Область техники
Настоящая заявка относится к области реакции аминирования, в частности, к катализатору, подходящему для производства органических аминов путем каталитического аминирования, к его получению и его применению.
Уровень техники
Амины являются очень важными промышленными органическими соединениями, они широко используются в различных областях, например, в качестве растворителей, медицинских промежуточных веществ, исходных материалов для смол, текстильных добавок, инсектицидов, стабилизаторов для каучуков, резистов, а также в очистке и в переработке пластмасс. Тремя основными способами получения аминов являются гидроаминирование карбонильных соединений, гидроаминирование спиртов и гидрирование нитрилов. Гидроаминирование карбонильных соединений представляет собой, например, реакцию ацетона, водорода и аммиака, ведущую к образованию изопропиламина. Гидроаминирование спиртов включает, например, гидроаминирование этанола аммиаком в присутствии водорода с образованием этиламина, гидроаминирование изопропанола аммиаком в присутствии водорода с образованием изопропиламина, гидроаминирование бутанола аммиаком в присутствии водорода с образованием бутиламина, гидроаминирование гександиола аммиаком в присутствии водорода с образованием гександиамина и т.д. Гидрирование нитрила включает, например, гидрирование ацетонитрила до этиламина и гидрирование адипонитрила до гександиамина.
В заявке на патент Китая CN102658162A описан катализатор синтеза этиленамина и способ получения этиленамина. Катализатор состоит из трех частей, а именно, основного активного компонента, вспомогательного агента и аминированной подложки, при этом основной активный компонент представляет собой один или более компонентов, выбранных из Ni и Со, и составляет 1-40% от полного веса катализатора, вспомогательный агент представляет собой один или более агентов, выбранных из группы, состоящей из Fe, Cu, Ru, Re, K, Zn и В и их оксидов, и составляет 0,1-20% от полного веса катализатора; аминированная подложка получена аминированием одной или более подложек, выбранных из группы, состоящей из SiO2 и Al2O3, и аминирующая обработка включает: контакт подложки с источником аммиака при температуре 150-400°С в течение 0,5-15 часов. Авторы настоящей заявки обнаружили, что материал подложки тесно связан с активностью катализатора: поскольку на поверхности подложки из SiO2 или Al2O3 имеется большое количество гидроксильных групп, подложка находится в кислой среде, которая способствует полимеризации промежуточного продукта имина, если подложка катализатора аминирована, большое количество гидроксильных групп на поверхности подложки будет превращаться в аминогруппы, так что подложка обнаруживает щелочное окружение, возможность полимеризации имина снижается, и активность, селективность и стабильность катализатора улучшаются.
В известном уровне техники обычно признается, что когда катализатор для получения аминов путем аминирования спиртов обладает щелочностью, это более благоприятно для улучшения активности и селективности катализатора, и повышение активности существующих катализаторов реакции аминирования имеет большой потенциал.
Сущность изобретения
Целью настоящей заявки является разработка катализатора, подходящего для получения органических аминов путем каталитического аминирования, а также его получение и применение, при этом указанный катализатор должен демонстрировать улучшенные характеристики при использовании для реакции аминирования, например, по меньшей мере одну характеристику из улучшенной каталитической активности, улучшенной конверсии реакции, улучшенной селективности по продукту и улучшенной стабильности катализатора.
Для достижения вышеуказанной цели в одном аспекте настоящей заявки предлагается катализатор, подходящий для получения органических аминов путем каталитического аминирования, имеющий неорганическую пористую подложку, содержащую алюминий и/или кремний, и активный металлический компонент, нанесенный на подложку, причем активный металлический компонент содержит по меньшей мере один металл, выбранных из металлов группы VIII и группы IB, при этом подложка имеет содержание L-кислоты 85% или более от суммарного содержания L-кислоты и В-кислоты.
Предпочтительно, подложка содержит матрицу и легирующий элемент, причем матрица выбрана из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций, а легирующий элемент является неметаллическим элементом.
Далее, катализатор предпочтительно содержит металл-промотор, нанесенный на подложку, и указанный металл-промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы ИВ и элементов ряда лантанидов, или содержит комбинацию по меньшей мере одного металла группы IIA, по меньшей мере одного металла группы ИВ и по меньшей мере одного металла группы VA.
В другом аспекте предлагается способ получения катализатора по настоящей заявке, включающий этапы:
1) подготовка неорганической пористой подложки, содержащей алюминий и/или кремний, причем подложка имеет содержание L-кислоты 8 5% или больше от суммарного содержания L-кислоты и В-кислоты;
2) нанесение активного металлического компонента и, необязательно, металла-промотора на подложку; и
3) проведение термообработки и, необязательно, восстановительной обработки материала, полученного на этапе 2), для получения катализатора.
В еще одном аспекте настоящей заявки предлагается способ получения органических аминов, включающий: контактирование исходного материала для аминирования и аминирующего реагента с катализатором согласно настоящей заявки для реакции аминирования в присутствии водорода, чтобы получить органический амин, причем исходный материал для аминирования выбран из группы, состоящей из спиртов, кетонов, аминоспиртов, альдегидов и их комбинаций, а аминирующий реагент выбран из группы, состоящей из аммиака, первичных аминов, вторичных аминов и их комбинаций.
Катализатор по настоящей заявке демонстрирует улучшенные характеристики, в частности, улучшенную каталитическую активность, конверсию реакции, селективность по продукту и/или стабильность катализатора, при использовании для получения органических аминов путем каталитического аминирования.
Другие характеристики и преимущества настоящей заявки будут подробно описаны в приводимом ниже подробном описании.
Подробное описание изобретения
Далее настоящая заявка будет более подробно описана ниже в отношении частных вариантов ее осуществления. Следует отметить, что эти частные варианты осуществления настоящей заявки приводятся исключительно в целях иллюстрации и не имеют ограничительного характера.
Любые конкретные численные величины, включая границы числового диапазона, описанные в контексте настоящей заявки, не ограничены их точным значением, но должны интерпретироваться как охватывающие все значения в окрестности указанной точной величины, например, все значения в пределах ±5% от указанной точной величины. Кроме того, что касается любого описанного в настоящем документе числового диапазона, можно выбрать произвольные комбинации между граничными точками диапазона, между каждой граничной точкой и любым частным значением внутри диапазона или любыми двумя частными значениями внутри диапазона, чтобы создать один или более новых числовых диапазонов, при этом указанные новые числовые диапазоны также следует считать явно описанными в настоящей заявке.
Если не указано иное, используемые здесь термины имеют те же значения, которые обычно понимаются специалистом в данной области, но если термины определяются в настоящем документе и их определения отличаются от обычного понимания в данной области, преимущественную силу имеет определение, даваемое в настоящем документе.
В настоящей заявке отношение содержания L-кислоты в подложке к суммарному содержанию L-кислоты и В-кислоты измеряется методом адсорбционной спектроскопии с пиридиновым зондом.
В настоящей заявке способность подложки и катализатора адсорбировать аммиак измеряется методом термо-программируемой десорбции NH3, при этом адсорбционная емкость по аммиаку выражается в измеренном количестве десорбированного аммиака.
В настоящей заявке удельная поверхность, объем пор объем пор и доля пор разного диаметра в подложке измеряются методом адсорбции-десорбции азота согласно GB/T6609.35-2009.
В настоящей заявке выражение "С2-20" означает наличие от 2 до 20 атомов углерода, например, наличие 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 атомов углерода. Аналогично, выражение "С1-12" означает наличие от 1 до 12 атомов углерода.
В настоящей заявке размеры зерен активного металлического компонента и металла-промотора измеряются методом рентгенодифракционного анализа.
В настоящей заявке изоэлектрическая точка подложки измеряется с помощью потенциометрического анализатора размера частиц.
В настоящей заявке давление, если не указано иное, является манометрическим давлением.
В контексте настоящей заявки, в дополнение к явно указанным предметам обсуждения, любой не упоминавшийся предмет или предметы обсуждения считаются такими же, как известно в уровне техники, без каких-либо изменений. Кроме того, любой из описанных здесь вариантов осуществления может свободно комбинироваться с другими, одним или несколькими, описанными в настоящем документе вариантами осуществления, и полученные таким образом технические решения или идеи считаются частью исходного раскрытия или исходного описания настоящей заявки и не должны рассматриваться как новый предмет, который не был раскрыт или не предполагался в настоящем документе, если только специалисту в данной области техники не очевидно, что такая комбинация явно нецелесообразна.
Все цитируемые здесь патентные и непатентные документы, включая, без ограничений, учебники и журнальные статьи, включены в настоящий документ ссылкой во всей их полноте.
Как упоминалось выше, в первом аспекте настоящей заявки предлагается катализатор, подходящий для получения органических аминов путем каталитического аминирования, имеющий неорганическую пористую подложку, содержащую алюминий и/или кремний, и активный металлический компонент, нанесенный на подложку, при этом активный металлический компонент содержит по меньшей мере один металл, выбранный из металлов группы VIII и группы IB, причем подложка имеет содержание L-кислоты 85% или более от суммарного содержания L- и В-кислоты.
Согласно настоящей заявке, металл группы VIII может представлять собой, например, кобальт, никель или палладий, а металл группы IB может представлять собой, например, медь. В предпочтительном варианте осуществления металл активного металлического компонента выбран из кобальта, никеля, палладия, меди или их комбинации, более предпочтительно он выбирается из кобальта, никеля или их комбинации.
В катализаторе по настоящей заявке в качестве активного металлического компонента можно использовать металл только группы IB, такой, как медь, в этом случае он обычно используется в относительно большем количестве; он может также использоваться в сочетании с металлом группы VIII, и в этом случае он обычно используется в относительно меньшем количестве. При использовании в комбинации с металлом группы VIII, таким как кобальт, никель и палладий, металл группы IB обычно называется здесь металлом-промотором.
В предпочтительном варианте осуществления содержание L-кислоты в подложке составляет 88% или более, более предпочтительно 90% или более и особенно предпочтительно 92% или более, от суммарного содержания L-кислоты и В-кислоты.
В предпочтительном варианте осуществления подложка содержит матрицу и легирующий элемент, причем матрица выбрана из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций, и легирующий элемент является неметаллическим элементом, предпочтительно по меньшей мере одним элементом, выбранным из группы, состоящей из неметаллических элементов группы IIIA, группы VA, группы VIA и группы VIIA, за исключением хлора, более предпочтительно он представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена. Более предпочтительно, легирующий элемент в подложке происходит из неметаллического ион-радикала кислоты, и неметаллический ион-радикал кислоты предпочтительно представляет собой по меньшей мере один ион-радикал, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
В предпочтительном варианте осуществления подложка имеет по меньшей мере одну из следующих характеристик:
- доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет более 65%, предпочтительно 70-90%, и доля объема пор диаметром меньше 7 нм в объема пор подложки составляет 0-10%, предпочтительно 0-8%, и если подложка имеет описанное выше распределение пор по диаметру, это выгодно для повышения коэффициента поверхностной диффузии катализатора и улучшения активности и селективности катализатора по продукту;
- доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет менее 20%, предпочтительно 5-17%, а доля объема пор диаметром меньше 9 нм в объеме пор подложки составляет менее 40%, доля объема пор диаметром больше 27 нм в объеме пор подложки составляет менее 5%, предпочтительно 0,5-5%, предпочтительно, доля объема пор диаметром больше или равным 7,5 нм и меньше 9 нм в объеме пор подложки составляет 5-17%, и доля объема пор диаметром больше или равным 9 нм и меньше или равным 27 нм в объеме пор подложки составляет 61-89,5%, и если подложка имеет описанное выше распределение пор по диаметру, это выгодно для повышения коэффициента поверхностной диффузии катализатора и улучшения активности катализатора и его селективности по продукту;
- подложка имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, предпочтительно 0,3-0,6 ммоль/г, более предпочтительно 0,3-0,5 ммоль/г;
- содержание оксида алюминия в подложке составляет 65 вес. % или более, предпочтительно 7 0 вес. % или более, более предпочтительно 75 вес. % или более, от полного количества матрицы;
- легирующий элемент присутствует в количестве от 0,05 до 6 вес. %, предпочтительно от 0,05 до 5 вес. %, более предпочтительно от 0,05 до 4,5 вес. %, особенно предпочтительно от 0,07 до 4 вес. %, например от 0,08 до 4 вес. % или от 0,1 до 3 вес. % (например, это может быть 0,1 вес. %, 0,5 вес. %, 1 вес. %, 1,5 вес. %, 2 вес. %, 2,5 вес. %, 3 вес. % или значение между любыми двумя из них), от полного веса матрицы;
- подложка имеет удельную поверхность 100-220 м2/г, предпочтительно 105-210 м2/г, более предпочтительно 110-210 м2/г и особенно предпочтительно 120-210 м2/г;
- подложка имеет объем пор 0,4-1,1 мл/г, предпочтительно 0,43-1,1 мл/г, более предпочтительно 0,45-1,1 мл/г и особенно предпочтительно 0,45-1 мл/г; и
- изоэлектрическая точка подложки составляет 3-6, предпочтительно 3,5-5,5.
В предпочтительном варианте осуществления катализатора по настоящей заявке активный металлический компонент присутствует в количестве от 5 до 45 г, предпочтительно от 8 до 44 г, более предпочтительно от 10 до 38 г и особенно предпочтительно от 15 до 37 г, на 100 г матрицы.
В предпочтительном варианте осуществления активный металлический компонент имеет размер зерна менее 10 нм, более предпочтительно от 3 до 8 нм, что может быть хорошо согласовано со свойствами подложки, так что можно достичь лучшей каталитической активности и селективности по продукту.
В предпочтительном варианте осуществления катализатор содержит также металл-промотор, нанесенный на подложку, и металл-промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантанидов, предпочтительно по меньшей мере один металл, выбранный из группы, состоящей из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се; более предпочтительно металл-промотор присутствует в количестве от 0 до 10 г, предпочтительно от 0,1 до 10 г, более предпочтительно от 0,5 до 8 г, на 100 г матрицы.
В некоторых более предпочтительных вариантах осуществления металл-промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, при этом весовое отношение металла группы VIIB к металлу группы IB, рассчитанное на элементарный металл, составляет (0,05-15):1, предпочтительно (0,1-12):1; или металл-промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB, при этом весовое отношение металла группы VIIB к металлу группы IIB, рассчитанное на элементарный металл, составляет (0,2-20):1, предпочтительно (0,3-6):1, или металл-промотор содержит комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB, при этом весовое отношение металла группы VIB, металла группы IB и металла группы IIB, рассчитанное на элементарный металл, составляет (0,1-10):(0,1-10):1, предпочтительно (0,2-8):(0,2-8):1. Особенно предпочтительно металл группы VIIB выбран из марганца и/или рения, металл группы IB представляет собой по меньшей мере один металл, выбранный из группы, состоящей из меди, серебра и золота, металл группы IIB является цинком, и металл группы VIB выбран из молибдена и/или вольфрама.
В других предпочтительных вариантах осуществления катализатор содержит также металл-промотор, нанесенный на подложку, и металл-промотор представляет собой комбинацию по меньшей мере одного металла группы IIA, по меньшей мере одного металла группы IIB и по меньшей мере одного металла группы VA, более предпочтительно металл-промотор присутствует в количестве от 0,1 до 10 г, предпочтительно от 0,5 до 6 г на 100 г матрицы. Еще более предпочтительно, весовое соотношение между металлом группы IIA, металлом группы IIB и металлом группы VA в металле-промоторе составляет (0,1-10):(0,1-10):1, предпочтительно (0,2-8):(0,2-8):1. Особенно предпочтительно, металл группы IIA является по меньшей мере одним металлом, выбранным из группы, состоящей из магния, кальция и бария, металл группы IIB является цинком, и/или металл группы VA является висмутом.
В соответствии с настоящей заявкой, подложку катализатора можно получить способами, известными в данной области и подходящими для получения подложек с вышеупомянутыми свойствами, так что на эти способы в настоящей заявке не накладывается никаких особых ограничений. Подложку можно с успехом приготовить способом, включающим следующие этапы: последовательное формование, сушка и обжиг смеси, содержащей легирующий элемент и матрицу или ее предшественник, с получением подложки, при этом матрица выбирается из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций. В качестве молекулярного сита можно использовать, например, молекулярные сита ZSM-5 или ZSM-11. Когда используется предшественник матрицы, в качестве предшественника оксида алюминия можно использовать псевдобемит, а в качестве предшественника оксида кремния кремниевую кислоту, ортокремниевую кислоту или силикагель.
В описанном выше способе получения подложки предшественник матрицы предпочтительно представляет собой псевдобемит. Псевдобемит может быть получен по меньшей мере одним способом из способа карбонизации, способа гидролиза алюминийорганических соединений, алюминийсульфатного способа и азотнокислотного способа. Удельная поверхность псевдобемита предпочтительно составляет 250-400 м2/г, предпочтительно 255-360 м2/г, более предпочтительно 255-340 м2/г и особенно предпочтительно 260-330 м2/г; псевдобемит предпочтительно имеет объем пор от 0,5 до 1,3 мл, предпочтительно 0,75-1,25 мл/г, более предпочтительно 0,78-1,2 мл/г, особенно предпочтительно 0,78-1,1 мл/г. Используя псевдобемит с особой структурой пор, можно получить катализатор с улучшенными характеристиками.
В описанном выше способе получения подложки, когда исходный материал, обеспечивающий предшественник матрицы, уже содержит желаемое количество легирующего элемента, формование может быть выполнено, просто используя такой исходный материал, а когда исходный материал, обеспечивающий предшественник матрицы, не содержит легирующего элемента или содержание легирующего элемента низкое (недостаточное), можно ввести дополнительный легирующий элемент.
В описанном выше способе получения подложки легирующий элемент можно обеспечить, используя модификатор подложки, содержащий по меньшей мере одно соединение, способное предоставить неметаллический ион-радикала кислоты, например, неорганическую кислоту и/или неорганическую соль, содержащую неметаллический кислотный радикал; неметаллический ион-радикала кислоты предпочтительно представляет собой по меньшей мере один ион-радикал, выбранный из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Более предпочтительно, модификатор подложки представляет собой по меньшей мере один материал, выбранный из группы, состоящей из борной кислоты, бората никеля, бората кобальта, бората калия, бората аммония, бората магния, фторида калия, фторида магния, фторида кобальта, фторида никеля, фтористоводородной кислоты, фторида аммония, фосфорной кислоты, фосфата алюминия, трикалийфосфата, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, фосфата аммония, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, сульфата калия, сульфата магния, фосфата стронция, сульфата стронция и селеновой кислоты.
В описанном выше способе получения подложки способ формования может быть выбран из замешивания, раскатывания, расслаивания или подобного.
В описанном выше способе получения подложки модификатор подложки используется в таком количестве, чтобы содержание легирующего элемента составляло от 0,05 до 6 вес. %, предпочтительно от 0,05 до 5 вес. %, более предпочтительно от 0,05 до 4,5 вес. %, особенно предпочтительно от 0,07 до 4 вес. %, например, от 0,08 до 4 вес. %, от полного веса матрицы. Специалист может определить количество исходного материала (например, модификатора подложки) для компонента, исходя из количества указанного компонента в конечной подложке, поэтому количества некоторых исходных материалов здесь не приводятся.
В описанном выше способе получения подложки условия сушки могут включать: температура 80-150°С (например, 80°С, 85°С, 90°С, 95°С, 100°С, 110°С, 115°С, 120°С, 125°С, 130°С, 140°С, 150°С или значение между любыми двумя из них), предпочтительно 85-130°С, и продолжительность 6-20 ч (например, 6 ч, 7 ч, 7,5 ч, 8 ч, 8,5 ч, 9 ч, 10 ч, 12 ч, 15 ч, 18 ч, 20 ч или значение между любыми двумя из них), предпочтительно 10-20 ч.
В описанном выше способе получения подложки условия обжига могут включать: температура 500-1120°С, например, 500-б50°С, предпочтительно 700-1100°С, более предпочтительно 800-1050°С (например, 800°С, 850°С, 860°С, 870°С, 880°С, 890°С, 900°С, 920°С, 950°С, 960°С, 980°С, 1000°С, 1050°С или значение между любыми двумя из них), и продолжительность 2-20 ч (например, 2 ч, 3 ч, 3,5 ч, 4 ч, 4,5 ч, 5 ч, 6 ч, 7 ч, 8 ч, 10 ч, 15 ч, 20 ч или значение между любыми двумя из них).
Катализатор по настоящей заявке можно использовать после восстановления, например, его можно восстановить с использованием водородсодержащего газа при температуре 350-500°С, предпочтительно 350-450°С. Водородсодержащий газ может быть чистым водородом или водородом, разбавленным инертным газом, как, например, смесь азота и водорода. Во время восстановления температуру восстановления постепенно повышают, и скорость повышения температуры предпочтительно не слишком высокая, например, не выше 20°С в час. Продолжительность восстановления можно определить путем отслеживания образования H2O в восстановительной системе, то есть восстановление заканчивается, когда в восстановительной системе новой H2O больше не образуется, и специалист может соответствующим образом выбрать время восстановления, подробное описание этого здесь для краткости опущено, например, продолжительность восстановления может составлять 2-5 ч при самой высокой температуре. Восстановление можно реализовать непосредственно в реакторе с последующей каталитической реакцией. Можно также проводить восстановление в отдельном реакторе, что называется также внереакторным восстановлением, а пассивацию можно проводить после восстановления смесью газов, содержащей кислород, перед выгрузкой катализатора из реактора, при этом температура пассивации составляет, например, 10-60°С, в частности, 20-40°С. Катализатор, подвергнутый внереакторному восстановлению и пассивации, перед применением можно активировать, используя водород или смесь водорода и азота, например, при температуре от 150°С до 250°С, предпочтительно от 170°С до 200°С. Продолжительность активации можно определить, отслеживая образование H2O в активируемой системе, то есть активация заканчивается, когда в активируемой системе больше не образуется новой H2O, и специалист может соответствующим образом выбрать продолжительность активации, подробное описание этого здесь для краткости опущено. Например, продолжительность активации при самой высокой температуре может составлять, например, от 1 до 5 часов, предпочтительно от 2 до 3 часов, или катализатор можно использовать без активации, в зависимости от степени, до которой были окислены активный металлический компонент и металл-промотор катализатора.
Во втором аспекте предлагается способ получения катализатора по настоящей заявке, включающий этапы:
1) подготовка неорганической пористой подложки, содержащей алюминий и/или кремний, причем подложка имеет содержание L-кислоты 8 5% или больше от суммарного содержания L-кислоты и В-кислоты;
2) нанесение активного металлического компонента и, необязательно, металла-промотора на подложку; и
3) проведение термообработки и, необязательно, восстановительной обработки материала, полученного на этапе 2), для получения катализатора.
В предпочтительном варианте осуществления содержание L-кислоты в подложке составляет 88% или более, более предпочтительно 90% или более и особенно предпочтительно 92% или более, от суммарного содержания L-кислоты и В-кислоты.
В предпочтительном варианте осуществления указанное "подготовка неорганической пористой подложки, содержащей алюминий и/или кремний," на этапе 1) включает последовательное формование, сушку и обжиг смеси, содержащей легирующий элемент и матрицу или их предшественник, чтобы получить подложку, при этом матрица выбирается из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций, предпочтительно предшественник оксида алюминия является псевдобемитом, имеющим удельную поверхность 250-4 00 м2/г, предпочтительно 255-360 м2/г, более предпочтительно 255-340 м2/г, особенно предпочтительно 260-330 м2/г, и объем пор 0,5-1,3 мл/г, предпочтительно 0,75-1,25 мл/г, более предпочтительно 0,78-1,2 мл/г, особенно предпочтительно 0,78-1,1 мл/г; легирующий элемент представляет собой неметаллический элемент, предпочтительно по меньшей мере один элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, исключая хлор, предпочтительно по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
В еще одном предпочтительном варианте осуществления легирующий элемент обеспечивается использованием модификатора подложки, и легирующий элемент и модификатор подложки могут выбираться как описано выше в первом аспекте, поэтому подробное описание этого здесь для краткости опущено. Еще более предпочтительно, модификатор подложки используется в таком количестве, чтобы получаемая подложка имела содержание легирующего элемента от 0,05 до 6 вес. %, предпочтительно от 0,05 до 5 вес. %, более предпочтительно от 0,05 до 4,5 вес. %, особенно предпочтительно от 0,07 до 4 вес. %, например, от 0,08 до 4 вес. % или от 0,1 до 3 вес. % (например, это может быть 0,1 вес. %, 0,5 вес. %, 1 вес. %, 1,5 вес. %, 2 вес. %, 2,5 вес. %, 3 вес. % или значение между любыми двумя из них), от полного веса матрицы.
В еще одном предпочтительном варианте осуществления способ формования может быть выбран из замешивания, раскатывания, расслаивания или подобного.
В еще одном предпочтительном варианте осуществления условия сушки на этапе 1) включают: температура 80-150°С, продолжительность сушки 6-20 ч; и/или условия обжига включают: температура 500-1120°С, например, 500-650°С, предпочтительно 700-1100°С, более предпочтительно 800-1050°С, и продолжительность обжига 2-20 ч.
В еще одном предпочтительном варианте осуществления этап 1) имеет характеристики, какие описаны выше для способа получения подложки в первом аспекте, и подробное описание этого здесь для краткости опущено.
В предпочтительном варианте осуществления указанный этап 2) введения включает пропитку подложки раствором, содержащим предшественник указанного активного металлического компонента и, необязательно, предшественник указанного металла-промотора, пропиточный раствор предпочтительно имеет рН в интервале 3,5-5,5. Удерживание рН пропиточного раствора в вышеуказанном диапазоне может дополнительно улучшить степень дисперсности активного металлического компонента.
В соответствии с настоящей заявкой пропитка проводится путем погружения подложки в подходящий раствор, содержащий предшественники указанного активного металлического компонента и металла-промотора, так что предшественник вводится на подложку путем адсорбции. Способы пропитки можно классифицировать как сухая пропитка, влажная пропитка, многократная пропитка, смешанная пропитка, пропитка распылением и т.д. Сухая пропитка и влажная пропитка означают соответственно пропитку подложки, которая находится в сухом состоянии или заранее смочена водой, предшественником активного металлического компонента.
Многократная пропитка означает пропитку смешанным раствором предшественника или предшественников одного или более компонентов за несколько раз или пропитку в несколько раз разными предшественника, и в случае многократной пропитки сушку и обжиг необходимо проводить после каждой отдельной пропитки, чтобы "закрепить" включенные компоненты. Смешанная пропитка означает совместную пропитку предшественниками активного металлического компонента и металла-промотора, при этом между указанными предшественниками не происходит реакции осаждения. Пропитка распылением означает распыление пропиточного раствора на непрерывно вращающуюся подложку посредством пистолета-распылителя таким образом, чтобы пропиточный раствор просто заполнял объем пор подложки до насыщения. Эти способы пропитки могут выбираться надлежащим образом в соответствии с фактическими условиями производства катализатора по настоящей заявке.
В предпочтительном варианте осуществления предшественники активного металлического компонента и металла-промотора представляют собой растворимые соли соответствующих металлов, такие как нитраты, формиаты, оксалаты, лактаты и т.д. В качестве растворителя для образования раствора соли металла для пропитки подложки предпочтительно используется вода, хотя можно использовать и некоторые органические растворители, например, этанол. Пропитку подложки раствором соли металла можно проводить в любой желаемой последовательности или непрерывно несколькими растворами, содержащими одну или более солей металла. Все этапы пропитки или единственный этап пропитки можно провести в несколько ступеней, и порядок пропитки также можно менять. Концентрация раствора выбирается так, чтобы ввести на подложку желаемое количество металла.
Согласно настоящей заявке, подложка, в которую введен активный металлический компонент и, необязательно, металл-промотор, подвергается термообработке на этапе 3), при этом указанная термообработка предпочтительно включает обжиг или комбинацию сушки и обжига. Например, термообработка может включать сушку пропитанной подложки при 80-150°С, более предпочтительно при 80-120°С. Продолжительность сушки можно выбирать надлежащим образом в соответствии с температурой сушки, количеством материала, подлежащего сушке, сушильным оборудованием и т.п., и она может составлять, например, от 6 до 20 часов, пока влагосодержание после сушки не будет влиять на последующий обжиг. Далее, после сушки может следовать обжиг при 150-500°С для удаления кристаллизационной воды из соли или для разложения соли на оксиды, предпочтительно при 300-500°С в течение 1-6 ч. В случае многократных пропиток предпочтительно проводить сушку и обжиг после каждой пропитки.
В настоящей заявке введение активного металлического компонента и металла-промотора не оказывает существенного влияния на микроструктуру катализатора, и, следовательно, полученный катализатор имеет такую же пористую структуру, что и подложка.
В третьем аспекте настоящая заявка предлагает подложку, которая является неорганическим пористым материалом, содержащим алюминий и/или кремний, причем подложка имеет содержание L-кислоты 8 5% или больше от суммарного содержания L-кислоты и В-кислоты.
В предпочтительном варианте осуществления содержание L-кислоты в подложке составляет 88% или более, более предпочтительно 90% или более и особенно предпочтительно 92% или более, от суммарного содержания L-кислоты и В-кислоты.
В предпочтительном варианте осуществления подложка содержит матрицу и легирующий элемент, причем матрица выбрана из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций, и легирующий элемент является неметаллическим элементом, предпочтительно по меньшей мере одним элементом, выбранным из группы, состоящей из неметаллических элементов группы IIIA, группы VA, группы VIA и группы VIIA, за исключением хлора, более предпочтительно по меньшей мере одним элементом, выбранным из группы, состоящей из бора, фтора, фосфора, серы и селена. Более предпочтительно, легирующий элемент в подложке происходит из неметаллического ион-радикала кислоты, и неметаллический ион-радикал кислоты предпочтительно представляет собой по меньшей мере один ион-радикал, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
В предпочтительном варианте осуществления подложка имеет по меньшей мере одну из следующих характеристик:
- доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки превышает 65%, предпочтительно составляет 70-90%, а доля объема пор диаметром меньше 7 нм в объеме пор подложки составляет 0-10%, предпочтительно 0-8%;
- доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет менее 20%, предпочтительно 5-17%, доля объема пор диаметром меньше 9 нм в объеме пор подложки составляет менее 40%, доля объема пор диаметром больше 27 нм в объеме пор подложки составляет менее 5%, предпочтительно 0,5-5%, предпочтительно, доля объема пор диаметром больше или равным 7,5 нм и меньше 9 нм в объеме пор подложки составляет 5-17%, и доля объема пор диаметром больше или равным 9 нм и меньше или равным 27 нм в объеме пор подложки составляет 61-8 9,5%;
- подложка имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, предпочтительно 0,3-0,6 ммоль/г, более предпочтительно 0,3-0,5 ммоль/г;
- содержание оксида алюминия в подложке составляет 65 вес. % или более, предпочтительно 7 0 вес. % или более, более предпочтительно 75 вес. % или более, от полного количества матрицы;
- содержание легирующего элемента составляет 0,05-6 вес. %, предпочтительно 0,05-5 вес. %, более предпочтительно 0,05-4,5 вес. %, особенно предпочтительно 0,07-4 вес. %, от полного количества матрицы;
- подложка имеет удельную поверхность 100-220 м2/г, предпочтительно 105-210 м2/г, более предпочтительно 110-210 м2/г и особенно предпочтительно 120-210 м2/г;
- подложка имеет объем пор 0,4-1,1 мл/г, предпочтительно 0,43-1,1 мл/г, более предпочтительно 0,45-1,1 мл/г и особенно предпочтительно 0,45-1 мл/г; и
- изоэлектрическая точка подложки составляет 3-6, предпочтительно 3,5-5,5.
В четвертом аспекте настоящая заявка предлагает применение катализатора по настоящей заявке или подложки по настоящей заявке для получения органических аминов путем каталитического аминирования.
В пятом аспекте настоящая заявка предлагает способ получения органических аминов, включающий контактирование исходного материала для аминирования и аминирующего реагента с катализатором согласно настоящей заявке в присутствии водорода для реакции аминирования, чтобы получить органический амин.
В предпочтительном варианте осуществления исходный материал для аминирования выбран из группы, состоящей из спиртов, кетонов, аминоспиртов, альдегидов или их комбинаций, предпочтительно он выбирается из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов, С2-20 альдегидов и их смесей. Более предпочтительно, исходный материал для аминирования выбирается из группы, состоящей из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, 1,12-додекандиола, 1,12-додекандиальдегида, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и их смесей.
В настоящей заявке под аминирующим реагентом понимается реагент, способный обеспечить аминогруппу и/или группу амина. Предпочтительно, аминирующий реагент выбран из группы, состоящей из аммиака, первичных аминов, вторичных аминов и их комбинаций, предпочтительно он выбран из группы, состоящей из аммиака, С1-12 первичных аминов, С2-12 вторичных аминов и их смесей, например, представляет собой по меньшей мере одно соединение из алкиламина, циклоалкиламина и аралкиламина, более предпочтительно С1-С4 алкиламина. Более предпочтительно, аминирующий реагент выбран из группы, состоящей из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина, диэтиламина и их смесей.
В предпочтительном варианте осуществления условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-6):(2-35):1, предпочтительно (1-6):(2-33):1, более предпочтительно (1-5):(3-33):1, температура 105-230°С, предпочтительно 110-220°С, более предпочтительно 110-210°С, давление 0,7-25 МПа, предпочтительно 1-25 МПа, более предпочтительно 1-22 МПа, особенно предпочтительно 1-17 МПа, и объемная скорость жидкого исходного материала для аминирования (далее сокращенно часовая объемная скорость) 0,06-1 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления исходный материал для аминирования представляет собой одноатомный спирт и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования предпочтительно (1-4):(2-9):1, более предпочтительно (1-4):(2-8):1, температура 130-210°С, предпочтительно 130-208°С, более предпочтительно 130-200°С, давление 0,8-3,5 МПа, предпочтительно 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления исходный материал для аминирования представляет собой кетон или альдегид и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-6):1, предпочтительно (1-4):(2-5):1, температура 105-180°С, предпочтительно 110-170°С, более предпочтительно 110-160°С, давление 0,7-2,5 МПа, предпочтительно 1-2,5 МПа, более предпочтительно 1-2 МПа и часовая объемная скорость 0,1-1 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления исходный материал для аминирования является аминоспиртом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-23):1, предпочтительно (1-4):(3-20):1, более предпочтительно (1-4):(3-10):1, температура 130-200°С, давление 1-16 МПа, предпочтительно 1-13 МПа, более предпочтительно 1-11 МПа, и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления исходный материал для аминирования является двухатомным спиртом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (0,3-5):(2-35):1, предпочтительно (1-4):(3-35):1, более предпочтительно (1-4):(3-33):1, особенно предпочтительно (1-4):(3-32):1, температура 130-230°С, предпочтительно 130-220°С, более предпочтительно 130-210°С, давление 1-25 МПа, предпочтительно 1-22 МПа, более предпочтительно 1-17 МПа, и часовая объемная скорость 0,1-0,9 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления исходный материал для аминирования представляет собой смесь 1,6-гександиола, циклогексимида и 6-амино-1-гексанола, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (0,3-4):(3-35):1, предпочтительно (1-4):(3-33):1, более предпочтительно (1-4):(3-32):1, температура 130-230°С, предпочтительно 130-220°С, более предпочтительно 130-210°С, давление 1-22 МПа, предпочтительно 1-17 МПа, и часовая объемная скорость 0,1-0,9 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
Первый тип вариантов осуществления
В первом типе вариантов осуществления настоящей заявки предлагается катализатор, выполняющий функцию катализа гидроаминирования спирта для получения органического амина, причем катализатор содержит подложку, а также активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, причем подложка содержит матрицу и легирующий элемент, матрица содержит оксид алюминия и необязательно, дополнительный носитель, причем указанный дополнительный носитель представляет собой по меньшей мере один материал, выбранный из группы, состоящей из оксида кремния, молекулярного сита и диатомита; доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет менее 20%, доля объема пор диаметром меньше 9 нм в объеме пор подложки составляет менее 40%, и доля объема пор диаметром больше 27 нм в объеме пор подложки составляет менее 5%; подложка имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г; подложка имеет содержание L-кислоты 90% или больше от суммарного содержания L-кислоты и В-кислоты; активный металлический компонент представляет собой кобальт и/или никель.
Катализатор по первому типу вариантов осуществления настоящей заявки имеет особую кислотность и пористую структуру, и при его использовании для гидроаминирования спиртов катализатор не только демонстрирует высокую каталитическую активность, но также имеет отличную селективность. Когда катализатор используется для гидроаминирования 1,3-пропандиола, образуется меньше 3-аминопропанола и других примесей; когда катализатор используется для гидроаминирования этанола, образуется меньше метилэтиламина, метилдиэтиламина, этил-н-пропиламина и этил-втор-бутиламина; а когда катализатор используется для гидроаминирования 1,6-гександиола, уменьшается образование тяжелых компонентов и других примесей. При оценке после длительного срока эксплуатации было обнаружено, что катализатор по первому типу вариантов осуществления настоящей заявки имеет стабильные каталитические характеристики, а в результате удерживания кислотности катализатора в подходящем диапазоне улучшаются абсорбционно-десорбционные характеристики катализатора, еще больше облегчается диффузия реакционной системы, повышается скорость реакции, уменьшается отложение углерода и замедляется закупоривание поровых каналов.
В соответствии с первым типом вариантов осуществления настоящей заявки подложка состоит в основном из (легированного) оксида алюминия и может дополнительно содержать (легированный) оксид кремния или подобное, тем самым еще больше улучшая свойства катализатора, такие как тип структуры поровых каналов и стабильность пористой структуры. Предпочтительно, матрица в подложке выбирается из оксида алюминия, легированного по меньшей мере одним из оксида кремния, молекулярного сита и диатомита, и из нелегированного оксида алюминия, и содержание алюмооксидного носителя в матрице составляет 65 вес. % или более, предпочтительно 75 вес. % или более, в расчете на суммарное количество алюмооксидного носителя и указанного дополнительного носителя.
Предпочтительно, легирующий элемент присутствует в подложке в количестве от 0,05 до 3 вес. %, более предпочтительно от 0,08 до 2 вес. % и еще более предпочтительно от 0,1 до 1,5 вес. %, от полного веса матрицы.
Предпочтительно, легирующий элемент в подложке происходит из ион-радикалов кислоты, за исключением хлорид-иона. Поскольку легирующий элемент вводится в процессе получения подложки, легирующий элемент находится в основном в объемной фазе подложки. Ион-радикал кислоты может представлять собой по меньшей мере один радикал, выбранный из неметаллических ион-радикалов кислоты, более предпочтительно по меньшей мере один, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Легирующий элемент предпочтительно представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
Предпочтительно, доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет 5-17%, более предпочтительно 5-10%, доля объема пор диаметром больше или равным 7,5 нм и меньше 9 нм в объеме пор подложки составляет 5-17%, доля объема пор диаметром больше или равным 9 нм и меньше 27 нм в объеме пор подложки составляет 61-8 9,5%, и доля объема пор диаметром больше 27 нм в объеме пор подложки составляет 0,5-5%, более предпочтительно 0,5-3%. Авторы настоящей заявки обнаружили, что катализатор, имеющий структуру поровых каналов, удовлетворяющую вышеуказанным требованиям, обладает лучшими каталитическими характеристиками.
Предпочтительно, подложка имеет адсорбционную емкость по аммиаку от 0,3 до 0,5 ммоль/г.
Предпочтительно, подложка имеет содержание L-кислоты 92-100%, предпочтительно 96-100%, от суммарного содержания L-кислоты и В-кислоты.
Предпочтительно, подложка имеет удельную поверхность 105-220 м2/г.
Предпочтительно, подложка имеет объем пор от 0,4 до 1,1 мл/г.
В соответствии с первым типом вариантов осуществления настоящей заявки активный металлический компонент может присутствовать в количестве от 5 до 42 г, предпочтительно от 10 до 35 г, на 100 г матрицы.
В соответствии с первым типом вариантов осуществления настоящей заявки для улучшения характеристик катализатора, оптимизации соотношения между продуктами реакции и уменьшения нежелательных побочных реакций катализатор может дополнительно содержать металл-промотор, который может представлять собой по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантанидов, предпочтительно по меньшей мере один металл из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се. Предпочтительно, металл-промотор может присутствовать в количестве от 0 до 10 г, предпочтительно от 0,5 до 6 г, на 100 г матрицы.
В первом типе вариантов осуществления настоящей заявки предлагается также способ получения органических аминов, включающий: контактирование исходного материала для аминирования и аминирующего реагента с вышеописанным катализатором в присутствии водорода для реакции аминирования.
В соответствии с первым типом вариантов осуществления настоящей заявки исходный материал для аминирования или аминирующий реагент могут выбираться как описано выше, и подробное описание этого здесь для краткости опущено.
В соответствии с первым типом вариантов осуществления настоящей заявки условия аминирования могут включать: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 110-220°С, давление 1-25 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-200°С, давление 1-3,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования является кетоном или альдегидом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 110-180°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования является аминоспиртом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-20):1, температура 130-200°С, давление 1-11 МПа, и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой двухатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 130-220°С, давление 1-25 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола (сокращенно называемого аминогексанолом), и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-35):1, температура 130-200°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Второй тип вариантов осуществления
Во втором типе вариантов осуществления настоящей заявки предлагается катализатор, функцией которого является катализ образования аминов из спиртов, причем катализатор содержит подложку, а также активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, подложка представляет собой по меньшей мере один материал, выбранный из группы, состоящей из легированного оксида алюминия, легированного оксида кремния, легированного молекулярного сита и легированного диатомита; подложка имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г; подложка имеет содержание L-кислоты не менее 88% от суммарного содержания L-кислоты и В-кислоты; активный металлический компонент представляет собой кобальт и/или никель, и размер зерна активного металлического компонента в катализаторе меньше 10 нм.
Катализатор по второму типу вариантов осуществления настоящей заявки имеет улучшенную кислотность подложки, высокую дисперсность активных металлических компонентов и размер зерна меньше 10 нм, и демонстрирует повышенную каталитическую активность и, в то же время, проявляет более высокую селективность при использовании в гидроаминировании спиртов. Когда катализатор используется в гидроаминирование этаноламина, образуется меньше компонентов, отличных от этилендиамина, таких как пиперазин и т.п., а когда катализатор используется для гидроаминирования 1,6-гександиола, образуется меньше тяжелых компонентов и других примесей. При оценке после длительного срока эксплуатации было обнаружено, что катализатор по второму типу вариантов осуществления настоящей заявки имеет стабильные каталитические характеристики, а благодаря удерживанию кислотности катализатора в подходящем диапазоне улучшаются абсорбционно-десорбционные характеристики катализатора, так что облегчается диффузия реакционной системы, повышается скорость реакции, уменьшается отложение углерода и замедляется закупоривание поровых каналов.
В соответствии со вторым типом вариантов осуществления настоящей заявки подложка содержит матрицу, выбранную из группы, состоящей из оксида алюминия, оксида кремния, молекулярных сит, диатомита и подобного, а также содержит легирующий элемент. Предпочтительно, легирующий элемент присутствует в подложке в количестве от 0,05 до 5 вес. %, более предпочтительно от 0,08 до 4 вес. %, от полного веса матрицы.
Предпочтительно, легирующий элемент в подложке происходит из ион-радикалов кислоты, за исключением хлорид-иона. Легирующий элемент предпочтительно представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена. Поскольку легирующий элемент вводится во время подготовки подложки, указанный легирующий элемент присутствует в объемной фазе подложки. Более предпочтительно, ион-радикал кислоты может представлять собой по меньшей мере один компонент, выбранный из группы, состоящей из неметаллических ион-радикалов кислот, таких как по меньшей мере один, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
Предпочтительно, подложка имеет адсорбционную емкость по аммиаку от 0,3 до 0,5 ммоль/г (например, 0,3 ммоль/г, 0,32 ммоль/г, 0,35 ммоль/г, 0,38 ммоль/г, 0,4 ммоль/г, 0,4 5 ммоль/г, 0,4 8 ммоль/г, 0,5 ммоль/г, или значение между любыми двумя из них).
Предпочтительно, подложка имеет содержание L-кислоты 90-100% от суммарного содержания L-кислоты и В-кислоты.
Предпочтительно, активный металлический компонент катализатора имеет размер зерна от 3 до 8 нм.
Предпочтительно, подложка имеет удельную поверхность 100-200 м2/г.
Предпочтительно, подложка имеет объем пор от 0,45 до 1 мл/г.
Предпочтительно, подложка имеет изоэлектрическую точку от 3 до 6, предпочтительно от 3,5 до 5,5.
Предпочтительно, содержание активного металлического компонента в подложке может составлять от 7 до 45 г, предпочтительно от 12 до 38 г, на 100 г матрицы.
В соответствии со вторым типом вариантов осуществления настоящей заявки катализатор может дополнительно содержать металл-промотор для улучшения характеристик катализатора, для оптимизации соотношения между продуктами реакции и для уменьшения нежелательных побочных реакций. Металл-промотор может представлять собой по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантанидов, предпочтительно по меньшей мере один металл из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се. Предпочтительно, металл-промотор может присутствовать в подложке в количестве от 0 до 10 г, предпочтительно от 0,5 до 6 г, на 100 г матрицы. Более предпочтительно, размер зерна металла-промотора меньше 10 нм.
Во втором типе вариантов осуществления настоящей заявки предлагается также способ получения органических аминов, включающий: контактирование исходного материала для аминирования и аминирующего реагента с вышеописанным катализатором в присутствии водорода для реакции аминирования.
В соответствии со вторым типом вариантов осуществления настоящей заявки исходный материал для аминирования или аминирующий реагент могут выбираться, как описано выше, и подробное описание этого здесь для краткости опущено.
В соответствии со вторым типом вариантов осуществления настоящей заявки условия аминирования могут включать: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(3-33):1, температура 110-210°С, давление 1-22 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-200°С, давление 0,8-2,5 МПа, и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой кетон или альдегид, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-6):1, температура 110-170°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой аминоспирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-10):1, температура 130-200°С, давление 1-11 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования является смесью 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола (сокращенно называемого аминогексанолом) или двухатомного спирта, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-33):1, температура 130-210°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Третий тип вариантов осуществления
В третьем типе вариантов осуществления настоящей заявки предлагается катализатор, функцией которого является получение аминов путем гидроаминирования спиртов, причем катализатор содержит подложку, а также активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, подложка содержит матрицу и легирующий элемент, матрица содержит оксид алюминия и, необязательно, дополнительный носитель, выбранный из оксида кремния и/или молекулярных сит, легирующий элемент представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена; доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет более 65%; подложка имеет содержание L-кислоты 85% или более, от суммарного содержания L-кислоты и В-кислоты; активный металлический компонент представляет собой кобальт и/или никель.
Благодаря легированию подложки бором, фтором, фосфором, серой и селеном, катализатор по третьему типу вариантов осуществления настоящей заявки демонстрирует улучшенные каталитические характеристики, например, для гидроаминирования этанола, и образуется меньше метилэтиламина, метилдиэтиламина, этил-н-пропиламина, этил-втор-бутиламина. Когда катализатор используется для гидроаминирования 1,6-гександиола, образуется меньше тяжелых компонентов и других примесей, и срок службы катализатора увеличивается.
Кроме того, катализатор по третьему типу вариантов осуществления настоящей заявки имеет особую структуру поровых каналов, и при использовании для гидроаминирования спиртов катализатор демонстрирует высокую каталитическую активность и имеет отличные селективность и стабильность, тем самым уменьшая осаждение углерода в поровых каналах и эффективно предотвращая закупоривание поровых каналов катализатора.
В соответствии с третьим типом вариантов осуществления настоящей заявки, подложка состоит в основном из легированного оксида алюминия и может дополнительно включать (быть легированной) оксид кремния или подобное, в результате дополнительно улучшаются структура поровых каналов и кислотно-основные свойства подложки катализатора. Предпочтительно, матрица в подложке представляет собой оксид алюминия, легированный оксидом кремния и/или молекулярным ситом, и содержание алюмооксидного носителя в матрице подложки составляет не менее 70 вес. %, предпочтительно 75-100 вес. %.
Предпочтительно, легирующий элемент присутствует в подложке в количестве от 0,05 до 6 вес. %, более предпочтительно от 0,08 до 4 вес. %, от полного веса матрицы.
Предпочтительно, легирующий элемент вводится в подложку в виде по меньшей мере одного элемента, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Поскольку легирующий элемент вводится во время подготовки подложки, легирующий элемент в основном присутствует в объемной фазе подложки.
Предпочтительно, доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет более 65%. Более предпочтительно, доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет 7 0-90%. Более предпочтительно, доля объема пор диаметром меньше 7 нм в объеме пор подложки составляет 0-10%; доля объема пор диаметром больше 27 нм в объеме пор подложки составляет 18-32%.
Предпочтительно, подложка имеет содержание L-кислоты 85-98%, от суммарного содержания L-кислоты и В-кислоты.
Предпочтительно, подложка имеет удельную поверхность 120-210 м2/г.
Предпочтительно, подложка имеет объем пор от 0,43 до 1,1 мл/г.
Предпочтительно, активный металлический компонент может присутствовать в количестве от 8 до 44 г, предпочтительно от 12 до 37 г, на 100 г матрицы.
Предпочтительно, металл-промотор может присутствовать в количестве от 0 до 10 г, предпочтительно от 0,5 до 6 г, на 100 г матрицы.
В соответствии с третьим типом вариантов осуществления настоящей заявки, катализатор может дополнительно содержать металл-промотор для улучшения характеристик катализатора, для оптимизации соотношения между продуктами реакции и для уменьшения нежелательных побочных реакций. Металл-промотор может представлять собой по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантанидов, предпочтительно по меньшей мере один металл из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се.
В третьем типе вариантов осуществления настоящей заявки предлагается также способ получения органических аминов, включающий: контактирование исходного материала для аминирования и аминирующего реагента с вышеописанным катализатором в присутствии водорода для реакции аминирования.
В соответствии с третьим типом вариантов осуществления настоящей заявки исходный материал для аминирования и аминирующий реагент могут выбираться, как описано выше, и подробное описание этого здесь для краткости опущено.
В соответствии с третьим типом вариантов осуществления настоящей заявки, условия аминирования могут включать: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-6):(2-32):1, температура 105-210°С, давление 1-17 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-9):1, температура 130-208°С, давление of 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3-ч).
Предпочтительно, исходный материал для аминирования представляет собой кетон или альдегид, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 105-160°С, давление 1-2 МПа и часовая объемная скорость 0,1-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой аминоспирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-20):1, температура 130-200°С, давление 1-13 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола (для краткости называемого аминогексанолом) или двухатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-32):1, температура 130-210°С, давление 1-17 МПа и часовая объемная скорость 0,1-0,9 м3/(м3⋅ч).
Четвертый тип вариантов осуществления
В четвертом типе вариантов осуществления настоящей заявки предлагается катализатор, функцией которого является катализ получения аминов из спиртов, при этом катализатор содержит подложку, а также активный металлический компонент и металл-промотор, нанесенные на подложку, причем подложка имеет содержание L-кислоты 85% или более от суммарного содержания L-кислоты и В-кислоты, и активный металлический компонент представляет собой кобальт и/или никель; металл-промотор является комбинацией по меньшей мере одного металла группы IIA, по меньшей мере одного металла группы IIB и по меньшей мере одного металла группы VA.
Катализатор по четвертому типу вариантов осуществления настоящей заявки содержит особый металл-промотор, имеет высокую каталитическую активность и одновременно обладает высокой селективностью и производит мало побочных продуктов.
Предпочтительно, подложка имеет содержание L-кислоты 88% или более, более предпочтительно 90% или более, особенно предпочтительно 92% или более, от суммарного содержания L-кислоты и В-кислоты.
Предпочтительно, подложка содержит матрицу и легирующий элемент, матрица содержит оксид алюминия и, необязательно, дополнительный носитель, содержащий оксид кремния и/или молекулярные сита; легирующий элемент представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена; доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет более 65%.
В соответствии с четвертым типом вариантов осуществления настоящей заявки, подложка состоит в основном из легированного оксида алюминия и может дополнительно включать (быть легированной) оксид кремния и подобное, в результате дополнительно улучшается структура поровых каналов катализатора, тем самым позволяя легкую диффузию реагентов и продуктов в поровых каналах и обеспечивая более стабильную структуру пор. Предпочтительно, содержание оксида алюминия в матрице подложки составляет не менее 70 вес. %, предпочтительно 75-100 вес. %, от суммарного количества оксида алюминия и дополнительного носителя.
Предпочтительно, легирующий элемент присутствует в подложке в количестве от 0,05 до 4,5 вес. %, более предпочтительно от 0,07 до 2,8 вес. %, от полного веса матрицы.
Предпочтительно, легирующий элемент вводится в подложку в виде по меньшей мере одного соединения, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Легирующий элемент находится в предшественнике оксида алюминия. Если легирующий элемент вводится во время подготовки предшественника, он будет заключен в кристаллической фазе предшественника, а если легирующий элемент вводится после подготовки подложки, он будет находиться в основном в объемной фазе подложки.
Доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки предпочтительно составляет от 70% до 90%. Предпочтительно, доля объема пор диаметром меньше 7 нм в объеме пор подложки составляет 0-8%. Предпочтительно, доля объема пор диаметром больше 27 нм в объеме пор подложки составляет 15-35%, более предпочтительно 20-29%.
Предпочтительно, подложка имеет удельную поверхность 110-210 м2/г.
Предпочтительно, подложка имеет объем пор от 0,45 до 1,1 мл/г.
Предпочтительно, активный металлический компонент может присутствовать в количестве 8-45 г, предпочтительно 15-38 г (например, в любом количестве из 15, 20, 25, 28, 30, 32, 35, 37, 38 г или в значении между любыми двумя из них) на 100 г матрицы.
Предпочтительно, металл-промотор может присутствовать в количестве 0,1-10 г, предпочтительно 0,5-6 г (например, в любом количестве из 0,5, 1, 2, 3, 3,5, 3,8, 4, 4,2, 4,5, 4,8, 5, 5,2, 5,5, 6 г или в количестве между любыми из этих двух значений) на 100 г матрицы.
В соответствии с четвертым типом вариантов осуществления настоящей заявки, катализатор может содержать вышеописанный металл-промотор для улучшения характеристик катализатора, для оптимизации соотношения между продуктами реакции и для уменьшения нежелательных побочных реакций. Весовое отношение металла группы IIA к металлу группы IIB и металлу группы VA в металле-промоторе предпочтительно составляет (0,1-10):(0,1-10):1, более предпочтительно (0,2-8): (0,2-8):1. Предпочтительно, металл группы IIA представляет собой по меньшей мере один металл, выбранный из группы, состоящей из магния, кальция и бария. Предпочтительно, металл группы IIB представляет собой цинк. Предпочтительно, металл группы VA представляет собой висмут.
В четвертом типе вариантов осуществления настоящей заявки использование подложки с особой структурой пор и легирующим элементом позволяет катализатору демонстрировать высокую каталитическую активность и одновременно иметь повышенную селективность при использовании для гидроаминирования спиртов. Когда катализатор используется для гидроаминирования этанола, образуется меньше метилэтиламина, метилдиэтиламина, этил-н-пропиламина, этил-втор-бутиламина. При использовании
катализатора для гидроаминирования 1, 6-гександиола образуется меньше тяжелых компонентов и других примесей. При оценке после длительного срока эксплуатации было обнаружено, что катализатор имеет стабильные каталитические характеристики, облегчается диффузия реакционной системы, повышается скорость реакции, уменьшается отложение углерода и замедляется закупоривание поровых каналов.
В соответствии с четвертым типом вариантов осуществления настоящей заявки, подложку можно приготовить, используя известные способы, позволяющие предоставить легирующий элемент, поровую структуру, удовлетворяющую вышеуказанным диапазонам, и т.п., и специалист в состоянии получить подложку с легирующим элементом и структурой пор, удовлетворяющей вышеуказанным диапазонам. Предпочтительно, подложку получают способом, включающим этапы:
(1) последовательное формование, первая сушка и первый обжиг смеси, содержащей легирующий элемент, предшественник оксида алюминия и, необязательно, предшественник дополнительного носителя, причем указанный предшественник дополнительного носителя содержит предшественник оксида кремния (например, силиказоль) и/или предшественник молекулярного сита (например, ZSM-5);
(2) смешение продукта первого обжига с раствором предшественника металла группы IIA, а затем проведение второй сушки и второго обжига. Процесс формования может включать замешивание, раскатку или расслаивание и т.д.
Специалисту в данной области техники понятно, что в описанном выше способе получения подложки, когда исходный материал, обеспечивающий предшественник носителя, уже содержит желаемое количество легирующего элемента, формование может быть выполнено просто, используя такой исходный материал, а когда исходный материал, обеспечивающий предшественник носителя, не содержит легирующего элемента или содержание легирующего элемента низкое (недостаточное), можно ввести дополнительный легирующий элемент.
В описанном выше способе получения подложки легирующий элемент может быть включен в исходный материал, обеспечивающий предшественник оксида алюминия, или можно напрямую использовать предшественник оксида алюминия и/или предшественник дополнительного носителя, модифицированный легирующим элементом, такой предшественник оксида алюминия или предшественник дополнительного носителя, модифицированный легирующим элементом, может быть приобретен на рынке или получен обычным способом, подробное описание которого здесь для краткости опущено.
В описанном выше способе получения подложки специалист может определить количество исходного материала (например, модификатора подложки) для компонента (например, легирующего элемента), исходя из количества указанного компонента в конечной подложке, поэтому количества некоторых исходных материалов здесь не приводятся.
В описанном выше способе получения подложки предшественник оксида алюминия предпочтительно представляет собой псевдобемит. Удельная поверхность псевдобемита предпочтительно составляет 250-330 м2/г. Объем пор псевдобемита предпочтительно составляет от 0,5 до 1,1. Псевдобемит может быть получен по меньшей мере одним способом из способа карбонизации, способа гидролиза алюминийорганических соединений, алюминийсульфатного способа и азотнокислотного способа, особенно предпочтительно его получают алюминийсульфатным способом. Используя псевдобемит с особой поровой структурой, можно получить катализатор с улучшенными характеристиками.
В описанном выше способе получения подложки условия первой сушки и второй сушки могут независимо друг от друга включать: температура 80-150°С и продолжительность сушки 6-20 ч, предпочтительно температура 100-120°С и продолжительность сушки 8-15 ч.
В описанном выше способе получения подложки условия первого обжига могут включать: температура 500-650°С и продолжительность обжига 2-20 ч, предпочтительно температура 520-620°С и продолжительность обжига 4-8 ч.
В описанном выше способе получения подложки условия второго обжига могут включать: температура 800-1100°С и продолжительность обжига 2-20 ч, предпочтительно температура 800-980°С и продолжительность обжига 5-10 ч.
В четвертом типе вариантов осуществления настоящей заявки предлагается также способ получения органических аминов, включающий: контактирование исходного материала для аминирования и аминирующего реагента с вышеописанным катализатором в присутствии водорода для реакции аминирования.
В соответствии с четвертым типом вариантов осуществления настоящей заявки, исходный материал для аминирования или аминирующий реагент могут выбираться, как описано выше, поэтому подробное описание этого для краткости опущено.
Согласно четвертому типу вариантов осуществления настоящей заявки, условия аминирования могут включать: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 110-230°С, давление 0,7-22 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-210°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования является кетоном или альдегидом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 110-180°С, давление 0,7-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования является аминоспиртом, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-23):1, температура 130-200°С, давление 1-16 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, циклогексимида и 6-амино-1-гексанола или двухатомного спирта, и условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-35):1, температура 130-230°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления настоящая заявка предлагает следующие технические решения:
А1. Катализатор, функцией которого является катализ гидроаминирования спиртов для получения органического амина, содержащий подложку, активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, причем подложка содержит матрицу и легирующий элемент, матрица содержит алюмооксидный носитель и, необязательно, дополнительный носитель, и указанная дополнительный носитель представляет собой по меньшей мере один материал, выбранный из оксида кремния, молекулярного сита и диатомита; доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет менее 20%, доля объема пор диаметром меньше 9 нм в объеме пор подложки составляет менее 40%, и доля объема пор диаметром больше 27 нм в объеме пор подложки составляет менее 5%; подложка имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г; подложка имеет содержание L-кислоты 90% или больше от суммарного содержания L-кислоты и В-кислоты; активный металлический компонент представляет собой кобальт и/или никель.
А2. Катализатор по пункту А1, причем содержание алюмооксидной подложки в матрице составляет 65 вес. % или более, предпочтительно 75 вес. % или более, от суммарного количества алюмооксидной подложки и указанной дополнительной подложки;
и/или содержание легирующего элемента составляет 0,05-3 вес. %, предпочтительно 0,08-2 вес. %, от полного веса матрицы;
и/или легирующий элемент происходит из ионов-радикалов кислот, отличных от хлорид-иона; и ион-радикал кислоты представляет собой по меньшей мере один ион, выбранный из группы, состоящей из неметаллических ион-радикалов кислот, предпочтительно по меньшей мере один, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или, доля объема пор диаметром меньше 7,5 нм в объеме пор подложки составляет 5-17%, доля объема пор диаметром больше или равным 7,5 нм и меньшим 9 нм в объеме пор подложки составляет 5-17%, доля объема пор диаметром больше или равным 9 нм и меньше или равным 27 нм в объеме пор подложки составляет 61-89,5%, и доля объема пор диаметром больше 27 нм в объеме пор подложки составляет 0,5-5%;
и/или подложка имеет адсорбционную емкость по аммиаку 0,3-0,5 ммоль/г;
и/или подложка имеет содержание L-кислоты 92-100%, от суммарного содержания L-кислоты и В-кислоты;
и/или подложка имеет удельную поверхность 105-220 м2/г; и/или подложка имеет объем пор 0,4-1,1 мл/г;
и/или содержание активного металлического компонента составляет 5-42 г, предпочтительно 10-35 г, на 100 г матрицы.
A3. Катализатор по пункту А1 или А2, причем подложка приготовлена способом, включающим этапы: последовательное формование, сушка и обжиг смеси модификатора подложки, псевдобемита и, необязательно, источника дополнительного носителя, причем указанный источник дополнительного носителя представляет собой по меньшей мере одно из предшественника оксида кремния, предшественника молекулярного сита и предшественника диатомита, и температура обжига составляет 800-1050°С.
А4. Катализатор по пункту A3, причем модификатор подложки представляет собой по меньшей мере одно соединение, выбранное из группы, состоящей из неметаллических ион-радикалов кислот, предпочтительно по меньшей мере одно, выбранное из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
А5. Катализатор по пункту A3 или А4, причем модификатор подложки представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из борной кислоты, бората никеля, бората кобальта, бората калия, бората аммония, фторида калия, фторида кобальта, фторида никеля, фтористоводородной кислоты, фторида аммония, фосфорной кислоты, фосфата алюминия, трикалий фосфата, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, фосфата аммония, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, сульфата калия, сульфата магния, фосфата стронция, сульфата стронция и селеновой кислоты;
и/или псевдобемит имеет удельную поверхность 255-360 м2/г и объем пор 0,75-1,3 мл/г.
А6. Катализатор по любому из пунктов А3-А5, причем условия сушки включают: температура 80-150°С и продолжительность сушки 6-20 ч;
и/или условия обжига включают: температура 800-1050°С и продолжительность обжига 2-20 ч.
А7. Способ получения катализатора по любому из пунктов А1-А6, включающий: введение активного металлического компонента и, необязательно, металла-промотора на подложку.
А8. Подложка, какая определена в любом из пунктов А1-А6.
А9. Применение катализатора по любому из пунктов А1-А6, или способа по пункту А7, или подложки по пункту А8 при получении органических аминов путем аминирования.
А10. Способ получения органического амина, включающий: контактирование исходного материала для аминирования и аминирующего реагента с катализатором по любому из пунктов А1-А6 в присутствии водорода для реакции аминирования;
или, альтернативно, способ включает: просеивание катализатора, содержащего подложку, какая определена в любом из пунктов А1-А6, и контактирование исходного материала для аминирования и аминирующего реагента с просеянным катализатором в присутствии водорода для реакции аминирования.
A11. Способ по пункту А10, причем условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 110-220°С, давление 1-25 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере одно из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или аминирующий реагент представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере одно из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
А12. Способ по пункту A11, причем, когда исходный материал для аминирования является одноатомным спиртом, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-200°С, давление 1-3,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 110-180°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-20):1, температура 130-200°С, давление 1-11 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой двухатомный спирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 130-220°С, давление 1-25 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-35):1, температура 130-200°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
81. Катализатор, несущий функцию катализа получения аминов из спиртов, содержащий подложку, активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, причем подложка представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из легированного оксида алюминия, легированного оксида кремния, легированного молекулярного сита и легированного диатомита; подложка имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г; подложка имеет содержание L-кислоты 88% или более от суммарного содержания L-кислоты и В-кислоты; активный металлический компонент представляет собой кобальт и/или никель, и размер зерна активного металлического компонента в катализаторе меньше 10 нм.
82. Катализатор по пункту В1, причем подложка имеет матрицу, выбранную из группы, состоящей из оксида алюминия, оксида кремния, молекулярных сит и диатомита, а легирующий элемент присутствует в количестве от 0,05 до 5 вес. %, предпочтительно от 0,08 до 4 вес. %, от полного веса матрицы;
и/или легирующий элемент в подложке происходит из ион-радикалов кислоты, за исключением хлорид-иона; ион-радикал кислоты представляет собой по меньшей мере один ион, выбранный из группы, состоящей из неметаллических ион-радикалов кислот, предпочтительно по меньшей мере один, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или подложка имеет адсорбционную емкость по аммиаку 0,3-0,5 ммоль/г;
и/или подложка имеет содержание L-кислоты 90-100% от суммарного содержания L-кислоты и В-кислоты;
и/или размер зерна активного металлического компонента в катализаторе составляет 3-8 нм;
и/или подложка имеет удельную поверхность 100-200 м2/г;
и/или подложка имеет объем пор 0,45-1 мл/г;
и/или подложка имеет изоэлектрическую точку 3-6, предпочтительно 3,5-5,5;
и/или содержание активного металлического компонента составляет от 7 до 45 г, предпочтительно от 12 до 38 г, на 100 г матрицы.
В3. Катализатор по пункту В1 или В2, причем подложка приготовлена способом, включающим этапы: последовательное формование, сушка и обжиг смеси, содержащей легирующий ион и предшественник матрицы, причем легирующий ион обеспечивается модификатором подложки, и модификатор подложки представляет собой по меньшей мере одно из неорганических кислот, содержащих легирующий ион, и неорганических солей, содержащих легирующий ион, и температура обжига составляет не менее 800°С.
В4. Катализатор по пункту В3, причем неорганическая кислота представляет собой по меньшей мере одну кислоту, выбранную из неорганических кислот, содержащих неметаллический ион-радикал кислоты, неорганическая соль представляет собой по меньшей мере одну соль, выбранную из неорганических солей, содержащих неметаллический ион-радикал кислоты, и модификатор подложки предпочтительно представляет собой по меньшей мере одно вещество, выбранное из борной кислоты, бората калия, бората магния, фтористоводородной кислоты, фторида калия, фторида магния, фосфорной кислоты, фосфата калия, фосфата магния, серной кислоты, сульфата калия, сульфата магния и селеновой кислоты.
85. Катализатор по пункту В3 или В4, причем предшественник матрицы представляет собой псевдобемит, псевдобемит имеет удельную поверхность 260-400 м2/г и объем пор 0,75-1,2 мл/г.
86. Катализатор по любому из пунктов В3-В5, причем условия сушки включают: температура 80-150°С и продолжительность сушки 6-20 ч;
и/или условия обжига включают: температура 820-1120°С и продолжительность обжига 2-20 ч.
87. Способ получения катализатора по любому из пунктов В1-В6, включающий: пропитка подложки пропиточным раствором, содержащим предшественник активного металлического компонента и, необязательно, предшественник металла-промотора, чтобы нанести активный металлический компонент и факультативный металл-промотор на подложку, причем пропиточный раствор имеет рН в интервале от 3,5 до 5,5.
88. Подложка, какая определена в любом из пунктов В1-В6.
89. Применение катализатора по любому из пунктов В1-В6, или способа по пункту В7, или подложки по пункту В8 для получения органических аминов путем аминирования.
В10. Способ получения органического амина, включающий: контактирование исходного материала для аминирования и аминирующего реагента с катализатором по любому из пунктов В1-В6 в присутствии водорода для реакции аминирования;
или, альтернативно, просеивание катализатора, содержащего подложку, какая определена в любом из пунктов B1-В6, и контактирование исходного материала для аминирования и аминирующего реагента с просеянным катализатором в присутствии водорода для реакции аминирования.
В11. Способ по пункту В10, причем условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5): (3-33):1, температура 110-210°С, давление 1-22 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере одно из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола, и 1,12-додекандиола;
и/или аминирующий реагент представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере одно из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
В12. Способ по пункту В11, причем, когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-200°С, давление 0,8-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-6):1, температура 110-170°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-10):1, температура 130-200°С, давление 1-11 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомного спирта, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-33):1, температура 130-210°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
С1. Катализатор, несущий функцию катализа получения аминов путем гидроаминирования спиртов, содержащий подложку, активный металлический компонент и, необязательно, металл-промотор, нанесенные на подложку, причем подложка содержит матрицу и легирующий элемент, матрица содержит алюмооксидный носитель и, необязательно, дополнительный носитель, причем указанный дополнительный носитель выбран из оксида кремния и/или молекулярного сита; легирующий элемент представляет собой по меньшей мере один элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена; доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки превышает 65%; активный металлический компонент является кобальтом и/или никелем.
С2. Катализатор по пункту С1, причем содержание алюмооксидного носителя в матрице подложки составляет не менее 70 вес. %, предпочтительно от 75 до 100 вес. %;
и/или содержание легирующего элемента в подложке составляет от 0,05 до 6 вес. %, предпочтительно от 0,08 до 4 вес. %, от полного веса матрицы;
и/или легирующий элемент вводится в виде по меньшей мере одного соединения, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки превышает 65%; предпочтительно, доля объема пор диаметром 7-27 нм в объеме пор подложки составляет 70-90%, и доля объема пор диаметром меньше 7 нм в объеме пор подложки составляет 0-10%;
и/или подложка имеет содержание L-кислоты 85% или более, предпочтительно 85-98%, от суммарного содержания L-кислоты и В-кислоты;
и/или подложка имеет удельную поверхность 120-210 м2/г; и/или подложка имеет объем пор 0,43-1,1 мл/г;
и/или содержание активного металлического компонента составляет 8-44 г, предпочтительно 12-37 г, на 100 г матрицы.
С3. Катализатор по пункту С1 или С2, причем подложка приготовлена способом, включающим: последовательное формование, сушка и обжиг смеси, содержащей легирующий элемент и предшественник подложки, причем предшественник подложки выбран из предшественников оксида алюминия и, необязательно, предшественников дополнительной матрицы, и предшественник дополнительной матрицы выбран из предшественников оксида кремния и/или предшественников молекулярных сит.
С4. Катализатор по пункту С3, причем легирующий элемент обеспечивается по меньшей мере одним соединением, выбранным из соединений, содержащих неметаллический ион-радикал кислоты, предпочтительно по меньшей мере одним, выбранным из соединений, содержащих борат-ион, соединений, содержащих фторид-ион, соединений, содержащих фосфат-ион, соединений, содержащих сульфат-ион, и соединений, содержащих селенат-ион.
С5. Катализатор по пункту С3 или С4, причем предшественник оксида алюминия представляет собой псевдобемит, и псевдобемит имеет удельную поверхность 255-340 м2/г и объем пор 0,78-1,25 мл/г.
С6. Катализатор по любому из пунктов С3-С5, причем условия сушки включают: температура 80-150°С и продолжительность сушки 6-20 ч;
и/или условия обжига включают: температура 700-1100°С и продолжительность обжига 2-20 ч.
С7. Способ получения катализатора по любому из пунктов С1-С6, включающий: нанесение активного металлического компонента и, необязательно, металла-промотора на подложку.
С8. Подложка, какая определена в любом из пунктов С1-С6.
С9. Применение катализатора по любому из пунктов С1-С6, или способа по пункту С7, или подложки по пункту С8 для получения органических аминов путем аминирования.
С10. Способ получения органического амина, включающий: контактирование исходного материала для аминирования и аминирующего реагента с катализатором по любому из пунктов С1-С6 в присутствии водорода для реакции аминирования;
или, альтернативно, просеивание катализатора, содержащего подложку, какая определена в любом из пунктов С1-С6, и контактирование исходного материала для аминирования и аминирующего реагента с просеянным катализатором в присутствии водорода для реакции аминирования.
С11. Способ по пункту С10, причем условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-6):(2-32):1, температура 105-210°С, давление 1-17 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере одно из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или аминирующий реагент представляет собой по меньшей мере одно вещество, выбранное из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере одно из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
С12. Способ по пункту С11, причем, когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-9):1, температура 130-208°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 105-160°С, давление 1-2 МПа и часовая объемная скорость 0,1-1 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-20):1, температура 130-200°С, давление 1-13 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомный спирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-32):1, температура 130-210°С, давление 1-17 МПа и часовая объемная скорость 0,1-0,9 м3/(м3⋅ч).
D1. Катализатор, функцией которого является катализ образования аминов из спиртов, содержащий подложку, активный металлический компонент и металл-промотор, нанесенные на подложку, причем активный металлический компонент представляет собой кобальт и/или никель; металл-промотор представляет собой комбинацию по меньшей мере одного металла группы IIA, по меньшей мере одного металла группы IIB и по меньшей мере одного металла группы VA.
D2. Катализатор по пункту D1, причем подложка содержит матрицу и легирующий элемент, матрица содержит алюмооксидный носитель и, необязательно, дополнительный носитель, и дополнительный носитель содержит оксид кремния и/или молекулярные сита; легирующий элемент является по меньшей мере одним элементом, выбранным из группы, состоящей из бора, фтора, фосфора, серы и селена; доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки превышает 65%.
D3. Катализатор согласно пункту D1 или D2, причем содержание алюмооксидного носителя в матрице подложки составляет 70 вес. % или более, предпочтительно 75-100 вес. %, от общего количества алюмооксидного носителя и указанного дополнительного носителя;
и/или содержание легирующего элемента в подложке составляет от 0,05 до 4,5 вес. %, предпочтительно от 0,07 до 2,8 вес. %, от полного веса матрицы;
и/или легирующий элемент введен в подложку в виде по меньшей мере одного соединения, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или доля объема пор диаметром в интервале 7-27 нм в объеме пор подложки составляет 70-90%, а доля объема пор диаметром менее 7 нм в объеме пор подложки составляет 0-8%;
и/или подложка имеет удельную поверхность 110-210 м2/г;
и/или подложка имеет объем пор 0,45-1,1 мл/г;
и/или содержание указанного активного металлического компонента составляет 8-45 г, предпочтительно 15-38 г, на 100 г матрицы;
и/или металл-промотор присутствует в количестве от 0,1 до 10 г, предпочтительно от 0,5 до 6 г, на 100 г матрицы;
и/или весовое соотношение между металлом группы IIA, металлом группы IIB и металлом группы VA в металле-промоторе составляет (0,1-10):(0,1-10):1, предпочтительно (0,2-8):(0,2-8):1;
и/или металл группы IIA представляет собой по меньшей мере один металл, выбранный из группы, состоящей из магния, кальция и бария;
и/или металл группы НВ представляет собой цинк;
и/или металл группы VA представляет собой висмут.
D4. Катализатор согласно пункту D2 или D3, причем подложка получена способом, включающим этапы:
(1) последовательное формование, первая сушка и первый обжиг смеси, содержащей легирующий элемент, предшественник оксида алюминия и, необязательно, предшественник дополнительного носителя, причем указанный предшественник дополнительного носителя содержит предшественник оксида кремния и/или предшественник молекулярного сита;
(2) смешение продукта первого обжига с раствором предшественника металла группы IIA, а затем проведение второй сушки и второго обжига.
D5. Катализатор согласно пункту D4, причем предшественник оксида алюминия представляет собой псевдобемит, и псевдобемит имеет удельную поверхность 250-330 м2/г и объем пор 0,5-1,1 мл/г;
и/или условия первого обжига включают: температура 500-650°С и продолжительность обжига 2-20 ч;
и/или условия второго обжига включают: температура 800-1100°С и продолжительность обжига 2-20 ч.
D6. Способ получения катализатор по любому из пунктов D1-D5, включающий:
(1) последовательное формование, первую сушку и первый обжиг смеси, содержащей легирующий элемент, предшественник оксида алюминия и, необязательно, предшественник дополнительного носителя, причем указанный предшественник дополнительного носителя содержит предшественник оксида кремния и/или предшественник молекулярного сита;
(2) смешение продукта первого обжига с раствором предшественника металла группы IIA, а затем проведение второй сушки и второго обжига;
(3) нанесение по меньшей мере одного металла группы IIB, по меньшей мере одного металла группы VA и активного металлического компонента на продукт второго обжига.
D7. Подложка, определенная в любом из пунктов D1-D5.
D8. Применение катализатора по любому из пунктов D1-D5, или способа по пункту D6, или подложки по пункту D7 для получения органических аминов путем аминирования.
D9. Способ получения органических аминов, включающий следующие этапы: контактирование исходного материала для аминирования и аминирующего реагента с катализатором по одному з пунктов D1-D5 в присутствии водорода для реакции аминирования;
или, альтернативно, способ включает: просеивание катализатора, содержащего подложку по любому из пунктов D1-D5, и контактирование исходного материала для аминирования и аминирующего реагента с просеянным катализатором в присутствии водорода для реакции аминирования.
D10. Способ по пункту D9, причем условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-5):(2-35):1, температура 110-230°С, давление 0,7-22 МПа и часовая объемная скорость 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере один материал, выбранный из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере один материал из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанол, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или аминирующий реагент представляет собой по меньшей мере один реагент, выбранный выбран из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере один из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
D11. Способ по пункту D10, причем, когда исходный материал для аминирования является одноатомным спиртом, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-8):1, температура 130-210°С, давление 1-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-5):1, температура 110-180°С, давление 0,7-2,5 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-23):1, температура 130-200°С, давление 1-16 МПа и часовая объемная скорость 0,06-0,8 м3/(м3⋅ч);
или, когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомного спирта, условия аминирования включают: мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-35):1, температура 130-230°С, давление 1-22 МПа и часовая объемная скорость 0,1-0,8 м3/(м3⋅ч).
Примеры
Настоящая заявка будет дополнительно проиллюстрирована на следующих примерах, но настоящая заявка ими не ограничивается.
В нижеследующих примерах использовались следующие приборы и методы:
1) Метод термопрограммированной десорбции NH3 (NH3-TPD)
Измерительный прибор: полностью автоматизированный прибор для исследования хемосорбции Autochem 2920 (автоматизированная система для определения характеристик катализаторов), производство MICROMERITICS, США.
Условия анализа: осторожно отвешивали около 0,1 г образца, помещали в пробирку для образцов, нагревали до температуры 600°С со скоростью 10°С/мин, одновременно продувая газообразным гелием, выдерживали 1 ч и охлаждали до температуры 120°С; газ меняли на смесь газов 10% NH3-He, адсорбировали 60 мин, затем газ был снова заменен на газообразный гелий для продувки в течение 1 ч, подсчет был начат после того, как базовая линия стабилизировалась; полученную смесь нагревали до температуры 600°С со скоростью 10°С/мин, выдерживали 30 мин, запись прекращали и эксперимент завершали. Интегрированием рассчитывали площадь пика, получая количество десорбированного NH3, и десорбированное количество использовали для характеризации адсорбционной емкости образца по аммиаку.
2) Метод БЭТ
Измерительный прибор: полностью автоматический анализатор физико-химической адсорбции (Automatic Micropore & Chemisorption Analyzer), производство MICROMERITICS, США.
Условия анализа: поверочный газ: N2 (чистота 99,999%); условия дегазации: нагрев до 350°С со скоростью 10°С/мин и вакуумирование в течение 4 ч; условия анализа: выполнение полного анализа на изотерме адсорбции на мезопорах с целью определения удельной поверхности и объема пор.
3) Зондовая адсорбционная спектрометрия
Измерительный прибор: инфракрасный спектрометр NICOLET 6700 от компании Thermo Scientific, с локальной приставкой на пропускание.
Условия анализа: образец точно взвешивали и записывали его массу, нагревали до 500°С со скоростью нагрева 10°С/мин в условиях вакуума, подложку предварительно выдерживали под нагревом в течение 2 часов и затем охлаждали до комнатной температуры. Предварительно обработанной подложке давали возможность адсорбировать пары пиридина до насыщения при комнатной температуре. Затем подложку подвергали статической десорбции до равновесного состояния в условиях вакуума при значениях температур: комнатная температура, 100°С, 150°С, 200°С, 300°С и 400°С, соответственно, при скорости нагрева между каждыми двумя температурными точками 10°С/мин.
4) Рентгенодифракционный анализ
Измерительный прибор: рентгеновский дифрактометр Empyrean производства PANalytical B.V. с анодной мишенью из меди и пиксельным 3D-детектором.
Условия анализа: напряжение трубки 40 кВ, ток трубки 40 мА, щель расхождения 1/4°, антирассеивающая щель 1/2°, высота приемной щели 7,5 мм, скорость сканирования 0,013°/шаг, диапазон сканирования 5°-90°.
Размер зерна активного металлического компонента и металла-промотора, когда таковой присутствует, получали расчетом по уравнению Шеррера.
5) Анализ на изоэлектрическую точку
Контрольно-измерительный прибор: потенциометрический анализатор размера частиц Zetasizer Nano ZSP от компании Malvern Panalytical.
Аналитический метод: образец измельчали в порошок и диспергировали в слабоконцентрированном растворе NaCl, дзета-потенциал образца измеряли с помощью потенциометрического анализатора размеров частиц при разных значениях рН и затем строили график его зависимости от рН. Значение рН при дзета-потенциале, равном 0, является изоэлектрической точкой образца.
В следующих примерах и сравнительных примерах, если не указано иное, используемые реактивы и исходные материалы представляют собой коммерчески доступные продукты, которые имеют степень чистоты "чистый для анализа (ч.д.а)".
Серия примеров I
Ниже первый тип вариантов осуществления настоящей заявки дополнительно подробно иллюстрируется на примерах серии примеров I. В следующих примерах серии примеров I порошок псевдобемита имел содержание Al2O3 72 вес. % в пересчете на сухое вещество, а силиказоль был приобретен у компании Qingdao Ocean Chemical Co., Ltd. под торговым наименованием JN-40.
Пример I-1
Порошок псевдобемита (удельная поверхность 315 м2/г, объем пор 0,91 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и борную кислоту, экструдировали с получением полосок диаметром 5 мм, нарезали на отрезки длиной 4 мм, сушили 10 ч при 100°С, обжигали при 850°С в течение 4 ч с получением желаемой подложки, и количество борной кислоты подбирали так, чтобы получить содержание бора в подложке, какое указано в таблице I-1.
В воде растворяли 151,2 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 184 мл раствора, и раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 4 часа при 120°С после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 430°С в течение 3 часов с получением катализатора C-I-1. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа (подробное описание которого можно найти в тестовом примере 1-1), составлял 20 нм.
Пример I-2
В смесителе добавляли силиказоль в порошок псевдобемита (удельная поверхность 322 м2/г, объем пор 0,93 мл/г), однородно перемешивали, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и фтористоводородную кислоту, экструдировали с получением гранул в форме трилистника толщиной 3 мм, сушили 6 ч при 120°С, затем обжигали при 820°С в течение 3,5 ч с получением желаемой подложки, при этом количество фтористоводородной кислоты подбирали так, чтобы получить содержание элемента F в подложке, какое указано в таблице I-1. Количество силиказоля подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 9:1.
В воде растворяли 177 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 172 мл раствора, и 3,7 г пятиводного молибдата аммония (ч.д.а.) растворяли в воде, получая 86 мл раствора; раствор нитрата никеля наносили на 100 г подложки путем двукратной пропитки распылением, раствор молибдата аммония наносили на подложку путем однократной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, обжигали при 390°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 440°С с получением катализатора C-I-2. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 22 нм.
Пример I-3
В смесителе добавляли силиказоль в порошок псевдобемита (удельная поверхность 345 м2/г, объем пор 1,12 мл/г), однородно перемешивали, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и фосфорную кислоту, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили 20 ч при 80°С, затем обжигали 4 ч при 800°С с получением желаемой подложки, и количество фосфорной кислоты подбирали так, чтобы получить содержание элемента Р в подложке, какое указано в таблице I-1. Количество силиказоля подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 3:1.
Растворяли в воде 50,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 19,5 г раствора нитрата марганца концентрацией 50 вес. % и 8,5 г нитрата меди тригидрата (ч.д.а.), получая 158 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 4 часа при 120°С после каждой пропитки распылением, затем обжигали 4 часа при 395°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали в течение 3 часов при 430°С с получением катализатора C-I-3. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 15 нм.
Пример I-4
Порошок псевдобемита (удельная поверхность 350 м2/г, объем пор 1,13 мл/г) смешивали с порошком диатомита (удельная поверхность 57 м2/г), замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и серную кислоту, экструдировали в полоски диаметром 5 мм, нарезали на отрезки длиной 4 мм, сушили 6 часов при 150°С, обжигали при 880°С 4 часа с получением желаемой подложки, и количество серной кислоты подбирали так, чтобы получить содержание элемента S в подложке, какое указано в таблице I-1. Количество диатомита подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 19:1.
Растворяли в воде 126,4 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 176 мл раствора, смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 4 часа при 120°С после каждой пропитки распылением, затем обжигали при 380°С в течение 4,5 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 430°С в течение 3 часов с получением катализатора C-I-4. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 22 нм.
Пример I-5
Порошок псевдобемита (удельная поверхность 320 м2/г, объем пор 0,9 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и серную кислоту, экструдировали в гранулы в форме трилистника толщиной 3 мм, сушили 8 ч при 120°С и затем обжигали при 890°С в течение 4,5 ч с получением желаемой подложки, при этом количество серной кислоты подбирали так, чтобы получить содержание элемента S в подложке, какое указано в таблице I-1.
В 186 мл воды растворяли 40,4 г шестиводного нитрата никеля (технический сорт, чистота 98%), 60,5 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 2,9 г перрената аммония (чистота 99, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 6 часов при 100°С после каждой пропитки распылением, затем обжигали при 390°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 440°С в течение 3 часов с получением катализатора C-I-5. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 16 нм.
Пример 1-6
Порошок псевдобемита (удельная поверхность 312 м2/г, объем пор 0,88 мл/г) смешивали с порошком молекулярного сита (модель ZSM-5, доступен для приобретения у предприятия по производству катализаторов при университете г. Нанкай), замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и селеновую кислоту, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили 8 ч при 120°С, затем обжигали при 810°С в течении 6 ч с получением желаемой подложки, при этом количество селеновой кислоты подбирали так, чтобы получить содержание элемента Se в подложке, какое указано в таблице I-1. Количество молекулярного сита подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 94:6.
Растворяли в воде 126 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 152 мл раствора, смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 5 часов при 120°С после каждой пропитки распылением, затем обжигали при 400°С в течение 3,5 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 430°С с получением катализатора C-I-6. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 25 нм.
Пример I-7
Порошок псевдобемита (удельная поверхность 34 8 м2/г, объем пор 1,13 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и борную кислоту, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили 8 ч при 100°С, затем обжигали при 950°С в течение 6,5 ч с получением желаемой подложки, при этом количество используемой борной кислоты подбирали так, чтобы получить содержание элемента В в подложке, какое указано в таблице I-1.
Растворяли в воде 100,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 1,3 г нитрата серебра (ч.д.а.), получая 170 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 4 часа при 120°С после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 410°С в течение 3 часов с получением катализатора C-I-7. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 11 нм.
Пример I-8
Порошок псевдобемита (удельная поверхность 356 м2/г, объем пор 1,2 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту, серную кислоту и фосфорную кислоту, экструдировали с получением зубчатых сфер диаметром 3 мм, сушили 8 ч при 100°С, затем обжигали при 860°С в течении 4 ч с получением желаемой подложки, и количества используемых фосфорной кислоты и серной кислоты подбирали так, чтобы получить содержание элемента Р и содержание элемента S в подложке, какие указаны в таблице I-1.
Растворяли в воде 141,6 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 3,1 г шестиводного нитрата церия (ч.д.а.), получая 184 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 5 часов при 120°С после каждой пропитки распылением, затем обжигали при 410°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 4 часа при 400°С с получением катализатора C-I-8. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 12 нм.
Пример I-9
В смесителе добавляли силиказоль в порошок псевдобемита (удельная поверхность 315 м2/г, объем пор 0,88 мл/г), однородно перемешивали, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и серную кислоту, экструдировали с получением зубчатых сфер диаметром 3 мм, сушили 8 ч при 100°С, затем обжигали при 900°С в течение 6 ч с получением желаемой подложки, и количество серной кислоты подбирали так, чтобы получить содержание элемента S в подложке, какое указано в таблице I-1. Количество силиказоля подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 72:28.
Растворяли в воде 100,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 50,6 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 14 6 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили 8 часов при 100°С после каждой пропитки распылением, затем обжигали при 420°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 410°С в течение 3 часов с получением катализатора C-I-9. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 18 нм.
Пример I-10
В смесителе добавляли силиказоль в порошок псевдобемита (удельная поверхность 292 м2/г, объем пор 0,82 мл/г), однородно перемешивали, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту, фосфорную кислоту и фтористоводородную кислоту, экструдировали с получением зубчатых сфер диаметром 3 мм, сушили 7 ч при 110°С, затем обжигали 7 ч при 970°С с получением желаемой подложки, и количества фосфорной кислоты и фтористоводородной кислоты подбирали так, чтобы получить содержание элемента Р и содержание элемента F в подложке, какие указаны в таблице I-1. Количество оксида кремния подбирали так, чтобы получить весовое отношение Al2O3 к SiO2 в подложке 66:34.
Растворяли в воде 201,6 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 6,2 г шестиводного нитрата лантана (ч.д.а.), получая 165 мл раствора, и смешанный раствор наносили на 100 г подложки путем трехкратной пропитки распылением, сушили при 120°С 6 часов после каждой пропитки распылением, затем обжигали при 420°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 410°С в течение 3 часов с получением катализатора C-I-10. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 13 нм.
Пример I-11
Порошок псевдобемита (удельная поверхность 276 м2/г, объем пор 0,79 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и серную кислоту, экструдировали с получением гранул в форме трилистника толщиной 4 мм, сушили 6 ч при 110°С, затем обжигали при 930°С в течение 6 ч с получением желаемой подложки, при этом количество серной кислоты подбирали так, чтобы получить содержание элемента S в подложке, какое приведено в таблице I-1.
Растворяли в воде 15,2 г шестиводного нитрата никеля (технический сорт, чистота 98%), 25,2 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 4,4 г перрената аммония (чистота 99%), получая 176 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 390°С 5 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 440°С в течение 3 часов, получая катализатор C-I-11. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 19 нм.
Пример I-12
Порошок псевдобемита (удельная поверхность 260 м2/г, объем пор 0,77 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и фосфорную кислоту, экструдировали с получением гранул в форме трилистника толщиной 4 мм, сушили 12 ч при 100°С, затем обжигали при 860°С в течение 9 ч с получением желаемой подложки и количество фосфорной кислоты подбирали так, чтобы получить содержание элемента Р в подложке, какое приведено в таблице I-1.
Растворяли в воде 100,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 14,1 г тригидрата нитрата меди (ч.д.а.), получая 180 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 390°С 5 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 440°С с получением катализатора C-I-12. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 21 нм.
Пример I-13
Порошок псевдобемита (удельная поверхность 257 м2/г и объем пор 0,76 мл/г) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и борную кислоту, экструдировали с получением гранул в форме трилистника толщиной 3 мм, сушили 8 ч при 120°С, затем обжигали при 850°С в течение 6 ч с получением желаемой подложки, при этом количество борной кислоты подбирали так, чтобы получить содержание элемента В в подложке, какое приведено в таблице I-1.
Растворяли в воде 151,7 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 12,5 г шестиводного нитрата лантана (ч.д.а.), получая 18 мл раствора, и смешанный раствор наносили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 370°С 5 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 4 часа при 430°С с получением катализатора C-I-13. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 12 нм.
Пример I-14
Катализатор C-I-14 был приготовлен, как описано в примере 1-5, за исключением того, что серную кислоту добавляли в таком количестве, чтобы содержание элемента S в подложке было таким, как показано в таблице I-1. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 16 нм.
Пример I-15
Катализатор C-I-15 был приготовлен, как описано в примере 1-5, за исключением того, что температура обжига подложки составляла 800°С, а продолжительность обжига 2 часа. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 14 нм.
Пример I-16
Катализатор был приготовлен, как описано в примере 1-5, за исключением того, что при замешивании добавляли силиказоль в таком количестве, чтобы весовое отношение Al2O3 к SiO2 в подложке составляло 66:34, получая катализатор C-I-16. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 16 нм.
Пример I-17
ZSM-5 (удельная поверхность 338 м2/г, объем пор 0,61 мл/г, степень кристалличности 97,9%, SiO2/Al2O3=60) замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту, серную кислоту и фосфорную кислоту, экструдировали с получением зубчатых сфер диаметром 3 мм, сушили 6 часов при 120°С и затем обжигали при 880°С в течение 4 часов с получением желаемой подложки, и количества фосфорной кислоты и серной кислоты подбирали так, чтобы получить содержание элемента Р и содержание элемента S в подложке, какие указаны в таблице I-1.
Пропитку катализатора проводили, как описано в примере 1-8, получая катализатор C-I-17. Размер зерна нанесенного компонента, определенный методом рентгенодифракционного анализа, составлял 14 нм.
Сравнительный Пример I-1
Катализатор был приготовлен, как описано в примере 1-12, за исключением того, что при получении подложки температура обжига составляла 700°С, а продолжительность обжига 5 часов, чтобы получить катализатор D-I-1.
Сравнительный Пример I-2
Катализатор был приготовлен, как описано в примере 1-12, за исключением того, что при получении подложки температура обжига составляла 550°С, а продолжительность обжига 3 часа, и количество используемой фосфорной кислоты подбирали так, чтобы получить содержание элемента Р в подложке, какое указано в таблице I-1, чтобы получить катализатор D-I-2.
Тестовый Пример I-1
Элементный состав подложек и катализаторов анализировали методом эмиссионной спектроскопии с индуктивно-связанной плазмой, содержание элемента (иона), отличного от подложки, выражали как содержание элемента в 100 г матрицы, т.е., подложка рассчитывалась на основе компонента или компонентов, отличных от легирующего элемента (например, рассчитывалась на основе Al2O3, если в качестве источника подложки использовался псевдобемит); полученные выше подложки анализировали методом зондовой адсорбционной спектроскопии (характеризующей пропорцию содержания L-кислоты в суммарном содержании L-кислоты и В-кислоты (т.е., долю L-кислоты)), методом NH3-TPD, методом адсорбции-десорбции азота по БЭТ, результаты приведены в таблице I-1.
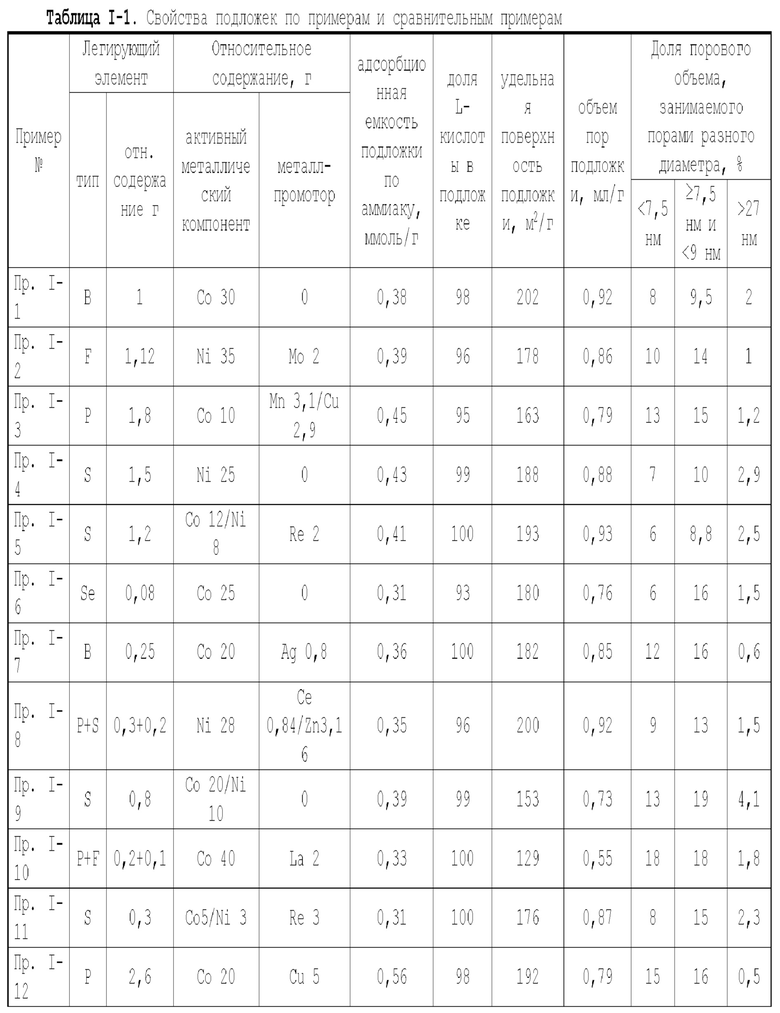
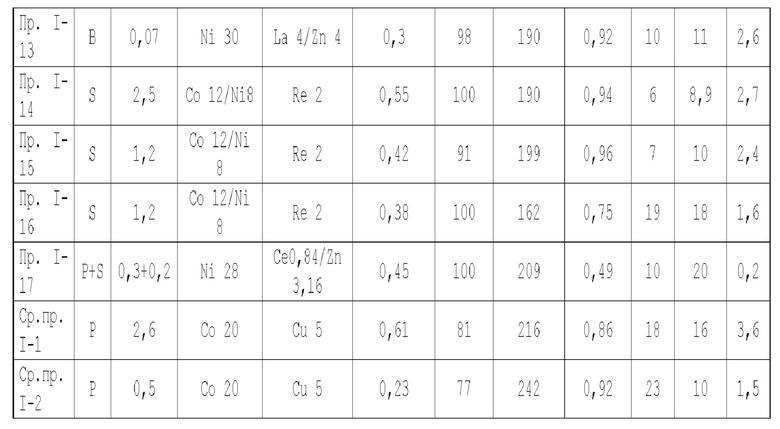
Тестовый Пример I-2
Этот тестовый пример иллюстрирует способ получения 1,6-гександиамина гидроаминированием 1,6-гександиола с использованием катализаторов по первому типу вариантов осуществления настоящей заявки.
Отмеряли соответственно по 100 мл полученных катализаторов, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 168°С, давление в системе повышали до 9,5 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 150°С и затем отправляли в верхний конец реактора; нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и 1,6-гександиолом составляло 3:12:1, объемная скорость жидкого 1,6-гександиола составляла 0,45 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 198°С и давление реакции 9,5 МПа, отбирали реакционный раствор и анализировали при времени реакции 200 часов. Результаты анализа представлены в таблице I-2.
Анализ образцов проводили методом газовой хроматографии, калибровку осуществляли с использованием поправочного коэффициента для рецептуры эталонного образца.
Конверсию и селективность рассчитывали, исходя из мольного содержания каждого компонента в реакционном растворе.

Селективность по гексаметиленимину рассчитывали, изменяя числитель в уравнении, приведенном выше для расчета селективности по гександиамину, на мольное содержание гексаметиленимина, селективность по аминогексанолу рассчитывали, изменяя числитель в уравнении, приведенном выше для расчета селективности по гександиамину, на мольное содержание аминогексанола, и т.д., а селективность по "другим" рассчитывали, изменяя числитель в уравнении, приведенном выше для расчета селективности по гександиамину, на мольное содержание димеров амин × 2, при этом димер амина относится к димеру 1,6-гександиамина (т.е., бис(гексаметилен)триамину, известному также как N-(6-аминогексил)-1,6-гександиамин), и к димеру 1,6-гександиамина с гексаметиленимином (т.е., к N-(6-аминогексил) гексаметиленимину).
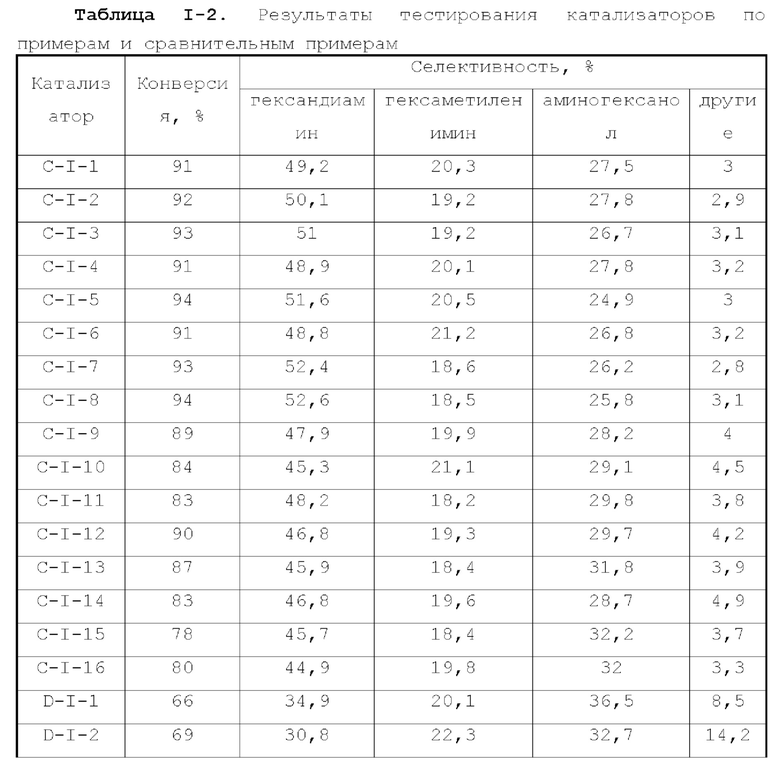
Как можно видеть из данных таблицы I-2, катализатор по настоящей заявке обеспечивает более высокую конверсию и имеет более высокую активность, чем сравнительные катализаторы, что указывает на то, что катализатор по настоящей заявке обеспечивает более высокую скорость реакции.
Тестовый Пример I-3
Этот тестовый пример иллюстрирует способ получения 1,3-пропандиамина путем гидроаминирования 1,3-пропандиола с использованием катализаторов по первому типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-I-3, полученного в примере 1-3, загружали в реактор с неподвижным слоем, активировали водородом 2 часа при 220°С, затем охлаждали до 165°С, давление в системе повышали до 8,8 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 120°С и затем отправляли в верхний конец реактора; 1,3-пропандиол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и 1,3-пропандиолом составляло 3:9:1, объемная скорость жидкого 1,3-пропандиола составляла 0,4 ч-1, каталитическую реакцию аминирования проводили в реакторе, и после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере 1-2). Результаты анализа представлены в таблице I-3.
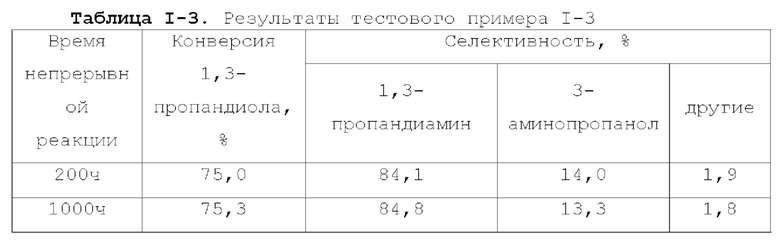
Тестовый Пример I-4
Этот тестовый пример иллюстрирует способ получения этиламина путем гидроаминирования этанола с использованием катализатора по первому типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-I-3, полученного в примере 1-3, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 170°С, давление в системе повышали до 1,8 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 125°С, затем отправляли в верхний конец реактора; этанол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и этанолом составляло 3:5:1, объемная скорость жидкого этанола составляла 0,6 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 180°С и давлении реакции 1,8 МПа, и после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере 1-2). Результаты анализа представлены в таблице I-4.
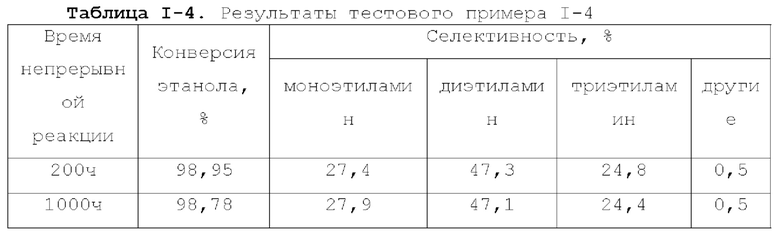
Катализаторы D-I-1 и D-I-2 испытывали в тех же условиях процесса, из анализа результатов было обнаружено, что при использовании сравнительных катализаторов D-I-1 и D-I-2, селективность которых составляла 0,8% и 1,2%, соответственно, образовывалось больше "других" компонентов, при оценке в течение длительного времени (период 200 ч) было установлено, что скорость дезактивации катализаторов D-I-1 и D-I-2 была относительно высокой. Через 200 часов оценки осаждение углерода в случае катализатора C-I-3 отличалось от такового для катализаторов D-I-1 и D-I-2, при этом осаждение углерода в двух последних случаях было явно больше, чем на катализаторе C-I-3, уменьшение удельной поверхности и объема пор катализатора C-I-3 было незначительным (менее 2%), тогда как уменьшение удельной поверхности катализаторов D-I-1 и D-I-2 составляло соответственно 7% и 9%, а уменьшение объема пор катализаторов D-1-1 и D-I-2 9% и 10%, соответственно, что указывает на блокирование поровых каналов вследствие осаждения углерода.
Серия примеров II
Ниже второй тип вариантов осуществления настоящей заявки дополнительно подробно иллюстрируется на примерах серии примеров II. В следующих примерах серии примеров II порошок псевдобемита имел содержание Al2O3 70 вес. % в пересчете на сухое вещество, а силиказоль был приобретен у компании Qingdao Ocean Chemical Co., Ltd. под торговым наименованием JN-40.
Пример II-l
Порошок псевдобемита (удельная поверхность 380 м2/г, объем пор 1,09 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3,5 об. % фосфорной кислоты, и экструдировали с получением полосок, сушили при 120°С в течение 10 ч, затем обжигали 4 ч при 850°С с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 141,56 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 166 мл раствора, рН раствора доводили до 4,3, раствор вводили на 100 г алюмооксидной подложки путем двукратной пропитки распылением, сушили 8 часов при 120°С после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 440°С, получая катализатор C-II-1.
Пример II-2
Порошок псевдобемита (удельная поверхность 400 м2/г, объем пор 1,15 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3 об. % борной кислоты, в процессе размешивания добавляли силиказоль, экструдировали с получением полосок, сушили при 120°С в течение 12 часов и затем обжигали при 900°С 4 часа с получением желаемой подложки, детальные параметры которой указаны в таблице II-1, при этом порошок псевдобемита и силиказоль использовали в таком количестве, чтобы весовое отношение Al2O3 к SiO2 в подложке составляло 4:1.
Растворяли в воде 176,38 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 160 мл раствора, рН раствора доводили до 3,9, раствор вводили на 100 г подложки путем двукратной влажной пропитки, сушили 6 часов при 120°С после каждой влажной пропитки, затем обжигали 4 часа при 390°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 460°С в течение 5 часов, получая катализатор C-II-2.
Пример II-3
Порошок псевдобемита (удельная поверхность 395 м2/г, объем пор 1,19 мл/г) замешивали разбавленной азотной кислотой концентрацией 2 об. % и разбавленным водным раствором кислоты, содержащим 2 об. % серной кислоты, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили при 150°С в течение 8 ч и затем обжигали при 950°С в течение 3,5 ч с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 75,59 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 27,36 г раствора нитрата марганца 50 вес. %, получая 138 мл раствора, рН раствора доводили до 4,4, раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С в течение 2 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 8 часов при 400°С, получая катализатор C-II-3.
Пример II-4
Порошок псевдобемита (удельная поверхность 395 м2/г, объем пор 1,05 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3 об. % борной кислоты, и экструдировали с получением полосок, сушили при 120°С в течение 18 ч и затем обжигали при 1010°С в течение 4,5 ч с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 60,47 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 50,56 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 0,73 г перрената аммония (чистота 99%), получая 120 мл раствора, рН раствора доводили до 4,3, и раствор вводили на 100 г подложки путем двукратной изоволюметрической пропитки, сушили при 120°С в течение 5 часов после каждой пропитки, затем обжигали при 390°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 450°С в течение 6 часов, получая катализатор C-II-4.
Пример II-5
Порошок псевдобемита (удельная поверхность 382 м2/г, объем пор 1,0 9 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 2 об. % серной кислоты, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили при 120°С в течение 10 ч и затем обжигали при 820°С в течение 10 ч с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 192,12 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 3,94 г нитрата серебра, получая 182 мл раствора, рН раствора доводили до 4,6, раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 480°С, получая катализатор C-II-5.
Пример II-6
Порошок псевдобемита (удельная поверхность 375 м2/г, объем пор 1,19 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3,5 об. % фосфорной кислоты, экструдировали с получением зубчатых сфер, сушили 15 ч при 120°С, затем обжигали 5,5 ч при 880°С с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
В воде растворяли 100,79 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 14,56 г нитрата цинка (ч.д.а.), получая 154 мл раствора, рН раствора доводили до 5,1, и раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 6 часов после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 450°С в течение 3 часов, получая катализатор C-II-6.
Пример II-7
Порошок псевдобемита (удельная поверхность 342 м2/г, объем пор 0,78 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3,5 об. % фосфорной кислоты, и экструдировали с получением зубчатых сфер, сушили 10 ч при 120°С, затем обжигали при 930°С в течение 4,5 ч с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 120,94 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 9,28 г пятиводного нитрата висмута (ч.д.а.), получая 102 мл раствора, рН раствора доводили до 4,9, и раствор вводили на 100 г подложки путем двукратной влажной пропитки, сушили при 120°С 4 часа после каждой влажной пропитки, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 4 часа при 430°С, получая катализатор C-II-7.
Пример II-8
Порошок псевдобемита (удельная поверхность 280 м2/г, объем пор 0,89 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, 2 об. % фтористоводородной кислоты и 0,5 вес. % нитрата селена, экструдировали с получением зубчатых сфер, сушили при 120°С в течение 20 ч, затем обжигали 12 ч при 980°С с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 202,23 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 13,65 г шестиводного нитрата цинка (ч.д.а.), получая 128 мл раствора, рН раствора доводили до 3,7, раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили 10 ч при 120°С после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 410°С в течение 6 часов, получая катализатор C-II-8.
Пример II-9
Порошок псевдобемита (удельная поверхность 380 м2/г, объем пор 1,0 9 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3,5 об. % фосфорной кислоты, в процессе замешивания добавляли силиказоль, экструдировали с получением полосок, сушили при 120°С в течение 10 часов, затем обжигали при 850°С с получением желаемой подложки, детальные параметры которой указаны в таблице II-1, при этом порошок псевдобемита и силиказоль использовали в таком количестве, чтобы весовое отношение Al2O3 к SiO2 в подложке составляло 9:1.
Растворяли в воде 75,59 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 50,56 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 29,31 г раствора нитрата марганца 50 вес. %, получая 106 мл раствора, рН раствора доводили до 4,0, раствор вводили на 100 г алюмооксидной подложки путем двукратной пропитки распылением, сушили при 120°С 8 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 440°С в течение 5 часов, получая катализатор C-II-9.
Пример I1-10
Порошок псевдобемита (удельная поверхность 369 м2/г, объем пор 1,15 мл/г) замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты и 3 об. % серной кислоты, и экструдировали с получением полосок, сушили 15 ч. при 120°С и затем обжигали при 1010°С в течение 6,5 ч с получением желаемой подложки, детальные параметры которой указаны в таблице II-1.
Растворяли в воде 35,39 г шестиводного нитрата никеля (технический сорт, чистота 98%) и 2,18 г перрената аммония (чистота 99%), получая 164 мл раствора, рН раствора доводили до 5,3, и раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 6 часов после каждой пропитки распылением, обжигали при 350°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 420°С в течение 5 часов, получая катализатор C-II-10.
Сравнительный пример II-1
Катализатор D-II-1 был приготовлен, как описано в примере II-3, за исключением того, что при получении подложки серную кислоту не добавляли, и рН пропиточного раствора не корректировали (собственный рН около 7).
Сравнительный пример II-2
Катализатор D-II-2 был приготовлен, как описано в примере II-3, за исключением того, что серную кислоту использовали в таком количестве, чтобы содержание S в подложке было таким, как указано в таблице II-1, и рН пропиточного раствора не корректировали (собственный рН около 7).
Сравнительный пример II-3
Катализатор был приготовлен, как описано в примере II-3, за исключением того, что серную кислоту использовали в таком количестве, чтобы содержание S в подложке было таким, как указано в таблице II-1, температура обжига при получении подложки составляла 650°С, продолжительность обжига составляла 4 часа, рН пропиточного раствора не корректировали (собственный рН около 7), в результате получали катализатор D-II-3.
Тестовый пример II-1
Элементный состав подложек и катализаторов анализировали методом эмиссионной спектроскопии с индуктивно-связанной плазмой, содержание элемента (иона), отличного от подложки, выражали как содержание элемента в 100 г матрицы; полученные выше подложки анализировали методом зондовой адсорбционной спектроскопии, методом NH3-TPD, методом адсорбции-десорбции азота по БЭТ, размер зерна активного металлического компонента в катализаторе измеряли методом рентгенодифракционного анализа, результаты приведены в таблице II-1.
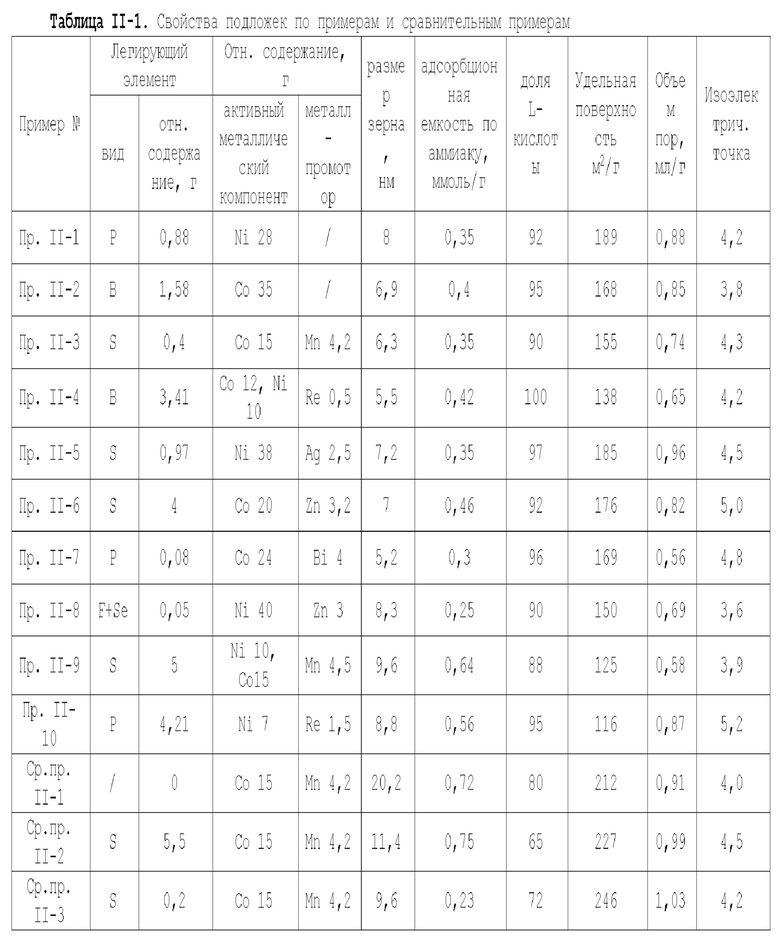
Тестовый пример II-2
Этот тестовый пример иллюстрирует способ получения 1,6-гександиамина путем гидроаминирования 1,6-гександиола с использованием катализатора по второму типу вариантов осуществления настоящей заявки.
Отмеряли соответственно по 100 мл катализаторов, полученных в примерах, загружали в реактор с неподвижным слоем, активировали водородом при 250°С в течение 4 часов, затем охлаждали до 160°С, давление в системе повышали до 11 МПа, используя водород, затем в реакционную систему вводили аммиак с помощью насоса-дозатора, предварительно нагревали до 170°С, затем отправляли в верхний конец реактора; нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора через насос-дозатор, и водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и 1, 6-гександиолом составляло 4:10:1, объемная скорость жидкого 1,6-гександиола составляла 0,4 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 185°С и давление реакции 11 МПа, отбирали образец реакционного раствора и анализировали при времени реакции 20 часов, результаты анализа приведены в таблице II-2.
Анализ образца проводили методом газовой хроматографии, калибровку осуществляли с использованием поправочного коэффициента для рецептуры эталонного образца.
Конверсию и селективность рассчитывали, исходя из мольного содержания каждого компонента в реакционном растворе, способы расчета были такими же, как описано в тестовом примере I-2.
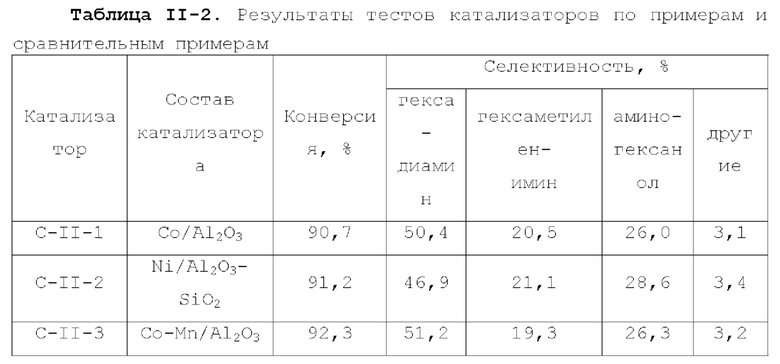
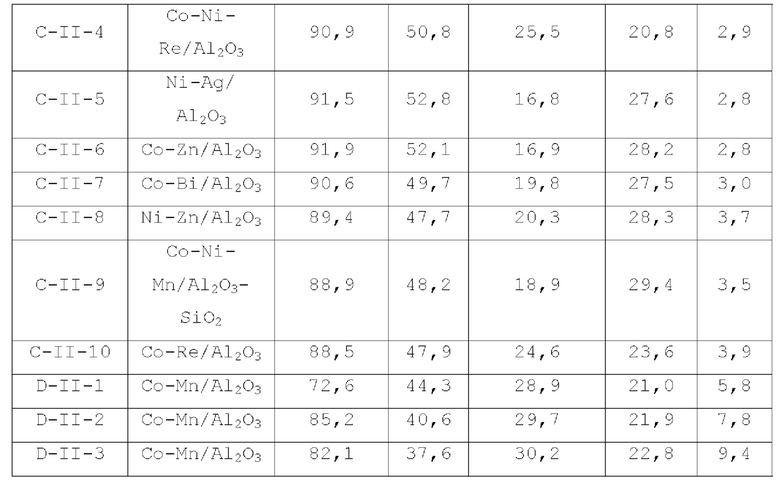
Как можно видеть из данных таблицы II-2, катализатор по настоящей заявке обеспечивает более высокую конверсию и имеет более высокую активность, чем сравнительные катализаторы, это говорит о том, что катализатор по настоящей заявке обеспечивает более высокую скорость реакции.
Тестовый пример II-3
Этот тестовый пример иллюстрирует способ получения этилендиамина путем гидроаминирования этаноламина с использованием катализатора по второму типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-II-3, полученного в примере II-3, загружали в реактор с неподвижным слоем, активировали 2 часа при 250°С водородом, затем охлаждали до 168°С, давление в системе повышали до 8 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 165°С и затем отправляли в верхний конец реактора; этаноламин подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и этаноламином составляло 3:10:1, объемная скорость жидкого этаноламина составляла 0,75 ч-1, каталитическое аминирование проводили в реакторе при температуре реакции 205°С и давлении реакции 8 МПа, после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере 1-2 II-2). Результаты анализа представлены в таблице II-3.
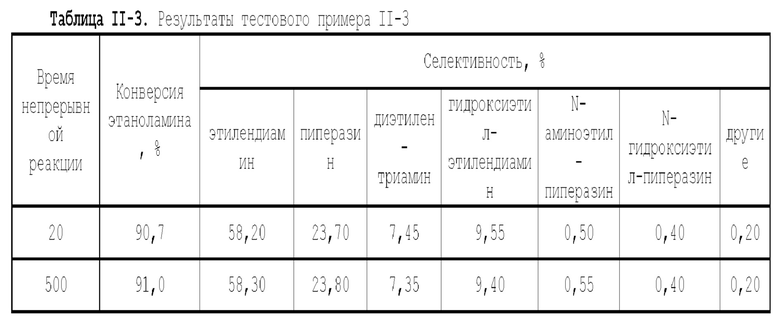
Катализаторы D-II-1 - D-II-4 испытывали в тех же условиях процесса, из анализа результатов было обнаружено, что при использовании сравнительных катализаторов D-II-1 - D-II-4 образовывалось больше "других" компонентов, при оценке в течение длительного времени было установлено, что катализаторы D-II-1 -D-II-4 имели сниженную селективность и конверсию и относительно высокую скорость дезактивации. Согласно оценке, после 500 часов осаждение углерода в случае катализатора C-II-3 отличалось от такового для катализаторов D-II-1 - D-II-4, при этом осаждение углерода в последних случаях было явно больше, чем на катализаторе C-II-3, уменьшение удельной поверхности и объема пор катализатора C-II-3 было незначительным (менее 2%), тогда как уменьшение удельной поверхности катализаторов D-II-1 - D-II-4 составляло соответственно 19%, 16%, 18% и 16%, а уменьшение объема пор катализаторов D-II-1 - D-II-4 соответственно 20%, 18%, 19% и 18%, это говорит о том, что осаждение углерода было довольно большим, что приводило к блокированию поровых каналов.
Серия примеров III
Ниже третий тип вариантов осуществления настоящей заявки дополнительно подробно иллюстрируется на примерах серии примеров III. В следующих примерах серии примеров III порошок псевдобемита имел содержание Al2O3 72 вес. % в пересчете на сухое вещество, а силиказоль был приобретен у компании Qingdao Ocean Chemical Co., Ltd. под торговым наименованием JN-40.
Пример III-1
Порошок псевдобемита (удельная поверхность 315 м2/г, объем пор 0,96 мл/г и содержание элемента Р в порошке 3,6 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением полосок диаметром 5 мм, нарезали на отрезки длиной 4 мм, сушили при 120°С 8 часов и обжигали 5 часов при 760°С, получая подложку.
В воде растворяли 186,5 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 142 мл раствора, раствор нитрата кобальта вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 430°С 3 часа с получением катализатора С-III-1.
Пример III-2
Порошок псевдобемита (удельная поверхность 295 м2/г, объем пор 0,93 мл/г и содержание элемента В в порошке 0,8 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, однородно перемешивали, экструдировали с получением гранул в форме трилистника толщиной 3 мм, сушили при 100°С в течение 15 ч и обжигали 4 ч при 930°С с получением подложки.
В воде растворяли 151,7 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 158 мл раствора, раствор нитрата никеля вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 100°С 8 часов после каждой пропитки распылением, затем обжигали при 390°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 440°С в течение 3 часов, получая катализатор C-III-2.
Пример III-3
Порошок псевдобемита (удельная поверхность 278 м2/г, объем пор 0,85 мл/г и содержание элемента S в порошке 0,4 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 3,5 об. % азотной кислоты, силиказоль добавляли в процессе размешивания, однородно перемешивали, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили при 150°С в течение 6 ч, обжигали при 980°С в течение 3 ч, получая подложку, причем количество используемого силиказоля подбирали так, чтобы получить весовое отношение оксида алюминия к оксиду кремния в подложке 4:1.
В воде растворяли 68,92 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 112 мл раствора, раствор нитрата кобальта вводили на 100 г алюмооксидной подложки путем двукратной пропитки распылением, сушили при 110°С 8 часов после каждой пропитки распылением, затем обжигали 4 часа при 390°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 2 часа при 460°С, получая катализатор C-III-3.
Пример III-4
Порошок псевдобемита (удельная поверхность 325 м2/г, объем пор 1,12 мл/г и содержание элемента F в порошке 2,2 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, однородно перемешивали с порошком молекулярного сита (ZSM-5, предприятие по производству катализаторов при университете г. Нанкай, мольное отношение SiO2/Al2O3=45), замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением полосок диаметром 5 мм, нарезали на отрезки длиной 4 мм, сушили при 80°С в течение 2 0 ч и обжигали при 730°С 8 ч с получением желаемой подложки, при этом количество порошка молекулярного сита ZSM-5 подбирали так, чтобы содержание в подложке оксида алюминия, происходящего из псевдобемита, составляло 90% от полного веса подложки.
В воде растворяли 126 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 202 мл раствора, раствор нитрата кобальта вводили на 100 г алюмооксидной подложки путем двукратной пропитки распылением, сушили при 90°С 20 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали часа при 430°С, получая катализатор С-III-4.
Пример III-5
Порошок псевдобемита (удельная поверхность 260 м2/г, объем пор 0,82 мл/г, содержание элемента Р в порошке 0,6 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и борную кислоту, экструдировали с получением гранул в форме трилистника толщиной 3 мм, сушили 8 часов при 100°С и затем обжигали 3 часа при 1005°С с получением желаемой подложки, при этом количество используемой борной кислоты составляло 2,2 9 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3, и концентрация азотной кислоты в разбавленном водном растворе кислоты составляла 5 об. %.
В воде растворяли 75,6 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 122 мл раствора, раствор нитрата кобальта вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, обжигали при 390°С 4 часа, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 440°С в течение 3 часов, получая катализатор C-III-5.
Пример III-6
Порошок псевдобемита (удельная поверхность 345 м2/г, объем пор 1,09 мл/г, содержание элемента В в порошке 0,02 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и серную кислоту, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили при 120°С в течение 10 ч и обжигали при 750°С в течение 12 ч с получением желаемой подложки, при этом количество используемой серной кислоты составляло 0,24 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3, и концентрация азотной кислоты в разбавленном водном растворе кислоты составляла 5 об. %.
В воде растворяли 126,4 г шестиводного нитрата никеля (технический сорт, чистота 98%), получая 184 мл раствора, раствор нитрата никеля вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 430°С в течение 3 часов, получая катализатор C-III-6.
Пример III-7
Порошок псевдобемита (удельная поверхность 320 м2/г, объем пор 0,93 мл/г, содержание элемента S в порошке 0,1 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим азотную кислоту и фосфорную кислоту, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили 15 ч при 100°С и обжигали при 780°С в течение 10 ч с получением желаемой подложки, при этом количество используемой фосфорной кислоты составляло 6,01 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3. Концентрация азотной кислоты в разбавленном водном растворе кислоты составляла 5 об. %.
В воде растворяли 296,85 г шестиводного нитрата кобальта (технический сорт, чистота 98%) получая 138 мл раствора, раствор нитрата кобальта вводили на 100 г алюмооксидной подложки путем двукратной пропитки распылением, сушили 5 часов при 120°С после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, обжигали 3 часа при 450°С, получая катализатор C-III-7.
Пример III-8
Порошок псевдобемита (удельная поверхность 295 м2/г, объем пор 0,88 мл/г, содержание элемента S в порошке 0,2 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением зубчатых сфер диаметром 4 мм, сушили при 90°С в течение 18 ч и обжигали при 810°С в течение 8 ч с получением желаемой подложки.
В воде растворяли 100,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%), получая 134 мл раствора, раствор вводили на 100 г подложки путем двукратной пропитки распылением, сушили при 100°С в течение 8 часов после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 часа при 420°С, получая катализатор C-III-8.
Пример III-9
Порошок псевдобемита (удельная поверхность 34 0 м2/г, объем пор 1,18 мл/г, содержание элемента F в порошке 0,12 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, силиказоль добавляли в процессе размешивания, экструдировали с получением цилиндрических стержней диаметром 3 мм, сушили 16 ч при 100°С, обжигали при 850°С в течение 6 ч с получением желаемой подложки, при этом количество силиказоля подбирали так, чтобы мольное отношение оксида алюминия к оксиду кремния в подложке составляло 73:27.
Растворяли в воде 176,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 1,3 г нитрата серебра (ч.д.а.), получая 178 мл раствора, и раствор вводили на 100 г полученной выше подложки путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 360°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 450°С в течение 3 часов, получая катализатор C-III-9.
Пример III-10
Порошок псевдобемита (удельная поверхность 310 м2/г, объем пор 0,92 мл/г, содержание элемента S в порошке 1,4 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением полосок диаметром 5 мм, сушили при 100°С в течение 12 ч и обжигали при 760°С в течение 10 ч с получением желаемой подложки.
Растворяли в воде 126 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 13,7 г шестиводного нитрата цинка (ч.д.а.), получая 172 мл раствора, раствор вводили на 100 г полученной выше подложки путем двукратной пропитки распылением, сушили при 120°С 4 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 420°С в течение 3 часов, получая катализатор C-III-10.
Пример III-11
Порошок псевдобемита (удельная поверхность 305 м2/г, объем пор 0,94 мл/г, содержание элемента Р в порошке 1,55 г на 100 г оксида алюминия), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением полосок диаметром 5 мм, сушили при 120°С в течение 6 ч и обжигали 5 ч при 860°С с получением желаемой подложки.
Растворяли в воде 141,1 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 19,5 г 50%-ного водного раствора нитрата марганца, получая 168 мл раствора, раствор вводили на 100 г полученной выше подложки путем двукратной пропитки распылением, сушили при 120°С в течение 4 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 420°С в течение 3 часов, получая катализатор C-III-11.
Сравнительный пример III-1
Катализатор D-III-1 был приготовлен, как описано в примере III-8, за исключением того, что использовали порошки псевдобемита с разными содержаниями элемента S, так что содержание элемента S было таким, как указано в таблице III-1, и при получении подложки температура обжига составляла 700°С, а продолжительность обжига 4 часа.
Сравнительный пример III-2
Катализатор D-III-2 был приготовлен, как описано в примере III-8, за исключением того, что использовали порошок псевдобемита, не содержащий легирующего элемента (удельная поверхность 224 м2/г, объем пор 0,95 мл/г).
Сравнительный пример III-3
Катализатор D-III-3 был приготовлен, как описано в примере III-8, за исключением того, что использовали порошки псевдобемита с разными содержаниями элемента S, так что содержание элемента S было таким, как указано в таблице III-1, и при получении подложки температура обжига составляла 500°С, а продолжительность обжига 4 часа.
Тестовый пример III-1
Элементный состав подложек и катализаторов анализировали методом эмиссионной спектроскопии с индуктивно-связанной плазмой, при этом содержания легирующего элемента, активного металлического компонента и металла-промотора выражали в весе на 100 г матрицы; полученные выше подложки анализировали методом зондовой адсорбционной спектроскопии, методом адсорбции-десорбции азота по БЭТ, результаты приведены в таблице III-1.
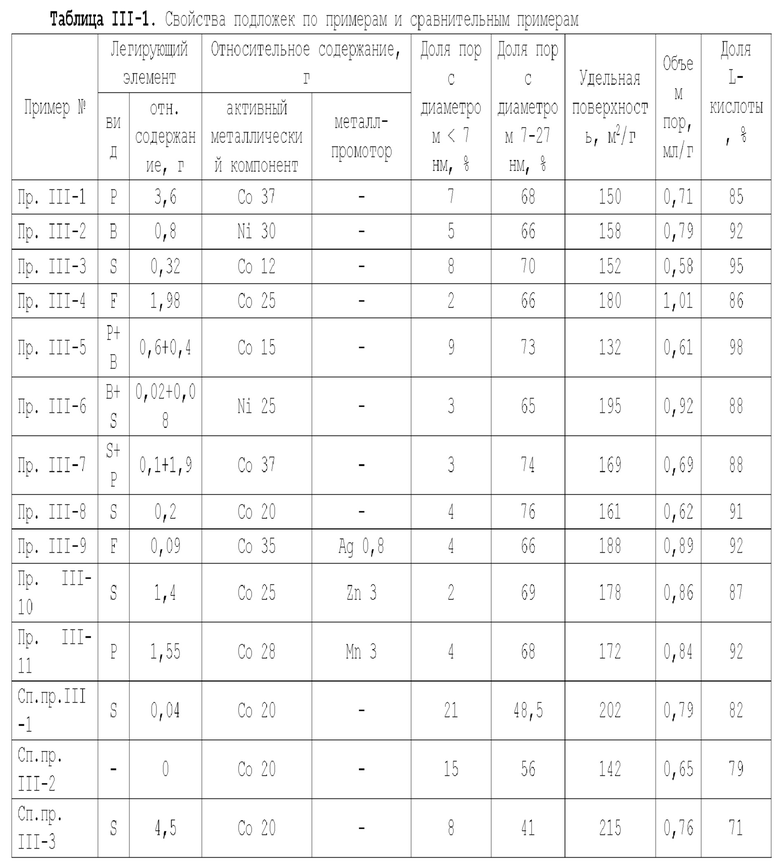
Тестовый пример III-2
Этот тестовый пример иллюстрирует способ получения 1,6-гександиамина путем гидроаминирования 1,6-гександиола с использованием катализатора по третьему типу вариантов осуществления настоящей заявки.
Отмеряли соответственно по 100 мл катализаторов, полученных в примерах, загружали в реактор с неподвижным слоем, активировали 2 часа при 220°С водородом, затем охлаждали до 170°С, давление в системе повышали до 10 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 150°С и затем отправляли в верхний конец реактора; нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и 1,6-гександиолом составляло 3:10:1, объемная скорость жидкого 1,6-гександиола составляла 0,45 ч-1, реакцию каталитического аминирования проводили в реакторе при температуре реакции 190°С и давлении реакции 10 МПа, отбирали образец реакционного раствора и анализировали при времени реакции 230 часов. Результаты анализа представлены в таблице III-2.
Анализ образца проводили методом газовой хроматографии, калибровку осуществляли с использованием поправочного коэффициента для рецептуры эталонного образца.
Конверсию и селективность рассчитывали, исходя из мольного содержания каждого компонента в реакционном растворе, способы расчета были такими же, как описано в тестовом примере I-2.
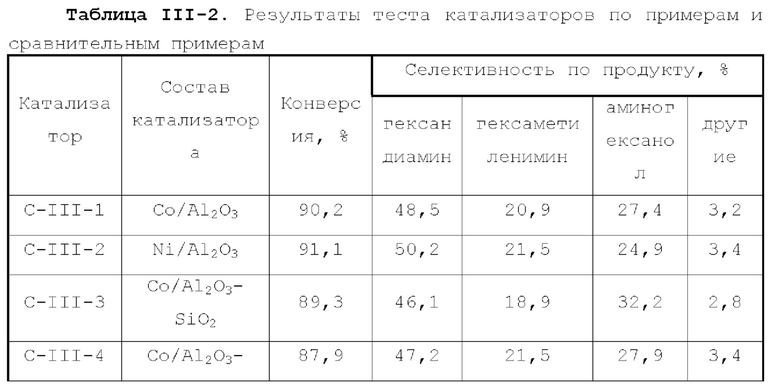
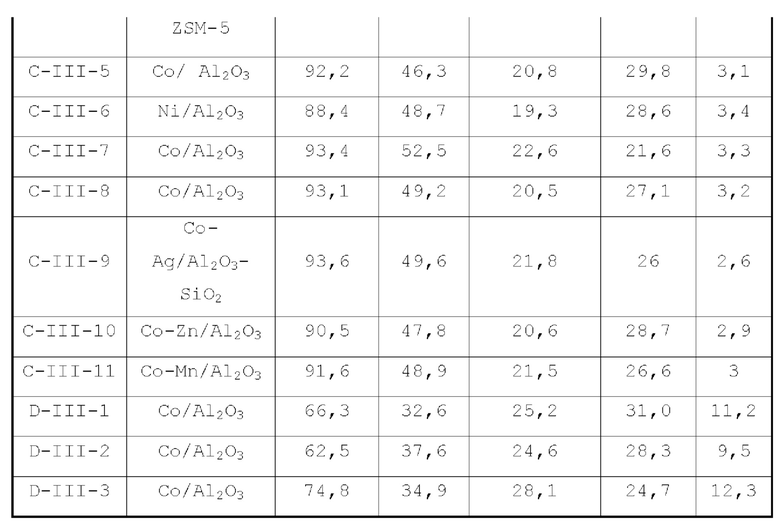
Выражение "другие" в таблице относится к аминам, содержащим 12 атомов углерода, которые трудно диффундируют из поровых каналов, где образуется относительно большое количество указанного амина, тем самым приводя к блокировке поровых каналов и, в свою очередь, к увеличению количества осажденного углерода, так что активность катализатора будет быстро снижаться. После оценки в течение 240 часов катализаторы выгружали и подвергали термогравиметрическому анализу, результаты которого показывают, что осаждение углерода на катализаторах D-III-1, D-III-2 и D-III-3 было по меньшей мере в два раза больше, чем на катализаторах C-III-1 - C-III-11.
После 1000 часов оценки результаты показывают, что снижение каталитической активности катализаторов C-III-1 - C-III-11 было пренебрежимо малым, а именно конверсия и селективность катализаторов, определенные после 1000 часов непрерывной эксплуатации, не снизились существенно по сравнению со значениями, определенными после 24 0 часов работы. В то же время конверсия в случае катализаторов D-III-1, D-III-2 и D-III-3 снизилась соответственно на 29%, 32% и 35%, а селективность этих катализаторов по гександиамину снизилась соответственно на 15,1%, 12,8% и 16,3%.
Тестовый пример III-3
Этот тестовый пример иллюстрирует способ получения этиламина путем гидроаминирования этанола с использованием катализатора по третьему типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-III-8, полученного в примере III-8, загружали в реактор с неподвижным слоем, активировали водородом 2 часа при 220°С, затем охлаждали до 160°С, давление в системе повышали до 1,6 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 135°С, затем отправляли в верхний конец реактора; этанол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и этанолом составляло 3:8:1, объемная скорость жидкого этанола составляла 0,45 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 170°С и давлении реакции 1,6 МПа, после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере III-2). Результаты анализа представлены в таблице III-3.
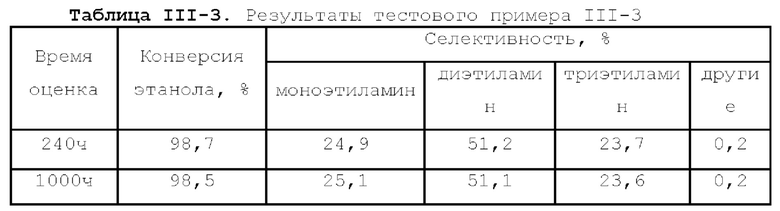
Тестовый пример III-4
Этот тестовый пример иллюстрирует способ получения изопропиламина гидроаминированием ацетона с использованием катализатора по третьему типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-III-10, полученного в примере III-10, загружали в реактор с неподвижным слоем, активировали водородом 2 часа при 200°С, затем охлаждали до 145°С, давление в системе повышали до 1,5 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 110°С, после чего отправляли в верхний конец реактора; ацетон подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и ацетоном составляло 3:6:1, объемная скорость жидкого ацетона составляла 0,4 ч-1, каталитическую реакцию аминирования проводили в реакторе, и после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа, способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере III-2). Результаты анализа приведены в таблице III-4.
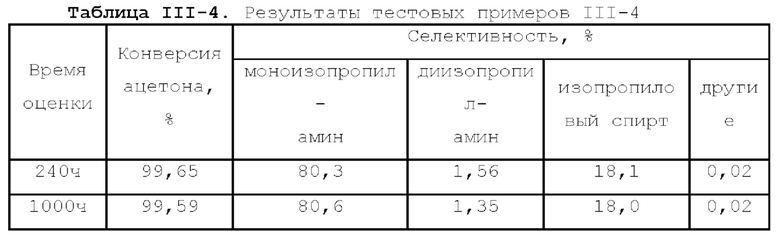
Серия примеров IV
Ниже четвертый тип вариантов осуществления настоящей заявки дополнительно подробно иллюстрируется на примерах серии примеров IV. В следующих примерах серии примеров IV порошок псевдобемита имел содержание Al2O3 70 вес. % в пересчете на сухое вещество, а силиказоль был приобретен у компании Qingdao Ocean Chemical Co., Ltd. под торговым наименованием JN-40.
Пример IV-1
Порошок псевдобемита (удельная поверхность 288 м2/г, объем пор 0,91 мл/г, легирующий элемент Р присутствует в порошке псевдобемита в количестве 0,22 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали с получением полосок диаметром 5 мм нарезали на отрезки длиной 4 мм, сушили при 120°С в течение 8 ч, затем обжигали 6 ч при 550°С. В воде растворяли 32,1 г шестиводного нитрата магния (ч.д.а.), получая 85 мл раствора, и водный раствор нитрата магния вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 100°С 10 часов и затем обжигали 5 часов при 820°С с получением желаемой подложки.
Растворяли в воде 186,5 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 6,83 г шестиводного нитрата цинка (ч.д.а.) и 1,16 г пятиводного нитрата висмута (ч.д.а.) получая 148 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 ч при 430°С, получая катализатор С-IV-1.
Пример IV-2
Порошок псевдобемита (удельная поверхность 285 м2/г, объем пор 0,93 мл/г, легирующий элемент В присутствует в порошке псевдобемита в количестве 0,53 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, экструдировали в гранулы в форме трилистника толщиной 3 мм, сушили при 120°С в течение 15 часов и затем обжигали 8 часов при 520°С. В воде растворяли 11,8 г пятиводного нитрата кальция (ч.д.а.), получая 91 мл раствора, и водный раствор нитрата кальция вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 110°С 10 часов и дополнительно обжигали при 800°С в течение 6 часов с получением желаемой подложки.
Растворяли в воде 151,7 г шестиводного нитрата никеля (технический сорт, чистота 98%), 6,83 г шестиводного нитрата цинка (ч.д.а.) и 1,16 г пятиводного нитрата висмута (ч.д.а.), получая 156 мл раствора, раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 ч при 430°С с получением катализатора C-IV-2.
Пример IV-3
Псевдобемит (удельная поверхность 285 м2/г, объем пор 0,9 мл/г, легирующий элемент Р присутствует в порошке псевдобемита в количестве 0,23 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, соединяли с разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, однородно перемешивали, экструдировали с получением полосок диаметром 5 мм, нарезали на отрезки длиной 4 мм, сушили 8 часов при 120°С и затем обжигали 6 ч при 550°С. Растворяли в воде 32,1 г шестиводного нитрата магния (ч.д.а.), получая 85 мл раствора, и водный раствор нитрата магния вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 100°С в течение 10 часов, после чего обжигали при 820°С 5 часов с получением желаемой подложки.
Растворяли в воде 45,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 6,83 г шестиводного нитрата цинка (ч.д.а.) и 1,16 г пятиводного нитрата висмута (ч.д.а.) получая 14 6 мл раствора, раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 ч при 430°С, получая катализатор С-IV-3.
Пример IV-4
Подложку готовили, как описано в примере IV-1, за исключением того, что порошок псевдобемита однородно смешивали с порошком молекулярного сита (ZSM-5, предприятие по производству катализаторов при университете г. Нанкай, мольное отношение SiO2/Al2O3=45), и количество порошка молекулярного сита ZSM-5 подбирали так, чтобы содержание в подложке оксида алюминия, происходящего из псевдобемита, составляло 85% от полного веса подложки.
Растворяли в воде 176,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 4,55 г шестиводного нитрата цинка (ч.д.а.) и 2,32 г пятиводного нитрата висмута (ч.д.а.) получая 178 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С 4 часа, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 ч при 430°С с получением катализатора C-IV-4.
Пример IV-5
Порошок псевдобемита (удельная поверхность 258 м2/г, объем пор 0,65 мл/г, легирующий элемент Р присутствует в порошке псевдобемита в количестве 0,18 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 4,5 об. % азотной кислоты, экструдировали в гранулы толщиной 4 мм в форме трилистника, сушили при 100°С 18 часов и затем обжигали при 560°С в течение 5 часов. Растворяли в воде 11,8 г пятиводного нитрата кальция (ч.д.а.), получая 73 мл раствора, и водный раствор нитрата кальция вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 120°С 10 часов и дополнительно обжигали 6 часов при 970°С с получением желаемой подложки.
Растворяли в воде 75,6 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 4,55 г шестиводного нитрата цинка (ч.д.а.) и 2,32 г пятиводного нитрата висмута (ч.д.а.) получая 124 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 100°С 10 часов после каждой пропитки распылением, затем обжигали при 388°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 420°С 4 ч, получая катализатор С-IV-5.
Пример IV-6
Псевдобемит (удельная поверхность 318 м2/г, объем пор 0,93 мл/г, легирующий элемент В присутствует в порошке псевдобемита в количестве 3,66 г на 100 г используемого порошка псевдобемита, рассчитанных как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5 об. % азотной кислоты, в процессе замешивания добавляли силиказоль (JN-40, доступен от Qingdao Ocean Chemical Co., Ltd.) однородно перемешивали, экструдировали с получением зубчатых сфер диаметром 3,5 мм, сушили 4 ч при 120°С и затем обжигали 5 ч при 630°С. Растворяли в воде 7,6 г нитрата бария (ч.д.а.), получая 88 мл раствора, и водный раствор нитрата бария вводили на 100 г обожженного продукта путем пропитки распылением, после чего сушили 12 часов при 100°С и дополнительно обжигали при 880°С в течение 3 часов с получением желаемой подложки. Количество силиказоля подбирали так, чтобы весовое отношение оксида алюминия к оксиду кремния в подложке составляло 4:1.
Растворяли в воде 126,4 г шестиводного нитрата никеля (технический сорт, чистота 98%), 4,55 г шестиводного нитрата цинка (ч.д.а.) и 2,32 г пятиводного нитрата висмута (ч.д.а.), получая 160 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С 4 часа, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3,5 ч при 420°С, получая катализатор С-IV-6.
Пример IV-7
Использовали подложку, полученную в примере IV-1.
Растворяли в воде 201,6 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 6,83 г шестиводного нитрата цинка (ч.д.а.) и 1,16 г пятиводного нитрата висмута (ч.д.а.), получая 150 мл раствора, раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С 4 часа после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 3 ч при 430°С с получением катализатора C-IV-7.
Пример IV-8
Псевдобемит (удельная поверхность 290 м2/г, объем пор 0,78 мл/г, легирующий элемент S присутствует в порошке псевдобемита в количестве 0,88 г в расчете на 100 г используемого порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 4,5 об. % азотной кислоты, силиказоль добавляли в процессе размешивания, однородно перемешивали, экструдировали в гранулы толщиной 4 мм в форме трилистника, сушили 20 ч при 80°С и затем обжигали 7 ч при 550°С. В воде растворяли 17,7 г пятиводного нитрата кальция (ч.д.а.), получая 84 мл раствора, и водный раствор нитрата кальция вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 120°С 10 часов и дополнительно обжигали при 900°С в течение 4 часов с получением желаемой подложки. Количество силиказоля подбирали так, чтобы мольное отношение оксида алюминия к оксиду кремния в подложке составляло 86:14.
Растворяли в воде 100,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 2,88 г шестиводного нитрата цинка (ч.д.а.) и 3,48 г пятиводного нитрата висмута (ч.д.а.), получая 152 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 100°С 10 часов после каждой пропитки распылением, затем обжигали при 400°С в течение 4 часов, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали при 430°С в течение 4 ч, получая катализатор C-IV-8.
Пример IV-9
Псевдобемит (удельная поверхность 320 м2/г, объем пор 0,95 мл/г, легирующий элемент F присутствует в порошке псевдобемита в количестве 0,82 г в расчете на 100 г порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 5,2 об. % азотной кислоты, силиказоль добавляли в процессе размешивания, однородно перемешивали, экструдировали в гранулы толщиной 3 мм в форме трилистника, сушили при 100°С в течение 8 ч и затем обжигали 4 часа при 620°С. В воде растворяли 3,8 г нитрата бария (ч.д.а.), получая 83 мл раствора, и водный раствор нитрата бария вводили на 100 г обожженного продукта путем пропитки распылением и затем сушили при 120°С 8 часов и обжигали при 950°С в течение 5 часов с получением желаемой подложки. Количество используемого силиказоля подбирали так, чтобы мольное отношение оксида алюминия к оксиду кремния в подложке составляло 89:11.
Растворяли в воде 176,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 2,88 г шестиводного нитрата цинка (ч.д.а.) и 3,48 г пятиводного нитрата висмута (ч.д.а.), получая 160 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 110°С в течение 8 часов после каждой пропитки распылением, затем обжигали 4 часа при 400°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 4,5 ч при 420°С, получая катализатор С-IV-9.
Пример IV-10
Порошок псевдобемита (удельная поверхность 291 м2/г, объем пор 0,93 мл/г, легирующий элемент S присутствует в порошке псевдобемита в количестве 0,95 г в расчете на 100 г порошка псевдобемита, выраженного как Al2O3), полученный алюминийсульфатным способом, замешивали разбавленным водным раствором кислоты, содержащим 4 об. % азотной кислоты, затем экструдировали в гранулы толщиной 3,5 мм в форме трилистника, сушили 8 часов при 100°С и затем обжигали при 600°С в течение 5 часов. В 92 мл воды растворяли 42,7 г пятиводного нитрата магния (ч.д.а.), водный раствор нитрата магния вводили на 100 г обожженного продукта путем пропитки распылением, сушили при 120°С 8 часов и дополнительно обжигали при 830°С в течение 8 часов с получением желаемой подложки.
Растворяли в воде 226,8 г шестиводного нитрата кобальта (технический сорт, чистота 98%), 2,88 г шестиводного нитрата цинка (ч.д.а.) и 4,64 г пятиводного нитрата висмута (ч.д.а.), получая 172 мл раствора, и раствор вводили на полученную выше подложку путем двукратной пропитки распылением, сушили при 120°С в течение 6 часов после каждой пропитки распылением, затем обжигали 4 часа при 4 00°С, после чего восстанавливали водородом при постепенном повышении температуры со скоростью 20°С/ч и, наконец, восстанавливали 4,5 ч при 420°С, получая катализатор С-IV-10.
Пример IV-11
Катализатор C-IV-11 был приготовлен, как описано в примере IV-1, за исключением того, что используемый порошок псевдобемита был легирован элементом Р, и элемент Р присутствовал в порошке псевдобемита в количестве 4,3 г в расчете на 100 г порошка псевдобемита, выраженного как Al2O3.
Пример IV-12
Катализатор C-IV-12 был приготовлен, как описано в примере IV-2, за исключением того, что используемый порошок псевдобемита не содержал легирующего элемента и имел удельную поверхность 286 м2/г и объем пор 0,93 мл/г.
Пример IV-13
Катализатор C-IV-13 был приготовлен, как описано в примере IV-2, за исключением того, что температура второго обжига составляла 1200°С.
Сравнительный пример IV-1
Катализатор D-IV-1 был приготовлен, как описано в примере IV-5, за исключением того, что водный раствор нитрата кальция был заменен равным объемом воды, и раствор, используемый для пропитки подложки распылением, готовили, растворяя 151,7 г шестиводного нитрата никеля (технический сорт, чистота 98%) в воде с получением 158 мл раствора, так что в катализатор в качестве активного металлического компонента вводился только никель.
Сравнительный пример IV-2
Катализатор D-IV-2 был приготовлен, как описано в примере IV-3, за исключением того, что раствор, используемый для пропитки подложки распылением, готовили, растворяя в воде 45,4 г шестиводного нитрата кобальта (технический сорт, чистота 98%) и 1,16 г пятиводного нитрата висмута (ч.д.а.) с получением 148 мл раствора.
Сравнительный пример IV-3
Катализатор D-IV-3 был приготовлен, как описано в примере IV-1, за исключением того, что нитрат цинка был заменен на 8,5 г нитрата меди тригидрата.
Тестовый пример IV-1
Элементный состав подложек и катализаторов анализировали методом эмиссионной спектроскопии с индуктивно-связанной плазмой, содержание элемента (иона), отличного от подложки, выражали в весе на 100 г матрицы, полученные выше подложки анализировали методом зондовой адсорбционной спектроскопии, методом адсорбции-десорбции азота по БЭТ, результаты приведены в таблице IV-1.
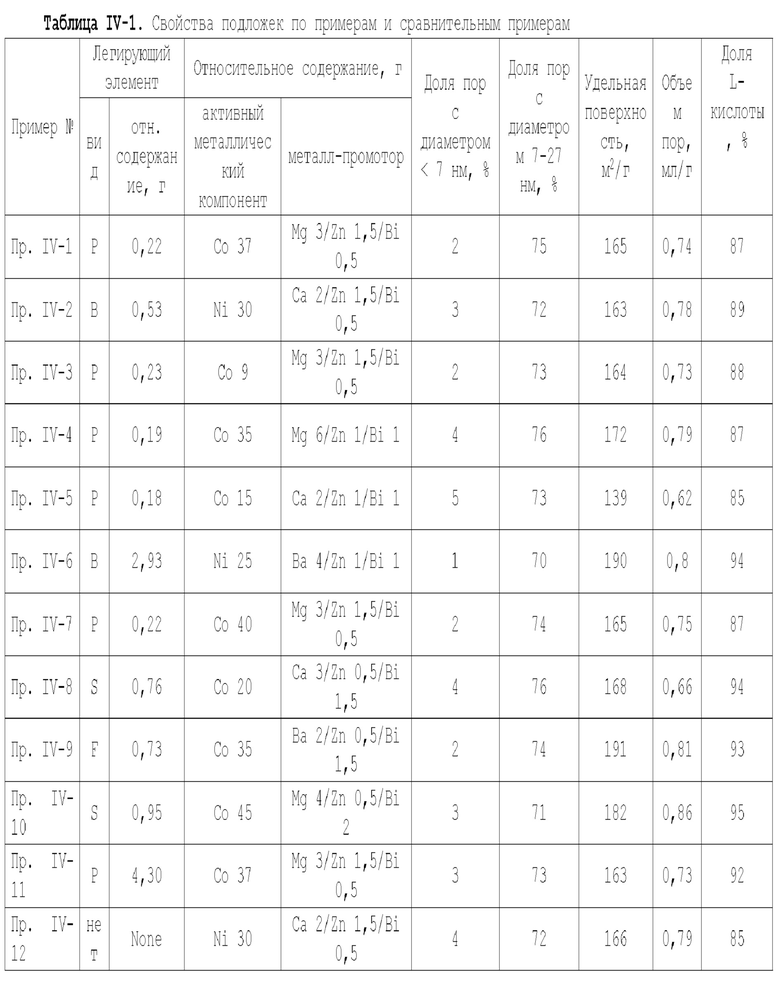
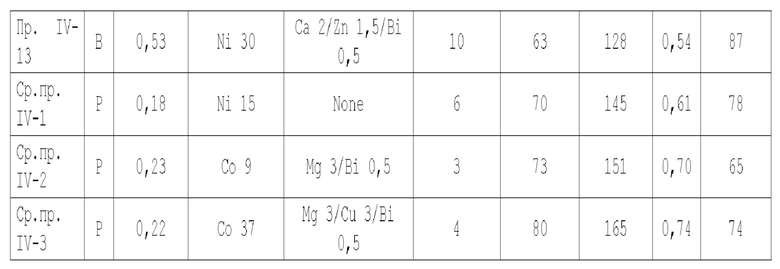
Тестовый пример IV-2
Этот тестовый пример иллюстрирует способ получения 1,6-гександиамина путем гидроаминирования 1,6-гександиола с использованием катализатора по четвертому типу вариантов осуществления настоящей заявки.
Отмеряли соответственно по 100 мл катализаторов, полученных в примерах и сравнительных примерах, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 172°С, давление в системе повышали до 12,5 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 150°С и отправляли в верхний конец реактора; затем нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и 1,6-гександиолом составляло 3:18:1, объемная скорость жидкого 1, 6-гександиола составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 195°С и давление реакции 12,5 МПа, после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали. Результаты анализа представлены в таблице IV-2.
Анализ образца проводили методом газовой хроматографии, калибровку осуществляли с использованием поправочного коэффициента для рецептуры эталонного образца.
Конверсию и селективность рассчитывали, исходя из мольного содержания каждого компонента в реакционном растворе, способы расчета были такими же, как описано в тестовом примере I-2.
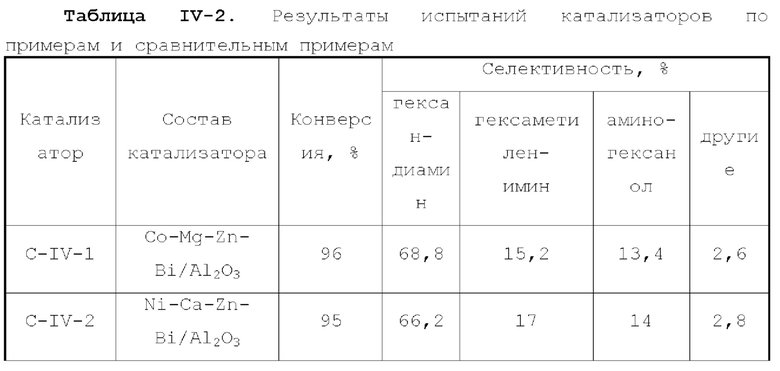
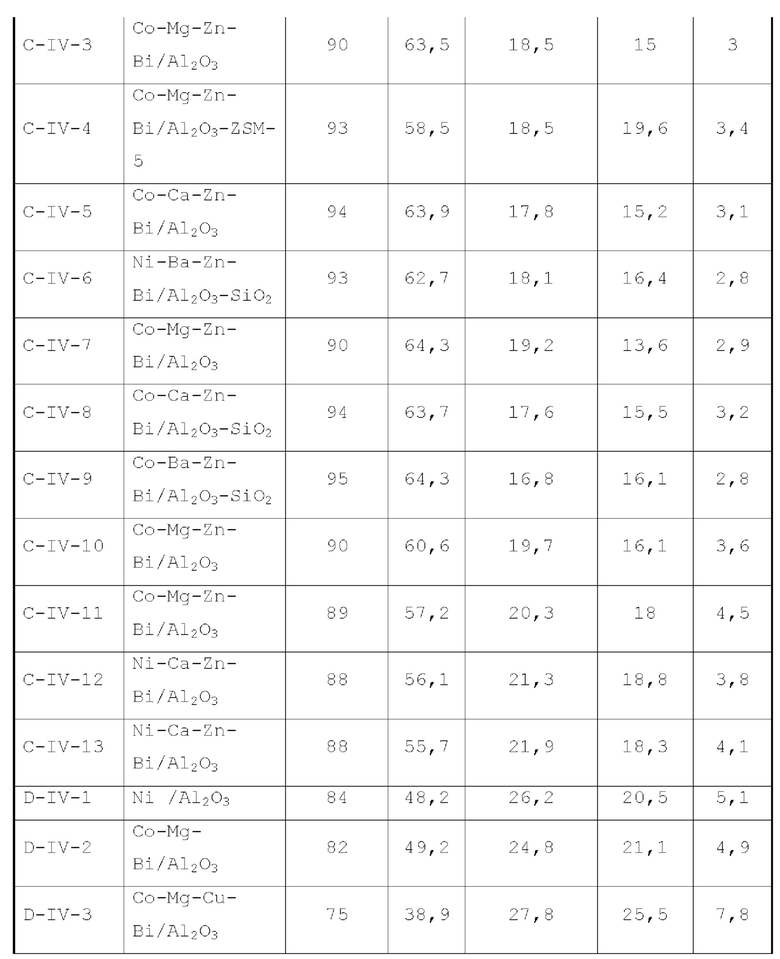
Как видно из таблицы, катализаторы D-IV-1 - D-IV-3 обеспечивают меньшую конверсию в идентичных условиях процесса, что указывает на более низкую активность, чем у катализаторов С-IV-1 - C-IV-13 по настоящей заявке; по сравнению с катализаторами C-IV-1 - C-IV-13 селективность катализаторов D-IV-1 - D-IV-3 по гександиамину ниже, тогда как чем селективность по "другим" компонентам выше, это указывает на то, что реакционный материал с трудом десорбируется из катализаторов D-IV-1 - D-IV-3 и, таким образом, подвергается дополнительным реакциям с образованием других побочных продуктов.
Тестовый пример IV-3
Этот тестовый пример иллюстрирует способ получения этиламина путем гидроаминирования этанола с использованием катализатора по четвертому типу вариантов осуществления настоящей заявки.
Отмеряли 100 мл катализатора C-IV-3, полученного в примере IV-3, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 173°С, давление в системе повышали до 1,65 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 110°С, затем отправляли в верхний конец реактора; этанол подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и этанолом составляло 3:5:1, объемная скорость жидкого этанола составляла 0,5 ч"1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 178°С и давление реакции 1,6 МПа, после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности аналогичны использовавшимся в тестовом примере IV-2). Результаты анализа представлены в таблице IV-3.
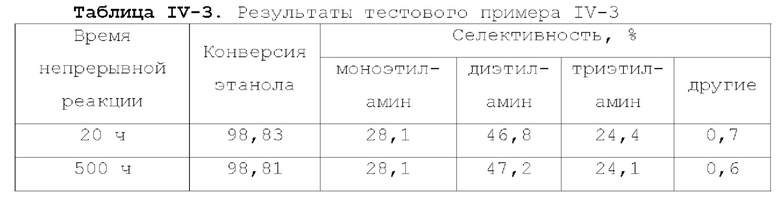
Тестовый пример IV-4
Катализаторы C-IV-2, C-IV-9, D-IV-1, D-IV-2, D-IV-3 загружали соответственно в реактор с неподвижным слоем в тех же условиях, что и в тестовом примере IV-2 с единственным отличием, что была увеличена продолжительность реакции и тест проводили в течение 500 часов. Анализ и сравнение проводили на реакционном растворе при времени реакции 20 часов (условия анализа и способы расчета конверсии и селективности идентичны тестовому примеру IV-2) и на реакционном растворе после 500 часов реакции (условия анализа и способы расчета конверсии и селективности идентичны тестовому примеру IV-2). Результаты анализа представлены в таблице IV-4.
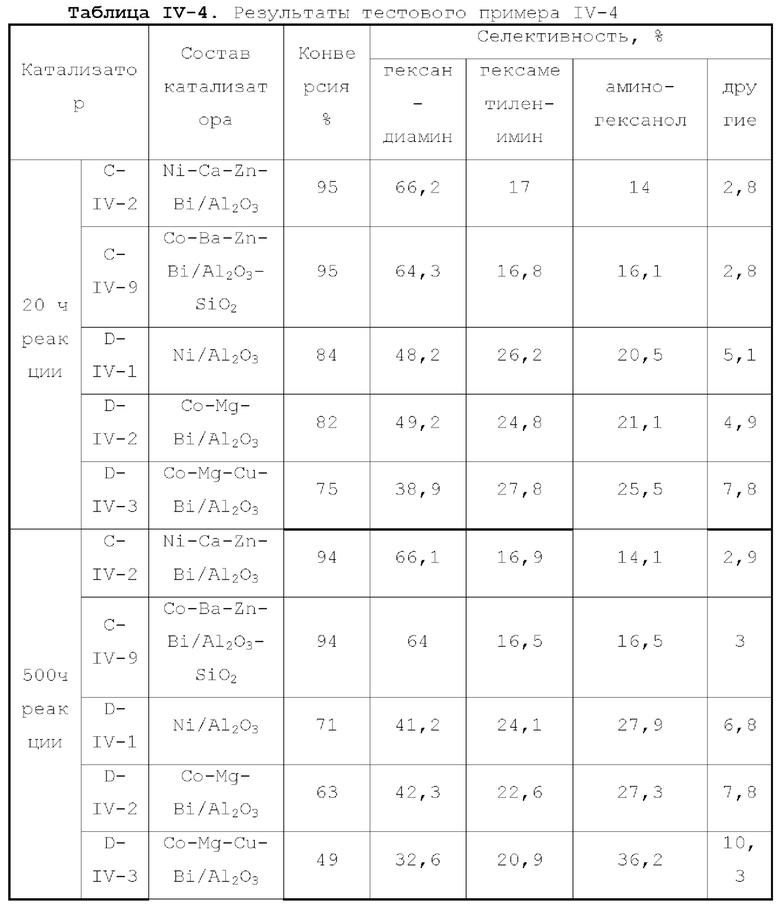
Согласно оценке после 500 часов, активность и селективность катализаторов C-IV-2 и C-IV-9 изменилась несущественно, тогда как активность и селективность сравнительных катализаторов D-IV-1, D-IV-2 и D-IV-3 явно уменьшились. Определяли удельную поверхность, объем пор и осаждение углерода для каждого катализатора, и было обнаружено, что удельная поверхность и объем пор катализаторов D-IV-1, D-IV-2 и D-IV-3 значительно уменьшились, тогда как удельная поверхность и объем пор катализаторов C-IV-2 и C-IV-9 изменились несущественно (степень уменьшения менее 2%), и осаждение углерода на катализаторах D-IV-1, D-IV-2 и D-IV-3 было более чем вдвое выше, чем на катализаторах C-IV-2 и C-IV-9.
Тестовый пример IV-5
Этот тестовый пример иллюстрирует способ получения гександиамина из смеси 1,6-гександиола, гексаметиленимина и аминогексанола с использованием катализатора по четвертому типу вариантов осуществления настоящей заявки.
Отвешивали 100 мл катализатора C-IV-3, полученного в примере IV-3, и загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 175°С, давление в системе повышали до 14 МПа, используя водород, затем в реакционную систему вводили аммиак, используя насос-дозатор, предварительно нагревали до 150°С и отправляли в верхний конец реактора; смешанный раствор, содержащий 53 вес. % 1,6-гександиола, 30 вес. % гексаметиленимина и 17 вес. % 6-амино-1-гексанола, подавали в верхний конец реактора через насос-дозатор, водород стабильно вводили через газовый массовый расходомер, при этом мольное соотношение между водородом, аммиаком и суммарным количеством трех веществ в смешанном растворе составляло 3:10:1, объемная скорость жидкого смешанного раствора составляла 0,5 ч"1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 180°С и давлении реакции 14 МПа; после того, как реакция стабилизировались, отбирали образец реакционного раствора и анализировали (условия анализа и способы расчета конверсии и селективности такие же, как в тестовом примере IV-2). Результаты анализа представлены в таблице IV-5.
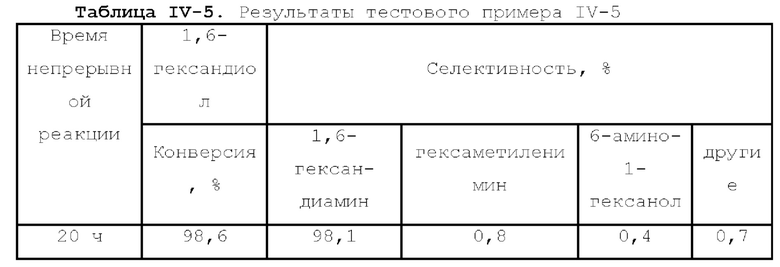

Настоящая заявка была подробно проиллюстрирована выше на предпочтительных вариантах осуществления, но она не ограничивается этими вариантами осуществления. Различные модификации могут быть выполнены в соответствии с изобретательской концепцией настоящей заявки, и эти модификации будут находиться в пределах объема настоящей заявки.
Следует отметить, что различные технические признаки, описанные в приведенных выше вариантах осуществления, могут комбинироваться любым подходящим образом без противоречия, и во избежание ненужного повторения различные возможные комбинации в настоящей заявке не описываются, но такие комбинации также будут охватываться объемом настоящей заявки.
Кроме того, различные варианты осуществления настоящей заявки могут произвольно комбинироваться, если только эта комбинация не противоречит сущности настоящей заявки, и такие комбинированные варианты осуществления следует рассматривать как раскрытие настоящей заявки.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР ДЛЯ АМИНИРОВАНИЯ И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2836173C1 |
| Катализатор для обессеривания жидких нефтепродуктов, его получение и применение | 2017 |
|
RU2745372C2 |
| СФЕРОПОДОБНЫЙ СУПЕРМАКРОПОРИСТЫЙ МЕЗОПОРИСТЫЙ МАТЕРИАЛ И СОДЕРЖАЩИЙ ЕГО ПОЛИОЛЕФИНОВЫЙ КАТАЛИЗАТОР | 2020 |
|
RU2829734C1 |
| КОМПОЗИЦИЯ, СПОСОБНАЯ СНИЖАТЬ ВЫБРОСЫ CO И NOX, ЕЕ СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ И СПОСОБ КАТАЛИТИЧЕСКОГО КРЕКИНГА В ПСЕВДООЖИЖЕННОМ СЛОЕ | 2018 |
|
RU2772281C2 |
| Материал-носитель из оксида алюминия и способ его получения, катализатор гидрирования и способ гидрирования остаточного масла | 2018 |
|
RU2753336C1 |
| КАТАЛИЗАТОР ДЕСУЛЬФУРИЗАЦИИ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2017 |
|
RU2749402C1 |
| АМИНОПОЛИМЕР, СПОСОБ ПОЛУЧЕНИЯ АМИНОПОЛИМЕРА И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2731560C2 |
| КАТАЛИЗАТОР ГИДРИРОВАНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2021 |
|
RU2836692C1 |
| КАТАЛИЗАТОР КАТАЛИТИЧЕСКОГО КРЕКИНГА И ЕГО ПОЛУЧЕНИЕ | 2018 |
|
RU2755891C2 |
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ДЛЯ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2832734C1 |
Изобретение может быть использовано в химической промышленности при получении аминов. Катализатор, пригодный для получения органических аминов путем каталитического аминирования, имеет неорганическую пористую подложку, содержащую алюминий и/или кремний, и активный металлический компонент, нанесенный на подложку. Активный металлический компонент содержит по меньшей мере один металл из группы, состоящей из металлов группы VIII и группы IB, причем подложка имеет содержание L-кислоты 85% или более в расчете на суммарное содержание L-кислоты и В-кислоты. Предложены также способ получения такого катализатора и способ получения органических аминов. Группа изобретений позволяет получить катализатор, обладающий улучшенной каталитической активностью, конверсией реакции, селективностью по продукту и стабильностью. 3 н. и 12 з.п. ф-лы, 16 табл., 62 пр.
1. Катализатор, пригодный для получения органических аминов путем каталитического аминирования, имеющий неорганическую пористую подложку, содержащую алюминий и/или кремний, и активный металлический компонент, нанесенный на подложку, при этом активный металлический компонент содержит по меньшей мере один металл из группы, состоящей из металлов группы VIII и группы IB, причем подложка имеет содержание L-кислоты 85% или более в расчете на суммарное содержание L-кислоты и В-кислоты.
2. Катализатор по п. 1, где подложка содержит матрицу и легирующий элемент, причем матрица выбрана из оксида алюминия, диоксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций и
легирующий элемент является неметаллическим элементом, предпочтительно выбранным из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA, неметаллических элементов группы VIIA и их комбинаций, за исключением хлора, более предпочтительно он выбирается из бора, фтора, фосфора, серы, селена или их комбинации;
предпочтительно легирующий элемент в подложке происходит из неметаллического ион-радикала кислоты, и неметаллический ион-радикал кислоты предпочтительно выбран из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона, селенат-иона и их комбинаций.
3. Катализатор по п. 2, причем подложка обладает по меньшей мере одной из следующих характеристик:
- доля объема пор с диаметром в интервале 7-27 нм в объеме пор подложки превышает 65%, предпочтительно составляет 70-90%, а доля объема пор с диаметром менее 7 нм в объеме пор подложки составляет 0-10%, предпочтительно 0-8%;
- доля объема пор с диаметром меньше 7,5 нм в объеме пор подложки составляет менее 20%, предпочтительно 5-17%, доля объема пор с диаметром меньше 9 нм в объеме пор подложки составляет менее 40%, доля объема пор с диаметром больше 27 нм в объеме пор подложки составляет менее 5%, предпочтительно 0,5-5%, предпочтительно доля объема пор с диаметром больше или равным 7,5 нм и меньше 9 нм в объеме пор подложки составляет 5-17% и доля объема пор с диаметром больше или равным 9 нм и меньше или равным 27 нм в объеме пор подложки составляет 61-89,5%;
- подложка имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, предпочтительно 0,3-0,6 ммоль/г, более предпочтительно 0,3-0,5 ммоль/г;
- содержание оксида алюминия в подложке составляет 65 вес. % или более, предпочтительно 70 вес. % или более, более предпочтительно 75 вес. % или более от полного количества матрицы;
- содержание легирующего элемента составляет от 0,05 до 6 вес. %, предпочтительно от 0,05 до 5 вес. %, более предпочтительно от 0,05 до 4,5 вес. % и особенно предпочтительно от 0,07 до 4 вес. % от полного количества матрицы;
- подложка имеет удельную поверхность 100-220 м2/г, предпочтительно 105-210 м2/г, более предпочтительно 110-210 м2/г и особенно предпочтительно 120-210 м2/г;
- подложка имеет объем пор 0,4-1,1 мл/г, предпочтительно 0,43-1,1 мл/г, более предпочтительно 0,45-1,1 мл/г и особенно предпочтительно 0,45-1 мл/г; и
- изоэлектрическая точка подложки составляет 3-6, предпочтительно 3,5-5,5.
4. Катализатор по п. 2 или 3, причем активный металлический компонент присутствует в количестве от 5 до 45 г, предпочтительно от 8 до 44 г, более предпочтительно от 10 до 38 г и особенно предпочтительно от 15 до 37 г на 100 г матрицы и активный металлический компонент предпочтительно имеет размер зерна меньше 10 нм, более предпочтительно от 3 до 8 нм.
5. Катализатор по любому из пп. 2-4, дополнительно содержащий металл-промотор, нанесенный на подложку, и металл-промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов групп VIB, VIIB, IB, IIB и металлов ряда лантаноидов, предпочтительно по меньшей мере один металл, выбранный из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се;
предпочтительно металл-промотор присутствует в количестве от 0 до 10 г, предпочтительно от 0,1 до 10 г, более предпочтительно от 0,5 до 8 г на 100 г матрицы.
6. Катализатор по п. 5, причем
металл-промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, причем весовое отношение металла группы VIIB к металлу группы IB, рассчитанное на элементарные металлы, составляет (0,05-15):1, предпочтительно (0,1-12):1; или альтернативно
металл-промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB, причем весовое отношение металла группы VIIB к металлу группы IIB, рассчитанное на элементарные металлы, составляет (0,2-20):1, предпочтительно (0,3-6):1; или альтернативно
металл-промотор содержит комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB, причем весовое соотношение между металлом группы VIB, металлом группы IB и металлом группы IIB, рассчитанное на элементарные металлы, составляет (0,1-10):(0,1-10):1, предпочтительно (0,2-8):(0,2-8):1,
предпочтительно металл группы VIIB выбран из марганца, рения или их комбинации, металл группы IB выбран из меди, серебра, золота или их комбинации, металл группы IIB является цинком и/или металл группы VIB выбран из молибдена, вольфрама или их комбинации.
7. Катализатор по любому из пп. 2-4, дополнительно содержащий металл-промотор, нанесенный на подложку, причем металл-промотор представляет собой комбинацию по меньшей мере одного металла группы IIA, по меньшей мере одного металла группы IIB и по меньшей мере одного металла группы VA,
предпочтительно металл-промотор присутствует в количестве от 0,1 до 10 г, предпочтительно от 0,5 до 6 г на 100 г матрицы;
предпочтительно весовое соотношение между металлом группы IIА, металлом группы IIB и металлом группы VA в металле-промоторе составляет (0,1-10):(0,1-10):1, предпочтительно (0,2-8): (0,2-8):1;
металл группы IIA предпочтительно выбран из магния, кальция, бария или их комбинации, металл группы IIB является цинком и/или металл группы VA является висмутом.
8. Способ получения катализатора по любому из пп. 1-7, включающий этапы:
1) подготовку неорганической пористой подложки, содержащей алюминий и/или кремний, причем подложка имеет содержание L-кислоты 85% или более от суммарного содержания L-кислоты и В-кислоты,
2) нанесение активного металлического компонента на подложку и
3) проведение термообработки материала, полученного на этапе 2), с получением катализатора,
причем термообработка включает обжиг или комбинацию сушки и обжига, где обжиг проводят при температуре 150-500°С в течение 1-6 ч.
9. Способ по п. 8, в котором стадия 2) дополнительно включает нанесение металла-промотора на подложку;
стадия 3) дополнительно включает проведение восстановительной обработки материала.
10. Способ по п. 8, причем указанный этап 1) "подготовка неорганической пористой подложки, содержащей алюминий и/или кремний," включает последовательное формование, сушку и обжиг смеси, содержащей легирующий элемент и матрицу или ее предшественник, с получением подложки,
причем матрица выбрана из оксида алюминия, оксида кремния, молекулярных сит, диатомита, алюмосиликатов или их комбинаций, предшественник оксида алюминия предпочтительно представляет собой псевдобемит с удельной поверхностью 250-400 м2/г, предпочтительно 255-360 м2/г, более предпочтительно 255-340 м2/г, особенно предпочтительно 260-330 м2/г и объемом пор 0,5-1,3 мл/г, предпочтительно 0,75-1,25 мл/г, более предпочтительно 0,78-1,2 мл/г, особенно предпочтительно 0,78-1,1 мл/г;
легирующий элемент является неметаллическим элементом, предпочтительно выбранным из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA и их комбинаций, за исключением хлора, более предпочтительно он выбирается из бора, фтора, фосфора, серы, селена или их комбинации;
условия сушки на этапе 1) предпочтительно включают температуру 80-150°С и продолжительность сушки 6-20 ч; и
условия обжига на этапе 1) предпочтительно включают температуру 500-1120°С, например 500-650°С, предпочтительно 700-1100°С, более предпочтительно 800-1050°С и продолжительность обжига 2-20 ч.
11. Способ по п. 10, где легирующий элемент получают с использованием модификатора подложки, содержащего по меньшей мере одно соединение, способное предоставить неметаллический ион-радикал кислоты, например неорганическую кислоту и/или неорганическую соль, содержащую неметаллический кислотный радикал, неметаллический ион-радикал кислоты выбран предпочтительно из борат-иона, фторид-иона, фосфат-иона, сульфат-иона, селенат-иона или их комбинации;
предпочтительно модификатор подложки выбран из борной кислоты, бората никеля, бората кобальта, бората калия, бората аммония, бората магния, фторида калия, фторида магния, фторида кобальта, фторида никеля, фтористоводородной кислоты, фторида аммония, фосфорной кислоты, фосфата алюминия, трикалийфосфата, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, фосфата аммония, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, сульфата калия, сульфата магния, фосфата стронция, сульфата стронция, селеновой кислоты или их комбинаций.
12. Способ по любому из пп. 8-11, где стадия 2) нанесения включает пропитку подложки раствором, содержащим предшественник активного металлического компонента и, необязательно, предшественник металла-промотора, пропиточный раствор предпочтительно имеет рН в интервале 3,5-5,5.
13. Способ получения органических аминов, включающий контактирование исходного материала для аминирования и аминирующего реагента с катализатором по любому из пп. 1-7 в присутствии водорода для реакции аминирования, чтобы получить органический амин,
причем исходный материал для аминирования выбран из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов или их комбинаций;
аминирующий реагент выбран из аммиака, С1-12 первичных аминов, С1-12 вторичных аминов или их комбинации.
14. Способ по п. 13, где условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1_6): (2-35):1, предпочтительно (1-6): (2-33):1, более предпочтительно (1-5): (3-33):1, температуру 105-230°С, предпочтительно 110-220°С, более предпочтительно 110-210°С, давление 0,7-25 МПа, предпочтительно 1-25 МПа, более предпочтительно 1-22 МПа, особенно предпочтительно 1-17 МПа и часовую объемную скорость 0,06-1 м3/(м3⋅ч).
15. Способ по п. 14, причем,
когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования предпочтительно (1-4):(2-9):1, более предпочтительно (1_4):(2-8):1, температуру 130-210°С, предпочтительно 130-208°С, более предпочтительно 130-200°С, давление 0,8-3,5 МПа, предпочтительно 1-2,5 МПа и часовую объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
когда исходный материал для аминирования представляет собой кетон, условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(2-6):1, предпочтительно (1-4):(2-5):1, температуру 105-180°С, предпочтительно 110-170°С, более предпочтительно 110-160°С, давление 0,7-2,5 МПа, предпочтительно 1-2,5 МПа, более предпочтительно 1-2 МПа и часовую объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч);
когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (1-4):(3-23):1, предпочтительно (1-4):(3-20): 1, более предпочтительно (1-4):(3-10):1, температуру 130-200°С, давление 1-16 МПа, предпочтительно 1-13 МПа, более предпочтительно 1-11 МПа и часовую объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
когда исходный материал для аминирования представляет собой двухатомный спирт, условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (0,3-5):(2-35):1, предпочтительно (1-4):(3-35):1, более предпочтительно (1-4):(3-33):1, особенно предпочтительно (1_4):(3-32):1, температуру 130-230°С, предпочтительно 130-220°С, более предпочтительно 130-210°С, давление 1-25 МПа, предпочтительно 1-22 МПа, более предпочтительно 1-17 МПа и часовую объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,9 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч); или,
когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, условия аминирования включают мольное соотношение между водородом, аминирующим реагентом и исходным материалом для аминирования (0,3-4): (3-35):1, предпочтительно (1-4): (3-33):1, более предпочтительно (1-4): (3-32):1, температуру 130-230°С, предпочтительно 130-220°С, более предпочтительно 130-210°С, давление 1-22 МПа, предпочтительно 1-17 МПа и часовую объемную скорость 0,1-0,9 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
| CN 101569862 A, 04.11.2009 | |||
| CN 101829581 A, 15.09.2010 | |||
| KR 20120103323 A, 19.09.2012 | |||
| СПОСОБ АМИНИРОВАНИЯ | 1998 |
|
RU2215734C2 |
| Способ получения диметиламина | 1984 |
|
SU1405699A3 |
| LU Y | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Халат для профессиональных целей | 1918 |
|
SU134A1 |

