ел го





ИЗОБРЕТЕНИЕ КАСАЕТСЯ ПРОИЗВОДНЫХ ЦЕФАЛОСПОРИНОВ, В ЧАСТНОСТИ СИН-ИЗОМЕРОВ СОЕДИНЕНИЙ ОБЩЕЙ Ф-ЛЫ N=C(NH2)-S-A=C-C(=N-OCH3)-C(O)NH- @ , где а) N=1 или 2
A- -CH*98 или -N*98
R-OH
б) N=2
A- -CH*98 или -N*98
R-карбомоил
в) N=2
A- -N*98
R-CH3, HOCH2, проявляющих антибактериальную активность, что может быть использовано в медицине. Цель - создание новых активных и малотоксичных веществ указанного класса. Синтез ведут из соответствующей цефемкарбоновой кислоты, которую переводят в целевой продукт обработкой сначала, например, N-метил-N-(триметилсилил)-трифторацетамидом, затем иодтриметилсиланом и далее 4-окси-1,4-метиленпиперидином. Новые вещества более эффективны и менее токсичны, чем известные антибиотики, и оказывают действие на широкий спектр микроорганизмов - грамположительных и грамотрицательных. 2 табл.
Изобретение относится к новым производным цефалоспорина в виде син- изомеров, которые проявляют антибактериальную активность и могут найти применение в медицине в качестве лекарственных средств.
Целью изобретения является создание новых антибиотиков цефалоспорино- вого ряда, обладающих антибактериальным действием против более широкого спектра как грамотрицательных, так и грамположительных микроорганизмов при низкой токсичности..
Пример 1. 7/3-t(Z)-2-(2-Aми- нoтиaзoл-4-ил)-2-мeтoкcииминoaцeт- : амидо}-3- (4-окси-1,4-метилен-1 -пипери- динио)метил-3-цефем-4-карбоксилат
N-J-C-CONH S H2N S N ..
сооо .®
(г)-2-(2-Аминотиазол-4-ил)-2- мeтoкcииминoaцeтaмидoJ-2-aцeтoкcимe- тил-З-цефем-4-карбоновую кислоту (240 мг) суспендируют в хлористом метилене (4 мл), после чего добаво
лятт N-мeтил-N-(тpимeтилcилил)тpи- фторацетамид (330 мкл). Полученную смесь перемешивают при комнатной температуре в течение 30 мин. После охлаждения льдом в раствор добавляют йодтриметилсилан (200 мкл), и полученную смесь перемешивают при комнатной температуре в течение 15 мин. Реакционную смесь концентрируют в вакууме, и в результате получают си лилированное производное 7p-(Z)-2- -(2-аминотиазол-4-ил)-2-метоксиимино ацетамидо -3-йодметил-З-цефем-4-кар- боновой кислоты.
Это силилированное производное растворяют в ацетонитриле (3 мл), после чего добавляют тетрагидрофу- ран (60 мкл). В полученньш таким образом раствор вводят 4-окси-1,4-ме- тиленпиперидин (72 мг) и полученную смесь перемешивают при комнатной температуре в течение 2ч. Затем в реакционную смесь добавляют метанол (0,3 мл) и перемешивают в тече- ние 15 мин. Полученньй осадок извлекают путем фильтрации и затем промывают ацетонитрилом. Этот осадок растворяют в 30%-ном этаноле. После концентрирования в вакууме оста- точньй продукт концентрирования растворяют в смеси растворителей ацетон- вода в соотношении 7:1. Полученньй раствор очищают в хроматографичес- кой колонке с сштикагелем (элюент: смешанный растворитель ацетон-вода в соотношении 9:1 и 7:1), и в результате получают желаемый продукт (36мг).
Пример 2. 7/3- (2)-2-(5-Ами- но-1,2,4-тиадиазол-3-ил)-2-метокси- иминоацетамидо -3-(4-окси-1,4-мети- лен-1-пиперидинио)метил-З-цефем-4-к боксилат
N-Tr-C-CONH S О II ,Ч
НгМ S N дД ЛсН2-К@-ОН
о-снз соое ®
7/i- (г)-2-(5-Амино-1, 2,4-тиaдиa- зoл-3-ил)-2-мeтoкcииминoaцeтaмидo - -3-ацетоксиметил-3-цефем-4 карбоно- вую кислоту (319 мг) суспендируют в хлористом метилене (4 мл), после чего добавляют N-мeтил-N-(тpимeтил- силил)трифторацетамид (877 мкл). Полученную смесь перемешивают при комнатной температуре в течение 1 ч После охлаждения льдом добавляют йодтриметилснлан (268 мкл),. и полу
0 5 5
5
0
ченную смесь перемешивают в течение 15 мин.Реакционную смесь концентрируют в вакууме, и в результате получают желаемое силилированное производное 7/3- (Z)-2- (5-амино-1,2, 4-тиадиазол-3-ил)-2-метоксиимино- ацетамидо -3-йодметил-3-цефем-4- карбоновой кислоты.
Это силилированное производное растворяют в ацетонитриле (3,6 мл). К полученному таким образом раствору добавляют 4-ОКСИ-1,4-метиленпи- перидин (71 мг) и смесь перемешивают в течение 2 ч при охлаждении льдом. Затем в реакционную смесь добавляют метанол (0,3 мл) и перемешивают в течение 15 мин. Полученный осадок извлекают путем фильтрации и затем промывают ацетонитрилом. Этот осадок растворяют в 30%-ном этаноле. После концентрирования в вакууме остаточньй продукт концентрирования растворяют в смеси растворителей ацетон-вода в соотношении 7:1. Полученный раствор очищают в хроматографической колонке, наполненной силикагелем (элюент: ацетон- вода в соотношении 7:1 и 5:1), и в результате получают желаемый продукт (29 мг).
Пример 3. 7/1-(г)-2-(2-Ами- нотиазол-4-ил)-2-метоксииминоацет- амидо -3-(4-окси-1-хинуклидинио)ме- тил-3-цефем-4-карбоксш1ат К-у-С -СОШ S
А-- - X5/CH,-g3-OH 0-сНз о 00© ®
7/J- С(г)-2-(2-Аминотиазол-4-ил)- 2-метоксииминоацетамидо -2-ацёто1 си- метил-З-цефем-4-карбоновую кислоту (977 мг) суспендируют в хлористом метилене (16 мл), после чего добавляют К-метил-Н-(триметилсилил) три- фторацетамид (1350 мкл). Полученную смесь перемешивают при комнатной температуре в течение I ч. После охлаждения льдом добавляют йодтриметилсилан (810 мкл), и полученную смесь перемешивают в течение 15 мин. Реакционную смесь концентрируют в вакуут е, в результате получают силилированное производное 7(J-(Z)-2- (2-аминотиазол-4-ил)-2-метоксиими- ноацетамидо -3-йодметил-3-цефем-4- карбоновой кислоты.
Это силилированное производное растворяют в ацетонктриле (12 мл), после чего добавляют тетрагидрофуНг S Ч
ран (240 мкл), К полученному таким образом раствору добавляют 4-окси- хинуклидин (300 мг) и полученную смесь перемешивают при комнатной температуре в течение полутора часо Затем в реакционную смесь добавляют метанол (1,2 мл) и перемешивают в течение 15 мин. Образовавшийся осадок извлекают путем фильтрации и затем промьгаают ацетонитрилом. Этот осадок растворяют в 30%-ном этаноле. После концентрирования в вакууме остаток концентрирования растворяют в смешанном растворителе ацетон-вода в соотношении 7:1. Образующийся раствор очищают в хромато- графической колонке с силикагелем (элюент; смешанный растворитель ацетон-вода в соотношении 7:), и в результате получают желаемый продукт (38 мг).
Пример 4. 7р-(г)-2-(5-Ами но-1,2,4-тиадиазол-3-ил)-2-метоксииминоацетамидоЗ-3-(4-окси-1-хинукли динио)метил-3-цёфем-4-карбоксилат
J- C-COMH S H,NV:1V ,-КЭ-ОН 0-СНз ° СОО©
7/1- (2)-2-(5-Амино-1 ,2,4-тиадиа- зoл-3-ил)-2-мeтoкcииминoaцeтaмидoJ- З-ацетоксиметип-З-цефем-4-карбоно- вую кислоту (486 мг) суспендируют в хлористом метилене (9 мл), после чего добавляют N-мeтил-N-(тpимeтшI- силил)трифторацетамид (980 мкл). Полученную смесь перемешивают -при комнатной температуре в течение 1 ч. После охлаждения льдом добавляют йодтриметилсилан (410 мкл), и полученную смесь перемешивают в течение 15 мин. Полученную реакционную смес концентрируют при пониженном давлении и в результате получают силили- рованное производное (Z)-2-(5- амино-1,2,4-тиадиазол-З-ил)-2-меток- сииминоацетамидо -3-йодметил-3-це- фем-4-карбоновой кислоты.
Это силилированное производное растворяют в ацетонитриле (6 мл), после чего добавляется тетрагидро- фуран (130 мкл). К полученному раст- вору добавляют 4-гидрохинуклидин . (150 мг), и полученную смесь перемешивают при комнатной температуре в течение 1 ч. Затем в реакционную смесь вводят метанол (0,6 мл) и перемешивают в течение 15 мин. 05разукнинйся осадок извлекают путем фильтрации и яатем промывают ацетонитрилом. Этот осадок растворянэт В: 30%-ном этаноле. После концентрирования в вакууме остаточный продукт концентрирования оч шпают в хрома- тографической колонке с силикагелем (элюент: смешанный растворитель
д ацетон-вода в соотношении 9:1), ив результате получают желаемый продукт (13 мг).
Пример 5. 7/э-(2)-2-( нотиазол-4-ил)-2-метоксии :иноа}дет5 амидо -3-(4-карбамоил-1-хинуклиди- нио)метил-3-цефем-4-карбоксш1ат
HiN S v QJ-N- CH -N(A)-CONl -2,
00-СНзCOO© ®
7p- (Z)-2-(2-Aминoтиaзoл-4-ил)- 2-мeтoкcииминoaцeтaмидo -3-aцeтoк cимeтил-3-цeфeм-4-кapбoнoвyю кислоту (240 мг) суспендируют в хлористом
5 метилене (4 мл), после чего добавляют К-метил-К-(триметилсилил)трифторацетамид (330 мкл). Полученную смесь перемешивают при комнатной температуре в течение 30 мин. После
0 охлаждения льдом добавляют йодтриметилсилан (200 мкл) и полученную смесь перемешивают в течение 15 мин. Затем реакционную смесь концентрируют в вакууме и в результате полус чают силилированное производное 7р- ((г)-2-(2-аминотиазол-4-ил)-2-меток- сииминоацетамидо -3-йодметил-З-це- фем-4-карбоновой кислоты.
Это силилированное производное
0 растворяют в ацетонитриле (3 мл), после чего вводят тетрагидрофуран (60 мкл). В полученный раствор вводят 4-карбамоилхинуклидин (98 мг) . и полученную смесь перемешивают при
с комнатной температуре в течение 2 ч. В реакционную смесь вводят метанол (0,3 мл) и перемешивают при комнатной температуре в течение 15 мин. Полученный осадок извлекают путем
0 фильтрации и затем промьшают ацетонитрилом. Этот осадок растворяют в 30%-ном зтаноле. После концентрирования в вакууме остаточный продукт концентрирования растворяют в смес шанном растворителе ацетон-вода в соотношении 7:1. Полученный таким образом раствор очищают в хромато- графической колонке с силикагелем (злюент: смешанный растворитель адетон-вода в соотношении 7:1), и в результате получают желаемый продукт (53 мг).
Пример 6. 7/1-(г)-2-(5-Ами- но-1,2,4-тиaдиaзoл-3-ил)-2-мeтoкcи- иминoaцeтaмидo J-3-(4 кapбaмoил-l xинyклидиииo)мeтил-3-цeфeм-A кap- боксилат
НпМ
t - -C-CONbl Д Л-V-r
N
®/
npu
иЬптI . COO®
7р- (Х)(.5-Амино-1,2,4 тиадиа- 3ол-3-ил)2-метоксииминоацетамидoj- 3-ацетоксиметил-3-цефем-4 карбонов5по кислоту (790 мг) суспендируют в хлористом метилене (10 мл), после чего добавляют Н-метил-М-(триметилсилил) трифторацетамид ( мл) Полученную смесь перемешивают при комнатной температуре в течение 1 ч. После охлаждения льдом добавляют йод- триметилсилан (660 мкл), и полученную смесь перемешивают в течение 15 мин. Реакционную смесь концентрируют в вакуу1ме, и в результате получают силилированное производное 7у5- (7)2-(5-амино-1,2,4-тиадиазол-З- ил)-2-метоксииминоацет.амидо 3-йод- метил-3-цефем-4-к;арбоновой кислоты
Это силилированное производное растворяют в ацетонитриле (9 мл), после чего добавляют 4-карбамонл- хинуклидин (240 мг), Полученную смесь перемешивают в течение 1 ч при охлаждении льдом. Затем в реакционную смесь вводят метанол (0,6 мл) и перемешивают в течение 15 мин. Полученный осадок извлекают путем фильтрации и промьшают ацетонитрилом Этот осадок растворяют в 30%-ном этаноле. После концентрирования при пониженном давлении остаточньй про- дзпкт концентрирования растворяют в смешанном растворителе ацетон-вода в соотношении 7:1. Раствор очищают в хроматографической колонке с силикагелем (элюент: смешанный растворитель ацетон-вода в соотношении 7:1 и 5:1), ив результате получают желаемый продукт (326 мг),
Пример 7, 7р-(г)-2-(5-Ами- но-192,4-тиадиазол-3-ил)2-метокси- иминоацетамидо-3-(4-оксиметил-1-хи нуклидинио) метил-3-цефем-4-карбокси лат N- C-COMH, S
HT,N S Noti CH - -CH Ol-i
O-CHj
coo®
Аналогично примерам 1-6 получают сили.пированное производное 7/3-(Z)2- (5-амино-,2,4-тиадиазол-3-ил)2-ме- токсииминоацетамидо-3-йодметил-3- цефем-4-карбоновой кислоты из 7/.3-(Z)- 2-(5-амино-15 2,4-тиадиазол-З-ил)-2- метоксииминоацетамидо-3-ацетоксиме- тил-3-цефем-4-карбоновой кислоты (460 мг)5 Ы-метил Н-(триметилсилил) трифторцетамида (640 мкл) и йодтри- метилсилана (390 мкл). Это силилированное производное химически взаимодействует с 4-оксиметш1хинуклидином (142 мг), и в результате получают желаемый продукт (8 мг)..
Пример 8. 7/ь-(7.)2-(5-Ами- но-15 2,4-тиадиазол-З-ил)-2 метокси- иминоацетамидо}-3-(4-метил-1-хинук- лидинио)-метил-3-цефем-карбоксилат
®/
S f то,л-л
-Ny -CH -N U-CHg
Joo®
5
0
5
0
5
0
5
пара-Метоксибензил (Z)-2-(5™ амино-1,254-тиадиазол-3-ил)-2-меток- сииминоацетамидо -3-йодметил-3-це- фем-4-карбоксилат (700 мг) растворяют в смешанном растворителе этил ацетата (50 мл) и метанола (1 мл). После охлаждения льдом к этому раствору добавляют этилацетатньм раствор (2,8 мл) 4-метилхинуклидина (114 мг), и смесь перемешивают в течение 15 мин. Полученный осадок извлекают путем фильтрации, затем его промьшают этил- ацетатом и получают йодид пара-меток- сибензил 7(i- (70-2-(3-амино-1,2,4- тиадиазол-3-ил)-2-метоксииминоацета- 4-метил-1-хинуклидинио)ме- :тил-З-цефем-4-карбоксилата (770 мг). Это соединение (770 мг) суспендируют в хлористом метилене (8 мл). После охлаждения льдом к раствору добавляют анизол (510 мкл) и трифтор- уксусную кислоту (730 мкл), Смесь перемешивают в течение 4ч, после чего ее дополнительно перемешивают в течение 2,5 ч при комнатной температуре. Полученный реакционный раствор по каплям вводят в диизопропило- простой эфир (30 мл) и образующийся осадок извлекают путем фильтрации. Осадок растворяют в воде (5 мл). Величину рН раствора доводят до 5,0 путем добавления бикарбоната натрияi Смесь подвергают хро- матографическому разделению с обратимой фазой в колонке, наполненной силикаг елем (элюент: раствор вода-5%- кый метанол) с целью ее очистки, и получают желаемый продукт (27 мг).
Пример 9. 7р.- (Z)-2(5-AMH- но-1,2,4-тиадиазол-3-ил)-2-метокси- имииоацетамидо -3(4-карбамоил 1-хи- нуклидино)-метил-3-цефем-4-карбокси- лат
Nи
H, N
-C-CONH
S
ОСИ-,
®/-л
Q i-N-ACH -N CONH
3COO©
Смесь, состояшую из 2-(5-амино- 1,2,4-тиадиазол-3-ил)-(г)-2-кето1 си- иминоуксусной кислоты (46 мг), 1-ок- си-Ш-бензотриазолгидрата (35 мг), N,N -дициклогексилкарбодиимида и (52 мг) N,N-димeтилфopмaмидa (1 мл) перемешивают при комнатной температуре в течение 3 ч, затем эту смесь фильтруют и фильтрат охлаждают до 0°С. Полученпый раствор вводят в охлажденный льдом раствор гидрохлори- да 7р-амино-3-(4-карбамоил-1-хинук- лидинио)метил-З-цефем-4-карбоновой кислоты (100 мг), К,Ы-диметилформ- амида (2 мл) и N,N-димeтилaнилинa (72 мкл), После перемешивания сне- шанного раствора при комнатной температуре в течение 14 ч реакционную смесь фильтруют и фильтрат вводят по каплям в диэтиловый эфир (100 мл) при одновременном перемешивании смеси. Вьтавший осадок фильтруют и про- мьшают диэтиловым эфиром. К промытому осадку добавляют воду (10 мл) и нерастворимое вещество отфильтро- вьшают. Полученный фильтрат очищают путем хроматографического разделения с обратимой фазой в колонке, напол 1енной силикагелем, и -в результате получают желаемый продукт (3 мг).
Пример 10. 7р-(Z)-.-(5- Амино-1,2,4-тиад,иазол-3-ил)-2-ме- токсииминоацетамидо -3-(4-карбамо- ил-1-хинуклидинио)метил-З-цефем-4- карбоксилат
N-TT-C-CONH H.
coo
Г идрохлоррщ 7й-амино-3-(4-карба- моил-1-хинуклидинио)-метил 3-цефем- 4-карбЬксилата (2 г) растворяют в смешанном растворе ацетокитрил - вода (1:1) (40 мл) и в раствор вводят триэтиламин (2,08 мл). Полученный раствор охлаждают льдом и в него
g
cH -N; coNHa
I с
0
5
вводят 2-(5-амино-1,2,4-тиадиаяол- 3-ил)-(7,)-2-метоксиимнноацетилхло- рид (2,55 г), и смесь перемешивают в течение 50 мин, В этанол (200 мл) вводят данный реакционный раствор, выпавшее в осадок твердое вещество фильтруют, твердый продукт промывают этанолом и изопропиловым эфиром, и в результате получают желаемый продукт (450 мг).
Пример 11. 7/5-Г(г)-2-(5-Ами- но-1,2,4-тиазол-3-ил)-2-метоксиими- ноацетамидо -3-(4-карбамоил-1-хинуклидинио )метил-3-цефем-4 -карбоксил ат
N-n-C-CONH. S.
Ч ,-N®-com,
° з оо©
0
5
0
0
5
0
7 5-Амино-3-(4-карбамоил-1-хинуклидинио)-не т ил-3-цефем-4-к арб ок си- лат перхлорат (300 мг) суспендируют в воде (1,5 мл). К суспензии добавляют ацетат натрия тригидрат (437 мл) и метанол (9 мл). К полученному продукту затем добавляют 2-(5-амино-1,2,4-тиадиазол-3-ил)-2- метоксииминоацетилхлорид гидрохлорид (180 мг) при . По окончании реакции через 1 ч реакционный продукт охлаждают льдом, чтобы получить кристаллическую массу, которую разделяют фильтрацией. Массу промывают метанолом и диизопропиловым эфиром соответственно, чтобы полу- 5 чить названный продукт (189 мг),
Пример 12. 7/1-(Z)-2-(5- Амино-1,2,4-тиадиазол-3-ил)-2-ме- токсииминоацетамидоj-3-(4-карбамо- Ш1-1-хинуклидинио) метил-3-цефем- 4-карбоксилат
N-irC-C01SlH S
д II ,- Vф
S N,4-к ЛсНг- |Я -СОННг.
0-%COO©
7/1-Амино-3-(4-карбамоил-1-хинуклидинио ) метил-З-цефем-4-карбокси- лат (750 мг) смешивают с водой (5 мл), метанолом (30 мл), ацетатом, натрия тригидратом (560 мг) и 2- (5-амино-1,2,4-тиадиаэол-3-ил)-2- метоксииминоацетилхлорид гвдрохло- ридом (796 мг).
Полученную смесь подвергают реакции в течение 2 ч при 28 С, ох
лаждают льдом и оставляют на ночь. Образовавшуюся кристаллическую массу подвергают фильтрации, затем промывают метанолом и диэтиловым эфиpo 4 соответственно, получая названный
продукт (275 мл). Фильтрат концентрируют с последующим прого- . ном полученной фазы на хроматогра- фичёской колонке с силикагелем, получая дополнительно 200 мг названного продукта, как вторая порция.
Предлагаемые соединения обладают сильным антибактериальным действием как против грамположительных, так и против грамотрицательных бактерий. Пределы их острых токсичностей (при внутривенной инъекции в органах мышей) составляют более 3 г/кг..
При использовании этик соедине- НИИ в качестве антибактериальных композиций их доза ввода в организм должна составлять 2 - 300 мг/кг/день или предпочтительно 10-100 мг/кг/ден
Антибактериальную композицию можно вводить в организм через рот (орально) в форме порошка, гранул, капсул, таблеток, и т.д., или парентерально в форме растворов, свечей и т.д. Эти композиции могут быть при готовлены с использованием эффективного количества соединения, отвечающего изобретению, и фармацевтически пригодных носителей.
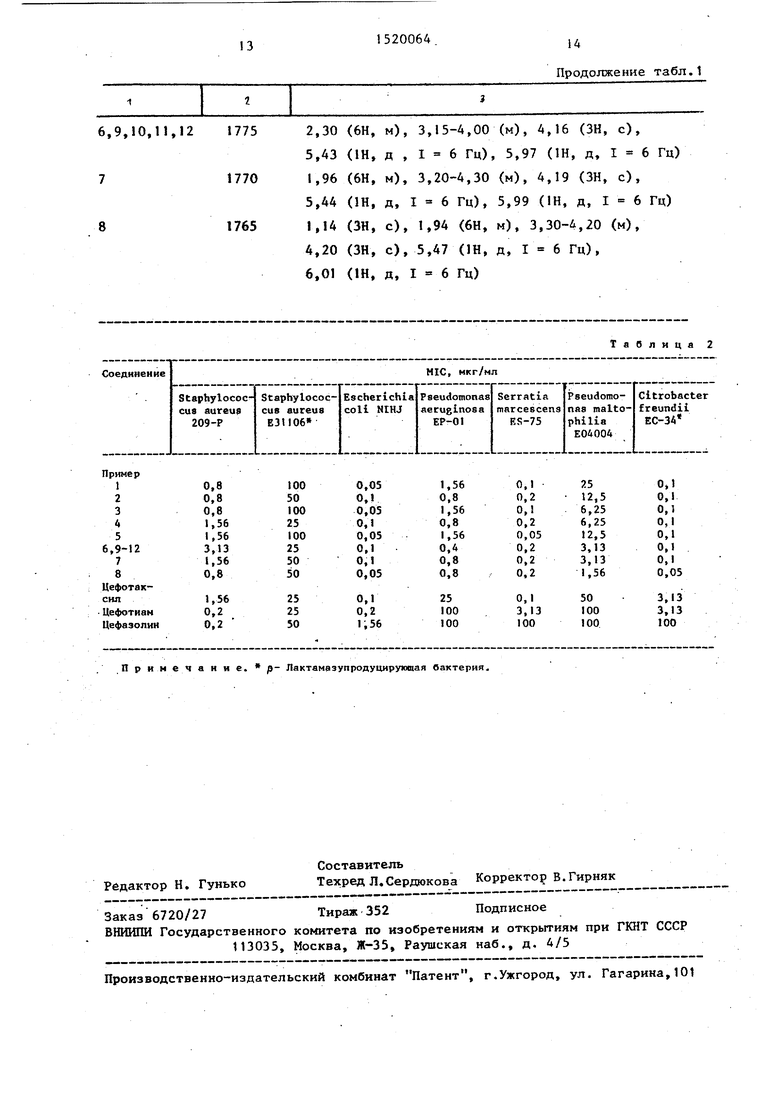
Данные спектральных параметров цёфалоспориновых соединений, полученных по примерам 1-12, приведены I
2 3
1765 2,30 (4Н, м), 3,20-4,40 (м), 4,08 (ЗН, с),
5,43 (1Н, д, Гц), 5,94 (1Н, Д, I 6 Гц),
1775 2,32 (4М, м), 3,30-4,00 (м), 4,18 (ЗН, с),
1765 2,20 (6Н, м), 3,30-4,00 (м), 4,18 (ЗН, с),
5,43 (Ш, д, I 6 Гц), 5,97 (1Н, д, 1 6 Гц) 1770 2,30 (6Н, м), 3,30-4,00 (м), 4,10 (ЗН, с),
в табл. 1; результаты изучения антибактериальной активности, выраженные в.величинах MIC (минимальных ингибирующих концентраций), представлены в табл. 2.
Предлагаемые производные цефало- спорина являются более эффективными, чем известные цефалоспориновые антибиотики, и представляют интерес для создания новых антибактериальных препаратов широкого спектра действия.
Формула изобретени
Производные цефалоспорина общей формулы
tsJ-jrC-GOTSIH S ,
оснз О Joo©
в виде син-изомеров, где при
п 1 или 2, АСН или -N J R -
оксигруппа; или
при п 9 2, А СН «или -N ,
R - карбамоил;
или при п 2, А - -К «, R - метил или оксиметнп,
проявляющие антибактериальную активность.
Таблица 1
6,9,10,П.2 1775
1770
1765
2,30(6Н, м), 3,15-4,00 (м), 4,16 (ЗН, с),
6,01(1Н, д, I 6 Гц)
Примечание. р- Лактамвзупродуцирующая бактерия.
Продолжение табл.1
Таблица 2
| Устройство для регистрации измеряемых величин | 1941 |
|
SU62321A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| Выложенная заявка ФРГ № 3316798, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Колосниковая решетка с чередующимися неподвижными и движущимися возвратно-поступательно колосниками | 1917 |
|
SU1984A1 |
