113642352
Изобретение относится к способу до . В полученный гомогенный
раствор.желтого цвета добавляют 136 г
ццолучения новых производных с.-токо;ферола или их ацетатов, которые могут быть использованы в качестве исходных соединений в синтезе витамина Е. Цель изобретения получение но:Вых производньк ot.-токоферола, позволяющих получить витамин Е более простым способом (с меньшим числом стадий),
Гидрированием продукта общей формулы I или ег,о ацетата, осуществляе- ФIM с помощью водорода в присутствии катализатора, такого как палладий на угле, в органическом растворителе, таком как уксусная кислота или этанол, при 50-100°С, получают Ы- -токоферол или ацетат с -ток оферола
(витамин Е),
Пример 1. В реактор объемом 250 см 3 вводят 8,4 г триметилгидрохинона, 22 г 1,7,11515-тетрахлор- 3,7,11,15-тетраметил-гексадецена-2 и ,30 см уксусной кислоты. Затем в течение 10 .мин добавляют раствор 1,5 г хлорида цинка в. 15 см безводной уксусной кислоты. Температура повышается до 25-30°С. Реакционную смесь перемешивают в течение 2 ч при , яатем ее вьшивают в смесь 100 см гексана и 100 см воды. Органическую фазу, отделенную декантацией, промывают 100 см смеси метанола с водой (50;50 по объему). В гексановой фазе образуется осадок белого цвета, который отделяют путем фильтрования и промывают 50 см смеси метанола с-водой (50:50 по объему) После сушки при пониженном давлении получают 14,3 г 2,5,758-тетраметил- 2-(,12- трихлор-4 , 8, 12- триметил-тридецил)-хроманола-6 в виде кристаллов белого цвета, т.пл. 102-104°С5 выход 62%.
Структура полученного продукта подтверждена масс-спектром и спектрами протонного ядерного магнитного резонанса и ЯМР- ..
1,,15-Тетрахлор-З,7,11,15- тетраметил-гексадецвн-2 может быть получен согласно одному из следующих способов,
а), В трехгорлую .колбу емкостью
мирцена (1 моль), чистота которого g вьше 95%, затем в течение 6 ч - 80 г безводной соляной кислоты. Полученный раствор вьщерживают при -25°С в течение 18 ч.
Реакционную смесь выпивают в смесь 10 400 см водного 10%-ного раствора хлорида аммония и 300 см пентана. После декантации органическую фазу промывают 3 раза 200 см воды, затем высушивают над карбонатом калия. Пос- 15 ле фильтрации и выпаривания растворителя получают 237,8 г масла.бледно- желтого цвета, содержащего главным образом 1,7-дихлор-З,7-диметил-октен- 2 в виде смеси изомеров Е и Zi
20
В реактор еьпсоствю 250 см вводят
12,15 г магния, 30 см тетрагидро- фурана и. кристаллический иод. Охлаждают до , затем в течение 5,5 ч добавляют раствор 20,9 г 1,7-дихлор25 3,7-диметилоктена-2, полученного ранее, в 85. см тетрагидрофурана. Продолжают перемешивание в течение 18 ч при . Избыток магния удаляют фильтрованием, затем полученный раст30 вор вводят в капельную воронку.
В реактор емкостью 250 см вводят 0,5 г иодида меди и 5 см тетрагидрофурана, добавляют 1,5 см раствора магнийорганического соединения. Заgg тем быстро добавляют 19,5 г 3-хлор- мирцена, чистота которого выше 87%, в 10 См тетрагидрофурана. Охлаждают до , затем в течение 3 ч добавляют остальной раствор магнийоргани40 ческого соединения. Температура повышается в течение 1 ч примерно до 20 с. К реакционной смеси добавляют воду (5 см) и пентан (100 см). Органическую фазу, отделенную деканта45 цией, высушивают над сульфатом маг-| ниЯо После фкльтрахцш и выпаривания растворителя получают 29,7 г масла. Согласно количественному анализу путем хроматографии в паровой фазе
50 с внутренним стандартом степень превращения 3-хлормирцена составляет 69%.
Полученное масло нагревают при 100-105 0 при пониженном давлении (0,1-1 мм рт.ст.-, 0,067-0,13 кПа),
500 см , снабженную магнитной мешал- gg чтобы удалить непрореагировавшце
кой, термометром, вводят в атмосфере аргона 3,4 г трнэтиламинохлогид- рата, 2,5 г хлорида меди (I) и 270 СМ метилешшорида. Охлаждают
продукты .
Полученный остаток (20 г) соде жит 85% 15-хлор-3-метш1ен-7,11,15 триметилгексадекатри.ена-1,6,10. В
мирцена (1 моль), чистота которого вьше 95%, затем в течение 6 ч - 80 г безводной соляной кислоты. Полученный раствор вьщерживают при -25°С в течение 18 ч.
Реакционную смесь выпивают в смесь 400 см водного 10%-ного раствора хлорида аммония и 300 см пентана. После декантации органическую фазу промывают 3 раза 200 см воды, затем высушивают над карбонатом калия. Пос- ле фильтрации и выпаривания растворителя получают 237,8 г масла.бледно- желтого цвета, содержащего главным образом 1,7-дихлор-З,7-диметил-октен- 2 в виде смеси изомеров Е и Zi
20
В реактор еьпсоствю 250 см вводят
12,15 г магния, 30 см тетрагидро- фурана и. кристаллический иод. Охлаждают до , затем в течение 5,5 ч добавляют раствор 20,9 г 1,7-дихлор25 3,7-диметилоктена-2, полученного ранее, в 85. см тетрагидрофурана. Продолжают перемешивание в течение 18 ч при . Избыток магния удаляют фильтрованием, затем полученный раст30 вор вводят в капельную воронку.
В реактор емкостью 250 см вводят 0,5 г иодида меди и 5 см тетрагидрофурана, добавляют 1,5 см раствора магнийорганического соединения. Заgg тем быстро добавляют 19,5 г 3-хлор- мирцена, чистота которого выше 87%, в 10 См тетрагидрофурана. Охлаждают до , затем в течение 3 ч добавляют остальной раствор магнийоргани40 ческого соединения. Температура повышается в течение 1 ч примерно до 20 с. К реакционной смеси добавляют воду (5 см) и пентан (100 см). Органическую фазу, отделенную деканта45 цией, высушивают над сульфатом маг-| ниЯо После фкльтрахцш и выпаривания растворителя получают 29,7 г масла. Согласно количественному анализу путем хроматографии в паровой фазе
50 с внутренним стандартом степень превращения 3-хлормирцена составляет 69%
Полученное масло нагревают при 100-105 0 при пониженном давлении (0,1-1 мм рт.ст.-, 0,067-0,13 кПа),
продукты .
Полученный остаток (20 г) содержит 85% 15-хлор-3-метш1ен-7,11,15- триметилгексадекатри.ена-1,6,10. Вы3 1364235 4
ход 82% по отношению к израсходован-в 50 см водного раствора хлорида
ному 3-хлормирцену.аммония с содержанием 100 г/л. ЭкстСтруктура полученного продуктарагируют 2 раза 50 см этилацетата,
подтверждена масс-спектром и спектромзатем сушат органические фазы над
протонного ядерного магнитного резо-сульфатом магния. После фильтрации
нанса.и выпаривания растворителя получают
В реактор емкостью 250 см вводят2,5,7,8-тетраметил-2-(4-,8 , 12 -три- в атмосфере аргона 0,48 г триэтил-хлор-4 , 8, 12 -триметилтридецил)- аминхлоридгидрата, 15 см метилен- Q хроманол-6 с выходом 43,5%. хлорида, 10 см уксусной кислоты иП р и м е р 3. В трехгорлую колбу 90 мг хлорида меди (I)о Реакционнуювводят в атмосфере аргона 2,1 г полусмесь перемешивают до получения го-ченного в примере 1 продукта, 150 мг могенного раствора. Охлаждают додиметиламинопиридина и 10 см три- -10 С, затем добавляют 10 г 15 -хлор-15 этиламина, затем при 25 С быстро до- 3-метилен-7,11,15-триметилгексадека-бавляют 6 см уксусного ангидрида при триена-1,6,10 и, в течение 1 ч 3,9 гперемеап вании. После переметтмвания газообразного сухого хлорводорода.в течение 1 ч добавля 20 см воды, Реакционную смесь вьшивают в 100 смпосле чего реакционную смесь нейтра- водного раствора хлорида аммония с со-20 лизуют постепенньм добавлением карбо- держанием 100 г/л. Органическую фазуната натрия до прекращения вьделения отделяют путем декантации, затем вод-углекислого газа. Реакционную смесь ную фазу экстрагируют 2 раза 100 см- экстрагируют 2 раза 50 см этилаце- метиленхлорида. Объединенные органи-тата. Органическую фазу промывают |ческие фазы промывают 100 см воды,25 3 раза 50 см водного 0,1 н.раствора (затем сушат над карбонатом калия. .соляной кислоты. (Органические фазы После фильтрования и выпаривания раст-высушивают над сульфатом магния. ворителя получают 13,1 г 1,7,11,15-После фильтрации и выпаривания раст- тетрахлор-3,7,11,15-тетраметнл-гекса-ворителя полученный остаток обрабаты- децена-2 с выходом 96,5%.ЗО вают гексаномо Образующийся осадок
Структура полученного продуктаотделяют путем фильтрования. Таким
подтверждена масс-спектром.образом, с выходом 93% получают ацеб). В реактор емкостью 250 см вво-тат 2,5,7,8-тетраметил-2-(4, 8,12 дят в атмосфере аргона 0,48 г три-трихлор-4 , 8, 12 -триметилтридецил)этиламинхлоргидрата, 15 см метилен-- хроманола-6, т.пл. 95-105 С.
хлорида, 10 см уксусной кислоты иСтруктура полученного продукта
90 мг хлорида меди (I). Реакционнуюподтверждена масс-спектром и ЯМР - Н
смесь перемешивают вплоть до получе-и ЯМР- Со
ния гомогенного раствора. Охлаждают. Пример 4.В реактор вводят
до , затем добавляют Ю г j3-40 атмосфере аргона 5 г полученного
спрингена и в течение, 1 ч - 5-2 г га-в примере 1 продукта, 20 см уксусной
зообразного сухого хлорводорода. Пос-кислоты и 320 мг безводного хлорида
ле обработки реакционной смеси в вы-цинка. Добавляют 5 см раствора соляшеописанных условиях, получают 14,2 гной кислоты в уксусной кислоте с
1,7,11,15-тетрахлор-8,7,11, 15-тетра-45 моль соляной кислоты на 1 л. Заметилгексадецена-2 с выходом 94%;тем добавляют в течение 15 мин
Структура полученного продукта2,7 см уксусного ангидрида. Темпераподтверждена после его гидрированиятура повьш1ается до 20-30°С. После пев фитан. . .ремешивания в течение 2 ч добавляют
Пример. 2. В реактор емкостьюgQ 10 см воды, 800 мг ацетата натрия
250 см вводят 0,22 г плавленногои 100 см этилацетата. После выпарихлорида цинка, 2,47 г триметилгидро-вания растворителей остаток обрабатыхинона и 10 см безводного диоксана.вают метиленхлоридом. После фипьтраНагревают до 40-45°С, затем добавляютции на силикагеле получают 4,99 г
в течение 20 мин раствор 6,8 г 1,7,gg ацетата 2,5,7,8-тетраметил-2-(4,8 ,
11,15-тетрахлор-8,7,11,15-тетраметил-12 -трихлор-4 ,8,12 -триметилтридегексадецена-2 в 7 см диоксана. Про-щш)-хроманола-6.
должают перемешивание в течение 1 чСтепень превращения 2,5,7,8-тетра30 мин. Реакционную смесь выпиваютметил-2-(4,8,12 -трихпор-4 ,8,
12 -триметилтридецил)-хроманола-6 составляет 100%, выход 92,5%о
Пример 5. В реактор вводят . в атмосфере аргона 186 мг хлорида цинка и 3 см уксусной-кислоты. Затем добавляют 1,85 г триметклгидрохинона.
13642356
генную смесь выпивают втечение 40 мин
,при 20-26°С 10 г смеси,1,7-дихлор3,7,11,15-гексадецена-2и 3,7-дихлор3,7,11,15-тетраметилгексадецена-1 в
1,5 см уксусной кислоты, и 4,5 см
виде раствора в 20 см уксусной кислоты. Смесь становится гомогенной и имеет коричнево-красное окрашивание, метиленхлорида. Затем добавляют в те- После перемешивания в течение 1 ч до- чение 15 мин при 23 С 5,1 г 1,7,11, ю бавляют 10 см- уксусного ангидрида, 15-тетрахлор-З,7,11,15-тетраметилгек- затем перемешивают в течение 2 ч. садецена-2 в виде раствора в 4 см После гидролиза водой, экстракции уксусной кислоты и 4 см метилеихло- эфиром и высушивания над сульфатом рида. После перемешивания в течение магния растворитель выпаривают при 2 ч при 22-25°С добавляют 3,5 см ук- 15 пониженном давлении. Таким образом, сусного ангидрида. Температура повы- получают 16,2 г масла желтого цвета, шается до 32°С. После выдерживания анализ которого с помощью масс-спект- в течение 15 ч при температуре, близ- рометрии, ЯМР-/ Н и HMP- С показы- кой к 25°С, добавляют 100 см воды, . вает, что оно образовано главным об- - затем бикарбонат натрия до нейтрали- 20 разом, aцeтaтo 2,5,7,8-тетраметил-2зации. Экстрагируют 2 раза 50 см этилацетата. Органические фазы сушат над карбонатом калия. После фильтрации и выпаривания растворителя полу(4 ,-хлор,4,8,12 -триметилтридецил) кроманола-6.
Степень превращения (определена количественным анализом рекупериро- чают 5,82 г масла, содержащего 64% 25 ванного диацетата триметилгидрохино- ацетата 2,5,7,8-тетраметил-2-(4.8 . на) составляет 80,4%.
Смесь 1,7-дихлор-3,7,11,15-тетра- метилгексадецена-2 и 3,7-дихлор-З,7, 11,15-тетраметилгексадецена-1 может 30 быть получена следуюшзим образом.
12 -трихлор-4 ,8 12 .-триметилтриде- цил)-хроманола-6. Выход 53%.
Пример 6.В трехгорлую колбу, снабженную магнитной мешалкой, термометром и восходящим холодильником головки, вводят 1 г полученного в примере 3 продукта, 20 см уксусной кислоты и 0,1 г палладия на угле
В трехгорлую колбу емкостью 250 см -вводят в атмосфере аргона 360,5 мг триэтиламинхлоргидрата (0,26x10 моль).
1-26 мг хлорида меди (I) (0,13х
с 10% палладия. Реакционную смесь на- gg-xlO моль), 9 см уксусной кислоты и гревают при 80°С под давлением водо- 9 см метиленхлорида. Перемешивают
рода. После охлаждения катализатор отфильтровывают. После вьтаривания растворителя получают 0,9 г масла бледно-желтого цвета, содержащего 89,5 мас.% ацетата C)L-токоферола.
Пример 7.Б автоклав вводят 2,04 г полученного в примере 1 продукта, 1,44 мг палладия на угле с 10% палладия и 25 см этанола. Уста- .навливают давление водорода 50 бар, затем нагревают при 80°С в течение 5ч, все время перемевшвая. После охлаждения, отделения катализатора путем фильтрации и выпаривания растворителя получают -токоферол с выходом 96%.
Пример 8. В трехгорлую колбу емкостью 250 см вводят в атмосфере аргона 990 мг безводного хлорида цинка (0,007 моль), которые растворяют в 20 см уксусной кислоты Затем добавляют 4,4 г триметилгидрохинона (0,0289 моль). В эту гетеро3,7,11,15-тетраметилгексадецена-1 в
виде раствора в 20 см уксусной кислоты. Смесь становится гомогенной и имеет коричнево-красное окрашивание, После перемешивания в течение 1 ч до бавляют 10 см- уксусного ангидрида, затем перемешивают в течение 2 ч. После гидролиза водой, экстракции эфиром и высушивания над сульфатом магния растворитель выпаривают при пониженном давлении. Таким образом, получают 16,2 г масла желтого цвета, анализ которого с помощью масс-спект рометрии, ЯМР-/ Н и HMP- С показы- вает, что оно образовано главным об- разом, aцeтaтo 2,5,7,8-тетраметил-2
В трехгорлую колбу емкостью 250 см -вводят в атмосфере аргона 360,5 мг триэтиламинхлоргидрата (0,26x10 моль).
до получения гомогенного желтого раствора. Охлаждают до 0°С, затем быстро добавляют 13,96 г 3-метилен40 7,11,15-триметш1гексадекадиена-1,6, чистота которого составляет 95%. Раствор охлаждают до температуры, близкой к -5 С, затем пропускают ток сухого газообразного хлорводорода в
45 течение 1 ч 20 мин, чтобы ввести 5 г (0,137 моль) соляной кислоты. После перемешивания в течение 30 мин при температуре, близкой к -5°С, реак- ционную смесь выливают в 20 см пенgQ тана и 20 см водного 10%-ного раствора хлорида аммония при температуре близкой к 20°С. Органическую фазу отделяют путем декантахщи, затем высушивают над сульфатом натрия. После
gg фильтрации и выпаривания растворителя получают 17,31 г сырого продукта, анализ которого с помощью масс-спект- роскопии и протонного ЯМР обнаруживает наличие 90% смеси 1,7-дихлор3,7,11,15-гексадецена-2 и 3,7-дихлор 3,7,11,15-тетраметилгексадецена-1.
Для подтверждения линейности скелета полученного продукта 1,7 г полученного выше продукта в виде раствора в 20 см этанола обрабатывают при 80°С под давлением 20 бар водородом в присутствии 170 мг 10%-ного палладия на угле (саже). После фильтрования катализатора и выпаривания растворителя, анализ путем хроматографии в паровой фазе с внутренним стандартом показывает, что выход фи- тана составляет 83,7% по отношению к использованному триену.
Селективность в фитан по отношени к другим изомерам составляет 98%.
Пример 9. Действуют как в примере 8, но используют следующие продукты: 10 г смеси 1,7,15-трихлор- 3,7,11,15-тетраметш1гексадецена-2 и 3,7,15-тршслор-3,7,11,15-тетраме- тш1гексадецена-1, 4 г триметилгидроСМ
хинона, 914 г хлорида 1щнка, 43 уксусной кислоты,10 см уксусного ангидрида.
После обработки реакционной смеси получают 16,63 г масла оранжевого цвета.
Степень превращения триметилгидрохинона составляет 81,3% (определено путем количественного анализа диаце- тата триметилгидрохинона).
Структура ацетата 2,5,7,8-тетра- ме.тш1-2-(4,12 -дихлор-4 ,. 8, триметш1тридецил);-хроманола-6 . подтверждена масс-спектром и и из очищенной фракции полученного масла.
Смесь 1,7,15-трихпор-3,7,11,15- тетраметилгексадецена-2 и 3,7,15- трихлор-3 7,11,15-тетраметилгексадецена-1 получают следующим образом.
Действуют как в примере 1, но используют следующие продукты: 14 г (5,1x10 моль) 2-метил-7,11,15-три- метилгексадекатриена-1,6-14, 370 мг тризтиламиихлоргидрата., 130м хлорида меди (1);, 9 см уксусной , кислоты, 9 см метилеихлорида.
В течение 1 ч пропускают ток сухого газообразного хлорводорода, чтобы ввести 7,3 г соляной кислоты.
После обработки реакционной смеси получают 19,31 г масла, анализ которого с помощью масс-спектрЪметрии и ПМР показывает, что оно образовано главным образом 1,7,15-трихлор-3,7,
-
.
ю
. получен-60 см
г
11,15-тетраметилгексадеценом-2 и 3,7, 15-трихлор-З,7,11,15-тетраметилгек- садеценом-1 и не содержит конъюгироg ванных диенов.
Гидрирование полученного продукта в условиях, описанных в примере 1, показывает, что согласно анализу путем хроматографии в паровой фазе с
10 внутренним стандартом выход фитана составляет 63% по отношению к использованному 2-метилен-7,11,15-гексаде- катриену-1,6,14.
Пример 10. Б аппарат для
15 14-/рирО лаккя вводят 6,67 г но г о в примере ггродуктя, уксусной кислс - ь; и 4С мг паллакия на саже (уг;:е) с 10 м.,с % палладия. Кагр;;ва 01 при 80 С в течение 2,5 ч
20 под давлением водорода 1 бар. После охпаяодения, фипьтгювания катализатора и выпаривания растворителя, получают 5,62 г прозрачного масла, содержащего 74,7% ацетата сА-токоферо25 ла.- х
Выход ацетата с.-токоферола составляет 93% по отношению к прореагировавшему триметилгидрохинону и 80% в расчете на прореагировавший тш1ен-7,11,15-триметш1гексадекади- ен-1,6.
Степень превращения 1,7-дихлор- 3,7,11,15-тетраметнлгексадецена-2 и 3,7-дихлор-З,7,11,15-тетраметил35 гексадецена-1 составляет 97%, причем определение осуществлено анализом .рекуперированного фитана. .П. ример 11. 2,9 г получеино- го в примере 9 масла растворяют в
40 30 см уксусной кислоты, содержащей 220 мг палладия на угле, содержащего IЮ мас.% палладия. Нагревают в течение 4,5 ч при 80°С под давлением водорода 1 бар. После обработки реак45 ционной смеси получают 2,17 г прозрачного масла, содержащего 62% ацетата сА-токоферола.
Вькод ацетата с --токоферола сос- 50 тавляет 76,7% в расчете на прореаги- р..вавший триметилгидрохинон и 65%. в расчете на прореагировавший 2-ме- гш1ен-7,11,15-триметш1гексадекатри- ен-1,6,14.
55 Степень превращения 1,7,15-трихлор-З, 7, 1 1,15-тетраметилгексадеце-. на-2 и 3,7 15-трихлор-З,7,11,15-тет- раметидгексадецена-1 составляет 97%j определение осуществлено количественным анализом рекуперированного фитанао
Предлагаемый способ позволяет получать продукты формулы I, которые превращают в d--токоферол гидрированием в присутствии палладия и которы значительно легче получать, чем продукты по известным.способам. Например, исходя из мирцена, продукты общей формулы I и, следовательно, витамин Е можно получить в 5 стадий. По известному способу витамин Е получают кокденсатдией изофитола, которьй в свою очередь, получают из фитона в две стадии, а фитон получают из мирдена в 5 стадий. Таким образом.
сн.
СНо
СН2
:о.
снг ,с.
С С - С-СНо
Л х /-к х хК хСНо, СН, СН СН СН СН СН С-ОН
2 9
где X и одинаковые или различные, водород или хлор.
СН
сн
,СНо
I
1 /ч с
/Ь.V riTА/у-ЬП- ч - x v-У
4. X: Х1 /,/
CHjl сн сн2. cH2,. сн он
где Х, и X й меют указанные значения, или смесь соединения формулы II с
GHa СЫп,
I л , S С СН.2 С СН2. СН
ч
Хг
где Х и Х имеют указанные значения, подвергают конденсации с триметилгид- рохиноном в среде уксусной кислоты или диоксана в присутствии хлористого цинка при 20-45 С и выделяют целевой продукт в свободном виде или подвергают его ацетштированию уксусным ангидридом в присутствии хлористого
4235,10
исходя из мирдена, витамин Е по из- .вестному способу получают в 8 стадий, а предлагаемый способ получения соединений формулы I позволяет сокра- . тить его получение до 5 стадий.
Кроме того, для осуществления предлагаемого способа требуется применение мирцена и триметилгидрохино Q на, для известного - мирцена, ацетилена, окиси мезитила, винилхлорида и триметилгидрохинона.
15
Формула изобретения
Способ получения производных d- токоферола общей формулы I
сн.
СНа
СН2
:о.
снг ,с.
С С - С-СНо,
К х СН СН С-ОН
сн
или их ацетатов, отличающийся тем, что соединение формулы II -
сн.
I
СН. С1
1 /ч сн.
Х1 /,/
2,. сн он
соединением формулы HI
СНа
СНз
СНо
50
цинка или смеси триэтиламина и диме- тиламинопиридина и выделяют целевой продукт в виде ацетата. Приоритет п. о п р и з н ак а м:
20„09о84 - при Х. и X -хлор, 15.03.85 - при X, и X 2-водород или хлор.



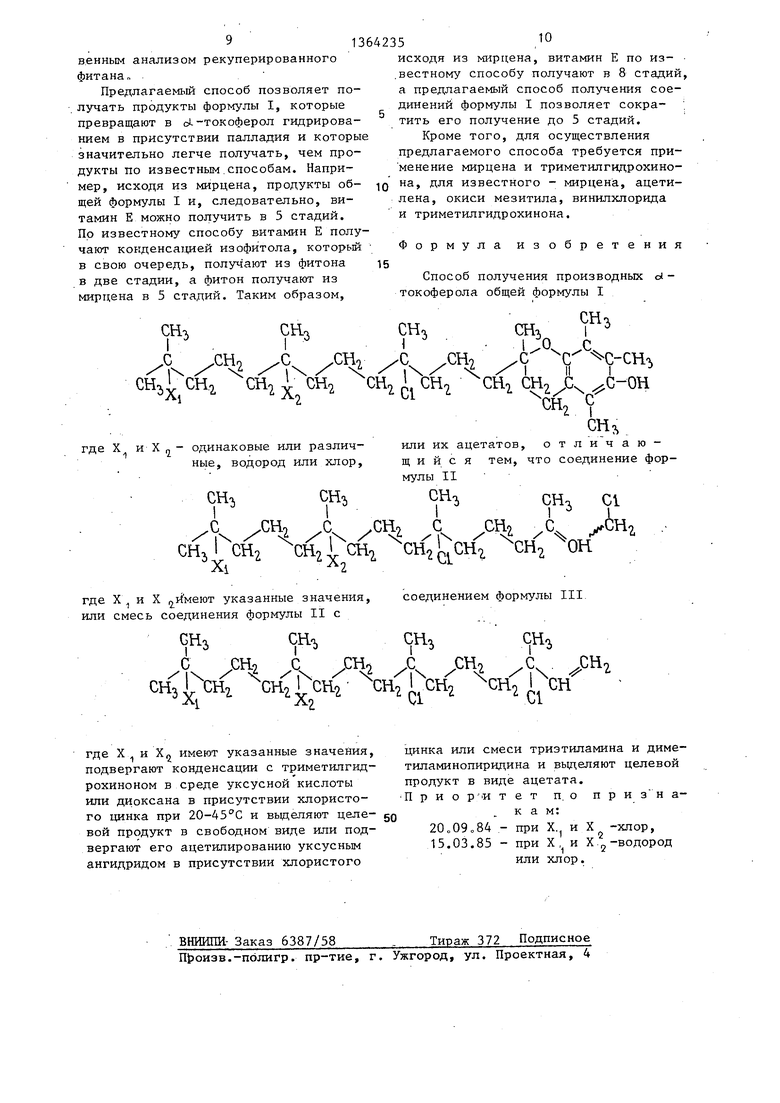
Изобретение касается кислородео-; держащих гетероциклических соедине-. НИИ, в частности производных d -токоферола (ПТ) общей формулы I ()СХ 1 -(СН2)з-СХ,(СНз)-(СН2)э-СС1(СНз) -(СН )-К, где К - группа -С(СН)-0-С-С(СН)С-СН, // // (СН)С-ОН X J и X Н или С1,, или их ацетатов, которые используют в синтезе витамина Е. Для получения последнего по уп- poKteHHONiy процессу созданы новые производные с.-токоферола. Их синтез ведут конденсацией соединений формул II и III (сНз)2 сх,-(сн,р5- cx,j(CHp- -(сН2),-сс1(сНз)-(сн)-с(снр сн-CH.Cl (II), (СНз)2СХ,-(СН2)з-( Х (CHJ -(СН)з-СС1(СН) -(CH)j-CCl (СН2).2(111), где X, и Х имеют указанные значения с триметилгидрохи- ноном в среде СН СООН или диоксана в присутствии ZnCl ,.j при 20-45°С. ПТ i затем вьщеляют или ацетилируют его уксусным ангидридом в присутствии ZnCl 2 или смеси триэтиламина и диме-, тиламиндипир1едина. Витамин Е получают гидрированием ПТ. Использование последних позволяет сократить продолжительность получения витамина Е из мирцена с 8 до 5 стадий. § О) с со tsS оо СП
| Патент Великобритании № 1568559, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Зубодолбежный станок | 1986 |
|
SU1465202A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Устройство станционной централизации и блокировочной сигнализации | 1915 |
|
SU1971A1 |
| Патент Франции № 1179011, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Pujcta I | |||
| et al | |||
| Syntheses | |||
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
