1
Изобретение от1носится к способу получе«ия новых производных 1,4-бензодиазепина, которые обладают Широким лекарствепным действием и могут иайти применение в фармацевти чеокой промышленности.
В литературе известен способ получения «роизводных бензодиазепииа общей формулы
где R -.водород, «изший алкил, со-держаЩИЙ до 3 атомов углерода, цпклоалкилмехил, содержащий 4-7 атомов углерода, X, R „ - водород, галоид.
Способ заключается в том, что соответствующий 2-аминометил-3-фенили.ндол подвергают обр аботке окислителем с .последующим выделением продукто а известным способом.
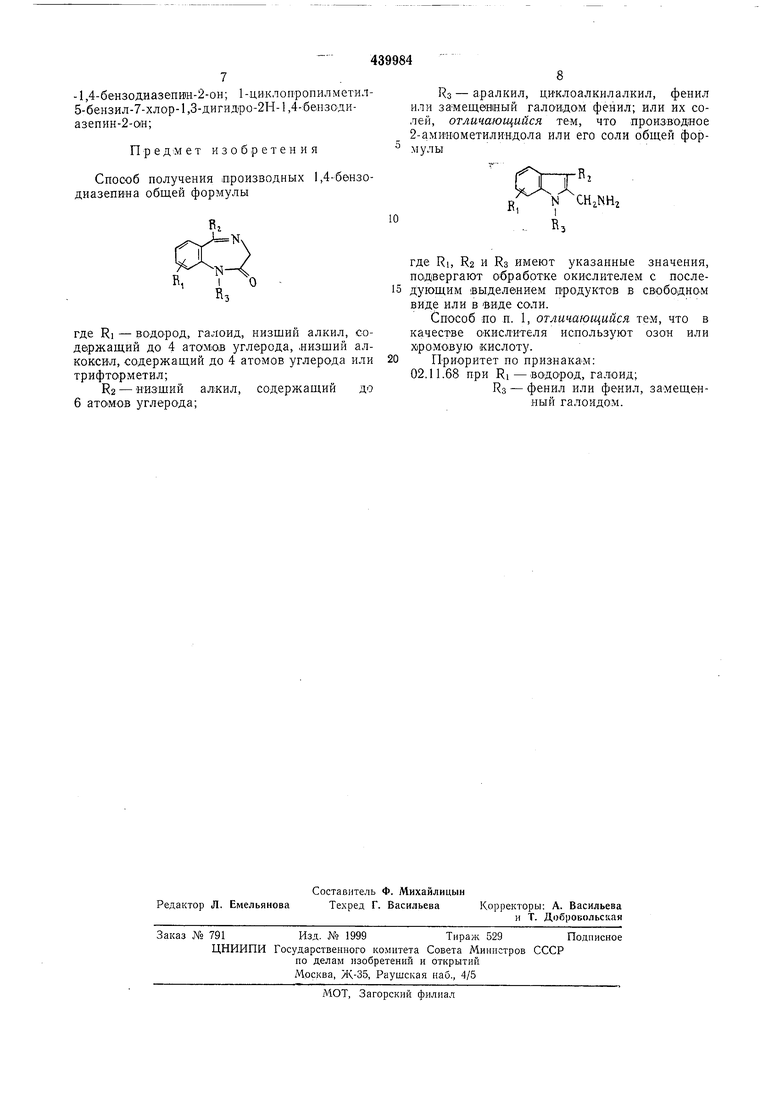
Предлагаемый, основаиный «а из.вестной реакции спо:соб получения произведных 1,4-бензодиазепина общей формулы
2 В.
.
R
3
где RI-1водор,од, галоид, «изший ал.кил, содержащий до 4 атомов углерода, низший алкоксйл, содержащий до 4 атомов углерода или трифторметил;
Ra -НИЗШИЙ алкил, содержащий до 6 атомов углерода;
RS - аралжил, циклоалкилалкил, фенил или замещенный галоидом фенил; или их солей, заклк чается .в том, что производное 2-аМИНометилиндола или его соли общей формулы П
R.
,
и
П)I
R,
где RI, Rz и RS имеют указанные значения, по.двергают взаимодейстВИЮ с окислителем.
В качестве окислителей для окисления соединений общей формулы П для получения целевых соединений общей фо.рмулы 1 можно
использовать озон, перекись водорода, надкислоты (например надмуравьиную, надуксуоную, надбеязойиую), хромовую 1 ислоту, перманганат калия, двуокись марганца, но этим список окислителей не ограничивается. Реа1КЦ:ия идет, как травило, легко при комнатной температуре, но адожяо вести процесс и при более высокой или более низкой температуре в заеисимоСти от задаедых условий регулирования реакции. Окислителем служит предпочтительно хромовая кислота иди озоад. Реакцию целесообразно вести в среде ipacTBoрителя. Выбор .растворителя зависит от используемого окислителя, его выбирают из группы, включающей воду, :ацетон, четыреххлористый углерод, уксуспую кислоту, серную кислоту и т. п. Окислитель берут в стехиометрическом количестве ил-и больше. Температура реакции зависит от взятого окислителя. При окислении хромовой кислотой в присутствии уксусной кислоты хромовую кислоту мож.но загрузить в трехкратиом размере против зквимолярного, а реакцию - вести при коМНатной температуре. Производное 2-а Минометилиндола формулы П растворяют или суспендируют в растворителе, затем в раствор или суспензию ЕнсУСят окислитель при перемешивании. Как правило, реакция закапчивается за 24 часа. При окислении озоном реакцию целесообразно вести при комнатной температуре. Производ ное 2-амйнометили«дола растворяют или суспендируют в растворителе, например в .муравьиной или уксусной кислоте, четы-реххлористом углероде или т. п., и через раствор или суспензию бар-ботируют озон при перемешивании. Целевое производное бензодиазепИНа можно выделить из реакционной смеси в Беочиш,енном виде путем экстрагировапия, с предварительной нейтрализацией или без нее и упариванием досуха. Продукт, если нужно, очиш,ают перекристаллизацией из соответствующего растворителя, например из этанола, изопропанола и т. п. известным способам. Полученное упомянутым способом производное 1,4-бензодиазепина можно также выделить в форме аддитивной соли обработкой кислотой, нащрилгер соЛЯ1НОЙ, серной, азотной, фосфорной или хромовой, или органической кислотой, например малеиновой, фумаровой, янтарной, муравьиной или уксусной.
Пример 1. В раствор 1 г этилового эфира о-хлор-3-1метилиндол-2-карбоно1Вой кислоты в 15 мл бромбепзола вносят 1 г безводного поташа и 0,1 г бромистой меди. Смесь нагревают с обратным холодильником 3,5 часа. Реакционную смесь охлаждают, отфильтровывают, остаток промывают бензолом. Объединяют фильтрат с промывной х идкостью, растворитель отгоняют под вакуумом. Остаток растворяют в 10%-ном этанольном растворе едкого кали и оставляют на ночь при комнатной температуре. Смесь разбавляют водой и промывают хлороформом. Водной слой подкисляют соляной кислотой. Осадок
отфильтровывают, получают 0,8 г 5-хлор-3-Метил-1-фенилиндол-2-КарбоноБОЙ кислоты. При
перекристаллизации из смеси эфира с бензо.лом получают бесцветные кубические кристаллы с т. пл. 241-242°С.
Используемый в качестве исходного материала этиловый эфир 5-хлор-3-1метилиндоЛ-2-карбоновой кислоты получают следующим образом.
Смесь 31 г /г-хлоранилина, 60 мл концентрированной соляной «ислоты и 60 мл воды нагревают, затем охлаждают до 0°. В смесь приливают по каплям при перемещивапии 18,2 г нитрита натрия в 45 мл воды при
3-5°С. Полученную смесь приливают в охлаждаемую смесь 31,6 г этилового эфира и этил а lieTO уксусной кислоты, 82 г ацетата натрия и 70%-го водного этанола при перемешивании и охлаждении. Перемешивание продолжают 4 часа при О--5°. Реакционную смесь экстрагируют эфиром. Эфирные экстракты объединяют и сушат над безводным сульфатом натрия, растворитель отгоняюг. Остаток нагревают с 20%-ным этанольным
pacTBiopoM серной кислоты с обратным холодильником в течение 5 час. Охлаждают, осадок отфильтровывают, промывают последовательно водой и небольшим количеством петролейного эфира, сушат, получают этиловый эфир 5-хлОр-3-метилиндол-2-карбоновой кислоты. При- перекристаллизации из бензола получают бесцветные иглы с т. пл. 162-163°С.
Пример 2. Смесь 1 г этилового эфира
5-хлор-3-метили1Ндол-2-карбоновой кислоты, 15 .мл бром беазола, 1 г безводного поташа и 0,1 г бромистой меди нагревают 3,5 часа с обратным холодильником. Реакционную смесь охлаждают, отфильтровывают, остаток промывают бензолом. Фильтрат объединяют с нромыдаюй укидкостью, растворитель отгоняют под вакуумом. Остаток растворяют в хЛорофор;ме и хроматографцруют на силикагеле, элюируют хлороформом и получагот
0,9 г этилового эфира 5-хлор-3-метил-1-фенилиндол-2-карбоновой кислоты в виде масла соломенно-желтого цвета, с характерным для карбонила (сложный эфир) поглощением црп 1710 см , но без полосы поглощения, характерной для водорода.
Этот маслянистый продукт растворяют Б 10%-ном этаноль1ном растворе едкого (кали и оставляют на ночь при комнатной температуре. Смесь разбавляют водой и промывают
хлороформом. Водный слой подкисляют соляной кислотой. Выпавший осадок отфильтровывают и получают 5-хлор-3-метил-1-фенили1Ндол-2-карбоноБую кислоту, которую перекристаллизовывают из смеси эфира с бензолом, т. пл. 241-242°С.
Аналогичным образом, пр,и замене соответг ствующих реагентов в примерах 1 и 2 получают следующие соединения: этиловый эфир 1-циклопропилметил-3-бензил-5-хлориндол-2-карбоновой кислоты; метиловый эфир 1-фенил-3-метил-5-хлори:ндол-2-карбоновон кислоты; 1-циклопрОПилмет1 л-3-бе1 зил-5-хлорИНдол-2-карбоновая кислота, 1-(/г-хлорфенил)-3-.метил-5-хлори ндол-2-карбоновая кислота, 1- (о-фторфенил)-3-метил-5-хлориндол-2-карбоиовая кислота.
Пример 3. Смесь 1 г 5-хлор-3-метил-1-фенили1Ндол-2-карбонОВой кислоты и 14 г тионилхлорида нагревают в течение 1 часа с обрат1ным холодильником. Избыток тионилхлорида отгоняют под вакуумом. Остаток обрабатывают 20 мл безводного бензола, растворитель отгоняют под вакуумом. Остаток растворяют в 70 мл безводного эфира и охлаждают до 0°С. В охлажденный раствор пропускают газообразный аммиак при перемешивании в течение 30 мин (охлаждение льдом). Смесь продолжают перемешивать 1-2 часа при комнатной температуре и упаривают до объема около 30 мл. После охлаждения отфильтровывают выпавший осадок, промывают водой, сушат и получают 0,9 г а;мида 5-хлс1р-3-метил-1-фенили1Ндол-2-карбоиовой кислоты в виде твердого белого веш,ества. При перекристаллизации из смеси метабола с ацетоном получают бесцветные иглы с т. пл. 245-247°С.
Аналогично описавиому в примере 3 получают следуюш,ие соединения: амид 1-фенил-3-метил-5-хлориндол-2-карбановой кислоты; амид-1-фенил-3-метил-5-хлориндол-2-карбоиовой кислоты; амид-Ьфсиил-З-пропил-б-хлориндол-2-|Карбоновой кислоты; амид-1- (о-фторфенил) -3-метил-5-хлорИ Ндол-2-«арбоновой кислоты; амид-1-(/г-хлорфенил)-3-метил-5-хл01риндол-2-карбо110вой кислоты; 1-фенил-3-метил-5-хлориндол-2-гидроксамовая кислота.
Пример 4. В суспензию 1 г амида 1-фенил-3-л1етил-5-хлориндол-2-карбоновой кислоты в 50 мл безводного эфира вносят 1 г литийалюминийгидрида. Смесь нагревают 8 час с обратным холодильником. В смесь по каплям приливают влажный эфир. Эфирный слой декантируют и сушат «ад безводным сульфатом «атрия, эфир ОТГО.НЯЮТ. Маслянистый остаток кристаллизуют обработкой соля«ой кислотой и получают гидрохлорид 1-фенил-2-ами1Нометил-3-:метил-5-хлориндола. При переК|ристаллизаци1и из смеси ад-етона с метанолом получают 0,6 г бесцветных игольчатых кристаллов, т. пл. 229°С.
Пример 5. Смесь 2 г амида 1-фенил-З-метил-5-хлориидол-2-карбо«овой кислоты и 10 мл хлорокиси фосфора .нагревают 2 часа с обратным холодильником. Реакционную смесь охлаждают, выливают в воду со и экстрагируют эфИ(ром. Эфириые экстракты объединяют, промывают насыщенным раствором хлористого натрия и сушат над безводным сульфатом натрия; растворитель отгоняют и получают 1-фенил-3-метил-5-Х1ЛОриндол-2-карбониррил. Инфракрасный спектр вещества показывает поглощение при 2220 , характердюе для нитрила.
Неочищаняый 1-фэнил-3-метил-5-хло,риндол-2-карбонитрил растворяют в 50 мл безводного эфира и в раствор вносят 2 г литийалюминийгидрида. Смесь нагревают 8 час с обратным холодильником. Реакционную смесь охлаждают, приливают по каплям эфир. Эфирный слой декандируют, сушат над
сульфато.м натрия, эфир отгоняют. Маслянистый остаток обрабатывают эта-нольным раствором хлористого водорода и получают хлоргидрат 1-фенил-2-аминометил-3-метил-5-хлориндола. При перекристаллизации из
смеси ацетона с метанолом получают бесцветные иглы с т. пл. 227-228° (при разложении).
Аналогично описанному в примерах 4 и 5 получают следующие соединения: 1-фенил-2-амино метил-З-метилиндол; 1-фе-нил-2-аМинометил-3-,метил-5-хлориндол; 1-фенил-2-аминометил-З-лропил-5-хлориндол;1- (о-фторфенил)-2-аминометил-3-,метил-5-хлориндол; 1 - (/г-хлорфенил)-2-ами«ометил-3-метил-5-хлориндол; 1-фенил-2-аминометил-3-,метил-5-броминдол; 1-циклопропилметил-2-а:мш1ометил-З-бензил-5-хлориндол и их хлоргидраты, гидробромиды и сульфаты.
ГЪример 6. В раствор 0,6 хлоргидратп
1-февил-2-амш1ометил-3-метил-5-хлориндола в 20 мл ледяной уксусной кислоты приливают 1 мл 30%-ной перекиси водорода и 0,5 мл 1%-ного водного раствора молибдата аммония. Смесь перемешивают при комнатной
температуре в течение ночи. Реакционную смесь подщелачивают водным раствором аммиака и экстрагируют метиленхлоридом. А етиленхлоридовый экстракт сушат над безводным сульфатом натрия и растворитель отгоняют под вакуумом. Остаток очищают хроматографически на силикагеле и получают 1-фенил-5-метил-7-хлор-1,3-дигидро-2П-1,4-бензодиазепин-2-он в виде масла, при обработке которого избытком хлористого водорода в эфире образуется хлоргидрат. При перекристаллизации из смеси Метанола с ацетоном получают бесцветные игольчатые кристаллы, т. пл. 202-204°С (разложение). Повторяют опыт примера 6, за исключением того, что гидрохлорид 1-фенил-2-аминометил-3-метил-5-хлори:ндола заменяют гидрохлоридом 1- (о-фторфенил) -2-аминОМетил-3-.метил-5-хлориндола. Получают 1-(о-фторфеиил) -5-1мет Ил-7-хлор-1,3-дигидро-2П-1,4-бензодиазепин-2-он.
Аналогично описанно.му в примере 6 получают следующие соединения: 1-феНИл-З-метил-1,3-дигидро-2Н-1,4-бензодиазепин-2-он; 1-фенил-5-этил-7-хлор-1,3-дигидро-2Н-1,4-бензодиазепип-2-он; 1-фенил-5-пропил-7-хлор-1,3-дигидро-2Н-1,4-бензодиазепин-2-он; 1-фвнил-5-метил-7-бром-1,3-дигидро-2Н-1,4-бензодиазепин-2-о.н; 1- (о-фторфенил) -5-метил-7-хлор-1,3-дигидро-2Н-1,4-бензодиазепин-2-он; 1 - (л-хлорфенил) -5-метил-7-хлор-1,3-дигидро-2Н-1,4-бензодиазепи1н-2-он; Ьциклопропилметил5-бензил-7-хлор-1,3-дигид|ро-2Н-1,4-:бензодиазепин-2-он;
изобретения
Способ получения производных 1,4-бензодиазепина общей формулы
где RI - водород, галоид, низший алкил, содержащий до 4 атомов углерода, .низщий алкоксИЛ, содержащий до 4 атомов углерода или трифторметил;
R2 - низший ал;кил, содержащий до 6 атомов углерода;
Ra - аралкил, циклоалкилалкил, фенил или замещенный гало-идоад феяил; или их солей, отличающийся тем, что производное 2-аминометили-ндола или его соли общей формулы
где RI, R2 и Ra имеют указанные значения, подвергают обработке окислителем с последующим выделением продуктов в свободно виде или в виде соли.
Способ по п. 1, отличающийся тем, что в качестве окислителя используют озон или Х1ромовую кислоту. Приоритет по признакаМ:
02.11.68 при RI-водород, галоид;
Ra - фенил или фенил, замещенный галоидом.


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных 1,4бензодиазепина или их солей | 1974 |
|
SU618042A3 |
| Способ получения соединенийиМидАзО (1,5-A)(1,4)диАзЕпиНАили иХ фАРМАцЕВТичЕСКи пРиМЕНи-МыХ СОлЕй | 1975 |
|
SU814278A3 |
| Способ получения 2-ациламинометил-1Н-2,3-дигидро-1,4-бензодиазепинов или их солей присоединения кислот | 1980 |
|
SU1253430A3 |
| Способ получения 2-ациламинометил-1 @ -2,3-дигидро-1,4-бензодиазепиновых соединений,а также их оптических изомеров и их солей кислотного присоединения | 1982 |
|
SU1245259A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 1970 |
|
SU453841A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА | 1971 |
|
SU436495A3 |
| АМИНОМЕТИЛХИНОЛОНЫ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2629111C2 |
| Способ получения производных бензодиазепина | 1970 |
|
SU497774A3 |
| ИМИДАЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2139873C1 |
| Способ получения производных бензодиазепина | 1971 |
|
SU498909A3 |
