гексил, феноксимётил, которые могут быть замещены дифенидметилом или фенилом, который может быть замещен одним или двумя заместителями, выбранными из группы, включаквдей хлор или циано-, ОКСИ-, фенил или диметял аминогруппу, или R и R , если они находятся в цис-положении, вместе с атомз ми углерода, с которьоми они связаны, образуют циклопропановое, циклобутановое, циклопентановое или циклогексановоё кольцо,или R -карбоксигруппа и , R. и R - водород, .или их солей с основаниями, или, если соединение общей формулы I содержит дополнительно свободную карбоксигруппу, их солей с основаниями и по этой группе, или, если соединение общей формулы I содержит свободную основную группу, их кислотно-аддитивных солей, отличающийс я тем, 4TQ от сложного дифенилметиловогоп-метоксибенэиловогоИЛитрет-бутилового эфира кисло№1 общей формулы 1 отщепляют сложноэфирнуто за щитную группу путем обработки сильной кислотой, такой как трифторуксус
ная кислота, или в случае tpijT-бутилового эфира, такой как муравьиная кислота, в присутствии избытка кислоты в качестве разбавителя или растворителя, или в присутствии анизола или толуола в качестве дополнительного разбавителя или растворителя, и .вы.деляют целевой продукт в виде свобрдг ной кислоты, свободного основания или амфотерного иона,или соединение общей формулы Г в виде свободной кис лоты KSIK амфотерного иона вводят во взаимодействие с основанием, или соединение общей 1 в виде свободного основания или с1мфотерного иона вводят во взаимодействие с кислотой и выделяют целевой продукт в виде соли с основанием или кислотноаддитивной сопи.
2. Способ по п. 1, отл ичаю щ и и с я тем, что в качестве исходного соединения используют сложный эфир кислоты общей формулы I, где 1Н-1,2,3-триазол-4-илтиометил; R - водород и А - остаток общей формулы } , где ft и R - водо род.






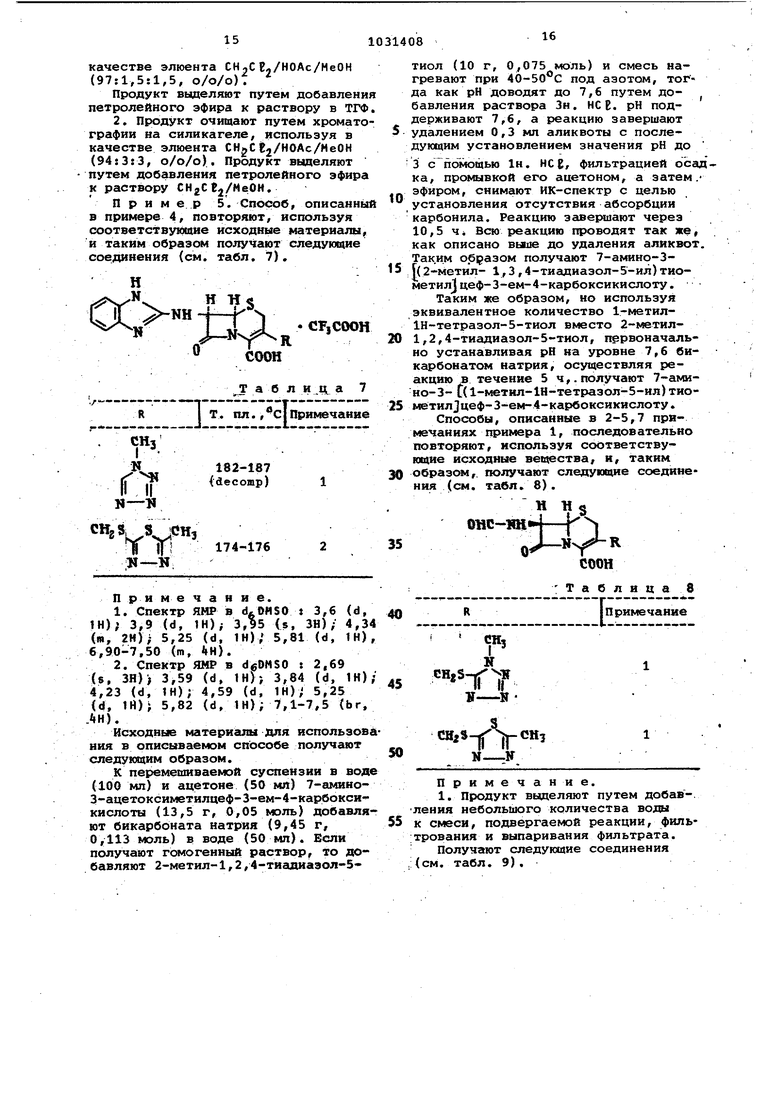








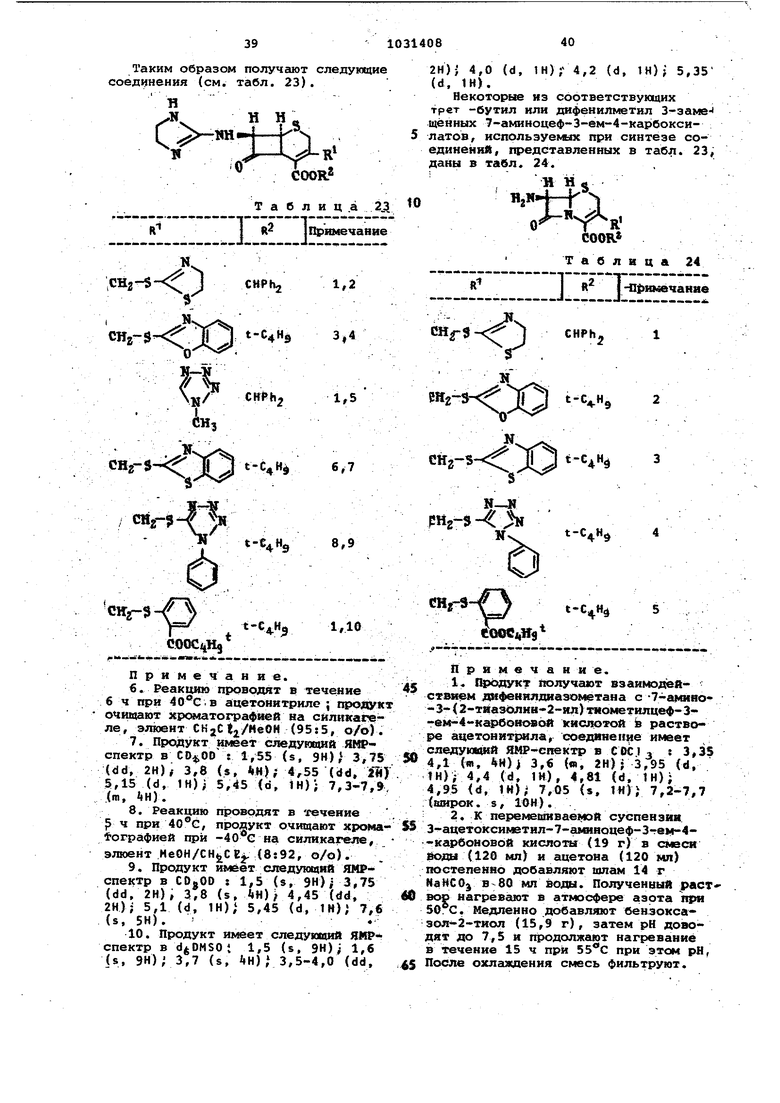








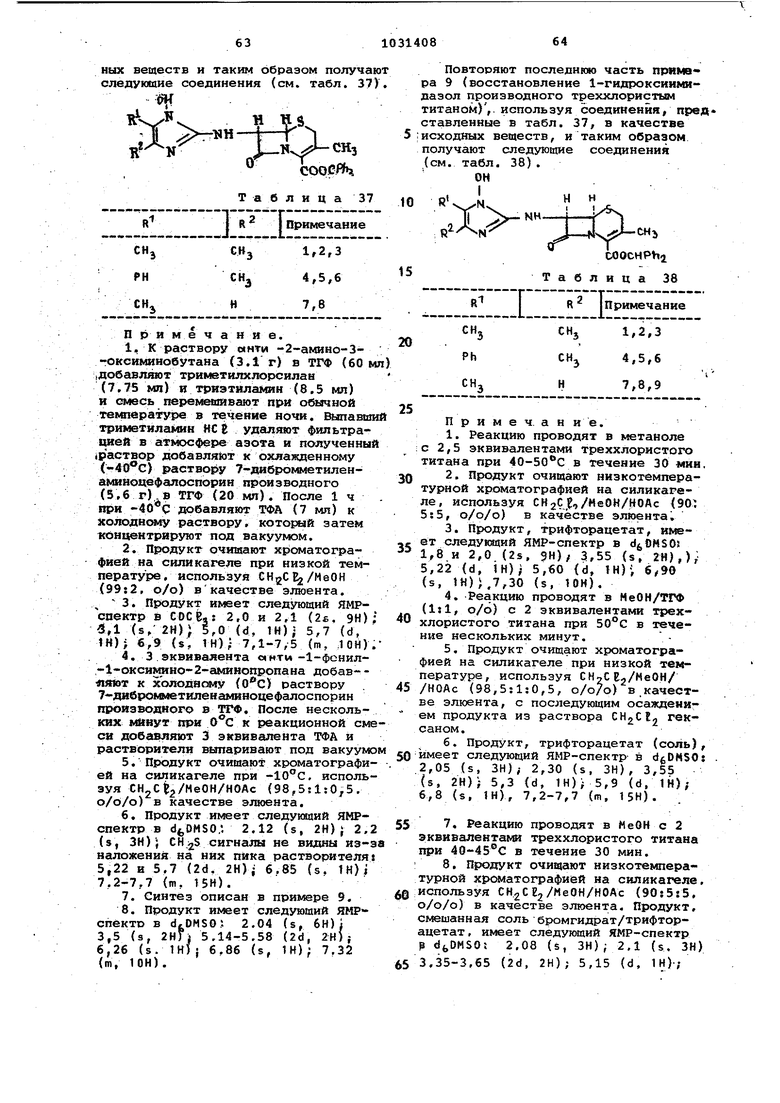














1. Способ получения производных цефалоспоршга общей формулы 1,2,З-тиадиазол-5-илтисметил, 1Н-1,2, З-триаэоп-4-илтиометйл, 5-трифтормаТИЛ-1Н-1,2,4-триазой-З-илтиометип, 4,6-диметиппиримид-2-иптиометип, 2-тиазолин-2-илтиометил, бензоксазол-2иптиометил, бензтиазол -2-илтиометил4 2-карбоксифенилтйометил, 
Изобретение относится к способу получения новых производных цеФалосп рина или их солей с основаниями или их кислотно-аддитивных селей, обладакадих антибактериальными свойства5ии, которые могут быть использованы в медицине. Известен способ псшучения обладв ющих антибактериальными свойствами производных 7-ацетиламидо-З-гетероциклический тиометил-З-цефем-4-карбо новой кислоты ацилированием 7-амино3-гетероциклический тиометил-3-цефем 4-карбоновой кислоты реакционноспособным производным соответствующей карбоновой кислоты 1 . Цель изобретения - получение новы антибиотиков цефалоспоринового ряда, расширяющих арсенал средств воздействия на живой с ганизм. Цель достигается тем, что на осно вании известной реакции кислотной дб этерификации кислот цефалоспоринового ряда f2 согласно способу получе ния цроизводных цефалоспорина общей формулы где R - метил,ацетоксиметил,1-метил-1Н-тетразап-5-илтиометил, 1-карт боксиметил-1Н-тетразол-1Н-тетразал-5илтиометил, 1-(2-диметиламино)этил1Н-тетразол-5-ш1тиометил, 1-сульфо1Н-тетразол-э-илтиометил, ±-сульфо1-1Н-тетразол-5-илтиометил, 1-изометИЛметил-хн-тетразсш-э-илтиометил, J.TJI-1 Н-т а тйчптт MnTMrtiutoTMnпрсм1Ил-1И-тетразал-5- илтиометил, 1(2,2,2-трифтор)этил-1Н-т&тразол5-илтиометш1, 1-фенил-1Н-тетраэол-5илтиометил, 1-{2-метилтио)этил-1Н-тетразал-5-илтиометил, 1,3,4-тиадиазал-2-илтиометил, 5-метиаг1-1,3,4-тиадиазол-2-илтиометил, 1,2,3-тиадиазал 5-илтиси 1етил, 1н-1,2,3-триазол-4-илтиометил, 5-трифторметил-1Н-1,2,4триазол-3-илтиометил, 4,6-диметилпиримид-2-илтиометил, 2--тиазолин-2-Ю1тиометвл, бензоксазол-2-илтиометил, бензтиазол-2-илтиометил, 2-карбоксифенилтиометил, {б-карбосшу1етил-7-окс пирроло l, 2-,1ij-пиридазин-2-ил) метил 14t TtJrTt R - водород, метил, окси-г, метОКСИ-, ацетиламино- или 4-метоксибен зильная группа) А - остаток формулы
Производные цефалоспорина представляют собой антибактериальные агенты, многие из которых имеют широкий спектр активности 1 п v i t го против стандартных лабораторных микро- организмов, как грам-отрицательных так и грам-положительных, которые применяются для проверки активности по сравнению с патогенными бактериями.
Характеристика спектров ЯМР даетс в величинах сГ относительно тетраметилсилана ( как внутренний стандарт, (s-синглет, d - дуплет, t -. триплет, m - мульТиплет, Ьг - уширенная полоса). Температуры даны в градусах Цельсия, а точка кипения петролейного эфира, если не будет указана другим образом, 47-61°С.
При этом используют трифторуксусную кислоту (ТФА), тетрагидрофуран (ТГФ), уксусную кислоту (ОАЦ), этилацетат (EtOAc), метанол (МеОН), диметилформамид (DMF), диметилсульфоксид (DMSO), диэтиловый эфир (si-Ъег жидкостную хроматографию высокого
давления {НР1С), ..
Согласно предлагаемому способу производное цефалоспорина вьоделяют в форме соли, амфиона, амфотерного соединения или соли с кислотой, как Н.В г или СРзСООН. Наиболее важная
соль, которую выделяют, зависит от оснойности продукта, условий реакции обработки и очистки и природы исходного материала (соль или свободное основание). Таким образом, например, в примерах 1-5 из-за рК бензимидазо,лоэого кольца выделенная кислая соль обычно представляет собой трифторацетат, но может быть и смесью амфиона и трифторацетата. В примере 6 из-за рК имидазолинового кольца продукт может быть выделен в форме амфиона, трифторацетата той же соли, что и исходное вещество (гидробромид) или . смеси двух или трёх компонентов, при веденных выше.
Пример 1. Раствор дйфенил-З-ацетоксимётил-7- (бензимидазол-2-ил аминоцёф-З-ем-4-карбоксилата (0,28 г О,5 ммоль) в ТФА (0,8 мл) перемешива ют в те1чение 20 мин при комнатной тем пературе. ТФА вьтаривают под вакуумо создаваемым масляным насосом, осадок растворяют в СН2СЕ2 раствор промывают водой. Органическую фазу высуши вают (((9504) и концентрируют, а затем к ней добавляют смесь равных частей толуола и эфира. Полученный в результате осадок собирают и высушивают до получения 0,05 т 3-ацетоксиметил-7-(бензимидазол-2-ил) аминоцеф-З-ем-4-карбоксикислоты трифторацетата, т. пл. 210-230°С (разл.), имеющего следующий с пектр ЯМР в dbOMSO: 2,04(s,3H); 3,82 (m,.2)i 4.76 ( d, 1Н) i 5,07 (d, ПО , 5,28
(d, IH); 5,84 (d, IH); 6,8-7,7 (m, ЦН) .
Дифенклметил- З-ацетоксиметил- 7-(бензимидазол-2-ил)аминоцеф-3-ем-4-карбоксилат, используемый в качестве исходного материала, получают следующим образом.
К перемешиваемой суспензии 7-амигно-З-ацетоксиметилцеф-З-ем-4-карбокскислоты толуол-р-сульфонат дигидрата (19,2 г, 40 ммоль) в безводном ТГФ (500мл) под азотом в литровую колбу добавляют триэтиламин, высушенный над калием (27,76 мл, 200 ммоль), затем триметилхлорсилан (20,18 мл, 17,36 г, 160 ммоль), поддерживая при этом температуру около с помощь охлаждающей бани. Через 30 мин добавляют дополнительно 10% триэтиламина и 10% триметилхлорсилана и полученную в результате смесь переманивают в течение 2,5 ч. К смеси добавляют Уксусно-муравьиный смешанный ангидри (7,04 г, 80 ммоль), охлажденный в ледяной бане. Дополнительные количесва 10% триметиламина, 10% триметилхлорсилана и 20% уксусно-муравьиного смаианного ангидр ща, а затем еще по 10% каждого из трех реагентов добавляют до полного растворения исходного материала ТСХ. Затем добавляют воду, фильтруют суспензию на стеклянном диске и твердый остаток высушивают до получения 3-ацетоксиметил-7-формиламиноцеф-З-ем-карб.оксикислоты
Привейенное соединение обрабатывают раствором дифенилдиазометана в петролейном эфире. Продукт рекристаллизуют из метанол/эфира (3:7%) до получения дифенипметил-3-ацетоксиметил-7-формиламиноцеф-3-ем-4-кар.боксилата, т. пл. 157-158°.
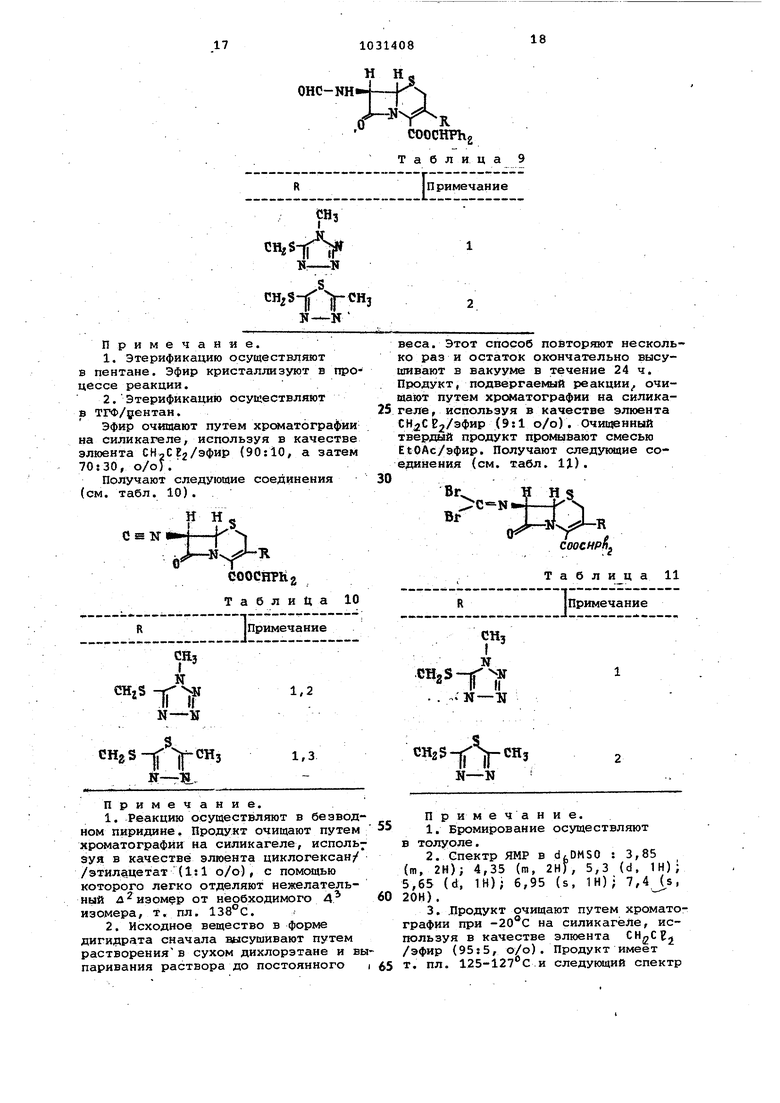
В 250 мл двугорлую колбу, снабженную магнитной моиалкой и капельницей,высушенную в печи, добавляют дифенилметан-З-ацетоксиметил-7-формилс1Миноцеф-3-ем-4-карбоксилат(9,22 г, 20 ммоль), а затем хлорметилен (120 мл) , высушенный над пятиокисью фосфора. Смесь помещают в атмосферу азота и охлаждают до -78с смесью СО /яцетон. Затем-добавляют ангидрид пиридина (3,2 мл, 3,12 г, 40 ммоль), а затем 20%-ного в/о раствора фосгена в толуоле (10,32 мл 20 ммоль). После взаимодействия добавляют воду (100 мл) и отделяют, высушивают над сульфатом магния и упаривают-до постоянного веса органическую фазу. Сырой продукт подвергают хроматографии на силикагеле (100 г), используя в качестве элюента эфир /СН 2,С 2(7 3%) , до получения 6,0 г дифенилметип-З-ацетоксиметип7-изоциано-цеф-3-ем-4-карбоксш1ата, который имеет следующий спектр ЯМР В COCBj: 1,97 (s, ЗН) 3,45 (m, 2Н); 4,75 (d, 1Н) ; 5,07 (d, IH) , 4,72 (d, IH); 5,05 (d, IH); 6,88 (s, H), 7,28 (m, 10H). К раствору ди({)енилметил-3-ацетоксиметил-7-йзоцианоцеф-3-ем-4-карбоксилата (0,080 г, 0,178 ммоль) в хлоряиметилене, охлажденном до -78°С смесью СО /ацетон, добавляют раствор брома (0,1)285 г, 0,178 ммоль) в CDCE Таким образом, получают раствор дифенилметил-З-ацетоксиметил-7-дибромметиленаминоцеф-3-ем-4-карбоксилата, который 1спользуют без последующей очистки.. Продукт имеет следующий спектр ЯМР в CDCE,: 2,02 (s, 3 И); 3,45 (го, 2Н); 4,73 (d, 1Н); 4,97 (d, IHj; 5,07 (d, IH); 5,25 (d, IH); 6,94 (s, IH); 7,32 (m, lOH). Продукт при необходимости очицают путем хроматографии на силикагеле при использоваНИИ в качестве элюента СН.2СЕ. Бромирование осуществляют также в толуоле при -78с, при этом получа ют целевой продукт в меньшем количестве.. Соответствующее дихлорсоединение получают путем хлорирования раствора иэоцианида раствором хлора в четырех хлористом углероде при -78°С. Продук очидают путем хроматографии на сипик геле, жпользуя в качестве элюента при -20 С. Продукт имеет следующий спектр ЯМР в COCta: 1,98 (s, ЗН)} 3,45 (т, 2Н); 4,70 (d, 1Н); 4,9 (d, 1H)i 5,02 (d, IH); 5.37 (d, 1H)j 6,92 (s, IH), 7,3 (m, 10H). К раствору д ифенилметйл-3-aцетоке имет ип-7 -д иброммет ипенс1минрцеф -3 ем-4-карбоксилата (0,608 г, 1 ммоль) в ТГФ, перемешиваемому под азотом при комнатной температуре, добавляют О -фенилендиамин (0,216 г, 2 ммоль) в ТГФ и реакцио затем продолжают в течение 4 ч. Раствор аьпаривают до постоянного веса, а остаток растворяют в СН2СЕ2 содержащем небольшое количество метанола. Этот раствор подвергают хроматографии на силикатгеле (50 г) при -40°С, используя CH-CJEj (МеОН(85:15%) в качестве элюента до получения дифенилметил-3-ацетоксиметил-7-(бенз1 1Кцазол-2-ил) ам иоцеф-3-ем-4-карбоксилата (0,39 г Продукт имеет следующий спектр ЯМР в СОСЕз (s, ЗН); 3,25 (т, 2Н); 4,57 (d, 1H)i 4,97 (d, 1Н) 5,15 (d, IH), 5,90 (d, IH), 6,87 (s, 1H)i 3,2 (m, 10H). Пример 2. Способ, описанный в примере 1,- повторяют , используя соответствующий исходиый получают соединения материал, (см. табл. 1). CHgQCOCH. оJSC-.. ° t а б л и ц а 1 (нумерация кольца Примечание бензимидазола) 5-Метил 1г2,3,4 4-Метил 5,3,6,7 5,6-Диметил 5,8,3,9 4-Амино 1,3,10 4-Ацетиламино 1,3,11 5-Нитро 5,12,3,13 4-КарбЬкси5,3,14 Примечание. 1.Реакшво осуществляют в ТФА/анизол в течение 30 мин. 2.Продукт очицают путем хроматографии на силикагеле, используя в . качеству эгаоента СНлСЕ,/МеОН/НОАс (96:2:2, о/о/о). 3.Продукт отделяют путем растворения в минимальном количестве и осаждения эфирс «. 4. Спектр 5ШР в dcOMSO/CD30D:2,02 (s, ЗН), 2,43 (5,ЗНГ{ 3,40-4,0 {т, 2Н) 4,0-6,0 (Вг, )6,70-7,70 (т, И). 5. Реакцио осуществляют в ТФА/то6.Т. пл. 240(разл.) после рекристалл гаации из CH2Ct2 /МеОН/ эфир, 7.Спектр ЯМР в d OMSO: 2,05 (s, Н), 2,47 (s,3H); 3,48, 3,72 (2d, 2Н), 4,75, 5,05 (2d, 2Н)/ 5;32, 5,96 (2d, 2); 7,05 (m, ЗН). 8. Продукт очищают путем хроматог; графии на сипикагеле (промывают 2н. НС 1 и реактивируют при 120 С inv««coo, используя в качестве злюента /НеОН/Ц)Лс (94:3:3 о/о/о). при . Фракции окисляются CFjCOOH., 9.Спектр ЯМР -в diDMSO/CD300:2,04 (s, ЗН)- 2,35 (s, 6Н), 3,43, 3,73 (2d, 2Н); 4,85, 5,15 (2d, 2H)i 5,30 (d, 1H)i 5,72 (d, 1H) 7,23 (s,2H). 10.Спектр ЯМР в. dt,OMSO:2,12 (s, ЗН); 3,7 (br, 2H); 4,77, 5,13 (2d. 2H)f 5,37 (d, IH), 5,9 (br, IH), 6,60 ( d, TH); 6,73 ( d., IN), 7,05 (t, IH), 6,90-7,80 (m, 3H). 8,35-8,80 (m, IH), 9,97 (br, H). 7,05 (t, IH), 6,90-7,80 (b r, IH 06менный), 10,15 (br, IH, обменный).
5-Метил
2,3 4-Метил
Примечание.
(нумерация кольца
Примечание бензимидазола)
Водород
1 5,6-Диметил
2,3 5-Метокси
4,2,5 5,6-Дихлоро
6,7,8 5.4-Амино
4,9,10
4-Аце тиламино
4,9,11
Примечание. 60 1. т. пл. 185-187 с (разл.).
Спектр ЯМР в О О/СОаОО/ТФА : 2,3 (s. ЗН); 3,24, 3,60 (2d, 2Н) 7,4 (т, 4Н). Другие протоны замаскированы резонансами растворителя. Соответствующую натриевую соль получают
путем обработки суспензии трифторацетатной соли в воде стехиометрическим количеством NaHCOj . Если реакционная смесь гомогенна, ее экстрагируют дважды СНдС , а водную фазу высушивают вымораживанием до получения гигроскопической натриевой соли. Спектр ЯМР в D О : 2,10 (s, ЗН), 3,35, 3,77 (2d, 2Н), 5,35 (d, 1Н), 16,80 (d, 1Н), 7,20-7,65 (т, kH) .
CHgC и осаждения избытком .,
(s. 3H)i 3,43, 3,56 (2d, 2Н); 5,20, 5,65 (2d, 2Н); 7,42 (s, 2Н).
Исходные вещества для использования в описанном способе получают следующим образом.
К суспензии 7-амино-З-метилцефЗ-ем-4-карбоксикислоты (7,76 г, 0,036 моль) в безводном ТГФ при температуре , охлажденной в ледяной бане, добавляют триметилхлорсилан (7,8 г, 9,07 МП, 2-х эквивалентный) и триэтиламин (7,3 г, 10 мл, 2-х эквивалентный) ..Через 10 мин удаляют ледяную баию и оставшуюся смесь выдерживают при комнатной температуре в течение 2ч. К,смеси добавляют 2 эквивалента ангидрида уксусномуравьиной смеси и смесь оставляют на 1,5 ч при комнатной температуре. Добавляют воду (5 мл), фильтруют осажденный триэтиламинхлоргидрат и фильтрат выпаривают на роторном испарителе.
ТГФ (50 мл) добавляют к оставшемуся маслу и раствор этерифицируют дифенилдиазометаном. Продукт очищают путем хроматографии .на силикагеле, используя в качестве элюента
5 СН2С 2/эФир (1;1, о/о), до получе.ния дифенилмётил-7-формиламино-Зметил-цеф-З-ем-4-карбоксилата.
Указанный формамид (1,02 г) растворяют в безводном CH CEjdO мл)
0 и пиридине (0,42 г, 2-х эквивалентном) под азотом при . К этому раствору по каплям добавляют фосген (0,272 г в виде раствора 20%-ного (в/о) в толуоле - 1 эквивалент).
5 Через 20 мин реакционную смесь обрабатывают водой и продукт очищают путем хроматографии на силикагеле, используя в качестве элюента. Таким образом получают
0 дифенилметил-7-изоциано-З-метилцеф-З-ем-4-карбоксилат.
К раствору изоцианида (0,10 г, 0,256 ммоль) в CHjCEjdO мл) под
с азотом при по каплям добавляют бром (0,041 г, 13 /сЕ) в (2 мл). Окончание реакции определяют по устойчивой окраске брома. Раствор выпаривают до постоянного веса и остаток очищают путем хроматог)а
0 фии на силикагеле, используя в качестве элюента ,, Таким образом получают 0,1 г дифенилметил-7-дибромметилен-З-метилцеф-З-ем-4-карбоксилата, устойчивого при температуре .
5 Он имеет следующий спектр ЯМР в CDCBj: 2,1 {s, ЗН), 3,07, 3,4 (2d, 2Н); 5,14, 4;92 (2d. 2Н) 6,9 (s, )Н) , 7,3 (т, ЮН).
0 Соответствукядий дихлоризоцианид получают идентичным способом, используя раствор хлора в СС1. Он имеет следующий спектр ЯМР в 2,1 (г, ЗН), 3,1, 3,35 (2d, 2Н); 4,92,
5 5,32 (2d, 2Н),- 6,9 (s, 1Н); 7,28 (т, 10Н).
Указанный дибромизоцианид получают из изоцианида, используя толуол
л вместо CHjCfj в качестве растворителя. К смеси, подвергаемой реакции, затем добавляют соответствующий ортодиамин в СН2СЕ2 Ри . Затем скюсь, подвергаемую реакции, нагревают до комнатной температуры и перемешивают от 4 до 24 ч. Продукт выделяют путем промывания органического слоя водой и хроматографии остатка, полученного при выпаривании сухого органического слоя (если ТГФ
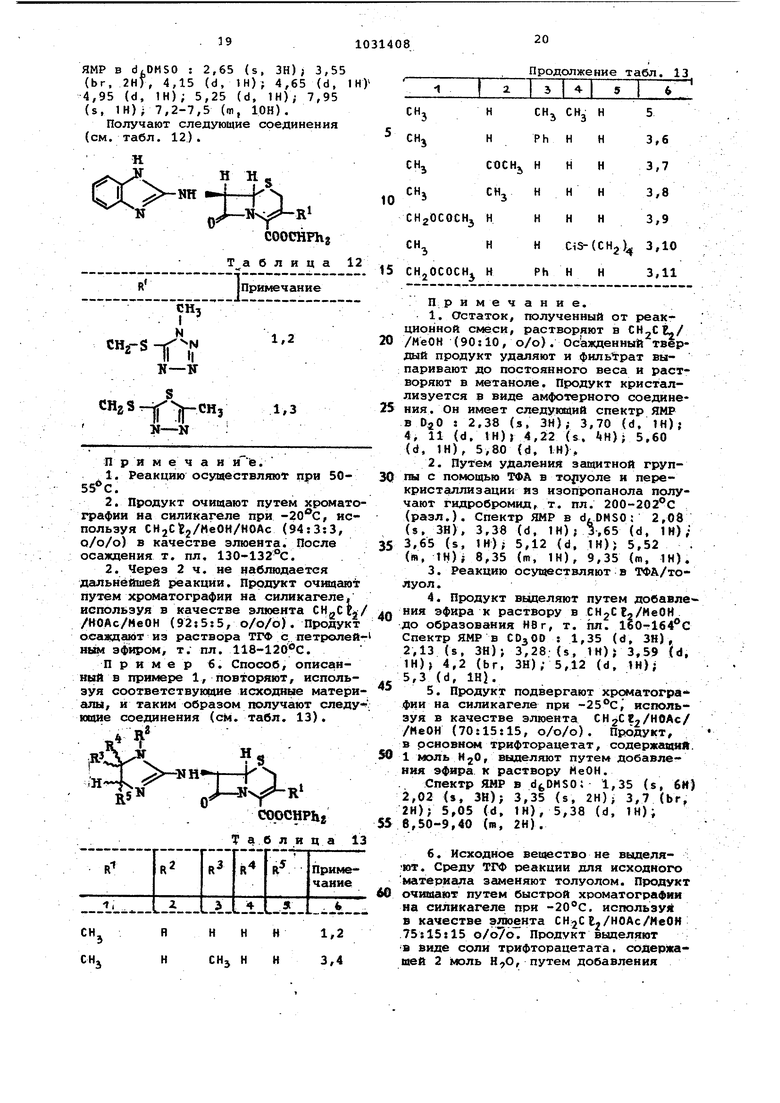
0 используют вместо CHjCEj в качестве растворителя, получают продукт, не содержащий и реакция завершает ся в более короткий срок). Таким образом получают следующие соединения (см. табл. 4). J NyСНзCOOHPh.2 Та. блица 4 Примечание (нумерация кольца бензимидазола) 5 ,6-Dimethyl 5-Methoxy 5 ,6-Dichloro 4-Amino Примечание, 1,Используют два эквивалента о -фенилендиамина. 2,Продукт очищают путем хроматографии на силикагеле, используя в качестве элюента CH CBj/MeOH (95;5, о/о) . 3,Используют 1,5 эквивалента диамина, а через 5 ч - 0,5 эквивалента диамина. 4,Продукт очищают путем хроматографии на силикагеле, используя в качестве элюента CHgCFj/EtOAc/MeOH (58:40s2, о/о/о). 5,Смесь, подвергаемую реакции, нагревают при. 50°С в течение 18 ч. 6,Продукт очищают путем хромато графии на силикагеле, используя сле дующие элюентн; СНтСЕЗ, затем смесь СЙаСгз/МеОН (95;5, о/о); СНгСЕз/НОА (96-85 : 10-15. о/о), 7,Смесь, подвергаемую реакции, нагревают при в течение 4 ч. 8,Продукт очищают путем хромато графии на силикагеле, используя в качестве элюента G НgC Ej/МеОН/НОАс КИСЛОТУ (94:3:3, о/о/о) при темпера туре ОС. Продукт кристаллизуют из толуола, содержащего небольшое коли чество и метанола. Дифенилме- (4-ацетиламинобензимидазол-2ил)-З-метилцеф-З-ем-4-карбоксилат, иепользуемый в качестве исходного материала, получают путем взаимодей соответствующего производного i-аминобенэиМидазола с одним эквива лентом хлорацетила в безводном СН„С 2 в токе азота и вьщеления про дукта осаждением его из раствора в смеси минимального количества CHgCE и метанола избытком эфира. Пример 4. Способ, описанны в пркмере 1, повторяют, используя соответствующие исходные материал;ы. еакцию осуществляют в растворе рифторуксусная кислота/анизол и родукт выделяют путем добавления фира к концентрированному раствору етанола. Получают следующие соединения (см. табл. 5). Н Н ., RNM «.. Г I Cf,COOH , о соон Таблица 5 RТ. пл.,с Примечание 220-225 Примечание. 1,СпектрЯМР в d OMSO:2,08 (s, ЗН); 3,35(d, 1Н);3,68 (d, 1H);5,25(d, 1Н); 5,85{d, 1Н);7,20-8,15 (т.бН), 2. Спектр ЯМР в dfcDMSO : 2,03 (s, ЗН) , 3,32 (s, fH),- 3,30 (d, VH); 3,60 (d, 1H); 5,22 {d, 1H-. 5,80 (d, i 1H); 4,30-6,70 (m, обменный); 7,15 (d, 1H); 7,30 (s, 1H); 77,44 (t, 1H) 7,87 (d, 1H); 8,57 (br, обменный). Исходные вещества для использования в описанном способе получают путем повторения последних двух частей примера 3, осуществляя бромирование в хлорметилене, используя соответствуняций диамин, используя последнюю часть в тетрагидрофуране, Смесь нагревают при 50°С в течение 2-4 ч. Таким образом получают следукадие соединения (см, табл. 6). Н HS , COOCHPhg , Таблиц а IТ. пл,,°С Тпримечание 141-143 .150-154 Примечание. 1. Продукт очищают путем хроматографии на силикагеле, используя в
качестве элюента СН,СЕ«/НОАс/НеОН (97:1,5:1,5, о/о/о)Т
Продукт вццеляют путем добавления петролейного эфира к раствору в ТГФ.
Пример 5. Способ, описанный в примере 4, повторяют, используя соответствующие исходные материалы, и таким образом получают следующие соединения (см. табл. 7).
Н
Н Не
Г кн -4-И
3
И I
174-176 «-Н
Примечание.
(б, ЗН) 3,59 (d, tH)j 3,84 (d, 1Н); 4,23 (d, 1Н); 4,59 (d. 1Н); 5,25 d, 1Н); 5,82 (d,1H); 7,1-7,5 (br, .H).
исходные материалы для использования в описываемом способе получают следующим образом.
К перемешиваемой суспензии в воде (100 мл) и ацетоне (50 мл) 7-аминоЗ-ацетоксиметилцеф-З-ем-4-карвоксикислоты (13,5 г, 0,05 моль) добавляют бикарбоната натрия (9,45 г, 0,113 моль) в воде (50 мл). Если получают гомогенный раствор, то добавляют 2-метил-1,2,4-тиадиаэол-5тиол (10 г, 0,075 моль) и смесь нагревают при 40-50 с под азотом, тогда как рН доводят до 7,6 путем добавления раствора Зн. НСЕ. рН поддерживают 7,6, а реакцию завершают удалением 0,3 мл аликвоты с последующим установлением значения рН до
3 с помощью 1н. НСЕ, фильтрацией осадка, промывкой его ацетоном, а затем/ эфиром, снимают ИК-спектр с целью установления отсутствия абсорбции карбонила. Реакцию згивершают через 10,5 ч i Всю реакцию проводят так же, как описано вьиие до удаления аликвот. Так.им образом получают 7-аминр-З5 Г(2-метил- 1,3,4-тиадиазол-5-ил)тиометил цеф-3-ем-4-карбоксикислоту. Таким же образом, но используя эквивалентное количество 1-метил1Н-тетразол-5-тиол вместо З-метил 1,2,4-тиадиазол-5-тиол, первоначально устанавливая рН на уровне 7,6 бикарбонатом натрия, ос ааествляя реакцию в течение 5 ч,, получают 7-амино-3- С(1-метил-1Н-тетразол-5-ил)тио5 метилДцеф-3-ем-4-карбоксикислоту.
Способы, описанные в 2-5,7 примечаниях примера 1, последовательно повторяют, используя соответствующие исходные вещества, и, таким
0 образом,, получают следующие соединения (см. табл. 8) .
И Н S
о соон
Таблица 8 {Примечание
, NК
Примечание, 1. Продукт выделяют путем добав-, ления небольшого количества воды 55 к смеси, подвергаемой реакции, фильтрования и выпаривания фильтрата. Получают следующие соединения ,:(см. табл. 9) .
OHO-NH«4--4
Сиз
CH,S-|fV
IS:-H
CH S-|fV 3
N-N
Примечание.
Эфир очищают путем хрсматографии на силикагеле, используя в качестве элюента СН2СЕ2/эфир (90; 10, а затем 70:30, о/о).
Получают следующие соединения (см. табл. 10).
Н HS С S К шН-1
-Nx...
COOCHPIig Таблица 10
1
Примечание
,s.
CH2S-| Y-СИЗ
1,3
N-ISLПримечание. 1.Реакцию осуществляют в безводном пиридине. Продукт очищают путем хроматографии на силикагеле, исполь зуя в качестве элюента циклогексан/ /этилацетат (1:1 о/о), с помощью которого легко отделяют нежелательный л изомер от необходимого 4 изомера, т. пл. 138°С. 2.Исходное вещество в форме дигидрата сначала высушивают путем растворенияв сухом дихлорзтане и вы паривания раствора до постоянного
Ь
COOCHPhg
Таблица 9
Примечание
веса. Этот способ повторяют несколько раз и остаток окончательно высушивают в вакууме в течение 24 ч. ПРОДУКТ, подвергаеАФлй реакции очишают путем хроматографии на силика5 геле, используя в качестве элюента СН5СВ2/ЭФИР (9:1 о/о). Очищенный твердаай продукт промывают смесью EtOAc/эФир. Получают следующие соединения (см. табл. 1J.) .
Вг.
9 S
-:
C--N Вг
COOCHP l т а б л а (примечание
СН,
f
c%s-y
. . - N-и
irV
СНгЗ
CHj
N-N Примечание. 1.Бромирование осуществляют в толуоле. 2.Спектр ЯМР в d/DMSO : 3,85 (m, 2Н); 4,35 (га, 2НЬ 5,3 (d, 1Н); 5,65 (d, 1Н); 6,95 (s, 1Н); 7,4 (s, 20Н). 3..Продукт очищают путем хроматографии при на силикагеле, используя в качестве злюента CHgCg /эфир (95:5, о/о). Продукт имеет т. пл. 125-127 с и следующий спектр
ЯМР в d/OMSO : 2,65 (s, ЗН); 3,55 (Ьг. 2НЬ 4,15 (d, 1Н); 4,65 (d, IH) 4,95 (d, 1Н); 5,25 (d, }Н)i 7,95 (s, IH); 7,2-7,5 (ffl, lOH).
Получают следующие соединения (см. табл. 12).
Л
Vv s CH28r-|Y-C 3 13 N--N- ; П р и м е ч а и и fe. 1.Реакцию осуществляю при 5 . 2.Продукт очищают путем хром графии на силикагёле при , пользуя CHjCtj/MeOH/HOAe (94:3: О/о/о) в качестве элюента. После осаждения т. пл. 130-132 С. 2. Через 2 ч. ие наблюдается дальнейшей реакции. Продукт оч путем хроматографии на силикагёл используя в качестве элюента CHg /НОА с/М е О Н (92:5:5, о/о/о). Прод осаясдают из раствора ТГФ с,, петро ным эфиром, т. пл. 118-120 С. Пример 6. Способ, описа м в щ)имере 1, повторяют, испо зуя соответствуюрше исходные мат алы, и таким образом получают сл ющие соединения (см. табл. 13). ин уз;нН /R COOCHPhz Таблица ИНН CHj Н Н
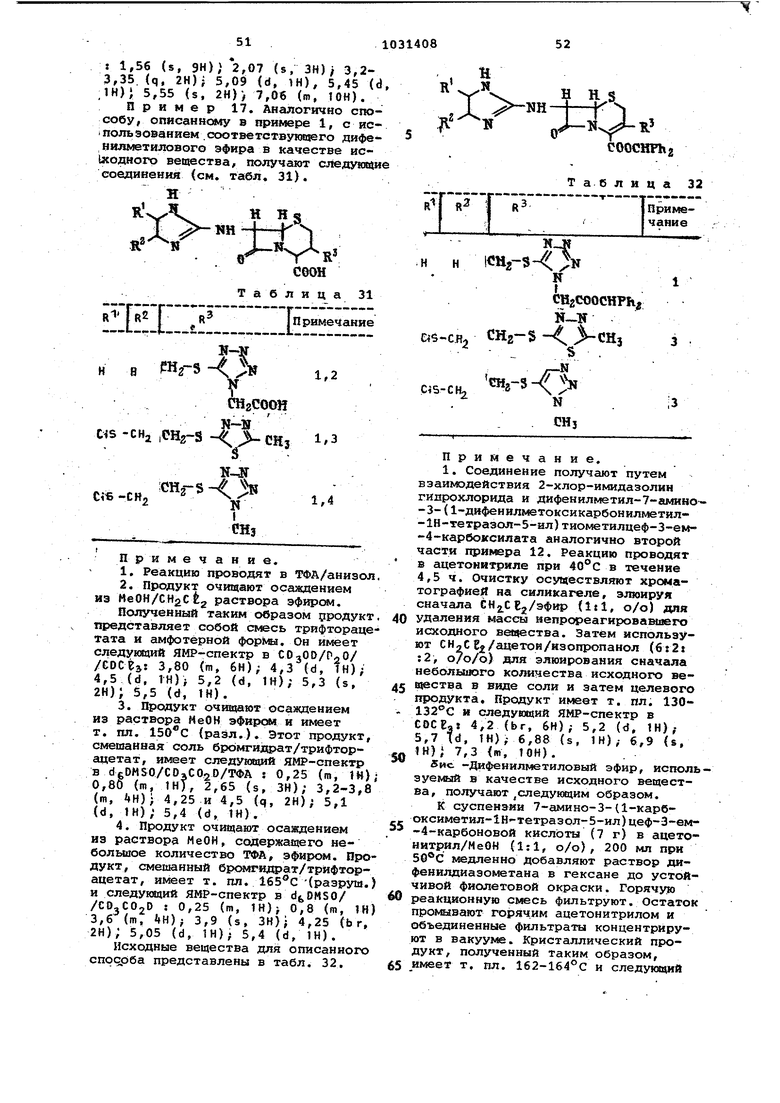
Продолжение табл. 13 Примечание. 1.Остаток, полученный от реакциомной смеси, растворяют в ,/ /МеОН (90:10, о/о). Осужденный твердый продукт удаляют и фильтрат выпаривают ло постоянного веса и растворяют в метаноле. Продукт кристаллизуется в виде амфотерного соединения. Он имеет следукпий спектр ЯМР в DaO : 2,38 (s, ЗН),- 3,70 (d, 1«)} 4, 11 (d, VH)j 4,22 (s. JjH); 5,60 (d, IH), 5,80 (d, IH), 2.Путем удалений защитной группы с помощью ТФА в то|1уоле и перекристаллизации ИЗ изопропанола получают гидробромид, т. пл. 200-202°С (разл.). Спектр ЯМР в d/DMSO: 2,08 (s. ЗН), 3,38 (d. 1H)j 3,65 (d, IH); 3,65 (s, IH); 5,12 (d, 1H)J 5,52 (m, TH); 8,35 (m, IH), 9,35 (m, IH). 3.Реакцию осуществляют в ТФА/толуол. 4.Продукт выделяют путем добавления эфира к раствору в СН2С до образования НВг, т. пл. 160-164°С Спектр ЯМР в СОзОО : 1,35 (d, ЗН), 2,13 (s, ЗН); 3,28 (s, 1Н)} 3,59 {d, 1Н), 4,2 (Ьг. ЗН),- 5,12 (d, 1Н); 5,3 (d, 1Н). 5.Продукт подвергают xpwaTorpa фии на силикагёле при используя в качестве элюента СН2СЕ2/НОАс/ /МеОН (70:15:15, о/о/о). Продукт, в рсновном трифторацетат, содержащий. 1 моль HjO, выделяют путем добавления эфира к раствору ИеОН. Спектр ЯМР в dt,DMSO; 1,35 (s, бН) 2,02 (s, ЗН); 3,35 (s, 2Н), 3,7 (br, 2Н); 5,05 (d, 1Н), 5,38 (d, IH); 8,50-9,40 (га, 2Н). 6.Исходное вещество не выделяют. Среду ТГФ реакции для исходного материала заменяют толуолом. Продукт очищают путем быстрой хроматографии на силикагёле при -20°С. используя в качестве элюеята CHjC Ej/HOAc/HeOH 75:15:15 . Продукт вьщеляют в виде соли трифторацетата. содержащей 2 моль Н,О, путем добавления эфира к раствору МеОН/ТГФ, т. пл. 175-177 С (разл.). Спектр ЯМР в d(,DMSO: 2,07 (s, З 3,30 (m, 2Н),- 3,50 (m, ГН) ; 4,15 ( 1Н) 5,15 (d, 1Н), 5,21 (t, IH); 5,51 (d, IH); 7,40 (s, 5H), 9,0 (b 1H обменный). 7.Продукт выделяют путем добав ления эфира к раствору в, ТГФ, в ос новном трифторацетат, т. пл, 110-115С. Спектр ЯМР в d/OMSO : 2,03 (s, ЗН), 2,26 (s, ЗН), 3,30 (d, IH); 3,62 (s, IH); 3,73 (m, 2H); 4,13 (m, 2H), 5,16 (d, IH), 5,51 (d, IH 7,22 (m, IH). 8.Продукт выделяют путем добавления эфира к раствору СН2СЕ,/НеОН в основном НВг соли. Т. пл, 153-15ь ЯМР в : 2,06 (s, ЗН); 2,97 Cs, ЗН); 3,49 (s, 2Н); 3,64 (s, 5,1 (d, 1Н), 5,44 (d, IH). 9.Продукт, соль трифторацетата выделяют путем добавления эфира к раствору CHjClj/MeOH. ЯМР в еОз007СРзСООО : 2,10 (s, ЗН) 3,44 (d, IH); 3,81 (d, 1H)v 3,81 (s, ); 4,86 (d, 1H)) 5,19 (d, IH); 5,20 (d, 1H) 5,51 (d, IH 10.Продукт выделяют путем яобав ления по каплям эфира к раствору CH CPj/MeOH, в основном в виде НВг соли, т. пл. 167-170 С. Спектр ЯМР в dfeOMSO : 1,52 (br, 8Н), 2,08 (s, ЗН) . 3,8 (d. 1Н); 3.65 (d. IH); 3.92 (s. 2Н), 5,12 (d. IH) ; 5.45 (d, IH). 11; Остаток, полученный в резуль тате реакции, распределяют между и водой, а водный слой выпаривают до получения такого продукта как соль трифторацетата. Спектр ЯМР в dgOMSO 2,0 (s, ЗН 4,75-,(d,. 1Н); 5,0 (d, IH), 5,1 (d, IH); 5,55 (m, IH) , 5,2 (m. 1H). Некоторые резонансы замаскировавы широким обменным резонансом. Исходные материалы для использования в указанном способе получают путем повторения последней части примера 1, используя соответствующи исходные материалы. Реакционную смесь первоначально обрабатывают путем добавлений небол шого избытка НВг, или предпочтитель но ТФА для полного завершения нейтрализации избытка диамина. Таким образом получают следующие соединения (см. табл. 14). R il Н Н -tS , 1-ж COOCHTll2i Таблица 14 CHjCHj н Phн н COCHj н нн . н СН, CHjOCOCH н нн и нCi5(CH2)4 1.8 CHjOCOCHj н Phн н 9 Примечание. 1.Продукт очищают путем хроматографии на силикагеле при -20с, используя ;В качестве элюента /МеОН (95:5, о/о), 2.Фочка плавления 156-158 С. 3.Продукт очищают путем хроматографии на силикагеле при -30°С, используя в качестве элюента /МеОН (92:8, о/о), 4.Продукт выделяют путем добавления эфира к раствору CHgCE,,, т, пл. 138-140С. 5.Исходный материал, полученный путем бромирования изоцианида, не вьщеляют, СН2СЕ„ растворитель, исполь зуемый для бромирования, заменяют ТГФ. 6.Исходный материал в форме свободного основания получают путем ацйлирования соответствующего NH-coединения уксусный ангидрид/триэтиламин в ТГФ, 7.Т, пл. при рекристаллизации из СН2Се2 143-145С. 8.Т. пл, при рекристаллизации из CHjCgg 142-145С. 9.4 моль ТФА, а не НВг, добавляют к смеси, подвергаемой реакции, р,о обработки. Продукт очищают путем хроматографии на силикагеле при -45®С, используя CHjCE /MeOH 97:3 о/о в качестве элюента. Продукт, т, пл. 107-112с, вьщеляют путем осаждения эфиром. П р и м е.р 7. Раствор t-бутил3-метил-7-(1-метилбензимидазол-2-илУ аминоцеф-З-ем-4-карбоксилата в двух объемах ТФА и одном объеме анизола оставляют на 1,5 ч при комнатной температуре, Растворитель удаляют выпариванием и продукт, З-метил-7. (1-метилбензимидазол-2-ил)аминоцеф-З-ем-4-карбоксикислоту, очищают путем рекристаллизации из изопропанола, т. пл. 181-182 С (разл.). Спектр ЯМР в : 2,05 (s, ЗН), 3,5 (m, 2Н); 3,7 (s, ЗН); 5,2 (d, 1Н); 5,7 (d, IH); 7,2-7,7 (m, 48).
Трет -Бутил-3-метил-7-(1-метилбензимидаэол-2-ил)-аминоцеф-З-ем-4карбоксилат, используемый в качестве исходного материала, получают следующим образом.
Раствор трет -бутил-7-амино-З-метилцеф-З-ем-4-карбоксилат 0,5 .4мoль в метаноле (0,5 мп) обрабатывают 1-метил-З-метоксибензимидазол-иодом (О,5 ммоль) и смесь, подвергаемую реакции, сбивают в течение 48 ч при комнатной температуре. Продукт очищают путем хроматографии на силикагеле, используя в качестве элюёнта СН.,се,/МеОН (97:3, о/о) с пocJrJeдyющей рекристаллизацией из изопропанол до получения З-меткл-7-(Х-метилбеизг имидазол-2-ил)аминоцеф-3-ем-4-карбоксилата,т.пл. ИЗ-Иб С, имеющего следункций спектр ЯМР в CDCBi: 1г (s. 9Н, 2,1 (s, ЗН); 3,17 td, 1Н); 3,5 (d, 1Н); 3,55 (s, ЗН); 5,1 (d, 1Н); 6,05 (d, 1Н); 6,9-7,6 (т, Н).
З-Метил-7-(1-метил 5ензимидазол2-ил)а миноцеф-3-ем-4-карбоксикислоту можно получить также путем прямой реакции при комнатной температуре между 7-амино-3-метилцеф-3-ем-4-карбоксикислотой и 1-метил-З-метоксибензимидазолиодом в водном буфере или в воде, содержащей 1 эквивалент NaHCOg,
П р и м е р 9. Способ, описан ный в примере 7, повторяют, используя соответствующее 3-ацетоксиметияпроизводное, и таким образом получают З-ацетоксиметил-7-(1-метилбензимидазол -2-ил )аминоцеф-3-ем-4-карб;рксикислоту, т.пл. (разл) , после осаждения из раствора /ИеОН эфирсял.
Продукт имеет следующий спектр ЯМР в : 2,0 (s, ЗН)j 3,58 (s, +m, 3H+2H)J 4,67 (d, tH) 4,95 (d, ГН) 5,2 (d, IH); 5,76 (d, IH), 6,95-7,42 (m, 4H).
Исходный материал получают путем повторения второй части примера 7, но используя при этом трет -бутил3-ацеток симетил-7-(1-метилбензимидазол-2-ил)аминоцеф-3-ем-4-карбоксилат вместо соответствующего производного 3-метила, 1-метил-З-метоксибенз:имидазол - метансульфонат вместо соответствующего иода, осуществляя реакцию в течение 3 дней. Продукт очищают путем хроматографии на силикагеле, используя в качестве элюёнта CHaCBg/MeOH/HOAc (98:1;1, о/о/о), до получения трет -бутил-3-ацетоксиметил-7- (1-метилбензимидаз6л-2-ил) .аминоцеф-З-ем-4-карбоксилата, т. пл, 98-103®С, И1«2ющего следукмаий спектр
ЯМР в d.DMSO : 1,5 (s, 9Н); 2,0 (s, ЗН); 3,38 (d, IH),- (d, IN); 3,53 (s, 3H); 4,56 (d, IH); 4,8 (d, IH), 5,17 (d, IH); 5,8 (m, IH); 6,75
7,26 (m, ).
Пример 9. Способ, описанный в примере 1, повторяют, используя соответствующие исходные материалы, и таким образом получают следукндие соединения (см. табл. 15) .
i
-R
U Н
Y
соон
т аб .л и ц а 15
R j R3 Примечание
СН. Н ОН Н1,2,3,4
Н СМ CN1,5,6,7
5
CHi
ОН CHj Н1,8,9,10
СМзОСОСНз Н ОН Н11,12,13
Примечание.
Н); 3,60 (d,1H), 3,60 (d, 2Н)} 4,95 (d, IH), 5,55 (d-d, IH), 6,40 (t, IH), 6,95-7,30 (m. 2H) .
. 6. Продукт выделяют выпариванием реакционной смеси и рекристаллизацией остатка из эфира.
1 Н) .
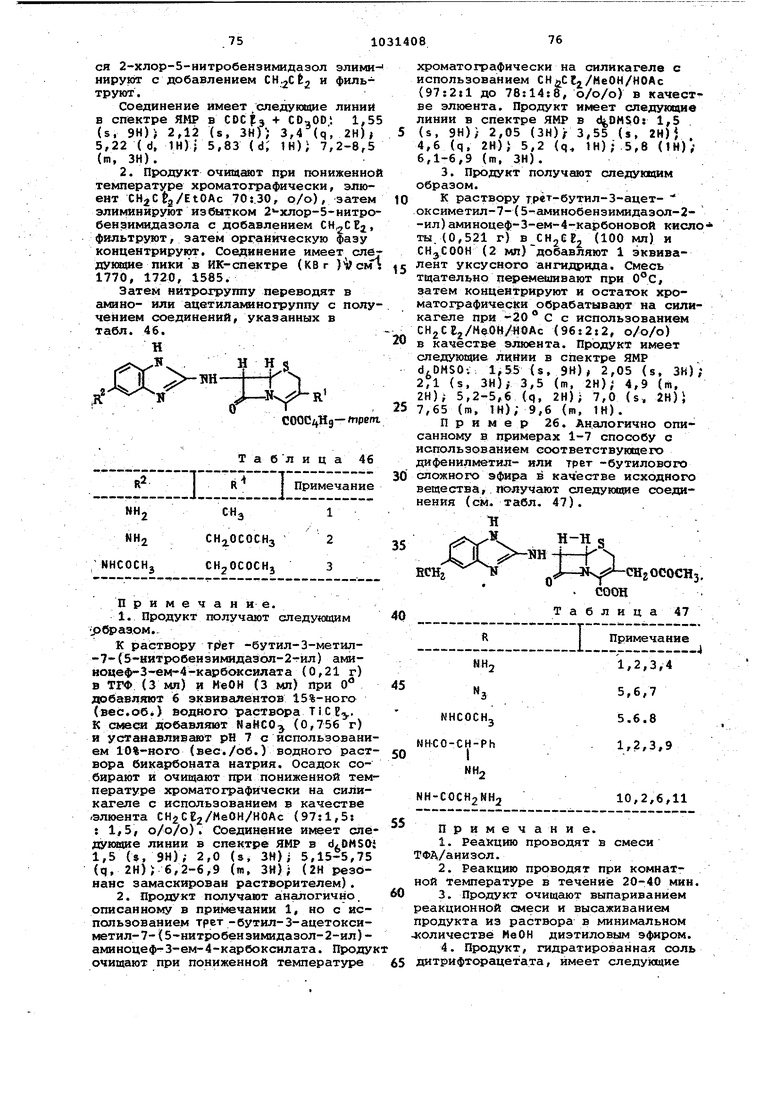
теютературе ниже -20(1, используя в качестве элюёнта CH2Ct2/MeOH/HOAc 88:8:4, о/о/о), .а затем осаждают из раствора СН2Сб2/МеОН с диизопропил эфиром. 10.Продукт имеет т. пл. (разл.) и следующий спектр ЯМР в d,DMSO.: 2,04 (s, 6Н) ; 3,44 (s, 2Н 5.04(d, 1Н); 5,48 (d, 1Н); 6,42 (s, 1Н). 11.Реакцию осуществляют в ТФА/ /толуол при комнатной температуре в течение 20 мин. 12.Продукт выделяют выпаривани ем реакционной смеси и рекристалли зацией осадка из изопропанола. 13.Продукт имеет т. пл. 220 С (разл.) и следующий спектр ЯМР в d DMSO: 2,0 (s, ЗН) , 4,65 (d, H) 5,0 (d, IH); 5,05 (d, 1Н), 5,65 (d JH); 6,35 (t, IH); 7,05 (d, IH); 6,95 (br, s, IH). 14.Реакцию осуществляют в ТФА при 0°С в течение 2ч. 15.Продукт, такой как соль ТФА выделяют тритурированием остатка из реакции с эфиром. 16.Продукт имеет следующий спе ЯМР в dbDMSO; 2,075 (s, ЗН) 3,48 (q, 2Н) ; 5,13 ( d, IH),- 5,5 (q. IH) 7.05(s, 2H); 9,45 (d, 1H). 17.Реакцию проводят в ТФА/анизол (0,85:2,8 о/о) сначала при температуре Ос, затем при комнатной температуре в течение 0,5 ч. 18.Продукт выделяют выпариванием реакционной смеси при и оса дением из раствора СН2СЕ92/МеОН с эфир/гексан (50:50, о/о). 19.Продукт имеет следующий спек ЯМР в dgDMSO : 2,08 (s, ЗН){ 2,10 (s, ЗН) ; 3,44 (d-d, 2Н); 6,10 (d, 6,48 (d, IH), 6,55 (br, s, IH) . Исходные материалы и промежуточные для использования в указанном способе получают путем повторения последней части примера 1 или после ней части примера 3, используя соот ветсхвуюЕчий исходный материал вместо ортодиамина. Таким образом получают следующие соединения (см. табл. 16). Т Н S « /-«txi. I ИJ-rl s;:; -R СГТ COOCHFtig Таблица 16 И ОН Н Н CN CN Продолжение табл. 16 Примечание. 1. Способ осуществляют путем добавления раствора 1 эквивалента 7-дибромметиленамино производного в ТГФ к раствору 2-х эквивалентов глицинамидо в ТФА, содержащему минимальное количество МеОН, до получения раствора при -78°С. Температуру повышают до комнатной и продукт очищают путем хроматографии на силикагеле, используя в качестве элюента EtOAc/eH,CE2 (20:80, о/о). Продукт рекристаллизуют из изопропанрл/СН2СЕ т. пл. 192-194с. 2. Реакцию осуществляют по примечанию 1, используя 2 эквивалента 1,2-диамино-1,2-дицианэтилена. Продукт рекристаллизуют из безводного , т. пл. 180-185°С. 3. Реакцию осуществляют путем добавления раствора 1-амино-2-оксиминопропана (528 мг) в ТГФ, содержащего минимальное количество МеОН, к раствору производного 7-дибромметиленамино (1,1 г) в безводном ТГФ (30 мл) при . Температуру реакции повышают до в течение 2 ч, затем добавляют ТФА (500 л), а растворитель удаляют выпариванием. Остаток очищают путем хроматографии на силикагеле при , используя в качестве элюента ./ (96:4, о/о), и окончательно продукт осаждают из раствора СН2СЕ путем добавления диизопропил эфира, т.пл. 140-145 С (разл.). 4.Продукт очищают путем хроматографии на силикагеле, используя в качестве элюента EtOAc/CH CEj(20:80, о/о) при температуре ниже -20°С. Продукт рекристаллизуют из изопропанола, т. пл. 176-179 С. Исходный материал формулы, првдеденной в табл. 16, где К - .Н R - СН, а Н, получают из соответствующего соединения, где ОН, путем реакции в метаноле с 1,7 эквивалента трихлортитана при 40-45°С в течение 30 мин с последующим добавлением дифенилдиазометана к переэтерифицированной любой свободной кислоте. Исходное вещество, соответствующее формуле, поиведенной в табл.16., где R-Cf а R, R, , получают взаимодействием 2-фторимидазола с трет -бутил-7-амино-З-метилцеф-Зем-4-карбоксилат толуол-р-сульфонатом в ацетонитриле при 70°С в течение 4 ч. Продукт очищают препаратив ной тех, используя систему раствори телей еН-еВ-ХМеОН (9:1, о/о). П р и ме р 10. Повторяют способ , описанный в примере 1 или в примере 7, используя соответствующий дифенилметиловый или грет -бу тиловый эфир в качестве исходного вещества, и таким образом получают следующие соединений (см. табл.17) ш Н-f N -HxjfJ CHzOCOC ® соон 8.Продукт имеет т. пл. (разя.) и следующий ЯМР-спектр : 2,05 (s, ЗН); 3,52 (d, 1И); 3,80 (d, 1Н),- 4,87 (d, 1И) ) 5,16 (d, 1Н)} 5,28 (d, 1Н), 5,74 (d, IH); 6,6-7,3 (m, ЗН). 9.Реакцию проводят в ТФА/анизол (5il,6 о/о) при обычной температуре в течение 20 мин в атмосфере азота. 10.Продукт выделяют выпариванием реакционной смеси до сухости с последующим осаждением из раствора НеОН эфиром. 11.Щюдукт имеет следующий i спектр ЯМР в d6DMSO:2,01 (s, ЗН)/ 3,55 (я, ЗН) ; 4,04 («я, 2Н) ,- 4,68 (s, К) 5,01 (s, 1Н); 5,22 (8, 1Н) 5,95 (s ТН); 7-7,4 {т, ЗН). исходные вещества, используемою в опкса1| ом способе, получают повторением дослед не и части примера 1 с применением соответствующего диамина вместо 6-фенилен диамина. Таким образом получают следующие соединёнхш (см. табл. 18).
9,10., 11
S-CHjNHj
Н HS
«H-f-rV
CHjOOOCKj иIIt
COOR
т a б л и ц a 18 Примечание. . 1. Реакцию проводят в ТФА/анизоя (1,5:1, о/о) при обычной температуре в течение 3,5 ч. 2. Продукт выдел51ют выпариванием реакционной смеси до сухости с последующим осаждением из раствора CHgGB2 эфире. 3.Продукт имеет т. пл. (разл..) и следующий ЯМР-спектр в dtDMSO:. 2,05 (s, ЭН) 3,42 (, 1 3,68 (d, 1Н)- 4,70 (d, tH),- 5,05 (d, ТН)Г5,18 (d, IH); 5,78 (d, IH) 6,4-7,1 (m, ItH). 4.Реакцию проводят в ТФА/анизол (1:1, о/о) при обычной температуре в течение 30 мин. 5.Продукт имеет т. пл. 160°С и следующий ЯЕМР-Спектр в CD.jOp, содержащем две капли d iONSO; 2,00 (s, ЗН) 3,70 (s, 387; 3,5 (d, IN); 3,75 (d, IH); 4,8 (m, 2H); 5,15 (d, IH),- 5,67 (d, IH), 5,31 (d, 2H) 6,66-7,66 (m, BH). 6.Реакцию проводят в ТФА/анизол (1:2, о/о) при обыпной тецтературе в течение 35 мин. 7.Продукт выделяют выпариванием реакционной смеси до сухости и до- бавлением к остатку ., содержащего небольшое количество МеОН, с кристаллизацией продукта. Примечание П р и м е ч а н- и е. 1.Прощгкт очищают хроматографией на сиянкагеле с использованием €Н2СЕ2/МеОН/НвАс (96:2:2, о/о/о) в качестве элюента. Продукт гюрекристаллизовывают из СНчСЕ т. пл. 160°С (разруш.). 2.Реакция) проводят в атосфере ооргона 1ФИ 45- С. 3.Нрояукт очищают хроматографией на сйликагеле при с исяользование СН СЕ2/зФир/НеОН (6-5: :35:2, о/о/о)в качестве элюеита с последующей хроматографией на силвкагеле при -40с с использованием etOAc/CHjCEj/MeOH (25:70:5, о/о/о) в качестве элюента. Этот продукт используют без дальнейшей .очистки. 4.Время реакции 18 ч. 5.Продукт очищают хроматографией на силикагеле при -40°под давлением 0,3 бар с использованием CHjCf / eOH/HOAc (98:1,3:0,67, о/о/о в качестве элюента. Исходное вещество, соответствующее формуле в начале табл. 18, где R - водород, R - 4-метоксибензил ий- трет -бутил, получают взаимодействием эквимолярных количеств трет -бутил-7-амино-З-ацетоксиметил цеф-3-ем-4-карбоксилата и l-MeTojfCH -(4-метоксибензил)имидаэолий иодида в безводном метаноле при комнатной температуре в течение 36 ч. Реакционную смесь выпаривают до сухости и продукт очищают хроматографией на силикагеле с использованием СН2СЕ2/эфир/НеОН (49,5:49,5:1, о/о/б в качестве элюента. грет -Бутил З-ацетоксиметил-7-дибромметиленаминоцеф-3-ем-4-карбрк си лат j используемый в качестве промежуточного продукта, получают следующим образом. К раствору трет -бутИл-7-амино-3-ацетоксиметилцеф-3-ем-4-карбоксилата {3,26 г) в еН;,СЕ, (50 мл) при добавляют смесь уксусного и муравьиного ангидрида (0,88 г).Реакционную смесь перемешивают в теченИе 10 мин .и затем выпаривают до сухости. Остаток растворяют в эфир/ /С},(2Л, о/о) и объем уменьшают без нагревания, при охлаждении раствора кристаллизуется 7-формиламиносоединение ,т. пл. 110-115 С. К раствору.этого продукта (3,65 мг) в СНдСВ (10 мл) добавляют пиридин (790 мг), а затем после охлаждения до -78С под азотом 20%-ный (вес./об раствор фосгена в толуоле (555мкл) Добавляют лед (5 г) и органическую фазу промывают 4 раза водой, сушат и выпаривают до сухости. Продукт, 7-йзоцианид, очищают хроматографией на силикагеле, используя /эфир в качестве элюента К раствору этого изоцианида (169 мг) в толуоле при -78С по каплям добавляют брсян (26 мкл) в СН2СЕ2. После исчезновения исходного BSaiecTBa (ТСХ) реак-цнонную смесь выпаривают до сухости без нагревания с получением трбт-бути л-З-ацетоксиметил-7-дибромметилен-аминоцеф-З-ем-4-карбоксилата, который используют без дальнейшей очистки., . , - - , 1,2-Диамино-4-( трет -бутоксикарбонйла шнс летил) -бензол, используелвдвй в качестве промежуточного продукта, получают следующим образом. Один эквивалент диборана в ТГФ добапляют к раствору 1-амино-2-нитро-4-циаН9бензола (500 мг) в безводйом ТГФ (20 мл) в атмосфере азота при . Через 2 избыток диборана разрушают, добавлением-Vie ОН и реакционную смесь выпаривает до сухости. Остаток растворяютЧ метаноле и добавляют не 2 в мэтанрле. Раств ритель выпаривают, остаток подШелачиваюТ| после чего продукт экстрагируют эфиром -(3 раза) . Объединенные экстракты cvmaT и выпаривают с получением чистого 1-амино-2-нитоо-4 -аминометилбензола. К оаствооу этого хдаамина (350 мг) в сухом диоксане (10 мл) добавляют (258 мг) Вос-0«( PMHch Cliem cae СО ) . Реакционную смесь перемешивают при кокшатной температуре в течение 18 ч концентрируют и dciaToK очищают хроматографией на силикагеле, исполь-зуя СН СБд/эфир в качестве элюента. Таким образом получают 1-амино-2-нитро-4-( -бутоксикарбониламинометид) бензол. Этот продукт гидрируют при атмосферном давлении в ТГФ/ /этанол-растворе, используя (10% вес./вес.) палладированный уголь,с получением 1,2-диа1 шно-4-(трвт бyтoкcикapбoнилa Q нoмeтия)бензола, который используют без дальнейшей очистки. П.р и м е р 11. Аналогично способу , описанному в примере 1 или прим е 7, с испоЯ1ьзованием соответстBywta/SFo дшфеиилметилового или трет бутилового эфира в качестве исхопно,; го вещества, получают следукяцие соединения (см. табл. 19). Н - н KV--H 0т Таблица 19 PhOCHgН 1,2,3 , И 4,5,6 Н 7,2,8 {СНз)2Н-/ Н 9,2,10 СНоОСОСН Н 11,2,12 Н 16,2,17 Н 16-, 2,18 19,20,21 Продолжение табл. 1 Примечание: 1.Реакцию проводят в ТФА/анизол. 2.Продукт очищают осаждением из CH2Ct2/MeOH-paeTBOpa эфиром. 3.Продукт имеет т. пл. 137-14 С (оазл.) и следующий ЯМР-спектр в dfeDMSO S 2,02 (s, ЗН); 3,2-4,5 (m, ЗН); 5,1 (2d, 1Н,- 4,45 (m, 1Н); 6,8-7,5 (m, 5Н). 4.Реакцию проводят в ТФА, используя то же исходное В аество, кЬторое.применяется JVIH получения первого соединения в таблице. При этих условиях .дифенилилетильвый ради кал мигрирует, присоединяясь к бензольному кольцу. 5.Продукт очищают хроматографией на силивагеле при , используя CHjCei/MeOH/HOAc , о/о/ в качестве элюента. 6.Продукт имеет т. пл. 172-18в (разл.) и следующий ЯМР-спектр в dgOHSO,:- -1,95 (s, 3H)j 2,7-4,5 (т, 7H)i 5,0 (m, IH); 5,5 (m, 1H); 7,1 (m/lliH). 7.Реакцию проводят в толуол/ТФА (20:3, о/о) в течение 30 мин при комнатной температуре,. 8.Продукт имеет т. пл. 1бО-1б4 и следующий ЯМР-спектр в .1,35 (d, ЗН); 2,14 (s, ЗН); 3,28 (d, IH); 3,59 (d, 1Н),- 4,2 (Br, ЭК) 5,12 (d, 2Н); 5,3 (d, 1HJ. 9.Реакцию проводят в ТФА/аниэоя (10:1, о/о) в течение 30 мин при комнатной температуре. 10.Продукт имеет следующий ЯМРспектр в t 2,0 (s., ЗН) 2,9 (s, 6Н) 3,0-3,8 (b г, ек.с.КаП«5а Ь6 ) 4,6-5,1 (br, ЗН)} 5,2 (s, IH); 5,45,6 (q, IH), 6,7 (d, 2H) : 7,2 (d, 2H). 11.Реакцию проводят в ТФА/анизо (3:1, о/о) при кo в{aтнoй температуре в течение 30 мин. 12.Продукт, соль ТФА, имеет сле дующий ЯМР-спектр в dtDMSO/ТФА : 2, (s, ЗН),- 3,45 (q, 2Н), 3,6-4,0 (т, 2Н), 4,55 (т, 1Н), 5,05 (d 1H)f 5,4 (dd, IH); 9,55 (d, IH). 13.Реакцию проводят в ТФА/анизол (20:3, о/о) при комнатной температуре в течение 30 мин. 14.Продукт выделяют выпариванием реакционной смеси и оса кдением из раствора МеОН эфиром. 15.Продукт имеет т. пл. и следукяаий ЯМР-спектр в d OMSOi : 2,05 (s, ЗН); 2,85 (s, 6Н), 3,253.6(m, ЗН); 4,05 (t, IH); 4,95 (s, IH); 5,12 (d, IH); 5,14-5,55 (q, IH) 6.7(d, 2H); 7,2 (d, 2H),8,4-8,8 ,(m, IH); 9,2 (m, IH); 9,75 (d, IH). 16.Реакцию проводят в ТФА/анизол (20:7 о/о) при комнатной температуре в течение 30 мин. 17.Продукт имеет следующий ЯМРспектр в d DMSO: 2,0 (s, 3,5 (dd, 2Н); 3,6 (s, k)) 4,85 (dd, 2Н),- 5,1 (d, IH); 5,55 (d, IH), 9,9 (ra, IH). 18.Продукт имеет следующий ЯМРспектр в d(,DMSb;: 2,07 (s, ЗН) ; 3,55 (dd, 2Н); 3,65 (s. ); 5,1 (d, tH); 5,45 (d, IH); 9,7 (m, IH). 19.Реакцию проводят в ТФА/анизол 6:1 о/о лри комнатной температуре в течение 30 мин. 20.Продукт выделяют нз /МеОН раствора EtOAc эфиром. 21.Продукт имеет следукяций ЯМРспектр в d OMSOi 2,35 (s, 6Н); 3,65 (m, бЯ)) 4,3 (dd, 2Н); 5,1 (d, 1Н), 5,45 (d, 1H)J 6,95 (s, IH). 22.Продукт имеет следующий ЯМРспектр в dbOHSOi 2,65 (s, 3H)i 3,65 (s, 4,75 (dd, 2H)J 4,4 (dd, 2H); 5,1 (d, IH), 5,5 (dd, IH),9,5 (d, tH). 23.Реакцию проводят в ТФА/толуол (3,6-2,3, о/о) при комнатной температуре в течение 40 мин. 24.Продукт выделяют ш:тариванием реакционной смеси и осаждением из раствора.эфиром. 25.Продукт имеет т. пл. 120-150 с (раэл.) и следующий ЯМР-спектр в СОзСО,0: 0,3 ,(т, 1Н); 0,9 (т, 1Н) 2.08Is, ЗН); 3,1-3,9 (т, 4Н) 5,1 d, 1Н); 5,34 (d. 1Н). 26.ИК-спектр. Продукт (KBг диск) имеет оюдующие полосы поглощения, , 1775 (СО-НН); 1730 (СООН), 1650 (гуанидиний). Исходные вещества, используемые в описание способе, получают повторением последней части примера 1 или примера 3 с применением соответствующего диамина. Таким образом получают следующие соединения (ем. табл. 20). соон
Т. а б л и ц а 20
(СН.)
СН,
3/2
Н CHPhj 5
CHjOCOCHj cis-(CHj) Примечание. 1.Продукт очищают хроматографией на силикагеле при -1б°С, используя CHj,CB2/MeOH 95:5, о/о) в качестве элюента. Продукт имеет т. пл. 111120°С (разлож.). 2.Продукт очищают хроматографией на силикагеле при -Э0 используя CHijiC Ej/MeOH (90;8, о/о) в качестве элюента. Продукт затем осаждают из раствора С Нп С ,, эфир ом, т. пл. 138-140®С. 3.Продукт очищают хроматографией на силикагеле, используя СН2СР,/ /МеОН (9:1, о/о) в качестве элюента. 4.Продукт очищают двойной хроматографией на силийагеле при -40°С, используя СН СЕ2/МеОН (92:8 о/о) в качестве элюента. 5.Продукт очищают хроматографией на силикагеле при , исполь -эуя CHjCtj/MeOH (9:1, о/о) в качестве элюента. 6.Продукт имеет т. пл. 120155 с (разл.). 7.Реакционную смесь нейтрализуют ТФА. Продукт очищают хроматографией на силикагеле при , используя в качестве элюента CHnCU/ /МеОН (95:5, о/о). ЯМР-спектр в COCCj: 1,1 (Ьг, 8Н); 2,0 (s, ЗН) ) 3,4.5 (s, 2Н); 3,8 (Ьг, 2Н),- 4,9 (т, 3H)j 5,3 (d, IH); 6,85 (s, tH)) 7,2 (s, lOH). Альтернативно (no Выбору) исходные вещества, используегше в описанг ном способе, получают следующим спбсобом, К раствору трет -бутил-3-ацетокси метил-7-аминоцеф-3-ен-4-карбоксилата (9,84 г) в ацетонитриле (350 мл) добавляют 2-хлоримидазолин гидрохлорид . (4,23 г) и CM€fCb перемешивают в атмосфере азота в течение б ч при температуре окружакяцей среды. Смесь
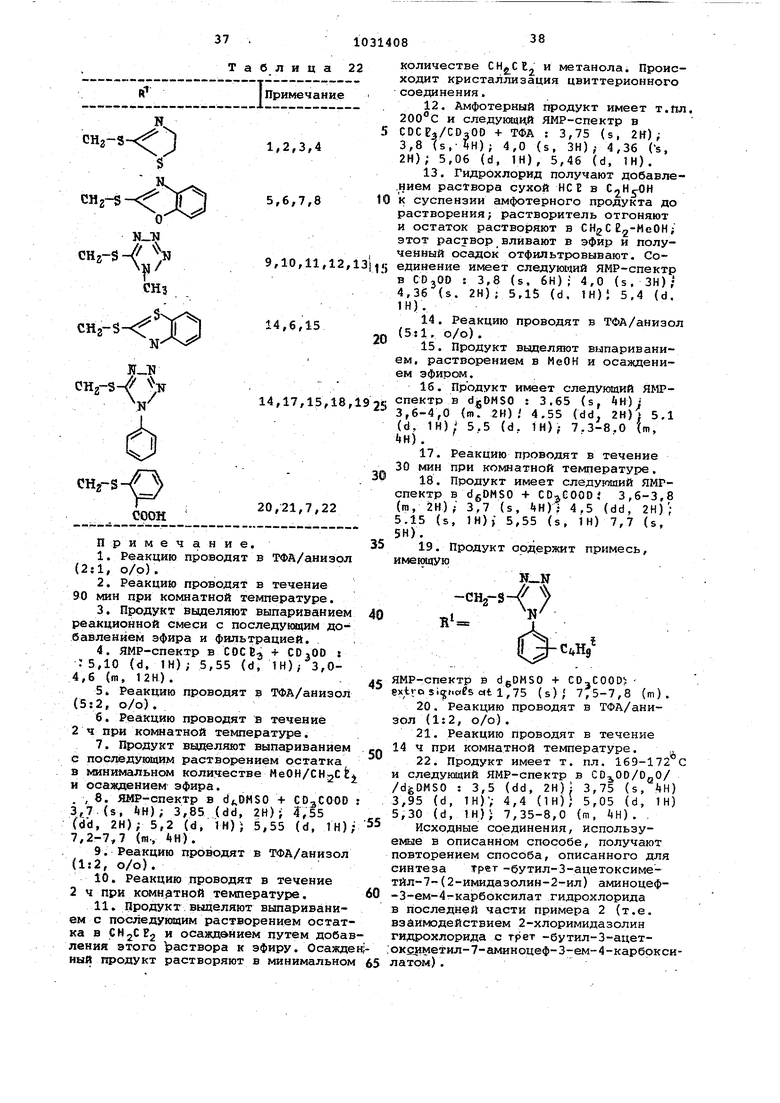
CHPh, фильтруют, осажденное твердое вещество промывают ацетонитрилом и объединенные фильтраты выпаривают. Остаток очищают хроматографией на силикагеле (600 г) при -40, используя CHjCEg/ /МеОН (9:1, о/о) в качестве элюента, с получением трет -бутил-3-ацетоксиметил-7- (2-й««мдазолин-2-ил) аминоцеф-3-ем 4-карбоксилат гидрохлорида, который применяют без дальнейшей очистки. Аналогичным способом, используя соответствуюпщй трет -бутил З-згииещенный 7-аминоцефг-3-ем-4-карбоксилат, получают следующие соединения (см. табл. 21): СН.- , Примечание. 1.Реакцию проводят в течение 4 ч 2.Реакцию проводят при . 3. Продукт очищают, хроматографией на силикагеле при , используя СН2СВ /МеОН(9б;4, о/о) в качестве элюента, с последующим осаждением из раствора СН5С 5Эфир/Е10Ас (2il, о/о). Дифенилметил 2,3-диаминопропиона используемый в качестве исходного вещества, получают следующим образо Суспензию бромгидрата 2,3-диаминопропионовой кислоты (3,7 г) в дис тиллированной воде (20 мл) перемеши вают с моногидратом толуол-р-сульф кислоты (3,8 г). Через 30 мин получаю прозрачный раствор. Воду удаля выпариванием и остаток после сушки над PjO5 суспендируют в DMf при 50 Постепенно добавляют раствор дифени диазометана в ДМФ до устойчивой фи Олетовой окраски. ОИР выпаривают при и остаток осаждают из СН2С12 раствора эфиром. перемешивают в метаноле с двумя эквивалентами КОН. Через 15 мин суспензию фильтруют через диатомовую землю и фильтрат выпаривают. Остаток растворяют в раствор фильтруют через стеклянный пористый фильтр № 4, фильтрат выпаривают до сухости. Остаток дифенилметилового эфира гфевращают в порошок с петролейным эфиром для удаления остаточного DMF. 1-(1,2-Диамииоэтил)-4-дщметилЗминобензол, нспольэуекФ в качестве исходного ве|цества, получают сле дующим образом. К раствору едкого натра (1,12 г) в метаноле (20 мя) при О добавляют тдроксиламин гндрохлорид (2,09 г), растворенный в минимальном количест ве водаа. После нескольких минут перемв1 Ш1вания NaCB отфильтровывают и фильтрат добавляют к раствору 1-нит рЬ-2-(4-дйметиламинофенил)-этилена (5,0 г) в метаноле. После перемешивания смеси в течение 2 ч при комнатной температуре добавляют 20%-ны избыток раствора гидроксиламина и . перемешивание продолжают еще в тече ние двух дней. Суспензию фильтруют с получением продукта, кото1зый гидрируют в вЁЗде суспензии в метаноле под давлением 4 бар в присутствии никелй Ренея в течение 18 ч с получением 1-(1,2-диа шноэтил)-4-диметиламинобензола в виде коричневого масла. 2-Хлоримидазолин гидрохлорид получают следующим образом. Райтвор дигидрата хлористого бария (33,8 г) в воде (120 мл) добавляют к раствору 2-хлоримидазолин сульфата (28,0 г) в воде (85 мл). Суспензию фильтруют через стеклянный пористый фильтр /W 4 и фильтрат выпаривсшт при ямдкой пасти которую несколько раз растирают в порошок с ацетоном с получением твердых гранул. Это твердое вещест:во сушат- над . с получением продукта, т. Ш1. которого 17g. - 180 «С. трет -Бутил-7-амино-З-(4,6-диметилпиримид-2-ил)тиометил-цеф-3-ем.-4-карбоксилат, используе1ъ«йв качестве промежуточного продукта, поглучают следующим образом. Раствор бикарбоната натрия (9, 45 г) в воде (100 мл) добавляют порциями к перемешиваемой суспензии 3-ацетоксиметил-7-аминоцеф-3-ем-4-карбоновой кислоты (13,5 г) в воде (100 мл) и ацетоне (50 мл). Твердое вещество растворяемся с бурным вьщелением газа. К этому раствору в атмосфере аз.ота при 55 быстро добавляют по каплям раствор 4,6-диметил-2-меркаптопиримидина (10,5 г) в воде (100 мл)и ацетоне (100 мл), рН поддер вают 7,4-7,8 с добавлением 6н. НСЁ или 5%-ного (об.) водного МаНСОз . После нагревания при в течение 23ч реакционную смесь OJuieasдают до 0 и подкисляют до рН 4,0. Полученный осадок фильтруют, промывают водой и ацетоном и сушат над Pg О с получением 7-амиио-З-(4,6-диметилпиримид- 2-ил)тиометилцеф-3-ем-4-карбоновой кислоты. Смесь этой кислоты (3,52 г) и 0-трет-бyтил-l,3-диизoпpoпилизсмочевины (6,0 г), в сухом (60 мп) перемешивают под азотом в течение 20 мин. Реакционную смесь фильтруют, фильтрат выпаривают и остаток очидают хроматографией на силикагеле (200 г) при -40°С, используя еИгС з/эФиР (бг4, о/о) в качестве элюента, с получением трет бутилового . Аналогичным способом, но используя 2-метил-5-мер.капто-1,3,4-тиадиазол вместо 4,6;-диметш1-2-меркаптопиримидина, по- 7-анино-З- (2-метил-1 ,;3 4-тиа№ азол-5-ил)хиометилцеф-3-ем-4-карбоновую кислоту и соответствующий fper -бутиловый эфир. Пример 12. Повторяют способ, используемый в примере 1 или 7, приА няя соответствующий дифенилметиловый или трет -бутиловый эфир в качестве исходного вещества, в результате получают следукяцие соединения (см. табл. 22).
Таким образом получают следующие соединения (см. табл. 23).
н н
V ч
,
COOR2
Т а б л и ц .а
Jilp-,
Пргаючание
. .
;CH2-S- JCHPhj1,2
2НИ 4,0 (d, IH) 4,2 (d, IH)- 5,35 (d. IHb
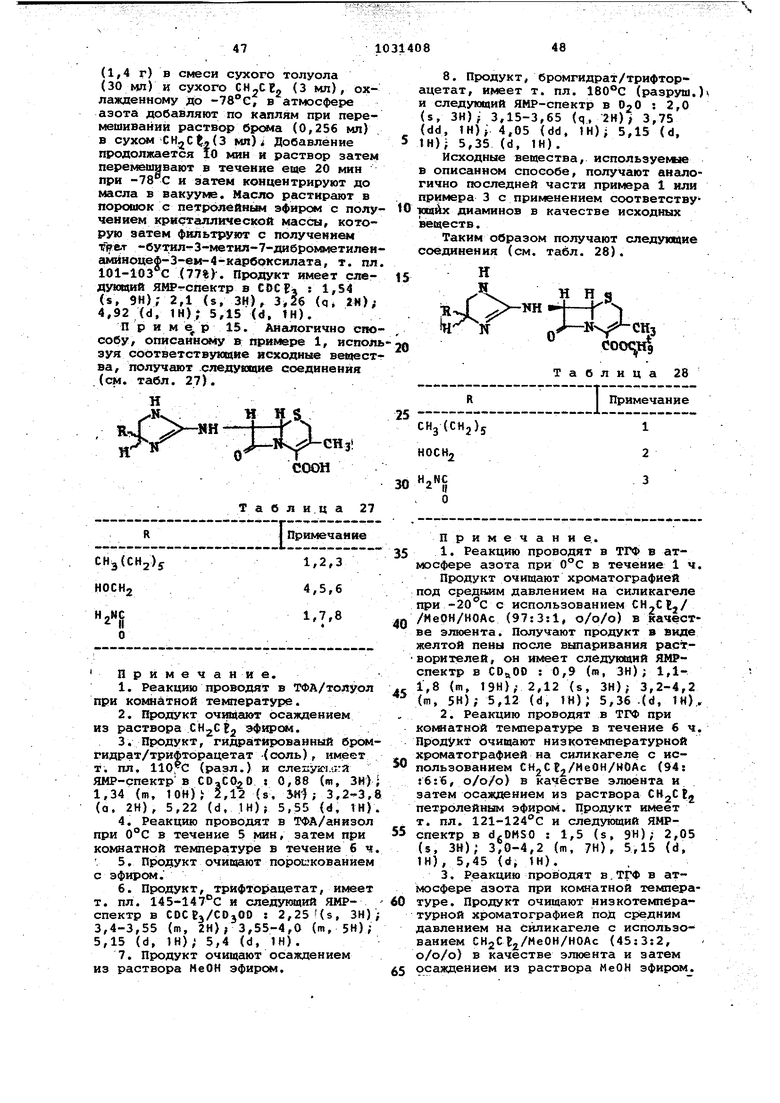
Некоторые из соответствукщих трет -бутил или дифенилметил З-замещённых -аминоцеф-3-ем-4-карбокси5 лдтов, используемых при синтезе соединений, представленных в табл. 23, даны в табл, 24.
И 5 10Я2Н«4г- N
О-N R
COOR
Т а б л и ц а 24
IZI
41| имечание
CffPh,
1,5
CHz-S
6,7
8,9
N-
Нг-$-О
-Ч«д
и
Примечание.
€. Реакцию проводят в течение 6ч при ,в а1цетонитриле ; продо кт очищают хрснлатографией на сйликах«- ле, элюент CKjCti/MeOH (95:5, о/о).
ч при , продукт очищают хрсжа ографией при -40 G на силикагеле, элюент ИеОН/CH CEj, (8:92, о/о).
Примечание. 1. { ixinyKT получают взаимодействием дифеннлдиазометан а с 7-амино - 3- ( 2-тйазС лив-2-ия) тнометилцеф-3-ем-4-к«й бонов6Й киспр«Л в растворе ацетонитрила,соеданеиие имеет
следугадай ЯМР-спектр в С ОС 13 s 3,35
4,1 (w, W)i 3,6 (я, 2Н); 3,95 (d, IH); 4,4 (d. 1И), 4,81 (d, IH); 4,95 d, IH), 7,05 s. tH),V 7,2-7,7 (широк, s, lOH). 2. К перемешиваемой суспензия
3-ацетоксиметил-7-аминоцеф-3-гем-4-карбоновбй кислота (19 г) в смгси ёоды (120 мл) и ацетона (120 мл) постепенно добавляют шлам 14 г НаНСО) в-80 мл воды. Полученный
вор нагревают в атмосфере азота 1фй 50 С. Медленно добавляют бенэоксаЗОЛ-2-ТИОЛ (15,9 г), затем рН доводят до 7,5 и продолжают нагревание в течение 15 ч при при этом рН,
После охлаждения смесь фильтруют. Фильтрат охлаждают на льду и доводят до РН 4,4. Полученный осадок про мывают водой и ацетоном и сушат над Pi-Oj с получением 12,6 г 7-амино-З-Гбенэоксазол-2-ил) тиометилцеф-3-ем--4-карбоновой кислоты. К суспен эии 5,45 г этого соединения в 90 мл ТГФ добавляют 9 г свежеперегнанной о-трег -бутил-М,М -бис-изопропилизомочевины. Через 24 ч при обычной темперетуре смесь фильтруют, фильтрат концентрируют и остаток очищают хроматографией. ИК-спектр (KBг); 3340 (w), 2980 (w),1775 (s), 1710 {s),1620. 3.Продукт получают ззаимодействием D-тp.eт-бyтил-N,N -диизопропилизомочейивы и 7-амино-3-(бензтиазол-2-ил)тиометилцеф-3-ем-4-карбановой кислотой в ТГФ (см. последнкио часть примечания 2 для аналогичного способа). Продукт имеет следующий ЯМР-спектр в COCFj 1,55 {s, 9Н)Г 1,8 (s, 2Н); 3,65 (s,.2H)i 4,45 {dd 2И), 4,7 {d. 1Н); 4,9 (d, 1Н); 7,25 7,95 (ffl, ). 4.К 13,6 г йуспензии 3-ацетокси метил-7-а ||иноцеф-3-ем-4-карбоновой кислоты в воде (100 мл) и ацетоне (50чмл) добавляют шлам 10 г МаНСО в 100 мл воды. Раствор нагревают до 55°С к порциями, добавляют 13,35 1-фенил-1й-тетразол-5-тиола. рН под держивают на уровне 7,5 в течение 15 ч. После охлаждения рН доводят до 2 и осадок отфильтровывают, промывают водой и ацетоном и сушат под вакуумом над с получением 9,36 г аминокислоты. К 4,22 г этой кислоты добавляют б,48 г О- третбутил-NiN -5мс-изопропилизомочеаи-г вы. Через 15 мин добавляют 65 мл СНрСЕз и смесь перемешивают в течение 18 ч. Реакционную смесь фильтруют, фильтрат концентрируют и остаток растворяют в EtOAc, затем промывают 5%-ным (вес./об.) водным RaHCej, а потом водой. После сушки над . раствор концентрируют и oceuttOK очищают хроматографией. Продукт имеет слеДУквдий ЯМР-спектр в C&Cta: 1,5 (s, 9Н); 3,65 (s, 2Н),4,4 tdd, 2Н)} 4,7 (d, 1Н)/ 4,9 (d-, 1Й)| 7,5 (s, 5Н). 5.боединение получают согласно оиособу, описанному в примечании 2. Сырой продукт обрабатывают растворся4 езгасой НЕЕ в Me ОН и кристалличес к йродукт (т. пл. ) полу чз1вт в виде гидрохло1жда без хромат . Оа имеет следующий ЯМР-сцек В CBsOD : 1,5 (8, 9Н 1,6 (s, И) 3,7 liis, 2Н); 3,95 (d, 1Н); 4,4 (d, 1Н), 5,0 (d, 1«); 5,2 (d, IH); 7,37,9 (т, М). Пример 13.«Аналогично способу, описанному в примере 7, испол зуя соответствующий дифенилметиловый эфир в качестве исходного вещества, получают следующее соединение: ОН И Н 5 ш Н-f S С.1 ii- ч CHgOCOCH j, COOH примечание: 1.Реакцию проводят в течение 5 мин при ксмнатной температуре в ТФА/анизол (40:60, о/о). 2.Продукт вьцделйют вьтариванием реакционной смеси с последующим растворением в CtigC / eOH и осаждением, гек саном.., 3.Продукт имеет т. пл. 230 С (разл.) и следующий ЯМР-спектр в dfcOHSO/CFiCOOO : 2,0 (s, ЗН); 3,6 (го, бН), 4,7 (d, 1Н), 5,0 (d, 1Н) ; 5,1 (d, 1Н); 5,5 (d, 1Н) Исходное вещество, используемое в описанном способе, получают следуиацим образом. К перемешиваемой суспензии 2,06 г М-гидоокси-1,2-диаминоэтан дигидообоомида (2,.Об) в метаноле (5 мл) добавляют два эквивалента КОН в метаноле. Реакционную смесь добавляют к перемеишваемому раствору 1,76 г дифенилметил З-ацетоксиметил-7-дибромметиленаминоцеф-З-ем-4-карбоксилата(1,76 г) в сухом ТГФ (80 мл) , охлажденному до . Через 2 ч добавляют ТФА (0,7 мл) . Смесь концентрируют до сухости и затем добавляют СН2С Ij. Смесь фильтруют и фильтрат концентрируют и сушат, после чего остаток очищают .хроматографией на силикагеле . элюируя содержащим 0,1 и 2% (о/о) ИеОН. Продукт имеет следующий Я1-1Р-спектр в dgOMSO + : 2,0 (s, ЗН)/ 3,6 (m, 6Н); 4,7 (d, IH)4,9 (d, IH); 5,2 (d, IH)) 5,6 (d, tH)) 6,9 (s, IH, 7,2-7,5 (m, 1 OH). П p И M e p 14. Повторяют способ, описанный в примере 1, используя соответствующие исходные вещест за, в рез57льтате получают следующие соединещ5я (см. табл. 25). Н 5 - V -1 JN-jA&Hs ° fcooH
Продолжение табл. 25
Примечание.
Выпавшую соль растворяют в МеОИ и обрабатывают эпоксипропаном с получением амфотерной формы в В1зде кристаллов. Лмфотериую форму превращают в гидрохлорид путем обработки раствором НС t в МеОН и осаждения эфирсж.
dgOMSO/COjjCOjD : 2,05 (s,3H),
3,4 (m, 3H)i 3,5 (о, 2Н); 4,12 (m, IN); 5,1 (d, 1H) 5,15 (m, lH)i 5,5 (d, IH); 7,45 (m. iH) .
0 ЯМР-спектр в diDMSO/COgCOoD : 2,05 (s, ЗН); 3,47 Та. 2Н); 3,5-3,7 (т, 1Н); 4,2 (t, tH); 5,08 и 5,1 {2d, IH); 5,25-5,6 (ш. 2Н); 5,4 (m, Н).
0 (s) и 2,05 (s) (totat ЗН) 3,23,6 (m, ЗН); 4,1 (t, IH)S 5,0 (m, IH); 5,1 (d, IH); 5,45 (d, IH), 6,77,4 (m, 4H).
ет следующий ЯМР-спектр в d.DHSO/ ; 2,3 (s. ЗН), 3.4-3,85 (m, ЗН)} 4,2 (t, IH),- 5,05-5,2(
,20
(m. iH)j 5,30 (d. 1H)J 5,7 (d. IN); 6,7-7,1 .(m, ЗИ).
0 Исходные setnecTBa, используеквле в описанt oM способе, получают повторением последней части приме а 1 или 3 с применением соответствующего арилэтилендиамина. Таким образом
5 получают следующие соединения (см. табл. 26).
, . Н s
-l sH-CHj
м К 600R2
Таблица 26
,
1,2,3,4,5,18 2,3,6,7,18
л /Г 8,9,10
CHPh11,3,6,12,18
2,3,6,13,18
(1,4 г) в смеси сухого толуола (30 мл) и сухого СН-СВп (3 мл), охлажденному до -78°е, в атмосфере азота добавляют по каплям при перемешивании раствор брсма (0,256 мл) в сухом мл) Добавление продолжается 10 мин и раствор затем перемешивают в течение еще 20 мни при и затем концентрируют до масла в вакууме. Масло растирают в порошок с петролёйным эфиром с получением кристаллической массы, которую затем фильтруют с получением Tfе.т -бутил-3-метил-7-да бр01 Л(етилвнги«иноцеф-3-ем-4-карбоксилата, т. пл. 101-103 С (77%). Продукт имеет слеПУКЯЦИЛ ЯМР-спектр в COCF : 1,54 (s, ЭН); 2,1 (s, ЗН), (q. 2Н); 4,92 (d, 1Н); 5,15 (d, 1Н).
П р и м р 15. Аналогично способу, описаинсту в примере 1, используя соответствукхкие исходные вей|естг ва, получают следующие соединения (см. табл, 27).
Н V Н Н S
/ ,
соон
Т а б л и.ц а 27
Примечание
Примечание.
и следующий ЯМР-спектр в OjO
2,0
(s, ЗН); 3,15-3,65 (q. 2Н) 3,75 (dd, 1H)j 4,05 (dd. tH)- 5,15 (d, IH)- 5,35 (d, 1H).
Исходные вещества, используемые в описанном способе, получают аналогично последней части примера 1 или примера 3 с применением соответству-кявйх диаминов в качестве исходных веществ.
Таким образом получают следующие соединен ия (см. табл. 28).
Н Я
Н Н а
«н4-г
Л
Л-Кх/-СНз
k
N СОО(Н|
Таблица 28
I Примечание
СНзССН2)5 1
НОСНо
H,NC
30 2 ц
Примечание.
1 Реакцию проводят в ТГФ в атмосфере азота при 0°С в течение 1 ч.
Продукт очищают хроматографией под среднем давлением на силикагеле при с использованием Ij/
/«ерН/НОАс (97:3:1, о/о/о) в качестве SJBoeHTa. Получают продукт в анде желтой пены после выпаривания раст в ори те лей, он имеет следукяций ЯМРспектр в СОдОО : 0,9 (га, ЗН); 1,11,8 (т, 19Н), 2,12 (s, ЗН); 3,2-4,2 (т, 5Н); 5,12 (d, 1Н); 5,36 .(d, 1Н)..
хроматографией на силикагеле с нспользованием СНзСf /MeOH/HOAc (94: :6:6, о/о/о) в качестве элюента и затем осаждением из раствора петролейным эфиром. Продукт имеет
т. пл. 121-124°С и следующий ЯМР1,5 (s, 9Н); 2,05
спектр в dgOHSO
(s, ЗН); 3,0-4,2 (m. 7Н), 5,15 (d, IH), 5,45 d, IH).
осаждением из раствора МеОН эфиром.
Продукт имеет т. пл. 190°С (разруш.) и следующий ЯМР-спектр в /CDaCOnD S 1,52 (s, 9Н); 2,08 (s, ЗН); 3,3-3,8 {q,2H}{ 5,15 (d, IH); 5,45 (d, IH).
Пример 16. Аналогично способу, описанному в примере 1, с использованием соответствующего грет бутилового эфира в качестве исходного вещества получают следующие соединения (см. табл. 29).
1 х.Н Н s
,н..т-
R .CH
э
ооои
Таблица 29
R IR|Пвимеч§ние
CiS-(GH2)21,2,3
1,2,4
CiS-CH2CH CH
CiS-COjCHjС З-СОзСН 1,2,5
CiS -С, Н..
CiS-C Hj 1,6
о э
Примечание,
5,8 .(brd, IH) , 6,05 .(brdjJH)) .
(q, 2Н); 5,19 (d, IH); 5,56 (d, IH) 7,05 (m, lOH).
Исходные вещества, используемые в описанном способе, получают аналогично последней части примера 1 или 3 с грименением соответствующих диаминов. Подобным путем получают следующие соединения (см. табл. 30).
н
-R /Н Н И
Х -«й-М,
R .
Т а б л и ц а 30
Примечание.
s : 1,52 (s,9H)i 2,1 (s, ЗН); 2,15-2,8 (m, Н); 3,12-3,8 (a, 2Н) ,4,5 (m, 2Н); 5,12 (d, 1Н); 5,35 (d, IH).
в d4,OMSO/D2.0 : 1,55 (s. 9Н) 2.1 (s, ЗН),- 2.6 (m, 2Н); 3.25-3.85 (а, 2НЬ 4.5-5.1 (т. 2Н) 5.18 (d, 1H)i 5.4 (d. IH) j 5,8 (b r, IH), 6,1 (br. f H) .
т. пл. (разруш.) и следующий 5 ЯМР-спектр в d,DMSO : 1.5 (s, 9Н) / 2.02 (s, ЗН) .- 3.2-3,8 (а, 2Н) , 3.7 (s. 6Н) , 4,9 (s, 2Н) ; 5,12 (d. 1Н),5,54 (d, IN).
давлением, элюируя .EtOAc/MeOH (95:5, о/о) с последующим осаждениемиз раствора tHgC to эфиром. Продукт имеет т. пл. . (разруш.) и сле5 дуняций ЯМР-спектр в CDCfj/CD OD :
j 1,56 (s, 9H); 2,07 (s, 3H) 3,23,35.(q. 2H); 5,09 id, IH), 5,45 (d,
1H)) 5,55 (s, 2H) 7,06 (m, ЮН).
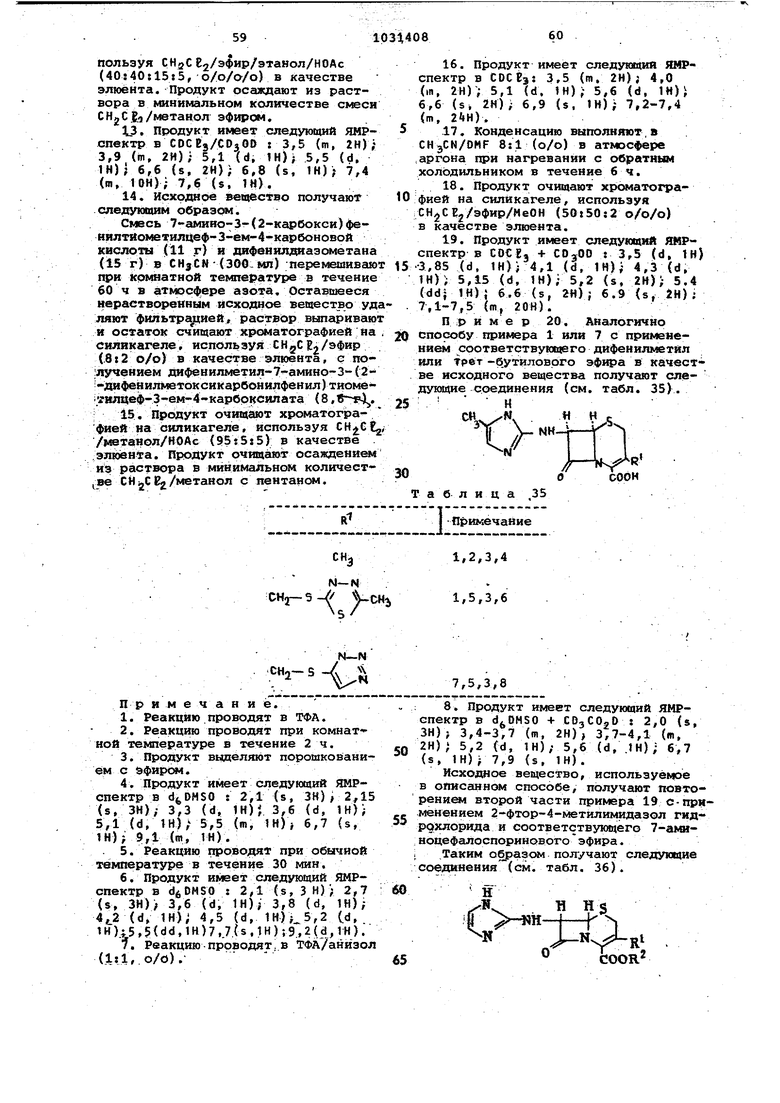
Пример 17. Аналогично способу, описанному в примере 1, с ясi ПОЛЬ зеванием . соответствукадего дифенилметилового эфира в качестве ис1ходногр вещества, получают следуихцие соединения (см. табл. 31).
R
1
И
Таблица 32
Н
|н и«
.
И С00Н
Таблица 31
I
ilH--t
Примечание
Н В
1,2
igCOOH
- . . -CH-i iCH2-S - 1-СЙЗ
К-ЗГ
;CH2-S- N
Ci€ -С «2
1,4
Примечание.
Полученный таким образом РОДУкт. представляет собой смесь трифторацетата и амфотёрной форил. Он имеет следукядий ЯМР-спектр в COjOD/OjO/ /CDCtj: 3,80 (m, 6Н); 4,3 (d, 1Н);
т. пл. {разл.). Этот продукт, смешанная соль бромгидрат/трифторацетат, имеет следующий ЯМР-спектр в сОМ&0/СО,С020/ТФА : 0,25 (т, 1И); 0,80 (т, 1Н), 2,65 (s, ЗН); 3,2-3,8 (т, itH); 4,25 и 4,5 (q, 2Н); 5,1 (d, fH); 5,4 (d, IH).
Исходные вещества для описанного спосрба представлены в табл. 32.
,3,
Примечание
Г8Ч
|С%
К
CHgCOOCHPh
N-ir
CHz-S Ч -СНз
Си:
«г
;3
N CHj
Примечание. 1. Соединение получают путем взаимодействия 2-хлор-имидазолин гидрохлорида и дифенилметил-7-амино-3-(1-дифеиилметоксикарбонилметил-1Н-тетразол-5-ил)тиометилцеф-3-ем-4-карбо1ссилата аналогично второй части примера 12. Реакцию проводят
в ацетонитриле при 40°С в течение 4,5 ч. Очистку ссучествляют хроматографиеЯ на снликагеле, злюируя сначала СНз СЕ /эфир (lil, о/о) для
удаления массьэ иепрореагировавшего исходного вей|ества. Затем используют СН С 2/эЦетои/изопропанол (6t2t ;2, о/о/о) для элюирования сначала неболыяого количества исходного веBtecTBa в виде соли и затем целевого продукта. Продукт имеет т. пл. 130132С и следующий ЯМР-спектр в eOCEai 4,2 (br, 6Н),- 5,2 (d, IN),5,7 td, fH),- 6,88 (s, IH),- 6,9 (s, IH); 7,3 m, 10H).
Swt -ДифенилметиловЕлй эфир, используешой в качестве исходного вещества, получаютJследующим образом. К суспензии 7-амино-3-(1-карбоксиметил-1Н-тетразол-5-ил)цеф-З-ем 4-карбоновой кислоты (7 г) в ацетонитрил/МеОН (1:1, о/о), 200 мл при медленно добавляют раствор дифенилдиазометана в гексане до устойчивой фиолетовой окраски. Горяч о
реакционную сьюсь фильтруют. Остаток промывают горячим ацетонитрилом и объединенные фильтраты концентрируют в вакууме. Кристаллический продукт, полученный таким образом,
,имеет т. пл. 162-164°С и следукадий ЯМР-спектр в dfcDMSO : 3,55 (s, 2Н) 4,05 (d, IH) ; 4,35 (d, 1Н); 4,8 (d, IH); 4,9 (d, IH); 5,5 (s, 2H.) , 6,85 (s, 2H}; 7,3 (s, 10H). 2. Соединение получают повторе-, нием способа, описанного в последней части примера 1, используя соответствующие .исходные вещества. Реакцию проводят в ТГФ при комнатной температуре. Продукт очищают низкотемпературной хроматографией на силикагеле, используя смесь качестве элюента. Полученный продукт растворяют в СНоСЕ, и осаждают эфиром. Продукт имеет т. пл.- 125°С (разл.) и следующий ЯМР-спектр в : 0,25 (m, 1Н); 0,85 (ffl, IH); 2,62 (s, ЗН); 3,4-3,9 (m, 2Н); 4,35 (q, 2Н) ; 5,19 (d, 1Н) 5,5 (m, IH 6,85 (s, IH); 7,3 (m, 10H); 9,4 (m, 3H) . 3. Соединение получают повторе-. нием способов, описанных в последней части примера 1, используя соответствукдцие исходные вещества. Реакцию проводят в ТГФ. Продукт очищают аналогично описанному способу в примечании 2. Он имеет т. пл. (разл.) и следующий ЯМР-спектр в dv DMSO/CDjCOjD : 0,3 (m, IH); 0,9 (m, IH); 3,4-4,0 (m, 8Н); 4,2 И 4,4 ад (q. 2Н); 5,2 (d, IH); 5,5 (d. IH); 6,9 (s, IH); 7,38 (m, 10H). Пример 18. щё (с г 15 CH CH 25 CH получают следующие соединения бл. 33). . «.S п П g « J RТаблица 33 Примечание И-N 1,2,3,4,5 гаг()г, 3N 1,6,3,7 СНз 8,9,3,10 Hj - VcHv8,9,3,11 «
Раствор пивалоилоксиметил-3-метил-7-аминоцеф-3-ем-4-карбсксилат тоЛуол-сульфоната (3 г) з воде (ЮО гт) обрабатывают избытком NaHCОд. Смесь экстрагируют три раза этилацетатом и объединенные экстракта промывают рассолом и концентрируют. Остаток (2 г) растворяют в. ацетонитриле {75 мл) и добавляют 2-хлор-2-имида- золин гидрохлорид (0,86 г). Смесь перемешивают при 40°С в течение 5ч. фильтруют и фильтрат концентрируют. Остаток перекристаллизовывают из смеси изопропанол/эфир с получением пивалоилоксиметил-3-метил-7- (2-имидазолин-2- ил) аминоцеф-З-ем-4-карбо- ксилат гидрохлорида (0,99 г), име- j надего следующий ЯМР-спектр в CD,OD : : 1,2 (s, 9Н); 2,15 (s, ЗН) 3,55 (dd, 2Н) 3,8 (s, ); 5,15 (d, IH); 5,4 (d, 1H)i 5,85 (dd, 2H).
np и M e p 19. Аналогично спо- : собу, используемому в примере 1 или 7, с применением соответствующего дифенилметилового или трет -бутилового эфира в качестве исходного веN-JT35 CHg-S1,9,3,12
CHg-S
1,13,3,14
еоон
: ; -кCHjrS-4,
1,13,3,15 N
СНгСООН
Примечание. , 1. Реакцию провопят в ТФА/аНизол (1:1, о/о).
с эфиром.
U-H
СНГ 5 К
е11гС1%11(енэ)г
CHj,OCOCHj
1,2,3
CHPh,
4,5,6 в ТГФ (50 мл). После перемешивания в течение ночи смесь вливают в эфир (2 л) и образовавшуюся смесь-фильтруют. Твердое вещество- обрабатывают избытком водного раствора NaHCO и тризвды экстрагируют . Объеди ненные экстракты сушат над MqSO, фильтруют и выпаривают с получением 19 г твердого вещества., которое очищают хрс {атрграфией на силикагеле (800 г), нетольэуя CH.jC В /эфир (5:5, о/о) в качестве элюента. Таким образом, получают дифенилметил-3- С(1 « Тил-1Н-тетраэол-5-ил)тиометил}цеф-3-ем-4-карбоксилат в виде белого кристаллического твердого вещества ( г). К раствору дифенилметил-3- (1-метил-1,2,3,4-тетраэол-5-ил)тиомеИ1л цеф-3-ем-4-карбоксилата (2,47 р| в ацетонитриле (30 мл) добавляют 2 -|| оримидаэол НС t 0,615 г). Сйесь перемешивают при нагревании с о Е раткым холоднльником в атмосфере аэоТа в течение 1,5 ч. Растворитель вьтаривают и остаток хроматографщ удат На силикагеле (150 г), используя СНаСг2/эт,анол/ /НОАс (90:10:5, о/о/о) в качестве элюента. Полученное таким очищенное соединение растворяют в минимальном количестве и осаждают эфиром с получением дифени.л-7-(имидазол-2-ил)-3- (1-метил-1Н-тетразол-5-ил)тиометнл цеф-З-ем-4-карбоксилата в виде бежевого твердого вещества (1 г) ,. которое имеет т. ал. 125-130 С и следующий ЯМР-спектр в CDjOD : 3,75 (га, 2Н); 3,85 (s, ЗН); 4,25 (т, 2Н); 5,1-5,7 (т, 2Н); 6,8 (s, 2Н); 6,85 (s, 1Н), (т, 10Н). Следующие исходные вещества получают аналогичным путем (см. табл.34) аблица
ООНРЬг, Примечание. 1.Исходиое соединение получают следующим образом. Суспензию 7-амино-3-(1-С2-диметил аминоэтил3-1Н-тетраз9л-5-ил)тиометилцеФ 3-ем-4-карбоновой кислоты . (5,0 г) в смесь метанола {170 мл) и fHCEj (80 мл) нагревают с обратным холодильником. Затем кюдленно добавляют СНСЕ, ,- раствор дифенилдиазометана до устойчивой красиой окраски, после чего добавляют 1н. неЕ {50 мл). Смесь выпаривают, к оставшейся водной фазе добавляЕют воду и полученную смесь экстрагируют этилацетатом {50 мл).Водную фазу отделяют, охлаждают до О С -ц нейтрализуют до рН 20%-мь м вес./об.) вод ным раствором аммиака. Осадок собир ют, промывают водой и МеОН с получе нием дифенилметил-:7-амино-3-{1-С2-Яиметиламиноэтил )-1.Н-тетразол-5-ил тиометилцеф-З-ём-4-карбоксилата {2,9 г), имеющего следунвдий ЯМРСПвктр в eOCfaf 2,3 (s, бН); 2,7 {m, 2И); 3,7 |т, 2Н),- 4,2 {т, ), 4,7 {q, 2Н). 2.Взаимодействие одного эквивалента с 2,2 эквивалентам 2гфторимидазол гидрохлорида. 3.Реакцию проводят в MeOH/CHCiy tl:l, о/о) при нагревании с обратны .холодильником в течение 15 ч. Проду; используют без очистки на следующей стадии. 4i Реакцию проводят в при нагревании с обратным холодильником в течение 2ч. 5. Продукт выдел}пот хрс атографией на силикагеле при -20°С, испо ьэуя {95t5 2, о/о/о) в качестве элюента.
Продолжение табл. 34
4,7,8,9
10,11,12,13
14,4,15,16
CHP.h
17,18,19
CHPhq 6.Продукт имеет следующий ЯМРспектр в еоеЕэ + СОзОО 4 1,6 {s, ЗН) 2,15 {s, ЗН); 3,3 {d, tH); 3,7 {d, 1Н); 4,8 И 5,15 {2d, 2Н) , 5,25 {d, 1Н); 5,7 Id, 1Н)) 6,8 {s, 2Н). 7.Продукт очищают хроматографией на силикагеле, используя /этанол/НбАс {85:15:5, о/о/о) в качестве элюента, и осаждают пентаном в виде смеси оолей. 8.Продукт имеет следующий ЯМРспектр в СОСе s 1,5 (s, 9Н); 2,95, (s, ЗН); 3,9 (m, 2Н); 4,45 {m, 2Н); 5,4 {d, 1H)V 6,0 (dd, IH); 6.9 {s, 2H); 8,6 {d, IH). 9.Коняенсацию выполняют в смеси GHjCR/DHF {3:1, о/о). 10.Исходное вещество получают следуге1 шм образом. 7-Аминр-З-{1,2,З-триазол-4-ил)тиометилцеф-З-ем-4-карбоновую кислоту {13,7 г) суспендируют в смеси {150 мл) и метанола {150 мл). Добавляют избыток дифенилдиазометана {18 г) в {100 мп) к смеси, которую затем нагревают до 40°С в течение 8 ч и перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют, выпаривают и остаток хроматографируют на сипикагеле {400 г), используя еН2 СЕ2/эфир/ИеОН {20:79:1, о/о/о) в качестве элюента, с получением дифен ил14е тил- 7- азмино- 3-{1,2,3- т и азол-4-ил)тиомятилцеф-3-ем-4-карб оксилата 8,3 г):11.Реакцию проводят в смеси СНзСН {30 мл) и ТГФ {50 мл) при нагревании с обратным холодильником в течение 2ч. 12.Продукт очищают хроматографией на силикагеле при -20С, используя Е2/эфир/этанол/НОАс (40;40 l5t5, о/о/о/о) в качестве элюента. Продукт осаждают из раствора в минимальном количестве смеси СHjС {л/метанол эфиром.
13,Продукт имеет следукидий ЯМРспектр в СОСВз/СРзОд : 3,5 (т, 2Н) ; 3,9 (т, 2И); 5,1 (d, 1Н); 5,5 (d. 1Н); 6,6 (s, 2Н),- 6,8 (s, 1Н); 7,4 (т, ЮН),- 7,6 U. 1Н).
Смесь 7 амиио-3-(2-карбокси)фенилтйометилцеф-3-ем-4каре4оновойкислоты (11 г) и дифеиилдиазометана (15 г) CHjCN(300-мл) перемешивают при комнатной температуре в течение 60 ч в атмосфере азота. Оставвюеся нераетворенным исходное вещество удаляют фальтра ией, раствор выпаривают и остаток счищают хроматографией:на , силикагеле, используя СН СЕ /эфир (.812 о/о) в качестве элюента, е получеиием дифенилметил-7-амино-3-(2: ;оифенилметоксикарбонилфенил)тиомеiгилцеф-З-ем-4-карбрксилата (8, . К 15. предукт очищают хроматогра - фией на силикагеле, испол ь з у я С Н .С /мета«ол/НОАе (95t5:5) в качестве элюента. Продукт очища ют осаждением из раствора в минимальном количест вe СHjjCB /метанол с пентаном.
СН
(vi-N CHj-S- CHj
N-N
П p и M e ч a H и e. 1. Реакцию.проводят в ТФА. 2. Реакцию проводят при комнатной температуре в э«чение 2ч.
. 5. Реакцию проводят при обычиой температуре в течение 3D мин.
f. Реакцию проводят, в ТФА/анизол (1:1, о/о).
СН СЕ /эфир/ИеОН (50t50:2 о/о/о) в качестве элюента.
П :p и M e p 20. Аналогично
0 способу примера 1 или 7 с примеиением соответствующего дифенил ютйл или трет -бутилового эф1фа в качестве исходного вещества получают следующие соединения (см. табл. 35).
Н N.
« Н
t
NH
J-M
R СООН
Таблица .35
-Примечание
1,2,3,4
1,5,3,6
7,5,3,8
: 8. Продукт имеет следующий 5ШРспектр в -f COjCOjO : 2,0 (s, ЗН), 3,4-3,7 (ffl. 2Н), 3,7-4,1 (m, 2Н); 5,2 (d, IH); 5,6 (d, .JH); 6,7 (s, IH)j 7,9 (s, IH).
Hcxonftoe вещество, используемое в опксгшном способе, получают повторени 4 второй части примера 19 с-применением 2-фтор-4-метилимидазол гидрохлорида и соответствующего 7-аминодефалоспориновогоэфира. i Таким образе получают следующие соединения (см. табл. 36).
Н
CHs.П
61
СН
5- Примечание. 1.Реакцию проводят в при нагревании с обратным холодильником в течение 2 ч 30 мин. 2.Соединение очищают хроматографией на силикагеле, используя СН СЕ /этанол/НОйс (90:5:5, о/о/о,, а затем 85510:5, о/о/о) в качестве элюентов. Продукт осаждают из раствора СН2СЕ2 эфиром. .3. Продукт имеет еледуняций ЯМРспектр в CDCEa: 1,4 (s, 9Н); 2,05 (з, ЗН); 2,1 (S, ЗН);- 3,0-3,6 (т, 2Н); 5,0 (d, 1Н) 5,5 (d, 1Н}; 6,25 (d, 1Н) . 4.Реакцию проводят в смеси СИзСМ/ОМР (2:1 о/о) при 80-9О С в течение 1,5 ч. 5.Продукт очищают хроматогра- , . фией на силикагеле, исполь зуя С Н С EJ /этанол/НОАс (85:15s5, о/о/о) в качестве элюента, с последуюц им растворением в минимальном количестве и осаждением пентаном. Свободное основание получают обработкой водным бикарбонатом экстракцией EtOAc и концентриробанием органи ческих экстрактов. 6.Свободное основание имеет следующий ЯМР-спектр в : 1,5 (s. 9Н) ; 2,0 (s, ЗН) 2,7 (s, ЗН)г 3.2-3,8 (m. 2Н) ; 4,0-4,6 (m, 2Н) ; 5,15 (d, IH) 5,6 .(d, 1Н),- 6,2 (s, 1Н) . 7.Реакцию проводят в CHjCN/DMF (4:1, о/о) при ВО-90 С. 8.Продукт очищают хрсялатографией на силикагеле, используя /этанол/НОАс (85:10:5, о/о/о) в качестве элюента. Продукт растворяют в СНгСВз/МеОН (97:3, о/о), раствор фильтруют и фильтрат выпаривают. Ос таток обрабатывают водным бикарбона том, экстрагируют EtOAc/этанол (97: о/о) к экстракт выпаривают до сухос ти с получением свободного основани имеющего следующий ЯМР-спектр в dfcDMSO + CDaCOgO : 2,0 (s, ЗН) ,3,4-4,0 (m.JfH); 5,2 (d, 1Н) ; 5,4 (d IH); 6,4 (s. 1H) ,- 6,8 (s. IH),- 7,3 (m, 10H) , 7,8 (s, IH) . П p и мер 21. Аналогично способу, описанному в примере 1 или 7,
103140862
Т а б л иц а 36
I R примечание
1,2,3
t-C.H
4,5,6
7,8,9
CHPh использованием соответствующего ифенилметилового эфира в качестве сходного вещества получают следущее соединение Н Т KH-4-f4 СООН . Примеча;ние. 1.Реакцию проводят в анизол/ТФА (5:2, о/о). 2.Реакцию проводят при комнатной температуре в течение 30 мин. 3.Продукт очищают растворением в минимальном количестве СНлС /MeОН и осаждением смесью эфир/гексан; 4.Продукт, трифторацетат (соль), имеет следующий ЯМР-спектр в dtDMSOt 2.05и 2,1 (2s, Н),- 3,40-3,60 (2d, 2Н) ; 5,1 (di IH); 5,4 (d, IH). 5.Реакцию проводят при комнат-, ной температуре в течение 1 ч в анизол/ТФА (5:1, о/о). 6.Продукт очищают растворением в СН2СЕ2 и осаждением диизопропиловым эфиром.: 7.Продукт, трифторацетат, имеет следующий ЯМР-спектр в dtPJM SOV. 2.06(s, ЗН); 2,28 (s, ЗН); 3,35-.3,60 (2d, 2Н)- 5,18 (d,;1H)- 5,65 (d, IH); 7,2-7,6 (m, 5H), 8,2 (m, IH). 8.Peакцию проводят при температуре и тё$шературе окружающей среды в течение 30 мин. 9.Продукт, смешанная соль бромгидрат/трифторацетат, имеет следующий ЯМР-спектр в dgDMSOi 2,08 (s, ЗН); 2,10 (s, ЗН),« 3,30-3,60 (2d, 2Н); 5,10 (d, IH); 5,48 (d, IN)/ 6,55 (s, IH). Исходные вещества, используемые в описанном способе, получают следующим образом. Повторяют вторую часть примера 9 с применением соответствующих исходных веществ и таким образом получают следующие соединения (см. табл. 37).
НИ-I-
СООСЯК
Таблица 37
I R I Примечание 1г2,3
СН,
СН,
Примечание.
1,К раствору «нти -2-амино-З-оксимииобутана (3.1 г) в ТГФ (60 мл)
Добавляют триметилхлорсилан (7.75 мл) и триэтиламин (8.5 мл) и смесь перемешивают при обачно1Л твАшературе в течение ночи. Выпавший триметиламин НС Е удаляют фильтрацией в атмосфере азота и полученный pacTBop добавляют к охлажденному ) раствору 7-дибро «||етиленаминоцефалоспорин производного (5,6 г), в ТГФ (20 мл). После 1ч . ПРИ добавляют ТФА (7 мл) к холодне раствору, который затем концентрируют под вакуумом.
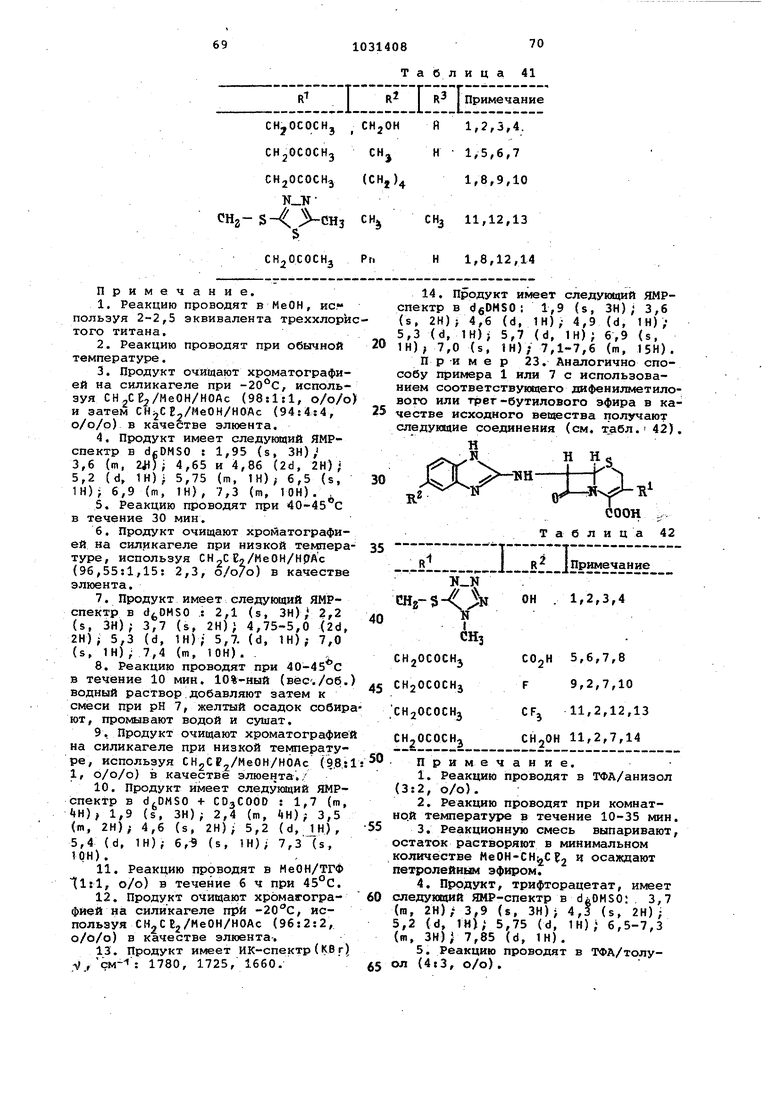
Повторяют последнкио часть примера 9 (восстановление 1-гидроксиимиазол производного треххлористым титаном),, используя соединения, пред ставленные в табл. 37, в качестве исходных веществ, и таким образом получают следующие соединения (см. табл. 38). ОН
. . I NHi-f
R LJs
CHj
r т
COOCHPtij Таблица 38
20
Примечание.
титана при 40-50 С в течение 30 мин,
/НОАс (98,5:1:0,5, о/о/о) в качесте элюента, с последующим осажденим продукта из раствора , геканом.
2,05 (s, ЗН),- 2,30 (s, ЗН), 3,55 (s. 2Н) 5,3 d, 1Н) 5,9 (d, 1Н), 6,8 (s, IH), 7,2-7,7 (tn, 15H).
55 7. Реакцию проводят в МеОН с 2 эквиваленташ треххлористого титана при 40-45°С в течение 30 мин. : 8. Продукт очищают низкотемпературной хроматографией на силикагеле,
@:используя CHgCEj/MeOH/HOAc (90s5i5, о/о/о) в качестве элюента. Продукт, смешанная соль бромгидрат/трифторацетат, имеет следующий ЯМР-спектр р dfcDMSOj 2,08 (s, ЗН); 2.1 (s, ЗН)
65 3,35-3,65 (2d, 2Н); 5,15 (d, 1Н)-;
5,65 (d, 1Н); 6,45 (s, 1Н); 6,88 (s, 1Н),- 7,15-7,6 (m, ЮН).
Пример 22. Аналогично СПО собу, описанному в примере 1, с использованием соответствующих исход-; ных веществ получают следующие соединения (см. табл. 39):
I R I RППpимeчaниc
t«
H CHjOH H 1,2,3
N-Л
CHr$- X 3 ,2,14
S . , .
CHjjOCOCHj
Примечание.
из раствора в минимальнсяи количестве СН2СЕ2/МеОИ гексаном.
Y -ш-f-f
..
Т а б л и ц а 39
H 4,5,2,6
H Me H (CH.2)4 4,7,8,9 Ph 1,10,11,12
H
H 4,,15
H Ph
+ CDjCOOD; 2,0 (s, ЗН) j 3,5-3,6
(го, 2Н), 4,.7 (d, IN); 5,0 (d 1Н);
5,2 (d, 1Н); 5,7 (d, 1Н); 7,0 (s,
IH), 7,1-7,7 (m, 5H|. 35 13. Реакцию проводят в ТФА/анизол (2:1, о/о) в течение 2 мин при
комнатной температуре.
40 2.,Qf (s, 6И),- 2,67 (s, ЗН); 3,68
(2d,2H), 4,26-4,56 (2d, 2Н); 5,16 5,48 (2d, 2Н).
45 (s, ЗН); 3,5-3,6 .(q, 2Н) ; 4,7 7 (d, IH); 5,0 (d, IH), 5,2 (d, 1H)J 5,8 (d, IH)/ 7,3 (s, 1H)i 7,2-7,8 (m, 5H),. 8,4-8,7 (m, IH).
Исходные вещества, используемые -л в описанном способе, получают следуWHM образом.
Повторяют вторую часть примера 9, используя соответствующие исходные вещества, и таким образом по(см.
лучают следукягше соединения табл. 40). S
ш-нН-Ч
COOCHPh
67
CHgOCOCHjСН ОМ W 1,2,3,4,5
CHgOCCCHjCHj
СНзОСОСНз(CH2 )
N-Jf
Hz-S- J CHj cHg
CH OCOCHgPh
Примечание.
в ТГФ/НеОН (10:1, о/о) при температуре между -35 С и в течение -2ч..: .
1031408€8
Таблица 40
y rjrjj {Примечание
H б ,.3,7 ,8 9,3,10,11
CHj 12,10,13
H 14,3,10,16,15
0 9. Конденсацию выполняют 1-триметилсилиламино-2-триметилсилилоксииминоциклогексаном (полученным из 1-амино-2-окси диноциклогексана) в безводном ТГФ Ъри комнатной температуре в течение 2,5ч.
(d, 1Н); 5,1 (d, 1Н) 5,2 (d, 1Н
(s, ЗН); 3,6 (s, 2rf); 4,6 (d,. 1Н), 4,9 (d, 1Н); 5,3 (d, 1 H) ; 5,7 (d, 1H); 6,8 (s, 1H); 6,0 (s, 1H); 7,17.7(m, 15H).
Затем повторяют вторую последнюю часть примера 9, используя соединения, представленные в таблице 40, в качестве исходных веществ, и таким образом получают следующие соединения (см. табл. 41).
(Ж
JT И li b-NH
-H сооснркг
69 R I Примечание
Примечание.
в течение 10 мин. 10%-ный (вёс./об. водный раствор добавляют затем к смеси при рН 7, желтый осадок собирют, промывают водой и сушат,
спектр в
Н) 1,9 (s, ЗН) ; 2,4 (т, 4Н) ,- 3,5 (т, 2Н); 4,6 (s, 2Н); 5,2 (d, ) , 5,4 (d, IN); 6, (s, IN); 7,з(з, 1 q H) .
1031408О
Таблица 41
Пример 23. Аналогично способу примера 1 или 7 с использованием соответствующего дифенилметилового или трет-бутилового эфира в качестве исходного вещества получают следующие соединения (см. . 42).
Н
Яз
COjH5,6,7,8
9,2,7,10
CF,11,2,12,13
CHjOH11,2,7,14
Примечание.
(га, 2Н)/ 3,9 (s, ЗН) 4,3 (s, 2Н); 5,2 (d, 1Н); 5,75 (d, 1Н); 6,5-7,3 (т, ЗН); 7,85 (d, 1Н).
в спектре ЯМР в смеси d.OMCOJ . 2,04 (s, ЗН), 3,6 (m, 2Н) 4,72-5,1 (о, 2Н); 5,28-5,82 {q, 2Н) 6,7-7,5 (т, ЗН); 9,05 (пг, 1Н).
в спектре ЯМР;.- в ; 2,1 (s, ЗИ)) 3,7 (о, 2Н); 4,80-5,12 (q, 2Н); 5,34-5,90 (q, 2Н); 7,4-7,8 (т, ЗН).
Исходные вещества, используемые в указанном процессе, получают аналогично описанному в конце примеров 1,5 и 10 (реакция соответствующего замещенного о -фениле.ндиамина с соотввтствунвдим дифенилметилом или fper -бутил-7-дибромметиленамин З замещен ный-гце ф- 3- ем- 4 - к арбок си л ). Таким образом получают следувхцие соединения (см. табл. 43);
Таблица 43
СН,ОСОСН CO,CvHq-TpeT трйт С Нэ 1,6,3,7,8
CH OCOCH FCHPh2l,6,3,7,9
Прод олжение тaбЛ..
CHjOCOCHj CFjCHPh2l,6,3,10,ll
CHjOCOCHj CH,OH CHPh,l,2,3,12,13
Примечание. Д. Реакцию проводят в безводном ТГФ.
.2. Реакцию проводят в течение 1 ч при комнатной температуре.
в dgOMSO J 3,7 (q, 2Н) ,- 3,96 (s, ЗН); 4,22 (m, 2Н)- 5,24 (d, 1Н) 5,9 (d, 1Н); 6,4-6,7 (т, ЗИ); 6,95 (s, l)f 7,2--,6 (m,OH); 8,7 (т. 1Н). .
(d, 1Н); 7,85 (m, 2Н).
до 7:3.
13Н)..
91:6:3.
(q, 2Н),- 5,1-5,93 (q, 2Н),- 6,92 (s, 1Н) 7,38 (m, ИН).
Используелйлй, как указано выше, 3,4-диаминобензол-1-карбоксилат получают следующим образом.
К суспензии 3,4-диаминобензойной кислоты (3,4 г) в диоксане (100 мл) и концентрированной (10 мл) при О С добавляют изобутилен (50 мл) Смесь вст6 яхивают в течение 20 ч в автоклаве при комнатной температуре, затем вливают в 200 мл воды. В полученной смеси устанавливают рН 10 и зкстрагируют три раза СИСЕ,. Объединенные экстракты сушат и концентрируют с получением 2,5 г тре-с -бутил-2,4-диаминобензол-1-карбоксилата, который используют без дальнейшей очистки.,
Пример 24. К перемешиваемой суспензии пивалоилоксиметил 7-амино-З-метилцеф-З-ем-4-карбоксилат толуол- ndpct -сульфоната в этилацетате до бавляют бикарбонат натрия (0,336 г) в воде. Отделяют органический слой, высушивают над MgS04 и добавляют 1 эквивалент НС С в диэтиловом эфире. Смесь выпаривают до сухого остатка, в который затем добавляют сухой ДМФ (3 мл) и 2-хлоробензимидазола (1,218 Смесь перемешивают при в течение 24 ч и затем выпаривают до сухого остатка. Остаток растворяют в . СН2СEj промывают водой, органический слой высушивают и концентрируют, продукт очищают хроматографически на силикагеле с использованием ,/ /МеОН/НОАс (98,5:1:0,5) в качестве элюента. Полученное масло затем очищают высаживанием из раствора диизопропиловым эфиром с получением пивалоилоксиметил-7-.(бензимилазол-2-ил)-амино-3-метилцеф-3-ем-4-кар оксилат гидрохлорида (14%), имеющего следующие линии в спектре ЯМР в d DMSOi 1,1 (s, 9Н),- 2,05 (s, ЗН); 3,65 (q, 2Н); 4,25-4,8 (q, 2Н); 4,75 Cq, 2Н); 6,9-7,4 (m, 2Н).
Описанный процесс повторяют с использованием эквивалентного количества 2-хлор-5-нитробензимидаэола вместо 2-хлорбензимидазола и при температуре реакции 50. Продукт очищают хроматографически при пониженной температуре на силикагеле с использованием CH2CZ,/EtOAc (70:30 о/о в качестве элюента, растворяют в СН2СВ2 и отфильтровывают, затем подвергают хроматографии при пониженной температуре с использованием в качестве элюента CHjCtj (диэтиловый : эфир) МеОН (69:30:1, о/о/о) с получением пивалоилоксиметил-7-(5-нитробензимидазол-2-ил)-3-метилцеф-3-ем-4-карбоксилата (25%), имеющегЬ следющие линии спектра ЯМР в d DMSO: J 1,15 (s. 9Н)- 2,05 (s, ЗН),- 3,5 (q, 2Н); 5,25-5,8 (q, 2Н),- 5,8 (m, 2H)j 7,35-8,0 (m. ЗН), 8,5 (m, 1Н).
Пример 25. Аналогично примерам 1-7 с использованием соответствующего сложного эфира трет -бутила в качестве исходного вещества получают следующие соединения (см. табл. 44).
Таблица 44
Примечание.
Исходные вещества, используемые в описанном процессе, получают следующим образом.
Аналогично описанному в примере 24, но с использованием соответствующих трёт -бутил-3-замещенный 7-аминоцеф-3-ем-4-карбоксилат толуол- пари-сульфоната и 2-хлор-5-нитробеизимидазола получают следующие хзоединения (см. табл. 45) .
ft
Примечание. 1. Продукт очищают при понижен,ных температурах хроматографически с элюированием смесью CH;2CE2/EtOAc/ 65 /НеОН 30:70:0,2 (о/о/о), остающийся 2-хлор-5-нитробензимидаэол элими- нируют с добавлением и фильтруют.
Соединение имеет сшедуюсцие линий в спектре ЯМР в CDCpj + 1,55 (s. 9Н); 2,12 (s, ЗН)) 3,4 (q, 2НЬ 5,22 ( d, 1Н)- 5,83 (d; 1Н); 7,2-8,5 (m, ЗН).
Затем нитрогруппу переводят в амино- или ацетиламиногруппу с получением соединений, указанных в табл. 46.Та б л и ц а 46
Примечание
хроматографнчески на силикагеле с использованием tj,/МеОН/НОАс (97:2:1 до 78:14:8, о/о/о) в качестве элюента. Продукт имеет следующие линии в спектре ЯМР в «kDHSO: 1,5 (s, 9Н);- 2,05 (3H)f 3,55 (s, 2И) 4,6 (q, 2Н)) 5,2 (q, 1Н),- 5,8 (IH)/ 6,1-6,9 (m, ЗН).
К раствору трвт-бутил-3-ацет- оксиметил-7- (5- аминобензимидазол-2-ил)аминоцеф-З-ем-4-карбоновой кисло ты. (0,521 г) B CHjeEj, (100 мл) и CHjCOOH (2 мл)добавляют 1 эквивалент уксусного ангидрида. Смесь тщательно перемешивают при 0°С, затем концентрируют и остаток хроматографически обрабатывают на силикагеле при -20 С с использованиш4 CH2CE2/MeOH/flOAc (96:2:2, о/о/о) в качестве элюента. Продукт имеет следуюгдае линии в спектре ЯМР d,DMSO-. 1,55 (s, 9Н), 2,05 (s, ЗН) ; 2,1 (s. ЗН); 3,5 (m, 2Н); 4,9 (m, 2Н); 5,2-5,6 (q, 2Н); 7,0 (s, 2Н) 7,65 (т, 1Н); 9,6 (т, 1Н).
Пример 26. Аналогично описанному в примерах 1-7 способу с использованием соответствующего дифенилметил- или трет -бутилового сложного эфира в качестве исходного вещества, получают следующие соединения (см. табл. 47). Примечание. 1.Продукт получают ешедующим рбраз.ом.. К раствору TfJer -бутил-3-метил-7- (5-нитробензимидазол-2-гил) аминоцеф-3-ем-4-карбоксилата (0,21 г) в ТГФ (3 мл) и НеОН (3 мл) при О добавляют б эквивалентов 15%-ного (вес.обЛ водного раствсфа Т i С Е, К смеси добавл5ШТ НаЙСО (0,756 г) и устанавливают рн 7 с использованием (вес./об.) водного раствора бикарбоната натрия. Осадок собирают и очищают при пониженной температуре хроматографически на силикагеле с использованием в качестве «люента СНгСЕг/ еОН/НОАс (97:1,5: : , о/о/о). Соединение имеет сшёдукщие линии в спектре ЯМР в d BHSOi 1,5 (s, ЭН); 2,0 (s, 3H)i 5,15-5,75 (q,2H)j 6,2-6,9 (m, ЗН); (2Н резонанс замаскирован растворителем). 2.Продукт получают аналогично, описанному в примечании 1, но с использованием трет -бутил-3-ацетоксиметил-7- 1(5-нитробензимидазол-2-ил) аминоцеф-З-ем-4-карбокснлата. Продукт очищают при пониженной температуре 65 ТФА ной реа про -кол дит примечание. 1.Реакцию проводят в смеси /анизол. 2.Реакцию проводят при комнаттемпературе в течение 20-40 мин. 3.Продукт очищают выпариванием кционной смеси и высаживанием дукта из раствора в минимальном ичестве ИеОН диэтиловым эфиром. 4.Продукт, гидратированная соль рифтсфацетата, имеет следующие
лики; э спектре ЯМР в drDMSO: 2,01 (s, ЗН); 3,55 (m, ЗН); 4,04 (m, 2Н)/ 4,68 и 5,01 q, 2Н),- 5,22 и 5,85 (q, 2Н); 7,4-7,7 т, ЗН).
Использованные в описанном выше процессе вещества получают следующим образом.
Вначале получают замещенные о -фенилендиамнны, указанные в табл. 48 (t - трет ).
КСНг
Таблица 48
2,3
Продолжение табл. 48
2,4,5,6
Н
0 -rNH-C-CHj Н-С-ОСдН 2,4,7,8
Прим е ч а н и е.
5 (вес./об.) NaOH и экстрагируют . (трижды) диэтиловым эфиром. После вырушивания и концентрирования получают 0,35 г 1-амиио-2-нитро-4-аминометил-бензола.
К переманиваемому раствору этого соединения в сухом диоксане (10 мл) добавляют 0,258 г 2-( -бутокси-карбонилоксиамино)-2-фенилацетонит-, рила. После выдержки в течение 20 ч при комнатной температуре смесь концентрируют и остаток очищают хроматографией на силикагеле, элюент C. (диэтиловый эфир (90:10, о/о), с получением 1-амино-2-нитро-4-трет -бутоксикарбониламинометилбензола.
0,2 г этого соединения восстанавливают водородом в ТГФ/диэтиловом эфире при атмосферном давлении на 10% (вес./вес.) Pd/C с получением 0,18 г 1,2-диамино-4-т;реГ -бутокси5 карбониламинометилбензола.
5 водного раствора. Смесь нагревают до комнатной температуры, концентрируют, в вaкyy fe, затем нейтрализуют при 0°С коицёнтрированным . TiO отфильтровывают, |1ильтрат экстрагируют EtOAc, сушат MgSO и концентрируют с .получением сирого образца 1,2-дяамино-4-азидометилбензола. ИКС (пленка) i), 3300-3450, 2100. 4.В реакторе, работающем под давлением/ суспендируют соединение, полученное по примечанию 2 (1о г), в 400 мл НеОНг насг ценного NH (400 мл); смесь встряхивают при О в течение 20. ч. После фильтрации и частичного концентрирования « шьтрат обрабатывают при 0°С раствором НС ЕВ диэтиловом эфире 12% (вес./од После фильтрации и прокшвки эфиром получают 5,2 г l,2-дHнитpo-4-aминoмeтилбeнзoл гидрохдорида (5,2 г). При обработке водного раствора гидрохлорида основанием до установления рН 10 получают свободное основание, экстракция этилацетатом, осушка органической фазы над К2СОз и концентрирование. 5.Свободное основание, полученное по примечанию 4 (1,2 г ), добавляют к перемешиваемой смеси 1,5 г N- (трдт -бутил- оксйкарбонил) -Д1-фенилглицина, сухого (Ю мл), сухого ТГФ (15 мл) и 1-ЭТ-ОКСМ-1,2-дйгидрохинолина.(1,5 г). Через 1,5 ч смесь концентрируют, остаток растворяют в CHjCEi и раствор промы вают водной 2н. НСЕ, водным 5%-ным (вес./об.) NaHCOj, С5яиат над и концентрируют. Остаток очищают хро матографически на силикагеле с исггрл зованием tOAc/циклогексан (1:1/ о/о в качестве элюента с получением продукта (2,1 г). 6.Раствор соединения, полученног по примечанию 5, (1,8 г) в этаноле (30 мл) восстанавливают водородом в течение 1 ч при атмосферном давлеНИИ на 0,1 г каташизатора Pt02. Смес фильтруют и фильтрат выпаривают с получением производного о-фениленди амина (1,4 г), который используют бе дальнейшей очистки. 7.Процесс, описанный в примечаНИИ 5, проводят с использованием Н-(трет -бутилрксикарбонил)-глицина вместо Н-(трет -бутилоксикарбонил)Д1-фенилглицина. 8.Продукт, полученный по примечанию 7 (2 г), в ТГФ и этаноле (15 м и 30 мл соответствеино), подвергают каталитическому, восстановлению водородом (10% Pd/C) при атмосферном давлении в течение 3ч. После фильтр ции и концентрирования получают 1,5 вязкого масла. Это соединение исполь зуют без дальнейшей очистки. Затем проводят процесс аналогично описанно му в конце примеров 1,5 или 10 с исользованием соответствующих замещен ных о-фенилендиаминов и соответственно дифёнилметил или грет-бутил-7-дибромметиленамино- 3-аце.токсиметилеф-З-ем-4-карбоксилат. Один из проуктов этой реакции подвергают дальнейшим хикшческим превращениям. По-, учают следукяцие соединения (см. табл. 48). н нШ-t-fVу - С1%ОООСИз feoOR а б л и ц а 48 ю Примечание «-С-ОС Н|СНРЬз 1,2,3,4 5,2,6,7 NHCOCH, CHPhj 8,9 NH-C-|H-CgH5CHPhj 1,2,3,10 О « -tfH-C-CHjNHC-OC Hlt-C H 1,11,12,13-,14 ОО. Примечание. 1.Реакцию проводят в сухом ТГФ. 2.Реакцию проводят 20 ч при комнатной температуре. 3.Продукт очищают при пониженных температурах хроматографически элюирование СН,С -/МеОН/НОА ; (98: А .1,3: 0,7, о/о/о) . 4.Продукт имеет следующие линии в спектре ЯМР в dxDMSO: 1,4 (s, 9) 1,98 (s, ЗН)} 3,64 (m, 2Н); 4,13 (m, 2Н); 4,66 и 4/92 (q, 2Н) 5,3 (d. 1Н); 6,93 (s, 1Н) 7,12 (s. 1НЬ 7,2-7,6 (т, 13 Н). 5.Реакцию проводят в смеси МеОН/ТГФ (1:10, о/о). 6.Аналогично примечанию 3 при соотношении растворителей 995:5:1, о/о/о). 7.Продукт имеет следупцие линни в спектре ЯМР в diOMSO; 2,0 (s, ЗН) 3,7 (m, 2H)i 4,51 (s, 2Н); 4,,97 (q, 2Н); 5,37-6,0 (q, 2Н); 6,98 (s, 1Н); 7,1-7,6 (го, 13Н). 8.К раствору соединения по примечанию 7 (0,47 г)в этаноле (20 мл) и диоксане (10 мл) добавляют окись платины (0,2 г) и сямесь гидрируют при комнатной температуре и атмосферном давлении в течение 3 ч. Катализатор удаляют и смесь концентрируют с получением 0,42 г промежуточного продукта, имеющего R NH R CHPh. Этот промежуточный продукт раств ряют в сухом и добавляют 1 э вивалент уксусного ангидрида. Реакция протекает при комнатной темпера туре под атмосферой азота. Затем ре акционную смесь обрабатывают аналогично описанному в примечании 6. Ко нечный продукт дальше очищается высаживанием из CH2CP2/MeOH с эфиром. 9.Продукт - смесь изомеров д . ЯМР-спектр для изомера л з dftOMSO-. 1,87 (s, ЗН); 2,0 (s, ЗН) j 3;67 (m, 2Н); 4,3 (q. 2Н); 4,7 И 4,93 (q. 2Н); 5,35 и 5,90 (q, 2Н) ; 6,95 (s, 1Н); 7,4 (s, 13Н); 8,35 (т, 1Н). 10.Продукт имеет следующие лини в ЯМР-спектре в .; 1,36 Cs, 9Н)/ 1,90-1,98 (2s, ЗН)) 3,62 (m, 2Н), 4,3 (d, 2Н), 4,68 и 4,93 (q, 2Н); 5,15 (d, 1Н); 5,30 и 5,88 (q, 2Н); 6,91 (т, 1Н); 7,1-7,5 (т, 18Н) 8,45 (т, 1Н), 11.Реакцию проводят 7 ч при ком натной температуре. 12.Аналогично примечанию 3 при соотношении растворителей 990:5;5 (о/о/о). 13.Соединение затем очищают высаживанием из раствора в минимаяь.ном количестве СН СЕг/МеОН эфиром. 14.Продукт характеризуется следующими линиями в спектре ЯМР в d.DMSpi 1,35 (s, ЗН),- 1,5 (s, ЗН) (s, ЗН); 3,6 (т, 4,3 (dd 2Н),- 4,63 и 4,94 (q, 2Н) ; 5,2 и 5,8 {q, 2Н), 6,7-7,1 (т, Н); 8,05 (т, 1Н). Пример 27. Процесс проводят аналогично примерам 1 или 7с использованием соответствуквдего трет -бутилового сложного эфира в качестве исходного вещества ,при эт получают следующие соединения (см. табл. 49) . Н -NV S j,H,ix% снгосос RCHj N Таблиц примечание. 1 Реакцию проводят 2,25 си ТГФ/анизол (1:1 о/о) при ной температуре. 4
{Примечание
R
1,2,3,4
CN СОМЩ 1,5,6,7,8 ч в сме-1. Реакцию проводят в сухом ТГФ. комнат-2. Реакцию проводят 5 ч при ком2.Реакционную смесь выпаривают, статок высаживают из раствора в СНАСЕ,,/МеОН эфиром. 3.Продукт, соль трифторацетата, характеризуется следующими линиями в спектре ЯМР в d OMSOi 2,03 (s, ЗН); 3,42 и 3,67 (q, 2Н); 4,01 Й-к 2Н)г 4,71 и 5,01 (q, 2Н); 5,22jd, . 1Н); 5,80 (d, 1Н); 6,90-7,45 (трЗН) 4.Реакцию проводят 1,5 ч при комнатной температуре в ТФА. 5.Продукт, соль трифторацетата, характеризуется следующими линиями в спектре ЯМР в d,.DMSO : 2,07 (s, , ЗН),- 3,39 (s, 2И)/ 3,0-4,0 (m, 2Н) ; 4,7 (d, 1Н); 5,15 (d, 1Н),- 5,24 (d. 1Н); 5,86 (т, 1Н); 6,65-7,45 (т, 7Н) Исходные вещества, использованные в указанном выше процессе, получают следующим образом. Раствор 1-амино-2-нитро-4-цианометилбензола (6 г) в этаноле (100 мл) гидрируют Ij5 ч при атмосферном давлении в присутствии 3,6 г 10% (вес./ вес.) Pd/C. Катализатор удаляют и растворитель выпаривают с получением 1,2-диамино-4-цианометилбензола, который используют без дальнейшей очистки. К 1,03 г 1,2-диамино-4-1дианометилбензола (1,03 г) медленно добавляют концентрированную серную.кислоту (7 мл). Смесь нагревают до 90°С и вБщерживают при этой температуре 90 мин, затем вливают в 50 мл со льдом и устанавливают рН 8 с помощью NaOH. Осадок, который образовывался при охлаждении, удаляют и водную фазу концентрируют до сухого остатка Остаток экстрагируют ацетоном, экстракты концентрируют путем последующей хроматографии на силикагеле с использованием СН СЕг/НеОН (9:1 до 8:2 о/о). Получают 1,2-диамино-4-карбамоилметилбензол (0,65 г), который используют без дальнейшей очистки. Аналогично описанному в конце примера 1 с использованием соответствующих о-фенилендиаминов и трет-бутил-7-дибромометиленамино-3-ацет,оксиметилцеф-3-ем-4-карбоксилата в качестве исходных продуктов получают следующие соединения (см, табл.50). Н Н, -1Цг -сн20С оссн т а б л и Примечание. 65 натной температуре.
на силикагеле с использованием ё качестве элюента смеси CHjCEo/MeOH/ /НОАс (95:4:1, затем 97:2,5:0,5, о/о/о). .
П р и м е р 28. Процесс проводят аналогично описанному- в примере 1 с использованием в качестве исходного вещества дифенйлметил-7-(имидазол-2-ил)амино-3-ацетоксиметилцеф-З-ем-4-карбоксилата. Получают следующее соединение:
/NHИ Н:|Г - н-4-ГЧчo - VСНгОСОСНз
СООН
Примечани е.
В описанном выше процессе в качестве исходного используют вещество полученное следующим образом.
К перемешиваемому раствору дифенилметил-7-(1-окси-2-имидазол-2-ил)-амино-3-ацетоксиметилцеф-3-ем-4-карбоксилата, полученного аналогично описанному в примере 13, (0,174 г в СН2СЕ2 ( мл) при -40°С добавляют 2-фтор- 1-метилпиридинийтолуол-пвра-сульфонат (98 мг) , затем свежего, отдистиллированного триэтиламина (147 мкл) в сухом 6,(1 мл) Через 1 ч при добавляют ТФА (90 мкл)о смесь разбавляют дважды промывают водой и органическую фазу сушат над MgS04. Раствор концентрируют и остаток хроматографируют при низкой температуре над силикагелем, элюирование с использованием CHgCl,/MeOH/HOAc (lOOiOiO до 92:4:4, о/о/о). Получают дифенилметил-7-( 2-имидазолил)-амино-3-ацетоксиметнл-цеф-3-ем-4-кар6оксилат(О,105 г), характеризуквдийся спедующими ЛИНИЯМИ в спектре ЯМР в
dfeDHSO 4 CD.COOD: 2,0 (s, зн); з,7
(s, 2И) 4,7 (d, 1Н), 4,9 (d, 1Н) k 5,4 (d, Н); 5,7 (d, 1Н); 6,9 (s, 1Н); 7.0 {s. 2Н); 7,3 {s, ЮН). Пример 29.л
н н s
П 3
NH-J--HN ,, N-Jt CH2S- - ОНз
Vт«
ш
COOCHjOCOCHj
Раствор ацетоксиметил-7-амино-З-(2-метил-1,3,4-тиадазол-5-нл)тиометилцеф-З-ем-4-карбоксилата (0,28 г) .и гидрохлорида 2гфторимидазола (0,1 г) в сухом ДМФ (1 мл) нагревают при 2 ч. Реакционную смесь концентрируют в BaKyj e и остаток
подвергают хроматографии на силикагеле, элюирование CH CEj/HeOH (95j 5, о/о). Очищенное соединение затем обрабатывают одним эквивалент(М«1 HCt в ИеОН,- добавлением полученного
раствора в сухой эфир получают высаживанием гидрохлорид. Продукт, ГИД
рохлорид ацетоксиметил-7-(имидaзpл-2-ил) -aминo-3-(2-мeтил-l,3,4-тиaдиaзoл-5-ил)тиoмeтилцeф-3-eм-4-кapбоксилата, характеризуется следующими
пиками в спектре ИК (KBr)v, 1780 (широкий), 1740 (расщеплен), 1655 (узкий) и следующими линиями в спектре ЯМР в dgpHSO + CF COOD: 2,15 (s, ЗН); 2,7 (s, ЗН); 3,8 (brs,
2Н); 5,3 и 5,8-(q, 2Н),- 7,1 (s, 2Н), другие резонансы маскируются растворителем или плохо разрешимы.
Исходный сложный эфир, используемый в описанном процессе, получают
следующим образом.
К раствору NaCE (3 г) в ацетоне (6 мл) добавляют хлорметилацетат (2,17 г). Смесь переманивают 1 ч при
комнатной температуре и затем добав ляют раствор 3-(2-метил-1,3,4-тиaдиa 3 ол- 5 - ил ) ацет и л ами ноцеф- 3- ем- 4 - карбоксилата натрия (4,7 г),в DMSO (6 мл). Смесь нагревают при в течение 5 ч, ацетон выпаривают и
остаток добавляют в 200 мл воды. После растирания получают порошок, который отфильтровывают и промывают эфиром, i Влажный продукт очищают путем хроматографии на силикагеле с эЛ)оиро.
ванием CHjCEj/HeOH (95:5, о/о) . Полученный ацето.ксиметиловый сложный эфир характеризуется следующими линиями в спектре ЯМР в d(,DMSO CD,00 2,1 (s, ЗН); 2,7 (s, ЗН) 3,8 (т,
2Н); 4,2 и 4,7 (q, 2Н),- 5,2 (d, 1Н).,
5,45 ($, 2Н); 5,80 (d, 1Н), 5,9 (m, IH); 9,3 (s, 1H).
К раствору РСЕу (2,08 г) в сухом СН2СЕ2 {12 мл) добавляют хинолин (1,93 г). Образующуюся суспензию охлаждают до и порциями добавляют указанный ацетоксиметиловый сложный эфир (2,63 г). Черную смесь нагревают до комнатной температуры и после одного часа перемешивания образующегося раствора его добавляют под азотом в раствор 1,3-бутандиола (3 г) в CHXEj (5 мл), охлажденного до -il5°C. Смесь перемешивают 2 ч при комнатной температуре и снова добавляют CH CEjdOOмл). Полученный осадок отфильтровывают с получением гидв охлорида ацетоксиметил-7амино-3-(2-метил-1,3,4-тиадиазол-5-ил)тиометилцеф-3-ем-4-карбоксилата (1,8 г), характеризующегося следующими частичными линиями в ЯМРспектре в + СОзСООО; 2,1 (s, ЗН); 2,7 (s. ЗН); 5,2 (brs, IH); 5,7 (brs, IH).
При добавлении триэтиламина к суспензии гидрохлорида в смеси води/ /СН2СЕ2 до установления рН 8 получают свободное основание. Органическую фазу затем отделяют и выпаривают
Пример 30. К раствору гидрата толуол-порч-сульфоновой кисло- ты (54 мг) в сухом даф (1 мл) добавляют 2-фтор-1-трифенилметилимидазол (110 мг) и раствор нагревают в бане предварительного нагревания до вО®С. Через 5 мин для завершения образования добавляют 2-фторимидазол-7-амино -3-(1,3,4-тиадиазол-2-ил)тиометилцеф-З-ем-4-карбоновую кислоту. Нагревание продолжают 2,5 ч. Затем добавляют следующую порцию фторимидазола (50 мг) , нагревание гтррдолжают следующие 30 мин, смесь охлаждают и выпаривают при комнатной температуре. К остатку добавляют воду (10 мл) и этилацетат (25 мл), смесь фильтруют и разделяют фазы. Водный слой конценрируют до 4 мл, фильтруют и подвергают препаративной жидкостной хроматографии под высоким давлением на Ватмане Партисил 10 с использрванием в качестве элюента смеси вода/ /МеОН/НОАс (80:20:1, о/о/о). Продукт кристаллизуют с добавлением ацетона. Затем его промывают ацетоном и эфиром и получают 7-(имидазол-2-ил)-aiuMHO-3-(l, 3,4-тиадиазол-2-ил)тиомётилцеф-З-ем-4-карбоновую кислоту (15 мг), характеризующуюся след у1ощи-. ми линиями в спектре ЯМР в + + еОзСООО; 3,52 (d, 1Н) 3,79 (d, 1Н),- 4,33 (d, 1Н); 4,6 (d, 1Н) 5,12 (d, 1Н) ; 5,58 (d, 1Н)/ 6,83 (s, 2Н)/ 9,49 (s, 1Н).
Аналогично описанному процессу с использованием 4-карбокси-2-фтор-1-трифенилметилимидазола, 4-этоксикарб6нил-2-фтор-1-трифенилметилимидазола и З-ацетоксиметил-7-аминоцеф-З-ем-4-карбоновой кислоты в качестве исходных веществ получают следующие соединения .(см. табл. 51).
Н Н
t
нн4-Г
-Мх СНгОСОСНз
соон
Таблица 51
Примечание.
Продукт характеризуется следующими линиями в ЯМР-спектре в d(,DMSO; 1,22 (t, ЗН); 2,03 (s, ЗН) 3,323,61 (q, 2Н),- 4,17 (q, 2Н) ; 4,564,93 (q, 2Н); 5,14 (d, 1Н); 5,67 (q. IH); 7,01 (d. IH); 7,28 (s, IH).
dfcOMSO + СОзСО О; 2,03 (s, ЗН); 3,43-3,6 (q, 2Н); 4,72-5,0 (q, 2Н); 5,16 (d, 1Н) ; 5,71 (d, 1Н); 7,24 (s, 1Н).
Исходный имидазол получают следующим образом.
К раствору 2-фторимидазола (4,45 г) в (100 мл) и триметиламине (7,93 мл) добавляют трифенилметилхлорид (14,4 г), смесь перемешивают 2,5 ч. Раствор промывают водой и рассолом,сушат (MgSO), обрабатывают обесцвечивающим активированным углем, фильтруют и выпаривают Сухой остаток растирают с эфиром и затем метанолс 1 с получением 2-фтор-1-трифенилметилимидазола (13,6 г), т. пл. 182-185°С.
Раствор 2-фтор-1-трифенилметилимидазола (3,28 г) в сухом ТГФ (33 мл) обрабатывают под аргоном при -75с двумя эквивалентами трет -бутилатом лития (10 мл, 1,93 М раствора в пентане). После перемешивания в течение 3 ч при -75°С добавляют даф (1,5 мл). Реакционную смесь выдерживают еще 1 ч при -15°С, затем медленно нагревают до комнатной температуры. Реакцию завершают разбавлением эфиром, промывкой 2н. НСЕ и затем рассолом. Эфирный слой концент рируют под потоком аргчэна с получением 4-формил-2-фтор-1-трифенилметил имидазола (2,2 г), т, пл. 177-179 с. Раствор 4-формил-2-фтор-1-трифенилметилимидазола (356 мг) в этаноле (5 мл) и , (3 мл), обра атывают нитратом . серебра {0,37 г) в воде (0,5 мл), затем по каплям добавляют 5 мл раствора гиадюокиси калия (5 МП раствора 2,1 КОН в 35 мл воды). Смесь перемешивают при комнатной температуре 2 ч, фильтруют и экстрагируют фильтрат эфиром. Водный слой обрабатывают концентрирован ной НС1 с образованием кислой среды и экстрагируют CHCI. Органический слой сушат над , фильтруют и растворитель выпаривают с получением 4-карбокси-2-фтор-1-трифенилметилимидазола (261 мг) в виде белого сухого вещества, характеризующегося следукидими линиями ЯМР-спектра в dfeDMSO:. 7,0-7,68 (m, 16Н); 11,5-12, (Ьг , ГН). Раствор 4-карбокси-2-фтор-1-трифеиилметилимидазола (280 г) в ТГФ (0,75 мл) . обрабатывают под аргоном 1,5-диаза-бицикло-5,4,О-ундец-5-еном (0,112 мл), затем йодистым этилом (0,069 мл). Смесь перемешивают 2 ч при комнатной температуре, затем добавляют воду и jCMecb экстрагируют эфиром. Эфирный экстракт сушат (MgSO) и выпаривают с получением 4-этоксикарбонил-2-фтор-1-трифенилметилимидазола (185.мг) в виде желтой пены, характеризующейсй следующими линиями в ЯМР-спектре в CDCEa 1,38 (t. ЗН); 4,36 (q, 2Н) ; 7.,0-t7,5 (m, 1бН). П р И м е р 31. Суспензию безвод ной толуол-паре -сульфоновой кислоты (0,74 г) и З-ацетометил-7-аминоцеф-З-ем-4-карбоновой кислоты (1,17 г) в сухом ДМФ (175 мл) перемешивают 15 мин при комнатной температуре.с образованием частичного раствора. Затем добавляют еще порцию 2-фтор|имидазола (0,74 г) и смесь перемешивают при 90°С 2 ч. Растворитель выпаривают При комнатной температуре, добавляют 2%-ного (об./об.) водного НОАс (20 мл) к осадку и смесь экстра гируют этилацетатом (20 мл). Водный слой концентрируют до 15 мл, фильтруют и фильтрат очищают препаративной жидкостной, хроматографией высокого давления на Ватмане Партисил 10 с использованием в качестве элюата смеси вода/МеОН/НОАс (о/о/о). Затем продукт очищают растиранием с ацетоном и промывкой ацетоном и эфиром с получением 3-ацетоксиметил-7-(имидазол-2-ил)аминоцеф-3-ем-4-карбоновой кислоты (0,42 г) в виде гидратированной смешанной соли ацетат/толуол-пора -сульфоиата, имещей т. пл. 160°С (разл.) и хаактеризующейся следующими линиями в ЯМР-спектре в 2,28 (s, ЗН); 3,58 (d, 1«); 3,89 ( d, 1Н); 4,92 (d, 1Н); 5,13 (d, 1Н); 5,42 (d, 1Н) 5,70 ( d, 1Н),- 7,08 (s, 2Н). Аналогично описанному процессу с использованием соответствухдаих 7-аминоцефалоспориновых производных получают следующие соединения см. табл . 52), н н н г f -ш Таблица 52 I Примечание снг-з - N М-и- К-СНгСООН H-N CH2-S У . Примечание. 1., Реакцию проводят при 85°С в течение 3ч. 2.Продукт очищают выделением остатка из реакционной смеси в дистиллированной воде, фильтрованием и экстрагированием этилацетатом. Водный слой осветляют активированным углем, устанавливают рН 6 водным раствором NaOH и снижают объем до 2 мл. Затем продукт кристаллизуют. 3.Продукт имеет т. пл. 203220°С (разл.).. 4.Продукт очищают препаративной жидкостной хроматографией высокого давления на Ватмане Партиснл 10 с использованием в качестве элюента вода/МеОН/НОАс (70:30:1, о/о/о). 5.Продукт, дигидрат, характеризуется следующими линиями в ЯМРспектре в 020 + ТФА: 3,3 (d, 1Н);. 3,64 (d, 1Н); 3,92 (d, 1Н), 4,26 (d 1Н); 4,59 (s. 1Н); 4,93 -(d. 1Н), 5,20 (d, 1Н); 6,56 (s, 2Н); 6,75 ( d, 1Н), 7,25 (d, 1Н). 6.Продукт очищают препаративной ЖХ ВД на Ватмане Партисил с использованием смеси вода/МеОН/НОАс . Продукт кристаллизуют при обработке ацетоном. 7.Продукт, соль толуол-noipaсульфоната, характеризуется следующими линиями в спектре ЯМР в dfiDMSO + CDjCOzDj 2,32 (s, ЗН)j ,3,64 (d, 1Н); 3,9 (d, 1Н); 4,19 4d, 1Н); 4,46 (d, 1Н); 5,05 (s, 2Н) 5,17 (d. 1Н); 5,57 (d,, 1Н); 7,06 (s, 2Н),- 7,14 (d, 2Н); 7,54 (d, 2Н) Пример 32. К суспензии 3-ацетоксиметил-7-(имидазол-2-ил)аминоцеф-З-ем-4-карбоновой кислоты (39,8 мг) в сухом ацетонитриле (200 мкл) добавляют 1-метил-1Н-тетразол-5-тиол (12 мг) и непосредст, венно далее трифторид-эфират бора в эфире 48% (вес./вес, 31 мкл). Образуется чистый желтый -раствор, кот рый пере; 1ешивают при комнатной температуре 0,5 ч и затем при 2,5 ч. Растворитель удаляют под по ниженным давлением, остаток растворяют в смеси воды (900 мкл) и элюен та (400 мкл), смесь фильтруют и очи щают препаративной ЖХВД на Ватмане Партисил 10 с использованием вода /МеОН/НОАс (80:20:1, о/о/о). Таким образом, получают 7-(имидазол-2-ил) -амино-3- (1-метил-1Н-тетразол-5 ил) тиометилцеф-3-ем-4-карбоновую киело ту в виде кристаллического твердого вещества, идентичного продукту, пол ченному по примеру 31. Аналогично описанному процессу с использованием соответствующих тиолов в качестве исходных веществ, получают следующие соединения ;(см. табл. 53) . Н Ч1Г Л Таблица 53
К-N
1,2,3
N
N-N CHz-S- 3
4,5,6
сн(сн,)г
используют 1-(2,2,2-тpифтopзтиJr)-lH-тeтpaзoл-5-тиoл, который получ«ют следующим образом.
ct Смесь 2,2,2-трифторэтилизотитцианата (1,8 г) и азида натрия (1,25г) в воде (9 мл) нагревают на паровой бане в течение 18 ч, после охлаждения смесь экстрагируют эфиром (20 мл) и устанавливают рН 2 конценУ
60 рированной HCt в присутствии EtOAc (20 мл). Экстракт EtOAc сушат (МдЗОц) и выпаривают с получением кристаллического сухого остатка, который растирают с петролейным эфиром (т. ккп. 60-80 0) с получением Продолжение табл. 53 CHg-S -. N CHjCFj 7,8,9,10 N-N CH2-S-, Jhr 11,12,13 CHgCHgSCHj Примечание. 1.Реакцию проводят при в течение 5ч. 2.Продукт очищают препаративной ЖХВД на Ватмане Партисил 10 с использованием вода/МеОН/НОАс (60:40:1, о/о/о). 3.Продукт, частично гидратированный, (37 мг) имеет т. пл. 244с и характеризуется следующими линиями в спектре ЯМР в ОьО + ТФА$ 3,03 (d, 1Н), 3,32 (d, 1Н)7 3,58 (d, 1Н) 3,78 (d, 1H)j 4,95 (d, 1Н) ; 6,32 (s, 2Н). (один -лактамный протон затемнен резонансом при 4,6-4,8) 4.Реакцию проводя при 50 С в течение 3 ч. 5.Продукт очищают ЖХВД на Ватмане Партисил 10 с использованием смеси вода/МеОН/НОАс (75:25:1, о/о/о затем 60:40:1 о/о/о). 6.Продукт, гидрат (51 мг), имеет т. пл. 219-220®С (разл.) и характеризуется следующими линиями в спектре ЯЙР в ОтО + TФAi 1,50 (d, 6Н); 3,6 (d, 1Н); 3,83 (d, lH)i 4,2 (s, 2Н), 4,6-5,1 (m, 1Н); 5,48 (d, 1Н)/ 6,82 (s, 2Н). (один р.лактамный протон затемнен резонансом HgO). 1- (2,2,2-трифторэтия} -IH-Teipaaofl-5-тиола ( г), т. пл. IIS-IIS C. 8.Реакцию проводят в течение 1 ч при . 9.Продукт очивоают ЖХВД на Ватма не Партисил 10, используя смесь вода/МеОН/НОАс (60:40:1, о/о/о). 10.Продукт (выход 68%) получают частично сольватированным в ацетоне он характеризуется следунщими линиямя в спектре ЯМР в О «О + ТФА, 3,3 (d, 1Н); 3,60 (d, 1НЬ 4,14 (s, 2Н) 4,98 (d, 1Н), 5,0 (q. 2Н); 5,26 (d, 1Н); 6,64 (s, 2Н). 11.Реакцию проводят 2 ч при 40 С, затем 2 ч при 60°С. 12.Продукт очищают ЖХВД на Ватмане Партисил 10 с использованием смеси вода/МеОИ/НОАс (60:40:1, о/о/о 13.Продукт (40%) кристаллизуют путем удаления растворителя. Он характеризуется сле кщими линиями в спектре ЯМР в dgOMSO + CD COgD: 2,05 (s, ЗН)) 2,97 (t. 2Н),- 3,52 (d, 1Н)/ 3,78 ( d, 1Н 4,36 (bs, 2Н); 4,51 (t, 2Н); 5,09 (d, 1Н); 5,52 (di, 1Н); 6,82 (s, 2Н). Пример 33. Раствор 0,18 г пивалоил-оксиметил-3-метил-7-аминоцеф-3-ем-4-кар6оксилата.(0,18 г) и гидрохлорида 2-фторимидазола
Таблица 54 (0,14 г)-В ДМФ (1 мл), и ацетонитри ле (1 мп) нахревают при 7 ч. После выпаривания остаток обрабатывают хроматографическина силикагеле с использованием в качестве элюента CHgCej/MeOH (95:5, о/о). Маслянистый продукт «обрабатывают одним эквивалентом ЙСЁ в НеОН. Раствор выпаривают и остаток растирают с эфиром и фильтруют с получением 1Ч5дрохлорида пивалоил-оксиметил-7- -(имидазол-2-ил)aминo-3-мeтилцeф-3-eм-4-кapбoкcилaтa, характеризующегося следующими линиями в спектре ЯМР в + COjCOOO: 1,2 (s, 9Н); 2,1 (s, ЗН); 3,6 (m, 2Н); 5,25 (d, IH); 5,7 (d, 1H) 5,85 (m, 2H), 7,05 (s, 2H). Пример 34. Аналогично процессу, описаннс 1у в примере 7, с использованием соответствукяцих трет-бутиловьос сложных эфиров в качестве исходных веществ получают следующие соединения (см. табл. 54) N S / -1ш4-4 Т5
СН,
CHjOCOCH-
. JN-CH U-CH;
4,2,5
Примечание. 1. Реакцию проводят в смеск ТФА/ /анизол (3:1 о/о) в течение 45 мин.
2,05 (s, ЗН); 3,2-3,8 (m, ЗН) ; . 4,0 (t, IH),- 4,9-5,1 (m, ЗН) , 5,45 (br, 1H) 6,8 (d, 2H); 7,2 (d, 2H).
Первое исходное вещество, используемое в указанном пр.оцессе, получают следующим ,
К пед)емешиваемой суспензии п-бен3ил-оксибензальдегида (21,2 г) в метаноле (400 мл) при 5с по каплям добавляют нитрометан(6,1 г). После перемешивания 45 мин при в реакционную смесь добавляют при перемешивании 5н. НСЕ (25 мл). Полученный осадок прерывают водой и перекристаллизоЕывают из этанола (400 мл) с получением 1-нитро-2-(п -бензилокси)-фенилэтилена (-18,7 г), характеризующегося следующими линиями в спектре ЯМР в dgDMSOt 5,2 (s, 2Н) , 7,15 (d, 2Н); 7,45 (s, 5Н);7,85 (d, 2Н); 8,15 (s, 2Н).
Суспензию указанного ничроэтилена (1,275 г) в метаноле (30 мл) добавляют к трем эквивалентам гидроксиламина (в виде гидрохлорида) в метаноле (10 мл). Смесь перемешивают 2ч, выпаривают до сухого остатка, котсрый затем вносят в 5 смесь СНСЕз/этанол (9:1 о/о). Раствор сушат (MgS04) выпаривают с получением 1-оксиамино-1-(п -бензилокси)-фенил-2-нитроэтана, имекядего в спектре ЯМР следукяцие линии в dfeOMSOi 4,8 (ffl, ЗН); 5,15 (s, 2Н) , 7,05 (d, 2Н),- 7,4 (d, 2Н); 7,5 (s, 5Н).
Суспензию указанного производного нитроэтана (0,56 г) в этаноле (30 мл гидрируют при давлении 3 атм на никеле Рэнея и 10% (вес/вес) Pd/C в течение 18 ч при комнатной темпег ратуре. -Смесь фильтруют, фильтрат выпаривают и остаток растворяют в смеси СН.СЕ2/ЭФИР, содержащей минимальное количество МеОН с образованием раствс а. К этому раствору добавляют сухую метанольную ИСК и получают гидрохлс ид 1,2-диамино-1-(4-окси)-фенилэтана. Он характеризуется следующими линиями в спектре ЯМР в d,DMSO + СОзСООО; 2,9 (т, 2Н) 4,1 (т, 1Н),- 6,55 (d, 2Н) 7,0 (d, 2Н). .
Указанный диамин, в виде свобод(Ного основания вводят в реакцию с трет -бутил-З-метил-7-дибромметилен-аминоцеф-З-ем-4-карбоксилатом аналогично процессу, описанному в щ имере 14 (втекая часть). Продукт очищают хроматографией на силийагеле с использованием смеси СН СЕо/МеОН . (3;97, 5:95 и затем. 10:90, о/о) в качестве элюента с получением трет -бутил-З-метил-7- -4-(4-окси)тфенил-2-имидазапин-2-ил аминоцеф-3-ем-4-карбоксилата, имеющего следующие линии в спектре ЯМР в dj DMSOJ . 1,45 (s, ЗН),- 2,0 (s, ЗН); 3,2-3,75 (m, ЗНЬ 4,0 {t, 1Н); 4,9-5,15 (m, ЗН) 5,55 (d, 1Н); 6,75 ( d, 2Н); 7,2 (d, 2Н).
Второй исходный реагент для указанного процесса получают следующим образом.
К раствору трет-бутил-3-ацетоксиметил-7-(5-аминометилбензимидазол-2-ил)аминоцеф-З-ем-4-карбоксилата (93 мг) в диоксане (2 мл) и метаноле (3 мл) при добавляют диметокси-гексагидро-1Н-азепин-1-ил-метан{30 мг) и затем триэтиламин. Смесь охлаждают до 0°С к перемешивают 30 мин. Добавляют два эквивалента НВг в метаноле и раствс итель выпаривают. Остаток растворяют в минимальном количестве СНлСЕ и гидробро лдад триэтиламина осаждают путем добавления эфира. Продукт очищают гфоматографией на силикагеле магния с использованием а качестве элюента смеси СН2СЕ2/НеОН (98:2, о/о) с получением трет-бутил-3-ацетоксиметил-7-(5-гексагидро-1Н-азепин-1-ил-метилен-аминометил-бен зимидазол-2-ил)аминоцеф-З-ем-4-карбоксилата, имеющего cлeдykll иe линии в спектре ЯМр в dfeOMSO. 1,63 (s, 9Н); 1,7 (s, 8Н
2,17(s, ЗН); 3,3-4,0 (br, 6Н) ; 4,67 (s, 2Н), 4,75 (d, IH); 5,1 (d, IH), 5,38 (d, IH); 6,0 (d, IN); 6,8-7,5 (m, 3H) 8,43 (s, IH).
Пример .35. Аналогично процессу, описанному в примере 30, с использованием Соответствующих исходных веществ получают следующие соединения (см. табл. 55).
s s
(CHa)- Vlw4iri
соон
Таблица 55
Примечание
1
.2,3
CH2
2,4
Ы
Примечание.
(m, ); 4,9 (q, 2Н) ; 5,17 (d, IH),5,63 (q, IH); 6,61 (s, IH); 7,28 (q, 2H), 8,8 (s, tH).
воду/МеОН/НОАс (70:30:1, о/о/о).
в спектре ЯМР следующие характерные линии в dfcDMSO; 1,6г-1,9 (т, 2Н) , 12,5 (t, 2Н); 3,3-3,7 (т, ) ; 3,95
(s, ЗН); 4,34 (s, 2Н) ,- 5,08 (d, IH); 5,70 (q, IH);. 6,58 (s, IH); 8,65 (s, IH).
следукадие характерные линии в спектре ЯМР в d4,DMSOf, 1,6-1,9 (m, 2Н); 2, 44 (t, 2Н),- 3,42 (t, 2Н) j 3,59 (d, 2Н); 4,04 (s, 2Н); 5,08 (d, IH); 5,50 (d, IH); 6,57 (s, IH); 7,84 s, IH).
Используемый в качестве исходного вещества 2-фтор-1-трифенилметил-4-(З-окси)-пропилимидазол, получают сяедукядим . К раствору 2-фтор-1-трифенилмети -имидаэола (1,31 г) в ТГФ (22 мл) при -70°С под аргоном добавляют трет -бутилат лития (4 мл 2М раство ра в пентане). Красный раствор пере мешивают при в течение 2ч и затем добавляют йодид меди (0,78 г).Образовавшийся раствор темно-красного цвета перемешивают при в течение 1 ч и затем добавляют аллил-бромид (1,8 мл). Смес нагревают до комнатной температуры в течение 18 ч и затем вносят в эфир (150 мл). Смесь прогнивают насы щенным водным раствс ом аммония (б раз по 50 мл), затем рассолом (50 мл) обра батывают активированным углем и сушат (MgSOi). Рас вор выпфивают с получением 4-аллил -2-фтор-1-трифенилметил-имидазола в виде слабо-желтого твердого вещес ва с т. пл. 136-138°С. К перемешиваемому раствс у этого производного алпила (3,68 г) в ТГФ под аргоном при 5с добавляют диборан,(40 мл 1М раствора ). Смесь перемешивгиот при 5 С в течение 15 мин, затем npit комнатной температуре 16 ч. К раствсч У добавляют воду (20 мп, затем через 15 . мин 2н. NaOH (20 мл водного раствора) и HjOj (6 МП 30 вёс.% водный раствор). Смесь олдерживают 2 ч при 50°С при энергичном перемешивании, затем охлаждают, насьоцают NaCt и слои разделяют. Водный слой экстрагируют эфиром (триждл по 75 мл) и объе;о1ненные экстракты промывают рассолом и высушивают (MgSO). Раст воритель выпаривают и остаток очищают хрс 4атографически на силикагеле с использованием в качестве элюента смеси СН2СЕ2/МеОН (40:1 о/о) с получением 2-фТ9Р-1-1рифенилметил-4-(3-окси)-п-пропилимидазола в виде кристаллического твердого вещества , имеющего в спектре ЯМР в следующие характерные линии; 1,5-1,9 (т, 2Н),- 2,47 (t, 2Н),- 3,54 (t, 2Н); 6,37 (s, 1Н),- 7,0-7,5 (т, 15Н). Пример 36. Процесс гфоводят аналогично описанному в примере 7 с использованием в качестве -исходного вещества трет -бутил-7- (4-аминометил-2-у мидазолин-2-ил)-амино-З-метилцеф-З-ем-4-карбоксилата. Щ одукт очищают высаживанием из CHg С Eg эфиром и таким образом получают дитрифторацетат 7-(4-г№Шнометил-2-имидазолин-2-ил)-амино-З-метилцеф-З-ем-4-карбоновой кислоты. Продукт характеризуется следующими линиями в спектре ЯМР в 020: 2,0 (s, ЗН); 3,2-4,4 (т, 6Н) 4,4-4,7 (т, 1Н); 5,15 (d, 1Н); 5,4 (d, 1Н). Используемый в качестве исходного вещества трег -бyтил-7-(4-aминoмeтил-2-имидaзoлин-2-ил)-aминo-3-мeтилцeф-3-eм-4-кapбoкcилaт получают след}ующим образом. К раствору 1,2-ди(трет -бутоксикарбонил-амино)-3-оксипропана (3 г) и триэтиламина (2 мл) в CHjCtj (30 мл) при добавляют метаносульфонил-хлорид (1,0 мл) при перемешивании. Перемешивание при этой температуре продолжают в течение 1 ч и затем смесь промывают водой. Органический слой отделяют, высушивают и растворитель выпаривают с получением 1,2-ди-(трет -бутокси-карбониламино)-3-метансульфонилокси-пропана (2,9 г), т. лл. 94-96. К раствору азида натрия (О,825 г) в воде (2,5 мл), добавляют раствор указанного метан-сульфоната (2,34 г) в диметилацетамиде (60 мл). Смесь перемешивают под аргоном при 60°С в течение 7,5 ч и затем растворитель выпаривают. Остаток растворяют в этилацетате, промывают водой и высушивают, затем растворитель выпаривают. Остаток очищают хроматографически на силикагеле с использованием в качестве элюёнта смеси СН СЕ2/эфир (50:50, о/о) с получением 1, -ди- (-грет -бутоксикарбонил-амино-)-3-азид-пропана, характеризукяцегося следующими линиями в спектре ЯМР в CDCS : 1,45 (s, 1вИ) 3,25 (m, 2Н); 3,45 (d, 2Н); 3,65-4, (m, 1Н); 4,75-5,3 (br, 2Н). К раствору указанного азида (1,15 г) в анизоле (10 мл) при по каплям добавляют ТФА (5 мп). Смесь перемешивают 15 мин при , затем при комнатной температуре 1 ч. Растворитель выпаривают и остаток растирают со смесью диэтиловый эфир/петролейный эфир и затем с петролейным эфиром с получением дитрифторацетата свободного диамина (1% г). К раствору указанной соли диамина (1,029 г) в метаноле (3 мл) добавляют метоксид натрия (0,324 г). После перемешивания добавляют ТГФ (37 мл), а затем трет -бутил-7-дибромометиленамино-3-метилцеф-З-ем-4-карбоксилат (1,32 г) . Смесь перемешивают 2 ч, растворитель вываривают, остаток растворяют в минимальном количестве С N2 С Ej. Продукт высаживают смесью диэтилового и петролейного эфиров. Затем продукт очищают хрюматографически на силикагеле с использованием сначала смеси СН-,СЕ /эфир (1:1 о/о) и затем этого растворителя, содержащего 1% с повышением до 6% метанола. Таким образом получают трет-бyтил-7-(4-aзидoмeтил-2-имидaзoлин-2-ил)-aминo-3-мeтилцeф-3-eм-4-карбоксилат в виде
гидратированного гидробромида, характеризующегося следующими лиииями в ЯМР-спектре в COCEj-f CD,00; 1,4 (s, 9Н); 2,15 (s, ЗН); ,4-4,1 (m, 7Н)-, 5,1 (d, IH); 5,35 (d, 1H).
Раствор описаиного производного азидометила (42 мг) в этаноле (2 мл и хлороформе (1 мл) гидрируют под давлением 3 атм в течение 20 ч при комнатной температуре в присутствии POj. Затем катализатор удал5пот декантацией, центрифугированием и фил трацией через тонкопористый фильтр, а затем растворитель выпаривают. Таким образом получают грег -бутил-7-(4-аминометил-2-имидазолин-2-ил)-амино-З-метилцеф-З-ем-4-карбоксилат в виде гидрохлорида-гидробромида (70%), имеющего в спектре ЯМР . в CDCEj + СОзОО следующие линии: 1,45 (s, 9Н); 2,05 (s,3H); 3,0-3,8 (m, 7Н) 5,1 (d, IH), 5,35 (d, IH).
Пример 37. Проводят процесс аналогично описанному в примере 30 с использованием в качестве исходного вещества 7-амино-З-(1,2,3-тиадиазол-5-ил)тиометилцеф-3-ем-4-карбоновой кислоты. Продукт очищают жидкостной хроматографией под высоким давлением с использованием в качестве элюента смеси вода/ /МеОН/НОАс (70:30:1, о/о/о). Получают 7-(имидазол-2-ил)-aминo-3- ( 1 / 2 , З-тиадиазол-5-ил)тиометилцеф-З-ем-4-карбоновую кислоту, имеющую в спектре ЯМР в + СОзС020 линии: 3,47 (d, 1Н); 3,71 (d, 1Н); 4,36 (s, 2Н); 5,13 (d, 1Н);.5,58 (d, 1Н) 6,81 (s, 2Н),- 8,88 (s, 1Н)
il р и м е р 38. Проводят процесс аналогично описанному в примере 7 с использованием только ТФА при и в качестве исходного соединения, трет -бутил-7-(l-мeтoкcибeнзимидaзoл-2-ил)-aминo-3-мeтилцeф-3-eм-4-кapбoкcилaтa. Получают 7-(15 -метоксибензимидазол-2-ил)-амино-3-метилцеф-З-ем-4-карбоновую кислоту в виде гидратированной трифторацетатной соли (69%). Он характеризуется следующими линиями в спектре
0 ЯМР в d,DMSO:. 2,10 (s, ЗН); 3,40 (d, IH), 3,60 (d, IH); 3,72 (s, 3H); 5,23 (d, IH); 5,67 (d, IH); 7,187,58 (m, IjH).
Используемый в качестве исходного
5 материала трет -бутил-7-(1-метокси6ензимидазол-2-ил)-амино-3-метилцеф-З-ем-4-карбоксилат получают следующим образом.
Смесь 1-метоксибензимидазола (1,48 г) и оС-бромизобутирофейона (2,27 г) в толуоле (6 мл) нагревают в течение ночи с обратным холодильником. Затем смесь охлаждают и собирают осадок (1,0 г). Смесь этого осадка и трет-бутил-7-амино-З-метилцеф-З-ем-4-карбоксилата (0,54 г) в метаноле (4 мл) перемешивают при комнатной температуре в течение 6 дней. Затем добавляют (25 мл) раствор промывают водным раствором NaHCO -J , водой и рассолом, затем сушат (HgS04). Растворитель выпаривают и остаток чистят хроматографически йа силикагеле с использованием в качестве элюента смеси
5 эфира/СНтС з (1:1 о/о). Получают трет -бутил-7-(1-метоксибензимидазол-2-ил)-амино-3-метилцеф-3-ем-4-карбоксилат, кото1Я:1й затем исполь|3уют без дальнейшей очистки.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ГЕНЕРАТОР ГУТИНА К.И. И ЦАГАРЕЙШВИЛИ С.А. ВВОДА ТОКОВ СИГНАЛОВ В ТРЕХФАЗНУЮ ЛИНИЮ ЭЛЕКТРОПЕРЕДАЧИ 0,4 кВ ПО СХЕМЕ "ФАЗА" - "ФАЗА" С ИСТОЧНИКОМ ПИТАНИЯ "ФАЗА" - "ФАЗА" | 2010 |
|
RU2421907C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Патент ОНА 3917587, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Сплав для отливки колец для сальниковых набивок | 1922 |
|
SU1975A1 |
