Изобретение относится к области получейия новых производных алкалоида спорыньи, которые могут найти применение в фармацевтической промышленности.
Известен способ получения алкалоидов общей формулы
R4 Н CONH-р уЧ
. .
,Уч N-CH R2 Ri
где Ri - метил, водород, Rj - алкил, содержащий от одного до четырех атомов углерода, Rg - алкил, содержащий от одного до четырех атомов углерода, бензил, либо R2 и Rs вместе с атомом азота образуют насыщенное карбоциклическое кольцо, содерл ащее четыре - семь атомов углерода, R4 - алкил, содержащий от одного до четырех атомов углерода, ХУ- группа -СНгСН или -СН С
Способ заключается в том, что соединение общей формулы
H.N-f-°4 Y
,
где Rs, Ra, R4 имеют вышеуказанные значения, нодвергают взаимодействию с соответствующим реакционноспособным функциональным производным лизергиновой кислоты таким, как хлорангидрид или ангидрид кислоты в присутствии основного конденсирующего агента в среде органического растворителя или без него. Продукты выделяют известным снособом в свободном виде или в виде солей.
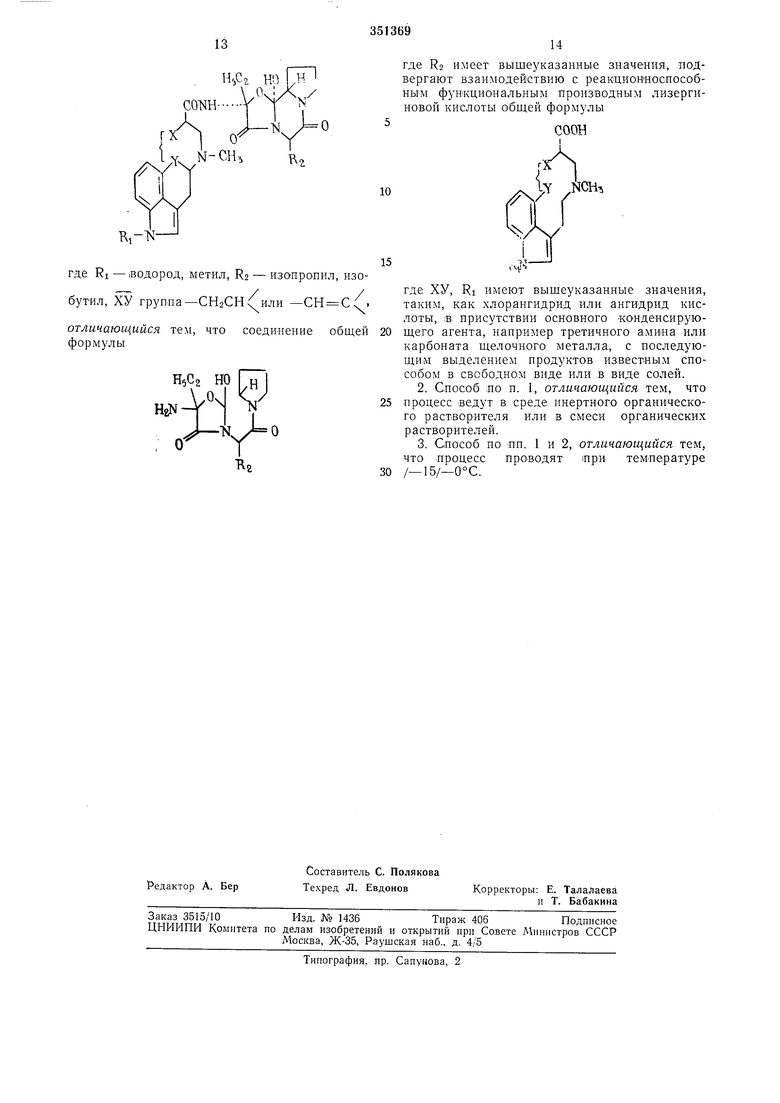
Предлагаемый способ получения алкалоидов общей формулы
T,-Nгде RI - водород, метил, R2 - изопронил,
бутил, ХУ группа -СН2СН( или -СН
Сх , основан на известной реакции. Способ
заключается в том, что соединение общей формулы
Н.С
HpN
где R2 имеет выщеуказанные значения, подвергают взаимодействию с соответствующим производным лизергиновой кислоты или ее реакционным производным, таким, как хлорангидрид или ангидрид кислоты, в присутствии основного конденсируюидего агента, например третичного амипа или карбоната щелочного металла. Процесс преимущественно проводят в среде инертного органического растворителя или в смеси органических растворителей при температуре -15 -0°С. Продукты выделяют известпым способом в свободном виде или в виде солей.
Полученные новые алкалоиды общей формулы 1 являются при комнатной температуре кристаллическими веществами, которые образуют с неорганическими или сильными органическими кислотами устойчивые кристаллические при комнатной температуре соли. В качестве кислот для солеобразования могут применяться минеральные кислоты (соляная, бромистоводородная, сериая кислоты) или
сильные органические кислоты (винная, щавелевая, метансульфоновая кислоты).
Определения рК были проведены в системе метилцеллюлозольва/вода в объемном соотнощении 80 : 20.
Пример 1. Эргонин, аргонинин.
В охлажденную до -10°С суспензию из 3,3 г хлорангидрида (2R, 5S, lOaS, 10bS)-2амино-2-этил-5 - изопропил - 3,6-диоксо - lOb оксиоктагидро-8Н-оксазоло - (3,2-а) - пирроло (2,1-е)-пиразина и 6 г хлоргидрата хлорангидрида (i-лизергиновой кислоты в 40 мл абсолютного метиленхлорида при размешивании
прикапывают в течение 15 мин 7,9 мл абсолютного пиридина так, чтобы температура оставалась постояпной. Затем размешивают бурую суспензию 30 мин при 0°С и еще 3 час при 20°С. После этого разбавляют реакционную смесь 100 мл метиленхлорида и взбалтывают со 100 мл 2 н. водного раствора соды. Затем дополнительно экстрагируют три раза при Помощи 50 мл метиленхлорида, содержащего 5% .пиридина, промывают соединенные
органические фазы один раз 50 мл 10%-ноге раствора поваренной соли и сущат над сульфатом натрия. После отгонки растворителя в вакууме при температуре 60°С в бане удаляют оставщийся пиридин промыванием остатка два раза по 40 мл теплым толуолом с последующей его отгопкой. Полученную светло-бурую пену сушат 1 час при 60°С в высоком вакууме и хроматографируют 50кратным количеством окиси алюминия (активность II-III). При помощи 0,1% метанола в метиленхло1риде элюируют эргонинин, который получается из метанола в виде бесцветных призм с т. пл. 206-207°С.
После двукратной перекристаллизации из
метиленхлорида/метанола точка плавления поднимается до 219-220°С, а ° +424° (, хлороформ), с помощью 0,5% метанола в метиленхлориде элюируют эргонин, который кристаллизуется из этанола. Т. пл. 207-
208°С, -182° (с I, хлороформ).
Бималеинат.
Пз этанола, т. пл. 183-185°С, +88° (с 1, пиридин).
Применяемый в качестве исходного продукта хлоргидрат (2R, 5S, lOaS, 10Ь5)-2-амиНО2-этил-5-изопроппл-3,6-диоксо-10Ь - оксиоктагидро-8П-оксазоло- (3,2-а) -пирроло- (2,1 -с) - пиразина получают так:
а) (3S, 8aS, aS)-2-(a - этоксикарбонил-абензилоксибутирил)-3-изопропил-1,4 - диоксооктагидропирроло-(1,2-а)-пиразин.
Смесь из 19,6 г (3S, 8а5)-1,4-диоксо-3 - изопропилоктагидропирроло - (1,2-а) - пиразина,
15 мл абсолютного пиридина, 28,5 г сложного этилового моноэфира хлорангидрида S( + )-2этил-2-бензилоксималоновой кислоты и 12 мл диоксана размешивают I час при комнатной тем.пературе и затем 1,5 час при 75°С. После
700 мл простого эфира, взбалтывают с ледяной 2 и. соляной кислотой, промывают водным раствором NaHCO:) и сушат над Na2S04. После отгонки растворителя при пониженном давлении и температуре 50°С в бане получают красноватое вязкое масло, которое можно сейчас же обрабатывать дальше.
б) (2R, 5S, lOaS, 10Ь5)-2-этоксикарбонил-2этил-5-изопропил-3,6-диоксо-10Ь - оксиоктагид ;опропил-3,6-диоксо-10Ь - оксиоктагидро-8Н-оксазоло-(3,2-а)-пирроло-(2,1-с) - пира
Я 9-я Vnunnnnn- 9 }-с - ггипяЗИН.
(3S, 8aS, а5)-2-(ос-этоксикарбонил-а - бензилoкcибyтиpl л)-3-изoпpoпил - 1,4 - диоксооктагидропирроло-(1,2-а)-Пиразин гидрируют в 800 мл 70%-ной водной уксусной кислоты на 10 г .предварительно гидрированного Pd (5% Pd в катализаторе) при комнатной температуре и нормальном давлении. Примерно через 20 час заканчивается поглощение водорода. Для дальнейшей обработки отфильтровывают катализатор и концентрируют фильтрат в вакууме при 50°С. Остаток хроматографируют на 20-кратном количестве силикагеля. С помощью 1 % метанола в метиленхлориде элюируют (2R, 5S, lOaS, 10Ь5)-2-этоксикарбонил-2-этил-5-изопропил-3,6-диоксо - lOb - оксиоктагидро - 8Н - оксазоло - (3,2-а) - пирроло (2,1-е)-пиразин и получают его после кристаллизации из простого изопропилового эфира в виде бесцветиы.х кристаллов с т. пл. 90-93°С. Чистый для анализа (2R, 5S, lOaS, 10bS)-2этоксикарбонил-2-этил-5-изопропил - 3,6-диоксо-ЮЬ - окси - октагидро-8П-оксазоло-(3,2-а)пирроло - (2,1-е)-пиразин плавится после однократной перекристаллизации при 94-95°С, 0,8° (с 2, этанол). рК„ез 11,1.
в) (2R, 5S, lOaS, 10Ь5)-2-карбокси-2-этил-5изопропил-3,6-диоксо-10Ь-оксиоктагидро - 8Ноксазоло- (3,2-а) -пирроло- (2,1-е) -пиразин.
35,4 3 (2R, 5S, lOaS, 10Ь5)-2-этоксикарбонил-2-этил-5-изопропил-3,б-диоксо-10Ь - оксиоктагидро-8П - оксазоло - (3,2 - а) - пирроло (2,1-е)-пиразина растворяют в 150 мл 1 н. водного раствора гидроокиси натрия и размешивают 2 час при комнатной температуре. Слегка мутный раствор взбалтывают один раз со сложным эфиром уксусной кислоты, затем подкисляют 2 н. ледяной соляной кислотой и сейчас же проводят экстракцию, исиользуя 4 ipasa по 300 м.л сложного эфира уксусной кислоты. Соединенные органические фазы сушат над сульфатом натрия и концентрируют при температуре 30°С в бане под вакуумом, причем получаетея (2R, 5S, lOaS, l6bS)-2карбоксил-2-этил-5-изопропил-3,6-диоксо - lObоктагидро - 8Н - оксазоло - (3,2-а) - пирроло ,1-е)-пиразин с 1 моль кристаллизационного ;ложного эфира уксусной кислоты в виде бесцветных кристаллов с т. разл. 154-155°С. Свободную от растворителя кислоту можно получить после однократного переосажденпя из смееи сложного эфира уксусной кислоты
(н-гептана; т. разл. 147-148 С, ,2° (с 2, пиридин), pKi 3,92 в MCS, ,4 в MCS.
г) (2R, 5S, lOaS, lObS) - 2-хлороформил-2этил-5-изопропил-3,6-диоксо-10Ь - оксиоктагидро-8Н-оксазоло-(3,2-а)-пирроло -(2,1-е) - пиразин. 32,6 г (2R, 5S, lOaS, 10Ь5)-2-карбокси-2этил-5-изопро.пил-3,б - диоксо - lOb - оксиоктагидро-8П-оксазоло-(3,2-а) - пирроло - (2,1-с)пиразина выливают в охлажденный до 0°С раствор из 23,1 г свежесублимированного пентахлорида фосфора в 500 м.л абсолютного иростого эфира и размеишвают 90 мин при комнатной температуре. После добавления 700 мл абсолютного петролейного эфипа оставляют смесь для дальнейшей кртсталлизации 1 час при О и -10°С. После отфчльтровьтвания получают кристаллический бесцветный (2R, 5S, lOaS, 10Ь51-2-хлопформил-2-этил-5-изопропил3,6-диокси-10Ь- оксиоктагидро - 8П - оксазоло (3,2-а)-пирроло-(2.1-е)-пиразин, который очень чувствнтелен к влаге и который нужно сейчас
же перерабатывать дальше,
д){2Р„ 5S, lOaS, 10Ь5)-2-азидокарбонпл-2этил-5-изопропил-3,6 - диоксо - lOb - оксиоктагидро-8П-оксазоло- (3,2-а) -пирроло- (2,1 -с) - пиразин,
34,4 г (2R, 5S, lOaS, 10Ь5)-2-хлорформил-2этил-5-изопропил-3,6-диоксо- lOb - оксоктагидро-8Н-оксазоло-(3,2-а)-пирроло-(2,1-с) - пиразина растворяют в 500 мл абсолютного метиленхлорида, охлаждают до и сильно
трясут 4 мин после отслаивания холодным оаствором 15 г азида натрия в 70 мл воды. После добавления 100 мл иасыщеииого раствора гидрокарбоната калия трясут enie 1 .««« прн 0°С и после разделения в делительной
воронке экстрагируют водную фазу еще два
раза при помощи 300 мл метиленхлорида,
Соединенные органические фазы сушат над
сульфатом натрия и концентрируют при 20°С
в бане в вакууме в желтую пену, которую
сейчае же перерабатывают дальше.
е)(2R, 5S, lOaS, 10Ь5)-2-бензилокеикарбониламино-2-этил - 5 - изопропил - 3,6 - диоксо10Ь-оксиоктагидро-8П-океазоло-(3,2-а)- пир:роло-(2,1-е)-пиразин.
Сырой (2R, 5S, lOaS, 10bS)-2 - азидокарбонил-2-этил-5-изопропил-3,6-диоксо - lOb - оксиоктагидро - 8Н - оксазоло - (3,2-а) - нирроло (2,1-е)-пиразин растворяют в 400 мл абсолютного хлороформа и разогревают его 15 мин с
обратным холодильником. После добав,тения 30 .ил абсолютного бензилового спирта нагревают еще 45 мпн с обратным холодильником и концентрнруют реакционную емееь при пониженном давлении, затем в высоком вакууме
при 80°С. Часть кристаллического остатка поглон ают 100 мл сложного эфира уксусной кислоты и фильтруют его после стояния 1 час нри 0°С, Получают чистый для ана,11за (2R, 5S, lOaS, 10bS)-2 - бензилокеикарбонилсиоктагидро-8Н - оксазоло - (3,2-а) - пирроло(2,1-е)-пиразин в форме мелкого, кристаллического, белого порошка с точкой разложения 216-218°С, 4-40,2° (, этанол), +41° , хлороформ). ж) Хлоргидрат (2R, 5S, lOaS, 10bS)-2амино-2-этил-5-изопр01ПИл - 3,6 - диоксо-lOb оксиоктагндро-8Н-оксазоло - (3,2-а - пирроло (2,1-е)-пиразина. 43,1 г (2R, 5S, lOaS, lObS) - 2 - бензилоксикарбаниламино-2-этил-5-изопропил-3,6 - диоксо-10Ь-оксиоктагидро-8Н - оксазоло - (3,2-а) (2,1-е)-пиразина растворяют в 800 мл абсолютного тетрагидрофурана, содержащего 4,2 г растворенного хлористоводородного газа, и гидрируют на 25 г предварительно гидрированного Pd (10% Pd в катализаторе) при комнатной температуре и нормальном давлении. Примерно через 40 мин водород больше не поглош,ается. Отфильтровывают катализатор (фильтрат можно уничтожить) и в нескольких порциях промывают его 1 л метиленхлорида/метанола (1 : 1). При температуре 20°С в бане сгущают элюат под вакуумом в желтоватую пену, из которой получают после поглощения ее в 300 мл абсолютного диметокеиэтана, кварцевания и стояния в течение 30 мин при температуре между О и --10°С гидрохлорид (2R, 5S, lOaS, 10bS)-2амино-2-этил - 5 - изо1пропил-3,6 - диоксо - lOb оксиоктагидро-8Н-оксазоло-(3,2-а) -пирроло (2,1-е)-пиразина в желтоватых мелких кристаллах с т. разл. 172-173°С. Для дальнейшей переработки соединение довольно чистое и оно 1не кристаллизуется без разложения. + 17° (,9, метиленхлорид/метанол 1:1). Пример 2. 9,10-Дигидроэргонин. В охлажденную до -15°С, размешанную суспензию 6 г гидрохлорида хлорангидрида 9,10-дигидролизврниновой кислоты в 30 мл абсолютного ацетонитрила и 30 мл диметилформамида прикапывают 20 мл абсолютного пиридина так, чтобы температура осталась поетоянной. Затем к образовавшейся, густой бурой кашице добавляют при хорошем перемешивании 3,3 г хлоргидрата (2R, 5S, lOaS, 10Ь5)-2-амино-2-этил-5-изопропил-3,6- диокео 10Ь-оксиоктатидро-8Н-оксазоло- (3,2-а) -пирроло - (2,1-е)-пиразина и повышают реакционную температуру в течение 90 мин до 0°С. Хорошо охлаждая, осторожно разлагают реакционную емесь при помощи 40 мл 2 н. соляной кислоты, выливают ее в 200 мл 2 н. водного раствора соды и экстрагируют выделившийся 9,10-дигидроэргонин пять раз, используя по 200 мл метиленхлорида, содержащего 10% этанола. Соединенные органические фазы промывают один раз 100 мл водного раствора аммиака и один раз водным раствором поваренной соли, сушат над Na2S04 и обесцвечивают 1 г активированного угля. После отгонки растворителя поглощают остаток еще два раза 40 мл толуола и азеотропной отгонкой удаляют оставшийся пиридин. Остаток хро,матографируют на 30-кратном количестве окиси алюминия (активность II-III), причем элюируют 9,10-дигидроэргонин при помощи 0,6% метанола в метиленхлориде. После кристаллизации из ацетона и трехчасовой сушки при 100°С в высоком вакууме он содержит еще 0,5 моль метиленхлорида; т. разл. 245-247°С, {а +3,8° (, метанол). Гидрохлорид. Из метанола (простого эфира, т. разл. 244- 245°С; +26,r (, метиленхлорид/метанол 1:1). Пример 3. 9,10-Дигидроэргоптин. К охлажденной до -15°С размешанной суспензии из 6 г хлоргидрата 9,10-дигидролизергиновой кислоты в 30 мл абсолютного ацетонитрила и 30 мл диметилформамида добавляют 20 мл абсолютного пиридина по каплям, так чтобы темнература оставалась постоянной. К Oiбpaзoвaв:шeйcя густой кашице прибавляют затем при хорошем перемешивании 3,44 г хлоргидрата (2R, 5S, lOaS, 10bS)-2амино-2-этил-5-изо,бутил-3,6-диоксо - lOb-оксигидро-8Н-оксазоло-(3,2-а)-пирроло - (2,1-е) пиразина, причем реакционная температура поднимается в течение 90 мин до 0°С. При хорошем охлаждении разлагают реакционную смесь при помощи 40 мл 2 н. соляной кислоты, выливают ее в 200 мл 2 н. водного раствора соды и экстрагируют выделившийся 9,10-дигидроэргоптин, используя пять раз по 200 мл метиленхлорида, содержащего 10% этанола. Соединенные органические фазы промывают один раз 100 мл водного раствора аммиака и один раз водным раствором поваренной соли, сушат над сульфатом натрия и обесцвечивают с помощью 1 г активированного угля. После отгонки растворителя поглощают остаток еще два раза в 40 мл толуола и после азеотропной отгонки удаляют оставшийся еще пиридин. Остаток хроматографируют на 30-кратном количестве окиси алюминия (активность II-III), причем элюируют 9,10-дигидроэргоптин при помощи 0,6% метанола и метиленхлорида. После кристаллизации из этанола и следующего осаждения из сложного эфира уксусной киелоты прибавлением простого эфира получают 9,10-дигидроэргонтин. Поеле четырехчасового высушивания в высоком вакууме при 100°С т. разл. 218-220°С, afo° -f4,5° (,75, метиленхлорид/метанол-1 : 1). Гидрохлорид. Из этанола (проетого эфира; т. разл. 225- 226°, af° + 17,6° (,5, метиленхлорид/метанол 1:1). П р И м е р 4. Эргоптин, эргоптинин.
В охлажденную до -10°С, размешанную суснензию из 3,44 г хлоргидрата (2R, 5S; lOaS, lObS) -2-амино-2 - этил - 5-изобутил-3,6 диоксо - lOb - оксиоктагидро - 8Н - оксазоло (3,2-а)-пирроло-(2,1-е)-пиразина и 6 г хлоргидрата хлорангидрида drлизepгинoвoй кислоты в 40 мл абсолютного метиленхлорида прикапывают в течение 15 мин 7,9 мл абсолютного так, чтобы температура осталась nocTOHBHoft. Образовавшуюся бурую реакционную смесь размешивают зател 30 мин при 0°С и 3 час при комнатпой температуре. Для дальнейшей обработки разбавляют реакционную смесь 100 мл метиленхлорида и взбалтывают со 100 л«уг 2 и. водного раствора соды. Затем дополнительно экстрагируют еще три раза 50 мл метиленхлорида, содержащего 5% пиридина, промывают соединенные органические фазы один раз 50 мл 10%-ного раствора поваренной соли и сушат над сульфатом патрия. После отгонки растворителя в вакууме при 60°С удаляют оставшийся пиридин диспергированием остатка, используя два раза по 40 мл теплого толуола, и последующей отгонкой. Оставшуюся бурую пену сушат 1 час при 60°С в высоком вакууме и хроматографируют на 50-кратном количестве окиси алюминия (активность II-III). При яомощи 0,1% метанола в метиленхлориде элюируют эргоптинин, который перекристаллизовывается из метанола в виде бесцветных плиток, с т. разл. 198-200°С (начиная с 193°С, наблюдается воронение), а 4-408° (,5, хлороформ). При помощи 0,5% метиленхлорида элюируют эргоптин, который выкристаллизовывается из 70%-iHoro водного ацетона в виде светло-бежевых Призм. После четырехчасовой сушки в высоком вакууме при 100°С, т. разл. 198- 200°С, alg -180° (с 1, хлороформ).
Бималеинат - моногидрат.
Из этанола, т. разл. 176-178°, а 4-100,5° (, метиленхлорид/метанол 1:1).
Применяемый в качестве исходного продукта хлоргидрат (2R, 5S, lOaS, 10Ь5)-2-амино-2этил-5-изобутил-3,6-диоксо-1 Ob -оксиоктагидро8Н-оксазоло-(3,2-а)-пирроло-(2,1-с) - пиразина получают следующим образом.
а) (3S, 8aS, а5)-2-(.(х-этоксикарбонил - абензилоксибутирил)-3-изобутил-1,4 - диоксооктагидропирроло-(1,2-а)-пиразин.
К разогретой до 70°С размешанной суспензии 21,0 г (3S, 8а5)-1,4-диоксо-3-изобутилоктагидропирроло-(1,2-а)-пиразина в 50 мл абсолютного диоксана и 17,4 г N-этил-ди-изоггропиламина прикапывают 5 мин 34 г сложного этилового моноэфнра хлораигидрида 5( + )-2-этил-2-бензилоксималоновой кислоты и размешивают реакционную смесь еще 2,5 час при этой температуре. Затем разбавляют при помощи 600 мл простого эфира, промывают два раза 100 мл ледяной 2 н. со10
ляной кислоты, один раз 100 мл ледяной воды и один раз 100 мл насыщенного водного раствора бикарбоната натрия. Соединенные органические фазы сушат над сульфатом натрия и после дистилляции растворителя в вакууме при 50°С получают (3S, 8aS, а5)-2-(а-этоксикарбонил-а-бензилоксибутирил)-3 - изобутил1,4-диоксооктагидро-пирроло-(1,2-а)-пиразин в виде масла, которое сейчас же перерабатывают дальше.
Экстракцией метиленхлоридом, сушкой над сульфатом натрия, отгонкой растворителя и по следующей кристаллизацией из ацетона получают из соединенных водных фаз чистый
(3S, 8а5)-1,4-диоксо - 3 - изобутилоктагидропирроло-(1.2-а)-пиразин с т. пл. 163-165°С.
б)(2R, 5S, lOaS, 10Ь5)-2-этоксикарбонил-2этил-5-изобутил-3,6 - диоксо-10Ь - окси - октагидро-8Н-оксазоло-(3,2-а) - пирроло - (2,1-с)ниоазин.
(3S, 8aS, а5)-2-(а-этоксикарбонил-а-бензилоксибутирил) -З-изобутил-1,4 -диоксооктатидропирроло-(1,2-а)-пиразин гидрируют пои 50°С при нормальном давлении на 20 г Pd (10%
Pd в катализаторе) в 800 мл неденатурированного спирта. Через 4-6 час поглощение водорода заканчивается. Отфильтровывают катализатор и выпаривают фильтоат в оотационном выпарном аппарате при 50°С. Остаток поглощают в 100 мл изонропилового эфира, причем получают уже часть чистого (2R, 5S, lOaS. 10Ь5)-2-этоксикарбонил-2-этил-5-изобутил-3,6-диоксо-10Ь-оксиоктагидро-8Н - оксазоло-(3,2-а)-пирроло-(2,1-с)-пиразина с т. пл.
94-95°С. Фильтрат концентрируют опять и хроматографиоуют на 20-кратном количестве силикагеля. При помощи 1 % метанола в метиленхлориде и путем следующей кристаллизации можно еще значительно повысить
выход чистого (2R, 5S, lOaS, 10Ь5)-2-этоксикарбонил-2-этил-5-изобутил - 3,6-диоксо - ЮЬоксиоктагидро-8Н-оксазоло-(3,2-а) - пирроло(2,1-е)-пиразина с т. пл. 94-96°С. a
-3,5° (, метиленхлорид), рКмс5 10,8.
в)(2R, 5S, lOaS, 10Ь5)-2-карбокси-2-этил5-изобутил-3,6-диоксо-10Ь-оксо-октагидро - 8Ноксазоло-(3,2-а)-пирроло-(2,1-е)-пиразин.
36,8 г (2R, 5S, lOaS, 10Ь5)-2-этоксика:рбонил-2-этил-5-изобутил-3,6 - ДИОКСО - ЮЬ-оксиоктагидро - 8Н - оксазоло - (3,2-а) - пирроло (2,1-е)-пиразина растворяют в 150 мл 1 н. водного раствора гидроокиси натрия и размешивают 2 час при комнатной температуре.
После охлаждения прозрачного раствора до 0°С подкисляют 2 н. ледяным раствором соляной кислоты и проводят исчерпывающую экстракцию при помощи сложного эфира уксусной кислоты. После промывания водой,
высушивания «ад сульфатом натрия и концентрирования растворителя при 30°С в вакууме примерно до 300 мл, пр,иба вляют 300 мл гексана и оставляют раствор на 1 час между О и -10°С для улучшения .крнсталли11
ния кристаллов В течение 16 час лри «омнаткой темлературе в высоком вакууме получают (2R, 5S, lOaS, 10Ь5)-2-карбокси-2-этил-5-изобут,ил-3,6 - диоксо - lOb - оксиоктагидро - 8Н оксазоло-(3,2-а)-пирроло- (2,1-е) - пиразин в виде моногидрата с т. разл. 138-140°С. Безводную кислоту можно получить растворением моиогидрата в абсолютном сложном эфире уксусной кислоты при двухчасовом
стоянии над молекулярным ситом 4А Линдэ и последующей кристаллизацией путем разба.вления н-гексаном, вследствие чего т. разл. поднимается до 142-143:, сс - 17° (с 1, лиридин), рКмс5 3,98 и 12,3.
-г) (2R, 5S, lOaS, lObS)-2-хлороформил - 2этил-5-изобутил-3,6-диоксо-1 ОЬ-оксиоктагидро 8Н-оксазоло-(3,2-а) - пи-рроло - (2,1-е) - пиразин.
К охлажденному до 0°С размешанному раствору 23 г свежесубл.имироваНного пентахлоряда фосфора в 1400 мл абсолютного простого эсЬиоа прибавляют 34,0 г (2R, 5S. lOaS, 10Ь5Ь2-каобокси-2-этил-5-изобутил - 3,6 - диоксо-10Ь-оксиоктагидро-8Н-оксазоло - (3,2-а)пи1рроло-(2,1-е)-пиразина, после чего размепливагот реакщионнуго смесь при комнатной температуре до получения прояоачного раствора (примерно через 1 час. После кондентрирования до половины при температуре 20°С осторожно прибавляют с мварцеванием абсолютный п,и)клогекса« пока «е начнется WPMсталлизация хлорангидрила кислоты. После одночасового стояния между О и -10°С получают чистый, чрезмерно чувствительный к влажности (2R, 5S, lOaS, 10Ь5)-2-хлорформил-2-этил-5-изобутил-3,6 - диоксо - 1 Ob - оксиоктагидро- 8Н - оксазоло - (3,2-а) - пирроло (2,1-с)-пиразин с т. разл. 113-115°С, который нужно сейчас же перерабатывать дальше.
д) (2R, 5S, lOaS, 10Ь5)-2-азидокарбонил-2этил-5-изобутил-3,6-диоксо - lOb - оксиоктагидро-8Н-оксазоло-(3,2-а)-пирроло - (2,1-с)-оиразин.
38,6 г СвежеперекристаллИзоВанного (2R, 5S, lOaS, 10Ь5)-2-хлороформил-2 - этил-5 - изобутил-3,6-диоксо-10Ь - о«сиокта1Гидро-8Н-оксазоло - (3,2-а)-пирроло-(2,1-с)-пиразина растворяют в 1000 мл абсолютного метиленхлорида при 0°С и интенсивно перемешивают смесь 4 мин между -5 и 0°С с раствором 55 г азида натрия в 250 мл воды. После добавления 300 мл насыш,енного водного раствора бикарбоната калия вибрируют еще 1 мин при этой температуре и после разделения фаз экстрагируют еще два раза при помощи 300 мл метиленхлорида. Соединенные органические экстракты промывают ледяной водой, сушат над сульфатом натрия и .выпаривают при 20°С в вакууме. Остаток перекристаллизовывается из смеси абсолютного простого эфира/петролейното эфира. Точка плавления нехарактерная, со вспышкой. Полученный так (2R, 5S, lOaS, 10Ь5)-2-азидо.карбонил-2этил-5-изобутил-3,6 - диоксо - lOb - оксиокта12
гидро-8Н-оксазоло - (3,2-а)-пирроло - (2,1-с) пиразин перерабатывают сейчас же дальше, е) (2R, 5S, lOaS, 10Ь5)-2-бензилоксикарбониламино-2-этил - 5 - изобутил - 3,6 - диоксо10Ь-оксиоктагидро-8Н - оксазоло-(3,2 - а) - пирроло- (2,1-с) -пиразин.
39,3 г Свежеперекристаллизованного (2R, 5S, lOaS, 10Ь5)-2-азидокарбонил - 2 - этил - 5изобутил-3,6-диоксо-10Ь-оксиоктагидро - 8Н оксазоло -(3,2-а) - пирроло - (2,1-с)-пиразина растворяют в 750 мл абсолютного .хлороформа, прибавляют 32 мл бензилового спирта и по возможности скорее погружают смесь в предварительно нагретую баню, так что сейчас же начинается выделение азота. После нагревания до кипения в течение 40 мииут с обратным холодильником отгоняют растворитель и освобождают остаток от излишнего бензилового спирта в высоком вакууме при
температуре 80°С. После кристаллизации из сложного эфира уксусной кислоты получают мелко кристаллический поронок с т. разл. 212-214°С. Па тонкослойной хроматографии получают чистый (2R, 5S, lOaS, 10Ь5)-2-бензилоксикаобониламино-2-этил-5-изобутил -3,6диоксо - lOb - окооктагндро - 8П - оксазоло (3,2-а)-пирроло-(2,1-е)-пиразин, alg -2,8°
(с 2, метиленхлорид).
ж) Хлоргидрат (2R, 5S, lOaS, 10Ь5)-2-амино-2-этил-5-изобутил - 3,6 - диоксо-1 Ob - оксиоктагидро - 8П - оксазоло - (3,2-а) - пирроло (2,1-е)-пиразина. В суспензию 30 г поедварительно гидрированного Pd (10% Pd в катализато-ре) в 400 мл абсолютного тетрагидрофураиа прикапывают раствоп 44,5 г (2R. 5S, lOaS, 10bS)-2бензилоксика.рбониламино-2-этил-5- изобутил 3.6-диоксо-10Ь- оксиоктагидро - 8Н - оксазоло (3,2-а)-пирроло-(2,1-е)-пиразина в 480 мл абсолютного тетра-гидрофурана, содержащего 120 ммоль растворенного хлористоводородного газа. Затем гидрируют при комнатной температуре и нормальном давлении. Через
40 мин заканчивается гидрирование, причем, поглощается примерно 2 л водорода. Затем фильтруют катализатор и уничтожают фильтрат. Катализатор хорошо промывают в неекольких фракциях метиле1нхлорида/метанола
f : 1) и концентрируют фильтрат при 20°С в вакууме, получая желтую пену. Из диметокси-этана кристаллизуется хлоргидрат (2R, 5S, lOaS, 10Ь5)-2-амино-2-этил - 5-изобутил - 3,6диоксо - 1 Ob - оксиоктагидро - 8Н - оксазоло (3,2-а)-пирроло-(2.1-с)-пиразина, который содержит еще примерно А моль кристаллизационного диметокси-этана после суШки в течение ночи в высоком вакууме. Т. разл. 181 - 182°С.
Предмет изобретения
Способ получения алкалоидов общей форМУЛЫсош-i---f N-CH.K, где RI - водород, метил, Rj - изопро бутил, ХУ группа-CH2CHv или -С отличающийся тем, что соединение формулы де R2 имеет вышеуказанные значения, подергают взаимодействию с реакционноспособым фун кциональным производным лизергиновой кислоты общей формулы где ХУ, RI имеют выщеуказанные значения, таким, как хлорангидрид или ангидрид кислоты, ;в присутствии основиого кОНденсирующего агента, например третичного амина или карбоната щелочного металла, с последующим выделением продуктов известным способом в свободном виде или в виде солей. 2.Способ :по п. I, отличающийся тем, что процесс ведут в среде ннертного органического растворителя или в смеси органических растворителей. 3.Способ по пп. 1 и 2, отличающийся тем, что .процесс проводят три температуре /-15/-0°С.





| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛКАЛОИДОВ | 1973 |
|
SU372813A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАЛОИДОВ | 1970 |
|
SU417944A3 |
| Способ получения алкалоидов спорыньи | 1970 |
|
SU542475A3 |
| Способ получения 5 @ -/2 @ -бутил/-пептидэрготалкалоида или его аддитивных солей с кислотами | 1983 |
|
SU1189351A3 |
| Способ получения производных эргопептина или их солей | 1980 |
|
SU953984A3 |
| Способ получения производных октагидрооксазоло/3,2-а/пирроло /2,1-с/пиразина | 1977 |
|
SU725565A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДСОДЕРЖАЩИХ ЭРГОАЛКАЛОИДОВ | 1966 |
|
SU423292A3 |
| Способ получения производных 9,10-дигидро-лизергиновой кислоты | 1974 |
|
SU516355A3 |
| Способ получения производных эргопептина или их солей | 1981 |
|
SU1022660A3 |
| Способ получения производных октагидрооксазоло (3,2-а) пирроло (2,1-с) пиразина | 1974 |
|
SU593664A3 |
