(54) СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГЛЮКОЗЫ
типовой кислоты. Предпочтительно проводпть способ с солью никотиновой кислоты в органическом растворителе. Обычно реакцию проводят в таких органических растворителях, как, например, ацетонитрил, диметилсульфоксид, диметилформамид, нитроалкан, галогепированный углеводород при температуре между 0°С м температурой кинення реакционной смеси. Соедннения формулы II являются известными или могут быть получены известным способом.
Пример 1. 2 г 2-дезокси-2-никотинамидо-1, 3,4,6-тетра-0-ацетил-|3,В-глюкоциранозы растворяют в 50 мл хлористого метилена и прибавляют 2 мл хлористого ацетила. В этот раствор вводят нрп перемешивании в течеиие 1 ч хлористый водород. Раствор выдерживают в течеиие 48 ч в холодильнике, затем добавляют 100 мл ледяной воды и доводят до нейтрального состояния при номощи 2 в. раствора карбоната натрия. Органическую фазу отделяют, суншт сульфатом магния и выпаривают. Остаток растворяют в 30 мл ацетонитрила, вводят раствор триэтилампновой соли никотиновой кислоты (полученной из 0,65 г ннкотниовой кислоты и 20 мл трнэтиламина) в 20мл ацетоннтрила, перемешивая в течение 10-15ч. Смесь выпаривают, добавляют 100 мл хлороформа и промывают три раза 50 мл воды. Органическую фазу отделяют, сушат сульфатом натрня, выпаривают и выкристаллизовывают из этанола. Получают 0,550 г (24,2%) 3,4,6три-0-ацетил-2-дезокси-2-никотинамидо - 1-0ннкотинонл-р,В-глюконнранозы. Т. пл. 172- 173°С.
Применяемая в качестве нсходного материала тетраацетилглюкопираноза может быть получена следующим образом.
24 г гидрохлорида 2-амино-2-дезокси-1,3,4,6тетра -О-ацетил-р,В-глюкониранозы растворяют в 150 мл пиридина и нрибавляют к смеси 20 г гидрохлорида хлорида никотиновой кислоты в 150 мл пиридина. Смесь перемешивают в течение 2 ч и выпаривают досуха. Остаток растворяют в 500 мл хлороформа и нромывают насыш,енными растворами карбоната натрия н хлористого натрия. Органическую фазу сушат сульфатом натрня и снова вынарнвают. После выкристаллизации нз этанола получают 29,5 г вешества. Т. пл. 218-219°С.
Прнмер 2. Раствор 3,4,6-три-0-ацетил-2дезокси-2-никотинамидо-1-хлор - р,В-глюкопиранозы, полученный из 100 г М-никотиноил-Оглюкозамнна и 300 мл ацетнлхлорида, разбавляют 1,2 л хлористого метилена и заливают 1,2 л ледяной воды. Смесь при перемешивании доводят до значения рП 5-6, применяя 2 н. раствор карбоната натрия. Органическую фазу отделяют, сушат сульфатом магния и выпаривают досуха. Остаток растворяют в 500 мл ацетонитрила, прибавляют раствор соли, полученной из 70 г никотиновой кислоты и 150 мл триэтиламина, и перемешивают в течение 10-15 ч. Раствор выпаривают, добавляют 1 л хлороформа, промывают три раза 1,5 л
воды, сушат сульфатом натрня и снова выпаривают. Остаток растворяют в 1,5 л этанола, обрабатывают 20 г активированного угля, фильтруют и выпаривают до одной трети иервоначального объема. После выкрнсталлизацин из спиртового раствора и двукратной перекристаллизации из этанола нолучают 48 г (26,5%) 3,4,6-трн-О-ацетил - 2-дезокси-2-никотинамидо-1-О-никотииоил - р,О-глюконнранозы. Т. нл. 175-176°С.
Пример 3. Суспензию 10 гN-никотнноилD-глюкозамина в 30 мл хлорангндрида пропионовой кислоты перемешивают в течение 18 ч при комнатной температуре, а полученный раствор выпарнвают досуха. Остаток растворяют в 150 мл хлористого метилена н заливают 150 мл ледяной воды. Реакционную смесь доводят до значения рН 7 при примененни 2 н. раствора бикарбоната натрия, затем органическую фазу сушат сульфатом магния, а раствор вынаривают досуха. К раствору остатка в 50 мл ацетонитрила прибавляют никотниат триэтиламмония (из 6,5 г никотиновой кислоты и 15 мл триэтиламина) и в течение 10-15 ч перемешивают при комнатной температуре. После вьшаривания реакционной смеси остаток вводят в 250 мл хлористого метилена, органическую фазу промывают два раза 100 мл воды, сушат сульфатом магния
и выпаривают. После выкристаллизации из сложного нзопропилового эфира этилацетата и перекристаллизации нз изопропанола получают 3,3 г (16,8%) 3,4,6-три-О-пронионнл-2дезокси-2-иикотинамндо - 1-О-никотиноил-р,Оглюкопнранозы. Т. пл. 161°С.
Пример 4. Хлористый метилен насыщают хлористым водородом, а в 5 мл полученного раствора растворяют 0,5 г 1,3,4,6-тетра-О-бензоил-2-дезокси - 2-никотинамидо-р,В-глюкопиранозы (т. пл. 195-196°С). Раствор выдерживают в течение 24 ч при комнатной температуре. После этого раствор выпаривают досуха, а остаток растворяют в 20 мл хлористого метилена н выливают в 10 мл ледяной воды. Реакционную смесь доводят до значения рН 7 при ирименении 2н. раствора бикарбоната натрия, органическую фазу сушат сульфатом магния, а раствор вынаривают досуха. В раствор остатка в 10 мл ацетонитрила вводят ннкотинат триэтиламмония (полученный из 0,15 г никотиновой кислоты н 3 мл триэтиламина) и перемешивают в течение 10-15 ч при комнатной температуре. После вьшарнвания реакционной смеси остаток вводят в 25мл хлористого метилена, а органическую фазу иромывают два раза 10 мл воды, сушат сульфатом магния и вынаривают. Смесь наносят на колонку снликагеля (150 г) и вымывают
при применении хлороформа - этанола (19:1). После выкристаллизации и перекристаллизации чистого иродукта из этанола иолучают 0,1 г (20%) 3,4,6-три-О-бензоил-2-дезокси-2-никотинамидо-1-0 - никотиноил - |3,Dглюкопиранозы. Т. пл. 178°С.
Формула изобретения
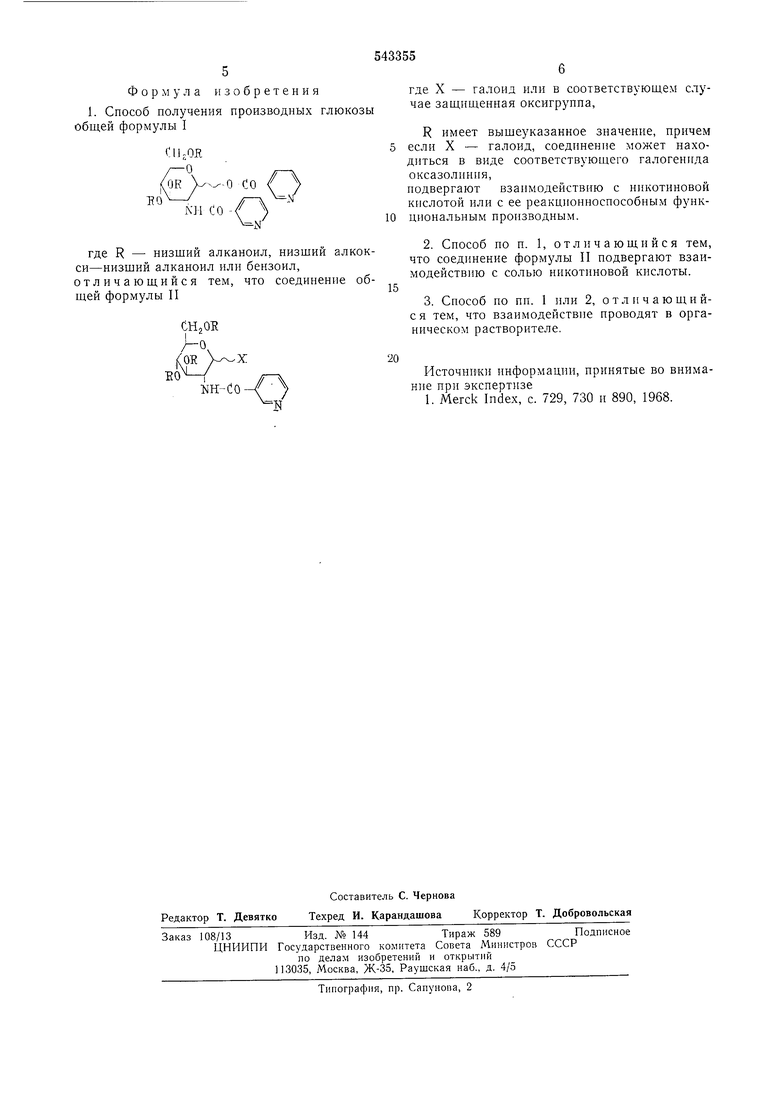
1. Способ получения производных глюкозы общей формулы I
ClljOR
-О OR
СО
ЕО
NH Со -/
где R - низший алканоил, низший алкокси-низший алканонл или бензоил, отличаюшийся тем, что соединение общей формулы II
СНгОК
OR
X
ЕО
кн-Со
где X - галоид или в соответствующем случае защищенная оксигруппа,
R имеет вышеуказанное значение, причем если X - галоид, соединение может находиться в виде соответствующего галогенида оксазолиния,
подвергают взаимодействию с никотиновой кислотой или с ее реакционноспособным функциональным производным.
2.Способ по п. 1, отличаюшнйся тем, что соединение формулы II подвергают взаимодействию с солью никотиновой кислоты.
3.Способ по пп. 1 или 2, отличающийся тем, что взаимодействие проводят в органическом растворителе.
Источники информации, принятые во внимание при экспертизе
1. Merck Index, с. 729, 730 и 890, 1968.