1
Изобретение относится к области получения новых алкалоидов, по своему действию отличающихся от действия известных аналогичных соединений, обладающих фармакологической активностью.
Известен способ получения алкалоидов общей формулы
CoHs п С1Н
У-О0 )
аключающийся в том, что соль соединения бщей формулы
Н-С СН,
Ч/
сн
.-.0
тт
H2N-/-N
О
л
н сн
CH,
I сн
3
подвергают взаимодействию с соответствующим реакционноспособным функциональным производным лизергиновой кислоты (II) или 1-метиллизергиновой кислоты, например ее смешанным ангидридом с трифторуксусной или серной кислотой, хлоргидратом хлорангидрида кислоты, их азидом или их продукт присоединения с имидогалогенидом N-ди-низший алкилзамещенного амида карбоновой кислоты, например диметилформамида или диметилацетамида. Целевой продукт выделяют в виде основания или переводят его в соли известными приемами.
Процесс обычно ведут в инертном органическом растворителе или смеси растворителей при температуре от - 20 до 0°С. Продукты выделяют известным способом в свободном виде или в виде соли.
При изготовлении смешанных ангидридов соединений приведенной формулы с трифторуксусной кислотой выбирают соотношение исходных продуктов так, что на 1 моль сухого соединения II применяют от 1 до 1,4 моль ангидрида трифторуксусной кислоты и 2 моль трифторуксусной кислоты.
в качестве инертного при реакционных условиях растворителя или смеси растворителей можно применять ацетонйтрил, диметилформамид, диметилацетамид, пропионитрил, N-метилпирролидон, метиленхлорид или их смеси.
Можно использовать в качестве исходного продукта смесь, содержащую кроме лизергиновой или 1-метиллизергиновой кислот, изолизергиновую или 1-метилизолизергиновую кислоту и 6-метил-Д -эрголен-8-карбоновую или 1-метил - 6-метил - А -эрголен-8 - карбоновую кислоту. Смесь из лизергиновой кислоты, изолизергиновой кислоты и 6-метил-А -эрголен8-карбоновой кислоты можно непосредственно получить сапрофитическим выращиванием штамма грибов NRRL 3080 рода Claviceps paspali Stevens et Halt. Эту смесь можно сушить нагреванием до 150°С в вакууме, и ее предпочтительно применяют в сухой форме.
Последовательность прибавления реагентов для изготовления смешанных ангидридов различна.
Так, например, можно соединения II в безводной форме суспендировать в одном из вышеуказанных органических растворителей и
доводить до растворения прибавлением от 1 до 5 моль, предпочтительно примерно 2 моль трифторуксусной кислоты, после чего добавляют 1,2 моль ангидрида трифторуксусной кислоты, или оба реагента одновременно прибавляют по каплям в полученную суспензию. Порядок прибавления трифторуксусной кислоты и ангидрида трифторуксусной кислоты можно менять.
Так как полученные таким образом смешанные ангидриды очень легко разлагаются, их сразу же используют в реакции. В раствор смешанных ангидридов прибавляют соединение формулы I в виде соли, например хлоргидрата. Прибавлением большого избытка третичного органического основания при температуре от -20 до - 10°С освобождают основание формулы I, которое сразу реагирует с соединениями общей формулы II. Порядок
прибавления основания и соединения формулы I в виде его соли, можно менять.
В случае использования смешанных ангидридов кислоты II с серной кислотой проводят конденсацию в инертном при реакционных условиях растворителе, таком как диметилформамид, в присутствии третичного органического
основания при температуре от - 10 до 0°С с
соединением формулы I в виде его соли.
В случае использования хлоргидридов хлоргидратов кислот II проводят конденсацию в инертном при реакционных условиях растворителе в присутствии третичного органического или слабого неорганического основания при - 10-0°С с соединением формулы II в форме его соли.
В случае использования азидов кислоты II проводят конденсацию в присутствии третичного органического основания при температурах примерно от 0°С до комнатной температуры в инертном при реакционных условиях растворителе с соединением формулы II в форме его солей.
Можно соль соединения I подвергнуть взаимодействию с соединением II в инертном при
реакционных условиях растворителе и в присутствии N-динизшего-алкилзамещенного амида алифатической монокарбоновой кислоты и средства хлорирования и бромирования, а также связывающего кислоту агента.
В случае, если R - водород, полученное соединение можно прометилировать в присутствии основного конденсируюшего агента, например алкоголята или амида щелочного металла.
Метилирование лучше проводить следующим образом.
К раствору низшего алифатического спирта, например этанола в жидком аммиаке, прибавляют по порциям металлический натрий или калий, и выдерживают до обесцвечивания раствора. К полученному таким образом раствору алкоголята металла прибавляют при примерно -40°С при хорошем перемешивании нужное соединение и после его растворения прибавляют метилйодид, аммиак
испаряют в вакууме и остаток распределяют между водным раствором карбопата щелочного металла и метиленхлоридом. органическую фазу промывают водой, сушат над сульфатом натрия и метиленхлорид удаляют. Для этой реакции применяют на моль соединения предпочтительно примерно 5 моль алкоголята щелочного металла и примерно тот же избыток метилйодида.
Пример 1. Эргоноркорнин (25-изопропил-5а-н-пропилэргопептин).
2,68 г (10 ммоль) безводной смеси 40% J-лизергиновой кислоты, 40% б-метил-Д - -эрголен-8-карбоновой кислоты и 20% изолизергиновой кислоты растворяют в 25 мл абсолютного диметилформамида прибавлением 2,28 г (20 ммоль) трифторуксусной кислоты и доводят до - 10°С перемешиванием. При этой температуре прибавляют по каплям в течепие 5 мин смеси 2,52 г (12 ммоль) ангидрида трифторуксусной кислоты в 12 мл абсолютного ацетопитрила и перемещивают раствор еще в течение 10 мин. После этого добавляют при сильном охлаждении 1,73 г (5 ммоль) (2R, 5S, lOaS, 10&5)-2-амино - 3,6-диоксо-10Ь - окси-2изопропил - 5-(пропил-1) - октагидро-8Н-оксазоло-(3,2-а) - пирроло-(2,1-с) - пиразингидрохлорида и 12 мл пиридина и перемешивают реакционную смесь еще в течение 1 час при температуре между - 10-0°С. Разбавляют 200 мл метиленхлорида и хорошо взбалтывают со 100 мл 2 н. раствора карбоната натрия. Водную фазу еще трижды дополнительно экстрагируют, употребляя но 100 мл метиленхлорида. Объединенные органические фазы сушат над сульфатом натрия и испаряют в вакууме. Остаток хроматографируют на силикагеле посредством сложного уксусного эфира. После перекристаллизации из сложного уксусного эфира получают аргоноркорнип с т. пл. 175- 177°С; - 75° (с 1,5, пиридин).
Пример 2. Эргоноркорнин (2В-изопропил-5а-к-пропилэргопептин).
37,2 г (115 ммоль) хлорангидридя-гидрохлорида й -лизергиновой кислоты н 20,8 г (60 ммоль) (2R, 5S, lOaS, 10&S)-2 - амино-3,6диоксо-ЮЬ-окси - 2-изопропил-5 - пропилоктагидро-8Н - оксазоло- 3,2-а -пироло-Г2,1-с -пиразингидрохлорида суспендирзют в 300 мл диметилформамида и перемешивают при -10°С. В течение 10 мин прибавляют по каплям 11,5 мл безводного пиридина, причем суспензия постепенно растворяется. Пере мешивают еще 90 мин при 25°С. После прибавления 60 мл 4 н. раствора карбоната натрия испаряют досуха при уменыненном давлении при 30°С, растворяют остаток в смеси 1000 мл метиленхлорида-метанола (8:2) и 200 мл 4 н. раствора карбоната натрия и разделяют фазы. Органическую фазу промывают трижды, употребляя по 200 мл 4 н. раствора карбоната натрия, объединенные водные фазы экстрагируют 4 раза, употребляя по 500 мл метиленхлорида-метанола (8:2), объединенные
органические фазы сушат над сульфатом натрия и животным углем и испаряют досуха. Остаток хрпматографпруют на силикагеле сложным уксусным . После перркристаллпзапип из сложного уксусного эфира получают Эргоноркорнин; т. пл. 175-177°С; afo -75° (с 1,5 пиридин).
Пример 3. 1-Метилэргоноркорнин (1-метил-2р-изопропил-5а-н-пропилэргопептин).
В раствор 0,49 г металлического натрия в 100 мл жидкого ам- гиака прибавляют по каплям при перемспп вании в течение 20 мин раствор 2 мл абсолютного этанола в 2,5 мл абсолютного эфира, причем глубоко синий в начале раствор обесцвечивается. После этого вносят 1,31 г сухого эргоноркорнина, перемешивают еще в течепие 5 мин до его полного растворения и прибавляют по каплям 1,65 г метилйодида в 2 .л абсолютного эфира при - 40°С в течение 5 мин. После перемешивания в течение 1/2 час при той же самой температуре осторожно нагревают прозрачный раствор и аммиак отсасывают в частичном вакууме. Остающийся желтый остаток растворяют в смеси 50 мл метиленхлорида и 50 мл концентрированного водного раствора бикарбоната натрия. Органическую фазу промывают водным раствором бикарбоната натрия, после этого 30%-ным раствором поваренной соли. После суппчи объединенных органических фаз и отгонки растворителя при 50°С получают после коисталлизапии остатка из сложного уксусного эфира уже чистый в тонкослойной :ро атогря1 пте 1-метилэргоноркорнин с т. рязл. 223°С: а -88° (с 1,1 пиридин) .
Употребленный как исходный (2R, 5S. . 10ftS)-2 - амино-З.б-диоксо-lOf)окси-2 - изопроптш - 5-(пропил-1) - октагидоо8Н-оксазоло - Г3,2-я1 - пирроло- 2Л-с - пиразингидрохлорид получают следующим образом.
А. f3S, 8г78Ы .4-1Тиоксо - 3-(пропил-1)-октагидооппрроло-Г 1.2-(7l-пиразин.
24.9 г (0,1 моль) N-Kap6o6eH3OKCH-L-npoлина и 13,3 г (0,1 Атоль) свежедистиллированного сложнот-о eтплoвoгo эАирл L-ноРвалина растворяют в 100 мл сложного лксуспого эфира и прибавляют по каплям при 5-10°С и перемепптяании 22,6 г (0,11 моль) дициклогексилкярбодиилтита в 25 тл сложного уксусного эфира. РеакциоппЮ смесь переметтп вают в течение 1 чяс при 40°С, осажлаюи уюся дициклогексилмочевину отфильтровывают и фильтрпт проуывают сначала 1 н, соляной кислотой и затем 1 н. гидроокт1сью аммония. Послр сгущения раствора сложного уксусного эфира прибавляют петролейпып ч(Ьир и отЛильтровычаютвыкристаллизовавшийся
сложный мет-иловыц эфир N-Kap6o6en30Kcn-Lпролил-1.-норвалина: т. пл. 98°С; ос о + 8°
(в уксуспой кислоте).
36,2 г (0,1 моль) сложного метилового эфира N-карбобензокси - Ь-пролил-Ь-норвалина
растворяют в 400 мл метанола и гидрируют с 2 г палладия (10%-ного) на активированном угле при комнатной температуре и нормальном давлении. После отфильтровывания катализатора удаляют растворитель в вакууме и маслянистый остаток растворяют в 100 мл ж-ксилола. К раствору прибавляют 40 мг бензойной кислоты, отгоняют примерно 20 мл растворителя и после этого кипятят реакционную смесь в течение 5 час с обратным холодильником. После охлаждения раствора его готовят к кристаллизации. В течение нескольких дней при примерно 0°С получают кристаллический (3S, 8aS) -1,4-диоксо-З- (пропил1)-октагидропирроло- 1,2-а -пиразин; т. пл.
134°С; - 135° + 2° (с 1 % в этаноле).
Б. (2R, 5S, lOaS, 106S)-2 - Кгрбокси-3,6-диокси-106-о:кси - 2-изопропил - 5-(пропил-1)-октагидро-8П-оксазоло- 3,2-а -пироло - 1,2-с пиразин.
78,4 г (400 ммоль) (3S, 8aS)-l,4-диoкco-3(пpoпил-l) - октагидропирроло- 2,1-а - пиразина растворяют в 200 мл диоксана, прибавляют 144 г N-этилдиизопропиламина и 120 г (400 ммоль) сложного моноэтилового эфира -2-бензилокси-2-ызо-пропил малоновой кислоты и нагревают в течение 3 час до 70°С при перемешивании. Полученную густую массу растворяют в 600 мл ледяной уксусной кислоты и ее гидрируют в присутствии 25 г 10%-ного палладинированного угля при 50°С и нормальном давлении. После окончания поглощения водорода прибавляют еще раз 5 г -катализатора и гидрируют дальше. Катализатор отфильтровывают, фильтрат сушат при 30°С и остаток растворяют в сложном уксусном эфире, промывают 1 н. соляной кислотой и после этого 1 н. раствором NaHCOs и органический раствор испаряют досуха. Полученный (2R, 5S, lOaS, 106S) - 2-этоксикарбонил-3,6-диОКСО-106-ОКСИ - 2-изопропил-5-(пропил-1)-октагидро-8Н-оксазоло- 3,2-а -пироло- 2,1-с - пиразин растворяют в смеси 100 мл диоксана и 550 мл 2 н. раствора едкого натра и хранят в течение 4 час при 25°С. После охлаждения до 0°С доводят до рН 7,5 посредством 4 н. серной кислоты, половину объема испаряют, промывают сложным уксусным эфиром и водную фазу подкисляют 4 н. серной кислотой ао рН 1 и экстрагируют сложным уксусным эфиром. Этот экстракт сложного уксусного эфира сушат над, сульфатом натрия, выпаривают досуха и остаток перекристаллизовывают из эфира. Полученный таким образом (2R. 5S, lOaS, 106S) - 2-карбокси-3,6-диоксо- 0&-окси2-изопропил - 5-(пропил-1) - октагидро-8Н-оксазоло- 3,2-а -пироло- 2,1 -с -пиразнн плавится при 152-153°С (разложение); 4-31° (с 2 в диметилформамнде).
В. (2R, 5S, lOaS, 10&S) - 2-Хлорформил-3,6ДИОКСО-10& - окси-2-изопропнл - 5-(пропил-1)октагидро - 8Н - оксазоло - 3,2-а - пироло 2,1-е -пиразин.
27 г (130 ммоль) фосфорпентахлорида суспендируют в смеси 320 мл безводного диэтилоБОго эфира и 320 мл петролейного эфира, перемешивают в течение 60 мин при 25°С, охлаждают до 10°С, прибавляют 34 г (100 ммоль) (2R, 5S, lOaS, 10&5)-2-карбокси3,6-диоксо-10Ь - окси-2 - изопропил-5-(пропйл1)-октггидро - 8Н-оксазоло- 3,2-а - пирроло 2,1-е -пиразина, и суспензию перемешивают
в течение 4 час при 25°С. После фильтрапии промывают кристаллическую массу эфиром- петролейным эфиром (1:1) н сушат в вакууме с исключением влажности.
Получают таким образом (2R, 5S, lOaS,
106S)-2 - хлорформил-3,6-днoкco-10&-oкcи-2изoпpoпил-5-(пpoпил-l) - октагидро - 8Н-оксазоло- 3,2-а -пирроло- 2,1-с -пиразин; т. пл.
115-117°С (разложение); +33° (в метиленхлориде). Вешество-неустойчиво, и его
как можно скорее перерабатывают для последующего синтеза. При применении фосфорпентабромида как средства галогенирования получают соответствующий (2R, 5S, lOaS, 1065)-2-бромформил - 3,6-диоксо - 10&-ОКСИ-2изопропил-5-(пропил-1) - октагидро-8Н - оксазоло-ГЗ,2-а -пирроло- 2,1-е -пиразин.
Г. (2R, 5S.lOaS,106S)-2 - Бензилоксикарбониламино-3,6 - диоксо-10{)-окси-2-изопропил-5(пропил-1)-октагидро - 8Н - оксазоло- 3,2-а пирроло- 2.1-е -пиразин.
К смеси 250 мл метиленхлорида, 34 мл воды и 11.3 г (173 ммоль) азида натрия прибавляют постепенно при - 5°С и очень сильном перемешивании 23,5 г (67 ммоль) (2R. 5S,
lOaS, 10&S)-2 - хлорформил-3,6-диоксо-10&-окси-2 - изопропил-5-(пропил-1) - октагидро-8Ноксазоло- 3,2-а - пирроло- 2,1- :1-пиразина и перемешивают enie в течение 6 мин. После разделения фаз экстрагируют водную фазу
100 V.. метиленхлорида, объединенные органические фазы промывают 1 н. раствором бикарбоната натрия, сушат над сульфатом натрия и выпаривают досуха. Остаток растворяют в 130 мл безводного и безсппртового хлороформа, прибавляют 10,3 г (96 ммоль) бензилового спирта, нагревают в течение 90 мин с обратным холодильником, испаряют и кристаллический остаток перекристаллизовывают из диэтилового эфира. Получают таким образом (2R, 5S. lOaS,I06S)-2- бензилоксикарбониламино-3.6 - диоксо-10Ь-окси-2-изоппопил5-(пропил-П-октагидро - 8Н-оксазоло- 3,2-а1пирроло- 2.1-е -пиразин с т. пл. 205-207°С;
,20
X)D +39° (с I в пиридине).
Д. (2R,5S. lOnS, )-2 - Амино-3.6 - диокСО-10& - оксп-2-изопропил - 5-(пропил-1)-октагидро - 8Н-оксазоло- 3,2-а - пирроло - 2,1-е пиразингидрохлорид.
49,5 г (111 ммоль) (2R, 5S, lOaS. 10ftS)-2бензилоксИКарбониламино - 3.6-диоксо-106-окси-2-нзопропил - 5-(пропил-1)-октагидро - 8Ноксазоло - 3,2-а -пирроло - 2,1-е - пиразина растворяют в смеси 200 мл диметилформамида и 500 мл диоксана, прибавляют 34 мл 4 н.
9
раствора соляной кислоты в диоксане и 12 г 10%-1-юго палладинированного угля и гидрируют при нормальном давлении и комнатной температуре. После окончания поглощения водорода фильтруют, катализатор промывают метилепхлоридом и фильтрат сушат. После кристаллизации остатка из 100 мл тетрагидрофурана получают (2R, 5S, lOaS, 10&S)-2амино-3,6 - диоксо - 10 -окси-2 - изопропил - 5(пропил-1) - октагидро-8Н - оксазоло- 3,2-а пирроло- 2,1-с -ииразингидрохлорид с т. пл. 142°С (разложение); y-fo +29° (с 2 в трифторуксусной кислоте). Употребленный исходный продукт на ступени Б сложный моноэтиловый эфир й -2-беизилокси-2-изонропилмалоновой кислоты можно получить следующим образом.
А. Сложный диэтиловый эфир 2-бензило,кси-2изопропилмалоновой кислоты.
К 133 г (0,5 моль) сложного диэтилового эфира 2-бензилоксималоновой кислоты и 110 г (0,6 моль) диизопропилсульфата прибавляют в течение 90 мин изготовленный из 15 г натрия и 300 мл абсолютного этанола раствора алкоголята натрия при перемешивании и легком охлаждении (примерно 35-45°С) по каплям. Перемешивают после этого в течение 2 час при 45°С и потом в течение 1 час при 60°С. Реакционную смесь охлаждают до комнатной температуры, нейтрализуют ледяной уксусной кислотой и прибавляют 1,5 л воды. Далее экстрагируют многократно эфиром, промывают объединенные эфирные фазы разбавленным водным раствором карбоната натрия и после этого водой. Эфирные вытяжки сушат над сульфатом натрия и растворитель выиаривают, остающееся желтое масло дистиллируют для очистки при 0,1 мм рт. ст. и при температуре ванны 200°С.
Б. Слол-сный моноэтиловый эфир 2-бензилокси-2-изопропилмалоновой кислоты растворяют в 2400 мл этанола, при перемешивании прибавляют 4400 мл (6,15 ммоль) 1,40 н. раствора гидроокиси калия в этаноле и реакционную смесь перемешивают в течение 16 час при 25°С. После прибавления 3000 г льда доводят до рН 8,0 посредством примерно 120 мл концентрированной фосфорной кислоты и этанол удаляют в вакууме при 30-40°С. После прибавления 3000 мл дистиллированной водой доводят до рН 8-9 с помощью примерно 180 мл 4 н. раствора едкого натра. Полученный таким образом светло-желтый раствор экстрагируют трижды, употребляя по 1000 мл эфира, причем эфирный экстракт каждый раз экстрагируют 60 мл 10%-ного раствора бикарбоната натрия и объединенные экстракты бикарбоната натрия прибавляют к водному раствору. Щелочной водный раствор охлаждают до -5°С, разделяют 3000 мл эфира и после этого подкисляют до рН 2 медленно при сильном перемешивании примерно 840 мл концентрированной фосфорной кислоты. Обе фазы разделяют, и водную фазу экстрагиру10
ют еще дважды по 600 мл эфира. Объедииенные эфирные растворы промывают по 600 мл воды до тех пор, пока промывная вода не достигла значение рН 4 (4-5 раз), причем промывную воду каждый раз экстрагируют вновь 100 мл эфира. Объединенные эфирные растворы промывают дважды по 600 мл 30%-ного раствора хлористого натрия, сушат над сульфатом натрия, отфильтровывают, сгущают и
сущат в глубоком вакууме до постоянного веса. Остается вязкотекущее, слегка желтоватое лгасло. которое оказывается однородным в тонкослойной хроматограмме (силикагель), растворитель метанол и хлороформ-метанол
(7:3); пв 1,4988.
В. Слолчпый моноэтиловый эфир К(+)-2-бензилокси-2-изопроиилмалоновой кислоты.
К раствору 2330 г (8,36 моль) рацемата
сложного моноэтилового эфира 2-бензилокси2-изопропилмалоновой кислоты в 15 мл выдерживаемого над натрием эфира прибавляют при сильном перемещивании при исключении влажности 1460 г (8,83 моль) высушенного в течение 16 час при 50°С в глубоком вакууме 1-псевдоэфидрина, заражают при помощи 1 г образованного из 1-псевдоэфидрина и сложного моноэтилового эфира (-)-2-бензилокси-2-изопропилмалоновой кислоты диастереомера и оставляют стоять в течение двух дней при 0°С. Образуется корка кристаллов, которую отделяют и промывают 1000 мл безводного эфира. К эфирному раствору прибавляют 5000 г льда и осторожно подкисляют
при очень сильном перемешивании концентрированной фосфорной кислотой. После разделения фаз экстрагируют водную фазу еще трижды, употребляя ио 1000 мл эфира. Объединенные эфирные фазы промывают 5 раз,
употребляя ио 2000 мл воды, и промывную воду каждый раз экстрагируют 500 мл эфира, который прибавляют к объединенной эфирной фазе; рН последней промывной воды должен составлять примерно 4. После иромывки эфирной фазы посредством 2000 мл 30%-ного раствора поваренной соли сушат над сульфатом натрия и раствор сгущают. Остается вязкотекучий, маслянистый остаток, который сушат при 30°С Б глубоком вакууме в течение
16 час в ротационном испарителе при медленном повороте до постоянного веса. Остающееся, обогащенное сложным моноэтиловым эфиром R (+) -2-бензилокси-2-изопропилмалоновой кислоты масло растворяют в стоящем над натрием эфире и прибавляют при отсутствии влажности и при сильном перемешивании 1127 г (6,81 моль) высушенного в течение 16 час при 50°С в глубоком вакууме /-псевдоэфедрина. После растворения (2-
3 мин) и заражения с образованным из d-псевдоэфедрина и сложного этилового эфира R( + ) -2-бензилокси-2-изопропилмалоновой кислоты диастереомером оставляют раствор стоять в течение 2 дней при 0°С. Корку кристаллов декантируют и кристаллическую мае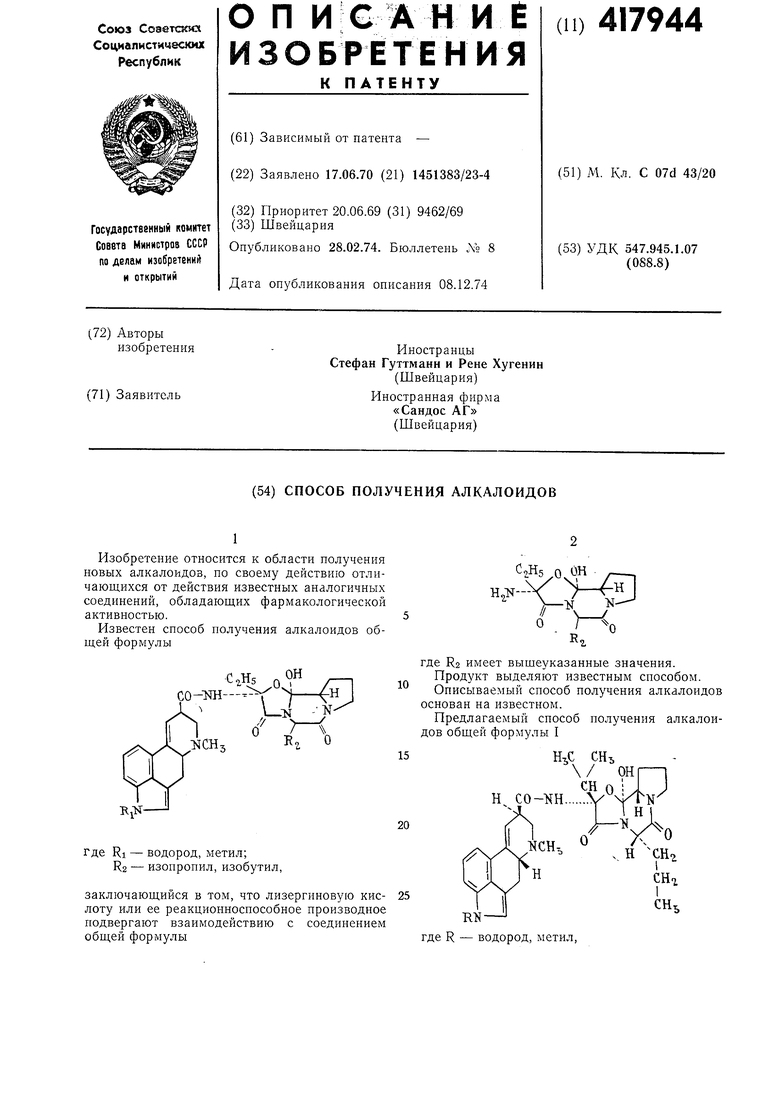




| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛКАЛОИДОВ | 1973 |
|
SU372813A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАЛОИДОВ | 1972 |
|
SU351369A1 |
| Способ получения 5 @ -/2 @ -бутил/-пептидэрготалкалоида или его аддитивных солей с кислотами | 1983 |
|
SU1189351A3 |
| Способ получения производных эргопептина или их солей | 1980 |
|
SU953984A3 |
| Способ получения алкалоидов спорыньи | 1970 |
|
SU542475A3 |
| Способ получения производных октагидрооксазоло/3,2-а/пирроло /2,1-с/пиразина | 1977 |
|
SU725565A3 |
| Способ получения производных октагидрооксазоло (3,2-а) пирроло (2,1-с) пиразина | 1974 |
|
SU593664A3 |
| НОВОЕ ПРОИЗВОДНОЕ ФЕНИЛПИРРОЛА | 2009 |
|
RU2470917C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| Способ получения производных эргопептина или их солей | 1981 |
|
SU1022660A3 |
