а 11 Изобретение относится к усовершен ствованному способу получения бенз (g)изатинов, которые могут быть использованы в качестве промежуточных продуктов синтеза гетероциклических соединений различных классов, в том числе лекарственных веществ и кубовых красителей. Известен способ получения бенз() изатина из этилового эфира 0 -нафтодиоксииндол-3-карбоновой кислоты с выходом до 95%. Исходный этиловый эфир ot -нафтодиоксиндол-3-карбоново кислоты получают взаимодействием 1-нафтиламина с мезоксалевым эфи ром l . Однако мезоксалевый эфир является труднодоступным и дорогим лабораторным реактивом, не производимым в промьшшенном масштабе. Поэтому способ получения бенз(d)изатина в общем является промьшшенно неосуществимым. Наиболее близким по технической сущности к предлагаемому является способ получения 4Н-6,7,8,9-тетрагидробенз()изатина, которьй заключается в том, что в на гретую до 90%-ную серную кислоту добавляют АН-оксиминоацет-(5,6,7,8-тeтpaгидpo-1-нaфтил)-aмид, вьщержнваю1Т, вьшивают на лед, вьщеляют серой продукт - 4Н-6,7,8,9-тетрагидробенз()изатин с выходом, не достига{рщим 40%. Его очищают исчерпывающей экстракцией эфиром в аппарате Сокслета, после упаривания эфира. перекристалли зовывают из этанола, вьщеляют оранже вые кристаллы с т.пл. 228 С L2J. Недостатком известного способа является низкий выход целевого продукта. Причиной этого является высокая температура реакционной смеси (кроме нагрева извн до 75 С она повышается и вследствие экзотермичности процесса),. В таких условиях соеди нения нафтиламинового ряда склонны к смолообразованию и полисульфонированию, что и предопределяет образование больших количеств побочных про дуктов различного строения. Целью изобретения является повыше ние выхода и расширение ассортимента целевых продуктов. Поставленная цель достигается спо собом получения бенз(Юизатинов формулы I или 11 , заключающимся в том, что при комнатной температуре готовя смесь соответствующего оксиминоацет62-1-нафтиламида постепенным добавлением серной кислоты и вьщержку осуществляют при 25-45°С. При этом используют 85-96%-ную серную кислоту. Продукт вьщеляют следующим образом. Охлажденную до комнатной температуры реакционную смесь вьтивают на лед, вьщеляют осадок. Сырой продукт очищают известными методами, используя те из них, которые не требуют превышения комнатной температуры. Для процесса использзпот 80-96%-ную серную кислоту, но предпочтительным является использование 85-96%-ной серной кислоты (табл. 2) То, что вместо воздействия на оксиминоацет-1-нафтиламид серной кислотой при 75 С приготавливают смесь этих двух компонентов при комнатной температуре, а в дальнейшем поддерживают температуру реакционной смеси 25-45 С, приводит к ослаблению протекания побочных процессов, и после первой стадии очистки выход целевого бенз()изатина, в зависимости от его структуры, составляет до 53,3-72,1%. Данные, полученные при осуществлении предлагаемого способа,, приведены Б табл. 1 и 2. Получение исходных оксиминоацет-1-нафтиламидов. 4Н-Оксиминоацет-(5,6,7,8-тетрагидро-1-нафтил)амид. В колбу емкостью 6000 мл помещают 4800 мл воды и в ней при нагревании последовательно растворяют 500 г кристаллического сульфата натрия, 145 г солянокислого гидроксиламина и 55 г технического (0,3 моль) гидрохлорида 1-аминотвтралина. (Вместо гидрохлорида 1-аминотетралина можно последовательно добавлять в смесь 27,5 г 40%-ной кислоты и 44 г 1-аминотетралина) . Температуру раствора нагреванием на бане доводят до 75 С и растворяют в нем 57 г хлоральгидрата. Затем, время от времени перемеивая, нагревают до 80°С, причем начинается вьщеление осадка. Нагревание прекращают и оставляют растор при комнатной температуре, дожиаясь самопроизвольного охлаждения. ьщелившийся осадок отсасывают, проывают 50 мл воды и высушивают. Выод сырого продукта 47 г (78%). Одноратной кристаллизацией из водного пирта с углем получают желтоватый
А11-оксиминоацет(5,6, 7,8-тетрагидро-1-нафтил)амид с т.пл. 175-177 С, пригодный для дальнейших превращений
Оксиминоацет-1-нафтиламид.
В колбу еюсостью 8 л помещают 3-50 мл воды температуры , растворяют в ней 1130 г кристаллического сульфата натрия, 218 г солянокислого гидроксиламина и добавляют раствор 63 г (0,37 моль) гидрохлорида 1-нафтиламина в 2150 мл этанола. Затем там же при растворяют 85 г хлоральгидрата. Вьздерживают смесь при 45°С 1,5 52°С 45 мин, затем нагревают до и оставляют колбу при комнатной температуре. На следующий день отсасывают выпавший буро-зеленый осадок, высушивают, хорошо перемешивают с 150 мл этанола, прибавляют 15 мл воды и фильтруют. Оставшийся на фильтре осадок снова обрабатывают 100 мл этанола, прибавляют 5 мл воды и фильтруют. Оставшиеся на фильтре загрязнения отбрасывают,,.а водно-эта- нольные фильтраты объединяют и тонкой струей при перемешивании выливают в 2 л воды. Спустя 1-2 ч отсасывают 45 г (выход 56%) зелено-желтоватого Оксиминоацет-1 - нафтилавида с т.пл. 178°С (разл.) пригодного для дальнейших превращений.
Пример 1. 4Н-6,7,8,8-тетрагвдробензСЛизатин.
В фарфоровой ступке емкостью 500 мл при комнатной температуре растирают 21,8 г (0,1 моль) желтоватого технического 4Н-оксиминоацет(5, 6,7,8-тетрагидро-1-нафтил)амида. Добавляют 50 мл 90%-ной водной серной кислоты комнатной температуры. Быстро и тщательно растирают пестиком до получения тонкоизмельченной смеси. Начинается самопроизвольное нагревание до 30-40°С. В течение 10-15 мин реакционную смесь время от времени еремешивают, причем она приобретает емно-фиолетовый цвет. Затем ее вылиают в колбу и, охладив до комнатной температуры, выливают на 250 г мелкораздробленного льда. Вьщелившийся осадок отсасывают на воронке Бюхнера и высушивают при комнатной температуре, получают 20 г сырого продукта в виде коричневатого порошка.
Сырой продукт растирают в фарфороой ступке, запивают последовательно З порциями по too мл ацетона комнатной температуры, каждый раз тщательно растирая, дожидаясь осалсдения и сливая получаемый раствор. После этого оставшиеся в ступке загрязнения
отбрасывают, растворы объединяют и прибавляют 280 мл воды. Фильтруют на воронке Бюхнера, оставшиеся на фильтре загрязнения отбрасьгеают, фильтрат осторожно упаривают в вакууме до объема 400 мл и добавляют
600 мл воды. Вьщелившийся осадок отфильтровывают на воронке Бюхнера и высушивают при комнатной температуре. Получают 14,5 г (выход 72,1%)
4Н-6,7,8,9-тетрагидробенз(Юизатина в виде оранжевого порошка с т.пл. 225-227°С. Для получения аналитически чистого продукта проводят кристаллизацию из водного спирта до получения оранжевых кристаллов с т.пл. 230231 С. Хроматография на пластинке Silufol UV 254, элюент бензол - 95% ный этанол (17:3), в видимом свете R{ 0,35.
Найдено, %: С 71,80; Н 5,66; N 6,80.
Вычислено, %: С 71,63; Н 5,51J N 6,96.
Пример 2. БеH3(g)изатин.
Синтез аналогичен примеру 1. Его осуществляют из 21,4 г (0,1 моль) зелено-желтоватого технического оксиминоацет-(1-нафтил)-амида, но со следующими отличиями: количество серной кислоты 200 мл, время вьщерживания смеси 5-10 мин, количество льда 1000 г. Выход сырого продукта 17 г в виде темно-красного порошка. Очистки аналогична примеру 1, но при этом везде Количество растворителей увеличивают в 1,5 раза. Получают 10,5 г (выход 53,3%) бенз()изатина в виде, красного порошка с т.пл. 250-252 С.
Аналитически чистый продукт получают кристаллизацией из водного спирта до получения красных кристаллов с т.пл. 254-256°С. Хроматография на пластинке Silufol UV 254, элюент .Сензол - 95%-ш,1йэтанол (17:3), в видимом свете RJ 0,34.
Найдено, %: С 72,98; Н 3,51; N 7,27.
С HjO.N
Вычислено, %: С 73,09; Н 3,58, N 7,10.
Влияние: температурного режима на выход 4Н-6,7,8,9-тетрагидробенз()изатина. S11 Синтез аналогичен примеру 1,но количество вещества и емкость аппаратуры уменьшают в 5 раз. После того, как получают тонкоиэмельченную смесь исходных продуктов, ее перепивают в колбу со вставленным термометром, нагревают на бане либо охлаждают проточной водой до 20-50°С и выдерживают ее при этой температуре в течение 10-f5 мин, время от времени перемешивая. После этого колбу охлаждают водой до комнатной температзгры. Далее поступают как в примере t,
Таблица 1 6 Влияние концентрации Серной кислоты на выход 4Н-6,7,8,9-тетрагидробенэ()изатина. Процесс получения аналогичен примеру 1, но количество веществ и емкость аппаратуры везде изменяют в О,2-2 раза и применяют серную кислоту концентрации 80-96 вес.%. Предпочтительным является использование серной кислоты 85-96%-ной концентрации. Температурные и хроматографические данные для полученных соединений (табл. 2) те , что и в табл. 1.

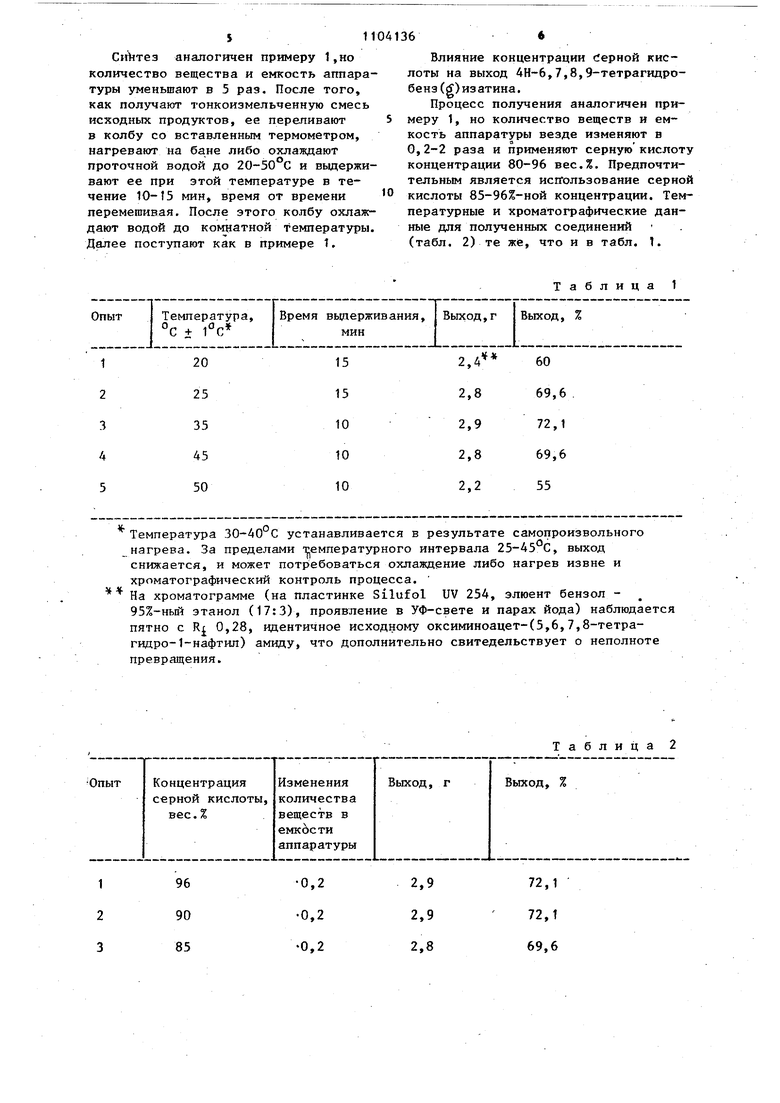
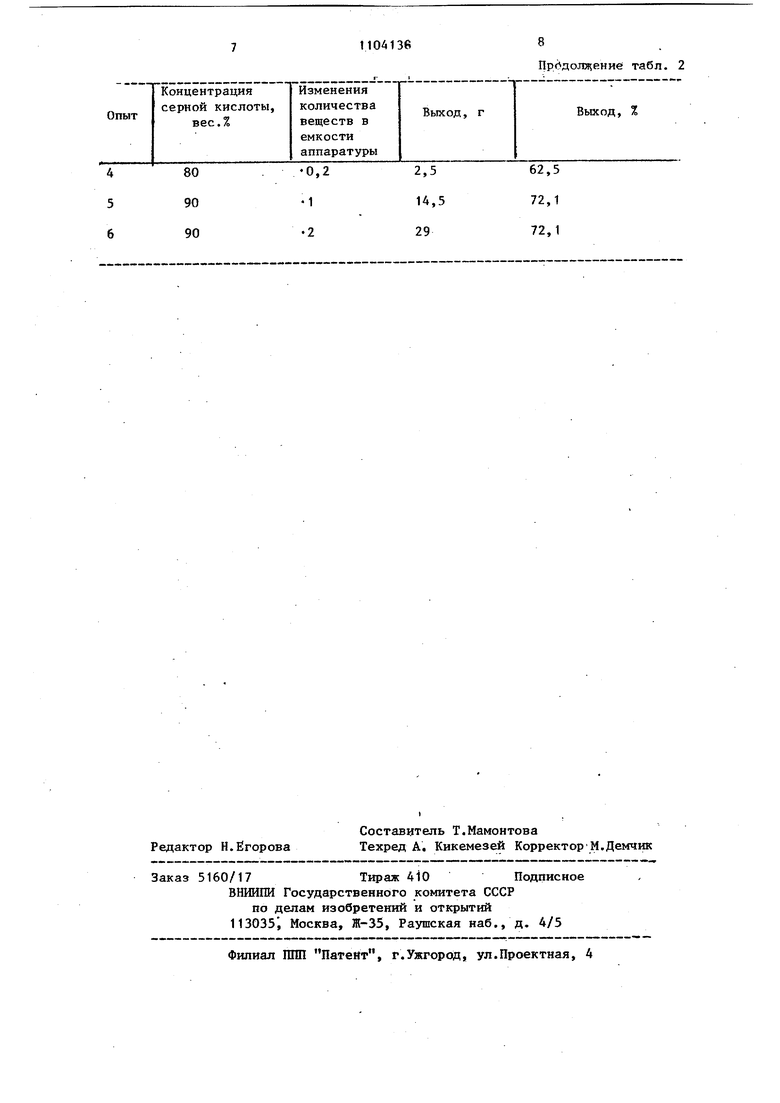

Температура 30-40°С устанавливается в результате самопроизвольного нагрева. За пределами температурного интервала 25-45 С, выход снижается, и может потр ебоваться охлаждение либо нагрев извне и хроматографический контроль процесса. На хроматограмме (на пластинке Silufol UV 254, элюент бензол 95%-ньй этанол (17:3), проявление в УФ-свете и парах йода) наблюдается пятно с R 0,28, идентичное исходному оксиминоацет-(5,6,7,8-тетрагидро-1-нафтил) амиду, что дополнительно свитедельствует о неполноте превращения.
0,2
80 90 90
1
2
8
Пр.(.ние табл. 2
62,5 72,1 72,1
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Martinet J | |||
| Synthesis dans la serie de l indol.- Annales de chimie, 11,9, 1919, 15-130 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Braun J | |||
| Шзег das Tetrophan und seine Derivate.- Ann, 451, 1927, 1-54 (прототип). | |||
