тестируемых соединений при комнатной температуре в течение 30 минут добавляли субстрат и аденозинтрифосфат (АТФ) для запуска реакции. После инкубации при комнатной температуре в течение 40 минут добавляли криптат антитела TK и стрептавидин-XL665, и тест проводили после инкубации при комнатной температуре в течение 45 минут. С группой растворителя (ДМСО) в качестве отрицательного контроля и буферной группой (без фермента RET) в качестве пустого контроля относительные проценты ингибирующей активности (т. е. степени ингибирования) различных концентраций соединений вычисляли по следующей формуле:
Процент относительной ингибирующей активности = 1- (группа соединений с разными концентрациями - пустой контроль)/(отрицательный контроль - пустой контроль)*100%
Проценты относительной ингибирующей активности различных концентраций соединений были нанесены на график относительно концентраций соединений, и кривая была построена в соответствии с четырехпараметрической моделью для вычисления значения IC50 по следующей формуле:
y=min+(max-min)/(1+(x/IC50)^(- угловой коэффициент Хилла))
где y - процент относительной ингибирующей активности, max - максимальное значение подобранной кривой, min - минимальное значение подобранной кривой, x - логарифмическая концентрация соединения, а угловой коэффициент Хилла - наклон кривой.
Экспериментальный пример 2: Эксперимент по ингибированию VEGFR2
Экспериментальный метод: В соответствии с инструкциями к набору HTRF KinEASE-TK (Cisbio) соединения по настоящему изобретению были протестированы относительно их ингибирующего эффекта на активность фермента VEGFR2. После предварительной инкубации фермента VEGFR2 и различных концентраций тестируемых соединений при комнатной температуре в течение 30 минут добавляли субстрат и аденозинтрифосфат (АТФ) для запуска реакции. После инкубации при комнатной температуре в течение 40 минут добавляли криптат антитела TK и стрептавидин-XL665, и тест проводили после инкубации при комнатной температуре в течение 45 минут. С группой растворителя (ДМСО) в качестве отрицательного контроля и буферной группой (без фермента VEGFR2) в качестве пустого контроля относительные проценты ингибирующей активности (т. е. степени ингибирования) различных концентраций соединений вычисляли по следующей формуле:
Процент относительной ингибирующей активности = 1- (группа соединений с разными концентрациями - пустой контроль)/(отрицательный контроль - пустой контроль)*100%
Проценты относительной ингибирующей активности различных концентраций соединений были нанесены на график относительно концентраций соединений, и кривая была построена в соответствии с четырехпараметрической моделью для вычисления значения IC50 по следующей формуле:
y=min+(max-min)/(1+(x/IC50)^(- угловой коэффициент Хилла))
где y - процент относительной ингибирующей активности, max - максимальное значение подобранной кривой, min - минимальное значение подобранной кривой, x - логарифмическая концентрация соединения, а угловой коэффициент Хилла - наклон кривой.
Результаты эксперимента
Результаты экспериментов показаны в таблицах 1-4.
Таблица 1. Степени ингибирования соединений по настоящему изобретению в концентрации 100 нМ активности мутантного фермента RET
Примечание: Н/Д означает «не протестировано».
Как видно из таблицы 1, соединения по настоящему изобретению обладают значительным ингибирующим действием на мутантный фермент RET.
Таблица 2. Степень ингибирования соединения по настоящему изобретению в отношении фермента RET-V804M
Как видно из таблицы 2, соединения по настоящему изобретению обладают значительным ингибирующим действием на фермент RET-V804M.
Таблица 3-1. Ингибирование IC50 (нМ) соединения по настоящему изобретению в отношении фермента RET-WT
Таблица 3-2. Ингибирование IC50 (нМ) соединения по настоящему изобретению в отношении фермента RET-CCDC6
Таблица 3-3. Ингибирование IC50 (нМ) соединения по настоящему изобретению в отношении фермента RET-V804L
Таблица 3-4. Ингибирование IC50 (нМ) соединения по настоящему изобретению в отношении фермента RET-V804M
Таблица 3-5. Ингибирование IC50 (нМ) соединения по настоящему изобретению в отношении фермента RET-M918T
Как видно из таблиц с 3-1 по 3-5, соединения по настоящему изобретению обладают значительным ингибирующим действием в отношении любого из ферментов RET-CCDC6, RET-M918T, RET-V804M, RET-V804L и RET- WT.
Таблица 4. Степень ингибирования соединений по настоящему изобретению в отношении VEGFR2
Кроме того, тесты показали, что IC50 ингибирования соединения 11 в отношении VEGFR2 составляет 158,02 ± 25,08 нМ, а IC50 ингибирования соединения 17 в отношении VEGFR2 составляет 62,97 ± 11,77 нМ. Приведенные выше результаты в комбинации с данными о степени ингибирования в таблице 4 показали, что соединения по настоящему изобретению обладают более слабым ингибированием VEGFR2 и обладают лучшим селективным ингибирующим действием в отношении фермента RET, а не VEGFR2.
Экспериментальный пример 3: Фармакокинетика и распределение в тканях соединений у крыс
Путем внутрижелудочного введения (РО) BLU-667 (полученного в соответствии с примером 5 в WO2017/079140A1) и соединения 17 самцам крыс SD соответственно, концентрации BLU-667 и соединения 17 в плазме крови и тканях в мозге, легких и щитовидной железе крыс были определены для исследования фармакокинетических характеристик. Дозировка для введения путем PO составляла 5 мг/кг, а растворитель представляли собой 0,5% MC (метилцеллюлоза). При пероральном введении образцы крови собирали в разные моменты времени (0 ч. до введения и 0,25 ч., 0,5 ч., 1 ч., 2 ч., 4 ч., 6 ч., 8 ч. и 24 ч. после введения). Образцы крови подвергали антикоагуляции с помощью дикалия эдетата и центрифугировали, чтобы получить образцы плазмы крови, которые хранили при -80°C. Крыс умерщвляли обескровливанием из брюшной аорты через 0,5 ч., 2 ч., 8 ч. и 24 ч. после перорального введения. Мозг, легкие и щитовидную железу собирали, промывали и гомогенизировали физиологическим раствором в определенном соотношении, чтобы получить образцы тканей, которые хранили при -80°C. Образцы плазмы и образцы тканей обрабатывали осажденным белком, а затем анализировали с помощью ЖХ-МС/МС. Фармакокинетические параметры были рассчитаны с использованием программного обеспечения WinNonlin 6.3 и некомпартментной модели. Результаты показаны в таблице 5.
Таблица 5. Фармакокинетические параметры соединений, вводимых путем PO в плазме крови и тканях крыс
AUClast, ч*нг/мл
Cmax, нг/мл
Tmax, ч.
Соотношение AUClast в ткани и плазме крови
Данные в таблице 5 показывают, что после внутрижелудочного введения 5 мг/кг BLU-667 и соединения 17 крысам SD экспериментальное количество соединения 17 в каждой ткани органа-мишени, такого как мозг, легкое и щитовидная железа, было лучше, чем экспериментальное количество BLU-667.
Экспериментальный пример 4: Тест эффективности соединения у мышей
Цель эксперимента Оценить in vivo эффективность соединения 17 и BLU-667 на мышиной модели Balb/c-nu с подкожной ксенотрансплантатной опухолью ТТ-клеток медуллярной карциномы щитовидной железы человека.
Получение лекарственного средства Соединение 17 растворяли в 0,1 M водном растворе H3PO4, и BLU-667 растворяли в 0,1 M водном растворе HOAc с получением прозрачных растворов (pH: около 4,0). Водный раствор H3PO4 при pH около 4,0 использовали для контрольной группы растворителя. Способ введения был PO, дважды в сутки.
Измерение опухоли. Диаметр опухоли измеряли штангенциркулем дважды в неделю. Формула вычисления объема опухоли: V=0,5×a×b2, где а представляет собой длинный диаметр опухоли, а b представляет собой короткий диаметр опухоли. Оценка степени ингибирования роста опухоли (TGI (%)) для эффективности ингибирования опухоли соединениями: TGI (%) = [(1- (средний объем опухоли в группе лечения в конце введения лекарственного средства - средний объем опухоли в группе лечения в начале введения лекарственного средства)) / (средний объем опухоли в контрольной группе растворителя в конце лечения - средний объем опухоли контрольной группы растворителя в начале лечения)] × 100%. Результаты показаны на фиг. 1.
Статистический анализ Программное обеспечение Prism Graphpad 5.0 использовали для статистического анализа на основе относительного объема опухоли в конце эксперимента. Сравнение между несколькими группами было проанализировано с помощью двухфакторного дисперсионного анализа, и p <0,05 считалось значимым различием.
Результаты эксперимента В ксенотрансплантатной модели бестимусных мышей TT медуллярной карциномы щитовидной железы человека соединение 17 имеет значительный дозозависимый противоопухолевый эффект при дозе 5 мг/кг. Противоопухолевый эффект соединения 17 в дозе 5 мг/кг (T/C = 17,44%, TGI = 131,36%, p <0,05) лучше, чем у группы BLU-667 в дозе 5 мг/кг (T/C = 33,62%, TGI = 88,82%, p <0,05). Относительная скорость роста опухоли T/C (%) = TRTV/ CRTV× 100%, где TRTV - средний относительный объем опухоли в группе тестируемого соединения, CRTV - относительный объем опухоли в группе контроля с растворителем; и относительный объем опухоли RTV = Vt/V0, где V0 - средний объем опухоли в начале введения, а Vt - средний объем опухоли, измеренный в день t после введения.
Приведенные выше примеры никоим образом не ограничивают решение настоящего изобретения. В дополнение к описанным в данном документе, различные модификации настоящего изобретения будут очевидны специалистам в данной области техники на основании предшествующего описания. Предполагается, что такие модификации также входят в объем прилагаемой формулы изобретения. Ссылки, процитированные в настоящем изобретении (включая все патенты, патентные заявки, журнальные статьи, книги и любые другие публикации), полностью включены в данный документ посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| ПРОИЗВОДНЫЕ 1,5-НАФТИРИДИНА И ИНГИБИТОРЫ MELK, СОДЕРЖАЩИЕ ИХ | 2012 |
|
RU2645339C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| ОКСАСПИРОПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2016 |
|
RU2733373C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| АНИЛИДЫ АМИНОКИСЛОТ В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ МОДУЛЯТОРОВ IL-17 | 2019 |
|
RU2815505C2 |
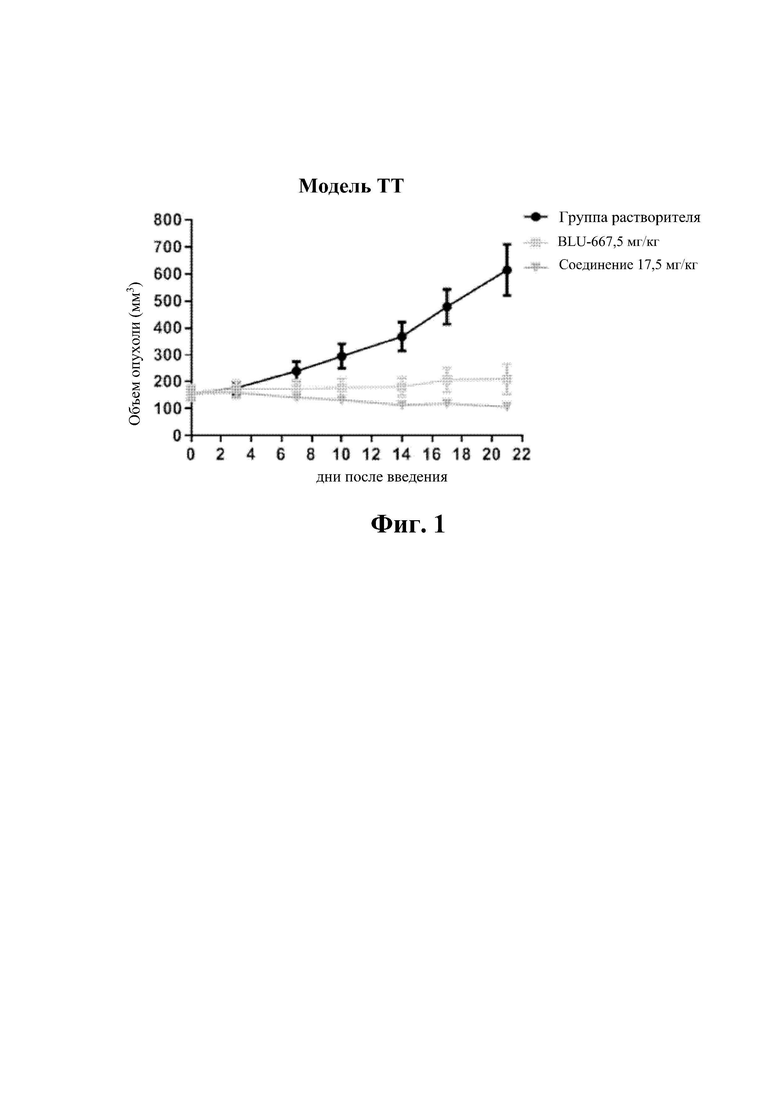
Изобретение относится к гетероциклическому соединению формулы I
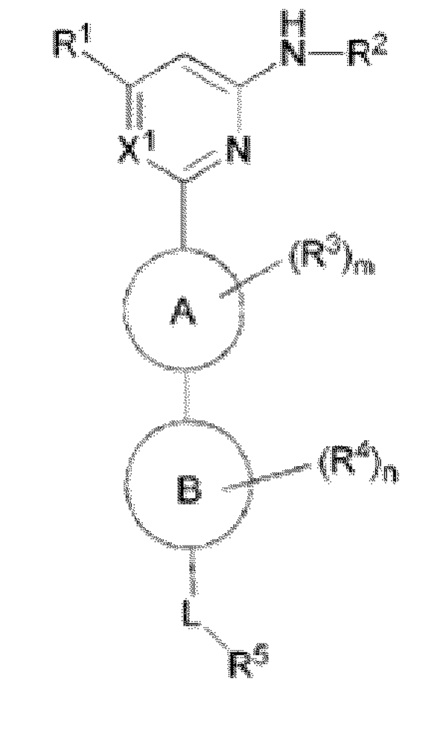
где кольцо А выбрано из 5-6-членного гетероароматического кольца, содержащего один гетероатом, выбранный из азота; кольцо В выбрано из 4-11-членного гетероциклила, содержащего один или два гетероатома, выбранные из азота; X1 выбран из СН и N; R1 выбран из C1-6 алкила; R2 выбран из 5-10-членного гетероарила, содержащего два гетероатома, выбранные из азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-4 алкила и С3-6 циклоалкила; R3 и R4 отсутствуют; L выбран из группы, состоящей из -О-, C1-6 алкилена и  и алкилен необязательно замещен одним или более заместителями, выбранными из C1-6 алкила; R5 выбран из С6-12 арила и 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, где каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси; каждый из R23a и R23b независимо представляет собой Н; R31 независимо выбран из C1-6 алкила и С3-8 циклоалкила; m равно 0; n равно 0; и t равно 0 или 1; при условии, что, если кольцо В представляет собой пиперазиновое кольцо и X1 представляет собой СН, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил, а также к способам его получения, фармацевтической композиции и применению в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET. Технический результат: получены новые соединения, которые могут быть применимы в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET, такого как рак, или опухоль, или синдром раздраженного кишечника. 11 н. и 35 з.п. ф-лы, 5 табл., 4 пр., 1 ил.
и алкилен необязательно замещен одним или более заместителями, выбранными из C1-6 алкила; R5 выбран из С6-12 арила и 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, где каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси; каждый из R23a и R23b независимо представляет собой Н; R31 независимо выбран из C1-6 алкила и С3-8 циклоалкила; m равно 0; n равно 0; и t равно 0 или 1; при условии, что, если кольцо В представляет собой пиперазиновое кольцо и X1 представляет собой СН, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил, а также к способам его получения, фармацевтической композиции и применению в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET. Технический результат: получены новые соединения, которые могут быть применимы в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET, такого как рак, или опухоль, или синдром раздраженного кишечника. 11 н. и 35 з.п. ф-лы, 5 табл., 4 пр., 1 ил.
1. Соединение, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, причем соединение имеет структуру формулы I
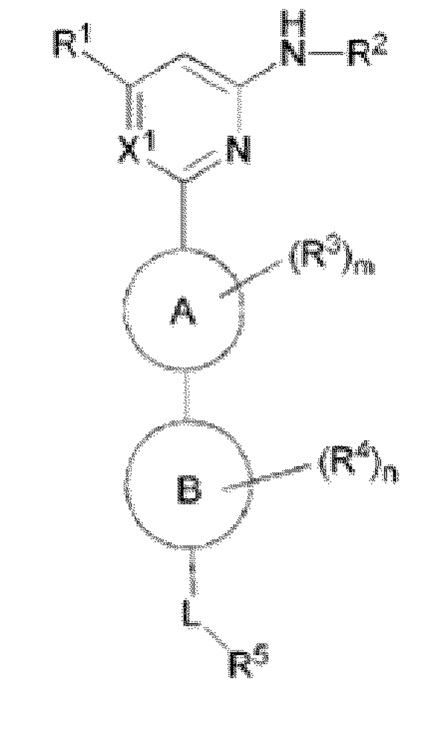
где кольцо А выбрано из 5-6-членного гетероароматического кольца, содержащего один гетероатом, выбранный из азота;
кольцо В выбрано из 4-11-членного гетероциклила, содержащего один или два гетероатома, выбранные из азота;
X1 выбран из СН и N;
R1 выбран из C1-6 алкила;
R2 выбран из 5-10-членного гетероарила, содержащего два гетероатома, выбранные из азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-4 алкила и С3-6 циклоалкила;
R3 и R4 отсутствуют;
L выбран из группы, состоящей из -О-, C1-6 алкилена и  и алкилен необязательно замещен одним или более заместителями, выбранными из C1-6 алкила;
и алкилен необязательно замещен одним или более заместителями, выбранными из C1-6 алкила;
R5 выбран из С6-12 арила и 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, где каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси;
каждый из R23a и R23b независимо представляет собой Н;
R31 независимо выбран из C1-6 алкила и С3-8 циклоалкила; m равно 0; n равно 0; и t равно 0 или 1;
при условии, что если кольцо В представляет собой пиперазиновое кольцо и X1 представляет собой СН, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил.
2. Соединение по п. 1, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где кольцо А представляет собой пиридиновое кольцо.
3. Соединение по п. 1, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где кольцо А представляет собой 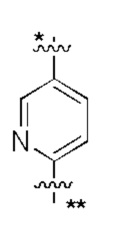 и соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом В в позиции, отмеченной **.
и соединено с кольцом, где X1 расположен в позиции, отмеченной *, и соединено с кольцом В в позиции, отмеченной **.
4. Соединение по любому из пп. 1-3, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где кольцо В представляет собой 5-7-членный гетероциклил, содержащий один или два гетероатома, выбранные из азота.
5. Соединение по любому из пп. 1-3, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где кольцо В представляет собой пиперидиновое кольцо, пиперазиновое кольцо, азациклогептановое мостиковое кольцо или диазациклогептановое мостиковое кольцо.
6. Соединение по любому из пп. 1-3, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где кольцо В представляет собой 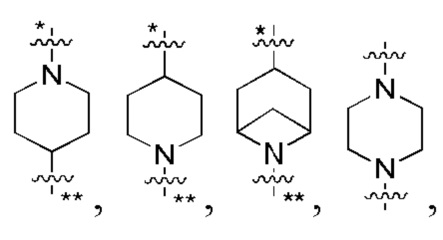
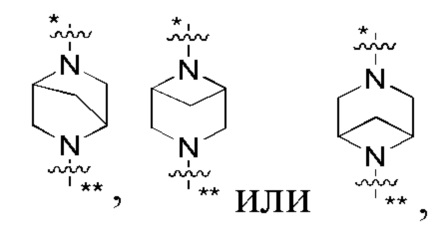 соединено с кольцом А в позиции, отмеченной *, и соединено с L в позиции, отмеченной **.
соединено с кольцом А в позиции, отмеченной *, и соединено с L в позиции, отмеченной **.
7. Соединение по любому из пп. 1-6, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где X1 представляет собой СН или N.
8. Соединение по любому из пп. 1-6, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где X1 представляет собой N.
9. Соединение по любому из пп. 1-8, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R1 выбран из С1-4 алкила.
10. Соединение по любому из пп. 1-8, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R1 выбран из С1-3 алкила.
11. Соединение по любому из пп. 1-10, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R1 представляет собой метил.
12. Соединение по любому из пп. 1-10, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 выбран из 5-6-членного гетероарила, содержащего два гетероатома, выбранные из азота, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-4 алкила и С3-6 циклоалкила.
13. Соединение по любому из пп. 1-10, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 выбран из 5-6-членного гетероарила содержащего два гетероатома, выбранные из азота, и гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-3 алкила и С3-6 циклоалкила.
14. Соединение по любому из пп. 1-10, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 выбран из пиразолила и имидазолила и каждый из пиразолила и имидазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-3 алкила и С3-6 циклоалкила.
15. Соединение по любому из пп. 1-10 или 14, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 выбран из пиразолила и имидазолила и каждый из пиразолила и имидазолила необязательно замещен метилом.
16. Соединение по любому из пп. 1-10, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 представляет собой метил-замещенный пиразолил или циклопропил-замещенный пиразолил.
17. Соединение по любому из пп. 1-10 или 16, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 представляет собой 5-метил-1H-пиразол-3-ил, 1-метил-1H-пиразол-4-ил или 5-циклопропил-1H-пиразол-3-ил.
18. Соединение по любому из пп. 1-17, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где L выбран из группы, состоящей из -О-, -С(О)-, C1-4 алкилена и 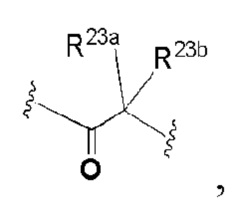 и алкилен необязательно замещен одним или более заместителями, выбранными из С1-4 алкила.
и алкилен необязательно замещен одним или более заместителями, выбранными из С1-4 алкила.
19. Соединение по любому из пп. 1-17, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где L выбран из группы, состоящей из -О-, -С(О)-, - C1-3 алкилена и  и алкилен необязательно замещен одним или более заместителями, выбранными из C1-3 алкила, где R23b представляет собой Н.
и алкилен необязательно замещен одним или более заместителями, выбранными из C1-3 алкила, где R23b представляет собой Н.
20. Соединение по любому из пп. 1-17, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где L представляет собой -СН2-, -СН(СН3)-, -О-, - С(О)- или 
21. Соединение по любому из пп. 1-20, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 выбран из группы, состоящей из С6-10 арила и 5-6-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, и каждый из арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, C1-3 алкила, C1-3 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-4 алкила, С1-4 галогеналкила, С1-4 гидроксиалкила, С1-4 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
22. Соединение по любому из пп. 1-20, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 выбран из фенила и 5-6-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, и каждый из фенила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-3 алкила, C1-3 галогеналкила, 5-8-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-3 алкила, C1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
23. Соединение по любому из пп. 1-20 или 22, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 представляет собой 5-6-членный гетероарил, содержащий один или два гетероатома, выбранные из азота и серы, который выбран из пиридила, пиримидила, пиразинила, пиридазинила, пиразолила, имидазолила или тиазолила, и гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-3 алкила, С1-3 галогеналкила, 5-8-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-3 алкила, C1-3 галогеналкила, С1-3 гидроксиалкила, С1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
24. Соединение по любому из пп. 1-20, 22 или 23, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 выбран из фенила и 5-6-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, и каждый из фенила и гетероарила необязательно замещен одной или более 5-8-членной гетероарильной группой, содержащей один или два гетероатома, выбранные из азота, кислорода и серы, которая выбрана из пиридила, пирролила, пиразолила, фурила, оксазолила, имидазолила, тиазолила или циклопентил-пиразолила, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-3 алкила, C1-3 галогеналкила, С1-3 гидроксиалкила, С1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
25. Соединение по любому из пп. 1-20, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 выбран из группы, состоящей из фенила, пиридила, пиразолила и тиазолила, и каждый из фенила, пиридила, пиразолила и тиазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-3 алкила, С1-3 галогеналкила, 5-8-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-3 алкила, С1-3 галогеналкила, С1-3 гидроксиалкила, С1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
26. Соединение по любому из пп. 1-20, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 представляет собой фенил, пиридил, пиразолил или тиазолил, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из фтора, CN, метила, этила, трифторметила, метокси, этокси и 5-6-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, где 5-6-членный гетероарил необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из фтора, хлора, метила, этила, изопропила, фторметила, гидроксиметила, гидроксипропила, метокси, циклопропила, циклопропокси и циклобутокси.
27. Соединение по любому из пп. 1-26, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R31 выбран из группы, состоящей из С1-4 алкила и С3-8 циклоалкила.
28. Соединение по любому из пп. 1-26, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R31 выбран из С1-4 алкила.
29. Соединение по п. 1, стереоизомер, или их смесь, или его фармацевтически приемлемая соль,
где соединение имеет структуру, показанную в одной из формул I-A, I-B, I-C, I-D, 1-Е, I-G:
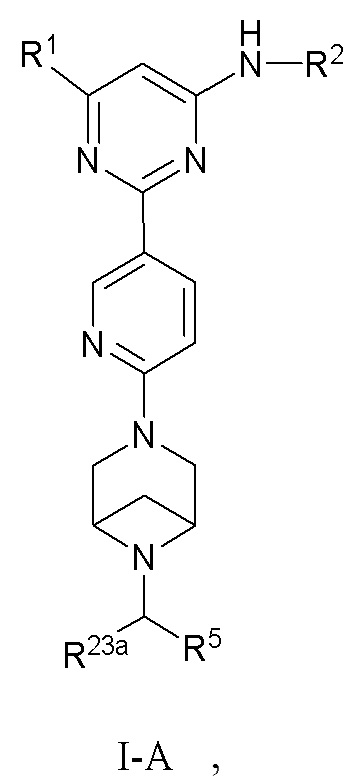
где R5 выбран из группы, состоящей из С6-12 арила и 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, где (1) С6-12 арил необязательно замещен одним или более 5-10-членными гетероарильными заместителями, содержащими один или два гетероатома, выбранные из азота, кислорода и серы, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-4 алкила, С1-4 галогеналкила, С1-4 гидроксиалкила и С3-6 циклоалкила; и (2) 5-10-членный гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-4 алкила, С1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси; и
R1, R2, R23a и R31 определены в любом из пп. 1-28;
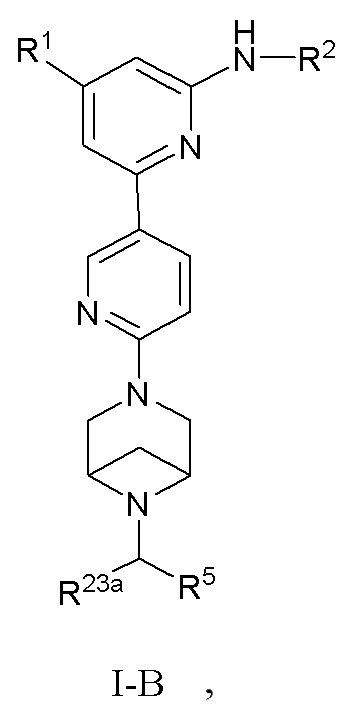
где R1, R2, R5 и R23a определены в любом из пп. 1-28;
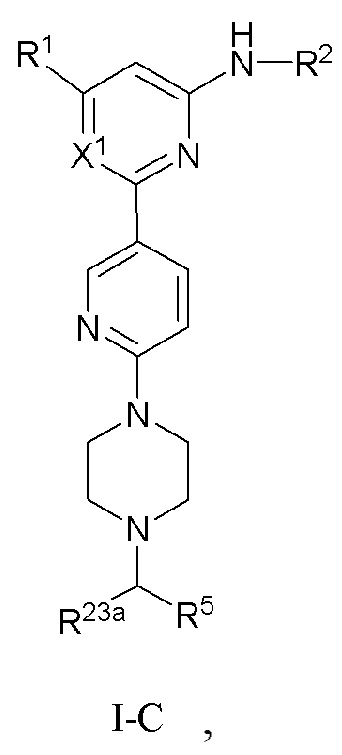
где, если X1 представляет собой СН, R1, R2, R5 и R23a определены в любом из пп. 1-28; и, если X1 представляет собой N, R1, R2, R5 и R23a определены выше в формуле I-A;
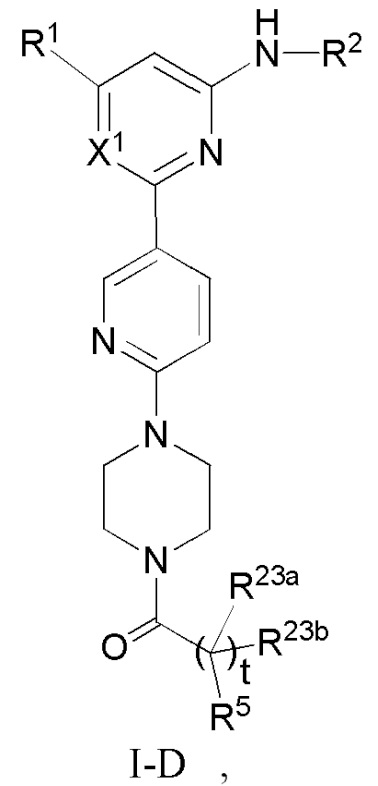
где R1, R2, R23a, R23b и t определены в любом из пп. 1-28;
если X1 представляет собой СН, R5 определен в любом из пп. 1-28; и, если X1 представляет собой N, R5 представляет собой С6-12 арил или 5-10-членный гетероарил, содержащий один или два гетероатома, выбранные из азота и серы, где
(i) если t равно 0, R5 представляет собой С6-12 арил или 5-10-членный гетероарил, содержащий один или два гетероатома, выбранные из азота и серы, где каждый из С6-12 арила и 5-10-членного гетероарила, содержащий один или два гетероатома, выбранные из азота и серы, необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, С1-4 алкокси и С3-6 циклоалкила,
(ii) если t равно 1, R5 представляет собой 5-10-членный гетероарил, содержащий один гетероатом, выбранный из азота, причем 5-10-членный гетероарил, содержащий один гетероатом, выбранный из азота, необязательно замещен -OR31; и
R31 определен в любом из пп. 1-28;
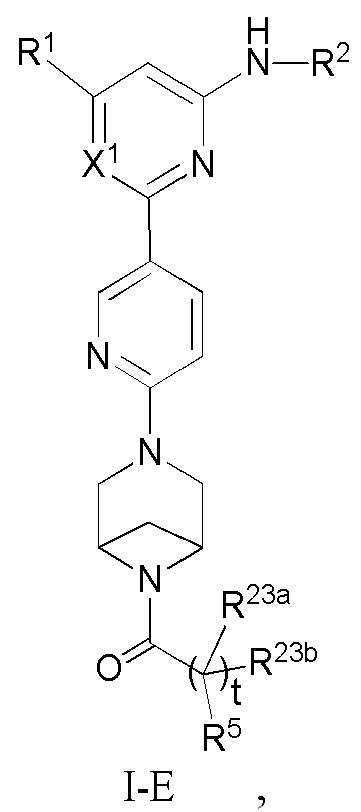
где R1, R2, R5, R23a, R23b, X1 и t определены выше в формуле I-D;
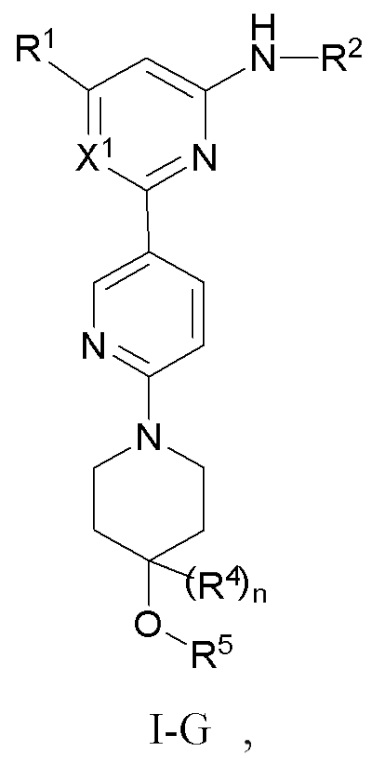
где X1 представляет собой СН или N;
R1, R2 и R4 определены в любом из пп. 1-28;
n равно 0;
R5 выбран из С6-12 арила и 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота и серы, где
(1) С6-12 арил необязательно замещен одним или более 5-10-членными гетероарильными заместителями, содержащими один или два гетероатома, выбранные из азота, кислорода и серы, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-4 алкила, С1-4 галогеналкила, С1-4 гидроксиалкила, С1-4 алкокси и С3-6 циклоалкила; и
(2) 5-10-членный гетероарил, содержащий один или два гетероатома, выбранные из азота и серы, необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-4 алкила, С1-4 галогеналкила, 5-10-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-4 алкила, C1-4 галогеналкила, C1-4 гидроксиалкила, C1-4 алкокси и С3-6 циклоалкила; и
R31 определен в любом из пп. 1-28.
30. Соединение по п. 1, стереоизомер, или их смесь, или его фармацевтически приемлемая соль,
где соединение выбрано из:

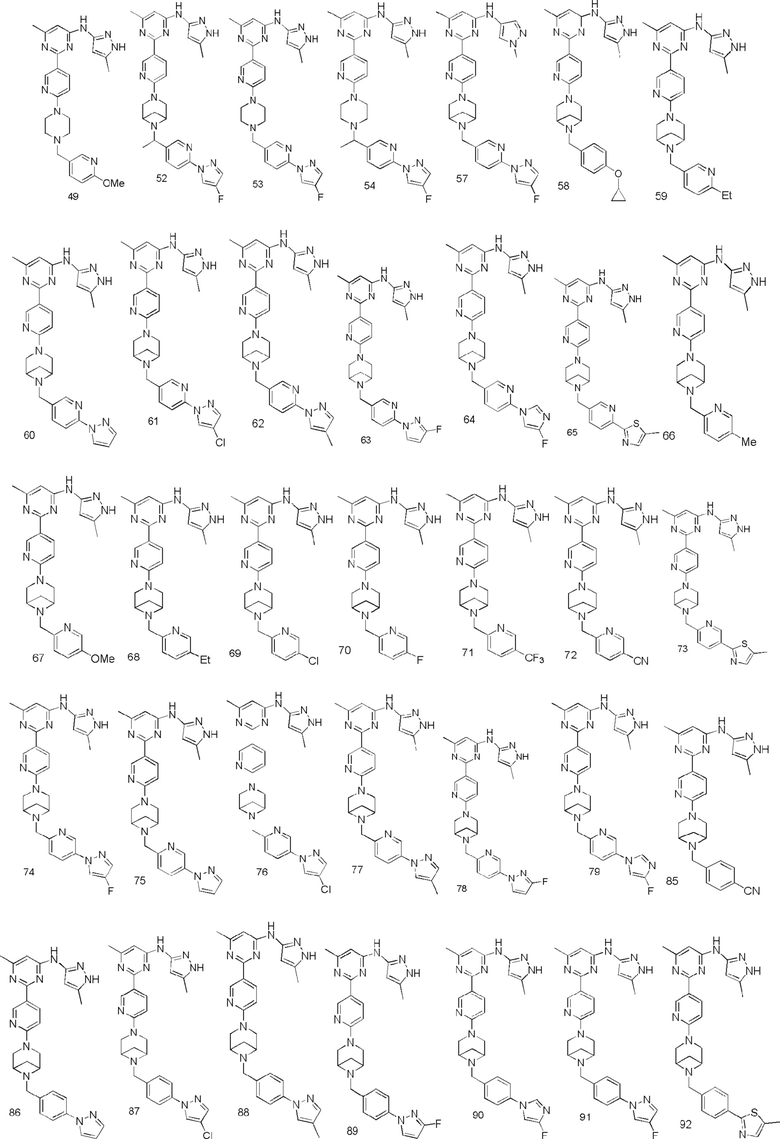
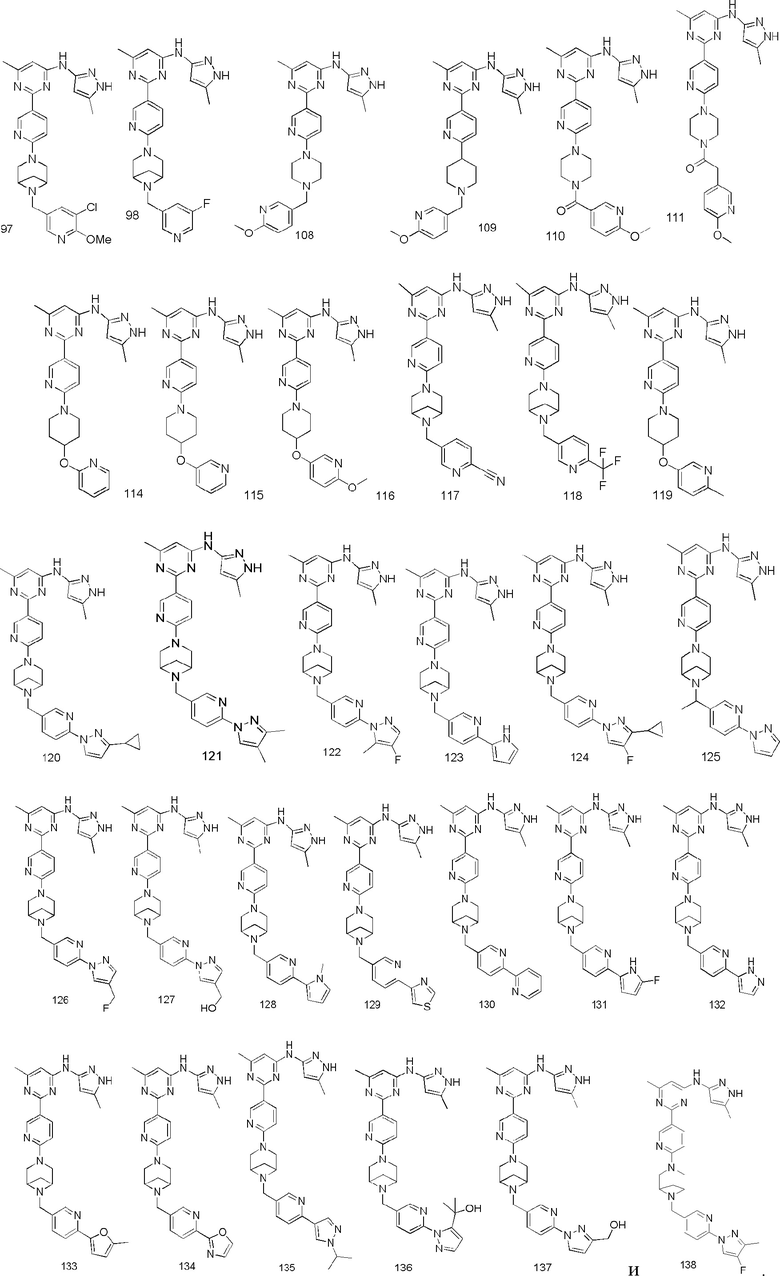
31. Соединение по п. 30, его стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где соединение представляет собой
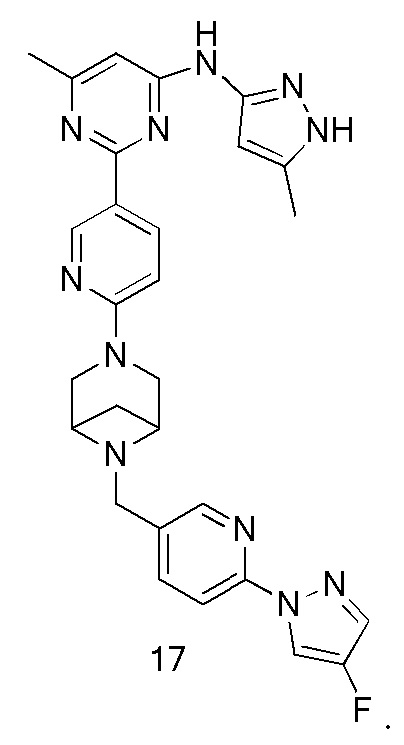
32. Соединение, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, причем соединение имеет структуру формулы I
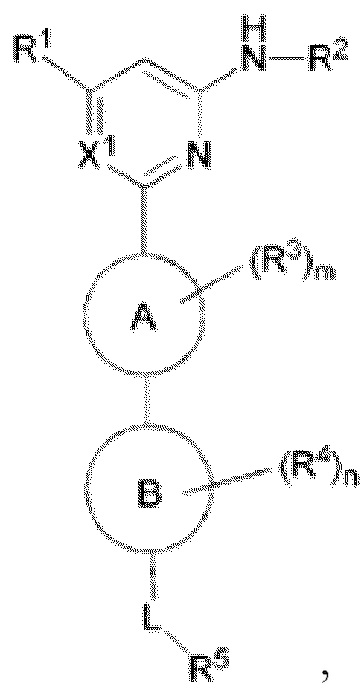
где кольцо А представляет собой пиридиновое кольцо;
кольцо В представляет собой пиперидиновое кольцо, пиперазиновое кольцо, азациклогептановое мостиковое кольцо или диазациклогептановое мостиковое кольцо;
X1 выбран из СН и N;
R1 выбран из C1-6 алкила;
R2 выбран из пиразолила и имидазолила и каждый из пиразолила и имидазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-3 алкила и С3-6 циклоалкила;
R3 и R4 отсутствуют;
L выбран из группы, состоящей из -О-, С1-6 алкилена и 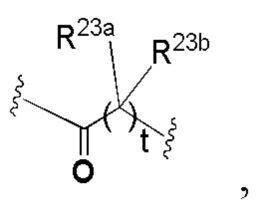 и алкилен необязательно замещен одним или более заместителями, выбранными из С1-6 алкила;
и алкилен необязательно замещен одним или более заместителями, выбранными из С1-6 алкила;
R5 выбран из группы, состоящей из фенила, пиридила, пиразолила и тиазолила, и каждый из фенила, пиридила, пиразолила и тиазолила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, CN, С1-3 алкила, С1-3 галогеналкила, 5-8-членного гетероарила, содержащего один или два гетероатома, выбранные из азота, кислорода и серы, и -OR31, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-3 алкила, С1-3 галогеналкила, С1-3 гидроксиалкила, С1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси;
каждый из R23a и R23b независимо представляет собой Н;
R31 независимо выбран из С1-6 алкила и С3-8 циклоалкила;
m равно 0;
n равно 0; и
t равно 0 или 1;
при условии, что, если кольцо В представляет собой пиперазиновое кольцо и X1 представляет собой СН, R2 не представляет собой 4-CF3-пиридин-2-ил или 4-CN-пиридин-2-ил.
33. Соединение по п. 32, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R2 представляет собой метил.
34. Соединение по п. 32 или 33, стереоизомер, или их смесь, или его фармацевтически приемлемая соль, где R5 выбран из группы, состоящей из фенила, пиридила, пиразолила и тиазолила, и каждый из фенила, пиридила, пиразолила и тиазолила необязательно замещен одной или более 5-8-членной гетероарильной группой, содержащей один или два гетероатома, выбранные из азота, кислорода и серы, где 5-8-членная гетероарильная группа выбрана из пиридила, пирролила, пиразолила, фурила, оксазолила, имидазолила, тиазолила или циклопентил-пиразолила, где гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-3 алкила, С1-3 галогеналкила, C1-3 гидроксиалкила, C1-3 алкокси, С3-6 циклоалкила и С3-6 циклоалкокси.
35. Способ получения соединения по п. 29, причем способ включает следующие стадии:
где каждый из Hal1 и Hal2 независимо представляет собой F, Cl, Br или I; и предпочтительно Hal1 представляет собой F, Cl, Br или I и Hal2 представляет собой Cl, Br или I;
R1 выбран из C1-6 алкила;
R2 представляет собой 5-6-членный гетероарил, содержащий два гетероатома, выбранные из азота, и гетероарил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из С1-4 алкила и С3-6 циклоалкила;
R23a представляет собой Н; и
R5 определен в формуле I-A по п. 29;
Стадия 1: введение в реакцию соединения I-A-1 с R2-NH2 в присутствии основания с образованием соединения I-A-2;
Стадия 2: введение в реакцию соединения I-A-3 с соединением I-A-4 в присутствии основания с образованием соединения I-A-5;
Стадия 3: введение в реакцию соединения I-A-5 с борсодержащим реагентом с образованием соединения I-A-6;
Стадия 4: введение в реакцию соединения I-A-2 с соединением I-A-6 с образованием соединения I-A-7;
Стадия 5: снятие защиты с соединения I-A-7 в кислых условиях с образованием соединения I-A-8; и
Стадия 6: введение в реакцию соединения I-A-8 с соединением I-A-9 с образованием соединения I-A.
36. Способ получения соединения по п. 29, причем способ включает следующие стадии:
где каждая группа определена выше в Пути синтеза А;
Стадия 1: снятие защиты с соединения I-A-5 в кислых условиях с образованием соединения I-A-10;
Стадия 2: введение в реакцию соединения I-A-10 с соединением I-A-9 с образованием соединения I-A-11;
Стадия 3: введение в реакцию соединения I-A-11 с борсодержащим реагентом с образованием соединения I-A-12; и
Стадия 4: введение в реакцию соединения I-A-12 с соединением I-A-2 с образованием соединения I-A.
37. Способ получения соединения по п. 29, причем способ включает следующие стадии:
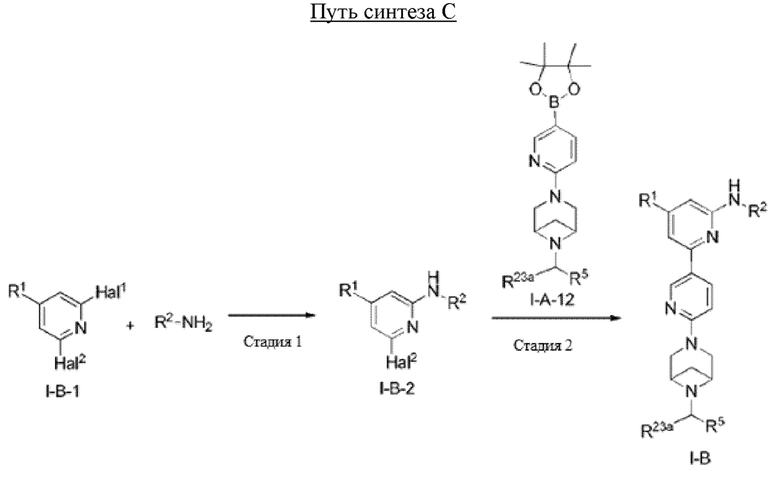
где каждая группа определена выше в Пути синтеза А;
Стадия 1: введение в реакцию соединения I-B-1 с R2-NH2 в присутствии основания с образованием соединения I-B-2; и
Стадия 2: введение в реакцию соединения I-B-2 с соединением I-A-12 с образованием соединения I-B.
38. Способ получения соединения по п. 29, причем способ включает следующие стадии:

где каждая группа определена выше в Пути синтеза А; и X1 выбран из СН и N;
Стадия 1: введение в реакцию соединения I-C-1 с R2-NH2 в присутствии основания с образованием соединения I-C-2;
Стадия 2: введение в реакцию соединения I-C-3 с борсодержащим реагентом с образованием соединения I-C-4;
Стадия 3: введение в реакцию соединения I-C-2 с соединением I-C-4 с образованием соединения I-C-5;
Стадия 4: снятие защиты с соединения I-C-5 в кислых условиях с образованием соединения I-C-6; и
Стадия 5: введение в реакцию соединения I-C-6 с соединением I-A-9 с образованием соединения I-C.
39. Способ получения соединения по п. 29, причем способ включает следующие стадии:
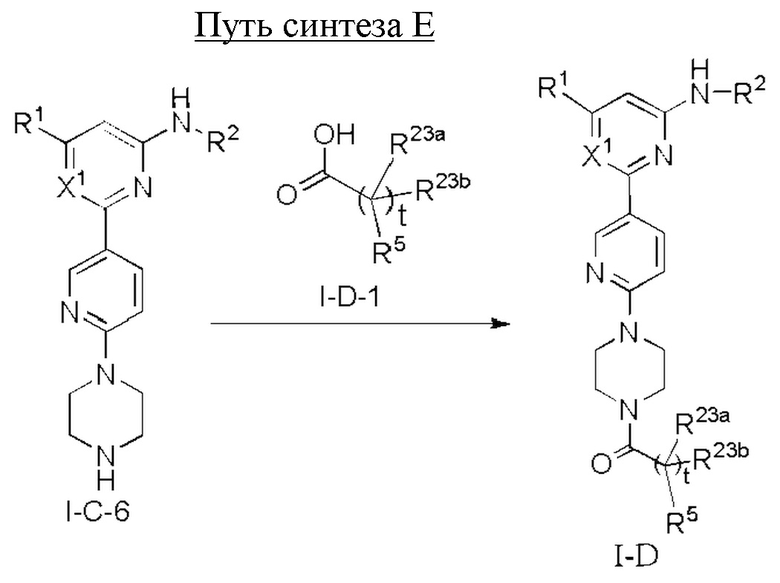
где R1 и R2 определены в Пути синтеза А;
R5 определен в формуле I-D по п. 29;
каждый из R23a и R23b независимо представляет собой Н;
X1 выбран из СН и N; и
t равно 0 или 1.
40. Способ получения соединения по п. 29, причем способ включает следующие стадии:
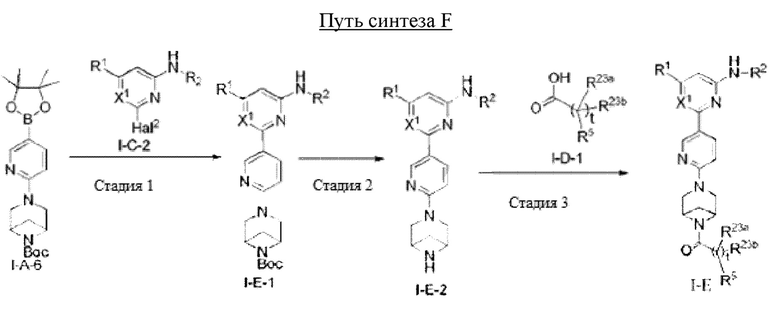
где R1, R2, R5, R23a, R23b и t определены в Пути синтеза Е; X1 выбран из СН и N; и
Hal2 представляет собой F, Cl, Вr или I; и предпочтительно Hal2 представляет собой С1, Вr или I;
Стадия 1: введение в реакцию соединения I-C-2 с соединением I-A-6 с образованием соединения I-Е-1;
Стадия 2: снятие защиты с соединения I-E-1 в кислых условиях с образованием соединения I-E-2; и
Стадия 3: введение в реакцию соединения I-E-2 с соединением I-D-1 посредством реакции конденсации с образованием соединения I-Е.
41. Способ получения соединения по п. 29, причем способ включает следующие стадии:
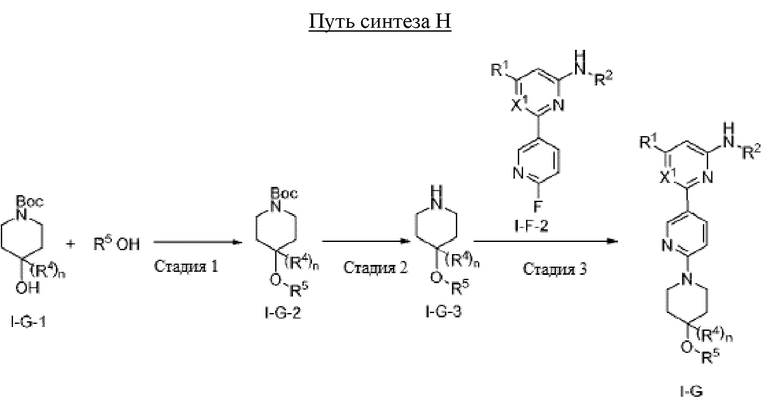
где R1 и R2 определены в Пути синтеза А; X1 выбран из СН и N; R4 отсутствует;
R5 определен выше в формуле I-G; и n равно 0;
Стадия 1: введение в реакцию соединения I-G-1 с R5-OH с образованием соединения I-G-2;
Стадия 2: снятие защиты с соединения I-G-2 в кислых условиях с образованием соединения I-G-3; и
Стадия 3: введение в реакцию соединения I-F-2 с соединением I-G-3 посредством реакции нуклеофильного замещения в присутствии основания с образованием соединения I-G.
42. Фармацевтическая композиция для предотвращения или лечения заболевания или состояния, связанного с активностью RET, содержащая профилактически или терапевтически эффективное количество соединения по любому из пп. 1-34, стереоизомера, или их смеси, или его фармацевтически приемлемой соли.
43. Фармацевтическая композиция по п. 42, где фармацевтическая композиция дополнительно содержит один или более фармацевтически приемлемых носителей.
44. Применение соединения по любому из пп. 1-34, стереоизомера, или их смеси, или его фармацевтически приемлемой соли, или фармацевтической композиции по п. 42 или 43 в получении лекарственного средства для предотвращения или лечения заболевания или состояния, связанного с активностью RET.
45. Применение по п. 44, где заболевание или состояние, связанное с активностью RET, представляет собой рак, или опухоль, или синдром раздраженного кишечника.
46. Применение по п. 45, где рак или опухоль представляют собой рак легких, немелкоклеточный рак легких, рак молочной железы, рак головы и шеи, рак прямой кишки, рак печени, лимфому, рак щитовидной железы, медуллярный рак щитовидной железы, папиллярный рак щитовидной железы, рак толстой кишки, множественную миелому, меланому, глиому, опухоль головного мозга или саркому.
| Система и способ обнаружения вредоносного приложения путем перехвата доступа к отображаемой пользователю информации | 2016 |
|
RU2634176C1 |
| WO 2012154518 A1, 15.11.2012 | |||
| НОВОЕ ПРОИЗВОДНОЕ АМИНОПИРИДИНА С ИНГИБИТОРНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ SYK. | 2006 |
|
RU2363699C2 |
| LI B-K | |||
| et al, In silico prediction of spleen tyrosine kinase inhibitors using machine learning approaches and an optimized molecular descriptor subset generated by recursive feature elimination method, Computers in Biology and Medicine, 2013, vol.45, no.4, | |||

