Родственные заявки
Настоящая заявка испрашивает приоритет по предварительной заявке на патент США под регистрационным № 62/944082, поданной 5 декабря 2019 года, полное раскрытие которой настоящим включено в данный документ посредством ссылки.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящая заявка содержит перечень последовательностей, который был подан в электронном виде в формате с кодировкой ASCII и настоящим включен посредством ссылки во всей своей полноте. Копия указанного файла с кодировкой ASCII, созданная 4 декабря 2020 года, имеет название 712533_SA9-295PC_ST25.txt, и ее размер составляет 9675 байта.
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к составам на основе антител, применимых для лечения заболевания. Более конкретно, оно относится к составам на основе антител к CD38, подходящим для применения при подкожном введении для лечения форм рака, включая множественную миелому, а также других заболеваний и состояний, в которых играют важную роль CD38+ клетки.
Предпосылки изобретения
CD38 представляет собой трансмембранный гликопротеин II типа массой 45 кДа с длинным С-концевым внеклеточным доменом и коротким N-концевым цитоплазматическим доменом. Белок CD38 представляет собой бифункциональный, выделяемый во внешнюю среду фермент, который может катализировать превращение NAD+ в циклическую ADP-рибозу (cADPR), а также гидролизует cADPR до ADP-рибозы.
Экспрессия CD38 повышена при многих гематологических злокачественных новообразованиях и у линий клеток, происходящих от различных гематологических злокачественных новообразований. Более того, наиболее примитивными плюрипотентными стволовыми клетками гематологической системы являются CD38- клетки. Экспрессия CD38 при гематологических злокачественных новообразованиях и ее корреляция с прогрессированием заболевания при хроническом лимфоцитарном лейкозе (CLL) делают CD38 привлекательной мишенью для терапии посредством антител.
Сообщается также, что CD38+ клетки ассоциированы с различными другими заболеваниями и состояниями, включая многие аутоиммунные заболевания, такие как ревматоидный артрит и красная волчанка, а также острое повреждение почек, вызванное липополисахаридом (LPS) или сепсисом (Shu B et al., Cell Signal (2018) 42: 249-58).
Антитела к CD38, которые специфически распознают CD38, были описаны ранее, например, в международной заявке на патент WO2006/099875. Однако данные антитела не смогли индуцировать апоптоз при применении в качестве монотерапии и инкубации с клетками, экспрессирующими CD38+.
Моноклональные антитела к CD38 были описаны в международной заявке на патент WO2008/047242.
О применении данных специфических антител к CD38 в комбинации с цитотоксическими средствами, такими как цитарабин, винкристин, циклофосфамид и мелфалан, сообщалось в международных заявках на патент WO2010/061357, WO2010/061358, WO2010/061359 и WO2010/061360.
В международных заявках на патент WO2015/066450, WO2012/076663 и WO2014/089416, WO2014/159911 также описано применение гуманизированного варианта 38SB19 (также известного в качестве SAR650984 или изатуксимаба). Клиническое исследование фазы 3 (NCT02990338) с применением изатуксимаба в комбинации с помалидомидом и дексаметазоном для лечения пациентов с рецидивирующей/рефрактерной множественной миеломой недавно достигло своей первичной конечной точки, продления выживаемости без прогрессирования.
Сущность изобретения
Аспект настоящего изобретения относится к составам на основе антитела к CD38, подходящим для подкожного введения субъекту. Преимуществом является то, что раскрытые в данном документе составы подходят для подкожного введения посредством либо инъекции, либо инфузии, включая подкожную инфузию большого объема. Составы можно применять для лечения заболевания или состояния, характеризующегося клетками, экспрессирующими CD38. Такие заболевания и состояния включают без ограничения солидные опухоли, экспрессирующие CD38, такие как рак предстательной железы, различные гематологические злокачественные новообразования, такие как неходжкинская лимфома (NHL), множественная миелома (ММ), острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (В-клеточный ALL) и/или хронический лимофцитарный лейкоз (CLL). Такие заболевания и состояния дополнительно включают без ограничения аутоиммунные заболевания, включая ревматоидный артрит и системную красную волчанку, и острое повреждение почек, вызванное липополисахаридом (LPS) или сепсисом.
В определенных вариантах осуществления настоящее изобретение относится к составу на основе антитела к CD38, характеризующемуся высокой концентрацией антитела, рН 5,9-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
В определенных вариантах осуществления настоящее изобретение относится к составу, содержащему по меньшей мере 100 мг/мл антитела к CD38, средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, где состав характеризуется рН 5,9-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
Аспект настоящего изобретения относится к составу, содержащему по меньшей мере 100 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR2-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, и
состав характеризуется рН 5,9-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления буферное средство представляет собой гистидин.
В определенных вариантах осуществления буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38.
В определенных вариантах осуществления стабилизатор представляет собой сахарозу.
В определенных вариантах осуществления стабилизатор представляет собой 2% (вес/об.) сахарозы.
В некоторых вариантах осуществления рН составляет 5,9-7,0.
В некоторых вариантах осуществления рН составляет 5,9-6,5.
Аспект настоящего изобретения относится к составу, содержащему 140 мг/мл антитела к CD38, где антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR- H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, и где
состав характеризуется рН 6,2 и вязкостью не более 14 мПа⋅с при 20°С.
Аспект настоящего изобретения относится к составу, содержащему 140 мг/мл антитела к CD38, где антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR- H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80, и
состав характеризуется рН 6,2 и вязкостью не более 14 мПа⋅с при 20°С.
В определенных вариантах осуществления состав подходит для подкожного введения посредством либо инъекции, либо инфузию, включая подкожную инфузию большого объема.
В определенных вариантах осуществления VH антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 7, и VL антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 8.
В определенных вариантах осуществления антитело к CD38 представляет собой изатуксимаб.
Аспект настоящего изобретения относится к упакованному фармацевтическому продукту, содержащему стерильный контейнер, содержащий терапевтически эффективное количество состава по настоящему изобретению.
Аспект настоящего изобретения относится к устройству, содержащему терапевтически эффективное количество состава по настоящему изобретению.
В определенных вариантах осуществления устройство может представлять собой, например, шприц, шприцевую помпу и инфузионный насос, содержащие состав.
В определенных вариантах осуществления шприц представляет собой предварительно заполненный шприц.
Аспект настоящего изобретения представляет собой способ лечения заболевания или состояния, характеризующегося наличием или активностью CD38+ клеток, включающий введение субъекту, нуждающемуся в этом, эффективного количества состава по настоящему изобретению, где состав вводят подкожно.
В определенных вариантах осуществления заболевание или состояние, характеризующееся наличием или активностью CD38+ клеток, представляет собой CD38+ гематологическое злокачественное новообразование.
В определенных вариантах осуществления заболевание или состояние, характеризующееся наличием или активностью CD38+ клеток, представляет собой аутоиммунное или воспалительное заболевание или состояние.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования, включающий введение субъекту, нуждающемуся в этом, эффективного количества состава по настоящему изобретению, где состав вводят подкожно.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования, включающий введение субъекту, нуждающемуся в этом, эффективного количества состава на основе антитела к CD38, содержащего по меньшей мере 100 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, и
состав характеризуется рН 5,5-7,0 и вязкостью не более 25 мПа⋅с при 20°С, где состав вводят подкожно.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования, включающий введение субъекту, нуждающемуся в этом, эффективного количества состава на основе антитела к CD38, содержащего 140 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, и где
состав характеризуется рН 6,2 и вязкостью не более 14 мПа⋅с при 20°С, где состав вводят подкожно.
В определенных вариантах осуществления способ включает введение субъекту, нуждающемуся в этом, эффективного количества состава, содержащего 140 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, и где
состав характеризуется рН 6,3 и вязкостью не более 14 мПа⋅с при 20°С, где состав вводят подкожно.
В определенных вариантах осуществления состав вводят посредством подкожной инфузии.
В определенных вариантах осуществления состав вводят посредством подкожной инфузии.
В определенных вариантах осуществления подкожная инфузия представляет собой подкожную инфузию большого объема, например, от >2 мл до 30 мл.
В определенных вариантах осуществления способ лечения CD38+ гематологического злокачественного новообразования дополнительно включает введение субъекту одного или нескольких дополнительных средств, подходящих для лечения CD38+ гематологического злокачественного новообразования. В некоторых вариантах осуществления другое средство представляет собой, например, кортикостероид (например, дексаметазон), химиотерапевтическое лекарственное средство, ингибитор протеасом, иммуномодулирующее лекарственное средство или их комбинацию.
В определенных вариантах осуществления химиотерапевтическое средство представляет собой, например, цитарабин, даунорубицин, дауномицин, доксорубицин, липосомальный доксорубицин, идарубицин, митоксантрон, гемтузумаб, клофарабин, кладрибин, гидроксимочевину, этопозид, амсакрин, ингибитор FLT3, такой как гилтеритиниб, 5-азацитидин, децитабин, мелфалан, циклофосфамид или винкристин или их комбинации.
В определенных вариантах осуществления иммуномодулирующее лекарственное средство представляет собой, например, талидомид, леналидомид или помалидомид или их комбинации.
В определенных вариантах осуществления ингибитор протеасом представляет собой, например, иксазомиб, карфилзомиб или бортезомиб или их комбинации.
В определенных вариантах осуществления способ лечения CD38+ гематологического злокачественного новообразования включает введение субъекту изатуксимаба, составленного для подкожного введения, как определено в данном документе, и двух или более дополнительных средств из разных классов соединений, таких как, например, иммуномодулирующее лекарственное средство или ингибитор протеасом.
В определенных вариантах осуществления CD38+ гематологическое злокачественное новообразование представляет собой множественную миелому.
В определенных вариантах осуществления множественная миелома представляет собой рецидивирующую/рефрактерную множественную миелому. В некоторых вариантах осуществления пациент получил по меньшей мере два предшествующих вида терапии в отношении множественной миеломы, включая леналидомид и ингибитор протеасом, и продемонстрировал прогрессирование заболевания в ходе последней терапии или после завершения последней терапии.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 представлена серия графиков, изображающих число довидимых частиц ≥10 мкм и ≥25 мкм после стрессового термического воздействия (верхние панели) и стрессового воздействия встряхивания (нижние панели) на изатуксимаб в указанных буферных системах. 2w 40°C, две недели при 40°С; 4w 40°C, 4 недели при 40°С; Cit, цитратный буфер; His, гистидиновый буфер; Pho, фосфатный буфер; Ace, ацетатный буфер.
На фиг. 2 представлен график, изображающий процент растворимых агрегатов (высокомолекулярные агрегаты (HMW), измеренные с применением эксклюзионной хроматографии (SE-HPLC)) после стрессового термического воздействия при 40°C. 2w 40°C, две недели при 40°С; 4w 40°C, 4 недели при 40°С; Cit, цитратный буфер; His, гистидиновый буфер; Pho, фосфатный буфер; Ace, ацетатный буфер.
На фиг. 3 представлен график, изображающий процент растворимых агрегатов (высокомолекулярные агрегаты (HMW), измеренные с применением эксклюзионной хроматографии (SE-HPLC)) после стрессового теплового воздействия при 40°C в гистидиновых буферах с указанными значениями pH и концентрации. 1M 40°C, один месяц при 40°C.
На фиг. 4 представлен график, изображающий процент кислотных форм изатуксимаба, измеренных с применением анализа слабого катионного обмена после стрессового воздействия встряхивания при 40°C в гистидиновых буферах с указанными значениями pH и концентрации. 1M 40°C, один месяц при 40°C.
На фиг. 5 представлен график, изображающий вязкость изатуксимаба в концентрации 200 мг/мл в аргинине-Cl, рН 6,0, при указанных концентрациях.
На фиг. 6А представлен график, изображающий вязкость изатуксимаба в концентрации 150 мг/мл в диапазоне концентраций буфера на основе аргинина-HCl, рН 6,3. На вставке более подробно показаны более высокие концентрации аргинина-HCl.
На фиг. 6В представлен график, изображающий вязкость изатуксимаба в концентрации 180 мг/мл в диапазоне концентраций буфера на основе аргинина-HCl, рН 6,3. На вставке более подробно показаны более высокие концентрации аргинина-HCl.
На фиг. 7А представлен график, изображающий вязкость изатуксимаба в концентрации 150 мг/мл в диапазоне рН в буфере на основе 150 мМ аргинина-HCl.
На фиг. 7В представлен график, изображающий вязкость изатуксимаба в концентрации 150 мг/мл в диапазоне рН в буфере на основе 200 мМ аргинина-HCl.
На фиг. 8 представлен график, изображающий вязкость в зависимости от концентрации mAb при pH 5,5, 5,9, 6,2 и 7,0 (T=20°C). Подгонка: на основе уравнения Муни.
На фиг. 9 представлен график, изображающий вязкость в зависимости от pH при концентрациях mAb 126, 140, 147 и 154 г/л (T=20°C).
На фиг. 10 представлен график, изображающий вязкость в зависимости от температуры при концентрациях mAb 126, 143 и 154 г/л (pH=6,2).
На фиг. 11 представлен график, изображающий вязкость в зависимости от температуры при концентрациях mAb 142 и 152 г/л (pH=5,9).
На фиг. 12 представлен график, изображающий вязкость в зависимости от концентрации аргинина при концентрации mAb 140 г/л (pH=6,2, T=20°C).
На фиг. 13 представлен график, изображающий относительное содержание мономеров, обнаруженное с применением анализа HP-SEC изатуксимаба в составах от F4-1 до F4-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 14 представлен график, изображающий относительное содержание мономеров, обнаруженное с применением анализа HP-SEC изатуксимаба в составах от F10-1 до F10-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 15 представлен график, изображающий относительное содержание всех агрегатов (HMWS), обнаруженных с применением анализа HP-SEC изатуксимаба в составах от F4-1 до F4-16 (прогон 1 - опыт 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 16 представлен график, изображающий относительное содержание всех агрегатов (HMWS), обнаруженных с применением анализа HP-SEC изатуксимаба в составах от F10-1 до F10-16 (прогон 1 - опыт 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 17 представлен график, изображающий относительное содержание всех фрагментов (LMWS), обнаруживаемых с помощью анализа HP-SEC изатуксимаба в составах от F4-1 до F4-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 18 представлен график, изображающий относительное содержание всех фрагментов (LMWS), обнаруживаемых с помощью анализа HP-SEC изатуксимаба в составах от F10-1 до F10-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, Т-mech: стрессовое механическое воздействие, T-5xFT: 5 циклов замораживания/оттаивания, T-1m_40°C: 1 месяц при 40°С.
На фиг. 19 представлен график, изображающий содержание кислотных пиков, полученное при анализе капиллярного изоэлектрического фокусирования (cIEF) составов на основе изатуксимаба от F4-1 до F4-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, T-1m_40°C: 1 месяц при 40°С.
На фиг. 20 представлен график, изображающий содержание кислотных пиков, полученное при анализе cIEF изатуксимаба в составах от F10-1 до F10-16 (прогон 1 - прогон 16 соответственно, n=2, среднее); T0: без обработки, T-1m_40°C: 1 месяц при 40°С.
На фиг. 21 представлен график, изображающий потерю относительной площади [%] содержания пика мономера, полученного в анализе cIEF изатуксимаба в составах от F4-1 до F4-16 и от F10-1 до F10-16 после хранения в течение одного месяца при 40°C/отн. влаж. 75% (прогон 1 - прогон 16 соответственно, n=2, среднее).
На фиг. 22 представлен график, изображающий уровень кортизола в плазме крови у мини-свиней, которым вводили подкожной инфузией указанные составы изатуксимаба или NaCl в качестве контроля, как описано в примере 5.
На фиг. 23 представлен график, изображающий уровень субстанции Р в плазме крови у мини-свиней, которым вводили подкожной инфузией указанные составы изатуксимаба или NaCl в качестве контроля, как описано в примере 5.
На фиг. 24 представлен график, изображающий концентрацию изатуксимаба в сыворотке крови в зависимости от времени у отдельных мини-свиней 1-5 в группе I, как описано в примере 6.
На фиг. 25 представлен график, изображающий концентрацию изатуксимаба в сыворотке крови в зависимости от времени у отдельных мини-свиней 6-10 в группе II, как описано в примере 6.
На фиг. 26 представлен график, изображающий концентрацию изатуксимаба в сыворотке крови в зависимости от времени у отдельных мини-свиней 11-15 в группе III, как описано в примере 6.
На фиг. 27 представлен график, изображающий концентрацию изатуксимаба в сыворотке крови в зависимости от времени у отдельных мини-свиней 16-20 в группе IV, как описано в примере 6.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В данном документе предусмотрены составы на основе антитела к CD38, подходящие для подкожного введения субъекту. Преимуществом является то, что раскрытые в данном документе составы подходят для подкожного введения посредством либо инъекции, либо инфузии, включая подкожную инфузию большого объема. Составы можно применять для лечения заболевания или состояния, характеризующегося клетками, экспрессирующими CD38. Такие заболевания и состояния включают без ограничения различные гематологические злокачественные новообразования, такие как неходжкинская лимфома (NHL), множественная миелома (ММ), острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (В-клеточный ALL) и/или хронический лимофцитарный лейкоз (CLL). Такие заболевания и состояния дополнительно включают без ограничения аутоиммунные заболевания, включая ревматоидный артрит и системную красную волчанку, и острое повреждение почек, вызванное липополисахаридом (LPS) или сепсисом. В определенных вариантах осуществления предусмотренные в данном документе составы характеризуются высокой концентрацией антитела, рН 5,5=7,0 и вязкостью не более 25 мПа⋅с при 20°С. В некоторых вариантах осуществления предусмотренный в данном документе состав представляет собой водный состав.
“Гематологические злокачественные новообразования” представляют собой типы рака, которые поражают кровь, костный мозг и лимфатические узлы. Поскольку эти три части организма тесным образом связаны посредством иммунной системы, болезнь, поражающая один из трех, может также поражать остальные. Гематологические злокачественные новообразования включают неходжкинскую лимфому (NHL) (включая, например, лимфому Беркитта (BL) и Т-клеточную лимфому (TCL)), множественную миелому (ММ), хронический лимфоцитарный лейкоз (CLL) (например, В-клеточный хронический лимфоцитарный лейкоз (B-CLL) и волосатоклеточный лейкоз (HCL)), В- и Т-клеточный острый лимфоцитарный лейкоз (ALL), острый миелоидный лейкоз (AML), лимфому Ходжкина (HL) и хронический миелоидный лейкоз (CML). В некоторых вариантах осуществления гематологическое злокачественное новообразование представляет собой CD38+ гематологическое злокачественное новообразование.
Таким образом, “CD38+ гематологическое злокачественное новообразование” представляет собой гематологическое злокачественное новообразование, как описано выше, где раковые клетки экспрессируют CD38. Сообщается также, что CD38+ клетки вовлечены во многие аутоиммунные и воспалительные заболевания и нарушения, включая ревматоидный артрит и системную красную волчанку, а также другие состояниях, включая острое повреждение почек, вызванное LPS или сепсисом.
CD38+ гематологические злокачественные новообразования включают В-клеточную неходжкинскую лимфому (NHL), множественную миелому (ММ), острый миелоидный лейкоз (AML), острый лимфобластный лейкоз (В-клеточный ALL) и/или хронический лимофцитарный лейкоз (CLL). В некоторых вариантах осуществления CD38+ гематологическое злокачественное новообразование представляет собой ММ. В некоторых вариантах осуществления CD38+ гематологическое злокачественное новообразование представляет собой рецидивирующую и/или рефрактерную множественную миелому.
“Антитело” может представлять собой природное или традиционное антитело, в котором две тяжелые цепи соединены друг с другом дисульфидными связями, и каждая тяжелая цепь соединена с легкой цепью дисульфидной связью. Существует два типа легких цепей, лямбда (λ) и каппа (κ). Существуют пять главных классов (или изотипов) тяжелых цепей, определяющих функциональную активность молекулы антитела: IgM, IgG, IgA, IgD и IgE. Дополнительно хорошо охарактеризованы подклассы (или подизотипы) иммуноглобулинов, например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2, и они, как известно, обеспечивают функциональную специализацию. Каждая цепь содержит домены с отличающейся последовательностью. Легкая цепь содержит два домена или области, вариабельный домен (VL) и константный домен (CL). Тяжелая цепь содержит четыре домена, вариабельный домен (VH) и три константных домена (CH1, CH2 и CH3, совместно обозначаемых CH). Вариабельные области как легких (VL), так и тяжелых (VH) цепей определяют распознавание при связывании и специфичность к антигену. Домены константных областей легких (CL) и тяжелых (CH) цепей обеспечивают важные биологические свойства, такие как объединение цепей антитела, секреция, перемещение через плаценту, связывание комплемента и связывание с Fc-рецепторами (FcR). Fv-фрагмент представляет собой N-концевую часть Fab-фрагмента иммуноглобулина, который включает вариабельные части одной легкой цепи и одной тяжелой цепи. Специфичность антитела заключается в структурной комплементарности между паратопом антитела и антигенной детерминантой. Паратопы антител составлены остатками главным образом из гипервариабельных или определяющих комплементарность областей (CDR). Остатки из негипервариабельных или каркасных областей (FR) иногда влияют на общую структуру домена и, следовательно, паратоп.
"Определяющие комплементарность области" или "CDR" относятся к аминокислотным последовательностям, которые совместно определяют специфичность связывания и аффинность природной Fv-области нативного связывающего участка иммуноглобулина. Каждая из легких и тяжелых цепей иммуноглобулина имеет три CDR, обозначенные как CDR-L1, CDR-L2, CDR-L3 и CDR-H1, CDR-H2, CDR-H3 соответственно. Следовательно, антигенсвязывающий участок традиционного антитела содержит шесть CDR, которые включают набор CDR из каждой из V-областей тяжелой и легкой цепей.
Определение CDR/FR, касающееся легкой или тяжелой цепи иммуноглобулина, приводится на основании определения по Kabat (worldwideweb.bioinf.org.uk/abs/).
Антитело также может являться антителом, не встречающимся в природе, например, моноклональным антителом, химерным антителом или гуманизированным антителом. Используемый в данном документе термин "моноклональное антитело" или "mAb" относится к молекуле антитела с единым аминокислотным составом, которая нацеливается на специфический антиген, и его не следует истолковывать как требующий получения антитела любым конкретным способом. Моноклональное антитело может продуцироваться одним клоном B-клеток или гибридомой, но также может быть рекомбинантным, т. е. полученным с помощью белковой инженерии.
Термин "гуманизированное антитело" относится к антителу, изначально полностью или частично происходящему не от человека, которое было модифицировано с заменой определенных аминокислот, например, в каркасных областях тяжелой и легкой цепей, чтобы избежать или свести к минимуму иммунного ответа у людей. Константными доменами гуманизированного антитела могут быть, например, человеческие домены CH и CL. В варианте осуществления гуманизированное антитело имеет константные домены, происходящие от человека.
В некоторых вариантах осуществления антитело к CD38 по настоящему изобретению содержит тяжелую цепь, содержащую CDR-H1, содержащую аминокислотную последовательность, указанную под SEQ ID NO: 1, CDR-H2, содержащую аминокислотную последовательность под SEQ ID NO: 2, и CDR-H3, содержащую аминокислотную последовательность под SEQ ID NO: 3, и легкую цепь, содержащую CDR-L1, содержащую аминокислотную последовательность, указанную под SEQ ID NO: 4, CDR-L2, содержащую аминокислотную последовательность, указанную под SEQ ID NO: 5, и CDR-L3, содержащую аминокислотную последовательность, указанную под SEQ ID NO: 6.
CDR-H1 DYWMQ (SEQ ID NO: 1)
CDR-H2 TIYPGDGDTGYAQKFQG (SEQ ID NO: 2)
CDR-H3 GDYYGSNSLDY (SEQ ID NO: 3)
CDR-L1 KASQDVSTVVA (SEQ ID NO: 4)
CDR-L2 SASYRYI (SEQ ID NO: 5)
CDR-L3 QQHYSPPYT (SEQ ID NO: 6)
В некоторых вариантах осуществления указанное антитело содержит вариабельный домен тяжелой цепи (VH), содержащий аминокислотную последовательность, указанную под SEQ ID NO: 7.
В некоторых вариантах осуществления указанное антитело содержит вариабельный домен легкой цепи (VL), содержащий аминокислотную последовательность, указанную под SEQ ID NO: 8.
В некоторых вариантах осуществления указанное антитело содержит вариабельный домен тяжелой цепи (VH), содержащий аминокислотную последовательность, указанную под SEQ ID NO: 7, и вариабельный домен легкой цепи (VL), содержащий аминокислотную последовательность, указанную под SEQ ID NO: 8.
QVQLVQSGAEVAKPGTSVKLSCKASGYTFTDYWMQWVKQRPGQGLEWIGTIYPGDGDTGYAQKFQGKATLTADKSSKTVYMHLSSLASEDSAVYYCARGDYYGSNSLDYWGQGTSVTVSS
(SEQ ID NO: 7)
DIVMTQSHLSMSTSLGDPVSITCKASQDVSTVVAWYQQKPGQSPRRLIYSASYRYIGVPDRFTGSGAGTDFTFTISSVQAEDLAVYYCQQHYSPPYTFGGGTKLEIKR
(SEQ ID NO: 8)
В некоторых вариантах осуществления антитело к CD38 по настоящему изобретению представляет собой изатуксимаб. Тяжелая цепь (HC) изатуксимаба содержит аминокислотную последовательность, указанную под SEQ ID NO: 9, и легкая цепь (LC) изатуксимаба содержит аминокислотную последовательность, указанную под SEQ ID NO: 10.
QVQLVQSGAEVAKPGTSVKLSCKASGYTFTDYWMQWVKQRPGQGLEWIGTIYPGDGDTGYAQKFQGKATLTADKSSKTVYMHLSSLASEDSAVYYCARGDYYGSNSLDYWGQGTSVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
(SEQ ID NO: 9)
DIVMTQSHLSMSTSLGDPVSITCKASQDVSTVVAWYQQKPGQSPRRLIYSASYRYIGVPDRFTGSGAGTDFTFTISSVQAEDLAVYYCQQHYSPPYTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
(SEQ ID NO: 10)
В некоторых вариантах осуществления антитело предназначено для применения в лечении CD38+ гематологического злокачественного новообразования, такого как множественная миелома (ММ), включая рецидивирующую и/или рефрактерную ММ, или пациентов с ММ, которые получали один или несколько видов предшествующей терапии в отношении ММ.
В контексте настоящего изобретения используемый в данном документе термин “осуществление лечения” или “лечение” означает устранение, облегчение, подавление прогрессирования или предупреждение нарушения или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого нарушения или состояния. В определенных вариантах осуществления используемый в данном документе термин “осуществление лечения” или “лечение” означает устранение, облегчение или подавление прогрессирования нарушения или состояния, к которому применяется такой термин, или одного или нескольких симптомов такого нарушения или состояния.
Под используемым в данном документе термином “осуществление лечения CD38+ гематологического злокачественного новообразования” подразумевается ингибирование роста злокачественных CD38+ клеток опухоли и/или прогрессирования метастазов из указанной CD38+ опухоли. Такое лечение также может приводить к регрессии роста опухоли, т.е. снижению размера измеряемой опухоли.
Под “терапевтически эффективным количеством” антитела в контексте настоящего изобретения подразумевается количество антитела, достаточное для лечения указанного CD38+ гематологического злокачественного новообразования, раскрытого в данном документе.
В определенных вариантах осуществления указанное терапевтически эффективное количество антитела, вводимого подкожно субъекту, представляет собой дозу, находящуюся в диапазоне от 500 мг до 2000 мг антитела на дозу.
В определенных вариантах осуществления указанное терапевтически эффективное количество антитела, вводимого субъекту, представляет собой 1000 мг антитела на дозу. В определенных вариантах осуществления указанное терапевтически эффективное количество антитела, вводимого субъекту, представляет собой 1400 мг антитела на дозу. В определенных вариантах осуществления указанное терапевтически эффективное количество антитела, вводимого субъекту, представляет собой 1600 мг антитела на дозу.
Используемый в данном документе термин “субъект” относится к млекопитающему. В определенных вариантах осуществления термин “субъект” относится к человеку.
Антитело по настоящему изобретению можно вводить один раз в неделю (QW), один раз в две недели (Q2W) или комбинацию один раз в неделю и один раз в две недели. В некоторых вариантах осуществления антитело вводят один раз в четыре недели.
Например, антитело можно вводить субъекту в дозе, находящейся в диапазоне от 500 мг до 1400 мг один раз в неделю в течение четырех недель (цикл 1), а затем один раз в две недели (например, в день 1 и день 15 каждого последующего четырехнедельного цикла).
В некоторых вариантах осуществления субъекту вводят 1000 мг антитела один раз в неделю в течение четырех недель (цикл 1), а затем субъекту вводят 1000 мг антитела в дни 1 и 15 каждого последующего четырехнедельного цикла.
В некоторых вариантах осуществления субъекту вводят 1000 мг антитела один раз в две недели.
В некоторых вариантах осуществления субъекту вводят 1400 мг антитела один раз в неделю в течение четырех недель (цикл 1), а затем вводят 1400 мг антитела в дни 1 и 15 каждого последующего четырехнедельного цикла.
В некоторых вариантах осуществления субъекту вводят 1400 мг антитела один раз в две недели. В некоторых вариантах осуществления антитело можно вводить в соответствии с прерывистой программой с интервалом после каждого введения, составляющим 1 неделю или 2 недели, который может быть удлинен на 1-2 недели в зависимости от переносимости предыдущего введения.
Используемый в данном документе термин “цикл” относится к 4 календарным неделям, т.е. 28 дням. Введение “один раз в неделю” означает один раз в каждые 7 дней. Введение “один раз в две недели” означает один раз в каждые 14 дней. Введение один раз в цикл или один раз в четыре недели означает один раз в 28 дней.
В некоторых вариантах осуществления число циклов введения антитела может составлять от 2 до 50, например 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, 25, 30, 35, 45, 50 циклов.
Составы по настоящему изобретению
В настоящем изобретении предусмотрены определенные составы на основе антител к CD38. В некоторых вариантах осуществления составы представляют собой жидкие составы. В определенных вариантах осуществления такие составы (составы на основе антитела) подходят для введения субъекту, нуждающемуся в лечении с помощью антитела к CD38.
Преимуществом является то, что составы на основе антител по настоящему изобретению можно вводить субъекту подкожно посредством либо инъекции, либо инфузии, включая подкожную инфузию большого объема.
В определенных вариантах осуществления настоящее изобретение относится к составу на основе антитела к CD38, характеризующемуся высокой концентрацией антитела, рН 5,5-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
В определенных вариантах осуществления настоящее изобретение относится к составу, содержащему по меньшей мере 100 мг/мл антитела к CD38, средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, где состав характеризуется рН 5,5-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
В некоторых вариантах осуществления предусмотрены составы на основе антитела, содержащие по меньшей мере 100 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, и
состав характеризуется рН 5,5-7,0 и вязкостью не более 25 мПа⋅с при 20°С.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-130 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl.
В определенных вариантах осуществления стабилизатор представляет собой сахарозу.
В определенных вариантах осуществления водный состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-130 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 110 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-130 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl и сахарозу.
В определенных вариантах осуществления состав содержит 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-130 мМ Arg-Cl и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит поверхностно-активное вещество.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы и поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-130 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы и поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой от 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и полоксамер 188.
В определенных вариантах осуществления состав содержит 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления состав содержит буферное средство.
В определенных вариантах осуществления буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой гистидин.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и гистидин.
В определенных вариантах осуществления буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-150 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой сахарозу, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой полоксамер 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-125 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl, стабилизатор представляет собой 2% (вес/об.) сахарозы, поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188, а буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 125-155 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 90-125 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления раствор содержит 140 мг/мл антитела к CD38, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы, 0,4% (вес/об.) полоксамера 188 и 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac и сахарозу.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac и сахарозу.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac и сахарозу.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac и сахарозу.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac и 2% (вес/об.) сахарозы.
В определенных вариантах осуществления состав содержит поверхностно-активное вещество.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, сахарозу и полисорбат 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, сахарозу и полисорбат 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, 2% (вес/об.) сахарозы и полисорбат 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, 2% (вес/об.) сахарозы и полисорбат 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac, сахарозу и полисорбат 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac, сахарозу и полисорбат 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и полисорбат 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и полисорбат 80.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, сахарозу и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, сахарозу и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 90-150 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac, стабилизатор представляет собой сахарозу, а поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac, сахарозу и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac, сахарозу и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac, стабилизатор представляет собой 2% (вес/об.) сахарозы, а поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38, 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38, 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления рН состава составляет 5,9-7,0.
В определенных вариантах осуществления рН состава составляет 5,9-6,5.
В определенных вариантах осуществления рН состава составляет 6,2.
В некоторых вариантах осуществления состав содержит 140 мг/мл антитела к CD38, где антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR- H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, и где
состав характеризуется рН 6,2-6,3 и вязкостью не более 14 мПа·с при 20°С.
Аспект настоящего изобретения относится к составу, содержащему 140 мг/мл антитела к CD38, где антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR- H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80, и
состав характеризуется рН 6,2 и вязкостью не более 14 мПа·с при 20°С.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления VH антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 7.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления VL антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 8.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления VH антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 7, и VL антитела к CD38 содержит аминокислотную последовательность, указанную под SEQ ID NO: 8.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления антитело к CD38 представляет собой изатуксимаб.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления состав дополнительно содержит воду, например воду для инъекций (WFI), в количестве, достаточном для достижения установленных концентраций других ингредиентов.
В соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления состав подходит для подкожного введения. Например, состав может быть стерильным. В определенных вариантах осуществления компоненты состава могут быть объединены с образованием раствора, а затем раствор можно подвергать стерильной фильтрации с получением стерильного состава.
В определенных вариантах осуществления готовый состав практически не содержит растворенного кислорода. Например, состав можно уравновешивать с помощью газообразного азота, а затем герметизировать в атмосфере азота.
При этом дополнительно в соответствии с каждым из вышеуказанных аспектов и вариантов осуществления в определенных вариантах осуществления состав может дополнительно содержать по меньшей мере одно дополнительное вспомогательное вещество или компонент для повышения стабильности, например, консервирующее средство.
Упакованные фармацевтические продукты
Аспект настоящего изобретения относится к упакованному фармацевтическому продукту, содержащему стерильный контейнер, содержащий однократную дозу состава по настоящему изобретению. Подходящие стерильные контейнеры включают без ограничения флаконы, ампулы, бутыли, мешки, пакеты, предварительно заполненные шприцы, шприцевые помпы, инфузионные насосы и контейнеры, приспособленные для применения со шприцевыми помпами и/или инфузионными насосами. Подходящие контейнеры включают контейнеры одноразового использования и контейнеры многократного использования. В определенных вариантах осуществления контейнер представляет собой контейнер одноразового использования, например, флакон, содержащий антитело в количестве, соответствующем однократной дозе.
Используемая в данном документе шприцевая помпа относится к механическому или пневматическому устройству, сконструированному и предназначенному для зацепления с поршнем шприца и перемещения его в осевом направлении вперед и/или назад, чтобы обеспечить доставку содержимого шприца с требуемой скоростью. Шприцевые помпы известны в данной области техники и включают, например, без ограничения устройства, раскрытые в патентах США №№ 5064413; 5449345; 5954695; 6428509; 6645177; 7195610; 8231576; 8814830, содержание каждого из которых включено в данный документ посредством ссылки.
Инфузионные насосы хорошо известны в данной области техники и включают, например, волюметрический инфузионный насос Baxter Colleague CXE и насос Cané Crono.
Аспект настоящего изобретения относится к устройству, содержащему терапевтически эффективное количество состава по настоящему изобретению. В определенных вариантах осуществления устройство может представлять собой, например, шприц, шприцевую помпу и инфузионный насос, содержащие состав. В определенных вариантах осуществления шприц представляет собой предварительно заполненный шприц.
В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению предусмотрен в формате с фиксированным объемом. Такой состав может быть представлен во флаконе или ампуле или в виде, например, флакона или ампулы. Например, в некоторых вариантах осуществления состав на основе антитела по настоящему изобретению предусмотрен в объеме, составляющем от приблизительно 10 мл до приблизительно 20 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению предусмотрен в объеме, составляющем от приблизительно 10 мл до приблизительно 15 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению предусмотрен в объеме от приблизительно 10 мл до приблизительно 12,5 мл. Например, в одном варианте осуществления состава, содержащего 140 мг/мл антитела, флакон, содержащий 10 мл такого состава, содержит 1400 мг антитела.
Способы лечения
Составы по настоящему изобретению можно применять в способе лечения заболевания или состояния, характеризующегося наличием или активностью CD38+ клеток. Такое заболевание или состояние может включать без ограничения CD38+ гематологическое злокачественное новообразование, аутоиммунное заболевание или состояние, воспалительное заболевание или состояние, и повреждение или дисфункцию почек, вызванное(-ую) LPS или сепсисом. Способ обычно включает введение субъекту, нуждающемуся в этом, эффективного количества антитела в готовом составе, представленного в данном документе, где введение осуществляют посредством подкожной инъекции или инфузии, необязательно посредством подкожной инфузии большого объема (например, 10 мл или более). В определенных вариантах осуществления субъектом является человек.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования у субъекта-человека, нуждающегося в этом, причем указанный способ включает введение указанному субъекту-человеку эффективного количества состава, содержащего по меньшей мере 100 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит средство для уменьшения вязкости, стабилизатор, буферное средство и поверхностно-активное вещество, и
состав характеризуется рН 5,7-7,0 и вязкостью не более 25 мПа⋅с при 20°С, и где введение представляет собой подкожное введение.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-130 мМ Arg-Cl.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 110 мМ Arg-Cl.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полоксамер 188.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой 0,4% (вес/об.) полоксамера 188.
В определенных вариантах осуществления буферное средство представляет собой гистидин.
В определенных вариантах осуществления буферное средство представляет собой 9 мМ гистидина.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 90-150 мМ Lys-Ac.
В определенных вариантах осуществления средство для уменьшения вязкости представляет собой 125 мМ Lys-Ac.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой полисорбат 80.
В определенных вариантах осуществления поверхностно-активное вещество представляет собой 0,04% (вес/об.) полисорбата 80.
В определенных вариантах осуществления состав содержит 125-155 мг/мл антитела к CD38.
В определенных вариантах осуществления состав содержит 140 мг/мл антитела к CD38.
В определенных вариантах осуществления стабилизатор представляет собой сахарозу.
В определенных вариантах осуществления стабилизатор представляет собой 2% (вес/об.) сахарозы.
В определенных вариантах осуществления рН состава составляет 5,9-7,0.
В определенных вариантах осуществления рН состава составляет 5,9-6,5.
В определенных вариантах осуществления рН состава составляет 6,2.
В определенных вариантах осуществления рН состава составляет 6,3.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования у субъекта-человека, нуждающегося в этом, причем указанный способ включает введение указанному субъекту-человеку эффективного количества состава, содержащего 140 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg-Cl, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, и
состав характеризуется рН 6,2 и вязкостью не более 14 мПа⋅с при 20°С, и
где введение представляет собой подкожное введение.
Аспект настоящего изобретения представляет собой способ лечения CD38+ гематологического злокачественного новообразования у субъекта-человека, нуждающегося в этом, причем указанный способ включает введение указанному субъекту-человеку эффективного количества состава, содержащего 140 мг/мл антитела к CD38, где
антитело к CD38 содержит вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 1-3 соответственно, и вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности, указанные под SEQ ID NO: 4-6 соответственно,
состав содержит 125 мМ Lys-Ac, 2% (вес/об.) сахарозы и 0,04% (вес/об.) полисорбата 80, и
состав характеризуется рН 6,2 и вязкостью не более 14 мПа·с при 20°С, и
где введение представляет собой подкожное введение.
В определенных вариантах осуществления в соответствии с каждым из вышеуказанных способов подкожное введение включает одну или несколько подкожных инъекций.
В определенных вариантах осуществления в соответствии с каждым из вышеуказанных способов подкожное введение включает одну или несколько подкожных инфузий.
В определенных вариантах осуществления в соответствии с каждым из вышеуказанных способов подкожное введение включает одну или несколько подкожных инфузий большого объема. Используемый в данном документе термин “инфузия большого объема” относится к объему инфузии, превышающему или равному 5 мл. В определенных вариантах осуществления “инфузия большого объема” относится к объему инфузии приблизительно 5-10 мл, приблизительно 10-15 мл, приблизительно 15-20 мл, приблизительно 20-25 мл или приблизительно 25-30 мл. В одном варианте осуществления “инфузия большого объема” относится к объему инфузии приблизительно 5-10 мл. В одном варианте осуществления “инфузия большого объема” относится к объему инфузии приблизительно 10-15 мл. В одном варианте осуществления “инфузия большого объема” относится к объему инфузии приблизительно 15-20 мл. В одном варианте осуществления “инфузия большого объема” относится к объему инфузии приблизительно 20-25 мл. В одном варианте осуществления “инфузия большого объема” относится к объему инфузии приблизительно 25-30 мл.
Неожиданно было обнаружено, что инфузии большого объема из композиций, предусмотренных в данном документе, являются эффективными для достижения системной доставки терапевтически эффективных количеств антитела к CD38, такого как изатуксимаб, для лечения заболевания или состояния, которые характеризуются наличием и/или активностью CD38+ клеток, включая CD38+ гематологическое злокачественное новообразование у человека. Неожиданно, состав на основе антитела, предусмотренный в данном документе, продемонстрировал биодоступность, составляющую по меньшей мере 89%, при подкожном введении мини-свиньям в отсутствие диспергирующего средства, такого как гиалуронидаза. Таким образом, в данном документе предусмотрен состав, который не включает биологических диспергирующих средств. В альтернативных вариантах осуществления в данном документе предусмотрен состав, который дополнительно включает одно или несколько биологических диспергирующих средств.
Как показано в примере 6, после однократной внутривенной (IV) инфузии мини-свиньям изатуксимаба в дозе 1800 мг/животное на протяжении 30-минутного периода средняя AUC за полный 672-часовой период сбора образцов после введения дозы (AUClast) составила 364000 ч*мкг/мл. После однократной подкожной (SC) инфузии мини-свиньям изатуксимаба в дозе 1806 мг/животное при скорости потока 0,5, 1 или 2 мл/мин средняя AUC за полный 672-часовой период сбора образцов после введения дозы (AUClast) составило 326000, 565000 и 369000 ч*мкг/мл соответственно. Более того, абсолютная SC биодоступность изатуксимаба у мини-свиней при введении в дозе 1806 мг/животное (раствор 140 мг/мл) посредством SC инфузии при скорости потока от 0,5 до 2 мл/мин составляла по меньшей мере 89%.
Используемый в данном документе термин “субъект” и/или “субъект, нуждающийся в этом” представляет собой индивидуума, у которого имеется CD38+ гематологическое злокачественное новообразование или у которого подозревается наличие CD38+ гематологического злокачественного новообразования. Используемый в данном документе термин “субъект” может также относиться к пациенту.
Субъект в соответствии с настоящим изобретением может быть мужчиной или женщиной.
В некоторых вариантах осуществления субъект ранее проходил лечение одним или несколькими средствами или видами терапии, подходящими для лечения гематологического злокачественного новообразования, экспрессирующего CD38. Предшествующая противоопухолевая терапия может представлять собой, например, кортикостероид (например, дексаметазон), химиотерапевтическое лекарственное средство, ингибитор протеасом, иммуномодулирующее лекарственное средство, лучевую терапию, трансплантацию костного мозга и/или стволовых клеток и иммунотерапию.
“Химиотерапевтические лекарственные средства” представляют собой цитотоксические средства, применяемые, например, для лечения гематологического злокачественного новообразования, включая без ограничения цитарабин (цитозинарабинозид или ara-C) и антрациклиновые лекарственные средства (такие как даунорубицин и/или дауномицин, доксорубицин и липосомальный доксорубицин, идарубицин и митоксантрон), гемтузумаб, клофарабин, кладрибин, гидроксимочевину, этопозид, амсакрин, ингибиторы FLT3 и деметилирующие средства (5-азацитидин и децитабин), мелфалан, циклофосфамид и винкристин.
Ингибиторы протеасом включают, например, бортезомиб, карфилзомиб и иксазомиб. Иммуномодулирующие лекарственные средства включают, например, талидомид, леналидомид и помалидомид.
“Лучевая терапия” или “облучение” относится к излучению высокой энергии, применяемому для удаления раковых клеток. Лучевая терапия может применяться перед трансплантацией костного мозга или стволовых клеток периферической крови.
“Трансплантация костного мозга и/или стволовых клеток” относится к трансплантации клеток, направленной на восстановление стволовых клеток, которые были разрушены высокими дозами химиотерапевтического(-их) лекарственного(-ых) средства(средств) и/или лучевой терапией. Источники стволовых клеток включают костный мозг, периферическую кровь или пуповинную кровь. В зависимости от источника стволовых клеток, подлежащих трансплантации, процедура может быть разделена на трансплантацию костного мозга (BMT), трансплантацию стволовых клеток периферической крови (PBSCT) или трансплантацию пуповинной крови (UCBT). Более того, трансплантация костного мозга и/или стволовых клеток может относиться к аутологичной трансплантации стволовых клеток и/или аллогенной трансплантации.
При “аутологичной трансплантации” собственные стволовые клетки субъекта извлекают из его или ее костного мозга или периферической крови, замораживают и хранят, в то время как человек получает лечение (высокая доза химиотерапевтического(-их) лекарственного(-ых) средства(средств) и/или облучение). Процесс, называемый “чисткой”, может применяться для попытки удаления любых раковых клеток из образцов. Затем стволовые клетки повторно вводят инфузией в кровь субъекта после лечения.
“Аллогенные трансплантаты” представляют собой трансплантаты от совместимого донора. Преимущество аллогенной трансплантации костного мозга заключается в том, что трансплантированные клетки от донора могут создать новую иммунную систему, которая может обнаруживать лейкозные клетки в качестве чужеродных и удалять их. Недостатком аллогенных трансплантатов являются ограничение в подборе доноров и побочные эффекты.
“Иммунотерапия” относится к стимуляции иммунной системы субъекта для атаки на злокачественные опухолевые клетки, которые ответственны за заболевание. Ее можно проводить либо посредством иммунизации субъекта, например, путем введения противораковой вакцины, и в этом случае собственная иммунная система субъекта обучается распознавать опухолевые клетки в качестве мишеней, подлежащих уничтожению, либо посредством введения терапевтических антител в качестве лекарственных средств, в этом случае иммунная система субъекта задействуется для уничтожения опухолевых клеток под действием терапевтических антител.
В контексте настоящего изобретения субъект мог ранее получать лечение в отношении гематологического злокачественного новообразования, но у него возник рецидив, и/или оно было рефрактерным.
В некоторых вариантах осуществления субъект страдает от множественной миеломы. В некоторых вариантах осуществления у субъекта имеется рецидивируюшая и/или рефрактерная множественная миелома.
“Рецидивирующее” относится к заболеванию или состоянию, такому как гематологическое злокачественное новообразование, которое ранее подвергалось лечению и которое прогрессирует и требует начала дополнительного лечения, но не соответствует критериям ни первичного рефрактерного, ни рецидивирующего и рефрактерного заболевания.
“Рефрактерное” относится к заболеванию или состоянию, которое не отвечает (не смогло достичь минимального ответа, или на фоне терапии развивается прогрессирование заболевания) во время первичной или спасительной терапии, или прогрессирует в пределах 60 дней после последней терапии.
Рецидивирующее и рефрактерное заболевание не отвечает на спасительную терапию (например, терапию, которая вводится после неэффективного лечения с помощью первой линии терапии) или является заболеванием, которое прогрессирует в пределах 60 дней после последней терапии у пациентов, которые достигли минимального ответа или лучше в некоторый момент перед прогрессирования в их текущее течение заболевания.
Первичное рефрактерное заболевание представляет собой заболевание, которое не отвечает на лечение, у пациентов, у которых никогда не достигался минимальный ответ или лучше с помощью любой терапии.
В некоторых вариантах осуществления субъект ранее получал лечение бортезомибом и/или леналидомидом.
В некоторых вариантах осуществления субъект ранее получил аутологичную трансплантацию стволовых клеток (ASCT).
В некоторых вариантах осуществления у субъекта возник рецидив в пределах 6 месяцев после аутологичной трансплантации.
Дозировка и введение
В некоторых вариантах осуществления антитело в готовом составе, предусмотренное в данном документе, вводится в качестве “фиксированной дозы”, так что количество антитела, вводимого пациенту, не регулируется на основании размера тела или веса тела. В некоторых вариантах осуществления фиксированная доза, вводимая пациенту, содержит 1000-1800 мг антитела. В некоторых вариантах осуществления фиксированная доза составляет 1000 мг. В некоторых вариантах осуществления фиксированная доза составляет 1400 мг.
В некоторых вариантах осуществления антитело в готовом составе, как например, фиксированная доза антитела, вводится пациенту в фиксированном объеме. Например, в некоторых вариантах осуществления состав на основе антитела по настоящему изобретению вводится в объеме от приблизительно 10 до приблизительно 20 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению вводится в объеме от приблизительно 10 до приблизительно 15 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению вводится в объеме от приблизительно 10 мл до приблизительно 12,5 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению вводится в объеме от приблизительно 10 мл до приблизительно 11 мл. В некоторых вариантах осуществления состав на основе антитела по настоящему изобретению вводится в объеме от приблизительно 10 мл до приблизительно 10,5 мл.
В некоторых вариантах осуществления доза антитела в готовом составе, предусмотренного в данном документе, вводится подкожно на протяжении от приблизительно 10 до приблизительно 60 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении от приблизительно 20 до приблизительно 40 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 10 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 20 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 30 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 40 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 50 минут. В некоторых вариантах осуществления доза антитела в готовом составе вводится подкожно на протяжении приблизительно 60 минут.
В некоторых вариантах осуществления подкожное введение происходит при определенной скорости инфузии антитела. Например, состав можно вводить подкожно со скоростью, подходящей для достижения полной доставки требуемой дозы, за минимальное время, без значительного подтекания или значительного дискомфорта. Такая скорость может находиться в диапазоне, например, от приблизительно 0,1 мл/мин до приблизительно 1,5 мл/мин. В некоторых вариантах осуществления скорость инфузии составляет приблизительно 0,8 мл/мин. В некоторых вариантах осуществления скорость составляет 1 мл/мин. В некоторых вариантах осуществления скорость составляет 1,2 мл/мин. В некоторых вариантах осуществления скорость составляет 1,5 мл/мин.
В некоторых вариантах осуществления начальную скорость инфузии можно поддерживать в течение всего периода инфузии. В других вариантах осуществления скорость инфузии можно увеличивать или уменьшать, или как увеличивать, так и уменьшать во время периода инфузии.
В некоторых вариантах осуществления антитело к CD38 вводится отдельно. В других вариантах осуществления антитело к CD38 вводится вместе с другим средством, подходящим для лечения CD38+ гематологического злокачественного новообразования. В некоторых вариантах осуществления другое средство представляет собой кортикостероид (например, дексаметазон), химиотерапевтическое лекарственное средство, ингибитор протеасом, иммуномодулирующее лекарственное средство или их комбинацию.
При введении с другим средством, подходящим для лечения CD38+ гематологического злокачественного новообразования, антитело к CD38 и другое(-ие) средство(-а) можно вводить либо одновременно, либо по отдельности (например, последовательно на протяжении периода времени). Антитело к CD38 и другое(-ие) средство(-а) можно вводить одним и тем же или разными путями введения. Если антитело к CD38 и другое(-ие) средство(-а) вводят одним и тем же путем введения, то их можно вводить в одни и те же или разные участки введения.
Кортикостероиды, такие как дексаметазон, применяются для лечения различных воспалительных, аутоиммунных и аллергических состояний. Он также применяется при лечении рака, либо в качестве средства прямого действия (например, при множественной миеломе), либо в комбинации с другими средствами (например, иммуномодулирующими лекарственными средствами, химиотерапевтическими лекарственными средствами и ингибиторами протеасом). Кортикостероиды, такие как дексаметазон, также можно применять для противодействия побочным эффектам химиотерапевтического(-их) лекарственного(-ых) средства(средств) (например, тошноты и воспаления). Кортикостероиды, такие как дексаметазон, также можно применять в качестве премедикации для уменьшения потенциального риска и/или тяжести инфузионных реакций (IR) вследствие инфузии антитела. Дексаметазон, как правило, вводится перорально.
Помалидомид является аналогом талидомида и иммуномодулирующим лекарственным средством с множественными эффектами в отношении клеток, которые подавляют рост и выживаемость клеток множественной миеломы, блокируя стромальную поддержку со стороны микроокружения костного мозга, которая может способствовать росту клеток миеломы; кроме того, помалидомид характеризуется выраженными иммуномодулирующими эффектами, которые усиливают иммунный ответ на клетки миеломы путем стимуляции естественных киллерных (NK) клеток и путем ингибирования регуляторных Т-клеток. Помалидомид, как правило, вводится перорально.
В контексте настоящего изобретения врач может оценить ответ заболевания и, таким образом, адаптировать режим введения.
В других вариантах осуществления антитело к CD38 вводят вместе с одним или несколькими биологическими диспергирующими средствами. При введении с биологическим диспергирующим средством антитело к CD38 и другое(-ие) средство(-а) могут вводиться либо одновременно, либо по отдельности (например, последовательно на протяжении периода времени). Антитело к CD38 и другое(-ие) средство(-а) можно вводить одним и тем же или разными путями введения. Если антитело к CD38 и другое(-ие) средство(-а) вводят одним и тем же путем введения, то их можно вводить в одни и те же или разные участки введения.
Перед введением антитела субъект получает премедикацию для уменьшения риска и/или тяжести инфузионных реакций (IR), как правило, наблюдаемых при введении моноклонального антитела. Премедикация может включать, например, монтелукаст, ацетаминофен, ранитидин, дифенилдрамамин, дексаметазон или их комбинации. В некоторых вариантах осуществления, если у субъекта не возникает IR после четырех последовательных введений антитела, описанного в данном документе, премедикация может быть прекращена.
“Ответ заболевания” может быть определен в соответствии со стандартными критериями для гематологических злокачественных новообразований и стадирования. Способы для оценки ответа заболевания в случае гематологического злокачественного новообразования, в частности CD38+ гематологического злокачественного новообразования, известны специалистам в данной области. Например, способы для оценки ответа заболевания включают оценки общего состояния, как например, оценку общего состояния по шкале Восточной кооперативной онкологической группы (ECOG) и по критериям ответа Международной группы по изучению миеломы (см. Oken, et al., Am. J. Clin. Oncol. 1982;5(6):649-655; и Kumar, et al., Lancet Oncol. 2016;17(8):328-346, соответственно). Способы для оценки ответа заболевания также могут включать количественную оценку маркеров заболевания, биопсию и/или аспирацию костного мозга, радиологическую визуализацию плазмоцитомы, исследование костного скелета, количественную оценку М-белка (в сыворотке крови и/или 24-часовой моче) и уровни свободных легких цепей в сыворотке крови или уровни легких цепей в моче, сывороточный β2-микроглобулин, биопсию лимфатических узлов, радиологическую оценку опухоли (с помощью рентгенографии, компьютерной томографии (CT), PET-сканирования или магнитно-резонансной томографии (MRI)) и анализ крови, включая количество бластов. Данный список методов оценки следует понимать как неограничивающий.
На основании результатов, полученных в результате оценки ответа заболевания, затем ответ заболевания можно стратифицировать в соответствии со стандартными критериями для основного заболевания и классифицировать на полный ответ или полную ремиссию (CR), частичный ответ (PR), стабильное заболевание (SD) или прогрессирование заболевания (PD).
“Маркеры”, применяемые в контексте оценки ответа, могут включать маркеры сыворотки и/или плазмы крови, такие как С-реактивный белок (CRP), фактор некроза опухоли альфа (TNF-α), IL-6, IL-1β или IFN. Маркеры также могут включать маркеры клеточной поверхности, такие как CD38.
Методы для оценки ответа заболевания у субъекта, у которого имеется множественная миелома, включают, например, биопсию и/или аспирацию костного мозга, радиологическую визуализацию плазмоцитомы, исследование костного скелета, количественный анализ М-белка и измерение сывороточного β2-микроглобулина.
Оценка ответа заболевания может дополнительно включать плотность рецепторов и занятость рецепторов на циркулирующих опухолевых клетках (в периферической крови), плотность рецепторов и занятость рецепторов на бластах и плазматических клетках в костном мозге, и уровень человеческих антител к лекарственному средству (ADA).
Полное содержание всех патентов и опубликованных патентных заявок, цитируемых в настоящем изобретении, включено в данный документ посредством ссылки.
Более полное понимание настоящего изобретения будет достигаться при обращении к следующим неограничивающим примерам. Примеры были изложены ниже с целью иллюстрации и для описания определенных вариантов осуществления настоящего изобретения. Объем формулы настоящего изобретения не должен каким-либо образом ограничиваться примерами, изложенными в данном документе.
ПРИМЕРЫ
Следующие примеры изложены для того, чтобы обеспечить специалистов средней квалификации в данной области полным раскрытием и описанием того, как осуществлять и применять способы и получать и композиции по настоящему изобретению, и они не предполагают ограничение объема того, что авторы настоящего изобретения рассматривают в качестве своего изобретения. Были предприняты усилия для обеспечения точности в отношении используемых чисел (например, количеств, температуры и т. д.)
Пример 1. - Начальный скрининг
Начальные действия по разработке состава включали скрининг систем буфер-pH, термостабилизаторов, поверхностно-активных веществ, а также средства уменьшения вязкости, чтобы идентифицировать вспомогательные вещества и их комбинации, которые совместимы с изатуксимабом и повышают его стабильность, сохраняя при этом осмоляльность и вязкость, подходящие для подкожной инъекции.
Выбор системы буфера и pH
Чтобы идентифицировать систему буфера и рН, стабильность изатуксимаба оценивали в 16 различных системах буфер-рН. Системы буфер-pH (таблица 1) тестировали на основе их буферной способности в представляющем интерес диапазоне pH.
Таблица 1. - Тестируемая система буфер-pH
Системы буфер-pH оценивали в отношении их влияния на агрегацию изатуксимаба с точки зрения образования видимых и довидимых частиц и растворимых агрегатов (высокомолекулярных разновидностей, HMW) при стрессовом воздействии встряхивания и температуры в жидком составе на основе изатуксимаба при концентрация 5 мг/мл.
Как показано в таблице 2, обнаружили, что агрегация изатуксимаба в видимые частицы зависит от системы рН и буфер. Буферная система на основе гистидина с рН, находящемся в диапазоне от 5,5 до 6,5, показала наибольшую стабильность при стрессовом воздействии встряхивания (демонстрируя меньше видимых частиц после воздействия), и буферная система на основе цитрата в пределах рН 5,0-7,0 и цитратный буфер при рН 5,5, 6,5 и 7,0 показали наибольшую стабильность, стабильность после стрессового термического воздействия (демонстрируя меньше видимых частиц после двух недель воздействия). Интересно, что буферная система на основе фосфата при рН, находящемся в диапазоне от 6,5 до 7,4, показала наибольшую термическую стабильность после одной недели стрессового термического воздействия, но имела несколько видимых частиц после двух недель стрессового термического воздействия и показала наименьшую стабильность с точки зрения изатуксимаба при стрессовом воздействии встряхивания из всех тестируемых буферных систем.
Таблица 2. - Визуальный осмотр
40°C
40°C
0 : отсутствие видимых частиц
+: отдельные видимые частицы
++: некоторое количество видимых частиц
+++: многочисленные видимые частицы.
Число довидимых частиц ≥10 мкм и ≥25 мкм после стрессового воздействия встряхивания и стрессового термического воздействия измеряли с помощью светоблокировки (LO). Как показано на фиг. 1, при стрессовом воздействии встряхивания и термическом воздействии фосфатные буферы демонстрировали наибольшие уровни довидимых частиц. Буферные системы на основе гистидина и ацетата обеспечивали наименьшие уровни довидимых частиц, что указывает на более высокую стабильность изатуксимаба.
Растворимые агрегаты (HMW) наблюдали с помощью эксклюзионной хроматографии (SE-HPLC) после стрессового термического воздействия в цитратном, гистидиновом, фосфатном, сукцинатном и ацетатном буферах при различных pH. Как показано на фиг. 2, что буферные системы на основе цитрата, фосфата и сукцината показали более высокое увеличение растворимых агрегатов, и наблюдали общую тенденцию, показывающую, что чем выше pH, тем выше содержание растворимых агрегатов (HMW). Это было особенно значимо при рН выше 7,0. Стрессовое воздействие встряхивания, по-видимому, не оказывает никакого влияния на растворимые агрегаты.
Влияние pH на стабильность изатуксимаба дополнительно изучали в гистидиновом буфере в присутствии сахарозы и полисорбата 80. Изатуксимаб в концентрации 5 мг/мл в составах, показанных в таблице 3, инкубировали при 40°C в течение 1 месяца и измеряли HMW с помощью SE-HPLC.
Таблица 3. - Тестируемая система буфер-pH
HMW наблюдали с помощью SE-HPLC. Как показано на фиг. 3, после 1 месяца стрессового термического воздействия при 40°C менее число растворимых агрегатов (HMW) отмечено в гистидиновых буферах при pH 6,0 по сравнению с pH 6,5.
Уровень кислотных изоформ изатуксимаба измеряли с помощью WCX (слабый катионный обмен) после инкубации 5 мг/мл изатуксимаба в составах, показанных в таблице 3, в течение 1 месяца при 40°C. Как показано на фиг. 4, составы на основе гистидина при pH 6,0 показали меньшее повышение уровня кислотных форм изатуксимаба по сравнению с рН 6,5 через 1 месяц при 40°С.
Эти результаты показали, что система рН и буфера влияли на стабильность изатуксимаба, и значение рН около pH 6,0 было более стабильным, чем рН 6,5.
Выбор вспомогательного вещества, уменьшающего вязкость
Чтобы обеспечить возможность подкожной доставки изатуксимаба, протестировали различные условия составления со вспомогательными снижающими вязкость веществами, чтобы определить, может ли быть получена высокая концентрация с вязкостью менее 25 сП при 20°C.
Антитело концентрировали и включали в состав тестируемых составов. Концентрацию белка в полученных составах подтверждали спектроскопически с помощью прибора SoloVPE и измеряли pH конечного раствора.
Таблица 4. - Составы, тестируемые в исследовании вязкости
% вес/об.
мМ
ацетата гистидина
ацетата гистидина
ацетата гистидина
сукцината
сукцината
фосфата
сукцината
фосфат-Трис
*: В составе применяли ацетат аргинина вместо аргинина-Cl.
Вязкость всех образцов измеряли на приборе RheoSense Initium с применением автоматического метода при 20°С. Он автоматически определяет скорость сдвига, подходящую для работы прибора в требуемом диапазоне давления для сенсоров. Поскольку скорость сдвига не превышала 10000 с-1, неньютоновские эффекты, такое как сдвиговое разжижение, считаются пренебрежимо малыми.
На фиг. 5 показана вязкость (сП) раствора изатуксимаба в концентрации 200 мг/мл при рН 6,0 в присутствии 0, 50, 100 и 200 мМ L-аргинина-Cl. Предварительные измерения показали, что аргинин-Cl был эффективным вспомогательным веществом, уменьшающим вязкость, действуя зависимым от концентрации образом; при этом увеличение концентрации аргинина-Cl коррелировало со снижением вязкости.
Протестировали более 40 различных составов с переменными значениями pH, средством для уменьшения вязкости для различных концентраций изатуксимаба. Результаты показаны в таблице 4.
Эффект концентрации аргинина-Cl на вязкость показан на фиг. 6A и 6B для двух концентраций изатуксимаба, 150 мг/мл (фиг. 6A) и 180 мг/мл (фиг. 6B) в присутствии 10 мМ гистидина и 2% сахарозы.
В дополнение к концентрации аргинина-Cl наблюдали выраженный эффект рН на вязкость изатуксимаба. На фиг. 7А и 7В показана зависимость вязкости от рН для 150 мМ аргинина-Cl (фиг. 7А) и 200 мМ аргинина-Cl (фиг. 7В).
Неожиданно, но результаты, показанные на фиг. 7А и 7В показали обратно пропорциональную зависимость между вязкостью и рН, а именно, увеличение вязкости было ассоциировано со снижением рН. При рН ≤ 5,7 наблюдалось резкое увеличение вязкости. Эта обратно пропорциональная зависимость противоположна тому, что, как правило, ожидается и наблюдается для белковых растворов. Кроме того, этот неожиданный эффект дополнительно усиливался с увеличением концентрации антитела (фиг. 8).
Было показано, что концентрация аргинина-Cl, составляющая по меньшей мере 100 мМ, уменьшает вязкость изатуксимаба в концентрации 150 мг/мл до менее 20 сП и вязкость изатуксимаба в концентрации 180 мг/мл до менее 40 сП. Кроме того, было показано, что рН оказывает значительный эффект на вязкость, о чем свидетельствует резкое увеличение вязкости при рН ниже 5,7. В то же время необходимо учитывать влияние рН на стабильность изатуксимаба. На фиг. 3 и 4 показано, что буферные системы на основе гистидина при значении рН, близком к 6,0, приводили к меньшему содержанию HMW после 1 месяца стрессового термического воздействия по сравнению с буферными системами при рН 6,5.
Пример 2. - Исследования вязкости
Располагая результатами исследований, описанных в примере 1, и с целью получения составов, содержащих высокую концентрацию (например, по меньшей мере 100 мг/мл) изатуксимаба, потенциально подходящих для подкожного введения, приготовили и исследовали более детально целый ряд составов. На вязкость белка в растворе могут влиять различные параметры, такие как концентрация белка, рН, концентрация средства для уменьшения вязкости и температура. Проводили несколько экспериментов для оценки влияния концентрации белка, pH, концентрации аргинина на вязкость состава. В таблице 5 показаны тестируемые параметры и диапазоны значений. Также проверяли эффект температуры, принимая во внимание, что лекарственный продукт будет храниться в охлажденном состоянии (5 ± 3°C).
Таблица 5. - Исследование вязкости
Целевую концентрацию устанавливали на уровне 140 мг/мл для достижения вязкости ниже 25 сП·с при 20°C, в том числе, когда растворы mAb демонстрируют колебания концентрации антитела или концентрации средства для уменьшения вязкости, или pH, присущие процессу производства. Действительно, такие различия между фактическими и целевыми композициями каждого вспомогательного вещества обычно наблюдаются при UF/DF из-за эффекта Доннана или во время стадий смешивания из-за точности взвешивания вспомогательных веществ или других производственных стадий (например, фильтрации), которые могут влиять на уровни вспомогательных веществ в конечном лекарственном продукте.
Растворы при высоких концентрациях mAb имеют тенденцию проявлять высокую вязкость. Аргинин выбрали в качестве средства уменьшения вязкости для определения возможности разработки жидкого состава изатуксимаба с высокой концентрацией, характеризующегося вязкостью ниже 25 мПа·с при 20°C.
Данное исследование проводили в трех группах. В первой группе проводили грубую оценку pH и концентрации. Во второй группе проводили тонкую оценку pH и концентрации. В третьей группе проверяли концентрацию аргинина.
В первой группе исследования изучали эффекты концентрации изатуксимаба, рН и температуры на вязкость. Для этой группы исследования приготовили семь составов. В таблице 6 представлены подробные сведения о составах с измеренными значениями.
Таблица 6. - Составы с измеренными значениями концентрации изатуксимаба (Cmab) и рН в составах
(мг/мл)
(мМ)
% (вес/об.)
% (вес/об.)
Концентрация Р188 на уровне 0,4% вес/об. показала положительный эффект в отношении стабильности по сравнению с составом без поверхностно-активного вещества. Сахароза в концентрации 2% обеспечивала достаточную стабильность при сохранении осмоляльности, близкой к изотоничности.
Во второй группе исследования изучали эффекты концентрации изатуксимаба и pH на вязкость при 20°C. Приготовили в общей сложности 32 состава. Растворы готовили при 4 целевых концентрациях антитела (126, 140, 147 и 154 г/л) и 8 целевых значениях рН (5,5, 5,7, 5,9, 6,2, 6,5, 6,7, 6,9, 7,0). В таблице 7 представлены подробные сведения о составах с измеренными значениями.
Таблица 7. - Составы с измеренными значениями концентрации изатуксимаба (Cmab) и рН в составе
(мг/мл)
(мМ)
% (вес/об.)
% (вес/об.)
В третьей группе исследования концентрация аргинина менялась в пределах 90-150 мМ. Подробная композиция 5 составов с измеренными значениями, если они были доступны, представлена в таблице 8.
Таблица 8. Составы для эталонных растворов (перед внесением) в группе исследования 3 - исследование влияния концентрации аргинина на стабильность и вязкость
(мг/мл)
% (вес/об.)
% (вес/об.)
Для группы 1 исследования вязкость всех образцов определяли с помощью вискозиметра Rheosense m-VROC при 5, 10, 15, 20, 25 и 30°C. Для каждого образца и каждого значения температуры скорости потока выбирали на уровне 50% от максимальной скорости потока, что приводило к скоростям сдвига от 250 до 2500 с-1. Для всех образцов предполагали ньютоновскую вязкость.
Для группы 2 исследования вязкость всех образцов определяли с помощью вискозиметра Rheosense m-VROC при 20°C. Для каждого образца скорость потока выбирали на уровне 50% от максимальной скорости потока, определенной с помощью прибора во время фазы заполнения. Это приводило к скоростям сдвига от 250 до 2500 с-1. Для всех образцов предполагали ньютоновскую вязкость.
Для группы 3 исследования вязкость всех образцов определяли с помощью вискозиметра Rheosense m-VROC при 20°C. Для каждого образца скорость потока выбирали на уровне 50% от максимальной скорости потока, определенной с помощью прибора во время фазы заполнения. Это приводило к скоростям сдвига от 1200 до 1600 с-1. Для всех образцов предполагали ньютоновскую вязкость.
Вязкость в зависимости от концентрации
При рН 5,5 вязкость увеличилась от 16 мПа·с при концентрации 126 мг/мл до 54 мПа·с при концентрации 154 мг/мл. При рН 7,0 вязкость увеличилась от 5,8 мПа·с до 11 мПа·с в том же диапазоне концентрации. Для всех условий данные подгоняли с применением уравнения Муни:
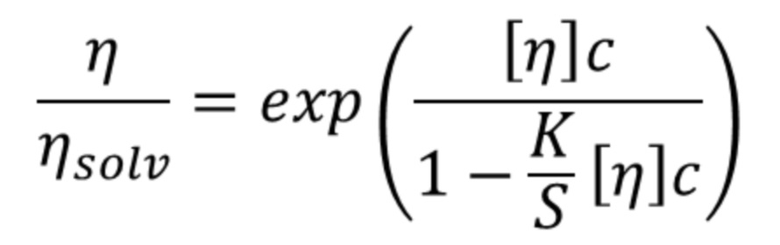
где ηsolv представляет собой вязкость растворителя, [η] представляет собой собственную вязкость белка, K представляет собой «фактор группирования» и S представляет собой «фактор формы». Подгоняемыми параметрами были [η] и K/S. Фиксированное значение использовали для ηsolv=1,26 мПа⋅с, что соответствует вязкости буфера для составления при 20°C.
Как показано на фиг. 8, вязкость растворов изатуксимаба увеличивалась с увеличением концентрации mAb.
Вязкость в зависимости от pH
На фиг. 9 показаны данные, полученные с помощью m-VROC в группе 2, зависимости вязкости от pH для 5 концентраций изатуксимаба (126, 140, 147 и 154 мг/мл). Как показано на фиг. 9, вязкость растворов изатуксимаба снижалось с увеличением pH.
При рН 6,2 вязкость составляла менее 25 мПа·с по всему диапазону концентраций тестируемого антитела. Вязкость составляла менее 25 мПа·с для всех значений рН, превышающих или равных 5,9. Однако при более низких исследованных значениях pH (5,5 и 5,7) вязкость была выше 25 мПа·с для концентраций mAb, превышающих или равных 147 г/л.
Вязкость в зависимости от температуры
В группе 1 вязкость исследуемых составов измеряли в зависимости от температуры от 5 до 30°C. Как показано на фиг. 10 и фиг. 11, вязкость снижалась по мере повышения температуры. Энергию активации составов определяли с применением подгонки Аррениуса (таблица 9). Значения использовали в комбинации с подгонкой данных при 20°C с помощью уравнения Муни для расчета теоретических значений. Как показано на фиг. 10, модель совпадала с экспериментальными данными.
Таблица 9. - Энергия активации для различных составов в группе 1
Энергия активации увеличивалась с увеличением концентрации изатуксимаба. Энергия активации снижалась при pH от 5,9 до 6,2. В меньшей степени энергия активации увеличивалась при рН от 6,2 до 6,8. Чтобы перемещаться в растворе, mAb должны избегать своих соседей и поэтому нуждаются в минимальной энергии, определенной в данном документе как Ea. Вероятность того, что mAb может получить данную энергию, пропорциональна exp(-Ea/RT) согласно закону Больцмана, и, таким образом, вязкость, которая обратно пропорциональна подвижности mAb, подчиняется уравнению вязкость=exp(+Ea/RT).
Для составов при рН 6,2 с концентраций mAb, составляющей 143 и 126 мг/мл, вязкость оставалась ниже 25 мПа·с на протяжении диапазона температур 5-30°С. При концентрации 154 мг/мл значение, зарегистрированное при 5°С, было слегка выше 25 мПа·с.
Для составов при pH 5,9 вязкость изатуксимаба при 142 мг/мл составляла приблизительно 25 мПа·с при 5°C.
Вязкость в зависимости от концентрации аргинина
Предварительные исследования показали, что для состава на основе изатуксимаба при концентрации 150 мг/мл (без поверхностно-активного вещества) вязкость снижалась от 60 мПа·с до 16 мПа·с при увеличении концентрации аргинина от 0 до 100 мМ и упала до 12 мПа·с только при увеличении концентрации аргинина от 100 до 200 мМ.
В настоящем исследовании тестировали концентрации аргинина от 90 до 150 мМ с точками 90, 100, 115, 125 и 150 мМ. Вязкость снижалась с увеличением концентрации аргинина (фиг. 12). Значения снижались от 11 до 9 мПа·с, подтверждая тенденцию, полученную в предварительных исследованиях.
В исследованном диапазоне концентраций аргинина (от 90 до 150 мМ) изменение вязкости составляло менее 2 мПа·с.
Пример 3. - Стабильность
В данном примере описывается серия исследований стабильности, в которых 2 набора составов подвергали исследованию стабильности при 5°C и ускоренному исследованию стабильности при 40°C/отн. влаж. 75%, а также исследованиям под стрессовым воздействием замораживания-оттаивания и встряхивания.
Тестируемые составы кратко изложены в таблице 10 и таблице 11.
Таблица 10. - F4-1 до F4-16
*1 тестировали в рамках ускоренного исследования.
Таблица 11 - F10-1 до F10-16
*1 тестировали в рамках ускоренного исследования.
Стрессовое воздействие замораживания-оттаивания выполняли с применением установки замораживания-оттаивания Epsilon1-6CC (Martin Christ GmbH, Остерод, Германия). Образцы подвергали замораживанию-оттаиванию.
Один флакон из каждого состава (F4 и F10) помещали в лиофилизатор и проводили циклы в соответствии со следующими параметрами: скорость 0,1°С/мин; температура замораживания -30°С; температура оттаивания 25°С; число циклов 5 и время выдержки при температуре, 60 мин. После пяти циклов образцы проверяли на наличие видимых частиц и гомогенизировали.
Один флакон с каждым составом устанавливали на горизонтальную платформу для встряхивания (IKA, KS4000 IC) и подвергали стрессовому воздействию при 25°C и 300 об/мин в течение 21 дня. Образцы T-mech анализировали вместе с моментом времени T-1, длительностью в один месяц.
Осмоляльность образцов измеряли с помощью способа снижения температуры замерзания с использованием Gonotec Osmomat 3000 (Gonotec, Берлин, Германия). Перед использованием прибора проводили калибровку по 3 точкам, которая включала воду Milli-Q и два стандарта осмоляльности при 300 и 400 мОсмоль/кг.
Концентрацию белка определяли с помощью УФ-спектроскопии, выполненной в 96-луночных планшетах (Corning Incorporation, Нью Йорк, США) на планшет-ридере Tecan Safire2 (Tecan Austria GmbH, Гредиг, Австрия). Образцы разбавляли гравиметрически от раствора с концентрацией 150 мг/мл до концентрации белка 1 мг/мл. Факторы разбавления рассчитывали из распечаток баланса. Каждую точку данных измеряли в трех лунках (n=3), заполненных 200 мкл раствора, для сведения к минимуму ошибки измерения. Температуру измерительной ячейки устанавливали на 25°С. Буфер для разведения измеряли в качестве холостого спектра. После измерения полученные при 280 нм значения поглощения корректировали с учетом длины оптического пути и вычитали с соответствующего холостой пробы Рассчитанный молярный коэффициент экстинкции (εмолярный) 224320 М-1см-1 при 280 нм применяли для расчета коэффициента экстинкции (ε). Рассчитанный коэффициент экстинкции (ε), составляющий 1,548 мл мг-1 см-1, применяли для определения концентрации белка на основе значений поглощения при 280 нм.
Флаконы проверяли на наличие или отсутствие видимых частиц при осторожном ручном радиальном помешивании в течение 5 секунд на белом фоне и в течение 5 секунд на черном фоне в соответствии с Европейской фармакопеей (8-е издание; монография 2.9.20) при примерно 3750 люкс. Осмотр проводили два обученные эксперты независимо друг от друга.
Для классификации наблюдаемых видимых частиц применяли как числовой показатель на основе “Deutscher Arzneimittel-Codex” (DAC 2006) (0, частицы не видны в течение 5 с; 1, несколько видимых частиц течение 5 с; 2, среднее число видимых частиц в течение 5 с; 10, большое число непосредственно видимых частиц). Волокноподобные структуры и частицы, которые, вероятно, не свойственны продукту, не учитывают в числовой оценке.
Высокоэффективная эксклюзионная хроматография (HP-SEC)
Перед анализом образцов характеристики тестировали с использованием стандарта гель-фильтрации BioRad (содержащего тироглобулин, гамма-глобулин, овальбумин, миоглобин и витамин B12). Стандарт готовили путем растворения лиофилизированного материала в 500 мкл воды Milli-Q с последующим 10-кратным разбавлением в подвижной фазе (конечная концентрация белка 3,6 мг/мл). Тест на пригодность системы проводили в начале каждой последовательности путем введения стандарта гель-фильтрации (BioRad) и оценивали посредством расчета разрешения USP между пиками гамма-глобулина и овальбумина. Исходные образцы с концентрацией 50 мг/мл разбавляли в 5 раз в полипропиленовых пробирках (Eppendorf) на 1,5 мл путем смешивания 40 мкл исходного раствора с 160 мкл раствора А (подвижная фаза без ацетонитрила), в результате чего концентрация белка составляла 10,0 мг/мл.
Чтобы избежать блокировку колонки HP-SEC более крупными нерастворимыми частицами, которые потенциально могли бы формироваться во время ускоренного тестирования стабильности/тестирования стрессового воздействия, образцы центрифугировали после разбавления при 18000 rcf в течение 5 мин, и надосадочную жидкость переносили во флаконы для HPLC. Образцы встряхивали на вортексе и хранили при 5°С в автоматическом пробоотборнике до проведения анализа.
Для анализа HP-SEC использовали следующие параметры:
Прибор: Ultimate 3000 (Dionex)
Колонка: колонка с аморфным диоксидом кремния ProSEC 300S; 300 мм
Security guard: предколонка 50 мм x 7,5 мм
Скорость потока: 0,3 мл/мин
Подвижная фаза: 90% 100 мМ фосфата, pH 7,2, 300 мМ NaClO4+10% ACN
Обнаружение: УФ при 280 нм и при 214 нм
Термостат колонок: 25 °C
Охлаждение образца: 5-8°C
Объем вводимой пробы: 10 мкл для образца с концентрацией 5,0 мг/мл, 10 мкл для холостых проб и стандартов SEC
Время анализа: 90 мин.
Капиллярное изоэлектрическое фокусирование
Капиллярное изоэлектрическое фокусирование (cIEF) с визуализацией выполняли на приборе iCE280, сопряженном с микроинжектором PrinCE (Convergent Bioscience, Торонто, Канада). Вместо элюирования сфокусированные молекулярные разновидности проходят фиксированную точку обнаружения, как это принято при традиционном cIEF, при этом в cIEF с визуализацией молекулы обнаруживают по всему капилляру IEF. Для этого УФ-свет с длиной волны 280 нм фокусировали на УФ-прозрачном капилляре, и изображения получали через равные промежутки времени с помощью камеры прибора с зарядовой связью (CCD).
Перед анализом в соответствии с инструкциями к прибору устанавливали картридж с покрытием из плавленного кварца (FC). Анодный резервуар заполняли 0,08 М фосфорной кислоты (в 0,1% метилцеллюлозе, набор электролитов, ProteinSimple), а катодный резервуар заполняли 0,1 М гидроксида натрия (в 0,1% метилцеллюлозе, электролитный набор, ProteinSimple). Производительность системы проверяли путем измерения стандарта гемоглобина (набор для проверки пригодности системы iCE280, ProteinSimple). Изоэлектрическую фокусировку раствора со стандартом гемоглобина проводили согласно инструкции производителя (предварительная фокусировка: 1 мин при 1500 В; фокусировка: 4,5 мин при 3000 В).
Мастер-микс для 20 образцов готовили путем смешивания 2360 мкл воды Milli-Q, 1400 мкл 1% метилцеллюлозы, 160 мкл фармалита, pH 3-10, 20 мкл маркера, pI 7,05, и 20 мкл маркера, pI 9,50. Мастер-микс гомогенизировали с помощью встряхивания на вортексе и быстро центрифугировали при 5000 rcf. Смесь фильтровали с помощью 0,45 мкм шприцевого фильтрующего элемента из PVDF (Millex-GV, Millipore).
Образцы предварительно разбавляли до концентрации белка 20,0 мг/мл путем смешивания 20 мкл исходных растворов (C=50 мг/мл) с 30 мкл соответствующих буферов для составления. Окончательные образцы для анализа cIEF готовили путем смешивания 2 мкл предварительно разведенных образцов с концентрацией 20,0 мг/мл со 198 мкл мастер-микса с получением общего объема 200 мкл и концентрацией белка 0,2 мг/мл.
Изоэлектрическое фокусирование DP проводили путем предварительной фокусировки в течение 1 мин при 1500 В с последующей фокусировкой в течение 8 мин при 3000 В. Изображения УФ-поглощения анализировали с применением программы ChromPerfect (версия 5.5.6).
Результаты визуального осмотра
Визуальный осмотр проводили два независимых оператора. Для растворов образцов после замораживания-оттаивания отмечали негомогенное состояние (S- Шлирен*, фазовое разделение). Для всех тестируемых составов во время исследования стабильности никаких существенных изменений в содержании видимых частиц и мутности не наблюдали.
Результаты измерений осмоляльности представлены в таблице 12. В Т0 осмоляльность находилась в диапазоне 275-418 мОсмоль/кг. Для составов F10 показаны более высокие значения осмоляльности в отличие от составов F4. Хранение при 40°C/отн. влаж. 75% не оказывало влияния на осмоляльность в тестируемых образцах.
Таблица 12.- Результаты измерения осмоляльности в T0 (n=1)
Таблица 13. - Результаты измерений осмоляльности в T-1w_40°C, T-2w_40°C и T-1m_40°C
Результаты измерений вязкости в Т0 указывают на то, что для составов F10 показана несколько более высокие значения вязкости, чем для составов F4. Наибольшее значение вязкости измеряли для состава F4-8 (30,03 сП при 1000 с-1 при 20°С) и F10-8 (34,13 сП при 1000 с-1 при 20°С). В прогоне 8 для обоих наборов составов (F4 и F10) содержалась наибольшая концентрация белка (154 мг/мл), наименьшее значение pH (5,9) и наибольшая концентрация сахарозы (24 мг/мл).
Результаты определения концентрации белка с помощью УФ-спектроскопии представлены в таблице 14 и таблице 15. В Т0 концентрация белка находилась в диапазоне от 124 до 156 мг/мл. Концентрация белка в тестируемых составах оставалась стабильной во время хранении при 40°C/отн. влаж. 75%.
Таблица 14. - Концентрация белка
Таблица 15 - Концентрация белка
Гель-электрофорез с додецилсульфатом натрия (SDS-PAGE) применяли для определения молекулярной массы и относительного количества разновидностей белков. Относительные количества разделенных разновидностей рассчитывали на основе гелей SDS-PAGE путем измерения оптической плотности обнаруженных белковых полос. Относительное количество и молекулярная масса всех обнаруженных разновидностей белков в невосстанавливающих и восстанавливающих условиях содержаться в таблице 16 и таблице 17 соответственно.
Таблица 16. - Относительное количество и расчетная молекулярная масса разновидностей, обнаруженных с помощью невосстанавливающего SDS-PAGE
T0
T0
T1w_40°C
T1w_40°C
T2w_40°C
T2w_40°C
T1m_40°C
T1m_40°C
Таблица 17. - Относительное количество и расчетная молекулярная масса разновидностей, обнаруженных с помощью восстанавливающего SDS-PAGE
Для оценки в отношении мономера, агрегатов и фрагментов выполняли высокоэффективную эксклюзионную хроматографию (HP-SEC). Были определены четыре разновидности агрегатов (HMW1, HMW2, HMW3 и HMW4) и две разновидности фрагментов (LMW1 и LMW2).
В Т0 относительное содержание мономеров в составах F4 и F10 находилось в диапазоне от 98,5 до 99,0%. Хранение при 40°С/отн. влаж. 75% приводило к снижению содержания мономера во всех тестируемых образцах. Содержание мономера в тестируемых составах после хранения при 40°С/отн. влаж. 75% в течение одного месяца находилось в диапазоне от 96,2 до 97,2%; при этом наименьшее содержание мономера в этот момент времени обнаружили в составе F4-2. Повторные циклы замораживания-оттаивания практически не оказали влияния на относительное содержание мономеров, в то же время стрессовое механическое воздействие приводило к небольшому снижению (от 97,7 до 98,2%). См. фиг. 13 и фиг. 14.
Почти для всех образцов и моментов времени относительное содержание HMW1 было ниже предела количественного определения, составлявшего 0,15%, и, аналогичным образом, почти для всех образцов и моментов времени относительное содержание HMW2 было ниже предела количественного определения, составлявшего 0,15%.
Относительное содержание HMW3 находилось в диапазоне 0,7-0,9% в Т0. После одного месяца хранения при 40°C/отн. влаж. 75% содержание HMW3 увеличилось до 1,1-1,5%. Наибольшее увеличение содержания HMW3 наблюдали в образцах F4-14, F10-5, F10-12 и F10-14, в то время как наименьшее увеличение наблюдали в образцах состава F4-1 и F4-7. Повторные циклы замораживания-оттаивания не приводили к увеличению относительного содержания HMW3, в то время как стрессовое механическое воздействие приводило к незначительному увеличению HMW3 до 0,9-1,2%.
Почти для всех образцов и моментов времени относительное содержание HMW4 было ниже предела количественного определения, составлявшего 0,15%.
Сумму всех агрегатов (HMWS) рассчитывали по пикам ≥0,15% относительной площади. В Т0 общее содержание агрегатов (HMWS) находилось в диапазоне 0,6-1,2%. Хранение при 40°С/отн. влаж. 75% приводило к заметному увеличению содержания HMWS во всех тестируемых составах. Относительное содержание HMWS после хранения при 40°С/отн. влаж. 75% в течение одного месяца находилось в диапазоне 1,1-1,9%. Наименьшее увеличение разновидностей HWM наблюдали в случае состава F4-7, а наибольшее увеличение наблюдали в случае составов F4-2 и F10-14. После замораживания-оттаивания существенных изменений содержания HMWS не наблюдали, и лишь небольшие увеличения обнаружили после воздействия механического стресса, приводящего к содержанию HWMS в диапазоне 0,9-1,3%. См., фиг. 15 и фиг. 16.
В Т0 содержание фрагмента LMW1 составляло приблизительно 0,3. Хранение при 40°С/отн. влаж. 75% приводило к увеличению содержания LMW1 во всех тестируемых образцах. Относительное содержание LMW1 было наибольшим после хранения при 40°С/отн. влаж. 75% в течение одного месяца и находилось в диапазоне 1,1-1,5%. Замораживание-оттаивание не оказало влияния на содержание LMW1. Стрессовое механическое воздействие приводило к небольшому увеличению содержания LMW1.
В Т0 относительное содержание фрагмента LMW2 было ниже предела количественного определения (≥0,15%) во всех тестируемых составах. Хранение при 40°С/отн. влаж. 75% в течение двух недель и одного месяца приводило к небольшому увеличению содержания LMW2, при этом наибольшие значения наблюдали при T-1m_40°С (0,3-0,5%).
Сумму содержания всех LMWS рассчитывали по пикам ≥ 0,15%. В Т0 общее содержание LMWS составляло приблизительно 0,3% во всех тестируемых составах. В то время как замораживание-оттаивание не оказывало влияния на содержание LMWS, стрессовое механическое воздействие приводило к небольшому увеличению содержания LMWS (0,7-0,9%). Хранение при 40°C/отн. влаж. 75% приводило к более заметному увеличению, особенно в более поздние моменты времени (фиг. 17 и фиг. 18). Наибольшее увеличение наблюдали в T-1m_40°С, при этом общее содержание LMWS находилось в диапазоне 1,4-1,8%.
В Т0 относительное содержание главного пика, измеренное с помощью капиллярного изоэлектрического фокусирования (cIEF), составляло 70,3-74,0%. Постепенное снижение относительного содержания главного пика обнаружили после хранения при 40°C/отн. влаж. 75% в течение одной недели, двух недель и одного месяца. Наибольшее снижение наблюдали для состава F10-8 (55,4% в T- 1m_40°C). pI главного пика оставалась стабильной на уровне приблизительно 8,2 во всех составах во все моменты времени проверки стабильности.
В T0 относительное содержание кислотных видов находилось в диапазоне приблизительно 17,9-20,9%. Хранение при 40°С/отн. влаж. 75% приводило к увеличению содержания кислотных видов. Наибольшее увеличение наблюдали в момент времени T-1m_40°C со значениями от 30,4 до 36,9%. Наибольшее увеличение наблюдали в случае состава F10-8, в то время как составе F4-7 продемонстрировал наименьшее увеличение. Графическое представление данных показано на фиг. 19 и фиг. 20.
В T0 наблюдали содержание основного пика в диапазоне от 8,1 до 9,5%. Во время исследования стабильности содержание главного пика оставалось относительно стабильным для всех составов в большинстве моментов времени. В случае T-1m_40°С содержание основного пика находилось в диапазоне 7,1-9,1%.
Данные капиллярного изоэлектрического фокусирования (cIEF) после хранения при 40°C/отн. влаж. 75% показали увеличение содержания кислотных видов за счет содержания главного пика для всех составов. Составы F4 продемонстрировали лучшую химическую стабильность, чем составы F10 при меньшем снижении содержания главного пика после одного месяца хранения при 40°C/отн. влаж. 75%, как показано на фиг. 21. После одного месяца хранения при 40°C/отн. влаж. 75% составы F4 продемонстрировали потерю пика мономера в диапазоне 9,5-13,9%, в то время как составы F10 продемонстрировали потерю пика мономера в диапазоне 14,7-17,3%.
Пример 4. - Стабильность
Представлены результаты исследований стабильности, проведенных для изатуксимаба в концентрации 140 мг/мл, составленного в 110 мМ аргинина-HCl, 9 мМ гистидина, 2% сахарозы, 0,4% полоксамера-188, рН 6,2.
РЕЗУЛЬТАТЫ
Стабильность при -20°C±5°C
Как показано в таблице 18, после хранения при -20°C±5°C в течение 1 месяца все тестируемые показатели качества (видимые частицы, цвет, степень опалесценции, чистота по SEC и cGE, концентрация белка по УФ, активность по биоанализам ADCC и CDC, твердые частицы по светоблокировке и pH) оставались стабильными.
Гетерогенность заряда согласно icIEF не показала каких-либо значительных изменений по сравнению с исходным материалом.
Стабильность при +5°С±3°С (условия долгосрочного хранения)
Как показано в таблице 19 и таблице 20, после хранения при температуре +5°C±3°C в течение 1, 3, 6, 9 и 12 месяцев все тестируемые показатели качества (видимые частицы, чистота по SEC и cGE, концентрация белка по УФ, активность по биоанализу CDC, твердые частицы по светоблокировке и pH) оставались стабильными в течение по меньшей мере 12 месяцев.
Цвет, степень опалесценции и гетерогенность заряда согласно icIEF не показали каких-либо значительных изменений по сравнению с исходным материалом в течение по меньшей мере 12 месяцев по сравнению с исходным материалом.
Стабильность при +25°C±2°C/60%±5% RH (условия хранения с "ускоренным старением")
Как показано в таблице 21, после хранения при температуре +25°C±2°C в течение 6 месяцев все тестируемые показатели качества (видимые частицы, чистота по SEC, концентрация белка по UV, активность по биоанализам ADCC и CDC, твердые частицы по светоблокировке и pH) оставались стабильными в течение по меньшей мере 6 месяцев.
После 6 месяцев при +25°С±2°С не наблюдали никаких изменений ни в цвете, ни в степени опалесценции.
После 6 месяцев при +25°С±2°С наблюдали следующее изменение профиля гетерогенности заряда по icIEF: наблюдали снижение содержания главной изоформы на 9% вместе с увеличением кислотных изоформ на 9%.
Наблюдали небольшое снижение чистоты главного пика (-2%) по cGE, коррелирующее с увеличением суммы низкомолекулярных разновидностей (+2%).
Наблюдали небольшое снижение чистоты мономера (-1,7%) по SEC.
Все остальные параметры не показали какого-либо значительного изменения по сравнению с исходным материалом.
Стабильность при +40°C±2°C/75%±5% RH (условия стрессового воздействия)
Как показано в таблице 22, в условиях стрессового воздействия через 1 месяц наблюдали следующие изменения.
Наблюдали следующее изменение профиля гетерогенности заряда по icIEF: снижение содержания главной изоформы на 14% в основном коррелировало с увеличением кислотных форм на 15%.
Наблюдали небольшое снижение чистоты главного пика (-2%) по cGE, коррелирующее с увеличением суммы низкомолекулярных разновидностей (+2%).
Процентное содержание агрегатов по SEC оставалось постоянным.
ВЫВОД
Результаты данного исследования стабильности показывают, что изатуксимаб - 140 мг/мл, составленный в 110 мМ аргинина-HCl, 9 мМ гистидина, 2% сахарозы, 0,4% полоксамера-188, рН 6,2, остается стабильным при -20°C в течение по меньшей мере 1 месяца и при +5°С±3°С в течение по меньшей мере 12 месяцев.
Таблица 18. - Изатуксимаб в концентрации 140 мг/мл - результаты исследования стабильности при -20°C±5°C
Таблица 19. - Раствор изатуксимаба для инъекций в концентрации 140 мг/мл -
результаты исследования стабильности при +5°C±3°C (от T1M до T6M)
Таблица 20. - Раствор изатуксимаба для инъекций в концентрации 140 мг/мл - результаты исследования стабильности при +5°C±3°C (от T9M до T12M)
Таблица 21. - Раствор изатуксимаба для инъекций в концентрации 140 мг/мл - результаты исследования стабильности при +25°C±2°C/60%±5% RH
Таблица 22. - Раствор изатуксимаба для инъекций в концентрации 140 мг/мл - результаты исследования стабильности при +40°C±2°C/75%±5% RH
Пример 5. - Исследование in vivo на мини-свиньях
В данном примере описано исследование, в котором мини-свиней обрабатывали составами на основе изатуксимаба, вводимыми посредством подкожной инъекции. Мини-свинью выбрали в качестве тестовой модели для данного исследования подкожной местной переносимости из-за ее общепризнанной пригодности и поскольку ее обычно применяют для неклинической оценки предполагаемого пути введения человеку.
Тестировали четыре состава (F1, F2, F4 и F10) и солевой раствор (0,9% хлорида натрия) в качестве отрицательного контроля на 8 мини-свиньях (самка возрастом 1 год, 20-25 кг), по 4 животных на группу тестирования. Каждому животному вводили инъекцией 2 состава и отрицательный контроль с 3-недельным периодом восстановления между каждой инъекцией. Введение осуществляли в область бока через катетер, оснащенный иглой типа "бабочка" калибром 27G, с применением шприцевого насоса (Harvard Apparatus Model '22'), оснащенного удержателем обратного давления (субминиатюрный датчик нагрузки RSB5 50LB/200N) для мониторинга обратного давления в тканях.
Во время инфузии за животными наблюдали, уделяя особое внимание общему поведению, голосовым сигналам и визуальным параметрам для оценки боли. Также отслеживали давление в инфузионном шприце и признаки подтекания. В местах инъекций наблюдали изменения кожи и выполняли анализ плазмы крови на субстанцию Р и кортизол. Биопсию кожи собрали на 5-й день в 10% NBF.
Симптомы боли регистрировали во время и после инъекции в течение 5 дней. Маркеры боли определяли в крови, образцы которой собирали на протяжении инъекции и в течение следующих 90 минут. Затем проводили гистопатологическое исследование образца кожи, взятого в месте инъекции. С точки зрения гистопатологии все составы хорошо переносились.
Все растворы изатуксимаба готовили из одной и той же партии предварительно составленного раствора изатуксимаба. Данной партия была составлена при концентрации 30 мг/мл, pH 6, с 20 мМ гистидина и 5% сахарозы.
Все составы, протестированные в поисковом исследовании на мини-свиньях, описаны в таблице 23.
Таблица 23. Составы для исследования на мини-свиньях
% (p/v)
** номинальные значения диафильтрационного буфера и CES без учета сдвига, вызванного эффектом Доннана во время; ультрафильтрации, но позже оцененные количественно в F4 и F10.
Для приготовления каждого состава с помощью диафильтрации проводили замену буфера (без сахарозы и поверхностно-активного вещества), и антитело концентрировали до более высокого значения, чем в целевом составе. После первой корректировки концентрации добавляли концентрированный раствор вспомогательного вещества (концентрированную сахарозу и поверхностно-активное вещество, растворенные в диафильтрационном буфере) для получения конечных составов.
Исследуемые диафильтрационные буферы представлены в таблице 24.
Таблица 24. - Композиция диафильтрационных буферов
Зарегистрированные значения конечного состава показаны в таблице 25.
Таблица 25. - Результаты анализа
(мМ)
(мМ)
(мМ)
** pH, измеренный после фильтрации с помощью микроэлектрода;
n.a.=недоступно (не измерено).
В исследовании использовали в общей сложности восемь самок мини-свиней возрастом 1 год. Каждый состав тестировали на 4 животных, каждое из которых получало объем, составляющий 18 мл, за 30 минут (0,6 мл/мин). Исследование продолжалось 5 дней, в течение которых регистрировали маркеры и симптомы боли. По истечении 5 дней собирали образец (биопсию), представляющий собой диск кожи диаметром 8 мм, для проведения гистопатологического исследования.
Составы F2 и F4 впервые тестировали на мини-свиньях. Затем после задержки в 21 день на тех же 8 мини-свиньях тестировали составы F1 и F10. Наконец, после второй задержки, на тех же 8 мини-свиньях тестировали физиологический раствор.
Для каждого состава/солевого раствора исследование проводили следующим образом:
• Введение раствора 4 мини-свиньям (8 для физиологического раствора)
• Во время инфузии: запись в режиме реального времени следующих параметров:
o Телеметрические измерения (ECG)
o Обратное давление шприца (для расчета тканевого обратного давления)
o Маркеры боли (кортизол, субстанция Р, определяемые в крови - требуется регулярный отбор образцов крови)
o Симптомы боли (почесывание, царапанье, шум, покраснение) и размер отека от инфузии.
• После инфузии:
o Маркеры боли в течение 90 минут после окончания инфузии
o Симптомы боли (почесывание, царапанье, покраснение) в течение 5 дней (визуальный осмотр, произвольные единицы)
o Гистопатологическое исследование образцов ткани (биопсия кожи через 5 дней)
Основные результаты в отношении боли обобщены в таблице 26.
Таблица 26. - Маркеры боли и симптомы боли
(максимум из среднего значения для 4 мини-свиней)*
**n.r.= не сообщается.
В таблице 26 в качестве маркера боли сообщали только о кортизоле плазмы крови. Уровни субстанции Р, измеренные для всех составов, не изменились с течением времени. После инъекции регистрировали повышение уровня кортизола в плазме крови. Максимум из среднего значения, рассчитанное для 4 мини-свиней для каждого состава, сообщается в таблице 26. Максимальное значение данного среднего получили для F10. В случае всех составов наблюдали изменчивость уровня кортизола в плазме крови между 4 тестируемыми животными.
Число симптомов болт в случае F10 также было наибольшим из 4 составов и характеризовалось наибольшими уровнями кортизола в плазме крови (фиг. 22). F2 характеризовался вторым местом по числу симптомов боли и наибольшими уровнями субстанции P (фиг. 23). В таблице 26 не показано, что оба F1 и F2 характеризовались неудовлетворительной стабильностью антител.
Во время инъекции 4 составов не наблюдали подтекания и отмечали почти постоянное обратное давление в шприце.
Основные результаты гистопатологического исследования кратко обобщены в таблице 27.
Таблица 27. - Результаты гистопатологического исследования
Для дермы и подкожной ткани не показано значимых различий между составами. Для состава F2 показана большая частота встречаемости (100%) изменений скелетных мышц по сравнению с составом F1 (25%), F4 (25%), F10 (0%) или солевым раствором (25%).
Пример 6. - Второе исследование in vivo на мини-свиньях
Цель исследования, описанного в данном примере, заключалась в оценке местной переносимости и фармакокинетики изатуксимаба в плазме крови после однократного введения путем подкожной инфузии мини-свиньям с применением трех различных скоростей потока. Кроме того, одной группе животных однократно вводили дозу изатуксимаба внутривенно, чтобы оценить его биодоступность после различных подкожных инфузий.
Использовали тестируемые образцы: изатуксимаб 500 мг/25 мл (20 мг/мл) для IV инфузии и изатуксимаб 140 мг/мл (состав F4 из примера 5) для SC инфузии. Солевой раствор (0,9% NaCl) для инъекций использовали в качестве продукта отрицательного контроля для групп 2, 3 и 4.
Исследование выполняли на 20 самках SPF (без специфических патогенов) мини-свиней Göttingen от Ellegaard Göttingen Minipigs A/S, DK-4261 Далмос, Дания. Заказывали животных с весом тела от 20 до 25 на момент поступления. Предусматривался предшествующий обработке период, составляющий 15 дней (включая период акклиматизации, составляющий 5 дней), во время которого животных ежедневно осматривали, чтобы отбраковать любое животное в неудовлетворительном состоянии. Все наблюдения регистрировались.
Животных рандомизировали в четыре группы обработки, как показано в таблице 28:
Таблица 28. - Группы обработки
концентрация (мг/мл)
(скорость инфузии)
(мл)
Доза
(мг/животное)
№
(20)
(3 мл/мин)
(140)
(0,5 мл/мин)
(140)
(1 мл/мин)
(140)
(2 мл/мин)
Для внутривенной инфузии 6 животным (5+1 запасное) имплантировали катетеры в ушные вены за 9 дней до начала обработки. Пять из этих животных распределяли в группу 1 (группа IV инфузии); последнее животное включали в одну из групп подкожной обработки.
Все введения выполняли с применением волюметрического инфузионного насоса Baxter Colleague CXE. С помощью насоса Baxter Colleague CXE можно выполнять инфузию из полужестких контейнеров, жестких контейнеров, гибких пакетов для IV вливаний и вентилируемых шприцев. Во время введений тестируемые продукты и продукты отрицательного контроля помещали в стерильные стеклянные инфузионные флаконы.
— Первый день обработки обозначили как день 1.
— В день 1 группе 1 однократную дозу изатуксимаба (1800 мг/животное) вводили посредством 30-минутной инфузии через имплантированный катетер в ушной вене при скорости потока 3 мл/мин.
— В день 1 группам 2, 3 и 4 однократную дозу изатуксимаба (1806 мг/животное) вводили через подкожный катетер с иглой-бабочкой при скорости потока 0,5, 1 и 2 мл/мин соответственно. Иглу-бабочку помещали в область левого нижнего бока прямо перед областью колена.
— В день 8 группам 2, 3 и 4 однократную дозу солевого раствора (отрицательный контроль) вводили через подкожный катетер с иглой-бабочкой при скорости потока 0,5, 1 и 2 мл/мин соответственно. Иглу-бабочку помещали в область правого нижнего бока прямо перед областью колена.
— Объем дозы составлял 90 мл в группе 1 и 12,9 мл в группах 2-4.
— Места подкожных инъекций в группах 2-4 наблюдали на предмет подтекания во время инфузии.
— Места подкожных инъекций маркировали вдоль края местного отека, который развивался во время процедуры инфузии, и при необходимости маркировали повторно.
— Калибр иглы для SC инфузии составлял 23G.
Ежедневно регистрировали все признаки ухудшения здоровья и любые изменения в поведении. Регистрировали любое отклонение от нормы. Во время введения дозы наблюдали за общим поведением животного и любым голосовым сигналом, уделяя особое внимание любым признакам стресса, дискомфорта или боли.
В случае групп 2-4 места подкожных инъекций осматривали ежедневно от дня введения дозы на наличие кровоизлияния, эритемы, припухлости (образование пузырьков с указанием размера) и уплотнения, но не исключая другие признаки. В дни введения дозы места инъекций осматривали перед инфузией и в конце инфузии, и затем через 15 мин (±2 мин), 30 мин (±2 мин), 1 (±3 мин), 2 (±6 мин) и 4 часа (±12 мин) после окончания введения дозы. После этого места инъекций осматривали ежедневно до дня 17, поскольку с дня 10 местных реакций не наблюдали. Параметры оценивали согласно следующей системе оценивания: 0 - не присутствует; 1 - минимальный; 2 - легкий; 3 - умеренный и 4 - выраженный.
Образцы крови для фармакокинетики
Начиная в день 1, у всех животных собирали образцы крови. Сбор образцов крови выполняли в следующие моменты времени: до обработки, в пределах 2 минут после окончания инфузии и через 1 (±3 мин), 4 (±12 мин), 24 (±1 час 12 мин), 48 ( ±2 часа 24 мин), 72, 96, 168, 192, 264, 336, 504 и 672 часа после окончания инфузии (с допуском ±3 часа от 72 часов).
Фармакокинетический (PK) анализ выполняли с применением программного обеспечения Phoenix WinNonlin версии 6.3 от Pharsight Corporation, Маунтин-Вью, Калифорния, США. При необходимости выполняли некомпартментный анализ с применением модели концентрации в плазме крови WinNonlin (модель внутривенной инфузии и экстраваскулярной дозы).
Данные зависимости концентрации в плазме крови от времени для каждого отдельного животного применяли для фармакокинетических расчетов. В дополнение к оценкам параметров для отдельных животных при необходимости сообщали описательные статистики (например, среднее, стандартное отклонение и коэффициент вариации). Все параметры для каждого животного получали на основе индивидуальных концентраций тестируемого продукта в плазме крови после осуществления обработки в день 1. Для определения индивидуальных фармакокинетических параметров концентрации, находившиеся ниже предела количественного определения, считали нулевыми. Для определения средних концентраций образцы, находившиеся ниже предела количественного определения, считали нулевыми.
Параметры оценивали с применением номинальных уровней дозы. Параметры оценивали с применением номинального времени сбора образцов, поскольку не задокументировано отклонений по времени более чем на 15% от номинального. Концентрации перед введением дозы в день 1 устанавливали равными нулю.
Описательные статистические данные (среднее, стандартное отклонение, если применимо) и фармакокинетические параметры сообщали в виде трехзначных разрядов. Коэффициент вариации сообщали без десятичного знака.
Для изатуксимаба оценивали следующие фармакокинетические параметры:
• Cmax - наблюдаемая максимальная концентрация,
• tmax - время до достижения максимальной концентрации,
• Clast - последняя измеряемая концентрация,
• tlast - время до достижения последней измеряемой концентрации,
• AUC0-24 ч - площадь под кривой зависимости концентрация в плазме крови-время от 0 до 24 часов, которую рассчитывали с помощью некомпартментного анализа с применением линейного правила трапеции,
• AUC0-72 ч - площадь под кривой зависимости концентрация в плазме крови-время от 0 до 72 часов, которую рассчитывали с помощью некомпартментного анализа с применением линейного правила трапеции,
• AUC0-168 ч - площадь под кривой зависимости концентрация в плазме крови-время от 0 до 168 часов, которую рассчитывали с помощью некомпартментного анализа с применением линейного правила трапеции,
• AUC0-t - площадь под кривой зависимости концентрация в плазме крови-время, рассчитанную для 0-t, где t представляет собой время до достижения последней измеряемой концентрации, которую рассчитывали с помощью некомпартментного анализа с применением линейного правила трапеции,
• Биодоступность SC инфузий по сравнению с IV инфузиями, которую оценивали с применением AUC0-24 ч, AUC0-72 ч, AUC0-168 ч и AUC0-t.
Дополнительные параметры (т. е. t1/2z, Vz., CL) рассчитывали для IV пути введения.
Сбор образцов биопсии кожи из мест SC инъекций (группы 2-4)
В оба день 8 и день 29 для гистопатологического исследования из места левой подкожной инфузии слева (место введения тестируемого продукта) у всех животных в группах 2-4 собрали три образца биопсии кожи глубиной приблизительно 7-10 мм с применением 6-мм иглы для биопсии. Сбор в день 8 выполняли после введения дозы солевого раствора (отрицательный контроль), чтобы иметь возможность отслеживать животных во время инфузий.
Образцы биопсии, собранные в день 8 (образцы биопсии № 1-3), отбирали из краниальной половины области инфузии. Образец биопсии № 1 собрали из дорсальной области, образец биопсии № 2 из средней области и образце биопсии № 3 из вентральной части. Образцы биопсии, собранные в день 29, собирали аналогичным образом из каудальной половины области инфузии. Кроме того, необработанный контроль (образец биопсии № 4) собирали за пределами области введения дозы из одной и той же области у всех животных в оба дня.
В день 15 и в день 36 аналогичным образом собирали образцы биопсии из места правой подкожной инфузии (место введения солевого раствора) у всех животных в группах 2-4.
Образцы биопсии, собранные в день 15 (образцы биопсии № 5-7), отбирали из краниальной половины области инфузии. Образец биопсии № 5 собрали из дорсальной области, образец биопсии № 6 из средней области и образце биопсии № 7 из вентральной части. Образцы биопсии, собранные в день 36, собирали аналогичным образом из каудальной половины области инфузии. Кроме того, необработанный контроль (образец биопсии № 8) собирали за пределами области введения дозы из одной и той же области у всех животных в оба дня.
Каждый образец биопсии от каждого животного помещали в отдельный контейнер и фиксировали в нейтральном 4% формальдегиде с фосфатным буфером.
Результаты
Ни у одного из животных не наблюдали клинических признаков, связанных с тестируемым продуктом.
У трех животных (№ 4, группа 1, и № 6 и 7, группа 2) кожа казалась теплой на ощупь в день 1 и день 2. Однако, поскольку количество пораженных животных было низким, это сочли случайной результатом.
Во всех группах балльную оценку местных реакций в местах инфузии выполняли в день инфузии. В остальные дни балльные оценки не начисляли.
В группе 2 (0,5 мл/мин) в день 1 (изатуксимаб) у всех животных в месте инфузии первоначально отмечали легкую эритему, и минимальный или умеренный отек (образование пузырьков) наблюдали у трех животных в пределах первых двух часов после окончания инфузии. Кроме того, у трех животных было минимальное кровотечение в пределах первых 15 минут после окончания инфузии.
В день 8 (солевой раствор при 0,5 мл/мин) у всех животных в группе 2 в месте инфузии первоначально отмечали минимальную эритему, и минимальный или умеренный отек (образование пузырьков) наблюдали у двух животных в пределах первых двух часов после окончания инфузии. Кроме того, минимальное кровотечение наблюдали у трех животных после окончания инфузии.
В группе 3 (1 мл/мин) в день 1 (изатуксимаб) у четырех животных в месте инфузии наблюдали минимальную или легкую эритему в пределах первых 4 часов после окончания инфузии, а у трех животных отметили минимальный или выраженный отек (образование пузырьков), уменьшавшийся в размерах через 4 часа после окончания инфузии. У трех животных наблюдали минимальное кровотечение, а у одного животного легкое кровотечение в пределах первых 30 минут после окончания инфузии.
В день 8 (солевой раствор при скорости 1 мл/мин) в группе 3 у всех животных в месте инфузии наблюдали минимальную или легкую эритему в пределах первых 4 часов после окончания инфузии, а у одного животного после введения дозы был минимальный или умеренный отек (образование пузырьков), уменьшавшийся в размере через 2 часа после окончания инфузии. У всех животных наблюдали минимальное кровотечение, а у одного животного легкое кровотечение в пределах первых 15 минут после окончания инфузии.
В группе 4 (2 мл/мин) в день 1 (изатуксимаб) у четырех животных в месте инфузии наблюдали минимальную или легкую эритему (умеренную у одного животного) в пределах первых 2 часов после окончания инфузии, а у трех животных после введения дозы отметили вплоть до выраженного отека (образование пузырьков), уменьшавшегося в размерах через 4 часа после окончания инфузии. У двух животных показано минимальное кровотечение в конце инфузии.
В день 8 (солевой раствор при скорости 2 мл/мин) в группе 4 у всех животных в месте инфузии первоначально наблюдали минимальную эритему (легкую эритему в двух случаях) в пределах первых 30 минут после окончания инфузии, а у двух животных после введения дозы отмечали вплоть до умеренного отека (образование пузырьков), уменьшавшегося в размерах через 1 час после окончания инфузии. Минимальные кровотечения наблюдали у двух животных в конце инфузии.
Во время инфузий признаков подтекания не наблюдали.
Таблица 29. - Краткий обзор данных о местных реакциях в местах SC инфузии
инфузии
(мл/мин)
эритемы
(степень тяжести)
кровотечения
(степень тяжести)
отека
(степень тяжести)
[максимальный размер]
(минимальная-умеренная)
(минимальная)
(минимальная-умеренная)
[50×35×10 мм]
(минимальная-легкая)
(минимальная)
(минимальная-умеренная)
[45×45×10 мм]
(минимальная-легкая)
(минимальная-легкая)
(минимальная-выраженная)
[50×40×8 мм]
(минимальная-легкая)
(минимальная-легкая)
(умеренная)
[35×35×12 мм]
(минимальная-умеренная)
(минимальная)
(минимальная-выраженная)
[55×40×10 мм]
(минимальная-легкая)
(минимальная)
(минимальная-умеренная)
[25×25×8 мм]
Фармакокинетика
Индивидуальные концентрации изатуксимаба в плазме с течением времени показаны на фиг. 24-27.
Некомпартментный фармакокинетический анализ данных об уровне в плазме крови проводили с применением программного обеспечения для фармакокинетического анализа Phoenix WinNonlin версии 6.3.
Таблица 30. - Средние (CV%, n=5) фармакокинетические параметры изатуксимаба в плазме крови мини-свиней после однократной внутривенной (IV) или подкожной (SC) инфузии
Скорость
(мл/мин)
(ч)
мкг/
мл)
(ч)
мкг/
мл)
ч*мкг/мл)
ч*мкг/мл)
ч*мкг/мл)
ч*мкг/мл)
3
(0)
(8)
(0)
(22)
(11)
(11)
(10)
(10)
0,5
[72-192]2
(50)
(8)
(0)
(24)
(13)
(48)
(20)
(11)
1
[48-264]2
(54)
(23)
(0)
(34)
(25)
(37)
(14)
(5)
2
[72-192]2
(45)
(12)
(0)
(36)
(23)
(19)
(10)
(10)
Конец=конец инфузии;
1 рассчитано с применением AUClast;
2 медиана [мин-макс];
3 одинаковое значение для медианы, мин и макс;
4 высокое F-значение связано с более высокой вариабельностью, отмеченной для данной группы; вклад 2/5 животных, которые показали явно более высокую экспозицию.
Изатуксимаб можно было количественно определить во всех образцах плазмы крови в исследовании, собранных от окончания инфузии и далее, за исключением одного образца, взятого в конце SC инфузии у самки из группы 4 и через 1 час после SC инфузии у самки из группы 3, которые были ниже нижнего предела количественного определения (LLOQ). Концентрации во всех образцах до введения дозы были ниже LLOQ. Профили соответствовали экстраваскулярному и внутривенному введению дозы для SC и IV путей введения соответственно. В целом наблюдаемая вариабельность PK-параметров была низкой для IV инфузии и от низкой до умеренной для SC инфузии.
После однократной IV инфузии мини-свиньям изатуксимаба в дозе 1800 мг/животное на протяжении 30-минутного периода максимальные уровни в плазме крови наблюдали в конце периода инфузии у всех животных. После однократной SC инфузии мини-свиньям изатуксимаба в дозе 1806 мг/животное при скорости потока 0,5, 1 или 2 мл/мин медианные максимальные уровни в плазме крови наблюдали через 96, 192 и 168 часов после окончания инфузии соответственно. Однако индивидуальные значения Tmax находились в диапазоне от 48 до 264 часов и не показали взаимосвязи со скоростью SC инфузии.
После однократной IV инфузии мини-свиньям изатуксимаба в дозе 1800 мг/животное на протяжении 30-минутного периода средняя AUC за полный 672-часовой период сбора образцов после введения дозы (AUClast) составила 364000 ч*мкг/мл. После однократной SC инфузии изатуксимаба в дозе 1806 мг/животное мини-свиньям при скорости потока 0,5, 1 или 2 мл/мин среднее значение AUC за полный 672-часовой период отбора проб после введения дозы (AUClast) составило 326000, 565000 и 369000 ч*мкг/мл соответственно. Значения AUClast были в целом сходными для каждой группы SC введения, что свидетельствует об отсутствии влияния скорости инфузии на экспозицию.
В условиях настоящего исследования абсолютная SC биодоступность изатуксимаба при SC инфузии мини-свиньям при скорости потока 0,5, 1 или 2 мл/мин была в целом подобной для 3 тестируемых скоростей потока в каждом из рассматриваемых интервалов AUC. Биодоступность увеличивалась с увеличением интервала AUC, достигая F-значений 0,89, 1,55 и 1,01 для скоростей потока 0,5, 1 и 2 мл/мин соответственно, при расчете с применением AUC для полного 672-часового периода сбора образцов после введения дозы (AUClast). Для группы, получавшей инфузию со скоростью 1 мл/мин, высокое рассчитанное F-значение было связано с более высокой вариабельностью, отмеченной для этой группы; вклад 2/5 животных, которые показали явно более высокую экспозицию.
В целом был сделан вывод, что абсолютная SC биодоступность изатуксимаба у мини-свиней при введении в дозе 1806 мг/животное (раствор 140 мг/мл) посредством SC инфузии при скорости потока от 0,5 до 2 мл/мин составляла по меньшей мере 89%.
Микроскопическое анализ образцов биопсии кожи
Никаких изменений, связанных с обработкой, не наблюдали ни в одной из групп подкожного введения при микроскопическом анализе образцов биопсии кожи с места инъекции. Микроскопическими реакциями в основном были минимальная очаговая инфильтрация мононуклеарных клеток и минимальные очаговые эпидермальные корки без различий в частоте возникновения и морфологии реакций между группами, получавшими подкожную дозу. Аналогичным образом, реакции считали подобными при сравнении мест инъекций, получавших солевой раствор (отрицательный контроль), с местами инъекций, получавших тестируемые образцы, в пределах группы определенной дозы (одинаковая скорость инфузии). Все микроскопические реакции считались хорошо известными случайными фоновыми изменениями в коже геттингенских мини-свиней.
Обсуждение
Изатуксимаб, вводимый путем внутривенной инфузии (в виде раствора с концентрацией 20 мг/мл) в дозе 1800 мг/животное и в условиях скорости потока 3 мл/мин или в виде подкожной инфузии (в виде раствора с концентрацией 140 мг/мл в составе F4 из примера 5) в дозе 1806 мг/животное и в условиях скорости потока 0,5, 1 или 2 мл/мин, хорошо переносился самками геттингенских мини-свиней как клинически, так и патогистологически.
Местные реакции в местах подкожных инъекций во всех группах наблюдали только в день инфузии в местах введения.
Частота возникновения и показатели тяжести эритемы и кровотечения были сопоставимы для трех тестируемых скоростей подкожного введения (0,5, 1 и 2 мл/мин) и подобны как для изатуксимаба, так и для отрицательного контроля с солевым раствором. Однако отек (образование пузырей) в месте инфузии был более выражен при инфузии изатуксимаба со скоростью 1 мл/мин и 2 мл/мин по сравнению со скоростью 0,5 мл/мин. Скорее всего, это было физическое явление, связанное с инъекцией большого объема в одно место введения, при этом тяжесть была обратно пропорциональна времени инфузии при постоянной скорости удаления жидкости из места инфузии. Кроме того, отек был более выражен после инфузии изатуксимаба, чем после инфузии солевого раствора. Отек после инфузии солевого раствора был сравнимым для всех 3 скоростей потока.
Никаких изменений, связанных с обработкой, не наблюдали ни в одной из групп подкожного введения при микроскопическом анализе образцов биопсии кожи с места инъекции.
После однократной IV инфузии мини-свиньям изатуксимаба в дозе 1800 мг/животное на протяжении 30-минутного периода максимальные уровни в плазме крови наблюдали в конце периода инфузии у всех животных. После однократной SC инфузии мини-свиньям изатуксимаба в дозе 1806 мг/животное при скорости потока 0,5, 1 или 2 мл/мин медианные максимальные уровни в плазме крови наблюдали через 96, 192 и 168 часов после окончания инфузии соответственно. Однако индивидуальные значения Tmax находились в диапазоне от 48 до 264 часов и не показали взаимосвязи со скоростью SC инфузии.
После однократной IV инфузии мини-свиньям изатуксимаба в дозе 1800 мг/животное на протяжении 30-минутного периода средняя AUC за полный 672-часовой период сбора образцов после введения дозы (AUClast) составила 364000 ч*мкг/мл. После однократной SC инфузии изатуксимаба в дозе 1806 мг/животное мини-свиньям при скорости потока 0,5, 1 или 2 мл/мин среднее значение AUC за полный 672-часовой период отбора проб после введения дозы (AUClast) составило 326000, 565000 и 369000 ч*мкг/мл соответственно. Значения AUClast были в целом сходными для каждой группы SC введения, что свидетельствует об отсутствии влияния скорости инфузии на экспозицию.
В условиях настоящего исследования абсолютная SC биодоступность изатуксимаба, составленного в виде F4 в примере 4, при SC инфузии мини-свиньям при скорости потока 0,5, 1 или 2 мл/мин была в целом подобной для 3 тестируемых скоростей потока в каждом из рассматриваемых интервалов AUC. Биодоступность увеличивалась с увеличением интервала AUC, достигая F-значений 0,89, 1,55 и 1,01 для скоростей потока 0,5, 1 и 2 мл/мин соответственно, при расчете с применением AUC для полного 672-часового периода сбора образцов после введения дозы (AUClast). Для группы, получавшей инфузию со скоростью 1 мл/мин, высокое рассчитанное F-значение было связано с более высокой вариабельностью, отмеченной для этой группы; вклад 2/5 животных, которые показали явно более высокую экспозицию.
Пример 7. - Исследование фазы 1b подкожного введения изатуксимаба у людей
В настоящем примере описано многоцентровое открытое исследование фазы 1b для оценки фармакокинетики, безопасности и эффективности подкожного и внутривенного введения изатуксимаба в комбинации с помалидомидом и дексаметазоном у пациентов с рецидивирующей/рефрактерной множественной миеломой (RRMM).
Настоящее исследование разработано для оценки SC введения изатуксимаба в первый раз. Кроме того, состав для SC введения отличается от состава, применяемого для IV введения дозы. Изатуксимаб для SC введения в комбинации с помалидомидом и дексаметазоном вводят той же популяции пациентов, что и в предыдущих исследованиях, упомянутых выше. Данное исследование также включает когорты с IV введением изатуксимаба для обеспечения возможности оценки безопасности и фармакокинетики в сравнении с изатуксимабом для SC введения.
Первичными конечными точками исследования являются (i) оценка безопасности и переносимости (включая местную переносимость в месте инъекции) изатуксимаба, вводимого подкожно (SC) с применением инфузионного насоса, в сравнении с изатуксимабом, вводимым внутривенно (IV); и (ii) оценка фармакокинетики изатуксимаба при SC и IV введении в комбинации с помалидомидом и дексаметазоном. Вторичные конечные точки исследования включают (i) оценку абсолютной биодоступности изатуксимаба для SC и IV введения; (ii) измерение занятости рецептора (RO) CD38 изатуксимабом в плазматических клетках из аспирата костного мозга после SC введения в сравнении с IV введением; и (iii) оценку эффективности SC/IV введения изатуксимаба.
В исследование включены 5 когорт участников. Пациентов рандомизируют в когорты 1a (SC доза 1000 мг) или 1b (IV) (соотношение рандомизации 2:1). После оценки данных безопасности PK и RO изатуксимаба для SC введения в когорте 1а дополнительных участников рандомизируют в когорты 2а или 2b (соотношение рандомизации 2:1) с более высокой дозой изатуксимаба для SC введения в когорте 2а (доза 1400 мг) и с такой же IV дозой в когорте 2b. Окончательный обзор данных по безопасности, PK и RO после введения изатуксимаба для SC и IV введения проводят после того, как все пациенты в когортах 2a и 2b завершили цикл 1 лечения. После подтверждения рекомендуемого уровня дозы фазы 2 (RP2D) в когорту 2с набирают дополнительных 22 пациента, которым вводят изатуксимаб для SC введения на данном уровне дозы. В таблице 31 более подробно описано лечение по когортам.
Таблица 31. - Подробное описание лечения по когортам
Изатуксимаб вводят еженедельно в течение 4 недель (цикл 1), а также в день 1 и день 15 день каждого последующего цикла в комбинации с помалидомидом и дексаметазоном. Каждый цикл длится 28 дней. Все участники в исследовании продолжают лечение до прогрессирования заболевания, неприемлемой нежелательной реакции или другой причины прекращения лечения.
Когорты 1a, 2a и 2c получают состав изатуксимаба, содержащий 140 мг/мл изатуксимаба, 9 мМ гистидина, 110 мМ моногидрохлорида аргинина, 2% (вес/об.) сахарозы и 0,4% (вес/об.) полоксамера 188, рН 6,2, путем подкожной (SC) инфузии. Когорты 1a, 2a и 2c также получают 4 мг помалидомида перорально (p.o.) в дни 1-21 каждого 28-дневного цикла; и дексаметазон 4 мг p.o. в дни 1, 8, 15 и 22 каждого 28-дневного цикла.
Когорты 1b и 2b получают отличающийся состав изатуксимаба, содержащий 20 мг/мл изатуксимаба, 20 мМ гистидина, 10% (вес/об.) сахарозы и 0,02% (вес/об.) полисорбата 80, рН 6,0, путем внутривенной (IV) инфузии. Когорты 1b и 2b также получают 4 мг помалидомида перорально (p.o.) в дни 1-21 каждого 28-дневного цикла; и дексаметазон 4 мг p.o. в дни 1, 8, 15 и 22 каждого 28-дневного цикла.
Данные по безопасности, PK и RO из цикла 1 (первые 4 недели) в когорте 1a и когорте 2a проверяют перед переходом к когорте 2a или 2c соответственно.
Обзор данных по безопасности, PK и RO, собранных в когортах 1(a/b) и 2(a/b), применяется для отбора наиболее подходящей дозы RP2D изатуксимаба для SC введения.
Дозу изатуксимаба 1000 мг выбрали в качестве начальной дозы на основании моделирования и симуляций PK, которые продемонстрировали, что даже при условии 80% SC биодоступности, концентрации изатуксимаба (остаточная концентрация в плазме крови [Ctrough] в день 28) будут находиться в диапазоне, подобном для концентраций в день 28, наблюдаемым после IV введения в дозе 10 мг/кг, и намного ниже, чем после IV введения в дозе 20 мг/кг, которая показала себя безопасной в клинических условиях. Дозу ниже 1000 мг не рассматривали в качестве начальной дозы из-за нелинейности PK изатуксимаба.
Выбор дозы изатуксимаба 1400 мг для SC введения основан на популяционной PK модели, построенной с помощью данных IV введения (n=127). Данная модель показала, что 1400 мг изатуксимаба для SC введения, вводимого QWx4/Q2W, будет поддерживать Ctrough выше уровней, достигаемых после IV введения дозы 10 мг/кг QWx4/Q2W, с гипотезой об абсолютной биодоступности 2:50%. Анализы PK/PD продемонстрировали, что Ctrough через 4 недели является значимым предиктором ответа (частота объективного ответа, IV введение).
Критерии включения включают следующее:
Пациенты, которым ранее был поставлен диагноз множественной миеломы (ММ) на основании стандартных критериев, и которым в настоящее время требуется лечение, поскольку возник рецидив ММ после ответа согласно критериям Международной группы по изучению миеломы (IMWG).
Пациенты, которые получили по меньшей мере 2 предшествующих вида терапии, включая леналидомид и ингибитор протеасом, и продемонстрировали прогрессирование заболевания в ходе последней терапии или после завершения последней терапии; и
Пациенты с поддающимся измерению заболеванием, которое определяется как по меньшей мере одно из следующего:
• Количество М-белка в сыворотке крови ≥0,5 г/дл (≥5 г/л);
• Количество М-белка в моче ≥200 мг/24 часа; и
• Анализ свободной легкой цепи в сыворотке крови (FLC): включал анализ FLC ≥10 мг/дл (≥100 мг/л) и отклоняющееся от нормы соотношение FLC в сыворотке крови (<0,26 или >1,65).
Образцы костного мозга и крови собирают для анализов следующих биомаркеров:
• Занятость рецептора CD38 изатуксимабом измеряют в плазматических клетках из аспирата костного мозга и устанавливают взаимосвязь с параметрами PK и клинического ответа. Образцы костного мозга собирают в момент скрининга и в день 1 цикла 2 (до введения дозы). Данный сбор образцов прекращают после того, как выбрали RP2D дозу изатуксимаба для SC введения (только в когортах 1a/b и когортах 2a/b).
• Минимальную остаточную болезнь (MRD) оценивают с помощью секвенирования нового поколения в аспиратах костного мозга и устанавливают взаимосвязь с параметрами клинического ответа. Образцы костного мозга собирают в момент скрининга у всех участников и во время максимального подтвержденного ответа, которым является либо полный ответ (CR), либо очень хороший частичный ответ (VGPR). Образцы, взятые в момент скрининга, анализируют только у участников, которым удастся достичь VGPR или лучше.
• Потенциальную интерференцию изатуксимаба с оценкой М-белка в иммуноэлектрофорезе и анализах иммунофиксации оценивают в образце сыворотки крови с помощью анализа, который устраняет интерференцию изатуксимаба.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Ballet, Thomas
Bangari, Kiran Chari, Ravi Huille, Sylvain Perez-Ramirez,
Bernardo Vasco, Filipe
<120> СОСТАВЫ НА ОСНОВЕ АНТИТЕЛ К CD38 ДЛЯ ПОДКОЖНОГО ВВЕДЕНИЯ
<130> 712533: SA9-295PC
<150> 62/944,082
<151> 2019-12-05
<160> 10
<170> PatentIn версия 3.5
<210> 1
<211> 5
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 1
Asp Tyr Trp Met Gln
1 5
<210> 2
<211> 17
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 2
Thr Ile Tyr Pro Gly Asp Gly Asp Thr Gly Tyr Ala Gln Lys Phe Gln
1 5 10 15
Gly
<210> 3
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 3
Gly Asp Tyr Tyr Gly Ser Asn Ser Leu Asp Tyr
1 5 10
<210> 4
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 4
Lys Ala Ser Gln Asp Val Ser Thr Val Val Ala
1 5 10
<210> 5
<211> 7
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 5
Ser Ala Ser Tyr Arg Tyr Ile
1 5
<210> 6
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 6
Gln Gln His Tyr Ser Pro Pro Tyr Thr
1 5
<210> 7
<211> 120
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 7
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Ala Lys Pro Gly Thr
1 5 10 15
Ser Val Lys Leu Ser Cys Lys Ala Ser Gly Tyr Thr Phe Thr Asp Tyr
20 25 30
Trp Met Gln Trp Val Lys Gln Arg Pro Gly Gln Gly Leu Glu Trp Ile
35 40 45
Gly Thr Ile Tyr Pro Gly Asp Gly Asp Thr Gly Tyr Ala Gln Lys Phe
50 55 60
Gln Gly Lys Ala Thr Leu Thr Ala Asp Lys Ser Ser Lys Thr Val Tyr
65 70 75 80
Met His Leu Ser Ser Leu Ala Ser Glu Asp Ser Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Gly Asp Tyr Tyr Gly Ser Asn Ser Leu Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Ser Val Thr Val Ser Ser
115 120
<210> 8
<211> 108
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 8
Asp Ile Val Met Thr Gln Ser His Leu Ser Met Ser Thr Ser Leu Gly
1 5 10 15
Asp Pro Val Ser Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Thr Val
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Arg Arg Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Ile Gly Val Pro Asp Arg Phe Thr Gly
50 55 60
Ser Gly Ala Gly Thr Asp Phe Thr Phe Thr Ile Ser Ser Val Gln Ala
65 70 75 80
Glu Asp Leu Ala Val Tyr Tyr Cys Gln Gln His Tyr Ser Pro Pro Tyr
85 90 95
Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys Arg
100 105
<210> 9
<211> 450
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 9
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Ala Lys Pro Gly Thr
1 5 10 15
Ser Val Lys Leu Ser Cys Lys Ala Ser Gly Tyr Thr Phe Thr Asp Tyr
20 25 30
Trp Met Gln Trp Val Lys Gln Arg Pro Gly Gln Gly Leu Glu Trp Ile
35 40 45
Gly Thr Ile Tyr Pro Gly Asp Gly Asp Thr Gly Tyr Ala Gln Lys Phe
50 55 60
Gln Gly Lys Ala Thr Leu Thr Ala Asp Lys Ser Ser Lys Thr Val Tyr
65 70 75 80
Met His Leu Ser Ser Leu Ala Ser Glu Asp Ser Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Gly Asp Tyr Tyr Gly Ser Asn Ser Leu Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Ser Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Asp Glu Leu Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 10
<211> 214
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Синтетический полипептид
<400> 10
Asp Ile Val Met Thr Gln Ser His Leu Ser Met Ser Thr Ser Leu Gly
1 5 10 15
Asp Pro Val Ser Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Thr Val
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Gln Ser Pro Arg Arg Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Ile Gly Val Pro Asp Arg Phe Thr Gly
50 55 60
Ser Gly Ala Gly Thr Asp Phe Thr Phe Thr Ile Ser Ser Val Gln Ala
65 70 75 80
Glu Asp Leu Ala Val Tyr Tyr Cys Gln Gln His Tyr Ser Pro Pro Tyr
85 90 95
Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ МНОЖЕСТВЕННОЙ МИЕЛОМЫ | 2020 |
|
RU2838203C2 |
| ПРЕПАРАТ, СОДЕРЖАЩИЙ АНТИТЕЛО | 2017 |
|
RU2748046C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2775944C2 |
| СОВМЕСТНЫЕ СОСТАВЫ АНТИ-LAG3 АНТИТЕЛА И АНТИ-PD-1 АНТИТЕЛА | 2019 |
|
RU2822192C2 |
| ПОДКОЖНОЕ ДОЗИРОВАНИЕ АНТИТЕЛ К CD38 | 2019 |
|
RU2810953C2 |
| АНТИТЕЛА К CD38 И СОСТАВЫ | 2020 |
|
RU2826957C2 |
| СОСТАВ, СОДЕРЖАЩИЙ АНТИТЕЛО | 2019 |
|
RU2806628C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АНТИТЕЛ К CD40 И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2778572C1 |
| СОСТАВЫ АНТИТЕЛ К RSV И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2807524C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ АНТИ-TRBV9 АНТИТЕЛА И ЕЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2826886C1 |

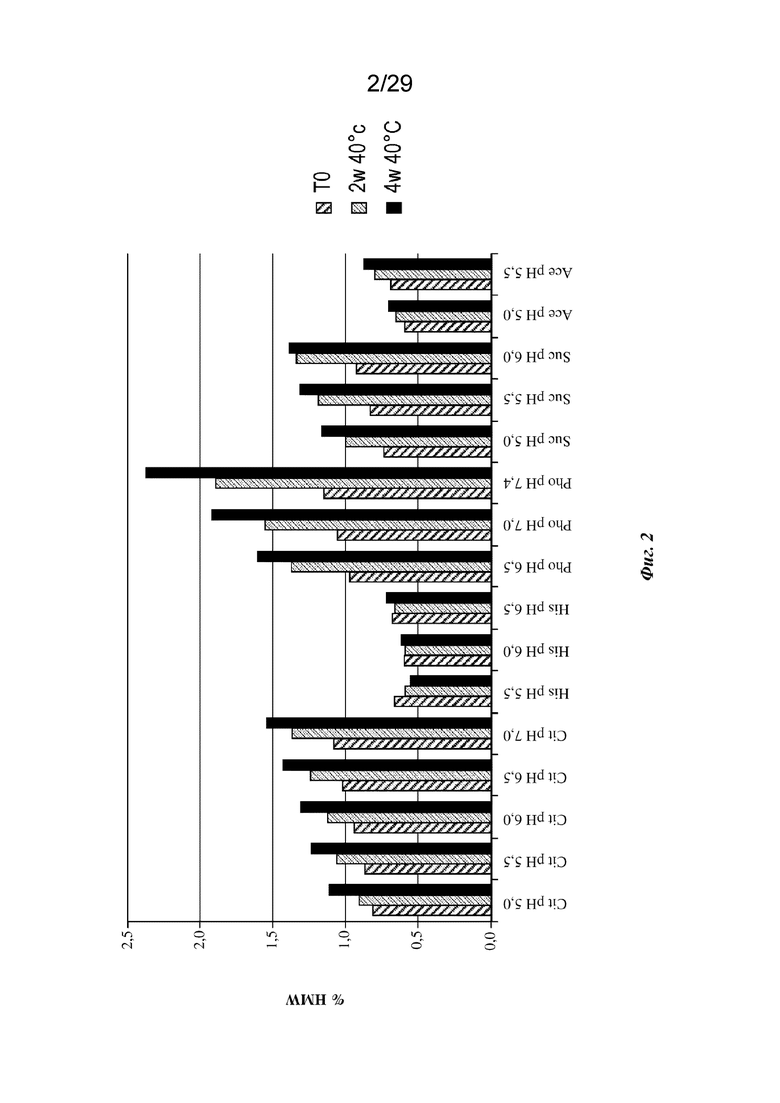
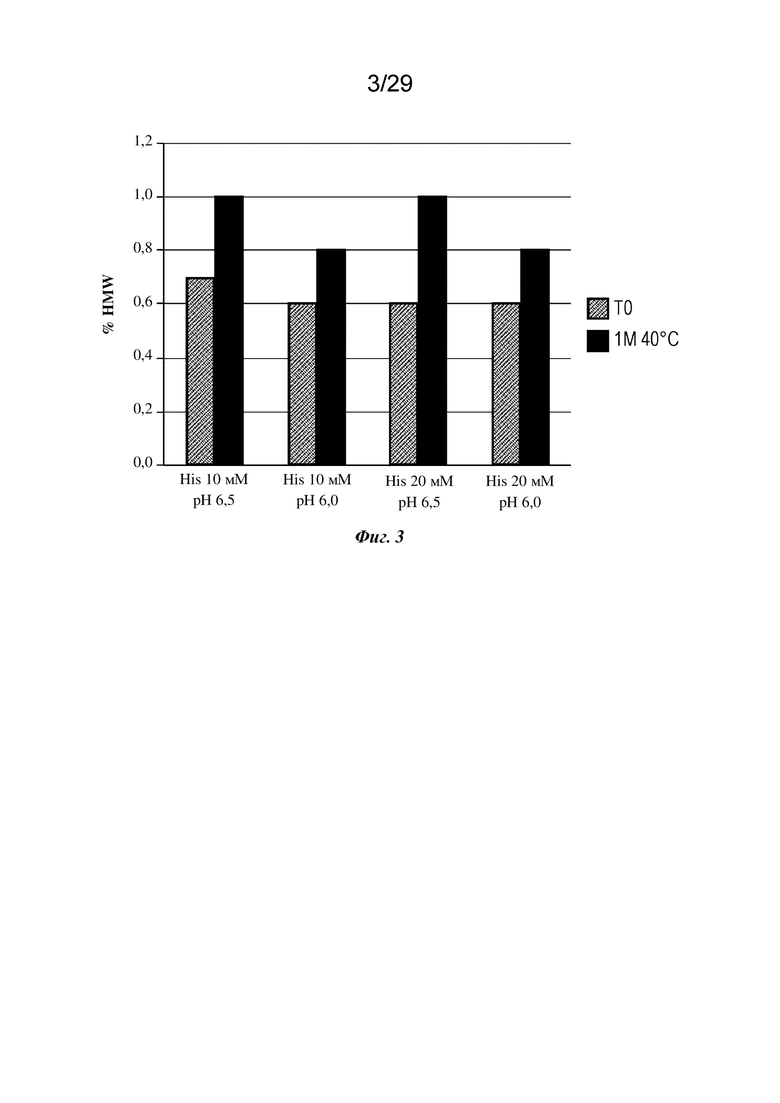
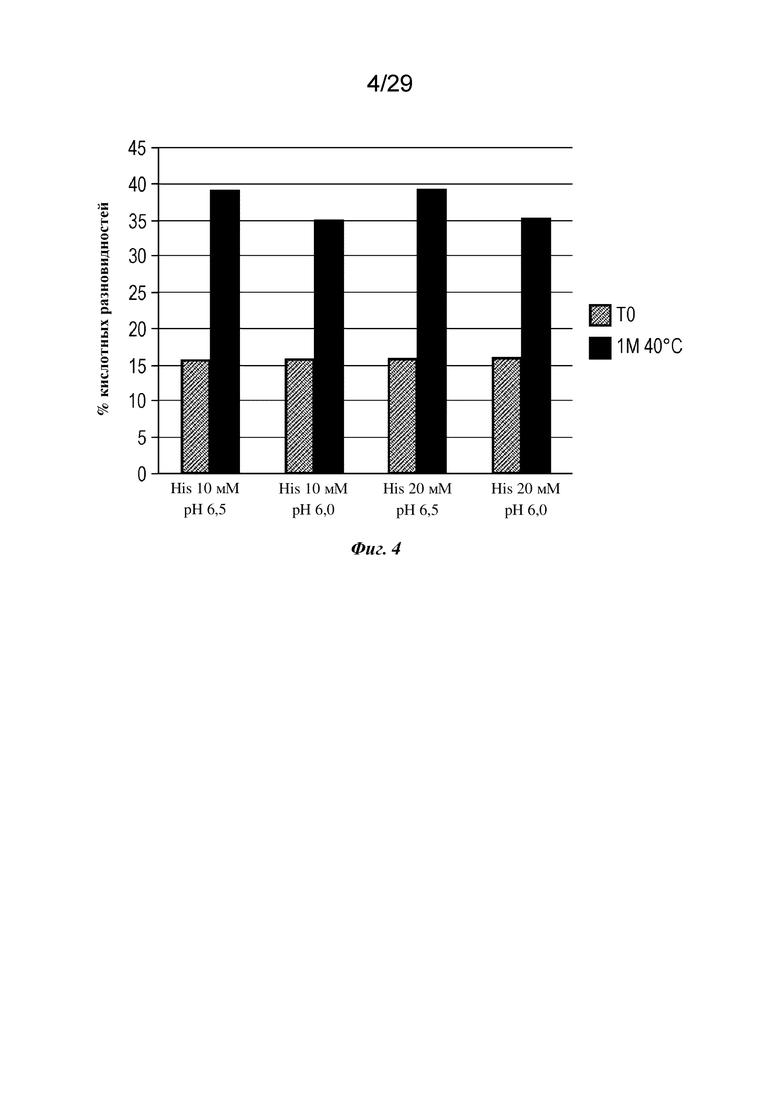
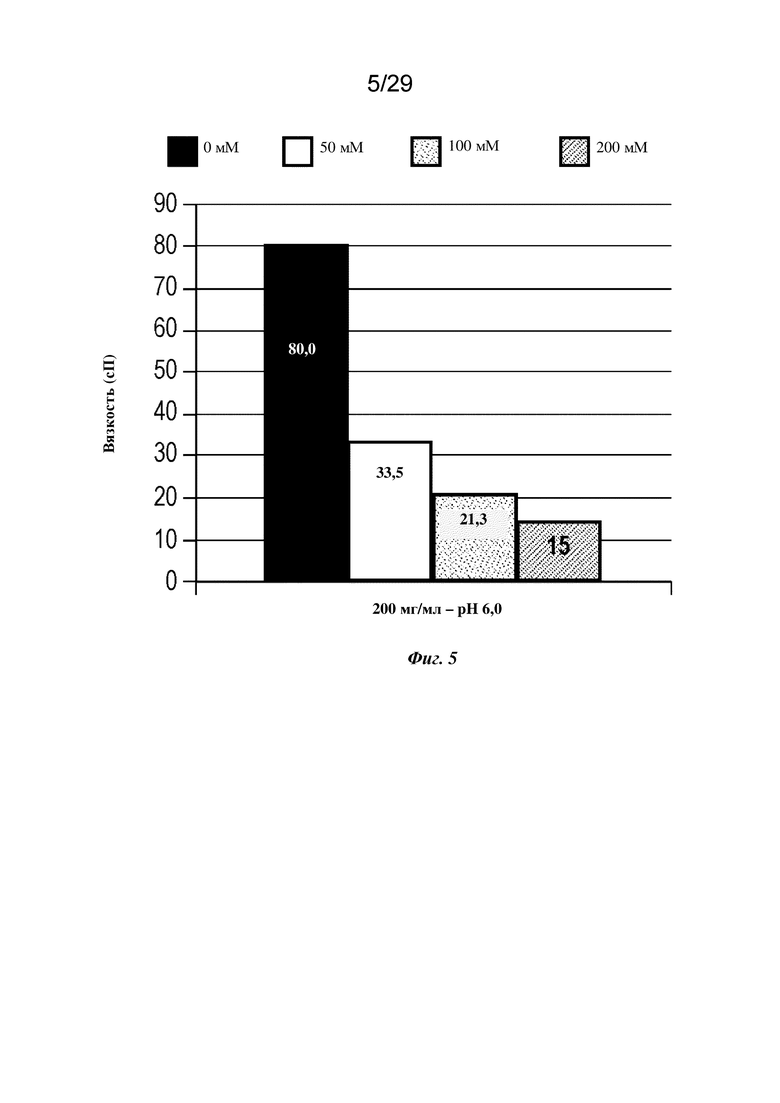
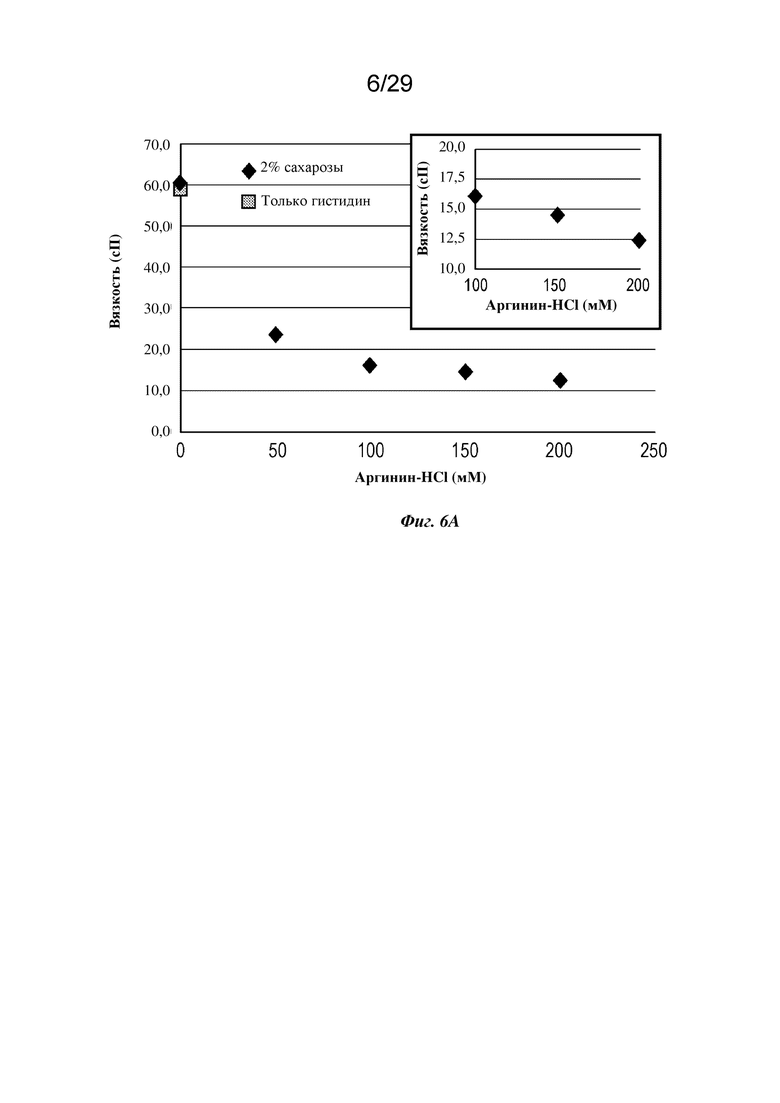
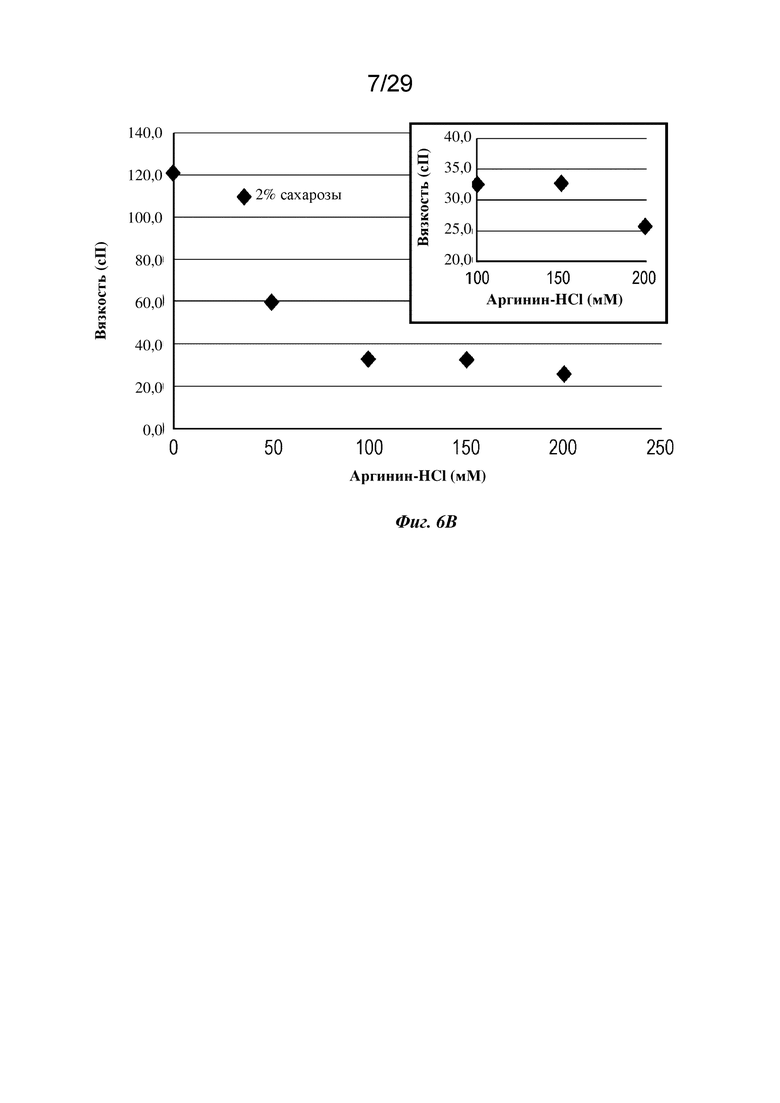
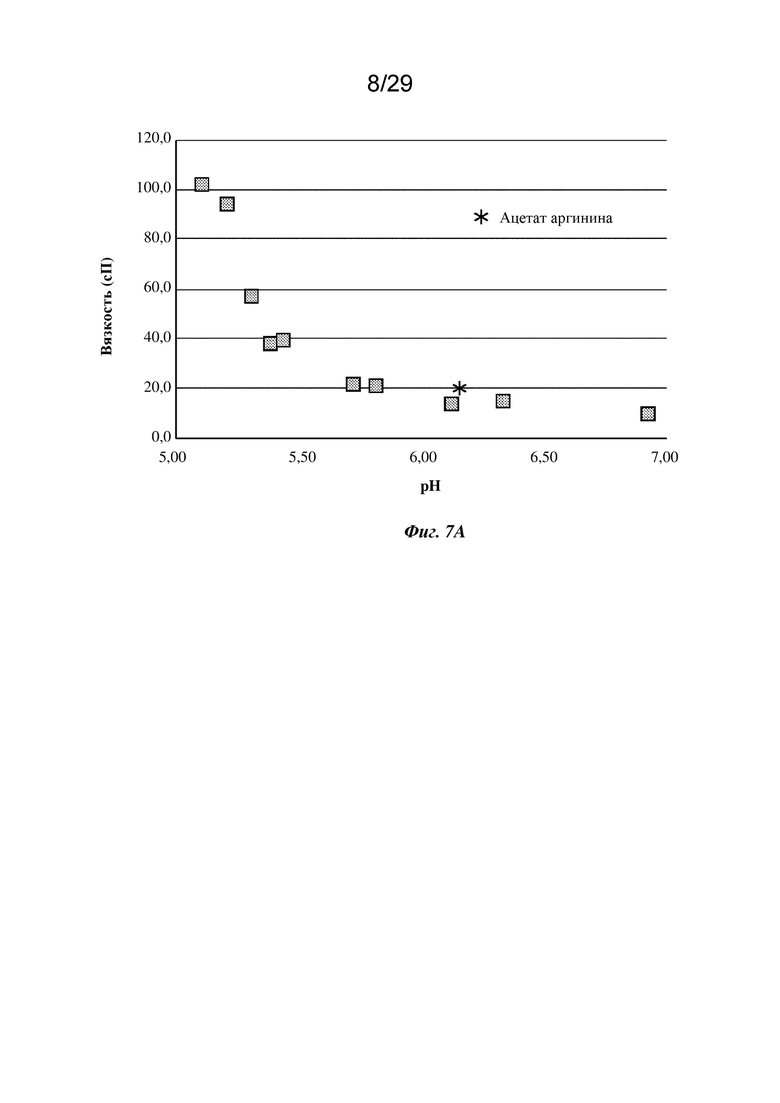
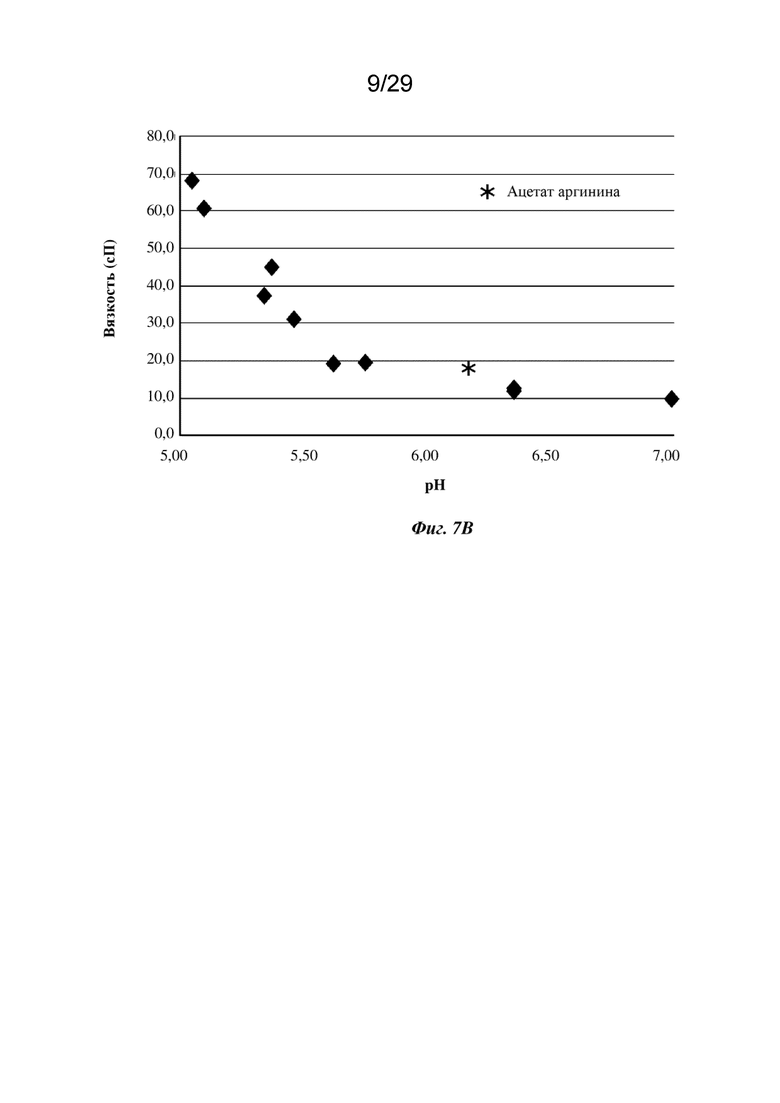
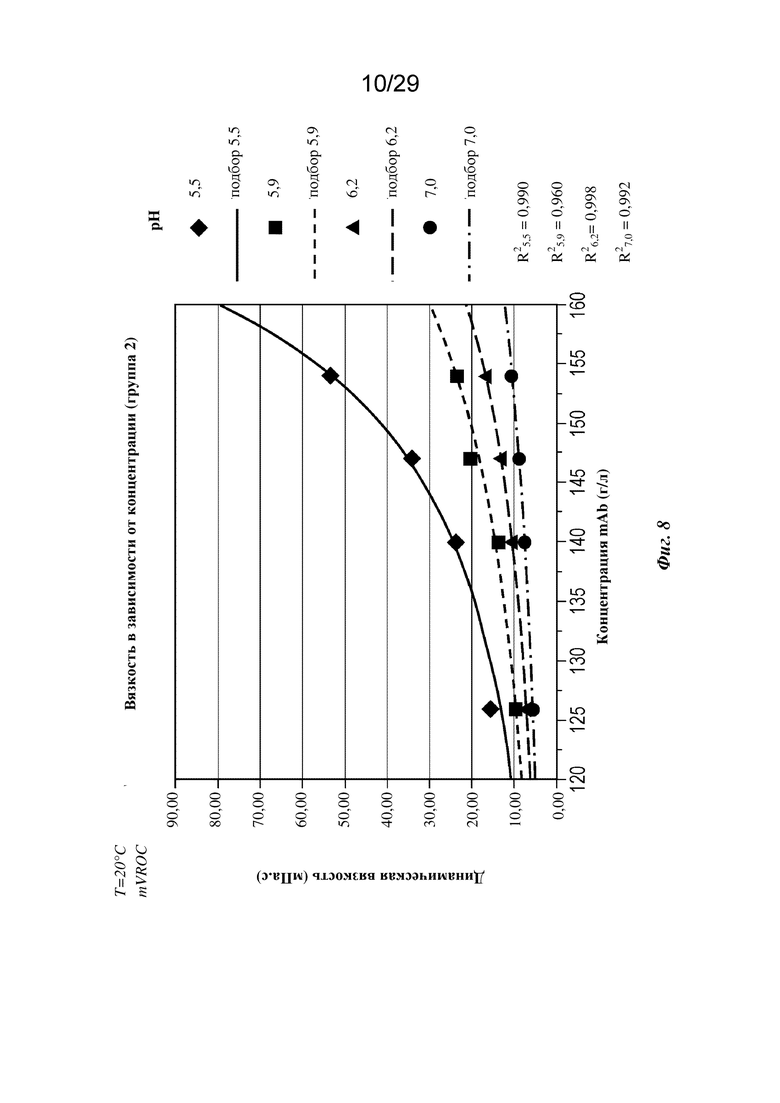
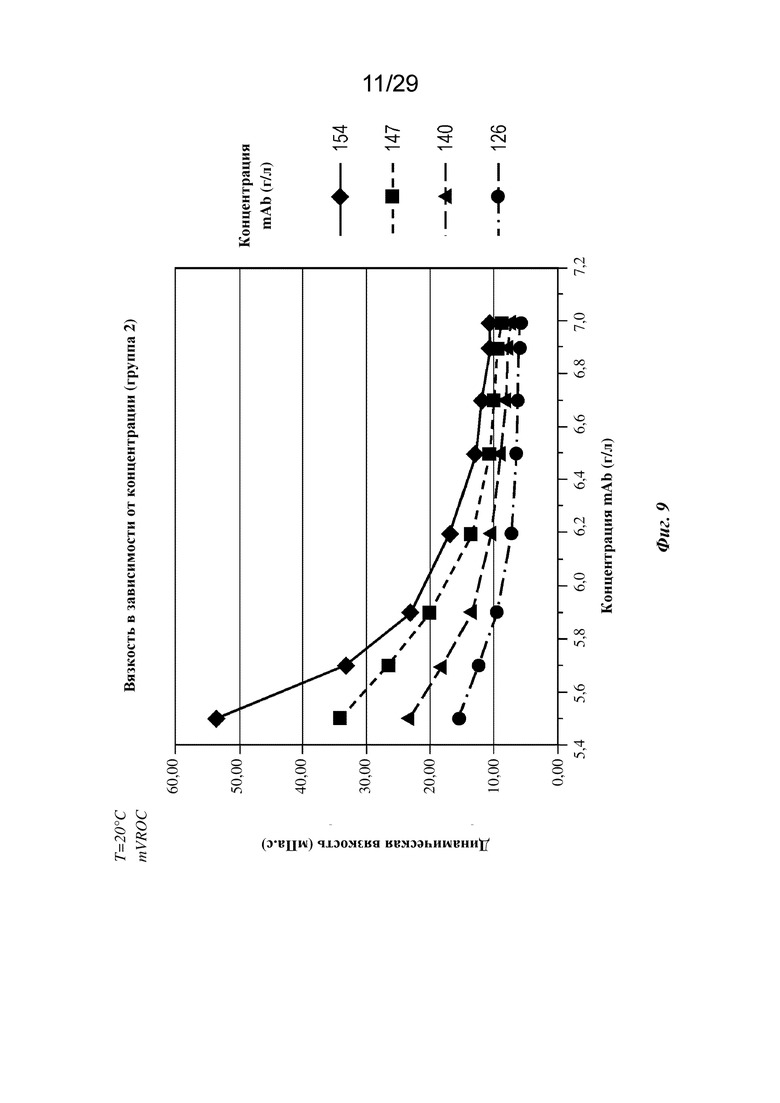
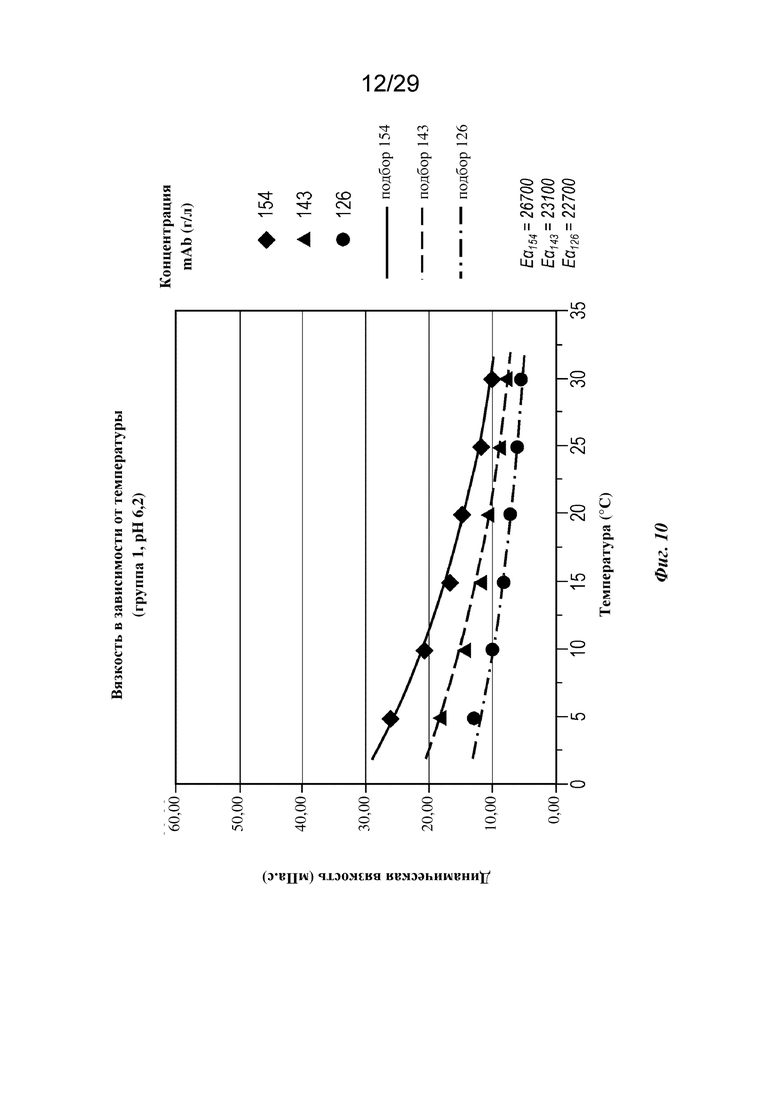
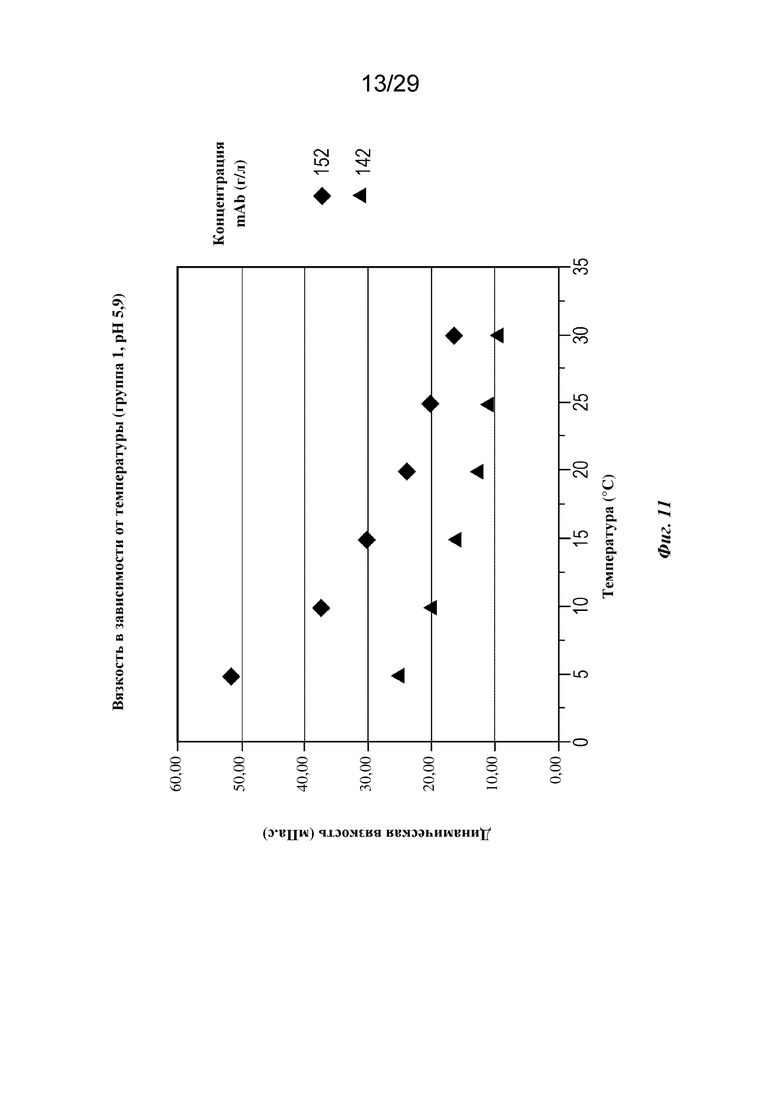
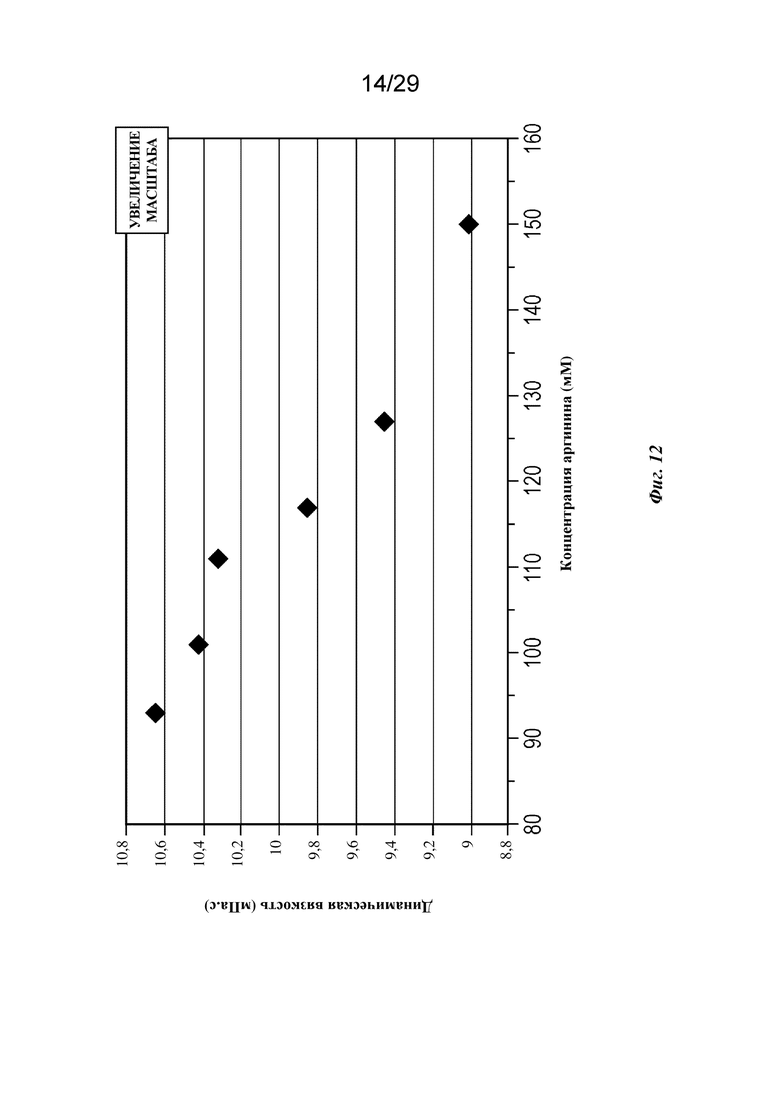
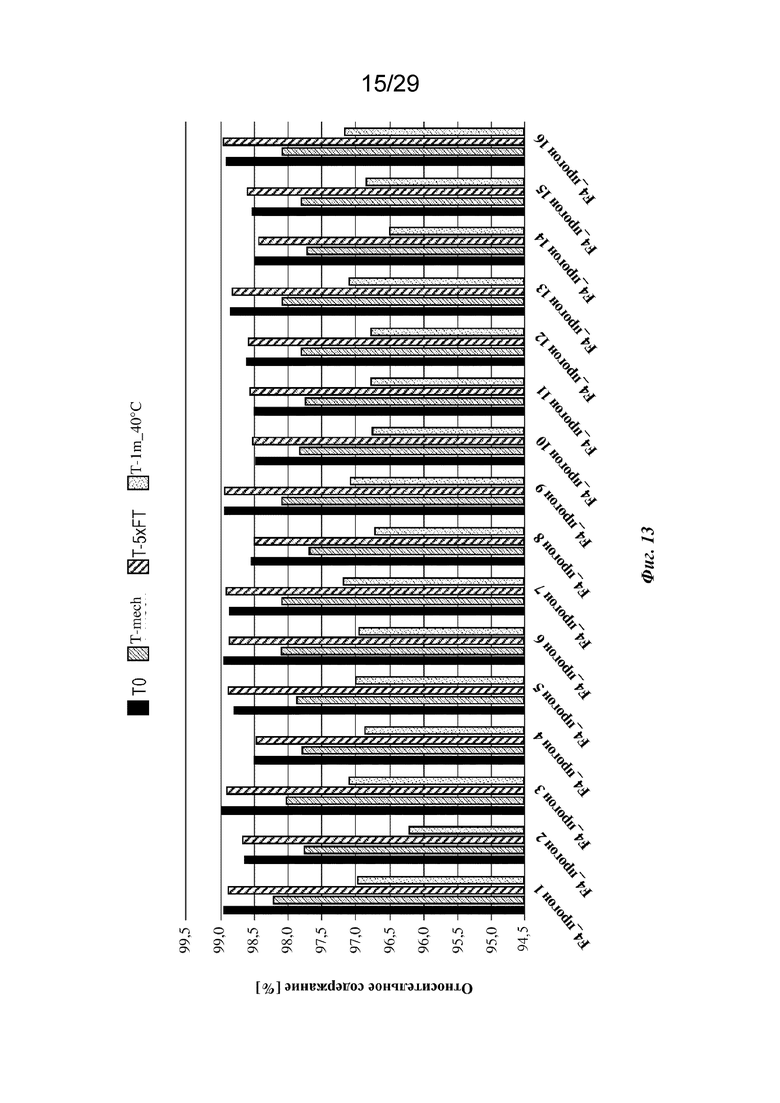
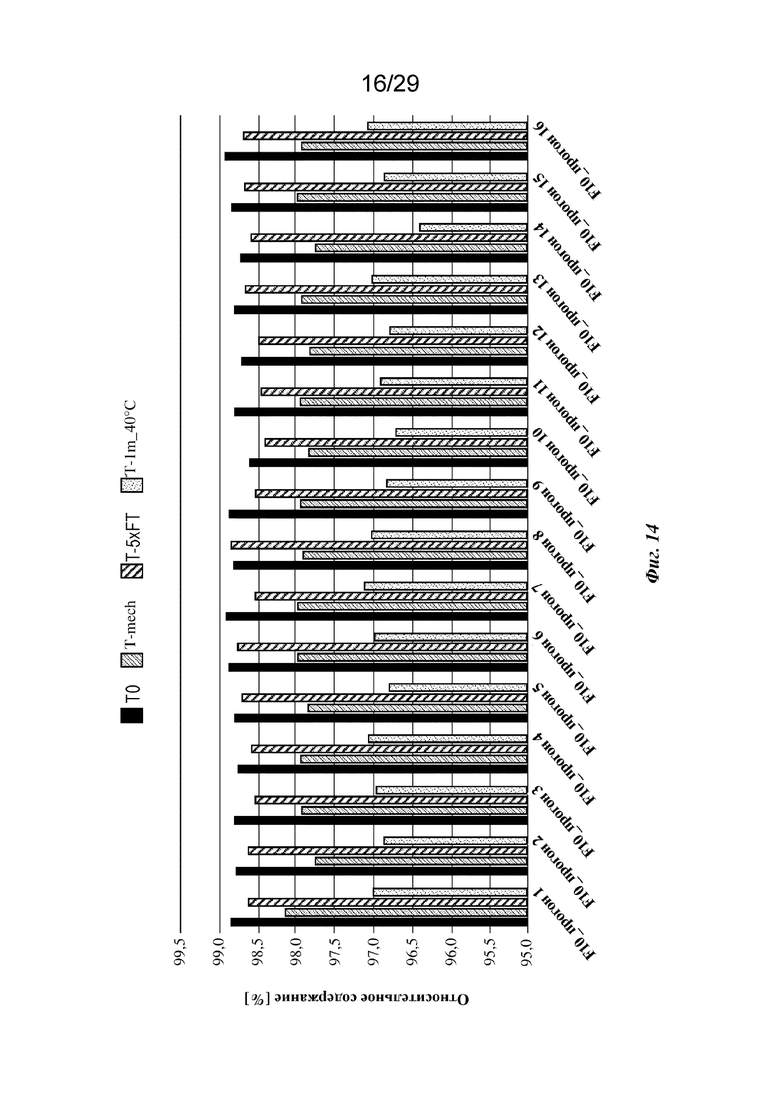
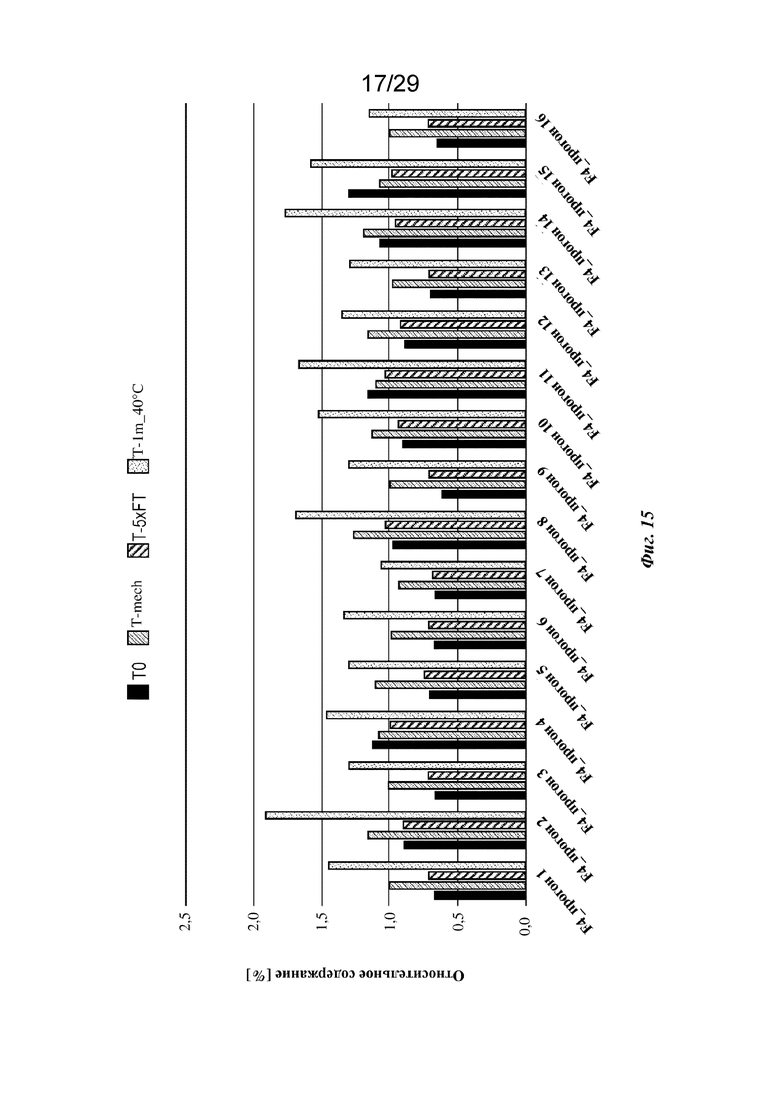
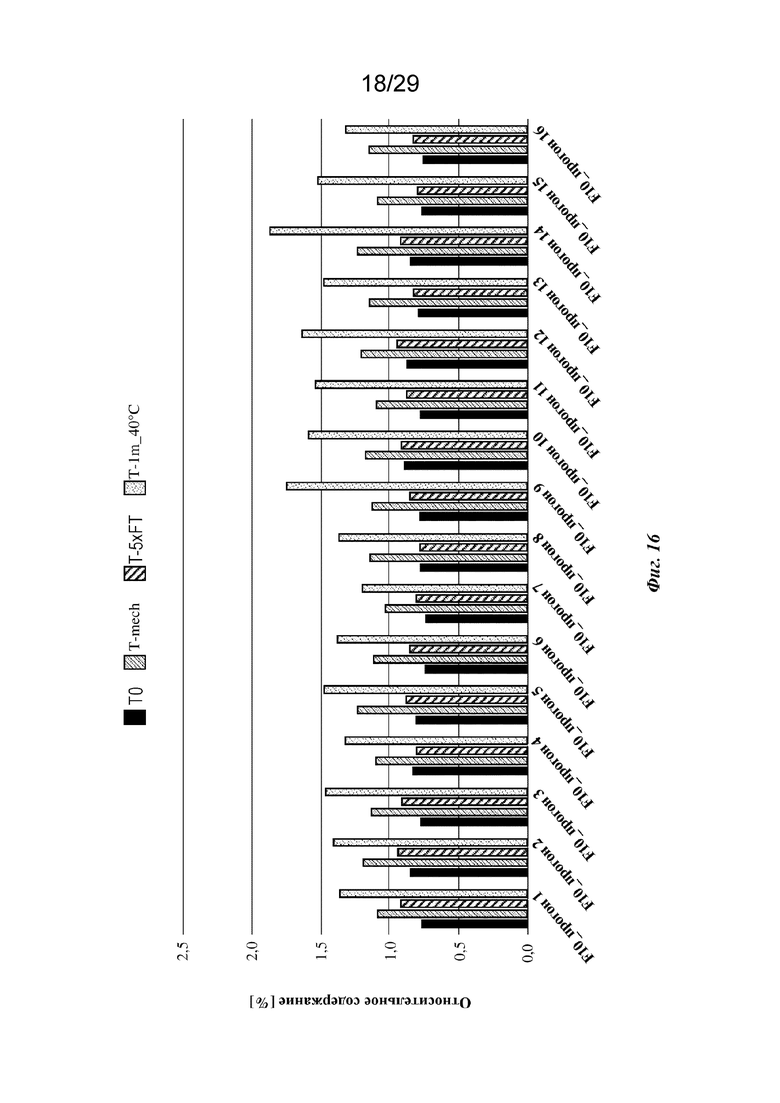
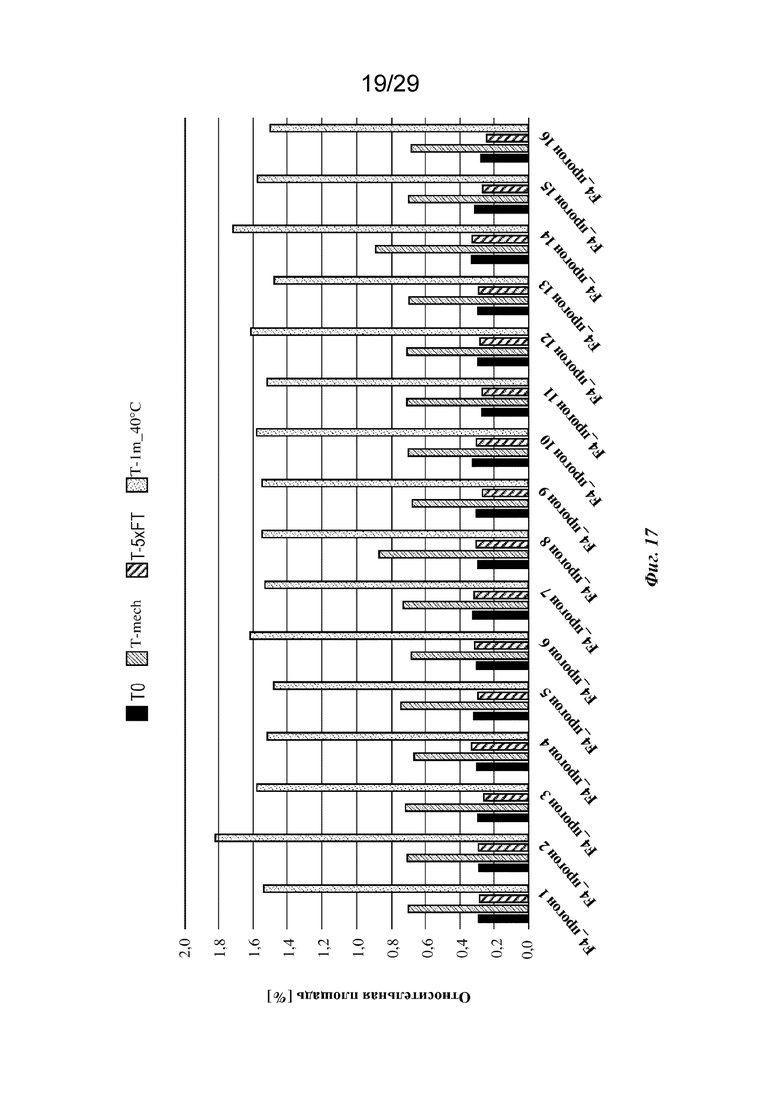
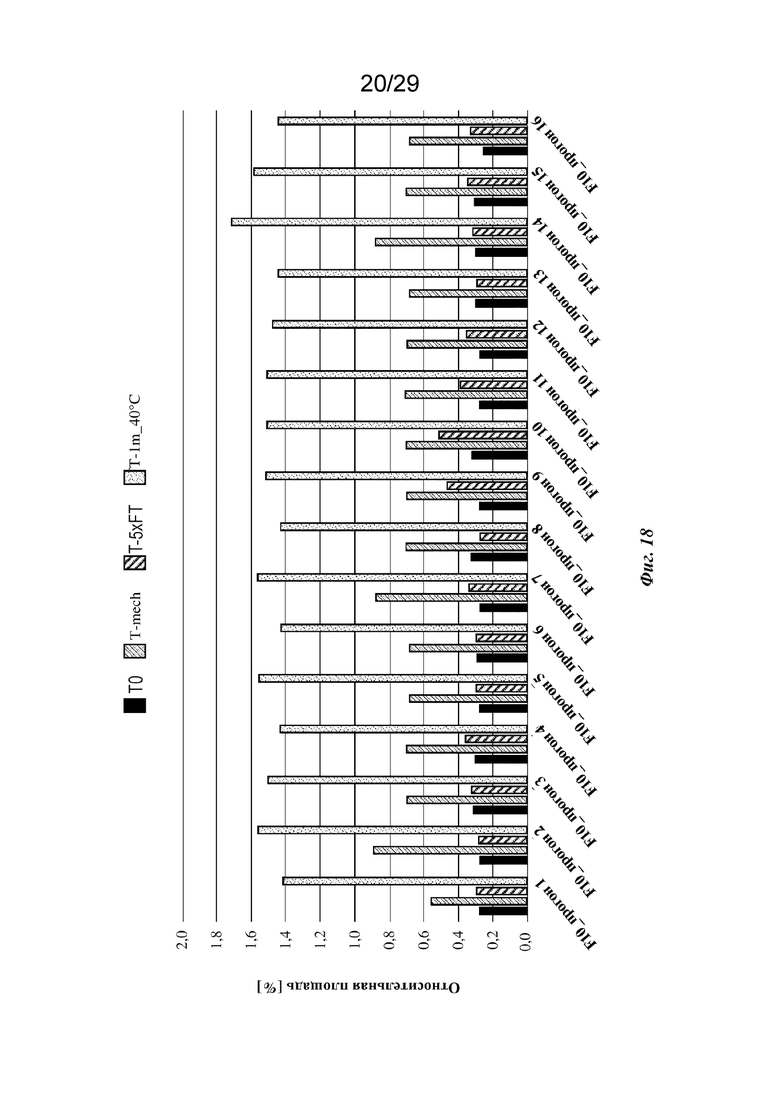
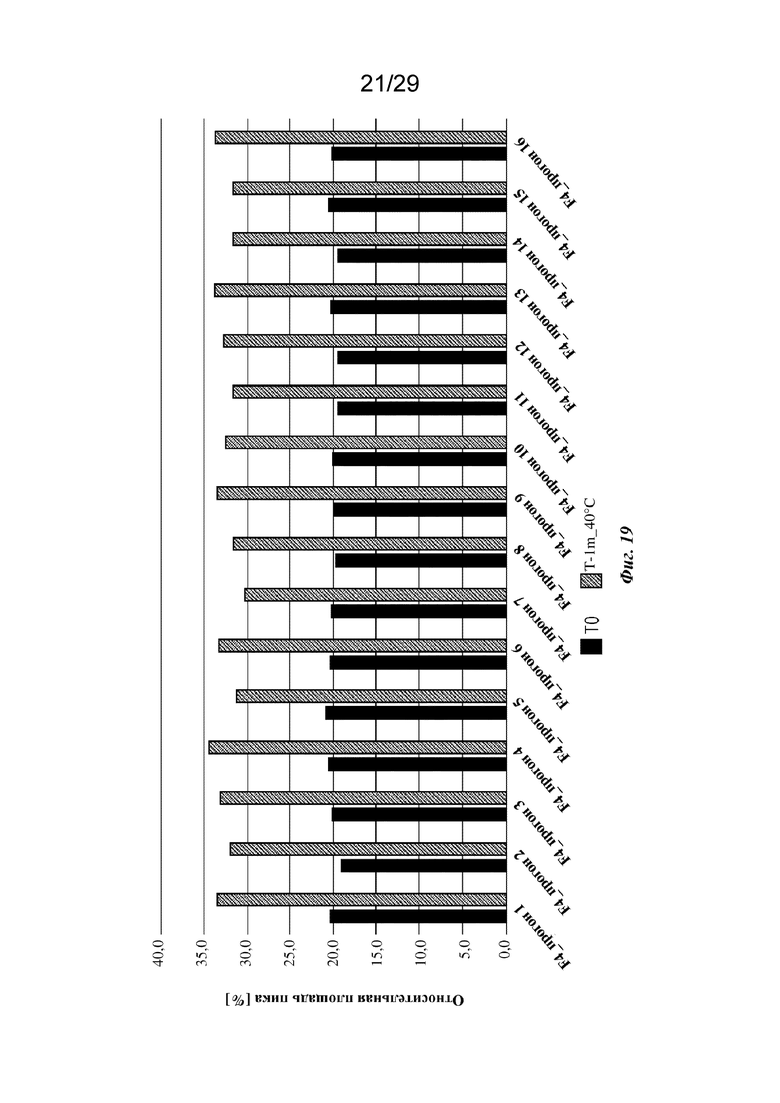
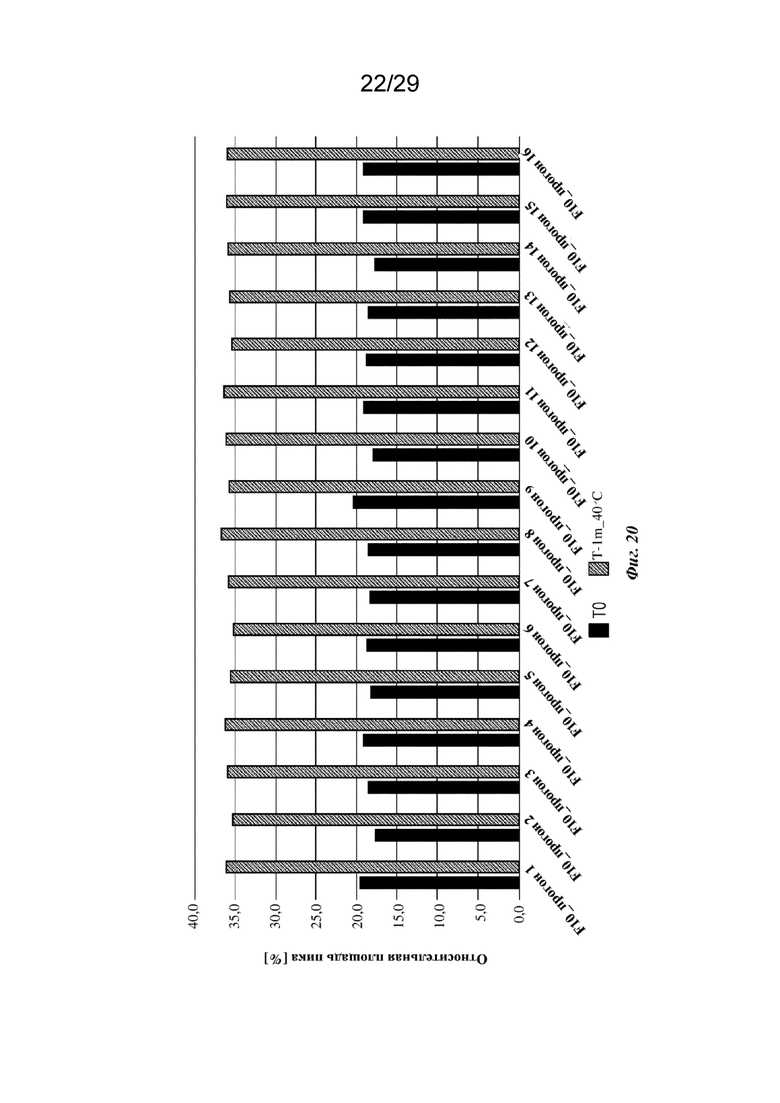
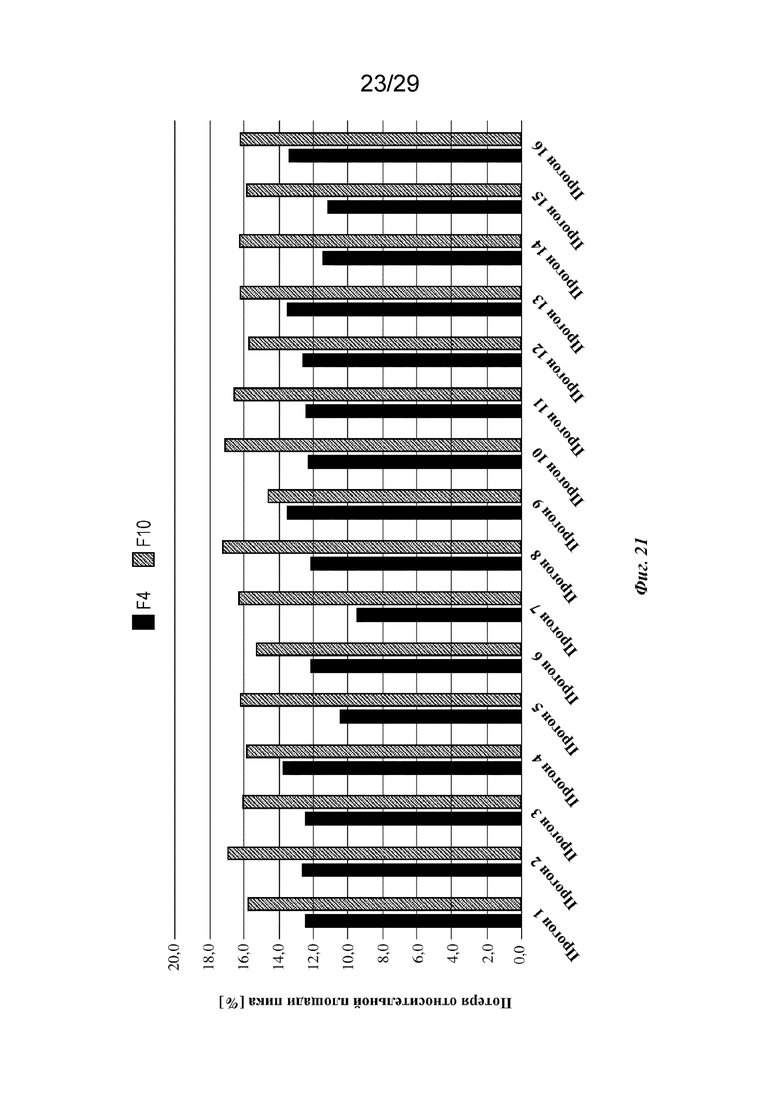
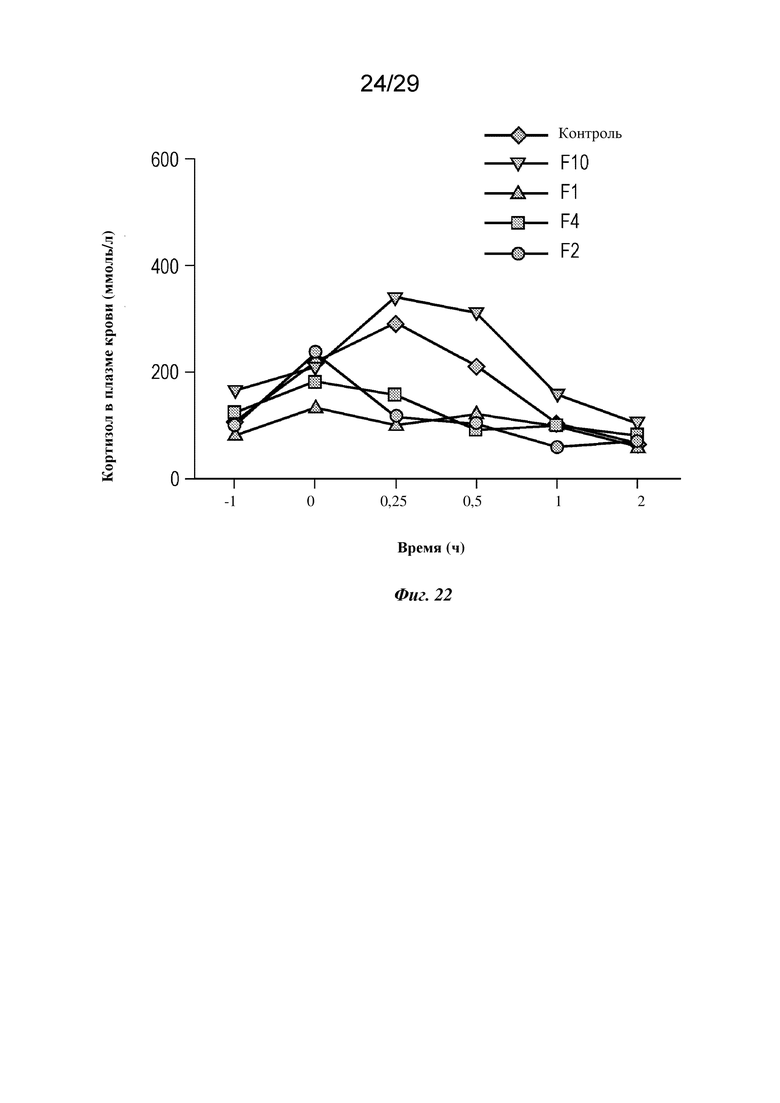
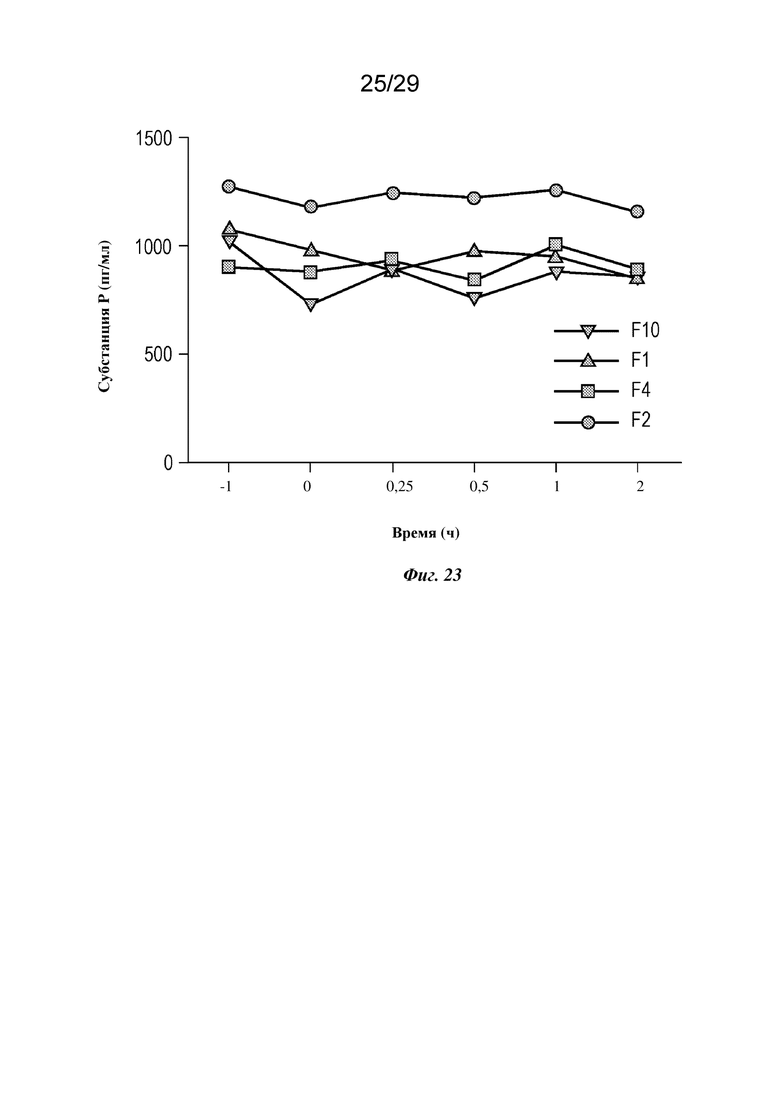
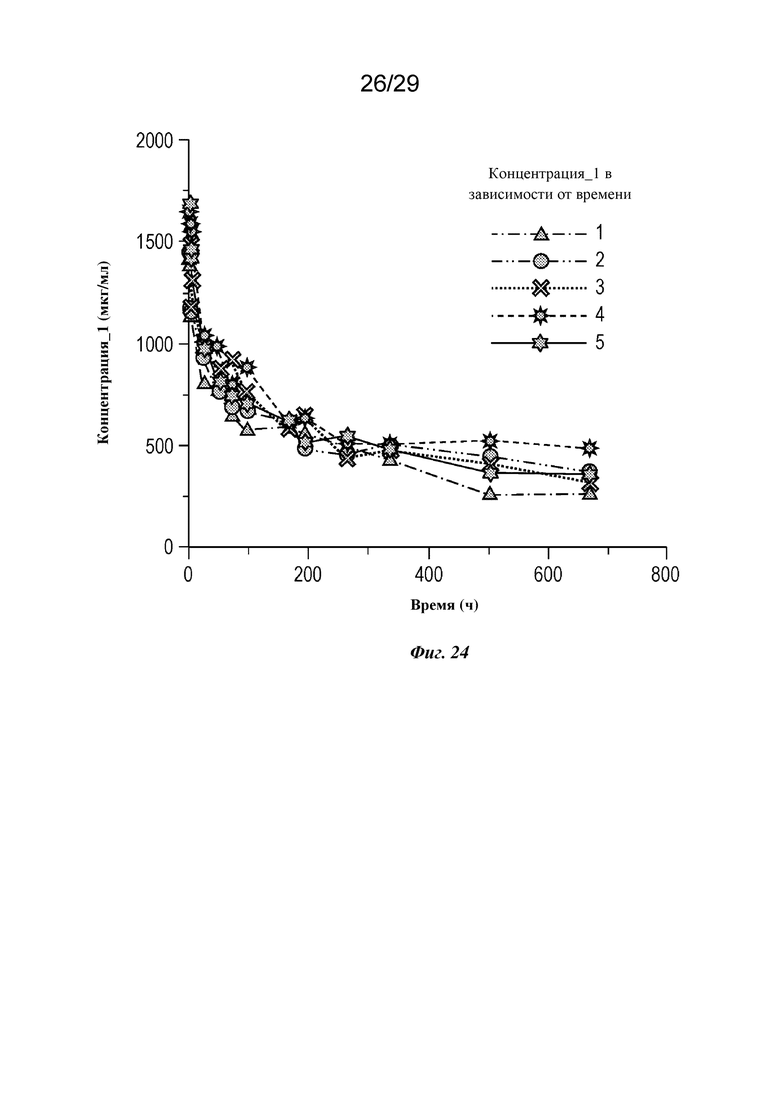
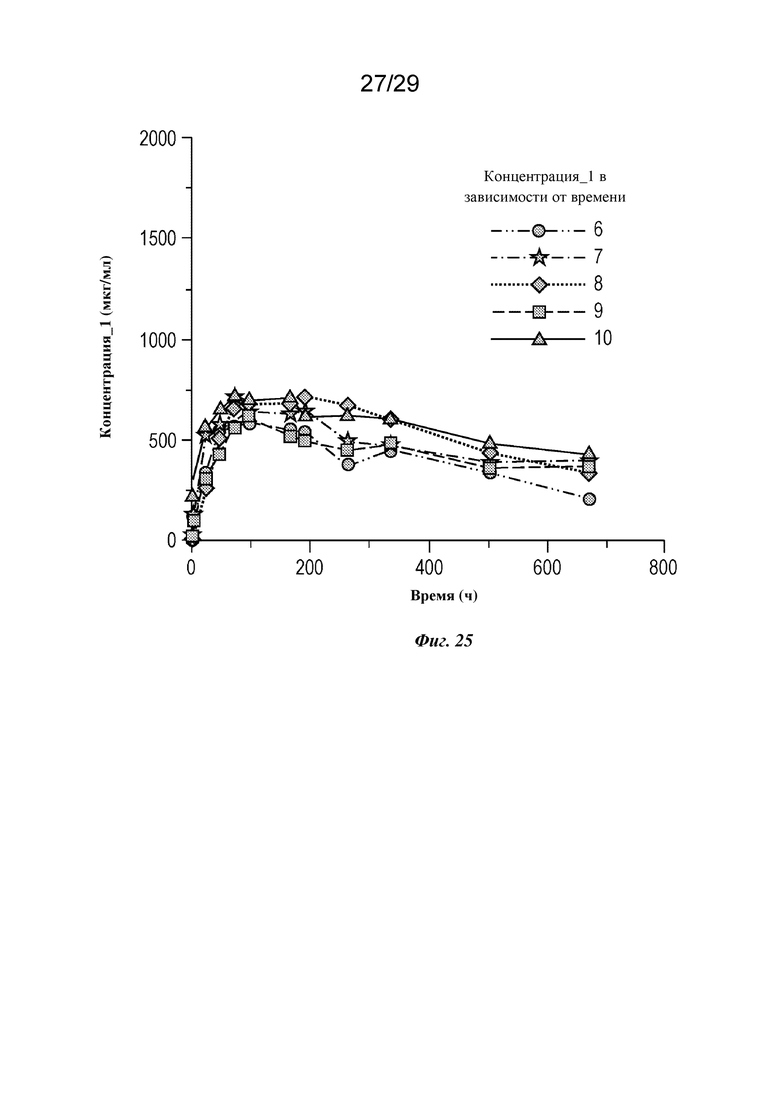
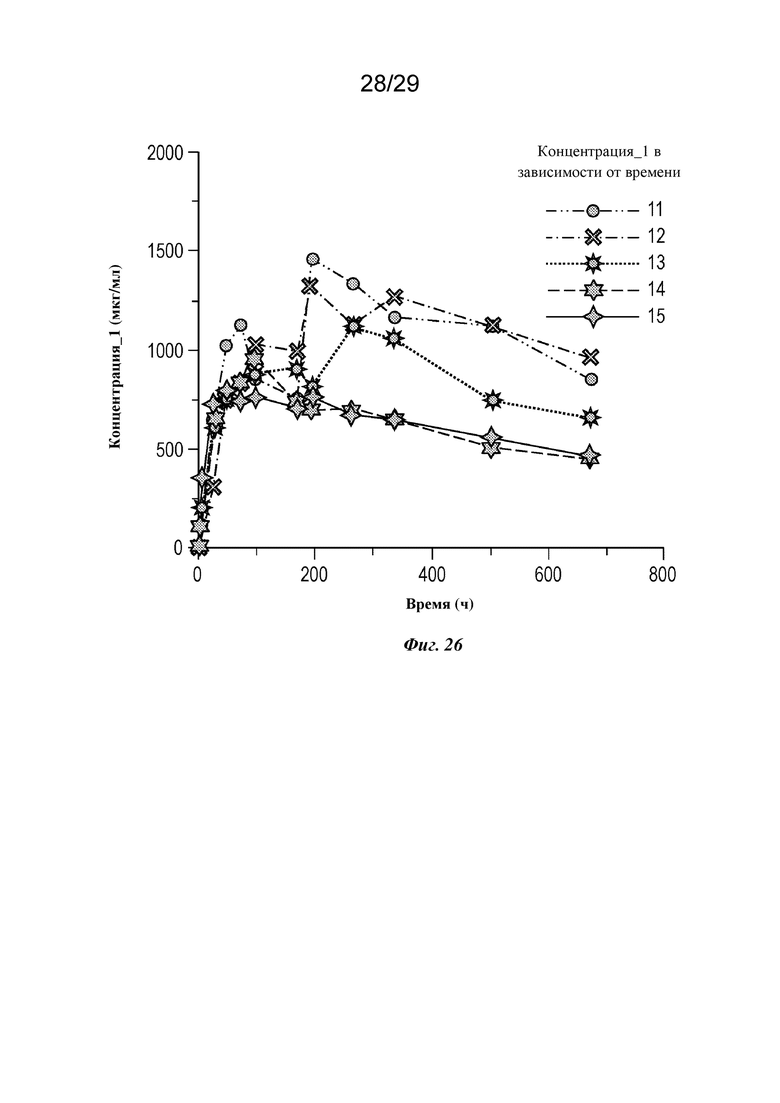
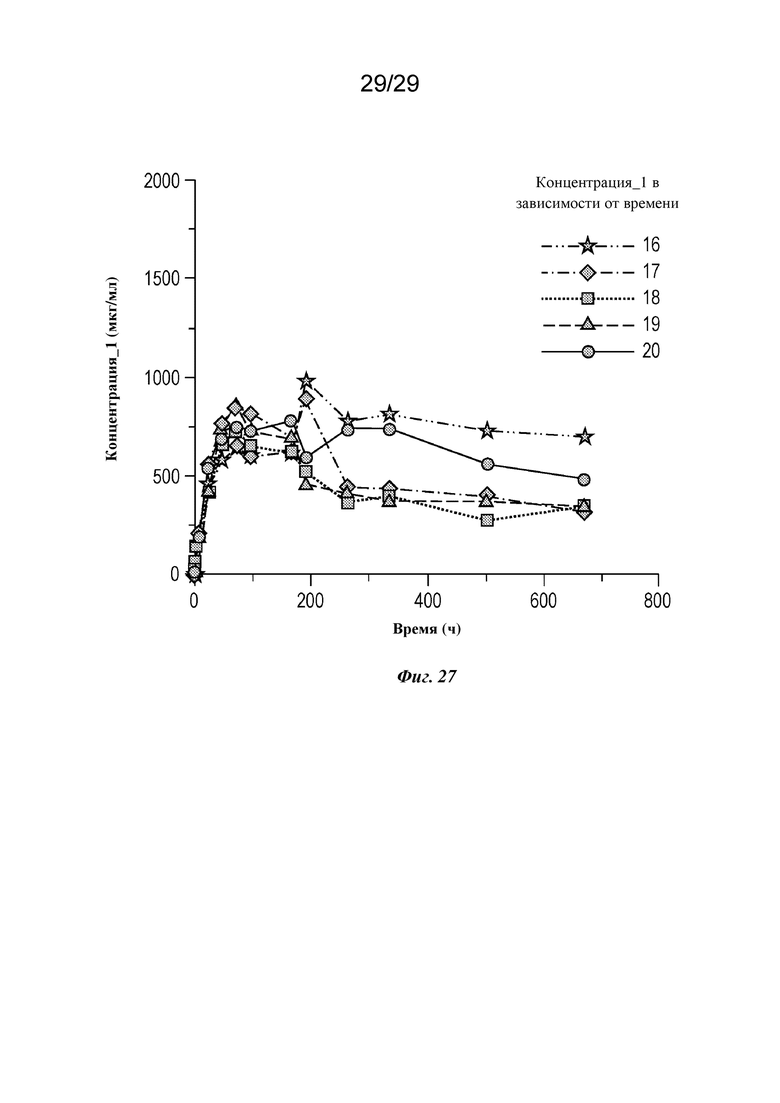
Группа изобретений относится к биотехнологии. Раскрыты составы для лечения заболевания, характеризующегося наличием CD38+ клеток, где состав подходит для подкожного введения и содержит 125-155 мг/мл антитела против CD38, а также их применение для лечения заболевания или состояния, характеризующегося наличием CD38+ клеток, которое представляет собой CD38+ гематологическое злокачественное новообразование, аутоиммунное или воспалительное заболевание или состояние. Кроме того, представлен упакованный фармацевтический продукт для лечения заболевания, характеризующегося наличием CD38+ клеток, содержащий стерильный контейнер или устройство, содержащие терапевтически эффективное количество состава, где состав содержит 125-155 мг/мл анти против CD38. Изобретение может быть использовано для лечения злокачественных новообразований, аутоиммунных или воспалительных заболеваний. 4 н. и 9 з.п. ф-лы, 29 ил., 31 табл., 7 пр.
1. Состав для лечения заболевания, характеризующегося наличием CD38+ клеток, где состав подходит для подкожного введения и содержит 125-155 мг/мл антитела против CD38,
где антитело против CD38 связывается с CD38 и содержит
вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности SEQ ID NO:1-3, соответственно, и
вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности SEQ ID NO:4-6, соответственно,
состав содержит
аргинин (Arg) в концентрации 90-150 мМ в качестве средства для уменьшения вязкости,
сахарозу в концентрации 2% (масс./об.) в качестве стабилизатора,
гистидин в концентрации 9-10 мМ в качестве буферного средства и
Полоксамер 188 в концентрации 0,4% (масс./об.) или Полисорбат 80 в концентрации 0,04% (масс./об.) в качестве поверхностно-активного вещества, и рН состава составляет 5,9-7,0 и вязкость составляет не более 25 мПа×с при 20°С.
2. Состав по п.1, где средство для уменьшения вязкости представляет собой 90-125 мМ Arg или 110 мМ Arg.
3. Состав для лечения заболевания, характеризующегося наличием CD38+ клеток, где состав подходит для подкожного введения и содержит 125-155 мг/мл антитела против CD38,
где антитело против CD38 связывается с CD38 и содержит
вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности SEQ ID NO:1-3, соответственно, и
вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности SEQ ID NO:4-6, соответственно,
состав содержит 9 мМ гистидина, 110 мМ Arg, 2% (масс./об.) сахарозы и 0,4% Полоксамера 188, и где рН состава составляет 6,2-6,3 и вязкость составляет не более 14 мПа×с при 20°С.
4. Состав по любому из предыдущих пунктов, где подкожным введением является подкожное введение большого объема.
5. Состав по любому из предыдущих пунктов, где
VH антитела против CD38 содержит аминокислотную последовательность SEQ ID NO:7, и
VL антитела против CD38 содержит аминокислотную последовательность SEQ ID NO:8.
6. Состав по любому из предыдущих пунктов, где антитело против CD38 представляет собой изатуксимаб.
7. Состав по любому из пп.1-6, где состав не содержит гиалуронидазу.
8. Упакованный фармацевтический продукт для лечения заболевания, характеризующегося наличием CD38+ клеток, содержащий стерильный контейнер или устройство, содержащие терапевтически эффективное количество состава, где состав содержит 125-155 мг/мл анти против CD38,
где антитело против CD38 связывается с CD38 и содержит
вариабельную область тяжелой цепи (VH), содержащую три определяющие комплементарность области (CDR): CDR-H1, CDR-H2 и CDR-H3, содержащие аминокислотные последовательности SEQ ID NO:1-3, соответственно, и
вариабельную область легкой цепи (VL), содержащую три CDR: CDR-L1, CDR-L2 и CDR-L3, содержащие аминокислотные последовательности SEQ ID NO:4-6, соответственно,
состав содержит
аргинин (Arg) в концентрации 90-150 мМ в качестве средства для уменьшения вязкости,
сахарозу в концентрации 2% (масс./об.) в качестве стабилизатора,
гистидин в концентрации 9-10 мМ в качестве буферного средства и
Полоксамер 188 в концентрации 0,4% (масс./об.) или Полисорбат 80 в концентрации 0,04% (масс./об.) в качестве поверхностно-активного вещества, и рН состава составляет 5,9-7,0 и вязкость составляет не более 25 мПа×с при 20°С.
9. Упакованный фармацевтический продукт по п.8, где устройство выбрано из группы, состоящей из шприца, шприцевой помпы, предварительно заполненного шприца и инфузионного насоса, содержащих состав.
10. Применение состава по любому из пп.1-8 для лечения заболевания или состояния, характеризующегося наличием CD38+ клеток, которое представляет собой CD38+ гематологическое злокачественное новообразование, аутоиммунное или воспалительное заболевание или состояние.
11. Применение по п.10, где CD38+ гематологическое злокачественное новообразование представляет собой множественную миелому.
12. Применение по п.11, где множественная миелома представляет собой рецидивирующую/рефрактерную множественную миелому.
13. Применение по любому из пп.10-12, дополнительно включающее введение субъекту одного или нескольких средств, выбранных из группы, состоящей из кортикостероида, ингибитора протеасом, иммуномодулирующего лекарственного средства, химиотерапевтического лекарственного средства и их комбинаций.
| WO 2017079150 A1, 11.05.2017 | |||
| Прибор для записи работы паровоза | 1931 |
|
SU26112A1 |
| Способ получения соединения висмута с винной или лимонной кислотой | 1932 |
|
SU31013A1 |
| KR 1020080098504 A, 10.11.2008 | |||
| CN 101678103 A, 24.03.2010 | |||
| CA 3011082 A1, 03.08.2017 | |||
| АНТИТЕЛО К CD38 И ЛЕНАЛИДОМИД ИЛИ БОРТЕЗОМИБ ДЛЯ ЛЕЧЕНИЯ МНОЖЕСТВЕННОЙ МИЕЛОМЫ И NHL | 2011 |
|
RU2595839C2 |

