ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области фармацевтики, в частности к солям бензотиопиранонового соединения, представляющим собой соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она, показанного в формуле (I), способу их получения, их фармацевтической композиции и применению при получении лекарственных препаратов для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis.
УРОВЕНЬ ТЕХНИКИ
Туберкулез (ТБ) представляет собой хроническое контагиозное инфекционное заболевание, вызываемое Mycobacterium tuberculosis (М. tuberculosis), а также серьезное инфекционное заболевание, угрожающее здоровью человека и приводящее к смерти. Согласно отчету Всемирной организации здравоохранения (ВОЗ), по оценкам в 2017 г. около 1,7 миллиарда человек, 23% населения мира, имели латентную ТБ инфекцию. Во всем мире у около 10,0 миллионов человек развилось заболевание ТБ. По оценкам, от ТБ умерло 1,4 миллиона человек. На 100000 населения приходилось 133 новых случая заболевания, в том числе на детей в возрасте до 15 лет и ВИЧ-инфицированных приходилось, соответственно, 10% и 9% новых случаев. У примерно 558000 человек развился лекарственно-устойчивый ТБ, из них 82% имели ТБ с множественной лекарственной устойчивостью. Между тем, уровень лекарственной устойчивости (XDR-TB) быстро увеличивался, а успех лечения лекарственно-устойчивого ТБ остается низким, составляя 55% во всем мире.
Уникальная клеточная стенка Mycobacterium tuberculosis характеризуется многослойной структурой. Биосинтетический путь данных уникальных компонентов является хорошим источником потенциальных мишеней для лекарственных препаратов. Например, лекарственные препараты первой линии, изониазид и этамбутол, оказывают мешающее влияние на образование клеточной стенки Mycobacterium tuberculosis путем ингибирования синтеза микотической кислоты и арабиногликанового слоя, соответственно. Основным компонентом арабиногалактанового слоя и арабиноманнанового слоя на наружной клеточной мембране Mycobacterium tuberculosis является сахар арабиноза, являющийся важным предшественником DPA (decaprenylphosphoryl-D-Ara - декапренилфосфорил-D-Ara). DPA в основном получают путем эпимеризации DPR (decaprenylphosphoryl ribose - декапренилфосфорил рибоза) при совместном действии DprE1 и DprE2. Следовательно, ингибирование активности DprE1 может блокировать синтез клеточной стенки и в конечном итоге уничтожить Mycobacterium tuberculosis (Decaprenylphosphoryl arabinoiuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. Journal of bacteriology 2005, 187 (23), 8020-8025).
В настоящее время не представлены ингибиторы DprE1 в качестве противотуберкулезных препаратов. Соединение PBTZ169 в качестве ковалентного ингибитора DprE1 было включено в фазу II клинического исследования. Недавно автор настоящего изобретения провел углубленное исследование в отношении перспективной мишени DprE1 и определил каркас бензотиопиранона в качестве доминирующего скелета путем оценки активности, токсичности и пригодности быть лекарственным препаратом. Поданы две патентных заявки (заявка №201810092333.Х и PCT/CN2018/080787). После комплексной оценки было получено бензотиопираноновое соединение 6b (2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он), которое показало значительную активность в отношении лекарственно-чувствительных и лекарственно-устойчивых штаммов ТБ и низкую токсичность (Identification of novel benzothiopyranone compounds against Mycobacterium tuberculosis through scaffold morphing from benzothiazinones[J]. Eur. J. Med. Chem., 2018, 160, 157-170).
В патентных заявках (заявка №201810092333.Х и PCT/CN2018/080787) раскрыто соединение 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он и его гидрохлоридная соль, но не приведены подробные примеры, относящиеся к другим приемлемым солям, и результаты экспериментов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является обеспечение соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она с высокой активностью in vitro и in vivo в отношении Mycobacterium tuberculosis, а также улучшенными фармакокинетическими и физико-химическими свойствами. Авторы изобретения обнаружили, что соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она обладают очень высокой активностью in vitro и in vivo в отношении Mycobacterium tuberculosis, которые можно применять для лечения или предотвращения инфекционных заболеваний, вызванных бактериями, в частности ТБ легких, вызванного Mycobacterium tuberculosis, а также демонстрируют значительно улучшенные фармакокинетические и физико-химические свойства по сравнению с его свободным основанием и гидрохлоридной солью. Авторы изобретения завершили настоящее изобретение на такой основе.
Для решения технических задач изобретения, изобретение обеспечивает следующие технические решения:
Первый аспект технических решений согласно настоящему изобретению заключается в обеспечении фармацевтически приемлемых солей 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она, представленного формулой (I),
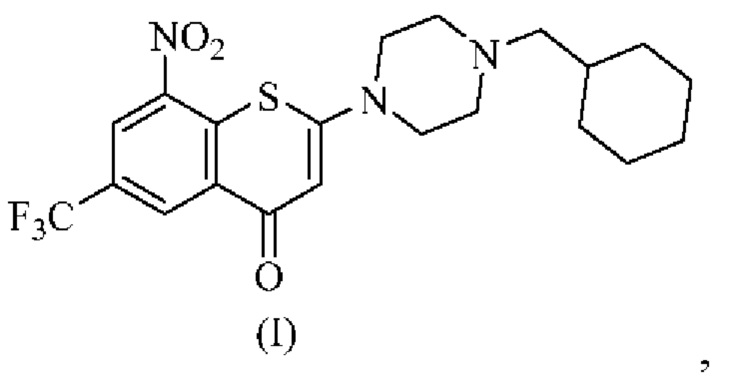
причем гидрохлоридная соль
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она исключена.
Для любой соли по первому аспекту соль представляет собой малеат, фумарат, цитрат или L-малат 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она.
Для любой соли согласно первому аспекту соль выбрана из 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1 малеата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1/2 малеата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅3/2 малеата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1 фумарата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1/2 фумарата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅3/2 фумарата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1 цитрата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1/2 цитрата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅3/2 цитрата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1 L-малата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅1/2 L-малата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он⋅3/2 L-малата.
Второй аспект технических решений согласно настоящему изобретению относится к способу получения солей по первому аспекту, включающему следующие стадии:
взаимодействие
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он с кислотой (например, фармацевтически традиционными кислотами, предпочтительно малеиновой кислотой, фумаровой кислотой, лимонной кислотой и L-яблочной кислотой) в подходящем растворителе (например, метаноле, этаноле, ацетоне, ацетонитриле, предпочтительно метаноле) при 20-140°С течение 2-8 ч, предпочтительно при 20-100°С в течение 2-5 ч с получением солей, показанных в формуле (I), путем реакции образования солей.
В третьем аспекте технических решений согласно настоящему изобретению предложен способ получения фармацевтически приемлемой соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она, который включает следующие стадии:
взаимодействие 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он с фармацевтически традиционной кислотой в растворителе, представляющем собой спирт, при комнатной температуре или в условиях кипячения с обратным холодильником в течение 2-8 ч с получением соли, показанной в формуле (I), посредством реакции образования соли.
В четвертом аспекте технических решений согласно настоящему изобретению предложена фармацевтическая композиция, содержащая эффективное количество фармацевтически приемлемой соли для лечения и/или профилактики в соответствии с первым аспектом, и один или более фармацевтически приемлемых носителей, вспомогательных веществ, разбавителей, адъювантов и сред.
Пяиый аспект технических решений согласно настоящему изобретению относится к применению фармацевтически приемлемой соли согласно первому аспекту или фармацевтической композиции согласно третьему аспекту для производства лекарственного препарата для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis.
Вышеизложенная сущность описывает только некоторые аспекты изобретения, но не ограничивается этими аспектами. Эти и другие аспекты будут описаны более конкретно и подробно ниже.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Различные аспекты и признаки настоящего изобретения дополнительно описаны ниже.
Все ссылки, цитируемые согласно настоящему изобретению, полностью включены в настоящую заявку посредством этих ссылок. Если значения, выраженные этими ссылками, не соответствуют значениям согласно настоящему изобретению, преимущественную силу имеет выражение согласно настоящему изобретению. Кроме того, различные термины и фразы, используемые в настоящем изобретении, имеют общие значения, понятные специалисту в данной области техники. Тем не менее, настоящее изобретение по-прежнему хочет описать и объяснить эти термины и фразы более подробно в настоящем документе. Если указанные термины и фразы не соответствуют известным значениям, значения, выраженные в настоящем изобретении, имеют преимущественную силу. Ниже приведены определения различных терминов, используемых в настоящем изобретении, которые применимы к терминам, используемым в описании настоящей заявки, если иное не указано в конкретных условиях.
Как описано в настоящей заявке, термин «комнатная температура» относится к диапазону температур от 10°С до 40°С. В некоторых вариантах реализации термин «комнатная температура» относится к диапазону температур от 20°С до 30°С. В других вариантах реализации комнатная температура относится к 25°С.
Как описано в настоящей заявке, термин «эффективная доза» относится к количеству лекарственного препарата, которое можно применять у субъекта для достижения желаемого лечения заболевания или симптомов, описанных в настоящей заявке.
Как описано в настоящей заявке, термин «фармацевтически приемлемый», например, при описании «фармацевтически приемлемой соли», указывает на то, что соль является не только физиологически приемлемой для субъекта, но также относится к синтетическим веществам, имеющим фармацевтическую ценность.
Как описано в настоящей заявке, термин «фармацевтическая композиция» также может относиться к «композиции», которую можно применять для лечения заболеваний или симптомов, описанных в настоящей заявке, у субъектов, в частности млекопитающих.
«Лечение» заболеваний включает:
(1) Предотвращение заболевания, то есть предотвращение возникновения клинических симптомов заболевания у млекопитающих, подверженных или восприимчивых к заболеванию, но не испытывающих или не проявляющих его симптомов.
(2) Ингибирование заболевания, то есть предупреждение или уменьшение прогрессирования заболевания или его клинических симптомов.
(3) Облегчение заболевания, то есть обеспечение излечения от заболевания или устранение его клинических симптомов.
Термин «терапевтическая эффективная доза» относится к количеству химических веществ, которое является достаточным для лечения заболевания при применении у млекопитающих. Эффективная доза лечения варьирует в зависимости от химического состава, заболевания, подлежащего лечению, и его тяжести, а также от возраста, веса и пола млекопитающих. Терапевтическая эффективная доза также относится к любому количеству соединения, достаточному для достижения желаемого благоприятного эффекта, который включает предотвращение заболевания, подавление заболевания или облегчение заболевания, как описано в пп. (1)-(3) выше. Например, количество соединения может составлять в диапазоне 0,1-250 мг/кг, или предпочтительно 0,5-100 мг/кг, или более предпочтительно 1-50 мг/кг, или даже 2 20 мг/кг. Предпочтительно, указанное количество соединения применяют у млекопитающих два раза в сутки. Более предпочтительно, указанное количество соединения применяют у млекопитающих один раз в сутки.
Как описано в настоящей заявке, термин «заболевание и/или симптомы» относится к физическому состоянию субъекта, связанному с заболеванием и/или симптомами, описанными в настоящем изобретении. Например, заболевание и/или симптомы, описанные в настоящем изобретении, относятся к инфекционным заболеваниям, вызванным туберкулезной палочкой.
Как описано в настоящей заявке, термин «субъект» может относиться к пациенту или другим животным, в частности, млекопитающему, такому как человек, собака, обезьяна, крупный рогатый скот, лошадь и т.д., которые получают соединение, представленное солью формулы (I), или его фармацевтическую композицию для лечения заболевания или симптомов, описанных в настоящей заявке.
С одной стороны, настоящее изобретение также относится к фармацевтическим композициям, в которых соединения согласно настоящему документу применяют в качестве активных ингредиентов. Фармацевтическая композиция может быть получена в соответствии со способом, известным в данной области техники. Любой состав, пригодный для человека или животного, может быть получен путем объединения соединения согласно настоящему изобретению с одним или более фармацевтически приемлемыми твердыми или жидкими вспомогательными веществами и/или адъювантами.
Соединения по настоящему изобретению или фармацевтические композиции, содержащие их, можно вводить в форме разовой дозы через кишечный или отличный от кишечного тракт, в частности пероральным, внутривенным, внутримышечным, подкожным, назальным путем, через слизистую оболочку полости рта, глаза, легкие, и дыхательные пути, кожу, влагалище, прямую кишку и т.п.
Лекарственные формы могут представлять собой жидкие лекарственные формы, твердые лекарственные формы или полутвердые лекарственные формы. Жидкие лекарственные формы могут представлять собой раствор (включая истинный раствор и коллоидный раствор), эмульсию (включая эмульсию типа масло в воде, эмульсию типа вода в масле и множественную эмульсию), суспензию, инъекцию (включая водную инъекцию, порошок для инъекций и инфузий), капли глазные, капли назальные, лосьон и линимент и т.д. Твердая лекарственная форма может представлять собой таблетку (включая обычные таблетки, таблетки, покрытые кишечнорастворимой оболочкой, таблетки защечные, таблетки диспергируемые, таблетки жевательные, таблетки шипучие, таблетки диспергируемые в полости рта), капсулы (включая капсулы твердые, капсулы мягкие, капсулы, покрытые кишечнорастворимой оболочкой), гранулы, порошки, пеллеты, капли, пилюли, суппозитории, мембраны, трансдермальные формы, аэрозоли, спреи и т.д. Полутвердые составы могут представлять собой мазь, желатин, пасту и т.д.
Соединения в настоящем изобретении могут быть получены в виде обычного препарата, препарата с замедленным высвобождением, препарата с контролируемым высвобождением, препарата для таргетной терапии и различных систем доставки частиц.
Для получения соединений согласно настоящему изобретению в виде таблеток можно широко применять различные вспомогательные вещества, известные в данной области, включая разбавители, адгезивные вещества, смачивающие вещества, разрыхлители, смазывающие вещества и сорастворители. Разбавители могут представлять собой крахмал, декстрин, сахарозу глюкозу лактозу маннит, сорбит, ксилит, целлюлозу микрокристаллическую, кальция сульфат, кальция гидрофосфат, кальция карбонат и т.д. Смачивающие вещества могут представлять собой воду, этанол, изопропанол и т.д. Адгезивные вещества могут представлять собой крахмальный сироп, декстрин, сироп, мед, раствор глюкозы, микрокристаллические волокна, аравийскую камедь, желатин, натрия карбоксиметилцеллюлозу, метилцеллюлозу гидроксипропилметилцеллюлозу этилцеллюлозу акриловую смолу, карбомер, поливинилпирролидон, полиэтиленгликоль и т.д. Разрыхлители могут представлять собой сухой крахмал, целлюлозу микрокристаллическую, гидроксипропилцеллюлозу с низкой степенью замещения, сшитый поливинилпирролидон, сшитую натрия карбоксиметилцеллюлозу, натрия карбоксиметилкрахмал, натрия гидрокарбонат и лимонную кислоту, сложный эфир полиоксиэтиленсорбита и жирной кислоты, натрия лаурилсульфонат и т.д. Смазывающими веществами и сорастворителями могут быть тальк, кремния диоксид, стеарат, винная кислота, жидкий парафин, полиэтиленгликоль и т.д.
Таблетки также могут быть дополнительно приготовлены в виде таблеток, покрытых оболочкой, таких как таблетки, покрытые сахарной оболочкой, таблетки, покрытые пленочной оболочкой, таблетки, покрытые кишечнорастворимой оболочкой, или двухслойные таблетки и многослойные таблетки.
Для получения единичной формы доставки лекарственного препарата в виде капсулы соединение, представляющее собой активный ингредиент, может быть смешано с разбавителями и сорастворителями, и смесь может быть непосредственно помещена в твердую капсулу или капсулу мягкую. Соединение согласно настоящему изобретению также может быть приготовлено в виде гранул или пеллет с разбавителями, адгезивными веществами и разрыхлителями, а далее помещено в капсулы твердые или капсулы мягкие. Различные разбавители, адгезивные вещества, смачивающие вещества, разрыхлители и растворители, используемые для получения таблеток соединения в настоящем документе, также могут быть использованы для получения капсул соединения в настоящем документе.
Для получения соединения согласно настоящему изобретению в виде инъекции в качестве растворителя могут быть использованы вода, этанол, изопропанол, пропиленгликоль или их смесь и может быть добавлено соответствующее количество обычно используемых в данной области растворителей, сорастворителей, регуляторов рН и регуляторов осмотического давления. Растворители или сорастворители могут представлять собой полоксамер, лецитин, гидроксипропил-бета-циклодекстрин и т.д. Регуляторы рН могут представлять собой фосфат, ацетат, хлористоводородную кислоту натрия гидроксид и т.д. Регуляторами осмотического давления могут быть натрия хлорид, маннит, глюкоза, фосфат, ацетат и т.д. Если готовят лиофилизированный порошок для приготовления раствора для инъекций, в качестве несущих агентов можно также добавить маннит и глюкозу.
Кроме того, в фармацевтические препараты при необходимости можно добавить красители, консерванты, ароматизирующие вещества, вкусоароматические агенты или другие добавки.
Для достижения назначения лекарственного средства и усиления терапевтического эффекта лекарственный препарат или лекарственную композицию согласно настоящему изобретению можно вводить любым известным способом введения.
Соединения или композиции согласно настоящему изобретению могут быть приняты отдельно или в сочетании с другими терапевтическими лекарственными препаратами или симптоматическими лекарственными препаратами. Когда соединение согласно настоящему изобретению обладает синергетическим эффектом с другими терапевтическими лекарственными препаратами, его дозировку следует корректировать в соответствии с фактической ситуацией.
ПОЛЕЗНЫЕ ТЕХНИЧЕСКИЕ ЭФФЕКТЫ
Авторы настоящего изобретения провели тщательное исследование и синтезировали соли соединения формулы (I) и измерили их МИК (минимальная ингибирующая концентрация) в отношении штамма H37RV М. tuberculosis с помощью метода МАВА (микропланшетный анализ с использованием аламарового синего). Соединения показывают высокую противотуберкулезную активность, при которой 5 солей с МИК < 0,016 мкг/мл проявляют более высокую противотуберкулезную активность, чем противотуберкулезный препарат первой линии изониазид. Малеат, фумарат, цитрат и L-малат соединения формулы (I) согласно настоящему изобретению показывают превосходную проницаемость в клетки по сравнению с гидрохлоридной солью соединения формулы (I), что указывает на то, что соли согласно настоящему изобретению обладают улучшенными свойствами абсорбции. Результаты фармакокинетического исследования на мышах показывают, что биодоступность малеата и L-малата соединения формулы (I) значительно выше, чем биодоступность соединения (I). Результаты исследования фармакокинетики у крыс показывают, что малеатная соль соединения формулы (I) характеризуется значительно улучшенными воздействием in vivo (AUC) и биодоступностью по сравнению с соединением формулы (I) и его гидрохлоридной солью, что указывает на то, что соль согласно настоящему изобретению обладает улучшенными фармакокинетическими свойствами по сравнению со свободным основанием и его гидрохлоридной солью. Результаты фармакодинамического исследования на мышах показывают, что малеатная соль соединения формулы (I) обладает более сильной противотуберкулезной активностью in vivo в той же дозе, что и соединение формулы (I). Результаты исследования в стрессовых условиях показывают, что малеатная соль соединения формулы (I) является очень стабильной после помещения в условия воздействия света, высокой температуры и высокой влажности в течение 10 дней, особенно в условиях воздействия света стабильность малеатной соли значительно лучше, чем у соединения (I), что указывает на то, что стабильность соли согласно изобретению при воздействии света была значительно улучшена. В настоящем изобретении предложены соли бензотиопиранонового соединения, обладающие высокой активностью в отношении Mycobacterium tuberculosis и улучшенными фармакокинетическими и физико-химическими свойствами, которые можно применять для лечения или предотвращения инфекционных заболеваний, вызванных бактериями, в частности туберкулезных (ТБ) заболеваний, вызванных Mycobacterium tuberculosis, а также применять для преодоления проблем, связанных с лекарственной устойчивостью Mycobacterium tuberculosis.
ПОДРОБНЫЕ ВАРИАНТЫ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть подробно описано следующими вариантами реализации изобретения, но это не подразумевает каких-либо неблагоприятных ограничений для изобретения. Настоящее изобретение было подробно описано, и его конкретные варианты реализации также раскрыты. Для специалистов в данной области техники очевидно внесение различных изменений и улучшений в конкретные варианты реализации настоящего изобретения, не отступая от сущности и объема настоящего изобретения.
Стандартные операции и способы очистки, известные специалистам в данной области техники, могут быть использованы для всех следующих вариантов реализации изобретения. Если не указано иное, все температуры выражены как °С (Цельсия). Структуры соединений были подтверждены методом ядерного магнитного резонанса (ЯМР).
Получение варианта реализации
Структуры соединений были подтверждены 1Н-ЯМР-спектроскопией. Химический сдвиг (δ) ядерного магнитного резонанса (ЯМР) задается в единицах миллионной доли (м.д.). Константы связи (J) указаны в герцах (Гц). ЯМР-спектры определяли на ЯМР-спектрометре Mercury-400, используя CD3OD или ДМСО-d6 в качестве растворителей и тетраметилсилан (ТМС) в качестве внутреннего стандарта.
Тип электронных весов японские Yanaco LY-300.
Безводные растворители были получены стандартными методами. Все остальные реактивы являются коммерчески доступными квалификации «чистый для анализа» (англ. analytical purity).
В настоящем изобретении используются следующие сокращения:
КОЕ - колониеобразующая единица.
МИК - минимальная ингибирующая концентрация.
Рарр - коэффициент кажущейся проницаемости.
ро - перорально.
в/в - внутривенная инъекция.
AUC - содержание вещества в плазме крови.
F - пероральная биодоступность.
t1/2β - период полувыведения из плазмы крови.
Cmax - максимальная концентрация в плазме крови.
Tmax - время достижения максимальной концентрации в плазме крови.
СРАВНИТЕЛЬНЫЕ ПРИМЕРЫ
Сравнительный пример 1
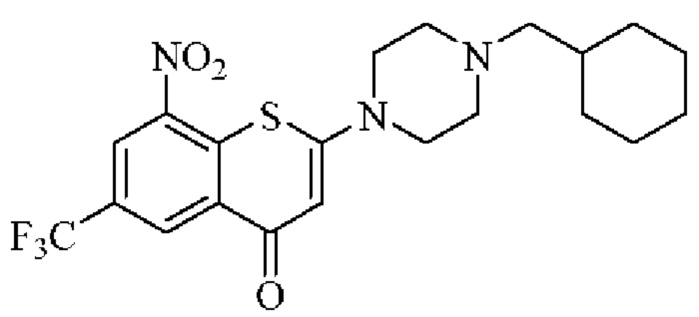
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он (соединение (I))
Соединение (I) получали согласно способу синтеза из примера 11 (соединение 11) в патентных документах 201810092333.X и PCT/CN2018/080787.
Сравнительный пример 2
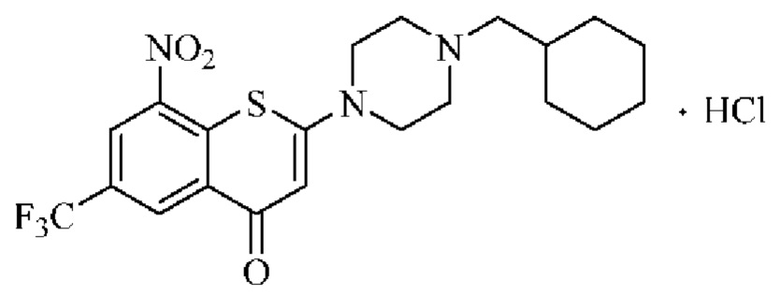
2-(4-(Циклогексилметил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она гидрохлорид (соединение (I) 1 гидрохлорид)
Соединение (I)⋅1 гидрохлорид получали согласно способу синтеза из примера 15 (соединение 22) в патентных документах 201810092333.Х и PCT/CN2018/080787.
ПРИМЕРЫ
Пример 1
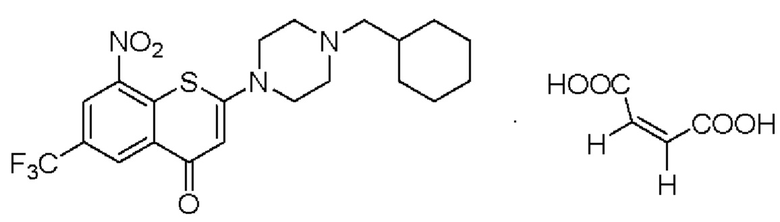
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она⋅1 малеат (соединение 1)
Способ синтеза:
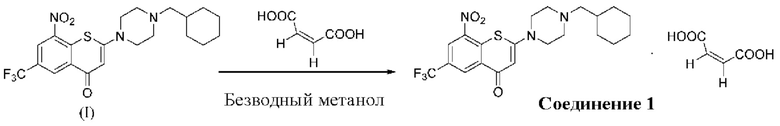
В трехгорлой колбе вместимостью 100 мл соединение (I) (1,14 г, 2,5 ммоль) суспендировали в метаноле безводном (21 мл) и хорошо перемешивали при комнатной температуре. К реакционной смеси медленно добавляли малеиновую кислоту (0,348 г, 3,0 ммоль) при комнатной температуре. Твердое вещество желтого цвета начинало осаждаться из смеси через 2-3 мин и смесь перемешивали при комнатной температуре в течение 3 ч, далее фильтровали. Полученное твердое вещество промывали метанолом (5 мл) и сушили с получением порошка желтого цвета, 1,23 г, выход продукта - 86%.
1Н-ЯМР-спектроскопия (400 МГц, CD3OD) δ: 9,02 (d, J=2,2 Гц, 1Н), 8,90 (d, J=2,2 Гц, 1Н), 6,40 (s, 1Н), 6,27 (s, 2Н), 3,98 (brs, 4Н), 3,32 (brs, 4Н), 2,94-2,92 (m, 2Н), 1,86-1,79 (m, 5Н), 1,76-1,72 (m, 1H), 1,39-1,21 (m, 3Н), 1,12-1,03 (m, 2Н).
Пример 2
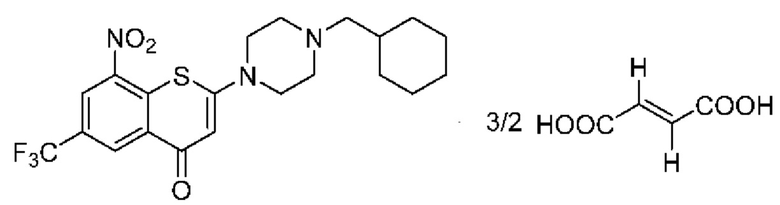
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она⋅3/2 фумарат (соединение 2)
Способ синтеза:
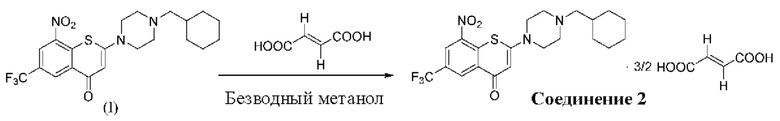
К суспензии соединения (I) (0,228 г, 0,5 ммоль) в метаноле безводном (6 мл) в колбе вместимостью 25 мл добавляли фумаровую кислоту (0,232 г, 2,0 ммоль) при комнатной температуре. Смесь нагревали до кипения с обратным холодильником при температуре 80°С в течение 3 ч. После охлаждения до комнатной температуры на ледяной бане в течение 10 мин осажденное твердое вещество фильтровали, промывали метанолом (1 мл) и сушили с получением порошка желтого цвета, 0,25 г, выход продукта - 79%.
1Н-ЯМР-спектроскопия (400 МГц, ДМСО-d6) δ: 13,08 (brs, 2Н), 8,85-8,83 (m, 2Н), 6,62 (s, 3Н), 6,30 (s, 1H), 3,66-3,64 (m, 4Н), 2,48 (brs, 4Н), 2,16-2,14 (m, 2Н), 1,76-1,65 (m, 5Н), 1,54-1,48 (m, 1Н), 1,27-1,12 (m, 3Н), 0,90-0,82 (m, 2Н).
Пример 3
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она⋅1 цитрат (соединение 3)
Способ синтеза:
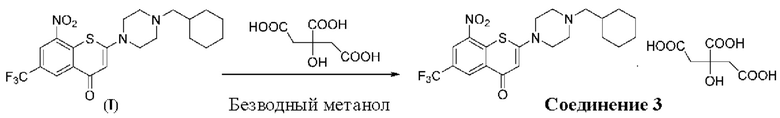
К суспензии соединения (I) (0,2 г, 0,44 ммоль) в метаноле безводном (5 мл) в колбе вместимостью 25 мл добавляли лимонную кислоту (0,127 г, 0,66 ммоль) при комнатной температуре. Смесь нагревали до кипения с обратным холодильником при температуре 80°С в течение 3 ч. После охлаждения до комнатной температуры и перемешивания в течение 5 мин осажденное твердое вещество фильтровали, промывали метанолом (1 мл) и сушили с получением порошка желтого цвета, 0,27 г, выход продукта - 83%.
1Н-ЯМР-спектроскопия (400 МГц, ДМСО-d6) δ: 8,85-8,84 (m, 2Н), 6,61 (s, 1Н), 3,69-3,66 (m, 4Н), 2,74 (d, J=15,4 Гц, 2Н), 2,64 (d, J=15,4 Гц, 2Н), 2,57 (brs, 4Н), 2,24-2,22 (m, 2H), 1,77-1,62 (m, 5H), 1,56-1,50 (m, 1H), 1,27-1,13 (m, 3H), 0,91-0,82 (m, 2H).
Пример 4
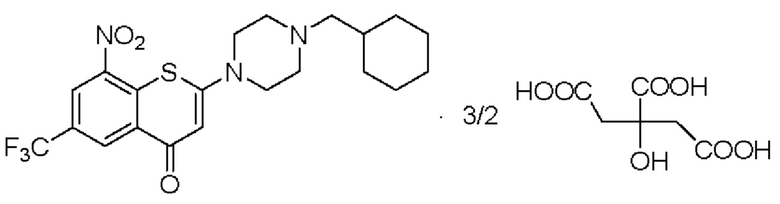
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она⋅3/2 цитрат (соединение 4)
Способ синтеза:
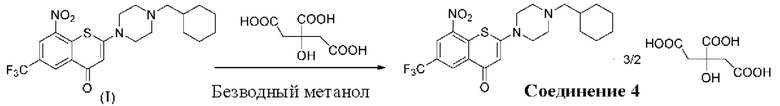
К суспензии соединения (I) (0,2 г, 0,44 ммоль) в метаноле безводном (5 мл) в колбе вместимостью 25 мл добавляли лимонную кислоту (0,42 г, 2,2 ммоль) при комнатной температуре. Смесь нагревали до кипения с обратным холодильником при температуре 80°С в течение 4 ч. После охлаждения до комнатной температуры в течение ночи осажденное твердое вещество фильтровали, промывали метанолом (1 мл) и сушили с получением порошка желтого цвета, 0,25 г, выход продукта 76%.
1Н-ЯМР-спектроскопия (400 МГц, ДМСО-d6) δ: 8,83 (brs, 2Н), 6,29 (s, 1Н), 3,67 (brs, 4Н), 2,74 (d, J=15,4 Гц, 3Н), 2,64 (d, J=15,4 Гц, 3Н), 2,58 (brs, 4Н), 2,24-2,22 (m, 2Н), 1,76-1,62 (m, 5Н), 1,55-1,50 (m, 1H), 1,24-1,12 (m, 3Н), 0,91-0,82 (m, 2Н).
Пример 5
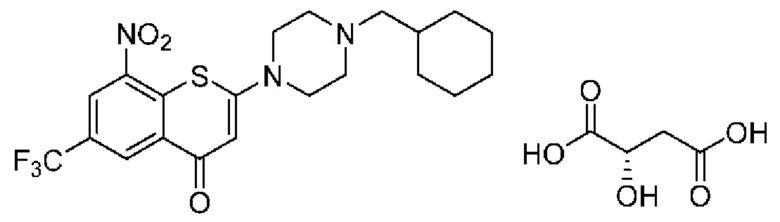
2-(4-(Циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она⋅1 L-малат (соединение 5)
Способ синтеза:
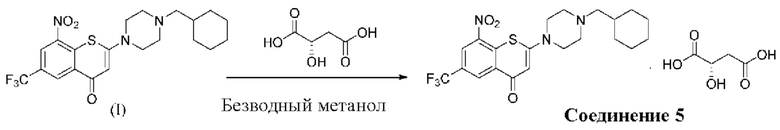
К суспензии соединения (I) (0,228 г, 0,5 ммоль) в метаноле безводном (6 мл) в колбе вместимостью 25 мл добавляли L-яблочную кислоту (0,268 г, 2,0 ммоль) при комнатной температуре. Смесь нагревали до кипения с обратным холодильником при температуре 80°С в течение 5 ч. После охлаждения до комнатной температуры смесь фильтровали. К фильтрату медленно добавляли ледяную воду (6 мл) и перемешивали в течение 30 мин на ледяной бане. Выпавшее в осадок твердое вещество фильтровали и сушили с получением твердого вещества желтого цвета, 0,1 г, выход продукта - 34%.
1Н-ЯМР-спектроскопия (400 МГц, CD3OD) δ: 9,00 (s, 1H), 8,87 (s, 1H), 6,33 (s, 1Н), 4,46-4,43 (m, 1Н), 3,81 (brs, 4Н), 2,82-2,77 (m, 5Н), 2,65-2,59 (m, 1Н), 2,40-2,38 (m, 2Н), 1,86-1,64 (m, 6Н), 1,37-1,21 (m, 3Н), 1,01-0,92 (m, 2Н).
Экспериментальный пример
Испытание на биологическую активность
Экспериментальный пример 1. Испытание на противотуберкулезную активность in vitro
Метод: Метод микропланшетного анализа с использованием аламарового синего (МАВА) применяли для определения противотуберкулезной активности in vitro.
Принцип эксперимента: Аламаровый синий можно использовать в качестве окислительно-восстановительного индикатора при добавлении в питательную среду. Наблюдается изменение цвета с синего на красный, что показывает потребление микроорганизмами молекул кислорода. Изменение цвета аламарового синего может быть измерено с помощью фотометра с длиной волны излучения 590 нм.
Экспериментальная методика: См. описанный в литературе метод (CN 201810092333.Х и Antimicrob. Agents and Chemother., 2011, 55, 5185-5193).
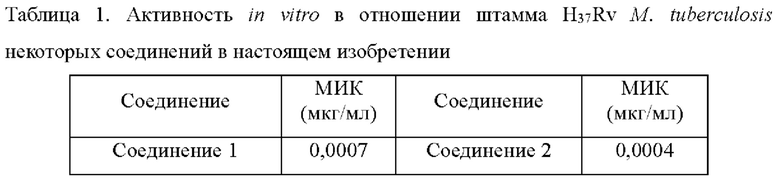
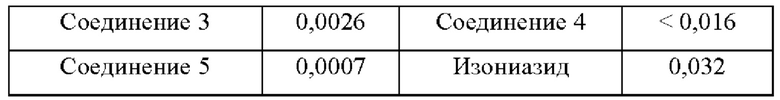
Согласно данным в табл. 1, соединения согласно настоящему изобретению проявляют очень высокую активность in vitro в отношении М. tuberculosis.
Экспериментальный пример 2. Испытание на проницаемость в клетки Сасо-2 Метод: См. описанный в литературе метод (Advanced drug delivery reviews, 2001, 46, 27-43.).
Клетки Сасо-2 представляют собой клонированные клетки аденокарциномы толстой кишки человека, сходные по структуре и функции с дифференцированными эпителиальными клетками, с микроворсинками и другими структурами, которые широко используются in vitro для имитации проникновения и всасывания лекарственных препаратов в кишечный тракт. Коэффициент кажущейся проницаемости (Рарр) соединения рассчитывают по следующей формуле:
Рарр=(dQ/dt)/(C0×А)
Где dQ/dt - скорость проникновения молекул лекарственного препарата через мембрану, С0 - начальная концентрация лекарственного препарата, а А - площадь монослоя.
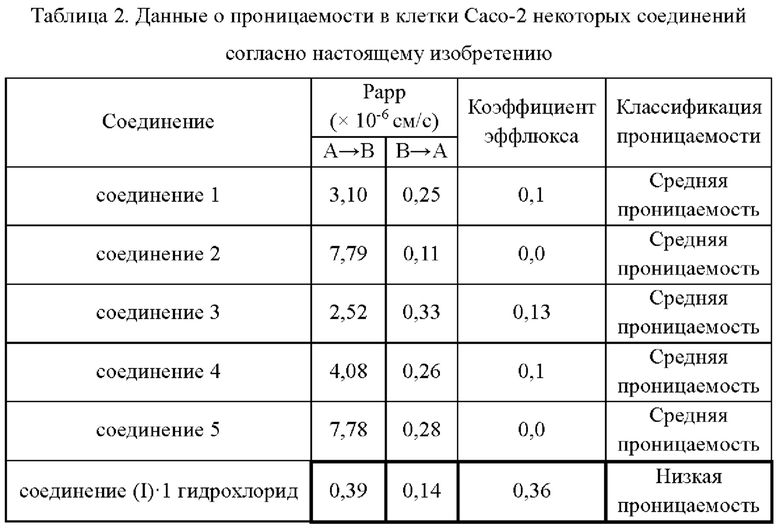
Согласно данным, приведенным в табл. 2, соединения согласно настоящему изобретению показывают улучшенную проницаемость по сравнению с соединение (I)⋅1 гидрохлорид, что указывает на то, что соединения согласно настоящему изобретению обладают превосходными свойствами абсорбции.
Экспериментальный пример 3. Фармакокинетические исследования на мышах
Экспериментальный метод:
Фармакокинетические исследования соединения 1, соединения 2, соединения 3 и соединения 5 проводили на мышах линии Balb/c (самцах) массой от 23 до 25 г с тремя мышами в каждой группе введения перорально или внутривенно. Соединение 1, соединение 2, соединение 3 и соединение 5 готовили в виде суспензии 5 мг/мл с 0,5% карбоксиметилцеллюлозы, соответственно, и вводили перорально в дозе 50 мг/кг. Соединение 1, соединение 2, соединение 3 и соединение 5 готовили в виде 1 мг/мл раствора с 20% HP-β-CD и 1 н. раствором хлористоводородной кислоты, соответственно, и вводили дозу 5 мг/кг внутривенно.
Образцы плазмы крови отбирали через 5, 15, 30 минут и через 1, 2, 4, 7, 12 и 24 ч после перорального и внутривенного введения. Отобранные образцы плазмы крови хранили при температуре -80°С до использования для анализа. Образцы плазмы крови экстрагировали ацетонитрилом, содержащим терфенадин в качестве внутреннего стандарта при соотношении экстрагента к плазме крови 20:1. Количественное определение анализируемого вещества проводили с помощью масс-спектрометра LC/TSQ Quantum Access (АВ Sciex 5500). Условия хроматографирования: колонка: Kinetex С18 100А (30 мм × 3,0 мм, 2,6 мкм); температура колонки: комнатная температура; подвижная фаза: ацетонитрил/вода (80:20, (об./об.)) (содержащая 0,1% муравьиной кислоты); скорость потока: 0,8 мл/мин. Обнаружение соединения на масс-спектрометре проводили в положительном режиме ионизации электрораспылением. Фармакокинетические параметры рассчитывали с помощью программного обеспечения WinNonlin (6.3 Pharsight Corporation, Mountain View, США).
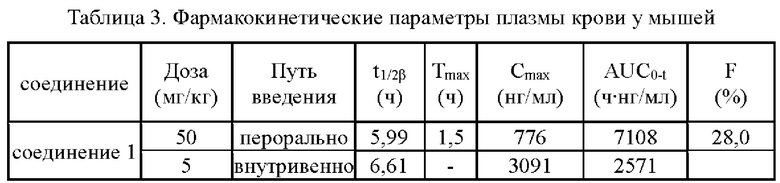
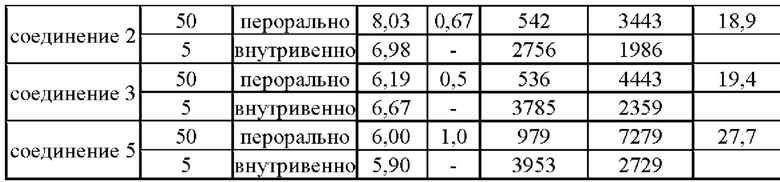
Согласно данным табл. 3 биодоступность (F) соединений 1, 2, 3 и 5 согласно настоящему изобретению находилась в диапазоне 18,9 28,0%, в то время как биодоступность свободного основания 6b (соединения (I)), указанная в сравнительном документе (Eur J. Med. Chem., 2018, 160, 157-170), составила 13,1%. По сравнению со свободным основанием биодоступность соединений 1, 2, 3 и 5 была повышена, в частности для соединений 1 и 5 она повысилась однократно. Вышеуказанные результаты показали, что соединения согласно настоящему изобретению обладают превосходными фармакокинетическими свойствами.
Экспериментальный пример 4. Фармакокинетические исследования на крысах
Экспериментальный метод
Фармакокинетические исследования соединения 1, соединения (I) и соединения (I) гидрохлорида проводили на крысах SD (самцах) массой от 223 до 245 г с тремя крысами в каждой группе введения перорально или внутривенно. Соединение 1, соединение (I) и соединения (I) гидрохлорид готовили в виде 5 мг/мл суспензии с 0,5% карбоксиметилцеллюлозы, соответственно, и вводили перорально в дозе 50 мг/кг. Соединение 1, соединение (I) и соединения (I) гидрохлорид готовили в виде 1 мг/мл раствора с 20% HP-β-CD (гидроксипропил-β-циклодекстрин) и 1 н. раствором хлористоводородной кислоты, соответственно, и вводили дозу 5 мг/кг внутривенно.
Образцы плазмы крови отбирали через 5, 15, 30 минут и через 1, 2, 4, 7, 12 и 24 ч после перорального и внутривенного введения. Отобранные образцы плазмы крови хранили при температуре -80°С до использования для анализа. Для расчета фармакокинетических параметров использовали программное обеспечение WinNonlin (6.3 Pharsight Corporation, Mountain View, США).
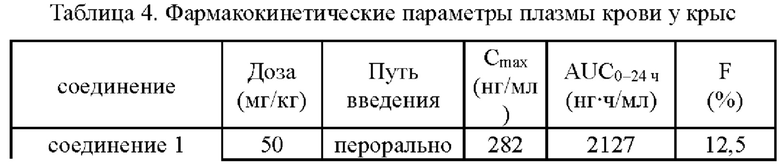
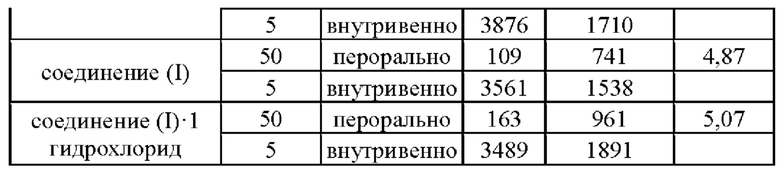
Согласно данным, приведенным в табл. 4, соединение 1 согласно настоящему изобретению демонстрирует значительно улучшенные характеристики Cmax, AUC и биодоступность (F) при пероральном приеме по сравнению с соединением (I) и его гидрохлоридной солью в той же дозе. Вышеуказанные результаты показали, что соединение 1 обладает превосходными фармакокинетическими свойствами.
Экспериментальный пример 5. Анализ противотуберкулезной активности in vivo у мышей
Экспериментальный метод
Мышей линии Balb/c инфицировали с помощью аэрозоля суспензией штамма H37Rv М. tuberculosis. Лекарственные препараты вводили на 10-й день после инфицирования (25, 50, 100 мг/кг), и мышам вводили препарат в течение 5 дней в неделю. Мышей умерщвляли после введения в течение трех недель, и значение КОЕ в легких было основным показателем оценки. Противотуберкулезную активность соединения (I) и соединения 1 in vivo оценивали с использованием 0,5% КМЦ в качестве контрольной группы и клинического лекарственного препарата первой линии изониазида в качестве положительного контроля.
Экспериментальные процедуры проводили в соответствии с литературными данными (Antimicrobial agents and chemotherapy 2011, 55 (11), 5185-5193).
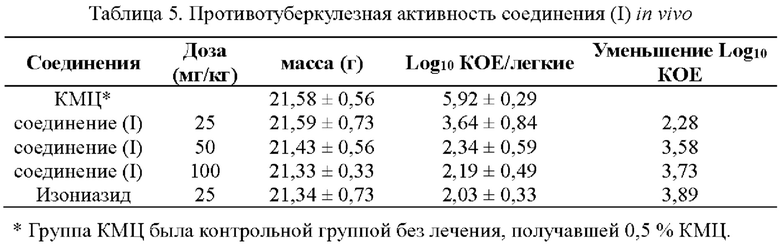
Экспериментальные результаты показали, что соединение (I) обладает очень высокой противотуберкулезной активностью in vivo в дозах 25, 50 и 100 мг/кг, соответственно. Количество жизнеспособных бактерий в легочной ткани мышей уменьшилось на 2,28, 3,58 и 3,73 log10KOE по сравнению с контрольной группой, не получавшей лечение, соответственно.
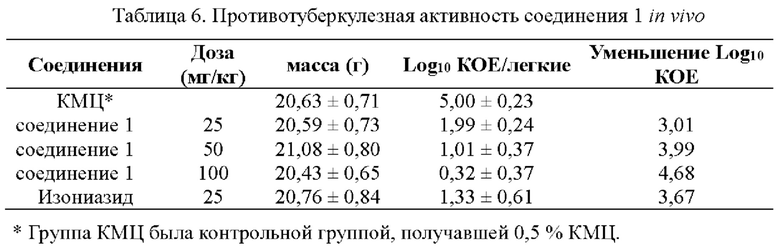
Экспериментальные результаты показали, что соединение 1 обладает очень высокой противотуберкулезной активностью in vivo с явной зависимостью доза-эффект при 25, 50 и 100 мг/кг, соответственно. Количество жизнеспособных бактерий в легочной ткани мышей уменьшилось на 3,01, 3,99 и 4,68 log10KOE по сравнению с контрольной группой, не получавшей лечение, соответственно.
Согласно данным в табл. 5 и 6, соединение 1 согласно настоящему изобретению может уменьшать Log10 КОЕ на большее значение в дозе 25, 50 и 100 мг/кг, соответственно. В частности, в дозе 100 мг/кг соединение 1, которое уменьшало бактериальную нагрузку в легких на 4,68 log10 КОЕ по сравнению с контрольной группой, не получавшей лечение, продемонстрировало превосходную противотуберкулезную активность in vivo по сравнению с соединением (I) со снижением на 3,73 log10 КОЕ. Вышеуказанные результаты показали, что соединение 1 проявляет более высокую противотуберкулезную активность in vivo.
Экспериментальный пример 6. Исследования стабильности
Стабильность соединения 1, соединения (I) и его гидрохлоридной соли после помещения в условия воздействия света, высокой температуры и высокой влажности в течение 10 дней исследовали методом ВЭЖХ, и результаты были показаны в табл. 7.
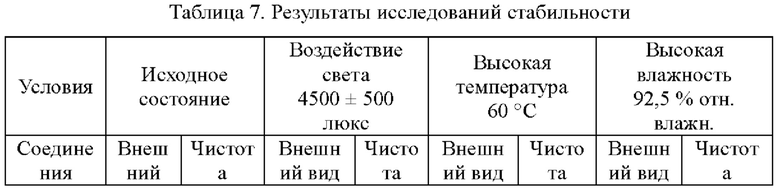
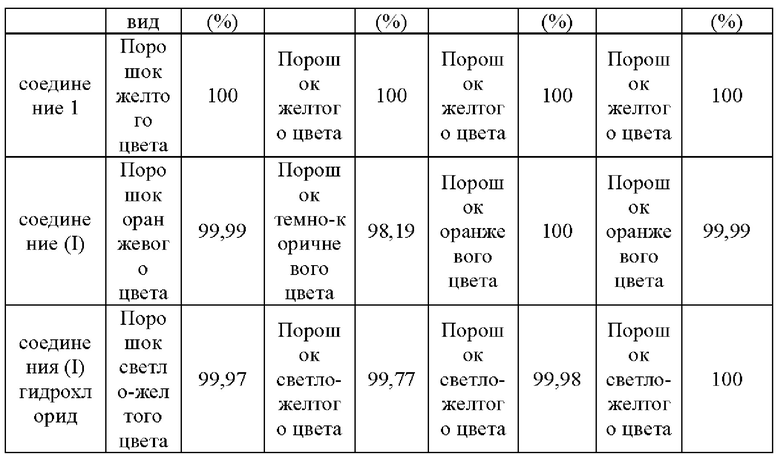
Чистоту соединений определяли с помощью системы ВЭЖХ Waters e2695-PDA. Хроматографические условия: колонка: Kromasil С18 (250 мм × 4,6 мм, 5 мкм); температура колонки: 30°С; подвижная фаза: ацетонитрил/вода (84:16, (об./об.)) при градиентном элюировании; скорость потока: 1,0 мл/мин. В табл. 7 показано, что соединение 1 согласно настоящему изобретению продемонстрировало существенную стабильность в условиях воздействия света, высокой температуры и высокой влажности. Однако для соединения 11 (соединение, представленное формулой (I)), описанное в патентном документе (201810092333.X), представляющего собой свободное основание соединения 1 согласно настоящему изобретению, изменился внешний вид, и снизилась чистота в условиях воздействия света. Следовательно, соединение 1 согласно настоящему изобретению обладает превосходными физико-химическими свойствами.
Хотя примеры настоящего изобретения были показаны и описаны выше, понятно, что приведенные выше примеры являются иллюстративными и не могут рассматриваться в качестве ограничений настоящего изобретения, и что специалисты в данной области техники могут изменять, модифицировать, заменять и трансформировать приведенные выше примеры в пределах объема настоящего изобретения.
Изобретение относится к фармацевтически приемлемой соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она, представленного формулой (I), где соль выбрана из 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1малеата, 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·3/2фумарата, 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1цитрата и 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1L-малата. Изобретение относится к способу получения фармацевтически приемлемой соли по изобретению, который включает стадии взаимодействия 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она с фармацевтически традиционной кислотой в растворителе, представляющей собой спирт, при комнатной температуре или в условиях кипячения с обратным холодильником в течение 2-8 ч с получением соли, показанной в формуле (I), посредством реакции образования соли. Также изобретение относится к фармацевтической композиции для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis, характеризующейся тем, что фармацевтическая композиция содержит эффективное количество фармацевтически приемлемой соли по изобретению и один или более фармацевтически приемлемых носителей, вспомогательных веществ, разбавителей, адъювантов и сред. Технический результат - фармацевтически приемлемые соли 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она для получения лекарственного препарата для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis. 4 н.п. ф-лы, 7 табл., 10 пр. 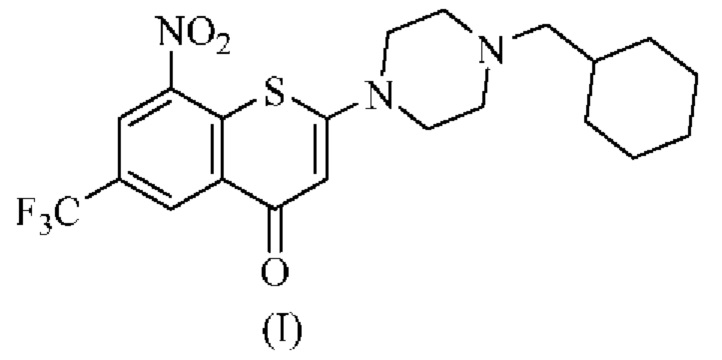
1. Фармацевтически приемлемая соль 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она, представленного формулой (I):
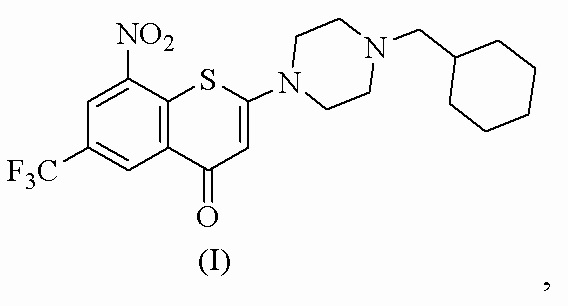
где соль выбрана из
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1малеата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·3/2фумарата,
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1цитрата, и
2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-он·1L-малата.
2. Способ получения фармацевтически приемлемой соли по п. 1, который включает следующие стадии:
взаимодействие 2-(4-(циклогексил)метил)пиперазин-1-ил)-8-нитро-6-(трифторметил)-4H-тиохромен-4-она с фармацевтически традиционной кислотой в растворителе, представляющей собой спирт, при комнатной температуре или в условиях кипячения с обратным холодильником в течение 2-8 ч с получением соли, показанной в формуле (I), посредством реакции образования соли.
3. Фармацевтическая композиция для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis, характеризующаяся тем, что фармацевтическая композиция содержит эффективное количество фармацевтически приемлемой соли по п. 1 и один или более фармацевтически приемлемых носителей, вспомогательных веществ, разбавителей, адъювантов и сред.
4. Применение фармацевтически приемлемой соли по п. 1 или фармацевтической композиции по п. 3 для получения лекарственного препарата для лечения и/или предотвращения микробного инфекционного заболевания, вызванного Mycobacterium tuberculosis.
| CN 108929329 A, 04.12.2018 | |||
| Richard J.Bastin et al | |||
| Salt selection and Optimisation Procedures for Pharmaceutical New Chemical Entities, ORGANIC PROCESS RESEARCH & DEVELOPMENT, 2000, v | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ уравновешивания движущихся масс поршневых машин | 1925 |
|
SU427A1 |
| Abu T.M.Serajuddin | |||
| Salt formation to improve drug solubility, ADVANCED DRUG DELIVERY REVIEWS, 2007, v | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| МЕТАЛЛИЧЕСКАЯ ШАРНИРНАЯ СЕТКА | 1922 |
|
SU603A1 |
| Peng Li, Bin Wang, et al | |||
