Изобретение относится к области аналитической химии, а именно к методам количественного определения «-парафиновых углеводородов в биоматериалах, пищевых продуктах, нефтепродуктах, сточных и природных водах.
Известен способ количественного определения углеводородов в биоматериале путем экстрагирования их растворителем, упаривания выделенной фракции, хроматографического выделения углеводородной фракции на силикагеле вымыванием ее, например, четыреххлористым углеродом и последующего определения количества углеводородов в упаренном фильтрате взвешиванием. Такой способ длителен; при использованиИ его не происходит полного выделения w-парафинов, так как клеточный материал перед экстракцией растворителем не разрущается. Кроме того, применение взвешивания при количественном определении углеводородов приводит к потере при высушивании углеводородов до 5,0 ± 0,9% за счет улетучивания низкомолекулярных углеводородов.
С целью интенсификации процесса и повышения точности определения предложено биоматериал предварительно обрабатывать азотной кислотой и полученный хроматографическим разделением слой «-парафиновых углеводородов элюировать ацетоном.
Количество н-парафиновых углеводородов может быть определено нефелометрическим способом.
Пример. 1 - 10 г биологического образца
помещают в коническую колбу на 150 мл с обратным воздуишым холодильником (100- 200X14 мм) на шлифе, затем добавляют 20- 25 мл 11,5 и. раствора азотной кислоты (уд. вес 1,35) и осторожно подогревают на водяной бане при периодическом перемешивании содержимого колбы вращен 1ем. В лервые .минуты наблюдается слабое вспенивание, которое далее становится бурным. При появлении вспенивания колбу снимают с водяной бани на
5-10 мин или даже охлаждают водой, а затем продолжают нагр.евание до просветления раствора и прекрандения бурного образования маленьких пузырьков на поверхности жидкости. Разрушение биоматериала продолжается 20-
40 мин.
Затем раствор охлаждают, разбавляют водой в 2-3 раза и экстрагируют 25 мл гексана (встряхивание в течение 3 мин). Водную фазу отбрасывают, а экстракт промывают 25 м.л воды (встряхивание в течение 3 мин). Аликвотную часть промытого экстракта или весь экстракт (в зависимости от ожидаемого абсолютного содержания «-иарафинов в навеске образца), предварительно упаренный до минивакууме, создаваемом водоструйным насосом, наносят на нластину (18X13 см) с силикагелем КСК, содержащим 5% очищенного гипса (пластину с силикагелем очищают предварительным пропусканием СС1.4 или смеси диэтиловый эфир-петролейный эфир (1:1) через слой адсорбента), и нроизводят распределение в ecu, за фронтом распространения которого только углеводороды. Проявленную в парах йода полосу углеводородов на хроматограмме очерчивают, а силикагель с углеводородами носле испарения йода соскабливают и смешивают с 5-10 мл ацетона, после чего слой органического растворителя отделяют фильтрованием через стеклянный фильтр № 3 или 4, промывают силикагель на фильтре ацетоном (2X2 мл), фильтрат переносят в мерную колбу на 10-25 мл, доводят объем раствора до метки ацетоном и нроизводят ieфелометрическое определение углеводородов по модифицированному методу Лурье и Щербакова, предложенному для определения нефтепродуктов.
В колбу объемом 10-15 мл, содержаидую соответственно 6 или 15 мл раствора пищевого желатина (1 мг/мл) в 0,5%-ном растворе СНзСООН, вводят 0,5 или 1,0 мл ацетонового испытуемого раствора и тем же раствором желатина доводят объем до метки. Далее содержимое колбы взбалтывают и через 15 мин производят измереиие иа ФЭК (светофильтр
№2, толщина кюветы 1 см). Концентрацию углеводородов находят по стандартной калибровочной кривой, построенной в координатах (100-Т) %-С. Калибровочную кривую получают для концентраций смеси углеводородов 25, 50, 100, 200, 300, 400, 500 лшг, вводимых в ацетоновом растворе в колбу на 25 мл в том же порядке, что и испытуемый раствор, причем содержание ацетона в каждом стандарте
должно быть одинаковым.
Найденное но стандартной калибровочной кривой значение концентрации углеводородов делят на 0,8 для того, чтобы учесть потери при хроматографическом отделении углеводородов и объективные 01нибки метода нефелометрии. Чувствительность нефелометрического метода онределення углеводородов 2 мкг1мл.
Продолжнтельность анализа по предлагаемому сиособу не больше 4 час. Нижний предел онределения абсолютного количества w-naрафинов в образце составляет 250 мк.г, если вымытую с пластины смесь н-нарафинов растверить в 10 мл ацетона, отобрать 1 мл этого раствора и приготовить исследуемый раствор с желатином в мерной колбе на 10 лы или пикнометре.
Результаты определения количества н-парафннов в искусственных смесях с кормовыми дрожжами представлены в табл. I.
Таблица 1

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения имидаклоприда в биологическом материале. | 2024 |
|
RU2835768C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ Н-БУТИЛОВОГО ЭФИРА 2-[4-(5-ТРИФТОРМЕТИЛПИРИДИЛ-2-ОКСИ)ФЕНОКСИ]ПРОПИОНОВОЙ КИСЛОТЫ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2005 |
|
RU2287812C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ N-(БЕНЗИМИДАЗОЛИЛ-2)-О-МЕТИЛКАРБАМАТА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2005 |
|
RU2300765C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ УГЛЕВОДОРОДОВ В СОДЕРЖАЩИХ ИХ ПРОДУКТАХ | 1970 |
|
SU265041A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ НИТРОСОЕДИНЕНИЙ В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 1996 |
|
RU2100808C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СЛОЖНОГО НИТРОФЕНОЛЬНОГО ПРЕПАРАТА "НИТРАФЕН" В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 1999 |
|
RU2153169C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ 2-ДИМЕТИЛАМИНО-1,3-БИС-(ФЕНИЛСУЛЬФОНИЛТИО)ПРОПАНА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2010 |
|
RU2426726C1 |
| Способ определения алкил-динитрофенолов в биологическом материале | 1990 |
|
SU1786426A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДЕЛЬТАМЕТРИНА И ЛЯМБДА-ЦИГАЛОТРИНА В БИОЛОГИЧЕСКОМ МАТЕРИАЛЕ | 2009 |
|
RU2413225C1 |
| Способ количественного определения кадмия в биологических объектах | 1981 |
|
SU1007003A1 |
Таким образом, в случае введения добавки - 0,3% смеси утлеводордов в кормовые дрожжи, содержащие 1,25 ± 0,03% углеводородов, ири разрушении кормовых дрожжей азотной кислотой обнаруживают 1,57 ± 0,02% углеводородов.
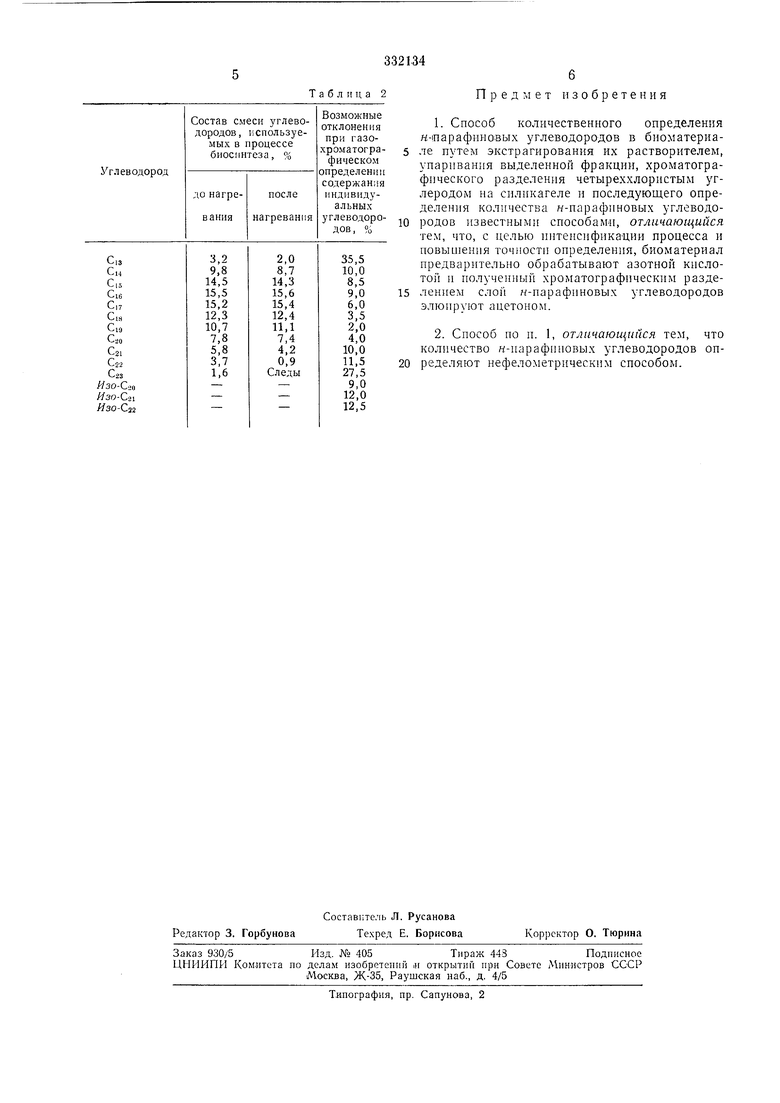
Газохроматографическое сравнение состава смеси углеводородов до и после нагревания с азотной кислотой даио в табл. 2.
Как видио из таблицы, при обработке азотиой кислотой разрушения отдельных углеводородов не происходит.
Таким образом, предлагаемый способ обеспечивает полное выделение «-парафиновых углеводородов из растительных и животных клеток, является быстрым, эффективным и чувствительным, не требует специальной иодготовки образца к анализу.
Таблица 2Предмет изобретения
