Изобретение относится к усовершенствованному способу получения аиенаф- тенхино и/или нафталезого ангидрида, которые используются как промежуточные продукты в синтезе органических красителей.
Цель изобретения - повы1пе ше сте- vieiiH чистоты и кьгхода Tienenoro продукта.
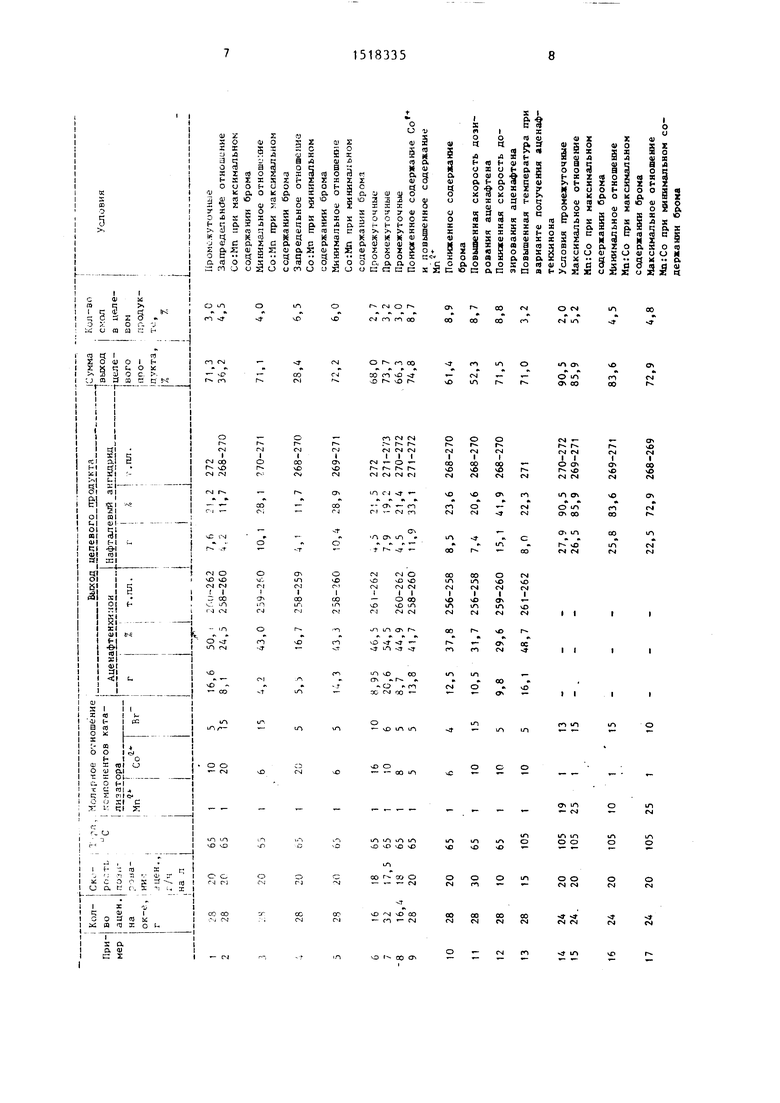
Пример 1, Смесь, состоящую из 2,1 г ацетата кобальта, 0,2 г апе- ата марггнца5 0,6 г КВч при MOjmpHON: отнс„:.лнии :Мп :Вг 10:1:5 i 100 1-гг ;-;едяной уксусной кислоты, заг- p isKaio ,--. реактор, обогреваемый через и- г.1хромовой проволоки и
оборудованный обратнь:м холодильником с разделителем конденсат.ч. контактным тepмo aтpoм, ст;/-.анным с реле, барбо- тером для подачи и дозатором, обогреваемым чсрег рубашку горячей водой, и оборудованиьъ -- обратным холодил 1)НИком.
В дозатор загр -жают 7 г аценафтена в 70 мл ледяной уксусной кислоты при 80°С. Температуру в дозаторе поддерживают постоянной. Перед загрузкой аценафтена в дозатор с небольшим расходом подавать азот, поскольку при контакте с воздухом аг енафтен Б растворе темнеет вследствие его осмоления.
сл
00 00
со ел
31
После загрузки реактора реакционную массу при размешивании нагревают до 65 С с одновременной подачей воздуха со скоростью 0,8-1,0 л/мин. По достижении температуры 65 С из дочатора в реакционную массу начинают подавать раствор аценафтена со скоростью 2 г/ч в пересчете на аце- нафтен. Объем реакционной массы поддерживают постоянным за счет отвода избыточной уксусной кислоты через разделитель конденсата.
После окончания подачи аценафтена реакционную массу выдерживают в вышеуказанном режиме еще в течение 20-30 мин и затем охлаждают в токе воздуха до 20-25°С. Затем твердый осадок отфильтровывают, промывают выделенной уксусной кислотой, сушат и отправляют на отделение нафталево- го ангидрида от аценафтенхинона посредством содовой разварки. Фильтрат, содержащий катализатор и оставшиеся растворенными продукты, объединенный с промьшкой уксусной кислотой, рециркулируют на следующее окисление Перед кажд,ым последующим окислением к фильтрату добавляют 0,25 v КВг (40% от исходного количества) для компенсац ги его потерь Один и тот раствор катализатор без заметного снижения селективности процесса по аценафтенхинону используют четыре раза.
В результате суммарная масса твердого осадка составляет 24,2 г.
После разделения осадка получают 16,6 г аценафтенхинона, 50,1% от теории с Т.Ш1. 260-262 С (по лит.данным т.ил. 261-263°С) и 7,6 г нафталевого ангидрида с т.пл, 272 С (по лит.данным т.пл. 272-274 С), 21,2% от теории. Продукты реакции определяют методам газожидкостной хроматографии
Пример 2. Смесь, состоящую из 4,28 г ацетата марганца, 0,2 г ацетата кобальта, 1,39 г бромида калия при их молярном отношении
19:1:13 и 100 мл ледяной уксусной кислоты, загружают в реактор. Реакционную массу при раз- мепгивании нагревают до 105 С с одновременной подачей воздуха со скоростью 0,8-1,0 л/мин.
При достижении температуры 105 с из дозатора начинают подавать в реакционную массу 6 г аценафтена в 60 МП уксусной кислоты со скоростью
354
2 г/ч в пересчете на аценафтен. Далее следуют те же операции, что и в примере 1.
В результате четьрех окислений масса твердого осадка составляет 27,9 г, 90,5% от теории, и который представляет собой практически чистый нафталевый ангидрид с т.пл.270- 272°С (по литературным данным т.пл.272-274°С).
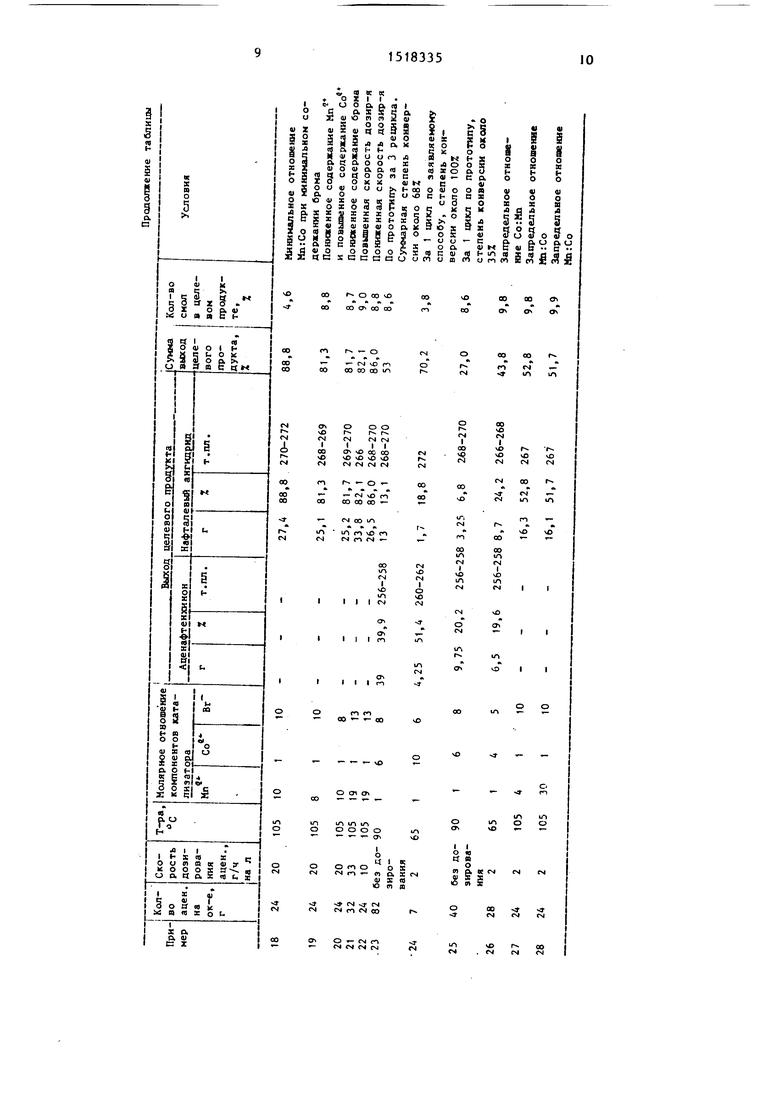
Примеры 3-28. Процесс проводят как и в примерах 1 и 2.
Условия ведения процесса и полученные результаты приведены в таб
лице.
Пример 29 (сравнительный). Процесс проводят аналогично примеру 1. Смесь, состоящую из 2,1 г ацетата кобальта. О,2 г ацетата марганца и 0,6 г бромида калия при молярном отношении 10:1:5 и 100 мл ледяной уксусной кислоты, загружают в реактор. Реакционную смесь нагревают при размешивании до 105 с с одновременной подачей воздуха со скоростью 0,8- I,О л/мин.
При достижении температуры 105 С из дозатора начинают подавать в реакционную массу 7 г аценафтена в 60 М.Ч уксусной кислоты со скоростью 1,5 г/ч в пересчете на аценафтен. Время выдержки после окончания дозирования 25 мин.
После окончания реакции с трехкратной рециркуляцией раствора, выделения осадка и разделения его компонентов получают 24,1 г твердого продукта, в том числе 16,1 г аценафтенхинона (48,7%) с т.пл. 261-262 0 и 8,0 г нафталевого ангидрида (22,3%) с т.ш. 271°С.
Как видно из приведенных примеров, выход за указанные в формуле
изобретения пределы варьирования концентрации катализатора ведет к нежелательным результатам.
При получении аценафтенхинона наиболее эффективная суммарная концентрагдая Со и Мп соответствует 0,08- 0,09 моль/л. Уменьшение ее до 0,07 моль/л и ниже ведет к увеличению в оксидате количества промежуточных продуктов, которые участвуют в смолообразовании. В результате вькод целевых продуктов падает и снижается их чистота. Увеличение суммарной кон центрации О, 1() моль/л .
5
и выше ведет к интенсификации процеса окисления и усилению осмоления образующихся продуктов, что приводи к снижению выхода и чистоты целевых веществ.
При получении нафталевого ангидрида наиболее эффективна суммарная концентрация Со и Мп 0,17 - 0,18 моль/л. Как и в случае с вариантом получения аценафтенхинона выход за границь указанного интервала до О,16 моль/л и ниже и до 0,19 моль и выше, ведет к снижению выхода и загрязнению целевых продуктов.
Из приведенных примеров видно,
2
ЧТО уменьшение концентрации Со и увеличение концентрации Мп вне указанных в формуле интервалов веде к образованию менее активного катализатора при низкой температуре и в то же время менее селективному по отношению к аценафтенхинону. Это ведет к увеличению выхода нафталевого ангидрида по отношению к аценафтенхинону, усилению осмоления в связи с увеличением количества промежуточных продуктов и загрязнению целевых веществ, уменьшению суммарного выхода аценафтенхинона и нафталевого ангидрида.
Кроме того увеличение концентрации Со и уменьшение концентращш вне указанных диапазонов ведет к преимущественному окислению через аценафтенхинон, к появлению его среди конечных продуктов, увеличению смолообразования и загрязнению целевых веществ, уменьшению суммарного их выхода.
Как видно из данных таблицы, суммарный выход целевых продуктов в расчете на загруженный аценафтен за 1 цикл по предлагаемому способу составляет 70,2% при конверсии 100%, а по прототипу - 27% при степени конверсии исходного аценафтена 35% (примеры 24 и 25 соответственно), что значительно ниже.
Тот факт, что степень конверсии исходного аценафтена за 1 цикл по
356
прототипу составляет всего 30-35%, в то время, как по предлагаемому способу степень конверсии близка к 100% означает, что использование предлагаемого способа позволяет значительно повысить удельную производительность процесса, т.е. увеличить съем продукта с единицы реакторного
объема.
Сравнивая окисление по предлагаемому способу с тремя рециклами (примеры 1-22) с окислением по прототипу с тремя рециклами (пример 23) видно,
что суммарный выход целевых продуктов по прототипу и за три рецикла невысок, степень конверсии всего около 68%, а содержание смолообралных продуктов высокое.
Формула изобретения
Способ получения аценафтенхинона и/или нафталевого ангидрида жидкофаз- ным окислением аценафтена кислородсодержащим газом при 65-105°С в присутствии катализатора - смеси ацетатов Со и Мп, промотированного броми- ДОМ щелочного металла в органическом растворителе, отличающий- с я тем, что, с целью повьпиения степени чистоты, выхода целевого продукта, окисление ведут воздухом при молярном соотношении компонентов ка- тализатора, равном Мп tCo : Вг - 1:(6-16):(5-15 ) в случае совместного получения ацекафтенхинона и нафталевого ангидрида или Мп :Вг (10-25):1:(10-15) в случае получения нафталеного ангидрида, при зтом процесс ведут в полупериодическом режиме с непрерьтным дозированием аценафтена в растворителе, в качестве которого используют ледяную уксусную кислоту, со скоростью подачи 15- 20 г/ч на 1 л релкююнно массы в пересчете на аценафтен с одновременным отбором избыточной уксусной кислоты для поддержания постоянного объема реакционной массы.


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НАФТАЛЕВОГО АНГИДРИДА | 1971 |
|
SU291910A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕПАФТЕНХИНОНА | 1970 |
|
SU280469A1 |
| Способ получения нафталевого ангидрида | 1975 |
|
SU535306A1 |
| Способ получения 4-ацетилнафталевого ангидрида | 1987 |
|
SU1659394A1 |
| СПОСОБ ПОЛУЧЕНИЯ НАФТАЛЕВЫХ КИСЛОТ ИЛИ ИХ 5 | 1967 |
|
SU202114A1 |
| Способ получения 4-бромнафталевого ангидрида | 1982 |
|
SU1084275A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАНГИДРИДА 4,4´-БИНАФТИЛ-1,1´,8,8´-ТЕТРАКАРБОНОВОЙ КИСЛОТЫ ИЗ ГАЛОГЕНАЦЕНАФТЕНОВ | 2017 |
|
RU2671579C1 |
| 4-(П-карбоксифенилсульфон)нафталевый ангидрид, как полупродукт при синтезе термостойких азотсодержащих гетероциклических полимеров, и способ его получения | 1976 |
|
SU598897A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕМИМЕЛЛИТОВОЙ | 1973 |
|
SU380638A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-НИТРОНАФТАЛЕВОЙ КИСЛОТЫ | 1973 |
|
SU374287A1 |
Изобретение касается ароматических кетонов и ангидридов кислот, в частности получения аценафтохинола и или нафталевого ангидрида. Процесс ведут жидкофазным окислением аценафтена O2 - содержащим газом или 65-105°С в присутствии смеси ацетатов CO и MP, промотированной бромидом щелочного металла в среде ледяной CH3C(O)OH. В случае совместного получения целевых продуктов используют катализатор, имеющий молярное соотношение MN2+:CO2+:BR- = 1:6-16:5-15, а для получения нафталевого ангидрида 102-5:1:10-15 при этом процесс ведут в полупериодическом режиме с непрерывным дозированием аценафтена в CH3C/O/OH со скоростью 15-20 г/ч.л реакционной массы (в пересчете на аценафтен) с одновременным отбором избыточной CH3C(O)OH для поддержания постоянного объема реакционной массы. Эти условия повышают степень чистоты целевого продукта при 100%-ной конверсии исходного аценафтена и суммарном выходе целевых продуктов 70,2 против 35 и 27%. 1 табл.
| СПОСОБ ПОЛУЧЕНИЯ АЦЕПАФТЕНХИНОНА | 0 |
|
SU280469A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
