Изобретение относится к способу получения производных пептидов - новых биологически активных соединений, которые могут найти применение в медицине.
Цель изобретения - способ получения новых производных пептидов - малотоксичных, высокоактивных в отношении противогипертонической активности соединений, устойчивых к гидролизу в биологических средах.
Системы растворителей, используемые при хроматографировании в тонком слое силикагеля.:



| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пептидов | 1986 |
|
SU1609455A3 |
| Способ получения N-[3-(N-циклопентакарбонил-D-аланилтио)-2-D-метилпропаноил]-L-пролина | 1981 |
|
SU1660578A3 |
| Способ получения производных пролина или их фармацевтически приемлемых солей | 1982 |
|
SU1272982A3 |
| Способ получения производных пролина или их фармацевтически приемлемых солей | 1982 |
|
SU1316556A3 |
| Способ получения производных 4(3Н)-оксо-5,6,7,8-тетрагидропиридо(2,3- @ )пиримидина или их таутомерных форм | 1987 |
|
SU1581222A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСОЛА | 1990 |
|
RU2017724C1 |
| КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНИ ГЛАЗ, СВЯЗАННОЙ С НУКЛЕИНОВЫМИ КИСЛОТАМИ | 2012 |
|
RU2642609C2 |
| Способ получения рекомбинантной ДНК,кодирующей нуклеотидную последовательность инсулина | 1978 |
|
SU1308199A3 |
| ЭФФЕКТИВНЫЕ АНАЛОГИ КОМПСТАТИНА | 2012 |
|
RU2656102C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ФОРМА ДЛЯ ВВЕДЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА В ОБОДОЧНУЮ КИШКУ, СПОСОБ ВВЕДЕНИЯ ЛЕКАРСТВЕННОГО ПРЕПАРАТА И СПОСОБ ПОЛУЧЕНИЯ МАТРИЦ ДЛЯ ТАКОЙ ФОРМЫ | 1991 |
|
RU2113221C1 |
Изобретение касается пептидов, в частности получения пентида общей ф-лы R-A-S-CH2-CH(CH3)-C(O)-R1, где R=H, бензоил, циклопентакарбонил, трет-бутилоксикарбонил, ацетил
A-D-аланил, D-лейцил
D -и L-фенилаланил
R1-L-пролил которые проявляют противогипертоническую активность, что может быть использовано в медицине. Цель - создание новых активных и малотоксичных пептидов. Последние получают реакцией соединений ф-л II и III H-S-CH2-CH(CH3)-C(O)-R1 (II) и R-A-H (III). Новые вещества по действию сравнимы с каптоприлом, но более устойчивы к гидролизу в биологических и других средах (6 ч против 30 мин), а при использовании N-[3-(N-циклопентанкарбонил-D-аланилтио-2-D-метилпропаноил)]-L-пролила - более длительны по действию (его доза 40 мг/кг против 100 мг/кг). 4 табл.
система 1 Rf (1)1 - метанол:хлороформ, 1:1 об.ч.;
система 2 Rf (2) - бензол:вода:бутанол, 9:1:9 об. ч;
система 3 Rf (3)1 - уксусная кислота:вода:н-бутанол, 26:24:150 об.ч.;
система 4 кЈ (4) - н-бутанол:пиридин:уксусная кислотагвода, 15:10:3:
:12 об.ч.; система 5 kf (5)3 - н-бутанол:уксусная кислота :этшшцетат: вода, 1:1:1:
:1 об.ч.;
система 6 Rf (6)3 система 7 Rf (/) система 8 Rf (8)
15671254
хлороформ:метинолГуксусная кислота, 2:1:0,003 об.ч.; н-бутанол:уксусная кислота:вода, 4:1:1 об.ч.; этилацетат:уксусная кислота, 38:2 об.ч.
ЯМР-слектральные данные получены на приборе 60 МГц.
Все соединения получены по методике, аналогичной описанной для N-Ј3- (М-бензоил-L-фенилаланилтио)- тилпропаношГ -1-.-пролина.
К раствору 67Ь мг N-бензоил-L-фе- нилаланина в 5 мл ДМФА при -13 С добавляют 406 мг М,М-карбонилдиими- дазола в 5 мл ДМФА и результирующий раствор перемешивают при -10 С в течение 1 ч, затем добавляют 543 мг N-(3-меркапто-2-D-метилпропаноил)-L- пролина и 0,35 мл триэтиламина и пе- ремешивают 10 мин при той же температуре и еще 50 мин при комнатной температуре.
Растворитель отгоняют при пониженном давлении при 30 С и к остатку добавляют 20 мл воды и 20 мл бензола.
Смесь доводят до рН 2 добавлением 4н. раствора НС1 при перемешивании и охлаждении на ледяной ванне.
Органический слой дважды промыва- ют насыщенным раствором NaCl и органический растворитель удаляют отгонкой на центробежном испарителе после осушения над безводным MgS04.
Остаток очищают хроматографией в колонке на SiOj. (2 см х 45 см) и элю- ируют смесью СНС1Э и МеОН (0,5-2% МеОН в СНС13).
Пиковые фракции собирают воедино и растворитель отгоняют при понижен- ном давлении с получением /05,5 мг (60,3%) N-Јj-(Г -бензоил-Ь-фенилала- нилтио)-2-D-метилпропаноил -L-проли- на в виде масла.
Физико-химические характеристики полученных соединений.
N-LJ-(N-циклопентакарбонил-D-ала- нилтио)-2-В-метилпропаношь-L-пролин выход 49,0%; (,2; Rf (6)0,62; Rf (7)0,74.
ЯМР В (в CDC13+CD3OD): 1,20 (ЗН, м.д., СН}); 1,38 (ЗН, д., СИ,); 1,5- 2,4 (12Н, м., СНг); 2,4-3,2 (4Н, м. , СН и СН2), 3,3-3,8 (2Н, м., СН2) и 4,2-4,8 (2Н, м., СН).
(Ы-трет-бутшюксикарбонил-D- аланилтио)-2-U-метилпроианошГ -L-про- лин - выход 82,0%; Wj,- -62,5; Rf (6) ,59; Rf (7) 0,73.
0
5 0
5
0
,.
5
0
5
5
Ы-р-(М-ацетил В-лейцилтио) метилпропаноилЗ-Ь-иролин - выход 56,0%; ),5; Rf (b)0,55; Rf 7)0,59.
М-(В-аланилтио-2-D-метилиропаноил)- L-пролин - выход 92,0%; (,9; Rf (b)0, W; Rf (7)0,49;
Ы-оЈ-Ј3-(Ы- -бензоил-2-фенилаланил- тио)-2-U-метилпропаношГ - -пролин - мол.вес 468; ( 1 b,8; выход 60,3%; Rf (1)0,52; Rf (2)0,/0; Rf (3) 0,56; Rf (4) 0,61; Rf (5) 0,75; Rf (b) 0,5b; Rf (7) 0,74.
ЯМР 8 (в C1)3C13 CD3OU): 1,19 (3H, б.д., CH3); 1,5-2,4 (4H, м., CH2) ; 2,5-3,3 (5H, м., СН и C1I2); 3,3-3,8 - (2H, м., СНг); 4,0-4,4 (Hi, м., СН); 4,3-5,2 (1Н, м., CH); 7,23 (5H, c., ароматич. H); 7,2-7,9 (5H, м., арома- тич. H).
, .
N- З-(N-бензоил-U-бенилаланилтио) - 2-D-метилпропаноил -L-пролин - выход 55,6%; (cO -25,1; Rt (b)0,56; Rf (7)0,74.
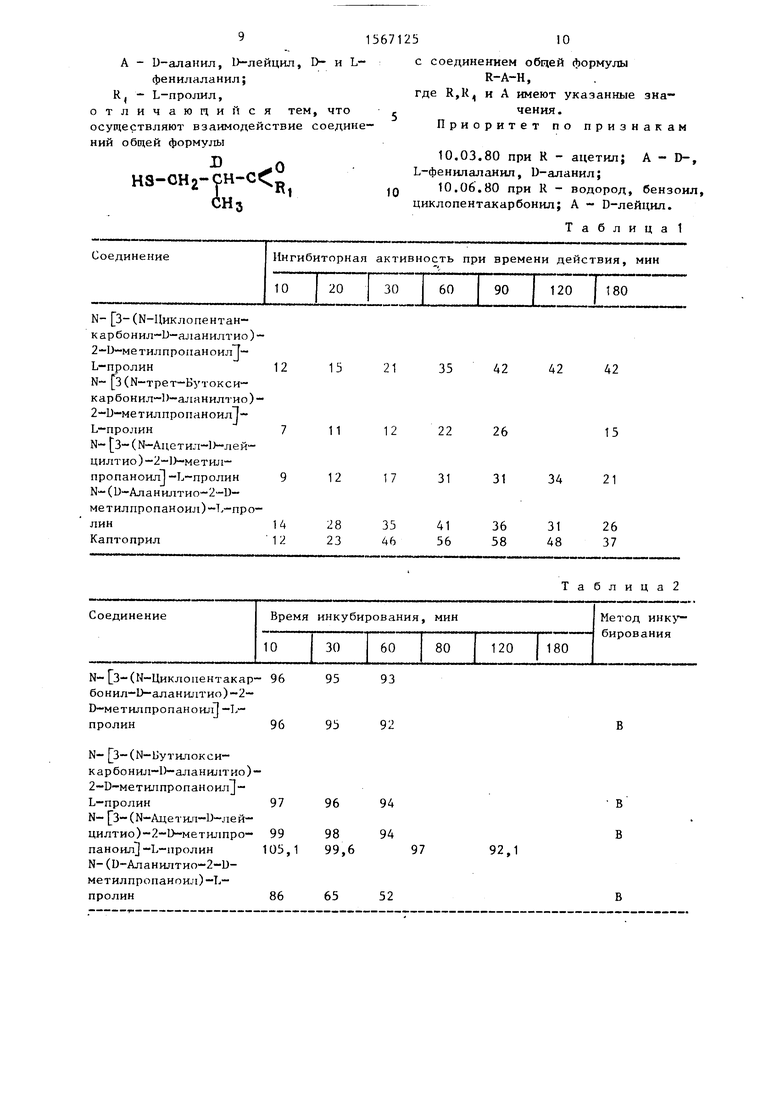
Проведены биологические испытания соединений, полученных в условиях предлагаемого способа.
Определена противогипертоническая активность предлагаемых соединений и известного - каптоприла.
Значение ,Т 50 - концентрация 50%-ной ингибиторной активности на ангиотензин преврацающий Лермент.
Система анализа: 1 мл инкубационной смеси, состоящей из 0,6 мл борат- ного буфера, 0,2 мл W,5 мМ бензоил- Гли-Гис-Лей боратного буЛера, смешивают боратный 6vAep и раствор Лермен- та, инкубируют при 37 С в течение 60 мин.
Боратный буфер: 0,1 М борная кислота, содержащая 0,8 М NaCl и 0,1 М , содеркащий 0,8 II NaCl, смешивают и рН доводят до 8,3.
АСЕ - сухой ацетоновый экстракт кроличьего легкого.
Измерение - Уо поглоцение при 228 нм после экстракции бензоил-Гли этилацетатом.
В табл. 1 приведен средний процент ингибирования вызванной гипертонии
51
в результате использования дозировок в 0,4 ммоль/кг каждого из перечисленных соединении.
Из данных табл. 1 видно, что несмотря на то, что эффективность предлагаемых соединений меняется в зависимости от времени и от конкретного применяемого лекарства, тем не менее действие этих соединений сопоставимо с действием каптоприла в выбранных дозировочных концентрациях.
При таких высоких концентрациях применение каптоприла приводит к достаточно высокому уровню возможных побочных эффектов у человека, что вынудило использовать гораздо меньшие дозировки в реальных клинических условиях лечения человека. Кроме того, по уровню длительности действия пер- вое из указанных в табл. 1 соединени превосходит каптоприл.
Предлагаемые соединения по сравнению с каптоприлом значительно боле устойчивы как в биологической, так и в других средах. Об этом свидетельствует их устойчивость к непосредственному гидролизу в сильно г«дролизую- щей среде, а также сравнительно долгое время хранения, которое они проявляют. Данные испытаний in vitro, представленные ниже, показывают присущую многим из этих соединений устойчивость в разрушающей среде плазмы и гомогенатов тканей.
A.Инкубируемая смесь: 1 мл 10%-ного гомогената печени крысы
(0,1 М буфера фосфата калия, рН 7,4) плюс 1 мл испытуемого соединения ( в том же буферном растворе фосфата). Метод испытания: из инкубируемой смеси отбирают 100 мкл, де- протонизируют добавлением 200- мкл этанола и центрифугируют. 20 мкл - центрифугата подвергают ВЭЖХ (высокоэффективной жидкостной хроматографии) .
B.Инкубируемая смесь: 1 мл 5%-но го гомогената печени крысы (0,1 М буфера фосфата калия, рН 7,4) плюс 1 мл раствора испытуемого соединения ( в том же буферном растворе фосфата). Метод испытания: из инкубируемой среды отбирают 100 мкл, после чего добавляют 6 капель I и НС1 и 5 мл этилацетата. После механического перемешивания в течение 10 мин
и 10 мин центрифугирования отобранный слой этилацетата испаряют. Оста-
56
ток растворяют в 1 мл воды и затем 0,5 мл 0,43 М фосфатного буфера (рН - 6,85) и 5 мкл БИФМ ((2- бензимидазолил)фенил малеимидл и оставляют на 40 мин при 0°С. Определения проводят при поглощении 310 нм и эмиссии 365 нм (предел измерения 0,14 г каптоприла).
С. 0,154 М фосфатный
буфер1,5 мл
2% центрифугата,
105000х0,5 мл
10 мМ испытуемого
соединения0,1 мл
0
5 Q
0 $
0
5
Всего: 2,1 мл 37°С; 0, 30, 60, 120, 180 мин 100 мкл инкубируемой смеси 200 мкл метанола
Встряхивание и центрифугирование 10 мкл центрифугата ВЭЖХ (колонка Бондаиак С18, смесь растворителей CH3CN - 0,1% тетраме- тиламмонийгидроксид, детектирование 220 нм, скорость потока 1 мл/мин).
D.Инкубируемая смесь: 1 мл плазмы крови крысы (0,1 М буфера фосфата калия, рН 7,4) плюс 1 мл раствора испытуемого соединения (400 мкг/мл
в том же буфере). Метод испытания: из реакционной смеси отбирают 100 мкл, депротонизируют добавлением 200 мкл EIOH и центрифугируют, 20 мкл центрифугата подвергают ВЭЖХ.
E.Икубируемая смесь: 1 мл 20%-но- го гомогената кишечника крысы (0,1 М буфера фосфата калия, рН 7,4) плюс
1 мл раствора испытуемого соединения (400 мкг/мл в том же буфере). Метод испытания: из реакционной смеси отбирают 100 мкл, деиротониэируют добавлением 20 мкл EIOH и центрифугируют. 20 мкл центрифугата подвергают ВЭЖХ.
F.Инкубируемая смесь: 1 мл 20%-ного гомогената печени крысы (0,1 М буфера фосфата калия, рН 7,4) плюс 1 мл раствора испытуемого соединения (400 мкг/мл в том же буфере); Метод испытания: из реакционной смеси отбирают 100 мкл, депротонизируют добавлением 200 мкл EIOH и центрифугируют. 20 мкл центрифугата подвергают ВЭЖХ. Данные приведены в табл.2%
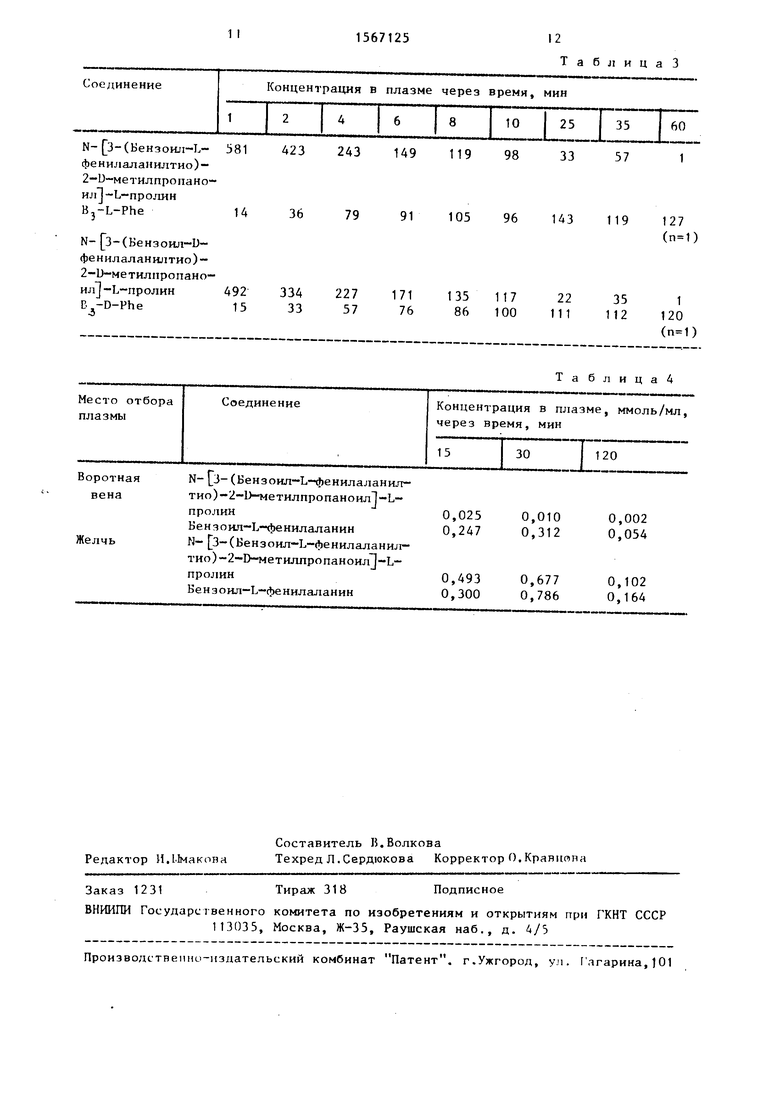
Присущая этим соединениям устойчивость подтверждена данными испытаний in vivo, показывающими, что в большинстве случаев значительный процент исходного соединения остается на многие минуты в кровеносной системе крыс после его введения. Эти данные для двух соединений приводятся ниже.
Внутривенное введение.
36 мг М- 3-(Ы-бензоил-Т,-фенилала- нилтио)- 2-1)-метилпропаноил - L- проли- на (B3-L-Phe-SO) и N- з- -бензоил- О-фенилаланилтио)-.)-метилпропано- RJij-L-пролина (B -D-Phe-SO) в 0,5 мл 0,1 М раствора NaHCOj вводят внутривенно двум крысам линии 5D. Вес каждой крысы 360 г, перед инъекцией крыс связывают на 21 ч. С помощью жидкостной хроматографии определяют концентрацию в плазме, введенного соединения и продуктов его гидролиза: бензоил-1)-фенилаланина (Вэ- D-Phe) и бензоил-1-фенилаланина (Bj-L-Phe). Данные приведены в табл. 3.
Пероральное введение. Связанным на ночь крысам весом 350 г вводят перорально 100 мг на 1 кг массы тела N- ГЗ-(бензоил-L-фенилаланилтио)-2-D- метилпропаноил -Ь-пролина. Испытуемый раствор содержит 20 мг N-Ј3-(бензоил- L-фенилаланилтио)-2-D-метилпро- паноил -Ь-пролина в 1%-ном водном растворе гумиарабика. Концентрацию N- 3(бензоил-1-фенилаланилтио)-2-D- метилпропаноил -L-пролина и продукта его гидролиза (бензоил-L-фенилалани- на) определяют в плазме крови, отбираемой из воротной вены, и в потоке желчи с помощью жидкостной хроматографии. Данные приведены в табл.4. Известно, что каптоп рил напротив быстро разрушается с образованием дисульфида и других продуктов метаболизма, каждый из которых неактивен в качестве ингибитора.
Через 60 мин инкубирования с плазмой крови крыс, человека, обезьян и собак 558-каптоприл, оставшийся без изменения, составляет соответственно О, 7, 10 и 25%, а основным продуктом разложения является дисульфид.
Продукт центрифугирования при 9000xQ гомогената печени, почек и кишечника крыс представляет собой чере 120 мин продукт почти полного превращения каптоприла в его дисульфид и другие соединения.
По меньшей мере одно из предлагаемых соединений, а именно (П-цик- лопентилкарбонил-)-аланилтио) метилпропаноил -L-пролин (cpc-D-ala- -SO), показал неуменычаюпуюся эффективность в течение от 90 до 180 мин
0
5
0
5
0
5
0
после перорального введения. Данные, подтверждающие это, приведены в табл.1. Показано, что каптолрил достигает пиковой эффективности при 90 мин, после чего следует падение эффективности. По истечении 180 мин N- 3-(Н-циклопентилкарбонил-Г)-аланил- тио)-2-1 метилпропаноил -L-пролин остается более эффективным ингибитором без всяких признаков того, что его действие ослабевает.
Сравнительные испытания показали, что действие на нормального человека 40 мг (0,096 ммоль) Н- 3-(М-циклогек- силлкарбонил-D-аланилтио)-2-D-метил- пропаношГ - L-пролина более длительно по сравнению со 100 мг (0,46 ммоль) каптоприла. Эти результаты дают подтверждение тому, что сложные тиоэфир- ные производные каптоирила с ацил- аминокислотами обладают улучшенным действием по сравнению с самим капто- прилом.
Предлагаемые соединения обладают большой химической устойчивостью. Кап1 оприл имеет необычайно короткий срок хранения, период полураспада его в водных растворах составляет примерно 30 мин. Сухой порошок разлагается более чем на 50% при выдерживании 3 недели при 85°С.
Нредлагемые соединения, не имея свободной меркаптогруппы, сами по себе устойчивы. Их растворы могут храниться в течение длительного периода времени, так раствор (М-бензоил- и-фенилаланилтио)-2-0-метилпропано- ,-пролина в смеси метанола с водой (1:1) в концентрации 1 мг/мл не проявляет никаких признаков разложения спустя 6 ч хранения. Кроме того, эти соединения не требуют подмешивания восстановителя к их порошкообразным формам с целью избежать их ди- меризации, характерной для капто- прила.
Формула изобретения
Способ получения пептидов обцей формулы
55
Я-А-8- СН2-СН-С ° СН5
где R - водород, бензоил, циклопента- карбонил, трет-бутилокси- карбонил, ацетил;
А - U-аланил, 1 -лейцил, D- и Lфенилаланил;R( - L-пролил,
отличающийся тем, что осуществляют взаимодействие соединений общей формулы
нз-он2- н-с
СН3
ч
N- 3-(Ы-Циклопентанк арбонил-и-аланилтио)2- D-метилпропаноил Т-
L-пролин12152135424242
N- 3(N-трет-Бутокси-
карбонил-1 -аланилтио) -
2-Ь-метилпропаноид |L-пролин71112222615
N- 3-(М-Адетил-1 -лей
цилтио)- 2- -метил-
пропаноил -L-пролин9121731313421
М-(и-Аланилтио-2-1 метилпропаноил)- Ь-проЫ- з-(Н-Циклопентакар- 96
бонил-О-аланилтио)-2-
D-метилпропаношт -L-
пролин96
N- p- (N- 1 утилокси-
карбонил-I)-ал анилтио) -
2-В-метилпропаноил -
L-пролин97
(N-Ацетил-U-леи-
цилтио)-2-D-метилпро- 99
паноилЗ-L-пролин105
N-(U-Аланилтио-2-Dметилпропаноил)-L-
пролин86
с соединением общей формулы
R-A-H,
где R,K,, и А имеют указанные зна- .чения.
Приоритет по признакам
JQ10.06.80 при R - водород, бензоил
циклопентакарбонил; А - D-лейцил.
Таблица 1
В В
92,1
ТаблицаЗ
| Шредер Э | |||
| , Любке К | |||
| Пептиды | |||
| - М.: Мир, 1.967, с | |||
| Способ получения бензидиновых оснований | 1921 |
|
SU116A1 |
