Изобретение относится к способу получения новых имидазолпроизводных или их фармацевтически приемлемых солей, которые могут найти применение для лечения и предупреждения воспалений и отеков.
Цель изобретения - синтез новых соединений в ряду имидазолов, которые обладают активностью,не характерной для данного ряда соединений.
Пример 1. а. 5 г гидроксил- амин-0-сульфокислоты растворяют в
30 мл воды и нейтрализуют 3,7 г бикарбоната натрия с охлаждением льдом. Затем этот раствор по каплям добавляют к раствору 3,63 г 2-метил- имидазола в 15 мл воды. Полученную перемешивают около 20 ч при комнатной температуре и подкисляют 13 мл 2н. соляной кислоты до тех пор, пока раствор не достигнет значения рН 1. Затем добавляют две порции активного угля (кончиком шпателя), перемешивают в течение 15 мин,
О
ел VI о ел VI
со
фильтруют и к полученному прозрачному фильтрату добавляют Ь мл бензаль- дегида и 10 мл диэтилового эфира. Затем перемешивают в течение 6ч,охлаждают в течение 15 мин на ледяной бане и фильтруют выпавшее твердое тело. Фильтрат подвергают дальнейшей обработке, как описано ниже.
После переосаждения твердого тела из смеси метанола и диэтилового эфира получают 1,3-бис(бензилиденамино)- -2-метилимидаэолийхлорид ст. пл. 244-249°С.
Полученный фильтрат три раза эк- страгируют встряхиванием с помощью диэтилового эфира (по 15 мл). Водную фазу с охлаждением нейтрализуют (рН 7) 9 мл 4 н. раствора едкого натра. Затем отфильтровывают полученный осадок, переосаждагат его из смеси метанола и дк тилового эфира, в результате чего получают 1-бензилиден- анино 2-метилимидазол ст.пл. 122- 124ЙС.
б.3,24 г 1,3-бис(бензилиденами но)-2-метилимидазолий-хлорида растворяют в 50 мл метанола. Полученный раствор охлаждают до внутренней температуры 0°С, после чего с перемеши- ваннем добавляют раствор 2,5 г цианида калия в 8 мл воды. При этом температура не должна значительно повышаться. Через 15 мин упаривают метанол
при минимальной температуре в вакууме. Полученный остаток разбавляют 30 мл воды, после чего проводят экстракцию хлороформом. Высушенный хлороформный экстракт выпаривают, в результате чего получают 1-бензилиденамино- -2-метилимидазол с т. пл. 12ЬвС.
в.7,4 г 1-бензилиденамино-2-ме- тилимндазола взвешивают в 50 мл воды. Взвесь подкисляют (до значения рН 1-2 25 мл 2 н. соляной кислоты, после че- го ее подвергают перегонке с водяным паром до прекращения перегонки бен- зальдегида. Оставшийся прозрачный раствор выпаривают досуха, а маслянистый остаток перекристаллизовывают при О С с помощью диэтилового(.
эфира. Полученные кристаллы переосаждают из смеси этанола и днэтипового эфира, i результате чего получают 1-амино-2-метилимидазол-гидрохлорид с т. пл. 139-К1 С.
г.5,35 г 1-амино-2-метилимидазол- -гидрохлорида растворяют в 300 мл
этанола, затем добавляют 9,3 г
д
$
5
0
5
4-окси-3.5-ди-трет-бутилбензальде- гида и перемешивают 1 ч при комнатной температуре. Массу затем выпаривают в вакууме. К остатку добавляют 150 мл ледяной воды и 200 мл метиле нхлорида, перемешивают и нейтрализуют (рН 7) насыщенным раствором бикарбоната натрия. Затем отделяется метиленхлоридная фаза, а водная фаза экстрагируется 100 мл метиленхлорида. Собранные метнленхлоридные фазы высушивают и выпаривают. Остаток размешивается в диэтиловом эфире, фильтруется и фильтрат высушивается в вакууме при 80вС. В результате получают 1- -(4-окси-3,5-ди-трет-бутилбензилиден- амино)-2-метилимидазол ст.пл. 206- 207ЛС.
Пример 2. 3,13 г 1-(4-окси- -3,5-ди-трет-бутилбензилиденамино)-2метилимидазола растворяют в 100 мл метанола, после чего добавляют 0,5 г палладиевого катализатора (5% на угле) и 10 мл 1 н. соляной кислоты и гидрируют под нормальном давлением и при комнатной температуре. После поглощения 240 мл водорода отфильтровывают катализатор и выпаривают фильтрат. Маслянистый остаток растворяют в 50 мл воды и добавляют насыщенный раствор бикарбоната натрия до достижения нейтральной реакции (рН 7). Выпавший остаток отфильтровывают и добавляют в 100 мл метиленхлорида. Метиленхлоридный раствор промывают 50 мл воды, высушивают и выпаривают. Остаток размешивают в диэтиловом эфире, отфильтровывают и получают неочищенный 1-(4-окси-3,5-ди-тре т-бутилбе нз ил- амино)-2-метилимидазол с т.пл. 181- . Перекристаллизацией из ацето- нитрила получают чистый продукт с т.пл. 182-183°С.
Пример 3. а. 22,6 г гидроксиламин-0-сульфокислоты растворяют в 60 мл ледяной воды и нейтрализуют (до рН 6) приблизительно 18 г бикарбоната натрия. Затем с перемешиванием в течение 45 мин по каплям добав4ляют раствор 27,2 г имидазола в 60 мл
воды. Затем полученную смесь перемешивают в течение около 20 ч при ком,натной температуре и подкисляют 2 н. соляной кислотой ( ft-160 мл) с охлаждением льдом до рН 1-2. Потом перемешивают темный раствор в течение 15 мин с активным углем, фильтруют
и к полученному прозрачному фильтрату добавляют 28 мл бензальдегида и 30 мл диэтилового эфира. Затем перемешивают массу в течение 18 ч, охлаждают, образовавшуюся взвесь в течение 15 мин и фильтруют выпавший 1,3-бис(бензилиденамино)имидаз олий- хлорид.
Фильтрат три раза экстрагируют встряхиванием с помощью диэтилового эфира (по АО мл). Потом нейтрализуют (до рН 7) водную кислую фазу, охлаждая ее льдом, 40 мл 4 н. раствора едкого натра. Выпавший осадок один раз экстрагируют встряхиванием 100мл хлоросрорма и еще два раза хлороформом (по 20 мл). Собранные хлороформные фазы высушивают сульфатом натрия и выпаривают. Полученный остаток с нагреванием растворяют в этаноле, после чего добавляют такое количество горячей воды, при котором не наблюдается помутнения. Затем быстро фильтруют и охлаждают. После охлаждения льдом в течение 1 ч отфильтровывают выпавший 1-бензилиденаминоимида- эол.
б.Аналогично описанному в примере .16 методу из 1, 3-бис-(бензилиденамино) имидаз олийхлорида получают 1-бензилиденаминоимидазол- с т. пл. 115вС.
в.8,68 г 1-бензилиденаминоимида- зола взвешивают в 50 мл воды. Полученную взвесь подкисляют (рН 1-2) 28 мл
2 н. раствора соляной кислоты и подвергают ее перегонке с водяным паром о прекращения перегонки бензальдегида. Остающийся прозрачный раствор в вакууме при 50-bOeC выпаривают досуха. Остаток от выпаривания выкристаллизовывают при в диэтиловом эфире. Переосаждением кристаллизата з смеси этанола и диэтилового эфира олучают 1-аминоимидазол-гидрохлорид с т. пл. .
г.0,48 г 1-аминоимидазол-гидро- хлорида взвешивают в 30 мл этанола, после чего добавляют 0,93 г 4-окси- -3,5-ди-трет-бутил-бе нз альде гида. Взвесь перемешивают 3 ч при комнатной
температуре. При этом образуется прозрачный раствор, который выпари- вают в вакууме. Остаток растворяют в 15 мл воды, нейтрализуют (рН 7) добавкой ледяного насыщенного раствора бикарбоната натрия и три раза быстро встряхивают метиленхлоридом
(по 30 мл). Собранные органические (разы высушивают сульфатом натрия, фильтруют и выпаривают в вакууме. Сублимацией остатка ( Торр) получают 1-(4-окси-3,5-ди-трет-бу тилбензилиденамино)имидазол с т. пл. 171-174 С.
Пример 4. 0,9 г 1-(4-окси-3,5-ди-трет-бутилбензилиденамино)
имидазола взвешивают в 10 мл ледяной . уксусной кислоты. В взвесь добавляют 1,Ы.г цианоборгидрида натрия, в результате чего образуется раствор,
5 который перемешивают в течение ночи, после чего его выпаривают в вакууме. К остатку добавляют 10 мл воды, нейтрализуют (рН 7) добавлением ледяного насыщенного раствора бикарбоната нат0 рия и встряхивают три раза метилен- хлоридом (по 30 мл). Собранные органические фазы высушивают сульфатом натрия, фильтруют и выпаривают в вакууме. Остаток выкристаллизовывают
5 в диэтиловом эфире. Продукт отфильтровывают и переосаждают из смеси этанола и воды, в результате получают 1-(4-окси-3,5-ди-трет-бутилбенз- иламино)имидазол с т. пл. 171-174 С.
0 Пример 5. 28,7 г 1-(4-окси- -3,5-ди-трет-бутилбензилиденамино) имидазола растворяют в 50 мл метанола, после чего добавляют 96 мл 1 н. соляной кислоты и 3 г Pd/C и гидри5 руют под нормальным давлением и при комнатной температуре. После поглощения 2,2 л водорода отфильтровывают катализатор и выпаривают полученный фильтрат. К остатку добавля0 ют 800 мл метиленхлорида и 300 мл воды со встряхиванием нейтрализуют (до рН 7) насыщенным раствором бикарбоната натрия, отделяют метилен- хлоридную фазу и два раза экстраги5 руют водную фазу метиленхлоридом
(по 100 мл). Затем собирают метилен- хлоридные фазы, высушивают и выпари- - вают их, после чего полученный остаток перекристаллиэовывают из смеси
0 этанола и воды (3:1). В результате получают 1-(4-окси-3,5-ди-трет-бу- тилбензиламино)-имидазол с. т. пл. 178-180сС.
Пример 6. а. Раствор 120 мл
е сероуглерода и 60,6 г дициклогексил- карбодиимида в 300 мл ТГФ охлаждают до с помощью смеси льда и соли. Затем с перемешиванием по каплям до- бавляют 39,9 г аминоацетальдегид
диэтилацеталя с такой скоростью, чтобы температур4 реакционной массы не поднималась выше (примерно 1ч). Потом смесь перемешиванием медленно нагревают до комнатной температуры (примерно 2 ч) и дают ей стоять в течение ночи. Образовавшийся в результате осадок (дицнклогексилтиомочеви- ну) отфильтровывают и интенсивно промывают n-гексаном. Фильтрат концентрируют в вакууме, а выпавшую ди- циклогексилмочевину снова отфильтровывают и интенсивно промывают п-гек- саном. Эту процедуру повторяют до тех пор, пока при концентрировании не прекратится выпадение дициклогек- силтиомочевины. Наконец из фильтрата полностью удаляют растворитель. Остаток перегоняют при 11 Торр (температура бани 130-134 0), в результате чего получают 2,2-диэтоксиэтилизотио- цианат в виде бесцветного масла с т. пл. 100йС/11 Торр.
б.2,5 г гидразингидрата (98- 100%-ного) растворяют в 10 мл 96%-но- го этанола. В этот раствор с перемешиванием по каплям добавляют 8,75 г 2,2-диэтоксиэтилизотиоцианата с такой скоростью, чтобы температура раствора не превышала . Когда добавление закончено, охлаждают содержимое колбы, которое при этом полностью затвердевает. Этанол упаривают в вакууме при температуре бани 30 40е С. Оставшийся в виде бесцветного кристаллического остатка 4-(2,2 -диэток,си- этил)тиосемикарбазид (т.пл. 92-97 С) без очистки пригоден для дальнейшей переработки. Проба, перекристалли- зоианная из воды, имеет т.пл. 95-97 С
в.К 2,07 г 4-(2г,2 -диэтоксиэтил) тиосемикарбазпда добавляют 10 мл 2 н. серной кислоты и смесь нагревают в течение 15 мин при температуре дефлегмации на бане с кипящей водой. Затем в желтоватый раствор при добавляют 1,06 г бензальдегида и все хорошо размешивают. После охлаждения до комнатной температуры отфильтро- ( вывают образовавшийся желтый осадок, который промывают водой и высушивают. В целях очистки сырой продукт взвешивают в горячей воде ( 15 мл) и в горячем состоянии к взвеси добавляют столько этанола, сколько нужно для получения полностью растворенного вещества. Этот раствор отфильтровывают в горячем состоянии, после чего его
6570Ь78
охлаждают. При этом выпадает 1-бен- зилиденамино-2-меркаптоимидазол в виде белых игол. Пробу этого продукта
0
5
0
5
Q
высушивают в течение 12 ч при 40 С иод давлением 10 Торр и она обнаруживает т. пл. 158-161°С.
г.10,16 г 1-бензилиденамино-2- -меркаптоимидазола взвешивают в 30мл абсолютного этанола, после чего к полученной взвеси добавляют раствор 1,15 г натрия в 70 мл абсолютного этанола. Затем добавляют 7,1 г метил- йодида и перемешивают реакционную массу в течение 3 ч при комнатной температуре. Потом образовавшийся желтый раствор концентрируют до четверти объема и доливают 100 мл воды. Полученный кристаллический сырой продукт отфильтровывают, три раза промывают водой и перекристаллизовывают из смеси метанола и воды. В результате получают 1-бензилиденамино-2- -меркаптоимидазол с т.пл. 95-97 С.
д.2,17 г1-бензилиденамино-2-меркаптоимидазола взвешивают в 50 мл воды и после добавления 6 мл 2 н. соляной кислоты полученную смесь подвергают перегонке с водяным паром. По окончании отщепления бензальдегида образовавшийся раствор концентрируют до объема в несколько миллилитров, после чего до достижения щелочной реакции добавляют 5 н. раствор едкого натра и пять раз экстрагируют метиленхлоридом (по 15 мл). Затем собранные метиленхлоридные фазы высушивают сульфатом натрия, фильтруют и выпаривают. Затем перекристаллизовывают кристаллический остаток из смеси n-гексана и этилацетата. В результате получают 1-амино-2-метил- меркаптоимидазол в виде бесцветных кристаллов с т. пл. 71-73е С (сублимация начинается с температуры ) .
е. 0,65 г 1-амино-2-метилмеркап- тоимидазола растворяют в 50 мл метанола, после чего добавляют 3 мл 2 н. соляной кислоты. По добавлении 1,05 г 4-окси-3,5-ди-трет-бутилбен- зальдегида перемешивают реакционную массу в течение ночи при комнатной температуре. Затем выпаривают растворитель в вакууме, взвешивают остаток в 30 мл воды и добавляют насыщенный раствор карбоната натрия до достижения щелочной реакции. Затем три раза экстрагируют метиленхлоридом
5
0
0
(по 20 мл), высушивают собранные органические фазы сульфатом натрия, фильтруют и выпаривают. Полученный кристаллический остаток перекристал- лизовыв ают из смеси воды и метанола. В результате получают 1-(4-окси-З,5- -ди-трет-бутилбензилиденамино)-2- -метилмеркаптоимидазол с т. пл. 181- .
Пример 7. 1,04 г 1-(4-ок- си-3,5-ди-трет-бутилбензилиденаминоО -2-метилмеркаптоимидазола растворяют в 20 мл ледяной уксусной кислоты. В раствор в течение 30 мин порциями добавляют 0,43 г 90%-ного цианобор- гидрида натрия. Реакционную массу перемешивают в течение ночи при комнатной температуре. Потом упаривают растворитель в вакууме, остаток раст воряют в 20 мл воды. Затем добавляют насыщенный раствор карбоната натрия до достижения щелочной реакции и экстрагируют три раза метиленхлоридом (по 15 мл). Собранные органические фазы высушивают сульфатом натрия, фильтруют и выпаривают. Остаток пе- рекристаллизовывают из смеси метанола и воды, в результате чего получаю 1-(4-окси-З,5-ди-трет-бутил-бензил- амино)-2-метилмеркаптоимидазол с т. пл. 110-113 С.
Пример 8. Вариант а. 200мг 1-(4-окси-З,5-ди-трет-бутилбензил- амино)-2 -метилмеркаптоимидазола вместе с 0,41 г хлорида никеля (II). 6 г растворяют в 8 мл ме танола. Полученный раствор охлаждают на ледяной бане и порциями в течение 1 ч. добавляют 0,7 г боргидрида натрия. Когда добавление закончено удаляют ледяную баню и перемешивают черную реакционную массу еще 4 ч при комнатной температуре. Для дальнейшей обработки массы ее снова охлаждают и добавляют 2 н. соляной кислоты до кислой реакции реакционной массы. Потом перемешивают еще несколько минут и добавляют концентрированный аммиак до достижения щелочной реакции Массу затем фильтруют через слой из силикагеля и промывают лепешку 5 раз метиленхлоридом. Затем отделяют органическую фазу фильтрата, промывают два раза водой, высушивают сульфатом натрия и выпаривают. Кристаллический остаток переосаждают смесью метанола и воды. Получают 1-(4-окси-3,5-ди- -трет-бутилбензиламино)имидазол в
0
5
Q
виде бесцветных кристаллов ст. пл. 171-174°С.-/
Вариант б. 600 мг 1-(4-окси-3,5- -ди-трет-бутилбензиламино)-2-метил- меркаптоимндазола, растворенного в 20 мл этанола, вместе с намоченным этанопом никелем Ранея W2 в количестве, превышающем количество имида- зола по весу в 5 раз, с перемешиванием в течение двух часов нагревают до 60-70СС. Затем отфильтровывают никель и промывают два раза этанолом (по 10 мл). Собранные спиртовые фильтраты выпаривают. В качестве остатка получают 1-(4-окси-3,5-ди- -трет-бутилбензиламино)-имидазол в виде бесцветных кристаллов с т. пл. 171-174°С.
Пример 9. 300 мг 1-(4-ок- си-3,5-ди-трет-бутилбензиламино)ими- дазола вместе с 10 каплями ледяной уксусной кислоты в 25 мл бензола на водяной бане нагревают до температуры 5л/60-70°С. После добавления по каплям раствора 250 мг дицианодихлорбензохи- нона в 10 мл бензола полученную реакционную массу нагревают еще 15 мин. Затем ее охлаждают на ледяной бане 0 и фильтруют выпавшее твердое вещество. Затем встряхивают фильтрат с 10%-ным раствором карбоната натрия. После отделения органической фазы водную фазу один раз экстрагируют бензолом. Собранные органические фазы высушивают безводным сульфатом магния, фильтруют и выпаривают под пониженным давлением. Затем перекрис- таллизовывают кристаллический остаток из смеси этилацетата и п-гекса- на, в результате чего получают 1-(4- -окси-3,5-ди-трет-бутилбензилиденами- но)имидазол с т. пл. 173.
Пример 10. а. 25 г 2-пропил- имидазола взвешивают в 70 мл воды. Затем в взвесь в течение 20 мин с перемешиванием добавляют приготовленный при раствор 25 г гид- роксиламино-0-сульфокислоты и 18,5 г бикарбоната натрия в 150 мл воды. После 20-часового перемешивания при комнатной температуре подкисляют 65 мл 2 н. соляной кислоты, добавляют раствор 15,5 г бензальдегида в г 50 мл диэтилового эфира и перемешивают 6 ч при комнатной температуре. Образовавшиеся кристаллы отфильтро- . вывают. Фильтрат перерабатывают, как описано ниже.
0
5
0
Кристаллы растворяют в метилен- хлориде. Раствор высушивают сульфатом натрия, фильтруют и концентрируют. Остаток выкристаллизовывают добавлением диэтилового эфира. После перекристаллизации из этанола получа- ют 1,3-бис(бензилиденамино)-2-пропил- имидазолий-бензальдоксим-0 -судьфонат с т. пл. 143-145СС.
Указанный фильтрат промывают диэти- ловым эфиром. Затем добавляют 60 мл 3 н. раствора едкого натра и экстрагируют метиленхлоридом. После высушивания экстракта сульфатом натрия, фильтрации и упаривания метиленхлорида получают красноватое масло, из которого в результате хроматографии на силнка- геле (крупность зерна 0,063-0,2 мм) с помощью смеси метиленхлорида и метанола (99:1%) получают 1-бензилиденами- но-2-пропилимидазол с т. пл. 61-62 С.
б.Взвесь 14 г 1-бензилиденамино- -2-пропилимидазола в 113 мл воды и 79 мл 2 н. соляной кислоты подвергают перегонке с водяным паром до тех пор, пока не прекратится перегонка бензальдегида. Оставшийся слегка путный раствор охлаждают и выпаривают в вакууме. К маслянистому остатку добавляют метанол и бензол, затем выпаривают полученную массу, после чего добавляют 30 мл этанола. В полу- ченный раствор добавляют диэтиловый эфир до тех пор, пока раствор не станет мутнеть. После охлаждения раст вора отфильтровывают выпавшие кристал лы, которые промывают диэтиловым эфиром и n-гексаном и высушивают в вакууме при 60°. Получают 1-амино- -2-пропшшмидазол-гидрохлорид с
т. пл. 108-109°С.
в.6,47 г 1-амино-2-пропилимида- зол-гидрохлорида растворяют в 300 мл этанола, после чего добавляют 3,5 г 4-окси-3,5-ди-трет-бутилбензальдегида, перемешивают в течение 3 ч при комнатной температуре и затем выпаривают полученный раствор. К остатку добавляют 150 мл воды и 150 мл метиленхлорида, после чего с перемешиванием добавляют насыщенный раствор бикарбоната натрия до достижения нейтральной реакции (рН 7). Метиленхлоридную фазу отделяют, а водную фазу экстрагируют один раз
50 мл метиленхлорида, после чего объединяют метиленхлоридные растворы и высушивают и выпаривают их. Оста0
5
0
5
30
55
40
45
50
5
ток с перемешиванием смешивают с диэтиловым эфиром, после чего отфильтровывают твердое тело и высушивают его при 60СС в вакууме. В результате по:гучают 1-(4-окси-3,5-ди трет- -бутил-бензилиденамино)-2-пропил- имидазол с т. пл. 160-161°С.
Пример 11. 9,9 г 1-(4-ок- си-3,5-ди-трет-бутилбензилиденами- но)-2-пропилимидазола гидрируют в 200 мл метанола и 29 мл 1 н. соляной кислоты в присутствии 1 г паллади- евого катализатора (5% на угле) под нормальным давлением при комнатной температуре. Когда поглощено 700 мл водорода,отфильтровывают от катализатора и выпаривают фильтрат. Остаток растворяют в 100 мл воды, после чего добавляют насыщенный раствор бикарбоната натрии до достижения нейтральной реакции (рН 7). Затем отфильтровывают выпавшие кристаллы и растворяют их в 300 мл метиленхлорида. Метиленхлоридный раствор промывают один раз водой, высушивают и выпаривают. Остаток растворяют в 50 мл горячего изопропилового эфира. Выпавшие во время охлаждения кристаллы отфильтровывают, промывают изо- пропиловым эфиром и n-гексаном и высушивают при 60 С в вакууме. В результате получают 1-(4-окси-3,5-ди- трет-бутилбензиламино)-2-пропил-ими- дазол с т. пл. 122-123dC.
Производные имидазола общей формулы (1) и их фармацевтически приемлемые соли с кислотами обладают ценны- йи фармакодинамическими свойствами.
В описанном опыте на животных испытывали противовоспалительные свойства представительных соединений формулы (1).
Крысам-самцам (120-140 г) впрыскивали в корень хвоста 0,1 мл 0,5%-ной (вес/объем) взвеси умерщвленной нагреванием и высушенной микробактерии Mycobacterium butyricum в тяжелом минеральном масле, содержащей 0,2% дигитонина. Животных держат отдельно, кормят и поят водой. Вызванному таким образом артриту дают развиваться в течение 21 сут, после чего определяют вес животных, с одной стороны, и объем обеих задних лап (погружением лап в ртутный плетисмограф до уровня лодыжки латеральной), с другой. Животных затем объединяют в группы по 6 особей, задние лапы которых имеют примерно одинаковый средний объем и в течение 7 суток им вводят испытуемое вещество интубацией. В конце лечебного цикла сно- ва определяют вес тела и объем задних лап и подсчитывают их изменение во время лечения. После этого умерщвляют животных, отбирают пробы из плазмы и по методу автора Exner (Amer. J. Cl in Path) определяют количество фибриногена, осадив пробы сульфатом аммония.
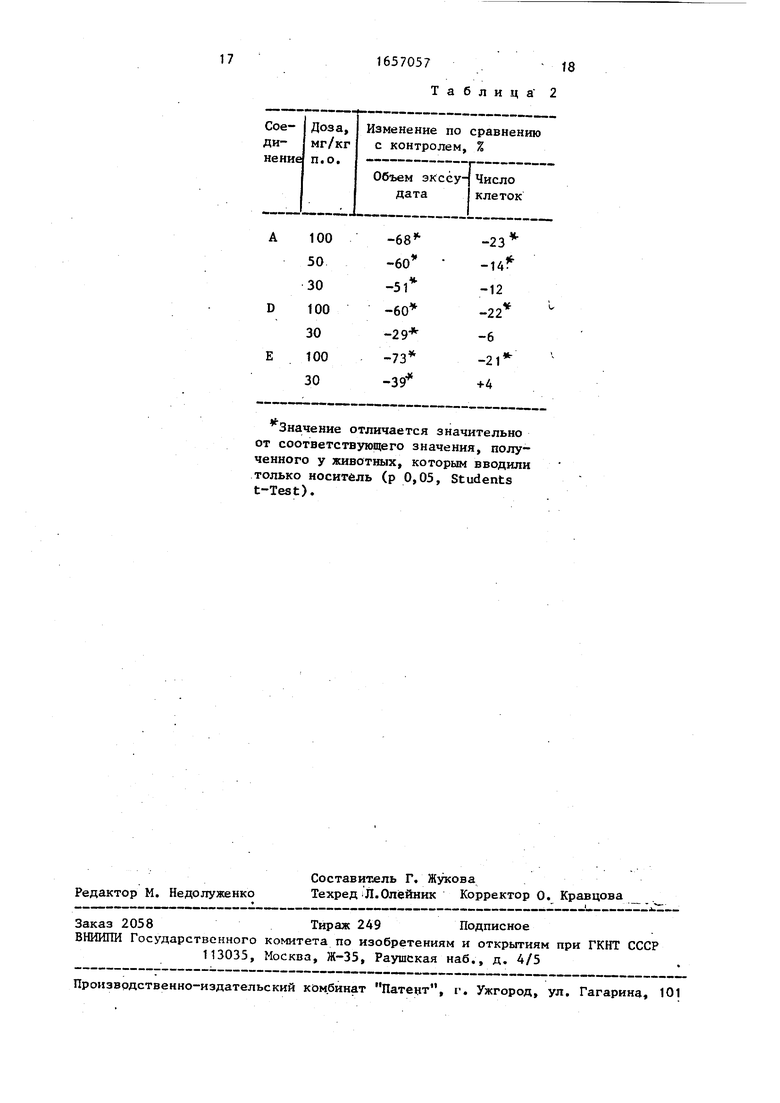
В нижеследующей табл. 1 приведены результаты, полученные в описанно опыте с тремя представленными соединениями указанной формулы vl).
Ниже приведены данные об острой токсичности приведенных трех соединений (ДЦSO ПРИ разовом оральном вве- дении у мышей). Соединение ABC
ЛДда, мг/кг, (перорально)
1000 1000
- Кроме того, в опыте на животных испытывали сдерживающее образование отеков действие указанного соединения А и соединений В-1-(4-окси-3,5- -ди-трет-бутилбензиламино)-2-метил- имидазола и Е-1-(4-окси-3,5-ди-трет- -бутилбензиламино)-2-пропилимидазола которые формула (I) тоже охватывает. У крыс-самцов (230-250 г), которые получают корм и воду, сколько им угодно, впрыскиванием 0,2 мл 1%-ного раствора каррагена (Carrageen) в стерильном беспироге ином растворе поваренной соли в правую плевральную полость вызывают плеврит. За 1 ч до впрыскивания каррагена и через 5 ч после этого животным интубацией вводят испытуемые вещества, взвшенные в водном носителе (содержащем 0,5% карбоксиметилцеллюлозы, 0,9% поваренной соли, 0,37% Tween 80 и 0,86% бензилового спирта), и носитель соответственно. Через 24 ч после впрыскивания каррагена умерщвляют животных, обезглавливая их, выпуска
ют всю кровь из туши животных и открывают плевральную полость, разделяя ребра с обеих сторон грудины. Пипеткой удаляют экссудат из плевральной полости и определяют его объем. Плевральную полость затем промывают один раз раствором поваренной соли, содержащим.фосфат в качестве буфера и фетальную бычью сыворотку (1:1) и
- Q
5
0
5
0 Q
объединяют промывной раствор с экссудатом. Применяя счетное устройство Coulter Counter, которое отрегулировано так, чтобы эритроциты не считались, подсчитывается общее количество клеток в плевральной полости. Мазки клеток на стеклянных пластинках можно получить непосредственно из экссудата. Их фиксируют метанолом и окрашивают для проведения дифференциального подсчета полимор- фоядерных лейкоцитов (ПМЛ) и макрофагов: подсчитывают всего 200 ПМЛ и макрофагов. Результат можно выражать в процентах для каждого типа клеток, имеющегося в плевральном экссудате.
Результаты описанного опыта приведены в табл. 2
Соединения формулы (I) и их фармацевтически приемлемые соли с кислотами находят применение как лечебные средства, например, в виде фармацевтических препаратов. Фармацевтические препараты можно вводить орально, например, в виде таблеток, в том числе и с лаковым покрытием, драже, капсул из мягкой или твердой желатины, растворов, эмульсий или взвесей. Одна-.- ко введение этих препаратов может быть осуществлено и ректально, например, в виде суппозиториев или парентерально в виде растворов для .впрыскивания.
Формула изобретения
Способ получения имилазолпроиз- водных общей формулы (I)
нэс /RI
J-N-CH
N-yN
«5 R4 R3
где К. и K. каждый низший алкил;
водород и Нц - водород или R-J и R ц. вместе означают дополнительную связь между углеродом и азотом; RC- - водород, низший алкил
или низший алкилтио
или их фармацевтически приемлемых солей с кислотами, отличающий с я тем, что соединение общей формулы (II)
г
R5 NH
где Кд, Rs имеют указанные значения,
выделяют или восстанавливают до получения соединений общей формулы (U






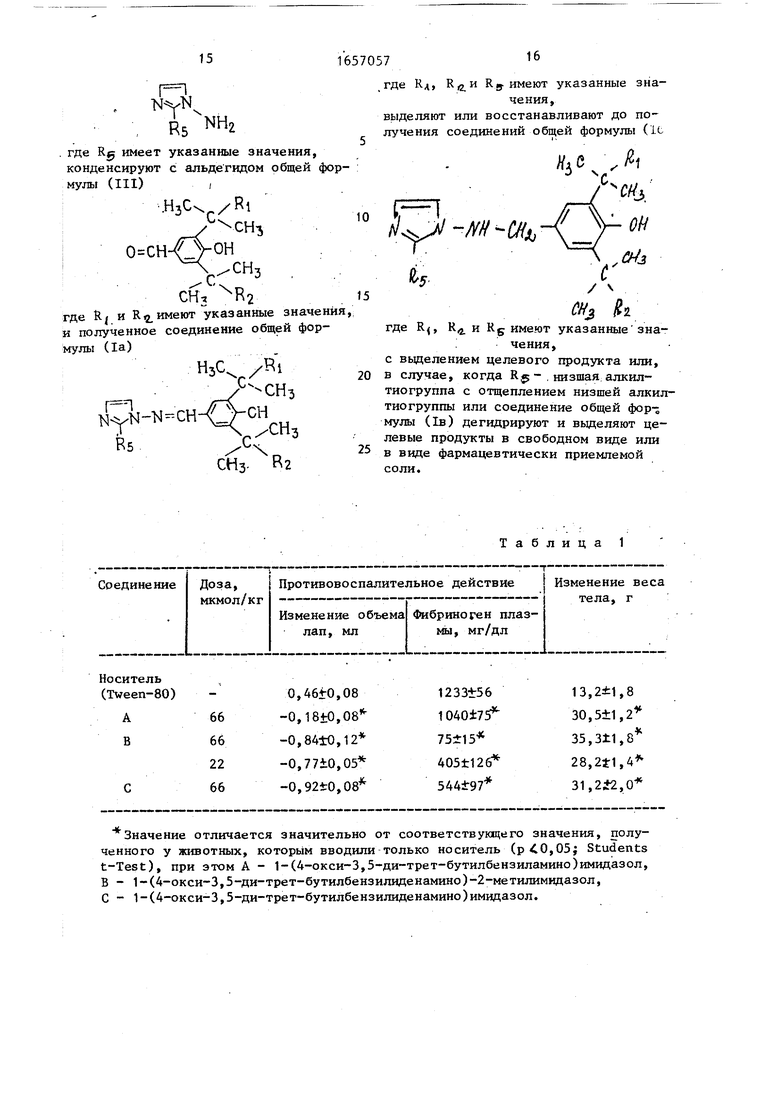
Изобретение относится к гетероциклическим соединениям, в частности к получению имидазолпроизводных ф-лы Н3С NYN-N-CH-C Rs «ч Кэ v -см3 он -СН-, Н3с ч. где R и RC каждый - низший ал кил, R-y-H и или RJ + 4 дополнительная связь между С и N, Rg-- H, низшие : алкил или алкилтио, или их фармацевтически приемлемых солей с кислотами, которые могут найти применение для лечения и предупреждения воспалении и отеков. Цель - разработка способа получения соединений, обладающих активностью, не характерной для данного ряда соединений. Получение ведут конденсацией соединения ф-лы (NH(2), где Rs - указано выше, с соответствующим альдегидом. Полученное соединение выделяют или восстанавливают. В случае, когда низшая алкилтиогруппа, процесс ведут с отщеплением низшей .;. алкнлтиогруппы или дегидрированием с получением целевых продуктов в свободном виде или в виде фармацевтически приемлемой соли. 2 табл. Ё
где Rg имеет указанные значения, конденсируют с альдегидом общей формулы (III)
СН,
где RJ и R имеют указанные значения, и полученное соединение общей формулы (1а)
NYN-M --CH Rs
СН3
Значение отличается значительно от соответствующего значения, полученного у животных, которым вводили только носитель (р.0,05; Students t-Test), при этом А - 1-(4-окси-3,5-ди-трет-бутилбензиламино)имидазол, В - 1-(4-окси-3,5-ди-трет-бутилбензилиденамино)-2-метилимидазол, С - 1-(4-окси-3,5-ди-трет-бутилбензилиденамино)имидазол.
il -M-Ub Ь5
С
/ v
СИ #2
где Rj, R4 и R6 имеют указанные значения,
с наделением целевого продукта или, в случае, когда R $-, низшая алкил- тиогруппа с отщеплением низшей алкил- тиогруппы или соединение общей формулы (1в) дегидрируют и выделяют целевые продукты в свободном виде или в виде фармацевтически приемлемой соли.
Таблица 1
17
L
Значение отличается значительно от соответствующего значения, полученного у животных, которым вводили только носитель (р 0,05, Students t-Test).
165705718
Таблица 2
| Бюлер К., Пирсон Д | |||
| Органические синтезы | |||
| Ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| М.: Мир, 1973, с | |||
| МАШИНА ДЛЯ ПРОИЗВОДСТВА ПОДЗЕМНЫХ РАБОТ | 1919 |
|
SU524A1 |
