со
DO
9
Изобретение относится к синтезу органических соединений и предназначено для получения Н,Ы-дизамещенных нитрозогидразинов общей формулы
RlN-NHRj
(I
т
где R - алкил, арилалкил,
Rn - Н и И -алкилы, алкенил,
апкинил, арилалкил. Эти соединения могут быть использованы как исходные для получения азосоединений Ri|N NR2 денитрозированием до NjN -дизамещенных гидCH N(NO)NH2 C HgCHaN NOlNHg
К производному нитрозогидразина в 8-10%-ном растворе едкого натра добавляют по каплям диметилсульфат при 30-40 С с последующим выделением продуктов. Выход целевых продуктов составляет 76% от теоретического L2.
Недостаток способа заключается в его ограниченных возможностях CH3N(
СбНбСИ се x CH NiNOlNHCH CfeMg ,H5CH2N(NO NHCH2C6Hg C HgCHgNlblOlNH Эти синтезы проводят при 60-80 С в течение 8-10 ч, выходы целевых продуктов составляют 62-76% от теоретических pi. Недостаток способа заключается в том, что невозможно использовать другие галоидсоединения, например галоидалкилы. Последние в сильно щелочной среде при повьщ1енной температуре легко дегидрогалогенируются и получить N,N-дизамещенные нит розопщразины не удается (у хлористого бензила возможность дегидрогалогенирования отсутствует). Целью изобретения является получение N,N-дизамещенных нитрозогидразиноз с разнообразными замес ТИТелями, Пост 1шенная цель достигается тем, что согласно способу получения N,N -дизамещенных нитрозогидразинов общей формулы (I) из нитроз о п-щразинов (NO)Nn2 галоидсоединений Rn Гал, нитрозогидразины обрабатывают амидом натрия в жидком аммиаке
разииов R NHNHR (гидразосоединеНИИ) и окислением последних П
И1в о . Азрсоединения нашли применение в прог-ышленности и исследовательской практике как инициаторы радикальной полимеризации, вулканизукжцие агенты, газообразователи и как исходные для получения азоксисоединений, интересных способнрстью к существованию в жидкокристаллическом состоянии.
Известен способ получения метильных производных Ы-метил-Н-нитрозогидразина и N-бeнзIШ-N-нитpoзoгидразина с помощью диметилсульфата:
CHjb.504yСИ3N(NO NНСН-j
C HgCHaNlNO NHCH.
только метилирование. Диэтилсульфат и другие алкилсульфаты, как известно, плохие алкилирующие агенты. Наиболее близким к изобретению
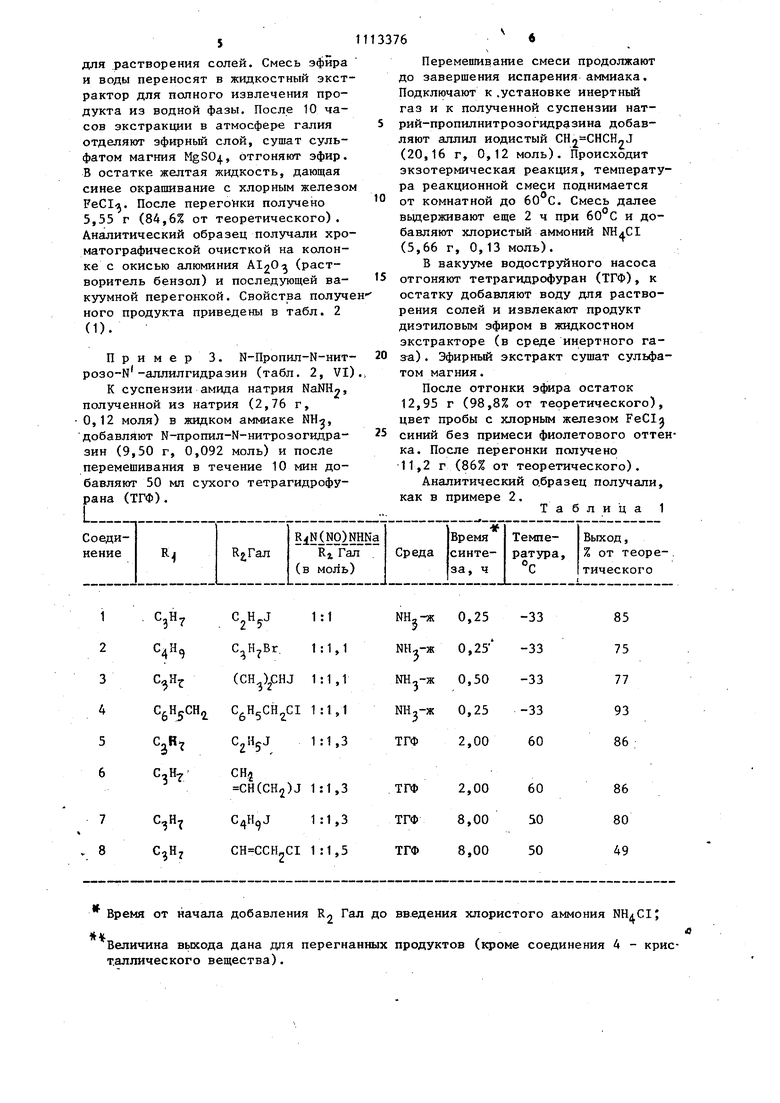
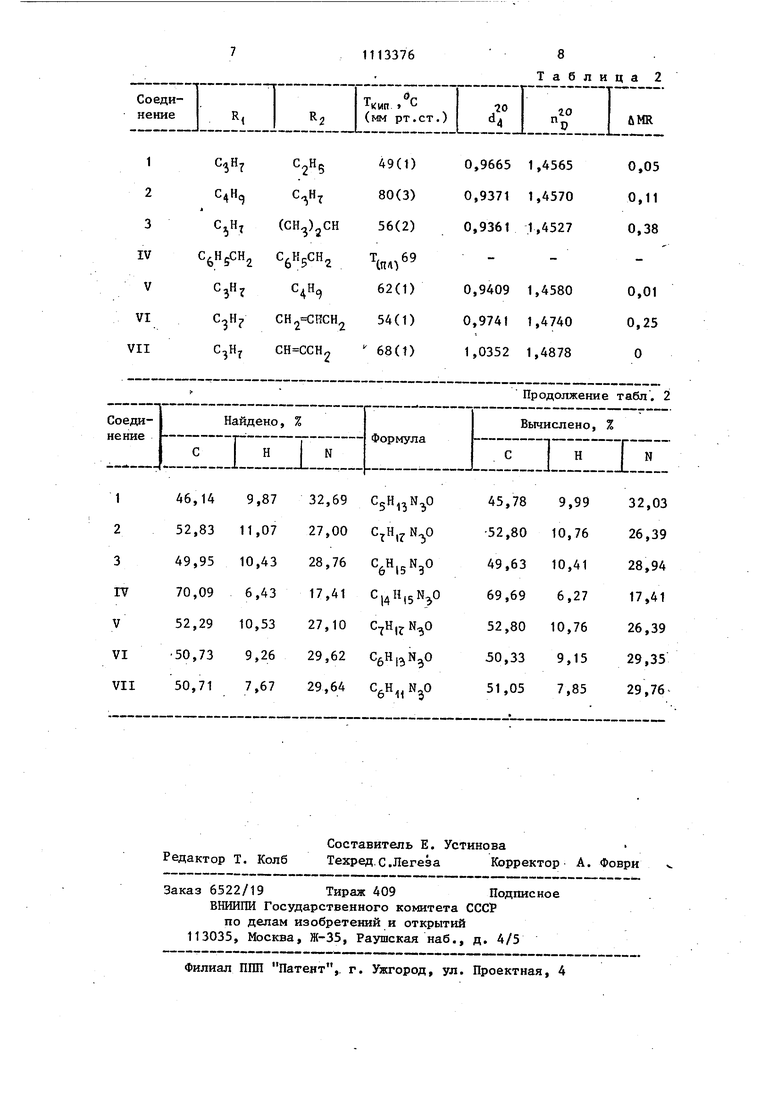
является способ получения бензильных производных Н-метил-Ы-нитрозогидразина и Ы-бензил-Ы-нитрозогидразина с помощью хлористого бензила в щелочной среде: а полученные при этом натрийнитро- зогидразины без выделения из аммиачной среды обрабатывают галоидсоединениями в среде жидкого аммиака или органического растворителя: ( (NOlNHNa RiNiNO NHMa RtN(NOlNHR Обработку галоидалкилом в среде жидкого аммиака проводят при температуре кипения жидкого аммиака в течение 15-30 мин, обработку галоидсоединениями в среде органического растворителя проводят при 50-60 С в течение 2-8 ч. Все синтезы заканчивают через 15-30 мин добавлением в реакционную смесь хлористого аммония для дезактивации натрийнитрозогидразина. Условия синтеза N,N-дизамещенных нитрозогидразинов (NO)NHR2 приведены в табл. 1. 3 Взаимодействие натрийнитрозогидразинов RjN(NO)NHNa и галоидсоединений проходит почти количественно. Приведенные в табл. 1 величины выходом меньше (в опытах 1-7 75-93%, в опыте 8 (получение ацетиленового производного) 50% от теоретического, но это связано с тем, что синтезы проводили с неболь шими количествами нитрозогидразинов и относительные потери при перегонках были ощутимы (например за счет кубового остатка). Характеристики N,N -дизамещенных нитрозогидразинов (NO)NHR2 представлены в табл. 2, При получении кристаллического N,N дибeнзшI-N-нитpoзoгидpaзинa (табл. 2, IV) выход составил 93% (табл. 1, 4). Для введения заместителя в R,N(NO)NH2 можно использовать соединения хлора, брома, иода (табл. 1). Если заместитель в галоидсоединении инертен относительно аммиака, то обработку галоидсоединением проводят в среде жидкого аммиака независимо от природы галоида (хлор, бром или иод) при температур кипения жидкого аммиака в течение 15-30 мин. Если заместитель в галоидсоедине нии может взаимодействовать с аммиаком, то до -введения в реакционную смесь галоидсоединения нужно произвести замену жидкого аммиака на дру гой растворитель. В опытах с аллилом йодистым и пропаргилом хлористым (табл. 1, 7 и 8), аммиак заменяли на тетрагидро фуран (ТГФ). Натрийнитрозогидразины нерастворимы в ТГФ, реакционные смеси получаются гетерофазными, и это удлиняет время синтеза до нескольких часов. При использова. НИИ тетрагидрофурана синтезы проводили при 50-бО С в течение 2-8 ч Все синтезированные соэдинения, за исключением IV, не описаны в литературе. Их структура подтверждена следующими данными. Все вещества имеют в ИК-спектре полосу поглощения при 3200 см (Н), с водным раствором хлорного железа дают сине окрашивание (эту качественную пробу на Н,Ы-дизамещенные нитрозогидразины описал И. Тиле), с реактивом Грисса дают малиновое окрашивание (проба на нитрозоаминогруппу), най764денный элементный состав соответствует вычисленному, отличается от MR 0уц в допустимых пределах (табл. 2). Нитрозогидразины (NO)NH, исходные для получения N,N -дизамещенных нитрозогидразинов R.N(NO)NHRrt, являются доступными соединениями. Они получаются при обработке гидразинов алкилнитритами с выходами 80-90% от теоретического. Пример 1. N,N-Дибензил-N-нитрозогидразин (табл. 2, IV). К суспензии амида натрия NaNH., полученной из натрия Na (0,50 г, 0,022 моль) в жидком аммиаке Nl.j-m добавляют Ы-бензил-Ы-нитрозогццраЗИН (3,00 г, 0,020 моль) и после перемешивания в течение 10 мин (за это время образуется раствор Na-бензилнитрозогидразина в жидком аммиаке серо-черного цвета) добавляют ко каплям xлopиcтьп бензил С НгСНоС (2,75 г, 0,022 моль). Реакционную смесь перемешивают 15 мин и добавляют кристаллический хлористый аммоний NH4CI (1,00 г, 0,023 моль). После испарения аммиака к остатку добавляют воду, кристаллы продукта (IV) отфильтровывают, трижды промывают водой и сушат в вакуум-эксикаторе. Выход 4,46 г (93% от теоретического). Проба с хлорным железом FeCI дает синий цвет (если в Н,М-дизамещенном нитрозогидразине есть примесь исходного нитрозогидразина (NO)NH2 проба имеет фиолетовый цвет) . Для получения аналитического образца вещество перекристал-лизовали из петролейного эфира. Свойства N,N -дибензил-Н-нитрозогидразина приведены в табл . 2. Пример 2. М-Пропил-Ы-нитрозо-N-этилгидразин (табл. 2, VT); К суспензии амида натрия NaNH-, полученной из натрия Na (1,15 г, 0,05 моль) в жидком аммиаке NH, добавляют N-пpoпил-N-нитpoзoгидРазин (5,10 г, 0,05 моль) и после 15-минутного перемешивания добавяют по каплям этил иодистьв1 (7,80 г, 0,05 моль). Реакционную смесь перемешивают 15 мин и вводят ристаллический хлористый аимо.щй (2,70 г, 0,06 моль). После испарения аммиака к остатку добавляют 50 мл эфира и воду
для растворения солей. Смесь эфира и воды переносят в жидкостный экстрактор для полного извлечения продукта из водной фазы. После 10 часов экстракции в атмосфере галия отделяют эфирный слой, сушат сульфатом магния MgS04, отгоняют эфир. В остатке желтая жидкость, дающая синее окрашивание с хлорным железом FeCI. После перегонки получено 5,55 г (84,6% от теоретического). Аналитический образец получали хроматографической очисткой на колонке с окисью алюминия (растворитель бензол) и последующей вакуумной перегонкой. Свойства полученого продукта приведены в табл. 2 (1).
П р и м е р 3. Н-Пропил-Ы-нитРОЗО-N-аллилгидразин (табл, 2, VI)
К суспензии амида натрия NaNH2, полученной из натрия (2,76 г, 0,12 моля) в жидком аммиаке NH, добавляют М-пропил-Ы-нитрозогидразин (9,50 г, 0,092 моль) и после перемешивания в течение 10 мин добавляют 50 МП сухого тетрагидрофурана (ТГФ).
L.
Перемешивание смеси продолжают до завершения испарения аммиака. Подключают к .установке инертньй газ и к полученной суспензии натрий-пропилнитрозогидразина добавляют аллил йодистый CH2-CHCH-J (20,16 г, 0,12 моль). Происходит экзотермическая реакция, температура реакционной смеси поднимается от комнатной до 60 С. Смесь далее вьщерживают еще 2 ч при 60°С и добавляют хлористый аммоний (5,66 г, 0,13 молъ).
В вакууме водоструйного насоса отгоняют тетрагидрофуран (ТГФ), к остатку добавляют воду для растворения солей и извлекают продукт диэтиловым эфиром в жидкостном экстракторе (в среде инертного газа) . Эфирный экстракт сушат сульфатом магния.
После отгонки эфира остаток 12,95 г (98,8% от теоретического), цвет пробы с хлорным железом FeCIa синий без примеси фиолетового оттека. После перегонки получено 11,2 г (86% от теоретического).
Аналитический образец получали, как в примере 2.
Таблица 1

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения N @ -алкил(арилалкил)-N @ -нитрозо-бензамидразонов | 1989 |
|
SU1664790A1 |
| Способ получения моно- или дизамещенных @ - или @ - карборанов | 1980 |
|
SU888493A1 |
| Способ получения N-нитрозо-N-(бэта-хлорэтил)-карбамоилпептидов или их кислотно-аддитивных солей | 1982 |
|
SU1424739A3 |
| Способ получения @ , @ -дизамещенных 1,8-диаминонафталинов | 1985 |
|
SU1318587A1 |
| Способ получения 1,3-дизамещенных нитрозомочевин | 1977 |
|
SU963464A3 |
| СПОСОБ ПОЛУЧЕНИЕ АМИНОЭПОКСИДА ПУТЕМ НЕПРЕРЫВНОГО ПРОЦЕССА СИНТЕЗА IN SITU | 1997 |
|
RU2194045C2 |
| Способ получения бензил-2,2-диметокси ацетамидов | 1976 |
|
SU663299A3 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРАНГИДРИДОВ N-3AMElUEHHbIX ИМИНОСУЛЬФОКИСЛОТ | 1967 |
|
SU191542A1 |
| СПОСОБ ПОЛУЧЕНИЯ N-ЗАМЕЩЕННЫХ ИМИДОВ ДИФЕНОВОЙ КИСЛОТЫ | 2005 |
|
RU2285694C1 |
| Способ получения N-(L-аспаргил)-N-(1-ациламино-)-алкиламинов | 1984 |
|
SU1494862A3 |
1. СПОСОБ ПОЛУЧЕШЯ Ы.Ы-ДИЗАМЕЩЕННЫХ НЙТРОЗОГИДРАЗИНОВ общей формулы RlN--NHR2 io где R - алкил, арилалкил, R- - Н и И -алкилы, алкенил, алкинил, арилалкил. из нитрозогидразинов (NO)N Но и галоидсоединений Rr,roiA, гдeR/ и Rj имеют указанные значения, о т личающийся тем, что, с целью создания возможностей для синтеза соединений с разнообразными заместителями, нитрозогидразины обрабатывают амидом натрия в жидком аммиаке с последующей обработкой полученных выше продуктов галоидсоединением в среде жидкого аммиака или органического растворителя. 2.Способ по п. 1, отличающийся тем, что обработку галоидсоединением в жидком аммиаке § ведут при температуре кипения жидкого аммиака 15-30 мин. (Л 3.Способ по п. 1, отличающийся тем, что обработку галоидсоединением в органическом растворителе ведут при 50-60 С в тече§ ние 2-8 ч.
Время от начала добавления R2 Гал до 1t Величина вькода дана для перегнанных д пер т.аллического вещества). введения хлористого аммония продуктов (кроме соединения 4 - крисТаблица 2
| Dimroth К., Tuncher W | |||
| Synthesis, 1977, № 5, 339 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Thiele J., Ann., 376 | |||
