шнх ,кетонов, таких, каж ацетон, метилэтилс етон или диэтилкетон.
Температура реакции 20-100°С.
Для связывания выделяющегося бромистого водорода используют обычно избыток исходного амина.
Примером радикала Rl, замещаемого ка водород при гидролизе, являются, например, ацил, низший алканоил, предпочтительно с -1 - 4 атомами углерода, такой, как ацетил, аренкарбонил, такой, как бензоил, радикалы мо1нсфу:нкцис1нальны1хпроизводных угольной кислоты, такие, как метоксикарбонил, этоксикарбонил или феноксикарбонил.
Гидролиз можно проводить в присутствии гидроокиси щелочного металла, например гидроокиси калия или натрия, предпочтительно при температуре кипения реакционной смеси в высококинящем органическом растворителе, содержащем гидроксильную группу, например в этиленгликоле, диэтиленгликоле, низшем локоалкиловом эфире такого гликоля, в низшем алканоле, таком, как метанол или эта-. НОЛ. Гидролиз можно проводить в кислой среде, например в спиртовом растворе соляной кислоты, в водном растворе бромистого водорода или в ледяной уксусной кислоте.
Полученные производные азепина могут быть переведены в их аддитивные соли с неорганическими или органическими кислотами в растворителе, в котором образующаяся соль труднорастворима, например в метаноле, эфире, ацетоне, метилэтилкетоне, смеси ацетонэтанол, метанол-Э|фИ|р, этанол-эфи.р или метллев.хлорид-этанол.
Пример 1. 20 г (0,088 моль} мелкоизмельченного 3-хлор-5Н-дибенз- 6, / -азенина в 600 мл 48%-ной бромистОБО(дорО)Д«ой кислоты нагревают 90 мин с обратным холодильником, о.хлаждают льдом, наблюдая частичное образование З-хлор-9-метилакридин-гидробромида. Охлаждая льдом, к нолученной суспензии норциями добавляют 450 мл концентрированного аммиака и экстрагируют эфиром. ЭфИ:рный экстракт промывают водой и экстрагируют 300 .мл 1 и. серной кислоты. Если сырой продукт выпадает как сульфат, то после добавки воды он опять растворяется. Эфирный экстракт трижды промывают водой и присоедини-: ют промывную воду к кислому экстракту.- Водный кислый раствор обрабатывают активированным углем, фильтруют и светло-желтый фил:ьтрат подщелачивают концентрированным аммиако.м. Выделившееся основание обрабатывают эфиром, эфирный раствор сущат над сульфатом магния, фильтруют и вынаривают в вакууме. Остаток растворяют в горячем гексане, добавляют активированный уголь, фильтруют и сгущают. Получают З-хлор-9-метилакридин, т. пл. П7-118°С.
22,7 г (0,100 моль) полученного производного акридина растворяют при нагревании в 120 лг.г 2 н. серной кислоты, охлаждают, разбавляют 120 мл ледяной воды, помещают в ванну из хлористого натрия и льда и при 9°С
добавляют 29 мл (0,512 моль) холодного, как лед, ацеталъдегида. Температура смеси повыщается до 15° С. Сильно размещивая, охлаждают до 8°С и одновременно прикапывают охлажденный до 4°С раствор 144 г (0,520 моль) гентагидрата сульфата железа в 480 мл воды, а также 60 мл (0,450 моль) охлажденноГт до Т 75%-ной гидроперекиси трег-бутила. Скорость прикапывания регулирзют таким
образом, чтобы температура в реакционном сосуде была 10-13°С. После добавления половины обоих реактивов на стенке сосуда образуется корка, которую удаляют. По окончании прикапывания размешивают 15 мин, причем
температура в реакционном сосуде снижается до 3°С. Г1олученную коричневую суспензию экстрагируют метиленхлоридом, промывают экстракт водой, сушат над сульфатом натрия и выпаривают в вакууме. Остаток растворяют в 72 мл теплого абсолютного бензола, охлаждают, фильтруют через колонку с 52 г силикагеля, элюируя 320 мл абсолютного бензола, и выпаривают элюат. Остаток перекристаллизовывают из смеси эфир - гексан и получают метил- (3-хлор-9-|МетилакрИ|Дан-9-ил) -хетоп, т. пл. 116-118°С.
27,4 г (0,10 моль) полученного кетона растворяют в 500 мл метанола, охлаждают до 10°С, при размешивании в ледяной ванне в течение
10 мин порциями добавляют 19,1 г (0,500 моль) боргидрида натрия, перемешивают 1 час при 5°С и концентрируют в вакууме до 70 г. При добавлении 66 мл воды и затравочных кристаллов после охлаждения льдом
начинается кристаллизация. Охлаждая льдо.м, медленно нрибавляют 100 мл воды и выдерживают 1 час при 0°С. Кристаллы отделяют на нутче, промывают водой до нейтрализации и сушат в важууме над гидроокисью кали-я.
Получают 27,4 г сырого 3-хлор-а, 9-диметил-9акридан.метанола.
В охлажденную до комнатной температуры смесь 200 мл концентрированной серной кислоты и воды (10 : 1 но объему), сильно размешивая, в течение 15 мин вносят 27,4 г (0,100 моль) полученного гидроксисоединения. При постепенном растворении гидроксисоединения раствор нагревается до 30°С. Через 90 мин после начала добавления образуется
прозрачный раствор, который перемешивают 45 мин. Затем раствор выливают на смесь 800 г льда, 1 л воды и 500 мл метиленхлорида. Отделяют органическую фазу, экстрагируют водную фазу метиленхлоридом, объединенные
метиленхлоридные экстракты промывают водой, сушат над сульфатом натрия и выпарива-ют в . Остаток растворяют в абсолютном бензоле, бензольный раствор выливают на 55 г силикагеля, отфильтровывают его
:на нутче, промывают омесью абсолютный бензол-этиладетат (10:1) и фильтрат .выпаривают в вакууме. Остаток выкристаллизовывают из смеси эфир-гексан и получают З-хлор-10, 11-диметил-5Н-дибенз- & / -азепин, т. пл. 137-
139°С.
10,0 г (0,039 люль) полученного производного азепина кипятят 10 мин в 100 мл ацетаигидрида с обратным холодильником, отгоняют избыток ацетангидрида в вакууме при 80°С, оставшееся красное масло растворяют в абсолютном бензоле и хроматографируют па колонке со 120 г силикагеля. Колонку промывают 300 мл абсолютного бепзола и элюируют смесью абсолютный бензол-этилацетат (10 : 1). При .вьшаривании элюата в BaiKyyiMe получают 11,3 3-хлор-5-ацетил-10,11-дн.метнл - 5Ндиюенз- 6, / -азвннна.
К .раствору 11,3 г (0,38 моль) полученного азепина в 110 мл четы.реххлористого углерода доба вляют 13,7 г (0,077 моль) N-6po.McyKцини:мида, освещают полученную суспензию лам1наМИ мощностью 200 вт и килятят 1 час с обратным холодильником. После охлажде,ния фильтруют, выпаривают фильтрат в вакууме, растворяют остаток в бензоле, отфильтровьгвают -сукщинимид и |Выпари1вают фильтрат в вакууме. Остаток раст1воряют в эфире, эфирный .pacTiBop обрабатывают активированным углем, фильтруют и .вы пари:вают в вакууме. Получают 18 г аморфного 3-.хлор-5-ацетил-10,11- биобрО|М(метил-5Ннди|бенз- 6, / -азсеи.на.
Раствор 17,4 г (0,036 моль) полученного соединения в 350 мл абсолютного бензола охлаждают, в течение 20 мин, размешивая, прикапывают ПО мл (0,051 моль) 21%-ного бензольного раствора этиламина, размешивают еще 10 мин и фильтруют через очищенную диатомитовую землю. Избыток этиламина в фильтрате выпаривают в вакууме, оставшийся бензольный раствор экстрагируют 1 н. соляной кислотой, обрабатывают экстракт активированным углем, фильтруют и подщелачивают концентрированным раствором едкого кали. Сырое основание выпадает в виде масла, которое экстрагируют эфиром, эфирный экстракт промывают водой до нейтрализации, сущат над карбонатом калия и выпаривают в вакууме. Получают 10,9 г сырого 2-этил-6-хлор-8а:цетил- 1,2,3,8-тетрагидради|бе1Нз- Ь, f -лирроло- 3,4- -азенина.
К раствору г10,9 г (0,032 моль) полученного соединения в 50 мл абсолютиого этанола добавляют 40 мл 20%-пого этанольного раствора едкого кали, кипятят 3 час с обратным холодильником, охлаждают, отсасывают 6,16 г оранжевого кристаллического нродукта. К фильтрату добавЛЯют 1 г паро-щ ксобразного едкого кали, кипятят 1 час, отгоняют 70 мл этанола и охлаждают. Выпадает еще 1,53 г сырого продукта, который отсасывают. Маточный раствор разбавляют водой, обрабатывают эфиром, эфирный раствор экстрагируют 2 н. соляной кислотой, солянокислый экстракт обрабатывают активированным углем, фильтруют и подщелачивают фильтрат концептрированным раствором едкого кали. Выделивщееся основание обрабатывают эфиром, органическую фазу сушат над карбонатом калия и выпаривают в вакууме. После кристаллизации остатка из небольнюго количества эфира иолучают еще 1,25 г продукта реакции. Соединенные продукты перекристаллизовывают из
бензола и получают 2-этил-6-хлор-1, 2, 3, 8тетра1Г1гдродибенз- &, ,рроло- 3,4-ё -азеп1П1, т. пл. 193-195°С.
7,57 г (0,025 моль) полученного основания растворяют в 300 мл теплого метиленхлорида,
0 добавляют 5,1 мл 5,2 п. абсолютного этаиольного раствора соляной кислоты, отсасывают гидрохлорид, промывают эфиром и сушат в вакууме, т. пл. 238-240°С.
Пример 2. 22,8 г (0,100 моль) 2-хлор-95 метилакридина в 35 мл 2 н. соляной кислоты и 65 мл ледяной воды обрабатывают 7,2 мл (0,128 моль) ацетальдегнда, 15 мл (0,114 моль) 75 о-яой гидроперекиси грег-бутила и раствором 36 г (0,130 .ноль) гептагиарата сульфата железа в 120 мл воды. Получают 13,20 г (51 %) .метил-(2-хлор-9-метилакридан-9-ил)кетона, т. пл. 134-135°С (эф1ф-гексан).
3,11 г (0,0114 моль) полученного кетона растворяют в 40 j/л метанола, добавляют 0,50 г
(0,013 моль) боргидрида натрия и размешивают 1 час при комнатной температуре. Реакционную смесь осторожно выпаривают в вакууме, обрабатывают остаток 100 мл метилеЕхлорИ1ла, прибавляют немного безводного
0 сульфата магния, фильтруют н выпаривают фильтрат в вакууме. Получают 3,18 г сырого 2-хлор-, 9-диметил-9-акридан метанол а.
Если полученное соединение применяют не сразу, то его хранят при 0°С.
5
15,1 г (0,055 моль) полученного гидроксисоединения раз.мещивают со смесью 300 мл концентрированной серной кислоты и воды (10:3 по объему) при комнатной тем пературе
f, до образования раствора, размешивают еще 30 мин 1, размешивая, вносят в смесь 700 мл 50%-ного раствора едкого кали и 2 кг льда. Полученную суспензию разбавляют водой, экстрагируют эфиром, промывают эфирный экстракт водой, сушат над сульфатом магния и выпаривают в вакууме. Перекристаллизовывают остаток из смеси эфир-reiKcan нсюлучают 11,47 г 2-хлор-10,11-диметил-5П-дибенз Ь, / -азсп:1на, т. пл. 137-.138°С.
9Аналогично примеру 1, 13,11 г (0,051 .ноль полученного соединеиия кииятят 15 мин с обратным холодильником в присутствии 100 мл ацетангидрида i получеп1 ый сырой продукт пропускают через силикагель, выделяя 14,66 г
5 2-хлор-5-ацетил-10,4 1 чдиметил-5П-дибенз- Ь, / -азвпина.
13,50 г (0,045 моль) получепного сырого нродукта аналогично примеру 1 обрабатывают 17,7 г (0,091 моль) N-бро.мсукциним.ида, обра60зующийся сырой продукт пропускают через 150 г силикагеля, элюируя сначала абсолютны: бензолом побочный продукт, а затем смесью абсолютного бепзола и этилацетата (10: 1) целевой продукт. Элюат выпаривают в вакууме и получают 20,74 г чистого желтоватого аморф65 ного 2-хлор-5-ацетил-10,1 -бисбромметил-5Ндибе113- 6, / -азег ииа.
Размешивая, 100 мл (0,32 моль) 10%-ного бензольного метиламина в течение QMUH вкапывают в раствор 19,0 г (0,040 моль) полученного соединения в 200 мл абсолютного бензола и охлаждают льдом (температура не выше 20°С). Затем размешивают 30 мин, отделяют па путче соль и вынаривают фильтрат в вакууме. Остаток растворяют в эфире, экстрагируют эфирный раствор 1 н. соляной кислотой, подщелачивают солянокислый экстракт концентрированным раствором едкого кали и экстрагируют выделившееся основание эфиром. Эфирный экстракт сушат над карбонатом калия, вынаривают в вакууме и получают аморфный желтоватый 2-метил-5-хлор-8-ацетил-1, 2, 3, 8-тетрагидродибенз- 6, / -пирроло-{3,4-й -азепин, который надо хранить ,на холоду под азотом.
К раствору 6,63 г (0,020 моль) полученного соединения в 50 мл абсолютиого этанола добавляют 4,60 г едкого кали, кипятят 5 час под азотом и с обратным холодильником, охлаждают до 0°С, выделившиеся оранжевые кристаллы (4,61 г) отсасывают, промывают пебольшим количеством холодног-, как лед, этанола и сушат. Фильтрат выпаривают в вакууме, остаток растворяют в эфи,ре, эфирный раствор экстрагируют 2 iH. соляной кислотой и подщелачивают экстракт концентрированным раствором едкого кали. Выделившееся свободное основание растворяют в метиленхлориде, сушат раствор над карбонатом калия и выпаривают. Кристаллический остаток (0,66 г) соединяют с первой фракцией кристаллов и растворяют в бензоле, обрабатывают активированным углем, фильтруют и сгущают. Получают 5,03 г 2-метил-5-хлор-1, 2, 3, 8-тет|ра Гид.р01Дибенз- 6, / -ии|р,роло- 3,4-й(-азе:Нина, т. пл. 210-212°С.
5,35 г полученного основания растворяют в 200 мл метиленхлорида, охлаждают и, размешивая, добавляют 3,60 мл 19,5%-ного этапольного раствора соляной кислоты.
После охлаждения льдом выделяют гидрохлорид, промывают его небольишм количеством метиленхлорида, сушат и растворяют в этаноле. Этанольный раствор обрабатывают активированным углем, сгущают и сушат полученные кристаллы при 100°С/0,01 мм. Выделяют красноватый гидрохлорид, т. пл. 230- 241°С.
Пример 3.10,0 г (0,038 .-иоль) 3-трифторметил-9- етилак,ридина растворяют в 200 мл ледяной уксусной кислоты и 150 мл 0,05 н. серной кислоты, охлаждают до 10°С, добавляют 11 мл (0,195 моль) ацетальдегида и охлаждают до 10°С. Размешивая, в течение 20 мин одновременно прикапывают раствор 55 г (0,198 моль) гептагидрата сульфата железа в 180 мл воды н 23 мл (0,175 моль) 75%-ной гидроперекисп трег-бутила, размешивают 1 час при комнатпой температуре, разбавляют 1 л воды и экстрагируют эфиром. Эфирный экстракт промывают до нейтрализации водой.
10%-ным раствором карбоната натрия н водой, сушат над сульфатом магния и выпаривают в вакууме Остаток растворяют в абсолютном бензоле, фильтруют через колон ку со 140 г силикагеля (размер зерен 0,05-0,2 мм) и промывают абсолютным бензолом. Фильтрат выпаривают в вакууме и перекристаллизовывают остаток из гексана. Получают желтоватый метил - (3 - трифторметил-9-метилакридан-9-ил)кетон, т. пл. 125-126°С.
К раствору 5,0 г (0,016 моль) полученного кетона в 50 мл метанола добавляют 0,760 г (0,020 моль) боргидрида натрия и размешивают 1 час при комнатной температуре, осторожно выпаривают в вакууме и растворяют остаток в 100 мл метиленхлорида. К раствору добавляют немного безводного сульфата магния, фильтруют и выпаривают фильтрат в вакууме. Получают 5,12 г сырого 3-трифторметил-а, 9-диметил-9-акрида нметанола, 5,00 л (0,016 моль) которого добавляют к охлажденной смеси 50 мл концентрированной серной кислоты и воды (40: 1 по объему). Полученную суспензию размешивают 30 мин. Образующийся красный раствор размешивают 30 мин, выливают в смесь 130 мл 50%-ного раствора едкого кали и 800 г льда, разбавляют для растворения выделившегося сульфата калия, экстрагируют полученный раствор эфиром, промывают экстракт водой, сущат над сульфатом магния и вынаривают в вакууме. Остаток растворяют в горячем гексане, горячий раствор обрабатывают активированным углем и фильтруют. Фильтрат сгущают и выделяют :3,07 г (65%) З-трифторметил-10,11-ди метил-5Ннди бенз- Ь, f -азепи.на, т. нл. 153- 155°С.
17,0 г (0,0588 моль) получен ното соединения и 116 мл ащетантидрида кипятят 0,5 час
с обратным холодильников, сгущают раствор IB iBaiKyyiMe. растворяют остаток -в 30 мл толуола и (вьшаривают раствор ,в .вакууме. Для полного удаления ацетангиариаа этот процесс 1повто.ряют. Получают 3-трифтарметил-5-ацетил - 10,1 Ьдиметил - 5П - дибенз, Ь, f - азепин, желтое аморфное вещес11во.
Смесь 20,0 г (0,0604 .моль) получепного соединения, 185 мл четыреххлористого углерода и 27,6 г (0,155 моль) N-бромсукцинимида кипятят 6 час с обратным холодильником при одновременном освещении двумя лампами накаливания мощностью 200 вт. После охлаждения фильтруют, сгущают фильтрат в вакууме н получают 31,0 2 красного масла, которое растворяют в бензоле и фильтруют через колонку с 300 г силикагеля (размер зерен 0,05-0,2 мм). После выпаривания растворителя 1нолучают чистый З-трифторметил-5-ацетил-10,11-бисбромметил - 5Н - дибенз- &,/ -азепин в виде желтоватой пены.
В охлажденн{з1й льдом раствор 20,0 г (0,0408 моль) полученного соединения в 145 мл бензола в течение 30 мин под азотом, размешивая, вкапывают 75 мл 19,6%-ного (по объему) бензольного раствора этиламина, размешивают 0,5 час, не охлаждая раствор, отфильтровывают гидробромид этиламина и выпаривают фильтрат в вакууме. Остаток растворяют в 250 мл эфира и 250 мл воды, водный слой отделяют после сильного встряхивания и экстрагируют эфиром. Эфирные экстракты соединяют и экстрагируют 3X150 мл 2 н. соляной кислоты. Объединенные кислые экстракты подщелачивают едким кали и экстрагируют 2X100 Л1л метиленхлорида. После сушки органического слоя над поташем и сгущения в вакууме получают 2-этил-6-трифторметил-8-ацетил-1, 2, 13, 8-тетрагидродибенз-{&, / пирроло- 3,4-й -азепин в виде желтоватой пены.
Раствор 11,0 г (0,0296 моль) полученного соединения в 55 мл 96%-ного этанола кипятят 2 час с обратным холодильником под азотом с 7,0 г (0,125 моль) едкого кали, охлаждают лыдом. отсасывают желтые кристаллы, промывают спиртом и эфиром и сушат. Получают 2-этил-6-трифторметил-1, 2, 3, 8-тетрагидродибенз- 6, / -пирроло- 3,4-й - азепнн, т. пл. 188-190°С.
При соответствующей переработке маточного раствора получают еще 0,6 г продукта.
Т. пл. чистого вещества 194°С.
Из 5,1 г основания и 3,0 мл 19,5%-ного раствора хлористого водорода в этаноле аналогично примеру 2 получают оранжевые кристаллы гидрохлорида, т. пл. 230-233°С.
Пример 4. Раствор 7,2 г (0,0147 моль) 3-трифторметил - 5 - ацетил-10,11 - бисбромметил-5Н-дибенз- 6, / -азепина в 70 мл метиленхлорида в атмосфере сухого азота охлаждают до-20° С, прикапывают к нему 45л«л 10%-ного бензольного раствора метиламина, размешивают 0,5 час при комнатной темлературе. Экстрагируют насыщенны.м раствором бикарбоната натрия, органическую фазу сушат над сульфатом магния и сгущают в вакууме. Остаток растворяют в 100 мл эфира, промывают раствор водой и экстрагируют 2X100 мл 2 н. соляной кислоты. Кислые экстракты подщелачивают концентрированным натровым щелоком, экстрагируют 2X100 мл метиленхлорида, сушат экстракты над сульфатом магния, выпаривают в вакууме и получают 2-метил-8-ацетил-6-трифторметил-1, 2, 3, 8-тетрагидродибенз- 6, / -пирроло- 3,4- -азепин в виде белой пены.
Раствор 4,9 г (0,0137 моль) полученного соединения в 26 мл 96%-ного этанола и 3,3 г (0,059 моль) едкого кали кипятят 1 час с обратны.м холодильником no/i азотом. После охлаждения льдом отсасывают кристаллы, промывают их небольшим количеством этано.ча и воды, сушат и получают 2-метил-6-трифторметил-1. 2, 3, 8-тетрагидродибенз-{6, / -пирроло- 3,4-й -азенин в вице желтых кристалл,, т. пл. 195-197 0.
К охлажденному льдом раствору 2,2 г (0,070 моль) этого соединения в 77 мл метиленхлорида добавляют 1,2 мл 19,5%-ного этанольного раствора хлористого .водорода, выдерживают 30 мин, отсасывают осадок, сушат
в вакууме и получают гидрохлорид в виде красно-коричневых кристаллов, т. пл. 230- 2ЖС.
Пример 5. К раствору 14,6 г 10,0307 моль) 2-.хлор-5-ацетил-10,11-бисбромметил-5Пдибенз- 6, / -азепина в 200 мл бензола при охлаждении и размешивании в атмосфере азота по каплям добавляют 93 мл 21%-ного бензольного раствора этиламина, проводят синтез,
как в примере 1, и получают 2-этил-5-хлор-8ацетил-1, 2, 3, 8-тетрагидродибенз- 6, / -пирроло- 3,4-с1 -азепии в виде бесцветной пены, кокоторый хра1нят под азотом.
5,0 г (0,01475 моль) полученного соединеНИН и 3,28 г едкого кали в 50 мл этанола кипятят 2,5 час с обратным холодильником под азотом, охлаждают и отсасывают кристаллы. К маточному раствору добавляют то же самое количество едкого кали и этанола и омыляют,
получая еще немного кристаллов. Объединенные кристаллы перекристаллизовывают из бензола и получают 2-этил-5-хлор-1, 2, 3, 8тетрагидродибенз- &, / -нирроло- 3,4- азепин в виде желтых кристаллов, т. пл. 202-204°С.
2,82 г основания растворяют в 100 мл метиленхлор да, добавляют 1,86 мл 19,5%-ного этанольного раствора хлористого водорода, выдерживают 30 м-ин, отсасывают кристаллы и под азотом перекристаллизовывают из этанола. Получают гидрохлорид в виде светлокрасных кристаллов, т. пл. 232-235°0.
При м ер 6. К раствору 25,0 г (0,0525 моль) 3-.хлор-5-ацетил-10,11-бисбромметил-5Ндибенз-- 6, / -азенина в 200 мл бензола при охлаждении льдом в течение 10 мин добавляют 112 мл 10%-(ного бензольного раствора метилламина, размешивают 30 мин при комнатной температуре, сгущают в вакууме, растворяют остаток в 200 мл эфира и 200 мл воды, органическую фазу экстрагируют 2X100 мл 0,5 н. соляной кислоты, кислые экстракты и водную фазу соединяют, фильтруют через активированный уголь и подщелачивают до рН 8 с помощью 2 н, натрового щелока и насыщенного
раствора бикарбоната. Образующуюся суспензию экстрагируют 3X200 мл метиленхлорида, экстракты сушат над карбонитом калия и сгушают в вакууме. Полученное сырое основание растворяют в хлороформе и хроматографируют на колонке с 5-кратным количеством силикагеля (размер зерен 0,05-0,2 мм), элюируя хлороформом, садержащи(.м 1-2% .метанола. После выпаривания элюата получают 2-метил6-хлор-8-ацетил-1, 2, 3, 8-тетрагидродибенз- 6,
f -1пирроло- 3,4-йГ -азепин в виде бесцветного аморфного веи1,ества.
7,50 г (0,0234.) (Полученного соединения и 5,0 г едкого кали в 50 мл этанола кипятят 2 час с обратным холодильником под азотом,
охлаждают, отсасывают н-селтые кристаллы, промывают их водой и сушат в вакууме. Т. пл. 196-198°С.
Фильтрат сгущают, экстрагируют остаток
эфиром, сгущают и получают дополнительное
количество 2-метнл-6-хлор-1, 2, 3, 8-тетрагидрод-ибен;з- 6, / -1п рроло- 3,4-й -азепина, т. пл. 196-198°С.
Из 5,69 г основания аналогично нрнмеру 5 получают гидрохлорид, носле перекристаллиза-ции которого из смеси метанол - этанол выделяют оранжевые кристаллы с т. пл. 228- 231°С (субл.).
Предмет изобретения
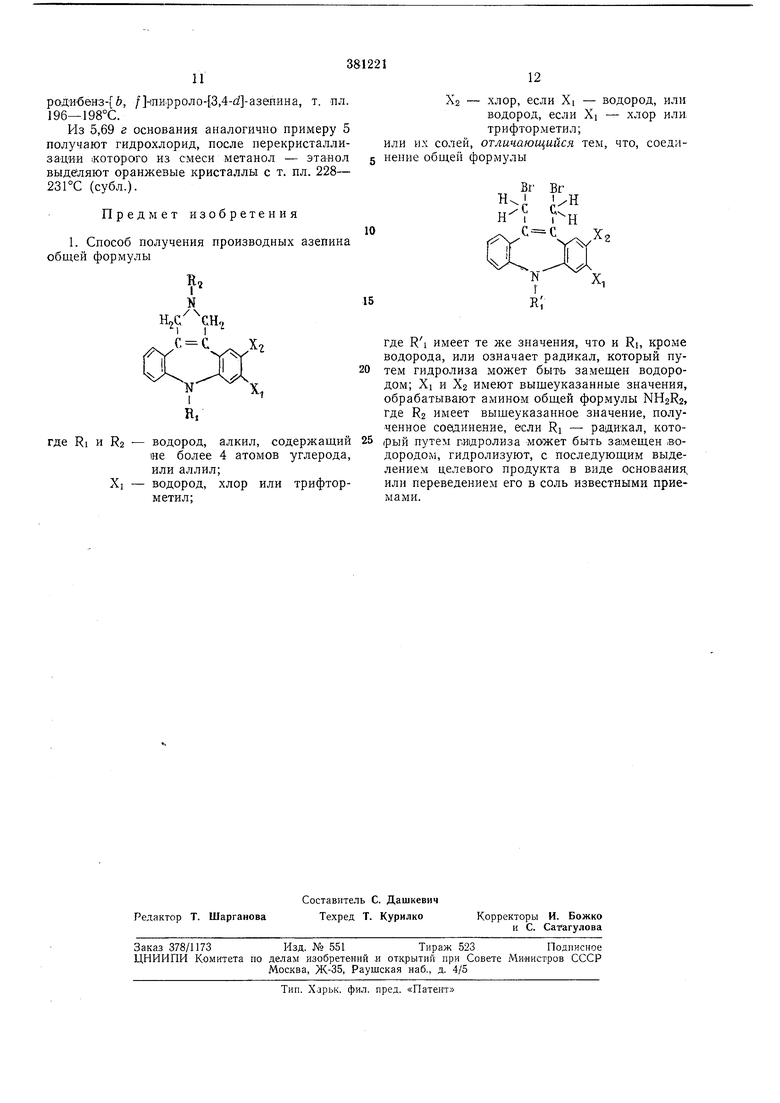
1. Способ получения производных азепина общей формулы
RI и R2 - водород, алкил, содержащий не более 4 атомов углерода, или аллил;
Х - водород, хлор или трифторметил;
Х2 - хлор, если Xi - водород, или водород, если Xj - хлор или трифторметил;
или их солей, отличающийся, тем, что, соединение обо;ей формулы
где Rl имеет те же значения, что и Ri, кроме водорода, или означает радикал, который путем гидролиза может быть замещен водородом; Xi и Хз имеют вышеуказанные значения, обрабатывают амином общей формулы NH2R2, где R2 имеет вышеуказанное значение, полученное соединение, если Rj - радикал, кото,рый путем гидролиза может быть замещен водородом, гидролизуют, с последующим выделением целевого продукта в виде основания, или переведением его в соль известными приемами.




| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЗЕПИНА ИЛИ ИХ СОЛЕИ | 1971 |
|
SU422149A3 |
| Способ получения производных азепина или их солей | 1971 |
|
SU507237A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЗЕПИНА | 1973 |
|
SU382283A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЗЕПИНА | 1971 |
|
SU321000A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 5-АРИЛ-1Н-1,5- БЕНЗОДИАЗЕПИН-2,4-(ЗН,5Н)-ДИОНОВ1 | 1973 |
|
SU361567A1 |
| Способ получения производных бензодиазепина | 1971 |
|
SU466660A3 |
| Способ получения производных диазепина | 1972 |
|
SU472505A3 |
| СССРОпубликовано 23.V. 1973. Бюллетень № 23 Дата онубликования описания 30.VIII.1973УДК 547.736'853Л.07 (088.8)(Федеративная Республика Германии)Иностранная фирма «Д-р Карл Томэ ГмбХ»(Федеративная Республика Германии) | 1973 |
|
SU383301A1 |
| Способ получения производных хинолина,их солей или их изомеров | 1974 |
|
SU535034A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-А!\\ИНО- АЛ КИЛАМИ НОТИЕНО-[3,2сг]-ПИРИЛ1ИДИ НА | 1970 |
|
SU419032A3 |
