Область техники
Заявленное изобретение относится к области аналитической химии, а именно к количественному определению микроколичеств воды в различных органических соединениях (различных органических растворителях, топливе для транспорта (бензин, биэтанол, дизельное топливо), растворах твердых органических соединений, реагентах для органического синтеза, реакционных смесях и др.) с целью определения их чистоты и пригодности для решения различных практических задач, когда содержание воды является критическим параметром качества продукта, включая коррозионную способность реагентов и химическую реакционноспобность в органическом синтезе, полноту экстракционного извлечения лекарственных препаратов из растительного сырья в фармацевтической промышленности, ингибирование каталитических процессов и отравление катализаторов в химической промышленности, формирование нежелательных азеотропных смесей при перегонке в нефтехимической промышленности.
Уровень техники
Во многих случаях высокая степень очистки и отсутствие нежелательных активных примесей в органических соединениях и, в частности, органических растворителях, являются важными условиями их использования, например, в различных технологических процессах тонкого органического синтеза. Одной из наиболее распространенных и нежелательных примесей в органических растворителях, особенно полярных, является вода, а микропримеси влаги присутствуют практически всюду. Определение примесей воды в органических соединениях является одной из наиболее важных и трудных аналитических задач и имеет огромное практическое значение, так как даже незначительные примеси воды могут приводить к деактивации катализаторов, изменению кинетики протекания реакций и состава получаемых соединений, коррозии металлических частей аппаратов и их ускоренному износу, возникновению побочных реакций с нежелательными эффектами и уменьшением выхода целевого продукта.
Известен способ титриметрического определения воды в органических растворителях по Карлу Фишеру. Способ основан на реакции окисления двуокиси серы раствором йода в спиртовой среде в присутствии воды и органического основания. Данная реакция позволяет проводить количественное определение воды в широком диапазоне содержаний в пробе от 0,001 до 100 масс.%. воды [«Water determination», N.L.T. Padivitage, J.P. Smuts, D.W. Armstrong, «Specification of Drug Substances and Products: Development and Validation of Analytical Methods», Elsevier, 2013, p. 223-241].
Недостатками данного способа являются высокая стоимость, токсичность и ограниченная стабильность реагентов, а также протекание побочных реакций ряда соединений (альдегидов, кетонов, сильных кислот, борной кислоты, перекисей металлов) с реагентами с образованием воды, что влияет на определение конечной точки титрования.
Существенными недостатками являются необходимость трудоемкой и требующей дополнительных временных затрат стандартизации растворов, окислительно-восстановительная активность соединений (например, диметилсульфоксида) по отношению к ключевому реагенту метода Карла Фишера - йоду, а также способность некоторых веществ (например, хлорида лития) связывать и удерживать воду.
Известен способ определения воды с применением эксклюзионной жидкостной хроматографии. Данный способ основан на разделении соединений, молекулы которых заметно отличаются по размеру или массе, и прямом определении воды в органических соединениях. Способ может быть использован для прямого анализа нелетучих и термически нестабильных соединений [«On the determination of water in hydrocarbons by gel chromatography», U. Fehrmann, W. Schnabel, «Fresenius Zeit. Anal. Chem.», 1974, v. 269, 116-118].
Недостатком данного способа является то, что селективность обычных хроматографических колонок недостаточна для разделения соединений близких по молекулярной массе, например, воды и метанола, воды и ацетонитрила, поэтому метод пригоден для определения воды только в органических растворителях с заметно более высокой молекулярной массой. Наличие других низкомолекулярных примесей, не воды, влияет на точность и правильность ее определения.
Наиболее близким к заявляемому решению является способ определения примесей воды в органических соединениях с использованием метода высокоэффективной жидкостной хроматографии (ВЭЖХ) в изократическом режиме с кондуктометрическим детектированием и использованием хроматографических колонок на основе силикагеля с привитыми фазами C18 Whatman Partisil 10-ODS-3, катион-обменных колонок на основе силикагеля с привитыми бензолсульфоновыми кислотными группами Whatman Partisil SCX, силикагелевых колонок Du Pont Zorbax SIL, колонок на основе катионообменника - сульфированного гидрофобного сополимера стирол и дивинилбензола Bio-Rad - Aminex 50WX4 в Na+ или H+ форме. В качестве элюентов используют метанол с добавками неорганических солей (NaCl или LiBr) или кислот (соляной, серной, пара-толуолсульфокислоты). Причем добавки кислот увеличивают чувствительность определения в 160 раз по сравнению с добавками солей (сильная кислота имеет примерно в 5 раз большую удельную проводимость, чем равная концентрация соли кислоты, образующейся при нейтрализации сильным основанием). Пределы обнаружения воды составляют 2.5*10-3 масс.%. Линейность наблюдается до 0,1 масс.% воды [«Аpparatus and method for the determination of water by liquid chromatography», Stevens T.S., Chritz K.M., Small H. US 4696904 A].
К недостаткам этого решения можно отнести способность катион-обменных колонок (колонки Aminex 50WX4, Whatman Partisil SCX) необратимо удерживать органические катионы, например амины и четвертичные аммониевые основания, а также неполярные гидрофобные соединения, что приводит к изменению хроматографических свойств колонки за счет постепенного отравления и связанному с этим изменению параметров удерживания и аналитических характеристик системы. Это существенно усложняет анализ из-за необходимости дополнительной трудоемкой калибровки системы. анализа. При использовании другого типа хроматографических колонок - неполярных Whatman Partisil 10-ODS-3, наблюдается слабое удерживание воды, что затрудняет анализ из-за возможного перекрывания с основными хроматографическими, элюируемыми в мертвом объеме колонки. К тому же линейность градуировочных зависимостей содержания воды соблюдается лишь до 0,1 масс.%, что существенно ограничивает применимость данного метода. Недостатком вышеупомянутого является невозможность использования неполярной колонки Whatman Partisil 10-ODS-3 для анализа гидрофобных полярных и неполярных соединений.
Техническая проблема, решаемая посредством заявляемого изобретения, заключается в необходимости преодоления недостатков, присущих аналогам и прототипу при количественном определении микропримесей воды в различных органических соединениях, таких как протекание побочных реакций с реагентами, вызывающих образование воды, низкая селективность разделения соединений, невозможность проведения анализов термолабильных, нелетучих и вязких соединений.
Раскрытие изобретения
Технический результат, на получение которого направлено изобретение, заключается в создании способа прямого количественного определения микропримесей воды, который характеризуется простотой, экспрессностью (время анализа 15-20 минут), высокой селективностью (сорбент селективно удерживает лишь воду, коэффициент селективности α=k2/k1 между водой и остальными соединениями ≥ 11,495). Кроме того, способ характеризуется возможностью проведения анализа широкого спектра полярных и нелетучих органических соединений. Заявляемый способ применим для анализа следов влаги в широком диапазоне концентраций 0,005-9,6 масс.% и обеспечивает хорошую воспроизводимость (RSD ≤ 3,0%) и высокую чувствительность определения (ПО=0,0005 масс.%).
Техническая проблема решается сорбентом, представляющим собой микропористый кристаллический алюмосиликат с площадью поверхности от 200 до 1200 м2/г, средним размером частиц от 1,5 до 20 мкм и диаметром пор от 0,3 до 1,2 нм, для определения микроколичеств воды в органических соединениях методом ВЭЖХ. Предпочтительно для определения микроколичеств воды использовать сорбент с площадью поверхности от 400 до 900 м2/г, средним размером частиц от 3,0 до 10 мкм и диаметром пор от 0,4 до 1,0 нм, наиболее предпочтительно - с площадью поверхности в диапазоне от 600 до 700 мкм, размером частиц от 4 до 6 мкм и диаметром пор от 0,5 до 0,8 нм.
Также техническая проблема решается способом определения микроколичеств воды в органических соединениях ВЭЖХ, включающим заполнение хроматографической колонки вышеописанным сорбентом, промывание сорбента элюентом до уравновешивания базовой линии рефрактометрического детектора с последующим введением пробы и определение концентрации воды в пробе с использованием предварительно построенной градуировочной зависимости. При этом используют хроматографическую колонку с длиной равной 25-300 мм и внутренним диаметром 0,2-10 мм. В качестве элюента используют безводные низшие спирты, такие как метанол, этанол, пропанол или изопропанол. Для детектирования используют спектрофотометрический или кондуктометрический детекторы. При этом градуировочную зависимость строят способом добавок как зависимость площади хроматографического пика воды от ее содержания в растворе.
Техническая проблема также решается применением вышеописанного сорбента для определения микроколичеств воды в органических соединениях методом ВЭЖХ.
Таким образом, предложенные параметры заявленного способа позволяют быстро и достоверно определить содержания воды в органических соединениях. Высокая селективность разделения остаточных количеств воды и матричных растворителей делает возможным проведение прямого анализа без предварительной или с минимальной подготовкой образца. Отсутствие побочных реакций при выбранных условиях методом ВЭЖХ делает возможным анализ различных полярных и неполярных органических растворителей, включая нелетучие.
Описание чертежей
Заявленное изобретение поясняется чертежами, где изображено следующее:
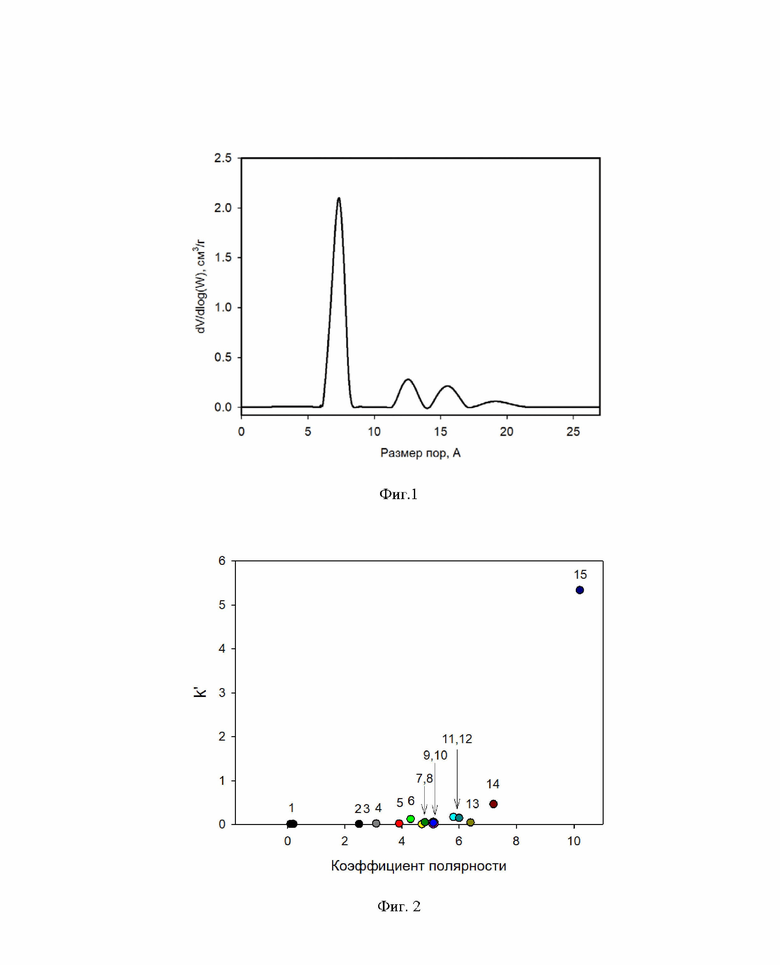
на фиг. 1 - распределение пор по размерам в цеолите 13Х;
на фиг. 2 - зависимость удерживания органических растворителей (k') от их полярности (P'). Условия проведения экспериментов: колонка 50×4,6 мм, заполненная цеолитом 13X, элюент: хроматографически чистый метанол, скорость потока: 0,5 мл/мин, температура колонки: 35°C, объем пробы: 20 мкл.
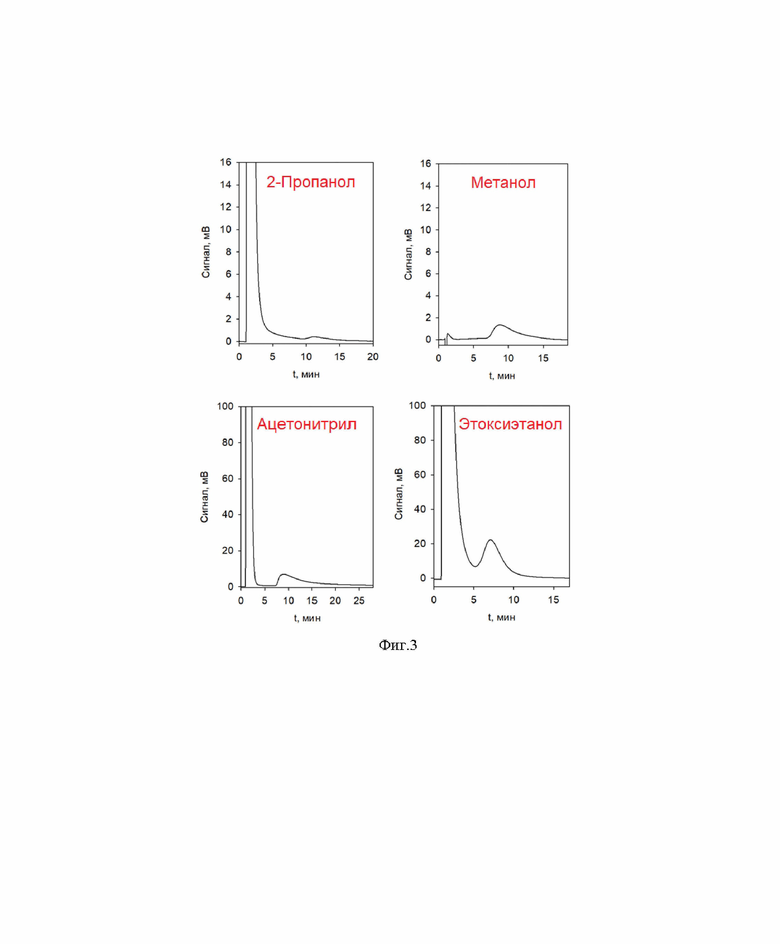
на фиг. 3 - хроматограммы, полученные прямым вводом чистых органических растворителей. Условия проведения: колонка 50×4,6 мм, заполненная цеолитом 13X, элюент: хроматографически чистый метанол, скорость потока: 0,5 мл/мин, температура колонки: 35°C, объем пробы: 20 мкл.
Позициями на фигуре 2 обозначены: 1 - гексан; 2 - п-ксилол; 3 - бензол; 4 - дихлорметан; 5 - 2-пропанол; 6 - этанол; 7 - метилэтилкетон; 8 - 1,4-диоксан; 9 - ацетон; 10 - метанол; 11 - ацетонитрил; 12 - нитрометан; 13 - ДМФА; 14 - ДМСО; 15 - вода.
Найденные значения примесей воды в органических растворителях представлены в таблице 2.
Осуществление изобретения
Метод ВЭЖХ имеет ряд существенных достоинств. В отличие от титрования по Карлу Фишеру используются более устойчивые и дешёвые реагенты. ВЭЖХ может быть использована для прямого анализа реакционноспособных соединений, однако для определения воды требуется высокая селективность хроматографического разделения воды и матричного соединения.
Большую роль в хроматографии играет пористость сорбента. Большой интерес уделяется микропористым сорбентам, таким как цеолиты, по причине того, что большинство молекул имеют размер, соизмеримый с диаметром их пор. Размер пор микропористых цеолитов примерно в два раза больше размера молекул большинства низкомолекулярных соединений, что позволяет использовать молекулярно-ситовой эффект для повышения селективности разделения компонентов смеси. Механизм удерживания соединений цеолитами определяется также наличием кислотных центров Льюиса и Бренстеда, отвечающих за ионный обмен и образование водородных связей.
Известно, что из-за перечисленных особенностей цеолиты обладают высоким сродством к воде, но это уникальное свойство не было ранее использовано для определения воды методом ВЭЖХ. Желательно, чтобы элюент не вступал в реакцию с другими компонентами образца с образованием значительного количества воды, мешающего его работе. Например, кетоны и альдегиды могут вступать в реакцию с метанолом с образованием кеталей и ацеталей, а также воды в качестве побочного продукта. Эти недостатки хорошо известны при использовании метода Карла Фишера для определения воды и устраняются заменой метанола в реагенте Карла Фишера другим растворителем, инертным к компонентам пробы.
Скорость потока элюента выбрана с учетом построения и изучения кривой Ван-Деемтера и зависимости объема удерживания от линейной скорости потока подвижной фазы. Минимум кривой Ван-Деемтера наблюдается при линейной скорости элюента 0,03 мм/сек, максимум объема удерживания достигается в промежутке от 0,3 мм/сек до 0,6 мм/сек, однако при таких скоростях подвижной фазы время анализа значительно увеличивается. В целях экономии времени анализа использовали компромисс - 1,02 мм/сек = 0,5 мл/мин.
Заявленный способ обеспечивает возможность использования колонок с различным значением длины (от 30 до 300 мм) с достижением технического результата - прямого определения содержания воды в органическом соединении. Однако при увеличении длины колонки увеличивается время удерживания и анализа в целом, так как соединения проходят через больший слой сорбента, что также приводит к уширению и размытию пиков и снижению точности способа соответственно.
Градуировочную зависимость получают методом добавок в тех же условиях. В заявленном методе используют заполненную микропористым цеолитом колонку длиной от 50 до 250 мм и внутренним диаметром от 3 до 10 мм. Предварительно колонку промывают метанолом со скоростью потока 0,5 мл/мин при 35°С до достижения динамического равновесия, после чего осуществляют введения в колонку пробы в количестве 20 мкл. Серию проб готовят разбавлением соединений в метаноле и добавлением рассчитанных количеств воды. Градуировочную зависимость строят в диапазоне концентраций 0,005-9,55 масс.%, что является наиболее оптимальным. Нижнее значение обусловлено пределом обнаружения (ПО) воды 0,0005 масс.% в заявленном способе. Верхнее значение не является конечным значением линейности градуировочной зависимости, однако определение более высоких концентраций воды не является целью заявленного способа.
Для подтверждения правильности полученных значений содержания воды используют независимый метод титрования по Карлу Фишеру. Измерения проводят волюметрически на приборе Metrohm 870 KF Titrino plus. Реактивы Фишера - рабочая среда № 817 (Акваметрия, Россия), композит титрант 5К (Акваметрия, Россия). Правильность полученных результатов также подтверждают способом «введено-найдено».
Заявленный способ можно осуществлять на хроматографической системе, снабженной насосом высокого давления, дегазатором подвижной фазы, термостатом, спектрофотометрическим и рефрактометрическим детекторами. Для проведения анализов используют колонку из нержавеющей стали, заполненную суспензионным способом микропористым цеолитом с размером частиц от 1,5 до 20 микрон, длиной от 2,5 до 300 мм и внутренним диаметром от 0,2 до 10 мм по известной методике.(Asshauer, J., Halász, I., Reproducibility and efficiency of columns packed with 10 μ silica in liquid chromatography. Journal of Chromatographic Science 1974, 12, (3), 139-147).
Все используемые реагенты являются коммерчески доступными.
Эффективность колонки практически не меняется при изменении температуры, поэтому в данном методе используют температуру термостата 35°С. Однако изобретение не ограничивается использованием данной конкретной температуры термостата. Анализы возможно проводить в диапазоне от комнатной температуры до 55°С.
Условия получения градуировочных зависимостей содержания воды: элюент - метанол, температура термостата = 35°С, объемная скорость элюента = 0,5 мл/мин, объем пробы - 20 мкл, детектор - рефрактометрический и спектрофотометрический.
Для приготовления растворов реагентов при осуществлении заявляемого способа используют деионизованную воду из системы очистки Milli Q (Merck, Германия) и хроматографически чистый метанол. Согласно заявляемому способу концентрацию воды в органических соединениях определяют следующим образом. Предварительно колонку промывают элюентом - метанолом со скоростью 0,5 мл/мин до уравновешивания базовой линии рефрактометрического детектора. Для определения концентрации воды в соединениях используют способ добавок. Для построения градуировочного графика готовят растворы анализируемых соединений без добавок воды и с рассчитанным содержанием добавленной воды. Далее осуществляют введение в колонку полученных проб соединения в количестве 20 мкл со скоростью потока 0,5 мл/мин и фиксируют значение площади пика воды в пробе. По имеющимся данным строят градуировочную зависимость и определяют начальное содержание воды в соединении.
Пределы обнаружения аналитов по дисперсии параметров градуировочной зависимости рассчитывают следующим образом.
Градуировочная зависимость имеет вид
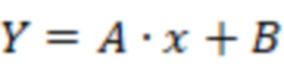 (1)
(1)
Где Y - значение площади пика воды, х - концентрация воды в анализируемом образце.
При этом дисперсия рассчитываемой по градуировочному уравнению величины Yk имеет следующий вид:
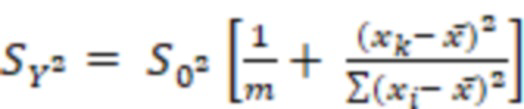 (2)
(2)
Где m - число экспериментальных измерений аналитического сигнала в ходе градуировки, а xk - концентрация, соответствующая Yk, s02 - дисперсия разности между экспериментальными (yk) и рассчитанными (Yk) значениями аналитического сигнала.
Доверительный интервал величины Yi рассчитывали по формуле
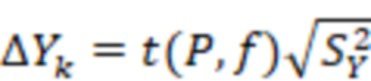
Где t(P,f) - коэффициент Стьюдента для вероятности P и числа степеней свободы f = m - 2. Вероятность принимали равной 0,95.
Для каждого Yk обозначили верхнюю и нижнюю границы доверительного интервала как Yk + ΔYk = Ykверхн, Yk - ΔYk = Ykнижн. Нашли Ykверхн для xk = 0 (то есть рассчитали максимальный аналитический сигнал, который возможно с заданной вероятностью P получить при отсутствии определяемого компонента в пробе):
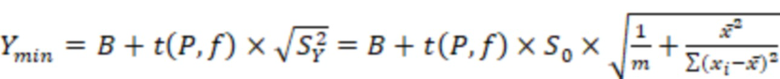 (3)
(3)
Величина Ymin является пороговым значением аналитического сигнала: если в результате анализа получен сигнал Yk < Ymin, то нельзя сделать вывод о наличии определяемого компонента в пробе так как такой сигнал можно получить от пробы, не содержащей определяемого компонента. Пороговая величина концентрации, связанная с Ymin градуировочным уравнением
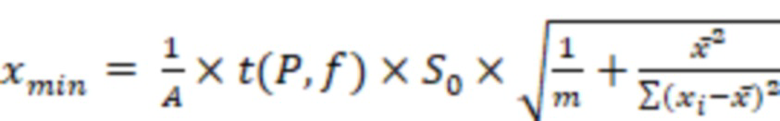 (4)
(4)
Если в результате анализа получен аналитический сигнал меньший, чем Ymin (уравнение 3), то концентрация определяемого компонента в пробе с заданной вероятностью (Р) не превышает xmin (уравнение 4). Пределом обнаружения метода является xmin, Ymin - критической минимальной величиной аналитического сигнала, ниже которой количественная интерпретация результатов анализа невозможна.
Возможность реализации заявляемого способа показана на следующих примерах.
Пример 1. Характеристика сорбента
Для проведения анализов используют колонку из нержавеющей стали длиной 50 мм и внутренним диаметром 4,6 мм, заполненную суспензионным способом широкопористым цеолитом 13Х.
По данным электронной микроскопии известно, что полученная после седиментации фракция сорбента 13Х однородна, с размером частиц 5 мкм (фиг. 1).
Методом низкотемпературной адсорбции азота (модель BET) было установлено, что исследуемый цеолит имеет микропористую структуру с преобладающими стандартными порами диаметра 0,73 нм и небольшим содержанием пор с диаметром 1,3 нм, 1,6 нм и 1,9 нм (фиг. 2). Измеренная удельная поверхность сорбента составила Sуд = 630 м2/г.
Методом рентгенофлуоресцентной спектроскопии (РФС) определен элементный состав цеолита и найдено, что цеолит находится в Na+ форме и имеет мольное соотношение Si/Al = 1,228.
Кристаллическая структура сорбента - 13X, изученная с помощью рентгеновской дифракции, соответствует фожазиту типа Х.
После набивки была оценена эффективность колонки в условиях нормально-фазовой ВЭЖХ: элюент изооктан, скорость потока подвижной фазы 1 мл/мин, адсорбат - бензол. Эффективность колонки составила 3200 ТТ/м. Невысокие значения характерны для нанопористых сорбентов.
Еще один важный параметр - мертвый объем колонки (V0). Для твердых сорбентов значение V0 можно представить как сумму объема алюмосиликатного каркаса (Vкар) и объема неподвижного слоя элюента возле поверхности и в малодоступных частях пористой структуры сорбента (Vпов). V0 измеряли по разности масс колонки, уравновешенной последовательно растворителями с различной плотностью, кинетический диаметр (dK) которых меньше эффективного размера пор фожазита. Для этой цели использовали метанол (dK = 0,36 нм, плотность ρ = 0,7866 г/см3) и хлористый метилен (dK = 0,31 нм, плотность ρ = 1,3165 г/см3).
Масса хроматографической колонки (mc) складывается из массы пустой колонки (mпуст), массы адсорбента (mads) и массы соединений в свободном для проникновения пространстве (ρV0): mc = mпуст + mads + ρV0. mпуст и mads остаются постоянными для одной колонки, поэтому V0 может быть найден по соотношению разности масс колонки, уравновешенной каждым растворителем, и разности плотностей используемых растворителей: V0 = (mC(CH2Cl2) - mC(CH3OH))/(ρ(CH2Cl2) - ρ(CH3OH)). Согласно уравнению, V0 исследуемой колонки, набитой фожазитом, составил (0,5037 ±0,0005) см3 (n = 3, P = 0,95).
Пример 2. Адсорбционные свойства фожазита
При хроматографии на цеолитах механизм удерживания включает 2 типа взаимодействий: молекулярно-ситовой эффект и кинетическую селективность, включающую адсорбцию сорбатов на внешней и внутренней поверхностях цеолита, взаимодействие сорбатов с элюентом, а также сродство сорбента к подвижной фазе.
Изучена зависимость удерживания соединений на цеолите от их кинетического диаметра и полярности. Установлено, что на цеолите удерживаются соединения с кинетическим диаметром (расстояние между центрами незаряженных частиц в состоянии соударения) ниже эффективного размера пор сольватированного элюентом цеолита (табица 1) и с полярностью выше полярности элюента по шкале Снайдера (Snyder, L.R., Kirkland, J.J. Introduction to Modern Liquid Chromatography. 2nd ed.; John Wiley and Sons Inc.: New York, 1979) или близкой к полярности элюента (фиг. 2). Практически для каждого соединения на хроматограмме появлялся характерный пик с большим временем удерживания, нежели пик растворителя (фиг. 3). Был сделан вывод, что данный пик принадлежит воде, которая наиболее сильно удерживалась цеолитом. В соответствии с результатами, при использовании метанола в качестве элюента на колонке удерживается лишь вода.
Таблица 1. Физические характеристики соединений, использованных для изучения адсорбционных свойств цеолита 13Х.
Пример 3. Построение градуировочных зависимостей для количественного определения воды в органических соединениях
Пробы органических растворителей готовят путем добавления расчетных количеств воды к растворителям без разбавления другими растворителями. Используют деионизированную воду из системы очистки Milli Q (Merck, Германия). Образцы твердых веществ готовят путем разбавления взвешенных количеств в хроматографически чистом метаноле. Осуществляют перемешивание.
Далее проводят ВЭЖХ анализ на хроматографической колонке, заполненной цеолитом 13Х, на любой хроматографической системе, снабженной насосом высокого давления, дегазатором подвижной фазы, термостатом, спектрофотометрическим и рефрактометрическим детекторами. Предварительно колонку 50×4,6 мм, заполненную фожазитом 13Х, промывают элюентом - метанолом до достижения динамического равновесия - выравнивания базовой линии детектора.
Условия проведения экспериментов: колонка 50×4,6 мм, заполненная цеолитом 13X, элюент: хроматографически чистый метанол, скорость потока: 0,5 мл/мин, температура колонки: 35°C, объем пробы: 20 мкл.
Для получения калибровочных кривых использовали аликвоты ацетонитрила и 2-пропанола с различными добавками воды. Однако изобретение не ограничивается использованием данных конкретных растворителей для построения градуировочных зависимостей. Детектирование проводили с помощью рефрактометрического детектора. Градуировочные графики были построены способом добавок как зависимость площади хроматографического пика воды от ее содержания в растворе. Измерения каждой пробы проводили трижды.
Полученные уравнения для градуировочных зависимостей:
S = (1521 ± 6)*w + (2050 ± 18) для ацетонитрила
S = (1519 ± 4)*w + (41 ± 29) для 2-пропанола,
где S - площадь пика, мВ*сек, w - содержание воды, масс.%. Хорошая линейность наблюдалась в диапазоне от 0,01 до 9,55 масс.% (R2 = 0,9998), а значение ПО составило 0,001 масс.%.
Одинаковый наклон градуировочных зависимостей указывает на незначительное влияние матрицы органического растворителя на проведение анализа и получение результатов. Различные значения свободных членов градуировочных уравнений соответствуют различному начальному содержанию воды в используемых органических растворителях.
Пример 4. Определение примесей воды в органических соединениях
На основании градуировочных зависимостей были определены содержания воды в нескольких органических растворителях (таблица 2). Было проведено сравнение полученных результатов с паспортными данными. Правильность полученных значений содержания воды в ацетонитриле и метаноле была подтверждена методом титрования по Карлу Фишеру.
Таблица 2. Содержание воды в органических растворителях, согласно паспортным данным, и найденных методом ВЭЖХ и титрованием по Карлу Фишеру (n = 3, P = 0,95)
Для проверки правильности метода была также проведена процедура «введено-найдено». Точно взвешенные количества воды добавляли к образцам растворителей (таблица 3). Полученные данные хорошо согласуются с ожидаемым содержанием воды.
Таблица 3. Содержание воды в органических соединениях, полученное способами градуировочного графика и «введено-найдено»
Таким образом, заявленный способ определения содержания примесей воды в органических соединениях с использованием метода ВЭЖХ на колонке с цеолитом 13Х обеспечивает простое, быстрое, селективное и достоверное определение содержания микропримесей воды в органических соединениях.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВЫСОКОЭФФЕКТИВНОГО ЖИДКОСТНО-ХРОМАТОГРАФИЧЕСКОГО ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИЙ АЛКИЛФОСФОНОВЫХ И/ИЛИ О-АЛКИЛАЛКИЛФОСФОНОВЫХ КИСЛОТ В ВОДНОМ РАСТВОРЕ | 2017 |
|
RU2653582C1 |
| Способ определения 2-диметиламино-1,3-бис-(фенил-сульфонилтио)пропана в биологическом материале | 2017 |
|
RU2647477C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 4,5,6-ТРИХЛОРБЕНЗОКСАЗОЛОНА-2 В ТЕХНИЧЕСКОМ ПРОДУКТЕ | 1991 |
|
RU2006859C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НЕОНИКОТИНОИДОВ В ПОДМОРЕ ПЧЕЛ С ИСПОЛЬЗОВАНИЕМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2019 |
|
RU2730399C1 |
| Способ определения нифедипина в биологическом материале | 2023 |
|
RU2812598C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРОКАИНА В ПЛАЗМЕ КРОВИ | 2013 |
|
RU2537179C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙЕССОТОКСИНОВ В МОЛЛЮСКАХ | 2018 |
|
RU2716233C1 |
| Способ определения N-(бензимидазолил-2)-О-метилкарбамата в биологическом материале | 2018 |
|
RU2692127C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОКСИУКСУСНОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2011 |
|
RU2453848C1 |
| Способ определения нифедипина в биологическом материале | 2018 |
|
RU2686741C1 |
Изобретение относится к области аналитической химии, а именно к количественному определению микроколичеств воды в различных органических соединениях (органических растворителях, соединениях для органического синтеза, реакционных смесях и др.) с целью определения их чистоты и пригодности для решения различных практических задач, когда содержание воды является критическим параметром качества продукта. Предложен сорбент, представляющий собой микропористый кристаллический алюмосиликат со структурой фожазита в форме, насыщенной катионами натрия, с соотношением Si/Al=1,228, с площадью поверхности от 200 до 1200 м2/г, средним размером частиц от 1,5 до 20 мкм и диаметром пор от 0,3 до 1,2 нм, для определения микроколичеств воды в органических соединениях методом ВЭЖХ. Также предложен способ определения микроколичеств воды ВЭЖХ с использованием предложенного сорбента. Технический результат - создание прямого метода количественного определения микропримесей воды, который характеризуется простотой оптимизации условий определения, минимальной пробоподготовкой, экспресcностью, высокой селективностью, возможностью анализа нелетучих, достаточно вязких и полярных соединений, устойчивостью стационарной фазы, широким диапазоном линейности градуировочной зависимости и хорошей воспроизводимостью. 2 н. и 5 з.п. ф-лы, 3 ил., 3 табл., 4 пр.
1. Сорбент, представляющий собой микропористый кристаллический алюмосиликат со структурой фожазита в форме, насыщенной катионами натрия, с соотношением Si/Al=1,228, с площадью поверхности от 200 до 1200 м2/г, средним размером частиц от 1,5 до 20 мкм и диаметром пор от 0,3 до 1,2 нм, для определения микроколичеств воды в органических соединениях методом ВЭЖХ.
2. Способ определения микроколичеств воды в органических соединениях ВЭЖХ, включающий заполнение хроматографической колонки сорбентом по п. 1, промывание сорбента элюентом с последующим введением пробы и определение концентрации воды в пробе с использованием предварительно построенной градуировочной зависимости.
3. Способ по п. 2, характеризующийся тем, что используют хроматографическую колонку с длиной, равной 25-300 мм, и внутренним диаметром 0,2-10 мм.
4. Способ по п. 2, характеризующийся тем, что в качестве элюента используют безводные низшие спирты.
5. Способ по п. 4, характеризующийся тем, что в качестве безводного низшего спирта используют метанол, этанол, пропанол или изопропанол.
6. Способ по п. 2, характеризующийся тем, что в качестве детектирования может быть также использован спектрофотометрический или кондуктометрический детектор.
7. Способ по п. 2, характеризующийся тем, что градуировочную зависимость строят способом добавок как зависимость площади хроматографического пика воды от ее содержания в растворе.
| Интернет-источник URL: https://silikagel.ru/katalog/tseolity/tseolit-nax/, опубликованный в Wayback Internet Archive Machine 28.05.2018 | |||
| Прибор для испытания психомоторной деятельности | 1932 |
|
SU36585A1 |
| US 4696904 A, 29.09.1987 | |||
| Fehrmann et al | |||
| On the determination of water in hydrocarbons by gel chromatography | |||
| Fresenius Zeit | |||
| Anal | |||
| Chem., 1974, v | |||
| Нож для надрезывания подошвы рантовой обуви | 1917 |
|
SU269A1 |
| Способ получения бензидиновых оснований | 1921 |
|
SU116A1 |
| АГЛОМЕРИРОВАННЫЕ ЦЕОЛИТНЫЕ АДСОРБЕНТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2008 |
|
RU2453364C2 |

