Область техники
Изобретение относится к области неорганической химии и касается способа получения висмут-натрий-кальций оксоапатита Ca8BiNa(PO4)6O. Внедрение в кристаллическую решетку апатита ионов натрия, как правило, способствует увеличению биорезорбции материала, тогда как висмут может придавать материалу бактерицидные свойства. Данное вещество ввиду своей биосовместимости с клетками человека может стать химической основой создания керамических материалов медицинского назначения.
Уровень техники
Известен способ получения магний-замещенного гидроксиапатита (RU 2617103 С1, кл. С01В 25/32, С01В 25/34, C01F 5/00, C01F 11/00, опубл. 20.04.2017). Способ включает синтез Mg-ГА с использованием водных растворов нитратов, диаммонийфосфата и аммиака, фильтрацию осадка и последующую сушку, причем синтез Mg-ГА осуществляют смешиванием в течение 1 ч водных растворов нитрата магния и кальция, взятых соотношении 9:1, при добавлении эквимолекулярного количества раствора диаммонийфосфата и 25%-ного водного раствора NH4OH до образования Mg-ГА в виде осадка. Mg-ГА выдерживают до созревания в течение 20-26 ч, фильтруют, сушат при температуре 90-95°С, затем в течение 100-130 ч при температуре 200-250°С. Прокаливают 6-8 ч при температуре 600-650°С, затем Mg-ГА охлаждают при комнатной температуре в течение 2-3 ч и измельчают в течение 15-20 мин.
Недостатком метода является длительность проведения процесса. Также метод подразумевает изовалентное замещение магния на кальций.
Известен способ получения гидроксиапатита, обладающего антимикробной активностью (RU 2026073 С1, кл. A61K 33/38, A61K 33/34, опубл. 09.01.1995). Изобретение решает задачу получения антимикробного гидроксиапатита, содержащего высокие концентрации бактерицидных веществ. Указанный результат достигается тем, что в способе получения гидроксиапатита, обладающего антимикробной активностью, включающем смешивание гидроксиапатита с неорганическими или органическими бактерицидными соединениями в воде, отделение осадка, его промывку водой и сушку, смешивание гидроксиапатита и бактерицидных соединений производят при их массовом соотношении 1:0,2-0,3, а перед смешиванием ингредиентов к воде добавляют динатриевую соль этилендиаминтетрауксусной кислоты и препарат энтеродез из расчета соответственно 0,03-0,05 и 0,08-0,1 мас.ч. на 1 мас.ч. гидроксиапатита. При этом в качестве неорганических бактерицидных соединений используют нитрат серебра или сульфат цинка, а в качестве органических бактерицидных соединений - лизоцим или хлоргексидин.
Недостатком является использование не изоморфно внедренных в кристаллическую структуру антимикробных агентов, а второй фазы с соответствующими свойствами.
В качестве прототипа взят способ получения биосовместимых висмут апатитов, описанный в патенте RU 2776293 С1, кл. С01В 24/455, С01В 25/45, A61K 6/838, A61L 27/12, опубликован 18.07.2022. Изобретение относится к неорганической химии и касается способа получения биосовместимых висмут-апатитов состава Ca10-2xBixNax(PO4)6F2, где х = 1, 2, 3, 4, которые могут быть использованы в медицине, в том числе в стоматологии, для производства медицинских керамических материалов, стимулирующих восстановление дефектов костной ткани, а также обеспечивающих защиту от развития бактериальных инфекций. Способ включает помещение в алундовый тигель стехиометричной смеси четырехводного нитрата кальция, пятиводного нитрата висмута, нитрата натрия, гидрофосфата диаммония и фторида аммония, после чего тигель помещают в муфельную печь с закрытой спиралью и нагревают до температуры 300-350°С со скоростью 3-5 град./мин до окончания дегидратации исходных кристаллогидратов и термического разложения используемых реагентов. Затем тигель охлаждают до комнатной температуры и шихту диспергируют в агатовой ступке с использованием этилового спирта для создания дополнительного расклинивающего давления. Полученный монодисперсный порошок помещают в тигель и прокаливают до 600-670°С со скоростью 5-7 град./мин с диспергированием каждый час в течение 10 минут. Далее смесь прокаливают в алундовом тигле при температуре 950-1100°С в течение 7-9 часов.
Недостатком описанного подхода является сложность в контроле количества фтора, вошедшего в кристаллическую структуру конечного продукта.
Краткое описание чертежей
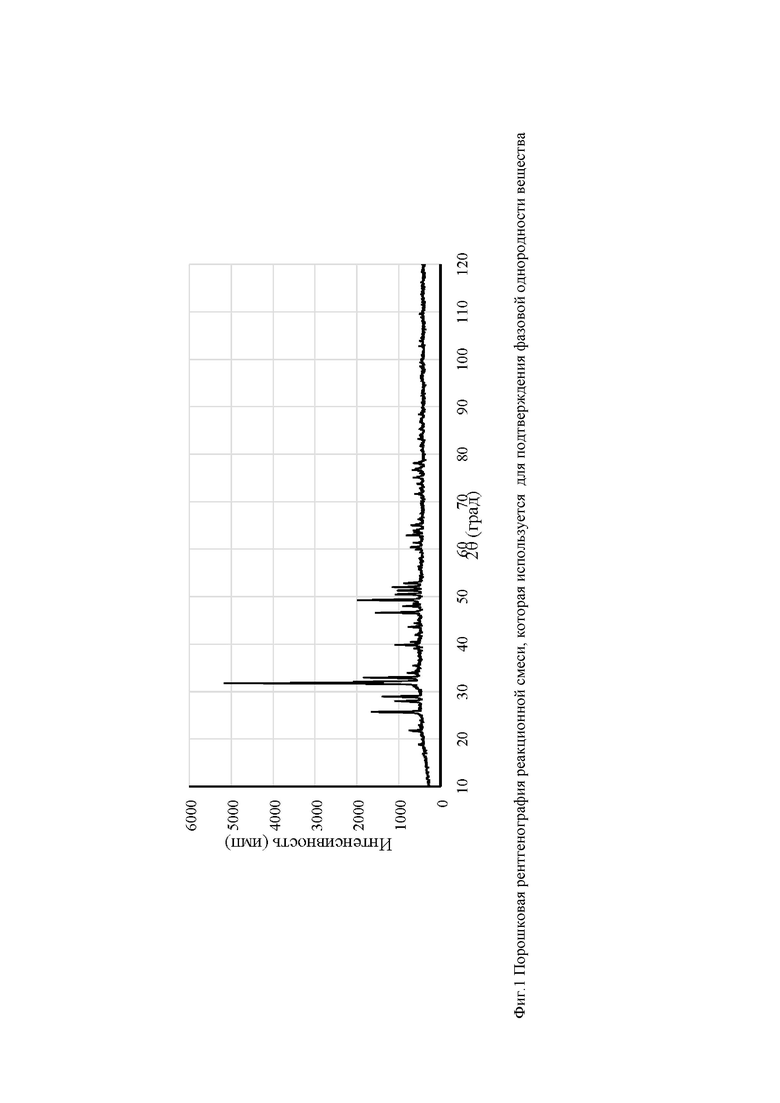
Фиг. 1. Порошковая рентгенография реакционной смеси, которая используется подтверждения фазовой однородности вещества.
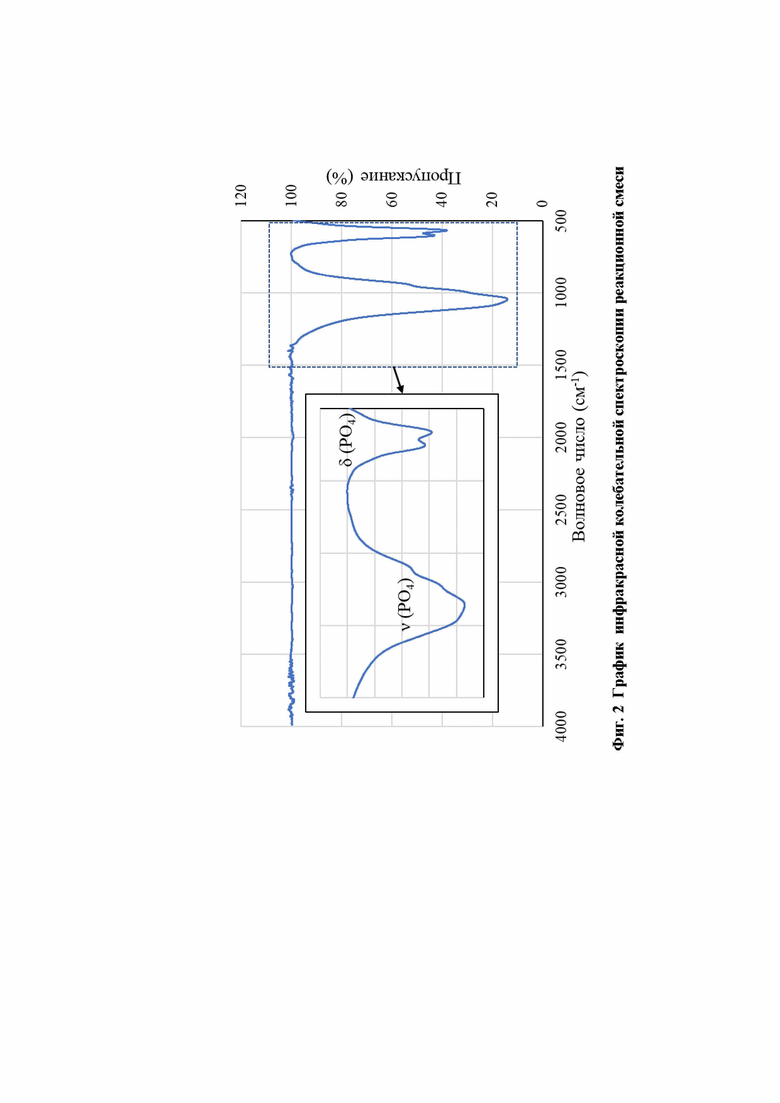
Фиг. 2. График инфракрасной колебательной спектроскопии реакционной смеси.
Фиг. 3. Фото частицы полученного апатита.
Сущность изобретения
Задачей данного изобретения является создание способа получения висмут-натрий-кальций оксоапатита состава Ca8BiNa(PO4)6O в форме кристаллического порошка. Описание способов получения висмут-содержащих оксоапатитов в охранных документах не встречается.
Техническим результатом от использования предлагаемого изобретения являйся создание оптимальной технологии изготовления конечного продукта - висмут-натрий-кальций оксоапатита состава Ca8BiNa(PO4)6O, подразумевающий использование доступных реагентов и относительно короткого промежутка времени.
Описание изобретения
Решение задачи достигается путем проведения процесса синтеза в твердой фазе между четырехводным нитратом кальция, пятиводным нитратом висмута, нитратом натрия и гидрофосфатом диаммония согласно представленной схеме реакции:
Bi(NO3)3⋅5H2O++NaNO3+8Ca(NO3)2⋅4H2O+6(NH4)2HPO4→Ca8BiNa(PO4)6O+
газообразные продукты реакции
Реакционную смесь реактивов в стехиометрическом количестве (мольное соотношение Ca/Bi/Na/P=8/1/1/6) помещают в алундовый тигель. Из-за высокого содержания воды в исходных кристаллогидратах первичное диспергирование стехиометрической смеси реагентов не проводится целью сохранения необходимого соотношения нужных атомов. Диспергирование шихты в агатовой ступке проводится перед каждым повышением температуры в процессе синтеза, который состоял из трех стадий.
На первом этапе «тигель помещали в муфельную печь и нагревали до температуры 573 K со скоростью 3-5 град./мин и прокаливали при температуре 573 K в течение 3 ч, до полного удаления гидратной воды и разложения нитрата висмута (348 K). Затем полученную шихту диспергировали и помещали в печь при температуре 873 K (6 часов) до разложения нитрата кальция (834 K). Использование четырехводного нитрата кальция позволяет значительно сократить время проведения процесса и снизить его температуру за счет ряда факторов. Во-первых, температура разложения нитрата кальция (834 K) значительно ниже температуры разложения других соединений кальция. Во-вторых, в результате разложения нитрата кальция образуется оксид кальция, обладающий повышенной поверхностной энергией за счет высокой дисперсности и дефектности поверхностного слоя частиц, что в свою очередь повышает его реакционноспособность. Заключительный этап синтеза проводился при температуре 1273 K до получения конечного продукта (в течение восьми часов).
Для подтверждения фазовой однородности вещества используется порошковая рентгенография (фиг. 1). Положения основных дифракционных максимумов (угол 2θ) должно совпадать с точностью до 0,5° с таковыми для рентгенограммы апатита состава BiCa4(РО4)3О из кристаллографической базы данных.
Для независимого подтверждения группового строения полученного вещества может быть использован метод инфракрасной колебательной спектроскопии (фиг. 2). При этом исследуемое вещество должно быть подготовлено к исследованию в виде таблетки из бромида калия (KBr), содержащей 4-5 мас. % исследуемого вещества. В спектре соединения будут наблюдаться характеристические полосы, отвечающие деформационным (550-640 см-1) и валентным (950-1200 см-1) колебаниям тетраэдрических групп PO4, а также отсутствовать полосы колебаний ОН-групп (ок. 3600 см-1), полосы колебаний пирофосфатной группы Р2О7 (~720 см-1).
Метод растровой электронной микроскопии используется для определения морфологии частиц вещества (фиг. 3). Частица полученного апатита должны иметь сферическую форму и средний размер 1-10 мкм.
Биологическая совместимость полученного вещества подтверждается стандартным МТТ-тестом с использованием культуры фибробластов человека.
Ниже представлены примеры конкретного осуществления предлагаемого изобретения:
Пример 1
В алундовый тигель объемом 40 мл помещается последовательно 9,96 г Ca(NO3)2⋅4H2O, 2,56 г Bi(NO3)3⋅5H2O, 0,45 г NaNO3 4,18 г (NH4)2HPO4. Тигель помещали в муфельную печь с закрытой спиралью и нагревали до температуры 573 K со скоростью 3-5 град./мин. Затем тигель охлаждали до комнатной температуры и шихту диспергировали в агатовой ступке с использованием этилового спирта для создания дополнительного расклинивающего давления. Полученный монодисперсный порошок помещали в тигель и нагревали до 873 K со скоростью 5-7 град./мин с диспергированием каждый час в течение 10 минут. При конечной температуре шихту выдерживали в течение 6 часов. Далее после диспергирования смесь прокаливается в алундовом тигле при температуре 1273 K в течение 6-8 часов.
Пример 2
Получение висмут-натрий-кальций апатита осуществляется аналогично примеру 1, но реакционная смесь гомогенизируется путем перетирания в ступке и разделяется на 5 частей, что позволяет проводить ее нагрев более равномерно и быстро, сокращая общее время синтеза.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения биосовместимых висмут-апатитов | 2021 |
|
RU2776293C1 |
| Способ синтеза фосфатов металлов в степени окисления III | 2020 |
|
RU2758257C1 |
| СЛОЖНЫЙ СИЛИКАТ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2008 |
|
RU2379328C2 |
| Способ получения наноразмерного гидроксиапатита | 2020 |
|
RU2736048C1 |
| СВЕТОТРАНСФОРМИРУЮЩИЙ МАТЕРИАЛ И КОМПОЗИЦИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 1997 |
|
RU2132856C1 |
| СПОСОБ ПОЛУЧЕНИЯ МАГНИЙ-ЗАМЕЩЕННОГО ГИДРОКСИАПАТИТА | 2015 |
|
RU2617103C1 |
| СПОСОБ ПОЛУЧЕНИЯ NaSn(PO) СО СТРУКТУРОЙ NASICON | 2021 |
|
RU2777643C1 |
| Способ получения аморфного гидроксиапатита | 2025 |
|
RU2839844C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИАПАТИТА | 2008 |
|
RU2391117C1 |
| Способ получения диопсида | 2022 |
|
RU2801146C1 |

Изобретение может быть использовано при создании керамических материалов медицинского назначения. Для получения висмут-натрий-кальций оксоапатита в алундовый тигель объемом 40 мл помещают последовательно 9,96 г Ca(NO3)2⋅4H2O, 2,56 г Bi(NO3)3⋅5H2O, 0,45 г NaNO3, 4,18 г (NH4)2HPO4. Тигель помещают в муфельную печь и нагревают до температуры 573 K со скоростью 3-5 град./мин. Затем проводят прокаливание при температуре 573 K в течение 3 ч, после чего тигель охлаждают до комнатной температуры и шихту диспергируют в агатовой ступке с использованием этилового спирта. Полученный монодисперсный порошок помещают в тигель и снова нагревают до 873 K со скоростью 5-7 град/мин с диспергированием каждый час в течение 10 мин. При конечной температуре шихту выдерживают в течение 6 ч. После диспергирования смесь вновь прокаливают в алундовом тигле при температуре 1273 K в течение 6-8 ч. Изобретение позволяет получить висмут-натрий-кальций оксоапатит состава Ca8BiNa(PO4)6O в форме кристаллического порошка из доступных реагентов в течение относительно короткого промежутка времени. 3 ил., 2 пр.
Способ получения висмут-натрий-кальций оксоапатита, включающий в себя следующие этапы: в алундовый тигель объемом 40 мл помещают последовательно 9,96 г Ca(NO3)2⋅4H2O, 2,56 г Bi(NO3)3⋅5H2O, 0,45 г NaNO3, 4,18 г (NH4)2HPO4, тигель помещают в муфельную печь и нагревают до температуры 573 K со скоростью 3-5 град/мин, прокаливают при температуре 573 К в течение 3 ч, после чего тигель охлаждают до комнатной температуры и шихту диспергируют в агатовой ступке с использованием этилового спирта, полученный монодисперсный порошок помещают в тигель и снова нагревают до 873 K со скоростью 5-7 град./мин с диспергированием каждый час в течение 10 мин, при конечной температуре шихту выдерживают в течение 6 ч, отличающийся тем, что после диспергирования смесь вновь прокаливают в алундовом тигле при температуре 1273 K в течение 6-8 ч.
| Способ получения биосовместимых висмут-апатитов | 2021 |
|
RU2776293C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОГИДРОКСИАПАТИТА | 2015 |
|
RU2614772C1 |
| US 5441717 A, 15.08.1995 | |||
| ГОЛИЦЫНА О.Н | |||
| и др | |||
| Синтез магний-замещенных биосовместимых висмут-фторапатитов, ХХV Всероссийская конференция молодых учёных-химиков (с международным участием), Тезисы докладов, Нижний Новгород, 19-21 апреля 2022 года, Национальный исследовательский Нижегородский | |||

