нитрозированием кторого получают оксииминовое соединение общей формулы обработкой которого агентом регенерация кетона из оксима получают диоксосоединение общей формулыкоторое подвергают взаимодействию с метилатом щзлочного металла,после вьщеляют целевсм продукт известными приемами. Соединение формулы I, в котором циклы Д и Я сочленены в Ч с-положении, т.е. водород в ггЬЛо се НИИ 3 и этильный радикал в положении 16 находятся г нс-положении один по отношению к лругому, соответствует dB- винкамину I а. Соединение формулы I, в котором циклы Д и Е сочленены в 7ас-положении, соответствует dl изовинкамину 16. Таким образом по этому способу получают (исходя и:з соединения II а, в котором атом водорода в положении 12 б и этильная группа в положении dt - винкамин, а исходя из цис - сочленены) соединения II б Н,СО,С
В котором атом водорода в положении 126 и этильная группа в положении 1 транс-сочленены, получают d 6-изовинкамин.
В дайной заявке производные индоло - (2, 3 а) - хкнолизииа, заместители которых 125 Н и 1 -этил находятся в положении цис один по отношений к другому, называются цис (2, 3 - а) хинолизин.
Аналогичным образом производные индоло- (2,3- а) - хинолизина, заместители которых 12 б Н,9 J - этил находятся в положении транс один по отношению к другому, иазьтаются тр ис-индоло - (2, 3 - а) 4ШНОЛИЗИН.
Производные збурнана или Е гомозбурнаиа с соедииением циклов Д (Е) - цис, назьшаются чис-збуриан или цис Е - гомозбуриаи.
Аналогичным образом производные збурнаиа или Е - гомозбурнаиа с соединением циклов ,Дf -транс, иазьшаются траяс-збурнан или транс - Е гомозбурнан.
Превращение диоксо соединения V в конечньш продукт I производят обработкой соединения V метилатом щелочного металла, например метилатом натрия или калия, при afoM превращают цикл Е - гомо в цикл Я с шестью цепями, имеющим в положении 14 желаемые функции ОН и СОзСНз со стереохимией соответствующей стереохимии естественного винкамина, в том случае, когда работают в серии Д (Ё) - цис, или со стереохимией соответствующей стереохимии изовинкамина в с,ггучае работы в серии Д (Е) - транс.
Образование в зтой фазе исключительно или преобладающе только одного изомера представляет большое преимущество, так как исчезает }ieoGходимость длинных и трудных очищений. Если обрабатьшать соединение V кислотным или (лелочиым агентом, образующим гидроксильные ноны ОН, например гидроокисью щелочного или щелочноземельного металла - едким кали или е,ч,кнм барием, то получают соединение общей фо1) VI Превращение четырехциклического соединения II в лактам III производится преимущественно в присутствии сильного щелочного основания, например гидрида, амидметалла или алкоголята щелочного металла, причем употребляют предпочтительно третичный алкоголят щелочного металла - триамилат натрия. Следующая стадия, состоящая из превращения лактама III в гидроксии ми новое производное IV, состоит в том, что метиленовая группа в а положении лактамного карЗонила может быть нитрозирована, что является неожиданным, так как лактамы с трудом образуют енольную форму, Нитрозирование проводят преимущественно действием алкилнитрита (алкил содержит от 1 до 5 атомов углерода), и реакодю проводят в присутствии щелочного агента, В качестве алкил нитрита употребляют нитрит н-пропила, Нитрит бутила, нитрит трибутила или нитрит изоамила. В качестве щелочного агента употребляют сильное основание, как то гидрид или третичный алкоголят щелочного металла, гидрид натрия, трибутилат или триамилат натрия. В этих условиях получают, исходя из 14 оксоЕомозбурнана, 14- оксо - 15- гидроксиимино - гомоэбурнан серии Д (Е),- цис. Переход от гидроксииминного производного IV к соответствующему диоксосоединению V произво,дится классической регенерацией кетона из оксима. Для этого обрабатьтают соединение IV альдегидным или кетонным реагентом - бензойным альдегидом, формальдегидом, пировиноградной кислотой, глиоксиловой кислотой или левулиновой кислотой. Г акцию проводят предпочтительно в присутствии кислоты, например соляной или серной кислоты. Из 14 - оксо - 15 - гидроксиимино Е - гомозбурнана таким образом получают 14,15 - диоксо Е - гомозбурнан. чД-к. но в виде кислоты, которую затем превращают в сложный эфир обычным способом, например действием диазометана, для получения метилового эфива формулы I. V v -г г J, Переход от соединения V к соепииеш ю Ь Си осуществляют либо прямо, либо посредством сое- - - , динешм VI. Это превращение цикла с 7 цепями в цикл сбцепямипроисходитблагодаряразрывулактамовой связи N-, а затем образованию новой связи меж дуиндольным азотом и углеродом кетонной групп или же благодаря перегруппировке бензилового типа Применение способа изобретения к оптически деятельным соединениям позволяет получить оптически активные конечные продукты и в частности (+) - винкамин, идентичный с алкалоидом, вьзделенньш, например из Vinca minor L. Этот способ позволяет также получить (-) - винкамин, являющийся оптическим антиподом (+) - винкамина. Применение способа изобретения к энантиомеру.чис- 1, 2,3,4,6,7, 12, 126-октагидро- 1 - зтил 1 - карбометоксиэтидиндоло - (2, 3 - а) хинолизина (На) позволяет получить оптически активный винкамин, причем получают в зависимости от выбранного энантиомера либо (+) - винкамин, исходя из зкантиомера 126 а - Н 1а - этил, либо (-) - винкамин, исходя из этантиомера 2б&Н 1 р - зтии Эпймерьрчыс- н транс - 1, 2, 3, 4, 6, 7,12, 126«октагидро - 1 - зтил - 1 - карбоксиэтил - 4 Ч1КСОИНДОЛО - (2,3 - а) - хинолизина получаются омылением смеси эпимеров 1, 2, 3,4, 6,7, 12,12 б - октагидро -1 - этил - 1 - карбометоксиэтил 4 - оксоиндоло - (2, 3 - а) - хинолизина и могут быть отделены, например простой дробной кристаллизацией. Разложение 1 мс-изомера производят образованием соли с оптически активным основанием, ди астереоизомерные соли разделяют обьпсновенны ми способами, в частности дробной кристаллизацией, выделением каждого из этих двух оптнческих антиподов цис - 1,2,3, 4, 6, 7, 12, 126 (ястагидро - 1 - этил - 1 - карбоксиэтял - 4 оксоиндоло - (2, 3 - а) - хинолнзина кислотной обработкой соответствующей соли. Сравнение кривых кругового дихроизма двух полученных знантиомеров с кривыми кругового дихроизма (+) - винкамина естественного происхождения позволило опознать в этой стадии, энантиомер, который имеет ту же конфигурацию, что и (+) винкамин. Этот энантиомер (певовращающий в диметилформамнде) в процессе синтеза приводит к (+) - винкамину. Так как абсолютная конфигурация (+) - винкамина известна, то можно сделать вьшод, что этим левйвращающим изомером является 1, 2, 3, 4, 6, 7, ,12, 12 б - октагидро - 1а этил - 1 (3 - карбоксизтил -4- оксо- 126а- индоло (2, 3 - а) - хинолизин. Другим энаятмомером, правовращающим в диметилформамиде, является соответствуюшее 1 - этил - 12 0 - Н - производное, и оно привопит при продолжении синтеза к (-) - винкамину. Разделеиие изомеров транс- и «ис- и разпвоение изомера цис в ранией (. 1зе синтеза позволяет получить оптически активный винкамин. Для этого ., л превращают сягпгчески активный цис 1, 2, 3, 4, 6, - ,- Г1,,, 1 1 12, 126 - октападро- 1 - этил- 1 - карбоксиэтил .,, , ч -4 - оксоиндоло - (2, 3 - а) - хинолизин в оптически , 2, 3, 4, 6, 7, 12, 12 б - октагмдро .j . ., . i . карбометоксиэтилиндоло - (2. 3 - а)«хинолиэйн. Применение способа по изобретению к последнему соединению приводит к оптически активному винкамину. Предметом заявки также является способ получения (+) - винкамина или (-) - винкамина по вьщ1еука:шгаому способу омылением 1,2,3, 4, 6, 7, Д2, 126 - октагидро - 1 - этил - J - карбометокси отил - 4 - оксоиндоло (2, 3 а) хинолизина разъеданением изомеров цис- и транс - (2, 3 а) хино1шэкна, раздвоением цис - 1, 2, 3, 4, 6, 7, 12, 126 - сястапифо - 1 - этил - 1 - карбоксиэтил - 4 гоксоиндоло (2, 3 - а) - хинолизина на его оптические антш10Ш 1 при помощи оптического активного основания, зтерификацией знантиомера цисИ, 2, 3, 4, 6, 7, 12, 126 . октагилро - 1 - этил 1 1карб(жсиэтил 4 - оксоиндоло - (2, 3 - а) хиноянзика в яинтиомер мыс - 1, 2, 3, 4, 6, 7, 12, ,12 6 - (жтапщро - - этил 1 карбометоксизтил - 4- ОКСОИНДОЛО (2, 3 а) хннолизина реакцией этш-о последнего с лятнсернистым фосфором лшя полу«ния соогветстеуюшего энантиомера цис - I, ,2, 3, 4, 6, 7, 12, 126 октагидро - 1 - этил - 1 исарбомет жситгнл - 4 тиокс(жндоло - (2, 3 а) чсинолизииа и обессеривакием этого последнего в энaнтиoмep чмс - 1, 2, 3, 4,6,7, 12, 12 6 - сястагидро- 1 - этил - 1 - карбо(етоксиэтялиидоло (2, 3 - а) .хинозилина. 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - la этил 13 - карбометоксиэтил - 12 6 а индоло (2, 3 - а) .хинотмн, употребляемый в качестве исходного продукта для приготсжления (4) - виикамсна, получают омылен|{ем 1, 2, 3, 4, 6, 7, 12, 126 октагищю - 1 - этил - 1 - карбометоксиэтнл 4 -юксонмдоло - (2,3 - а) - хинолизина, разъединеюмм изомеров цис- и гране - 1, 2, 3, 4, 6, 7, 12, 25 чжтагищ - 1 - карбоксиэтил - 4 - оксоиндоло - (2, |3 - а) - хннолизина раздвоением цис - изомера на оптические антиподы при помощи оптически активного основания, выделением эншггаомера 1, 2, 3,4, ,6, 7, 12, 126 - октагидро - 1а - этил - 1|3 г карбоксиэтил - 4 - оксо- 126а-индоло- (2,3-а)хинолмзина, этерификацией этого последнего в 1, 2, ,3, 4, 6, 7, 12, 126 - октагидро - 1 а - этил - 1 карбометоксизгил - 4 - оксо - 12 б а - индоло - (2, 3 - а) - хннолизин, реакцией этого последнего с пятисернистым фосфором для образова1шя 1, 2, 3, ,4, 6, 7, 12, 12 б октагидро - 1а этил - 1 )3 -карбометоксиэтил 4 - тиоксо 126 а- индоло (2, 3 - а) хинолизнна и обработка этого последнего аген том обессеривания. Способ предпочтительно выполняется следующим образом. A.Омыле1ше производ т обычным способом, действием гидроокиси щелочного металла, например едким натром или едким кали. Б. Разделение эпимеров цис и транс производят, например простой дробной кристаллизацией. Это разделение может быть произведено с помощью смеси щелочньрс солей, пол чеш1ых омылением, до освобожде1Ш Г| карбоксильной группы кислотной обработкой. Если употребляют едкий натр в качестве омыляющсго средства, то получештые эпимерные соли натрия могут быть разделены кристалшоацией одного иззпимеров в этаноле или в смеси этанола воды, как то в 95%НОМ этаноле. B.В. роли оптически акт1шного основания используют, например, & - зфедрин, d - эфедрин, хинин, (d) а - фенилэтиламин, хииолин,Л (-)-или Z (+) - трео 1 - и - нитрофе шл - 2 - N, N -даметиламинопропан - 1, 3 - диол,2 (+) трео- 1 -п нитрофениламинопропан - 1, 3 - даолшшг (-) -трео - 5 - амино 6 - фенил - I, 3 - диоксан. Г, Этерификацию полученной оптически активной кислоты производят метанолом в присутствии минеральной кислоты - соляной или серной в качестве катализатора. Эта этерификация может быть также произведена диазометаном. Д. Ееакдая обессердаа1шя производится при помощи никеля Рёнея. Опыт I. 1, 2, 3, 4, 6, 7, 12, 120 - октагидро - 1 - 1 - карбометоксизтилиндоло - (2, 3 - а) -хинолизин. Стадия А. 1,, 2, 3, 4, 6, 7,12,12 б октагидро 1 -этил - карбометоксиэтил - 4 - оксоиндоло (2, 3 -а) - хинспизин. Нагревают с обратным холодильником смесь, состоящую из 231,5 г триптамина, 371 г диметилового зфира 4 ,- этил 4 - формнлпимелиновой кислоты, полученного известным способом и 1160 см бензола, нагревают с обратным холодкльником в течеше 1 чаС( удалйя образовавшуюся воду азеотропной перегонкой и перегоняют раствор досуха, в вакууме при 50°С, забирают остаток 463 смуксусной кислоты, нагревают с обратным холодильником в течение 1,5 час и.; перегоняют досуха. Вьшивают остаток в смесь ЗОСЮ см воды-льда и 231 см раствора едкого натра. Экстрагируют хлористым метиленом, промьтают водой органические слои, заново экстрагируют хлористы метиленом промывные маточные растворы, сушат объединенные экстракты над сернокислым магнием, фильтруют и перегоняют досуха. Забирают остатокдтиловым эфиром уксусной кислоты, замортживают льдом в течение одной ночи, фильтруют под разрежением, промываня ледяным этиловым эфиром уксусной кислоты и сушат. Получают 366,2 г 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1 - этил 1 карбометоксиэт ш 4 - оксоивдоло - (2, 3 - а) хинолизина; т.пл. 135-140°С. Срльватащ1Я 5,3%, . Маточные растворы этилового эфира уксусной кислоты вьтарт1вают досуха, забирают остаток 200 см уксусной кислоты, нагревают с обратным холодильником в тече1ше 2 час, вьтаривают досуха и Бышшают остаток в смесь 2000 см воды - льда и 50 см раствора едкого натра. Эктрагируют хлористым метиленом, промьшают водой С|рга1шческие слои, сушат над сернокислым мапшем и перегоняют досуха. Забирают остаток этилацетатом, замораживают льдом в тече1ше 4 час, фильтруют под разреже1шем, промывают ледяным этиладетатом и сушат. Получают еще 85,4 г. Стадия Б. 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - Ь - этил - 1 - карЬометоксиэтилиндоло - (2, 3 - а) хинолизин. При перемешива ши в атмосфере азота вводят в суспензию 250 г 1, 2, 3, 4, 6, 7, 12, 126 октагидро I - этил - I - карбометоксиэтил - 4 оксоиндоло - (2, 3 - а) - хинолизина в 2500 см тетрагидрофурана, прибавляют 172,5 г пятисер1гастого фосфора и продолжают перемешивание в тече1Ше 4 час при 25-27° С, фильтруют, прополаскивают фильтр тетрагидрофураном и получают таким образом раствор А, . Промьгоают тетрагидрофураном 1,250 кг никеля Ренея для удаления воды при перемешивании, дают декантировать и удаляют жидкость, находящуюся на поверхности. К приготовленной таким образом суспензии никеля Рвнея прибавляют, при перемешива1ши и в атмосфере азота, предьщущий тетрагидрофураиовый раствор А, вьщерживая температуру при 25° С. Оставляют в контакте 1,5 час, декантируют тетрагидрофуранойый слой, промьшаKfT никель тетрагидрофураном, перегоняют в вакууме объединенные слои и сушат остаток при 60° С. Собирают 1S1 г смеси двух изомеров. Перекристаллизовьшают 176,5гзтой смесив 3150 см кипящего метанола, фильтруют, охлаждают до 20° С при перемешившши и оставляют смесь в течение ПЯ1И часов при 20° С, Фильтруют под разряжением и сушат. Получают 68 г транс производаого 1, 2, 3, 4, 6, 7, 12, 126 - октагнщю - 1 - этил - 1 i-карбометоксиэтилиндоло - (2, 3 - а) - хинолизина, плавящегося при 149° С, который употребляют для приготовления изовинкамина. Концентрируют маточные растворы и охлаждают до 20° С, оставляют на 2 час при этой температуре, фильтруют под разрежением, перекристаллизуют осадск в бОО.см кипящего метанола, доводят до комнатной. Температуры, фильтруют под разрежением и сушат при 40°С. Получают 45,8 г цис пронзво;ЕЦЮГо 1, 2,3i 4, 6,7,12,126 - октагидро - 1- этил - 1 - карбометокснэтшшндоло - (2, 3 - а) -хинолизиш, плавящегося при 140° С, который употребляют для приготовления винкамина.
Для анализа пе скгриста.ч;п1:юпыватт оба проукта в циклогексанс и яссольватируют в кипящей оде.
Транс - произвс)Ш1ые I. 2. . 4, 6, 7, 12, 2fi
ктагидро 1 этил - 1 - ка Зомстокси.)Т11;пиию;1о (2, 3 - а) - хинолнзина прелсталляст собой гверлый
бесцветный продукт, плавящийся при 149 С.
Мол. вес 340,45.
Вычислено,; С 74,0«; И 8,;S; N ,23. oCj.HjeO N:НайденоД. С 73,9: И S,3; N 8,4. В ИК-спектре присутствуют полосы при 17 1 И и 1740см , характерные для СО-группы и полосы 3495, 3436 и 3355 см , характерные ;и1я NH.
Цис произвоцное 1, 2, 3, 4, 6, 7, 12, 12 ( октагидро 1 - этил - 1 - карбометоксиэтилиндоло {2, 3 - а) - хинояизина также представляет собой твердый прог;укт, плавяищйся при 140° С. Вычислено,: С 74,08; И 8,28; N 8,23. CjiHjeOjNj (340,45), Найдено,%: С 74,3; И 8,4; N 8,5. В ИК-спектре присутствуют полосы при 1727 и 1736см- (С О) и 3498 (NH).
Опыт II. 1, 2, 3, 4, 6, 7, 12, 12 - октагидро - IQ тил 1(3 - карбометокси - 1 2 а - индоло (2,3- а)хинолизин.
Стадия А, 1,. 2, 3, 4, 6, 7, 12, 12 б октагидро - 1-этил - 1 - карбокси - 4 - оксои1щоло - (2, 3 - а) хинолизин. Разделение изомеров цчс- и гране.
Нагревают с обратным холодиль(даком при перемешивании в течение 1 час смесь, состоящую из 700 г 1, 2, 3, 4, 6, 7, 12, 2 б - октагидро 1 - этил - Ь
карбометоксютил - 4 - оксоиндоло - (2, 3 - а) хинолизина (полученного по способу, описанному в стадии А при мера 1), 158 г едкого натра (таблетки) и 2,8л 95%-него этанола. Фильтруют кипящую суспензию и промьшают два раза осадок при помощи 350 см кипящего 959 -ного этанола.
Обработка фильтрата. Получение ццс - изомера. Растворитель удаляют перегонкой. Прибавляют 2,8л воды к полученному маслянистому остатку. Перегоняют около 300 см смеси для полного удаления этанола. Полученный раствор охлаждают до 20° С. Прибавляют 1,975л 2 н. соляной кислоты и перемещивают в течение 2 час при 20-24° С.
Осадок отделяют фильтрацией, промьшакгг водой и сушат.
Перекристаллизацией из метанола получают цис - 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1 - этил - 1-карбоксизтил - 4 - оксоиндоло - (2, 3 - а) хинолизин с выходом около 45л, т.е. 90% по
отношению к цис - изомеру, находящемуся в исходном продукте; т.пл. 264° С.
Вычислено,%: С 70,56; II 7,10; N 8,23.
CjoH.NjOs (340,41).
Найдено,%:С70,6; Н7,2; N8,3.
Обработка осадка. транс изомера. Осадок забирается водой и подкисляется 1 н. соляной кислотой дорН1. Выделяют фильтра|щей и перекристаллизацией , из метаио.та гринс -1.2, 3, 4,
2б
- этил - 1
А 7,
октагидро (2, 3 - а)
-кариоксиэтил - 4 - оксоиндоло -.хииплизин, т.пл. 254С.
Вьгп1слено,%; С70,56; Н7,10; N8,23. (2oH24N203 (340,41).
Иайдено,%: С70,6; Н7,2; N8,4. Б. Раздвоетше цис - 1, 2, 3, 4, 6, 7, 12, ,1 2 б - октагидро - 1 )тил - 1 карбоксиэтнл 4 оксоиняоло - (2, 3 - а) - хннолизина. К раствору, coдepжaшe 1y 263 г 1- эфендрина в
1,45 л дихлорэтана, прибавляют 525 г цис - изомера, полуюнного на пре;гыдутей стадии, а затем 380 см дихлорэтана. Нагревают с обратным холодильником при перемешивании и перегоняют около 380 см дихлорэтана. Охлаждают до 25°С, возбуждают кристаллизацию скоблением и оставляют в течение 20 час при 20° С. Полученный осадок выделяют фильтрацией.
Обработка осадка. Осадок - это правовращаюцияся соль а. + 137 ± 3° (с 17г, /шметилформамия), которую обработкой раствором соляной кислоты приводят к соответствующей правоврашаюикй кислоте; (a.i° -f 235 ±3° (с 17г, диметилформамид); т.пл. около 293° С.
Обработка фильтрата. К фильтрату прибавляют
390 см водного раствора, содержащего 130см концентрированной соляной кислоты. Смесь пере eшивaют в течение 2,5 час при температуре около 20°С. Отделяют образовавшийся осадок фильтрашей и получают 157л левовращающей кислоты, т.е. (-) - цис - 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1-этил - 1 - карбоксиэтил - 4 - оксоиндоло - (2, 3 .-а) - хинолиэин; т.пл.около 293°С (термический
дифференциальный анализ); W - 235° (с , диметилформамид).
Сравнение кривых кротового дихроизма с кривыми (+) - ви п амина (оптически актнвньш винкамин, природного прюисхождения) позволило заключить, что этот левовращающий изомер имеет конфигурацию, аналогичную с конфигурацией (+) винкамина.
Вьплеполученный левоврашаюший изолгер - это 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1 а - этил - 1 /3 -карбоксиэтил - 4 - оксо - 12 б о - индоло (2,3- а)- хинолизин и правовращающий изомер (М -235°) - это 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1 этил - 1 а - карбоксизтил - 4 - оксо - индоло
-(2, 3 - а) - хинолизин.
Маточные растворы после фильтрации левовращающего изомера вьтариваются досуха. Остаток,
перектристаллизова1шый нз этанола, состоит из
159 г исходной, рацемической кислоты, которую
можно снова раздвоить.
Вращение плоскости поляризации соли 6- -эфедрина 1, 2, 3, 4, 6, 7, 12, 12 б октагидро -la- этил -113 - карбоксизгил - 4 - оксо - 12 б а - индолй - (2, 3-а) - хинолизина характеризуется величиной (о -154° (с 1%,диметилформамид).
Если вместо Е - эфедрина раздвоение производят с помощью а - эфедрина, то на этот раз менее растворимой солью окажется соль левовращагощейся кислоты, т.е. соль а - эфедрина 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро - 1 а - этил - 1 j3 - карбоксиэтил 4 - оксо - 2б а - иидоло - (2, 3 - а) хинолизина, отличающаяся своей способностью вращать плоскость поляризации «1 - 137° ±3° (с 1%, ди йетилформамид), . Сйработка этой соли раствором соляной кислоты приводит к 1, 2, 3, 4, 6, 7, 12, 12 б - октагидро -1 а - этил - 113 - ка1Йоксиэтил - 4 - оксо - 12 б а -индоло - (2, 3 - а) - Хйнолизину, который идетичен с вьпиеполученным продуктом. Стадия В. 1,,2, 3,4, 6, 7,12, 12 б - октагидро - 1о - этил - 1 - карбометоксиэтил - 4 - 12 б а - ивдоло (2, 3 - а) хинолизин. 32,4 г левовращающего изомера, получетюго в предьвдущей стадии, вводят в 130 см метанола, содержащего 4 г/л серной кислоты. Нагревают в атмосфере азота и при перемешивании с обратным холодильником в течение 2 час, после чего охлаждают до 25° С и нейтрализуют 1,2 см пиридина. Щ)ибавляют при перемешивании и медле шо 1300 см воды, осадок отделяют фильтрацией, промьтают водой и сушат. Получают 33,35 г 1, 2, 3, 4, j6, 7, 12, 126 - октагидро - 1а - этил - 10 -карбометоксиэтил - 4 - оксо - I2ffa- индоло - (2, 3 - 205 ± 3,5° (с 0,5%, а) - хинолизина; этанол); а - 212 ± 1,5 (с 1%, диметилформамид); т.пл. 152° С, а затем 16ГС (Коффлер), , ВычислЕно,%:С71,17; Н7,39; N7,90. CjiHjeNjOs (354,44). Найдено,%: С 71,1; Н7,4; N7,9. Стадия Г. 1,.2, 3, 4, 6, 7, 12, 126 - октагидро - la этил- 1|3- карбометоксиэтил- 125а:- индоло- (2,3 хинолизин. 200 г продукта, получегаюго в предыдущей ст дии, вводят в суспензию в 2л тетрагидрофурана, прибавляют 138 г Ш1тисерш1сного фосфора и перелюшивают в течение 5 час в атмосфере азота, выдерживая температуру около 25°С. Фильтруют и nji поласкивают фильтр тетрагидрофураном. Получе1шьй фильтрат - это раствор 1, 2, 3, 4, 6, ,7, 12, 126 - октагидро - 1 а - этил - 1Д - карбомв токсиэтил - 4 - тиоксо - 126 а- индоло - (2,3 - а) ошнолизина; (alj - 204° (с 1%, диметилформамид) Число омыления 157 мг К(Ш/г по теории 1511. Содержание серы 8,75% по теории 8,65%). Этот продукт характеризуется при хроматографированки в тонком слое Rf 0,7, Фильтрат медленно прибавляют к 1 кг никеля Режя (предварительно промытого тетрагидрофураном), при переметивший и в атмосфере азота приблизительно при 25° С. Оставляют контактировать в течение 1,5 «ис после окончания прибавления фильтрата. Никель отделяют фильтрацией, фильтрат упар1тают досуха в вакууме. Получают 173 г 1, 2, 3, 4,6,7, 12, 126- октагидро- 1 а-этил 1 0 - карбометоксиэтил - 126 а- индоло - (2, 3 - а) инолизина. Этот продукт характеризуется при хроитографиравании в тонком слое Rf - 0,39. Опыт 111.1,2,3,4,6,7, 12, 12 б-октагидро- 1 Дтил - 1 а - карбометоксиэтил - 126/3 - индоле - (2, 3а) - хинолизин. Исходя из 1, 2, 3, 4, 6, 7, 12, 126 - октагидро 1 - этид - 1 а - карбоксиэтил - 4 - оксо - 126 ндоло - (2, 3 - а) - хинолизина, полученного в тадии Б примера 2 и применяя способ, описанный стадиях В и Г. примера 2, получают последоваельно;1, 2, 3, 4, 6, 7, 12, 126 - октагидро - Ij3 - этил la- карбометоксиэтил - 4 - оксо- 12 5 - индоло (2,3 - а) - хинолизин; 1, 2, 3, 4, 6, 7, 12, 126 - октагидро - 1 /3 - этил 1 а - карбометоксиэтил - 4 - тиоксо - 1260 - индоло-(2, 3 - а) - хинолизин; 1, 2, 3, 4, 6, 7, 12, 12 6 - октагидро - 113 - этил I tt - карбометоксиэтил - 126/3 - индоло - (2, 3 - а) хинолизин. Пример 1. dB- Виикамин. Стадия А. 14 - оксо - Е - гомоэбурнан, dl изомер - цис. Растворяют 10 г цис - производного 1, 2, 3, 4, 6, 7, 12, i26 - октагидро - 1 - этил - 1 - карбометоксиэтилиндоло - (2, 3 - а) - хинолизина, полученного по способу, описанному в стадии Б опыта I, в 140 см толуольного раствора триамилата натрия, содержащего 1,45 г натрия на 100 см, при перемешивании и в атмосфере азота. Перемешивают в течение 10 мин при 21-22° С, выливают в раствор Юг хлористого аммония в 300 см воды. Экстрагируют толуолом, промьшают органические слои водой, сушат на сернокислом натрие и перегоняют досуха. Остаток забирают 30 см эфира, фильтруют при разрежений,. проМ.шагот эфиром и сушат. Получают 6,43 г 14 - оксо - Я - гомоэбурнана . изомера - цас,й виде твердого продукта, бесцветного, плавящегося при 164° С. Вшод 71%. Вьгшслено,%:С77,88; И 7,84; N9,08. CjoH240Ni (308,4), Найдено,%: С77,7; Н7,8; N9,0. ИК-спектр показывает присутствие С-О. УФ-спектр в этаноле:}% прт 242-ммкЕ 538, Шкс пря 268-269 ммк Е -361, 1% Перегиб у 273 ммк Е 337, 1% Макс, при 292 ммк Е 163, 1% Е 153, Макс, при 301 ммк
13
Стадия Б. 14 - оксо - 15 - гидроксиимино - Е -гомоэбуриан dp, изомер - цмс.
Смешивают 12,2 г 14 - оксо Е - гомоэбурнана «dS; изомера -. цис, 80,5 толуола и 36,6 см нитрита трибутила, прибавляют 80,5 см толуольного раствора триамилата натрия й юшрго титр в 1,7 г натрия на 100 см , и оставляют в контакте в течение 1 час при 24-26° С в атмосфере азота. Реакцко1шую смесь выливают в раствор 25 г хлористого аммония в 300 см воды, экстрагируют толуолом, промьшают экстракт водой, сушат над сернокислым натрием и перегоняют досуха в вакууме. Остаток превращают в кашу при помощи эфира, фильтруют при разрежении, промьтают эфиром и сушат. Получают 7,8 г - изомера 14 - оксо15 - гидроксиимино - Е гомоэбурнана - dt Упариванием маточных растворов и перекристаллизацией из эфира получают вторично 0,375 г.
Это соединение имеет вид бесцветного твердого продукта, плавящегося при 260° С, .
Вычислено,%: С71,19; Н6,87; N 12,44.
CioHjjOjNs (337,4). .
Найдено,%:С71,1; Н6,8; N12,8.
УФ-шектр в этаноле: %
Макс при 217 ммкЕ 507,
1см
1%
Е 556, 1см
1% Е 133,
1см
0,1 н.
1% Е 551,
1см
1%
к
Е 509, 1см
1%
Е 588, 1см
1%
Е 165, 1см
Стадия В. 14,15 диоксо - Е - гомоэбурнан - dl, цис изомер.
Растворяют 6,78 г чис - изомера 14 - оксо - 15 чтщроксиимино - Е - гомоэбурнана dtB 34см формальдегида, 17см воды и 17 см соляной кислоты, нагревают раствор при 75 С в течение 15 мин и охлаждают, подщелачивают прибавлением аммиака, экстрагируют хлористым метиленом, органический слой промьгеают водой, сущат над сернокислым натрием и перегоняют досуха. Остаток очищают хроматографическим способом и перекристаллизацией в эфире, полушют цисизомер 1,38 г 14,15 - диоксо - Е гомоэбурнана - djB ввде твердного продукта желтого цвета, плавящегося прт158°С.
14
Вычислено,%; С 74,50; Н 6,87; N 8,69. CjoHjjOjNj (322,50). На{щено,%: С 74,3; Н 7,1; N 8,5. УФ-спектр в этаноле: %
Перегиб у 224 ммкЕ 343,
1см
1%
Макс при 255 ммк
Е 459, 1см
1%
Макс при 305 ммк
Е
123. 1см
В ИК-спектре отсутствует полоса, характерная ДШ1 ОН, имеются полосы 1728 и 1690 (С 0). Стадия Г. d -Винкамин.
Растворяют 0,25 г натрия в 50 см метанола, доводят раствор до 25° С и прибавляют 0,50 г цис изомера 14,15 - диоксо - Е - гомоэбурнана - dt в атмосфере азота, оставляют в контакте в течение 1 час при комнатной температуре, нейтрализуют прибавленнем 0,65 см уксусной кислоты, перегоняют лктанол в вакууме и забнрают остаток водой, затем фильтруют под разрежением, промьтают водой и сущат г.ри . Получают 0,471 г dt инкамина в виде твердого, бесцветного продукта, лавящегося при 265° С (блок Коффлера) и при 239,5° С по термическому дифференциальнс- у анаизу.
Вычислено,%: С71,15; Н7,39; N7,90. CiiHjiNj (354,44). .
Найдено,%: С 70,9; Н 7,4; N 7,9. ЯМР-спектр показывает: Триплет эт№.а при 46; 53 и 61 гц; СООСНз при 229,5 гц; Угловой прогон при 234 гц; Шпри275пц.
Ароматическое соедияение при 426, 429 я 449 ГЦ. Масс-спектр и инфракрасный-спектр соответствуют спектрам естественного винкамина. Пример 2.
tit- Изовинкамин.
Стадия А, 14 - сяссо - Е - гомоэбурная - dt изомер - транс.
Шодят в суспензию в 200см тетрвгидрофурана 4,2 г 50%-ного гидрида натрня в минеральном масле и перемешивают в течение 10 мин при комнатнсж телтературе, прибавляют 20 г транс прояэводного 1, 2, 3, 4, 6, 7,12, 12 5 - октагндро - 1 - этил - 1 - карбометоксиэтилиндоло - (2, 3 - а) хинолизина, полученного по способу, описанному в стадии Б шьгга 1, н 400см тетрвшдрофурана н перемешивают в течение 15 мин при 25°С. Получают раствор 14 - оксо - Е гомоэбурнана, транс изомер, который употребляют в данном виде в следующей стадии.
Для выделения продукта выливают полученный вьшк раствор в водный раствор 4G%-saro хлористого аммония, выпаривгют тетрагидрофуран в вакууме, экстрагируют хлористым метиленом, сушат органические слои над сернокислым натрием и
15
перегоняют досуха, остаток забирают 50 см метанола, затем вьшаривают досуха и остаток тфевращают в кашу при помощи 60 см метанола, оставляют в контакте в течение 2 час, фильтруют под разрежением и сушат при 60° С. Продукт очищают растворением в хлористом метилене и осаждением прибавлением метанола н после высутинвания при 40° С, получают гране изомер 13,4 г 14 - оксо - Е гoмoз5ypнaнa - d6 в виде бесцветного продукта, плавящегося при 132° С. .
Вычислено,%:С77,88; Н7,84; N9,08,
CaoHj40jN (308,41, ,
Найдено,%: С 77,6; Н 7,8;
N9,1.
УФ-спектр в этаноле:
1% Макс при 242 ммк
Е 563, 1см
1%
Макс при 267 ммк
Е - 351, 1см
h 327, 1см
1%
Е 154, 1см
1%
Е 150,
ItM
н.
1%
Е 565, 1см
1%
Е 349, 1см
1%
Е 323, 1см
Макс при 290 ммк
182,
1см
1%
Макс при 299 ммк
Е 181, 1см
15
Стадия Б: транс - Изомер 14 - оксо гидроксиамино Е - гомозбурнан - d&.
К тетрапщрофурановому раствору транс
изомера 14 - оксо - Е гомозбурнана - dP, полученному в стадии А, прибавляют 60 см нитрита трибутила и оставляют в контакте в течение 1 час 15 мин в атмосфере азота при 25 ° С. №акционную смесь выпивают в раствор 40 г хлористого аммония в 1500см воды, перемешивают в течение нескольких минут и вьшаривают тетрагидрофуран в вакууме, после чего экстрагируют хлористым метиленом, экстракт промьтают водой, сушат над сернокислым магнием и перегоняют досуха в вакумме. Остаток забирают 50 см метанола, выпаривают досуха, остаток превращают в кащу с помощью 100 см метанола,оставляют на два дня в леднике, фильтруют под разрежением, промьтают холодным
505365
16
лктанолом и сушат. Собирают 0,45 г сырого продукта, крторьте очищают перекристаллизацией этанола при 24°С. После высушивания получают транс -изомер 14 - оксо - 15 - гидроксимино - Е - гомоэбурнан - df, с выходом 57%. Соединете имеет вид твердого продукта желтого, плавящегося при 226°С. Ёычислено,%; С 71,19; Н6,87; N 12,44, CioHjsOzNs (337,4). , Найдено,%: С70,9; Н6,8; N 12,4.
Стадия В. 14,15 - диоксо - Е гомоэбурнан - dt,
транс изомер.
Нагревают при 85° С смесь, состоящую из 2 г транс изомер 14 - оксо - 15- гидроксиимино-fi гомоэбурнана - dt; 10 см 5 н. соляной кислоты и 10см 40%-ного формальдегида в течение 15 мин. 1 акционную смесь выливают на лед, дoвoдяt до рН 10 прибавлением аммиака и экстрагируют хлористым метиленом, после чего фильтруют, сушат над сернокнслым магнием и вьшаривают досуха. Остаток хроматографируют на силикагеле, смесь злюируют хлористым метиленом - ацетоном (10-2) и вьщаривают досуха. Получают 1,15 г 14,15 - диоксо - Е - гомоэбурнана - dj, транс изомера, представляющего собой твердый продукт ЖЕЛТОГО цвета, плавящегося при 143° С. . УФ-спектр, в зтаноле - НСб 0,1 н.
1%
Е 361, Перегиб у 214 ммк
1см
1%
Е 354, 1см
Л% .., Е 125,
1%
Е 266, 1см
1%
Е 124, 1см
OH 0,1 н.:
1%
Е 811, 1см
1%
Е 194, 1см
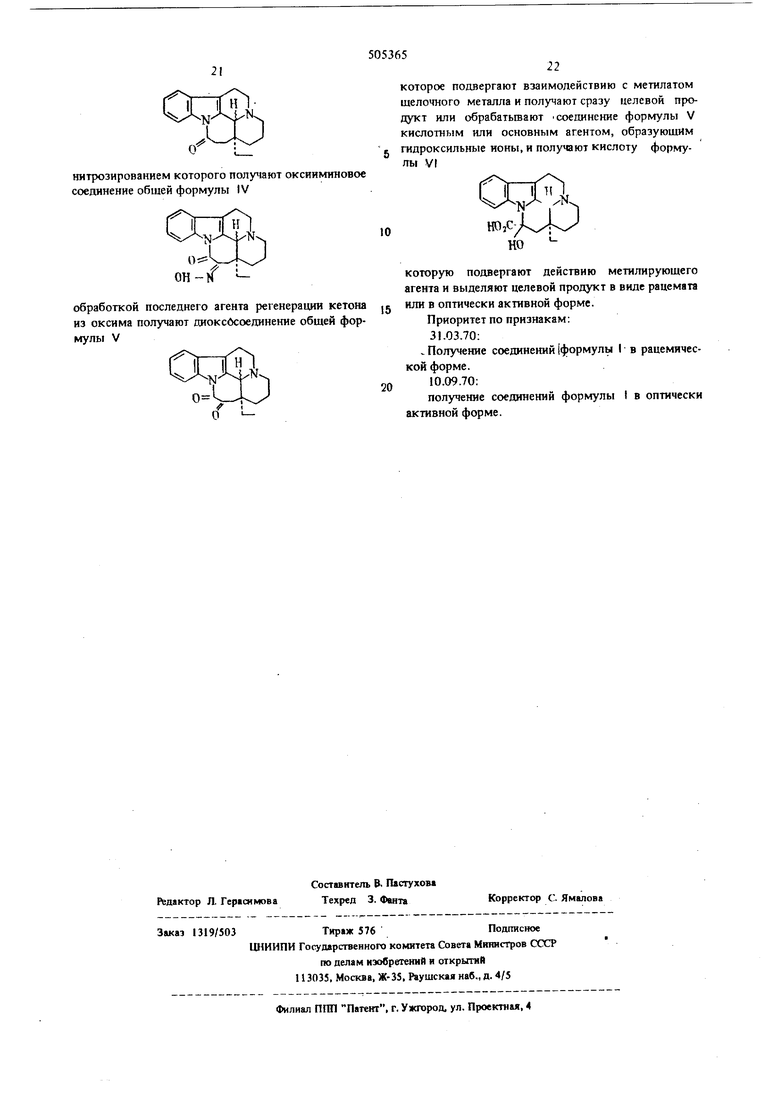
Е - 199, 1см 1% . Перегиб у 290 ммк Е 164, 1см Макс при 335 ммк Е 15. Стадия Г, 3. - Иэовннкаминовая кислота db. Нагревают с обратным холодильником в течение 8 час в атмосфере азота 1,1 г транс - изомера 14, 15 диоксо - Е - гомоэбурнана - At, 100 см 95%-ноге спирта и 10 г едкого кали в таблетках, упаривают досуха в вакууме. К остатку прибавляют 60 г льда, нейтрализуют 12см соляной кислотой и 0,4 см уксусной кислоты и оставляют на 2 час, затем фильтруют при разрежении, промьшают остаток водой и сушат при 40° С, Получают 0,61 г 3 изовинкаминовой кислоты - виде бесцветного твердого продукта, плавящегося при 247° С. , ИК-спектр (вазелиновое масло): поглощение характерное ОН, N Н и ОН - кислоты, присутствует С-О-группа при 1630 . Стадия Д. dt- Изовинкамин, К 2см раствора диазометаиа в хлористом метилене прибавляют 30 мг 3 - изовинкаминовой кислоты - dt.H оставляют в контакте в течение 15 мин при комнатной температуре. Избыток диазометана разлагают уксусной кислотой. Упаривают досуха в вакууме и получают 3 - изовинкамин - dEB виде бесцветного твердого продукта. ИК-спектр показьшает присутствие С-О при 1730 и 1755 см-,С-С при 1638см-, ОН и -Ni. . d2- Изовинкамин может быть получен также следующим образом. Нагревают при 85° С в течение 15 мин в атмосфере азота 0,50 г транс - изомера 14 - оксо - 15 гадроксимино . Е :гомоэбурнана - dj, 2,5 см 5 и. соляной киелоты и 2,5 см 40%-ного формальдегида прибавляют. лед и доводят рН до 10 аммиаком, пос ле чего Экстрагируют хлористым метиленом, фшатруют, экстракт сушат над сернокислым магнием и перегоняют досуха, остаток забирают 25 см летаиольного раствора метилата натрия, содержащего 5 г натрия на 100 см и оставляют в контакте в течение 1 час в атмосфере азота при 25-27° С. Избьтгшс метилата натрия разлагают прибавлением уксуснш кислоты, фильтруют при разрежении, остаток щюмывают водой и сушат при 80° С. Получают 0,21 г dE - изовинкамина, плавящегося при 229° С. , Вьиисяено,%:С71,15; Н7,39; N7,90. CjiHjeOjNz (354,44), . Найдено,%:С70,5; Н7,2; N7,9. В ИК-спектре присутствуют полосы ОН, карбонила. С-С, третичного амииа. Пример 3. () - Винкамин. Стадия А, 14 - оксо - За, 16а (20) Е гомоэбурнан. К 173 г 1, 2, 3, 4,6,7, 12, 126- окта1-идро - 1а - 113 - карбометоксиэтил - 12 5 а - индоло - (2, 3 - а) - хинолизина, полученного по способу, описанному в стадии г опыта II, прибавляют 1025см раствора триамилата натрия в толуоле, содержащего 19 г натрия на i л. Раствор перемешивают в тече1{ие 15 мин при 24-25° С в атмосфере азота. Затем вьишвают раствор в 1 л воды, содержащий 200 г хлористого аммония. Отделяют органический слой и экстрагируют водный слой толуолом. Экстракты объединяют, лромьюают «ПС водой до нейтральности, сушат над сернокислым магнием и перегоняют досуха в вакууме. Остаток перекристаллизовывают в этиловом эфире. Получают 89,4 г оптически активного Е - гомоэбурнамонина, соответствующего изомеру 14ксо - 3 а, 16а (20) - Е - гомоэбурнана. Маточные растворы вьшаривают досуха иподвергают новой обработке триамилатом натрия. Таким образом получают второй выход 23,25 г 14 ксо - За, 16а (20) - Е гомоэбурана, идентичного с продуктом первого выхода; тлл. 151°С; .° + 17,5 ± 1° (с 1% метилформамид). Стадия Б. 14 - оксо - 15 - гидроксиимино За, 16 о: (20) - Е - гомоэбурнан, Перемешивают 110 г 14 - оксо - За, 16а- (20) В - гомоэбурнана, 660 см толуола и 440 см нитрита трибутила. К полученной суспензии прибавляют 670 см раствора триамилата натрия в толуоле, содержащего 19 г натрия на литр. Оставляют в контакте в течение одного часа при 21-22°С, а затем выливают смесь в 5,5 л воды, содержащей , 138 г хлористого аммония. Перемешивают в течение 15 мин, отделяют толуольный слой и экстрагируют водный слой толуолом. Экстракты объединяют, промывают водой до нейтральности, сушат их над сернокислым магнием и перегоняют досуха в вакууме. Получают 14 - оксо - 15 - гидроксиимино- 3 а, 16 а- (20) - Е - гомоэбурнан. Этот продукт характеризуют хроматографированием в тонком слое Rf 0,22. Основа кизельгель F 254, растворитель для элюирования хлористый метилен - ацетон (5:1). Для очищенногоПродукта. +55° (с ,диметилформамид). ИК-спектр показьтает присутствие С-О, ОН и ароматической С-С - связи. УФ-спектр. Этанол - HCtO,l н.: Перегиб у 220 ммк Е 470, Макс 253,5 ммк 1см Е 139, Макс 307 ммк 1см Круговой дихроизм: ДЕ 260-262 ДЕ 222-220 Стадия В. 14, IS даоксо - За, 16а (20) - Е -гомоэбурнан. Продукт, полученный в предыдущей стадии, смешивают с 600 см 40%-ного формалина, 300 см воды и 300 см соляной кислоты и нагревают при 75С в атмосфере азота, перемешивая в течение 20 мин. Затем раствор выливают на 2 кг льда, нейтрализуют медленным прибавлением 300 г кислого углекислого натрия. Прибавляют 500 см хлористого метилена, а затем 100 г кислого углекислого натрия, перемешивают в течение 15 мин, н экстрагируют хлористым метиленом, экстракты промьшают водой и вьшаривают досуха. Остаток хроматографируют на снликагеле магняя и злюируют хлористым метиленом. Получают 30,8 г 14, 15 - диоксо - За, 16 а (20 -Б гомоэбурнана. Элюированием ацетоном рекуперирзоот затем 21,8 г исходного продукта. 14, 15 - диоксо 3о, 16а(20) -Е- гомозбурнан имеет Т.Ш1. . ИК-спектр показьшает присутствие С-О - группы. УФ-спектр: этанол - ,1 н.: 1% Maxd 244 ммк Е 477, 1см Макс 262ммк Е 374. 1см Е 163. Макс 293-294 ммк 1см Макс 301-302 ммк Е 170, 1см Круговой дихроизм: -0,9(+2Q%); + 2;9(±l№fe): + 4,1 (±30%). ДЕ 200-208 Стадия Г. (+) Винкамин. Продукт, полученньш в предыдущей стадии, ввод в суспенэтио в 100 см метанола, и смесь выливают в раствор 15 г натрия в 2,5 л метанола. Перемешивают в течение 1 час приблизитешло при 25 С в атмосфере азота. Избыток реагевгов разлагают 37 см уксусной кислоты, а затем к гацентрируют перегонков приблизительно до 300 см . Охлаждают до 25° С и оставляют в контакте в течение 30 мин при этой температуре. Образовавшийся осадок фнныруют, промывают и сушат. Полученный сьфоа виякамии очищают путем образования эфира уксусной кислоты, а затем разложением этого эфира триэтиламином. Получают 22,8 г (+) - винкамина: т.пл.(моментальное) 272°С; а +41° (с1%,пиридин). Вычислено,%:СГ1,15; Н 7,39; . CjiHjbNiOa (354,44). Найдено,%:С70,9; Н7,1; N7,9. УФ-спектр: Е 893, Макс 220 ммк 1см 1% Е 242. Макс 268 ммк Это соединение идентично с вянкамнном, выделен- ным из Vinca Minor., (+) Винкамин может быть также получен следующим образом. 14,15 - диоксо - За; 16а (20) - Б-гомоэбурнан обрабатывают смесью едкого кали и 95%-ного этилового спирта. После семичасового нагревания с обратным холодильником смесь вьшаривают досуха, забирают водой и нейтрализуют при 0°С соляной кислотой и оставляют на несколько часов, а затем отделяют фильтрованием осадок 14J3 идрокси 14 а- карбокси - 3 о, 16 а (20) - эбурнана, или винкамнновой кислоты, промьшают его водой и сушат. Винкаминовзоо кислоту затем этерифицируют диазометаном и получают (+) - винкамин, идентичный с Продуктом, получешп.1М выше. Пример 4. (-) - Вкнкашн. Исходя из 1,2,3,4, 6, 7, 12, 126 октагндро 1Р - этил - 1а карбометоксиэтил - 126 - индоло - (2, 3 - а) -хянолизина, полученного в опыте .111 и применяя способ, описанный в стадиях А, Б, В, Г примера 3, получают последовательно: . 14 - оксо - 3 ft 16 (20) - Е- гомоэбурнан; 14 - оксо - 15 - оксиимино - 3 0, 16 (20) - Е гомоэбурнан; -14, 15 - диоксо - 3,160 (20) Е гомоэбуран(-) - винкамин.. Формула изобрётения -Способ получения пятициклическнх алкалоидов общей формулы -Отличающийся тем, что, с целью упрощения процесса, четырехцикяическое соединение общей формулы И обрабатывают щелочным агентом для получения лактама общей формулы HI
нитрозированием которого полуиют оксииминовое соединение общей формулы IV
ОН
обработкой последнего агента регенерации кетона из оксима получают диоксбсоединение общей формулы V
(
1 1 J
0
которое погшергают взаимодействию с метилатом щелочного металла и получают сразу целевой продукт или обрабатьтают соединение формулы V кислотным или основным агентом, образующим гидроксильные ионы, и получают кислоту формулы VI
которую подвергают действию метилирующего агента и выделяют целевой продукт в виде рацемата или в оптически активной форме.
Приоритет по признакам:
31.03.70:
Получение соединений (формулы I в рацемической форме.
10.09.70:
получение соединений формулы I в оптически активной форме.