Настоящее изобретение относится к области синтеза неорганических материалов, а именно мелкокристаллического станната кальция CaSnO3, в дальнейшем используемого как сырье для изготовления диэлектрической керамики для керамических конденсаторов.
В статье (Mar’ina U.A., Vorob’ev V.A., Mar’in A.P. CaSnO3 : Yb3+, Er3+, Ho3+ system synthesis and study of its luminescence under IR excitation // Modern Electronic Materials. – 2018. – Vol. 4(2). – P. 71–75. https://doi.org/10.3897/j.moem.4.2.38545) представлен способ синтеза станната кальция путем твердофазной реакции между карбонатом кальция CaCO3 и гидроксидом олова (II) Sn(OH)2. Указанные реагенты были механически смешаны в мольном отношении 1:1 и просеяны через сито (100 меш). Подготовленную смесь перенесли в тигель, выполненный из алунда, и подвергли прокаливанию при температуре 1250 °С в течение 18 ч. Способ прост в исполнении, предусматривает использование доступных реагентов и позволяет получить однофазный станнат кальция, однако требует проведения длительной высокотемпературной обработки реакционной смеси, что приводит к высоким энергетическим затратам.
В статье (Moshtaghi S., Gholamrezaei S., Niasari M. S. Nano cube of CaSnO3: Facile and green co-precipitation synthesis, characterization and photocatalytic degradation of dye //Journal of Molecular Structure. – 2017. – Vol. 1134. – P. 511-519. https://doi.org/10.1016/j.molstruc.2016.12.098) для синтеза станната кальция использовали салицилат кальция и хлорид олова (II) SnCl2 в качестве реагентов. Реагенты отдельно растворяли: салицилат кальция в метаноле, SnCl2 – в дистиллированной воде. Раствор салицилата кальция нагревали до 60 °С в течение 10 мин. К этому раствору по каплям добавляли раствор SnCl2. Затем в полученную смесь прикапывали щелочной агент, в качестве которого могут быть использованы аммиак, этилендиамин, пропилендиамин, триэтилентетрамин или тетраэтиленпентамин, до достижения pH 13. Полученный раствор выдерживали при 60 °C в течение 1 ч при постоянном перемешивании со скоростью 300 об/мин. Далее образовавшийся осадок отделяли путем центрифугирования, промывали последовательно дистиллированной водой и этанолом и сушили в вакууме при 60 °С. Затем высушенный порошок нагревали на воздухе со скоростью 30 °С/мин до достижения 900 °С и прокаливали при достигнутой температуре в течение 5 ч. По завершении прокаливания порошок охлаждали естественным путем. В результате был получен кристаллический станнат кальция с примесью оксида олова (IV) SnO2. Размер частиц продукта проявлял чувствительность к природе щелочного агента и увеличивался от нановеличин до нескольких микрон при увеличении молекулярной массы агента. К достоинствам представленного способа синтеза относится возможность контроля размера частиц продукта путем выбора щелочного агента, а также преимущественное использование не опасных для окружающей среды реагентов и вспомогательных веществ. Недостатками способа являются его многостадийность и необходимость предварительного синтеза части реагентов (салицилата кальция), что может негативно влиять на воспроизводимость процесса и чистоту продукта.
В статье (Alves M. et al. Influence of the precursor salts in the synthesis of CaSnO3 by the polymeric precursor method //Journal of Thermal Analysis and Calorimetry. – 2007. – Vol. 87. – No. 3. – P. 763-766. https://doi.org/10.1007/s10973-006-7853-2) исходными веществами для синтеза станната кальция являлись цитрат кальция и цитрат олова, которые предварительно получали из коммерчески доступных реагентов. Цитрат кальция получали растворением лимонной кислоты в дистиллированной воде при температуре 70 °C с последующим медленным добавлением гидрата ацетата кальция Ca(CH3COO)2·H2O до его полного растворения. Отдельно аналогичным образом готовили цитрат олова с использованием дигидрата хлорида олова (II) SnCl2⋅2H2O. Мольное отношение лимонной кислоты и ионов металла (для кальция и олова) составляло 3:1. Полученные растворы цитратов соединяли при перемешивании и затем добавляли этиленгликоль до достижения массового соотношения этиленгликоль : лимонная кислота, равного 40:60. Для проведения реакции этерификации раствор нагревали до температуры 100 °C. Образовавшийся полимерный прекурсор прогревали на воздухе при температуре 350 °С. Полученные порошкообразные прекурсоры механически перетирали, просеивали через сито (120 меш) и далее последовательно прокаливали в атмосфере кислорода при 250 °С в течение 12 часов и на воздухе при температуре 700 °С в течение 2 часов. Достоинство описанного способа заключается в возможности получения однофазного станната кальция при сравнительно низкой температуре из коммерчески доступных реагентов и вспомогательных веществ. Недостатками способа являются его многостадийность, использование больших избытков вспомогательных веществ, необходимость длительного прогрева прекурсора в атмосфере кислорода, а также высокий выброс углекислого газа при удалении полимерной составляющей прекурсоров.
В статье (Cheng H., Lu Z. Synthesis and gas-sensing properties of CaSnO3 microcubes //Solid state sciences. – 2008. – Vol. 10. – No. 8. – P. 1042-1048. https://doi.org/10.1016/j.solidstatesciences.2007.11.001) исходными веществами для синтеза станната кальция служили хлорид кальция CaCl2 и станнат натрия Na2SnO3. Навески исходных веществ, соответствующие 2,2 ммоль CaCl2 и 2 ммоль Na2SnO3, растворяли каждую в 10 мл дистиллированной воды, соответственно, для получения двух водных растворов. Затем к раствору CaCl2 при перемешивании добавляли 1–5 мл 0,2 М водного раствора поливинилпирролидона (ПВП, средняя молекулярная масса составляет 1300000 г/моль). Раствор Na2SnO3 прикапывали к раствору CaCl2, содержащему ПВП, при интенсивном перемешивании. После образования суспензии доводили ее рН до 7-12 с использованием растворов 1 М КОН и 0,2 М HCl. После этого полученную суспензию переносили в автоклав объемом 50 мл с внутренней осносткой из тефлона, и дистиллированной водой доводили его заполнение до 80% от общей емкости. Автоклав герметично закрывали, нагревали до температуры 140 °C и выдерживали при этой температуре в течение 10 часов. Затем автоклав естественным образом охлаждали до комнатной температуры. Полученный продукт прекурсор отфильтровывали, последовательно промывали этанолом и дистиллированной водой, затем сушили в вакууме при температуре 80 °C в течение 3 часов и прокаливали при 500 °С в течение 5 часов в атмосфере аргона. Полученный продукт состоял из однофазного станната кальция и представлял собой дисперсный порошок из частиц кубической формы со средним размером 900 нм. Достоинства представленного способа заключаются в мягких условиях синтеза однофазного станната кальция и возможности управления средним размером частиц синтезированного порошка за счет количества ПВП, добавленного в реакционную смесь. Вместе с тем, метод обладает рядом недостатков, таких как многостадийность, использование коммерчески малодоступных исходных веществ (Na2SnO3), избытка агрессивных вспомогательных веществ (щелочь), а также специальных условий сушки и прокаливания продукта (вакуум, атмосфера аргона).
В статье (Shojaei S., Hassanzadeh-Tabrizi S. A., Ghashang M. Reverse microemulsion synthesis and characterization of CaSnO3 nanoparticles // Ceramics International. – 2014. – Vol. 40. – No. 7. – P. 9609-9613. https://doi.org/10.1016/j.ceramint.2014.02.037) реагентами для синтеза станната кальция служили дигидрат хлорида кальция (CaCl2·2H2O) и тетрахлорид олова SnCl4, вспомогательными веществами – гидроксид аммония NH4OH, циклогексан, бутанол и ПАВ (бромид гексадецилтриметиламмония или Triton X-100). Реагенты растворяли в дистиллированной воде и полученный раствор прикапывали к смеси циклогексана, бутанола и ПАВ при непрерывном перемешивании в течение 30 минут. Водный раствор NH4OH аналогичным образом прикапывали к смеси циклогексана, бутанола и ПАВ при непрерывном перемешивании в течение 30 минут. Полученные эмульсии соединяли и перемешивали в течение 1 часа. Образовавшийся осадок отделяли, промывали дистиллированной водой, высушивали на воздухе и затем прокаливали при температуре 600-700 °С в течение 3 часов. Синтезированный продукт являлся однофазным станнатом кальция с размером частиц 1-3 мкм при использовании Triton X-100 в качестве ПАВ или обладал средним размером 20 нм при использовании бромида гексадецилтриметиламмония. Таким образом, описанный способ позволяет получить однофазный порошок станната кальция с контролируемым размером частиц (наноразмерный / микронный) в мягких условиях из коммерчески доступных реагентов. Однако данный способ синтеза также предусматривает использование ряда вспомогательных веществ, что приводит к удорожанию процесса и может оказывать негативное влияние на окружающую среду.
В статье (Bogdan T. V. et al. Formation of the CaSnO3 crystalline phase during catalytic aldol condensation of acetone under supercritical conditions //Russian Chemical Bulletin. – 2022. – Vol. 71. – No. 9. – P. 1930-1939. https://doi.org/10.1007/s11172-022-3611-2) реагентами для получения станната кальция являлись тетрагидрат нитрата кальция Ca(NO3)2·4H2O и пентагидрат хлорида олова SnCl4·5H2O, гидроксид натрия NaOH использовался в качестве вспомогательного вещества. Готовили водный раствор реагентов с мольным отношением ионов металлов 1:1 и их общей концентрацией 1,1 М. К приготовленному раствору при перемешивании прикапывали водный раствор NaOH концентрацией 3,4 М при комнатной температуре. Образовавшийся осадок перемешивали в течение 1 часа и затем выдерживали при комнатной температуре в течение 24 часов, трижды промывали избытком дистиллированной воды. Осадок высушивали на воздухе при температуре 120 °C в течение 8 часов, прокаливали на воздухе при температуре 750 °C в течение 4 часов для получения однофазного станната кальция с размером частиц 1-3 мкм. Описанный метод синтеза предусматривает использование коммерчески доступных реагентов и мягких условий для проведения реакции, однако требует применения агрессивного вспомогательного вещества (щелочи) и включает выдержку прекурсора при повышенной температуре.
Наиболее близким к представленному способу синтеза мелкокристаллического станната кальция является способ синтеза станната кальция, описанный в статье (A.A. Bhat, R. Tomar, Mn and Ce doping in hydrothermally derived CaSnO3 nanostructure. A facile way to enhance optical, magnetic and electrochemical properties // J. Alloys and Compounds. – 2021. – Vol. 876. – № 160043. https://doi.org/10.1016/j.jallcom.2021.160043). Способ получения станната кальция включает приготовление водного раствора, содержащего хлорид кальция CaCl2 и хлорид олова SnCl2 в мольном отношении 1:1, гидроксид натрия для поддержания pH > 10 и полиэтиленгликоль, путем механического перемешивания в течение 2 часов, обработку полученного раствора в реакторе (автоклаве) при температуре 180 °С в течение 24 часов, отделение полученного осадка от раствора путем фильтрования и промывание осадка этиловым спиртом, дистиллированной водой и ацетоном. Промытый осадок прокаливают на воздухе при температуре 950 °С в течение 6 часов. В результате был получен агломерированный кристаллический порошок, состоящий из частиц субмикронных и микронных размеров. Недостатками предложенного метода является его многостадийность, энергозатратность в связи с высокотемпературным прокаливанием продукта, а также необходимость тщательного промывания полученного порошка станната кальция от хлорид-ионов, содержащихся в реагентах.
Задачей заявляемого технического решения является разработка экономичного, энергосберегающего и безопасного для окружающей среды способа синтеза мелкокристаллического станната кальция со средним размером первичных частиц, не превышающим 20 мкм.
Поставленная задача решается способом, заключающимся в обработке в реакторе (автоклаве) смеси порошков оксида кальция и диоксида олова с предварительно нанесенным на его поверхность гидроксидом натрия паром воды в до- или сверхкритических условиях, после чего реактор охлаждают до комнатной температуры, полученный станнат кальция промывают последовательно водным раствором уксусной кислоты с концентрацией 5-10 масс. % и дистиллированной водой до нейтральной реакции промывных вод и высушивают на воздухе при температуре 70 ± 10 °С в течение 12 часов.
Предпочтительно нанесение гидроксида натрия на поверхность частиц диоксида олова проводят путем высушивания суспензии диоксида олова в водном растворе гидроксида натрия на воздухе при температуре от 50 до 70 °С.
Предпочтительно введение гидроксида натрия в смесь реагентов в количестве 0,5-1,5 масс. % по отношению к общей массе оксида кальция и оксида олова.
Предпочтительно нагрев реакционной смеси проводят со скоростью 70 °С/ч.
Предпочтительно обработку смеси проводят при температуре от 400 ± 10 °С и давлении от 26,5 МПа в течение 20-50 часов.
Предпочтительно промывание полученного станната кальция водным раствором уксусной кислоты и затем дистиллированной водой проводят методом декантации.
Технический результат представленного изобретения заключается в сокращении числа стадий синтеза станната кальция с использованием водной среды. Замена хлорида кальция и дихлорида олова в качестве реагентов на оксид кальция и диоксид олова повышают атомную эффективность синтеза станната кальция и снимают необходимость промывания продукта рядом различных растворителей для удаления ионов хлора. В предложенном способе происходит близкое к полному превращение исходных оксидов в готовый продукт – сложный оксид, что указывает на высокие показатели экологической чистоты процесса. Синтезированный станнат кальция может использоваться в качестве сырья для изготовления керамических конденсаторов с низкочастотным рабочим диапазоном.
Существенным отличительным признаком предлагаемого способа синтеза станната кальция является использование пара воды и/или сверхкритического водного флюида для обработки реагентов – оксида кальция и диоксида олова. По сравнению с жидкой реакционной средой, используемой при традиционном исполнении гидротермального синтеза, в частности, в приведенных выше аналогах и прототипе, молекулы воды в паровой фазе и в сверхкритическом флюиде образуют между собой меньшее количество водородных связей. Благодаря этому молекулы воды более активно взаимодействуют с твердой фазой оксидов, что приводит к образованию гидроксильных групп на их поверхности, а также в объеме. Гидроксилирование сопровождается разрывом связей металл-кислород в кристаллической решетке оксидов и тем самым снижает энергию активации их взаимодействия по твердофазному механизму. Таким образом, использование пара воды и/или сверхкритического водного флюида в качестве реакционной среды позволяет понизить температуру образования фазы станната кальция из ее компонентов (оксида кальция и диоксида олова) не менее чем на 800 °С по сравнению с превращением тех же реагентов в станнат кальция на воздухе. В традиционных гидротермальных условиях, основывающихся на проведении реакции в жидкой водной среде, растворимость реагентов значительно выше, чем в паре воды и/или сверхкритическом флюиде. В этом случае взаимодействие реагентов идет по другому механизму, включающему их растворение с последующей кристаллизацией продукта из раствора. Однако реализация этого механизма приводит к осаждению гексагидроксистанната кальция CaSn(OH)6, для превращения которого в станнат кальция требуются дополнительные стадии по выделению и прокаливанию.
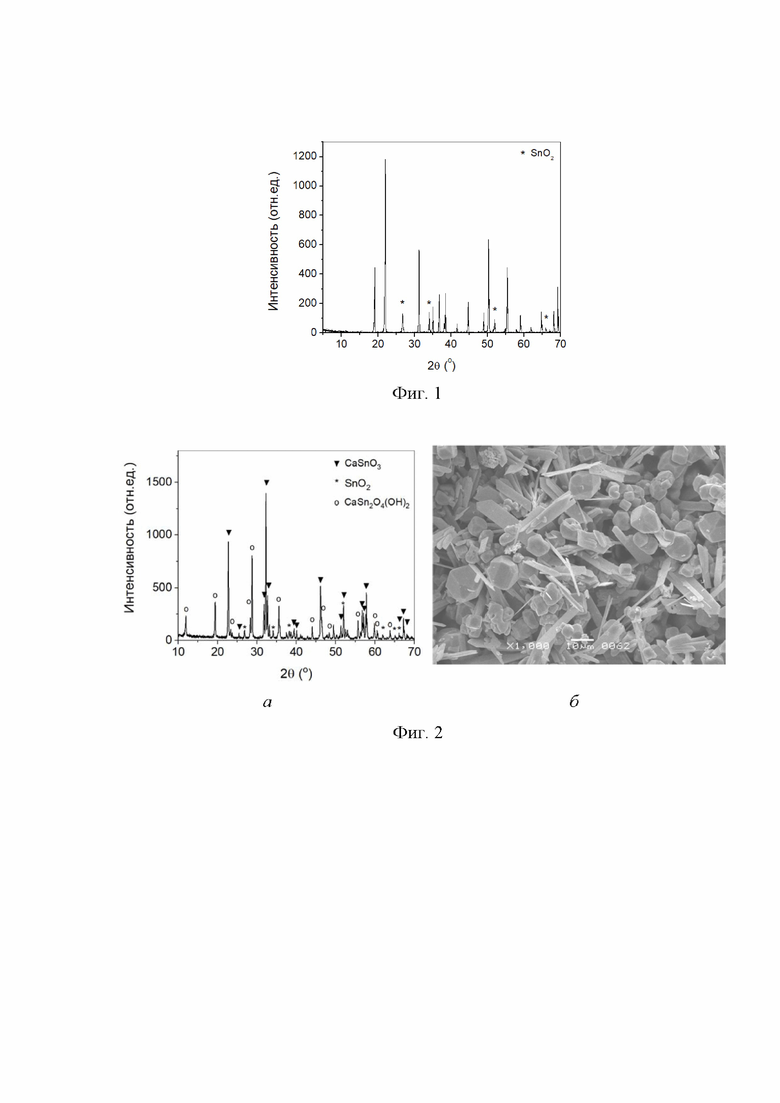
На фиг. 1 представлена дифрактограмма образца, полученного при 277 °С и давлении 6,13 МПа в течение 48 ч из смеси с мольным отношением CaO : SnO2 = 0,90:1.
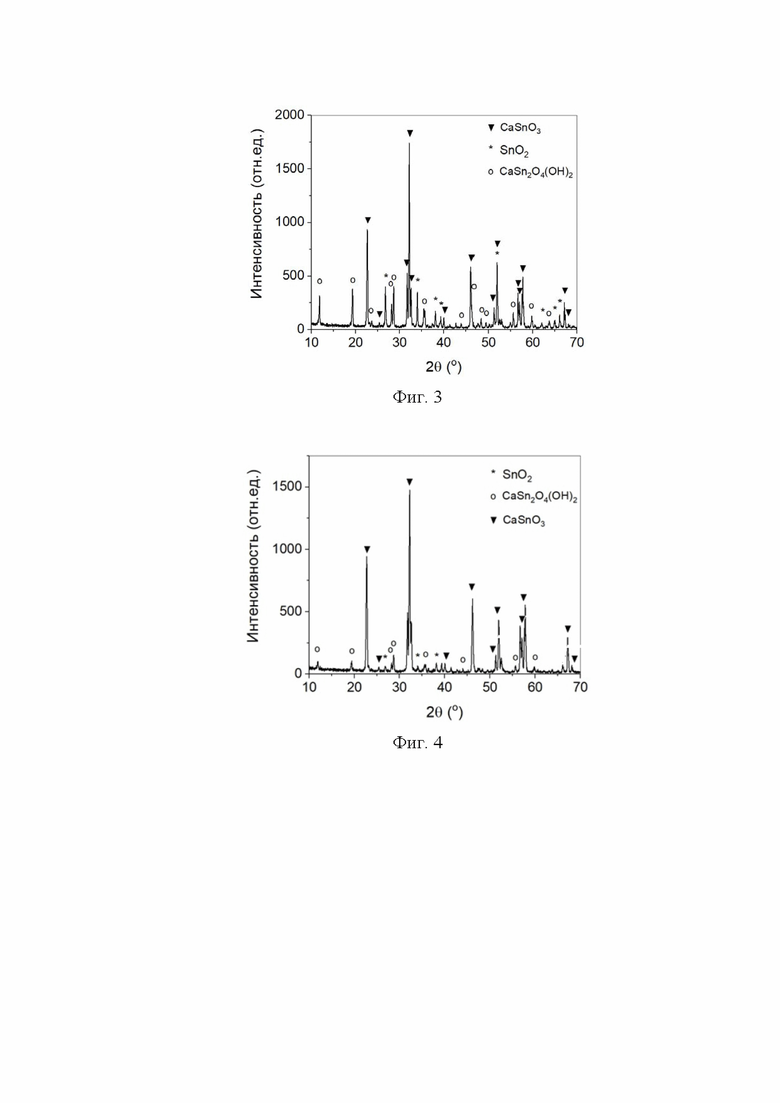
На фиг. 2 представлена дифрактограмма (а) и снимок СЭМ (б) образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,00 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 0,90:1.
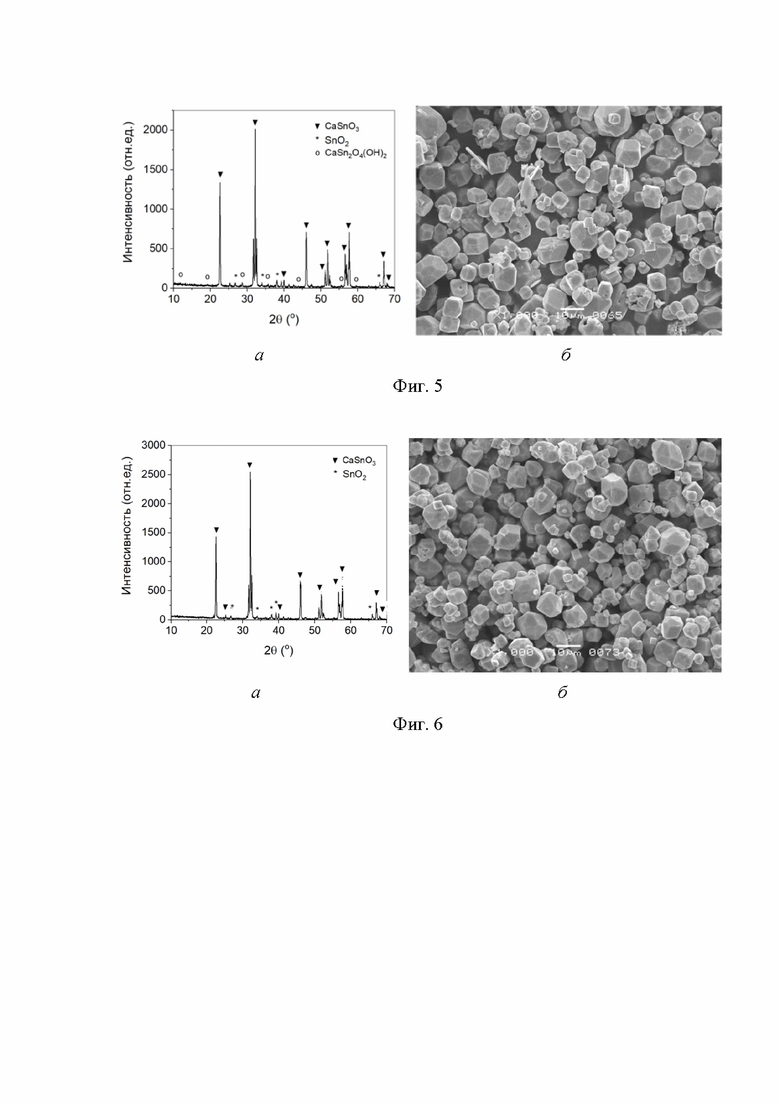
На фиг. 3 представлена дифрактограмма образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,50 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 0,81:1.
На фиг. 4 представлена дифрактограмма образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,50 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 1,26:1.
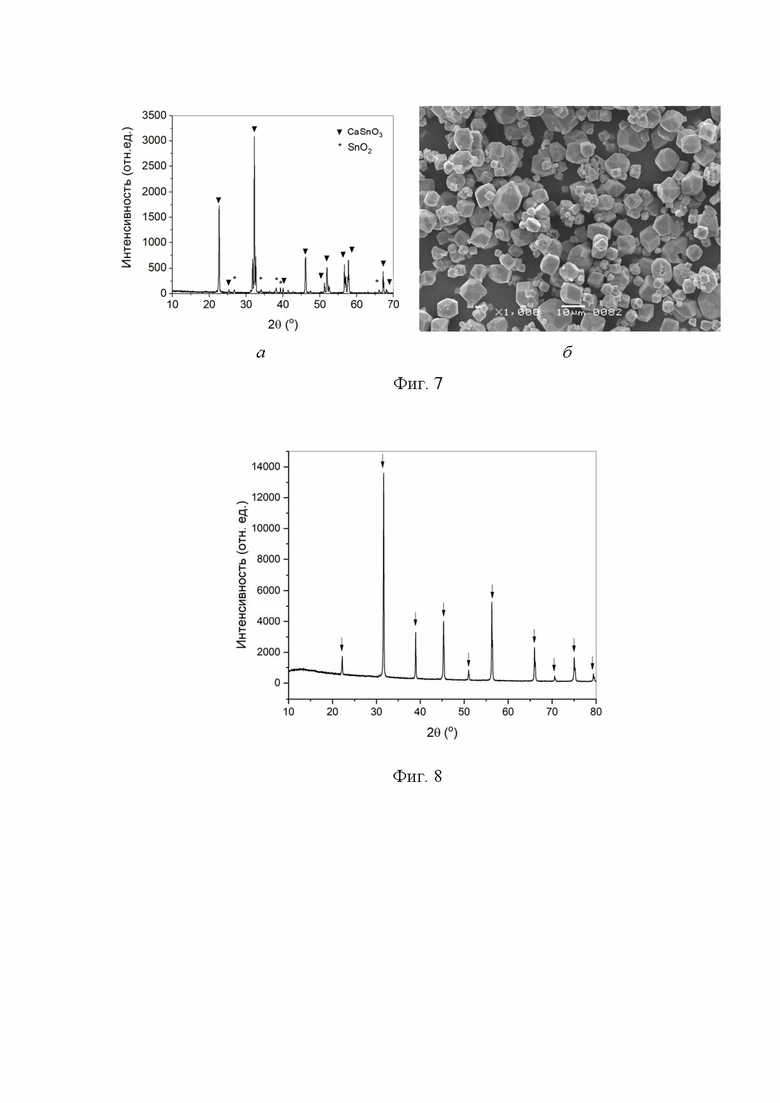
На фиг. 5 представлена дифрактограмма (а) и снимок СЭМ (б) образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,50 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 1,44:1.
На фиг. 6 представлена дифрактограмма (а) и снимок СЭМ (б) образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,50 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 1,80:1.
На фиг. 7 представлена дифрактограмма (а) и снимок СЭМ (б) образца, полученного в среде сверхкритического водного флюида при 400 °С и давлении 26,50 МПа в течение 38 ч из смеси с мольным отношением CaO : SnO2 = 2,16:1.
Предлагаемый способ синтеза станната кальция включает обработку смеси порошков оксида кальция (CaO) и диоксида олова (SnO2), с нанесенным на его поверхность в качестве вспомогательного вещества гидроксидом натрия (NaOH) в количестве 0,5-1,5 масс. % по отношению к смеси оксидов в воде в сверхкритических условиях при температурах 400 ± 10 °С. Синтез проводят в автоклаве из материала, химически устойчивого к действию воды в любом агрегатном состоянии и в сверхкритических условиях. Навеску диоксида олова предварительно пропитывают гидроксидом натрия следующим образом. В химически стойкой емкости готовят водный раствор гидроксида натрия концентрацией от 0,29 до 0,31 М, содержащий количество растворенного вещества, соответствующее 0,5-1,5 масс. % гидроксида натрия в смеси реагентов для синтеза станната кальция. В приготовленный раствор помещают навеску оксида олова, тщательно перемешивают и оставляют при температуре 70 ± 10 °С на 48 ч. Навески исходных порошков оксида кальция и диоксида олова с нанесенным гидроксидом натрия, взятые в мольном отношении в интервале от 1,8 до 3,25, смешивают механически. На дно автоклава вливают дистиллированную воду в таком количестве, что она заполняет внутренний объем автоклава на 20%. Смесь навесок исходных порошков помещают в открытый сверху контейнер из материала, химически устойчивого к действию воды в любом агрегатном состоянии. Контейнер со смесью порошков внутри автоклава размещают на опоре так, чтобы дно контейнера было выше уровня налитой на дно автоклава воды, и автоклав герметично закрывают. Далее смесь порошков и воду внутри автоклава нагревают со скоростью 70 °С/ч до заданной температуры в интервале 400 ± 10 °С и выдерживают в изотермических условиях при заданной температуре в течение периода времени 20-50 ч. Затем горячий закрытый автоклав в вертикальном положении охлаждают следующим образом: донную часть автоклава погружают в охлаждающий агент, например, воду, на глубину не выше уровня дна контейнера с порошком; охлаждающий агент должен обеспечивать температуру наружной поверхности погруженной в него части автоклава не выше 100 °С при атмосферном давлении. Охлажденный до комнатной температуры автоклав открывают, вынимают контейнер и продукт реакции извлекают из контейнера. Проводят однократную или многократную процедуру отмывки продукта реакции от примесей. Для этого применяют дистиллированную воду и/или водный раствор той или иной кислоты с водородным показателем pH не выше 7. Извлеченный из контейнера порошок тщательно перемешивают с водой или кислым раствором в химически стойкой емкости и затем отделяют порошок от промывающей жидкости декантацией или фильтрованием. Влажным порошок высушивают в воздушной атмосфере при температуре 70 ± 10 °С.
Рентгенофазовый анализ (РФА) синтезированных порошков станната кальция проводили с помощью рентгеновского дифрактометра Rigaku D/Max-2500 (Rigaku Corp., Япония) с CuKα-излучением в диапазоне 20° < 2θ < 70° с шагом 0,02°. Для определения фазового состава экспериментально полученные дифрактограммы сопоставляли с данными из базы ICDD PDF2.
Размер и форму частиц синтезированных порошков определяли с помощью сканирующего электронного микроскопа JSM-6390 LA (JEOL Ltd., Япония).
Ниже приведены примеры конкретного осуществления изобретения - способа получения мелкокристаллического станната кальция, для предоставления специалистам в данной области техники полного описания проведения способа по изобретению, и подразумевают, что приведенные примеры не ограничивают предлагаемый авторами объем изобретения.
Все используемые реагенты являются коммерчески доступными, все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25°C.
Химические символы имеют свои обычные значения: мкм (микрометр(ы)), мкл (микролитр(микролитры)), мкг (микрограмм(микрограммы)), М (моль(моли) на литр), л (литр(литры)), мл (миллилитр(миллилитры)), г (грамм(граммы)), мг (миллиграмм(миллиграммы)), моль(моли), ммоль (миллимоль(миллимоли)).
Примеры
Пример 1
Навеску 6,000 г диоксида олова поместили в раствор гидроксида натрия объемом 6,67 мл и концентрацией 1,235 масс. %, суспензию механически перемешали и высушили на воздухе при температуре 65 °С 48 ч. Полученный диоксид олова, содержащий 1,35 масс. % гидроксида натрия, смешали с навеской оксида кальция массой 2,012 г (мольное отношение CaO : SnO2 = 0,90:1) путем пятикратного совместного просеивания через капроновое сито с размером ячеек 300 мкм. Смесь поместили в контейнер внутри реактора из нержавеющей стали со свободным объемом 12,00 мл без учета объема смеси. На дно реактора, вне контейнера со смесью, налили 1,00 мл дистиллированной воды. Реактор герметизировали и нагревали со скоростью 70 °/ч до температуры 277 °С и выдерживали при достигнутой температуре 48 ч. Равновесное давление пара внутри реактора во время изотермической выдержки составило 6,13 МПа. Реактор охлаждали, полученный образец выгружали из реактора и промывали последовательно раствором уксусной кислоты концентрацией 10 масс. % и дистиллированной водой до достижения рН7 промывных вод. Осадок высушивали на воздухе при 65 °С до постоянной массы. По данным рентгенофазового анализа (РФА) (Фиг. 1) получили продукт, содержащий гексагидроксистаннат кальция CaSn(OH)6 с примесью диоксида олова.
Пример 2
По методике, приведенной в Примере 1, при температуре 400 °С и давлении сверхкритического водного флюида 26,00 МПа был проведен синтез продолжительностью 38 ч. РФА показал, что был получен станнат кальция с примесью диоксида олова и оксигидроксида станната кальция CaSn2O4(OH)2 (Фиг. 2а). На снимках сканирующей электронной микроскопии (СЭМ) (Фиг. 2б) станнат кальция имеет форму изометричных кристаллов, а оксигидроксида станната кальция – форму тонких пластинчатых полос или тонких удлинённых пластинок.
Пример 3
Навеску 3,000 г диоксида олова поместили в раствор гидроксида натрия объемом 3,34 мл и концентрацией 1,235 масс. %, суспензию механически перемешали и высушили на воздухе при температуре 70 °С 48 ч. Полученный порошок смешали с навеской оксида кальция массой 0,906 г (мольное отношение CaO : SnO2 = 0,81:1) путем пятикратного совместного просеивания через капроновое сито с размером ячеек 300 мкм. Смесь поместили в контейнер внутри реактора из нержавеющей стали свободным объемом 12,80 мл без учета объема смеси. Вне контейнера внутрь реактора налили 2,62 мл дистиллированной воды. Реактор герметизировали и нагревали со скоростью 70°/ч до температуры 400 °С. Проводили изотермическую выдержку 38 ч при достигнутой температуре. Давление сверхкритического водного флюида внутри реактора составило 26,50 МПа. Затем реактор охлаждали, образец извлекали и промывали последовательно раствором уксусной кислоты концентрацией 10 масс. % и дистиллированной водой до достижения рН7 промывных вод. Образец высушивали на воздухе при 70 °С до постоянной массы. По данным РФА (Фиг. 3) образец состоял из станната кальция с примесью оксигидроксида станната кальция CaSn2O4(OH)2 и диоксида олова.
Пример 4
По методике, приведенной в Примере 3, был проведен синтез из смеси 3,000 г диоксида олова с содержанием 1,35 масс. % гидроксида натрия и 1,409 г оксида кальция (мольное отношение CaO : SnO2 = 1,26:1). По результатам РФА (Фиг. 4) полученный образец состоял из станната кальция с примесью диоксида олова и оксигидроксида станната кальция CaSn2O4(OH)2.
Пример 5
По методике, приведенной в Примере 3, был проведен синтез из смеси 3,000 г диоксида олова с содержанием 1,35 масс. % гидроксида натрия и 1,610 г оксида кальция (мольное отношение CaO : SnO2 = 1,44:1). По данным РФА (Фиг. 5а) был получен станнат кальция со следовыми примесями диоксида олова и оксигидроксида станната кальция CaSn2O4(OH)2. Присутствие в образце CaSn2O4(OH)2 проявлялось на снимках СЭМ (Фиг. 5б) в виде редких пластинчатых частиц.
Пример 6
По методике, приведенной в Примере 3, был проведен синтез из смеси 3,000 г диоксида олова с содержанием 1,35 масс. % гидроксида натрия и 2,012 г оксида кальция (мольное отношение CaO : SnO2 = 1,80:1). По данным РФА (Фиг. 6а) был получен станнат кальция со следовой примесью диоксида олова. На снимке СЭМ образца (Фиг. 6б) наблюдались изометричные частицы, характерные для станната кальция.
Пример 7
По методике, приведенной в Примере 3, был проведен синтез из смеси 3,000 г диоксида олова с содержанием 1,35 масс. % гидроксида натрия и 2,414 г оксида кальция (мольное отношение CaO : SnO2 = 2,16:1). По данным РФА (Фиг. 7а) был получен станнат кальция со следовой примесью диоксида олова. На снимке СЭМ образца (Фиг. 6б) были обнаружены только изометричные частицы, характерные для станната кальция.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МЕЛКОКРИСТАЛЛИЧЕСКОГО ТИТАНАТА БАРИЯ | 2016 |
|
RU2637907C1 |
| Способ получения особочистого мелкокристаллического титаната бария | 2019 |
|
RU2713141C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ ПОРОШКОВОЙ ШИХТЫ Ag/SnO ДЛЯ РАЗРЫВНЫХ ЭЛЕКТРОКОНТАКТОВ | 2010 |
|
RU2442835C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕЛКОКРИСТАЛЛИЧЕСКОГО АЛЮМИНАТА МАГНИЯ | 2016 |
|
RU2630112C1 |
| Способ получения синтетического цеолита | 2022 |
|
RU2787819C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТАННАТОВ ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ | 1992 |
|
RU2049064C1 |
| НАНОПОРИСТЫЙ МАТЕРИАЛ ДЛЯ ЧУВСТВИТЕЛЬНЫХ ЭЛЕМЕНТОВ ГАЗОВЫХ ДАТЧИКОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2725031C1 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ УВЕЛИЧЕНИЯ НЕФТЕОТДАЧИ НА ОСНОВЕ СВЕРХКРИТИЧЕСКОГО ДИОКСИДА УГЛЕРОДА И НЕИОНОГЕННОГО ПОВЕРХНОСТНО-АКТИВНОГО ВЕЩЕСТВА | 2013 |
|
RU2635307C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛИРОВАННОГО НЕОРГАНИЧЕСКОГО СОРБЕНТА | 2020 |
|
RU2756163C1 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ МЕТИЛМЕРКАПТАНА | 2008 |
|
RU2497588C2 |
Изобретение относится к области синтеза порошковых оксидных материалов, а именно станната кальция, который может быть использован как компонент при изготовлении конденсаторной керамики. Для получения мелкокристаллического станната кальция проводят обработку смеси оксида кальция CaO и диоксида олова SnO2, взятых в мольном отношении 1,44-2,16 и смешанных многократным совместным просеиванием через полимерное сито с размером ячеек не более 500 мкм, в присутствии гидроксида натрия NaOH, введенного в смесь высушиванием на воздухе водной суспензии, содержащей частицы SnO2 и растворенный NaOH, в закрытом реакторе при температуре 400 ± 10°С со скоростью нагрева 70°C/ч и давлении сверхкритического водного флюида 26,5 МПа. Продукт последовательно промывают водным раствором уксусной кислоты концентрацией 5-10 масс. %, дистиллированной водой до нейтральной реакции промывных вод и высушивают на воздухе при температуре 70 ± 10°С в течение 12 ч. Заявленный способ позволяет получить кристаллический порошок станната кальция с размером частиц не более 20 мкм при сокращении числа стадий синтеза. 3 з.п. ф-лы, 8 ил., 8 пр.
1. Способ получения мелкокристаллического станната кальция, включающий обработку смеси кальций- и оловосодержащих реагентов в присутствии гидроксида натрия в водной среде при повышенных температуре и давлении, отличающийся тем, что в качестве кальцийсодержащего реагента используется оксид кальция CaO, в качестве оловосодержащего реагента используется диоксид олова SnO2, и обработку реагентов ведут в среде пара воды или сверхкритического водного флюида; при этом термообработку смеси реагентов ведут в течение времени от 20 до 50 часов в изотермических условиях при температуре, выбранной в интервале 400 ± 10°С со скоростью нагрева 70°C/ч и давлении пара воды или сверхкритического водяного флюида 26,5 МПа.
2. Способ по п. 1, отличающийся тем, что введение гидроксида натрия в реакционную смесь проводят путем высушивания на воздухе водной суспензии, содержащей частицы диоксида олова и растворенный гидроксид натрия.
3. Способ по п. 1, отличающийся тем, что смешение исходных веществ осуществляют путем их многократного просеивания через полимерное сито с размером ячеек не более 500 мкм.
4. Способ по п. 1, отличающийся тем, что полученный продукт последовательно промывают водным раствором уксусной кислоты концентрацией 5-10 масс. %, дистиллированной водой до нейтральной реакции промывных вод и высушивают на воздухе при температуре 70 ± 10°С в течение 12 часов.
| BHAT A.A | |||
| et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| A facile way to enhance jptical, magnetic and electrochemical properties, J | |||
| Alloys and Compounds, 1021, v | |||
| Соединение металлических труб раструбом | 1925 |
|
SU876A1 |
| СПОСОБ ПОЛУЧЕНИЯ СТАННАТОВ ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ | 1992 |
|
RU2049064C1 |
| ЭЛЕКТРОПРОВОД, ПРИКРЕПЛЕННЫЙ К ВНЕШНЕЙ ДЕТАЛИ, ЖГУТ ПРОВОДОВ, ВКЛЮЧАЮЩИЙ В СЕБЯ ТАКОЙ ЭЛЕКТРОПРОВОД, ПРИКРЕПЛЕННЫЙ К ВНЕШНЕЙ ДЕТАЛИ, И СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ | 2011 |
|
RU2552840C2 |
| Электропривод | 1978 |
|
SU748768A1 |
| JP 59156915 A, 06.09.1984. | |||

