(54) СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЦЕФ.АЛОС ПОРИНА
3
пособом. Получаемый при этом продукт при :еобходимости разделяют па оптические alпиодь: путем обработки огт}чески-активпо11 Р г а и IF; ос кой карбоновой или сул15фокис;10 ();;,
о ;л;ой как винная, дибсизллвипная, глутамиО;а;:, кам(5оарсульфок11слота, с последующей бработкой по.тучеыиой соли минеральным ,Л1 огранпческим оспованкем, таким как гидоокнсь калия или натрия, триэтиламин. И пользусмый п качест;1С неходкого соеднслня треонзомер формулы II может быть иоучсн следующим бразом, Амннокнслоту бн.ей формулы
R;NHCH2CH2COOI-I
подвергают 13зал1модейстиню с бснзиловы.м спнртом в присутствии кислого агеита ХН, где Ii имеет указанные значения, X - -анион галогена, сульфо- нли серной кислоть; и иолучалот СЛОЖНЫ эфнр (Ьорму.чы
©
R,NH ClbC}l2COOCH,C6ll,
110. J
yf
О
где Ас - ацильный остаток, который подвергают кислому алкоголизу.
Получаемый при этом соотяетствуюи1ий меркаптан подверггп./т взаимодейстзпю г енамкном формулы
Y-Cr
.OOR
где - ИИ31НИЙ алккл или ара.лкил, Yкмндо- нли ациламиногрунпа, в которой анил - остаток органнчес: ой иизиС) кярбонозой кислоты. При этом получа.от у-лактям 2A«R - оксикарбонил-я -1-мет ;л) - 5-ам1})юмстил-2,3-дигндро - КЗ-тиазин-i - карбо;и)иой к;1с,;огы (}10рм лы
который конденсируют с а:и:иловым или ар-алкиловым эфиром н,авелевой кислоты. Получают при зтом бензи.1овый эфир 2,3-дноксонярролидии-4 - карбопово KHc.ioTi)i ,i енольной форме o6Hi,eii формулы
COOOii CnHj;
но .1
который гндрогенолизом переводят в соответствующее 4-карбоксинронз1Юдное, которое подвергают амннометнлнро1 а1;ню в условиях реакцпн Манииха получают соответствующпй 2,3-дпоксо-4- (R, 1) -амииометилп 1рр()лиди 1, в которо.м R, R-алкил илн аралкнл ,11лп вместе группа - XRR образует г етероциклический остаток. В получепном соедииеннн замео 1,ают известны П1 прнема: п; ;;.миногруппу на тнольную и получают 2,3-дноксо 4-ацилтиометилпирролидии обндей формулв
Y- tlC R О О О,
.
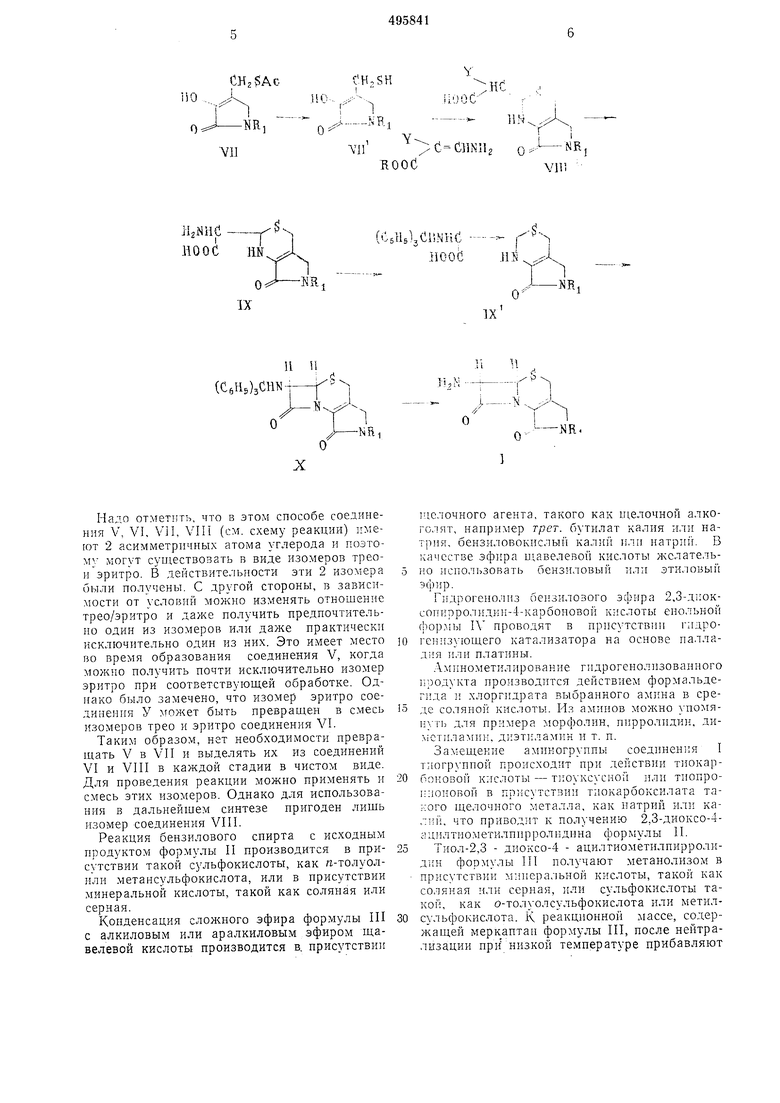
где R, RI и Y имеют указаннв:е значения, ко.горый обрабат1з1ва от гидразином в х ювиях кнс,:о1о гидролиза илн в с,И) гидрогеполиза, получая еоотвстств 01ций -;-. 1:- (R-oкc; кapбoнил -1.3-тиазин-4 - клрбоново .к;ть, лО)орв)й обрабатывают KHCHIJIM аген :о.м. При этом получают у-лактам 2-(7,-карбо ;чс;|-а-амм11омет|;л )-о - аммпометпл-2,о - )()- i ,3-71П13ин-4-карб(;;1О1из11 кпслс ты п подвергакуг ei о ipnTiKiHpOiiaHHio хлорнст1) |р; тплом 3 прпсхтствии oci:.OBHOi(; агента, HanpjiMep i i)ii-,i}i;iaMiiHa., получая треои.зомер 2- (а-|;арбо1чси-с - три гпла ,i ;ii«MeTi:;i) -5-aмnпo eтllЛ2,o-;i,пгилpo-1 ,.i - - ca|)6oHoBoii 1..:сло1-В общей cijopAiy;iiJ Ji. П;1же приведена обн(ая схе.ма синтеза.






IV
Н
VI
СНг $АС
0. 0
Л/и
Y
НС
J, .J 4J
г
Yлп c criNii2
ROOCY ;
iijNHCj N
НО О С т о
IX
11 И
(C6H5)3CHNНадо отметить, что в этом способе соединения V, VI, VII, VIII (см. схему реакции) имеют 2 асимметричных атома углерода и поэтому могут существовать в виде изомеров треон эритро. В действительности эти 2 изомера были получены. С другой стороны, в зависимости от условий можно изменять отношение трео/эритро и даже получить предпочтительно один из изомеров или даже практически исключительно один из иих. Это имеет место во время образования соединения V, когда можно получить почти исключительно изомер эритро при соответствующей обработке. Однако было замечено, что изомер эритро соединения У может быть превращен в смесь изомеров трео и эритро соединения VI.
Таким образом, нет необходимости превращать V в VII и выделять их из соединений VI и VIII в каждой стадии в чистом виде. Для проведения реакции можно применять и смесь этих изомеров. Однако для использования в дальнейшем синтезе пригоден лишь изомер соединения VIII.
Реакция бензилового спирта с исходным продуктом формулы II производится в присутствии такой сульфокислоты, как п-толуолили метансульфокислота, или в присутствии минеральной кислоты, такой как соляная или серная.
Конденсация сложного эфира формулы III с алкиловым или аралкиловым эфиром щавелевой кислоты производится в. присутствии
.
J-NR,
IX
l м
тьн----г
1
о
.
щелочного агента, такого как щелочной алкоголят, например трет, бутилат калия или натрия, бензиловокислый калий или натрий. В качестве эфира щавелевой кислоты лчслательно использовать бензиловый или этиловый эфир.
Гидрогенолиз беизилозого эфира 2,3-.Д|Юксопирролидии-4-карбоновой кислоты енольной фор.мы IV проводят в присутствии гпдрогекизующего катализатора на основе паллаД1 я платины.
.минометилирование гидрогенолизованного продукта производится действием формальдеrn.:ia I хлоргидрата выбранного амина в среде соляной кислоты. Из аминов можно лпомяиуть для примера морфолин, пирролидин, димстиламин, диэтиламин и т. п.
Замещение аминогруппы соединения I тиогрупной происходит при действии тиокарбоковой кислоты - тиоуксусной или тиопрокпоноБОЙ в пр11сутстБии тиокарбоксилата такого щелочиого металла, как натрий или калий, что приводит к получению 2,3-диоксо-4а цнлтиометилпирроледина формулы 11.
Тиол-2,3 - диоксо-4 - ацилтиометилпирролидин формулы 1П получают метанолизом в присутствии мииера.тьной кислоты, такой как соляная или серная, или сульфокислоты такой, как о-толуолсульфокислота или метилсульфокислота. К реакционной лгассе, co.:tepжащей меркаптан формулы III, после нейтрализации при низкой температуре прибавляют
7
еиамин формулы IV, - еналгин фталимида малональдегида трет, бутила. Раствор упаривают в вакууме досуха ; полл чаемый остаток растворяют в сухом сензолс, нагревают с обратным холодильником или в случае необходимости азеотропной отгонкой образующейся воды в течение 12 ч, и получают, таким образом тиазин формулы V, который в этих условиях иолучается главным образом в виде изомера эритро.
Удаляют фталимидную группу у-лик.та.жг, 2-(rz-R-оксикарбонил-а - фталилгидо) - -лгетил5-аминометил-2,3-дигидро-1,3 - тиазин-4 - карбоновой кислоты форм} лы V при номогци гидрогенолиза в присутствии огранического растворителя типа двузамещенпого амида, как например диметилфор: 1амида, или циклического эфира, как например диоксапа.
Омыление карбоксильной группы у-лзктама 2-(a-R-оксикарбонил-а - амннометил)-5 - аминометил-2,3-дигидро-1,3-тиазин-4 - карбоновой кислоты формулы VI нроводят минеральной или органической кислотой, например соляной, бромистоводородной, я-толуолсульфокислотой, смесью бромистоводородной и уксусной кислот, или трифторуксусной кислотой в безводном ограническом растворителе, как бензол, толуол, эфир, диоксан, нитрометан, или полигалоидпый углеводород, как например хлороформ или хлористый метилен.
Тритилирование улзктама 2-(«-карбоксиа-аминометил)-5 - аминометил-2,3 - дигидро1,3-тиазин-4-карбоновой кислоты формулы VII проводят действием хлористого тритила в присутствии щелочного агента, как например триэтиламина.
Превращение формулы эритро у-лактама 2(сс-карбокси-сс - тритиламинометил)-5 - аминометил-2,3 - дигидро-1,3-тиазин-4 - карбоновой кислоты формулы VIII в форму трео проводят обработкой щелочного агента, как гидроокись щелочного металла, например, гидроокись натрия или лития, в среде снирта как метанол или этанол.
Пример 1. Приготовление у-л ктама 6Н, 7Н-бг с-7-амиио-3 - аминометил - цеф-3 - ий-4карбоновой кислоты
Фаза А. rt-Толуолсульфонат бензилового эфира аланина.
В аппарате, снабженно:. устройством для отделения воды образовавщейся во время реакции с помощью азеотропного растворителя, патревают с обратным холодильником в течение 5 ч следующую смесь:
(3-Аланин, г 89
Моногидрат п-толуолсульфокислоты, г210
Бензнловый спирт, мл450
Четыреххлористый углерод, мл500
В течение этого времени отделяется около 45 мл воды. Затем реакционную массу упаривают в вакууме, охлаждают и образовавщийся продукт перекристаллизовывают из эфира. Замораживают, фильтруют под вакуумом, сушат и собирают 350 г (количественным выход) кристаллов с.т. пл. 142°С.
По,л Чеиный продукт идентичен с тем, который был описан раиее.
Ф а 3 а Б. Бензиловый эфир 2,3-диоксопирролидин-4-карбоновой кислоты.
Добавляют 225 г трет, бутилата калия в 900 с.М безводного бензола прибавляют 500 см- бензилового спирта, охлаждают смесь на бане из льда-метаиола, и при температуре не выше 30°С добавляют 351 г и-толуолсзльфоната бензилового эфира р-аланина.
Растворяют 300 г бензилового эфира щавелевой кислоты в 600 СМ горячего бензола, оставляют охладиться до комнатной температуры и слабокислый раствор нейтрализуют добавлением 0,4 см триэтиламина. Этот раствор прибавляют к пол -ченной и выдержанной на охлаждающей бане смеси. Нагревают до комнатной температуры, а затем нагревают с обратным холодильником в течение 5 ч.
Отгоняют бензол в вакууме, нрибавляют последовательно сначала 2 л воды, содержащей 15 см уксусной кислоты, затем 1,5 л изопропилового эфира и, наконец, ПО см концентрированной соляной кислоты (до получения рН 1). Полученную массу охлаждают при неремещивании в течение 2,5 ч отсасывают, промывают водой, изопроии.човым эфиром и нерекристаллизовывают, растворяя в диметилформамиде и осаждая в воде. Получают 130,5 г (т. е. 56%) продукта с т. нл. 186°С, который растворим в спиртах, эфире и ацетоне, нерастворим в бензоле и воде. Найдено, %: С 62; Н 5,1; N 6,3
Ci2HnO4N (233,24)
Вычислено, %: С 61,8; Н 4,76; N 6,01
Р1нфракрасный спектр:
Два максимума в области карбонила 1729 и 1693 см-.
Поглощение в области обт единенных OH/NH.
Присутствие монозамещенного ароматического соединения. Это соединение в литературе не описано.
Ф а 3 а В: 2,3-Диоксонирролидин-4-карбоновая кислота.
а)Приготовление катализатора гидрогенизации.
В атмосфере водорода перемешивают 0,8 г суспензии активированного угля в 4 см 2%пого водного раствора хлористого палладия.
После насыпдения катализатора, его фильтруют без достуна воздуха и ополаскивают
несколько раз безводным диметилформамидом.
б)Гидрогенизация.
Растворяют 9,32 г бензилового эфира 2,3диоксопирролидин 4-карбоновой кислоты в
50 см безводного диметилформамида и прибавляют приготовленный катализатор. Смееь помещают в атмосферу водорода, затем время от времени перемещивают при охлаждении, чтобы избежать какого-либо заметного повышения температуры. Фильтруют, прибавляют
к фильтрату 500 см- изопропилового эфира, фильтруют под разрежением и сушат. Получают4,618 г (т. е. 96%) продукта, который используют без очистки далее в синтезе. Для анализа их заново растворяют в 6 объемах диметилсульфоксида и 4 объемах метанола. Фильтруют и прибавляют 4 объема метанола. Образуется белый осадок, который отсасывают и сушат. Выход после очистки 60%.
Получаемый в виде белых кристаллов продукт мало устойчив из-за декарбоксилирования. Он растворим в диметилсульфоксиде и диметилформамиде, нерастворим в изопропиловом эфире и в воде.
Инфракрасный спектр (в вазелиновом масле). Поглощение в области объединенных OH/NH.
Сложное и сильное поглощение в области карбоксила: выступ 1708 см- макс. 1677 .
Пайдено, %: С 41,7; Н 3,8; N 9,9
C5H504N
Вычислено,%: С 41,96; Н 3,52; N 9,79;
Это соединение в литературе не описано.
Фаза Г. Хлоргидрат 2,3-диоксо-4-морфолинометилпирролидина.
Прибавляют 2 капли соляной кислоты к 10 с.м- раствора хлоргидрата морфолина, приготовленного нейтрализацией 8,71 г морфолина концентрированной соляной кислотой и прибавке 50 см воды. Прибавляют 2 см формальдегида (30%-го), затем добавляют 2,83 г 2,3-диоксопирролидин-4-карбоновой кислоты. Реакционную смесь нагревают до 60-65°С при перемешивании в течение 30 ч. Выпаривают досуха и перекристаллизовывают остаток в этаноле. Получают 2,986 г продукта, который может быть использован прямо для продолжения синтеза.
Для анализа растворяют продукт в одном объеме горячей воды и прибавляют три объема этанола. Замораживают, фильтруют нод разрен ением и получают продукт с выходом 80%, в виде белых кристаллов, малорастворимых в этаноле и эфире и растворимых в воде.
Найдено, %: С 45,8; Н 6,4; N 11,8; С1 15,2.
CgHisOsNaCl: 234,7
Вычислено, %: С 46,06; П 6,44; N 11,94; С1 15,11;
Пнф|ракрасный спектр (в вазелиновом масле).
Полосы поглощения при 3210 и от 3.6-4,1 мкм.
Триплет в области карбонила 1 711 см1 691 см-1 1 664 см-1
Это соединение в литературе не описано.
Хлоргидрат 2,3-диоксо-4-морфолинометилпирролидина может быть получен, исходя из бензилового эфира 2,3-диоксопирролидин-4карбоновой кислоты без выделения промел уточной свободной кислоты следующим образом.
Добавляют 30,33 г бензилового эфира 2,3диоксопирролидин-4-карбоновой кислоты Б
300 см диоксана с 10% воды, немного нагревают чтобы растворить продукт, прибавляют 3 г активированного угля и 1 см 20%-ного водного раствора хлористого палладия, помещают в атмосферу водорода и перемешивают очень энергично. Через 1 ч 40 мин поглощается 2700 см водорода (объем, рассчитанный теоретически, равен 2912 см). Охлаждают, продувают азотом и затем добавляют 130 смсмеси следующего состава, мл:
Морфолин, г43,5
Вода100
Концентрированная соляная кислота40
Соляная кислота15
Формальдегид50
ВодаДо 500
Реакционную смесь нагревают до 50°С и собирают за 1 ч 2325 см углекислого газа. Теоретически количество выделенного углекислого газа равно количеству водорода, первоначально поглощенному в реакции. Следовательно, в лучшем случае надо здесь ож.идать выделение 2700 см- углекислого газа. В течение нескольких минут легко перемешивают, а
затем фильтруют и выпаривают досуха в ваку; -л1е. Остаток может быть использован без
очистки для продолжения синтеза.
Фаза Д. 2,3-Диоксо-4-ацетилтиометилцирролидик.
В сосуд, охлаждаемый иа бане со льдом п метанолом, вводят 140 см воды. 24 г первичного кислого фосфорнокислого натрия, 60 см т:10}-ксусной кислоты и 67,2 г кислого углеклслого натрия, перемещивают в течение 5 мин, а затем прибавляют 46,8 г хлоргидрата 2,3-дноксо-4-морфолинометилпирролидина. Реакционную смесь перемешивают при комнатной температуре в течение 3,5ч, прибавляя
через полчаса небольшие количества эфира для уничтожения образующей иены. Затем добавлением 80 см концентрированной соляной кислоты подкисляют до рП 1, отгоняют в вакууме избыточную тибуксусную кислоту,
фильтруют и сохраняют 4зильтрат. Отфильтрованный на воронке Бюхнера продукт промыпают водой, растворяют в 150 см горячего хлороформа, отделивщуюся воду декантируют и водный слой экстрагируют хлороформом. Соединенные органические вытяжки суHiaT над сернокислым натрием и выпаривают дослха в вакууме. Остаток растирают в эфире до тестообразного состояния и получают 25 г (67%) продукта с т. пл. 136°С.
Полученный фильтрат экстрагируют хлороформом, сушат над сернокислым натрием и выпаривают досуха в вакууме. Остаток растворяют в смеси 10 см этилового эфира уксусной кислоты и 10 см эфира. Оставляют иа
ночь в холодильнике, фильтруют под разрежением и получают 2,5 г продукта, идентичного нолученному при первом выделенин. Общий выход 72,5%. Продукт можно использовать без дополнительной ачистки для продолжения синтеза.
11
Д;;Я анализа продукт перскристаллизовывают в этпловом эфире уксусной кислоты до постоянной температуры плавления. Он получается, в виде белых кристаллов, растворимых 3 поде и малорастворимых г; эфире и этиловом эфире уксусной кислоты.
Найдено, %: С 44,7; Н 4,9; N 7,6; S 17,1.
CjHgOsNS (187,21)
Вычислено, %: С 44,91; Н 4,85; N 7,49; S 17,13.
Ультрафиолетовый спектр а) в этаноле - 0.1 Н. соляной кислоте. Максимум 225 ммкм E/L : 666
б) 3 этаноле - 0,1 и. едком натре.
Максимум 251 м.мкм Ej°,, : 564
Инфракрасный спектр (хлороформе). Поглощение карбонила - макс. 1689 NH тонкая полоса при 3 460 . Он макс, у 3210 см-.
Фаза Е: у-Лактау 2- (сс-трет. бутоксикарбопил-а-фталимидометил) -5 - аминометил-2,3дигидро-1,3-тиазин-4-карбоновой кислоты.
Растворяют 18,72 г 1,3-диоксо-4-адетилтиометил- 5Н -нирролидина в 300 см 5%-него метанольного раствора г-толуолсульфокислоты и нагревают с обратным холодильником в тс еиие 2,5 ч. Затем образовавшейся смеси дают остыть до комнатной температуры, ох лаждают до -50° и прикапывают 39,5 см2 Н. раствора уксуснокислого а.ммония в метаноле. Затем в OTMoccjiepe азота прибан/итют 30 г анаминафтолимндомалоиальдсгида грет. бутила и оставляют при перемешивании ;; течение нескольких минут при комнатной температуре. Растворители выпаривают в вакууме перегонкой, а остаток растворяют в сухом бензоле п 11агревают с обратным холодильником в течение 12 ч с отделением воды. Остаток, который состоит главным образом из изомера эритро, растворяют в 200 см метанола и выпавшие кристаллы отфильтровывают в вакууме. Очиндают последозательпым кони.еитрированпем в воде, метаиоле, эфире, растзорением в диметилформамиде и добавлением метанола.
Получают 16,6 г (40%) продукта с т. пл. 250° в в::дс белых кристаллов, растворимых в смеси метанол-хлороформ, малорастворимых в чистом метаноле и нерастворимых в бензоле и эфире.
Найдено, %; С 57,7; Н 5,4; N 9,9; S 7,9.
C.oHoiOsNaS (415.47)
Вычислено, %: С 57,8; Н 5,10. M0.11; S 7,72.
Фаза Ж: Хлоргидрат у-лактама 2-(а,-трет. бутоксикарбонил-а-аминометил)-5 - аминометил-2,3-дигидро-1,3-тиазин-4-карбоновой кислоты.
Вводят 16,6 г у-лактама 2-(а-трет. бутоксикарбонил-а-фталимидометил)-5 - аминометил2,3-дигидро-1,3-тиазин-4-карбоновой кислоты ;з 32 см днметилформамида. затем очень медленно, при перемешивании в атмосфере азота прибавляют 22 с:,г 2 и. раствора гидразингид12
рата в диметилфорламиде и перемеш1-шают в течение 30 мин при комнатной температуре. Затем прибавляют в течение 30 мин 44 см соляной кислоты, перемешивают в течение ЗЭ мин, затем замораживают п с|)ильтруют. Фл.ллрат шлпаривают досуха и , а остаток, растворенный в 30 см воды, обрабатывают животиы.м углем. Фильтруют, выпаривают досуха и кристаллизуют в .метаноле нолученный продукт. Промывают эфиром и получают 12,5 г (97%) нродукта, который, нес.мотря на то, что он состоит из смеси изомеров трео и эритро, может быть использован в таком виде для продолжения синтеза.
В случае необходимости компоненты смеси можно разделить дробной кристяиплизацией из смеси метанола с 20% воды, в котором нзомер э|)итро менее раетвори.м. Оба clepeoизо.мерных хлоргидрата, трео- и эритро-, едва
могут бьпъ различи.мы по их пн(1)ракрасным или ультрафиолетовым спектральным характеристикам, но 3 хроматографии в тонком слое форма эритро оказывается наиболее подвижной (основа Кизельгель gF 254 3/10 мм.
растворитель для элюирования смесь; этиловый эфир укс С110Й кислоты 60%. этанол 20%, пода 20%).
При этом из каждого чистого стереоизомерного хлоргидрата (или их смо.си) нри )ас-творении в маленьком количестве водного раствора кислого углекислого натрия по.гучают свободное основание (или смесь свободных ocHOi;a)), которое .можно экстрагировать
ЭТПЛОВЫ.М эфиром уксусной КИС.ГГ)ТЫ.
Фаза 3: iJ-Лактам 2-(а-карбокси-а-тритиламино)-метил-5-аминометил-2,3 - дигидро1.3-тиазин-4-карбоновой кислоты.
12,88 г смеси изомеров трео и эритро, соотБетствуюш;ей у-лактаму 2-{а-трсг. б токси -.:-.рбонилхлоргидратаминометила) - 5-а.миномет; л-2,3-дигидро-1,3 - тиазин-4 - карбоновой
к;;слоты, добавляют в 320 см нитрометана, который насышен газообразной соляной кислотой и охлажден смесью льда с метанолом. В образовавшуюся суспензию пропускают газообразную соляную кислоту в течение 50 мин,
а затем отгоняют соляную кислоту в закуу те. Это соединение в литературе не описано.
Полученную смесь помеш;ают в .Т.мосферу азота, охлаждают на ледяной бане и вводят 28 см триэтиламина и 24 г хлористого тритила в ВО см хлористого метилена. Оставляют стоять в атмосфере азота при комнатной температуре на ночь, и затем выпаривают досуха п вакууме. Остаток растворяют в смеси 200 метанола и 200 см хлористого метилена и прибавляют 16 см уксусной кислоты. Кои-центрируют до иолуобъема, перемешивают в течение получаса при комнатной температуре, фильтруют под разрежением, сушат я
собирают 7,161 г (38%) тритилироза шого
продукта, практически в чистой форме трео.
Это соединение не описано в литературе.
и выпаривают досуха в вакууме на водяной бане. Оставшееся масло растворяют в 200 см эфира, прикапывают 20 см воды и перемешивают в атмосфере азота в течение 4 ч при комнатной температуре. Фильтруют под разрежением, промывают эфиром, затем водой и получают 8,368 г тритилированного продукта (форма эритро, содержаш,ая немного изомеpa трео). Это соединение в литературе не описано.
Приводят в суспендированное состояние 8,368 г тритилированного производного- (форма эритро) в 170 см метанола, охлаждают до 10°С, нрибавляют 11,8 см водного гидрата окиси лития 3,4 н. и выдерживают в течение 3 мин при комнатной температуре. Затем прибавляют около 2,5 см уксусной кислоты до получения слегко кислого рН, нагревают на бане при 60°С в течение 10 мин, фильтруют под разрежением, промывают метанолом и собирают 4,728 г тритилированного произподного (форма трео).
Маточный раствор обрабатывают активированным углем; нерастворимую часть промывают метанолом, прибавляют к фильтрату и выпаривают досуха в вакууме. Остаток растворяют в 10 см эфира, прибавляют 1 см уксусной кислоты и I см воды. Оставляют на 2 ч при комнатной температуре, фильтруют под разрежением и получают 1,673 г тритилиропанного производного (форма эритро), которое изомеризируют, как описано выше, для получения, таким образом, 0,887 г производного формы трео.
Разные количества продукта формы трео соединяют и получают 12,77 г; прибавляют 10 см- метанола и нагревают с обратным холодильннко.м. Фильтруют, сун1ат после промывания эфиром и получают 12,13 г (95%) продукга, который может быт|, использован для продолжения синтеза.
Получают продукт в виде бесцветных кристаллов в т. пл. (разл). 240°С, изомер эритро плавится с разложением при - 220°С.
Он растворим в водном снирте, малорастворпм в диметилформамиде и диметилсульфоксиде, нерастворим в органических гидрофобных раство Я телях.
Пайдено, %: С 69; N 8,6; S 7,1; Н 5,3.
CavHssNsOsS (471,55)
Вычислено, %: С 68,77; N 8,91; S 6,80; П 5,35.
Фаза И; -Лактам DL-6H, 7Н-цис-7-трнтиламино-3-аминометил цефалоспорановой кислоты.
Приводя.- к суспендированное состояние ;4,1Б г |-ллктама 2-(а-карбоксн-я-тритиламино) метил-5-аминометил-2,3 - дигндро-1,3-тиазин-4-карбоновой кислоты в виде грео-изомера в 140 см безводного пиридина, помещенного в атмосферу азота. Прибавляют 0.,2 г днциклогексилкарбодиимида, перемешивают в течение 5 мин, li добавляют 300 смбеззодиого хлористого лгстнлена. а затем 300 см безводного нитрометана. Полученную белую суспензию оставляют при комнатной температуре, защищая от света в течение 65ч; дициклогексилмочезину в виде кристаллов фильтруют под разрежением, кристаллы прополаскивают хлор:;стым метиленом, фильтрат и промывные воды концентрируют под вакуумом до 1/4 объема, обрабатывают животным углем, фильтруют и выпаривают досуха в вакууме. Остаток кристаллизуют растиранием в эфире, фильтруют под разрежением, а маточные растворы от промывания сохраняют.
Перекристаллпзозанный продукт растворяют в 30 см- этилового эфира уксусной кислоты, осаждают добавлением 2 см воды, разбавляют 35 см- эф1фа и фильтруют под разрожение.м 9,3 г бесцветных кристаллов, сольна гированиых с половиной молекулы воды и плавящихся при 200 (с разложением).
Добавлением воды к эфирному раствору, KOToiJbn нолучс зыше, выделяют еще 0,18 г 1;1к думта, который одинаков с полученным ii.; lIopвo i выоросе.
Полученный продукт может быть использован для продолжения синтеза.
Для анализа его кристаллизуют в метиловом эфире уксусной кислогы и получают безводную пробу с т. пл. 240°С (разложение) .
Лпалпз продукта, сольватированного с 1/2 молекулы воды.
Пяйдено, %: С 69,9; Н 5.5; X 9.1; S 0,8.
С,7П,з02Кз8-1/2Н2О (462,5)
Вычислено, %: С 70,16; П 5,23; N 9,08; S 6,93.
Инфракрасный снектр в хлороформе:
Свободный КП 3 440 + объед ненный Nn
Р-Лактам 1777
-у-лактам 1698 смС С1663 см-1
Ароматический
Ультрафиолетовый спектрз этаноле
Изгиб около 226 ммкмEi y:42t
254 MiK.iЕ{..: 25
259-260 ммк.мEj j,: 121
в этаноле с 0,1 i;. соляиоГ; кпс,1ото| ;
.юг, 1см- -р1. . 1 .-,С.; (,,,,. I.., . loq Cj ,.
этаноле с 0,1 н. едким натром
ММК.
Ic
